User login
A young woman with a breast mass: What every internist should know
A 40-year-old premenopausal woman presents with a palpable lump in her left breast. She first noted it 2 months ago on self-examination, and it has steadily grown in size regardless of the phase of her menstrual cycle.
The patient has never undergone mammography. Her menarche was at age 12. At age 35, she had one child (whom she breastfed) after a normal first full-term pregnancy. She took oral contraceptives for 10 years before her pregnancy. She has no other medical problems. She has no family history of breast or ovarian cancer.
On examination, her breasts are slightly asymmetric, without skin discoloration, tenderness, swelling, nipple retraction, or discharge. A 1.5- to 2-cm, rubbery, mobile lump can be felt in the left breast at about the 2 o’clock position. No axillary lymph nodes can be palpated. The rest of her examination is normal.
BREAST CANCER MUST BE RULED OUT
Benign breast disease is found in approximately 90% of women 20 to 50 years of age who come to a physician with a breast problem.1
Nevertheless, breast cancer is of major concern. It is the most common type of cancer in women in the United States, responsible for an estimated 194,440 new cases and 40,610 deaths in 2009. It is also the leading cause of cancer-related death in women age 45 to 55 years in this country.2,3
Breast cancer is most common in postmenopausal women, its incidence rising sharply after the age of 45 and leveling off at age 75. The median age at diagnosis is 61 years. Still, 1.9% of breast cancers in women are diagnosed at age 20 to 34, 10.6% at age 35 to 44, and 22.4% at age 45 to 54.4
Thus, it is paramount to perform a thorough assessment and workup of women who have breast lumps, regardless of their age. Doing so allows breast cancer to be detected at an early stage. The 5-year survival rate is 98.0% for women with localized disease, 83.6% with regional disease, and 23.4% with distant disease.4
WHAT IS THE APPROPRIATE WORKUP?
1. Which of the following are appropriate in the workup of this patient?
- Mammography
- Ultrasonography
- Percutaneous needle biopsy of the lesion
- Magnetic resonance imaging (MRI) of the brain
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Positron emission tomography (PET)
She should undergo mammography, ultrasonography, and percutaneous needle biopsy.
Physical findings that suggest breast cancer include a hard, isolated, sometimes nonmobile lump, serosanguinous nipple discharge, and unilateral nipple retraction. Peau d’orange skin discoloration can occur. A scaly, vesicular, or ulcerated rash with or without pruritus, burning, irritation, or pain of the nipple or skin (Paget disease of the breast) is found in 1% to 3% of breast cancers and may be initially dismissed as mastitis.5,6 Palpable enlarged axillary lymph nodes can suggest invasive breast cancer.
Mammography is recommended in all cases of suspicious breast lumps. In a patient with a palpable lump, diagnostic mammography has a positive predictive value of 21.8%, a specificity of 85.8%, and a sensitivity of 87.7%, which are higher values than in a patient without signs or symptoms.7
The BIRADS score. Mammographic findings are summarized using a scoring system devised by the American College of Radiology called BIRADS (Breast Imaging Reporting and Data System). This system is based on mass irregularity, density, spiculation, and presence or absence of microcalcifications. It standardizes the results of mammography, gives an estimate of the risk of breast cancer, and recommends the frequency of follow-up examinations.8 Scores range from 0 to 6:
- 0—Incomplete assessment warranting additional evaluation
- 1—Completely negative mammogram
- 2—Benign lesion
- 3—Requires follow-up mammogram at 6 months
- 4—Risk of cancer is 2% to 95%; core biopsy needed
- 5—Risk of cancer is more than 95%; core biopsy needed
- 6—Cases that have already been proven to be malignant.
Ultrasonography is also done if a suspicious lesion is found on mammography or physical examination. It helps differentiate between solid and cystic masses. If a mass is identified as a cyst, ultrasonography can further characterize it as simple, complicated-simple, or complex. Simple cysts and complicated-simple cysts are unlikely to be malignant.9,10 Complex cysts or cysts associated with solid tissue are evaluated by biopsy.
Percutaneous needle biopsy should be done for a definitive diagnosis of most suspicious breast masses.
MRI can sometimes provide more accurate information about the possibility of multifocal breast cancer by revealing additional lesions missed on mammography or ultrasonography. It is also useful in determining more accurately the size of the breast tumor and looking for any possible contralateral lesions. In addition, it can sometimes detect enlarged axillary lymph nodes. However, it has poor specificity for breast cancer and may lead to additional and sometimes unnecessary diagnostic tests, which can delay treatment.
MRI’s role is therefore not clearly established, but it is commonly used in clinical practice. It is argued that workup of MRI findings may help in planning more accurate surgical procedures and may prevent reoperations. Based on retrospective analyses, results of breast MRI may lead to altered surgical treatment in approximately 13% of patients.11
Interestingly, a recent randomized trial showed no difference in reoperation rates between patients who underwent MRI before surgery vs those who did not. However, diagnostic workup of new MRI findings was not mandated by the study protocol, making the results of this trial difficult to interpret.12
DIFFERENTIAL DIAGNOSIS
2. Which of the following is in the differential diagnosis of a woman presenting with a breast abnormality?
- Fibrocystic changes
- Breast cyst
- Ductal ectasia
- Simple fibroadenoma
- Intraductal papilloma
- Ductal carcinoma in situ
- Mastitis
- Infiltrating ductal carcinoma
- Phyllodes tumor
All of these choices are part of the differential diagnosis.
Benign breast lesions

Simple fibroadenoma, one of the most common proliferative lesions, is not associated with a higher risk of developing breast cancer.
Fibrocystic changes are the most common nonproliferative lesions. Occasionally breast pain, nipple discharge, or significant lumpiness that varies during the course of the menstrual cycle can occur. The nipple discharge in women with fibrocystic changes is physiologic and pale green to brown in color. It can also be yellow, whitish, clear, or bloody. Bloody nipple discharge is considered pathologic and suggests a process other than fibrocystic changes, necessitating further workup. However, bloody discharge is not always a sign of malignancy, as it can have a benign cause as well.
Ductal ectasia, another nonproliferative lesion, is a result of dilation of subareolar ducts that contain fluid with a crystalline material. It can penetrate the duct, forming a nodule, which causes pain and occasionally fever.
Precancerous and cancerous lesions
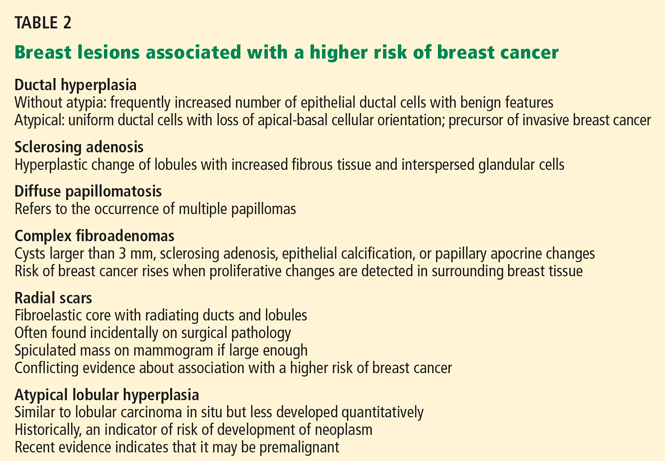
Ductal carcinoma in situ is a true neoplasm that has not yet developed the ability to invade through the basement membrane of the ducts. The likelihood of progression to invasive breast cancer depends on the histologic grade, the tumor size, and the patient’s age.
Lobular carcinoma in situ arises from lobules and terminal ducts of breast tissue. Much controversy surrounds this type of tumor, which was thought to be a marker of increased risk of developing ipsilateral and contralateral breast cancer and not to be a malignant lesion itself.15 However, there is emerging evidence to suggest that a pleomorphic variant of lobular carcinoma in situ is associated with development of breast cancer in the same site as the lesion, whereas a nonpleomorphic form is a marker of increased risk of ipsilateral and contralateral breast cancer.16
Invasive ductal and lobular carcinomas are the true invasive breast cancers, with a potential to metastasize.
Phyllodes tumors are uncommon fibroepithelial lesions that account for less than 1% of all breast neoplasms. The median age at presentation is 45 years.17 Despite the historical name “cystosarcoma phyllodes,” these lesions are not true sarcomas and have stromal and epithelial components.
These tumors display very heterogeneous behavior and, based on predefined histologic criteria, are often classified as benign, borderline, or malignant. Benign phyllodes tumors are similar to fibroadenomas in both histology and prognosis, making their diagnosis challenging. The most aggressive phyllodes tumors lose their epithelial component and have high metastatic potential. These tumors often have a biphasic growth pattern, and women may present with a smooth, round, well-defined breast lump that was stable for many years but then started to grow rapidly.17
Surgical resection with wide margins is the primary management of these tumors.18
Mastitis, ie, inflammation of the breast tissue, often presents with symptoms of breast erythema, swelling, tenderness, and nipple discharge. It may be secondary to infection (most often in lactating women) or other causes such as radiation or underlying malignancy. A complication of infectious mastitis is formation of a breast abscess. Underlying malignancy, especially inflammatory breast cancer, is a common cause of noninfectious mastitis and is very important to recognize.19
RISK FACTORS FOR BREAST CANCER
3. Which of the following are risk factors for breast cancer?
- Menarche before age 12
- Female sex
- Personal history of breast cancer
- Obesity
- Never having had children, or having given birth for the first time at an older age
- Older age
- History of hormone replacement therapy with estrogen and progesterone
- Family history of breast cancer
All of these choices are risk factors for breast cancer.
Family history
The overall relative risk of developing breast cancer in a woman with a first-degree relative with the disease is 1.7. However, the relative risk is about 3 if the first-degree relative developed breast cancer before menopause, and 9 if the first-degree relative developed bilateral breast cancer before menopause.5

Estrogen exposure
The duration and amount of estrogen exposure are also risk factors. For example, menarche before age 12 and menopause after age 55 are associated with a higher risk. Women who go through menopause after age 55 have a twofold higher risk of breast cancer compared with women who go through menopause at an early age. Pregnancy before age 30 lowers the risk of breast cancer; late first full-term pregnancy or nulliparity increases it. Lactation, on the other hand, has a protective effect.5
Oral contraceptives have traditionally been thought to increase the risk of breast cancer. In the 1990s, a meta-analysis involving 153,506 women found that those who had used oral contraceptives had a 24% higher risk of developing breast cancer.26 However, this association has come into question since newer oral contraceptive pills containing different progestins and lower amounts of estrogen have become available. In fact, recent studies showed no link between oral contraceptive use and breast cancer.27,28 Nevertheless, women at higher risk of developing breast cancer are advised not to use oral contraceptives.
Hormone replacement therapy with estrogen and progesterone was found to increase the risk of breast cancer by 26% in the Women’s Health Initiative (WHI) study, which involved 16,608 healthy women followed for a median of 5.6 years.29
In a study reported separately, the WHI investigators randomized 10,739 women who had undergone hysterectomy to receive either hormone replacement therapy with unopposed estrogen (which is feasible only in women without a uterus) or placebo. They found no increase in the risk of invasive breast cancer in women on hormone replacement therapy with estrogen alone. In fact, the study showed a trend towards a modest reduction of this risk (odds ratio 0.77; 95% confidence interval 0.59–1.01).30
After the results of the WHI were published, the use of hormone replacement therapy in postmenopausal women declined significantly. And in 2003—1 year later—the incidence of breast cancer had dropped by 6.7%.31
Most experts now recommend that estrogen-progestin combinations be used only selectively to treat the symptoms of menopause, and only for the short term.
Other risk factors
Other factors found to modestly increase the risk of breast cancer include:
- Alcohol use
- Obesity
- Radiation exposure. Patients are at higher risk of breast cancer 15 to 20 years after receiving upper-mantle radiotherapy for Hodgkin lymphoma.5
Case continues: Bad news on mammography, ultrasonography, biopsy
The patient undergoes mammography, which shows a 2.5-cm spiculated lesion with areas of calcifications (BIRADS score of 5). Subsequently, ultrasonography confirms that the suspicious mass is not a cyst. Ultrasound-guided core needle biopsy reveals that the lesion is a high-grade invasive ductal carcinoma. The tumor is positive for both estrogen and progesterone receptors and negative for HER2/neu overexpression.
STAGING EVALUATION
4. Given these findings, what is the next step to take?
- CT of the chest, abdomen, and pelvis
- MRI of the brain
- PET
- Referral to a surgeon for a possible mastectomy with sentinel lymph node dissection
- Referral to a surgeon for a possible lumpectomy with sentinel lymph node dissection
At this point, the patient should be referred to a surgeon for possible mastectomy or lumpectomy.
Women who appear clinically to have early breast cancer, such as in this case, should have a complete blood count, comprehensive metabolic panel, and chest x-ray as their initial staging evaluation. No further studies are recommended unless the findings on history, physical examination, or the above testing suggest possible metastases.
Mastectomy vs lumpectomy
Early-stage breast cancer is managed with definitive surgery. The two options are mastectomy and breast conservation therapy, the latter involving lumpectomy followed by breast radiation therapy.
Multiple randomized studies comparing mastectomy and lumpectomy showed no difference in survival rates, but patients in the lumpectomy groups had higher rates of local recurrence.32 Breast radiation therapy after lumpectomy lowered the rates of local recurrence and breast cancer death.33 Therefore, most patients can opt to undergo either lumpectomy with radiation or mastectomy, depending on personal preference.
However, mastectomy rather than breast conservation therapy is still recommended in cases of prior radiation therapy, inability to achieve negative surgical margins (as in cases of large tumors), multicentric disease (cancer in separate breast quadrants), or multiple areas of calcifications. Mastectomy is also preferred in most pregnant women unless the diagnosis of breast cancer is made in the third trimester and radiation therapy can be given after delivery. Patients who have large lesions in a small breast may also choose mastectomy with breast reconstruction rather than breast conservation therapy. Patients with a history of scleroderma are encouraged to undergo mastectomy because of increased toxicity from radiation treatment.
Sentinel vs axillary lymph node dissection
Knowledge of axillary lymph node involvement is important because it determines the stage in the tumor-node-metastasis (TNM) system, and it influences the choice of further therapy. Therefore, all patients with nonmetastatic invasive breast cancer must have their axillary lymph nodes sampled.
Conventionally, this involves axillary lymph node dissection. Unfortunately, upper extremity lymphedema develops in 6% to 30% of patients within the first 3 years, and in 49% of patients after 20 years following axillary lymph node dissection.34
Sentinel lymph node dissection was developed to minimize this complication. This procedure involves the injection of a blue dye, isosulfan blue (Lymphazurin), around the edge of the tumor or in the dermis overlying the tumor. The most proximal axillary lymph nodes that stain blue are dissected. Alternatively, a radioactive colloid (most commonly technetium sulfur colloid agents) may be injected, allowing sentinel lymph nodes to be identified by lymphoscintigraphy. If no metastases are found in the sentinel lymph nodes, axillary lymph node dissection is not performed.
A prospective study in 536 women found that at 5 years of follow-up, lymphedema developed in only 5% of patients after sentinel lymph node dissection compared with 16% of those who underwent axillary lymph node dissection (P < .001), with comparable outcomes in terms of disease recurrence.35
Case continues: Patient undergoes surgery
The patient elects to undergo lumpectomy with sentinel lymph node dissection. Pathologic review of the resection specimen reveals a 2.5-cm poorly differentiated invasive ductal carcinoma. Sentinel lymph node dissection shows metastases, and therefore axillary lymph node dissection is performed. One of eight lymph nodes removed is positive for metastases. All surgical margins are negative.
POSTOPERATIVE CARE
5. What would be the next step for our patient?
- Radiation followed by observation
- Tamoxifen (Nolvadex) for 5 years
- Observation only
- Chemotherapy followed by radiation therapy and 5 years of tamoxifen
She should receive chemotherapy, followed by radiation therapy and then tamoxifen for 5 years.
Chemotherapy. Almost all patients who have lymph-node-positive disease are advised to undergo chemotherapy.
The Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) performed a metaanalysis of 194 randomized trials that compared adjuvant chemotherapy and no treatment in early-stage breast cancer. Chemotherapy led to a 10% absolute improvement in survival at 15 years for women younger than 50 years and 3% in women age 51 to 69.36
Indications for chemotherapy include axillary lymph node involvement, locally advanced disease, and other risk factors for recurrence such as young age at diagnosis, strong positive family history of breast cancer, prior history of breast cancer, or lymph-node-negative, estrogen-receptor-negative tumors that are larger than 1 cm in diameter.
The Oncotype DX assay is a new tool to help oncologists decide whether to use chemotherapy in cases of estrogen-receptor-positive breast cancer, in which the benefit of chemotherapy is uncertain. It is a polymerase chain reaction assay that measures the expression of 16 cancer-specific genes and five reference genes within the breast tumor. Based on the pattern of expression of these genes, breast cancer can be characterized as low-risk, intermediate-risk, or high-risk. Patients in the high-risk group have a high chance of cancer recurrence and benefit from chemotherapy. Patients in the low-risk group are unlikely to have a recurrence or to benefit from chemotherapy.37 It is far less clear if patients in the intermediate-risk group benefit from chemotherapy, but this assay might eventually prove useful in deciding for or against chemotherapy in this group of patients as well.38 The Oncotype DX assay is presently being studied in a clinical trial.
Radiation therapy after mastectomy is recommended in patients who have breast tumors larger than 5 cm or metastases to more than three axillary lymph nodes.39
Antiestrogen therapy. After chemotherapy, patients with estrogen-receptor-positive cancers also receive 5 years of antiestrogen therapy. Available antiestrogen agents for such patients include tamoxifen, which is a selective estrogen receptor modulator, and drugs called aromatase inhibitors that block conversion of androgens to estrogens in peripheral tissues. Anastrozole (Arimidex), letrozole (Femara), and exemestane (Aromasin) are examples of available aromatase inhibitors. Premenopausal women are treated with tamoxifen, and postmenopausal women are offered aromatase inhibitors.
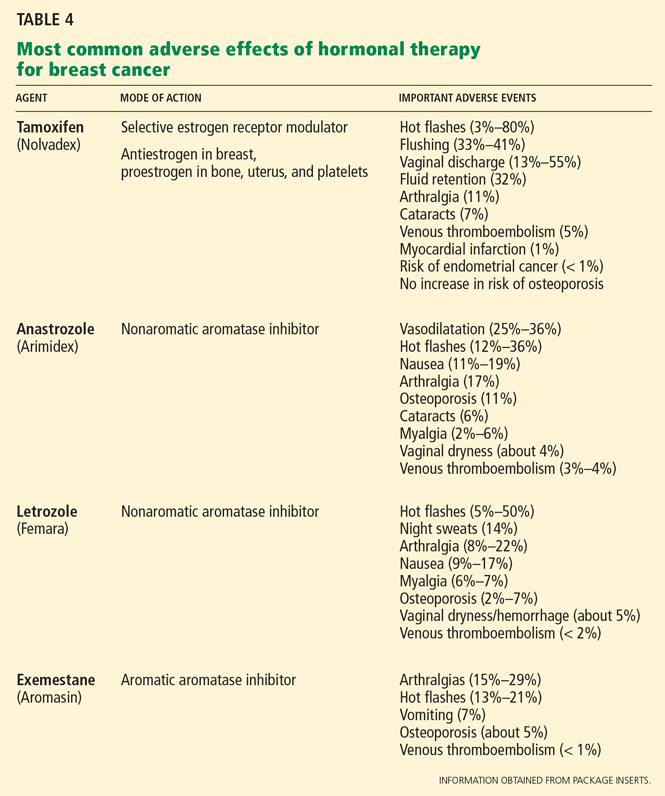
Table 4 lists the most common adverse effects of these agents. Aromatase inhibitors are associated with a higher risk of osteoporosis and arthralgia, while tamoxifen increases the risks of thromboembolism, endometrial cancer, and vaginal discharge. Both agents may produce menopausal symptoms such as hot flashes and mood swings.
Case continues: Seven years later, metastases in the spine
The patient achieves a complete remission. She is seen for a routine visit 7 years after diagnosis. She now reports mid-back pain that has worsened over the last 2 months. A bone scan reveals diffuse metastatic disease in the spine and in both humeral bones. CT of the chest, abdomen, and pelvis is negative for visceral metastases. Bone marrow aspiration and biopsy study show marrow infiltration by adenocarcinoma that stains positive for estrogen receptors and negative for HER2. The patient otherwise feels well and has no other symptoms.
WHAT TREATMENT FOR METASTATIC BREAST CANCER?
6. What should you now do for our patient?
- Discuss end-of-life care and refer her to a hospice program
- Educate the patient that no options for treatment exist and recommend enrolling in a phase I clinical trial
- Refer her to an oncologist for consideration of chemotherapy
- Refer her to an oncologist for consideration of endocrine treatment
She should be referred to an oncologist for consideration of endocrine treatment.
The most common sites of breast cancer metastases are the bones, followed by the liver and lungs. Metastatic breast cancer almost always is incurable. However, treatment can palliate symptoms.
Although a randomized trial of treatment vs best supportive care has never been done, many believe that treatment may improve survival. 40 The median survival of patients treated with standard therapy is about 3 years if the breast cancer is estrogen-receptor-positive and 2 years if it is estrogen-receptor-negative, but survival rates vary widely from patient to patient.41,42
Standard therapy or enrollment in a clinical phase II or III trial is indicated for this patient before considering enrollment in a phase I clinical trial or supportive care alone.
Endocrine therapy is the first-line therapy in women with estrogen-receptor-positive metastatic breast cancer. Postmenopausal women usually receive an aromatase inhibitor first.43,44 Response to endocrine therapy usually takes weeks to months but may last for several years.
Premenopausal women with estrogen-receptor-positive breast cancer also receive ovarian ablation therapy (oophorectomy or chemical ovarian ablation) with gonadotropin-releasing hormone agonists.
In addition, most patients with bone involvement are treated with high doses of intravenous bisphosphonates, which can reduce skeletal complications.45
Chemotherapy is reserved for patients with estrogen-receptor-negative breast cancer and those with cancer that progresses despite treatment with multiple antiestrogen agents. The time to response when chemotherapy is used is quicker, but the duration of response is usually shorter, lasting on average less than 1 year.37
Trastuzumab (Herceptin), a monoclonal humanized murine antibody to the extracellular domain of the HER2 protein, is indicated in patients with HER2-overexpressing tumors.46,47
STABLE 2 YEARS LATER
The patient was started on letrozole and a bisphosphonate, zolendronic acid (Zometa). Ovarian ablation was initiated with goserelin (Zoladex) given monthly. A bone scan performed 2 months after starting treatment showed improvement in bony metastases. She also noted significant improvement in pain. Her disease remains stable 2 years after starting endocrine therapy.
- Barton MB, Elmore JG, Fletcher SW. Breast symptoms among women enrolled in a health maintenance organization: frequency, evaluation, and outcome. Ann Intern Med 1999; 130:651–657.
- Petrelli NJ, Winer EP, Brahmer J, et al. Clinical cancer advances 2009: major research advances in cancer treatment, prevention, and screening—a report from the American Society of Clinical Oncology. J Clin Oncol 2009; 27:6052–6069.
- Jemal A, Siegel R, Ward E, et al. Cancer statistics 2008. CA Cancer J Clin 2008; 58:71–96.
- National Cancer Institute. SEER Stat Fact Sheets. www.seer.cancer.gov/statfacts/html/breast.html#ref09. Accessed June 7, 2010.
- Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ; the publishers of the journal Oncology. Cancer Management: A multidisciplinary Approach. Medical, Surgical & Radiation Oncology. 11th ed. CMP Medica; 2008.
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE. Paget’s disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187:171–177.
- Barlow WE, Lehman CD, Zheng Y, et al. Performance of diagnostic mammography for women with signs or symptoms of breast cancer. J Natl Cancer Inst 2002; 94:1151–1159.
- American College of Radiology. Breast Imaging Reporting and Data System: BIRADS Atlas. 4th ed. Reston, VA: American College of Radiology; 2003.
- Hong AS, Rosen EL, Soo MS, Baker JA. BI-RADS for sonography: positive and negative predictive values of sonographic features. AJR Am J Roentgenol 2005; 184:1260–1265.
- Berg WA, Campassi CI, Ioffe OB. Cystic lesions of the breast: sonographic-pathologic correlation. Radiology 2003; 227:183–191.
- Schell AM, Rosenkranz K, Lewis PJ. Role of breast MRI in the preoperative evaluation of patients with newly diagnosed breast cancer. AJR Am J Roentgenol 2009; 192:1438–1444.
- Turnbull L, Brown S, Harvey I, et al. Comparative effectiveness of MRI in breast cancer (COMICE) trial: a randomised controlled trial. Lancet 2010; 375:563–571.
- Worsham MJ, Abrams J, Raju U, et al. Breast cancer incidence in a cohort of women with benign breast disease from a multiethnic, primary health care population. Breast J 2007; 13:115–121.
- Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985; 312:146–151.
- Page DL, Kidd TE, Dupont WD, Simpson JF, Rogers LW. Lobular neoplasia of the breast: higher risk for subsequent invasive cancer predicted by more extensive disease. Hum Pathol 1991; 22:1232–1239.
- Sneige N, Wang J, Baker BA, Krishnamurthy S, Middleton LP. Clinical, histopathologic, and biologic features of pleomorphic lobular (ductallobular) carcinoma in situ of the breast: a report of 24 cases. Mod Pathol 2002; 15:1044–1050.
- Telli ML, Horst KC, Guardino AE, Dirbas FM, Carlson RW. Phyllodes tumors of the breast: natural history, diagnosis, and treatment. J Natl Compr Canc Netw 2007; 5:324–330.
- Reinfuss M, Mitus J, Duda K, Stelmach A, Rys J, Smolak K. The treatment and prognosis of patients with phyllodes tumor of the breast: an analysis of 170 cases. Cancer 1996; 77:910–916.
- Kamal RM, Hamed ST, Salem DS. Classification of inflammatory breast disorders and step by step diagnosis. Breast J 2009; 15:367–380.
- Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am J Hum Genet 1999; 64:963–970.
- Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med 2003; 348:2339–2347.
- Clarke-Pearson DL. Clinical practice. Screening for ovarian cancer. N Engl J Med 2009; 361:170–177.
- Hisada M, Garber JE, Fung CY, Fraumeni JF, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 1998; 90:606–611.
- Bell DW, Varley JM, Szydlo TE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999; 286:2528–2531.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 1996; 347:1713–1727.
- Hankinson SE, Colditz GA, Manson JE, et al. A prospective study of oral contraceptive use and risk of breast cancer (Nurses’ Health Study, United States). Cancer Causes Control 1997; 8:65–72.
- Marchbanks PA, McDonald JA, Wilson HG, et al. Oral contraceptives and the risk of breast cancer. N Engl J Med 2002; 346:2025–2032.
- Rossouw JE, Anderson GL, Prentice RL, et al; Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004; 291:1701–1712.
- Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 2007; 356:1670–1674.
- Fisher B, Anderson S, Redmond CK, Wolmark N, Wickerham DL, Cronin WM. Reanalysis and results after 12 years of follow-up in a randomized clinical trial comparing total mastectomy with lumpectomy with or without irradiation in the treatment of breast cancer. N Engl J Med 1995; 333:1456–1461.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Petrek JA, Senie RT, Peters M, Rosen PP. Lymphedema in a cohort of breast carcinoma survivors 20 years after diagnosis. Cancer 2001; 92:1368–1377.
- McLaughlin SA, Wright MJ, Morris KT, et al. Prevalence of lymphedema in women with breast cancer 5 years after sentinel lymph node biopsy or axillary dissection: objective measurements. J Clin Oncol 2008; 26:5213–5219.
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687–1717.
- Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351:2817–2826.
- Albain KS, Barlow WE, Shak S, et al; Breast Cancer Intergroup of North America. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010; 11:55–65.
- Harris JR, Halpin-Murphy P, McNeese M, Mendenhall NP, Morrow M, Robert NJ. Consensus Statement on postmastectomy radiation therapy. Int J Radiat Oncol Biol Phys 1999; 44:989–990.
- Gennari A, Conte P, Rosso R, Orlandini C, Bruzzi P. Survival of metastatic breast carcinoma patients over a 20-year period: a retrospective analysis based on individual patient data from six consecutive studies. Cancer 2005; 104:1742–1750.
- Mouridsen H, Gershanovich M, Sun Y, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol 2003; 21:2101–2109.
- Gamucci T, D’Ottavio AM, Magnolfi E, et al. Weekly epirubicin plus docetaxel as first-line treatment in metastatic breast cancer. Br J Cancer 2007; 97:1040–1045.
- Bonneterre J, Thürlimann B, Robertson JF, et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the Tamoxifen or Arimidex Randomized Group Efficacy and Tolerability study. J Clin Oncol 2000; 18:3748–3757.
- Nabholtz JM, Buzdar A, Pollak M, et al. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol 2000; 18:3758–3767.
- Hortobagyi GN, Theriault RL, Porter L, et al. Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. Protocol 19 Aredia Breast Cancer Study Group. N Engl J Med 1996; 335:1785–1791.
- Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673–1684.
- Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783–792.
A 40-year-old premenopausal woman presents with a palpable lump in her left breast. She first noted it 2 months ago on self-examination, and it has steadily grown in size regardless of the phase of her menstrual cycle.
The patient has never undergone mammography. Her menarche was at age 12. At age 35, she had one child (whom she breastfed) after a normal first full-term pregnancy. She took oral contraceptives for 10 years before her pregnancy. She has no other medical problems. She has no family history of breast or ovarian cancer.
On examination, her breasts are slightly asymmetric, without skin discoloration, tenderness, swelling, nipple retraction, or discharge. A 1.5- to 2-cm, rubbery, mobile lump can be felt in the left breast at about the 2 o’clock position. No axillary lymph nodes can be palpated. The rest of her examination is normal.
BREAST CANCER MUST BE RULED OUT
Benign breast disease is found in approximately 90% of women 20 to 50 years of age who come to a physician with a breast problem.1
Nevertheless, breast cancer is of major concern. It is the most common type of cancer in women in the United States, responsible for an estimated 194,440 new cases and 40,610 deaths in 2009. It is also the leading cause of cancer-related death in women age 45 to 55 years in this country.2,3
Breast cancer is most common in postmenopausal women, its incidence rising sharply after the age of 45 and leveling off at age 75. The median age at diagnosis is 61 years. Still, 1.9% of breast cancers in women are diagnosed at age 20 to 34, 10.6% at age 35 to 44, and 22.4% at age 45 to 54.4
Thus, it is paramount to perform a thorough assessment and workup of women who have breast lumps, regardless of their age. Doing so allows breast cancer to be detected at an early stage. The 5-year survival rate is 98.0% for women with localized disease, 83.6% with regional disease, and 23.4% with distant disease.4
WHAT IS THE APPROPRIATE WORKUP?
1. Which of the following are appropriate in the workup of this patient?
- Mammography
- Ultrasonography
- Percutaneous needle biopsy of the lesion
- Magnetic resonance imaging (MRI) of the brain
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Positron emission tomography (PET)
She should undergo mammography, ultrasonography, and percutaneous needle biopsy.
Physical findings that suggest breast cancer include a hard, isolated, sometimes nonmobile lump, serosanguinous nipple discharge, and unilateral nipple retraction. Peau d’orange skin discoloration can occur. A scaly, vesicular, or ulcerated rash with or without pruritus, burning, irritation, or pain of the nipple or skin (Paget disease of the breast) is found in 1% to 3% of breast cancers and may be initially dismissed as mastitis.5,6 Palpable enlarged axillary lymph nodes can suggest invasive breast cancer.
Mammography is recommended in all cases of suspicious breast lumps. In a patient with a palpable lump, diagnostic mammography has a positive predictive value of 21.8%, a specificity of 85.8%, and a sensitivity of 87.7%, which are higher values than in a patient without signs or symptoms.7
The BIRADS score. Mammographic findings are summarized using a scoring system devised by the American College of Radiology called BIRADS (Breast Imaging Reporting and Data System). This system is based on mass irregularity, density, spiculation, and presence or absence of microcalcifications. It standardizes the results of mammography, gives an estimate of the risk of breast cancer, and recommends the frequency of follow-up examinations.8 Scores range from 0 to 6:
- 0—Incomplete assessment warranting additional evaluation
- 1—Completely negative mammogram
- 2—Benign lesion
- 3—Requires follow-up mammogram at 6 months
- 4—Risk of cancer is 2% to 95%; core biopsy needed
- 5—Risk of cancer is more than 95%; core biopsy needed
- 6—Cases that have already been proven to be malignant.
Ultrasonography is also done if a suspicious lesion is found on mammography or physical examination. It helps differentiate between solid and cystic masses. If a mass is identified as a cyst, ultrasonography can further characterize it as simple, complicated-simple, or complex. Simple cysts and complicated-simple cysts are unlikely to be malignant.9,10 Complex cysts or cysts associated with solid tissue are evaluated by biopsy.
Percutaneous needle biopsy should be done for a definitive diagnosis of most suspicious breast masses.
MRI can sometimes provide more accurate information about the possibility of multifocal breast cancer by revealing additional lesions missed on mammography or ultrasonography. It is also useful in determining more accurately the size of the breast tumor and looking for any possible contralateral lesions. In addition, it can sometimes detect enlarged axillary lymph nodes. However, it has poor specificity for breast cancer and may lead to additional and sometimes unnecessary diagnostic tests, which can delay treatment.
MRI’s role is therefore not clearly established, but it is commonly used in clinical practice. It is argued that workup of MRI findings may help in planning more accurate surgical procedures and may prevent reoperations. Based on retrospective analyses, results of breast MRI may lead to altered surgical treatment in approximately 13% of patients.11
Interestingly, a recent randomized trial showed no difference in reoperation rates between patients who underwent MRI before surgery vs those who did not. However, diagnostic workup of new MRI findings was not mandated by the study protocol, making the results of this trial difficult to interpret.12
DIFFERENTIAL DIAGNOSIS
2. Which of the following is in the differential diagnosis of a woman presenting with a breast abnormality?
- Fibrocystic changes
- Breast cyst
- Ductal ectasia
- Simple fibroadenoma
- Intraductal papilloma
- Ductal carcinoma in situ
- Mastitis
- Infiltrating ductal carcinoma
- Phyllodes tumor
All of these choices are part of the differential diagnosis.
Benign breast lesions
Simple fibroadenoma, one of the most common proliferative lesions, is not associated with a higher risk of developing breast cancer.
Fibrocystic changes are the most common nonproliferative lesions. Occasionally breast pain, nipple discharge, or significant lumpiness that varies during the course of the menstrual cycle can occur. The nipple discharge in women with fibrocystic changes is physiologic and pale green to brown in color. It can also be yellow, whitish, clear, or bloody. Bloody nipple discharge is considered pathologic and suggests a process other than fibrocystic changes, necessitating further workup. However, bloody discharge is not always a sign of malignancy, as it can have a benign cause as well.
Ductal ectasia, another nonproliferative lesion, is a result of dilation of subareolar ducts that contain fluid with a crystalline material. It can penetrate the duct, forming a nodule, which causes pain and occasionally fever.
Precancerous and cancerous lesions
Ductal carcinoma in situ is a true neoplasm that has not yet developed the ability to invade through the basement membrane of the ducts. The likelihood of progression to invasive breast cancer depends on the histologic grade, the tumor size, and the patient’s age.
Lobular carcinoma in situ arises from lobules and terminal ducts of breast tissue. Much controversy surrounds this type of tumor, which was thought to be a marker of increased risk of developing ipsilateral and contralateral breast cancer and not to be a malignant lesion itself.15 However, there is emerging evidence to suggest that a pleomorphic variant of lobular carcinoma in situ is associated with development of breast cancer in the same site as the lesion, whereas a nonpleomorphic form is a marker of increased risk of ipsilateral and contralateral breast cancer.16
Invasive ductal and lobular carcinomas are the true invasive breast cancers, with a potential to metastasize.
Phyllodes tumors are uncommon fibroepithelial lesions that account for less than 1% of all breast neoplasms. The median age at presentation is 45 years.17 Despite the historical name “cystosarcoma phyllodes,” these lesions are not true sarcomas and have stromal and epithelial components.
These tumors display very heterogeneous behavior and, based on predefined histologic criteria, are often classified as benign, borderline, or malignant. Benign phyllodes tumors are similar to fibroadenomas in both histology and prognosis, making their diagnosis challenging. The most aggressive phyllodes tumors lose their epithelial component and have high metastatic potential. These tumors often have a biphasic growth pattern, and women may present with a smooth, round, well-defined breast lump that was stable for many years but then started to grow rapidly.17
Surgical resection with wide margins is the primary management of these tumors.18
Mastitis, ie, inflammation of the breast tissue, often presents with symptoms of breast erythema, swelling, tenderness, and nipple discharge. It may be secondary to infection (most often in lactating women) or other causes such as radiation or underlying malignancy. A complication of infectious mastitis is formation of a breast abscess. Underlying malignancy, especially inflammatory breast cancer, is a common cause of noninfectious mastitis and is very important to recognize.19
RISK FACTORS FOR BREAST CANCER
3. Which of the following are risk factors for breast cancer?
- Menarche before age 12
- Female sex
- Personal history of breast cancer
- Obesity
- Never having had children, or having given birth for the first time at an older age
- Older age
- History of hormone replacement therapy with estrogen and progesterone
- Family history of breast cancer
All of these choices are risk factors for breast cancer.
Family history
The overall relative risk of developing breast cancer in a woman with a first-degree relative with the disease is 1.7. However, the relative risk is about 3 if the first-degree relative developed breast cancer before menopause, and 9 if the first-degree relative developed bilateral breast cancer before menopause.5
Estrogen exposure
The duration and amount of estrogen exposure are also risk factors. For example, menarche before age 12 and menopause after age 55 are associated with a higher risk. Women who go through menopause after age 55 have a twofold higher risk of breast cancer compared with women who go through menopause at an early age. Pregnancy before age 30 lowers the risk of breast cancer; late first full-term pregnancy or nulliparity increases it. Lactation, on the other hand, has a protective effect.5
Oral contraceptives have traditionally been thought to increase the risk of breast cancer. In the 1990s, a meta-analysis involving 153,506 women found that those who had used oral contraceptives had a 24% higher risk of developing breast cancer.26 However, this association has come into question since newer oral contraceptive pills containing different progestins and lower amounts of estrogen have become available. In fact, recent studies showed no link between oral contraceptive use and breast cancer.27,28 Nevertheless, women at higher risk of developing breast cancer are advised not to use oral contraceptives.
Hormone replacement therapy with estrogen and progesterone was found to increase the risk of breast cancer by 26% in the Women’s Health Initiative (WHI) study, which involved 16,608 healthy women followed for a median of 5.6 years.29
In a study reported separately, the WHI investigators randomized 10,739 women who had undergone hysterectomy to receive either hormone replacement therapy with unopposed estrogen (which is feasible only in women without a uterus) or placebo. They found no increase in the risk of invasive breast cancer in women on hormone replacement therapy with estrogen alone. In fact, the study showed a trend towards a modest reduction of this risk (odds ratio 0.77; 95% confidence interval 0.59–1.01).30
After the results of the WHI were published, the use of hormone replacement therapy in postmenopausal women declined significantly. And in 2003—1 year later—the incidence of breast cancer had dropped by 6.7%.31
Most experts now recommend that estrogen-progestin combinations be used only selectively to treat the symptoms of menopause, and only for the short term.
Other risk factors
Other factors found to modestly increase the risk of breast cancer include:
- Alcohol use
- Obesity
- Radiation exposure. Patients are at higher risk of breast cancer 15 to 20 years after receiving upper-mantle radiotherapy for Hodgkin lymphoma.5
Case continues: Bad news on mammography, ultrasonography, biopsy
The patient undergoes mammography, which shows a 2.5-cm spiculated lesion with areas of calcifications (BIRADS score of 5). Subsequently, ultrasonography confirms that the suspicious mass is not a cyst. Ultrasound-guided core needle biopsy reveals that the lesion is a high-grade invasive ductal carcinoma. The tumor is positive for both estrogen and progesterone receptors and negative for HER2/neu overexpression.
STAGING EVALUATION
4. Given these findings, what is the next step to take?
- CT of the chest, abdomen, and pelvis
- MRI of the brain
- PET
- Referral to a surgeon for a possible mastectomy with sentinel lymph node dissection
- Referral to a surgeon for a possible lumpectomy with sentinel lymph node dissection
At this point, the patient should be referred to a surgeon for possible mastectomy or lumpectomy.
Women who appear clinically to have early breast cancer, such as in this case, should have a complete blood count, comprehensive metabolic panel, and chest x-ray as their initial staging evaluation. No further studies are recommended unless the findings on history, physical examination, or the above testing suggest possible metastases.
Mastectomy vs lumpectomy
Early-stage breast cancer is managed with definitive surgery. The two options are mastectomy and breast conservation therapy, the latter involving lumpectomy followed by breast radiation therapy.
Multiple randomized studies comparing mastectomy and lumpectomy showed no difference in survival rates, but patients in the lumpectomy groups had higher rates of local recurrence.32 Breast radiation therapy after lumpectomy lowered the rates of local recurrence and breast cancer death.33 Therefore, most patients can opt to undergo either lumpectomy with radiation or mastectomy, depending on personal preference.
However, mastectomy rather than breast conservation therapy is still recommended in cases of prior radiation therapy, inability to achieve negative surgical margins (as in cases of large tumors), multicentric disease (cancer in separate breast quadrants), or multiple areas of calcifications. Mastectomy is also preferred in most pregnant women unless the diagnosis of breast cancer is made in the third trimester and radiation therapy can be given after delivery. Patients who have large lesions in a small breast may also choose mastectomy with breast reconstruction rather than breast conservation therapy. Patients with a history of scleroderma are encouraged to undergo mastectomy because of increased toxicity from radiation treatment.
Sentinel vs axillary lymph node dissection
Knowledge of axillary lymph node involvement is important because it determines the stage in the tumor-node-metastasis (TNM) system, and it influences the choice of further therapy. Therefore, all patients with nonmetastatic invasive breast cancer must have their axillary lymph nodes sampled.
Conventionally, this involves axillary lymph node dissection. Unfortunately, upper extremity lymphedema develops in 6% to 30% of patients within the first 3 years, and in 49% of patients after 20 years following axillary lymph node dissection.34
Sentinel lymph node dissection was developed to minimize this complication. This procedure involves the injection of a blue dye, isosulfan blue (Lymphazurin), around the edge of the tumor or in the dermis overlying the tumor. The most proximal axillary lymph nodes that stain blue are dissected. Alternatively, a radioactive colloid (most commonly technetium sulfur colloid agents) may be injected, allowing sentinel lymph nodes to be identified by lymphoscintigraphy. If no metastases are found in the sentinel lymph nodes, axillary lymph node dissection is not performed.
A prospective study in 536 women found that at 5 years of follow-up, lymphedema developed in only 5% of patients after sentinel lymph node dissection compared with 16% of those who underwent axillary lymph node dissection (P < .001), with comparable outcomes in terms of disease recurrence.35
Case continues: Patient undergoes surgery
The patient elects to undergo lumpectomy with sentinel lymph node dissection. Pathologic review of the resection specimen reveals a 2.5-cm poorly differentiated invasive ductal carcinoma. Sentinel lymph node dissection shows metastases, and therefore axillary lymph node dissection is performed. One of eight lymph nodes removed is positive for metastases. All surgical margins are negative.
POSTOPERATIVE CARE
5. What would be the next step for our patient?
- Radiation followed by observation
- Tamoxifen (Nolvadex) for 5 years
- Observation only
- Chemotherapy followed by radiation therapy and 5 years of tamoxifen
She should receive chemotherapy, followed by radiation therapy and then tamoxifen for 5 years.
Chemotherapy. Almost all patients who have lymph-node-positive disease are advised to undergo chemotherapy.
The Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) performed a metaanalysis of 194 randomized trials that compared adjuvant chemotherapy and no treatment in early-stage breast cancer. Chemotherapy led to a 10% absolute improvement in survival at 15 years for women younger than 50 years and 3% in women age 51 to 69.36
Indications for chemotherapy include axillary lymph node involvement, locally advanced disease, and other risk factors for recurrence such as young age at diagnosis, strong positive family history of breast cancer, prior history of breast cancer, or lymph-node-negative, estrogen-receptor-negative tumors that are larger than 1 cm in diameter.
The Oncotype DX assay is a new tool to help oncologists decide whether to use chemotherapy in cases of estrogen-receptor-positive breast cancer, in which the benefit of chemotherapy is uncertain. It is a polymerase chain reaction assay that measures the expression of 16 cancer-specific genes and five reference genes within the breast tumor. Based on the pattern of expression of these genes, breast cancer can be characterized as low-risk, intermediate-risk, or high-risk. Patients in the high-risk group have a high chance of cancer recurrence and benefit from chemotherapy. Patients in the low-risk group are unlikely to have a recurrence or to benefit from chemotherapy.37 It is far less clear if patients in the intermediate-risk group benefit from chemotherapy, but this assay might eventually prove useful in deciding for or against chemotherapy in this group of patients as well.38 The Oncotype DX assay is presently being studied in a clinical trial.
Radiation therapy after mastectomy is recommended in patients who have breast tumors larger than 5 cm or metastases to more than three axillary lymph nodes.39
Antiestrogen therapy. After chemotherapy, patients with estrogen-receptor-positive cancers also receive 5 years of antiestrogen therapy. Available antiestrogen agents for such patients include tamoxifen, which is a selective estrogen receptor modulator, and drugs called aromatase inhibitors that block conversion of androgens to estrogens in peripheral tissues. Anastrozole (Arimidex), letrozole (Femara), and exemestane (Aromasin) are examples of available aromatase inhibitors. Premenopausal women are treated with tamoxifen, and postmenopausal women are offered aromatase inhibitors.
Table 4 lists the most common adverse effects of these agents. Aromatase inhibitors are associated with a higher risk of osteoporosis and arthralgia, while tamoxifen increases the risks of thromboembolism, endometrial cancer, and vaginal discharge. Both agents may produce menopausal symptoms such as hot flashes and mood swings.
Case continues: Seven years later, metastases in the spine
The patient achieves a complete remission. She is seen for a routine visit 7 years after diagnosis. She now reports mid-back pain that has worsened over the last 2 months. A bone scan reveals diffuse metastatic disease in the spine and in both humeral bones. CT of the chest, abdomen, and pelvis is negative for visceral metastases. Bone marrow aspiration and biopsy study show marrow infiltration by adenocarcinoma that stains positive for estrogen receptors and negative for HER2. The patient otherwise feels well and has no other symptoms.
WHAT TREATMENT FOR METASTATIC BREAST CANCER?
6. What should you now do for our patient?
- Discuss end-of-life care and refer her to a hospice program
- Educate the patient that no options for treatment exist and recommend enrolling in a phase I clinical trial
- Refer her to an oncologist for consideration of chemotherapy
- Refer her to an oncologist for consideration of endocrine treatment
She should be referred to an oncologist for consideration of endocrine treatment.
The most common sites of breast cancer metastases are the bones, followed by the liver and lungs. Metastatic breast cancer almost always is incurable. However, treatment can palliate symptoms.
Although a randomized trial of treatment vs best supportive care has never been done, many believe that treatment may improve survival. 40 The median survival of patients treated with standard therapy is about 3 years if the breast cancer is estrogen-receptor-positive and 2 years if it is estrogen-receptor-negative, but survival rates vary widely from patient to patient.41,42
Standard therapy or enrollment in a clinical phase II or III trial is indicated for this patient before considering enrollment in a phase I clinical trial or supportive care alone.
Endocrine therapy is the first-line therapy in women with estrogen-receptor-positive metastatic breast cancer. Postmenopausal women usually receive an aromatase inhibitor first.43,44 Response to endocrine therapy usually takes weeks to months but may last for several years.
Premenopausal women with estrogen-receptor-positive breast cancer also receive ovarian ablation therapy (oophorectomy or chemical ovarian ablation) with gonadotropin-releasing hormone agonists.
In addition, most patients with bone involvement are treated with high doses of intravenous bisphosphonates, which can reduce skeletal complications.45
Chemotherapy is reserved for patients with estrogen-receptor-negative breast cancer and those with cancer that progresses despite treatment with multiple antiestrogen agents. The time to response when chemotherapy is used is quicker, but the duration of response is usually shorter, lasting on average less than 1 year.37
Trastuzumab (Herceptin), a monoclonal humanized murine antibody to the extracellular domain of the HER2 protein, is indicated in patients with HER2-overexpressing tumors.46,47
STABLE 2 YEARS LATER
The patient was started on letrozole and a bisphosphonate, zolendronic acid (Zometa). Ovarian ablation was initiated with goserelin (Zoladex) given monthly. A bone scan performed 2 months after starting treatment showed improvement in bony metastases. She also noted significant improvement in pain. Her disease remains stable 2 years after starting endocrine therapy.
A 40-year-old premenopausal woman presents with a palpable lump in her left breast. She first noted it 2 months ago on self-examination, and it has steadily grown in size regardless of the phase of her menstrual cycle.
The patient has never undergone mammography. Her menarche was at age 12. At age 35, she had one child (whom she breastfed) after a normal first full-term pregnancy. She took oral contraceptives for 10 years before her pregnancy. She has no other medical problems. She has no family history of breast or ovarian cancer.
On examination, her breasts are slightly asymmetric, without skin discoloration, tenderness, swelling, nipple retraction, or discharge. A 1.5- to 2-cm, rubbery, mobile lump can be felt in the left breast at about the 2 o’clock position. No axillary lymph nodes can be palpated. The rest of her examination is normal.
BREAST CANCER MUST BE RULED OUT
Benign breast disease is found in approximately 90% of women 20 to 50 years of age who come to a physician with a breast problem.1
Nevertheless, breast cancer is of major concern. It is the most common type of cancer in women in the United States, responsible for an estimated 194,440 new cases and 40,610 deaths in 2009. It is also the leading cause of cancer-related death in women age 45 to 55 years in this country.2,3
Breast cancer is most common in postmenopausal women, its incidence rising sharply after the age of 45 and leveling off at age 75. The median age at diagnosis is 61 years. Still, 1.9% of breast cancers in women are diagnosed at age 20 to 34, 10.6% at age 35 to 44, and 22.4% at age 45 to 54.4
Thus, it is paramount to perform a thorough assessment and workup of women who have breast lumps, regardless of their age. Doing so allows breast cancer to be detected at an early stage. The 5-year survival rate is 98.0% for women with localized disease, 83.6% with regional disease, and 23.4% with distant disease.4
WHAT IS THE APPROPRIATE WORKUP?
1. Which of the following are appropriate in the workup of this patient?
- Mammography
- Ultrasonography
- Percutaneous needle biopsy of the lesion
- Magnetic resonance imaging (MRI) of the brain
- Computed tomography (CT) of the chest, abdomen, and pelvis
- Positron emission tomography (PET)
She should undergo mammography, ultrasonography, and percutaneous needle biopsy.
Physical findings that suggest breast cancer include a hard, isolated, sometimes nonmobile lump, serosanguinous nipple discharge, and unilateral nipple retraction. Peau d’orange skin discoloration can occur. A scaly, vesicular, or ulcerated rash with or without pruritus, burning, irritation, or pain of the nipple or skin (Paget disease of the breast) is found in 1% to 3% of breast cancers and may be initially dismissed as mastitis.5,6 Palpable enlarged axillary lymph nodes can suggest invasive breast cancer.
Mammography is recommended in all cases of suspicious breast lumps. In a patient with a palpable lump, diagnostic mammography has a positive predictive value of 21.8%, a specificity of 85.8%, and a sensitivity of 87.7%, which are higher values than in a patient without signs or symptoms.7
The BIRADS score. Mammographic findings are summarized using a scoring system devised by the American College of Radiology called BIRADS (Breast Imaging Reporting and Data System). This system is based on mass irregularity, density, spiculation, and presence or absence of microcalcifications. It standardizes the results of mammography, gives an estimate of the risk of breast cancer, and recommends the frequency of follow-up examinations.8 Scores range from 0 to 6:
- 0—Incomplete assessment warranting additional evaluation
- 1—Completely negative mammogram
- 2—Benign lesion
- 3—Requires follow-up mammogram at 6 months
- 4—Risk of cancer is 2% to 95%; core biopsy needed
- 5—Risk of cancer is more than 95%; core biopsy needed
- 6—Cases that have already been proven to be malignant.
Ultrasonography is also done if a suspicious lesion is found on mammography or physical examination. It helps differentiate between solid and cystic masses. If a mass is identified as a cyst, ultrasonography can further characterize it as simple, complicated-simple, or complex. Simple cysts and complicated-simple cysts are unlikely to be malignant.9,10 Complex cysts or cysts associated with solid tissue are evaluated by biopsy.
Percutaneous needle biopsy should be done for a definitive diagnosis of most suspicious breast masses.
MRI can sometimes provide more accurate information about the possibility of multifocal breast cancer by revealing additional lesions missed on mammography or ultrasonography. It is also useful in determining more accurately the size of the breast tumor and looking for any possible contralateral lesions. In addition, it can sometimes detect enlarged axillary lymph nodes. However, it has poor specificity for breast cancer and may lead to additional and sometimes unnecessary diagnostic tests, which can delay treatment.
MRI’s role is therefore not clearly established, but it is commonly used in clinical practice. It is argued that workup of MRI findings may help in planning more accurate surgical procedures and may prevent reoperations. Based on retrospective analyses, results of breast MRI may lead to altered surgical treatment in approximately 13% of patients.11
Interestingly, a recent randomized trial showed no difference in reoperation rates between patients who underwent MRI before surgery vs those who did not. However, diagnostic workup of new MRI findings was not mandated by the study protocol, making the results of this trial difficult to interpret.12
DIFFERENTIAL DIAGNOSIS
2. Which of the following is in the differential diagnosis of a woman presenting with a breast abnormality?
- Fibrocystic changes
- Breast cyst
- Ductal ectasia
- Simple fibroadenoma
- Intraductal papilloma
- Ductal carcinoma in situ
- Mastitis
- Infiltrating ductal carcinoma
- Phyllodes tumor
All of these choices are part of the differential diagnosis.
Benign breast lesions
Simple fibroadenoma, one of the most common proliferative lesions, is not associated with a higher risk of developing breast cancer.
Fibrocystic changes are the most common nonproliferative lesions. Occasionally breast pain, nipple discharge, or significant lumpiness that varies during the course of the menstrual cycle can occur. The nipple discharge in women with fibrocystic changes is physiologic and pale green to brown in color. It can also be yellow, whitish, clear, or bloody. Bloody nipple discharge is considered pathologic and suggests a process other than fibrocystic changes, necessitating further workup. However, bloody discharge is not always a sign of malignancy, as it can have a benign cause as well.
Ductal ectasia, another nonproliferative lesion, is a result of dilation of subareolar ducts that contain fluid with a crystalline material. It can penetrate the duct, forming a nodule, which causes pain and occasionally fever.
Precancerous and cancerous lesions
Ductal carcinoma in situ is a true neoplasm that has not yet developed the ability to invade through the basement membrane of the ducts. The likelihood of progression to invasive breast cancer depends on the histologic grade, the tumor size, and the patient’s age.
Lobular carcinoma in situ arises from lobules and terminal ducts of breast tissue. Much controversy surrounds this type of tumor, which was thought to be a marker of increased risk of developing ipsilateral and contralateral breast cancer and not to be a malignant lesion itself.15 However, there is emerging evidence to suggest that a pleomorphic variant of lobular carcinoma in situ is associated with development of breast cancer in the same site as the lesion, whereas a nonpleomorphic form is a marker of increased risk of ipsilateral and contralateral breast cancer.16
Invasive ductal and lobular carcinomas are the true invasive breast cancers, with a potential to metastasize.
Phyllodes tumors are uncommon fibroepithelial lesions that account for less than 1% of all breast neoplasms. The median age at presentation is 45 years.17 Despite the historical name “cystosarcoma phyllodes,” these lesions are not true sarcomas and have stromal and epithelial components.
These tumors display very heterogeneous behavior and, based on predefined histologic criteria, are often classified as benign, borderline, or malignant. Benign phyllodes tumors are similar to fibroadenomas in both histology and prognosis, making their diagnosis challenging. The most aggressive phyllodes tumors lose their epithelial component and have high metastatic potential. These tumors often have a biphasic growth pattern, and women may present with a smooth, round, well-defined breast lump that was stable for many years but then started to grow rapidly.17
Surgical resection with wide margins is the primary management of these tumors.18
Mastitis, ie, inflammation of the breast tissue, often presents with symptoms of breast erythema, swelling, tenderness, and nipple discharge. It may be secondary to infection (most often in lactating women) or other causes such as radiation or underlying malignancy. A complication of infectious mastitis is formation of a breast abscess. Underlying malignancy, especially inflammatory breast cancer, is a common cause of noninfectious mastitis and is very important to recognize.19
RISK FACTORS FOR BREAST CANCER
3. Which of the following are risk factors for breast cancer?
- Menarche before age 12
- Female sex
- Personal history of breast cancer
- Obesity
- Never having had children, or having given birth for the first time at an older age
- Older age
- History of hormone replacement therapy with estrogen and progesterone
- Family history of breast cancer
All of these choices are risk factors for breast cancer.
Family history
The overall relative risk of developing breast cancer in a woman with a first-degree relative with the disease is 1.7. However, the relative risk is about 3 if the first-degree relative developed breast cancer before menopause, and 9 if the first-degree relative developed bilateral breast cancer before menopause.5
Estrogen exposure
The duration and amount of estrogen exposure are also risk factors. For example, menarche before age 12 and menopause after age 55 are associated with a higher risk. Women who go through menopause after age 55 have a twofold higher risk of breast cancer compared with women who go through menopause at an early age. Pregnancy before age 30 lowers the risk of breast cancer; late first full-term pregnancy or nulliparity increases it. Lactation, on the other hand, has a protective effect.5
Oral contraceptives have traditionally been thought to increase the risk of breast cancer. In the 1990s, a meta-analysis involving 153,506 women found that those who had used oral contraceptives had a 24% higher risk of developing breast cancer.26 However, this association has come into question since newer oral contraceptive pills containing different progestins and lower amounts of estrogen have become available. In fact, recent studies showed no link between oral contraceptive use and breast cancer.27,28 Nevertheless, women at higher risk of developing breast cancer are advised not to use oral contraceptives.
Hormone replacement therapy with estrogen and progesterone was found to increase the risk of breast cancer by 26% in the Women’s Health Initiative (WHI) study, which involved 16,608 healthy women followed for a median of 5.6 years.29
In a study reported separately, the WHI investigators randomized 10,739 women who had undergone hysterectomy to receive either hormone replacement therapy with unopposed estrogen (which is feasible only in women without a uterus) or placebo. They found no increase in the risk of invasive breast cancer in women on hormone replacement therapy with estrogen alone. In fact, the study showed a trend towards a modest reduction of this risk (odds ratio 0.77; 95% confidence interval 0.59–1.01).30
After the results of the WHI were published, the use of hormone replacement therapy in postmenopausal women declined significantly. And in 2003—1 year later—the incidence of breast cancer had dropped by 6.7%.31
Most experts now recommend that estrogen-progestin combinations be used only selectively to treat the symptoms of menopause, and only for the short term.
Other risk factors
Other factors found to modestly increase the risk of breast cancer include:
- Alcohol use
- Obesity
- Radiation exposure. Patients are at higher risk of breast cancer 15 to 20 years after receiving upper-mantle radiotherapy for Hodgkin lymphoma.5
Case continues: Bad news on mammography, ultrasonography, biopsy
The patient undergoes mammography, which shows a 2.5-cm spiculated lesion with areas of calcifications (BIRADS score of 5). Subsequently, ultrasonography confirms that the suspicious mass is not a cyst. Ultrasound-guided core needle biopsy reveals that the lesion is a high-grade invasive ductal carcinoma. The tumor is positive for both estrogen and progesterone receptors and negative for HER2/neu overexpression.
STAGING EVALUATION
4. Given these findings, what is the next step to take?
- CT of the chest, abdomen, and pelvis
- MRI of the brain
- PET
- Referral to a surgeon for a possible mastectomy with sentinel lymph node dissection
- Referral to a surgeon for a possible lumpectomy with sentinel lymph node dissection
At this point, the patient should be referred to a surgeon for possible mastectomy or lumpectomy.
Women who appear clinically to have early breast cancer, such as in this case, should have a complete blood count, comprehensive metabolic panel, and chest x-ray as their initial staging evaluation. No further studies are recommended unless the findings on history, physical examination, or the above testing suggest possible metastases.
Mastectomy vs lumpectomy
Early-stage breast cancer is managed with definitive surgery. The two options are mastectomy and breast conservation therapy, the latter involving lumpectomy followed by breast radiation therapy.
Multiple randomized studies comparing mastectomy and lumpectomy showed no difference in survival rates, but patients in the lumpectomy groups had higher rates of local recurrence.32 Breast radiation therapy after lumpectomy lowered the rates of local recurrence and breast cancer death.33 Therefore, most patients can opt to undergo either lumpectomy with radiation or mastectomy, depending on personal preference.
However, mastectomy rather than breast conservation therapy is still recommended in cases of prior radiation therapy, inability to achieve negative surgical margins (as in cases of large tumors), multicentric disease (cancer in separate breast quadrants), or multiple areas of calcifications. Mastectomy is also preferred in most pregnant women unless the diagnosis of breast cancer is made in the third trimester and radiation therapy can be given after delivery. Patients who have large lesions in a small breast may also choose mastectomy with breast reconstruction rather than breast conservation therapy. Patients with a history of scleroderma are encouraged to undergo mastectomy because of increased toxicity from radiation treatment.
Sentinel vs axillary lymph node dissection
Knowledge of axillary lymph node involvement is important because it determines the stage in the tumor-node-metastasis (TNM) system, and it influences the choice of further therapy. Therefore, all patients with nonmetastatic invasive breast cancer must have their axillary lymph nodes sampled.
Conventionally, this involves axillary lymph node dissection. Unfortunately, upper extremity lymphedema develops in 6% to 30% of patients within the first 3 years, and in 49% of patients after 20 years following axillary lymph node dissection.34
Sentinel lymph node dissection was developed to minimize this complication. This procedure involves the injection of a blue dye, isosulfan blue (Lymphazurin), around the edge of the tumor or in the dermis overlying the tumor. The most proximal axillary lymph nodes that stain blue are dissected. Alternatively, a radioactive colloid (most commonly technetium sulfur colloid agents) may be injected, allowing sentinel lymph nodes to be identified by lymphoscintigraphy. If no metastases are found in the sentinel lymph nodes, axillary lymph node dissection is not performed.
A prospective study in 536 women found that at 5 years of follow-up, lymphedema developed in only 5% of patients after sentinel lymph node dissection compared with 16% of those who underwent axillary lymph node dissection (P < .001), with comparable outcomes in terms of disease recurrence.35
Case continues: Patient undergoes surgery
The patient elects to undergo lumpectomy with sentinel lymph node dissection. Pathologic review of the resection specimen reveals a 2.5-cm poorly differentiated invasive ductal carcinoma. Sentinel lymph node dissection shows metastases, and therefore axillary lymph node dissection is performed. One of eight lymph nodes removed is positive for metastases. All surgical margins are negative.
POSTOPERATIVE CARE
5. What would be the next step for our patient?
- Radiation followed by observation
- Tamoxifen (Nolvadex) for 5 years
- Observation only
- Chemotherapy followed by radiation therapy and 5 years of tamoxifen
She should receive chemotherapy, followed by radiation therapy and then tamoxifen for 5 years.
Chemotherapy. Almost all patients who have lymph-node-positive disease are advised to undergo chemotherapy.
The Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) performed a metaanalysis of 194 randomized trials that compared adjuvant chemotherapy and no treatment in early-stage breast cancer. Chemotherapy led to a 10% absolute improvement in survival at 15 years for women younger than 50 years and 3% in women age 51 to 69.36
Indications for chemotherapy include axillary lymph node involvement, locally advanced disease, and other risk factors for recurrence such as young age at diagnosis, strong positive family history of breast cancer, prior history of breast cancer, or lymph-node-negative, estrogen-receptor-negative tumors that are larger than 1 cm in diameter.
The Oncotype DX assay is a new tool to help oncologists decide whether to use chemotherapy in cases of estrogen-receptor-positive breast cancer, in which the benefit of chemotherapy is uncertain. It is a polymerase chain reaction assay that measures the expression of 16 cancer-specific genes and five reference genes within the breast tumor. Based on the pattern of expression of these genes, breast cancer can be characterized as low-risk, intermediate-risk, or high-risk. Patients in the high-risk group have a high chance of cancer recurrence and benefit from chemotherapy. Patients in the low-risk group are unlikely to have a recurrence or to benefit from chemotherapy.37 It is far less clear if patients in the intermediate-risk group benefit from chemotherapy, but this assay might eventually prove useful in deciding for or against chemotherapy in this group of patients as well.38 The Oncotype DX assay is presently being studied in a clinical trial.
Radiation therapy after mastectomy is recommended in patients who have breast tumors larger than 5 cm or metastases to more than three axillary lymph nodes.39
Antiestrogen therapy. After chemotherapy, patients with estrogen-receptor-positive cancers also receive 5 years of antiestrogen therapy. Available antiestrogen agents for such patients include tamoxifen, which is a selective estrogen receptor modulator, and drugs called aromatase inhibitors that block conversion of androgens to estrogens in peripheral tissues. Anastrozole (Arimidex), letrozole (Femara), and exemestane (Aromasin) are examples of available aromatase inhibitors. Premenopausal women are treated with tamoxifen, and postmenopausal women are offered aromatase inhibitors.
Table 4 lists the most common adverse effects of these agents. Aromatase inhibitors are associated with a higher risk of osteoporosis and arthralgia, while tamoxifen increases the risks of thromboembolism, endometrial cancer, and vaginal discharge. Both agents may produce menopausal symptoms such as hot flashes and mood swings.
Case continues: Seven years later, metastases in the spine
The patient achieves a complete remission. She is seen for a routine visit 7 years after diagnosis. She now reports mid-back pain that has worsened over the last 2 months. A bone scan reveals diffuse metastatic disease in the spine and in both humeral bones. CT of the chest, abdomen, and pelvis is negative for visceral metastases. Bone marrow aspiration and biopsy study show marrow infiltration by adenocarcinoma that stains positive for estrogen receptors and negative for HER2. The patient otherwise feels well and has no other symptoms.
WHAT TREATMENT FOR METASTATIC BREAST CANCER?
6. What should you now do for our patient?
- Discuss end-of-life care and refer her to a hospice program
- Educate the patient that no options for treatment exist and recommend enrolling in a phase I clinical trial
- Refer her to an oncologist for consideration of chemotherapy
- Refer her to an oncologist for consideration of endocrine treatment
She should be referred to an oncologist for consideration of endocrine treatment.
The most common sites of breast cancer metastases are the bones, followed by the liver and lungs. Metastatic breast cancer almost always is incurable. However, treatment can palliate symptoms.
Although a randomized trial of treatment vs best supportive care has never been done, many believe that treatment may improve survival. 40 The median survival of patients treated with standard therapy is about 3 years if the breast cancer is estrogen-receptor-positive and 2 years if it is estrogen-receptor-negative, but survival rates vary widely from patient to patient.41,42
Standard therapy or enrollment in a clinical phase II or III trial is indicated for this patient before considering enrollment in a phase I clinical trial or supportive care alone.
Endocrine therapy is the first-line therapy in women with estrogen-receptor-positive metastatic breast cancer. Postmenopausal women usually receive an aromatase inhibitor first.43,44 Response to endocrine therapy usually takes weeks to months but may last for several years.
Premenopausal women with estrogen-receptor-positive breast cancer also receive ovarian ablation therapy (oophorectomy or chemical ovarian ablation) with gonadotropin-releasing hormone agonists.
In addition, most patients with bone involvement are treated with high doses of intravenous bisphosphonates, which can reduce skeletal complications.45
Chemotherapy is reserved for patients with estrogen-receptor-negative breast cancer and those with cancer that progresses despite treatment with multiple antiestrogen agents. The time to response when chemotherapy is used is quicker, but the duration of response is usually shorter, lasting on average less than 1 year.37
Trastuzumab (Herceptin), a monoclonal humanized murine antibody to the extracellular domain of the HER2 protein, is indicated in patients with HER2-overexpressing tumors.46,47
STABLE 2 YEARS LATER
The patient was started on letrozole and a bisphosphonate, zolendronic acid (Zometa). Ovarian ablation was initiated with goserelin (Zoladex) given monthly. A bone scan performed 2 months after starting treatment showed improvement in bony metastases. She also noted significant improvement in pain. Her disease remains stable 2 years after starting endocrine therapy.
- Barton MB, Elmore JG, Fletcher SW. Breast symptoms among women enrolled in a health maintenance organization: frequency, evaluation, and outcome. Ann Intern Med 1999; 130:651–657.
- Petrelli NJ, Winer EP, Brahmer J, et al. Clinical cancer advances 2009: major research advances in cancer treatment, prevention, and screening—a report from the American Society of Clinical Oncology. J Clin Oncol 2009; 27:6052–6069.
- Jemal A, Siegel R, Ward E, et al. Cancer statistics 2008. CA Cancer J Clin 2008; 58:71–96.
- National Cancer Institute. SEER Stat Fact Sheets. www.seer.cancer.gov/statfacts/html/breast.html#ref09. Accessed June 7, 2010.
- Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ; the publishers of the journal Oncology. Cancer Management: A multidisciplinary Approach. Medical, Surgical & Radiation Oncology. 11th ed. CMP Medica; 2008.
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE. Paget’s disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187:171–177.
- Barlow WE, Lehman CD, Zheng Y, et al. Performance of diagnostic mammography for women with signs or symptoms of breast cancer. J Natl Cancer Inst 2002; 94:1151–1159.
- American College of Radiology. Breast Imaging Reporting and Data System: BIRADS Atlas. 4th ed. Reston, VA: American College of Radiology; 2003.
- Hong AS, Rosen EL, Soo MS, Baker JA. BI-RADS for sonography: positive and negative predictive values of sonographic features. AJR Am J Roentgenol 2005; 184:1260–1265.
- Berg WA, Campassi CI, Ioffe OB. Cystic lesions of the breast: sonographic-pathologic correlation. Radiology 2003; 227:183–191.
- Schell AM, Rosenkranz K, Lewis PJ. Role of breast MRI in the preoperative evaluation of patients with newly diagnosed breast cancer. AJR Am J Roentgenol 2009; 192:1438–1444.
- Turnbull L, Brown S, Harvey I, et al. Comparative effectiveness of MRI in breast cancer (COMICE) trial: a randomised controlled trial. Lancet 2010; 375:563–571.
- Worsham MJ, Abrams J, Raju U, et al. Breast cancer incidence in a cohort of women with benign breast disease from a multiethnic, primary health care population. Breast J 2007; 13:115–121.
- Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985; 312:146–151.
- Page DL, Kidd TE, Dupont WD, Simpson JF, Rogers LW. Lobular neoplasia of the breast: higher risk for subsequent invasive cancer predicted by more extensive disease. Hum Pathol 1991; 22:1232–1239.
- Sneige N, Wang J, Baker BA, Krishnamurthy S, Middleton LP. Clinical, histopathologic, and biologic features of pleomorphic lobular (ductallobular) carcinoma in situ of the breast: a report of 24 cases. Mod Pathol 2002; 15:1044–1050.
- Telli ML, Horst KC, Guardino AE, Dirbas FM, Carlson RW. Phyllodes tumors of the breast: natural history, diagnosis, and treatment. J Natl Compr Canc Netw 2007; 5:324–330.
- Reinfuss M, Mitus J, Duda K, Stelmach A, Rys J, Smolak K. The treatment and prognosis of patients with phyllodes tumor of the breast: an analysis of 170 cases. Cancer 1996; 77:910–916.
- Kamal RM, Hamed ST, Salem DS. Classification of inflammatory breast disorders and step by step diagnosis. Breast J 2009; 15:367–380.
- Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am J Hum Genet 1999; 64:963–970.
- Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med 2003; 348:2339–2347.
- Clarke-Pearson DL. Clinical practice. Screening for ovarian cancer. N Engl J Med 2009; 361:170–177.
- Hisada M, Garber JE, Fung CY, Fraumeni JF, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 1998; 90:606–611.
- Bell DW, Varley JM, Szydlo TE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999; 286:2528–2531.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 1996; 347:1713–1727.
- Hankinson SE, Colditz GA, Manson JE, et al. A prospective study of oral contraceptive use and risk of breast cancer (Nurses’ Health Study, United States). Cancer Causes Control 1997; 8:65–72.
- Marchbanks PA, McDonald JA, Wilson HG, et al. Oral contraceptives and the risk of breast cancer. N Engl J Med 2002; 346:2025–2032.
- Rossouw JE, Anderson GL, Prentice RL, et al; Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004; 291:1701–1712.
- Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 2007; 356:1670–1674.
- Fisher B, Anderson S, Redmond CK, Wolmark N, Wickerham DL, Cronin WM. Reanalysis and results after 12 years of follow-up in a randomized clinical trial comparing total mastectomy with lumpectomy with or without irradiation in the treatment of breast cancer. N Engl J Med 1995; 333:1456–1461.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Petrek JA, Senie RT, Peters M, Rosen PP. Lymphedema in a cohort of breast carcinoma survivors 20 years after diagnosis. Cancer 2001; 92:1368–1377.
- McLaughlin SA, Wright MJ, Morris KT, et al. Prevalence of lymphedema in women with breast cancer 5 years after sentinel lymph node biopsy or axillary dissection: objective measurements. J Clin Oncol 2008; 26:5213–5219.
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687–1717.
- Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351:2817–2826.
- Albain KS, Barlow WE, Shak S, et al; Breast Cancer Intergroup of North America. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010; 11:55–65.
- Harris JR, Halpin-Murphy P, McNeese M, Mendenhall NP, Morrow M, Robert NJ. Consensus Statement on postmastectomy radiation therapy. Int J Radiat Oncol Biol Phys 1999; 44:989–990.
- Gennari A, Conte P, Rosso R, Orlandini C, Bruzzi P. Survival of metastatic breast carcinoma patients over a 20-year period: a retrospective analysis based on individual patient data from six consecutive studies. Cancer 2005; 104:1742–1750.
- Mouridsen H, Gershanovich M, Sun Y, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol 2003; 21:2101–2109.
- Gamucci T, D’Ottavio AM, Magnolfi E, et al. Weekly epirubicin plus docetaxel as first-line treatment in metastatic breast cancer. Br J Cancer 2007; 97:1040–1045.
- Bonneterre J, Thürlimann B, Robertson JF, et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the Tamoxifen or Arimidex Randomized Group Efficacy and Tolerability study. J Clin Oncol 2000; 18:3748–3757.
- Nabholtz JM, Buzdar A, Pollak M, et al. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol 2000; 18:3758–3767.
- Hortobagyi GN, Theriault RL, Porter L, et al. Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. Protocol 19 Aredia Breast Cancer Study Group. N Engl J Med 1996; 335:1785–1791.
- Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673–1684.
- Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783–792.
- Barton MB, Elmore JG, Fletcher SW. Breast symptoms among women enrolled in a health maintenance organization: frequency, evaluation, and outcome. Ann Intern Med 1999; 130:651–657.
- Petrelli NJ, Winer EP, Brahmer J, et al. Clinical cancer advances 2009: major research advances in cancer treatment, prevention, and screening—a report from the American Society of Clinical Oncology. J Clin Oncol 2009; 27:6052–6069.
- Jemal A, Siegel R, Ward E, et al. Cancer statistics 2008. CA Cancer J Clin 2008; 58:71–96.
- National Cancer Institute. SEER Stat Fact Sheets. www.seer.cancer.gov/statfacts/html/breast.html#ref09. Accessed June 7, 2010.
- Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ; the publishers of the journal Oncology. Cancer Management: A multidisciplinary Approach. Medical, Surgical & Radiation Oncology. 11th ed. CMP Medica; 2008.
- Kollmorgen DR, Varanasi JS, Edge SB, Carson WE. Paget’s disease of the breast: a 33-year experience. J Am Coll Surg 1998; 187:171–177.
- Barlow WE, Lehman CD, Zheng Y, et al. Performance of diagnostic mammography for women with signs or symptoms of breast cancer. J Natl Cancer Inst 2002; 94:1151–1159.
- American College of Radiology. Breast Imaging Reporting and Data System: BIRADS Atlas. 4th ed. Reston, VA: American College of Radiology; 2003.
- Hong AS, Rosen EL, Soo MS, Baker JA. BI-RADS for sonography: positive and negative predictive values of sonographic features. AJR Am J Roentgenol 2005; 184:1260–1265.
- Berg WA, Campassi CI, Ioffe OB. Cystic lesions of the breast: sonographic-pathologic correlation. Radiology 2003; 227:183–191.
- Schell AM, Rosenkranz K, Lewis PJ. Role of breast MRI in the preoperative evaluation of patients with newly diagnosed breast cancer. AJR Am J Roentgenol 2009; 192:1438–1444.
- Turnbull L, Brown S, Harvey I, et al. Comparative effectiveness of MRI in breast cancer (COMICE) trial: a randomised controlled trial. Lancet 2010; 375:563–571.
- Worsham MJ, Abrams J, Raju U, et al. Breast cancer incidence in a cohort of women with benign breast disease from a multiethnic, primary health care population. Breast J 2007; 13:115–121.
- Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985; 312:146–151.
- Page DL, Kidd TE, Dupont WD, Simpson JF, Rogers LW. Lobular neoplasia of the breast: higher risk for subsequent invasive cancer predicted by more extensive disease. Hum Pathol 1991; 22:1232–1239.
- Sneige N, Wang J, Baker BA, Krishnamurthy S, Middleton LP. Clinical, histopathologic, and biologic features of pleomorphic lobular (ductallobular) carcinoma in situ of the breast: a report of 24 cases. Mod Pathol 2002; 15:1044–1050.
- Telli ML, Horst KC, Guardino AE, Dirbas FM, Carlson RW. Phyllodes tumors of the breast: natural history, diagnosis, and treatment. J Natl Compr Canc Netw 2007; 5:324–330.
- Reinfuss M, Mitus J, Duda K, Stelmach A, Rys J, Smolak K. The treatment and prognosis of patients with phyllodes tumor of the breast: an analysis of 170 cases. Cancer 1996; 77:910–916.
- Kamal RM, Hamed ST, Salem DS. Classification of inflammatory breast disorders and step by step diagnosis. Breast J 2009; 15:367–380.
- Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am J Hum Genet 1999; 64:963–970.
- Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med 2003; 348:2339–2347.
- Clarke-Pearson DL. Clinical practice. Screening for ovarian cancer. N Engl J Med 2009; 361:170–177.
- Hisada M, Garber JE, Fung CY, Fraumeni JF, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst 1998; 90:606–611.
- Bell DW, Varley JM, Szydlo TE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999; 286:2528–2531.
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297:2360–2372.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 1996; 347:1713–1727.
- Hankinson SE, Colditz GA, Manson JE, et al. A prospective study of oral contraceptive use and risk of breast cancer (Nurses’ Health Study, United States). Cancer Causes Control 1997; 8:65–72.
- Marchbanks PA, McDonald JA, Wilson HG, et al. Oral contraceptives and the risk of breast cancer. N Engl J Med 2002; 346:2025–2032.
- Rossouw JE, Anderson GL, Prentice RL, et al; Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- Anderson GL, Limacher M, Assaf AR, et al; Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004; 291:1701–1712.
- Ravdin PM, Cronin KA, Howlader N, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 2007; 356:1670–1674.
- Fisher B, Anderson S, Redmond CK, Wolmark N, Wickerham DL, Cronin WM. Reanalysis and results after 12 years of follow-up in a randomized clinical trial comparing total mastectomy with lumpectomy with or without irradiation in the treatment of breast cancer. N Engl J Med 1995; 333:1456–1461.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Petrek JA, Senie RT, Peters M, Rosen PP. Lymphedema in a cohort of breast carcinoma survivors 20 years after diagnosis. Cancer 2001; 92:1368–1377.
- McLaughlin SA, Wright MJ, Morris KT, et al. Prevalence of lymphedema in women with breast cancer 5 years after sentinel lymph node biopsy or axillary dissection: objective measurements. J Clin Oncol 2008; 26:5213–5219.
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687–1717.
- Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351:2817–2826.
- Albain KS, Barlow WE, Shak S, et al; Breast Cancer Intergroup of North America. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010; 11:55–65.
- Harris JR, Halpin-Murphy P, McNeese M, Mendenhall NP, Morrow M, Robert NJ. Consensus Statement on postmastectomy radiation therapy. Int J Radiat Oncol Biol Phys 1999; 44:989–990.
- Gennari A, Conte P, Rosso R, Orlandini C, Bruzzi P. Survival of metastatic breast carcinoma patients over a 20-year period: a retrospective analysis based on individual patient data from six consecutive studies. Cancer 2005; 104:1742–1750.
- Mouridsen H, Gershanovich M, Sun Y, et al. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol 2003; 21:2101–2109.
- Gamucci T, D’Ottavio AM, Magnolfi E, et al. Weekly epirubicin plus docetaxel as first-line treatment in metastatic breast cancer. Br J Cancer 2007; 97:1040–1045.
- Bonneterre J, Thürlimann B, Robertson JF, et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the Tamoxifen or Arimidex Randomized Group Efficacy and Tolerability study. J Clin Oncol 2000; 18:3748–3757.
- Nabholtz JM, Buzdar A, Pollak M, et al. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol 2000; 18:3758–3767.
- Hortobagyi GN, Theriault RL, Porter L, et al. Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. Protocol 19 Aredia Breast Cancer Study Group. N Engl J Med 1996; 335:1785–1791.
- Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673–1684.
- Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783–792.
Difficulty swallowing solid foods; food ‘getting stuck in the chest’
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
A 40-year-old man with spells of generalized weakness and paresthesias
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
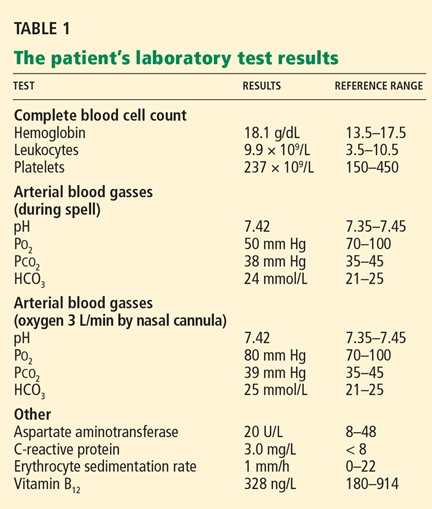
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
Abdominal pain in a 20-year-old woman
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
A 35-year-old Asian man with jaundice and markedly high aminotransferase levels
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).

- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.

HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18

Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
A 35-year-old man who was born in Vietnam presents to the emergency department of a local hospital because he has had jaundice for 5 days and fatigue, malaise, and anorexia for 2 weeks. He also has nausea and mild epigastric and right upper quadrant abdominal pain. He denies having fevers, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss.
His medical history is remarkable only for perinatally acquired hepatitis B virus (HBV) infection, for which he never received antiviral therapy. He does not take any prescribed, over-the-counter, or herbal medications.
He lives in the Midwest region of the United States and works full-time as a physician in private practice. He is married and has two children.
He has not travelled recently. He has no pets at home and has not been exposed to any.
He has never smoked. He drinks alcohol socially but has never used recreational drugs.
In a laboratory evaluation performed a year ago for insurance purposes, his liver function tests—serum albumin, total bilirubin, alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase levels—were all normal. He was positive for HBV surface antigen and HBV e antigen and negative for antibodies against these antigens.
PHASES OF HBV INFECTION
1. Which of the following best describes the status of HBV infection in this patient before his current symptoms developed?
- Resolved HBV infection
- Chronic inactive HBV infection
- Chronic active HBV infection
- Immune-tolerant chronic HBV infection
The correct answer is immune-tolerant chronic HBV infection.
Resolved infection. In immunocompetent adults, most primary HBV infections are self-limited: people clear the virus and gain lasting immunity (defined as the loss of HBV surface antigen, the development of antibody against surface antigen, no detectable HBV DNA in the serum, and normal alanine and aspartate aminotransferase levels). However, a minority of primary HBV infections persist and become chronic.
Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients with chronic HBV infection can be broadly classified as having either inactive disease (the inactive surface antigen carrier state) or chronic active hepatitis B (Figure 1).1–9
Chronic inactive HBV infection. Carriers of inactive HBV infection have low serum levels of HBV DNA (< 2,000 IU/mL), persistently normal aminotransferase levels, and no HBV e antigen; if a liver biopsy is performed, no significant hepatitis is found.
Chronic active HBV infection. Patients with chronic active HBV infection, in contrast, have high serum HBV DNA levels (> 20,000 IU/mL) and persistently or intermittently high aminotransferase levels; they do have HBV e antigen, and a liver biopsy shows moderate or severe necroinflammation.
A small group of patients with chronic active hepatitis B may be negative for e antigen but still have high aminotransferase levels, high HBV DNA levels, and continued necroinflammation in the liver.4 The virus in these patients has a mutation in its precore or core promoter gene that prevents the production of e antigen.
Patients with chronic active HBV infection (whether positive or negative for e antigen) are at a significantly greater risk of progressive liver injury and developing cirrhosis and hepatocellular carcinoma than are inactive carriers of HBV.
Immune-tolerant chronic HBV infection. Patients who acquired HBV at birth (eg, our patient) may have immune-tolerant HBV infection, which is characterized by significant HBV replication manifested by the presence of HBV e antigen and high levels of HBV DNA in the serum. However, these patients have no clinical or pathologic evidence of active liver disease (no symptoms, normal serum alanine aminotransferase levels, and minimal changes on liver biopsy).5 This was obviously the case in our patient, based on his history and laboratory results before his current symptoms developed.
Case continues: Liver function abnormalities
On physical examination, the patient’s temperature is 99.9°F (37.7°C), heart rate 106 per minute, blood pressure 98/54 mm Hg, respiratory rate 18 per minute, and oxygen saturation 100% while breathing ambient air. He is alert and oriented to time, place, and person.
He has icteric sclera, and his skin is jaundiced. His lymph nodes are not palpable. His cardiac examination is normal except for tachycardia. His lungs are clear to auscultation and percussion. He has mild epigastric and right upper quadrant abdominal tenderness with no peritoneal signs, hepatosplenomegaly, or masses.
His basic laboratory values on admission are listed in Table 1. His amylase and lipase levels are normal. A urine dipstick test is positive for bilirubin.
WHAT IS THE LEAST LIKELY DIAGNOSIS?
2. Which one of the following is the least likely diagnosis in this patient?
- Reactivation of hepatitis B
- Drug-associated liver injury
- Acute viral hepatitis
- Acute alcoholic hepatitis
- Ischemic hepatitis
The degree and pattern of liver function abnormalities in our patient reflect hepatocellular injury rather than cholestatic liver disease, because his aminotransferase levels are elevated much higher than his alkaline phosphatase level (Table 1). Bilirubin elevation does not help differentiate the two conditions.
The degree and pattern of aminotransferase elevations are also helpful in narrowing the differential diagnosis. Serum aminotransferase levels of more than 1,000 U/L are mainly seen in patients with ischemic, viral, and toxininduced liver injury. Other rare causes of such high levels include Budd-Chiari syndrome, Wilson disease, and autoimmune hepatitis.
Ischemic hepatitis. Our patient has mild hypotension, but it does not seem to have been severe enough or of long enough duration to have caused ischemic hepatitis.
Drug-associated liver injury. Hepatotoxicity associated with drugs (most commonly acetaminophen [Tylenol]), herbal therapy, or mushroom poisoning should be considered in any patient whose aminotransferase levels are this high. However, our patient denies taking any medications (prescribed or over-the-counter), herbal remedies, or illicit drugs.
Acute viral hepatitis can certainly explain the patient’s clinical picture. Infection with hepatitis A virus, hepatitis D virus, hepatitis E virus, cytomegalovirus, Epstein-Barr virus, herpes simplex viruses types 1 and 2, and varicella zoster virus have all been implicated in severe acute hepatitis. Although hepatitis E virus infection is more common in developing countries, it has been reported in the United States.6 It is unlikely that acute hepatitis C virus infection is producing this degree of elevation in aminotransferase levels.
Reactivation of the patient’s chronic HBV infection can also account for his clinical presentation.
Acute alcoholic hepatitis should be suspected clinically if a patient has a history of heavy alcohol use and clinical and laboratory findings that are compatible with the diagnosis. However, the absolute values of serum aspartate aminotransferase and alanine aminotransferase in acute alcoholic hepatitis are almost always less than 500 IU/L (and typically less than 300 IU/L). Our patient’s values are much higher, and he says he does not drink very much. Although people sometimes underestimate their alcohol intake, alcoholic hepatitis is the least likely diagnosis in our patient.
Case continues: The patient is hospitalized
The patient is admitted with a diagnosis of acute hepatitis. Given his history of chronic hepatitis B, he is empirically started on lamivudine (Epivir-HBV).
- Antinuclear antibody negative
- Autoimmune liver disease panel negative
- Serum ceruloplasmin 30 mg/dL (normal range 15–60)
- Alpha fetoprotein 35.1 μg/L (< 10).
Abdominal ultrasonography is performed and reveals a small stone in the gallbladder with no evidence of biliary dilatation; otherwise, the gallbladder appears normal. Doppler ultrasonography shows the liver vessels to be patent; the liver is normal in appearance. The abdomen and pelvis appear to be normal on computed tomography without intravenous contrast.
On the third hospital day, the patient’s blood test results are:
- Aspartate aminotransferase 199 U/L (normal range 7–40)
- Alanine aminotransferase 735 U/L (0–45)
- Total bilirubin 22.9 mg/dL (0–1.5)
- International normalized ratio 6.0 (0.77–1.17)
- White blood cell count 5.1 × 109/L (4–11)
- Hemoglobin 11.7 g/dL (12–16)
- Platelet count 166 × 109/L (150–400)
- Blood and urine cultures negative.
WHAT IS CAUSING HIS ACUTE HEPATITIS?
3. On the basis of the new data, which of the following statements about the cause of acute hepatitis in this patient is the most accurate?
- Herpetic hepatitis is the most likely cause, given his positive test for immunoglobulin M (IgM) against herpes simplex virus
- Hepatitis C cannot be excluded with the available data
- Negative HBV e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
- Hepatocellular carcinoma is the likely diagnosis, given the elevated alpha fetoprotein level
The third answer above is correct: a negative test for hepatitis B e antigen does not exclude the diagnosis of acute exacerbation of HBV infection
Herpetic hepatitis. Although not common, hepatitis due to herpes simplex virus infection should be considered in the differential diagnosis of any patient presenting with severe acute hepatitis, particularly when fever is present. Common features of herpetic hepatitis on presentation include high fever, leukopenia, markedly elevated aminotransferases, and mild cholestasis. Vesicular rash occurs in only less than half of cases of herpetic hepatitis.10
Serologic testing is of limited value because it has high rates of false-positive and false-negative results. The diagnosis can be confirmed only by viral polymerase chain reaction testing or by identifying herpes simplex viral inclusions in the liver biopsy.
However, the death rate is high in this disease, and since herpetic hepatitis is one of the few treatable causes of acute liver failure, parenteral acyclovir (Zovirax) should be considered empirically in patients presenting with acute liver failure. Our patient was started on acyclovir when his tests for IgM against herpes simplex virus came back positive.
Hepatitis C. Antibodies against hepatitis C virus do not develop immediately after this virus is contracted; they may take up to 12 weeks to develop after exposure. For this reason, about 30% to 50% of patients with acute hepatitis C virus infection are negative for these antibodies initially. In those patients, hepatitis C virus RNA in the blood is the most sensitive test to detect acute hepatitis C virus infection.
Our patient has neither antibodies against hepatitis C virus nor hepatitis C virus RNA by polymerase chain reaction testing, which rules out hepatitis C virus infection.
Disappearance of e antigen in HBV infection. The disappearance of HBV e antigen is usually associated with a decrease in serum HBV DNA and remission of liver disease. However, some patients continue to have active liver disease and high levels of HBV DNA despite e antigen seroconversion. This is due to a stop codon mutation in the precore region of the viral genome that decreases or prevents production of HBV e antigen.4 In other words, even though HBV e antigen is a good marker of HBV replication in general, a subgroup of patients with chronic HBV infection are negative for e antigen but still have a high rate of viral replication as evidenced by high serum HBV DNA levels.
Patients with perinatally acquired chronic HBV infection most often have immune-tolerant chronic HBV infection. Among those patients (mostly Asian),5,7 the virus is spontaneously cleared at a rate of approximately 2% to 3% per year,8 most often during the second and third decades of age.
Transition from the immune-tolerant phase to the immune clearance phase is frequently associated with mild transient worsening of the liver function profile.9,11,12 However, in a small percentage of patients, hepatic decompensation and even (rarely) death from hepatic failure may occur secondary to a sudden activation of the immune system as it attempts to clear the virus. This may result in an increase in immune-mediated lysis of infected hepatocytes.
Hepatocellular carcinoma. Exacerbation of hepatitis B may be associated with an elevation of alpha fetoprotein, which may falsely raise concerns about the possibility of hepatocellular carcinoma. However, our patient had abdominal imaging with both ultrasonography and computed tomography, which showed no evidence of hepatocellular carcinoma.
Comment. The most likely cause of the patient’s acute liver failure is an acute exacerbation of hepatitis B. However, herpetic hepatitis should be ruled out by testing for herpes simplex virus by polymerase chain reaction, performing a liver biopsy, or both.
Case continues: His condition worsens
A transjugular liver biopsy shows changes associated with chronic hepatitis B, severe acute hepatitis with extensive confluent and submassive hepatic necrosis, and no intracellular viral inclusions. Subsequently, acyclovir is stopped.
On the 6th hospital day, he develops progressive metabolic acidosis and hypotension, with worsening hypoxemia. A chest radiograph is obtained to look for pneumonia, but it is indeterminate; computed tomography of the chest without contrast medium is likewise unremarkable. Duplex ultrasonography of the four extremities is negative for venous thrombosis.
The patient becomes more lethargic and difficult to arouse. He is transferred to the intensive care unit and intubated. His prothrombin and partial thromboplastin times continue to rise, the prothrombin time reaching values of more than 50 seconds. In addition, progressive renal insufficiency develops.
WHAT IS THE NEXT STEP?
4. Which of the following is the most appropriate next step in the management of this patient?
- Liver transplantation
- HBV immunoglobulin only
- Interferon and a nucleoside analogue
- Liver-assist devices
- Continue supportive care only
Liver transplantation. Since the patient’s severe acute hepatitis is accompanied by coagulopathy and encephalopathy, he meets the definition of having acute liver failure. Liver transplantation remains the only definitive therapy.
HBV immune globulin immunoprophylaxis is indicated in patients with HBV infection undergoing liver transplantation, to prevent recurrence of hepatitis B after the transplant, particularly in those with a high pretransplant viral load.17 The use of pretransplant antiviral therapy and the posttransplant combination of antiviral therapy and HBV immune globulin has reduced the rate of hepatitis B recurrence to less than 10%. However, immune globulin is by no means the best single next step in managing this patient, who clearly needs a new liver.
Interferon, nucleoside analogues. Options for antiviral treatment are interferon alfa and nucleoside analogues. Interferon therapy is contraindicated in patients such as ours, who have decompensated liver disease, because it can exacerbate the disease.18
Liver-assist devices. Because liver allografts are in short supply, there has been a strong interest in developing a device that would provide the same benefits for patients with liver failure as hemodialysis does for patients with renal failure. Trials are under way to determine the efficacy and safety of these devices.20
Case continues: He receives a liver
The patient undergoes liver transplantation. He is given HBV immune globulin during and after the surgery.
Pathologic review. Under the microscope, his old liver has widespread necrosis and hemorrhage as well as inflammatory changes suggesting a chronic viral process. Regenerative nodules are present in the small amount of surviving liver parenchyma, consistent with early cirrhosis. Iron staining shows +3 depositions in areas of hepatic collapse (a nonspecific finding). Periodic acid-Schiff staining after diastase (used to detect alpha-1 antitrypsin deficiency) is negative. Herpetic viral inclusions are not present.
An immunoassay for herpes simplex virus antigen is negative. Immunostaining with antibodies to the HBV core antigen is negative. HBV surface antigen is strongly and diffusely positive in the cytoplasm of 80% to 90% of hepatocytes. The immunohistologic staining pattern is consistent with integration of HBV DNA into the DNA of hepatic tissue.
Postoperative course. Lamivudine is continued after surgery, and the patient is sent home. He has resumed the level of functioning he had before becoming ill.
Comment. The outcome of liver transplantation for hepatitis B has notably improved since HBV immune globulin and nucleoside analogues were introduced. The results of liver transplantation for hepatitis B, particuarly patient and graft survival rates, are now better than those in transplant patients with hepatitis C and similar to those in transplant patients with other types of liver disease.21 The combination of HBV immune globulin and lamivudine has cut the rate of HBV reinfection after liver transplantation to approximately 10% and increased the 5-year survival rate after transplantation to about 80%.17,22
KEY POINTS
- In immunocompetent adults, most primary HBV infections are self-limited.
- Chronic HBV infection is defined as the persistence of HBV surface antigen in the serum for at least 6 months. Patients having chronic HBV infection can be broadly classified as inactive carriers or having chronic active disease.
- Most patients with chronic active HBV infection are positive for HBV e antigen, except patients in whom the virus has a mutation in the precore or core region of its genome that prevents the production of e antigen.
- Patients who carry inactive HBV or who are immune-tolerant require serial measurements of aminotransferase and HBV DNA levels. Treatment can be considered if the patient has a high viral load (> 2,000 IU/mL), elevated aminotransferases, or active disease on liver biopsy.
- Carriers of chronic active HBV (whether positive or negative for HBV e antigen) should be referred to a hepatologist for consideration of liver biopsy and treatment.
- Interferon should not be used in immunocompromised patients or those with decompensated liver disease because it can further exacerbate the liver disease.
- Liver transplantation should be considered in patients with acute liver failure who have a poor prognosis according to the King’s College Hospital criteria.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
- Dusheiko G. Hepatitis B. In:Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd ed. Oxford, UK: Oxford University Press; 1999:876–896.
- Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 2002; 36:1408–1415.
- Pawlotsky JM, Bastie A, Hezode C, et al. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J Virol Methods 2000; 85:11–21.
- Brunetto MR, Giarin MM, Oliveri F, et al. Wild-type and e-antigen-minus hepatitis viruses and course of chronic hepatitis. Proc Natl Acad Sci USA 1991; 88:4186–4190.
- Lok AS, Lai CL. A longitudinal follow-up of asymptomatic hepatitis B surface antigen-positive Chinese children. Hepatology 1988; 5:1130–1133.
- Hsu HY, Chang MH, Hsieh KH, et al. Cellular immune response to HBcAg in mother-to-infant transmission of hepatitis B virus. Hepatology 1992; 15:770–776.
- Chang MH, Hsu HY, Hsu HC, Ni YH, Chen JS, Chen DS. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995; 22:1387–1392.
- Ruiz-Moreno M, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999; 29:572–575.
- Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983; 84:216–219.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transplant 2007; 13:1428–1434,
- Liaw YF, Pao CC, Chu CM, Sheen IS, Huang MJ. Changes of serum hepatitis B virus DNA in two types of clinical events preceding spontaneous hepatitis B e antigen seroconversion in chronic type B hepatitis. Hepatology 1987; 7:1–3.
- Maruyama T, Iino S, Koike K, Yasuda K, Milich DR. Serology of acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 1993; 105:1141–1151.
- O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Shakil AO, Kramer D, Mazariegos GV, Fung JJ, Rakela J. Acute liver failure: clinical features, outcome analysis, and applicability of prognostic criteria. Liver Transplant 2000; 6:163–169.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King’s criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Samuel D, Muller R, Alexander G, et al. Liver transplantation in European patients with the hepatitis B surface antigen. N Engl J Med 1993; 329:1842–1847.
- Lok A, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Sorren MF, Belangia EA, Costa J, et al. National Institutes of Health consensus development conference statement: management of hepatitis B. Ann Intern Med 2009; 150:104–110.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Kim WR, Poterucha JJ, Kremers WK, Ishitani MB, Dickson ER. Outcome of liver transplantation for hepatitis B in the United States. Liver Transplant 2004; 10:968–974.
- Terrault NA, Zhou S, Combs C, et al. Prophylaxis in liver transplant recipients using a fixed dosing schedule of hepatitis B immunoglobulin. Hepatology 1996; 24:1327–1333.
A 48-year-old man with uncontrolled diabetes
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
A 48-year-old white man who has had diabetes mellitus for 6 years presents to the outpatient clinic because his blood sugar levels have been rising for the past week.
Both his parents had diabetes, and at the time of his diagnosis he weighed 278 pounds, all of which supported a diagnosis of type 2 diabetes mellitus. His disease was initially managed with diet, exercise, and metformin (Glucophage). Four months later, with weight loss and exercise, his blood sugar levels were consistently under 100 mg/dL, and metformin was discontinued.
He did well until 1 week ago, when he noted polyuria, polydipsia, and rising fingerstick glucose values, higher than 200 mg/dL. He has been eating well, with no nausea, vomiting, or symptoms of dehydration. He denies having any fever, chills, cough, nasal congestion, chest pain, abdominal pain, or dysuria.
In addition to his type 2 diabetes, he has hypertension, for which he takes losartan (Cozaar); hyperlipidemia, for which he takes atorvastatin (Lipitor); and gout, for which he takes allopurinol (Zyloprim).
His blood pressure is 148/70 mm Hg, pulse 100, and weight 273 pounds, and he is afebrile. On examination, his skin, head, eyes, ears, nose, throat, lungs, heart, and abdomen are normal. Urinalysis in the clinic shows large amounts of glucose and ketones.
WHAT IS THE LEAST LIKELY CAUSE OF HIS POOR CONTROL?
1. Which of the following is the least likely cause of his poorly controlled diabetes?
- Occult infection
- Poor adherence to diet and exercise
- Diabetic ketoacidosis
- Pancreatitis
Until 1 week ago, this patient’s diabetes had been well controlled for several years. Pancreatitis is the least likely cause of his uncontrolled diabetes, because he has no history of pancreatitis and has none of the symptoms of acute pancreatitis (fever, vomiting, or severe midepigastric pain radiating into the back).
Poor adherence to medication and lifestyle issues are very common in patients with poorly controlled diabetes and should always be included in the differential diagnosis.
Occult infection should also be considered in a patient with uncontrolled diabetes. Although this patient had no symptoms or signs of infection, urinalysis was done to look for an occult urinary tract infection and, surprisingly, it showed a large amount of ketones.
Case continued: He is treated for diabetic ketoacidosis
Diabetic ketoacidosis in ‘atypical diabetes’
Diabetic ketoacidosis is one of the most serious complications of diabetes. Many patients present with nausea, vomiting, and abdominal pain. Dehydration is often present because hyperglycemia leads to glucosuria and volume depletion. Interestingly, our patient showed none of these symptoms or signs.
Diabetic ketoacidosis is increasingly being recognized as a complication in patients with type 2 diabetes mellitus.1–4 Since the mid-1990s, clinicians have become increasingly aware of a condition variably termed “atypical diabetes,” “Flatbush diabetes,” “diabetes type 1B,” and “ketosis-prone type 2 diabetes mellitus,” in which patients, usually obese, present with diabetic ketoacidosis as their first manifestation, but are subsequently found to have type 2 diabetes mellitus. These patients typically are African American or of African, Hispanic, or Caribbean descent.
Ketoacidosis results from transient suppression of beta-cell function, the cause of which is unknown. A recent study comparing patients who have type 2 diabetes mellitus with and without diabetic ketoacidosis presenting with decompensated diabetes suggested insulinopenia was the predominant mechanism.5 For many of these patients, insulinopenia is transient: as the ketoacidosis resolves, betacell function improves and, with adequate insulin, lipolysis is reduced.
WHAT CAUSES DIABETIC KETOACIDOSIS?
2. Which of the following hormonal changes underlies the development of diabetic ketoacidosis?
- Insulin resistance
- Insulin deficiency
- Glucagon excess
- Glucagon deficiency
- Insulin deficiency and glucagon excess
- Insulin deficiency and glucagon deficiency
Diabetic ketoacidosis can occur when there is too much glucagon and not enough insulin. Insulin lowers the serum glucose level by promoting glucose uptake in peripheral tissues and by inhibiting gluconeogenesis and glycogenolysis in the liver. Insulin is also anabolic: it inhibits lipolysis in adipocytes and thus decreases the amount of substrate for ketogenesis.
Glucagon is the primary counterregulatory hormone responsible for ketogenesis.6 In the presence of glucagon excess, malonyl CoA production decreases, causing unblocking of carnitine acyltransferase I (CAT I) and allowing beta-oxidation to occur.6
Therefore, the sequence initiating ketogenesis begins with a shift in the ratio of glucagon to insulin, so that there is a relative or absolute excess of glucagon and a deficiency of insulin. A deficiency of insulin accelerates lipolysis, providing more substrate for ketogenesis, while excess glucagon turns on the oxidative sequence for fatty acids in the liver.
Three ketone bodies are produced in diabetic ketoacidosis: two ketoacids (beta-hydroxybutyric acid and acetoacetic acid), and one neutral ketone (acetone). The concentration of insulin required to suppress lipolysis is only one-tenth of that required to promote glucose utilization.7 Diabetic ketoacidosis is uncommon in patients with type 2 diabetes because they typically have enough insulin to inhibit lipolysis (and therefore ketoacid formation) but not enough to promote glucose utilization.
RISK FACTORS FOR DIABETIC KETOACIDOSIS
3. Which of the following is not a risk factor for diabetic ketoacidosis in type 2 diabetes mellitus?
- Acute illness
- Age > 65
- Inadequate insulin doses
- Antipsychotic drugs
- Ethnicity
Diabetic ketoacidosis is often precipitated by an acute illness such as an infection, cerebrovascular accident, myocardial infarction, or acute pancreatitis.8–12 These acute illnesses induce stress in the body and elevate counterregulatory hormones.
Inadequate insulin doses can also lead to diabetic ketoacidosis.
Drugs that affect carbohydrate metabolism are also risk factors. These include glucocorticoids, thiazide diuretics in high doses (> 50 mg daily), sympathomimetic agents, and second-generation antipsychotic agents (also called “atypical” antipsychotics) such as clozapine (Clozaril) and olanzapine (Zyprexa), although some are worse than others.13,14
Ketosis-prone type 2 diabetes mellitus is more prevalent in African Americans and Hispanics.8,15,16
Age is not a risk factor for developing diabetic ketoacidosis. In fact, diabetic ketoacidosis is the leading cause of morbidity and death in children with type 1 diabetes and can also occur in children with type 2 diabetes, particularly in obese African American adolescents.2
DISTINGUISHING TYPE 1 FROM TYPE 2
4. Which of the following is most specific in distinguishing type 1 from type 2 diabetes mellitus?
- C-peptide levels
- Islet cell antibodies
- Body mass index
- Family history
- Hemoglobin A1c level
Type 1 diabetes is characterized by destruction of pancreatic beta cells, leading to absolute insulin deficiency. The process is usually mediated by autoimmunity; therefore, testing for antibodies to islet cells, glutamic acid decarboxylase, insulin, and tyrosine phosphatase is the most specific way to distinguish type 1 from type 2 diabetes mellitus.
The hemoglobin A1c level correlates with the mean blood glucose level over the previous 8 to 12 weeks. The hemoglobin A1c is typically elevated in both type 1 and type 2 diabetes mellitus and therefore is not a useful distinguishing feature.
C-peptide is made when proinsulin is cleaved into insulin and C-peptide. It is released from endocytic vesicles with insulin in a one-to-one molar ratio. Thus, the level of C-peptide in the blood can show how much insulin is being made by the pancreas. C-peptide levels can help distinguish between type 1 and type 2 diabetes mellitus later in the course of the disease (levels are usually lower in a patient with type 1 diabetes), but they are not as useful early on because they can be normal early in the course of type 1 diabetes.17
A family history of diabetes is more common in type 2 diabetes, but patients with either type 1 or type 2 can have an affected close relative.
Patients with type 2 diabetes are generally overweight, with a body mass index greater than the 85th percentile for their age and sex. In contrast, patients with type 1 diabetes are usually not overweight and often have a recent history of weight loss. There are exceptions, however, and some patients with type 1 diabetes have an elevated body mass index, while some patients with type 2 diabetes are thin.
Although individually, C-peptide, family history, and body mass index are not very specific in distinguishing type 1 from type 2 diabetes mellitus, together they often give the clinician a good idea of the type of diabetes the patient has. In our case, although islet cell antibodies were not drawn, the normal C-peptide level, high body mass index, and family history all support a diagnosis of type 2 diabetes mellitus.
THE PATIENT CONTINUES TO DO WELL
The patient is discharged from the hospital on an insulin regimen. His blood sugar levels are closely monitored and remain near normal. Six months after the episode of diabetic ketoacidosis, his insulin is discontinued.
TAKE-HOME POINTS
Diabetic ketoacidosis is not unique to type 1 diabetes mellitus. It can occur in type 2, more commonly in patients who are nonwhite and who have precipitating factors such as acute illness, inadequate insulin treatment, or newly diagnosed diabetes. Clinicians should be aware of the possibility of diabetic ketoacidosis even in patients with type 2 diabetes who may not have these risk factors.
One approach to recognizing diabetic ketoacidosis better in patients with type 2 diabetes mellitus would include checking urine for ketones and serum electrolytes for high anion gap acidosis when patients with type 2 diabetes present with uncontrolled blood sugar levels. If ketonuria or acidosis is present, serum ketone and beta-hydroxybutyrate levels should be obtained to evaluate for diabetic ketoacidosis.
Patients should take insulin for an indeterminate period of time after initial treatment of diabetic ketoacidosis. As our case illustrates, in many cases, beta-cell function will return sufficiently to allow insulin to be discontinued. There are no clear guidelines for how long to continue insulin, but most practitioners continue it for weeks to months and discontinue it when glucose levels are stable and remain so with tapering doses. Sometimes oral agents need to be added as insulin is tapered.
Insulin therapy is tailored to the individual patient on the basis of blood glucose values. There are no data on which type of insulin is the most effective, and there are no data on whether these patients are at greater risk of hypoglycemia than other patients taking insulin. In general, there is no evidence that “prophylactic” insulin (ie, giving insulin to prevent diabetic ketoacidosis during times of illness or stress) is required. However, blood glucose monitoring is appropriate during infection or stress, and if hyperglycemia occurs in these situations, insulin use is prudent to reduce the risks of recurrent diabetic ketoacidosis.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44:790–795.
- Valabhji J, Watson M, Cox J, Poulter C, Elwig C, Elkeles RS. Type 2 diabetes presenting as diabetic ketacidosis in adolescence. Diabet Med 2003; 20:416–417.
- Westphal SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med 1996; 101:19–24.
- Welch B, Zib I. Case study: diabetic ketoacidosis in type 2 diabetes: “look under the sheets.” Clin Diabetes 2004; 22:198–200.
- Linfoot P, Bergstrom C, Ipp E. Pathophysiology of ketoacidosis in type 2 diabetes mellitus. Diabet Med 2005; 22:1414–1419.
- Foster DW, McGarry JD. The regulation of ketogenesis. Ciba Found Symp 1982; 87:120–131.
- Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism: persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest 1964; 43:950–962.
- Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med 2004; 164:1925–1931.
- Umpierrez GE, Kelly JP, Navarrete JE, Casals MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med 1997; 157:669–675.
- Jabbour SA, Miller JL. Uncontrolled diabetes mellitus. Clin Lab Med 2001; 21:99–110.
- Ennis ED, Kreisberg RA. Diabetic ketoacidosis and the hyperglycemic hyperosmolar syndrome. In: Leroith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott-Raven Publishers; Philadelphia, 1996:276–286.
- Case CC, Maldonado M. Diabetic ketoacidosis associated with Metabolife: a report of two cases. Diabetes Obes Metab 2002; 4:402–406.
- Kitabchi AE, Umpierrez GE, Murphy MB. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. In:DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International Textbook of Diabetes Mellitus, 3rd ed. John Wiley and Sons, Ltd: Chichester, UK, 2004:1101–1119.
- Newcomer JW. Second generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005; 19( suppl 1):1–93.
- Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs. type 2 diabetes and the effect of ethnicity. Arch Intern Med 1999; 159:2317–2322.
- Davis SN, Umpierrez GE. Diabetic ketoacidosis in type 2 diabetes mellitus—pathophsyiology and clinical presentation. Nat Clin Pract Endocrinol Metab 2007; 3:730–731.
- Hoogwerf B, Rich S, Barbosa J. Meal-stimulated Cpeptide and insulin antibodies in type I diabetic subjects and their nondiabetic siblings characterized by HLA-DR antigens. Diabetes 1985; 34:440–445.
A 72-year-old man with a purpuric rash
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
A 43-year-old woman with chest pressure
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma

The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4

At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.

DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A young pregnant woman with shortness of breath
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
A 21-year-old woman who is 12 weeks pregnant according to the date of her last menstrual period comes to the emergency department with shortness of breath and chest pain.
One week ago she began experiencing pre-syncope and shortness of breath on minimal exertion and then even at rest on most days. The shortness of breath worsened throughout the week, eventually limiting her daily activities to such a degree that she restricted herself to bed rest.
Her chest pain started today while she was sitting in church, without any apparent provocation. It is right-sided, sharp, and focal, and it does not radiate. At the same time, her shortness of breath was more severe than before, so she immediately came to the emergency department.
This is her third pregnancy; she has had one live birth and one abortion. Her last pregnancy was full-term, with routine prenatal care and no complications. However, so far during this pregnancy, she has had no prenatal care, she has not taken prenatal vitamins, and she has been unable to maintain adequate nutrition because of persistent emesis, which began early in her pregnancy and continues to occur as often as two or three times daily. She has lost 20 pounds over the past 12 weeks.
She says she has no close contacts who are sick, and she has had no fever, diarrhea, dysuria, urinary frequency or urgency, palpitations, swelling of the legs or feet, blurry vision, or increase in neck girth. She says she does not smoke or use alcohol or illicit substances. Her only previous surgery was laser-assisted in situ keratoplasty (LASIK) eye surgery in 1998. She is allergic to seafood only. She has not eaten at any new places recently. She is up to date with her childhood vaccinations. She has no family history of hypercoagulability or venous thrombotic events.
PHYSICAL EXAMINATION
She is breathing rapidly—as fast as 45 breaths per minute. Her temperature is 37.2°C (98.9°F), blood pressure 95/60 mm Hg, oxygen saturation 100% while on 10 L of oxygen using a nonrebreather mask, pulse 102 beats per minute, and weight 55.9 kg (123.2 pounds). She appears alert, oriented, and comfortable, with a thin body habitus. She has no jugular venous distention, neck mass, or thyromegaly. Her lungs are clear to auscultation, with no wheezes or rales. The cardiovascular examination is normal. She has a regular heart rate and rhythm, normal S1 and S2 sounds, and no rubs, clicks, or murmurs. Pulses in the extremities are normal, and she has no peripheral edema. The neurologic examination is normal.
Electrocardiography shows sinus tachycardia with first-degree atrioventricular block.
DIFFERENTIAL DIAGNOSIS
1. At this point, which is the most probable cause of her symptoms?
- Pulmonary embolism
- Peripartum cardiomyopathy
- Acute coronary syndrome
- Aortic dissection
- Expected physiologic changes of pregnancy
Pulmonary embolism would be the most probable diagnosis, given the patient’s pregnancy, shortness of breath, and tachycardia and the pleuritic quality of her chest pain.
Peripartum cardiomyopathy is also a possible cause, as it may present with profound shortness of breath and markedly decreased cardiac function. But it is much less likely in this patient because she is early in her pregnancy, and peripartum cardiomyopathy usually is seen during the last month of gestation or the first months after delivery.
Acute coronary syndrome is unlikely, given her young age and the lack of significant risk factors or a supporting history.
Aortic dissection is unlikely in view of her medical history.
Physiologic changes of pregnancy. Many pregnant women experience a sensation of not being able to catch their breath or expand their lungs fully, as the diaphragm is limited by the gravid abdomen. They also present with dyspnea, fatigue, reduced exercise capacity, peripheral edema, or volume overload.1 However, these changes tend to occur gradually and worsen over time. This patient’s degree of shortness of breath and its sudden onset do not seem like normal physiologic changes of pregnancy.
Other possible causes of dyspnea in a pregnant woman include asthma, pleural empyema, pneumonia, and severe anemia. Asthma should be considered in anyone with a history of wheezing, cough, and dyspnea. Fever and sputum production would support a diagnosis of pneumonia or empyema. In addition, maternal heart disease (eg, endocarditis, pulmonary hypertension) complicates 0.2% to 3% of pregnancies.1
CASE CONTINUED
The emergency department staff decide to evaluate the patient for heart failure and pulmonary embolism.
Bedside echocardiography reveals an ejection fraction of 55% (normal range 50%–75%), normal heart function and size, and no valvular abnormalities.
Chest radiography is normal.
Lower-extremity duplex ultrasonography is negative for deep-vein thrombosis.
The D-dimer level is 380 ng/mL (normal range < 500 ng/mL).
The medical intensive care unit is consulted about the patient’s continued tachypnea and the possible need for intubation. A ventilation-perfusion scan is performed to screen for pulmonary embolism, and it is negative.
An obstetric team performs Doppler ultrasonography at the bedside; a fetal heartbeat can be heard, thus confirming a viable pregnancy.
The patient has normal serum levels of the cardiac enzymes troponin T and creatine kinase-MB fraction, thus all but ruling out myocardial ischemia.
The patient is admitted to the hospital the next day, and a cardiology consult is obtained.
RULING OUT PULMONARY EMBOLISM
2. Has pulmonary embolism been definitively ruled out at this point?
- Yes
- No
The answer is no. The negative ventilation-perfusion scan and normal D-dimer test in this patient are not enough to rule out pulmonary embolism. The diagnosis of pulmonary embolism should be based on the clinician’s estimation of the pretest probability of pulmonary embolism (which is based on presenting signs and symptoms), as well as on a variety of tests, including spiral computed tomography (CT), ventilation-perfusion lung scanning, and serum D-dimer testing. Signs and symptoms that may guide the clinician are chest pain (present in 70% of patients with pulmonary embolism), tachypnea (70%), cough (40%), shortness of breath (25%), and tachycardia (33%).2 A history of pregnancy, malignancy, immobility, or recent surgery may also increase the pretest probability of pulmonary embolism. In many cases, one’s clinical suspicion is highly predictive and is useful in diagnosing pulmonary embolism.
The accuracy of the tests varies widely, depending on the pretest probability of pulmonary embolism. For instance, in a patient with a high pretest probability but a low-probability ventilation-perfusion scan, the true probability of pulmonary embolism is 40%, but in a patient with a low pretest probability and a low-probability scan, the probability is only 4%.
The Wells criteria can be used to calculate the pretest probability of pulmonary embolism. Given this patient’s tachycardia and clinical presentation, her pretest probability according to the Wells criteria indicates increased risk. However, because her D-dimer test, lower-extremity Doppler test, and ventilation-perfusion scan were normal, pulmonary embolism is less likely.3
However, if one’s clinical suspicion is high enough, further investigation of pulmonary embolism would proceed despite the encouraging test results.
CASE CONTINUED
The cardiology consult team notes that her beta human chorionic gonadotropin (beta-hCG) level is much higher than would be expected at 12 weeks of pregnancy, and so they are concerned about the possibility of a molar pregnancy. In addition, her level of thyroid-stimulating hormone (TSH, or thyrotropin) is markedly low.
HYPERTHYROIDISM IN PREGNANCY
3. Which of the following would not explain this patient’s markedly low TSH level?
- Graves disease
- Molar pregnancy
- TSH-secreting pituitary adenoma
- Gestational transient thyrotoxicosis
- Twin pregnancy
Hyperthyroidism (also called thyrotoxicosis) has many causes, including but not limited to Graves disease, pituitary adenoma, struma ovarii (teratoma), hCG-secreting hydatidiform mole, and thyroid carcinoma (which is rare).4 In most of these disorders, the TSH level is low while the levels of thyroxine (T4), triiodothyronine (T3), or both are high.
Symptoms of hyperthyroidism are the effect of elevated T4 and T3 levels on the target organs themselves. Common symptoms include fever, tachycardia, tremor, stare, sweating, and lid lag. Other symptoms include nervousness, delirium, hypersensitivity to heat, flushing, palpitations, fatigue, weight loss, dyspnea, weakness, increased appetite, swelling of the legs, nausea, vomiting, diarrhea, goiter, tremor, atrial fibrillation, and cardiac failure.4 In its extreme form, called thyroid storm, thyrotoxicosis can be life-threatening. The likelihood of an impending thyroid storm can be assessed by clinical variables such as the patient’s temperature and heart rate and whether he or she has heart failure or gastrointestinal manifestations.5
Graves disease, the most common cause of hyperthyroidism in pregnancy, is due to stimulation of TSH receptors by antibodies against these receptors. Graves disease is possible in this patient, but a subsequent TSH receptor antibody test is negative.
Pituitary adenomas are one of the few causes of hyperthyroidism in which the TSH level is high, not low. Therefore, this is the correct answer.
Gestational transient thyrotoxicosis is a nonautoimmune condition that results in transient hyperthyroidism of variable severity.6 Usually, it occurs in otherwise normal pregnancies without complications, but the initial manifestation is hyper- emesis.6 It can be differentiated from Graves disease by the absence of TSH receptor antibodies and by no history of thyroid disorder.7 Common symptoms of gestational transient thyrotoxicosis include weight loss (or failure to gain weight), tachycardia, and fatigue.
The reason for the transient rise in T4 may be that beta-hCG is structurally similar to TSH (and also to luteinizing hormone and follicle-stimulating hormone), so that it has mild thyroid-stimulating effects.7 Sustained high levels of beta-hCG may in time give rise to the manifestations of thyrotoxicosis.
Molar pregnancy also can cause hyper-thyroidism via elevated levels of beta-hCG. However, twin pregnancy is more common and can produce sustained levels of beta-hCG above 100,000 IU/L. In most cases of twin pregnancy, the TSH level is decreased and the T4 level transiently elevated.6 The elevated beta-hCG and the subsequent thyrotropic manifestations are thought to be directly related, and symptoms resolve when beta-hCG levels go down.6
In most cases of hyperthyroidism in pregnancy, the acute condition can be managed by a short (≤ 2-month) course of a beta-blocker. In rare cases, propylthiouracil treatment may be required. Gestational transient thyrotoxicosis is not associated with detrimental outcomes.
Case continued
Our patient’s TSH level is low and her free T4 and T3 levels are elevated. Her high beta-hCG level may be stimulating the thyroid gland and may account for the low TSH value, as well as for her tachycardia, emesis, shortness of breath, and weight loss.
After an obstetric consult, it is determined that our patient has a viable pregnancy. However, further investigation with transvaginal ultrasonography reveals that she has two viable, single-placenta, intrauterine gestations, separated by a thin chorionic membrane.
Beta-hCG and free T4 levels are significantly higher in twin pregnancies than in single pregnancies, especially in the early stages.6 In our patient, the twin pregnancy led to the elevated beta-hCG, which eventually manifested as thyrotoxicosis, which caused the shortness of breath, hyperemesis, weight loss, tachycardia, and nausea.
Shortness of breath in patients with thyrotoxicosis is well recognized but not well explained. It may be caused by decreased lung compliance, engorged capillaries in the lung, or left ventricular failure, as well as by chest pain due to increased myocardial demand or coronary artery vasospasm.4 The dyspnea is present at rest and during exertion, and the high metabolic rate is thought to lead to an inappropriate response of the ventilatory system.3,8
WHAT TREATMENT?
4. How would you treat this patient at this point?
- No drug therapy, just supportive care
- Propranolol (Inderal)
- Levothyroxine
- Propylthiouracil
Several types of drugs are used to manage hyperthyroidism.
Antithyroid drugs such as propylthiouracil, methimazole (Northyx, Tapazole), and carbimazole block thyroid hormone synthesis by inhibiting thyroid peroxidase. Propylthiouracil also blocks peripheral conversion of T4 to T3. Side effects of these agents include abnormal sense of taste, pruritus, urticaria, agranulocytosis, and hepatotoxicity.4
Usually, hyperthyroidism is treated with propylthiouracil at the smallest effective dose. This has been proven to be safe to the fetus and mother during pregnancy.9 Propylthiouracil and the other drugs in its class cross the placenta, but propylthiouracil crosses at one-quarter the rate of the other two.9
Beta-blockers are effective in the acute phase of thyrotoxicosis against tachycardia, hypertension, and atrial fibrillation. They also decrease conversion of T4 to T3, which is an added benefit. Beta-blockers can be tapered as thyroid hormone levels decrease.
A short course of a short-acting beta-blocker would be an option for our patient and would decrease her symptoms, although she does not have the typical markedly elevated T4 or T3 levels. In the long term, a beta-blocker would present a fetal risk, but short courses can be tolerated without incident.9
Radioactive iodine 131 is used in patients with Graves disease. 131Iodine therapy is safe for most adults, but in pregnancy its use is contraindicated. Fetal thyroid tissue is thought to be present after 10 weeks of gestation and could be damaged by the use of radioactive iodine. Another warning with the use of radioactive iodine is that patients should avoid close contact with other adults for a few days after treatment, and should avoid close contact with children and pregnant women for 2 to 3 weeks after treatment because of the risk of exposure to radiation emanating from the thyroid gland.
Levothyroxine is a treatment for hypothyroidism, not hyperthyroidism.
CASE CONTINUED
Our patient is treated with propranolol and monitored for several days in the hospital, during which her symptoms markedly improve. She is discharged without complications.
TAKE-HOME POINTS
The evaluation of shortness of breath in adult patients can be difficult, given the many possible causes. It is especially challenging in pregnant patients, since normal physiologic changes of pregnancy may produce these symptoms.
In many instances, cardiomyopathy must be suspected if a pregnant patient complains of shortness of breath. However, it is not the only possible cause.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
- Dobbenga-Rhodes YA, Prive AM. Assessment and evaluation of the woman with cardiac disease during pregnancy. J Perinat Neonatal Nurs 2006; 20:295–302.
- Carman TL, Deitcher SR. Advances in diagnosing and excluding pulmonary embolism: spiral CT and D-dimer measurement. Cleve Clin J Med 2002; 69:721–729.
- Wells PS, Anderson DR, Rodger M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost 2000; 83:416–420.
- Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am 2006; 35:663–686.
- Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am 1993; 22:263–277.
- Grün JP, Meuris S, De Nayer P, Glinoer D. The thyrotrophic role of human chorionic gonadotrophin (hCG) in the early stages of twin (versus single) pregnancies. Clin Endocrinol (Oxf) 1997; 46:719–725.
- Glinoer D, De Nayer P, Robyn C, Lejeune B, Kinthaert J, Meuris S. Serum levels of intact human chorionic gonadotropin (HCG) and its free alpha and beta subunits, in relation to maternal thyroid stimulation during normal pregnancy. J Endocrinol Invest 1993; 16:881–888.
- Small D, Gibbons W, Levy RD, de Lucas P, Gregory W, Cosio MG. Exertional dyspnea and ventilation in hyper-thyroidism. Chest 1992; 101:1268–1273.
- Atkins P, Cohen SB, Phillips BJ. Drug therapy for hyper-thyroidism in pregnancy: safety issues for mother and fetus. Drug Saf 2000; 23:229–244.
A case of refractory diarrhea
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
A 68-year-old white woman with irritable bowel syndrome has had worsening symptoms of right-sided abdominal pain, excessive bloating, and loose stools. Her bowel movements have increased from one a day to two or three a day. She has not noted any mucus or blood in the stool. She cannot identify any alleviating or aggravating factors, and the pain is not related to eating.
She consumes a normal diet, including meat and dairy. Over-the-counter antidiarrheal medications do not relieve the symptoms. She has had no fevers, chills, or night sweats, and she has not lost weight over the past year.
Her medical history includes breast cancer (in remission), alcohol abuse (in remission), and hypothyroidism, osteoporosis, and supraventricular tachycardia, all controlled with treatment as noted below. She has never undergone abdominal surgery.
A general review of systems is normal. Her current medications include oxybutynin (available as Ditropan, others), calcium polycarbophil (FiberCon, others), risedronate (Actonel), levothyroxine (Synthroid, others), simethicone (Maalox Anti-Gas, others), atenolol (Tenormin), trazodone (Desyrel), a calcium supplement, and aspirin. She began taking duloxetine (Cymbalta) 18 months ago, and the dose was increased from 60 mg to 90 mg 1 week before this visit.
She has never smoked, and she has abstained from alcohol for 10 years. She has no family history of colon cancer, celiac disease, or inflammatory bowel disease. She has not traveled outside the country in the past several years, and she notes no change in her source of drinking water.
On physical examination, she does not appear to be in acute distress. Her pulse is 64 and her blood pressure is 112/78 mm Hg. The cardiopulmonary examination is normal. Her abdomen is soft, symmetrical, nondistended, and nontender. Bowel sounds are normal. No abdominal masses, palpable organomegaly, or abdominal bruits are noted.
Results of basic laboratory tests, including thyroid-stimulating hormone (TSH), complete blood count, blood chemistries, renal function, and liver function, are normal. Colonoscopy shows normal mucosa as far as the cecum.
DIFFERENTIAL DIAGNOSIS
1. In addition to irritable bowel syndrome, which of these can explain her symptoms?
- Ulcerative colitis
- Celiac disease
- Microscopic colitis
- Hyperthyroidism
- Lactase deficiency
Ulcerative colitis typically presents with blood and mucus in the stool and gross abnormalities on colonoscopy, none of which is present in this patient.
Hyperthyroidism can be ruled out by the normal TSH level.
Lactase deficiency or lactose intolerance is unlikely because it is present in only 15% of people of northern European descent (compared with 80% of blacks and Hispanics and up to 100% of Native Americans and Asians).1 Furthermore, her pain is apparently not related to consuming dairy products.
The hydrogen breath test can aid in the diagnosis of lactase deficiency. This test relies on the breakdown of malabsorbed lactose by colonic flora. This is the most widely used test for this deficiency, but its high false-negative rate of 25% means that a negative result does not exclude the diagnosis and should not be relied on in working up a patient with chronic diarrhea.2 Simply noting whether symptoms develop after ingesting 50 g of lactose is clinically useful when lactase deficiency is suspected.
Based on the information so far, it is reasonable in this patient to evaluate for celiac disease and for microscopic colitis.
Celiac disease, also called gluten-sensitive enteropathy, has a varied presentation that includes nonspecific symptoms such as those in this patient. Classically, it causes diarrhea, but patients may present with a single nutrient deficiency and no diarrhea.
This patient lacks the elevated alkaline phosphatase or evidence of vitamin deficiencies characteristic of malabsorption in celiac disease (ie, vitamins A, B12, D, K, and folate)3. She also lacks evidence of malnutrition, such as iron deficiency anemia, weight loss, or low serum albumin. Finally, she does not have the dermatitis herpetiformis rash to suggest autoimmune gluten-sensitive enteropathy, nor does she have evidence of follicular hyperplasia or petechiae due to vitamin malabsorption.3
Because no single serologic test is ideal for diagnosing gluten-sensitive enteropathy, several tests are typically used: immunoglobulin A (IgA) antigliadin antibody, IgG antigliadin antibody, IgA antitransglutaminase antibody, and IgA antiendomysial antibody. IgA antitransglutaminase antibody is 92% to 98% sensitive and 91% to 100% specific for celiac disease. IgG antigliadin antibody is 92% to 97% sensitive and 99% specific. The positive predictive value of the IgA and IgG antigliadin antibody tests is less than 2% in the general population, whereas the positive predictive value for antiendomysial antibody and antitransglutaminase antibody are 15.7% and 21.8%, respectively.4 A positive serologic test for antiendomysial antibody is nearly 100% specific.
Our patient’s entire celiac antibody panel is negative, and thus celiac disease is unlikely.
Case continued: Features of microscopic colitis
In our patient, colonic biopsy reveals a mildly expanded lamina propria, intraepithelial lymphocytes, and a patchy but prominent thickening of the subepithelial collagen table. This set of features is consistent with collagenous colitis, a variant of microscopic colitis. Histologic signs on biopsy specimens are fairly specific for the disease.5
Chronic, intermittent, secretory diarrhea without bleeding is the hallmark of microscopic colitis. Associated symptoms may include abdominal pain, weight loss, and fatigue. If biopsies are not taken at the time of the initial evaluation, and the colonic pathology is overlooked, patients with collagenous colitis may be diagnosed with irritable bowel syndrome with diarrhea.6 The sedimentation rate is often elevated, and the antinuclear antibody test can be positive.7 Steatorrhea or protein-losing enteropathy can occur, and fecal leukocytes are present in more than 50% of patients.8
This patient fits well the demographics of the typical collagenous colitis patient: ie, a middle-aged woman in her 6th decade in otherwise good general health. The female-to-male ratio is 15:1 overall, although the relative frequency of collagenous colitis in women is greater than that of lymphocytic colitis.9 In a population-based study, the incidence of collagenous colitis was 5.1 per 100,000 per year, with a prevalence of 36 per 100,000; the incidence of lymphocytic colitis was 9.8 per 100,000 per year, with a prevalence of 64 per 100,000.10
Symptoms are typically vague and range from an annoyance to more than 20 non-bloody stools per day. The course of the disease also varies. Case series have reported a spontaneous remission rate of 15% to 20%,11 though flare-ups are common. Microscopic colitis is largely a benign disease. It does not increase a person’s risk of colon cancer.
CAUSES OF COLLAGENOUS COLITIS
2. What causes of collagenous colitis have been identified?
- Alcohol abuse
- Previous gastrointestinal surgery
- Drug-induced injury to colon
Neither alcohol use nor previous gastrointestinal surgery has been associated with the development of collagenous colitis.
Collagenous colitis has, however, been linked to several causes. Abnormal collagen metabolism has been demonstrated in patients as a result of increased expression of procollagen I and metalloproteinase inhibitor TIMP-1.12 Bacterial toxins and a bile-acid malabsorption defect in the terminal ileum and subsequent exposure of the colon to high concentrations of bile acids have also been linked to the development of collagenous colitis.
Many drugs have been linked to the development of collagenous colitis. Damage to the large intestine related to the use of non-steroidal anti-inflammatory drugs has been attributed to the blockage of prostaglandin synthesis.13 Simvastatin (Zocor), lansoprazole (Prilosec), and ticlopidine (Ticlid) have been linked to collagenous colitis; ticlopidine, flutamide (Eulexin), gold salts, lansoprazole, and sertraline (Zoloft) have been linked to the development of lymphocytic colitis.14 In one small series, patients developed colitis after switching from omeprazole (Prevacid) to lansoprazole. All patients had their symptoms and biopsy findings resolve within 1 week of stopping the drug.15
WHICH DRUG IS BEST?
3. Which drug is best for microscopic colitis, based on the current evidence?
- Bismuth (eg, Kaopectate, Pepto-Bismol)
- Sulfasalazine (Sulfazine)
- Budesonide (Entocort)
- Prednisolone
Studies have evaluated bismuth subsalicylate, Boswellia serrata extract, probiotics, prednisolone, budesonide, and other drugs for treating collagenous colitis.16
Bismuth trials have been small. In an open-label study of bismuth,17 symptoms improved in 11 of 12 patients.
Prednisolone recipients had a trend towards clinical response with treatment vs placebo, but it was not statistically significant, and there was incomplete remission of disease.18
Boswellia serrataextract19 and probiotics20 showed no clinical improvement.
Cholestyramine has been shown to be helpful when used in conjunction with an anti-inflammatory agent,21 and it may be helpful when used alone.
Aminosalicylate compounds have not been tested in prospective randomized trials, even though they are the cornerstone of treatment for ulcerative colitis. Retrospective trials have been equivocal.22
Budesonide currently has the best evidence of efficacy in collagenous colitis,23,24 and some evidence suggests it is also effective for other variants of microscopic colitis.
A total of 94 patients were enrolled in three placebo-controlled trials of budesonide at 9 mg daily or on a tapering schedule for 6 to 8 weeks. The pooled odds ratio for clinical response to treatment with budesonide was 12.32 (95% confidence interval 5.53–27.46), with a number needed to treat of 1.58. Significant histologic improvement with treatment was noted in all three trials.23
Quality of life has also been studied in patients with microscopic colitis who take budesonide. Symptoms, emotional functioning, and physical functioning are improved. Budesonide also improved stool consistency and significantly reduced the mean stool frequency compared with placebo.24
Compared with cortisol, budesonide has a 200 times greater affinity for the glucocorticoid receptor, and a 1,000 times greater topical anti-inflammatory potency. It is also well absorbed in the gastrointestinal tract but is substantially modified into very weak metabolites as a result of first-pass metabolism in the liver.25 This localized effect further supports the use of budesonide in patients with any form of microscopic colitis.
Although studies have shown budesonide to be effective, not every patient with a histologic diagnosis of microscopic colitis needs it. It is reasonable to try antidiarrheal agents, bismuth, or both as a first step because they are inexpensive and have few side effects. If budesonide is used, it should be given for 6 to 8 weeks, then stopped, and the patient should then be monitored for symptom recurrence. If a flare does occur, budesonide can be restarted and continued as maintenance therapy.
KEY CONSIDERATIONS
Microscopic colitis is diagnosed histologically, while irritable bowel syndrome is a clinical diagnosis. In population-based cohorts of histologically confirmed microscopic colitis, 50% to 70% met symptom-based Rome criteria for the diagnosis of irritable bowel syndrome. The clinical symptom-based criteria for irritable bowel syndrome are not specific enough to rule out the diagnosis of microscopic colitis. Therefore, patients with suspected diarrhea-predominant irritable bowel syndrome should undergo colonoscopy with biopsy to investigate microscopic colitis if symptoms are not well controlled by antidiarrheal therapy.26 The patient’s management may be very different depending on whether colonoscopy is done.
Management of microscopic colitis should include stopping any drugs associated with it. Simple antidiarrheal agents should be tried first to manage symptoms. If symptoms persist, patients can be treated with budesonide (Entocort EC) 9 mg by mouth daily for 8 weeks to induce remission, or 6 mg by mouth daily for 3 months as maintenance therapy.
OUR PATIENT’S COURSE
Our patient’s medication list includes duloxetine, a serotonin-norepinephrine reuptake inhibitor related to drugs that have been associated with the development of microscopic colitis. We tapered the duloxetine, and her symptoms improved by 50%. Her symptoms were eventually controlled after an 8-week course of oral budesonide 9 mg and ongoing intermittent use of loperamide (Imodium).
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.
- Swagerty DL, Walling AD, Klein RM. Lactose intolerance. Am Fam Physician 2002; 65:1845–1856.
- Thomas PD, Forbes A, Green J, et al. Guidelines for the investigation of chronic diarrhea, 2nd edition. Gut 2003; 52(suppl 5):1–5.
- Nelsen DA. Gluten-sensitive enteropathy (celiac disease): more common than you think. Am Fam Physician 2002; 66:2259–2266.
- Bardella MT, Trovato C, Cesana BM, Pagliari C, Gebbia C, Peracchi M. Serological markers for coeliac disease: is it time to change? Dig Liver Dis 2001; 33:426–431.
- Barta Z, Mekkel G, Csipo I, et al. Micropscopic colitis: a retrospective study of clinical presentation in 53 patients. World J Gastroenterol 2005; 11:1351–1355.
- Tremaine WJ. Diagnosing collagenous colitis: does it make a difference? Eur J Gastroenterol Hepatol 1999; 11:477–479.
- Bohr J, Tysk C, Yang P, Danielsson D, Järnerot G. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996; 39:77–81.
- Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc 1995; 70:430–433.
- Olsen M, Eriksson S, Bohr J, Järnerot G, Tysk C. Lymphocytic colitis: a retrospective clinical study of 199 Swedish patients. Gut 2004; 53:536–541.
- Pardi DS. Microscopic colitis: an update. Inflamm Bowel Dis 2004; 10:860–870.
- Fernandez-Banares F, Salas A, Esteve M, Espinos J, Forne M, Viver JM. Collagenous and lymphocytic colitis: evaluation of clinical and histological features, response to treatment, and long-term follow-up. Am J Gastroenterol 2003; 98:340–347.
- Aignet T, Neureiter D, Müller S, Küspert G, Belke J, Kirchner T. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology 1997; 113:136–143.
- Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol 2007; 38:527–536.
- Fernández-Bañares F, Esteve M, Espinós JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol 2007; 102:324–330.
- Thomson RD, Lestine LS, Bensen SP, et al. Lansoprazole-associated microscopic colitis: a case series. Am J Gastroenterol 2002; 97:2908–2913.
- Chande N, McDonald JWD, MacDonald JK. Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Group. Interventions for treating collagenous colitis. Cochrane Database Syst Rev 2007 Jan 24;(1):CD006096.
- Fine K, Lee E. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology 1998; 114:29–36.
- Munck LK, Kjeldsen J, Philipsen E, Fscher Hansen B. Incomplete remission with short-term prednisolone treatment in collagenous colitis: a randomized study. Scand J Gastroenterol 2003; 38:606–610.
- Madisch A, Miehlke S, Eichele E, et al. Boswellia serrata extract for the treatment of collagenous colitis: a randomized, double-blind, placebo-controlled, multicenter trial. Int J Colorectal Dis 2007; 22:1445–1451.
- Wildt S, Munck LK, Vinter-Jensen L, et al. Probiotic treatment of collagenous colitis: a randomized, double-blind, placebo-controlled trial with Lactobacillus acidophilus and Bifidobacterium animalis subsp. lactis. Inflamm Bowel Dis 2006; 12:395–401.
- Calabrese C, Fabbri A, Areni A, Zahlane D, Scialpi C, Di Febo G. Mesalazine with or without cholestyramine in the treatment of microscopic colitis: randomized controlled trial. J Gastroenterol Hepatol 2007; 22:809–814.
- Wall GC, Schirmer LL, Page MJ. Pharmacotherapy for microscopic colitis. Pharmacotherapy 2007; 27:425–433.
- Feyen B, Wall GC, Finnerty EP, DeWitt JE, Reyes RS. Meta-analysis: budesonide treatment for collagenous colitis. Aliment Pharmacol Ther 2004; 20:745–749.
- Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. Int J Colorectal Dis 2005; 20:312–316.
- Craig CR, editor. Modern Pharmacology With Clinical Application. 6th edition. Philadelphia: Lippincott Williams and Wilkins, 2003:481.
- Limsui D, Pardi DS, Camilleri M, et al. Symptomatic overlap between irritable bowel syndrome and microscopic colitis. Inflamm Bowel Dis 2007; 13:175–181.