User login
Is chest radiography routinely needed after thoracentesis?
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
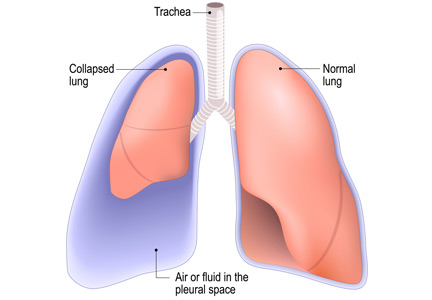
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
A woman, age 35, with new-onset ascites
A 35-year-old woman is admitted to the hospital with a 5-day history of abdominal distention and jaundice. She reports no history of fever, chills, night sweats, abdominal pain, nausea, vomiting, diarrhea, changes in urine color, change in stool color, weight loss, weight gain, or loss of appetite.
She is petite, with a body mass index of 19.4 kg/m2. She has no known history of medical conditions or surgery and is not taking any medications. Her family history is unremarkable, and she denies current or past tobacco, alcohol, or illicit drug use.
RECENT TRAVEL
She says that during a trip to Central America several months ago, she had suffered a seizure and was taken to a local hospital, where laboratory testing revealed elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. She says that the rest of the workup at that time was normal.
About 1 week after that incident, she returned home and saw her primary care physician, who ordered further testing, which showed mild hyperbilirubinemia and mild elevation of AST and ALT levels. Her physician attributed the elevations to atovaquone, which she had been taking for malaria prophylaxis, as repeat testing 2 weeks later showed improvement in AST and ALT levels.
The patient says she returned to her normal state of health until about 5 days ago, when she noticed jaundice and abdominal distention, but without abdominal pain, dark urine, or clay-colored stools. She became concerned and went to her local hospital. Testing there noted mild elevation of AST and ALT, as well as an elevated international normalized ratio (INR) and hyperbilirubinemia. Computed tomography of the abdomen and pelvis showed hepatomegaly with possible fatty liver. Because of these results, the patient was transferred to our institution for further evaluation.
EVALUATION AT OUR INSTITUTION
On examination at our institution, she is afebrile, and vital signs are within normal ranges. She has bilateral scleral icterus and diffuse jaundice, but no other skin finding such as rash or spider angioma. She has no lymphadenopathy. Her abdomen is distended, with tense ascites, and her liver is tender to palpation. The tip of the spleen is not palpable.

The cardiovascular examination reveals no murmurs, rubs, or gallops, but she has jugular venous distention and +2 pitting edema of both lower extremities.
On respiratory examination, there is dullness to percussion, with slight crackles on auscultation at the right lung base. The neurologic examination is normal.
Table 1 shows the results of initial laboratory testing.
1. Which study would provide the most information on the cause of ascites?
- Abdominal ultrasonography
- Abdominal paracentesis with ascitic fluid analysis
- Chest radiography
- Echocardiography
- Urine protein-to-creatinine ratio
Abdominal paracentesis with ascitic fluid analysis is the essential study for any patient with clinically apparent new-onset ascites.1–3 It is the study that provides the most information on the cause of ascites.
In our patient, abdominal paracentesis yields 1,000 mL of straw-colored ascitic fluid, and analysis shows 86 nucleated cells, 28 of which are polymorphonuclear cells, and 0 red blood cells, with negative Gram stain and culture. The ascitic albumin level is 0.85 g/dL, with an ascitic protein of 1.1 g/dL.
Abdominal ultrasonography shows a diffusely echogenic liver, no focal lesions, moderate ascites, normal portal vein flow, no intrahepatic or extrahepatic biliary duct dilation, normal kidney sizes, no hydronephrosis, and no intra-abdominal mass. Chest radiography is clear with no sign of consolidation, edema, or effusion. Echocardiography shows a normal left ventricular ejection fraction with no valvular disease or pericardial effusion. A random urine protein-creatinine ratio is normal at 0.1 (reference range < 0.2).
2. What is the most likely cause of her ascites based on the workup to this point?
- Cirrhosis
- Heart failure
- Nephrotic syndrome
- Portal vein thrombus
- Abdominal malignancy
- Malaria
 and ascitic protein levels.")
An initial approach to ascitic fluid analysis is to calculate the serum-ascites albumin gradient (SAAG). The SAAG is calculated as the serum albumin level minus the ascitic fluid albumin level.4,5 This is useful in determining the cause of the ascites (Figure 1).4,5 A gradient of 1.1 g/dL or higher indicates portal hypertension.4,5
Common causes of portal hypertension include cirrhosis, alcoholic hepatitis, heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome, portal vein thrombosis), idiopathic portal fibrosis, and metastatic liver disease.5,6
If portal hypertension is present based on the SAAG, the next step is to review the ascitic protein level to help distinguish between a hepatic and a cardiac etiology of the ascites. An ascitic protein level less than 2.5 g/dL indicates a primary liver pathology (eg, cirrhosis). An ascitic protein level of 2.5 g/dL or greater typically indicates a cardiac condition (eg, heart failure, pericardial disease) with secondary congestive hepatopathy.5,6
If the SAAG is less than 1.1 g/dL, the ascites is likely not from portal hypertension. Typical causes of a low SAAG include infection, malignancy, pancreatic ascites, and nephrotic syndrome.5,6
In our patient, the SAAG is 1.35 g/dL (2.2 g/dL minus 0.85 g/dL), ie, elevated and due to portal hypertension. With an SAAG of 1.1 g/dL or greater and an ascitic fluid protein level less than 2.5 g/dL, as in our patient, the most likely cause is cirrhosis.
Heart failure is unlikely based on her normal brain natriuretic peptide level, an ascitic fluid protein level below 2.5 g/dL, and normal results on echocardiography. Nephrotic syndrome is also very unlikely based on the patient’s normal random urine protein-creatinine ratio. Portal vein thrombus and abdominal malignancy are essentially ruled out by the negative results of Doppler abdominal ultrasonography, with normal venous flow and no intra-abdominal mass and coupled with an elevated SAAG.
Although the patient has a history of travel, the incubation period for malaria would not fit the time frame of presentation. Also, she did not have typical malarial symptoms, her rapid malaria test was negative, and a peripheral blood smear for blood parasites was negative. It should be noted, however, that Plasmodium malariae infection classically presents with flulike symptoms and can resemble nephrotic syndrome, including peripheral edema, ascites, heavy proteinuria, hypoalbuminemia, and hyperlipidemia.7
3. In which patients is antibiotic prophylaxis against spontaneous bacterial peritonitis (SBP) appropriate?
- Any patient with cirrhosis
- Any patient with cirrhosis who is hospitalized
- Any patient with cirrhosis and an ascitic fluid protein level below 2.0 g/dL
- Any patient with cirrhosis and a history of SBP
Any patient with cirrhosis and a history of SBP should receive prophylactic antibiotics,8 as should any patient deemed at high risk of SBP. It is indicated in the following patients:
- Patients with cirrhosis and gastrointestinal bleeding9,10
- Patients with cirrhosis and a previous episode of SBP8
- Patients with cirrhosis and an ascitic fluid protein level less than 1.5 g/dL with either impaired renal function (creatinine ≥ 1.2 mg/dL, blood urea nitrogen level ≥ 25 mg/dL, or serum sodium ≤ 130 mmol/L) or liver failure (Child-Pugh score ≥ 9 and a bilirubin ≥ 3 mg/dL)9
- Patients with cirrhosis who are hospitalized for other reasons and have an ascitic protein level < 1.0 g/dL.9
Our patient has no signs or symptoms of gastrointestinal bleeding and no history of SBP. Her ascitic fluid protein level is 1.1 g/dL, and she has normal renal function. However, her Child-Pugh score is 12 (3 points for total bilirubin > 3 mg/dL, 3 points for serum albumin < 2.8 g/dL, 2 points for an INR 1.7 to 2.2, 3 points for moderate ascites, and 1 point for no encephalopathy), with a bilirubin of 17.0 mg/dL. Based on this, she is placed on antibiotic prophylaxis for SBP.
Our patient then undergoes an extensive workup for liver disease. Results of tests for toxins, autoimmune diseases, and inheritable diseases are all within normal limits. At this point, despite the patient’s reported negative alcohol history, our leading diagnosis is alcoholic hepatitis.
 shows severe steatosis from lipid accumulation in hepatocytes (arrow). Higher magnification (bottom) shows inflammatory cells (neutrophils, arrows) surrounding Mallory-Denk bodies.")
To confirm this diagnosis, she subsequently undergoes transjugular liver biopsy, considered the gold standard for the diagnosis of alcoholic hepatitis. During the procedure, the hepatic venous pressure gradient is measured at 18 mm Hg (reference range 1–5 mm Hg), suggestive of portal hypertension. The pathology study shows severe fatty change, active steatohepatitis with ballooning degeneration, easily identifiable Mallory-Denk bodies, and prominent neutrophilic infiltration, as well as extensive bridging fibrosis (Figure 2). These findings point to alcoholic hepatitis.
After the biopsy results, we speak with the patient further about her alcohol habits. At this point, she informs us that she has consumed significant amounts of alcohol since the age of 18 (6 to 12 alcoholic beverages per day, including beer and hard liquor). Therefore, based on this new information, on her jaundice and ascites, and on results of laboratory testing and biopsy, we confirmed our diagnosis of alcoholic hepatitis.
4. When is drug treatment appropriate for alcoholic hepatitis?
- Model for End-stage Liver Disease (MELD) score greater than 12
- MELD score greater than 15
- Maddrey Discriminant Function score greater than 25
- Maddrey Discriminant Function score greater than 32
- Glasgow score greater than 5
- Glasgow score greater than 7
The best answer is a Maddrey Discriminant Function score greater than 32. A variety of scoring systems have been used to assess the severity of alcoholic hepatitis and to guide treatment, including the Maddrey Discriminant Function score, the MELD score, and the Glasgow score.11–16 They share similar laboratory values in their calculations, including prothrombin time (or INR) and total bilirubin.11–16 Typically, a Maddrey Discriminant Function score greater than 32, a Glasgow score of greater than 9, or a MELD score greater than 21 is used to determine whether pharmacologic treatment is indicated.11–16

The typical treatment is prednisolone or pentoxifylline.11,17–21 The Lille score is designed to help decide whether to stop corticosteroids after 1 week of administration due to lack of treatment response.22 It predicts mortality rates within 6 months; a score of 0.45 or less indicates a good prognosis, and corticosteroid therapy should continue for 28 days (Figure 3).22
Our patient’s discriminant function score is 50, her Glasgow score is 10, and her MELD score is 28; thus, she begins treatment with oral prednisolone. Her Lille score at 1 week is 0.119, indicating a good prognosis, and her corticosteroids are continued for a total of 28 days.
It should be highlighted that the most important treatment is abstinence from alcohol.11 Recent literature suggests that any benefit of prednisolone or pentoxifylline in terms of mortality rates is questionable,19–20 and there is evidence that giving both drugs simultaneously may improve mortality rates,11,21 but the evidence remains conflicting at this time.
ALCOHOLIC HEPATITIS
Alcoholic hepatitis is a clinical syndrome of jaundice and liver failure, often in the setting of heavy alcohol use for decades.11,12 The incidence is unknown, but the typical age of presentation is between 40 and 50.11,12 The chief sign is a rapid onset of jaundice (< 3 months); common signs and symptoms include fever, ascites, proximal muscle loss, and an enlarged, tender liver.12 Encephalopathy may be seen in severe alcoholic hepatitis.12
Our patient is 35 years old. She has jaundice with rapid onset, as well as ascites and a tender liver.
The diagnosis of alcoholic hepatitis must take into account the patient’s history, physical examination, and laboratory findings. Until proven otherwise, the diagnosis should be presumed in the following scenario: ascites and jaundice on examination (usually with a duration < 3 months); a history of heavy alcohol use; neutrophilic leukocytosis; an AST level that is elevated but below 300 U/L; an ALT level above the normal range but below 300 U/L; an AST-ALT ratio greater than 2; a total serum bilirubin level above 5 mg/dL; and an elevated INR.11,12 Liver biopsy is the gold standard for diagnosis. Though not routinely done because of risks associated with the procedure, it may help confirm the diagnosis if it is in question.
CASE CONCLUDED
We start our patient on oral prednisolone 40 mg daily for alcoholic hepatitis. Her symptoms and laboratory testing results including bilirubin improve. Her Lille score at 7 days indicates a good prognosis, prompting continuation of corticosteroid treatment for the full 28 days.
She is referred to an outpatient alcohol rehabilitation program and has remained sober as of the last outpatient note.
Alcoholic hepatitis is extremely difficult to diagnose, and no single blood test or imaging study confirms the diagnosis. The history, physical examination findings, and laboratory findings are crucial. If the diagnosis is still in doubt, liver biopsy may help confirm the diagnosis.
- Ruyon BA; AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009; 49(6):2087–2107. doi:10.1002/hep.22853
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2(4):399–407. pmid:7095741
- Ginès P, Cárdenas A, Arroyo V, Rodés J. Management of cirrhosis and ascites. N Engl J Med 2004; 350(16):1646–1654. doi:10.1056/NEJMra035021
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117(3):215–220. pmid:1616215
- Hernaez R, Hamilton JP. Unexplained ascites. Clin Liver Dis 2016; 7(3):53–56. https://aasldpubs.onlinelibrary.wiley.com/doi/epdf/10.1002/cld.537
- Huang LL, Xia HH, Zhu SL. Ascitic fluid analysis in the differential diagnosis of ascites: focus on cirrhotic ascites. J Clin Transl Hepatol 2014; 2(1):58–64. doi:10.14218/JCTH.2013.00010
- Bartoloni A, Zammarchi L. Clinical aspects of uncomplicated and severe malaria. Mediterr J Hematol Infect Dis 2012; 4(1):e2012026. doi:10.4084/MJHID.2012.026
- Titó L, Rimola A, Ginès P, Llach J, Arroyo V, Rodés J. Recurrence of spontaneous bacterial peritonitis in cirrhosis: frequency and predictive factors. Hepatology 1988; 8(1):27–31. pmid:3257456
- Fernández J, Ruiz del Arbol L, Gómez C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology 2006; 131(4):1049–1056. doi:10.1053/j.gastro.2006.07.010
- Runyon B; The American Association for the Study of Liver Diseases (AASLD). Management of adult patients with ascites due to cirrhosis: update 2012. https://www.aasld.org/sites/default/files/guideline_documents/141020_Guideline_Ascites_4UFb_2015.pdf. Accessed September 4, 2018.
- Sidhu SS, Goyal O, Kishore H, Sidhu S. New paradigms in management of alcoholic hepatitis: a review. Hepatol Int 2017; 11(3):255–267. doi:10.1007/s12072-017-9790-5
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360(26):2758–2769. doi:10.1056/NEJMra0805786
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75(2):193–199. pmid:352788
- Forrest EH, Evans CD, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005; 54(8):1174–1179. doi:10.1136/gut.2004.050781
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2):353–358. doi:10.1002/hep.20503
- Sheth M, Riggs M, Patel T. Utility of the Mayo end-stage liver disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2. pmid:11835693
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119(6):1637–1648. pmid:11113085
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60(2):255–260. doi:10.1136/gut.2010.224097
- Thursz MR, Richardson P, Allison M, et al; STOPAH Trial. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372(17):1619–1628. doi:10.1056/NEJMoa1412278
- Thursz M, Forrest E, Roderick P, et al. The clinical effectiveness and cost-effectiveness of steroids or pentoxifylline for alcoholic hepatitis (STOPAH): a 2 × 2 factorial randomised controlled trial. Health Technol Assess 2015; 19(102):1–104. doi:10.3310/hta191020
- Lee YS, Kim HJ, Kim JH, et al. Treatment of severe alcoholic hepatitis with corticosteroid, pentoxifylline, or dual therapy: a systematic review and meta-analysis. J Clin Gastroenterol 2017; 51(4):364–377. doi:10.1097/MCG.0000000000000674
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45(6):1348–1354. doi:10.1002/hep.21607
A 35-year-old woman is admitted to the hospital with a 5-day history of abdominal distention and jaundice. She reports no history of fever, chills, night sweats, abdominal pain, nausea, vomiting, diarrhea, changes in urine color, change in stool color, weight loss, weight gain, or loss of appetite.
She is petite, with a body mass index of 19.4 kg/m2. She has no known history of medical conditions or surgery and is not taking any medications. Her family history is unremarkable, and she denies current or past tobacco, alcohol, or illicit drug use.
RECENT TRAVEL
She says that during a trip to Central America several months ago, she had suffered a seizure and was taken to a local hospital, where laboratory testing revealed elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. She says that the rest of the workup at that time was normal.
About 1 week after that incident, she returned home and saw her primary care physician, who ordered further testing, which showed mild hyperbilirubinemia and mild elevation of AST and ALT levels. Her physician attributed the elevations to atovaquone, which she had been taking for malaria prophylaxis, as repeat testing 2 weeks later showed improvement in AST and ALT levels.
The patient says she returned to her normal state of health until about 5 days ago, when she noticed jaundice and abdominal distention, but without abdominal pain, dark urine, or clay-colored stools. She became concerned and went to her local hospital. Testing there noted mild elevation of AST and ALT, as well as an elevated international normalized ratio (INR) and hyperbilirubinemia. Computed tomography of the abdomen and pelvis showed hepatomegaly with possible fatty liver. Because of these results, the patient was transferred to our institution for further evaluation.
EVALUATION AT OUR INSTITUTION
On examination at our institution, she is afebrile, and vital signs are within normal ranges. She has bilateral scleral icterus and diffuse jaundice, but no other skin finding such as rash or spider angioma. She has no lymphadenopathy. Her abdomen is distended, with tense ascites, and her liver is tender to palpation. The tip of the spleen is not palpable.
The cardiovascular examination reveals no murmurs, rubs, or gallops, but she has jugular venous distention and +2 pitting edema of both lower extremities.
On respiratory examination, there is dullness to percussion, with slight crackles on auscultation at the right lung base. The neurologic examination is normal.
Table 1 shows the results of initial laboratory testing.
1. Which study would provide the most information on the cause of ascites?
- Abdominal ultrasonography
- Abdominal paracentesis with ascitic fluid analysis
- Chest radiography
- Echocardiography
- Urine protein-to-creatinine ratio
Abdominal paracentesis with ascitic fluid analysis is the essential study for any patient with clinically apparent new-onset ascites.1–3 It is the study that provides the most information on the cause of ascites.
In our patient, abdominal paracentesis yields 1,000 mL of straw-colored ascitic fluid, and analysis shows 86 nucleated cells, 28 of which are polymorphonuclear cells, and 0 red blood cells, with negative Gram stain and culture. The ascitic albumin level is 0.85 g/dL, with an ascitic protein of 1.1 g/dL.
Abdominal ultrasonography shows a diffusely echogenic liver, no focal lesions, moderate ascites, normal portal vein flow, no intrahepatic or extrahepatic biliary duct dilation, normal kidney sizes, no hydronephrosis, and no intra-abdominal mass. Chest radiography is clear with no sign of consolidation, edema, or effusion. Echocardiography shows a normal left ventricular ejection fraction with no valvular disease or pericardial effusion. A random urine protein-creatinine ratio is normal at 0.1 (reference range < 0.2).
2. What is the most likely cause of her ascites based on the workup to this point?
- Cirrhosis
- Heart failure
- Nephrotic syndrome
- Portal vein thrombus
- Abdominal malignancy
- Malaria
An initial approach to ascitic fluid analysis is to calculate the serum-ascites albumin gradient (SAAG). The SAAG is calculated as the serum albumin level minus the ascitic fluid albumin level.4,5 This is useful in determining the cause of the ascites (Figure 1).4,5 A gradient of 1.1 g/dL or higher indicates portal hypertension.4,5
Common causes of portal hypertension include cirrhosis, alcoholic hepatitis, heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome, portal vein thrombosis), idiopathic portal fibrosis, and metastatic liver disease.5,6
If portal hypertension is present based on the SAAG, the next step is to review the ascitic protein level to help distinguish between a hepatic and a cardiac etiology of the ascites. An ascitic protein level less than 2.5 g/dL indicates a primary liver pathology (eg, cirrhosis). An ascitic protein level of 2.5 g/dL or greater typically indicates a cardiac condition (eg, heart failure, pericardial disease) with secondary congestive hepatopathy.5,6
If the SAAG is less than 1.1 g/dL, the ascites is likely not from portal hypertension. Typical causes of a low SAAG include infection, malignancy, pancreatic ascites, and nephrotic syndrome.5,6
In our patient, the SAAG is 1.35 g/dL (2.2 g/dL minus 0.85 g/dL), ie, elevated and due to portal hypertension. With an SAAG of 1.1 g/dL or greater and an ascitic fluid protein level less than 2.5 g/dL, as in our patient, the most likely cause is cirrhosis.
Heart failure is unlikely based on her normal brain natriuretic peptide level, an ascitic fluid protein level below 2.5 g/dL, and normal results on echocardiography. Nephrotic syndrome is also very unlikely based on the patient’s normal random urine protein-creatinine ratio. Portal vein thrombus and abdominal malignancy are essentially ruled out by the negative results of Doppler abdominal ultrasonography, with normal venous flow and no intra-abdominal mass and coupled with an elevated SAAG.
Although the patient has a history of travel, the incubation period for malaria would not fit the time frame of presentation. Also, she did not have typical malarial symptoms, her rapid malaria test was negative, and a peripheral blood smear for blood parasites was negative. It should be noted, however, that Plasmodium malariae infection classically presents with flulike symptoms and can resemble nephrotic syndrome, including peripheral edema, ascites, heavy proteinuria, hypoalbuminemia, and hyperlipidemia.7
3. In which patients is antibiotic prophylaxis against spontaneous bacterial peritonitis (SBP) appropriate?
- Any patient with cirrhosis
- Any patient with cirrhosis who is hospitalized
- Any patient with cirrhosis and an ascitic fluid protein level below 2.0 g/dL
- Any patient with cirrhosis and a history of SBP
Any patient with cirrhosis and a history of SBP should receive prophylactic antibiotics,8 as should any patient deemed at high risk of SBP. It is indicated in the following patients:
- Patients with cirrhosis and gastrointestinal bleeding9,10
- Patients with cirrhosis and a previous episode of SBP8
- Patients with cirrhosis and an ascitic fluid protein level less than 1.5 g/dL with either impaired renal function (creatinine ≥ 1.2 mg/dL, blood urea nitrogen level ≥ 25 mg/dL, or serum sodium ≤ 130 mmol/L) or liver failure (Child-Pugh score ≥ 9 and a bilirubin ≥ 3 mg/dL)9
- Patients with cirrhosis who are hospitalized for other reasons and have an ascitic protein level < 1.0 g/dL.9
Our patient has no signs or symptoms of gastrointestinal bleeding and no history of SBP. Her ascitic fluid protein level is 1.1 g/dL, and she has normal renal function. However, her Child-Pugh score is 12 (3 points for total bilirubin > 3 mg/dL, 3 points for serum albumin < 2.8 g/dL, 2 points for an INR 1.7 to 2.2, 3 points for moderate ascites, and 1 point for no encephalopathy), with a bilirubin of 17.0 mg/dL. Based on this, she is placed on antibiotic prophylaxis for SBP.
Our patient then undergoes an extensive workup for liver disease. Results of tests for toxins, autoimmune diseases, and inheritable diseases are all within normal limits. At this point, despite the patient’s reported negative alcohol history, our leading diagnosis is alcoholic hepatitis.
To confirm this diagnosis, she subsequently undergoes transjugular liver biopsy, considered the gold standard for the diagnosis of alcoholic hepatitis. During the procedure, the hepatic venous pressure gradient is measured at 18 mm Hg (reference range 1–5 mm Hg), suggestive of portal hypertension. The pathology study shows severe fatty change, active steatohepatitis with ballooning degeneration, easily identifiable Mallory-Denk bodies, and prominent neutrophilic infiltration, as well as extensive bridging fibrosis (Figure 2). These findings point to alcoholic hepatitis.
After the biopsy results, we speak with the patient further about her alcohol habits. At this point, she informs us that she has consumed significant amounts of alcohol since the age of 18 (6 to 12 alcoholic beverages per day, including beer and hard liquor). Therefore, based on this new information, on her jaundice and ascites, and on results of laboratory testing and biopsy, we confirmed our diagnosis of alcoholic hepatitis.
4. When is drug treatment appropriate for alcoholic hepatitis?
- Model for End-stage Liver Disease (MELD) score greater than 12
- MELD score greater than 15
- Maddrey Discriminant Function score greater than 25
- Maddrey Discriminant Function score greater than 32
- Glasgow score greater than 5
- Glasgow score greater than 7
The best answer is a Maddrey Discriminant Function score greater than 32. A variety of scoring systems have been used to assess the severity of alcoholic hepatitis and to guide treatment, including the Maddrey Discriminant Function score, the MELD score, and the Glasgow score.11–16 They share similar laboratory values in their calculations, including prothrombin time (or INR) and total bilirubin.11–16 Typically, a Maddrey Discriminant Function score greater than 32, a Glasgow score of greater than 9, or a MELD score greater than 21 is used to determine whether pharmacologic treatment is indicated.11–16
The typical treatment is prednisolone or pentoxifylline.11,17–21 The Lille score is designed to help decide whether to stop corticosteroids after 1 week of administration due to lack of treatment response.22 It predicts mortality rates within 6 months; a score of 0.45 or less indicates a good prognosis, and corticosteroid therapy should continue for 28 days (Figure 3).22
Our patient’s discriminant function score is 50, her Glasgow score is 10, and her MELD score is 28; thus, she begins treatment with oral prednisolone. Her Lille score at 1 week is 0.119, indicating a good prognosis, and her corticosteroids are continued for a total of 28 days.
It should be highlighted that the most important treatment is abstinence from alcohol.11 Recent literature suggests that any benefit of prednisolone or pentoxifylline in terms of mortality rates is questionable,19–20 and there is evidence that giving both drugs simultaneously may improve mortality rates,11,21 but the evidence remains conflicting at this time.
ALCOHOLIC HEPATITIS
Alcoholic hepatitis is a clinical syndrome of jaundice and liver failure, often in the setting of heavy alcohol use for decades.11,12 The incidence is unknown, but the typical age of presentation is between 40 and 50.11,12 The chief sign is a rapid onset of jaundice (< 3 months); common signs and symptoms include fever, ascites, proximal muscle loss, and an enlarged, tender liver.12 Encephalopathy may be seen in severe alcoholic hepatitis.12
Our patient is 35 years old. She has jaundice with rapid onset, as well as ascites and a tender liver.
The diagnosis of alcoholic hepatitis must take into account the patient’s history, physical examination, and laboratory findings. Until proven otherwise, the diagnosis should be presumed in the following scenario: ascites and jaundice on examination (usually with a duration < 3 months); a history of heavy alcohol use; neutrophilic leukocytosis; an AST level that is elevated but below 300 U/L; an ALT level above the normal range but below 300 U/L; an AST-ALT ratio greater than 2; a total serum bilirubin level above 5 mg/dL; and an elevated INR.11,12 Liver biopsy is the gold standard for diagnosis. Though not routinely done because of risks associated with the procedure, it may help confirm the diagnosis if it is in question.
CASE CONCLUDED
We start our patient on oral prednisolone 40 mg daily for alcoholic hepatitis. Her symptoms and laboratory testing results including bilirubin improve. Her Lille score at 7 days indicates a good prognosis, prompting continuation of corticosteroid treatment for the full 28 days.
She is referred to an outpatient alcohol rehabilitation program and has remained sober as of the last outpatient note.
Alcoholic hepatitis is extremely difficult to diagnose, and no single blood test or imaging study confirms the diagnosis. The history, physical examination findings, and laboratory findings are crucial. If the diagnosis is still in doubt, liver biopsy may help confirm the diagnosis.
A 35-year-old woman is admitted to the hospital with a 5-day history of abdominal distention and jaundice. She reports no history of fever, chills, night sweats, abdominal pain, nausea, vomiting, diarrhea, changes in urine color, change in stool color, weight loss, weight gain, or loss of appetite.
She is petite, with a body mass index of 19.4 kg/m2. She has no known history of medical conditions or surgery and is not taking any medications. Her family history is unremarkable, and she denies current or past tobacco, alcohol, or illicit drug use.
RECENT TRAVEL
She says that during a trip to Central America several months ago, she had suffered a seizure and was taken to a local hospital, where laboratory testing revealed elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. She says that the rest of the workup at that time was normal.
About 1 week after that incident, she returned home and saw her primary care physician, who ordered further testing, which showed mild hyperbilirubinemia and mild elevation of AST and ALT levels. Her physician attributed the elevations to atovaquone, which she had been taking for malaria prophylaxis, as repeat testing 2 weeks later showed improvement in AST and ALT levels.
The patient says she returned to her normal state of health until about 5 days ago, when she noticed jaundice and abdominal distention, but without abdominal pain, dark urine, or clay-colored stools. She became concerned and went to her local hospital. Testing there noted mild elevation of AST and ALT, as well as an elevated international normalized ratio (INR) and hyperbilirubinemia. Computed tomography of the abdomen and pelvis showed hepatomegaly with possible fatty liver. Because of these results, the patient was transferred to our institution for further evaluation.
EVALUATION AT OUR INSTITUTION
On examination at our institution, she is afebrile, and vital signs are within normal ranges. She has bilateral scleral icterus and diffuse jaundice, but no other skin finding such as rash or spider angioma. She has no lymphadenopathy. Her abdomen is distended, with tense ascites, and her liver is tender to palpation. The tip of the spleen is not palpable.
The cardiovascular examination reveals no murmurs, rubs, or gallops, but she has jugular venous distention and +2 pitting edema of both lower extremities.
On respiratory examination, there is dullness to percussion, with slight crackles on auscultation at the right lung base. The neurologic examination is normal.
Table 1 shows the results of initial laboratory testing.
1. Which study would provide the most information on the cause of ascites?
- Abdominal ultrasonography
- Abdominal paracentesis with ascitic fluid analysis
- Chest radiography
- Echocardiography
- Urine protein-to-creatinine ratio
Abdominal paracentesis with ascitic fluid analysis is the essential study for any patient with clinically apparent new-onset ascites.1–3 It is the study that provides the most information on the cause of ascites.
In our patient, abdominal paracentesis yields 1,000 mL of straw-colored ascitic fluid, and analysis shows 86 nucleated cells, 28 of which are polymorphonuclear cells, and 0 red blood cells, with negative Gram stain and culture. The ascitic albumin level is 0.85 g/dL, with an ascitic protein of 1.1 g/dL.
Abdominal ultrasonography shows a diffusely echogenic liver, no focal lesions, moderate ascites, normal portal vein flow, no intrahepatic or extrahepatic biliary duct dilation, normal kidney sizes, no hydronephrosis, and no intra-abdominal mass. Chest radiography is clear with no sign of consolidation, edema, or effusion. Echocardiography shows a normal left ventricular ejection fraction with no valvular disease or pericardial effusion. A random urine protein-creatinine ratio is normal at 0.1 (reference range < 0.2).
2. What is the most likely cause of her ascites based on the workup to this point?
- Cirrhosis
- Heart failure
- Nephrotic syndrome
- Portal vein thrombus
- Abdominal malignancy
- Malaria
An initial approach to ascitic fluid analysis is to calculate the serum-ascites albumin gradient (SAAG). The SAAG is calculated as the serum albumin level minus the ascitic fluid albumin level.4,5 This is useful in determining the cause of the ascites (Figure 1).4,5 A gradient of 1.1 g/dL or higher indicates portal hypertension.4,5
Common causes of portal hypertension include cirrhosis, alcoholic hepatitis, heart failure, vascular occlusion syndromes (eg, Budd-Chiari syndrome, portal vein thrombosis), idiopathic portal fibrosis, and metastatic liver disease.5,6
If portal hypertension is present based on the SAAG, the next step is to review the ascitic protein level to help distinguish between a hepatic and a cardiac etiology of the ascites. An ascitic protein level less than 2.5 g/dL indicates a primary liver pathology (eg, cirrhosis). An ascitic protein level of 2.5 g/dL or greater typically indicates a cardiac condition (eg, heart failure, pericardial disease) with secondary congestive hepatopathy.5,6
If the SAAG is less than 1.1 g/dL, the ascites is likely not from portal hypertension. Typical causes of a low SAAG include infection, malignancy, pancreatic ascites, and nephrotic syndrome.5,6
In our patient, the SAAG is 1.35 g/dL (2.2 g/dL minus 0.85 g/dL), ie, elevated and due to portal hypertension. With an SAAG of 1.1 g/dL or greater and an ascitic fluid protein level less than 2.5 g/dL, as in our patient, the most likely cause is cirrhosis.
Heart failure is unlikely based on her normal brain natriuretic peptide level, an ascitic fluid protein level below 2.5 g/dL, and normal results on echocardiography. Nephrotic syndrome is also very unlikely based on the patient’s normal random urine protein-creatinine ratio. Portal vein thrombus and abdominal malignancy are essentially ruled out by the negative results of Doppler abdominal ultrasonography, with normal venous flow and no intra-abdominal mass and coupled with an elevated SAAG.
Although the patient has a history of travel, the incubation period for malaria would not fit the time frame of presentation. Also, she did not have typical malarial symptoms, her rapid malaria test was negative, and a peripheral blood smear for blood parasites was negative. It should be noted, however, that Plasmodium malariae infection classically presents with flulike symptoms and can resemble nephrotic syndrome, including peripheral edema, ascites, heavy proteinuria, hypoalbuminemia, and hyperlipidemia.7
3. In which patients is antibiotic prophylaxis against spontaneous bacterial peritonitis (SBP) appropriate?
- Any patient with cirrhosis
- Any patient with cirrhosis who is hospitalized
- Any patient with cirrhosis and an ascitic fluid protein level below 2.0 g/dL
- Any patient with cirrhosis and a history of SBP
Any patient with cirrhosis and a history of SBP should receive prophylactic antibiotics,8 as should any patient deemed at high risk of SBP. It is indicated in the following patients:
- Patients with cirrhosis and gastrointestinal bleeding9,10
- Patients with cirrhosis and a previous episode of SBP8
- Patients with cirrhosis and an ascitic fluid protein level less than 1.5 g/dL with either impaired renal function (creatinine ≥ 1.2 mg/dL, blood urea nitrogen level ≥ 25 mg/dL, or serum sodium ≤ 130 mmol/L) or liver failure (Child-Pugh score ≥ 9 and a bilirubin ≥ 3 mg/dL)9
- Patients with cirrhosis who are hospitalized for other reasons and have an ascitic protein level < 1.0 g/dL.9
Our patient has no signs or symptoms of gastrointestinal bleeding and no history of SBP. Her ascitic fluid protein level is 1.1 g/dL, and she has normal renal function. However, her Child-Pugh score is 12 (3 points for total bilirubin > 3 mg/dL, 3 points for serum albumin < 2.8 g/dL, 2 points for an INR 1.7 to 2.2, 3 points for moderate ascites, and 1 point for no encephalopathy), with a bilirubin of 17.0 mg/dL. Based on this, she is placed on antibiotic prophylaxis for SBP.
Our patient then undergoes an extensive workup for liver disease. Results of tests for toxins, autoimmune diseases, and inheritable diseases are all within normal limits. At this point, despite the patient’s reported negative alcohol history, our leading diagnosis is alcoholic hepatitis.
To confirm this diagnosis, she subsequently undergoes transjugular liver biopsy, considered the gold standard for the diagnosis of alcoholic hepatitis. During the procedure, the hepatic venous pressure gradient is measured at 18 mm Hg (reference range 1–5 mm Hg), suggestive of portal hypertension. The pathology study shows severe fatty change, active steatohepatitis with ballooning degeneration, easily identifiable Mallory-Denk bodies, and prominent neutrophilic infiltration, as well as extensive bridging fibrosis (Figure 2). These findings point to alcoholic hepatitis.
After the biopsy results, we speak with the patient further about her alcohol habits. At this point, she informs us that she has consumed significant amounts of alcohol since the age of 18 (6 to 12 alcoholic beverages per day, including beer and hard liquor). Therefore, based on this new information, on her jaundice and ascites, and on results of laboratory testing and biopsy, we confirmed our diagnosis of alcoholic hepatitis.
4. When is drug treatment appropriate for alcoholic hepatitis?
- Model for End-stage Liver Disease (MELD) score greater than 12
- MELD score greater than 15
- Maddrey Discriminant Function score greater than 25
- Maddrey Discriminant Function score greater than 32
- Glasgow score greater than 5
- Glasgow score greater than 7
The best answer is a Maddrey Discriminant Function score greater than 32. A variety of scoring systems have been used to assess the severity of alcoholic hepatitis and to guide treatment, including the Maddrey Discriminant Function score, the MELD score, and the Glasgow score.11–16 They share similar laboratory values in their calculations, including prothrombin time (or INR) and total bilirubin.11–16 Typically, a Maddrey Discriminant Function score greater than 32, a Glasgow score of greater than 9, or a MELD score greater than 21 is used to determine whether pharmacologic treatment is indicated.11–16
The typical treatment is prednisolone or pentoxifylline.11,17–21 The Lille score is designed to help decide whether to stop corticosteroids after 1 week of administration due to lack of treatment response.22 It predicts mortality rates within 6 months; a score of 0.45 or less indicates a good prognosis, and corticosteroid therapy should continue for 28 days (Figure 3).22
Our patient’s discriminant function score is 50, her Glasgow score is 10, and her MELD score is 28; thus, she begins treatment with oral prednisolone. Her Lille score at 1 week is 0.119, indicating a good prognosis, and her corticosteroids are continued for a total of 28 days.
It should be highlighted that the most important treatment is abstinence from alcohol.11 Recent literature suggests that any benefit of prednisolone or pentoxifylline in terms of mortality rates is questionable,19–20 and there is evidence that giving both drugs simultaneously may improve mortality rates,11,21 but the evidence remains conflicting at this time.
ALCOHOLIC HEPATITIS
Alcoholic hepatitis is a clinical syndrome of jaundice and liver failure, often in the setting of heavy alcohol use for decades.11,12 The incidence is unknown, but the typical age of presentation is between 40 and 50.11,12 The chief sign is a rapid onset of jaundice (< 3 months); common signs and symptoms include fever, ascites, proximal muscle loss, and an enlarged, tender liver.12 Encephalopathy may be seen in severe alcoholic hepatitis.12
Our patient is 35 years old. She has jaundice with rapid onset, as well as ascites and a tender liver.
The diagnosis of alcoholic hepatitis must take into account the patient’s history, physical examination, and laboratory findings. Until proven otherwise, the diagnosis should be presumed in the following scenario: ascites and jaundice on examination (usually with a duration < 3 months); a history of heavy alcohol use; neutrophilic leukocytosis; an AST level that is elevated but below 300 U/L; an ALT level above the normal range but below 300 U/L; an AST-ALT ratio greater than 2; a total serum bilirubin level above 5 mg/dL; and an elevated INR.11,12 Liver biopsy is the gold standard for diagnosis. Though not routinely done because of risks associated with the procedure, it may help confirm the diagnosis if it is in question.
CASE CONCLUDED
We start our patient on oral prednisolone 40 mg daily for alcoholic hepatitis. Her symptoms and laboratory testing results including bilirubin improve. Her Lille score at 7 days indicates a good prognosis, prompting continuation of corticosteroid treatment for the full 28 days.
She is referred to an outpatient alcohol rehabilitation program and has remained sober as of the last outpatient note.
Alcoholic hepatitis is extremely difficult to diagnose, and no single blood test or imaging study confirms the diagnosis. The history, physical examination findings, and laboratory findings are crucial. If the diagnosis is still in doubt, liver biopsy may help confirm the diagnosis.
- Ruyon BA; AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009; 49(6):2087–2107. doi:10.1002/hep.22853
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2(4):399–407. pmid:7095741
- Ginès P, Cárdenas A, Arroyo V, Rodés J. Management of cirrhosis and ascites. N Engl J Med 2004; 350(16):1646–1654. doi:10.1056/NEJMra035021
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117(3):215–220. pmid:1616215
- Hernaez R, Hamilton JP. Unexplained ascites. Clin Liver Dis 2016; 7(3):53–56. https://aasldpubs.onlinelibrary.wiley.com/doi/epdf/10.1002/cld.537
- Huang LL, Xia HH, Zhu SL. Ascitic fluid analysis in the differential diagnosis of ascites: focus on cirrhotic ascites. J Clin Transl Hepatol 2014; 2(1):58–64. doi:10.14218/JCTH.2013.00010
- Bartoloni A, Zammarchi L. Clinical aspects of uncomplicated and severe malaria. Mediterr J Hematol Infect Dis 2012; 4(1):e2012026. doi:10.4084/MJHID.2012.026
- Titó L, Rimola A, Ginès P, Llach J, Arroyo V, Rodés J. Recurrence of spontaneous bacterial peritonitis in cirrhosis: frequency and predictive factors. Hepatology 1988; 8(1):27–31. pmid:3257456
- Fernández J, Ruiz del Arbol L, Gómez C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology 2006; 131(4):1049–1056. doi:10.1053/j.gastro.2006.07.010
- Runyon B; The American Association for the Study of Liver Diseases (AASLD). Management of adult patients with ascites due to cirrhosis: update 2012. https://www.aasld.org/sites/default/files/guideline_documents/141020_Guideline_Ascites_4UFb_2015.pdf. Accessed September 4, 2018.
- Sidhu SS, Goyal O, Kishore H, Sidhu S. New paradigms in management of alcoholic hepatitis: a review. Hepatol Int 2017; 11(3):255–267. doi:10.1007/s12072-017-9790-5
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360(26):2758–2769. doi:10.1056/NEJMra0805786
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75(2):193–199. pmid:352788
- Forrest EH, Evans CD, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005; 54(8):1174–1179. doi:10.1136/gut.2004.050781
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2):353–358. doi:10.1002/hep.20503
- Sheth M, Riggs M, Patel T. Utility of the Mayo end-stage liver disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2. pmid:11835693
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119(6):1637–1648. pmid:11113085
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60(2):255–260. doi:10.1136/gut.2010.224097
- Thursz MR, Richardson P, Allison M, et al; STOPAH Trial. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372(17):1619–1628. doi:10.1056/NEJMoa1412278
- Thursz M, Forrest E, Roderick P, et al. The clinical effectiveness and cost-effectiveness of steroids or pentoxifylline for alcoholic hepatitis (STOPAH): a 2 × 2 factorial randomised controlled trial. Health Technol Assess 2015; 19(102):1–104. doi:10.3310/hta191020
- Lee YS, Kim HJ, Kim JH, et al. Treatment of severe alcoholic hepatitis with corticosteroid, pentoxifylline, or dual therapy: a systematic review and meta-analysis. J Clin Gastroenterol 2017; 51(4):364–377. doi:10.1097/MCG.0000000000000674
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45(6):1348–1354. doi:10.1002/hep.21607
- Ruyon BA; AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009; 49(6):2087–2107. doi:10.1002/hep.22853
- Hoefs JC, Canawati HN, Sapico FL, Hopkins RR, Weiner J, Montgomerie JZ. Spontaneous bacterial peritonitis. Hepatology 1982; 2(4):399–407. pmid:7095741
- Ginès P, Cárdenas A, Arroyo V, Rodés J. Management of cirrhosis and ascites. N Engl J Med 2004; 350(16):1646–1654. doi:10.1056/NEJMra035021
- Runyon BA, Montano AA, Akriviadis EA, Antillon MR, Irving MA, McHutchison JG. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med 1992; 117(3):215–220. pmid:1616215
- Hernaez R, Hamilton JP. Unexplained ascites. Clin Liver Dis 2016; 7(3):53–56. https://aasldpubs.onlinelibrary.wiley.com/doi/epdf/10.1002/cld.537
- Huang LL, Xia HH, Zhu SL. Ascitic fluid analysis in the differential diagnosis of ascites: focus on cirrhotic ascites. J Clin Transl Hepatol 2014; 2(1):58–64. doi:10.14218/JCTH.2013.00010
- Bartoloni A, Zammarchi L. Clinical aspects of uncomplicated and severe malaria. Mediterr J Hematol Infect Dis 2012; 4(1):e2012026. doi:10.4084/MJHID.2012.026
- Titó L, Rimola A, Ginès P, Llach J, Arroyo V, Rodés J. Recurrence of spontaneous bacterial peritonitis in cirrhosis: frequency and predictive factors. Hepatology 1988; 8(1):27–31. pmid:3257456
- Fernández J, Ruiz del Arbol L, Gómez C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology 2006; 131(4):1049–1056. doi:10.1053/j.gastro.2006.07.010
- Runyon B; The American Association for the Study of Liver Diseases (AASLD). Management of adult patients with ascites due to cirrhosis: update 2012. https://www.aasld.org/sites/default/files/guideline_documents/141020_Guideline_Ascites_4UFb_2015.pdf. Accessed September 4, 2018.
- Sidhu SS, Goyal O, Kishore H, Sidhu S. New paradigms in management of alcoholic hepatitis: a review. Hepatol Int 2017; 11(3):255–267. doi:10.1007/s12072-017-9790-5
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009; 360(26):2758–2769. doi:10.1056/NEJMra0805786
- Maddrey WC, Boitnott JK, Bedine MS, Weber FL Jr, Mezey E, White RI Jr. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978; 75(2):193–199. pmid:352788
- Forrest EH, Evans CD, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005; 54(8):1174–1179. doi:10.1136/gut.2004.050781
- Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2):353–358. doi:10.1002/hep.20503
- Sheth M, Riggs M, Patel T. Utility of the Mayo end-stage liver disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002; 2:2. pmid:11835693
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000; 119(6):1637–1648. pmid:11113085
- Mathurin P, O’Grady J, Carithers RL, et al. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut 2011; 60(2):255–260. doi:10.1136/gut.2010.224097
- Thursz MR, Richardson P, Allison M, et al; STOPAH Trial. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015; 372(17):1619–1628. doi:10.1056/NEJMoa1412278
- Thursz M, Forrest E, Roderick P, et al. The clinical effectiveness and cost-effectiveness of steroids or pentoxifylline for alcoholic hepatitis (STOPAH): a 2 × 2 factorial randomised controlled trial. Health Technol Assess 2015; 19(102):1–104. doi:10.3310/hta191020
- Lee YS, Kim HJ, Kim JH, et al. Treatment of severe alcoholic hepatitis with corticosteroid, pentoxifylline, or dual therapy: a systematic review and meta-analysis. J Clin Gastroenterol 2017; 51(4):364–377. doi:10.1097/MCG.0000000000000674
- Louvet A, Naveau S, Abdelnour M, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007; 45(6):1348–1354. doi:10.1002/hep.21607
When does S aureus bacteremia require transesophageal echocardiography?
Staphylococcus aureus is the most common infective agent in native and prosthetic valve endocarditis, and 13% to 22% of patients with S aureus bacteremia have infective endocarditis.1
Transthoracic echocardiography (TTE) is a good starting point in the workup of suspected infective endocarditis, but transesophageal echocardiography (TEE) plays a key role in diagnosis and is indicated in patients with a high pretest probability of infective endocarditis, as in the following scenarios:
- Clinical picture consistent with infective endocarditis
- Presence of previously placed port or other indwelling vascular device
- Presence of a prosthetic valve or other prosthetic material
- Presence of a pacemaker
- History of valve disease
- Injection drug use
- Positive blood cultures after 72 hours despite appropriate antibiotic treatment
- Abnormal TTE result requiring better visualization of valvular anatomy and function and confirmation of local complications
- Absence of another reasonable explanation for S aureus bacteremia.
Forgoing TEE is reasonable in patients with normal results on TTE, no predisposing risk factors, a reasonable alternative explanation for S aureus bacteremia, and a low pretest probability of infective endocarditis.1 TEE may also be unnecessary if there is another disease focus requiring extended treatment (eg, vertebral infection) and there are no findings suggesting complicated infective endocarditis, eg, persistent bacteremia, symptoms of heart failure, and conduction abnormality.1
TEE also may be unnecessary in patients at low risk who have identifiable foci of bacteremia due to soft-tissue infection or a newly placed vascular catheter and whose bacteremia clears within 72 hours of the start of antibiotic therapy. These patients may be followed clinically for the development of new findings such as metastatic foci of infection (eg, septic pulmonary emboli, renal infarction, splenic abscess or infarction), the new onset of heart failure or cardiac conduction abnormality, or recurrence of previously cleared S aureus bacteremia. If these should develop, then a more invasive study such as TEE may be warranted.
INFECTIVE ENDOCARDITIS: EPIDEMIOLOGY AND MICROBIOLOGY
The US incidence rate of infective endocarditis has steadily increased, with an estimated 457,052 hospitalizations from 2000 to 2011. During that period, from 2000 to 2007, there was a marked increase in valve replacement surgeries.2 This trend is likely explained by an increase in the at-risk population—eg, elderly patients, patients with opiate dependence or diabetes, and patients on hemodialysis.
Although S aureus is the predominant pathogen in infective endocarditis,2–5S aureus bacteremia is often observed in patients with skin or soft-tissue infection, prosthetic device infection, vascular graft or catheter infection, and bone and joint infections. S aureus bacteremia necessitates a search for the source of infection.
S aureus is a major pathogen in bloodstream infections, and up to 14% of patients with S aureus bacteremia have infective endocarditis as the primary source of infection.3 The pathogenesis of S aureus infective endocarditis is thought to be mediated by cell-wall factors that promote adhesion to the extracellular matrix of intravascular structures.3
A new localizing symptom such as back pain, joint pain, or swelling in a patient with S aureus bacteremia should trigger an investigation for metastatic infection.
Infectious disease consultation in patients with S aureus bacteremia is associated with improved outcomes and, thus, should be pursued.3
A cardiac surgery consult is recommended early on in cases of infective endocarditis caused by vancomycin-resistant enterococci, Pseudomonas aeruginosa, and fungi, as well as in patients with complications such as valvular insufficiency, perivalvular abscess, conduction abnormalities, persistent bacteremia, and metastatic foci of infection.6
RISK FACTORS
Risk factors for infective endocarditis include injection drug abuse, valvular heart disease, congenital heart disease (unrepaired, repaired with residual defects, or fully repaired within the past 6 months), previous infective endocarditis, prosthetic heart valve, and cardiac transplant.2–4,6 Other risk factors are poor dentition, hemodialysis, ventriculoatrial shunts, intravascular devices including vascular grafts, and pacemakers.2,3 Many risk factors for infective endocarditis and S aureus bacteremia overlap.3
DIAGNOSTIC PRINCIPLES
The clinical presentation of infective endocarditis can vary from a nonspecific infectious syndrome, to overt organ failure (heart failure, kidney failure), to an acute vascular catastrophe (arterial ischemia, cerebrovascular accidents, myocardial infarction). Patients may present with indolent symptoms such as fever, fatigue, and weight loss,6 or they may present at an advanced stage, with fulminant acute heart failure due to valvular insufficiency or with arrhythmias due to a perivalvular abscess infiltrating the conduction system. Extracardiac clinical manifestations may be related to direct infective metastatic foci such as septic emboli or to immunologic phenomena such as glomerulonephritis or Osler nodes.


ECHOCARDIOGRAPHY’S ROLE IN DIAGNOSIS
TTE plays an important role in diagnosis and risk stratification of infective endocarditis.6 TTE is usually done first because of its low cost, wide availability, and safety; it has a sensitivity of 70% and a specificity over 95%.8 While a normal result on TTE does not completely rule out infective endocarditis, completely normal valvular morphology and function on TTE make the diagnosis less likely.8,9
If suspicion remains high despite a normal study, repeating TTE at a later time may result in a higher diagnostic yield because of growth of the suspected vegetation. Otherwise, TEE should be considered.
TEE provides a higher spatial resolution and diagnostic yield than TTE, especially for detecting complex pathology such as pseudoaneurysm, valve perforation, or valvular abscess. TEE has a sensitivity and specificity of approximately 95% for infective endocarditis.8 It should be performed early in patients with preexisting valve disease, prosthetic cardiac material (eg, valves), or a pacemaker or implantable cardioverter-defibrillator.6,7
Detecting valve vegetation provides answers about the cause of S aureus bacteremia with its complications (eg, septic emboli, mycotic aneurysm) and informs decisions about the duration of antibiotic therapy and the need for surgery.3,6
As with any diagnostic test, it is important to compare the results of any recent study with those of previous studies whenever possible to differentiate new from old findings.
WHEN TO FORGO TEE IN S AUREUS BACTEREMIA
Because TEE is invasive and requires the patient to swallow an endoscopic probe,10 it is important to screen patients for esophageal disease, cervical spine conditions, and baseline respiratory insufficiency. Complications are rare but include esophageal perforation, esophageal bleeding, pharyngeal hematoma, and reactions to anesthesia.10
As with any diagnostic test, the clinician first needs to consider the patient’s pretest probability of the disease, the diagnostic accuracy, the associated risks and costs, and the implications of the results.
While TEE provides better diagnostic images than TTE, a normal TEE study does not exclude the diagnosis of infective endocarditis: small lesions and complications such as paravalvular abscess of a prosthetic aortic valve may still be missed. In such patients, a repeat TEE examination or additional imaging study (eg, gated computed tomographic angiography) should be considered.6
Noninfective sterile echodensities, valvular tumors such as papillary fibroelastomas, Lambl excrescences, and suture lines of prosthetic valves are among the conditions and factors that can cause a false-positive result on TEE.
- Young H, Knepper BC, Price CS, Heard S, Jenkins TC. Clinical reasoning of infectious diseases physicians behind the use or nonuse of transesophageal echocardiography in Staphylococcus aureus bacteremia. Open Forum Infect Dis 2016; 3(4):ofw204. doi:10.1093/ofid/ofw204
- Pant S, Patel NJ, Deshmukh A, et al. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J Am Coll Cardiol 2015; 65(19):2070–2076. doi:10.1016/j.jacc.2015.03.518
- Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 2015; 28(3):603–661. doi:10.1128/CMR.00134-14
- Palraj BR, Baddour LM, Hess EP, et al. Predicting risk of endocarditis using a clinical tool (PREDICT): scoring system to guide use of echocardiography in the management of Staphylococcus aureus bacteremia. Clin Infect Dis 2015; 61(1):18–28. doi:10.1093/cid/civ235
- Barton T, Moir S, Rehmani H, Woolley I, Korman TM, Stuart RL. Low rates of endocarditis in healthcare-associated Staphylococcus aureus bacteremia suggest that echocardiography might not always be required. Eur J Clin Microbiol Infect Dis 2016; 35(1):49–55. doi:10.1007/s10096-015-2505-8
- Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi10.1161/CIR.0000000000000296
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30(4):633–638. doi:10.1086/313753
- Habib G, Badano L, Tribouilloy C, et al; European Association of Echocardiography. Recommendations for the practice of echocardiography in infective endocarditis. Eur J Echocardiogr 2010; 11(2):202–219. doi:10.1093/ejechocard/jeq004
- Irani WN, Grayburn PA, Afridi I. A negative transthoracic echocardiogram obviates the need for transesophageal echocardiography in patients with suspected native valve active infective endocarditis. Am J Cardiol 1996; 78(1):101–103. pmid:8712097
- Hahn RT, Abraham T, Adams MS, et al. Guidelines for performing a comprehensive transesophageal echocardiographic examination: recommendations from the American Society of Echocardiography and the Society of Cardiovascular Anesthesiologists. J Am Soc Echocardiogr 2013; 26(9):921–964. doi:10.1016/j.echo.2013.07.009
Staphylococcus aureus is the most common infective agent in native and prosthetic valve endocarditis, and 13% to 22% of patients with S aureus bacteremia have infective endocarditis.1
Transthoracic echocardiography (TTE) is a good starting point in the workup of suspected infective endocarditis, but transesophageal echocardiography (TEE) plays a key role in diagnosis and is indicated in patients with a high pretest probability of infective endocarditis, as in the following scenarios:
- Clinical picture consistent with infective endocarditis
- Presence of previously placed port or other indwelling vascular device
- Presence of a prosthetic valve or other prosthetic material
- Presence of a pacemaker
- History of valve disease
- Injection drug use
- Positive blood cultures after 72 hours despite appropriate antibiotic treatment
- Abnormal TTE result requiring better visualization of valvular anatomy and function and confirmation of local complications
- Absence of another reasonable explanation for S aureus bacteremia.
Forgoing TEE is reasonable in patients with normal results on TTE, no predisposing risk factors, a reasonable alternative explanation for S aureus bacteremia, and a low pretest probability of infective endocarditis.1 TEE may also be unnecessary if there is another disease focus requiring extended treatment (eg, vertebral infection) and there are no findings suggesting complicated infective endocarditis, eg, persistent bacteremia, symptoms of heart failure, and conduction abnormality.1
TEE also may be unnecessary in patients at low risk who have identifiable foci of bacteremia due to soft-tissue infection or a newly placed vascular catheter and whose bacteremia clears within 72 hours of the start of antibiotic therapy. These patients may be followed clinically for the development of new findings such as metastatic foci of infection (eg, septic pulmonary emboli, renal infarction, splenic abscess or infarction), the new onset of heart failure or cardiac conduction abnormality, or recurrence of previously cleared S aureus bacteremia. If these should develop, then a more invasive study such as TEE may be warranted.
INFECTIVE ENDOCARDITIS: EPIDEMIOLOGY AND MICROBIOLOGY
The US incidence rate of infective endocarditis has steadily increased, with an estimated 457,052 hospitalizations from 2000 to 2011. During that period, from 2000 to 2007, there was a marked increase in valve replacement surgeries.2 This trend is likely explained by an increase in the at-risk population—eg, elderly patients, patients with opiate dependence or diabetes, and patients on hemodialysis.
Although S aureus is the predominant pathogen in infective endocarditis,2–5S aureus bacteremia is often observed in patients with skin or soft-tissue infection, prosthetic device infection, vascular graft or catheter infection, and bone and joint infections. S aureus bacteremia necessitates a search for the source of infection.
S aureus is a major pathogen in bloodstream infections, and up to 14% of patients with S aureus bacteremia have infective endocarditis as the primary source of infection.3 The pathogenesis of S aureus infective endocarditis is thought to be mediated by cell-wall factors that promote adhesion to the extracellular matrix of intravascular structures.3
A new localizing symptom such as back pain, joint pain, or swelling in a patient with S aureus bacteremia should trigger an investigation for metastatic infection.
Infectious disease consultation in patients with S aureus bacteremia is associated with improved outcomes and, thus, should be pursued.3
A cardiac surgery consult is recommended early on in cases of infective endocarditis caused by vancomycin-resistant enterococci, Pseudomonas aeruginosa, and fungi, as well as in patients with complications such as valvular insufficiency, perivalvular abscess, conduction abnormalities, persistent bacteremia, and metastatic foci of infection.6
RISK FACTORS
Risk factors for infective endocarditis include injection drug abuse, valvular heart disease, congenital heart disease (unrepaired, repaired with residual defects, or fully repaired within the past 6 months), previous infective endocarditis, prosthetic heart valve, and cardiac transplant.2–4,6 Other risk factors are poor dentition, hemodialysis, ventriculoatrial shunts, intravascular devices including vascular grafts, and pacemakers.2,3 Many risk factors for infective endocarditis and S aureus bacteremia overlap.3
DIAGNOSTIC PRINCIPLES
The clinical presentation of infective endocarditis can vary from a nonspecific infectious syndrome, to overt organ failure (heart failure, kidney failure), to an acute vascular catastrophe (arterial ischemia, cerebrovascular accidents, myocardial infarction). Patients may present with indolent symptoms such as fever, fatigue, and weight loss,6 or they may present at an advanced stage, with fulminant acute heart failure due to valvular insufficiency or with arrhythmias due to a perivalvular abscess infiltrating the conduction system. Extracardiac clinical manifestations may be related to direct infective metastatic foci such as septic emboli or to immunologic phenomena such as glomerulonephritis or Osler nodes.
ECHOCARDIOGRAPHY’S ROLE IN DIAGNOSIS
TTE plays an important role in diagnosis and risk stratification of infective endocarditis.6 TTE is usually done first because of its low cost, wide availability, and safety; it has a sensitivity of 70% and a specificity over 95%.8 While a normal result on TTE does not completely rule out infective endocarditis, completely normal valvular morphology and function on TTE make the diagnosis less likely.8,9
If suspicion remains high despite a normal study, repeating TTE at a later time may result in a higher diagnostic yield because of growth of the suspected vegetation. Otherwise, TEE should be considered.
TEE provides a higher spatial resolution and diagnostic yield than TTE, especially for detecting complex pathology such as pseudoaneurysm, valve perforation, or valvular abscess. TEE has a sensitivity and specificity of approximately 95% for infective endocarditis.8 It should be performed early in patients with preexisting valve disease, prosthetic cardiac material (eg, valves), or a pacemaker or implantable cardioverter-defibrillator.6,7
Detecting valve vegetation provides answers about the cause of S aureus bacteremia with its complications (eg, septic emboli, mycotic aneurysm) and informs decisions about the duration of antibiotic therapy and the need for surgery.3,6
As with any diagnostic test, it is important to compare the results of any recent study with those of previous studies whenever possible to differentiate new from old findings.
WHEN TO FORGO TEE IN S AUREUS BACTEREMIA
Because TEE is invasive and requires the patient to swallow an endoscopic probe,10 it is important to screen patients for esophageal disease, cervical spine conditions, and baseline respiratory insufficiency. Complications are rare but include esophageal perforation, esophageal bleeding, pharyngeal hematoma, and reactions to anesthesia.10
As with any diagnostic test, the clinician first needs to consider the patient’s pretest probability of the disease, the diagnostic accuracy, the associated risks and costs, and the implications of the results.
While TEE provides better diagnostic images than TTE, a normal TEE study does not exclude the diagnosis of infective endocarditis: small lesions and complications such as paravalvular abscess of a prosthetic aortic valve may still be missed. In such patients, a repeat TEE examination or additional imaging study (eg, gated computed tomographic angiography) should be considered.6
Noninfective sterile echodensities, valvular tumors such as papillary fibroelastomas, Lambl excrescences, and suture lines of prosthetic valves are among the conditions and factors that can cause a false-positive result on TEE.
Staphylococcus aureus is the most common infective agent in native and prosthetic valve endocarditis, and 13% to 22% of patients with S aureus bacteremia have infective endocarditis.1
Transthoracic echocardiography (TTE) is a good starting point in the workup of suspected infective endocarditis, but transesophageal echocardiography (TEE) plays a key role in diagnosis and is indicated in patients with a high pretest probability of infective endocarditis, as in the following scenarios:
- Clinical picture consistent with infective endocarditis
- Presence of previously placed port or other indwelling vascular device
- Presence of a prosthetic valve or other prosthetic material
- Presence of a pacemaker
- History of valve disease
- Injection drug use
- Positive blood cultures after 72 hours despite appropriate antibiotic treatment
- Abnormal TTE result requiring better visualization of valvular anatomy and function and confirmation of local complications
- Absence of another reasonable explanation for S aureus bacteremia.
Forgoing TEE is reasonable in patients with normal results on TTE, no predisposing risk factors, a reasonable alternative explanation for S aureus bacteremia, and a low pretest probability of infective endocarditis.1 TEE may also be unnecessary if there is another disease focus requiring extended treatment (eg, vertebral infection) and there are no findings suggesting complicated infective endocarditis, eg, persistent bacteremia, symptoms of heart failure, and conduction abnormality.1
TEE also may be unnecessary in patients at low risk who have identifiable foci of bacteremia due to soft-tissue infection or a newly placed vascular catheter and whose bacteremia clears within 72 hours of the start of antibiotic therapy. These patients may be followed clinically for the development of new findings such as metastatic foci of infection (eg, septic pulmonary emboli, renal infarction, splenic abscess or infarction), the new onset of heart failure or cardiac conduction abnormality, or recurrence of previously cleared S aureus bacteremia. If these should develop, then a more invasive study such as TEE may be warranted.
INFECTIVE ENDOCARDITIS: EPIDEMIOLOGY AND MICROBIOLOGY
The US incidence rate of infective endocarditis has steadily increased, with an estimated 457,052 hospitalizations from 2000 to 2011. During that period, from 2000 to 2007, there was a marked increase in valve replacement surgeries.2 This trend is likely explained by an increase in the at-risk population—eg, elderly patients, patients with opiate dependence or diabetes, and patients on hemodialysis.
Although S aureus is the predominant pathogen in infective endocarditis,2–5S aureus bacteremia is often observed in patients with skin or soft-tissue infection, prosthetic device infection, vascular graft or catheter infection, and bone and joint infections. S aureus bacteremia necessitates a search for the source of infection.
S aureus is a major pathogen in bloodstream infections, and up to 14% of patients with S aureus bacteremia have infective endocarditis as the primary source of infection.3 The pathogenesis of S aureus infective endocarditis is thought to be mediated by cell-wall factors that promote adhesion to the extracellular matrix of intravascular structures.3
A new localizing symptom such as back pain, joint pain, or swelling in a patient with S aureus bacteremia should trigger an investigation for metastatic infection.
Infectious disease consultation in patients with S aureus bacteremia is associated with improved outcomes and, thus, should be pursued.3
A cardiac surgery consult is recommended early on in cases of infective endocarditis caused by vancomycin-resistant enterococci, Pseudomonas aeruginosa, and fungi, as well as in patients with complications such as valvular insufficiency, perivalvular abscess, conduction abnormalities, persistent bacteremia, and metastatic foci of infection.6
RISK FACTORS
Risk factors for infective endocarditis include injection drug abuse, valvular heart disease, congenital heart disease (unrepaired, repaired with residual defects, or fully repaired within the past 6 months), previous infective endocarditis, prosthetic heart valve, and cardiac transplant.2–4,6 Other risk factors are poor dentition, hemodialysis, ventriculoatrial shunts, intravascular devices including vascular grafts, and pacemakers.2,3 Many risk factors for infective endocarditis and S aureus bacteremia overlap.3
DIAGNOSTIC PRINCIPLES
The clinical presentation of infective endocarditis can vary from a nonspecific infectious syndrome, to overt organ failure (heart failure, kidney failure), to an acute vascular catastrophe (arterial ischemia, cerebrovascular accidents, myocardial infarction). Patients may present with indolent symptoms such as fever, fatigue, and weight loss,6 or they may present at an advanced stage, with fulminant acute heart failure due to valvular insufficiency or with arrhythmias due to a perivalvular abscess infiltrating the conduction system. Extracardiac clinical manifestations may be related to direct infective metastatic foci such as septic emboli or to immunologic phenomena such as glomerulonephritis or Osler nodes.
ECHOCARDIOGRAPHY’S ROLE IN DIAGNOSIS
TTE plays an important role in diagnosis and risk stratification of infective endocarditis.6 TTE is usually done first because of its low cost, wide availability, and safety; it has a sensitivity of 70% and a specificity over 95%.8 While a normal result on TTE does not completely rule out infective endocarditis, completely normal valvular morphology and function on TTE make the diagnosis less likely.8,9
If suspicion remains high despite a normal study, repeating TTE at a later time may result in a higher diagnostic yield because of growth of the suspected vegetation. Otherwise, TEE should be considered.
TEE provides a higher spatial resolution and diagnostic yield than TTE, especially for detecting complex pathology such as pseudoaneurysm, valve perforation, or valvular abscess. TEE has a sensitivity and specificity of approximately 95% for infective endocarditis.8 It should be performed early in patients with preexisting valve disease, prosthetic cardiac material (eg, valves), or a pacemaker or implantable cardioverter-defibrillator.6,7
Detecting valve vegetation provides answers about the cause of S aureus bacteremia with its complications (eg, septic emboli, mycotic aneurysm) and informs decisions about the duration of antibiotic therapy and the need for surgery.3,6
As with any diagnostic test, it is important to compare the results of any recent study with those of previous studies whenever possible to differentiate new from old findings.
WHEN TO FORGO TEE IN S AUREUS BACTEREMIA
Because TEE is invasive and requires the patient to swallow an endoscopic probe,10 it is important to screen patients for esophageal disease, cervical spine conditions, and baseline respiratory insufficiency. Complications are rare but include esophageal perforation, esophageal bleeding, pharyngeal hematoma, and reactions to anesthesia.10
As with any diagnostic test, the clinician first needs to consider the patient’s pretest probability of the disease, the diagnostic accuracy, the associated risks and costs, and the implications of the results.
While TEE provides better diagnostic images than TTE, a normal TEE study does not exclude the diagnosis of infective endocarditis: small lesions and complications such as paravalvular abscess of a prosthetic aortic valve may still be missed. In such patients, a repeat TEE examination or additional imaging study (eg, gated computed tomographic angiography) should be considered.6
Noninfective sterile echodensities, valvular tumors such as papillary fibroelastomas, Lambl excrescences, and suture lines of prosthetic valves are among the conditions and factors that can cause a false-positive result on TEE.
- Young H, Knepper BC, Price CS, Heard S, Jenkins TC. Clinical reasoning of infectious diseases physicians behind the use or nonuse of transesophageal echocardiography in Staphylococcus aureus bacteremia. Open Forum Infect Dis 2016; 3(4):ofw204. doi:10.1093/ofid/ofw204
- Pant S, Patel NJ, Deshmukh A, et al. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J Am Coll Cardiol 2015; 65(19):2070–2076. doi:10.1016/j.jacc.2015.03.518
- Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 2015; 28(3):603–661. doi:10.1128/CMR.00134-14
- Palraj BR, Baddour LM, Hess EP, et al. Predicting risk of endocarditis using a clinical tool (PREDICT): scoring system to guide use of echocardiography in the management of Staphylococcus aureus bacteremia. Clin Infect Dis 2015; 61(1):18–28. doi:10.1093/cid/civ235
- Barton T, Moir S, Rehmani H, Woolley I, Korman TM, Stuart RL. Low rates of endocarditis in healthcare-associated Staphylococcus aureus bacteremia suggest that echocardiography might not always be required. Eur J Clin Microbiol Infect Dis 2016; 35(1):49–55. doi:10.1007/s10096-015-2505-8
- Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi10.1161/CIR.0000000000000296
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30(4):633–638. doi:10.1086/313753
- Habib G, Badano L, Tribouilloy C, et al; European Association of Echocardiography. Recommendations for the practice of echocardiography in infective endocarditis. Eur J Echocardiogr 2010; 11(2):202–219. doi:10.1093/ejechocard/jeq004
- Irani WN, Grayburn PA, Afridi I. A negative transthoracic echocardiogram obviates the need for transesophageal echocardiography in patients with suspected native valve active infective endocarditis. Am J Cardiol 1996; 78(1):101–103. pmid:8712097
- Hahn RT, Abraham T, Adams MS, et al. Guidelines for performing a comprehensive transesophageal echocardiographic examination: recommendations from the American Society of Echocardiography and the Society of Cardiovascular Anesthesiologists. J Am Soc Echocardiogr 2013; 26(9):921–964. doi:10.1016/j.echo.2013.07.009
- Young H, Knepper BC, Price CS, Heard S, Jenkins TC. Clinical reasoning of infectious diseases physicians behind the use or nonuse of transesophageal echocardiography in Staphylococcus aureus bacteremia. Open Forum Infect Dis 2016; 3(4):ofw204. doi:10.1093/ofid/ofw204
- Pant S, Patel NJ, Deshmukh A, et al. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J Am Coll Cardiol 2015; 65(19):2070–2076. doi:10.1016/j.jacc.2015.03.518
- Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 2015; 28(3):603–661. doi:10.1128/CMR.00134-14
- Palraj BR, Baddour LM, Hess EP, et al. Predicting risk of endocarditis using a clinical tool (PREDICT): scoring system to guide use of echocardiography in the management of Staphylococcus aureus bacteremia. Clin Infect Dis 2015; 61(1):18–28. doi:10.1093/cid/civ235
- Barton T, Moir S, Rehmani H, Woolley I, Korman TM, Stuart RL. Low rates of endocarditis in healthcare-associated Staphylococcus aureus bacteremia suggest that echocardiography might not always be required. Eur J Clin Microbiol Infect Dis 2016; 35(1):49–55. doi:10.1007/s10096-015-2505-8
- Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132(15):1435–1486. doi10.1161/CIR.0000000000000296
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30(4):633–638. doi:10.1086/313753
- Habib G, Badano L, Tribouilloy C, et al; European Association of Echocardiography. Recommendations for the practice of echocardiography in infective endocarditis. Eur J Echocardiogr 2010; 11(2):202–219. doi:10.1093/ejechocard/jeq004
- Irani WN, Grayburn PA, Afridi I. A negative transthoracic echocardiogram obviates the need for transesophageal echocardiography in patients with suspected native valve active infective endocarditis. Am J Cardiol 1996; 78(1):101–103. pmid:8712097
- Hahn RT, Abraham T, Adams MS, et al. Guidelines for performing a comprehensive transesophageal echocardiographic examination: recommendations from the American Society of Echocardiography and the Society of Cardiovascular Anesthesiologists. J Am Soc Echocardiogr 2013; 26(9):921–964. doi:10.1016/j.echo.2013.07.009
Near and Far
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
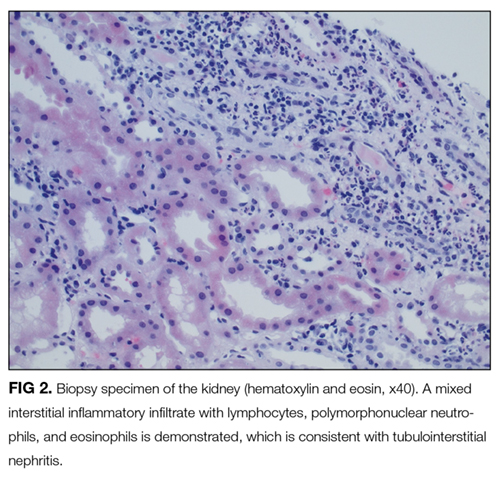
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
A previously healthy 30-year-old woman presented to the emergency department with 2 weeks of weakness.
True muscle weakness must be distinguished from the more common causes of asthenia. Many systemic disorders produce fatigue, with resulting functional limitation that is often interpreted by patients as weakness. Initial history should focus on conditions producing fatigue, such as cardiopulmonary disease, anemia, connective tissue disease, depression or cachexia related to malignancy, infection, or other inflammatory states. Careful questioning may reveal evidence of dyspnea, poor exercise tolerance, or joint pain as an alternative to actual loss of muscle power. If true weakness is still suspected, attention should be focused on the pattern, onset, anatomic site, and progression of weakness. Muscle weakness is often characterized by difficulty with specific tasks, such as climbing stairs, rising from a chair, raising a hand, or using cutlery. The physical examination is critical in determining whether weakness is due to true loss of motor power. The differential diagnosis of weakness is broad and includes neurologic, infectious, endocrine, inflammatory, genetic, metabolic, and drug-induced etiologies.
She initially experienced 3 days of mild cramps and soreness in her thighs. She then developed weakness that began in her thighs and progressed to involve her lower legs and upper and lower arms. She had difficulty combing her hair. She required the use of her arms to get up from a chair. She grasped onto objects to aid in ambulation around the house. In addition, she described 1 year of moderate fatigue but no fever, weight loss, dyspnea, dysphagia, visual changes, paresthesias, bowel or bladder incontinence, back pain, or preceding gastrointestinal or respiratory illness. She had experienced diffuse intermittent hives, most prominent in her chest and upper arms, for the past several weeks.
History certainly supports true weakness but will need to be confirmed on examination. The distribution began as proximal but now appears diffuse. The presence of myalgia and cramping raises the possibility of noninflammatory myopathies, which are usually more insidious in onset. A severe electrolyte disturbance would be possible, based on the diffuse nature of weakness that was preceded by cramping. The distribution of weakness and lack of bowel or bladder incontinence is reassuring and suggests against a spinal cord disorder; however, a high index of suspicion must be maintained for myelopathy because delayed treatment might result in irreversible paralysis.
The patient’s course also includes hives. Common causes of hives include infections and allergic reactions to medications, foods, and insect stings. Urticaria may also result from systemic disorders, such as vasculitis, lupus, lymphoma, mastocytosis, and paraproteinemias, which can be associated with weakness and fatigue. Although severe weakness in combination with hives makes an infectious and allergic reaction less likely, we still seek to ascertain if the evolving chief complaints of weakness and hives are the result of a single unifying and evolving multisystem disorder or are distinct and unrelated processes.
Her past medical history included fibromyalgia, kidney stones, and gastroesophageal reflux disease. One week prior to presentation, she was prescribed prednisone 60 mg daily for the treatment of hives; the dose had been tapered to 40 mg at presentation, with mild improvement of hives. She recently started doxepin for fibromyalgia and insomnia. She lived at home with her husband and 8-year-old child. She worked as a clerk in a pest control office and denied any pesticide exposure. She denied tobacco, alcohol, or illicit drug use. Her family history included systemic lupus erythematosus (SLE) in her mother and maternal aunt.
Glucocorticoids are associated with myopathy; however, the weakness preceded steroid therapy. Thus, unless there was unknown exposure to high-dose steroid medication to treat recurrent episodes of urticaria earlier in her course, glucocorticoid-related myopathy is unlikely. Fibromyalgia might cause the perception of weakness from pain. However, the history of difficulty combing her hair and rising from a chair suggests actual loss of motor power. The side effects of her medications, such as newly started doxepin, must be reviewed. A family history of SLE raises concern for rheumatologic conditions; however, one might expect improvement with steroid therapy.
On physical examination, her temperature was 36.9 °C, blood pressure 126/93 mmHg, pulse 81 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 100% on ambient air. Her cardiopulmonary examination was normal. Her abdomen was nontender and without hepatosplenomegaly. Her strength was 2 out of 5 in proximal and distal legs, bilaterally, and 4 out of 5 in proximal and distal upper extremities. She had normal muscle tone without fasciculations or atrophy. Her joints were without edema, erythema, or impaired range of motion. She had normal sensation to light touch in arms and legs. Her reflexes were 2+ in the patellar, Achilles, and brachioradialis tendons. She had no lymphadenopathy, mucosal ulcerations, or alopecia. A skin examination revealed smooth, slightly elevated, and faded pink wheals that were diffuse but most prominent in upper arms and chest.
Physical examination confirms the presence of true muscle weakness. The differential diagnosis is narrowed by several findings, both positive and negative, elicited in the examination. The diffuse nature of the weakness eliminates focal central nervous system lesions, such as stroke, intracranial mass lesions, or demyelinating white matter foci. Combining this finding with normal reflexes and history of preceding myalgias makes electrolyte-induced and inflammatory (eg, polymyositis) myopathies more likely. The normal deep tendon reflexes and the absence of a delayed relaxation phase lower the likelihood of hypothyroidism.
Diseases originating from the neuromuscular junction, such as myasthenia gravis, may also present with weakness and normal reflexes, although this pattern of weakness would be atypical; myasthenia gravis classically presents with fatigable weakness and ocular findings of diplopia and/or ptosis. First-tier testing should include a complete blood count to evaluate for eosinophilia, comprehensive metabolic panel, and urinalysis for myoglobinuria, thyroid stimulating hormone, and muscle enzymes.
Results of a complete blood count demonstrated a leukocyte count of 16.1 k/uL with 82% neutrophils, 13% lymphocytes, 5% monocytes, and 0% eosinophils. Hemoglobin was 13.2 g/dL, and platelet count 226 k/uL. Sodium was 136 mmol/L, potassium 1.5 mmol/L, chloride 115 mmol/L, bicarbonate 12 mmol/L, blood urea nitrogen 26 mg/dL, creatinine 1.0 mg/dL (baseline creatinine: 0.6), and glucose 102 mg/dL. Calcium was 9.4 mg/dL, magnesium 2.6 mg/dL, phosphorus 1.8 mg/dL, CK 501 U/L (normal: 40-230), and TSH 5.48 uIU/mL (normal: 0.5-4). Aspartate aminotransferase was 64 U/L, alanine aminotransferase 23 U/L, alkaline phosphatase 66 U/L, bilirubin 0.9 mg/dL, albumin 3.8 g/dL, and total protein 8.7 g/dL (normal: 6.2-7.8). Human immunodeficiency virus antibody screen was negative. An electrocardiogram revealed normal sinus rhythm, flattened T waves, and prominent U waves.
Potassium losses are classically categorized into 1 of 3 groups: renal losses, gastrointestinal losses, or transcellular shifts. Without a clear history of diuretic use, renal losses may not be apparent on history and examination. In contrast, gastrointestinal losses are almost always evidenced by a history of vomiting and/or diarrhea, with rare exceptions, including unreported laxative abuse or surreptitious vomiting. Transcellular potassium shifts can be seen in states of increased insulin or beta-adrenergic activity and alkalosis and result from both primary and secondary causes of hypokalemic periodic paralysis.
The presence of a reduced serum bicarbonate and elevated chloride concentration suggests a normal anion gap metabolic acidosis. Many conditions associated with normal anion gap metabolic acidosis are evident by history, such as diarrhea. In enigmatic cases such as this, it will be important to take a stepwise approach that includes an evaluation for urinary potassium losses and assessment of acid-base status. An unexplained normal anion gap metabolic acidosis combined with hypokalemia raises suspicion for a distal renal tubular acidosis (RTA). Additional testing to evaluate for a possible RTA should include the assessment of urinary electrolytes and urinary pH. The hypokalemia explains her weakness, but the etiology of such profound hypokalemia is not evident, nor is it clear how it relates to her hives.
The severity of the hypokalemia, combined with electrocardiogram changes, necessitates rapid intravenous potassium repletion, telemetry monitoring, and frequent serum potassium measurement. Treatment of her metabolic acidosis is more nuanced and depends upon both the severity of disturbance and the suspicion of whether the etiology is transcellular shift, potassium depletion, or both.
Urine studies demonstrated a urine specific gravity of 1.006 (normal: 1.001-1.030), urine pH was 6.5 (normal: 5-6.5), trace leukocyte esterase, negative nitrite, 30 mg/dL of protein (normal: <15), sodium 64 mmol/L (normal: 40-220), potassium 17 mmol/L (normal: 25-125), and chloride 71 mmol/L (normal: 110-250). Urine microscopy demonstrated 3 red blood cells per high power field (normal: 0-1), 4 white blood cells per high power field (normal: 0-4), 4+ bacteria per high power field, and no red blood cell casts. Urine protein-to-creatinine ratio was 1.6. C3 and C4 complement levels were 53 mg/dL (normal: 80-165) and 12 mg/dL (normal: 15-49), respectively. C-reactive protein was <0.5 (normal: 0-0.9), and erythrocyte sedimentation rate was 16 mm/hour (normal: 0-20).
A calculation of the urine anion gap (UAG; [urine sodium + urine potassium] – urine chloride) yields a UAG of 10 mq/L. A positive UAG, together with a nongap metabolic acidosis, should prompt the consideration of RTA. The normal renal response to acidosis is to reduce the urine pH to less than 5.3 through an increase in hydrogen ion excretion in the form of ammonium. A urine pH of 6.5 is highly suggestive of type 1 (distal) RTA and its associated impairment of distal acidification. Treatment with sodium bicarbonate to correct the acidosis and associated complications is warranted.
A distal RTA would account for her past medical history of renal stones. Acidemia promotes both increased calcium phosphate release from bone (with subsequent hypercalciuria) and enhanced citrate reabsorption in the proximal renal tubules, leading to decreased urinary citrate. Citrate inhibits calcium stone formation. The increased calcium load to renal tubules in addition to decreased urinary citrate both lead to increased precipitation of calcium stones in the genitourinary tract.
A diagnosis of distal RTA should prompt evaluation for specific etiologies, such as
Her antinuclear antibody titer was >1:1280 (normal: <80). Anti-SSA and -SSB antibodies were both positive, with a titer >100 (normal: <20). Rheumatoid factor was positive at 22 IU/mL (normal: 0-14). Anti-smith, anti-double stranded DNA, and anti-ribonucleoprotein antibodies were negative.
Sjögren’s syndrome appears to be the ultimate etiology of this patient’s distal RTA. The diagnosis of Sjögren’s is more classically made in the presence of lacrimal and/or salivary dysfunction and confirmed with compatible autoantibodies. In the absence of dry eyes or dry mouth, attention should be focused on her skin findings. Cutaneous vasculitis does occur in a small percentage of Sjögren’s syndrome cases. Urticarial lesions have been reported in this subset, and skin biopsy would further support the diagnosis.
Treatment of Sjögren’s syndrome with immunosuppressive therapy may ameliorate renal parenchymal pathology and improve her profound metabolic disturbances.
On further questioning, she described several months of mild xerostomia, which resulted in increased consumption of fluids. She did not have keratoconjunctivitis sicca. Biopsy of her urticarial rash demonstrated a leukocytoclastic vasculitis with eosinophilic infiltration (Figure 1). Renal biopsy with hematoxylin and eosin staining, immunofluorescence, and electron microscopy demonstrated an immune complex-mediated glomerulonephritis and moderate tubulointerstitial nephritis (Figure 2). A diagnosis of Sjögren’s syndrome was made based on the patient’s xerostomia, high titers of antinuclear antibodies, SSA and SSB antibodies, positive rheumatoid factor, hypocomplementemia, and systemic manifestations associated with Sjögren’s syndrome, including distal RTA, nephrolithiasis, and hives, with histologic evidence of leukocytoclastic vasculitis.
She received aggressive potassium and bicarbonate repletion and, several days later, had normalization of both. Her weakness and myalgia rapidly improved concomitantly with the correction of her hypokalemia. Five days later she was ambulating independently and discharged with potassium citrate and prednisone therapy. She had improved fatigue and rash at a 1-month follow-up with rheumatology. As an outpatient, she was started on azathioprine and slowly tapered off her steroids. Over the next several months, she had normal potassium, bicarbonate, and renal function, although she did require lithotripsy for an obstructive renal stone.
COMMENTARY
RTA should be considered in the differential diagnosis of an unexplained normal anion gap metabolic acidosis. There are 3 major types of RTAs, with different characteristics. In type 1 (distal) RTA, the primary defect is impaired distal acidification of the urine. Distal RTA commonly presents with hypokalemia, calciuria (often presenting as renal stones), and a positive UAG.1 In type 2 (proximal) RTA, the primary defect is impaired bicarbonate reabsorption, leading to bicarbonate wasting in the urine. Proximal RTAs can be secondary to an isolated defect in bicarbonate reabsorption or generalized proximal renal tubule dysfunction (Fanconi syndrome).1 A type 4 RTA is characterized by hypoaldosteronism, presenting usually with a mild nonanion gap metabolic acidosis and hyperkalemia. This patient’s history of renal stones, hypokalemia, and positive UAG supported a type 1 (distal) RTA. Distal RTA is often idiopathic, but initial evaluation should include a review of medications and investigation into an underlying systemic disorder (eg, plasma cell dyscrasia or autoimmune disease). This would include eliciting a possible history of xerostomia and xerophthalmia, together with testing of SSA (Ro) and SSB (La) antibodies, to assess for Sjögren’s syndrome. In addition, checking serum calcium to assess for hyperparathyroidism or familial idiopathic hypercalciuria and a review of medications, such as lithium and amphotericin,1 may uncover other secondary causes of distal RTA.
While Sjögren’s syndrome primarily affects salivary and lacrimal glands, leading to dry mouth and dry eyes, respectively, extraglandular manifestations are common, with fatigue and arthralgia occurring in half of patients. Extra-glandular involvement also often includes the skin and kidneys but can affect several other organ systems, including the central nervous system, heart, lungs, bone marrow, and lymph nodes.2
There are many cutaneous manifestations of Sjögren’s syndrome.3 Xerosis, or xeroderma, is the most common and is characterized by dry, scaly skin. Cutaneous vasculitis can occur in 10% of patients with Sjögren’s syndrome and often presents with palpable purpura or diffuse urticarial lesions, as in our patient.4 Erythematous maculopapules and cryoglobulinemic vasculitis may also occur.4 A less common skin manifestation is annular erythema, presenting as an indurated, ring-like lesion.5
Chronic tubulointerstitial nephritis is the most common renal manifestation of Sjögren’s syndrome.6 This often pre-sents with a mild elevated serum creatinine and a distal RTA, leading to hypokalemia, as in the case discussed. Distal RTA is well described, occurring in one-quarter of patients with Sjögren’s syndrome.7 The pathophysiology leading to distal RTA in Sjögren’s syndrome is thought to arise from autoimmune injury to the H(+)-ATPase pump in the renal collecting tubules, leading to decreased distal proton secretion.8,9 Younger adults with Sjögren’s syndrome, in the third and fourth decades of life, have a predilection to develop tubulointerstitial inflammation, distal RTA, and nephrolithiasis, as in the present case.6 Sjögren’s syndrome less commonly presents with membranoproliferative glomerulonephritis or membranous nephropathy.10,11 Cryoglobulinemia-associated hypocomplementemia and glomerulonephritis may also occur with Sjögren’s syndrome, yet glomerular lesions are less common than is tubulointerstitial inflammation. The patient discussed had proteinuria and evidence of immune complex-mediated glomerulonephritis.
Treatment of sicca symptoms is generally supportive. It includes artificial tears, encouragement of good hydration, salivary stimulants, and maintaining good oral hygiene. Pilocarpine, a cholinergic parasympathomimetic agent, is approved by the Food and Drug Administration to treat dry mouth associated with Sjögren’s syndrome. The treatment of extraglandular manifestations depends on the organ(s) involved. More severe presentations, such as vasculitis and glomerulonephritis, often require immunosuppressive therapy with systemic glucocorticoids, cyclophosphamide, azathioprine, or other immunosuppressive agents,12 including rituximab. RTA often necessitates treatment with oral bicarbonate and supplemental potassium repletion.
The base rate of disease (ie, prevalence of disease) influences a diagnostician’s pretest probability of a given diagnosis. The discussant briefly considered rare causes of hives (eg, vasculitis) but appropriately fine-tuned their differential for the patient’s hypokalemia and RTA. Once the diagnosis of Sjögren’s syndrome was made with certainty, the clinician was able to revisit the patient’s rash with a new lens. Urticarial vasculitis suddenly became a plausible consideration, despite its rarity (compared to allergic causes of hives) because of the direct link to the underlying autoimmune condition, which affected both the proximal muscles and distal nephrons.
TEACHING POINTS
- Evaluation of patients with weakness starts with determining true muscle weakness (ie, pathology involving the brain, spinal cord, peripheral nerve, neuromuscular junction, and/or muscle) from asthenia.
- Distal RTA should be considered in patients with a nonanion gap metabolic acidosis and hypokalemia.
- Sjögren’s syndrome has many extraglandular clinical manifestations, including vasculitis, urticaria, tubulointerstitial renal inflammation, glomerulonephritis, and lymphoma.
Acknowledgment
The authors thank Virgilius Cornea, MD, for his interpretation of the pathologic images.
Disclosure
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
1. Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160-2170. PubMed
2. Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: evaluation in a retrospective long-term study. J Intern Med. 1996;239(6):475-482. PubMed
3. Kittridge A, Routhouska SB, Korman NJ. Dermatologic manifestations of Sjögren syndrome. J Cutan Med Surg. 2011;15(1):8-14. PubMed
4. Ramos-Casals M, Anaya JM, García-Carrasco M, et al. Cutaneous vasculitis in primary Sjögren syndrome: classification and clinical significance of 52 patients. Medicine (Baltimore). 2004; 83(2):96-106. PubMed
5. Katayama I, Kotobuki Y, Kiyohara E, Murota H. Annular erythema associated with Sjögren’s syndrome: review of the literature on the management and clinical analysis of skin lesions. Mod Rheumatol. 2010;20(2):123-129. PubMed
6. Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4(9):1423-1431. PubMed
7. Pun KK, Wong CK, Tsui EY, Tam SC, Kung AW, Wang CC. Hypokalemic periodic paralysis due to the Sjögren syndrome in Chinese patients. Ann Intern Med. 1989;110(5):405-406. PubMed
8. Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3(2):264-271. PubMed
9. Bastani B, Haragsim L, Gluck S, Siamopoulos KC. Lack of H-ATPase in distal nephron causing hypokalaemic distal RTA in a patient with Sjögren’s syndrome. Nephrol Dial Transplant. 1995;10(6):908-909. PubMed
10. Cortez MS, Sturgill BC, Bolton WK. Membranoproliferative glomerulonephritis with primary Sjögren’s syndrome. Am J Kidney Dis. 1995;25(4):632-636. PubMed
11. Baba A, Hara S, Sato Y, Yamada K, Fujimoto S, Eto T. [Three patients with nephrotic syndrome due to membranous nephropathy complicated by Sjögren’s syndrome]. Nihon Jinzo Gakkai Shi. 2005;47(8):882-886. PubMed
12. Thanou-Stavraki A, James JA. Primary Sjogren’s syndrome: current and prospective therapies. Semin Arthritis Rheum. 2008;37(5):273-292. PubMed
© 2017 Society of Hospital Medicine
What Can Be Done to Maintain Positive Patient Experience and Improve Residents’ Satisfaction? In Reference to: “Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial”
We read the article by Monash et al.1 published in the March 2017 issue with great interest. This randomized study showed a discrepancy between patients’ and residents’ satisfaction with standardized rounds; for example, residents reported less autonomy, efficiency, teaching, and longer time of rounds.
We agree that letting residents lead the rounds with minimal participation of an attending (only when needed) may improve resident satisfaction. Other factors, such as quality of teaching, positive comments to learners during bedside rounds (whenever appropriate), and a positive attending attitude, might be helpful.2,3 We believe that the adaptation of such a model through the prism of residents’ benefit will lead to better satisfaction among trainees.
On the other hand, we note that the nature of the study might have exaggerated patient satisfaction when compared with real-world surveys.4 The survey appears to focus only on attending rounds and did not consider other factors like hospitality, pain control, etc. A low patient census and lack of double blinding are other potential factors.
In conclusion, we want to congratulate the authors for raising this important topic and showing positive patients’ satisfaction with standardized rounds on teaching services. Further research should focus on improving residents’ satisfaction without compromising patients’ experiences.
1. Monash B, Najafi N, Mourad M, et al. Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial. J Hosp Med. 2017;12(3):143-149. PubMed
2. Williams KN, Ramani S, Fraser B, Orlander JD. Improving bedside teaching: findings from a focus group study of learners. Acad Med. 2008;83(3):257-264. PubMed
3. Castiglioni A, Shewchuk RM, Willett LL, Heudebert GR, Centor RM. A pilot study using nominal group technique to assess residents’ perceptions of successful attending rounds. J Gen Intern Med. 2008;23(7):1060-1065. PubMed
4. Siddiqui ZK, Wu AW, Kurbanova N, Qayyum R. Comparison of Hospital Consumer Assessment of Healthcare Providers and Systems patient satisfaction scores for specialty hospitals and general medical hospitals: confounding effect of survey response rate. J Hosp Med. 2014;9(9):590-593. PubMed
We read the article by Monash et al.1 published in the March 2017 issue with great interest. This randomized study showed a discrepancy between patients’ and residents’ satisfaction with standardized rounds; for example, residents reported less autonomy, efficiency, teaching, and longer time of rounds.
We agree that letting residents lead the rounds with minimal participation of an attending (only when needed) may improve resident satisfaction. Other factors, such as quality of teaching, positive comments to learners during bedside rounds (whenever appropriate), and a positive attending attitude, might be helpful.2,3 We believe that the adaptation of such a model through the prism of residents’ benefit will lead to better satisfaction among trainees.
On the other hand, we note that the nature of the study might have exaggerated patient satisfaction when compared with real-world surveys.4 The survey appears to focus only on attending rounds and did not consider other factors like hospitality, pain control, etc. A low patient census and lack of double blinding are other potential factors.
In conclusion, we want to congratulate the authors for raising this important topic and showing positive patients’ satisfaction with standardized rounds on teaching services. Further research should focus on improving residents’ satisfaction without compromising patients’ experiences.
We read the article by Monash et al.1 published in the March 2017 issue with great interest. This randomized study showed a discrepancy between patients’ and residents’ satisfaction with standardized rounds; for example, residents reported less autonomy, efficiency, teaching, and longer time of rounds.
We agree that letting residents lead the rounds with minimal participation of an attending (only when needed) may improve resident satisfaction. Other factors, such as quality of teaching, positive comments to learners during bedside rounds (whenever appropriate), and a positive attending attitude, might be helpful.2,3 We believe that the adaptation of such a model through the prism of residents’ benefit will lead to better satisfaction among trainees.
On the other hand, we note that the nature of the study might have exaggerated patient satisfaction when compared with real-world surveys.4 The survey appears to focus only on attending rounds and did not consider other factors like hospitality, pain control, etc. A low patient census and lack of double blinding are other potential factors.
In conclusion, we want to congratulate the authors for raising this important topic and showing positive patients’ satisfaction with standardized rounds on teaching services. Further research should focus on improving residents’ satisfaction without compromising patients’ experiences.
1. Monash B, Najafi N, Mourad M, et al. Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial. J Hosp Med. 2017;12(3):143-149. PubMed
2. Williams KN, Ramani S, Fraser B, Orlander JD. Improving bedside teaching: findings from a focus group study of learners. Acad Med. 2008;83(3):257-264. PubMed
3. Castiglioni A, Shewchuk RM, Willett LL, Heudebert GR, Centor RM. A pilot study using nominal group technique to assess residents’ perceptions of successful attending rounds. J Gen Intern Med. 2008;23(7):1060-1065. PubMed
4. Siddiqui ZK, Wu AW, Kurbanova N, Qayyum R. Comparison of Hospital Consumer Assessment of Healthcare Providers and Systems patient satisfaction scores for specialty hospitals and general medical hospitals: confounding effect of survey response rate. J Hosp Med. 2014;9(9):590-593. PubMed
1. Monash B, Najafi N, Mourad M, et al. Standardized Attending Rounds to Improve the Patient Experience: A Pragmatic Cluster Randomized Controlled Trial. J Hosp Med. 2017;12(3):143-149. PubMed
2. Williams KN, Ramani S, Fraser B, Orlander JD. Improving bedside teaching: findings from a focus group study of learners. Acad Med. 2008;83(3):257-264. PubMed
3. Castiglioni A, Shewchuk RM, Willett LL, Heudebert GR, Centor RM. A pilot study using nominal group technique to assess residents’ perceptions of successful attending rounds. J Gen Intern Med. 2008;23(7):1060-1065. PubMed
4. Siddiqui ZK, Wu AW, Kurbanova N, Qayyum R. Comparison of Hospital Consumer Assessment of Healthcare Providers and Systems patient satisfaction scores for specialty hospitals and general medical hospitals: confounding effect of survey response rate. J Hosp Med. 2014;9(9):590-593. PubMed
When should brain imaging precede lumbar puncture in cases of suspected bacterial meningitis?
Brain imaging should precede lumbar puncture in patients with focal neurologic deficits or immunodeficiency, or with altered mental status or seizures during the previous week. However, lumbar puncture can be safely done in most patients without first obtaining brain imaging. Empiric antibiotic and corticosteroid therapy must not be delayed; they should be started immediately after the lumber puncture is done, without waiting for the results. If the lumbar puncture is going to be delayed, these treatments should be started immediately after obtaining blood samples for culture.
A MEDICAL EMERGENCY
Bacterial meningitis is a medical emergency and requires prompt recognition and treatment. It is associated with a nearly 15% death rate as well as neurologic effects such as deafness, seizures, and cognitive decline in about the same percentage of patients.1 Microbiologic information from lumbar puncture and cerebrospinal fluid analysis is an essential part of the initial workup, whenever possible. Lumbar puncture can be done safely at the bedside in most patients and so should not be delayed unless certain contraindications exist, as discussed below.2
INDICATIONS FOR BRAIN IMAGING BEFORE LUMBAR PUNCTURE

Table 1 lists common indications for brain imaging before lumbar puncture. However, there is a lack of good evidence to support them.
Current guidelines on acute bacterial meningitis from the Infectious Diseases Society of America recommend computed tomography (CT) of the brain before lumbar puncture in patients presenting with:
- Altered mental status
- A new focal neurologic deficit (eg, cranial nerve palsy, extremity weakness or drift, dysarthria, aphasia)
- Papilledema
- Seizure within the past week
- History of central nervous system disease (eg, stroke, tumor)
- Age 60 or older (likely because of the association with previous central nervous system disease)
- Immunocompromised state (due to human immunodeficiency virus infection, chemotherapy, or immunosuppressive drugs for transplant or rheumatologic disease)
- A high clinical suspicion for subarachnoid hemorrhage.3–5
However, a normal result on head CT does not rule out the possibility of increased intracranial pressure and the risk of brain herniation. Actually, patients with acute bacterial meningitis are inherently at higher risk of spontaneous brain herniation even without lumbar puncture, and some cases of brain herniation after lumbar puncture could have represented the natural course of disease. Importantly, lumbar puncture may not be independently associated with the risk of brain herniation in patients with altered mental status (Glasgow Coma Scale score ≤ 8).6 A prospective randomized study is needed to better understand when to order brain imaging before lumbar puncture and when it is safe to proceed directly to lumbar puncture.
CONTRAINDICATIONS TO LUMBAR PUNCTURE

General contraindications to lumbar puncture are listed in Table 2.
Gopal et al3 analyzed clinical and radiographic data for 113 adults requiring urgent lumbar puncture and reported that altered mental status (likelihood ratio [LR] 2.2), focal neurologic deficit (LR 4.3), papilledema (LR 11.1), and clinical impression (LR 18.8) were associated with abnormalities on CT.
Hasbun et al4 prospectively analyzed whether clinical variables correlated with abnormal results of head CT that would preclude lumbar puncture in 301 patients requiring urgent lumbar puncture. They found that age 60 and older, immunodeficiency, a history of central nervous system disease, recent seizure (within 1 week), and neurologic deficits were associated with abnormal findings on head CT (eg, lesion with mass effect, midline shift). Importantly, absence of these characteristics had a 97% negative predictive value for abnormal findings on head CT. However, neither a normal head CT nor a normal clinical neurologic examination rules out increased intracranial pressure.4,7
CHIEF CONCERNS ABOUT LUMBAR PUNCTURE
Lumbar puncture is generally well tolerated. Major complications are rare2 and can be prevented by checking for contraindications and by using appropriate procedural hygiene and technique. Complications include pain at the puncture site, postprocedural headache, epidural hematoma, meningitis, osteomyelitis or discitis, bleeding, epidermoid tumor, and, most worrisome, brain herniation.
Brain herniation
Concern about causing brain herniation is the reason imaging may be ordered before lumbar puncture. Cerebral edema and increased intracranial pressure are common in patients with bacterial meningitis, as well as in other conditions such as bleeding, tumor, and abscess.1 If intracranial pressure is elevated, lumbar puncture can cause cerebral herniation with further neurologic compromise and possibly death. Herniation is believed to be due to a sudden decrease in pressure in the spinal cord caused by removal of cerebrospinal fluid. However, the only information we have about this complication comes from case reports and case series, so we don’t really know how often it happens.
On the other hand, ordering ancillary tests before lumbar puncture and starting empiric antibiotics in patients with suspected bacterial meningitis may delay treatment and lead to worse clinical outcomes and thus should be discouraged.8
Also important to note is the lack of good data regarding the safety of lumbar puncture in patients with potential hemostatic problems (thrombocytopenia, coagulopathy). The recommendation not to do lumbar puncture in these situations (Table 1) is taken from neuraxial anesthesia guidelines.9 Further, a small retrospective study of thrombocytopenic oncology patients requiring lumbar puncture did not demonstrate an increased risk of complications.10
ADDITIONAL CONSIDERATIONS
In a retrospective study in 2015, Glimåker et al6 demonstrated that lumbar puncture without prior brain CT was safe in patients with suspected acute bacterial meningitis with moderate to severe impairment of mental status, and that it led to a shorter “door-to-antibiotic time.” Lumbar puncture before imaging was also associated with a concomitant decrease in the risk of death, with no increase in the rate of complications.6
If brain imaging is to be done before lumbar puncture, then blood cultures (and cultures of other fluids, whenever appropriate) should be collected and the patient should be started on empiric management for central nervous system infection first. CT evidence of diffuse cerebral edema, focal lesions with mass effect, and ventriculomegaly should be viewed as further contraindications to lumbar puncture.1
Antibiotic therapy
When contraindications to lumbar puncture exist, the choice of antibiotic and the duration of therapy should be based on the patient’s history, demographics, risk factors, and microbiologic data from blood culture, urine culture, sputum culture, and detection of microbiological antigens.1 The choice of antibiotic is beyond the scope of this article. However, empiric antibiotic therapy with a third-generation cephalosporin (eg, ceftriaxone) and vancomycin and anti-inflammatory therapy (dexamethasone) should in most cases be started immediately after collecting samples for blood culture and must not be delayed by neuroimaging and lumbar puncture with cerebrospinal fluid sampling, given the high rates of mortality and morbidity if treatment is delayed.5,8
Consultation with the neurosurgery service regarding alternative brain ventricular fluid sampling should be considered.11
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2016–2025.
- Ellenby MS, Tegtmeyer K, Lai S, Braner DA. Videos in clinical medicine. Lumbar puncture. N Engl J Med 2006; 355: e12.
- Gopal AK, Whitehouse JD, Simel DL, Corey GR. Cranial computed tomography before lumbar puncture: a prospective clinical evaluation. Arch Intern Med 1999; 159:2681–2685.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Glimåker M, Johansson B, Grindborg Ö, Bottai M, Lindquist L, Sjölin J. Adult bacterial meningitis: earlier treatment and improved outcome following guideline revision promoting prompt lumbar puncture. Clin Infect Dis 2015; 60:1162–1169.
- Baraff LJ, Byyny RL, Probst MA, Salamon N, Linetsky M, Mower WR. Prevalence of herniation and intracranial shift on cranial tomography in patients with subarachnoid hemorrhage and a normal neurologic examination. Acad Emerg Med 2010; 17:423–428.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition). Reg Anesth Pain Med 2010; 35:64–101.
- Ning S, Kerbel B, Callum J, Lin Y. Safety of lumbar punctures in patients with thrombocytopenia. Vox Sang 2016; 110:393–400.
- Joffe AR. Lumbar puncture and brain herniation in acute bacterial meningitis: a review. J Intensive Care Med 2007; 22:194–207.
Brain imaging should precede lumbar puncture in patients with focal neurologic deficits or immunodeficiency, or with altered mental status or seizures during the previous week. However, lumbar puncture can be safely done in most patients without first obtaining brain imaging. Empiric antibiotic and corticosteroid therapy must not be delayed; they should be started immediately after the lumber puncture is done, without waiting for the results. If the lumbar puncture is going to be delayed, these treatments should be started immediately after obtaining blood samples for culture.
A MEDICAL EMERGENCY
Bacterial meningitis is a medical emergency and requires prompt recognition and treatment. It is associated with a nearly 15% death rate as well as neurologic effects such as deafness, seizures, and cognitive decline in about the same percentage of patients.1 Microbiologic information from lumbar puncture and cerebrospinal fluid analysis is an essential part of the initial workup, whenever possible. Lumbar puncture can be done safely at the bedside in most patients and so should not be delayed unless certain contraindications exist, as discussed below.2
INDICATIONS FOR BRAIN IMAGING BEFORE LUMBAR PUNCTURE
Table 1 lists common indications for brain imaging before lumbar puncture. However, there is a lack of good evidence to support them.
Current guidelines on acute bacterial meningitis from the Infectious Diseases Society of America recommend computed tomography (CT) of the brain before lumbar puncture in patients presenting with:
- Altered mental status
- A new focal neurologic deficit (eg, cranial nerve palsy, extremity weakness or drift, dysarthria, aphasia)
- Papilledema
- Seizure within the past week
- History of central nervous system disease (eg, stroke, tumor)
- Age 60 or older (likely because of the association with previous central nervous system disease)
- Immunocompromised state (due to human immunodeficiency virus infection, chemotherapy, or immunosuppressive drugs for transplant or rheumatologic disease)
- A high clinical suspicion for subarachnoid hemorrhage.3–5
However, a normal result on head CT does not rule out the possibility of increased intracranial pressure and the risk of brain herniation. Actually, patients with acute bacterial meningitis are inherently at higher risk of spontaneous brain herniation even without lumbar puncture, and some cases of brain herniation after lumbar puncture could have represented the natural course of disease. Importantly, lumbar puncture may not be independently associated with the risk of brain herniation in patients with altered mental status (Glasgow Coma Scale score ≤ 8).6 A prospective randomized study is needed to better understand when to order brain imaging before lumbar puncture and when it is safe to proceed directly to lumbar puncture.
CONTRAINDICATIONS TO LUMBAR PUNCTURE
General contraindications to lumbar puncture are listed in Table 2.
Gopal et al3 analyzed clinical and radiographic data for 113 adults requiring urgent lumbar puncture and reported that altered mental status (likelihood ratio [LR] 2.2), focal neurologic deficit (LR 4.3), papilledema (LR 11.1), and clinical impression (LR 18.8) were associated with abnormalities on CT.
Hasbun et al4 prospectively analyzed whether clinical variables correlated with abnormal results of head CT that would preclude lumbar puncture in 301 patients requiring urgent lumbar puncture. They found that age 60 and older, immunodeficiency, a history of central nervous system disease, recent seizure (within 1 week), and neurologic deficits were associated with abnormal findings on head CT (eg, lesion with mass effect, midline shift). Importantly, absence of these characteristics had a 97% negative predictive value for abnormal findings on head CT. However, neither a normal head CT nor a normal clinical neurologic examination rules out increased intracranial pressure.4,7
CHIEF CONCERNS ABOUT LUMBAR PUNCTURE
Lumbar puncture is generally well tolerated. Major complications are rare2 and can be prevented by checking for contraindications and by using appropriate procedural hygiene and technique. Complications include pain at the puncture site, postprocedural headache, epidural hematoma, meningitis, osteomyelitis or discitis, bleeding, epidermoid tumor, and, most worrisome, brain herniation.
Brain herniation
Concern about causing brain herniation is the reason imaging may be ordered before lumbar puncture. Cerebral edema and increased intracranial pressure are common in patients with bacterial meningitis, as well as in other conditions such as bleeding, tumor, and abscess.1 If intracranial pressure is elevated, lumbar puncture can cause cerebral herniation with further neurologic compromise and possibly death. Herniation is believed to be due to a sudden decrease in pressure in the spinal cord caused by removal of cerebrospinal fluid. However, the only information we have about this complication comes from case reports and case series, so we don’t really know how often it happens.
On the other hand, ordering ancillary tests before lumbar puncture and starting empiric antibiotics in patients with suspected bacterial meningitis may delay treatment and lead to worse clinical outcomes and thus should be discouraged.8
Also important to note is the lack of good data regarding the safety of lumbar puncture in patients with potential hemostatic problems (thrombocytopenia, coagulopathy). The recommendation not to do lumbar puncture in these situations (Table 1) is taken from neuraxial anesthesia guidelines.9 Further, a small retrospective study of thrombocytopenic oncology patients requiring lumbar puncture did not demonstrate an increased risk of complications.10
ADDITIONAL CONSIDERATIONS
In a retrospective study in 2015, Glimåker et al6 demonstrated that lumbar puncture without prior brain CT was safe in patients with suspected acute bacterial meningitis with moderate to severe impairment of mental status, and that it led to a shorter “door-to-antibiotic time.” Lumbar puncture before imaging was also associated with a concomitant decrease in the risk of death, with no increase in the rate of complications.6
If brain imaging is to be done before lumbar puncture, then blood cultures (and cultures of other fluids, whenever appropriate) should be collected and the patient should be started on empiric management for central nervous system infection first. CT evidence of diffuse cerebral edema, focal lesions with mass effect, and ventriculomegaly should be viewed as further contraindications to lumbar puncture.1
Antibiotic therapy
When contraindications to lumbar puncture exist, the choice of antibiotic and the duration of therapy should be based on the patient’s history, demographics, risk factors, and microbiologic data from blood culture, urine culture, sputum culture, and detection of microbiological antigens.1 The choice of antibiotic is beyond the scope of this article. However, empiric antibiotic therapy with a third-generation cephalosporin (eg, ceftriaxone) and vancomycin and anti-inflammatory therapy (dexamethasone) should in most cases be started immediately after collecting samples for blood culture and must not be delayed by neuroimaging and lumbar puncture with cerebrospinal fluid sampling, given the high rates of mortality and morbidity if treatment is delayed.5,8
Consultation with the neurosurgery service regarding alternative brain ventricular fluid sampling should be considered.11
Brain imaging should precede lumbar puncture in patients with focal neurologic deficits or immunodeficiency, or with altered mental status or seizures during the previous week. However, lumbar puncture can be safely done in most patients without first obtaining brain imaging. Empiric antibiotic and corticosteroid therapy must not be delayed; they should be started immediately after the lumber puncture is done, without waiting for the results. If the lumbar puncture is going to be delayed, these treatments should be started immediately after obtaining blood samples for culture.
A MEDICAL EMERGENCY
Bacterial meningitis is a medical emergency and requires prompt recognition and treatment. It is associated with a nearly 15% death rate as well as neurologic effects such as deafness, seizures, and cognitive decline in about the same percentage of patients.1 Microbiologic information from lumbar puncture and cerebrospinal fluid analysis is an essential part of the initial workup, whenever possible. Lumbar puncture can be done safely at the bedside in most patients and so should not be delayed unless certain contraindications exist, as discussed below.2
INDICATIONS FOR BRAIN IMAGING BEFORE LUMBAR PUNCTURE
Table 1 lists common indications for brain imaging before lumbar puncture. However, there is a lack of good evidence to support them.
Current guidelines on acute bacterial meningitis from the Infectious Diseases Society of America recommend computed tomography (CT) of the brain before lumbar puncture in patients presenting with:
- Altered mental status
- A new focal neurologic deficit (eg, cranial nerve palsy, extremity weakness or drift, dysarthria, aphasia)
- Papilledema
- Seizure within the past week
- History of central nervous system disease (eg, stroke, tumor)
- Age 60 or older (likely because of the association with previous central nervous system disease)
- Immunocompromised state (due to human immunodeficiency virus infection, chemotherapy, or immunosuppressive drugs for transplant or rheumatologic disease)
- A high clinical suspicion for subarachnoid hemorrhage.3–5
However, a normal result on head CT does not rule out the possibility of increased intracranial pressure and the risk of brain herniation. Actually, patients with acute bacterial meningitis are inherently at higher risk of spontaneous brain herniation even without lumbar puncture, and some cases of brain herniation after lumbar puncture could have represented the natural course of disease. Importantly, lumbar puncture may not be independently associated with the risk of brain herniation in patients with altered mental status (Glasgow Coma Scale score ≤ 8).6 A prospective randomized study is needed to better understand when to order brain imaging before lumbar puncture and when it is safe to proceed directly to lumbar puncture.
CONTRAINDICATIONS TO LUMBAR PUNCTURE
General contraindications to lumbar puncture are listed in Table 2.
Gopal et al3 analyzed clinical and radiographic data for 113 adults requiring urgent lumbar puncture and reported that altered mental status (likelihood ratio [LR] 2.2), focal neurologic deficit (LR 4.3), papilledema (LR 11.1), and clinical impression (LR 18.8) were associated with abnormalities on CT.
Hasbun et al4 prospectively analyzed whether clinical variables correlated with abnormal results of head CT that would preclude lumbar puncture in 301 patients requiring urgent lumbar puncture. They found that age 60 and older, immunodeficiency, a history of central nervous system disease, recent seizure (within 1 week), and neurologic deficits were associated with abnormal findings on head CT (eg, lesion with mass effect, midline shift). Importantly, absence of these characteristics had a 97% negative predictive value for abnormal findings on head CT. However, neither a normal head CT nor a normal clinical neurologic examination rules out increased intracranial pressure.4,7
CHIEF CONCERNS ABOUT LUMBAR PUNCTURE
Lumbar puncture is generally well tolerated. Major complications are rare2 and can be prevented by checking for contraindications and by using appropriate procedural hygiene and technique. Complications include pain at the puncture site, postprocedural headache, epidural hematoma, meningitis, osteomyelitis or discitis, bleeding, epidermoid tumor, and, most worrisome, brain herniation.
Brain herniation
Concern about causing brain herniation is the reason imaging may be ordered before lumbar puncture. Cerebral edema and increased intracranial pressure are common in patients with bacterial meningitis, as well as in other conditions such as bleeding, tumor, and abscess.1 If intracranial pressure is elevated, lumbar puncture can cause cerebral herniation with further neurologic compromise and possibly death. Herniation is believed to be due to a sudden decrease in pressure in the spinal cord caused by removal of cerebrospinal fluid. However, the only information we have about this complication comes from case reports and case series, so we don’t really know how often it happens.
On the other hand, ordering ancillary tests before lumbar puncture and starting empiric antibiotics in patients with suspected bacterial meningitis may delay treatment and lead to worse clinical outcomes and thus should be discouraged.8
Also important to note is the lack of good data regarding the safety of lumbar puncture in patients with potential hemostatic problems (thrombocytopenia, coagulopathy). The recommendation not to do lumbar puncture in these situations (Table 1) is taken from neuraxial anesthesia guidelines.9 Further, a small retrospective study of thrombocytopenic oncology patients requiring lumbar puncture did not demonstrate an increased risk of complications.10
ADDITIONAL CONSIDERATIONS
In a retrospective study in 2015, Glimåker et al6 demonstrated that lumbar puncture without prior brain CT was safe in patients with suspected acute bacterial meningitis with moderate to severe impairment of mental status, and that it led to a shorter “door-to-antibiotic time.” Lumbar puncture before imaging was also associated with a concomitant decrease in the risk of death, with no increase in the rate of complications.6
If brain imaging is to be done before lumbar puncture, then blood cultures (and cultures of other fluids, whenever appropriate) should be collected and the patient should be started on empiric management for central nervous system infection first. CT evidence of diffuse cerebral edema, focal lesions with mass effect, and ventriculomegaly should be viewed as further contraindications to lumbar puncture.1
Antibiotic therapy
When contraindications to lumbar puncture exist, the choice of antibiotic and the duration of therapy should be based on the patient’s history, demographics, risk factors, and microbiologic data from blood culture, urine culture, sputum culture, and detection of microbiological antigens.1 The choice of antibiotic is beyond the scope of this article. However, empiric antibiotic therapy with a third-generation cephalosporin (eg, ceftriaxone) and vancomycin and anti-inflammatory therapy (dexamethasone) should in most cases be started immediately after collecting samples for blood culture and must not be delayed by neuroimaging and lumbar puncture with cerebrospinal fluid sampling, given the high rates of mortality and morbidity if treatment is delayed.5,8
Consultation with the neurosurgery service regarding alternative brain ventricular fluid sampling should be considered.11
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2016–2025.
- Ellenby MS, Tegtmeyer K, Lai S, Braner DA. Videos in clinical medicine. Lumbar puncture. N Engl J Med 2006; 355: e12.
- Gopal AK, Whitehouse JD, Simel DL, Corey GR. Cranial computed tomography before lumbar puncture: a prospective clinical evaluation. Arch Intern Med 1999; 159:2681–2685.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Glimåker M, Johansson B, Grindborg Ö, Bottai M, Lindquist L, Sjölin J. Adult bacterial meningitis: earlier treatment and improved outcome following guideline revision promoting prompt lumbar puncture. Clin Infect Dis 2015; 60:1162–1169.
- Baraff LJ, Byyny RL, Probst MA, Salamon N, Linetsky M, Mower WR. Prevalence of herniation and intracranial shift on cranial tomography in patients with subarachnoid hemorrhage and a normal neurologic examination. Acad Emerg Med 2010; 17:423–428.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition). Reg Anesth Pain Med 2010; 35:64–101.
- Ning S, Kerbel B, Callum J, Lin Y. Safety of lumbar punctures in patients with thrombocytopenia. Vox Sang 2016; 110:393–400.
- Joffe AR. Lumbar puncture and brain herniation in acute bacterial meningitis: a review. J Intensive Care Med 2007; 22:194–207.
- Thigpen MC, Whitney CG, Messonnier NE, et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med 2011; 364:2016–2025.
- Ellenby MS, Tegtmeyer K, Lai S, Braner DA. Videos in clinical medicine. Lumbar puncture. N Engl J Med 2006; 355: e12.
- Gopal AK, Whitehouse JD, Simel DL, Corey GR. Cranial computed tomography before lumbar puncture: a prospective clinical evaluation. Arch Intern Med 1999; 159:2681–2685.
- Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001; 345:1727–1733.
- Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004; 39:1267–1284.
- Glimåker M, Johansson B, Grindborg Ö, Bottai M, Lindquist L, Sjölin J. Adult bacterial meningitis: earlier treatment and improved outcome following guideline revision promoting prompt lumbar puncture. Clin Infect Dis 2015; 60:1162–1169.
- Baraff LJ, Byyny RL, Probst MA, Salamon N, Linetsky M, Mower WR. Prevalence of herniation and intracranial shift on cranial tomography in patients with subarachnoid hemorrhage and a normal neurologic examination. Acad Emerg Med 2010; 17:423–428.
- Proulx N, Fréchette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM 2005; 98:291–298.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition). Reg Anesth Pain Med 2010; 35:64–101.
- Ning S, Kerbel B, Callum J, Lin Y. Safety of lumbar punctures in patients with thrombocytopenia. Vox Sang 2016; 110:393–400.
- Joffe AR. Lumbar puncture and brain herniation in acute bacterial meningitis: a review. J Intensive Care Med 2007; 22:194–207.
Ring-enhancing cerebral lesions
A 39-year-old woman with a history of human immunodeficiency virus (HIV) and hepatitis B virus infection was brought to the emergency department for evaluation of seizures, which had started a few days earlier. She was born and raised in a state bordering the Ohio River, an area where Histoplasma capsulatum is endemic. She denied any recent travel.

Her vital signs and neurologic examination were normal. Computed tomography of the head showed two areas of increased attenuation anterior to the frontal horns. To better characterize those lesions, magnetic resonance imaging (MRI) with contrast was done, which showed about a dozen 1-cm ring-enhancing lesions in the right cerebellum and both cerebral hemispheres (Figure 1).
Results of a complete blood cell count, metabolic profile, and chest radiography were normal. Her CD4 count was 428/μL (reference range 533–1,674) and 20% (60%–89%); her HIV viral load was 326,000 copies/mL.
She was initially treated empirically with sulfadiazine, pyrimethamine, and leukovorin for possible toxoplasmosis, which is the most common cause of ring-enhancing brain lesions in HIV patients. In the meantime, cerebrospinal fluid, blood, and urine were sent for a detailed workup for fungi, including Histoplasma. Results of the Histoplasma antibody and antigen studies of the serum, urine, and cerebrospinal fluid were positive, while cerebrospinal fluid testing for Toxoplasma by polymerase chain reaction testing was negative. Empirical treatment for toxoplasmosis was stopped and amphotericin B was started to treat disseminated histoplasmosis.
During her hospital course, she underwent brain biopsy via right frontotemporal craniotomy with resection of right frontal lesions. Pathologic study showed partially organizing abscesses with central necrosis (Figure 2), microscopy with Grocott-Gomori methenamine silver stain was positive for budding yeast forms consistent with H capsulatum (Figure 3), and special stain for acid-fast bacilli was negative for mycobacteria. Cultures of the brain biopsy specimen, blood, and cerebrospinal fluid for fungi, acid-fast bacilli, and bacteria did not reveal any growth after 28 days.
The patient was discharged home with instructions to take amphotericin B for a total of 6 weeks and then itraconazole. About 1 year later, she remained free of symptoms, although repeat MRI did not show any significant change in the size or number of histoplasmomas.
She did not comply well with her HIV treatment, and her immune status did not improve, so we decided to continue her itraconazole treatment for more than 1 year.
CEREBRAL HISTOPLASMOMA
The term “histoplasmoma” was introduced by Shapiro et al1 in 1955, when they first described numerous focal areas of softening, up to 1 cm in diameter, scattered throughout the brain at autopsy in a 41-year-old man who had died of disseminated histoplasmosis. They coined the word to describe these discrete areas of necrosis that might resemble tumors on the basis of their size, location, and capability of causing increased intracranial pressure.
Central nervous system involvement can either be a manifestation of disseminated disease or present as an isolated illness.2 It occurs in 5% to 10% of cases of disseminated histoplasmosis.3 Histoplasmosis of the central nervous system can have different manifestations; the most common presentation is chronic meningitis.4
Laboratory diagnosis is based on detecting H capsulatum antigen and antibody in the urine, blood, and cerebrospinal fluid. Tissue biopsy (histopathology) as well as cultures of tissue samples or body fluids may also establish the diagnosis.4
Toxoplasmosis and primary central nervous system lymphoma are the most common causes of brain ring-enhancing lesions in HIV patients in developed countries, while in the developing world neurocysticercosis and tuberculomas are more common.5,6 Much less common causes include brain abscesses secondary to bacterial infections (pyogenic abscess),7 cryptococcomas,8 syphilitic cerebral gummata,9 primary brain tumors (gliomas), and metastases.10
Compared with other forms of the disease, histoplasmosis of the central nervous system has higher rates of treatment failure and relapse, so treatment should be prolonged and aggressive.2,3 The cure rate with amphotericin B ranges from 33% to 61%, and higher doses produce better response rates.3
Current treatment recommendations are based on 2007 guidelines of the Infectious Diseases Society of America.11 Liposomal amphotericin B is the drug of choice because it achieves higher concentrations in the central nervous system than other drugs and is less toxic. It is given for 4 to 6 weeks, followed by itraconazole for at least 1 year and until the cerebrospinal fluid Histoplasma antigen test is negative and other cerebrospinal fluid abnormalities are resolved.
In patients who have primary disseminated histoplasmosis that includes the central nervous system, itraconazole can be given for more than 1 year or until immune recovery is achieved—or lifelong if necessary.2,12 Long-term suppressive antifungal therapy also should be considered in patients for whom appropriate initial therapy fails.2
Nephrotoxicity (acute kidney injury, hypokalemia, and hypomagnesemia), infusion-related drug reactions, and rash are among the well-described side effects of amphotericin B. Maintenance of intravascular volume and replacement of electrolytes should be an integral part of the amphotericin B treatment regimen.13
TAKE-AWAY POINTS
- Histoplasmomas should be considered in the differential diagnosis of ring-enhancing lesions of the central nervous system, along with toxoplasmosis and primary central nervous system lymphoma. This will allow timely initiation of the diagnostic workup, avoiding unnecessary and potentially risky interventions and delays in starting targeted antifungal therapy.
- There is no single gold standard test for central nervous system histoplasmosis. Rather, the final diagnosis is based on the combination of clinical, laboratory, and radiologic findings.
Acknowledgment: Library research assistance provided by HSHS St. John’s Hospital Health Sciences Library staff.
- Shapiro JL, Lux JJ, Sprofkin BE. Histoplasmosis of the central nervous system. Am J Pathol 1955; 31:319–335.
- Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis 2005; 40:844–852.
- Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system: a clinical review. Medicine (Baltimore) 1990; 69:244–260.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Modi M, Mochan A, Modi G. Management of HIV-associated focal brain lesions in developing countries. QJM 2004; 97:413–421.
- Miller RF, Hall-Craggs MA, Costa DC, et al. Magnetic resonance imaging, thallium-201 SPET scanning, and laboratory analyses for discrimination of cerebral lymphoma and toxoplasmosis in AIDS. Sex Transm Infect 1998; 74:258–264.
- Cohen WA. Intracranial bacterial infections in patients with AIDS. Neuroimaging Clin N Am 1997; 7:223–229.
- Troncoso A, Fumagalli J, Shinzato R, Gulotta H, Toller M, Bava J. CNS cryptococcoma in an HIV-positive patient. J Int Assoc Physicians AIDS Care (Chic) 2002; 1:131–133.
- Land AM, Nelson GA, Bell SG, Denby KJ, Estrada CA, Willett LL. Widening the differential for brain masses in human immunodeficiency virus-positive patients: syphilitic cerebral gummata. Am J Med Sci 2013; 346:253–255.
- Balsys R, Janousek JE, Batnitzky S, Templeton AW. Peripheral enhancement in computerized cranial tomography: a non-specific finding. Surg Neurol 1979; 11:207–216.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Wheat J, Hafner R, Wulfsohn M, et al; National Institute of Allergy and Infectious Diseases Clinical Trials and Mycoses Study Group Collaborators. Prevention of relapse of histoplasmosis with itraconazole in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1993; 118:610–616.
- Saccente M. Central nervous system histoplasmosis. Curr Treat Options Neurol 2008; 10:161–167.
A 39-year-old woman with a history of human immunodeficiency virus (HIV) and hepatitis B virus infection was brought to the emergency department for evaluation of seizures, which had started a few days earlier. She was born and raised in a state bordering the Ohio River, an area where Histoplasma capsulatum is endemic. She denied any recent travel.
Her vital signs and neurologic examination were normal. Computed tomography of the head showed two areas of increased attenuation anterior to the frontal horns. To better characterize those lesions, magnetic resonance imaging (MRI) with contrast was done, which showed about a dozen 1-cm ring-enhancing lesions in the right cerebellum and both cerebral hemispheres (Figure 1).
Results of a complete blood cell count, metabolic profile, and chest radiography were normal. Her CD4 count was 428/μL (reference range 533–1,674) and 20% (60%–89%); her HIV viral load was 326,000 copies/mL.
She was initially treated empirically with sulfadiazine, pyrimethamine, and leukovorin for possible toxoplasmosis, which is the most common cause of ring-enhancing brain lesions in HIV patients. In the meantime, cerebrospinal fluid, blood, and urine were sent for a detailed workup for fungi, including Histoplasma. Results of the Histoplasma antibody and antigen studies of the serum, urine, and cerebrospinal fluid were positive, while cerebrospinal fluid testing for Toxoplasma by polymerase chain reaction testing was negative. Empirical treatment for toxoplasmosis was stopped and amphotericin B was started to treat disseminated histoplasmosis.
During her hospital course, she underwent brain biopsy via right frontotemporal craniotomy with resection of right frontal lesions. Pathologic study showed partially organizing abscesses with central necrosis (Figure 2), microscopy with Grocott-Gomori methenamine silver stain was positive for budding yeast forms consistent with H capsulatum (Figure 3), and special stain for acid-fast bacilli was negative for mycobacteria. Cultures of the brain biopsy specimen, blood, and cerebrospinal fluid for fungi, acid-fast bacilli, and bacteria did not reveal any growth after 28 days.
The patient was discharged home with instructions to take amphotericin B for a total of 6 weeks and then itraconazole. About 1 year later, she remained free of symptoms, although repeat MRI did not show any significant change in the size or number of histoplasmomas.
She did not comply well with her HIV treatment, and her immune status did not improve, so we decided to continue her itraconazole treatment for more than 1 year.
CEREBRAL HISTOPLASMOMA
The term “histoplasmoma” was introduced by Shapiro et al1 in 1955, when they first described numerous focal areas of softening, up to 1 cm in diameter, scattered throughout the brain at autopsy in a 41-year-old man who had died of disseminated histoplasmosis. They coined the word to describe these discrete areas of necrosis that might resemble tumors on the basis of their size, location, and capability of causing increased intracranial pressure.
Central nervous system involvement can either be a manifestation of disseminated disease or present as an isolated illness.2 It occurs in 5% to 10% of cases of disseminated histoplasmosis.3 Histoplasmosis of the central nervous system can have different manifestations; the most common presentation is chronic meningitis.4
Laboratory diagnosis is based on detecting H capsulatum antigen and antibody in the urine, blood, and cerebrospinal fluid. Tissue biopsy (histopathology) as well as cultures of tissue samples or body fluids may also establish the diagnosis.4
Toxoplasmosis and primary central nervous system lymphoma are the most common causes of brain ring-enhancing lesions in HIV patients in developed countries, while in the developing world neurocysticercosis and tuberculomas are more common.5,6 Much less common causes include brain abscesses secondary to bacterial infections (pyogenic abscess),7 cryptococcomas,8 syphilitic cerebral gummata,9 primary brain tumors (gliomas), and metastases.10
Compared with other forms of the disease, histoplasmosis of the central nervous system has higher rates of treatment failure and relapse, so treatment should be prolonged and aggressive.2,3 The cure rate with amphotericin B ranges from 33% to 61%, and higher doses produce better response rates.3
Current treatment recommendations are based on 2007 guidelines of the Infectious Diseases Society of America.11 Liposomal amphotericin B is the drug of choice because it achieves higher concentrations in the central nervous system than other drugs and is less toxic. It is given for 4 to 6 weeks, followed by itraconazole for at least 1 year and until the cerebrospinal fluid Histoplasma antigen test is negative and other cerebrospinal fluid abnormalities are resolved.
In patients who have primary disseminated histoplasmosis that includes the central nervous system, itraconazole can be given for more than 1 year or until immune recovery is achieved—or lifelong if necessary.2,12 Long-term suppressive antifungal therapy also should be considered in patients for whom appropriate initial therapy fails.2
Nephrotoxicity (acute kidney injury, hypokalemia, and hypomagnesemia), infusion-related drug reactions, and rash are among the well-described side effects of amphotericin B. Maintenance of intravascular volume and replacement of electrolytes should be an integral part of the amphotericin B treatment regimen.13
TAKE-AWAY POINTS
- Histoplasmomas should be considered in the differential diagnosis of ring-enhancing lesions of the central nervous system, along with toxoplasmosis and primary central nervous system lymphoma. This will allow timely initiation of the diagnostic workup, avoiding unnecessary and potentially risky interventions and delays in starting targeted antifungal therapy.
- There is no single gold standard test for central nervous system histoplasmosis. Rather, the final diagnosis is based on the combination of clinical, laboratory, and radiologic findings.
Acknowledgment: Library research assistance provided by HSHS St. John’s Hospital Health Sciences Library staff.
A 39-year-old woman with a history of human immunodeficiency virus (HIV) and hepatitis B virus infection was brought to the emergency department for evaluation of seizures, which had started a few days earlier. She was born and raised in a state bordering the Ohio River, an area where Histoplasma capsulatum is endemic. She denied any recent travel.
Her vital signs and neurologic examination were normal. Computed tomography of the head showed two areas of increased attenuation anterior to the frontal horns. To better characterize those lesions, magnetic resonance imaging (MRI) with contrast was done, which showed about a dozen 1-cm ring-enhancing lesions in the right cerebellum and both cerebral hemispheres (Figure 1).
Results of a complete blood cell count, metabolic profile, and chest radiography were normal. Her CD4 count was 428/μL (reference range 533–1,674) and 20% (60%–89%); her HIV viral load was 326,000 copies/mL.
She was initially treated empirically with sulfadiazine, pyrimethamine, and leukovorin for possible toxoplasmosis, which is the most common cause of ring-enhancing brain lesions in HIV patients. In the meantime, cerebrospinal fluid, blood, and urine were sent for a detailed workup for fungi, including Histoplasma. Results of the Histoplasma antibody and antigen studies of the serum, urine, and cerebrospinal fluid were positive, while cerebrospinal fluid testing for Toxoplasma by polymerase chain reaction testing was negative. Empirical treatment for toxoplasmosis was stopped and amphotericin B was started to treat disseminated histoplasmosis.
During her hospital course, she underwent brain biopsy via right frontotemporal craniotomy with resection of right frontal lesions. Pathologic study showed partially organizing abscesses with central necrosis (Figure 2), microscopy with Grocott-Gomori methenamine silver stain was positive for budding yeast forms consistent with H capsulatum (Figure 3), and special stain for acid-fast bacilli was negative for mycobacteria. Cultures of the brain biopsy specimen, blood, and cerebrospinal fluid for fungi, acid-fast bacilli, and bacteria did not reveal any growth after 28 days.
The patient was discharged home with instructions to take amphotericin B for a total of 6 weeks and then itraconazole. About 1 year later, she remained free of symptoms, although repeat MRI did not show any significant change in the size or number of histoplasmomas.
She did not comply well with her HIV treatment, and her immune status did not improve, so we decided to continue her itraconazole treatment for more than 1 year.
CEREBRAL HISTOPLASMOMA
The term “histoplasmoma” was introduced by Shapiro et al1 in 1955, when they first described numerous focal areas of softening, up to 1 cm in diameter, scattered throughout the brain at autopsy in a 41-year-old man who had died of disseminated histoplasmosis. They coined the word to describe these discrete areas of necrosis that might resemble tumors on the basis of their size, location, and capability of causing increased intracranial pressure.
Central nervous system involvement can either be a manifestation of disseminated disease or present as an isolated illness.2 It occurs in 5% to 10% of cases of disseminated histoplasmosis.3 Histoplasmosis of the central nervous system can have different manifestations; the most common presentation is chronic meningitis.4
Laboratory diagnosis is based on detecting H capsulatum antigen and antibody in the urine, blood, and cerebrospinal fluid. Tissue biopsy (histopathology) as well as cultures of tissue samples or body fluids may also establish the diagnosis.4
Toxoplasmosis and primary central nervous system lymphoma are the most common causes of brain ring-enhancing lesions in HIV patients in developed countries, while in the developing world neurocysticercosis and tuberculomas are more common.5,6 Much less common causes include brain abscesses secondary to bacterial infections (pyogenic abscess),7 cryptococcomas,8 syphilitic cerebral gummata,9 primary brain tumors (gliomas), and metastases.10
Compared with other forms of the disease, histoplasmosis of the central nervous system has higher rates of treatment failure and relapse, so treatment should be prolonged and aggressive.2,3 The cure rate with amphotericin B ranges from 33% to 61%, and higher doses produce better response rates.3
Current treatment recommendations are based on 2007 guidelines of the Infectious Diseases Society of America.11 Liposomal amphotericin B is the drug of choice because it achieves higher concentrations in the central nervous system than other drugs and is less toxic. It is given for 4 to 6 weeks, followed by itraconazole for at least 1 year and until the cerebrospinal fluid Histoplasma antigen test is negative and other cerebrospinal fluid abnormalities are resolved.
In patients who have primary disseminated histoplasmosis that includes the central nervous system, itraconazole can be given for more than 1 year or until immune recovery is achieved—or lifelong if necessary.2,12 Long-term suppressive antifungal therapy also should be considered in patients for whom appropriate initial therapy fails.2
Nephrotoxicity (acute kidney injury, hypokalemia, and hypomagnesemia), infusion-related drug reactions, and rash are among the well-described side effects of amphotericin B. Maintenance of intravascular volume and replacement of electrolytes should be an integral part of the amphotericin B treatment regimen.13
TAKE-AWAY POINTS
- Histoplasmomas should be considered in the differential diagnosis of ring-enhancing lesions of the central nervous system, along with toxoplasmosis and primary central nervous system lymphoma. This will allow timely initiation of the diagnostic workup, avoiding unnecessary and potentially risky interventions and delays in starting targeted antifungal therapy.
- There is no single gold standard test for central nervous system histoplasmosis. Rather, the final diagnosis is based on the combination of clinical, laboratory, and radiologic findings.
Acknowledgment: Library research assistance provided by HSHS St. John’s Hospital Health Sciences Library staff.
- Shapiro JL, Lux JJ, Sprofkin BE. Histoplasmosis of the central nervous system. Am J Pathol 1955; 31:319–335.
- Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis 2005; 40:844–852.
- Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system: a clinical review. Medicine (Baltimore) 1990; 69:244–260.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Modi M, Mochan A, Modi G. Management of HIV-associated focal brain lesions in developing countries. QJM 2004; 97:413–421.
- Miller RF, Hall-Craggs MA, Costa DC, et al. Magnetic resonance imaging, thallium-201 SPET scanning, and laboratory analyses for discrimination of cerebral lymphoma and toxoplasmosis in AIDS. Sex Transm Infect 1998; 74:258–264.
- Cohen WA. Intracranial bacterial infections in patients with AIDS. Neuroimaging Clin N Am 1997; 7:223–229.
- Troncoso A, Fumagalli J, Shinzato R, Gulotta H, Toller M, Bava J. CNS cryptococcoma in an HIV-positive patient. J Int Assoc Physicians AIDS Care (Chic) 2002; 1:131–133.
- Land AM, Nelson GA, Bell SG, Denby KJ, Estrada CA, Willett LL. Widening the differential for brain masses in human immunodeficiency virus-positive patients: syphilitic cerebral gummata. Am J Med Sci 2013; 346:253–255.
- Balsys R, Janousek JE, Batnitzky S, Templeton AW. Peripheral enhancement in computerized cranial tomography: a non-specific finding. Surg Neurol 1979; 11:207–216.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Wheat J, Hafner R, Wulfsohn M, et al; National Institute of Allergy and Infectious Diseases Clinical Trials and Mycoses Study Group Collaborators. Prevention of relapse of histoplasmosis with itraconazole in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1993; 118:610–616.
- Saccente M. Central nervous system histoplasmosis. Curr Treat Options Neurol 2008; 10:161–167.
- Shapiro JL, Lux JJ, Sprofkin BE. Histoplasmosis of the central nervous system. Am J Pathol 1955; 31:319–335.
- Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis 2005; 40:844–852.
- Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system: a clinical review. Medicine (Baltimore) 1990; 69:244–260.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Modi M, Mochan A, Modi G. Management of HIV-associated focal brain lesions in developing countries. QJM 2004; 97:413–421.
- Miller RF, Hall-Craggs MA, Costa DC, et al. Magnetic resonance imaging, thallium-201 SPET scanning, and laboratory analyses for discrimination of cerebral lymphoma and toxoplasmosis in AIDS. Sex Transm Infect 1998; 74:258–264.
- Cohen WA. Intracranial bacterial infections in patients with AIDS. Neuroimaging Clin N Am 1997; 7:223–229.
- Troncoso A, Fumagalli J, Shinzato R, Gulotta H, Toller M, Bava J. CNS cryptococcoma in an HIV-positive patient. J Int Assoc Physicians AIDS Care (Chic) 2002; 1:131–133.
- Land AM, Nelson GA, Bell SG, Denby KJ, Estrada CA, Willett LL. Widening the differential for brain masses in human immunodeficiency virus-positive patients: syphilitic cerebral gummata. Am J Med Sci 2013; 346:253–255.
- Balsys R, Janousek JE, Batnitzky S, Templeton AW. Peripheral enhancement in computerized cranial tomography: a non-specific finding. Surg Neurol 1979; 11:207–216.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Wheat J, Hafner R, Wulfsohn M, et al; National Institute of Allergy and Infectious Diseases Clinical Trials and Mycoses Study Group Collaborators. Prevention of relapse of histoplasmosis with itraconazole in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1993; 118:610–616.
- Saccente M. Central nervous system histoplasmosis. Curr Treat Options Neurol 2008; 10:161–167.
Wilson disease
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
Alcoholic hepatitis: An important consideration
To the Editor: I read with keen interest the high-quality review of the pathogenesis, diagnosis, and management of alcoholic hepatitis by Dugum et al.1 They clearly emphasized the high morbidity and mortality rates associated with this condition.
An important consideration for healthcare practitioners is that the presentation of alcoholic hepatitis can mimic an infectious process, eg, presenting with fever and an elevated white blood cell count. Indeed, clinicians should be vigilant and should routinely evaluate for an underlying infection in patients with suspected alcoholic hepatitis, because patients with liver disease are immunocompromised and several problems can potentially coexist in any given patient.
Therefore, clinicians should focus on the clinical history and examination (vital signs, mental status examination, presence of ascites) and should screen for common coinfections such as urinary tract infection and pneumonia with a white blood cell count with differential and other tests. Of particular importance, patients with ascites should undergo diagnostic abdominal paracentesis,2 and empiric antimicrobial therapy for spontaneous bacterial peritonitis should be considered on a case-by-case basis.3
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Runyon BA. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013; 57:1651–1653.
- Lutz P, Nischalke HD, Strassburg CP, Spengler U. Spontaneous bacterial peritonitis: the clinical challenge of a leaky gut and a cirrhotic liver. World J Hepatol 2015; 7:304–314.
To the Editor: I read with keen interest the high-quality review of the pathogenesis, diagnosis, and management of alcoholic hepatitis by Dugum et al.1 They clearly emphasized the high morbidity and mortality rates associated with this condition.
An important consideration for healthcare practitioners is that the presentation of alcoholic hepatitis can mimic an infectious process, eg, presenting with fever and an elevated white blood cell count. Indeed, clinicians should be vigilant and should routinely evaluate for an underlying infection in patients with suspected alcoholic hepatitis, because patients with liver disease are immunocompromised and several problems can potentially coexist in any given patient.
Therefore, clinicians should focus on the clinical history and examination (vital signs, mental status examination, presence of ascites) and should screen for common coinfections such as urinary tract infection and pneumonia with a white blood cell count with differential and other tests. Of particular importance, patients with ascites should undergo diagnostic abdominal paracentesis,2 and empiric antimicrobial therapy for spontaneous bacterial peritonitis should be considered on a case-by-case basis.3
To the Editor: I read with keen interest the high-quality review of the pathogenesis, diagnosis, and management of alcoholic hepatitis by Dugum et al.1 They clearly emphasized the high morbidity and mortality rates associated with this condition.
An important consideration for healthcare practitioners is that the presentation of alcoholic hepatitis can mimic an infectious process, eg, presenting with fever and an elevated white blood cell count. Indeed, clinicians should be vigilant and should routinely evaluate for an underlying infection in patients with suspected alcoholic hepatitis, because patients with liver disease are immunocompromised and several problems can potentially coexist in any given patient.
Therefore, clinicians should focus on the clinical history and examination (vital signs, mental status examination, presence of ascites) and should screen for common coinfections such as urinary tract infection and pneumonia with a white blood cell count with differential and other tests. Of particular importance, patients with ascites should undergo diagnostic abdominal paracentesis,2 and empiric antimicrobial therapy for spontaneous bacterial peritonitis should be considered on a case-by-case basis.3
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Runyon BA. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013; 57:1651–1653.
- Lutz P, Nischalke HD, Strassburg CP, Spengler U. Spontaneous bacterial peritonitis: the clinical challenge of a leaky gut and a cirrhotic liver. World J Hepatol 2015; 7:304–314.
- Dugum M, Zein N, McCullough A, Hanouneh I. Alcoholic hepatitis: challenges in diagnosis and management. Cleve Clin J Med 2015; 82:226–236.
- Runyon BA. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013; 57:1651–1653.
- Lutz P, Nischalke HD, Strassburg CP, Spengler U. Spontaneous bacterial peritonitis: the clinical challenge of a leaky gut and a cirrhotic liver. World J Hepatol 2015; 7:304–314.