User login
Pseudo-Ludwig angina
An 83-year-old woman with hypertension, hypothyroidism, and a history of depression presented to the emergency department with acute shortness of breath and hypoxia. She was found to have submassive pulmonary embolism, and a heparin infusion was started immediately.
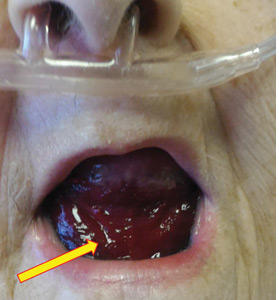
 After 48 hours of heparin infusion, the patient developed violaceous swelling at the floor of the oral cavity. (B) At 2 months after anticoagulation was stopped, the sublingual hematoma had completely resolved.")
Urgent nasopharyngeal laryngoscopy revealed a hematoma at the base of her tongue that extended into the vallecula, piriform sinuses, and aryepiglottic fold, causing acute airway obstruction. These features combined with the supratherapeutic aPTT led to the diagnosis of pseudo-Ludwig angina.
DANGER OF RAPID AIRWAY COMPROMISE
Pseudo-Ludwig angina is a rare condition in which over-anticoagulation causes sublingual swelling leading to airway obstruction, whereas true Ludwig angina is an infectious regional suppuration of the neck.
Most reported cases of pseudo-Ludwig angina have resulted from overanticogulation with warfarin or warfarin-like substances (rodenticides), or from coagulopathy due to liver disease.1–3 Early recognition is essential to avoid airway compromise.
In our patient, all anticoagulation was discontinued, and she was intubated until the hematoma began to resolve, the aPTT returned to normal, and respiratory compromise improved. At follow-up 2 months later, the sublingual hematoma had completely resolved (Figure 1). And at a 6-month follow-up visit, the pulmonary embolism had resolved, and pulmonary pressures by 2-dimensional echocardiography were normal.
- Lovallo E, Patterson S, Erickson M, Chin C, Blanc P, Durrani TS. When is “pseudo-Ludwig’s angina” associated with coagulopathy also a “pseudo” hemorrhage? J Investig Med High Impact Case Rep 2013; 1(2):2324709613492503. doi:10.1177/2324709613492503
- Smith RG, Parker TJ, Anderson TA. Noninfectious acute upper airway obstruction (pseudo-Ludwig phenomenon): report of a case. J Oral Maxillofac Surg 1987; 45(8):701–704. pmid:3475442
- Zacharia GS, Kandiyil S, Thomas V. Pseudo-Ludwig's phenomenon: a rare clinical manifestation in liver cirrhosis. ACG Case Rep J 2014; 2(1):53–54. doi:10.14309/crj.2014.83
An 83-year-old woman with hypertension, hypothyroidism, and a history of depression presented to the emergency department with acute shortness of breath and hypoxia. She was found to have submassive pulmonary embolism, and a heparin infusion was started immediately.
Urgent nasopharyngeal laryngoscopy revealed a hematoma at the base of her tongue that extended into the vallecula, piriform sinuses, and aryepiglottic fold, causing acute airway obstruction. These features combined with the supratherapeutic aPTT led to the diagnosis of pseudo-Ludwig angina.
DANGER OF RAPID AIRWAY COMPROMISE
Pseudo-Ludwig angina is a rare condition in which over-anticoagulation causes sublingual swelling leading to airway obstruction, whereas true Ludwig angina is an infectious regional suppuration of the neck.
Most reported cases of pseudo-Ludwig angina have resulted from overanticogulation with warfarin or warfarin-like substances (rodenticides), or from coagulopathy due to liver disease.1–3 Early recognition is essential to avoid airway compromise.
In our patient, all anticoagulation was discontinued, and she was intubated until the hematoma began to resolve, the aPTT returned to normal, and respiratory compromise improved. At follow-up 2 months later, the sublingual hematoma had completely resolved (Figure 1). And at a 6-month follow-up visit, the pulmonary embolism had resolved, and pulmonary pressures by 2-dimensional echocardiography were normal.
An 83-year-old woman with hypertension, hypothyroidism, and a history of depression presented to the emergency department with acute shortness of breath and hypoxia. She was found to have submassive pulmonary embolism, and a heparin infusion was started immediately.
Urgent nasopharyngeal laryngoscopy revealed a hematoma at the base of her tongue that extended into the vallecula, piriform sinuses, and aryepiglottic fold, causing acute airway obstruction. These features combined with the supratherapeutic aPTT led to the diagnosis of pseudo-Ludwig angina.
DANGER OF RAPID AIRWAY COMPROMISE
Pseudo-Ludwig angina is a rare condition in which over-anticoagulation causes sublingual swelling leading to airway obstruction, whereas true Ludwig angina is an infectious regional suppuration of the neck.
Most reported cases of pseudo-Ludwig angina have resulted from overanticogulation with warfarin or warfarin-like substances (rodenticides), or from coagulopathy due to liver disease.1–3 Early recognition is essential to avoid airway compromise.
In our patient, all anticoagulation was discontinued, and she was intubated until the hematoma began to resolve, the aPTT returned to normal, and respiratory compromise improved. At follow-up 2 months later, the sublingual hematoma had completely resolved (Figure 1). And at a 6-month follow-up visit, the pulmonary embolism had resolved, and pulmonary pressures by 2-dimensional echocardiography were normal.
- Lovallo E, Patterson S, Erickson M, Chin C, Blanc P, Durrani TS. When is “pseudo-Ludwig’s angina” associated with coagulopathy also a “pseudo” hemorrhage? J Investig Med High Impact Case Rep 2013; 1(2):2324709613492503. doi:10.1177/2324709613492503
- Smith RG, Parker TJ, Anderson TA. Noninfectious acute upper airway obstruction (pseudo-Ludwig phenomenon): report of a case. J Oral Maxillofac Surg 1987; 45(8):701–704. pmid:3475442
- Zacharia GS, Kandiyil S, Thomas V. Pseudo-Ludwig's phenomenon: a rare clinical manifestation in liver cirrhosis. ACG Case Rep J 2014; 2(1):53–54. doi:10.14309/crj.2014.83
- Lovallo E, Patterson S, Erickson M, Chin C, Blanc P, Durrani TS. When is “pseudo-Ludwig’s angina” associated with coagulopathy also a “pseudo” hemorrhage? J Investig Med High Impact Case Rep 2013; 1(2):2324709613492503. doi:10.1177/2324709613492503
- Smith RG, Parker TJ, Anderson TA. Noninfectious acute upper airway obstruction (pseudo-Ludwig phenomenon): report of a case. J Oral Maxillofac Surg 1987; 45(8):701–704. pmid:3475442
- Zacharia GS, Kandiyil S, Thomas V. Pseudo-Ludwig's phenomenon: a rare clinical manifestation in liver cirrhosis. ACG Case Rep J 2014; 2(1):53–54. doi:10.14309/crj.2014.83
FDA update: Higher late mortality with paclitaxel-coated devices
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.

This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Infective endocarditis: Beyond the usual tests
Prompt diagnois of infective endocarditis is critical. Potential consequences of missed or delayed diagnosis, including heart failure, stroke, intracardiac abscess, conduction delays, prosthesis dysfunction, and cerebral emboli, are often catastrophic. Echocardiography is the test used most frequently to evaluate for infective endocarditis, but it misses the diagnosis in almost one-third of cases, and even more often if the patient has a prosthetic valve.

But now, several sophisticated imaging tests are available that complement echocardiography in diagnosing and assessing infective endocarditis; these include 4-dimensional computed tomography (4D CT), fluorodeoxyglucose positron emission tomography (FDG-PET), and leukocyte scintigraphy. These tests have greatly improved our ability not only to diagnose infective endocarditis, but also to determine the extent and spread of infection, and they aid in perioperative assessment. Abnormal findings on these tests have been incorporated into the European Society of Cardiology’s 2015 modified diagnostic criteria for infective endocarditis.1
This article details the indications, advantages, and limitations of the various imaging tests for diagnosing and evaluating infective endocarditis (Table 1).
INFECTIVE ENDOCARDITIS IS DIFFICULT TO DIAGNOSE AND TREAT
Infective endocarditis is difficult to diagnose and treat. Clinical and imaging clues can be subtle, and the diagnosis requires a high level of suspicion and visualization of cardiac structures.
Further, the incidence of infective endocarditis is on the rise in the United States, particularly in women and young adults, likely due to intravenous drug use.2,3
ECHOCARDIOGRAPHY HAS AN IMPORTANT ROLE, BUT IS LIMITED
Echocardiography remains the most commonly performed study for diagnosing infective endocarditis, as it is fast, widely accessible, and less expensive than other imaging tests.
Transthoracic echocardiography (TTE) is often the first choice for testing. However, its sensitivity is only about 70% for detecting vegetations on native valves and 50% for detecting vegetations on prosthetic valves.1 It is inherently constrained by the limited number of views by which a comprehensive external evaluation of the heart can be achieved. Using a 2-dimensional instrument to view a 3-dimensional object is difficult, and depending on several factors, it can be hard to see vegetations and abscesses that are associated with infective endocarditis. Further, TTE is impeded by obesity and by hyperinflated lungs from obstructive pulmonary disease or mechanical ventilation. It has poor sensitivity for detecting small vegetations and for detecting vegetations and paravalvular complications in patients who have a prosthetic valve or a cardiac implanted electronic device.
Transesophageal echocardiography (TEE) is the recommended first-line imaging test for patients with prosthetic valves and no contraindications to the test. Otherwise, it should be done after TTE if the results of TTE are negative but clinical suspicion for infective endocarditis remains high (eg, because the patient uses intravenous drugs). But although TEE has a higher sensitivity than TTE (up to 96% for vegetations on native valves and 92% for those on prosthetic valves, if performed by an experienced sonographer), it can still miss infective endocarditis. Also, TEE does not provide a significant advantage over TTE in patients who have a cardiac implanted electronic device.1,4,5
Regardless of whether TTE or TEE is used, they are estimated to miss up to 30% of cases of infective endocarditis and its sequelae.4 False-negative findings are likelier in patients who have preexisting severe valvular lesions, prosthetic valves, cardiac implanted electronic devices, small vegetations, or abscesses, or if a vegetation has already broken free and embolized. Furthermore, distinguishing between vegetations and thrombi, cardiac tumors, and myxomatous changes using echocardiography is difficult.
CARDIAC CT
For patients who have inconclusive results on echocardiography, contraindications to TEE, or poor sonic windows, cardiac CT can be an excellent alternative. It is especially useful in the setting of a prosthetic valve.
Synchronized (“gated”) with the patient’s heart rate and rhythm, CT machines can acquire images during diastole, reducing motion artifact, and can create 3D images of the heart. In addition, newer machines can acquire several images at different points in the heart cycle to add a fourth dimension—time. The resulting 4D images play like short video loops of the beating heart and allow noninvasive assessment of cardiac anatomy with remarkable detail and resolution.
4D CT is increasingly being used in infective endocarditis, and growing evidence indicates that its accuracy is similar to that of TEE in the preoperative evaluation of patients with aortic prosthetic valve endocarditis.6 In a study of 28 patients, complementary use of CT angiography led to a change in treatment strategy in 7 (25%) compared with routine clinical workup.7 Several studies have found no difference between 4D CT and preoperative TEE in detecting pseudoaneurysm, abscess, or valve dehiscence. TEE and 4D CT also have similar sensitivities for detecting infective endocarditis in native and prosthetic valves.8,9

 angiography with iodinated contrast, including 4D reconstruc-tion, in the same patient, however, shows an 11-mm vegetation on the bioprosthetic aortic valve leaflets (arrow).")
 in the same patient confirms the diagnosis, showing a 13-mm hypermetabolic focus on the prosthetic valve (arrow), yielding the diagnosis of infectious endocarditis.")
Coupled with CT angiography, 4D CT is also an excellent noninvasive way to perioperatively evaluate the coronary arteries without the risks associated with catheterization in those requiring nonemergency surgery (Figure 1A, B, and C).
4D CT performs well for detecting abscess and pseudoaneurysm but has slightly lower sensitivity for vegetations than TEE (91% vs 99%).9
Gated CT, PET, or both may be useful in cases of suspected prosthetic aortic valve endocarditis when TEE is negative. Pseudoaneurysms are not well visualized with TEE, and the atrial mitral curtain area is often thickened on TEE in cases of aortic prosthetic valve infective endocarditis that do not definitely involve abscesses. Gated CT and PET show this area better.8 This information is important in cases in which a surgeon may be unconvinced that the patient has prosthetic valve endocarditis.
Limitations of 4D cardiac CT
4D CT with or without angiography has limitations. It requires a wide-volume scanner and an experienced reader.
Patients with irregular heart rhythms or uncontrolled tachycardia pose technical problems for image acquisition. Cardiac CT is typically gated (ie, images are obtained within a defined time period) to acquire images during diastole. Ideally, images are acquired when the heart is in mid to late diastole, a time of minimal cardiac motion, so that motion artifact is minimized. To estimate the timing of image acquisition, the cardiac cycle must be predictable, and its duration should be as long as possible. Tachycardia or irregular rhythms such as frequent ectopic beats or atrial fibrillation make acquisition timing difficult, and thus make it nearly impossible to accurately obtain images when the heart is at minimum motion, limiting assessment of cardiac structures or the coronary tree.4,10
Extensive coronary calcification can hinder assessment of the coronary tree by CT coronary angiography.
Contrast exposure may limit the use of CT in some patients (eg, those with contrast allergies or renal dysfunction). However, modern scanners allow for much smaller contrast boluses without decreasing sensitivity.
4D CT involves radiation exposure, especially when done with angiography, although modern scanners have greatly reduced exposure. The average radiation dose in CT coronary angiography is 2.9 to 5.9 mSv11 compared with 7 mSv in diagnostic cardiac catheterization (without angioplasty or stenting) or 16 mSv in routine CT of the abdomen and pelvis with contrast.12,13 In view of the morbidity and mortality risks associated with infective endocarditis, especially if the diagnosis is delayed, this small radiation exposure may be justifiable.
Bottom line for cardiac CT
4D CT is an excellent alternative to echocardiography for select patients. Clinicians should strongly consider this study in the following situations:
- Patients with a prosthetic valve
- Patients who are strongly suspected of having infective endocarditis but who have a poor sonic window on TTE or TEE, as can occur with chronic obstructive lung disease, morbid obesity, or previous thoracic or cardiovascular surgery
- Patients who meet clinical indications for TEE, such as having a prosthetic valve or a high suspicion for native valve infective endocarditis with negative TTE, but who have contraindications to TEE
- As an alternative to TEE for preoperative evaluation in patients with known infective endocarditis.
Patients with tachycardia or irregular heart rhythms are not good candidates for this test.
FDG-PET AND LEUKOCYTE SCINTIGRAPHY
FDG-PET and leukocyte scintigraphy are other options for diagnosing infective endocarditis and determining the presence and extent of intra- and extracardiac infection. They are more sensitive than echocardiography for detecting infection of cardiac implanted electronic devices such as ventricular assist devices, pacemakers, implanted cardiac defibrillators, and cardiac resynchronization therapy devices.14–16
The utility of FDG-PET is founded on the uptake of 18F-fluorodeoxyglucose by cells, with higher uptake taking place in cells with higher metabolic activity (such as in areas of inflammation). Similarly, leukocyte scintigraphy relies on the use of radiolabeled leukocytes (ie, leukocytes previously extracted from the patient, labelled, and re-introduced into the patient) to allow for localization of inflamed tissue.
The most significant contribution of FDG-PET may be the ability to detect infective endocarditis early, when echocardiography is initially negative. When abnormal FDG uptake was included in the modified Duke criteria, it increased the sensitivity to 97% for detecting infective endocarditis on admission, leading some to propose its incorporation as a major criterion.17 In patients with prosthetic valves and suspected infective endocarditis, FDG-PET was found in one study to have a sensitivity of up to 91% and a specificity of up to 95%.18
Both FDG-PET and leukocyte scintigraphy have a high sensitivity, specificity, and negative predictive value for cardiac implanted electronic device infection, and should be strongly considered in patients in whom it is suspected but who have negative or inconclusive findings on echocardiography.14,15
In addition, a common conundrum faced by clinicians with use of echocardiography is the difficulty of differentiating thrombus from infected vegetation on valves or device lead wires. Some evidence indicates that FDG-PET may help to discriminate between vegetation and thrombus, although more rigorous studies are needed before its use for that purpose can be recommended.19
Limitations of nuclear studies
Both FDG-PET and leukocyte scintigraphy perform poorly for detecting native-valve infective endocarditis. In a study in which 90% of the patients had native-valve infective endocarditis according to the Duke criteria, FDG-PET had a specificity of 93% but a sensitivity of only 39%.20
Both studies can be cumbersome, laborious, and time-consuming for patients. FDG-PET requires a fasting or glucose-restricted diet before testing, and the test itself can be complicated by development of hyperglycemia, although this is rare.
While FDG-PET is most effective in detecting infections of prosthetic valves and cardiac implanted electronic devices, the results can be falsely positive in patients with a history of recent cardiac surgery (due to ongoing tissue healing), as well as maladies other than infective endocarditis that lead to inflammation, such as vasculitis or malignancy. Similarly, for unclear reasons, leukocyte scintigraphy can yield false-negative results in patients with enterococcal or candidal infective endocarditis.21
FDG-PET and leukocyte scintigraphy are more expensive than TEE and cardiac CT22 and are not widely available.
Both tests entail radiation exposure, with the average dose ranging from 7 to 14 mSv. However, this is less than the average amount acquired during percutaneous coronary intervention (16 mSv), and overlaps with the amount in chest CT with contrast when assessing for pulmonary embolism (7 to 9 mSv). Lower doses are possible with optimized protocols.12,13,15,23
Bottom line for nuclear studies

FDG-PET and leukocyte scintigraphy are especially useful for patients with a prosthetic valve or cardiac implanted electronic device. However, limitations must be kept in mind.
A suggested algorithm for testing with nuclear imaging is shown in Figure 2.1,4
CEREBRAL MAGNETIC RESONANCE IMAGING
Cerebral magnetic resonance imaging (MRI) is more sensitive than cerebral CT for detecting emboli in the brain. According to American Heart Association guidelines, cerebral MRI should be done in patients with known or suspected infective endocarditis and neurologic impairment, defined as headaches, meningeal symptoms, or neurologic deficits. It is also often used in neurologically asymptomatic patients with infective endocarditis who have indications for valve surgery to assess for mycotic aneurysms, which are associated with increased intracranial bleeding during surgery.
MRI use in other asymptomatic patients remains controversial.24 In cases with high clinical suspicion for infective endocarditis and no findings on echocardiography, cerebral MRI can increase the sensitivity of the Duke criteria by adding a minor criterion. Some have argued that, in patients with definite infective endocarditis, detecting silent cerebral complications can lead to management changes. However, more studies are needed to determine if there is indeed a group of neurologically asymptomatic infective endocarditis patients for whom cerebral MRI leads to improved outcomes.
Limitations of cerebral MRI
Cerebral MRI cannot be used in patients with non-MRI-compatible implanted hardware.
Gadolinium, the contrast agent typically used, can cause nephrogenic systemic fibrosis in patients who have poor renal function. This rare but serious adverse effect is characterized by irreversible systemic fibrosis affecting skin, muscles, and even visceral tissue such as lungs. The American College of Radiology allows for gadolinium use in patients without acute kidney injury and patients with stable chronic kidney disease with a glomerular filtration rate of at least 30 mL/min/1.73 m2. Its use should be avoided in patients with renal failure on replacement therapy, with advanced chronic kidney disease (glomerular filtration rate < 30 mL/min/1.73 m2), or with acute kidney injury, even if they do not need renal replacement therapy.25
Concerns have also been raised about gadolinium retention in the brain, even in patients with normal renal function.26–28 Thus far, no conclusive clinical adverse effects of retention have been found, although more study is warranted. Nevertheless, the US Food and Drug Administration now requires a black-box warning about this possibility and advises clinicians to counsel patients appropriately.
Bottom line on cerebral MRI
Cerebral MRI should be obtained when a patient presents with definite or possible infective endocarditis with neurologic impairment, such as new headaches, meningismus, or focal neurologic deficits. Routine brain MRI in patients with confirmed infective endocarditis without neurologic symptoms, or those without definite infective endocarditis, is discouraged.
CARDIAC MRI
Cardiac MRI, typically obtained with gadolinium contrast, allows for better 3D assessment of cardiac structures and morphology than echocardiography or CT, and can detect infiltrative cardiac disease, myopericarditis, and much more. It is increasingly used in the field of structural cardiology, but its role for evaluating infective endocarditis remains unclear.
Cardiac MRI does not appear to be better than echocardiography for diagnosing infective endocarditis. However, it may prove helpful in the evaluation of patients known to have infective endocarditis but who cannot be properly evaluated for disease extent because of poor image quality on echocardiography and contraindications to CT.1,29 Its role is limited in patients with cardiac implanted electronic devices, as most devices are incompatible with MRI use, although newer devices obviate this concern. But even for devices that are MRI-compatible, results are diminished due to an eclipsing effect, wherein the device parts can make it hard to see structures clearly because the “brightness” basically eclipses the surrounding area.4
Concerns regarding use of gadolinium as described above need also be considered.
The role of cardiac MRI in diagnosing and managing infective endocarditis may evolve, but at present, the 2017 American College of Cardiology and American Heart Association appropriate-use criteria discourage its use for these purposes.16
Bottom line for cardiac MRI
Cardiac MRI to evaluate a patient for suspected infective endocarditis is not recommended due to lack of superiority compared with echocardiography or CT, and the risk of nephrogenic systemic fibrosis from gadolinium in patients with renal compromise.
- Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Durante-Mangoni E, Bradley S, Selton-Suty C, et al; International Collaboration on Endocarditis Prospective Cohort Study Group. Current features of infective endocarditis in elderly patients: results of the International Collaboration on Endocarditis Prospective Cohort Study. Arch Intern Med 2008; 168(19):2095–2103. doi:10.1001/archinte.168.19.2095
- Wurcel AG, Anderson JE, Chui KK, et al. Increasing infectious endocarditis admissions among young people who inject drugs. Open Forum Infect Dis 2016; 3(3):ofw157. doi:10.1093/ofid/ofw157
- Gomes A, Glaudemans AW, Touw DJ, et al. Diagnostic value of imaging in infective endocarditis: a systematic review. Lancet Infect Dis 2017; 17(1):e1–e14. doi:10.1016/S1473-3099(16)30141-4
- Cahill TJ, Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Fagman E, Perrotta S, Bech-Hanssen O, et al. ECG-gated computed tomography: a new role for patients with suspected aortic prosthetic valve endocarditis. Eur Radiol 2012; 22(11):2407–2414. doi:10.1007/s00330-012-2491-5
- Habets J, Tanis W, van Herwerden LA, et al. Cardiac computed tomography angiography results in diagnostic and therapeutic change in prosthetic heart valve endocarditis. Int J Cardiovasc Imaging 2014; 30(2):377–387. doi:10.1007/s10554-013-0335-2
- Koneru S, Huang SS, Oldan J, et al. Role of preoperative cardiac CT in the evaluation of infective endocarditis: comparison with transesophageal echocardiography and surgical findings. Cardiovasc Diagn Ther 2018; 8(4):439–449. doi:10.21037/cdt.2018.07.07
- Koo HJ, Yang DH, Kang J, et al. Demonstration of infective endocarditis by cardiac CT and transoesophageal echocardiography: comparison with intra-operative findings. Eur Heart J Cardiovasc Imaging 2018; 19(2):199–207. doi:10.1093/ehjci/jex010
- Feuchtner GM, Stolzmann P, Dichtl W, et al. Multislice computed tomography in infective endocarditis: comparison with transesophageal echocardiography and intraoperative findings. J Am Coll Cardiol 2009; 53(5):436–444. doi:10.1016/j.jacc.2008.01.077
- Castellano IA, Nicol ED, Bull RK, Roobottom CA, Williams MC, Harden SP. A prospective national survey of coronary CT angiography radiation doses in the United Kingdom. J Cardiovasc Comput Tomogr 2017; 11(4):268–273. doi:10.1016/j.jcct.2017.05.002
- Mettler FA Jr, Huda W, Yoshizumi TT, Mahesh M. Effective doses in radiology and diagnostic nuclear medicine: a catalog. Radiology 2008; 248(1):254–263. doi:10.1148/radiol.2481071451
- Smith-Bindman R, Lipson J, Marcus R, et al. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med 2009; 169(22):2078–2086. doi:10.1001/archinternmed.2009.427
- Ploux S, Riviere A, Amraoui S, et al. Positron emission tomography in patients with suspected pacing system infections may play a critical role in difficult cases. Heart Rhythm 2011; 8(9):1478–1481. doi:10.1016/j.hrthm.2011.03.062
- Sarrazin J, Philippon F, Tessier M, et al. Usefulness of fluorine-18 positron emission tomography/computed tomography for identification of cardiovascular implantable electronic device infections. J Am Coll Cardiol 2012; 59(18):1616–1625. doi:10.1016/j.jacc.2011.11.059
- Doherty JU, Kort S, Mehran R, Schoenhagen P, Soman P; Rating Panel Members; Appropriate Use Criteria Task Force. ACC/AATS/AHA/ASE/ASNC/HRS/SCAI/SCCT/SCMR/STS 2017 Appropriate use criteria for multimodality imaging in valvular heart disease: a report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and Society of Thoracic Surgeons. J Nucl Cardiol 2017; 24(6):2043–2063. doi:10.1007/s12350-017-1070-1
- Saby L, Laas O, Habib G, et al. Positron emission tomography/computed tomography for diagnosis of prosthetic valve endocarditis: increased valvular 18F-fluorodeoxyglucose uptake as a novel major criterion. J Am Coll Cardiol 2013; 61(23):2374–2382. doi:10.1016/j.jacc.2013.01.092
- Swart LE, Gomes A, Scholtens AM, et al. Improving the diagnostic performance of 18F-fluorodeoxyglucose positron-emission tomography/computed tomography in prosthetic heart valve endocarditis. Circulation 2018; 138(14):1412–1427. doi:10.1161/CIRCULATIONAHA.118.035032
- Graziosi M, Nanni C, Lorenzini M, et al. Role of 18F-FDG PET/CT in the diagnosis of infective endocarditis in patients with an implanted cardiac device: a prospective study. Eur J Nucl Med Mol Imaging 2014; 41(8):1617–1623. doi:10.1007/s00259-014-2773-z
- Kouijzer IJ, Vos FJ, Janssen MJ, van Dijk AP, Oyen WJ, Bleeker-Rovers CP. The value of 18F-FDG PET/CT in diagnosing infectious endocarditis. Eur J Nucl Med Mol Imaging 2013; 40(7):1102–1107. doi:10.1007/s00259-013-2376-0
- Wong D, Rubinshtein R, Keynan Y. Alternative cardiac imaging modalities to echocardiography for the diagnosis of infective endocarditis. Am J Cardiol 2016; 118(9):1410–1418. doi:10.1016/j.amjcard.2016.07.053
- Vos FJ, Bleeker-Rovers CP, Kullberg BJ, Adang EM, Oyen WJ. Cost-effectiveness of routine (18)F-FDG PET/CT in high-risk patients with gram-positive bacteremia. J Nucl Med 2011; 52(11):1673–1678. doi:10.2967/jnumed.111.089714
- McCollough CH, Bushberg JT, Fletcher JG, Eckel LJ. Answers to common questions about the use and safety of CT scans. Mayo Clin Proc 2015; 90(10):1380–1392. doi:10.1016/j.mayocp.2015.07.011
- Duval X, Iung B, Klein I, et al; IMAGE (Resonance Magnetic Imaging at the Acute Phase of Endocarditis) Study Group. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: a prospective study. Ann Intern Med 2010; 152(8):497–504, W175. doi:10.7326/0003-4819-152-8-201004200-00006
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media: 2018. www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf. Accessed July 19, 2019.
- Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology 2015; 276(1):228–232. doi:10.1148/radiol.2015142690
- McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial gadolinium deposition after contrast-enhanced MR imaging. Radiology 2015; 275(3):772–782. doi:10.1148/radiol.15150025
- Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology 2014; 270(3):834–841. doi:10.1148/radiol.13131669
- Expert Panel on Pediatric Imaging; Hayes LL, Palasis S, Bartel TB, et al. ACR appropriateness criteria headache-child. J Am Coll Radiol 2018; 15(5S):S78–S90. doi:10.1016/j.jacr.2018.03.017
Prompt diagnois of infective endocarditis is critical. Potential consequences of missed or delayed diagnosis, including heart failure, stroke, intracardiac abscess, conduction delays, prosthesis dysfunction, and cerebral emboli, are often catastrophic. Echocardiography is the test used most frequently to evaluate for infective endocarditis, but it misses the diagnosis in almost one-third of cases, and even more often if the patient has a prosthetic valve.
But now, several sophisticated imaging tests are available that complement echocardiography in diagnosing and assessing infective endocarditis; these include 4-dimensional computed tomography (4D CT), fluorodeoxyglucose positron emission tomography (FDG-PET), and leukocyte scintigraphy. These tests have greatly improved our ability not only to diagnose infective endocarditis, but also to determine the extent and spread of infection, and they aid in perioperative assessment. Abnormal findings on these tests have been incorporated into the European Society of Cardiology’s 2015 modified diagnostic criteria for infective endocarditis.1
This article details the indications, advantages, and limitations of the various imaging tests for diagnosing and evaluating infective endocarditis (Table 1).
INFECTIVE ENDOCARDITIS IS DIFFICULT TO DIAGNOSE AND TREAT
Infective endocarditis is difficult to diagnose and treat. Clinical and imaging clues can be subtle, and the diagnosis requires a high level of suspicion and visualization of cardiac structures.
Further, the incidence of infective endocarditis is on the rise in the United States, particularly in women and young adults, likely due to intravenous drug use.2,3
ECHOCARDIOGRAPHY HAS AN IMPORTANT ROLE, BUT IS LIMITED
Echocardiography remains the most commonly performed study for diagnosing infective endocarditis, as it is fast, widely accessible, and less expensive than other imaging tests.
Transthoracic echocardiography (TTE) is often the first choice for testing. However, its sensitivity is only about 70% for detecting vegetations on native valves and 50% for detecting vegetations on prosthetic valves.1 It is inherently constrained by the limited number of views by which a comprehensive external evaluation of the heart can be achieved. Using a 2-dimensional instrument to view a 3-dimensional object is difficult, and depending on several factors, it can be hard to see vegetations and abscesses that are associated with infective endocarditis. Further, TTE is impeded by obesity and by hyperinflated lungs from obstructive pulmonary disease or mechanical ventilation. It has poor sensitivity for detecting small vegetations and for detecting vegetations and paravalvular complications in patients who have a prosthetic valve or a cardiac implanted electronic device.
Transesophageal echocardiography (TEE) is the recommended first-line imaging test for patients with prosthetic valves and no contraindications to the test. Otherwise, it should be done after TTE if the results of TTE are negative but clinical suspicion for infective endocarditis remains high (eg, because the patient uses intravenous drugs). But although TEE has a higher sensitivity than TTE (up to 96% for vegetations on native valves and 92% for those on prosthetic valves, if performed by an experienced sonographer), it can still miss infective endocarditis. Also, TEE does not provide a significant advantage over TTE in patients who have a cardiac implanted electronic device.1,4,5
Regardless of whether TTE or TEE is used, they are estimated to miss up to 30% of cases of infective endocarditis and its sequelae.4 False-negative findings are likelier in patients who have preexisting severe valvular lesions, prosthetic valves, cardiac implanted electronic devices, small vegetations, or abscesses, or if a vegetation has already broken free and embolized. Furthermore, distinguishing between vegetations and thrombi, cardiac tumors, and myxomatous changes using echocardiography is difficult.
CARDIAC CT
For patients who have inconclusive results on echocardiography, contraindications to TEE, or poor sonic windows, cardiac CT can be an excellent alternative. It is especially useful in the setting of a prosthetic valve.
Synchronized (“gated”) with the patient’s heart rate and rhythm, CT machines can acquire images during diastole, reducing motion artifact, and can create 3D images of the heart. In addition, newer machines can acquire several images at different points in the heart cycle to add a fourth dimension—time. The resulting 4D images play like short video loops of the beating heart and allow noninvasive assessment of cardiac anatomy with remarkable detail and resolution.
4D CT is increasingly being used in infective endocarditis, and growing evidence indicates that its accuracy is similar to that of TEE in the preoperative evaluation of patients with aortic prosthetic valve endocarditis.6 In a study of 28 patients, complementary use of CT angiography led to a change in treatment strategy in 7 (25%) compared with routine clinical workup.7 Several studies have found no difference between 4D CT and preoperative TEE in detecting pseudoaneurysm, abscess, or valve dehiscence. TEE and 4D CT also have similar sensitivities for detecting infective endocarditis in native and prosthetic valves.8,9
Coupled with CT angiography, 4D CT is also an excellent noninvasive way to perioperatively evaluate the coronary arteries without the risks associated with catheterization in those requiring nonemergency surgery (Figure 1A, B, and C).
4D CT performs well for detecting abscess and pseudoaneurysm but has slightly lower sensitivity for vegetations than TEE (91% vs 99%).9
Gated CT, PET, or both may be useful in cases of suspected prosthetic aortic valve endocarditis when TEE is negative. Pseudoaneurysms are not well visualized with TEE, and the atrial mitral curtain area is often thickened on TEE in cases of aortic prosthetic valve infective endocarditis that do not definitely involve abscesses. Gated CT and PET show this area better.8 This information is important in cases in which a surgeon may be unconvinced that the patient has prosthetic valve endocarditis.
Limitations of 4D cardiac CT
4D CT with or without angiography has limitations. It requires a wide-volume scanner and an experienced reader.
Patients with irregular heart rhythms or uncontrolled tachycardia pose technical problems for image acquisition. Cardiac CT is typically gated (ie, images are obtained within a defined time period) to acquire images during diastole. Ideally, images are acquired when the heart is in mid to late diastole, a time of minimal cardiac motion, so that motion artifact is minimized. To estimate the timing of image acquisition, the cardiac cycle must be predictable, and its duration should be as long as possible. Tachycardia or irregular rhythms such as frequent ectopic beats or atrial fibrillation make acquisition timing difficult, and thus make it nearly impossible to accurately obtain images when the heart is at minimum motion, limiting assessment of cardiac structures or the coronary tree.4,10
Extensive coronary calcification can hinder assessment of the coronary tree by CT coronary angiography.
Contrast exposure may limit the use of CT in some patients (eg, those with contrast allergies or renal dysfunction). However, modern scanners allow for much smaller contrast boluses without decreasing sensitivity.
4D CT involves radiation exposure, especially when done with angiography, although modern scanners have greatly reduced exposure. The average radiation dose in CT coronary angiography is 2.9 to 5.9 mSv11 compared with 7 mSv in diagnostic cardiac catheterization (without angioplasty or stenting) or 16 mSv in routine CT of the abdomen and pelvis with contrast.12,13 In view of the morbidity and mortality risks associated with infective endocarditis, especially if the diagnosis is delayed, this small radiation exposure may be justifiable.
Bottom line for cardiac CT
4D CT is an excellent alternative to echocardiography for select patients. Clinicians should strongly consider this study in the following situations:
- Patients with a prosthetic valve
- Patients who are strongly suspected of having infective endocarditis but who have a poor sonic window on TTE or TEE, as can occur with chronic obstructive lung disease, morbid obesity, or previous thoracic or cardiovascular surgery
- Patients who meet clinical indications for TEE, such as having a prosthetic valve or a high suspicion for native valve infective endocarditis with negative TTE, but who have contraindications to TEE
- As an alternative to TEE for preoperative evaluation in patients with known infective endocarditis.
Patients with tachycardia or irregular heart rhythms are not good candidates for this test.
FDG-PET AND LEUKOCYTE SCINTIGRAPHY
FDG-PET and leukocyte scintigraphy are other options for diagnosing infective endocarditis and determining the presence and extent of intra- and extracardiac infection. They are more sensitive than echocardiography for detecting infection of cardiac implanted electronic devices such as ventricular assist devices, pacemakers, implanted cardiac defibrillators, and cardiac resynchronization therapy devices.14–16
The utility of FDG-PET is founded on the uptake of 18F-fluorodeoxyglucose by cells, with higher uptake taking place in cells with higher metabolic activity (such as in areas of inflammation). Similarly, leukocyte scintigraphy relies on the use of radiolabeled leukocytes (ie, leukocytes previously extracted from the patient, labelled, and re-introduced into the patient) to allow for localization of inflamed tissue.
The most significant contribution of FDG-PET may be the ability to detect infective endocarditis early, when echocardiography is initially negative. When abnormal FDG uptake was included in the modified Duke criteria, it increased the sensitivity to 97% for detecting infective endocarditis on admission, leading some to propose its incorporation as a major criterion.17 In patients with prosthetic valves and suspected infective endocarditis, FDG-PET was found in one study to have a sensitivity of up to 91% and a specificity of up to 95%.18
Both FDG-PET and leukocyte scintigraphy have a high sensitivity, specificity, and negative predictive value for cardiac implanted electronic device infection, and should be strongly considered in patients in whom it is suspected but who have negative or inconclusive findings on echocardiography.14,15
In addition, a common conundrum faced by clinicians with use of echocardiography is the difficulty of differentiating thrombus from infected vegetation on valves or device lead wires. Some evidence indicates that FDG-PET may help to discriminate between vegetation and thrombus, although more rigorous studies are needed before its use for that purpose can be recommended.19
Limitations of nuclear studies
Both FDG-PET and leukocyte scintigraphy perform poorly for detecting native-valve infective endocarditis. In a study in which 90% of the patients had native-valve infective endocarditis according to the Duke criteria, FDG-PET had a specificity of 93% but a sensitivity of only 39%.20
Both studies can be cumbersome, laborious, and time-consuming for patients. FDG-PET requires a fasting or glucose-restricted diet before testing, and the test itself can be complicated by development of hyperglycemia, although this is rare.
While FDG-PET is most effective in detecting infections of prosthetic valves and cardiac implanted electronic devices, the results can be falsely positive in patients with a history of recent cardiac surgery (due to ongoing tissue healing), as well as maladies other than infective endocarditis that lead to inflammation, such as vasculitis or malignancy. Similarly, for unclear reasons, leukocyte scintigraphy can yield false-negative results in patients with enterococcal or candidal infective endocarditis.21
FDG-PET and leukocyte scintigraphy are more expensive than TEE and cardiac CT22 and are not widely available.
Both tests entail radiation exposure, with the average dose ranging from 7 to 14 mSv. However, this is less than the average amount acquired during percutaneous coronary intervention (16 mSv), and overlaps with the amount in chest CT with contrast when assessing for pulmonary embolism (7 to 9 mSv). Lower doses are possible with optimized protocols.12,13,15,23
Bottom line for nuclear studies
FDG-PET and leukocyte scintigraphy are especially useful for patients with a prosthetic valve or cardiac implanted electronic device. However, limitations must be kept in mind.
A suggested algorithm for testing with nuclear imaging is shown in Figure 2.1,4
CEREBRAL MAGNETIC RESONANCE IMAGING
Cerebral magnetic resonance imaging (MRI) is more sensitive than cerebral CT for detecting emboli in the brain. According to American Heart Association guidelines, cerebral MRI should be done in patients with known or suspected infective endocarditis and neurologic impairment, defined as headaches, meningeal symptoms, or neurologic deficits. It is also often used in neurologically asymptomatic patients with infective endocarditis who have indications for valve surgery to assess for mycotic aneurysms, which are associated with increased intracranial bleeding during surgery.
MRI use in other asymptomatic patients remains controversial.24 In cases with high clinical suspicion for infective endocarditis and no findings on echocardiography, cerebral MRI can increase the sensitivity of the Duke criteria by adding a minor criterion. Some have argued that, in patients with definite infective endocarditis, detecting silent cerebral complications can lead to management changes. However, more studies are needed to determine if there is indeed a group of neurologically asymptomatic infective endocarditis patients for whom cerebral MRI leads to improved outcomes.
Limitations of cerebral MRI
Cerebral MRI cannot be used in patients with non-MRI-compatible implanted hardware.
Gadolinium, the contrast agent typically used, can cause nephrogenic systemic fibrosis in patients who have poor renal function. This rare but serious adverse effect is characterized by irreversible systemic fibrosis affecting skin, muscles, and even visceral tissue such as lungs. The American College of Radiology allows for gadolinium use in patients without acute kidney injury and patients with stable chronic kidney disease with a glomerular filtration rate of at least 30 mL/min/1.73 m2. Its use should be avoided in patients with renal failure on replacement therapy, with advanced chronic kidney disease (glomerular filtration rate < 30 mL/min/1.73 m2), or with acute kidney injury, even if they do not need renal replacement therapy.25
Concerns have also been raised about gadolinium retention in the brain, even in patients with normal renal function.26–28 Thus far, no conclusive clinical adverse effects of retention have been found, although more study is warranted. Nevertheless, the US Food and Drug Administration now requires a black-box warning about this possibility and advises clinicians to counsel patients appropriately.
Bottom line on cerebral MRI
Cerebral MRI should be obtained when a patient presents with definite or possible infective endocarditis with neurologic impairment, such as new headaches, meningismus, or focal neurologic deficits. Routine brain MRI in patients with confirmed infective endocarditis without neurologic symptoms, or those without definite infective endocarditis, is discouraged.
CARDIAC MRI
Cardiac MRI, typically obtained with gadolinium contrast, allows for better 3D assessment of cardiac structures and morphology than echocardiography or CT, and can detect infiltrative cardiac disease, myopericarditis, and much more. It is increasingly used in the field of structural cardiology, but its role for evaluating infective endocarditis remains unclear.
Cardiac MRI does not appear to be better than echocardiography for diagnosing infective endocarditis. However, it may prove helpful in the evaluation of patients known to have infective endocarditis but who cannot be properly evaluated for disease extent because of poor image quality on echocardiography and contraindications to CT.1,29 Its role is limited in patients with cardiac implanted electronic devices, as most devices are incompatible with MRI use, although newer devices obviate this concern. But even for devices that are MRI-compatible, results are diminished due to an eclipsing effect, wherein the device parts can make it hard to see structures clearly because the “brightness” basically eclipses the surrounding area.4
Concerns regarding use of gadolinium as described above need also be considered.
The role of cardiac MRI in diagnosing and managing infective endocarditis may evolve, but at present, the 2017 American College of Cardiology and American Heart Association appropriate-use criteria discourage its use for these purposes.16
Bottom line for cardiac MRI
Cardiac MRI to evaluate a patient for suspected infective endocarditis is not recommended due to lack of superiority compared with echocardiography or CT, and the risk of nephrogenic systemic fibrosis from gadolinium in patients with renal compromise.
Prompt diagnois of infective endocarditis is critical. Potential consequences of missed or delayed diagnosis, including heart failure, stroke, intracardiac abscess, conduction delays, prosthesis dysfunction, and cerebral emboli, are often catastrophic. Echocardiography is the test used most frequently to evaluate for infective endocarditis, but it misses the diagnosis in almost one-third of cases, and even more often if the patient has a prosthetic valve.
But now, several sophisticated imaging tests are available that complement echocardiography in diagnosing and assessing infective endocarditis; these include 4-dimensional computed tomography (4D CT), fluorodeoxyglucose positron emission tomography (FDG-PET), and leukocyte scintigraphy. These tests have greatly improved our ability not only to diagnose infective endocarditis, but also to determine the extent and spread of infection, and they aid in perioperative assessment. Abnormal findings on these tests have been incorporated into the European Society of Cardiology’s 2015 modified diagnostic criteria for infective endocarditis.1
This article details the indications, advantages, and limitations of the various imaging tests for diagnosing and evaluating infective endocarditis (Table 1).
INFECTIVE ENDOCARDITIS IS DIFFICULT TO DIAGNOSE AND TREAT
Infective endocarditis is difficult to diagnose and treat. Clinical and imaging clues can be subtle, and the diagnosis requires a high level of suspicion and visualization of cardiac structures.
Further, the incidence of infective endocarditis is on the rise in the United States, particularly in women and young adults, likely due to intravenous drug use.2,3
ECHOCARDIOGRAPHY HAS AN IMPORTANT ROLE, BUT IS LIMITED
Echocardiography remains the most commonly performed study for diagnosing infective endocarditis, as it is fast, widely accessible, and less expensive than other imaging tests.
Transthoracic echocardiography (TTE) is often the first choice for testing. However, its sensitivity is only about 70% for detecting vegetations on native valves and 50% for detecting vegetations on prosthetic valves.1 It is inherently constrained by the limited number of views by which a comprehensive external evaluation of the heart can be achieved. Using a 2-dimensional instrument to view a 3-dimensional object is difficult, and depending on several factors, it can be hard to see vegetations and abscesses that are associated with infective endocarditis. Further, TTE is impeded by obesity and by hyperinflated lungs from obstructive pulmonary disease or mechanical ventilation. It has poor sensitivity for detecting small vegetations and for detecting vegetations and paravalvular complications in patients who have a prosthetic valve or a cardiac implanted electronic device.
Transesophageal echocardiography (TEE) is the recommended first-line imaging test for patients with prosthetic valves and no contraindications to the test. Otherwise, it should be done after TTE if the results of TTE are negative but clinical suspicion for infective endocarditis remains high (eg, because the patient uses intravenous drugs). But although TEE has a higher sensitivity than TTE (up to 96% for vegetations on native valves and 92% for those on prosthetic valves, if performed by an experienced sonographer), it can still miss infective endocarditis. Also, TEE does not provide a significant advantage over TTE in patients who have a cardiac implanted electronic device.1,4,5
Regardless of whether TTE or TEE is used, they are estimated to miss up to 30% of cases of infective endocarditis and its sequelae.4 False-negative findings are likelier in patients who have preexisting severe valvular lesions, prosthetic valves, cardiac implanted electronic devices, small vegetations, or abscesses, or if a vegetation has already broken free and embolized. Furthermore, distinguishing between vegetations and thrombi, cardiac tumors, and myxomatous changes using echocardiography is difficult.
CARDIAC CT
For patients who have inconclusive results on echocardiography, contraindications to TEE, or poor sonic windows, cardiac CT can be an excellent alternative. It is especially useful in the setting of a prosthetic valve.
Synchronized (“gated”) with the patient’s heart rate and rhythm, CT machines can acquire images during diastole, reducing motion artifact, and can create 3D images of the heart. In addition, newer machines can acquire several images at different points in the heart cycle to add a fourth dimension—time. The resulting 4D images play like short video loops of the beating heart and allow noninvasive assessment of cardiac anatomy with remarkable detail and resolution.
4D CT is increasingly being used in infective endocarditis, and growing evidence indicates that its accuracy is similar to that of TEE in the preoperative evaluation of patients with aortic prosthetic valve endocarditis.6 In a study of 28 patients, complementary use of CT angiography led to a change in treatment strategy in 7 (25%) compared with routine clinical workup.7 Several studies have found no difference between 4D CT and preoperative TEE in detecting pseudoaneurysm, abscess, or valve dehiscence. TEE and 4D CT also have similar sensitivities for detecting infective endocarditis in native and prosthetic valves.8,9
Coupled with CT angiography, 4D CT is also an excellent noninvasive way to perioperatively evaluate the coronary arteries without the risks associated with catheterization in those requiring nonemergency surgery (Figure 1A, B, and C).
4D CT performs well for detecting abscess and pseudoaneurysm but has slightly lower sensitivity for vegetations than TEE (91% vs 99%).9
Gated CT, PET, or both may be useful in cases of suspected prosthetic aortic valve endocarditis when TEE is negative. Pseudoaneurysms are not well visualized with TEE, and the atrial mitral curtain area is often thickened on TEE in cases of aortic prosthetic valve infective endocarditis that do not definitely involve abscesses. Gated CT and PET show this area better.8 This information is important in cases in which a surgeon may be unconvinced that the patient has prosthetic valve endocarditis.
Limitations of 4D cardiac CT
4D CT with or without angiography has limitations. It requires a wide-volume scanner and an experienced reader.
Patients with irregular heart rhythms or uncontrolled tachycardia pose technical problems for image acquisition. Cardiac CT is typically gated (ie, images are obtained within a defined time period) to acquire images during diastole. Ideally, images are acquired when the heart is in mid to late diastole, a time of minimal cardiac motion, so that motion artifact is minimized. To estimate the timing of image acquisition, the cardiac cycle must be predictable, and its duration should be as long as possible. Tachycardia or irregular rhythms such as frequent ectopic beats or atrial fibrillation make acquisition timing difficult, and thus make it nearly impossible to accurately obtain images when the heart is at minimum motion, limiting assessment of cardiac structures or the coronary tree.4,10
Extensive coronary calcification can hinder assessment of the coronary tree by CT coronary angiography.
Contrast exposure may limit the use of CT in some patients (eg, those with contrast allergies or renal dysfunction). However, modern scanners allow for much smaller contrast boluses without decreasing sensitivity.
4D CT involves radiation exposure, especially when done with angiography, although modern scanners have greatly reduced exposure. The average radiation dose in CT coronary angiography is 2.9 to 5.9 mSv11 compared with 7 mSv in diagnostic cardiac catheterization (without angioplasty or stenting) or 16 mSv in routine CT of the abdomen and pelvis with contrast.12,13 In view of the morbidity and mortality risks associated with infective endocarditis, especially if the diagnosis is delayed, this small radiation exposure may be justifiable.
Bottom line for cardiac CT
4D CT is an excellent alternative to echocardiography for select patients. Clinicians should strongly consider this study in the following situations:
- Patients with a prosthetic valve
- Patients who are strongly suspected of having infective endocarditis but who have a poor sonic window on TTE or TEE, as can occur with chronic obstructive lung disease, morbid obesity, or previous thoracic or cardiovascular surgery
- Patients who meet clinical indications for TEE, such as having a prosthetic valve or a high suspicion for native valve infective endocarditis with negative TTE, but who have contraindications to TEE
- As an alternative to TEE for preoperative evaluation in patients with known infective endocarditis.
Patients with tachycardia or irregular heart rhythms are not good candidates for this test.
FDG-PET AND LEUKOCYTE SCINTIGRAPHY
FDG-PET and leukocyte scintigraphy are other options for diagnosing infective endocarditis and determining the presence and extent of intra- and extracardiac infection. They are more sensitive than echocardiography for detecting infection of cardiac implanted electronic devices such as ventricular assist devices, pacemakers, implanted cardiac defibrillators, and cardiac resynchronization therapy devices.14–16
The utility of FDG-PET is founded on the uptake of 18F-fluorodeoxyglucose by cells, with higher uptake taking place in cells with higher metabolic activity (such as in areas of inflammation). Similarly, leukocyte scintigraphy relies on the use of radiolabeled leukocytes (ie, leukocytes previously extracted from the patient, labelled, and re-introduced into the patient) to allow for localization of inflamed tissue.
The most significant contribution of FDG-PET may be the ability to detect infective endocarditis early, when echocardiography is initially negative. When abnormal FDG uptake was included in the modified Duke criteria, it increased the sensitivity to 97% for detecting infective endocarditis on admission, leading some to propose its incorporation as a major criterion.17 In patients with prosthetic valves and suspected infective endocarditis, FDG-PET was found in one study to have a sensitivity of up to 91% and a specificity of up to 95%.18
Both FDG-PET and leukocyte scintigraphy have a high sensitivity, specificity, and negative predictive value for cardiac implanted electronic device infection, and should be strongly considered in patients in whom it is suspected but who have negative or inconclusive findings on echocardiography.14,15
In addition, a common conundrum faced by clinicians with use of echocardiography is the difficulty of differentiating thrombus from infected vegetation on valves or device lead wires. Some evidence indicates that FDG-PET may help to discriminate between vegetation and thrombus, although more rigorous studies are needed before its use for that purpose can be recommended.19
Limitations of nuclear studies
Both FDG-PET and leukocyte scintigraphy perform poorly for detecting native-valve infective endocarditis. In a study in which 90% of the patients had native-valve infective endocarditis according to the Duke criteria, FDG-PET had a specificity of 93% but a sensitivity of only 39%.20
Both studies can be cumbersome, laborious, and time-consuming for patients. FDG-PET requires a fasting or glucose-restricted diet before testing, and the test itself can be complicated by development of hyperglycemia, although this is rare.
While FDG-PET is most effective in detecting infections of prosthetic valves and cardiac implanted electronic devices, the results can be falsely positive in patients with a history of recent cardiac surgery (due to ongoing tissue healing), as well as maladies other than infective endocarditis that lead to inflammation, such as vasculitis or malignancy. Similarly, for unclear reasons, leukocyte scintigraphy can yield false-negative results in patients with enterococcal or candidal infective endocarditis.21
FDG-PET and leukocyte scintigraphy are more expensive than TEE and cardiac CT22 and are not widely available.
Both tests entail radiation exposure, with the average dose ranging from 7 to 14 mSv. However, this is less than the average amount acquired during percutaneous coronary intervention (16 mSv), and overlaps with the amount in chest CT with contrast when assessing for pulmonary embolism (7 to 9 mSv). Lower doses are possible with optimized protocols.12,13,15,23
Bottom line for nuclear studies
FDG-PET and leukocyte scintigraphy are especially useful for patients with a prosthetic valve or cardiac implanted electronic device. However, limitations must be kept in mind.
A suggested algorithm for testing with nuclear imaging is shown in Figure 2.1,4
CEREBRAL MAGNETIC RESONANCE IMAGING
Cerebral magnetic resonance imaging (MRI) is more sensitive than cerebral CT for detecting emboli in the brain. According to American Heart Association guidelines, cerebral MRI should be done in patients with known or suspected infective endocarditis and neurologic impairment, defined as headaches, meningeal symptoms, or neurologic deficits. It is also often used in neurologically asymptomatic patients with infective endocarditis who have indications for valve surgery to assess for mycotic aneurysms, which are associated with increased intracranial bleeding during surgery.
MRI use in other asymptomatic patients remains controversial.24 In cases with high clinical suspicion for infective endocarditis and no findings on echocardiography, cerebral MRI can increase the sensitivity of the Duke criteria by adding a minor criterion. Some have argued that, in patients with definite infective endocarditis, detecting silent cerebral complications can lead to management changes. However, more studies are needed to determine if there is indeed a group of neurologically asymptomatic infective endocarditis patients for whom cerebral MRI leads to improved outcomes.
Limitations of cerebral MRI
Cerebral MRI cannot be used in patients with non-MRI-compatible implanted hardware.
Gadolinium, the contrast agent typically used, can cause nephrogenic systemic fibrosis in patients who have poor renal function. This rare but serious adverse effect is characterized by irreversible systemic fibrosis affecting skin, muscles, and even visceral tissue such as lungs. The American College of Radiology allows for gadolinium use in patients without acute kidney injury and patients with stable chronic kidney disease with a glomerular filtration rate of at least 30 mL/min/1.73 m2. Its use should be avoided in patients with renal failure on replacement therapy, with advanced chronic kidney disease (glomerular filtration rate < 30 mL/min/1.73 m2), or with acute kidney injury, even if they do not need renal replacement therapy.25
Concerns have also been raised about gadolinium retention in the brain, even in patients with normal renal function.26–28 Thus far, no conclusive clinical adverse effects of retention have been found, although more study is warranted. Nevertheless, the US Food and Drug Administration now requires a black-box warning about this possibility and advises clinicians to counsel patients appropriately.
Bottom line on cerebral MRI
Cerebral MRI should be obtained when a patient presents with definite or possible infective endocarditis with neurologic impairment, such as new headaches, meningismus, or focal neurologic deficits. Routine brain MRI in patients with confirmed infective endocarditis without neurologic symptoms, or those without definite infective endocarditis, is discouraged.
CARDIAC MRI
Cardiac MRI, typically obtained with gadolinium contrast, allows for better 3D assessment of cardiac structures and morphology than echocardiography or CT, and can detect infiltrative cardiac disease, myopericarditis, and much more. It is increasingly used in the field of structural cardiology, but its role for evaluating infective endocarditis remains unclear.
Cardiac MRI does not appear to be better than echocardiography for diagnosing infective endocarditis. However, it may prove helpful in the evaluation of patients known to have infective endocarditis but who cannot be properly evaluated for disease extent because of poor image quality on echocardiography and contraindications to CT.1,29 Its role is limited in patients with cardiac implanted electronic devices, as most devices are incompatible with MRI use, although newer devices obviate this concern. But even for devices that are MRI-compatible, results are diminished due to an eclipsing effect, wherein the device parts can make it hard to see structures clearly because the “brightness” basically eclipses the surrounding area.4
Concerns regarding use of gadolinium as described above need also be considered.
The role of cardiac MRI in diagnosing and managing infective endocarditis may evolve, but at present, the 2017 American College of Cardiology and American Heart Association appropriate-use criteria discourage its use for these purposes.16
Bottom line for cardiac MRI
Cardiac MRI to evaluate a patient for suspected infective endocarditis is not recommended due to lack of superiority compared with echocardiography or CT, and the risk of nephrogenic systemic fibrosis from gadolinium in patients with renal compromise.
- Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Durante-Mangoni E, Bradley S, Selton-Suty C, et al; International Collaboration on Endocarditis Prospective Cohort Study Group. Current features of infective endocarditis in elderly patients: results of the International Collaboration on Endocarditis Prospective Cohort Study. Arch Intern Med 2008; 168(19):2095–2103. doi:10.1001/archinte.168.19.2095
- Wurcel AG, Anderson JE, Chui KK, et al. Increasing infectious endocarditis admissions among young people who inject drugs. Open Forum Infect Dis 2016; 3(3):ofw157. doi:10.1093/ofid/ofw157
- Gomes A, Glaudemans AW, Touw DJ, et al. Diagnostic value of imaging in infective endocarditis: a systematic review. Lancet Infect Dis 2017; 17(1):e1–e14. doi:10.1016/S1473-3099(16)30141-4
- Cahill TJ, Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Fagman E, Perrotta S, Bech-Hanssen O, et al. ECG-gated computed tomography: a new role for patients with suspected aortic prosthetic valve endocarditis. Eur Radiol 2012; 22(11):2407–2414. doi:10.1007/s00330-012-2491-5
- Habets J, Tanis W, van Herwerden LA, et al. Cardiac computed tomography angiography results in diagnostic and therapeutic change in prosthetic heart valve endocarditis. Int J Cardiovasc Imaging 2014; 30(2):377–387. doi:10.1007/s10554-013-0335-2
- Koneru S, Huang SS, Oldan J, et al. Role of preoperative cardiac CT in the evaluation of infective endocarditis: comparison with transesophageal echocardiography and surgical findings. Cardiovasc Diagn Ther 2018; 8(4):439–449. doi:10.21037/cdt.2018.07.07
- Koo HJ, Yang DH, Kang J, et al. Demonstration of infective endocarditis by cardiac CT and transoesophageal echocardiography: comparison with intra-operative findings. Eur Heart J Cardiovasc Imaging 2018; 19(2):199–207. doi:10.1093/ehjci/jex010
- Feuchtner GM, Stolzmann P, Dichtl W, et al. Multislice computed tomography in infective endocarditis: comparison with transesophageal echocardiography and intraoperative findings. J Am Coll Cardiol 2009; 53(5):436–444. doi:10.1016/j.jacc.2008.01.077
- Castellano IA, Nicol ED, Bull RK, Roobottom CA, Williams MC, Harden SP. A prospective national survey of coronary CT angiography radiation doses in the United Kingdom. J Cardiovasc Comput Tomogr 2017; 11(4):268–273. doi:10.1016/j.jcct.2017.05.002
- Mettler FA Jr, Huda W, Yoshizumi TT, Mahesh M. Effective doses in radiology and diagnostic nuclear medicine: a catalog. Radiology 2008; 248(1):254–263. doi:10.1148/radiol.2481071451
- Smith-Bindman R, Lipson J, Marcus R, et al. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med 2009; 169(22):2078–2086. doi:10.1001/archinternmed.2009.427
- Ploux S, Riviere A, Amraoui S, et al. Positron emission tomography in patients with suspected pacing system infections may play a critical role in difficult cases. Heart Rhythm 2011; 8(9):1478–1481. doi:10.1016/j.hrthm.2011.03.062
- Sarrazin J, Philippon F, Tessier M, et al. Usefulness of fluorine-18 positron emission tomography/computed tomography for identification of cardiovascular implantable electronic device infections. J Am Coll Cardiol 2012; 59(18):1616–1625. doi:10.1016/j.jacc.2011.11.059
- Doherty JU, Kort S, Mehran R, Schoenhagen P, Soman P; Rating Panel Members; Appropriate Use Criteria Task Force. ACC/AATS/AHA/ASE/ASNC/HRS/SCAI/SCCT/SCMR/STS 2017 Appropriate use criteria for multimodality imaging in valvular heart disease: a report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and Society of Thoracic Surgeons. J Nucl Cardiol 2017; 24(6):2043–2063. doi:10.1007/s12350-017-1070-1
- Saby L, Laas O, Habib G, et al. Positron emission tomography/computed tomography for diagnosis of prosthetic valve endocarditis: increased valvular 18F-fluorodeoxyglucose uptake as a novel major criterion. J Am Coll Cardiol 2013; 61(23):2374–2382. doi:10.1016/j.jacc.2013.01.092
- Swart LE, Gomes A, Scholtens AM, et al. Improving the diagnostic performance of 18F-fluorodeoxyglucose positron-emission tomography/computed tomography in prosthetic heart valve endocarditis. Circulation 2018; 138(14):1412–1427. doi:10.1161/CIRCULATIONAHA.118.035032
- Graziosi M, Nanni C, Lorenzini M, et al. Role of 18F-FDG PET/CT in the diagnosis of infective endocarditis in patients with an implanted cardiac device: a prospective study. Eur J Nucl Med Mol Imaging 2014; 41(8):1617–1623. doi:10.1007/s00259-014-2773-z
- Kouijzer IJ, Vos FJ, Janssen MJ, van Dijk AP, Oyen WJ, Bleeker-Rovers CP. The value of 18F-FDG PET/CT in diagnosing infectious endocarditis. Eur J Nucl Med Mol Imaging 2013; 40(7):1102–1107. doi:10.1007/s00259-013-2376-0
- Wong D, Rubinshtein R, Keynan Y. Alternative cardiac imaging modalities to echocardiography for the diagnosis of infective endocarditis. Am J Cardiol 2016; 118(9):1410–1418. doi:10.1016/j.amjcard.2016.07.053
- Vos FJ, Bleeker-Rovers CP, Kullberg BJ, Adang EM, Oyen WJ. Cost-effectiveness of routine (18)F-FDG PET/CT in high-risk patients with gram-positive bacteremia. J Nucl Med 2011; 52(11):1673–1678. doi:10.2967/jnumed.111.089714
- McCollough CH, Bushberg JT, Fletcher JG, Eckel LJ. Answers to common questions about the use and safety of CT scans. Mayo Clin Proc 2015; 90(10):1380–1392. doi:10.1016/j.mayocp.2015.07.011
- Duval X, Iung B, Klein I, et al; IMAGE (Resonance Magnetic Imaging at the Acute Phase of Endocarditis) Study Group. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: a prospective study. Ann Intern Med 2010; 152(8):497–504, W175. doi:10.7326/0003-4819-152-8-201004200-00006
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media: 2018. www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf. Accessed July 19, 2019.
- Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology 2015; 276(1):228–232. doi:10.1148/radiol.2015142690
- McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial gadolinium deposition after contrast-enhanced MR imaging. Radiology 2015; 275(3):772–782. doi:10.1148/radiol.15150025
- Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology 2014; 270(3):834–841. doi:10.1148/radiol.13131669
- Expert Panel on Pediatric Imaging; Hayes LL, Palasis S, Bartel TB, et al. ACR appropriateness criteria headache-child. J Am Coll Radiol 2018; 15(5S):S78–S90. doi:10.1016/j.jacr.2018.03.017
- Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015; 36(44):3075–3128. doi:10.1093/eurheartj/ehv319
- Durante-Mangoni E, Bradley S, Selton-Suty C, et al; International Collaboration on Endocarditis Prospective Cohort Study Group. Current features of infective endocarditis in elderly patients: results of the International Collaboration on Endocarditis Prospective Cohort Study. Arch Intern Med 2008; 168(19):2095–2103. doi:10.1001/archinte.168.19.2095
- Wurcel AG, Anderson JE, Chui KK, et al. Increasing infectious endocarditis admissions among young people who inject drugs. Open Forum Infect Dis 2016; 3(3):ofw157. doi:10.1093/ofid/ofw157
- Gomes A, Glaudemans AW, Touw DJ, et al. Diagnostic value of imaging in infective endocarditis: a systematic review. Lancet Infect Dis 2017; 17(1):e1–e14. doi:10.1016/S1473-3099(16)30141-4
- Cahill TJ, Baddour LM, Habib G, et al. Challenges in infective endocarditis. J Am Coll Cardiol 2017; 69(3):325–344. doi:10.1016/j.jacc.2016.10.066
- Fagman E, Perrotta S, Bech-Hanssen O, et al. ECG-gated computed tomography: a new role for patients with suspected aortic prosthetic valve endocarditis. Eur Radiol 2012; 22(11):2407–2414. doi:10.1007/s00330-012-2491-5
- Habets J, Tanis W, van Herwerden LA, et al. Cardiac computed tomography angiography results in diagnostic and therapeutic change in prosthetic heart valve endocarditis. Int J Cardiovasc Imaging 2014; 30(2):377–387. doi:10.1007/s10554-013-0335-2
- Koneru S, Huang SS, Oldan J, et al. Role of preoperative cardiac CT in the evaluation of infective endocarditis: comparison with transesophageal echocardiography and surgical findings. Cardiovasc Diagn Ther 2018; 8(4):439–449. doi:10.21037/cdt.2018.07.07
- Koo HJ, Yang DH, Kang J, et al. Demonstration of infective endocarditis by cardiac CT and transoesophageal echocardiography: comparison with intra-operative findings. Eur Heart J Cardiovasc Imaging 2018; 19(2):199–207. doi:10.1093/ehjci/jex010
- Feuchtner GM, Stolzmann P, Dichtl W, et al. Multislice computed tomography in infective endocarditis: comparison with transesophageal echocardiography and intraoperative findings. J Am Coll Cardiol 2009; 53(5):436–444. doi:10.1016/j.jacc.2008.01.077
- Castellano IA, Nicol ED, Bull RK, Roobottom CA, Williams MC, Harden SP. A prospective national survey of coronary CT angiography radiation doses in the United Kingdom. J Cardiovasc Comput Tomogr 2017; 11(4):268–273. doi:10.1016/j.jcct.2017.05.002
- Mettler FA Jr, Huda W, Yoshizumi TT, Mahesh M. Effective doses in radiology and diagnostic nuclear medicine: a catalog. Radiology 2008; 248(1):254–263. doi:10.1148/radiol.2481071451
- Smith-Bindman R, Lipson J, Marcus R, et al. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med 2009; 169(22):2078–2086. doi:10.1001/archinternmed.2009.427
- Ploux S, Riviere A, Amraoui S, et al. Positron emission tomography in patients with suspected pacing system infections may play a critical role in difficult cases. Heart Rhythm 2011; 8(9):1478–1481. doi:10.1016/j.hrthm.2011.03.062
- Sarrazin J, Philippon F, Tessier M, et al. Usefulness of fluorine-18 positron emission tomography/computed tomography for identification of cardiovascular implantable electronic device infections. J Am Coll Cardiol 2012; 59(18):1616–1625. doi:10.1016/j.jacc.2011.11.059
- Doherty JU, Kort S, Mehran R, Schoenhagen P, Soman P; Rating Panel Members; Appropriate Use Criteria Task Force. ACC/AATS/AHA/ASE/ASNC/HRS/SCAI/SCCT/SCMR/STS 2017 Appropriate use criteria for multimodality imaging in valvular heart disease: a report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, and Society of Thoracic Surgeons. J Nucl Cardiol 2017; 24(6):2043–2063. doi:10.1007/s12350-017-1070-1
- Saby L, Laas O, Habib G, et al. Positron emission tomography/computed tomography for diagnosis of prosthetic valve endocarditis: increased valvular 18F-fluorodeoxyglucose uptake as a novel major criterion. J Am Coll Cardiol 2013; 61(23):2374–2382. doi:10.1016/j.jacc.2013.01.092
- Swart LE, Gomes A, Scholtens AM, et al. Improving the diagnostic performance of 18F-fluorodeoxyglucose positron-emission tomography/computed tomography in prosthetic heart valve endocarditis. Circulation 2018; 138(14):1412–1427. doi:10.1161/CIRCULATIONAHA.118.035032
- Graziosi M, Nanni C, Lorenzini M, et al. Role of 18F-FDG PET/CT in the diagnosis of infective endocarditis in patients with an implanted cardiac device: a prospective study. Eur J Nucl Med Mol Imaging 2014; 41(8):1617–1623. doi:10.1007/s00259-014-2773-z
- Kouijzer IJ, Vos FJ, Janssen MJ, van Dijk AP, Oyen WJ, Bleeker-Rovers CP. The value of 18F-FDG PET/CT in diagnosing infectious endocarditis. Eur J Nucl Med Mol Imaging 2013; 40(7):1102–1107. doi:10.1007/s00259-013-2376-0
- Wong D, Rubinshtein R, Keynan Y. Alternative cardiac imaging modalities to echocardiography for the diagnosis of infective endocarditis. Am J Cardiol 2016; 118(9):1410–1418. doi:10.1016/j.amjcard.2016.07.053
- Vos FJ, Bleeker-Rovers CP, Kullberg BJ, Adang EM, Oyen WJ. Cost-effectiveness of routine (18)F-FDG PET/CT in high-risk patients with gram-positive bacteremia. J Nucl Med 2011; 52(11):1673–1678. doi:10.2967/jnumed.111.089714
- McCollough CH, Bushberg JT, Fletcher JG, Eckel LJ. Answers to common questions about the use and safety of CT scans. Mayo Clin Proc 2015; 90(10):1380–1392. doi:10.1016/j.mayocp.2015.07.011
- Duval X, Iung B, Klein I, et al; IMAGE (Resonance Magnetic Imaging at the Acute Phase of Endocarditis) Study Group. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: a prospective study. Ann Intern Med 2010; 152(8):497–504, W175. doi:10.7326/0003-4819-152-8-201004200-00006
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media: 2018. www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf. Accessed July 19, 2019.
- Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology 2015; 276(1):228–232. doi:10.1148/radiol.2015142690
- McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial gadolinium deposition after contrast-enhanced MR imaging. Radiology 2015; 275(3):772–782. doi:10.1148/radiol.15150025
- Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology 2014; 270(3):834–841. doi:10.1148/radiol.13131669
- Expert Panel on Pediatric Imaging; Hayes LL, Palasis S, Bartel TB, et al. ACR appropriateness criteria headache-child. J Am Coll Radiol 2018; 15(5S):S78–S90. doi:10.1016/j.jacr.2018.03.017
KEY POINTS
- Echocardiography can produce false-negative results in native-valve infective endocarditis and is even less sensitive in patients with a prosthetic valve or cardiac implanted electronic device.
- 4D CT is a reasonable alternative to transesophageal echocardiography. It can also be used as a second test if echocardiography is inconclusive. Coupled with angiography, it also provides a noninvasive method to evaluate coronary arteries perioperatively.
- Nuclear imaging tests—FDG-PET and leukocyte scintigraphy—increase the sensitivity of the Duke criteria for diagnosing infective endocarditis. They should be considered for evaluating suspected infective endocarditis in all patients who have a prosthetic valve or cardiac implanted electronic device, and whenever echocardiography is inconclusive and clinical suspicion remains high.
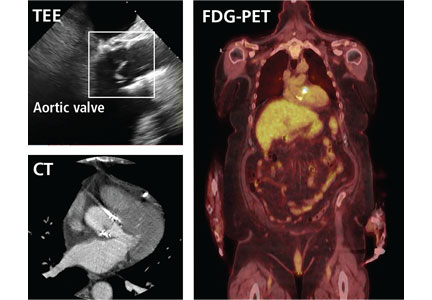
Giant cell arteritis: An updated review of an old disease
Giant cell arteritis (GCA) is a systemic vasculitis involving medium-sized and large arteries, most commonly the temporal, ophthalmic, occipital, vertebral, posterior ciliary, and proximal vertebral arteries. Moreover, involvement of the ophthalmic artery and its branches results in loss of vision. GCA can also involve the aorta and its proximal branches, especially in the upper extremities.
GCA is the most common systemic vasculitis in adults. It occurs almost exclusively in patients over age 50 and affects women more than men. It is most frequent in populations of northern European ancestry, especially Scandinavian. In a retrospective cohort study in Norway, the average annual cumulative incidence rate of GCA was 16.7 per 100,000 people over age 50.1 Risk factors include older age, history of smoking, current smoking, early menopause, and, possibly, stress-related disorders.2
PATHOGENESIS IS NOT COMPLETELY UNDERSTOOD
The pathogenesis of GCA is not completely understood, but there is evidence of immune activation in the arterial wall leading to activation of macrophages and formation of multinucleated giant cells (which may not always be present in biopsies).
The most relevant cytokines in the ongoing pathogenesis are still being defined, but the presence of interferon gamma and interleukin 6 (IL-6) seem to be critical for the expression of the disease. The primary immunogenic triggers for the elaboration of these cytokines and the arteritis remain elusive.
A SPECTRUM OF PRESENTATIONS
The initial symptoms of GCA may be vague, such as malaise, fever, and night sweats, and are likely due to systemic inflammation. Features of vascular involvement include headache, scalp tenderness, and jaw claudication (cramping pain in the jaw while chewing).
A less common but serious feature associated with GCA is partial or complete vision loss affecting 1 or both eyes.3 Some patients suddenly go completely blind without any visual prodrome.
Overlapping GCA phenotypes exist, with a spectrum of presentations that include classic cranial arteritis, extracranial GCA (also called large-vessel GCA), and polymyalgia rheumatica.2
Cranial GCA, the best-characterized clinical presentation, causes symptoms such as headache or signs such as tenderness of the temporal artery. On examination, the temporal arteries may be tender or nodular, and the pulses may be felt above the zygomatic arch, above and in front of the tragus of the ear. About two-thirds of patients with cranial GCA present with new-onset headache, most often in the temporal area, but possibly anywhere throughout the head.
Visual disturbance, jaw claudication, and tongue pain are less common but, if present, increase the likelihood of this diagnosis.2
Large-vessel involvement in GCA is common and refers to involvement of the aorta and its proximal branches. Imaging methods used in diagnosing large-vessel GCA include color Doppler ultrasonography, computed tomography with angiography, magnetic resonance imaging with angiography, and positron emission tomography. In some centers, such imaging is performed in all patients diagnosed with GCA to survey for large-vessel involvement.
Depending on the imaging study, large-vessel involvement has been found in 30% to 80% of cases of GCA.4,5 It is often associated with nonspecific symptoms such as fever, weight loss, chills, and malaise, but it can also cause more specific symptoms such as unilateral extremity claudication. In contrast to patients with cranial GCA, patients with large-vessel GCA were younger at onset, less likely to have headaches, and more likely to have arm claudication at presentation.6 Aortitis of the ascending aorta can occur with a histopathologic pattern of GCA but without the clinical stigmata of GCA.
The finding of aortitis should prompt the clinician to question the patient about other symptoms of GCA and to order imaging of the whole vascular tree. Ultrasonography and biopsy of the temporal arteries can be considered. Whether idiopathic aortitis is part of the GCA spectrum remains to be seen.
Laboratory tests often show anemia, leukocytosis, and thrombocytosis. Acute-phase reactants such as C-reactive protein and the erythrocyte sedimentation rate are often elevated. The sedimentation rate often exceeds 50 mm/hour and sometimes 100 mm/hour.
In 2 retrospective studies, the number of patients with GCA whose sedimentation rate was less than 50 mm/hour ranged between 5% and 11%.7,8 However, a small percentage of patients with GCA have normal inflammatory markers. Therefore, if the suspicion for GCA is high, treatment should be started and biopsy pursued.9 In patients with paraproteinemia or other causes of a spuriously elevated or low erythrocyte sedimentation rate, C-reactive protein is a more reliable test.
Polymyalgia rheumatica is another rheumatologic condition that can occur independently or in conjunction with GCA. It is characterized by stiffness and pain in the proximal joints such as the hips and shoulders, typically worse in the morning and better with activity. Although the patient may subjectively feel weak, a close neurologic examination will reveal normal muscle strength.
Polymyalgia rheumatica is observed in 40% to 60% of patients with GCA at the time of diagnosis; 16% to 21% of patients with polymyalgia rheumatica may develop GCA, especially if untreated.2,10
Differential diagnosis
Other vasculitides (eg, Takayasu arteritis) can also present with unexplained fever, anemia, and constitutional symptoms.
Infection should be considered if fever is present. An infectious disease accompanied by fever, headache, and elevated inflammatory markers can mimic GCA.
Nonarteritic anterior ischemic optic neuropathy can present with sudden vision loss, prompting concern for underlying GCA. Risk factors include hypertension and diabetes mellitus; other features of GCA, including elevated inflammatory markers, are generally absent.
TEMPORAL ARTERY BIOPSY: THE GOLD STANDARD FOR DIAGNOSIS
Temporal artery biopsy remains the standard to confirm the diagnosis. However, because inflammation in the temporal arteries can affect some segments but not others, biopsy results on conventional hematoxylin and eosin staining can be falsely negative in patients with GCA. In one study,11 the mean sensitivity of unilateral temporal artery biopsy was 86.9%.
Typical positive histologic findings are inflammation with panarteritis, CD4-positive lymphocytes, macrophages, giant cells, and fragmentation of the internal elastic lamina.12
When GCA is suspected, treatment with glucocorticoids should be started immediately and biopsy performed as soon as possible. Delaying biopsy for 14 days or more may not affect the accuracy of biopsy study.13 Treatment should never be withheld while awaiting the results of biopsy study.
Biopsy is usually performed unilaterally, on the same side as the symptoms or abnormal findings on examination. Bilateral temporal artery biopsy is also performed and compared with unilateral biopsy; this approach increases the diagnostic yield by about 5%.14
IMAGING
In patients with suspected GCA, imaging is recommended early to complement the clinical criteria for the diagnosis of GCA.15 Positron emission tomography, computed tomography angiography, magnetic resonance angiography, or Doppler ultrasonography can reveal inflammation of the arteries in the proximal upper or lower limbs or the aorta.2
In patients with suspected cranial GCA, ultrasonography of the temporal and axillary arteries is recommended first. If ultrasonography is not available or is inconclusive, high-resolution magnetic resonance imaging of the cranial arteries can be used as an alternative. Computed tomography and positron emission tomography of the cranial arteries are not recommended.
In patients with suspected large-vessel GCA, ultrasonography, positron emission tomography, computed tomography, and magnetic resonance imaging may be used to screen for vessel wall inflammation, edema, and luminal narrowing in extracranial arteries. Ultrasonography is of limited value in assessing aortitis.
Color duplex ultrasonography can be applied to assess for vascular inflammation of the temporal or large arteries. The typical finding of the “halo” sign, a hypoechoic ring around the arterial lumen, represents the inflammation-induced thickening of the arterial wall. The “compression sign,” the persistence of the “halo” during compression of the vessel lumen by the ultrasound probe, has high specificity for the diagnosis.16
Ultrasonography of suspected GCA has yielded sensitivities of 55% to 100% and specificities of 78% to 100%. However, its sensitivity depends on the user’s level of expertise, so it should be done only in medical centers with a high number of GCA cases and with highly experienced sonographers. High-resolution magnetic resonance imaging is an alternative to ultrasonography and has shown similar sensitivity and specificity.3
TREATMENT WITH GLUCOCORTICOIDS
Glucocorticoids remain the standard for treatment of GCA. The therapeutic effect of glucocorticoids in GCA has been established by years of clinical experience, but has never been proven in a placebo-controlled trial. When started appropriately and expeditiously, glucocorticoids produce exquisite resolution of signs and symptoms and prevent the serious complication of vision loss. Rapid resolution of symptoms is so typical of GCA that if the patient’s symptoms persist more than a few days after starting a glucocorticoid, the diagnosis of GCA should be reconsidered.
In a retrospective study of 245 patients with biopsy-proven GCA treated with glucocorticoids, 34 had permanent loss of sight.17 In 32 (94%) of the 34, the vision loss occurred before glucocorticoids were started. Of the remaining 2 patients, 1 lost vision 8 days into treatment, and the other lost vision 3 years after diagnosis and 1 year after discontinuation of glucocorticoids.
In a series of 144 patients with biopsy-proven GCA, 51 had no vision loss at presentation and no vision loss after starting glucocorticoids, and 93 had vision loss at presentation. In the latter group, symptoms worsened within 5 days of starting glucocorticoids in 9 patients.18 If vision was intact at the time of presentation, prompt initiation of glucocorticoids reduced the risk of vision loss to less than 1%.
High doses, slowly tapered
The European League Against Rheumatism recommends early initiation of high-dose glucocorticoids for patients with large-vessel vasculitis,19 and it also recommends glucocorticoids for patients with polymyalgia rheumatica.20 The optimal initial and tapering dosage has never been formally evaluated, but regimens have been devised on the basis of expert opinion.21
For patients with GCA who do not have vision loss at the time of diagnosis, the initial dose is prednisone 1 mg/kg or its equivalent daily for 2 to 4 weeks, after which it is tapered.21 If the initial dosage is prednisone 60 mg orally daily for 2 to 4 weeks, our practice is to taper it to 50 mg daily for 2 weeks, then 40 mg daily for 2 weeks. Then, it is decreased by 5 mg every 2 weeks until it is 20 mg daily, and then by 2.5 mg every 2 weeks until it is 10 mg orally daily. Thereafter, the dosage is decreased by 1 mg every 2 to 4 weeks.
For patients with GCA who experience transient vision loss or diplopia at the time of diagnosis, intravenous pulse glucocorticoid therapy should be initiated to reduce the risk of vision loss as rapidly as possible.22 A typical pulse regimen is methylprednisolone 1 g intravenously daily for 3 days. Though not rigorously validated in studies, such an approach is used to avoid vision impairment due to GCA, which is rarely reversible.
RELAPSE OF DISEASE
Suspect a relapse of GCA if the patient’s initial symptoms recur, if inflammatory markers become elevated, or if classic symptoms of GCA or polymyalgia rheumatica occur. Elevations in inflammatory markers do not definitely indicate a flare of GCA, but they should trigger close monitoring of the patient’s symptoms.
Relapse is treated by increasing the glucocorticoid dosage as appropriate to the nature of the relapse. If vision is affected or the patient has symptoms of GCA, then increments of 30 to 60 mg of prednisone are warranted, whereas if the patient has symptoms of polymyalgia rheumatica, then increments of 5 to 10 mg of prednisone are usually used.
The incidence of relapses of GCA in multiple tertiary care centers has been reported to vary between 34% and 75%.23,24 Most relapses occur at prednisone dosages of less than 20 mg orally daily and within the first year after diagnosis. The most common symptoms are limb ischemia, jaw claudication, constitutional symptoms, headaches, and polymyalgia rheumatica. In a review of 286 patients,25 213 (74%) had at least 1 relapse. The first relapse occurred in the first year in 50%, by 2 years in 68%, and by 5 years in 79%.
ADVERSE EFFECTS OF GLUCOCORTICOIDS
In high doses, glucocorticoids have well-known adverse effects. In a population-based study of 120 patients, each patient treated with glucocorticoids experienced at least 1 adverse effect (cataract, fracture, infection, osteonecrosis, diabetes, hypertension, weight gain, capillary fragility, or hair loss).26 The effects were related to aging and cumulative dosage of prednisone but not to the initial dosage.
Glucocorticoids can affect many organs and systems:
- Eyes (cataracts, increased intraocular pressure, exophthalmos)
- Heart (premature atherosclerotic disease, hypertension, fluid retention, hyperlipidemia, arrhythmias)
- Gastrointestinal system (ulcer, gastrointestinal bleeding, gastritis, visceral perforation, hepatic steatosis, acute pancreatitis)
- Bone and muscle (osteopenia, osteoporosis, osteonecrosis, myopathy)
- Brain (mood disorder, psychosis, memory impairment)
- Endocrine system (hyperglycemia, hypothalamic-pituitary-adrenal axis suppression)
- Immune system (immunosuppression, leading to infection and leukocytosis).
Patients receiving a glucocorticoid dose equivalent to 20 mg or more of prednisone daily for 1 month or more who also have another cause of immunocompromise need prophylaxis against Pneumocystis jirovecii pneumonia.27 They should also receive appropriate immunizations before starting glucocorticoids. Live-virus vaccines should not be given to these patients until they have been off glucocorticoids for 1 month.
Glucocorticoids and bone loss
Glucocorticoids are associated with bone loss and fracture, which can occur within the first few months of use and with dosages as low as 2.5 to 7.5 mg orally daily.28 Therefore, glucocorticoid-induced bone loss has to be treated aggressively, particularly in patients who are older and have a history of fragility fracture.
For patients with GCA who need glucocorticoids in doses greater than 5 mg orally daily for more than 3 months, the following measures are advised to decrease the risk of bone loss:
- Weight-bearing exercise
- Smoking cessation
- Moderation in alcohol intake
- Measures to prevent falls29
- Supplementation with 1,200 mg of calcium and 800 IU of vitamin D.30
Pharmacologic therapy should be initiated in men over age 50 who have established osteoporosis and in postmenopausal women with established osteoporosis or osteopenia. For men over age 50 with established osteopenia, risk assessment with the glucocorticoid-corrected FRAX score (www.sheffield.ac.uk/FRAX) should be performed to identify those at high risk in whom pharmacologic therapy is warranted.31
Bisphosphonates are the first-line therapy for glucocorticoid-induced osteoporosis.32
Teriparatide is the second-line therapy and is used in patients who cannot tolerate bisphosphonates or other osteoporosis therapies, and in those who have severe osteoporosis, with T scores of –3.5 and below if they have not had a fracture, and –2.5 and below if they have had a fragility fracture.33
Denosumab, a monoclonal antibody to an osteoclast differentiating factor, may be beneficial for some patients with glucocorticoid-induced osteoporosis.34
To assess the efficacy of therapy, measuring bone mineral density at baseline and at 1 year of therapy is recommended. If density is stable or improved, then repeating the measurement at 2- to 3-year intervals is suggested.
TOCILIZUMAB: A STEROID-SPARING MEDICATION
Due to the adverse effects of long-term use of glucocorticoids and high rates of relapse, there is a pressing need for medications that are more efficacious and less toxic to treat GCA.
The European League Against Rheumatism, in its 2009 management guidelines for large-vessel vasculitis, recommend using an adjunctive immunosuppressant agent.19 In the case of GCA, they recommend using methotrexate 10 to 15 mg/week, which has shown modest evidence of reducing the relapse rate and lowering the cumulative doses of glucocorticoids needed.35,36
Studies of tumor necrosis factor inhibitors and abatacept have not yielded significant reductions in the relapse rate or decreased cumulative doses of prednisone.37,38
Advances in treatment for GCA have stagnated, but recent trials39,40 have evaluated the IL-6 receptor alpha inhibitor tocilizumab, given the central role of IL-6 in the pathogenesis of GCA. Case reports have revealed rapid induction and maintenance of remission in GCA using tocilizumab.41,42
Villiger et al39 performed a randomized, placebo-controlled trial to study the efficacy and safety of tocilizumab in induction and maintenance of disease remission in 30 patients with newly diagnosed GCA. The primary outcome, complete remission at 12 weeks, was achieved in 85% of patients who received tocilizumab plus tapered prednisolone, compared with 40% of patients who received placebo plus tapering prednisolone. The tocilizumab group also had favorable results in secondary outcomes including relapse-free survival at 52 weeks, time to first relapse after induction of remission, and cumulative dose of prednisolone.
The GiACTA trial. Stone et al40 studied the effect of tocilizumab on rates of relapse during glucocorticoid tapering in 251 GCA patients over the course of 52 weeks. Patients were randomized in a 2:1:1:1 ratio to 4 treatment groups:
- Tocilizumab weekly plus prednisone, with prednisone tapered over 26 weeks
- Tocilizumab every other week plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 52 weeks.
The primary outcome was the rate of sustained glucocorticoid-free remission at 52 weeks. Secondary outcomes included the remission rate, the cumulative glucocorticoid dose, and safety measures. At 52 weeks, the rates of sustained remission were:
- 56% with tocilizumab weekly
- 53% with tocilizumab every other week
- 14% with placebo plus 26-week prednisone taper
- 18% with placebo plus 52-week taper.
Differences between the active treatment groups and the placebo groups were statistically significant (P < .001).
The cumulative dose of prednisone in tocilizumab recipients was significantly less than in placebo recipients. Rates of adverse events were similar. Ultimately, the study showed that tocilizumab, either weekly or every other week, was more effective than prednisone alone at sustaining glucocorticoid-free remission in patients with GCA.
However, the study also raised questions about tocilizumab’s toxic effect profile and its long-term efficacy, as well as who are the optimal candidates for this therapy. Data on long-term use of tocilizumab are primarily taken from its use in rheumatoid arthritis.43 As of this writing, Stone et al are conducting an open-label trial to help provide long-term safety and efficacy data in patients with GCA. In the meantime, we must extrapolate data from the long-term use of tocilizumab in rheumatoid arthritis.
Tocilizumab and lower gastrointestinal tract perforation
One of the major adverse effects of long-term use of tocilizumab is lower gastrointestinal tract perforation.
Xie et al,44 in 2016, reported that the risk of perforation in patients on tocilizumab for rheumatoid arthritis was more than 2 times higher than in patients taking a tumor necrosis factor inhibitor. However, the absolute rates of perforation were low overall, roughly 1 to 3 per 1,000 patient-years in the tocilizumab group. Risk factors for perforation included older age, history of diverticulitis or other gastrointestinal tract condition, and prednisone doses of 7.5 mg or more a day.
Does tocilizumab prevent blindness?
Another consideration is that tocilizumab may not prevent optic neuropathy. In the GiACTA trial, 1 patient in the group receiving tocilizumab every other week developed optic neuropathy.40 Prednisone had been completely tapered off at the time, and the condition resolved when glucocorticoids were restarted. Thus, it is unknown if tocilizumab would be effective on its own without concomitant use of glucocorticoids.
Vision loss is one of the most severe complications of GCA, and it is still unclear whether tocilizumab can prevent vision loss in GCA. Also, we still have no data on the effect of tocilizumab on histopathologic findings, and whether biopsy yield diminishes over time. We hope future studies will help guide us in this regard.
No guidelines on tocilizumab yet
Clinical guidelines on the appropriate use of tocilizumab in GCA are lacking. The American College of Rheumatology and the European League Against Rheumatism have yet to publish updated guidelines with comments on use of tocilizumab. Therefore, it is unclear if tocilizumab is a first-line treatment in GCA, as its efficacy alone without glucocorticoids and its long-term safety in GCA patients have not been studied.
Treatment with tocilizumab should be individualized; it should be considered in patients who have had adverse effects from glucocorticoids, and in patients who experience a flare or cannot have their glucocorticoid dose lowered to an appropriate range.
The optimal duration of tocilizumab therapy is also unknown. However, using the GiACTA study as a rough guide, we try to limit its use to 1 year until additional data are available.
Patients on IL-6 inhibition may have suppressed C-reactive protein regardless of disease activity.43 Therefore, this laboratory value may not be reliable in determining active disease in patients on tocilizumab.
The GiACTA trial has shown an impressive improvement in the relapse-free remission period in patients with GCA taking tocilizumab. However, much work needs to be done to define the safety of this medication and determine which patients should be started on it. In the meantime, we recommend starting high-dose glucocorticoid therapy as soon as the diagnosis of GCA is suspected. In patients who do not tolerate glucocorticoids or whose disease flares during glucocorticoid taper, we recommend starting treatment with tocilizumab either once a week or every other week for at least 1 year.
- Brekke LK, Diamantopoulos AP, Fevang BT, Aßmus J, Esperø E, Gjesdal CG. Incidence of giant cell arteritis in Western Norway 1972–2012: a retrospective cohort study. Arthritis Res Ther 2017; 19(1):278. doi:10.1186/s13075-017-1479-6
- Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology (Oxford) 2017; 56(4):506–515. doi:10.1093/rheumatology/kew273
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med 2014; 371(17):1653. doi:10.1056/NEJMc1409206
- Ghinoi A, Pipitone N, Nicolini A, et al. Large-vessel involvement in recent-onset giant cell arteritis: a case-control colour-Doppler sonography study. Rheumatology (Oxford) 2012; 51(4):730–734. doi:10.1093/rheumatology/ker329
- Prieto-González S, Depetris M, García-Martínez A, et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: a prospective, case-control study. Ann Rheum Dis 2014; 73(7):1388–1392. doi:10.1136/annrheumdis-2013-204572
- Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum 1999; 42(2):311–317. doi:10.1002/1529-0131(199902)42:2<311::AID-ANR14>3.0.CO;2-F
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45(2):140–145. doi:10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
- Liozon E, Jauberteau-Marchan MO, Ly K, Loustaud-Ratti V, Soria P, Vidal E. Giant cell arteritis with a low erythrocyte sedimentation rate: comments on the article by Salvarani and Hunder. Arthritis Rheum 2002; 47(6):692–694. doi:10.1002/art.10809
- Yu-Wai-Man P, Dayan MR. Giant cell arteritis with normal inflammatory markers. Acta Ophthalmol Scand 2007; 85(4):460. doi:10.1111/j.1600-0420.2006.00864.x
- Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA 2016; 315(22):2442–2458. doi:10.1001/jama.2016.5444
- Niederkohr RD, Levin LA. Management of the patient with suspected temporal arteritis a decision-analytic approach. Ophthalmology 2005; 112(5):744–756. doi:10.1016/j.ophtha.2005.01.031
- Bowling K, Rait J, Atkinson J, Srinivas G. Temporal artery biopsy in the diagnosis of giant cell arteritis: does the end justify the means? Ann Med Surg (Lond) 2017; 20:1–5. doi:10.1016/j.amsu.2017.06.020
- Daily B, Dassow P, Haynes J, Nashelsky J. Giant cell arteritis: biopsy after corticosteroid initiation. Am Fam Physician 2017; 95(2):116–117. pmid:28084703
- Durling B, Toren A, Patel V, Gilberg S, Weis E, Jordan D. Incidence of discordant temporal artery biopsy in the diagnosis of giant cell arteritis. Can J Ophthalmol 2014; 49(2):157–161. doi:10.1016/j.jcjo.2013.12.008
- Dejaco C, Ramiro S, Duftner C, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis 2018; 77(5):636–643. doi:10.1136/annrheumdis-2017-212649
- Aschwanden M, Imfeld S, Staub D, et al. The ultrasound compression sign to diagnose temporal giant cell arteritis shows an excellent interobserver agreement. Clin Exp Rheumatol 2015; 33(2 suppl 89):S-113–S-115. pmid:26016760
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100(4):550–555. pmid:8479714
- Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology 2003; 110(6):1204–1215. doi:10.1016/S0161-6420(03)00228-8
- Mukhtyar C, Guillevin L, Cid MC, et al; European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009; 68(3):318–323. doi:10.1136/ard.2008.088351
- Dejaco C, Singh YP, Perel P, et al; European League Against Rheumatism; American College of Rheumatology. 2015 recommendations for the management of polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Ann Rheum Dis 2015; 74(10):1799–1807. doi:10.1136/annrheumdis-2015-207492
- Bienvenu B, Ly KH, Lambert M, et al; Groupe d’Étude Français des Artérites des gros Vaisseaux, under the Aegis of the Filière des Maladies Auto-Immunes et Auto-Inflammatoires Rares. Management of giant cell arteritis: recommendations of the French Study Group for Large Vessel Vasculitis (GEFA). Rev Med Interne 2016; 37(3):154–165. doi:10.1016/j.revmed.2015.12.015
- Hayreh SS, Biousse V. Treatment of acute visual loss in giant cell arteritis: should we prescribe high-dose intravenous steroids or just oral steroids? J Neuroophthalmol 2012; 32(3):278–287. doi:10.1097/WNO.0b013e3182688218
- Restuccia G, Boiardi L, Cavazza A, et al. Flares in biopsy-proven giant cell arteritis in Northern Italy: characteristics and predictors in a long-term follow-up study. Medicine (Baltimore) 2016; 95(19):e3524. doi:10.1097/MD.0000000000003524
- Kermani TA, Warrington KJ, Cuthbertson D, et al; Vasculitis Clinical Research Consortium. Disease relapses among patients with giant cell arteritis: a prospective, longitudinal cohort study. J Rheumatol 2015; 42(7):1213–1217. doi:10.3899/jrheum.141347
- Labarca C, Koster MJ, Crowson CS, et al. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology (Oxford) 2016; 55(2):347–356. doi:10.1093/rheumatology/kev348
- Proven A, Gabriel SE, Orces C, O’Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum 2003; 49(5):703–708. doi:10.1002/art.11388
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34(8):1098–1107. doi:10.1086/339548
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13(10):777–787. doi:10.1007/s001980200108
- Heffernan MP, Saag KG, Robinson JK, Callen JP. Prevention of osteoporosis associated with chronic glucocorticoid therapy. JAMA 2006; 295(11):1300–1303. pmid:16541489
- Buckley L, Guyatt G, Fink HA, et al. 2017 American College of Rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2017; 69(8):1095–1110. doi:10.1002/acr.23279
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res 201; 62(11):1515–1526. doi:10.1002/acr.20295
- Allen CS, Yeung JH, Vandermeer B, Homik J. Bisphosphonates for steroid-induced osteoporosis. Cochrane Database Syst Rev 2016; 10:CD001347. doi:10.1002/14651858.CD001347.pub2
- Carpinteri R, Porcelli T, Mejia C, et al. Glucocorticoid-induced osteoporosis and parathyroid hormone. J Endocrinol Invest 2010; 33(suppl 7):16–21. pmid:20938221
- Saag KG, Wagman RB, Geusens P, et al. Denosumab versus risedronate in glucocorticoid-induced osteoporosis: a multicentre, randomised, double-blind, active-controlled, double-dummy, non-inferiority study. Lancet Diabetes Endocrinol 2018; 6(6):445–454. doi:10.1016/S2213-8587(18)30075-5
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46(5):1309–1318. doi:10.1002/art.10262
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19(5):495–501. pmid:11579707
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146(9):621–630. pmid:17470830
- Langford CA, Cuthbertson D, Ytterberg SR, et al; Vasculitis Clinical Research Consortium. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol 2017; 69(4):837–845. doi:10.1002/art.40044
- Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016; 387(10031):1921–1927. doi:10.1016/S0140-6736(16)00560-2
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Oliveira F, Butendieck RR, Ginsburg WW, Parikh K, Abril A. Tocilizumab, an effective treatment for relapsing giant cell arteritis. Clin Exp Rheumatol 2014; 32(3 suppl 82):S76–S78. pmid:24854376
- Loricera J, Blanco R, Hernández JL, et al. Tocilizumab in giant cell arteritis: multicenter open-label study of 22 patients. Semin Arthritis Rheum 2015; 44(6):717–723. doi:10.1016/j.semarthrit.2014.12.005
- Tamaki H, Hajj-Ali RA. Tocilizumab for giant cell arteritis—a new giant step in an old disease. JAMA Neurol 2018; 75(2):145–146. doi:10.1001/jamaneurol.2017.3811
- Xie F, Yun H, Bernatsky S, Curtis JR. Risk for gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologics. Arthritis Rheumatol 2016; 68(11):2612–2617. doi:10.1002/art.39761
Giant cell arteritis (GCA) is a systemic vasculitis involving medium-sized and large arteries, most commonly the temporal, ophthalmic, occipital, vertebral, posterior ciliary, and proximal vertebral arteries. Moreover, involvement of the ophthalmic artery and its branches results in loss of vision. GCA can also involve the aorta and its proximal branches, especially in the upper extremities.
GCA is the most common systemic vasculitis in adults. It occurs almost exclusively in patients over age 50 and affects women more than men. It is most frequent in populations of northern European ancestry, especially Scandinavian. In a retrospective cohort study in Norway, the average annual cumulative incidence rate of GCA was 16.7 per 100,000 people over age 50.1 Risk factors include older age, history of smoking, current smoking, early menopause, and, possibly, stress-related disorders.2
PATHOGENESIS IS NOT COMPLETELY UNDERSTOOD
The pathogenesis of GCA is not completely understood, but there is evidence of immune activation in the arterial wall leading to activation of macrophages and formation of multinucleated giant cells (which may not always be present in biopsies).
The most relevant cytokines in the ongoing pathogenesis are still being defined, but the presence of interferon gamma and interleukin 6 (IL-6) seem to be critical for the expression of the disease. The primary immunogenic triggers for the elaboration of these cytokines and the arteritis remain elusive.
A SPECTRUM OF PRESENTATIONS
The initial symptoms of GCA may be vague, such as malaise, fever, and night sweats, and are likely due to systemic inflammation. Features of vascular involvement include headache, scalp tenderness, and jaw claudication (cramping pain in the jaw while chewing).
A less common but serious feature associated with GCA is partial or complete vision loss affecting 1 or both eyes.3 Some patients suddenly go completely blind without any visual prodrome.
Overlapping GCA phenotypes exist, with a spectrum of presentations that include classic cranial arteritis, extracranial GCA (also called large-vessel GCA), and polymyalgia rheumatica.2
Cranial GCA, the best-characterized clinical presentation, causes symptoms such as headache or signs such as tenderness of the temporal artery. On examination, the temporal arteries may be tender or nodular, and the pulses may be felt above the zygomatic arch, above and in front of the tragus of the ear. About two-thirds of patients with cranial GCA present with new-onset headache, most often in the temporal area, but possibly anywhere throughout the head.
Visual disturbance, jaw claudication, and tongue pain are less common but, if present, increase the likelihood of this diagnosis.2
Large-vessel involvement in GCA is common and refers to involvement of the aorta and its proximal branches. Imaging methods used in diagnosing large-vessel GCA include color Doppler ultrasonography, computed tomography with angiography, magnetic resonance imaging with angiography, and positron emission tomography. In some centers, such imaging is performed in all patients diagnosed with GCA to survey for large-vessel involvement.
Depending on the imaging study, large-vessel involvement has been found in 30% to 80% of cases of GCA.4,5 It is often associated with nonspecific symptoms such as fever, weight loss, chills, and malaise, but it can also cause more specific symptoms such as unilateral extremity claudication. In contrast to patients with cranial GCA, patients with large-vessel GCA were younger at onset, less likely to have headaches, and more likely to have arm claudication at presentation.6 Aortitis of the ascending aorta can occur with a histopathologic pattern of GCA but without the clinical stigmata of GCA.
The finding of aortitis should prompt the clinician to question the patient about other symptoms of GCA and to order imaging of the whole vascular tree. Ultrasonography and biopsy of the temporal arteries can be considered. Whether idiopathic aortitis is part of the GCA spectrum remains to be seen.
Laboratory tests often show anemia, leukocytosis, and thrombocytosis. Acute-phase reactants such as C-reactive protein and the erythrocyte sedimentation rate are often elevated. The sedimentation rate often exceeds 50 mm/hour and sometimes 100 mm/hour.
In 2 retrospective studies, the number of patients with GCA whose sedimentation rate was less than 50 mm/hour ranged between 5% and 11%.7,8 However, a small percentage of patients with GCA have normal inflammatory markers. Therefore, if the suspicion for GCA is high, treatment should be started and biopsy pursued.9 In patients with paraproteinemia or other causes of a spuriously elevated or low erythrocyte sedimentation rate, C-reactive protein is a more reliable test.
Polymyalgia rheumatica is another rheumatologic condition that can occur independently or in conjunction with GCA. It is characterized by stiffness and pain in the proximal joints such as the hips and shoulders, typically worse in the morning and better with activity. Although the patient may subjectively feel weak, a close neurologic examination will reveal normal muscle strength.
Polymyalgia rheumatica is observed in 40% to 60% of patients with GCA at the time of diagnosis; 16% to 21% of patients with polymyalgia rheumatica may develop GCA, especially if untreated.2,10
Differential diagnosis
Other vasculitides (eg, Takayasu arteritis) can also present with unexplained fever, anemia, and constitutional symptoms.
Infection should be considered if fever is present. An infectious disease accompanied by fever, headache, and elevated inflammatory markers can mimic GCA.
Nonarteritic anterior ischemic optic neuropathy can present with sudden vision loss, prompting concern for underlying GCA. Risk factors include hypertension and diabetes mellitus; other features of GCA, including elevated inflammatory markers, are generally absent.
TEMPORAL ARTERY BIOPSY: THE GOLD STANDARD FOR DIAGNOSIS
Temporal artery biopsy remains the standard to confirm the diagnosis. However, because inflammation in the temporal arteries can affect some segments but not others, biopsy results on conventional hematoxylin and eosin staining can be falsely negative in patients with GCA. In one study,11 the mean sensitivity of unilateral temporal artery biopsy was 86.9%.
Typical positive histologic findings are inflammation with panarteritis, CD4-positive lymphocytes, macrophages, giant cells, and fragmentation of the internal elastic lamina.12
When GCA is suspected, treatment with glucocorticoids should be started immediately and biopsy performed as soon as possible. Delaying biopsy for 14 days or more may not affect the accuracy of biopsy study.13 Treatment should never be withheld while awaiting the results of biopsy study.
Biopsy is usually performed unilaterally, on the same side as the symptoms or abnormal findings on examination. Bilateral temporal artery biopsy is also performed and compared with unilateral biopsy; this approach increases the diagnostic yield by about 5%.14
IMAGING
In patients with suspected GCA, imaging is recommended early to complement the clinical criteria for the diagnosis of GCA.15 Positron emission tomography, computed tomography angiography, magnetic resonance angiography, or Doppler ultrasonography can reveal inflammation of the arteries in the proximal upper or lower limbs or the aorta.2
In patients with suspected cranial GCA, ultrasonography of the temporal and axillary arteries is recommended first. If ultrasonography is not available or is inconclusive, high-resolution magnetic resonance imaging of the cranial arteries can be used as an alternative. Computed tomography and positron emission tomography of the cranial arteries are not recommended.
In patients with suspected large-vessel GCA, ultrasonography, positron emission tomography, computed tomography, and magnetic resonance imaging may be used to screen for vessel wall inflammation, edema, and luminal narrowing in extracranial arteries. Ultrasonography is of limited value in assessing aortitis.
Color duplex ultrasonography can be applied to assess for vascular inflammation of the temporal or large arteries. The typical finding of the “halo” sign, a hypoechoic ring around the arterial lumen, represents the inflammation-induced thickening of the arterial wall. The “compression sign,” the persistence of the “halo” during compression of the vessel lumen by the ultrasound probe, has high specificity for the diagnosis.16
Ultrasonography of suspected GCA has yielded sensitivities of 55% to 100% and specificities of 78% to 100%. However, its sensitivity depends on the user’s level of expertise, so it should be done only in medical centers with a high number of GCA cases and with highly experienced sonographers. High-resolution magnetic resonance imaging is an alternative to ultrasonography and has shown similar sensitivity and specificity.3
TREATMENT WITH GLUCOCORTICOIDS
Glucocorticoids remain the standard for treatment of GCA. The therapeutic effect of glucocorticoids in GCA has been established by years of clinical experience, but has never been proven in a placebo-controlled trial. When started appropriately and expeditiously, glucocorticoids produce exquisite resolution of signs and symptoms and prevent the serious complication of vision loss. Rapid resolution of symptoms is so typical of GCA that if the patient’s symptoms persist more than a few days after starting a glucocorticoid, the diagnosis of GCA should be reconsidered.
In a retrospective study of 245 patients with biopsy-proven GCA treated with glucocorticoids, 34 had permanent loss of sight.17 In 32 (94%) of the 34, the vision loss occurred before glucocorticoids were started. Of the remaining 2 patients, 1 lost vision 8 days into treatment, and the other lost vision 3 years after diagnosis and 1 year after discontinuation of glucocorticoids.
In a series of 144 patients with biopsy-proven GCA, 51 had no vision loss at presentation and no vision loss after starting glucocorticoids, and 93 had vision loss at presentation. In the latter group, symptoms worsened within 5 days of starting glucocorticoids in 9 patients.18 If vision was intact at the time of presentation, prompt initiation of glucocorticoids reduced the risk of vision loss to less than 1%.
High doses, slowly tapered
The European League Against Rheumatism recommends early initiation of high-dose glucocorticoids for patients with large-vessel vasculitis,19 and it also recommends glucocorticoids for patients with polymyalgia rheumatica.20 The optimal initial and tapering dosage has never been formally evaluated, but regimens have been devised on the basis of expert opinion.21
For patients with GCA who do not have vision loss at the time of diagnosis, the initial dose is prednisone 1 mg/kg or its equivalent daily for 2 to 4 weeks, after which it is tapered.21 If the initial dosage is prednisone 60 mg orally daily for 2 to 4 weeks, our practice is to taper it to 50 mg daily for 2 weeks, then 40 mg daily for 2 weeks. Then, it is decreased by 5 mg every 2 weeks until it is 20 mg daily, and then by 2.5 mg every 2 weeks until it is 10 mg orally daily. Thereafter, the dosage is decreased by 1 mg every 2 to 4 weeks.
For patients with GCA who experience transient vision loss or diplopia at the time of diagnosis, intravenous pulse glucocorticoid therapy should be initiated to reduce the risk of vision loss as rapidly as possible.22 A typical pulse regimen is methylprednisolone 1 g intravenously daily for 3 days. Though not rigorously validated in studies, such an approach is used to avoid vision impairment due to GCA, which is rarely reversible.
RELAPSE OF DISEASE
Suspect a relapse of GCA if the patient’s initial symptoms recur, if inflammatory markers become elevated, or if classic symptoms of GCA or polymyalgia rheumatica occur. Elevations in inflammatory markers do not definitely indicate a flare of GCA, but they should trigger close monitoring of the patient’s symptoms.
Relapse is treated by increasing the glucocorticoid dosage as appropriate to the nature of the relapse. If vision is affected or the patient has symptoms of GCA, then increments of 30 to 60 mg of prednisone are warranted, whereas if the patient has symptoms of polymyalgia rheumatica, then increments of 5 to 10 mg of prednisone are usually used.
The incidence of relapses of GCA in multiple tertiary care centers has been reported to vary between 34% and 75%.23,24 Most relapses occur at prednisone dosages of less than 20 mg orally daily and within the first year after diagnosis. The most common symptoms are limb ischemia, jaw claudication, constitutional symptoms, headaches, and polymyalgia rheumatica. In a review of 286 patients,25 213 (74%) had at least 1 relapse. The first relapse occurred in the first year in 50%, by 2 years in 68%, and by 5 years in 79%.
ADVERSE EFFECTS OF GLUCOCORTICOIDS
In high doses, glucocorticoids have well-known adverse effects. In a population-based study of 120 patients, each patient treated with glucocorticoids experienced at least 1 adverse effect (cataract, fracture, infection, osteonecrosis, diabetes, hypertension, weight gain, capillary fragility, or hair loss).26 The effects were related to aging and cumulative dosage of prednisone but not to the initial dosage.
Glucocorticoids can affect many organs and systems:
- Eyes (cataracts, increased intraocular pressure, exophthalmos)
- Heart (premature atherosclerotic disease, hypertension, fluid retention, hyperlipidemia, arrhythmias)
- Gastrointestinal system (ulcer, gastrointestinal bleeding, gastritis, visceral perforation, hepatic steatosis, acute pancreatitis)
- Bone and muscle (osteopenia, osteoporosis, osteonecrosis, myopathy)
- Brain (mood disorder, psychosis, memory impairment)
- Endocrine system (hyperglycemia, hypothalamic-pituitary-adrenal axis suppression)
- Immune system (immunosuppression, leading to infection and leukocytosis).
Patients receiving a glucocorticoid dose equivalent to 20 mg or more of prednisone daily for 1 month or more who also have another cause of immunocompromise need prophylaxis against Pneumocystis jirovecii pneumonia.27 They should also receive appropriate immunizations before starting glucocorticoids. Live-virus vaccines should not be given to these patients until they have been off glucocorticoids for 1 month.
Glucocorticoids and bone loss
Glucocorticoids are associated with bone loss and fracture, which can occur within the first few months of use and with dosages as low as 2.5 to 7.5 mg orally daily.28 Therefore, glucocorticoid-induced bone loss has to be treated aggressively, particularly in patients who are older and have a history of fragility fracture.
For patients with GCA who need glucocorticoids in doses greater than 5 mg orally daily for more than 3 months, the following measures are advised to decrease the risk of bone loss:
- Weight-bearing exercise
- Smoking cessation
- Moderation in alcohol intake
- Measures to prevent falls29
- Supplementation with 1,200 mg of calcium and 800 IU of vitamin D.30
Pharmacologic therapy should be initiated in men over age 50 who have established osteoporosis and in postmenopausal women with established osteoporosis or osteopenia. For men over age 50 with established osteopenia, risk assessment with the glucocorticoid-corrected FRAX score (www.sheffield.ac.uk/FRAX) should be performed to identify those at high risk in whom pharmacologic therapy is warranted.31
Bisphosphonates are the first-line therapy for glucocorticoid-induced osteoporosis.32
Teriparatide is the second-line therapy and is used in patients who cannot tolerate bisphosphonates or other osteoporosis therapies, and in those who have severe osteoporosis, with T scores of –3.5 and below if they have not had a fracture, and –2.5 and below if they have had a fragility fracture.33
Denosumab, a monoclonal antibody to an osteoclast differentiating factor, may be beneficial for some patients with glucocorticoid-induced osteoporosis.34
To assess the efficacy of therapy, measuring bone mineral density at baseline and at 1 year of therapy is recommended. If density is stable or improved, then repeating the measurement at 2- to 3-year intervals is suggested.
TOCILIZUMAB: A STEROID-SPARING MEDICATION
Due to the adverse effects of long-term use of glucocorticoids and high rates of relapse, there is a pressing need for medications that are more efficacious and less toxic to treat GCA.
The European League Against Rheumatism, in its 2009 management guidelines for large-vessel vasculitis, recommend using an adjunctive immunosuppressant agent.19 In the case of GCA, they recommend using methotrexate 10 to 15 mg/week, which has shown modest evidence of reducing the relapse rate and lowering the cumulative doses of glucocorticoids needed.35,36
Studies of tumor necrosis factor inhibitors and abatacept have not yielded significant reductions in the relapse rate or decreased cumulative doses of prednisone.37,38
Advances in treatment for GCA have stagnated, but recent trials39,40 have evaluated the IL-6 receptor alpha inhibitor tocilizumab, given the central role of IL-6 in the pathogenesis of GCA. Case reports have revealed rapid induction and maintenance of remission in GCA using tocilizumab.41,42
Villiger et al39 performed a randomized, placebo-controlled trial to study the efficacy and safety of tocilizumab in induction and maintenance of disease remission in 30 patients with newly diagnosed GCA. The primary outcome, complete remission at 12 weeks, was achieved in 85% of patients who received tocilizumab plus tapered prednisolone, compared with 40% of patients who received placebo plus tapering prednisolone. The tocilizumab group also had favorable results in secondary outcomes including relapse-free survival at 52 weeks, time to first relapse after induction of remission, and cumulative dose of prednisolone.
The GiACTA trial. Stone et al40 studied the effect of tocilizumab on rates of relapse during glucocorticoid tapering in 251 GCA patients over the course of 52 weeks. Patients were randomized in a 2:1:1:1 ratio to 4 treatment groups:
- Tocilizumab weekly plus prednisone, with prednisone tapered over 26 weeks
- Tocilizumab every other week plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 52 weeks.
The primary outcome was the rate of sustained glucocorticoid-free remission at 52 weeks. Secondary outcomes included the remission rate, the cumulative glucocorticoid dose, and safety measures. At 52 weeks, the rates of sustained remission were:
- 56% with tocilizumab weekly
- 53% with tocilizumab every other week
- 14% with placebo plus 26-week prednisone taper
- 18% with placebo plus 52-week taper.
Differences between the active treatment groups and the placebo groups were statistically significant (P < .001).
The cumulative dose of prednisone in tocilizumab recipients was significantly less than in placebo recipients. Rates of adverse events were similar. Ultimately, the study showed that tocilizumab, either weekly or every other week, was more effective than prednisone alone at sustaining glucocorticoid-free remission in patients with GCA.
However, the study also raised questions about tocilizumab’s toxic effect profile and its long-term efficacy, as well as who are the optimal candidates for this therapy. Data on long-term use of tocilizumab are primarily taken from its use in rheumatoid arthritis.43 As of this writing, Stone et al are conducting an open-label trial to help provide long-term safety and efficacy data in patients with GCA. In the meantime, we must extrapolate data from the long-term use of tocilizumab in rheumatoid arthritis.
Tocilizumab and lower gastrointestinal tract perforation
One of the major adverse effects of long-term use of tocilizumab is lower gastrointestinal tract perforation.
Xie et al,44 in 2016, reported that the risk of perforation in patients on tocilizumab for rheumatoid arthritis was more than 2 times higher than in patients taking a tumor necrosis factor inhibitor. However, the absolute rates of perforation were low overall, roughly 1 to 3 per 1,000 patient-years in the tocilizumab group. Risk factors for perforation included older age, history of diverticulitis or other gastrointestinal tract condition, and prednisone doses of 7.5 mg or more a day.
Does tocilizumab prevent blindness?
Another consideration is that tocilizumab may not prevent optic neuropathy. In the GiACTA trial, 1 patient in the group receiving tocilizumab every other week developed optic neuropathy.40 Prednisone had been completely tapered off at the time, and the condition resolved when glucocorticoids were restarted. Thus, it is unknown if tocilizumab would be effective on its own without concomitant use of glucocorticoids.
Vision loss is one of the most severe complications of GCA, and it is still unclear whether tocilizumab can prevent vision loss in GCA. Also, we still have no data on the effect of tocilizumab on histopathologic findings, and whether biopsy yield diminishes over time. We hope future studies will help guide us in this regard.
No guidelines on tocilizumab yet
Clinical guidelines on the appropriate use of tocilizumab in GCA are lacking. The American College of Rheumatology and the European League Against Rheumatism have yet to publish updated guidelines with comments on use of tocilizumab. Therefore, it is unclear if tocilizumab is a first-line treatment in GCA, as its efficacy alone without glucocorticoids and its long-term safety in GCA patients have not been studied.
Treatment with tocilizumab should be individualized; it should be considered in patients who have had adverse effects from glucocorticoids, and in patients who experience a flare or cannot have their glucocorticoid dose lowered to an appropriate range.
The optimal duration of tocilizumab therapy is also unknown. However, using the GiACTA study as a rough guide, we try to limit its use to 1 year until additional data are available.
Patients on IL-6 inhibition may have suppressed C-reactive protein regardless of disease activity.43 Therefore, this laboratory value may not be reliable in determining active disease in patients on tocilizumab.
The GiACTA trial has shown an impressive improvement in the relapse-free remission period in patients with GCA taking tocilizumab. However, much work needs to be done to define the safety of this medication and determine which patients should be started on it. In the meantime, we recommend starting high-dose glucocorticoid therapy as soon as the diagnosis of GCA is suspected. In patients who do not tolerate glucocorticoids or whose disease flares during glucocorticoid taper, we recommend starting treatment with tocilizumab either once a week or every other week for at least 1 year.
Giant cell arteritis (GCA) is a systemic vasculitis involving medium-sized and large arteries, most commonly the temporal, ophthalmic, occipital, vertebral, posterior ciliary, and proximal vertebral arteries. Moreover, involvement of the ophthalmic artery and its branches results in loss of vision. GCA can also involve the aorta and its proximal branches, especially in the upper extremities.
GCA is the most common systemic vasculitis in adults. It occurs almost exclusively in patients over age 50 and affects women more than men. It is most frequent in populations of northern European ancestry, especially Scandinavian. In a retrospective cohort study in Norway, the average annual cumulative incidence rate of GCA was 16.7 per 100,000 people over age 50.1 Risk factors include older age, history of smoking, current smoking, early menopause, and, possibly, stress-related disorders.2
PATHOGENESIS IS NOT COMPLETELY UNDERSTOOD
The pathogenesis of GCA is not completely understood, but there is evidence of immune activation in the arterial wall leading to activation of macrophages and formation of multinucleated giant cells (which may not always be present in biopsies).
The most relevant cytokines in the ongoing pathogenesis are still being defined, but the presence of interferon gamma and interleukin 6 (IL-6) seem to be critical for the expression of the disease. The primary immunogenic triggers for the elaboration of these cytokines and the arteritis remain elusive.
A SPECTRUM OF PRESENTATIONS
The initial symptoms of GCA may be vague, such as malaise, fever, and night sweats, and are likely due to systemic inflammation. Features of vascular involvement include headache, scalp tenderness, and jaw claudication (cramping pain in the jaw while chewing).
A less common but serious feature associated with GCA is partial or complete vision loss affecting 1 or both eyes.3 Some patients suddenly go completely blind without any visual prodrome.
Overlapping GCA phenotypes exist, with a spectrum of presentations that include classic cranial arteritis, extracranial GCA (also called large-vessel GCA), and polymyalgia rheumatica.2
Cranial GCA, the best-characterized clinical presentation, causes symptoms such as headache or signs such as tenderness of the temporal artery. On examination, the temporal arteries may be tender or nodular, and the pulses may be felt above the zygomatic arch, above and in front of the tragus of the ear. About two-thirds of patients with cranial GCA present with new-onset headache, most often in the temporal area, but possibly anywhere throughout the head.
Visual disturbance, jaw claudication, and tongue pain are less common but, if present, increase the likelihood of this diagnosis.2
Large-vessel involvement in GCA is common and refers to involvement of the aorta and its proximal branches. Imaging methods used in diagnosing large-vessel GCA include color Doppler ultrasonography, computed tomography with angiography, magnetic resonance imaging with angiography, and positron emission tomography. In some centers, such imaging is performed in all patients diagnosed with GCA to survey for large-vessel involvement.
Depending on the imaging study, large-vessel involvement has been found in 30% to 80% of cases of GCA.4,5 It is often associated with nonspecific symptoms such as fever, weight loss, chills, and malaise, but it can also cause more specific symptoms such as unilateral extremity claudication. In contrast to patients with cranial GCA, patients with large-vessel GCA were younger at onset, less likely to have headaches, and more likely to have arm claudication at presentation.6 Aortitis of the ascending aorta can occur with a histopathologic pattern of GCA but without the clinical stigmata of GCA.
The finding of aortitis should prompt the clinician to question the patient about other symptoms of GCA and to order imaging of the whole vascular tree. Ultrasonography and biopsy of the temporal arteries can be considered. Whether idiopathic aortitis is part of the GCA spectrum remains to be seen.
Laboratory tests often show anemia, leukocytosis, and thrombocytosis. Acute-phase reactants such as C-reactive protein and the erythrocyte sedimentation rate are often elevated. The sedimentation rate often exceeds 50 mm/hour and sometimes 100 mm/hour.
In 2 retrospective studies, the number of patients with GCA whose sedimentation rate was less than 50 mm/hour ranged between 5% and 11%.7,8 However, a small percentage of patients with GCA have normal inflammatory markers. Therefore, if the suspicion for GCA is high, treatment should be started and biopsy pursued.9 In patients with paraproteinemia or other causes of a spuriously elevated or low erythrocyte sedimentation rate, C-reactive protein is a more reliable test.
Polymyalgia rheumatica is another rheumatologic condition that can occur independently or in conjunction with GCA. It is characterized by stiffness and pain in the proximal joints such as the hips and shoulders, typically worse in the morning and better with activity. Although the patient may subjectively feel weak, a close neurologic examination will reveal normal muscle strength.
Polymyalgia rheumatica is observed in 40% to 60% of patients with GCA at the time of diagnosis; 16% to 21% of patients with polymyalgia rheumatica may develop GCA, especially if untreated.2,10
Differential diagnosis
Other vasculitides (eg, Takayasu arteritis) can also present with unexplained fever, anemia, and constitutional symptoms.
Infection should be considered if fever is present. An infectious disease accompanied by fever, headache, and elevated inflammatory markers can mimic GCA.
Nonarteritic anterior ischemic optic neuropathy can present with sudden vision loss, prompting concern for underlying GCA. Risk factors include hypertension and diabetes mellitus; other features of GCA, including elevated inflammatory markers, are generally absent.
TEMPORAL ARTERY BIOPSY: THE GOLD STANDARD FOR DIAGNOSIS
Temporal artery biopsy remains the standard to confirm the diagnosis. However, because inflammation in the temporal arteries can affect some segments but not others, biopsy results on conventional hematoxylin and eosin staining can be falsely negative in patients with GCA. In one study,11 the mean sensitivity of unilateral temporal artery biopsy was 86.9%.
Typical positive histologic findings are inflammation with panarteritis, CD4-positive lymphocytes, macrophages, giant cells, and fragmentation of the internal elastic lamina.12
When GCA is suspected, treatment with glucocorticoids should be started immediately and biopsy performed as soon as possible. Delaying biopsy for 14 days or more may not affect the accuracy of biopsy study.13 Treatment should never be withheld while awaiting the results of biopsy study.
Biopsy is usually performed unilaterally, on the same side as the symptoms or abnormal findings on examination. Bilateral temporal artery biopsy is also performed and compared with unilateral biopsy; this approach increases the diagnostic yield by about 5%.14
IMAGING
In patients with suspected GCA, imaging is recommended early to complement the clinical criteria for the diagnosis of GCA.15 Positron emission tomography, computed tomography angiography, magnetic resonance angiography, or Doppler ultrasonography can reveal inflammation of the arteries in the proximal upper or lower limbs or the aorta.2
In patients with suspected cranial GCA, ultrasonography of the temporal and axillary arteries is recommended first. If ultrasonography is not available or is inconclusive, high-resolution magnetic resonance imaging of the cranial arteries can be used as an alternative. Computed tomography and positron emission tomography of the cranial arteries are not recommended.
In patients with suspected large-vessel GCA, ultrasonography, positron emission tomography, computed tomography, and magnetic resonance imaging may be used to screen for vessel wall inflammation, edema, and luminal narrowing in extracranial arteries. Ultrasonography is of limited value in assessing aortitis.
Color duplex ultrasonography can be applied to assess for vascular inflammation of the temporal or large arteries. The typical finding of the “halo” sign, a hypoechoic ring around the arterial lumen, represents the inflammation-induced thickening of the arterial wall. The “compression sign,” the persistence of the “halo” during compression of the vessel lumen by the ultrasound probe, has high specificity for the diagnosis.16
Ultrasonography of suspected GCA has yielded sensitivities of 55% to 100% and specificities of 78% to 100%. However, its sensitivity depends on the user’s level of expertise, so it should be done only in medical centers with a high number of GCA cases and with highly experienced sonographers. High-resolution magnetic resonance imaging is an alternative to ultrasonography and has shown similar sensitivity and specificity.3
TREATMENT WITH GLUCOCORTICOIDS
Glucocorticoids remain the standard for treatment of GCA. The therapeutic effect of glucocorticoids in GCA has been established by years of clinical experience, but has never been proven in a placebo-controlled trial. When started appropriately and expeditiously, glucocorticoids produce exquisite resolution of signs and symptoms and prevent the serious complication of vision loss. Rapid resolution of symptoms is so typical of GCA that if the patient’s symptoms persist more than a few days after starting a glucocorticoid, the diagnosis of GCA should be reconsidered.
In a retrospective study of 245 patients with biopsy-proven GCA treated with glucocorticoids, 34 had permanent loss of sight.17 In 32 (94%) of the 34, the vision loss occurred before glucocorticoids were started. Of the remaining 2 patients, 1 lost vision 8 days into treatment, and the other lost vision 3 years after diagnosis and 1 year after discontinuation of glucocorticoids.
In a series of 144 patients with biopsy-proven GCA, 51 had no vision loss at presentation and no vision loss after starting glucocorticoids, and 93 had vision loss at presentation. In the latter group, symptoms worsened within 5 days of starting glucocorticoids in 9 patients.18 If vision was intact at the time of presentation, prompt initiation of glucocorticoids reduced the risk of vision loss to less than 1%.
High doses, slowly tapered
The European League Against Rheumatism recommends early initiation of high-dose glucocorticoids for patients with large-vessel vasculitis,19 and it also recommends glucocorticoids for patients with polymyalgia rheumatica.20 The optimal initial and tapering dosage has never been formally evaluated, but regimens have been devised on the basis of expert opinion.21
For patients with GCA who do not have vision loss at the time of diagnosis, the initial dose is prednisone 1 mg/kg or its equivalent daily for 2 to 4 weeks, after which it is tapered.21 If the initial dosage is prednisone 60 mg orally daily for 2 to 4 weeks, our practice is to taper it to 50 mg daily for 2 weeks, then 40 mg daily for 2 weeks. Then, it is decreased by 5 mg every 2 weeks until it is 20 mg daily, and then by 2.5 mg every 2 weeks until it is 10 mg orally daily. Thereafter, the dosage is decreased by 1 mg every 2 to 4 weeks.
For patients with GCA who experience transient vision loss or diplopia at the time of diagnosis, intravenous pulse glucocorticoid therapy should be initiated to reduce the risk of vision loss as rapidly as possible.22 A typical pulse regimen is methylprednisolone 1 g intravenously daily for 3 days. Though not rigorously validated in studies, such an approach is used to avoid vision impairment due to GCA, which is rarely reversible.
RELAPSE OF DISEASE
Suspect a relapse of GCA if the patient’s initial symptoms recur, if inflammatory markers become elevated, or if classic symptoms of GCA or polymyalgia rheumatica occur. Elevations in inflammatory markers do not definitely indicate a flare of GCA, but they should trigger close monitoring of the patient’s symptoms.
Relapse is treated by increasing the glucocorticoid dosage as appropriate to the nature of the relapse. If vision is affected or the patient has symptoms of GCA, then increments of 30 to 60 mg of prednisone are warranted, whereas if the patient has symptoms of polymyalgia rheumatica, then increments of 5 to 10 mg of prednisone are usually used.
The incidence of relapses of GCA in multiple tertiary care centers has been reported to vary between 34% and 75%.23,24 Most relapses occur at prednisone dosages of less than 20 mg orally daily and within the first year after diagnosis. The most common symptoms are limb ischemia, jaw claudication, constitutional symptoms, headaches, and polymyalgia rheumatica. In a review of 286 patients,25 213 (74%) had at least 1 relapse. The first relapse occurred in the first year in 50%, by 2 years in 68%, and by 5 years in 79%.
ADVERSE EFFECTS OF GLUCOCORTICOIDS
In high doses, glucocorticoids have well-known adverse effects. In a population-based study of 120 patients, each patient treated with glucocorticoids experienced at least 1 adverse effect (cataract, fracture, infection, osteonecrosis, diabetes, hypertension, weight gain, capillary fragility, or hair loss).26 The effects were related to aging and cumulative dosage of prednisone but not to the initial dosage.
Glucocorticoids can affect many organs and systems:
- Eyes (cataracts, increased intraocular pressure, exophthalmos)
- Heart (premature atherosclerotic disease, hypertension, fluid retention, hyperlipidemia, arrhythmias)
- Gastrointestinal system (ulcer, gastrointestinal bleeding, gastritis, visceral perforation, hepatic steatosis, acute pancreatitis)
- Bone and muscle (osteopenia, osteoporosis, osteonecrosis, myopathy)
- Brain (mood disorder, psychosis, memory impairment)
- Endocrine system (hyperglycemia, hypothalamic-pituitary-adrenal axis suppression)
- Immune system (immunosuppression, leading to infection and leukocytosis).
Patients receiving a glucocorticoid dose equivalent to 20 mg or more of prednisone daily for 1 month or more who also have another cause of immunocompromise need prophylaxis against Pneumocystis jirovecii pneumonia.27 They should also receive appropriate immunizations before starting glucocorticoids. Live-virus vaccines should not be given to these patients until they have been off glucocorticoids for 1 month.
Glucocorticoids and bone loss
Glucocorticoids are associated with bone loss and fracture, which can occur within the first few months of use and with dosages as low as 2.5 to 7.5 mg orally daily.28 Therefore, glucocorticoid-induced bone loss has to be treated aggressively, particularly in patients who are older and have a history of fragility fracture.
For patients with GCA who need glucocorticoids in doses greater than 5 mg orally daily for more than 3 months, the following measures are advised to decrease the risk of bone loss:
- Weight-bearing exercise
- Smoking cessation
- Moderation in alcohol intake
- Measures to prevent falls29
- Supplementation with 1,200 mg of calcium and 800 IU of vitamin D.30
Pharmacologic therapy should be initiated in men over age 50 who have established osteoporosis and in postmenopausal women with established osteoporosis or osteopenia. For men over age 50 with established osteopenia, risk assessment with the glucocorticoid-corrected FRAX score (www.sheffield.ac.uk/FRAX) should be performed to identify those at high risk in whom pharmacologic therapy is warranted.31
Bisphosphonates are the first-line therapy for glucocorticoid-induced osteoporosis.32
Teriparatide is the second-line therapy and is used in patients who cannot tolerate bisphosphonates or other osteoporosis therapies, and in those who have severe osteoporosis, with T scores of –3.5 and below if they have not had a fracture, and –2.5 and below if they have had a fragility fracture.33
Denosumab, a monoclonal antibody to an osteoclast differentiating factor, may be beneficial for some patients with glucocorticoid-induced osteoporosis.34
To assess the efficacy of therapy, measuring bone mineral density at baseline and at 1 year of therapy is recommended. If density is stable or improved, then repeating the measurement at 2- to 3-year intervals is suggested.
TOCILIZUMAB: A STEROID-SPARING MEDICATION
Due to the adverse effects of long-term use of glucocorticoids and high rates of relapse, there is a pressing need for medications that are more efficacious and less toxic to treat GCA.
The European League Against Rheumatism, in its 2009 management guidelines for large-vessel vasculitis, recommend using an adjunctive immunosuppressant agent.19 In the case of GCA, they recommend using methotrexate 10 to 15 mg/week, which has shown modest evidence of reducing the relapse rate and lowering the cumulative doses of glucocorticoids needed.35,36
Studies of tumor necrosis factor inhibitors and abatacept have not yielded significant reductions in the relapse rate or decreased cumulative doses of prednisone.37,38
Advances in treatment for GCA have stagnated, but recent trials39,40 have evaluated the IL-6 receptor alpha inhibitor tocilizumab, given the central role of IL-6 in the pathogenesis of GCA. Case reports have revealed rapid induction and maintenance of remission in GCA using tocilizumab.41,42
Villiger et al39 performed a randomized, placebo-controlled trial to study the efficacy and safety of tocilizumab in induction and maintenance of disease remission in 30 patients with newly diagnosed GCA. The primary outcome, complete remission at 12 weeks, was achieved in 85% of patients who received tocilizumab plus tapered prednisolone, compared with 40% of patients who received placebo plus tapering prednisolone. The tocilizumab group also had favorable results in secondary outcomes including relapse-free survival at 52 weeks, time to first relapse after induction of remission, and cumulative dose of prednisolone.
The GiACTA trial. Stone et al40 studied the effect of tocilizumab on rates of relapse during glucocorticoid tapering in 251 GCA patients over the course of 52 weeks. Patients were randomized in a 2:1:1:1 ratio to 4 treatment groups:
- Tocilizumab weekly plus prednisone, with prednisone tapered over 26 weeks
- Tocilizumab every other week plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 26 weeks
- Placebo plus prednisone tapered over 52 weeks.
The primary outcome was the rate of sustained glucocorticoid-free remission at 52 weeks. Secondary outcomes included the remission rate, the cumulative glucocorticoid dose, and safety measures. At 52 weeks, the rates of sustained remission were:
- 56% with tocilizumab weekly
- 53% with tocilizumab every other week
- 14% with placebo plus 26-week prednisone taper
- 18% with placebo plus 52-week taper.
Differences between the active treatment groups and the placebo groups were statistically significant (P < .001).
The cumulative dose of prednisone in tocilizumab recipients was significantly less than in placebo recipients. Rates of adverse events were similar. Ultimately, the study showed that tocilizumab, either weekly or every other week, was more effective than prednisone alone at sustaining glucocorticoid-free remission in patients with GCA.
However, the study also raised questions about tocilizumab’s toxic effect profile and its long-term efficacy, as well as who are the optimal candidates for this therapy. Data on long-term use of tocilizumab are primarily taken from its use in rheumatoid arthritis.43 As of this writing, Stone et al are conducting an open-label trial to help provide long-term safety and efficacy data in patients with GCA. In the meantime, we must extrapolate data from the long-term use of tocilizumab in rheumatoid arthritis.
Tocilizumab and lower gastrointestinal tract perforation
One of the major adverse effects of long-term use of tocilizumab is lower gastrointestinal tract perforation.
Xie et al,44 in 2016, reported that the risk of perforation in patients on tocilizumab for rheumatoid arthritis was more than 2 times higher than in patients taking a tumor necrosis factor inhibitor. However, the absolute rates of perforation were low overall, roughly 1 to 3 per 1,000 patient-years in the tocilizumab group. Risk factors for perforation included older age, history of diverticulitis or other gastrointestinal tract condition, and prednisone doses of 7.5 mg or more a day.
Does tocilizumab prevent blindness?
Another consideration is that tocilizumab may not prevent optic neuropathy. In the GiACTA trial, 1 patient in the group receiving tocilizumab every other week developed optic neuropathy.40 Prednisone had been completely tapered off at the time, and the condition resolved when glucocorticoids were restarted. Thus, it is unknown if tocilizumab would be effective on its own without concomitant use of glucocorticoids.
Vision loss is one of the most severe complications of GCA, and it is still unclear whether tocilizumab can prevent vision loss in GCA. Also, we still have no data on the effect of tocilizumab on histopathologic findings, and whether biopsy yield diminishes over time. We hope future studies will help guide us in this regard.
No guidelines on tocilizumab yet
Clinical guidelines on the appropriate use of tocilizumab in GCA are lacking. The American College of Rheumatology and the European League Against Rheumatism have yet to publish updated guidelines with comments on use of tocilizumab. Therefore, it is unclear if tocilizumab is a first-line treatment in GCA, as its efficacy alone without glucocorticoids and its long-term safety in GCA patients have not been studied.
Treatment with tocilizumab should be individualized; it should be considered in patients who have had adverse effects from glucocorticoids, and in patients who experience a flare or cannot have their glucocorticoid dose lowered to an appropriate range.
The optimal duration of tocilizumab therapy is also unknown. However, using the GiACTA study as a rough guide, we try to limit its use to 1 year until additional data are available.
Patients on IL-6 inhibition may have suppressed C-reactive protein regardless of disease activity.43 Therefore, this laboratory value may not be reliable in determining active disease in patients on tocilizumab.
The GiACTA trial has shown an impressive improvement in the relapse-free remission period in patients with GCA taking tocilizumab. However, much work needs to be done to define the safety of this medication and determine which patients should be started on it. In the meantime, we recommend starting high-dose glucocorticoid therapy as soon as the diagnosis of GCA is suspected. In patients who do not tolerate glucocorticoids or whose disease flares during glucocorticoid taper, we recommend starting treatment with tocilizumab either once a week or every other week for at least 1 year.
- Brekke LK, Diamantopoulos AP, Fevang BT, Aßmus J, Esperø E, Gjesdal CG. Incidence of giant cell arteritis in Western Norway 1972–2012: a retrospective cohort study. Arthritis Res Ther 2017; 19(1):278. doi:10.1186/s13075-017-1479-6
- Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology (Oxford) 2017; 56(4):506–515. doi:10.1093/rheumatology/kew273
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med 2014; 371(17):1653. doi:10.1056/NEJMc1409206
- Ghinoi A, Pipitone N, Nicolini A, et al. Large-vessel involvement in recent-onset giant cell arteritis: a case-control colour-Doppler sonography study. Rheumatology (Oxford) 2012; 51(4):730–734. doi:10.1093/rheumatology/ker329
- Prieto-González S, Depetris M, García-Martínez A, et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: a prospective, case-control study. Ann Rheum Dis 2014; 73(7):1388–1392. doi:10.1136/annrheumdis-2013-204572
- Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum 1999; 42(2):311–317. doi:10.1002/1529-0131(199902)42:2<311::AID-ANR14>3.0.CO;2-F
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45(2):140–145. doi:10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
- Liozon E, Jauberteau-Marchan MO, Ly K, Loustaud-Ratti V, Soria P, Vidal E. Giant cell arteritis with a low erythrocyte sedimentation rate: comments on the article by Salvarani and Hunder. Arthritis Rheum 2002; 47(6):692–694. doi:10.1002/art.10809
- Yu-Wai-Man P, Dayan MR. Giant cell arteritis with normal inflammatory markers. Acta Ophthalmol Scand 2007; 85(4):460. doi:10.1111/j.1600-0420.2006.00864.x
- Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA 2016; 315(22):2442–2458. doi:10.1001/jama.2016.5444
- Niederkohr RD, Levin LA. Management of the patient with suspected temporal arteritis a decision-analytic approach. Ophthalmology 2005; 112(5):744–756. doi:10.1016/j.ophtha.2005.01.031
- Bowling K, Rait J, Atkinson J, Srinivas G. Temporal artery biopsy in the diagnosis of giant cell arteritis: does the end justify the means? Ann Med Surg (Lond) 2017; 20:1–5. doi:10.1016/j.amsu.2017.06.020
- Daily B, Dassow P, Haynes J, Nashelsky J. Giant cell arteritis: biopsy after corticosteroid initiation. Am Fam Physician 2017; 95(2):116–117. pmid:28084703
- Durling B, Toren A, Patel V, Gilberg S, Weis E, Jordan D. Incidence of discordant temporal artery biopsy in the diagnosis of giant cell arteritis. Can J Ophthalmol 2014; 49(2):157–161. doi:10.1016/j.jcjo.2013.12.008
- Dejaco C, Ramiro S, Duftner C, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis 2018; 77(5):636–643. doi:10.1136/annrheumdis-2017-212649
- Aschwanden M, Imfeld S, Staub D, et al. The ultrasound compression sign to diagnose temporal giant cell arteritis shows an excellent interobserver agreement. Clin Exp Rheumatol 2015; 33(2 suppl 89):S-113–S-115. pmid:26016760
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100(4):550–555. pmid:8479714
- Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology 2003; 110(6):1204–1215. doi:10.1016/S0161-6420(03)00228-8
- Mukhtyar C, Guillevin L, Cid MC, et al; European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009; 68(3):318–323. doi:10.1136/ard.2008.088351
- Dejaco C, Singh YP, Perel P, et al; European League Against Rheumatism; American College of Rheumatology. 2015 recommendations for the management of polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Ann Rheum Dis 2015; 74(10):1799–1807. doi:10.1136/annrheumdis-2015-207492
- Bienvenu B, Ly KH, Lambert M, et al; Groupe d’Étude Français des Artérites des gros Vaisseaux, under the Aegis of the Filière des Maladies Auto-Immunes et Auto-Inflammatoires Rares. Management of giant cell arteritis: recommendations of the French Study Group for Large Vessel Vasculitis (GEFA). Rev Med Interne 2016; 37(3):154–165. doi:10.1016/j.revmed.2015.12.015
- Hayreh SS, Biousse V. Treatment of acute visual loss in giant cell arteritis: should we prescribe high-dose intravenous steroids or just oral steroids? J Neuroophthalmol 2012; 32(3):278–287. doi:10.1097/WNO.0b013e3182688218
- Restuccia G, Boiardi L, Cavazza A, et al. Flares in biopsy-proven giant cell arteritis in Northern Italy: characteristics and predictors in a long-term follow-up study. Medicine (Baltimore) 2016; 95(19):e3524. doi:10.1097/MD.0000000000003524
- Kermani TA, Warrington KJ, Cuthbertson D, et al; Vasculitis Clinical Research Consortium. Disease relapses among patients with giant cell arteritis: a prospective, longitudinal cohort study. J Rheumatol 2015; 42(7):1213–1217. doi:10.3899/jrheum.141347
- Labarca C, Koster MJ, Crowson CS, et al. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology (Oxford) 2016; 55(2):347–356. doi:10.1093/rheumatology/kev348
- Proven A, Gabriel SE, Orces C, O’Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum 2003; 49(5):703–708. doi:10.1002/art.11388
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34(8):1098–1107. doi:10.1086/339548
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13(10):777–787. doi:10.1007/s001980200108
- Heffernan MP, Saag KG, Robinson JK, Callen JP. Prevention of osteoporosis associated with chronic glucocorticoid therapy. JAMA 2006; 295(11):1300–1303. pmid:16541489
- Buckley L, Guyatt G, Fink HA, et al. 2017 American College of Rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2017; 69(8):1095–1110. doi:10.1002/acr.23279
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res 201; 62(11):1515–1526. doi:10.1002/acr.20295
- Allen CS, Yeung JH, Vandermeer B, Homik J. Bisphosphonates for steroid-induced osteoporosis. Cochrane Database Syst Rev 2016; 10:CD001347. doi:10.1002/14651858.CD001347.pub2
- Carpinteri R, Porcelli T, Mejia C, et al. Glucocorticoid-induced osteoporosis and parathyroid hormone. J Endocrinol Invest 2010; 33(suppl 7):16–21. pmid:20938221
- Saag KG, Wagman RB, Geusens P, et al. Denosumab versus risedronate in glucocorticoid-induced osteoporosis: a multicentre, randomised, double-blind, active-controlled, double-dummy, non-inferiority study. Lancet Diabetes Endocrinol 2018; 6(6):445–454. doi:10.1016/S2213-8587(18)30075-5
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46(5):1309–1318. doi:10.1002/art.10262
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19(5):495–501. pmid:11579707
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146(9):621–630. pmid:17470830
- Langford CA, Cuthbertson D, Ytterberg SR, et al; Vasculitis Clinical Research Consortium. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol 2017; 69(4):837–845. doi:10.1002/art.40044
- Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016; 387(10031):1921–1927. doi:10.1016/S0140-6736(16)00560-2
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Oliveira F, Butendieck RR, Ginsburg WW, Parikh K, Abril A. Tocilizumab, an effective treatment for relapsing giant cell arteritis. Clin Exp Rheumatol 2014; 32(3 suppl 82):S76–S78. pmid:24854376
- Loricera J, Blanco R, Hernández JL, et al. Tocilizumab in giant cell arteritis: multicenter open-label study of 22 patients. Semin Arthritis Rheum 2015; 44(6):717–723. doi:10.1016/j.semarthrit.2014.12.005
- Tamaki H, Hajj-Ali RA. Tocilizumab for giant cell arteritis—a new giant step in an old disease. JAMA Neurol 2018; 75(2):145–146. doi:10.1001/jamaneurol.2017.3811
- Xie F, Yun H, Bernatsky S, Curtis JR. Risk for gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologics. Arthritis Rheumatol 2016; 68(11):2612–2617. doi:10.1002/art.39761
- Brekke LK, Diamantopoulos AP, Fevang BT, Aßmus J, Esperø E, Gjesdal CG. Incidence of giant cell arteritis in Western Norway 1972–2012: a retrospective cohort study. Arthritis Res Ther 2017; 19(1):278. doi:10.1186/s13075-017-1479-6
- Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology (Oxford) 2017; 56(4):506–515. doi:10.1093/rheumatology/kew273
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med 2014; 371(17):1653. doi:10.1056/NEJMc1409206
- Ghinoi A, Pipitone N, Nicolini A, et al. Large-vessel involvement in recent-onset giant cell arteritis: a case-control colour-Doppler sonography study. Rheumatology (Oxford) 2012; 51(4):730–734. doi:10.1093/rheumatology/ker329
- Prieto-González S, Depetris M, García-Martínez A, et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: a prospective, case-control study. Ann Rheum Dis 2014; 73(7):1388–1392. doi:10.1136/annrheumdis-2013-204572
- Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum 1999; 42(2):311–317. doi:10.1002/1529-0131(199902)42:2<311::AID-ANR14>3.0.CO;2-F
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45(2):140–145. doi:10.1002/1529-0131(200104)45:2<140::AID-ANR166>3.0.CO;2-2
- Liozon E, Jauberteau-Marchan MO, Ly K, Loustaud-Ratti V, Soria P, Vidal E. Giant cell arteritis with a low erythrocyte sedimentation rate: comments on the article by Salvarani and Hunder. Arthritis Rheum 2002; 47(6):692–694. doi:10.1002/art.10809
- Yu-Wai-Man P, Dayan MR. Giant cell arteritis with normal inflammatory markers. Acta Ophthalmol Scand 2007; 85(4):460. doi:10.1111/j.1600-0420.2006.00864.x
- Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA 2016; 315(22):2442–2458. doi:10.1001/jama.2016.5444
- Niederkohr RD, Levin LA. Management of the patient with suspected temporal arteritis a decision-analytic approach. Ophthalmology 2005; 112(5):744–756. doi:10.1016/j.ophtha.2005.01.031
- Bowling K, Rait J, Atkinson J, Srinivas G. Temporal artery biopsy in the diagnosis of giant cell arteritis: does the end justify the means? Ann Med Surg (Lond) 2017; 20:1–5. doi:10.1016/j.amsu.2017.06.020
- Daily B, Dassow P, Haynes J, Nashelsky J. Giant cell arteritis: biopsy after corticosteroid initiation. Am Fam Physician 2017; 95(2):116–117. pmid:28084703
- Durling B, Toren A, Patel V, Gilberg S, Weis E, Jordan D. Incidence of discordant temporal artery biopsy in the diagnosis of giant cell arteritis. Can J Ophthalmol 2014; 49(2):157–161. doi:10.1016/j.jcjo.2013.12.008
- Dejaco C, Ramiro S, Duftner C, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis 2018; 77(5):636–643. doi:10.1136/annrheumdis-2017-212649
- Aschwanden M, Imfeld S, Staub D, et al. The ultrasound compression sign to diagnose temporal giant cell arteritis shows an excellent interobserver agreement. Clin Exp Rheumatol 2015; 33(2 suppl 89):S-113–S-115. pmid:26016760
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100(4):550–555. pmid:8479714
- Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology 2003; 110(6):1204–1215. doi:10.1016/S0161-6420(03)00228-8
- Mukhtyar C, Guillevin L, Cid MC, et al; European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009; 68(3):318–323. doi:10.1136/ard.2008.088351
- Dejaco C, Singh YP, Perel P, et al; European League Against Rheumatism; American College of Rheumatology. 2015 recommendations for the management of polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Ann Rheum Dis 2015; 74(10):1799–1807. doi:10.1136/annrheumdis-2015-207492
- Bienvenu B, Ly KH, Lambert M, et al; Groupe d’Étude Français des Artérites des gros Vaisseaux, under the Aegis of the Filière des Maladies Auto-Immunes et Auto-Inflammatoires Rares. Management of giant cell arteritis: recommendations of the French Study Group for Large Vessel Vasculitis (GEFA). Rev Med Interne 2016; 37(3):154–165. doi:10.1016/j.revmed.2015.12.015
- Hayreh SS, Biousse V. Treatment of acute visual loss in giant cell arteritis: should we prescribe high-dose intravenous steroids or just oral steroids? J Neuroophthalmol 2012; 32(3):278–287. doi:10.1097/WNO.0b013e3182688218
- Restuccia G, Boiardi L, Cavazza A, et al. Flares in biopsy-proven giant cell arteritis in Northern Italy: characteristics and predictors in a long-term follow-up study. Medicine (Baltimore) 2016; 95(19):e3524. doi:10.1097/MD.0000000000003524
- Kermani TA, Warrington KJ, Cuthbertson D, et al; Vasculitis Clinical Research Consortium. Disease relapses among patients with giant cell arteritis: a prospective, longitudinal cohort study. J Rheumatol 2015; 42(7):1213–1217. doi:10.3899/jrheum.141347
- Labarca C, Koster MJ, Crowson CS, et al. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology (Oxford) 2016; 55(2):347–356. doi:10.1093/rheumatology/kev348
- Proven A, Gabriel SE, Orces C, O’Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum 2003; 49(5):703–708. doi:10.1002/art.11388
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34(8):1098–1107. doi:10.1086/339548
- van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002; 13(10):777–787. doi:10.1007/s001980200108
- Heffernan MP, Saag KG, Robinson JK, Callen JP. Prevention of osteoporosis associated with chronic glucocorticoid therapy. JAMA 2006; 295(11):1300–1303. pmid:16541489
- Buckley L, Guyatt G, Fink HA, et al. 2017 American College of Rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2017; 69(8):1095–1110. doi:10.1002/acr.23279
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res 201; 62(11):1515–1526. doi:10.1002/acr.20295
- Allen CS, Yeung JH, Vandermeer B, Homik J. Bisphosphonates for steroid-induced osteoporosis. Cochrane Database Syst Rev 2016; 10:CD001347. doi:10.1002/14651858.CD001347.pub2
- Carpinteri R, Porcelli T, Mejia C, et al. Glucocorticoid-induced osteoporosis and parathyroid hormone. J Endocrinol Invest 2010; 33(suppl 7):16–21. pmid:20938221
- Saag KG, Wagman RB, Geusens P, et al. Denosumab versus risedronate in glucocorticoid-induced osteoporosis: a multicentre, randomised, double-blind, active-controlled, double-dummy, non-inferiority study. Lancet Diabetes Endocrinol 2018; 6(6):445–454. doi:10.1016/S2213-8587(18)30075-5
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46(5):1309–1318. doi:10.1002/art.10262
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19(5):495–501. pmid:11579707
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146(9):621–630. pmid:17470830
- Langford CA, Cuthbertson D, Ytterberg SR, et al; Vasculitis Clinical Research Consortium. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol 2017; 69(4):837–845. doi:10.1002/art.40044
- Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016; 387(10031):1921–1927. doi:10.1016/S0140-6736(16)00560-2
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Oliveira F, Butendieck RR, Ginsburg WW, Parikh K, Abril A. Tocilizumab, an effective treatment for relapsing giant cell arteritis. Clin Exp Rheumatol 2014; 32(3 suppl 82):S76–S78. pmid:24854376
- Loricera J, Blanco R, Hernández JL, et al. Tocilizumab in giant cell arteritis: multicenter open-label study of 22 patients. Semin Arthritis Rheum 2015; 44(6):717–723. doi:10.1016/j.semarthrit.2014.12.005
- Tamaki H, Hajj-Ali RA. Tocilizumab for giant cell arteritis—a new giant step in an old disease. JAMA Neurol 2018; 75(2):145–146. doi:10.1001/jamaneurol.2017.3811
- Xie F, Yun H, Bernatsky S, Curtis JR. Risk for gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologics. Arthritis Rheumatol 2016; 68(11):2612–2617. doi:10.1002/art.39761
KEY POINTS
- Giant cell arteritis can present with cranial symptoms, extracranial large-vessel involvement, or polymyalgia rheumatica.
- Temporal artery biopsy is the standard for diagnosis.
- Adverse effects of glucocorticoid treatment, particularly bone loss, need to be managed.
- In patients treated with glucocorticoids alone, the relapse rate is high when the drugs are tapered; thus, prolonged treatment is required.

Clinical trials: More to learn than the results

The clinical update of giant cell arteritis (GCA) by Rinden et al in this issue of the Journal reminded me of just how much of our management of this disease has, for decades, been based on retrospective studies (we owe a lot to clinicians from the Mayo Clinic for their compiled observations) tempered by our own recalled experiences, which we may at times twist a bit to fit prevailing paradigms. Several prospective interventional studies, perhaps most importantly the Giant-Cell Arteritis Actemra (GIACTA) trial,1 evaluated the ability of the interleukin 6 (IL-6) antagonist tocilizumab to supplant the protracted use of glucocorticoids in the treatment of GCA. But I learned much more from this trial, in the form of collected clinical tidbits, than just the bottom-line abstract conclusion that IL-6 antagonism is at least a promising approach in many patients with GCA.
As teachers, we tell students to read the entire published clinical trial report, not just the abstract and conclusions. Over the years, I have been impatient with those who violated this dictum, but I now often find myself among the ranks of those who would have been targets of my disapproval. Usually, the articles that I merely skim lie outside my subsubspecialty areas of interest, as time constraints make this abridged reading a necessity for survival, but that excuse does not diminish the self-recognition of my often less-than-complete understanding of the clinical condition being reported. Delving into the nuances of GIACTA truly emphasized that point.
The external validity of any trial rests on understanding the trial’s methods. In the case of GIACTA, there was much more to be learned and affirmed from the trial1 than that 1 year of tocilizumab treatment met the primary end point of increasing the percent of patients achieving sustained remission at week 52 after a rapid 26-week tapering off of prednisone compared with placebo.
One treatment group in the GIACTA trial underwent an aggressive 6-month tapering of prednisone, while another underwent a more protracted tapering over 12 months (more in line with common practice). Patients tapered over 6 months also received either the IL-6 antagonist or placebo for the full year. The concept was that if IL-6 blockade is a correct approach, then it will maintain remission in more patients, and significantly reduce the total amount of steroid needed to control the disease, despite rapid, aggressive steroid tapering. This turned out to be correct, although more than 20% of the drug-treated patients still experienced a flare of GCA (vs 68% of the placebo-treated group).
Somewhat surprising was that almost 20% of the entered patients did not achieve an initial remission despite receiving high-dose prednisone. The traditional teaching is that if a patient diagnosed with GCA does not respond to high-dose steroids, the diagnosis should be questioned.
Another interesting facet of the study relates to the diagnosis. We are becoming more aware of the different GCA phenotypes, which include prominent polymyalgia rheumatica or constitutional features, “classic” GCA with cranial symptoms, and dominant large-vessel vasculitis (aortitis and major aortic branch disease). In GIACTA, even though imaging was not mandated, 37% of participants were enrolled based in part on imaging results (CT, MRI, angiography, or PET-CT), not on the results of temporal artery biopsy. This forces us to think more broadly about diagnosing and staging GCA, and to wonder if we should even modify our approach to other clinical challenges, including unexplained fever and wasting in older patients.
Another tidbit that came out of the study relates to the relationship between the acute-phase response and clinical flares. We already knew that a rise in the erythrocyte sedimentation rate is a nonspecific sign and does not equate with a flare. In this trial one-third of patients in the placebo group who had a flare had a normal sedimentation rate or C-reactive protein during the flare, and about one-third of patients in the placebo group were receiving more than 10 mg of prednisone. In preliminary reports of follow-up after 52 weeks of treatment,2 patients who had achieved complete remission with the IL-6 antagonist and were off of prednisone were still not out of the woods; when the drug was discontinued, many flares continued to occur over the 2-year study extension. We have more to learn about what triggers and drives flares in this disease.
Thus, in addition to informing us of a successful “steroid-sparing” and rescue drug option for our patients with GCA, the details of this well-conducted trial both challenge and reaffirm some of our clinical impressions. Clearly, GCA must be defined for many patients as a very chronic disease, perhaps with occult vascular reservoirs, the biologic basis of which remains to be defined.
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Stone JH, Bao M, Han J, et al. Long-term outcome of tocilizumab for patients with giant cell arteritis: results from part 2 of the GIACTA trial (abstract). Ann Rheum Dis 2019; 78:145–146. doi:10.1136/annrheumdis-2019-eular.2099
The clinical update of giant cell arteritis (GCA) by Rinden et al in this issue of the Journal reminded me of just how much of our management of this disease has, for decades, been based on retrospective studies (we owe a lot to clinicians from the Mayo Clinic for their compiled observations) tempered by our own recalled experiences, which we may at times twist a bit to fit prevailing paradigms. Several prospective interventional studies, perhaps most importantly the Giant-Cell Arteritis Actemra (GIACTA) trial,1 evaluated the ability of the interleukin 6 (IL-6) antagonist tocilizumab to supplant the protracted use of glucocorticoids in the treatment of GCA. But I learned much more from this trial, in the form of collected clinical tidbits, than just the bottom-line abstract conclusion that IL-6 antagonism is at least a promising approach in many patients with GCA.
As teachers, we tell students to read the entire published clinical trial report, not just the abstract and conclusions. Over the years, I have been impatient with those who violated this dictum, but I now often find myself among the ranks of those who would have been targets of my disapproval. Usually, the articles that I merely skim lie outside my subsubspecialty areas of interest, as time constraints make this abridged reading a necessity for survival, but that excuse does not diminish the self-recognition of my often less-than-complete understanding of the clinical condition being reported. Delving into the nuances of GIACTA truly emphasized that point.
The external validity of any trial rests on understanding the trial’s methods. In the case of GIACTA, there was much more to be learned and affirmed from the trial1 than that 1 year of tocilizumab treatment met the primary end point of increasing the percent of patients achieving sustained remission at week 52 after a rapid 26-week tapering off of prednisone compared with placebo.
One treatment group in the GIACTA trial underwent an aggressive 6-month tapering of prednisone, while another underwent a more protracted tapering over 12 months (more in line with common practice). Patients tapered over 6 months also received either the IL-6 antagonist or placebo for the full year. The concept was that if IL-6 blockade is a correct approach, then it will maintain remission in more patients, and significantly reduce the total amount of steroid needed to control the disease, despite rapid, aggressive steroid tapering. This turned out to be correct, although more than 20% of the drug-treated patients still experienced a flare of GCA (vs 68% of the placebo-treated group).
Somewhat surprising was that almost 20% of the entered patients did not achieve an initial remission despite receiving high-dose prednisone. The traditional teaching is that if a patient diagnosed with GCA does not respond to high-dose steroids, the diagnosis should be questioned.
Another interesting facet of the study relates to the diagnosis. We are becoming more aware of the different GCA phenotypes, which include prominent polymyalgia rheumatica or constitutional features, “classic” GCA with cranial symptoms, and dominant large-vessel vasculitis (aortitis and major aortic branch disease). In GIACTA, even though imaging was not mandated, 37% of participants were enrolled based in part on imaging results (CT, MRI, angiography, or PET-CT), not on the results of temporal artery biopsy. This forces us to think more broadly about diagnosing and staging GCA, and to wonder if we should even modify our approach to other clinical challenges, including unexplained fever and wasting in older patients.
Another tidbit that came out of the study relates to the relationship between the acute-phase response and clinical flares. We already knew that a rise in the erythrocyte sedimentation rate is a nonspecific sign and does not equate with a flare. In this trial one-third of patients in the placebo group who had a flare had a normal sedimentation rate or C-reactive protein during the flare, and about one-third of patients in the placebo group were receiving more than 10 mg of prednisone. In preliminary reports of follow-up after 52 weeks of treatment,2 patients who had achieved complete remission with the IL-6 antagonist and were off of prednisone were still not out of the woods; when the drug was discontinued, many flares continued to occur over the 2-year study extension. We have more to learn about what triggers and drives flares in this disease.
Thus, in addition to informing us of a successful “steroid-sparing” and rescue drug option for our patients with GCA, the details of this well-conducted trial both challenge and reaffirm some of our clinical impressions. Clearly, GCA must be defined for many patients as a very chronic disease, perhaps with occult vascular reservoirs, the biologic basis of which remains to be defined.
The clinical update of giant cell arteritis (GCA) by Rinden et al in this issue of the Journal reminded me of just how much of our management of this disease has, for decades, been based on retrospective studies (we owe a lot to clinicians from the Mayo Clinic for their compiled observations) tempered by our own recalled experiences, which we may at times twist a bit to fit prevailing paradigms. Several prospective interventional studies, perhaps most importantly the Giant-Cell Arteritis Actemra (GIACTA) trial,1 evaluated the ability of the interleukin 6 (IL-6) antagonist tocilizumab to supplant the protracted use of glucocorticoids in the treatment of GCA. But I learned much more from this trial, in the form of collected clinical tidbits, than just the bottom-line abstract conclusion that IL-6 antagonism is at least a promising approach in many patients with GCA.
As teachers, we tell students to read the entire published clinical trial report, not just the abstract and conclusions. Over the years, I have been impatient with those who violated this dictum, but I now often find myself among the ranks of those who would have been targets of my disapproval. Usually, the articles that I merely skim lie outside my subsubspecialty areas of interest, as time constraints make this abridged reading a necessity for survival, but that excuse does not diminish the self-recognition of my often less-than-complete understanding of the clinical condition being reported. Delving into the nuances of GIACTA truly emphasized that point.
The external validity of any trial rests on understanding the trial’s methods. In the case of GIACTA, there was much more to be learned and affirmed from the trial1 than that 1 year of tocilizumab treatment met the primary end point of increasing the percent of patients achieving sustained remission at week 52 after a rapid 26-week tapering off of prednisone compared with placebo.
One treatment group in the GIACTA trial underwent an aggressive 6-month tapering of prednisone, while another underwent a more protracted tapering over 12 months (more in line with common practice). Patients tapered over 6 months also received either the IL-6 antagonist or placebo for the full year. The concept was that if IL-6 blockade is a correct approach, then it will maintain remission in more patients, and significantly reduce the total amount of steroid needed to control the disease, despite rapid, aggressive steroid tapering. This turned out to be correct, although more than 20% of the drug-treated patients still experienced a flare of GCA (vs 68% of the placebo-treated group).
Somewhat surprising was that almost 20% of the entered patients did not achieve an initial remission despite receiving high-dose prednisone. The traditional teaching is that if a patient diagnosed with GCA does not respond to high-dose steroids, the diagnosis should be questioned.
Another interesting facet of the study relates to the diagnosis. We are becoming more aware of the different GCA phenotypes, which include prominent polymyalgia rheumatica or constitutional features, “classic” GCA with cranial symptoms, and dominant large-vessel vasculitis (aortitis and major aortic branch disease). In GIACTA, even though imaging was not mandated, 37% of participants were enrolled based in part on imaging results (CT, MRI, angiography, or PET-CT), not on the results of temporal artery biopsy. This forces us to think more broadly about diagnosing and staging GCA, and to wonder if we should even modify our approach to other clinical challenges, including unexplained fever and wasting in older patients.
Another tidbit that came out of the study relates to the relationship between the acute-phase response and clinical flares. We already knew that a rise in the erythrocyte sedimentation rate is a nonspecific sign and does not equate with a flare. In this trial one-third of patients in the placebo group who had a flare had a normal sedimentation rate or C-reactive protein during the flare, and about one-third of patients in the placebo group were receiving more than 10 mg of prednisone. In preliminary reports of follow-up after 52 weeks of treatment,2 patients who had achieved complete remission with the IL-6 antagonist and were off of prednisone were still not out of the woods; when the drug was discontinued, many flares continued to occur over the 2-year study extension. We have more to learn about what triggers and drives flares in this disease.
Thus, in addition to informing us of a successful “steroid-sparing” and rescue drug option for our patients with GCA, the details of this well-conducted trial both challenge and reaffirm some of our clinical impressions. Clearly, GCA must be defined for many patients as a very chronic disease, perhaps with occult vascular reservoirs, the biologic basis of which remains to be defined.
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Stone JH, Bao M, Han J, et al. Long-term outcome of tocilizumab for patients with giant cell arteritis: results from part 2 of the GIACTA trial (abstract). Ann Rheum Dis 2019; 78:145–146. doi:10.1136/annrheumdis-2019-eular.2099
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377(4):317–328. doi:10.1056/NEJMoa1613849
- Stone JH, Bao M, Han J, et al. Long-term outcome of tocilizumab for patients with giant cell arteritis: results from part 2 of the GIACTA trial (abstract). Ann Rheum Dis 2019; 78:145–146. doi:10.1136/annrheumdis-2019-eular.2099

Anti-Xa assays: What is their role today in antithrombotic therapy?
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.

Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.

The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.
Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.
The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.
Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.
The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
KEY POINTS
- Intravenous unfractionated heparin treatment is typically monitored by the activated partial thromboplastin time (aPTT), with a therapeutic target defined as the range that corresponds to an anti-Xa level of 0.3 to 0.7 U/mL.
- Monitoring unfractionated heparin is important to achieve a therapeutic target within the first 24 hours and to maintain therapeutic levels thereafter.
- The heparin anti-Xa assay is unreliable for unfractionated heparin monitoring when switching from oral factor Xa inhibitor therapy to intravenous unfractionated heparin. In such cases, the aPTT is preferred.
- Most patients receiving low-molecular-weight heparin do not need monitoring, but monitoring should be considered for pregnant women with prosthetic heart valves, using an anti-Xa assay specific for low-molecular-weight heparin.
Respiratory effects may account for worse survival in women undergoing DTA and TAAA repair
Women undergoing open descending thoracic aortic aneurysm (DTA) and open thoracoabdominal aortic aneurysm (TAAA) repair are not at greater risk for operative mortality than their male counterparts. However, they are at significantly greater risk for major adverse events and have significantly lower 5-year survival, according to the results of a single institution database review of 738 surgery patients.
From May 1997 to June 2017, there were 462 men (59%) and 321 women (41%) who underwent open repair of DTA or TAAA, according to Leonard N. Girardi, MD, and colleagues from Weill Cornell Medicine, New York, who performed the study published in the Journal of Vascular Surgery. The researchers used logistic regression and Cox regression analyses to assess the effect of sex on perioperative and long-term outcomes.
Demographically, women were significantly older (67.6 years vs. 62.6 years), with a significantly higher incidence of chronic obstructive pulmonary disease (47.0% vs. 35.7%) and a significantly greater percentage of patients with a forced expiratory volume in 1 second less than 50% (28.3% vs 18.2%). Degenerative aneurysms were significantly more common in women (61.7% vs. 41.6%), whereas chronic dissections significantly predominated in men (42.4% vs. 23.1%). Operative mortality was not significantly different between women and men (5.6% vs. 6.2%); however, women were significantly more likely to require a tracheostomy after surgery (10.6% vs. 5.0%).
Logistic regression found that being a woman was an independent risk factor for a composite of major adverse events (odds ratio, 2.68) and need for tracheostomy (OR, 3.73). In addition, women had significantly worse 5-year survival than men undergoing DTA or TAAA repair (59.7% vs. 66.2%, P =.025). There was no difference in overall survival between 1997-2007 and 2008-2017.
“Women and men undergoing TAAA repair have significant and consistent differences in preoperative characteristics. Despite these differences, operative mortality is similar between the two groups. However, women are at significantly increased risk of [major adverse events], especially respiratory failure, because of those differences in risk factors, including age, pulmonary function, and aneurysm etiology,” the researchers concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Girardi LN et al. J Vasc Surg 2019;69:1028-35.
Women undergoing open descending thoracic aortic aneurysm (DTA) and open thoracoabdominal aortic aneurysm (TAAA) repair are not at greater risk for operative mortality than their male counterparts. However, they are at significantly greater risk for major adverse events and have significantly lower 5-year survival, according to the results of a single institution database review of 738 surgery patients.
From May 1997 to June 2017, there were 462 men (59%) and 321 women (41%) who underwent open repair of DTA or TAAA, according to Leonard N. Girardi, MD, and colleagues from Weill Cornell Medicine, New York, who performed the study published in the Journal of Vascular Surgery. The researchers used logistic regression and Cox regression analyses to assess the effect of sex on perioperative and long-term outcomes.
Demographically, women were significantly older (67.6 years vs. 62.6 years), with a significantly higher incidence of chronic obstructive pulmonary disease (47.0% vs. 35.7%) and a significantly greater percentage of patients with a forced expiratory volume in 1 second less than 50% (28.3% vs 18.2%). Degenerative aneurysms were significantly more common in women (61.7% vs. 41.6%), whereas chronic dissections significantly predominated in men (42.4% vs. 23.1%). Operative mortality was not significantly different between women and men (5.6% vs. 6.2%); however, women were significantly more likely to require a tracheostomy after surgery (10.6% vs. 5.0%).
Logistic regression found that being a woman was an independent risk factor for a composite of major adverse events (odds ratio, 2.68) and need for tracheostomy (OR, 3.73). In addition, women had significantly worse 5-year survival than men undergoing DTA or TAAA repair (59.7% vs. 66.2%, P =.025). There was no difference in overall survival between 1997-2007 and 2008-2017.
“Women and men undergoing TAAA repair have significant and consistent differences in preoperative characteristics. Despite these differences, operative mortality is similar between the two groups. However, women are at significantly increased risk of [major adverse events], especially respiratory failure, because of those differences in risk factors, including age, pulmonary function, and aneurysm etiology,” the researchers concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Girardi LN et al. J Vasc Surg 2019;69:1028-35.
Women undergoing open descending thoracic aortic aneurysm (DTA) and open thoracoabdominal aortic aneurysm (TAAA) repair are not at greater risk for operative mortality than their male counterparts. However, they are at significantly greater risk for major adverse events and have significantly lower 5-year survival, according to the results of a single institution database review of 738 surgery patients.
From May 1997 to June 2017, there were 462 men (59%) and 321 women (41%) who underwent open repair of DTA or TAAA, according to Leonard N. Girardi, MD, and colleagues from Weill Cornell Medicine, New York, who performed the study published in the Journal of Vascular Surgery. The researchers used logistic regression and Cox regression analyses to assess the effect of sex on perioperative and long-term outcomes.
Demographically, women were significantly older (67.6 years vs. 62.6 years), with a significantly higher incidence of chronic obstructive pulmonary disease (47.0% vs. 35.7%) and a significantly greater percentage of patients with a forced expiratory volume in 1 second less than 50% (28.3% vs 18.2%). Degenerative aneurysms were significantly more common in women (61.7% vs. 41.6%), whereas chronic dissections significantly predominated in men (42.4% vs. 23.1%). Operative mortality was not significantly different between women and men (5.6% vs. 6.2%); however, women were significantly more likely to require a tracheostomy after surgery (10.6% vs. 5.0%).
Logistic regression found that being a woman was an independent risk factor for a composite of major adverse events (odds ratio, 2.68) and need for tracheostomy (OR, 3.73). In addition, women had significantly worse 5-year survival than men undergoing DTA or TAAA repair (59.7% vs. 66.2%, P =.025). There was no difference in overall survival between 1997-2007 and 2008-2017.
“Women and men undergoing TAAA repair have significant and consistent differences in preoperative characteristics. Despite these differences, operative mortality is similar between the two groups. However, women are at significantly increased risk of [major adverse events], especially respiratory failure, because of those differences in risk factors, including age, pulmonary function, and aneurysm etiology,” the researchers concluded.
The authors reported that they had no conflicts of interest.
SOURCE: Girardi LN et al. J Vasc Surg 2019;69:1028-35.
FROM THE JOURNAL OF VASCULAR SURGERY
Dabigatran-induced esophagitis
A 74-year-old man presented to the gastroenterology clinic with a 2-day history of retrosternal discomfort. His vital signs were normal, and laboratory testing showed a normal leukocyte count.
Esophagogastroduodenoscopy (EGD) revealed longitudinal sloughing mucosal casts in the middle and lower esophagus (Figure 1).

The patient had been taking dabigatran 110 mg twice daily for 2 years because of nonvalvular atrial fibrillation. He was also taking amlodipine 2.5 mg/day for hypertension.

DABIGATRAN-INDUCED ESOPHAGITIS

Although no study has investigated the overall prevalence of dabigatran-induced esophagitis, a retrospective database review of 91 patients taking dabigatran and undergoing upper-gastrointestinal endoscopy reported that 19 (20.9%) had endoscopic signs of dabigatran-induced esophagitis.2
Typical symptoms are the acute onset of chest pain, epigastralgia, odynophagia, and dysphagia. But patients can also have no symptoms or only mild symptoms.2,3
Despite dabigatran’s anticoagulant activity, there have been few reports of bleeding, perhaps because the lesions tend to be superficial on the surface of the esophageal mucosa.
Symptoms usually resolve within 1 week after stopping dabigatran and starting a proton pump inhibitor. To prevent mucosal injury, patients should be instructed to take dabigatran with sufficient water and to remain in an upright position for at least 30 minutes afterward.4
- Baehr PH, McDonald GB. Esophageal infections: risk factors, presentation, diagnosis, and treatment. Gastroenterology 1994; 106(2):509–532. pmid:7980741
- Toya Y, Nakamura S, Tomita K, et al. Dabigatran-induced esophagitis: the prevalence and endoscopic characteristics. J Gastroenterol Hepatol 2016; 31(3):610–614. doi:10.1111/jgh.13024
- Ueta E, Fujikawa T, Imagawa A. A case of a slightly symptomatic exfoliative oesophagitis. BMJ Case Rep 2015; pii:bcr2015211925. doi:10.1136/bcr-2015-211925
- Ootani A, Hayashi Y, Miyagi Y. Dabigatran-induced esophagitis. Clin Gastroenterol Hepatol 2014; 12(7):e55–e56. doi:10.1016/j.cgh.2013.09.010
A 74-year-old man presented to the gastroenterology clinic with a 2-day history of retrosternal discomfort. His vital signs were normal, and laboratory testing showed a normal leukocyte count.
Esophagogastroduodenoscopy (EGD) revealed longitudinal sloughing mucosal casts in the middle and lower esophagus (Figure 1).
The patient had been taking dabigatran 110 mg twice daily for 2 years because of nonvalvular atrial fibrillation. He was also taking amlodipine 2.5 mg/day for hypertension.
DABIGATRAN-INDUCED ESOPHAGITIS
Although no study has investigated the overall prevalence of dabigatran-induced esophagitis, a retrospective database review of 91 patients taking dabigatran and undergoing upper-gastrointestinal endoscopy reported that 19 (20.9%) had endoscopic signs of dabigatran-induced esophagitis.2
Typical symptoms are the acute onset of chest pain, epigastralgia, odynophagia, and dysphagia. But patients can also have no symptoms or only mild symptoms.2,3
Despite dabigatran’s anticoagulant activity, there have been few reports of bleeding, perhaps because the lesions tend to be superficial on the surface of the esophageal mucosa.
Symptoms usually resolve within 1 week after stopping dabigatran and starting a proton pump inhibitor. To prevent mucosal injury, patients should be instructed to take dabigatran with sufficient water and to remain in an upright position for at least 30 minutes afterward.4
A 74-year-old man presented to the gastroenterology clinic with a 2-day history of retrosternal discomfort. His vital signs were normal, and laboratory testing showed a normal leukocyte count.
Esophagogastroduodenoscopy (EGD) revealed longitudinal sloughing mucosal casts in the middle and lower esophagus (Figure 1).
The patient had been taking dabigatran 110 mg twice daily for 2 years because of nonvalvular atrial fibrillation. He was also taking amlodipine 2.5 mg/day for hypertension.
DABIGATRAN-INDUCED ESOPHAGITIS
Although no study has investigated the overall prevalence of dabigatran-induced esophagitis, a retrospective database review of 91 patients taking dabigatran and undergoing upper-gastrointestinal endoscopy reported that 19 (20.9%) had endoscopic signs of dabigatran-induced esophagitis.2
Typical symptoms are the acute onset of chest pain, epigastralgia, odynophagia, and dysphagia. But patients can also have no symptoms or only mild symptoms.2,3
Despite dabigatran’s anticoagulant activity, there have been few reports of bleeding, perhaps because the lesions tend to be superficial on the surface of the esophageal mucosa.
Symptoms usually resolve within 1 week after stopping dabigatran and starting a proton pump inhibitor. To prevent mucosal injury, patients should be instructed to take dabigatran with sufficient water and to remain in an upright position for at least 30 minutes afterward.4
- Baehr PH, McDonald GB. Esophageal infections: risk factors, presentation, diagnosis, and treatment. Gastroenterology 1994; 106(2):509–532. pmid:7980741
- Toya Y, Nakamura S, Tomita K, et al. Dabigatran-induced esophagitis: the prevalence and endoscopic characteristics. J Gastroenterol Hepatol 2016; 31(3):610–614. doi:10.1111/jgh.13024
- Ueta E, Fujikawa T, Imagawa A. A case of a slightly symptomatic exfoliative oesophagitis. BMJ Case Rep 2015; pii:bcr2015211925. doi:10.1136/bcr-2015-211925
- Ootani A, Hayashi Y, Miyagi Y. Dabigatran-induced esophagitis. Clin Gastroenterol Hepatol 2014; 12(7):e55–e56. doi:10.1016/j.cgh.2013.09.010
- Baehr PH, McDonald GB. Esophageal infections: risk factors, presentation, diagnosis, and treatment. Gastroenterology 1994; 106(2):509–532. pmid:7980741
- Toya Y, Nakamura S, Tomita K, et al. Dabigatran-induced esophagitis: the prevalence and endoscopic characteristics. J Gastroenterol Hepatol 2016; 31(3):610–614. doi:10.1111/jgh.13024
- Ueta E, Fujikawa T, Imagawa A. A case of a slightly symptomatic exfoliative oesophagitis. BMJ Case Rep 2015; pii:bcr2015211925. doi:10.1136/bcr-2015-211925
- Ootani A, Hayashi Y, Miyagi Y. Dabigatran-induced esophagitis. Clin Gastroenterol Hepatol 2014; 12(7):e55–e56. doi:10.1016/j.cgh.2013.09.010
New vascular 0 + 5 training programs yield fewer PAD cases
The new vascular surgery training protocol may not reflect what is currently occurring in practice for peripheral artery occlusive disease, and may warrant further investigation, according to a study performed by John Phair, MD, and his colleagues.

“Peripheral arterial occlusive disease constitutes a substantial portion of clinical practice in vascular surgery and, as such, trainees must graduate with proficiency in endovascular and open procedures to become capable vascular surgeons,” they stated, but the new paradigm does not appear to be reflecting that reality.
The researchers compared case volume for 0 + 5 integrated vascular surgery residents (IVSR) in the chief and junior years with their 5 + 2 fellowship counterparts (VSF) for the treatment of peripheral arterial occlusive disease. An aggregate of 887 residents and fellows from 137 programs were identified. From 2012 to 2018, VSFs performed significantly more total peripheral procedures than IVSR surgeon chiefs in the treatment of peripheral arterial occlusive disease, with VSFs consistently performing 1.7-fold (P less than .001) and 1.6-fold (P less than .001) more total peripheral cases than their integrated vascular surgery residents chief and junior counterparts, respectively. When stratified by anatomic location, VSFs were found to have performed more peripheral arterial procedures in the aortoiliac, femoropopliteal, and infrapopliteal arteries, compared with IVSR chiefs, and they also performed more total peripheral procedures than IVSR surgeon juniors, the authors reported online in the journal Surgery (2019 E-pub; doi/10.1016/j.surg.2019.02.021).
“Although both training paradigms offer operative experiences that meet criteria for graduation, it seems that VSFs currently perform more endovascular and open cases than their IVSR counterparts for peripheral arterial occlusive disease during the final phase of training. These findings may be contradictory to current thinking regarding the vascular surgery training paradigms,” the researchers concluded.
There were no disclosures reported.
SOURCE: Phari J et al. Surgery 2019 E-pub; doi/10.1016/j.surg.2019.02.021).
The new vascular surgery training protocol may not reflect what is currently occurring in practice for peripheral artery occlusive disease, and may warrant further investigation, according to a study performed by John Phair, MD, and his colleagues.
“Peripheral arterial occlusive disease constitutes a substantial portion of clinical practice in vascular surgery and, as such, trainees must graduate with proficiency in endovascular and open procedures to become capable vascular surgeons,” they stated, but the new paradigm does not appear to be reflecting that reality.
The researchers compared case volume for 0 + 5 integrated vascular surgery residents (IVSR) in the chief and junior years with their 5 + 2 fellowship counterparts (VSF) for the treatment of peripheral arterial occlusive disease. An aggregate of 887 residents and fellows from 137 programs were identified. From 2012 to 2018, VSFs performed significantly more total peripheral procedures than IVSR surgeon chiefs in the treatment of peripheral arterial occlusive disease, with VSFs consistently performing 1.7-fold (P less than .001) and 1.6-fold (P less than .001) more total peripheral cases than their integrated vascular surgery residents chief and junior counterparts, respectively. When stratified by anatomic location, VSFs were found to have performed more peripheral arterial procedures in the aortoiliac, femoropopliteal, and infrapopliteal arteries, compared with IVSR chiefs, and they also performed more total peripheral procedures than IVSR surgeon juniors, the authors reported online in the journal Surgery (2019 E-pub; doi/10.1016/j.surg.2019.02.021).
“Although both training paradigms offer operative experiences that meet criteria for graduation, it seems that VSFs currently perform more endovascular and open cases than their IVSR counterparts for peripheral arterial occlusive disease during the final phase of training. These findings may be contradictory to current thinking regarding the vascular surgery training paradigms,” the researchers concluded.
There were no disclosures reported.
SOURCE: Phari J et al. Surgery 2019 E-pub; doi/10.1016/j.surg.2019.02.021).
The new vascular surgery training protocol may not reflect what is currently occurring in practice for peripheral artery occlusive disease, and may warrant further investigation, according to a study performed by John Phair, MD, and his colleagues.
“Peripheral arterial occlusive disease constitutes a substantial portion of clinical practice in vascular surgery and, as such, trainees must graduate with proficiency in endovascular and open procedures to become capable vascular surgeons,” they stated, but the new paradigm does not appear to be reflecting that reality.
The researchers compared case volume for 0 + 5 integrated vascular surgery residents (IVSR) in the chief and junior years with their 5 + 2 fellowship counterparts (VSF) for the treatment of peripheral arterial occlusive disease. An aggregate of 887 residents and fellows from 137 programs were identified. From 2012 to 2018, VSFs performed significantly more total peripheral procedures than IVSR surgeon chiefs in the treatment of peripheral arterial occlusive disease, with VSFs consistently performing 1.7-fold (P less than .001) and 1.6-fold (P less than .001) more total peripheral cases than their integrated vascular surgery residents chief and junior counterparts, respectively. When stratified by anatomic location, VSFs were found to have performed more peripheral arterial procedures in the aortoiliac, femoropopliteal, and infrapopliteal arteries, compared with IVSR chiefs, and they also performed more total peripheral procedures than IVSR surgeon juniors, the authors reported online in the journal Surgery (2019 E-pub; doi/10.1016/j.surg.2019.02.021).
“Although both training paradigms offer operative experiences that meet criteria for graduation, it seems that VSFs currently perform more endovascular and open cases than their IVSR counterparts for peripheral arterial occlusive disease during the final phase of training. These findings may be contradictory to current thinking regarding the vascular surgery training paradigms,” the researchers concluded.
There were no disclosures reported.
SOURCE: Phari J et al. Surgery 2019 E-pub; doi/10.1016/j.surg.2019.02.021).
FROM SURGERY
Ask patients, not devices, about impact of PAD
The ankle-brachial index (ABI) is a poor indicator of patient-centered and clinician-based evaluations of functional status in patients with intermittent claudication, according to the results of PORTRAIT, a prospective observational study of patients with newly diagnosed or an exacerbation of nonlimb-threatening peripheral arterial disease (PAD).
PORTRAIT studied 1,251 patients with intermittent claudication enrolled at 16 sites. Researchers studied the correlation of ABI values and Rutherford symptom classification with PAD-specific health status as measured by the Peripheral Artery Questionnaire (PAQ).
ABI values were categorized as mild (greater than 0.80), moderate (0.40-0.79), and severe (less than 0.40). Spearman rank correlation coefficients were calculated between raw ABI values and PAQ scores and between the Rutherford classification and PAQ scores.
ABI explained only 0.1%-2.1% of the variation in PAQ scores and the Rutherford classification had stronger but still modest associations with PAQ scores, according to the researchers.
“This large study of IC patients found that the PAQ offers a unique and complementary
measure of disease burden that is not captured by physiologic or clinician-observed classifications. The findings from this study highlight the clinical complexity of PAD
and the difficulty in using common hemodynamic and symptom measures to classify the impact of this disease on patients’ health status,” the researchers concluded.
Several authors reported serving as consultant for and/or receiving grants from various device and pharmaceutical companies involved with PAD. The senior author. owns the copyright to the Peripheral Artery Questionnaire that formed the basis for the study.
SOURCE: Johnston A et al. J Vasc Surg. 2018;69(3):906-12.
The ankle-brachial index (ABI) is a poor indicator of patient-centered and clinician-based evaluations of functional status in patients with intermittent claudication, according to the results of PORTRAIT, a prospective observational study of patients with newly diagnosed or an exacerbation of nonlimb-threatening peripheral arterial disease (PAD).
PORTRAIT studied 1,251 patients with intermittent claudication enrolled at 16 sites. Researchers studied the correlation of ABI values and Rutherford symptom classification with PAD-specific health status as measured by the Peripheral Artery Questionnaire (PAQ).
ABI values were categorized as mild (greater than 0.80), moderate (0.40-0.79), and severe (less than 0.40). Spearman rank correlation coefficients were calculated between raw ABI values and PAQ scores and between the Rutherford classification and PAQ scores.
ABI explained only 0.1%-2.1% of the variation in PAQ scores and the Rutherford classification had stronger but still modest associations with PAQ scores, according to the researchers.
“This large study of IC patients found that the PAQ offers a unique and complementary
measure of disease burden that is not captured by physiologic or clinician-observed classifications. The findings from this study highlight the clinical complexity of PAD
and the difficulty in using common hemodynamic and symptom measures to classify the impact of this disease on patients’ health status,” the researchers concluded.
Several authors reported serving as consultant for and/or receiving grants from various device and pharmaceutical companies involved with PAD. The senior author. owns the copyright to the Peripheral Artery Questionnaire that formed the basis for the study.
SOURCE: Johnston A et al. J Vasc Surg. 2018;69(3):906-12.
The ankle-brachial index (ABI) is a poor indicator of patient-centered and clinician-based evaluations of functional status in patients with intermittent claudication, according to the results of PORTRAIT, a prospective observational study of patients with newly diagnosed or an exacerbation of nonlimb-threatening peripheral arterial disease (PAD).
PORTRAIT studied 1,251 patients with intermittent claudication enrolled at 16 sites. Researchers studied the correlation of ABI values and Rutherford symptom classification with PAD-specific health status as measured by the Peripheral Artery Questionnaire (PAQ).
ABI values were categorized as mild (greater than 0.80), moderate (0.40-0.79), and severe (less than 0.40). Spearman rank correlation coefficients were calculated between raw ABI values and PAQ scores and between the Rutherford classification and PAQ scores.
ABI explained only 0.1%-2.1% of the variation in PAQ scores and the Rutherford classification had stronger but still modest associations with PAQ scores, according to the researchers.
“This large study of IC patients found that the PAQ offers a unique and complementary
measure of disease burden that is not captured by physiologic or clinician-observed classifications. The findings from this study highlight the clinical complexity of PAD
and the difficulty in using common hemodynamic and symptom measures to classify the impact of this disease on patients’ health status,” the researchers concluded.
Several authors reported serving as consultant for and/or receiving grants from various device and pharmaceutical companies involved with PAD. The senior author. owns the copyright to the Peripheral Artery Questionnaire that formed the basis for the study.
SOURCE: Johnston A et al. J Vasc Surg. 2018;69(3):906-12.
FROM THE JOURNAL OF VASCULAR SURGERY