User login
Not acne, but what?
AN OTHERWISE HEALTHY
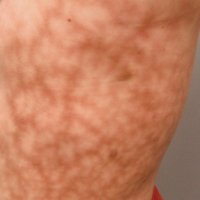
Scattered papules and pustules were present on the forehead, nose, and cheeks, with background erythema and telangiectasias (FIGURE 1). A few pinpoint crusted excoriations were noted. A sample was taken from the papules and pustules using a #15 blade and submitted for examination.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea with Demodex mites
Under light microscopy, the scraping revealed Demodex mites (FIGURE 2). It has been proposed that these mites play a role in the inflammatory process seen in rosacea, although studies have yet to determine whether the inflammatory symptoms of rosacea cause the mites to proliferate or if the mites contribute to the initial inflammatory process.1,2

Demodex folliculorum and D brevis are part of normal skin flora; they are found in about 12% of all follicles and most commonly involve the face.3 They often become abundant in the presence of numerous sebaceous glands. Men have more sebaceous glands than women do, and thus run a greater risk for infestation with mites. An abnormal proliferation of Demodex mites can lead to demodicosis.
Demodex mites can be examined microscopically via the skin surface sampling technique known as scraping, which was done in this case. Samples taken from the papules and pustules utilizing a #15 blade are placed in immersion oil on a glass slide, cover-slipped, and examined by light microscopy.
Rosacea is thought to be an inflammatory disease in which the immune system is triggered by a variety of factors, including UV light, heat, stress, alcohol, hormonal influences, and microorganisms.1,4 The disease is found in up to 10% of the population worldwide.1
The diagnosis of rosacea requires at least 1 of the 2 “core features”—persistent central facial erythema or phymatous changes—or 2 of 4 “major features”: papules/pustules, ocular manifestation, flushing, and telangiectasias. There are 3 phenotypes: ocular, papulopustular, and erythematotelangiectatic.5,6
Continue to: The connection
The connection. Papulopustular and erythematotelangiectatic rosacea may be caused by a proliferation of Demodex mites and increased vascular endothelial growth factor production.2 In fact, a proliferation of Demodex is seen in almost all cases of papulopustular rosacea and more than 60% of cases of erythematotelangiectatic rosacea.2
Patient age and distribution of lesions narrowed the differential
Acne vulgaris is an inflammatory disease of the pilosebaceous units caused by increased sebum production, inflammation, and bacterial colonization (Propionibacterium acnes) of hair follicles on the face, neck, chest, and other areas. Both inflammatory and noninflammatory lesions can be present, and in serious cases, scarring can result.7 The case patient’s age and accompanying broad erythema were more consistent with rosacea than acne vulgaris.
Seborrheic dermatitis is a common skin condition usually stemming from an inflammatory reaction to a common yeast. Classic symptoms include scaling and erythema of the scalp and central face, as well as pruritus. Topical antifungals such as ketoconazole 2% cream and 2% shampoo are the mainstay of treatment.8 The broad distribution and papulopustules in this patient argue against the diagnosis of seborrheic dermatitis.
Systemic lupus erythematosus is a systemic inflammatory disease that often has cutaneous manifestations. Acute lupus manifests as an erythematous “butterfly rash” across the face and cheeks. Chronic discoid lupus involves depigmented plaques, erythematous macules, telangiectasias, and scarring with loss of normal hair follicles. These findings classically are photodistributed.9 The classic broad erythema extending from the cheeks over the bridge of the nose was not present in this patient.
Treatment is primarily topical
Mild cases of rosacea often can be managed with topical antibiotic creams. More severe cases may require systemic antibiotics such as tetracycline or doxycycline, although these are used with caution due to the potential for antibiotic resistance.
Ivermectin 1% cream is a US Food and Drug Administration–approved medication that is applied once daily for up to a year to treat the inflammatory pustules associated with Demodex mites. Although it is costly, studies have shown better results with topical ivermectin than with other topical medications (eg, metronidazole 0.75% gel or cream). However, metronidazole 0.75% gel applied twice daily and oral tetracycline 250 mg or doxycycline 100 mg daily or twice daily for at least 2 months often are utilized when the cost of topical ivermectin is prohibitive.10
Our patient was treated with a combination of doxycycline 100 mg daily for 30 days and
1. Forton FMN. Rosacea, an infectious disease: why rosacea with papulopustules should be considered a demodicosis. A narrative review. J Eur Acad Dermatol Venereol. 2022;36:987-1002. doi: 10.1111/jdv.18049
2. Forton FMN. The pathogenic role of demodex mites in rosacea: a potential therapeutic target already in erythematotelangiectatic rosacea? Dermatol Ther (Heidelb). 2020;10:1229-1253. doi: 10.1007/s13555-020-00458-9
3. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504. doi: 10.1016/j.clindermatol.2010.03.006
4. Erbağci Z, OzgöztaŞi O. The significance of demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425. doi: 10.1046/j.1365-4362.1998.00218.x
5. Tan J, Almeida LMC, Criber B, et al. Updating the diagnosis, classification and assessment of rosacea: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:431-438. doi: 10.1111/bjd.15122
6. Gallo RL, Granstein RD, Kang S, et al. Standard classification and pathophysiology of rosacea: the 2017 update by the National Rosacea Society Expert Committee. J Am Acad Dermatol. 2018;78:148-155. doi: 10.1016/j.jaad.2017.08.037
7. Williams HC, Dellavalle RP, Garner S. Acne vulgaris. Lancet. 2012;379:361-372. doi: 10.1016/S0140-6736(11)60321-8.
8. Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
9. Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
10. Raedler LA. Soolantra (ivermectin) 1% cream: a novel, antibiotic-free agent approved for the treatment of patients with rosacea. Am Health Drug Benefits. 2015;8(Spec Feature):122-125.
AN OTHERWISE HEALTHY
Scattered papules and pustules were present on the forehead, nose, and cheeks, with background erythema and telangiectasias (FIGURE 1). A few pinpoint crusted excoriations were noted. A sample was taken from the papules and pustules using a #15 blade and submitted for examination.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea with Demodex mites
Under light microscopy, the scraping revealed Demodex mites (FIGURE 2). It has been proposed that these mites play a role in the inflammatory process seen in rosacea, although studies have yet to determine whether the inflammatory symptoms of rosacea cause the mites to proliferate or if the mites contribute to the initial inflammatory process.1,2
Demodex folliculorum and D brevis are part of normal skin flora; they are found in about 12% of all follicles and most commonly involve the face.3 They often become abundant in the presence of numerous sebaceous glands. Men have more sebaceous glands than women do, and thus run a greater risk for infestation with mites. An abnormal proliferation of Demodex mites can lead to demodicosis.
Demodex mites can be examined microscopically via the skin surface sampling technique known as scraping, which was done in this case. Samples taken from the papules and pustules utilizing a #15 blade are placed in immersion oil on a glass slide, cover-slipped, and examined by light microscopy.
Rosacea is thought to be an inflammatory disease in which the immune system is triggered by a variety of factors, including UV light, heat, stress, alcohol, hormonal influences, and microorganisms.1,4 The disease is found in up to 10% of the population worldwide.1
The diagnosis of rosacea requires at least 1 of the 2 “core features”—persistent central facial erythema or phymatous changes—or 2 of 4 “major features”: papules/pustules, ocular manifestation, flushing, and telangiectasias. There are 3 phenotypes: ocular, papulopustular, and erythematotelangiectatic.5,6
Continue to: The connection
The connection. Papulopustular and erythematotelangiectatic rosacea may be caused by a proliferation of Demodex mites and increased vascular endothelial growth factor production.2 In fact, a proliferation of Demodex is seen in almost all cases of papulopustular rosacea and more than 60% of cases of erythematotelangiectatic rosacea.2
Patient age and distribution of lesions narrowed the differential
Acne vulgaris is an inflammatory disease of the pilosebaceous units caused by increased sebum production, inflammation, and bacterial colonization (Propionibacterium acnes) of hair follicles on the face, neck, chest, and other areas. Both inflammatory and noninflammatory lesions can be present, and in serious cases, scarring can result.7 The case patient’s age and accompanying broad erythema were more consistent with rosacea than acne vulgaris.
Seborrheic dermatitis is a common skin condition usually stemming from an inflammatory reaction to a common yeast. Classic symptoms include scaling and erythema of the scalp and central face, as well as pruritus. Topical antifungals such as ketoconazole 2% cream and 2% shampoo are the mainstay of treatment.8 The broad distribution and papulopustules in this patient argue against the diagnosis of seborrheic dermatitis.
Systemic lupus erythematosus is a systemic inflammatory disease that often has cutaneous manifestations. Acute lupus manifests as an erythematous “butterfly rash” across the face and cheeks. Chronic discoid lupus involves depigmented plaques, erythematous macules, telangiectasias, and scarring with loss of normal hair follicles. These findings classically are photodistributed.9 The classic broad erythema extending from the cheeks over the bridge of the nose was not present in this patient.
Treatment is primarily topical
Mild cases of rosacea often can be managed with topical antibiotic creams. More severe cases may require systemic antibiotics such as tetracycline or doxycycline, although these are used with caution due to the potential for antibiotic resistance.
Ivermectin 1% cream is a US Food and Drug Administration–approved medication that is applied once daily for up to a year to treat the inflammatory pustules associated with Demodex mites. Although it is costly, studies have shown better results with topical ivermectin than with other topical medications (eg, metronidazole 0.75% gel or cream). However, metronidazole 0.75% gel applied twice daily and oral tetracycline 250 mg or doxycycline 100 mg daily or twice daily for at least 2 months often are utilized when the cost of topical ivermectin is prohibitive.10
Our patient was treated with a combination of doxycycline 100 mg daily for 30 days and
AN OTHERWISE HEALTHY
Scattered papules and pustules were present on the forehead, nose, and cheeks, with background erythema and telangiectasias (FIGURE 1). A few pinpoint crusted excoriations were noted. A sample was taken from the papules and pustules using a #15 blade and submitted for examination.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Rosacea with Demodex mites
Under light microscopy, the scraping revealed Demodex mites (FIGURE 2). It has been proposed that these mites play a role in the inflammatory process seen in rosacea, although studies have yet to determine whether the inflammatory symptoms of rosacea cause the mites to proliferate or if the mites contribute to the initial inflammatory process.1,2
Demodex folliculorum and D brevis are part of normal skin flora; they are found in about 12% of all follicles and most commonly involve the face.3 They often become abundant in the presence of numerous sebaceous glands. Men have more sebaceous glands than women do, and thus run a greater risk for infestation with mites. An abnormal proliferation of Demodex mites can lead to demodicosis.
Demodex mites can be examined microscopically via the skin surface sampling technique known as scraping, which was done in this case. Samples taken from the papules and pustules utilizing a #15 blade are placed in immersion oil on a glass slide, cover-slipped, and examined by light microscopy.
Rosacea is thought to be an inflammatory disease in which the immune system is triggered by a variety of factors, including UV light, heat, stress, alcohol, hormonal influences, and microorganisms.1,4 The disease is found in up to 10% of the population worldwide.1
The diagnosis of rosacea requires at least 1 of the 2 “core features”—persistent central facial erythema or phymatous changes—or 2 of 4 “major features”: papules/pustules, ocular manifestation, flushing, and telangiectasias. There are 3 phenotypes: ocular, papulopustular, and erythematotelangiectatic.5,6
Continue to: The connection
The connection. Papulopustular and erythematotelangiectatic rosacea may be caused by a proliferation of Demodex mites and increased vascular endothelial growth factor production.2 In fact, a proliferation of Demodex is seen in almost all cases of papulopustular rosacea and more than 60% of cases of erythematotelangiectatic rosacea.2
Patient age and distribution of lesions narrowed the differential
Acne vulgaris is an inflammatory disease of the pilosebaceous units caused by increased sebum production, inflammation, and bacterial colonization (Propionibacterium acnes) of hair follicles on the face, neck, chest, and other areas. Both inflammatory and noninflammatory lesions can be present, and in serious cases, scarring can result.7 The case patient’s age and accompanying broad erythema were more consistent with rosacea than acne vulgaris.
Seborrheic dermatitis is a common skin condition usually stemming from an inflammatory reaction to a common yeast. Classic symptoms include scaling and erythema of the scalp and central face, as well as pruritus. Topical antifungals such as ketoconazole 2% cream and 2% shampoo are the mainstay of treatment.8 The broad distribution and papulopustules in this patient argue against the diagnosis of seborrheic dermatitis.
Systemic lupus erythematosus is a systemic inflammatory disease that often has cutaneous manifestations. Acute lupus manifests as an erythematous “butterfly rash” across the face and cheeks. Chronic discoid lupus involves depigmented plaques, erythematous macules, telangiectasias, and scarring with loss of normal hair follicles. These findings classically are photodistributed.9 The classic broad erythema extending from the cheeks over the bridge of the nose was not present in this patient.
Treatment is primarily topical
Mild cases of rosacea often can be managed with topical antibiotic creams. More severe cases may require systemic antibiotics such as tetracycline or doxycycline, although these are used with caution due to the potential for antibiotic resistance.
Ivermectin 1% cream is a US Food and Drug Administration–approved medication that is applied once daily for up to a year to treat the inflammatory pustules associated with Demodex mites. Although it is costly, studies have shown better results with topical ivermectin than with other topical medications (eg, metronidazole 0.75% gel or cream). However, metronidazole 0.75% gel applied twice daily and oral tetracycline 250 mg or doxycycline 100 mg daily or twice daily for at least 2 months often are utilized when the cost of topical ivermectin is prohibitive.10
Our patient was treated with a combination of doxycycline 100 mg daily for 30 days and
1. Forton FMN. Rosacea, an infectious disease: why rosacea with papulopustules should be considered a demodicosis. A narrative review. J Eur Acad Dermatol Venereol. 2022;36:987-1002. doi: 10.1111/jdv.18049
2. Forton FMN. The pathogenic role of demodex mites in rosacea: a potential therapeutic target already in erythematotelangiectatic rosacea? Dermatol Ther (Heidelb). 2020;10:1229-1253. doi: 10.1007/s13555-020-00458-9
3. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504. doi: 10.1016/j.clindermatol.2010.03.006
4. Erbağci Z, OzgöztaŞi O. The significance of demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425. doi: 10.1046/j.1365-4362.1998.00218.x
5. Tan J, Almeida LMC, Criber B, et al. Updating the diagnosis, classification and assessment of rosacea: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:431-438. doi: 10.1111/bjd.15122
6. Gallo RL, Granstein RD, Kang S, et al. Standard classification and pathophysiology of rosacea: the 2017 update by the National Rosacea Society Expert Committee. J Am Acad Dermatol. 2018;78:148-155. doi: 10.1016/j.jaad.2017.08.037
7. Williams HC, Dellavalle RP, Garner S. Acne vulgaris. Lancet. 2012;379:361-372. doi: 10.1016/S0140-6736(11)60321-8.
8. Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
9. Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
10. Raedler LA. Soolantra (ivermectin) 1% cream: a novel, antibiotic-free agent approved for the treatment of patients with rosacea. Am Health Drug Benefits. 2015;8(Spec Feature):122-125.
1. Forton FMN. Rosacea, an infectious disease: why rosacea with papulopustules should be considered a demodicosis. A narrative review. J Eur Acad Dermatol Venereol. 2022;36:987-1002. doi: 10.1111/jdv.18049
2. Forton FMN. The pathogenic role of demodex mites in rosacea: a potential therapeutic target already in erythematotelangiectatic rosacea? Dermatol Ther (Heidelb). 2020;10:1229-1253. doi: 10.1007/s13555-020-00458-9
3. Elston DM. Demodex mites: facts and controversies. Clin Dermatol. 2010;28:502-504. doi: 10.1016/j.clindermatol.2010.03.006
4. Erbağci Z, OzgöztaŞi O. The significance of demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421-425. doi: 10.1046/j.1365-4362.1998.00218.x
5. Tan J, Almeida LMC, Criber B, et al. Updating the diagnosis, classification and assessment of rosacea: recommendations from the global ROSacea COnsensus (ROSCO) panel. Br J Dermatol. 2017;176:431-438. doi: 10.1111/bjd.15122
6. Gallo RL, Granstein RD, Kang S, et al. Standard classification and pathophysiology of rosacea: the 2017 update by the National Rosacea Society Expert Committee. J Am Acad Dermatol. 2018;78:148-155. doi: 10.1016/j.jaad.2017.08.037
7. Williams HC, Dellavalle RP, Garner S. Acne vulgaris. Lancet. 2012;379:361-372. doi: 10.1016/S0140-6736(11)60321-8.
8. Clark GW, Pope SM, Jaboori KA. Diagnosis and treatment of seborrheic dermatitis. Am Fam Physician. 2015;91:185-190.
9. Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol. 1996;135:355-362.
10. Raedler LA. Soolantra (ivermectin) 1% cream: a novel, antibiotic-free agent approved for the treatment of patients with rosacea. Am Health Drug Benefits. 2015;8(Spec Feature):122-125.

Facial Follicular Spicules: A Rare Cutaneous Presentation of Trichodysplasia Spinulosa
To the Editor:
A 57-year-old man with hypertension, dyslipidemia, and congestive heart failure presented with a disfiguring eruption comprised of asymptomatic papules on the face that appeared 12 months post–heart transplantation. Immunosuppressive medications included mycophenolic acid and tacrolimus ointment (FK506). The pinpoint papules spread from the central face to the ears, arms, and legs. Physical examination revealed multiple 0.5- to 1-mm flesh-colored papules over the glabella, nose, nasolabial folds, philtrum, chin, ears, arms, and legs sparing the trunk. The initial appearance of the facial rash resembled the surface of a nutmeg grater with central white spiny excrescences overlying fine papules (spinulosism)(Figure 1). In addition, eyebrow alopecia was present.
 on the central face")
A 3-mm punch biopsy of a papule with a central spine was performed on the left thigh. Microscopic examination revealed marked dilatation of anagen hair follicles with a proliferation of haphazard inner root sheath cells replacing the follicular lumen. Hair shafts were absent, and plugged infundibula were observed (Figure 2). The inner root sheath keratinocytes were enlarged and dystrophic with deeply eosinophilic trichohyalin granules (Figure 3). The epidermis, outer root sheath epithelium, and eccrine structures were unremarkable.

Transmission electron microscopy (TEM) confirmed the presence of intranuclear viral inclusions within affected inner root sheath keratinocytes composed of nonenveloped icosahedral viral particles measuring 33 to 38 nm in diameter (Figure 4). These findings morphologically were consistent with a polyomavirus. No intracytoplasmic or extracellular viral particles were identified. The clinical history, physical examination, histopathology, and electron microscopy features strongly supported the diagnosis of trichodysplasia spinulosa (TS) despite insufficient material being retrieved for polymerase chain reaction identification.

Trichodysplasia spinulosa was first described by Haycox et al1 in 1999. The authors suggested a viral etiology. Eleven years later, TS-associated polyomavirus (TSPyV) was identified by van der Meijden et al.2 Follicular keratinocytes are the specific target for TSPyV.3 Evidence has been presented suggesting that TS is caused by a primary infection or reactivation of TSPyV in the setting of immunosuppression.4,5

Patients with TS present with papular eruptions that appear on the central face with spiny excrescences and various degrees of alopecia involving the eyebrows or eyelashes. Histopathologic features include distended hair follicles with expansion of inner root sheath cells, eosinophilic trichohyalin granules, and the absence of hair shafts. The viral protein can be verified through immunohistochemistry TSPyV VP1 staining that demonstrates co-localization with trichohyalin. Viral particles also can be visualized as 35- to 38-nm intranuclear particles with an organized crystalloid morphology on TEM.6,7 The negative polymerase chain reaction in our patient could be the result of suboptimal template DNA concentration extracted from the limited amount of tissue remaining in the block after hematoxylin and eosin staining.
The clinical differential diagnosis of central facial spinulosism includes the follicular spicules of multiple myeloma (FSMM). In fact, FSMM and TS can only be differentiated after obtaining a blood profile and bone marrow biopsy that excludes the diagnosis of FSMM. A history of immunosuppression typically suggests TS. Histopathology often is equivocal in FSMM8; however, TEM reveals viral particles (TSPyV) in TS. Transmission electron microscopy in FSMM demonstrates fibrillary structures arranged in a paracrystalline configuration with unknown significance instead of viral particles. Despite the absence of viral particles on TEM, a low mean copy number of Merkel cell polyomavirus was isolated from a patient with FSMM who responded dramatically to treatment with topical cidofovir gel 1%.8 In addition to treating the underlying multiple myeloma in FSMM, topical cidofovir gel 1% also may have a role in treatment of these patients, suggesting a possible viral rather than simply paraneoplastic etiology of FSMM. Therefore, polyomavirus infection should be considered in the initial workup of any patient with fine facial follicular spicules.
The most effective management of TS in transplant recipients is to reduce immunosuppression to the lowest level possible without jeopardizing the transplanted organ.9 In our case, reduction of immunosuppressive drugs was not possible. In fact, immunosuppression in our patient was increased following evidence of early rejection of the heart transplant. Although manual extraction of the keratin spicules resulted in considerable improvement in a similar facial eruption in a patient with pediatric pre–B-cell acute lymphoblastic leukemia developing TS,10 it is impossible to apply this approach to patients such as ours who have thousands of tiny lesions. Fortunately, custom-compounded cidofovir gel 1% applied twice daily to the patient’s face and ears for 4 weeks led to near-complete clearance at follow-up (Figure 5). Due to the high cost of the medication (approaching $700 for one tube), our patient applied this medication to the face only several times weekly with excellent improvement. Thus, it appears that it is possible to suppress this virus with topical medication alone.

Polyomavirus infection should be considered in patients presenting with fine follicular spiny papules, especially those who are immunosuppressed. The possibility of coexisting multiple myeloma should be excluded.
Acknowledgment—We sincerely thank Glenn A. Hoskins (Jackson, Mississippi), the electron microscopy technologist, for the detection of viral particles and the electron microscope photographs.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa: a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:E1001024.
- Rouanet J, Aubin F, Gaboriaud P, et al. Trichodysplasia spinulosa: a polyomavirus infection specifically targeting follicular keratinocytes in immunocompromised patients. Br J Dermatol. 2016;174:629-632.
- van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- van der Meijden E, Horváth B, Nijland M, et al. Primary polyomavirus infection, not reactivation, as the cause of trichodysplasia spinulosa in immunocompromised patients. J Infect Dis. 2017;215:1080-1084.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2012;148:726-733.
- Kazem S, van der Meijden E, Feltkamp MC. The trichodysplasia spinulosa-associated polyomavirus: virological background and clinical implications. APMIS. 2013;121:770-782.
- van Boheemen S, Jones T, Muhlemann B, et al. Cidofovir gel as treatment of follicular spicules in multiple myeloma. JAMA Dermatol. 2015;151:82-84.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Barton M, Lockhart S, Sidbury R, et al. Trichodysplasia spinulosa in a 7-year-old boy managed using physical extraction of keratin spicules. Pediatr Dermatol. 2017;34:E74-E76.
To the Editor:
A 57-year-old man with hypertension, dyslipidemia, and congestive heart failure presented with a disfiguring eruption comprised of asymptomatic papules on the face that appeared 12 months post–heart transplantation. Immunosuppressive medications included mycophenolic acid and tacrolimus ointment (FK506). The pinpoint papules spread from the central face to the ears, arms, and legs. Physical examination revealed multiple 0.5- to 1-mm flesh-colored papules over the glabella, nose, nasolabial folds, philtrum, chin, ears, arms, and legs sparing the trunk. The initial appearance of the facial rash resembled the surface of a nutmeg grater with central white spiny excrescences overlying fine papules (spinulosism)(Figure 1). In addition, eyebrow alopecia was present.
A 3-mm punch biopsy of a papule with a central spine was performed on the left thigh. Microscopic examination revealed marked dilatation of anagen hair follicles with a proliferation of haphazard inner root sheath cells replacing the follicular lumen. Hair shafts were absent, and plugged infundibula were observed (Figure 2). The inner root sheath keratinocytes were enlarged and dystrophic with deeply eosinophilic trichohyalin granules (Figure 3). The epidermis, outer root sheath epithelium, and eccrine structures were unremarkable.
Transmission electron microscopy (TEM) confirmed the presence of intranuclear viral inclusions within affected inner root sheath keratinocytes composed of nonenveloped icosahedral viral particles measuring 33 to 38 nm in diameter (Figure 4). These findings morphologically were consistent with a polyomavirus. No intracytoplasmic or extracellular viral particles were identified. The clinical history, physical examination, histopathology, and electron microscopy features strongly supported the diagnosis of trichodysplasia spinulosa (TS) despite insufficient material being retrieved for polymerase chain reaction identification.
Trichodysplasia spinulosa was first described by Haycox et al1 in 1999. The authors suggested a viral etiology. Eleven years later, TS-associated polyomavirus (TSPyV) was identified by van der Meijden et al.2 Follicular keratinocytes are the specific target for TSPyV.3 Evidence has been presented suggesting that TS is caused by a primary infection or reactivation of TSPyV in the setting of immunosuppression.4,5
Patients with TS present with papular eruptions that appear on the central face with spiny excrescences and various degrees of alopecia involving the eyebrows or eyelashes. Histopathologic features include distended hair follicles with expansion of inner root sheath cells, eosinophilic trichohyalin granules, and the absence of hair shafts. The viral protein can be verified through immunohistochemistry TSPyV VP1 staining that demonstrates co-localization with trichohyalin. Viral particles also can be visualized as 35- to 38-nm intranuclear particles with an organized crystalloid morphology on TEM.6,7 The negative polymerase chain reaction in our patient could be the result of suboptimal template DNA concentration extracted from the limited amount of tissue remaining in the block after hematoxylin and eosin staining.
The clinical differential diagnosis of central facial spinulosism includes the follicular spicules of multiple myeloma (FSMM). In fact, FSMM and TS can only be differentiated after obtaining a blood profile and bone marrow biopsy that excludes the diagnosis of FSMM. A history of immunosuppression typically suggests TS. Histopathology often is equivocal in FSMM8; however, TEM reveals viral particles (TSPyV) in TS. Transmission electron microscopy in FSMM demonstrates fibrillary structures arranged in a paracrystalline configuration with unknown significance instead of viral particles. Despite the absence of viral particles on TEM, a low mean copy number of Merkel cell polyomavirus was isolated from a patient with FSMM who responded dramatically to treatment with topical cidofovir gel 1%.8 In addition to treating the underlying multiple myeloma in FSMM, topical cidofovir gel 1% also may have a role in treatment of these patients, suggesting a possible viral rather than simply paraneoplastic etiology of FSMM. Therefore, polyomavirus infection should be considered in the initial workup of any patient with fine facial follicular spicules.
The most effective management of TS in transplant recipients is to reduce immunosuppression to the lowest level possible without jeopardizing the transplanted organ.9 In our case, reduction of immunosuppressive drugs was not possible. In fact, immunosuppression in our patient was increased following evidence of early rejection of the heart transplant. Although manual extraction of the keratin spicules resulted in considerable improvement in a similar facial eruption in a patient with pediatric pre–B-cell acute lymphoblastic leukemia developing TS,10 it is impossible to apply this approach to patients such as ours who have thousands of tiny lesions. Fortunately, custom-compounded cidofovir gel 1% applied twice daily to the patient’s face and ears for 4 weeks led to near-complete clearance at follow-up (Figure 5). Due to the high cost of the medication (approaching $700 for one tube), our patient applied this medication to the face only several times weekly with excellent improvement. Thus, it appears that it is possible to suppress this virus with topical medication alone.
Polyomavirus infection should be considered in patients presenting with fine follicular spiny papules, especially those who are immunosuppressed. The possibility of coexisting multiple myeloma should be excluded.
Acknowledgment—We sincerely thank Glenn A. Hoskins (Jackson, Mississippi), the electron microscopy technologist, for the detection of viral particles and the electron microscope photographs.
To the Editor:
A 57-year-old man with hypertension, dyslipidemia, and congestive heart failure presented with a disfiguring eruption comprised of asymptomatic papules on the face that appeared 12 months post–heart transplantation. Immunosuppressive medications included mycophenolic acid and tacrolimus ointment (FK506). The pinpoint papules spread from the central face to the ears, arms, and legs. Physical examination revealed multiple 0.5- to 1-mm flesh-colored papules over the glabella, nose, nasolabial folds, philtrum, chin, ears, arms, and legs sparing the trunk. The initial appearance of the facial rash resembled the surface of a nutmeg grater with central white spiny excrescences overlying fine papules (spinulosism)(Figure 1). In addition, eyebrow alopecia was present.
A 3-mm punch biopsy of a papule with a central spine was performed on the left thigh. Microscopic examination revealed marked dilatation of anagen hair follicles with a proliferation of haphazard inner root sheath cells replacing the follicular lumen. Hair shafts were absent, and plugged infundibula were observed (Figure 2). The inner root sheath keratinocytes were enlarged and dystrophic with deeply eosinophilic trichohyalin granules (Figure 3). The epidermis, outer root sheath epithelium, and eccrine structures were unremarkable.
Transmission electron microscopy (TEM) confirmed the presence of intranuclear viral inclusions within affected inner root sheath keratinocytes composed of nonenveloped icosahedral viral particles measuring 33 to 38 nm in diameter (Figure 4). These findings morphologically were consistent with a polyomavirus. No intracytoplasmic or extracellular viral particles were identified. The clinical history, physical examination, histopathology, and electron microscopy features strongly supported the diagnosis of trichodysplasia spinulosa (TS) despite insufficient material being retrieved for polymerase chain reaction identification.
Trichodysplasia spinulosa was first described by Haycox et al1 in 1999. The authors suggested a viral etiology. Eleven years later, TS-associated polyomavirus (TSPyV) was identified by van der Meijden et al.2 Follicular keratinocytes are the specific target for TSPyV.3 Evidence has been presented suggesting that TS is caused by a primary infection or reactivation of TSPyV in the setting of immunosuppression.4,5
Patients with TS present with papular eruptions that appear on the central face with spiny excrescences and various degrees of alopecia involving the eyebrows or eyelashes. Histopathologic features include distended hair follicles with expansion of inner root sheath cells, eosinophilic trichohyalin granules, and the absence of hair shafts. The viral protein can be verified through immunohistochemistry TSPyV VP1 staining that demonstrates co-localization with trichohyalin. Viral particles also can be visualized as 35- to 38-nm intranuclear particles with an organized crystalloid morphology on TEM.6,7 The negative polymerase chain reaction in our patient could be the result of suboptimal template DNA concentration extracted from the limited amount of tissue remaining in the block after hematoxylin and eosin staining.
The clinical differential diagnosis of central facial spinulosism includes the follicular spicules of multiple myeloma (FSMM). In fact, FSMM and TS can only be differentiated after obtaining a blood profile and bone marrow biopsy that excludes the diagnosis of FSMM. A history of immunosuppression typically suggests TS. Histopathology often is equivocal in FSMM8; however, TEM reveals viral particles (TSPyV) in TS. Transmission electron microscopy in FSMM demonstrates fibrillary structures arranged in a paracrystalline configuration with unknown significance instead of viral particles. Despite the absence of viral particles on TEM, a low mean copy number of Merkel cell polyomavirus was isolated from a patient with FSMM who responded dramatically to treatment with topical cidofovir gel 1%.8 In addition to treating the underlying multiple myeloma in FSMM, topical cidofovir gel 1% also may have a role in treatment of these patients, suggesting a possible viral rather than simply paraneoplastic etiology of FSMM. Therefore, polyomavirus infection should be considered in the initial workup of any patient with fine facial follicular spicules.
The most effective management of TS in transplant recipients is to reduce immunosuppression to the lowest level possible without jeopardizing the transplanted organ.9 In our case, reduction of immunosuppressive drugs was not possible. In fact, immunosuppression in our patient was increased following evidence of early rejection of the heart transplant. Although manual extraction of the keratin spicules resulted in considerable improvement in a similar facial eruption in a patient with pediatric pre–B-cell acute lymphoblastic leukemia developing TS,10 it is impossible to apply this approach to patients such as ours who have thousands of tiny lesions. Fortunately, custom-compounded cidofovir gel 1% applied twice daily to the patient’s face and ears for 4 weeks led to near-complete clearance at follow-up (Figure 5). Due to the high cost of the medication (approaching $700 for one tube), our patient applied this medication to the face only several times weekly with excellent improvement. Thus, it appears that it is possible to suppress this virus with topical medication alone.
Polyomavirus infection should be considered in patients presenting with fine follicular spiny papules, especially those who are immunosuppressed. The possibility of coexisting multiple myeloma should be excluded.
Acknowledgment—We sincerely thank Glenn A. Hoskins (Jackson, Mississippi), the electron microscopy technologist, for the detection of viral particles and the electron microscope photographs.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa: a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:E1001024.
- Rouanet J, Aubin F, Gaboriaud P, et al. Trichodysplasia spinulosa: a polyomavirus infection specifically targeting follicular keratinocytes in immunocompromised patients. Br J Dermatol. 2016;174:629-632.
- van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- van der Meijden E, Horváth B, Nijland M, et al. Primary polyomavirus infection, not reactivation, as the cause of trichodysplasia spinulosa in immunocompromised patients. J Infect Dis. 2017;215:1080-1084.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2012;148:726-733.
- Kazem S, van der Meijden E, Feltkamp MC. The trichodysplasia spinulosa-associated polyomavirus: virological background and clinical implications. APMIS. 2013;121:770-782.
- van Boheemen S, Jones T, Muhlemann B, et al. Cidofovir gel as treatment of follicular spicules in multiple myeloma. JAMA Dermatol. 2015;151:82-84.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Barton M, Lockhart S, Sidbury R, et al. Trichodysplasia spinulosa in a 7-year-old boy managed using physical extraction of keratin spicules. Pediatr Dermatol. 2017;34:E74-E76.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa: a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- van der Meijden E, Janssens RWA, Lauber C, et al. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:E1001024.
- Rouanet J, Aubin F, Gaboriaud P, et al. Trichodysplasia spinulosa: a polyomavirus infection specifically targeting follicular keratinocytes in immunocompromised patients. Br J Dermatol. 2016;174:629-632.
- van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- van der Meijden E, Horváth B, Nijland M, et al. Primary polyomavirus infection, not reactivation, as the cause of trichodysplasia spinulosa in immunocompromised patients. J Infect Dis. 2017;215:1080-1084.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2012;148:726-733.
- Kazem S, van der Meijden E, Feltkamp MC. The trichodysplasia spinulosa-associated polyomavirus: virological background and clinical implications. APMIS. 2013;121:770-782.
- van Boheemen S, Jones T, Muhlemann B, et al. Cidofovir gel as treatment of follicular spicules in multiple myeloma. JAMA Dermatol. 2015;151:82-84.
- DeCrescenzo AJ, Philips RC, Wilkerson MG. Trichodysplasia spinulosa: a rare complication of immunosuppression. JAAD Case Rep. 2016;2:307-309.
- Barton M, Lockhart S, Sidbury R, et al. Trichodysplasia spinulosa in a 7-year-old boy managed using physical extraction of keratin spicules. Pediatr Dermatol. 2017;34:E74-E76.
Practice Points
- Trichodysplasia spinulosa (TS) is a rare skin disease caused by primary TS-associated polyomavirus (TSPyV) infecting follicular keratinocytes in immunocompromised patients.
- Trichodysplasia spinulosa typically presents with papular eruptions that appear on the central face with spiny excrescences and various degrees of alopecia involving the eyebrows or eyelashes.
- The viral protein can be verified through immunohistochemistry TSPyV major capsid protein VP1 staining or can be visualized on transmission electron microscopy.
- Follicular spicules of multiple myeloma should be ruled out before initiating treatment with cidofovir gel 1% for TS.
 on the central face")
Authors’ response
My co-authors and I appreciate the excellent comments regarding our Photo Rounds column, “Foot rash and joint pain,” and would like to provide some additional detail.
After our patient’s 27-day hospital stay, he was admitted to a rehabilitation center for continued inpatient physical therapy for 14 days due to weakness and deconditioning. Following his discharge from the rehabilitation center, the patient was still confined to a wheelchair. He was prescribed an oral prednisone taper (as mentioned in our article) and celecoxib 200 mg bid and referred for outpatient physical therapy. At a follow-up appointment with the rheumatologist, he received adalimumab 80 mg followed by 40 mg every other week, which led to improvement in his range of motion and pain. Two months after outpatient physical therapy, the patient was lost to follow-up.
We agree with Dr. Hahn et al that many of these patients with chlamydia-associated ReA become “long-haulers.” In medicine—especially when rare diseases are considered—we must often make decisions without perfect science. The studies referenced by Dr. Hahn et al suggest that combinations of doxycycline and rifampin or azithromycin and rifampin may treat not only chlamydial infection, but ReA and associated cutaneous disease, as well.1,2 While these studies are small in size, larger studies may never be funded. We agree that combination therapy should be considered in this population of patients.
Hannah R. Badon, MD
Ross L. Pearlman, MD
Robert T. Brodell, MD
Jackson, MS
1. Carter JD, Valeriano J, Vasey FB. Doxycycline versus doxycycline and rifampin in undifferentiated spondyloarthropathy, with special reference to chlamydia-induced arthritis. A prospective, randomized 9-month comparison. J Rheumatol. 2004;31:1973-1980.
2. Carter JD, Espinoza LR, Inman RD, et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 2010;62:1298-1307. doi: 10.1002/art.27394
My co-authors and I appreciate the excellent comments regarding our Photo Rounds column, “Foot rash and joint pain,” and would like to provide some additional detail.
After our patient’s 27-day hospital stay, he was admitted to a rehabilitation center for continued inpatient physical therapy for 14 days due to weakness and deconditioning. Following his discharge from the rehabilitation center, the patient was still confined to a wheelchair. He was prescribed an oral prednisone taper (as mentioned in our article) and celecoxib 200 mg bid and referred for outpatient physical therapy. At a follow-up appointment with the rheumatologist, he received adalimumab 80 mg followed by 40 mg every other week, which led to improvement in his range of motion and pain. Two months after outpatient physical therapy, the patient was lost to follow-up.
We agree with Dr. Hahn et al that many of these patients with chlamydia-associated ReA become “long-haulers.” In medicine—especially when rare diseases are considered—we must often make decisions without perfect science. The studies referenced by Dr. Hahn et al suggest that combinations of doxycycline and rifampin or azithromycin and rifampin may treat not only chlamydial infection, but ReA and associated cutaneous disease, as well.1,2 While these studies are small in size, larger studies may never be funded. We agree that combination therapy should be considered in this population of patients.
Hannah R. Badon, MD
Ross L. Pearlman, MD
Robert T. Brodell, MD
Jackson, MS
My co-authors and I appreciate the excellent comments regarding our Photo Rounds column, “Foot rash and joint pain,” and would like to provide some additional detail.
After our patient’s 27-day hospital stay, he was admitted to a rehabilitation center for continued inpatient physical therapy for 14 days due to weakness and deconditioning. Following his discharge from the rehabilitation center, the patient was still confined to a wheelchair. He was prescribed an oral prednisone taper (as mentioned in our article) and celecoxib 200 mg bid and referred for outpatient physical therapy. At a follow-up appointment with the rheumatologist, he received adalimumab 80 mg followed by 40 mg every other week, which led to improvement in his range of motion and pain. Two months after outpatient physical therapy, the patient was lost to follow-up.
We agree with Dr. Hahn et al that many of these patients with chlamydia-associated ReA become “long-haulers.” In medicine—especially when rare diseases are considered—we must often make decisions without perfect science. The studies referenced by Dr. Hahn et al suggest that combinations of doxycycline and rifampin or azithromycin and rifampin may treat not only chlamydial infection, but ReA and associated cutaneous disease, as well.1,2 While these studies are small in size, larger studies may never be funded. We agree that combination therapy should be considered in this population of patients.
Hannah R. Badon, MD
Ross L. Pearlman, MD
Robert T. Brodell, MD
Jackson, MS
1. Carter JD, Valeriano J, Vasey FB. Doxycycline versus doxycycline and rifampin in undifferentiated spondyloarthropathy, with special reference to chlamydia-induced arthritis. A prospective, randomized 9-month comparison. J Rheumatol. 2004;31:1973-1980.
2. Carter JD, Espinoza LR, Inman RD, et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 2010;62:1298-1307. doi: 10.1002/art.27394
1. Carter JD, Valeriano J, Vasey FB. Doxycycline versus doxycycline and rifampin in undifferentiated spondyloarthropathy, with special reference to chlamydia-induced arthritis. A prospective, randomized 9-month comparison. J Rheumatol. 2004;31:1973-1980.
2. Carter JD, Espinoza LR, Inman RD, et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 2010;62:1298-1307. doi: 10.1002/art.27394
Erythematous ear with drainage
A 6-year-old boy was seen in the hospital in consultation for a 3-week history of suspected cellulitis of the right ear. Drainage from the right ear was refractory to treatment with a 7-day course of cephalexin 15 mL po bid of 250 mg/5 mL solution and clindamycin 24.4 mL po tid of 75 mg/5 mL solution. Treatment was followed by admission to the hospital for treatment with intravenous (IV) cefazolin 1000 mg q6h and IV vancomycin 825 mg q6h for 1 week.
The patient had a significant past medical history for asthma, allergic rhinitis, and severe atopic dermatitis that had been treated with methotrexate 10 mg per week for 6 months beginning when the child was 5 years of age. When the methotrexate proved to be ineffective, the patient was started on Aquaphor and mometasone 0.1% ointment. A 6-month trial of these agents failed as well.
Physical examination revealed that the right ear and skin around it were edematous, erythematous, pruritic, and tender. There was also purulent drainage coming from the ear (FIGURE 1).

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Infectious eczematoid dermatitis
The patient was referred to a dermatologist after seeing an ear, nose, and throat (ENT) specialist who made the diagnosis of otitis externa when the rash failed to respond to topical and systemic antibiotics. The patient’s tender, pruritic, oozing, edematous eruption was recognized as an infectious eczematoid dermatitis (IED).
Although it is not an uncommon condition, IED may be underrecognized. It accounted for 2.9% of admissions to a dermatology-run inpatient service between 2000 and 2010.1 IED results from cutaneous sensitization to purulent drainage secondary to acute otitis externa or another primary infection.2 In fact, cultures from the purulent drainage in this patient grew methicillin-resistant Staphylococcus aureus. The patient’s right otitis externa drainage may have been associated with the previous history of atopic dermatitis. Atopic dermatitis is associated with an increased risk of skin infections due to decreased inflammatory mediators (defensins).
Cellulitis and herpes zoster oticus are part of the differential
The differential diagnosis in this case includes bacterial cellulitis, acute otitis media, and herpes zoster oticus.
Bacterial cellulitis manifests with erythema, edema, and tenderness with blistering when associated with bullous impetigo rather than pruritus. The clinical appearance of the patient’s diffuse, weeping, edematous external ear, the lack of response to guided antibiotic therapy, and the pruritus experienced by the patient argue against the diagnosis of bacterial cellulitis.
Acute otitis media, like otitis externa, produces ear discharge usually associated with significant pain. Thus, it is important when working through the differential to define the source of the ear discharge. In this case, a consultation with an ENT specialist confirmed that there was an intact tympanic membrane with no middle ear involvement, ruling out the diagnosis of acute otitis media.
Continue to: Herpes zoster oticus
Herpes zoster oticus. The absence of grouped vesicles at any point during the eruption, itching rather than pain, and negative viral culture and polymerase chain reaction studies for herpes simplex and varicella zoster virus excluded the diagnosis of herpes zoster oticus.
Diagnostic criteria were met
This case was compatible with the characterizations of IED as initially described by Engman3 in 1902 and further detailed by Sutton,4 who provided the following criteria for diagnosis:
- an initial eczematous or pustular lesion
- extension peripherally by autoinoculation
- an absence of central clearing
- Staphylococcus on culture of the initial lesion
- a history of infection.
Case reports have added to our understanding of the mechanism of autosensitization of surrounding skin.5 Yamany and Schwartz have proposed the diagnostic criteria summarized in the TABLE.2

Age factors into location. The ears, nose, and face are predominantly involved in cases of IED in the pediatric population, while the lower extremities are predominantly involved in adults.6 Laboratory tests and imaging may aid in excluding other potential diagnoses or complications, but the diagnosis remains clinical and requires the clinician to avoid jumping to the conclusion that every moist, erythematous crusting eruption is purely infectious in nature.
Tx and prevention hinge on a combination of antibiotics, steroids
The management of IED should be aimed at fighting the infection, eliminating the allergic contact dermatitis associated with infectious products, and improving barrier protection. Topical and/or systemic antibiotics guided by culture focus on killing bacteria. The allergic immune response is dampened by systemic steroids. Topical steroids, however, are difficult to utilize on moist, draining skin. In the case of otitis externa, a combination topical antibiotic and steroid otic drop can be utilized. As healing begins, emollients are applied to aid in skin repair.2 Topical antibiotics containing neomycin or polymyxin should be avoided to eliminate the possibility of developing contact sensitivity to these agents.
For our patient, inpatient wound cultures demonstrated methicillin-resistant S aureus, and empiric treatment with IV cefepime and vancomycin was transitioned to IV clindamycin based on sensitivities and then transitioned to a 12-day course of oral clindamycin 150 mg bid. In addition, the patient received ciprofloxacin/dexamethasone otic drops 3 times/d to treat his otitis externa. After initiating prednisone 30 mg (1 mg/kg/d) for 10 days to cover the allergic component, the patient showed prompt clinical improvement. Gentle cleansing of the right ear with hypoallergenic soap and water followed by application of petrolatum ointment 4 times/d was used to promote healing and improve barrier function (FIGURE 2). The patient’s mother indicated during a follow-up call that the affected area had dramatically improved.

1. Storan ER, McEvoy MT, Wetter DA, et al. Experience with the dermatology inpatient hospital service for adults: Mayo Clinic, 2000–2010. J Eur Acad Dermatol Venereol. 2013;27:1360-1365. doi: 10.1111/jdv.12010
2. Yamany T, Schwartz RA. Infectious eczematoid dermatitis: a comprehensive review. J Eur Acad Dermatol Venereol. 2015;29:203-208. doi: 10.1111/jdv.12715
. An infectious form of an eczematoid dermatitis. St. Louis Courier of Med. 1902;27:401414.
4. Infectious eczematoid dermatitis. J Am Med Assoc. 1920;75:976-979.
, , . Autosensitization dermatitis: report of five cases and protocol of an experiment. Arch Derm Syphilol. 1949;59:68-77. doi: 10.1001/archderm.1949.01520260072010
, . Autosensitization in infectious eczematoid dermatitis. AMA Arch Derm Syphilol. 1950;62:703-704. doi: 10.1001/archderm.1950.01530180092021
A 6-year-old boy was seen in the hospital in consultation for a 3-week history of suspected cellulitis of the right ear. Drainage from the right ear was refractory to treatment with a 7-day course of cephalexin 15 mL po bid of 250 mg/5 mL solution and clindamycin 24.4 mL po tid of 75 mg/5 mL solution. Treatment was followed by admission to the hospital for treatment with intravenous (IV) cefazolin 1000 mg q6h and IV vancomycin 825 mg q6h for 1 week.
The patient had a significant past medical history for asthma, allergic rhinitis, and severe atopic dermatitis that had been treated with methotrexate 10 mg per week for 6 months beginning when the child was 5 years of age. When the methotrexate proved to be ineffective, the patient was started on Aquaphor and mometasone 0.1% ointment. A 6-month trial of these agents failed as well.
Physical examination revealed that the right ear and skin around it were edematous, erythematous, pruritic, and tender. There was also purulent drainage coming from the ear (FIGURE 1).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Infectious eczematoid dermatitis
The patient was referred to a dermatologist after seeing an ear, nose, and throat (ENT) specialist who made the diagnosis of otitis externa when the rash failed to respond to topical and systemic antibiotics. The patient’s tender, pruritic, oozing, edematous eruption was recognized as an infectious eczematoid dermatitis (IED).
Although it is not an uncommon condition, IED may be underrecognized. It accounted for 2.9% of admissions to a dermatology-run inpatient service between 2000 and 2010.1 IED results from cutaneous sensitization to purulent drainage secondary to acute otitis externa or another primary infection.2 In fact, cultures from the purulent drainage in this patient grew methicillin-resistant Staphylococcus aureus. The patient’s right otitis externa drainage may have been associated with the previous history of atopic dermatitis. Atopic dermatitis is associated with an increased risk of skin infections due to decreased inflammatory mediators (defensins).
Cellulitis and herpes zoster oticus are part of the differential
The differential diagnosis in this case includes bacterial cellulitis, acute otitis media, and herpes zoster oticus.
Bacterial cellulitis manifests with erythema, edema, and tenderness with blistering when associated with bullous impetigo rather than pruritus. The clinical appearance of the patient’s diffuse, weeping, edematous external ear, the lack of response to guided antibiotic therapy, and the pruritus experienced by the patient argue against the diagnosis of bacterial cellulitis.
Acute otitis media, like otitis externa, produces ear discharge usually associated with significant pain. Thus, it is important when working through the differential to define the source of the ear discharge. In this case, a consultation with an ENT specialist confirmed that there was an intact tympanic membrane with no middle ear involvement, ruling out the diagnosis of acute otitis media.
Continue to: Herpes zoster oticus
Herpes zoster oticus. The absence of grouped vesicles at any point during the eruption, itching rather than pain, and negative viral culture and polymerase chain reaction studies for herpes simplex and varicella zoster virus excluded the diagnosis of herpes zoster oticus.
Diagnostic criteria were met
This case was compatible with the characterizations of IED as initially described by Engman3 in 1902 and further detailed by Sutton,4 who provided the following criteria for diagnosis:
- an initial eczematous or pustular lesion
- extension peripherally by autoinoculation
- an absence of central clearing
- Staphylococcus on culture of the initial lesion
- a history of infection.
Case reports have added to our understanding of the mechanism of autosensitization of surrounding skin.5 Yamany and Schwartz have proposed the diagnostic criteria summarized in the TABLE.2
Age factors into location. The ears, nose, and face are predominantly involved in cases of IED in the pediatric population, while the lower extremities are predominantly involved in adults.6 Laboratory tests and imaging may aid in excluding other potential diagnoses or complications, but the diagnosis remains clinical and requires the clinician to avoid jumping to the conclusion that every moist, erythematous crusting eruption is purely infectious in nature.
Tx and prevention hinge on a combination of antibiotics, steroids
The management of IED should be aimed at fighting the infection, eliminating the allergic contact dermatitis associated with infectious products, and improving barrier protection. Topical and/or systemic antibiotics guided by culture focus on killing bacteria. The allergic immune response is dampened by systemic steroids. Topical steroids, however, are difficult to utilize on moist, draining skin. In the case of otitis externa, a combination topical antibiotic and steroid otic drop can be utilized. As healing begins, emollients are applied to aid in skin repair.2 Topical antibiotics containing neomycin or polymyxin should be avoided to eliminate the possibility of developing contact sensitivity to these agents.
For our patient, inpatient wound cultures demonstrated methicillin-resistant S aureus, and empiric treatment with IV cefepime and vancomycin was transitioned to IV clindamycin based on sensitivities and then transitioned to a 12-day course of oral clindamycin 150 mg bid. In addition, the patient received ciprofloxacin/dexamethasone otic drops 3 times/d to treat his otitis externa. After initiating prednisone 30 mg (1 mg/kg/d) for 10 days to cover the allergic component, the patient showed prompt clinical improvement. Gentle cleansing of the right ear with hypoallergenic soap and water followed by application of petrolatum ointment 4 times/d was used to promote healing and improve barrier function (FIGURE 2). The patient’s mother indicated during a follow-up call that the affected area had dramatically improved.
A 6-year-old boy was seen in the hospital in consultation for a 3-week history of suspected cellulitis of the right ear. Drainage from the right ear was refractory to treatment with a 7-day course of cephalexin 15 mL po bid of 250 mg/5 mL solution and clindamycin 24.4 mL po tid of 75 mg/5 mL solution. Treatment was followed by admission to the hospital for treatment with intravenous (IV) cefazolin 1000 mg q6h and IV vancomycin 825 mg q6h for 1 week.
The patient had a significant past medical history for asthma, allergic rhinitis, and severe atopic dermatitis that had been treated with methotrexate 10 mg per week for 6 months beginning when the child was 5 years of age. When the methotrexate proved to be ineffective, the patient was started on Aquaphor and mometasone 0.1% ointment. A 6-month trial of these agents failed as well.
Physical examination revealed that the right ear and skin around it were edematous, erythematous, pruritic, and tender. There was also purulent drainage coming from the ear (FIGURE 1).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Infectious eczematoid dermatitis
The patient was referred to a dermatologist after seeing an ear, nose, and throat (ENT) specialist who made the diagnosis of otitis externa when the rash failed to respond to topical and systemic antibiotics. The patient’s tender, pruritic, oozing, edematous eruption was recognized as an infectious eczematoid dermatitis (IED).
Although it is not an uncommon condition, IED may be underrecognized. It accounted for 2.9% of admissions to a dermatology-run inpatient service between 2000 and 2010.1 IED results from cutaneous sensitization to purulent drainage secondary to acute otitis externa or another primary infection.2 In fact, cultures from the purulent drainage in this patient grew methicillin-resistant Staphylococcus aureus. The patient’s right otitis externa drainage may have been associated with the previous history of atopic dermatitis. Atopic dermatitis is associated with an increased risk of skin infections due to decreased inflammatory mediators (defensins).
Cellulitis and herpes zoster oticus are part of the differential
The differential diagnosis in this case includes bacterial cellulitis, acute otitis media, and herpes zoster oticus.
Bacterial cellulitis manifests with erythema, edema, and tenderness with blistering when associated with bullous impetigo rather than pruritus. The clinical appearance of the patient’s diffuse, weeping, edematous external ear, the lack of response to guided antibiotic therapy, and the pruritus experienced by the patient argue against the diagnosis of bacterial cellulitis.
Acute otitis media, like otitis externa, produces ear discharge usually associated with significant pain. Thus, it is important when working through the differential to define the source of the ear discharge. In this case, a consultation with an ENT specialist confirmed that there was an intact tympanic membrane with no middle ear involvement, ruling out the diagnosis of acute otitis media.
Continue to: Herpes zoster oticus
Herpes zoster oticus. The absence of grouped vesicles at any point during the eruption, itching rather than pain, and negative viral culture and polymerase chain reaction studies for herpes simplex and varicella zoster virus excluded the diagnosis of herpes zoster oticus.
Diagnostic criteria were met
This case was compatible with the characterizations of IED as initially described by Engman3 in 1902 and further detailed by Sutton,4 who provided the following criteria for diagnosis:
- an initial eczematous or pustular lesion
- extension peripherally by autoinoculation
- an absence of central clearing
- Staphylococcus on culture of the initial lesion
- a history of infection.
Case reports have added to our understanding of the mechanism of autosensitization of surrounding skin.5 Yamany and Schwartz have proposed the diagnostic criteria summarized in the TABLE.2
Age factors into location. The ears, nose, and face are predominantly involved in cases of IED in the pediatric population, while the lower extremities are predominantly involved in adults.6 Laboratory tests and imaging may aid in excluding other potential diagnoses or complications, but the diagnosis remains clinical and requires the clinician to avoid jumping to the conclusion that every moist, erythematous crusting eruption is purely infectious in nature.
Tx and prevention hinge on a combination of antibiotics, steroids
The management of IED should be aimed at fighting the infection, eliminating the allergic contact dermatitis associated with infectious products, and improving barrier protection. Topical and/or systemic antibiotics guided by culture focus on killing bacteria. The allergic immune response is dampened by systemic steroids. Topical steroids, however, are difficult to utilize on moist, draining skin. In the case of otitis externa, a combination topical antibiotic and steroid otic drop can be utilized. As healing begins, emollients are applied to aid in skin repair.2 Topical antibiotics containing neomycin or polymyxin should be avoided to eliminate the possibility of developing contact sensitivity to these agents.
For our patient, inpatient wound cultures demonstrated methicillin-resistant S aureus, and empiric treatment with IV cefepime and vancomycin was transitioned to IV clindamycin based on sensitivities and then transitioned to a 12-day course of oral clindamycin 150 mg bid. In addition, the patient received ciprofloxacin/dexamethasone otic drops 3 times/d to treat his otitis externa. After initiating prednisone 30 mg (1 mg/kg/d) for 10 days to cover the allergic component, the patient showed prompt clinical improvement. Gentle cleansing of the right ear with hypoallergenic soap and water followed by application of petrolatum ointment 4 times/d was used to promote healing and improve barrier function (FIGURE 2). The patient’s mother indicated during a follow-up call that the affected area had dramatically improved.
1. Storan ER, McEvoy MT, Wetter DA, et al. Experience with the dermatology inpatient hospital service for adults: Mayo Clinic, 2000–2010. J Eur Acad Dermatol Venereol. 2013;27:1360-1365. doi: 10.1111/jdv.12010
2. Yamany T, Schwartz RA. Infectious eczematoid dermatitis: a comprehensive review. J Eur Acad Dermatol Venereol. 2015;29:203-208. doi: 10.1111/jdv.12715
. An infectious form of an eczematoid dermatitis. St. Louis Courier of Med. 1902;27:401414.
4. Infectious eczematoid dermatitis. J Am Med Assoc. 1920;75:976-979.
, , . Autosensitization dermatitis: report of five cases and protocol of an experiment. Arch Derm Syphilol. 1949;59:68-77. doi: 10.1001/archderm.1949.01520260072010
, . Autosensitization in infectious eczematoid dermatitis. AMA Arch Derm Syphilol. 1950;62:703-704. doi: 10.1001/archderm.1950.01530180092021
1. Storan ER, McEvoy MT, Wetter DA, et al. Experience with the dermatology inpatient hospital service for adults: Mayo Clinic, 2000–2010. J Eur Acad Dermatol Venereol. 2013;27:1360-1365. doi: 10.1111/jdv.12010
2. Yamany T, Schwartz RA. Infectious eczematoid dermatitis: a comprehensive review. J Eur Acad Dermatol Venereol. 2015;29:203-208. doi: 10.1111/jdv.12715
. An infectious form of an eczematoid dermatitis. St. Louis Courier of Med. 1902;27:401414.
4. Infectious eczematoid dermatitis. J Am Med Assoc. 1920;75:976-979.
, , . Autosensitization dermatitis: report of five cases and protocol of an experiment. Arch Derm Syphilol. 1949;59:68-77. doi: 10.1001/archderm.1949.01520260072010
, . Autosensitization in infectious eczematoid dermatitis. AMA Arch Derm Syphilol. 1950;62:703-704. doi: 10.1001/archderm.1950.01530180092021

Foot rash and joint pain
A 21-year-old man presented to the emergency department (ED) with a 2-month history of joint pain, swelling, and difficulty walking that began with swelling of his right knee (FIGURE 1A). The patient said that over the course of several weeks, the swelling and joint pain spread to his left knee, followed by bilateral elbows and ankles. Nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin produced only modest improvement.
Two weeks prior to presentation, the patient also experienced widespread pruritus and conjunctivitis. His past medical history was significant for a sexual encounter that resulted in urinary tract infection (UTI)–like symptoms approximately 1 month prior to the onset of his joint symptoms. He did not seek care for the UTI-like symptoms.
In the ED, the patient was febrile (102.1 °F) and tachycardic. Skin examination revealed erythematous papules, intact vesicles, and pustules with background hyperkeratosis and desquamation on his right foot (FIGURE 1B). The patient had spotty erythema on his palate and a 4-mm superficial erosion on the right penile shaft. Swelling and tenderness were noted over the elbows, knees, hands, and ankles. No inguinal lymphadenopathy was noted.

An arthrocentesis was performed on the right knee that demonstrated no organisms on Gram stain and a normal joint fluid cell count. A complete blood count (CBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and urinalysis were ordered. A punch biopsy was performed on a scaly patch on the right elbow.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Keratoderma blenorrhagicum
The patient’s history, clinical findings, and lab results, including a positive Chlamydia trachomatis polymerase chain reaction (PCR) test from a urethral swab, pointed to a diagnosis of keratoderma blenorrhagicum in association with reactive arthritis (following infection with C trachomatis).
Relevant diagnostic findings included an elevated CRP of 26.5 mg/L (normal range, < 10 mg/L), an elevated ESR of 116 mm/h (normal range, < 15 mm/h) and as noted, a positive C trachomatis PCR test. The patient’s white blood cell count was 9.7/μL (normal range, 4.5-11 μL) and the rest of the CBC was within normal limits. Urinalysis was positive for leukocytes and rare bacteria. A treponemal antibody test was negative.
Additionally, the punch biopsy from the right elbow revealed acanthosis, intercellular spongiosis, and subcorneal pustules consistent with localized pustular psoriasis or keratoderma blenorrhagicum. After the diagnosis was made, human leukocyte antigen B27 allele (HLA-B27) testing was conducted and was positive.
A predisposition exacerbates the infection
Reactive arthritis, a type of spondyloarthropathy, features a triad of conjunctivitis, urethritis, and arthritis that follows either gastrointestinal or urogenital infection.1 Reactive arthritis occurs with a male predominance of 3:1, and the worldwide prevalence is 1 in 3000.1 Causative bacteria include C trachomatis, Yersinia, Salmonella, Shigella, and Campylobacter, Escherichia coli, Clostridioides (formerly Clostridium) difficile, and C pneumoniae.2 Patients with the HLA-B27 allele are 50 times more likely to develop reactive arthritis following infection with the aforementioned bacteria.1
Findings consistent with a diagnosis of reactive arthritis include a recent history of gastrointestinal or urogenital illness, joint pain, conjunctivitis, oral lesions, cutaneous changes, and genital lesions.3 Diagnostic tests should include arthrocentesis with cultures or PCR and cell count, ESR, CRP, CBC, and urinalysis. HLA-B27 can be used to support the diagnosis but is not routinely recommended.2
Pustules and psoriasiform scaling characterize this diagnosis
The differential diagnosis for the signs and symptoms seen in this patient include disseminated gonococcal arthritis, psoriatic arthritis, rheumatoid arthritis, and secondary syphilis.
Gonococcal arthritis manifests with painful, sterile joints as well as pustules on the palms and soles, but not with the psoriasiform scaling and desquamation that was seen in this case. A culture or PCR from urethral discharge or pustules on the palms and soles could be used to confirm this diagnosis.3
Continue to: Psoriasis in association with psoriatic arthritis
Psoriasis in association with psoriatic arthritis and the psoriasiform rashing of reactive arthritis (keratoderma blenorrhagicum) show similar histopathology; however, patients with psoriatic arthritis generally exhibit fewer constitutional symptoms.4
Rheumatoid arthritis also manifests with joint pain and swelling, especially in the hands, wrists, and knees. This diagnosis was unlikely in this patient, where small joints were largely uninvolved.4
Secondary syphilis also manifests with papular, scaly, erythematous lesions on the palms and soles along with pityriasis rosea–like rashing on the trunk. However, it rarely produces pustules or hyperkeratotic keratoderma.5 As noted earlier, a treponemal antibody test in this patient was negative.
Drug therapy is the best option
First-line therapy for reactive arthritis consists of NSAIDs. If the patient exhibits an inadequate response after a 2-week trial, intra-articular or systemic glucocorticoids may be considered.3 If the patient fails to respond to the steroids, disease-modifying antirheumatic drugs (DMARDs) may be considered. Reactive arthritis is considered chronic if the disease lasts longer than 6 months, at which point, DMARDs or tumor necrosis factor-α inhibitors may be utilized.3 For cutaneous manifestations, such as keratoderma blenorrhagicum, topical glucocorticoids twice daily may be used along with keratolytic agents.
Our patient received 2 doses of azithromycin (500 mg IV) and 1 dose of ceftriaxone (2 g IV) to treat his infection while in the ED. Over the course of his hospital stay, he received ceftriaxone (1 g IV daily) for 6 days and naproxen (500 mg tid po) which was tapered. Additionally, he received a week of methylprednisolone (60 mg IM daily) before tapering to oral prednisone. His taper consisted of 40 mg po for 1 week and was decreased by 10 mg each week. Augmented betamethasone dipropionate 0.05% cream and urea 20% cream were prescribed for twice-daily application for the hyperkeratotic scale on both of his feet.
1. Hayes KM, Hayes RJP, Turk MA, et al. Evolving patterns of reactive arthritis. Clin Rheumatol. 2019;38:2083-2088. doi: 10.1007/s10067-019-04522-4
2. Duba AS, Mathew SD. The seronegative spondyloarthropathies. Prim Care. 2018;45:271-287. doi: 10.1016/j.pop.2018.02.005
3. Yu DT, van Tubergen A. Reactive arthritis. In: Joachim S, Romain PL, eds. UpToDate. Updated April 28, 2021. Accessed June 3, 2021. https://www.uptodate.com/contents/reactive-arthritis?search=reactive%20arthritis&topicRef=5571&source=see_link#H9
4. Barth WF, Segal K. Reactive arthritis (Reiter’s Syndrome). Am Fam Physician. 1999;60:499-503, 507.
5. Coleman E, Fiahlo A, Brateanu A. Secondary syphilis. Cleve Clin J Med. 2017;84:510-511. doi: 10.3949/ccjm.84a.16089
A 21-year-old man presented to the emergency department (ED) with a 2-month history of joint pain, swelling, and difficulty walking that began with swelling of his right knee (FIGURE 1A). The patient said that over the course of several weeks, the swelling and joint pain spread to his left knee, followed by bilateral elbows and ankles. Nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin produced only modest improvement.
Two weeks prior to presentation, the patient also experienced widespread pruritus and conjunctivitis. His past medical history was significant for a sexual encounter that resulted in urinary tract infection (UTI)–like symptoms approximately 1 month prior to the onset of his joint symptoms. He did not seek care for the UTI-like symptoms.
In the ED, the patient was febrile (102.1 °F) and tachycardic. Skin examination revealed erythematous papules, intact vesicles, and pustules with background hyperkeratosis and desquamation on his right foot (FIGURE 1B). The patient had spotty erythema on his palate and a 4-mm superficial erosion on the right penile shaft. Swelling and tenderness were noted over the elbows, knees, hands, and ankles. No inguinal lymphadenopathy was noted.
An arthrocentesis was performed on the right knee that demonstrated no organisms on Gram stain and a normal joint fluid cell count. A complete blood count (CBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and urinalysis were ordered. A punch biopsy was performed on a scaly patch on the right elbow.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Keratoderma blenorrhagicum
The patient’s history, clinical findings, and lab results, including a positive Chlamydia trachomatis polymerase chain reaction (PCR) test from a urethral swab, pointed to a diagnosis of keratoderma blenorrhagicum in association with reactive arthritis (following infection with C trachomatis).
Relevant diagnostic findings included an elevated CRP of 26.5 mg/L (normal range, < 10 mg/L), an elevated ESR of 116 mm/h (normal range, < 15 mm/h) and as noted, a positive C trachomatis PCR test. The patient’s white blood cell count was 9.7/μL (normal range, 4.5-11 μL) and the rest of the CBC was within normal limits. Urinalysis was positive for leukocytes and rare bacteria. A treponemal antibody test was negative.
Additionally, the punch biopsy from the right elbow revealed acanthosis, intercellular spongiosis, and subcorneal pustules consistent with localized pustular psoriasis or keratoderma blenorrhagicum. After the diagnosis was made, human leukocyte antigen B27 allele (HLA-B27) testing was conducted and was positive.
A predisposition exacerbates the infection
Reactive arthritis, a type of spondyloarthropathy, features a triad of conjunctivitis, urethritis, and arthritis that follows either gastrointestinal or urogenital infection.1 Reactive arthritis occurs with a male predominance of 3:1, and the worldwide prevalence is 1 in 3000.1 Causative bacteria include C trachomatis, Yersinia, Salmonella, Shigella, and Campylobacter, Escherichia coli, Clostridioides (formerly Clostridium) difficile, and C pneumoniae.2 Patients with the HLA-B27 allele are 50 times more likely to develop reactive arthritis following infection with the aforementioned bacteria.1
Findings consistent with a diagnosis of reactive arthritis include a recent history of gastrointestinal or urogenital illness, joint pain, conjunctivitis, oral lesions, cutaneous changes, and genital lesions.3 Diagnostic tests should include arthrocentesis with cultures or PCR and cell count, ESR, CRP, CBC, and urinalysis. HLA-B27 can be used to support the diagnosis but is not routinely recommended.2
Pustules and psoriasiform scaling characterize this diagnosis
The differential diagnosis for the signs and symptoms seen in this patient include disseminated gonococcal arthritis, psoriatic arthritis, rheumatoid arthritis, and secondary syphilis.
Gonococcal arthritis manifests with painful, sterile joints as well as pustules on the palms and soles, but not with the psoriasiform scaling and desquamation that was seen in this case. A culture or PCR from urethral discharge or pustules on the palms and soles could be used to confirm this diagnosis.3
Continue to: Psoriasis in association with psoriatic arthritis
Psoriasis in association with psoriatic arthritis and the psoriasiform rashing of reactive arthritis (keratoderma blenorrhagicum) show similar histopathology; however, patients with psoriatic arthritis generally exhibit fewer constitutional symptoms.4
Rheumatoid arthritis also manifests with joint pain and swelling, especially in the hands, wrists, and knees. This diagnosis was unlikely in this patient, where small joints were largely uninvolved.4
Secondary syphilis also manifests with papular, scaly, erythematous lesions on the palms and soles along with pityriasis rosea–like rashing on the trunk. However, it rarely produces pustules or hyperkeratotic keratoderma.5 As noted earlier, a treponemal antibody test in this patient was negative.
Drug therapy is the best option
First-line therapy for reactive arthritis consists of NSAIDs. If the patient exhibits an inadequate response after a 2-week trial, intra-articular or systemic glucocorticoids may be considered.3 If the patient fails to respond to the steroids, disease-modifying antirheumatic drugs (DMARDs) may be considered. Reactive arthritis is considered chronic if the disease lasts longer than 6 months, at which point, DMARDs or tumor necrosis factor-α inhibitors may be utilized.3 For cutaneous manifestations, such as keratoderma blenorrhagicum, topical glucocorticoids twice daily may be used along with keratolytic agents.
Our patient received 2 doses of azithromycin (500 mg IV) and 1 dose of ceftriaxone (2 g IV) to treat his infection while in the ED. Over the course of his hospital stay, he received ceftriaxone (1 g IV daily) for 6 days and naproxen (500 mg tid po) which was tapered. Additionally, he received a week of methylprednisolone (60 mg IM daily) before tapering to oral prednisone. His taper consisted of 40 mg po for 1 week and was decreased by 10 mg each week. Augmented betamethasone dipropionate 0.05% cream and urea 20% cream were prescribed for twice-daily application for the hyperkeratotic scale on both of his feet.
A 21-year-old man presented to the emergency department (ED) with a 2-month history of joint pain, swelling, and difficulty walking that began with swelling of his right knee (FIGURE 1A). The patient said that over the course of several weeks, the swelling and joint pain spread to his left knee, followed by bilateral elbows and ankles. Nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin produced only modest improvement.
Two weeks prior to presentation, the patient also experienced widespread pruritus and conjunctivitis. His past medical history was significant for a sexual encounter that resulted in urinary tract infection (UTI)–like symptoms approximately 1 month prior to the onset of his joint symptoms. He did not seek care for the UTI-like symptoms.
In the ED, the patient was febrile (102.1 °F) and tachycardic. Skin examination revealed erythematous papules, intact vesicles, and pustules with background hyperkeratosis and desquamation on his right foot (FIGURE 1B). The patient had spotty erythema on his palate and a 4-mm superficial erosion on the right penile shaft. Swelling and tenderness were noted over the elbows, knees, hands, and ankles. No inguinal lymphadenopathy was noted.
An arthrocentesis was performed on the right knee that demonstrated no organisms on Gram stain and a normal joint fluid cell count. A complete blood count (CBC), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and urinalysis were ordered. A punch biopsy was performed on a scaly patch on the right elbow.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Keratoderma blenorrhagicum
The patient’s history, clinical findings, and lab results, including a positive Chlamydia trachomatis polymerase chain reaction (PCR) test from a urethral swab, pointed to a diagnosis of keratoderma blenorrhagicum in association with reactive arthritis (following infection with C trachomatis).
Relevant diagnostic findings included an elevated CRP of 26.5 mg/L (normal range, < 10 mg/L), an elevated ESR of 116 mm/h (normal range, < 15 mm/h) and as noted, a positive C trachomatis PCR test. The patient’s white blood cell count was 9.7/μL (normal range, 4.5-11 μL) and the rest of the CBC was within normal limits. Urinalysis was positive for leukocytes and rare bacteria. A treponemal antibody test was negative.
Additionally, the punch biopsy from the right elbow revealed acanthosis, intercellular spongiosis, and subcorneal pustules consistent with localized pustular psoriasis or keratoderma blenorrhagicum. After the diagnosis was made, human leukocyte antigen B27 allele (HLA-B27) testing was conducted and was positive.
A predisposition exacerbates the infection
Reactive arthritis, a type of spondyloarthropathy, features a triad of conjunctivitis, urethritis, and arthritis that follows either gastrointestinal or urogenital infection.1 Reactive arthritis occurs with a male predominance of 3:1, and the worldwide prevalence is 1 in 3000.1 Causative bacteria include C trachomatis, Yersinia, Salmonella, Shigella, and Campylobacter, Escherichia coli, Clostridioides (formerly Clostridium) difficile, and C pneumoniae.2 Patients with the HLA-B27 allele are 50 times more likely to develop reactive arthritis following infection with the aforementioned bacteria.1
Findings consistent with a diagnosis of reactive arthritis include a recent history of gastrointestinal or urogenital illness, joint pain, conjunctivitis, oral lesions, cutaneous changes, and genital lesions.3 Diagnostic tests should include arthrocentesis with cultures or PCR and cell count, ESR, CRP, CBC, and urinalysis. HLA-B27 can be used to support the diagnosis but is not routinely recommended.2
Pustules and psoriasiform scaling characterize this diagnosis
The differential diagnosis for the signs and symptoms seen in this patient include disseminated gonococcal arthritis, psoriatic arthritis, rheumatoid arthritis, and secondary syphilis.
Gonococcal arthritis manifests with painful, sterile joints as well as pustules on the palms and soles, but not with the psoriasiform scaling and desquamation that was seen in this case. A culture or PCR from urethral discharge or pustules on the palms and soles could be used to confirm this diagnosis.3
Continue to: Psoriasis in association with psoriatic arthritis
Psoriasis in association with psoriatic arthritis and the psoriasiform rashing of reactive arthritis (keratoderma blenorrhagicum) show similar histopathology; however, patients with psoriatic arthritis generally exhibit fewer constitutional symptoms.4
Rheumatoid arthritis also manifests with joint pain and swelling, especially in the hands, wrists, and knees. This diagnosis was unlikely in this patient, where small joints were largely uninvolved.4
Secondary syphilis also manifests with papular, scaly, erythematous lesions on the palms and soles along with pityriasis rosea–like rashing on the trunk. However, it rarely produces pustules or hyperkeratotic keratoderma.5 As noted earlier, a treponemal antibody test in this patient was negative.
Drug therapy is the best option
First-line therapy for reactive arthritis consists of NSAIDs. If the patient exhibits an inadequate response after a 2-week trial, intra-articular or systemic glucocorticoids may be considered.3 If the patient fails to respond to the steroids, disease-modifying antirheumatic drugs (DMARDs) may be considered. Reactive arthritis is considered chronic if the disease lasts longer than 6 months, at which point, DMARDs or tumor necrosis factor-α inhibitors may be utilized.3 For cutaneous manifestations, such as keratoderma blenorrhagicum, topical glucocorticoids twice daily may be used along with keratolytic agents.
Our patient received 2 doses of azithromycin (500 mg IV) and 1 dose of ceftriaxone (2 g IV) to treat his infection while in the ED. Over the course of his hospital stay, he received ceftriaxone (1 g IV daily) for 6 days and naproxen (500 mg tid po) which was tapered. Additionally, he received a week of methylprednisolone (60 mg IM daily) before tapering to oral prednisone. His taper consisted of 40 mg po for 1 week and was decreased by 10 mg each week. Augmented betamethasone dipropionate 0.05% cream and urea 20% cream were prescribed for twice-daily application for the hyperkeratotic scale on both of his feet.
1. Hayes KM, Hayes RJP, Turk MA, et al. Evolving patterns of reactive arthritis. Clin Rheumatol. 2019;38:2083-2088. doi: 10.1007/s10067-019-04522-4
2. Duba AS, Mathew SD. The seronegative spondyloarthropathies. Prim Care. 2018;45:271-287. doi: 10.1016/j.pop.2018.02.005
3. Yu DT, van Tubergen A. Reactive arthritis. In: Joachim S, Romain PL, eds. UpToDate. Updated April 28, 2021. Accessed June 3, 2021. https://www.uptodate.com/contents/reactive-arthritis?search=reactive%20arthritis&topicRef=5571&source=see_link#H9
4. Barth WF, Segal K. Reactive arthritis (Reiter’s Syndrome). Am Fam Physician. 1999;60:499-503, 507.
5. Coleman E, Fiahlo A, Brateanu A. Secondary syphilis. Cleve Clin J Med. 2017;84:510-511. doi: 10.3949/ccjm.84a.16089
1. Hayes KM, Hayes RJP, Turk MA, et al. Evolving patterns of reactive arthritis. Clin Rheumatol. 2019;38:2083-2088. doi: 10.1007/s10067-019-04522-4
2. Duba AS, Mathew SD. The seronegative spondyloarthropathies. Prim Care. 2018;45:271-287. doi: 10.1016/j.pop.2018.02.005
3. Yu DT, van Tubergen A. Reactive arthritis. In: Joachim S, Romain PL, eds. UpToDate. Updated April 28, 2021. Accessed June 3, 2021. https://www.uptodate.com/contents/reactive-arthritis?search=reactive%20arthritis&topicRef=5571&source=see_link#H9
4. Barth WF, Segal K. Reactive arthritis (Reiter’s Syndrome). Am Fam Physician. 1999;60:499-503, 507.
5. Coleman E, Fiahlo A, Brateanu A. Secondary syphilis. Cleve Clin J Med. 2017;84:510-511. doi: 10.3949/ccjm.84a.16089

Rural Residency Curricula: Potential Target for Improved Access to Care?
To the Editor:
There is an irrefutable trend toward urban dermatology practice in the United States, leading to growing problems with rural access to care. The provision of rural clinical experiences and telehealth in dermatology residency training might increase the likelihood of trainees establishing a rural practice.
In 2017, the American Academy of Dermatology released an updated statement supporting direct patient access to board-certified dermatologists in an effort to reduce morbidity and mortality associated with skin disease.1 Twenty percent of the US population lives in a rural and medically underserved location, yet these areas remain largely underserved, in part because of an irrefutable trend toward urban dermatology practice.2-4 Successful approaches to improving rural access to dermatology care are poorly defined in the literature.
Several variables have been shown to influence a young physician’s decision to establish a clinical practice in geographically isolated areas, including rural upbringing, longitudinal rural clinical experiences during medical training, and family influences.5 Location of residency training is an additional variable that impacts practice location, though migration following dermatology residency is a complex phenomenon. However, training location does not guarantee retention of dermatology graduates in any particular geographic area.6 Practice incentives and stipends might encourage rural dermatology practice, yet these programs are underfunded. Last, telemedicine in dermatology (including teledermatology and teledermoscopy), though not always an ideal substitute for a live visit, can improve access to care in geographically isolated or underserved areas in general.7-9
Focused recruitment of medical students interested in rural dermatology practice to accredited dermatology residency programs aligned with this goal represents another approach to improve geographic diversity in the field of dermatology. Online access to this information would be useful for both applicants and their mentors.
We assessed viewable online curricula related to rural dermatology and telemedicine experiences at all Accreditation Council for Graduate Medical Education (ACGME)–accredited residency programs. Telemedicine experiences at Veterans Health Administration (VHA) health systems also were assessed.
Methods
This study was exempt from review by the institutional review board at the University of Minnesota (Minneapolis, Minnesota)(IRB #STUDY00004915) because no human subjects were involved. Online curricula of all ACGME-accredited dermatology residency programs in the United States and Puerto Rico were reviewed from November to December 2018. The following information was recorded: specialized “rural-track” training; optional elective time in rural settings; teledermatology training; and teledermoscopy training.
Additionally, population density at each program’s primary location was determined using US Census Bureau data and with consideration to communities contained within particular Metropolitan Statistical Areas (MSAs)(eTable). Data were obtained from the VHA system to assess teledermatology services at VHA locations affiliated with residency programs.
Results
Of 154 dermatology residency programs identified in the United States and Puerto Rico, 142 were accredited at the time of data collection. Fifteen (10%) were based in communities of 50,000 individuals or fewer that were not near a large metropolitan area. One program (<1%) offered a specific rural track. Fifty-six programs (39%) cited optional rotations or clinical electives, or both, that could be utilized for a rural experience. Eighteen (12%) offered teledermatology experiences and 1 (<1%) offered teledermoscopy during training. Fifty-three programs (37%) offered a rotation at a VHA hospital that had an active teledermatology service.
Comment
Program websites are a free and easily accessible means of acquiring relevant information. The paucity of readily available data on rural dermatology and teledermatology opportunities is unfortunate and a detriment to dermatology residency applicants interested in rural practice, which may result in a missed opportunity to foster a true passion for rural medicine. A brief comment on a website can be impactful, leading to a postgraduate year 4 dermatology elective rotation at a prospective fellowship training site or a rural dermatology experience.
The paucity of dermatologists working directly in rural areas has led to development of teledermatology initiatives to reach deeply into underserved regions. One of the largest providers of teledermatology is the VHA, which standardized its teledermatology efforts in 2012 and provides remarkable educational opportunities for dermatology residents. However, many residency program and VHA websites provide no information about the participation of dermatology residents in the provision of teledermatology services.
A limitation of this study is that it is based on online published curricula. Dermatology residency programs with excellent rural curricula that are not published online might exist.
Residency program directors with an interest in geographic diversity are encouraged to provide rural and teledermatology opportunities and to update these offerings on their websites, which is a simple modifiable strategy that can impact the rural dermatology care gap by recruiting students interested in filling this role. These efforts should be studied to determine whether this strategy impacts resident selection as well as whether focused rural and telemedicine exposure during training increases the likelihood of establishing a rural dermatology practice in the future.
- American Academy of Dermatology. Position statement on access to specialty care and direct access to dermatologic care. Revised May 20, 2017. Accessed December 13, 2020. https://server.aad.org/forms/Policies/Uploads/PS/PS-Access%20to%20Specialty%20Care%20and%20Direct%20Access%20to%20Dermatologic%20Care.pdf
- Dill MJ, Salsberg ES. The Complexities of Physician Supply and Demand: Projections Through 2025. Center for Workforce Studies, Association of American Medical Colleges (AAMC); November 2008. Accessed December 13, 2020. http://innovationlabs.com/pa_future/1/background_docs/AAMC%20Complexities%20of%20physician%20demand,%202008.pdf
- Glazer AM, Rigel DS. Analysis of trends in geographic distribution of US dermatology workforce density. JAMA Dermatol. 2017;153:472-473.
- Yoo JY, Rigel DS. Trends in dermatology: geographic density of US dermatologists. Arch Dermatol. 2010;146:779.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Landow SM, Oh DH, Weinstock MA. Teledermatology within the Veterans Health Administration, 2002-2014. Telemed J E Health. 2015;21:769-773.
- Armstrong AW, Kwong MW, Ledo L, et al. Practice models and challenges in teledermatology: a study of collective experiences from teledermatologists. PloS One. 2011;6:e28687.
- Lewis H, Becevic M, Myers D, et al. Dermatology ECHO—an innovative solution to address limited access to dermatology expertise. Rural Remote Health. 2018;18:4415.
- Edison KE, Dyer JA, Whited JD, et al. Practice gaps. the barriers and the promise of teledermatology. JAMA Dermatol. 2012:148:650-651.
To the Editor:
There is an irrefutable trend toward urban dermatology practice in the United States, leading to growing problems with rural access to care. The provision of rural clinical experiences and telehealth in dermatology residency training might increase the likelihood of trainees establishing a rural practice.
In 2017, the American Academy of Dermatology released an updated statement supporting direct patient access to board-certified dermatologists in an effort to reduce morbidity and mortality associated with skin disease.1 Twenty percent of the US population lives in a rural and medically underserved location, yet these areas remain largely underserved, in part because of an irrefutable trend toward urban dermatology practice.2-4 Successful approaches to improving rural access to dermatology care are poorly defined in the literature.
Several variables have been shown to influence a young physician’s decision to establish a clinical practice in geographically isolated areas, including rural upbringing, longitudinal rural clinical experiences during medical training, and family influences.5 Location of residency training is an additional variable that impacts practice location, though migration following dermatology residency is a complex phenomenon. However, training location does not guarantee retention of dermatology graduates in any particular geographic area.6 Practice incentives and stipends might encourage rural dermatology practice, yet these programs are underfunded. Last, telemedicine in dermatology (including teledermatology and teledermoscopy), though not always an ideal substitute for a live visit, can improve access to care in geographically isolated or underserved areas in general.7-9
Focused recruitment of medical students interested in rural dermatology practice to accredited dermatology residency programs aligned with this goal represents another approach to improve geographic diversity in the field of dermatology. Online access to this information would be useful for both applicants and their mentors.
We assessed viewable online curricula related to rural dermatology and telemedicine experiences at all Accreditation Council for Graduate Medical Education (ACGME)–accredited residency programs. Telemedicine experiences at Veterans Health Administration (VHA) health systems also were assessed.
Methods
This study was exempt from review by the institutional review board at the University of Minnesota (Minneapolis, Minnesota)(IRB #STUDY00004915) because no human subjects were involved. Online curricula of all ACGME-accredited dermatology residency programs in the United States and Puerto Rico were reviewed from November to December 2018. The following information was recorded: specialized “rural-track” training; optional elective time in rural settings; teledermatology training; and teledermoscopy training.
Additionally, population density at each program’s primary location was determined using US Census Bureau data and with consideration to communities contained within particular Metropolitan Statistical Areas (MSAs)(eTable). Data were obtained from the VHA system to assess teledermatology services at VHA locations affiliated with residency programs.
Results
Of 154 dermatology residency programs identified in the United States and Puerto Rico, 142 were accredited at the time of data collection. Fifteen (10%) were based in communities of 50,000 individuals or fewer that were not near a large metropolitan area. One program (<1%) offered a specific rural track. Fifty-six programs (39%) cited optional rotations or clinical electives, or both, that could be utilized for a rural experience. Eighteen (12%) offered teledermatology experiences and 1 (<1%) offered teledermoscopy during training. Fifty-three programs (37%) offered a rotation at a VHA hospital that had an active teledermatology service.
Comment
Program websites are a free and easily accessible means of acquiring relevant information. The paucity of readily available data on rural dermatology and teledermatology opportunities is unfortunate and a detriment to dermatology residency applicants interested in rural practice, which may result in a missed opportunity to foster a true passion for rural medicine. A brief comment on a website can be impactful, leading to a postgraduate year 4 dermatology elective rotation at a prospective fellowship training site or a rural dermatology experience.
The paucity of dermatologists working directly in rural areas has led to development of teledermatology initiatives to reach deeply into underserved regions. One of the largest providers of teledermatology is the VHA, which standardized its teledermatology efforts in 2012 and provides remarkable educational opportunities for dermatology residents. However, many residency program and VHA websites provide no information about the participation of dermatology residents in the provision of teledermatology services.
A limitation of this study is that it is based on online published curricula. Dermatology residency programs with excellent rural curricula that are not published online might exist.
Residency program directors with an interest in geographic diversity are encouraged to provide rural and teledermatology opportunities and to update these offerings on their websites, which is a simple modifiable strategy that can impact the rural dermatology care gap by recruiting students interested in filling this role. These efforts should be studied to determine whether this strategy impacts resident selection as well as whether focused rural and telemedicine exposure during training increases the likelihood of establishing a rural dermatology practice in the future.
To the Editor:
There is an irrefutable trend toward urban dermatology practice in the United States, leading to growing problems with rural access to care. The provision of rural clinical experiences and telehealth in dermatology residency training might increase the likelihood of trainees establishing a rural practice.
In 2017, the American Academy of Dermatology released an updated statement supporting direct patient access to board-certified dermatologists in an effort to reduce morbidity and mortality associated with skin disease.1 Twenty percent of the US population lives in a rural and medically underserved location, yet these areas remain largely underserved, in part because of an irrefutable trend toward urban dermatology practice.2-4 Successful approaches to improving rural access to dermatology care are poorly defined in the literature.
Several variables have been shown to influence a young physician’s decision to establish a clinical practice in geographically isolated areas, including rural upbringing, longitudinal rural clinical experiences during medical training, and family influences.5 Location of residency training is an additional variable that impacts practice location, though migration following dermatology residency is a complex phenomenon. However, training location does not guarantee retention of dermatology graduates in any particular geographic area.6 Practice incentives and stipends might encourage rural dermatology practice, yet these programs are underfunded. Last, telemedicine in dermatology (including teledermatology and teledermoscopy), though not always an ideal substitute for a live visit, can improve access to care in geographically isolated or underserved areas in general.7-9
Focused recruitment of medical students interested in rural dermatology practice to accredited dermatology residency programs aligned with this goal represents another approach to improve geographic diversity in the field of dermatology. Online access to this information would be useful for both applicants and their mentors.
We assessed viewable online curricula related to rural dermatology and telemedicine experiences at all Accreditation Council for Graduate Medical Education (ACGME)–accredited residency programs. Telemedicine experiences at Veterans Health Administration (VHA) health systems also were assessed.
Methods
This study was exempt from review by the institutional review board at the University of Minnesota (Minneapolis, Minnesota)(IRB #STUDY00004915) because no human subjects were involved. Online curricula of all ACGME-accredited dermatology residency programs in the United States and Puerto Rico were reviewed from November to December 2018. The following information was recorded: specialized “rural-track” training; optional elective time in rural settings; teledermatology training; and teledermoscopy training.
Additionally, population density at each program’s primary location was determined using US Census Bureau data and with consideration to communities contained within particular Metropolitan Statistical Areas (MSAs)(eTable). Data were obtained from the VHA system to assess teledermatology services at VHA locations affiliated with residency programs.
Results
Of 154 dermatology residency programs identified in the United States and Puerto Rico, 142 were accredited at the time of data collection. Fifteen (10%) were based in communities of 50,000 individuals or fewer that were not near a large metropolitan area. One program (<1%) offered a specific rural track. Fifty-six programs (39%) cited optional rotations or clinical electives, or both, that could be utilized for a rural experience. Eighteen (12%) offered teledermatology experiences and 1 (<1%) offered teledermoscopy during training. Fifty-three programs (37%) offered a rotation at a VHA hospital that had an active teledermatology service.
Comment
Program websites are a free and easily accessible means of acquiring relevant information. The paucity of readily available data on rural dermatology and teledermatology opportunities is unfortunate and a detriment to dermatology residency applicants interested in rural practice, which may result in a missed opportunity to foster a true passion for rural medicine. A brief comment on a website can be impactful, leading to a postgraduate year 4 dermatology elective rotation at a prospective fellowship training site or a rural dermatology experience.
The paucity of dermatologists working directly in rural areas has led to development of teledermatology initiatives to reach deeply into underserved regions. One of the largest providers of teledermatology is the VHA, which standardized its teledermatology efforts in 2012 and provides remarkable educational opportunities for dermatology residents. However, many residency program and VHA websites provide no information about the participation of dermatology residents in the provision of teledermatology services.
A limitation of this study is that it is based on online published curricula. Dermatology residency programs with excellent rural curricula that are not published online might exist.
Residency program directors with an interest in geographic diversity are encouraged to provide rural and teledermatology opportunities and to update these offerings on their websites, which is a simple modifiable strategy that can impact the rural dermatology care gap by recruiting students interested in filling this role. These efforts should be studied to determine whether this strategy impacts resident selection as well as whether focused rural and telemedicine exposure during training increases the likelihood of establishing a rural dermatology practice in the future.
- American Academy of Dermatology. Position statement on access to specialty care and direct access to dermatologic care. Revised May 20, 2017. Accessed December 13, 2020. https://server.aad.org/forms/Policies/Uploads/PS/PS-Access%20to%20Specialty%20Care%20and%20Direct%20Access%20to%20Dermatologic%20Care.pdf
- Dill MJ, Salsberg ES. The Complexities of Physician Supply and Demand: Projections Through 2025. Center for Workforce Studies, Association of American Medical Colleges (AAMC); November 2008. Accessed December 13, 2020. http://innovationlabs.com/pa_future/1/background_docs/AAMC%20Complexities%20of%20physician%20demand,%202008.pdf
- Glazer AM, Rigel DS. Analysis of trends in geographic distribution of US dermatology workforce density. JAMA Dermatol. 2017;153:472-473.
- Yoo JY, Rigel DS. Trends in dermatology: geographic density of US dermatologists. Arch Dermatol. 2010;146:779.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Landow SM, Oh DH, Weinstock MA. Teledermatology within the Veterans Health Administration, 2002-2014. Telemed J E Health. 2015;21:769-773.
- Armstrong AW, Kwong MW, Ledo L, et al. Practice models and challenges in teledermatology: a study of collective experiences from teledermatologists. PloS One. 2011;6:e28687.
- Lewis H, Becevic M, Myers D, et al. Dermatology ECHO—an innovative solution to address limited access to dermatology expertise. Rural Remote Health. 2018;18:4415.
- Edison KE, Dyer JA, Whited JD, et al. Practice gaps. the barriers and the promise of teledermatology. JAMA Dermatol. 2012:148:650-651.
- American Academy of Dermatology. Position statement on access to specialty care and direct access to dermatologic care. Revised May 20, 2017. Accessed December 13, 2020. https://server.aad.org/forms/Policies/Uploads/PS/PS-Access%20to%20Specialty%20Care%20and%20Direct%20Access%20to%20Dermatologic%20Care.pdf
- Dill MJ, Salsberg ES. The Complexities of Physician Supply and Demand: Projections Through 2025. Center for Workforce Studies, Association of American Medical Colleges (AAMC); November 2008. Accessed December 13, 2020. http://innovationlabs.com/pa_future/1/background_docs/AAMC%20Complexities%20of%20physician%20demand,%202008.pdf
- Glazer AM, Rigel DS. Analysis of trends in geographic distribution of US dermatology workforce density. JAMA Dermatol. 2017;153:472-473.
- Yoo JY, Rigel DS. Trends in dermatology: geographic density of US dermatologists. Arch Dermatol. 2010;146:779.
- Feng H, Berk-Krauss J, Feng PW, et al. Comparison of dermatologist density between urban and rural counties in the United States. JAMA Dermatol. 2018;154:1265-1271.
- Landow SM, Oh DH, Weinstock MA. Teledermatology within the Veterans Health Administration, 2002-2014. Telemed J E Health. 2015;21:769-773.
- Armstrong AW, Kwong MW, Ledo L, et al. Practice models and challenges in teledermatology: a study of collective experiences from teledermatologists. PloS One. 2011;6:e28687.
- Lewis H, Becevic M, Myers D, et al. Dermatology ECHO—an innovative solution to address limited access to dermatology expertise. Rural Remote Health. 2018;18:4415.
- Edison KE, Dyer JA, Whited JD, et al. Practice gaps. the barriers and the promise of teledermatology. JAMA Dermatol. 2012:148:650-651.
Practice Points
- Access to dermatologic care in rural areas is a growing problem.
- Dermatology residency programs can influence medical students and resident dermatologists to provide care in rural and geographically isolated areas.
- Presenting detailed curricula that impact access to care on residency program websites could attract applicants with these career goals.
Painful ulcers on gingiva, tongue, and buccal mucosa
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.

Persistent facial hyperpigmentation
A 59-year-old woman presented to a dermatology clinic with an asymptomatic brown facial hyperpigmentation that had developed several years earlier, and had persisted, despite regular face washing. Physicians who previously treated this patient interpreted this as melasma and advised her to wear sunscreen. The condition was not aggravated by sun exposure. The patient reported that she was otherwise healthy.
Physical examination revealed a brown discoloration with a slightly rough texture. Upon rubbing the affected area with a 70% isopropyl alcohol pad, normal skin was revealed (FIGURE 1A) and brown flakes were apparent on the gauze (FIGURE 1B).

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Terra firma-forme dermatosis
The physician diagnosed terra firma-forme dermatosis (TFFD) in this patient, noting the “dirty brown coloration” and distribution that did not suggest post-inflammatory hyperpigmentation or melasma. TFFD is a rare and benign form of acquired hyperpigmentation characterized by “velvety, pigmented patches or plaques.”1 A simple bedside test, known as an “alcohol wipe test,” both confirms and treats TFFD; it involves rubbing the affected area with a 70% isopropyl alcohol pad.1
TFFD typically affects the face, neck, trunk, or ankles, but the scalp, axilla, back, and pubis also can be affected.1 Histopathology will show negligible amounts of dermal inflammation, hyperkeratosis with mild acanthosis, and hyperkeratosis and papillomatosis.1 Most patients diagnosed with TFFD report that the hyperpigmentation does not improve despite washing with soap and water.2
Hygiene is not a factor
In 2015, Greywal and Cohen followed the case presentations of 10 Caucasian patients with TFFD who presented with “brown and/or black plaques or papules or both.”2 Many of the individuals followed in this case series reported “[practicing] good hygiene and showered a minimum of every other day or daily.”2 The same was reported by the patient in this case. This suggests that TFFD is not a consequence of poor hygiene but perhaps a result of “sticky” sebum that produces a buildup of keratin debris, sebum, and bacteria on the skin.3 This produces the hyperpigmentation seen clinically.
Differential includes post-inflammatory hyperpigmentation
Several other hyperpigmentation disorders were considered on the initial differential diagnosis for this case, including melasma and post-inflammatory hyperpigmentation. However, these 2 conditions are macular, whereas this hyperpigmented condition had a rough, mildly papular texture. Additionally, melasma flares up in the summer with UV exposure, and post-inflammatory hyperpigmentation presents with pruritus and/or a pre-existing rash.4 This patient reported that the condition did not itch nor change with increased sunlight, thus making melasma and post-inflammatory hyperpigmentation unlikely diagnoses.
Acanthosis nigricans also was considered because it presents with a velvety brown pigmentation similar to what was seen with this patient. Acanthosis nigricans, however, primarily affects flexural areas, not the face, making it improbable.
Continue to: Our patient
Our patient. A “wipe test” was performed on the patient. This removed the brown flaky scaling and revealed the underlying normal skin. We instructed the patient to wash daily with a soapy wash cloth and scrub with 70% isopropyl alcohol should the hyperpigmentation recur. The patient did not return.
CORRESPONDENCE
Robert T. Brodell, MD, Department of Dermatology, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; rbrodell@umc.edu
1. Lunge S, Supraja C. Terra firma-forme dermatosis—a dirty dermatosis: report of two cases. Our Dermatol Online. 2016;7:338-340.
2. Greywal T, Cohen PR. Terra firma-forme dermatosis: A report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
3. Alonso-Usero V, Gavrilova M, et al. Dermatosis neglecta or terra firma-forme dermatosis. Actas Dermosifiliogr. 2012;103:932-934.
4. Lucas J, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J. 2006;12:5.
A 59-year-old woman presented to a dermatology clinic with an asymptomatic brown facial hyperpigmentation that had developed several years earlier, and had persisted, despite regular face washing. Physicians who previously treated this patient interpreted this as melasma and advised her to wear sunscreen. The condition was not aggravated by sun exposure. The patient reported that she was otherwise healthy.
Physical examination revealed a brown discoloration with a slightly rough texture. Upon rubbing the affected area with a 70% isopropyl alcohol pad, normal skin was revealed (FIGURE 1A) and brown flakes were apparent on the gauze (FIGURE 1B).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Terra firma-forme dermatosis
The physician diagnosed terra firma-forme dermatosis (TFFD) in this patient, noting the “dirty brown coloration” and distribution that did not suggest post-inflammatory hyperpigmentation or melasma. TFFD is a rare and benign form of acquired hyperpigmentation characterized by “velvety, pigmented patches or plaques.”1 A simple bedside test, known as an “alcohol wipe test,” both confirms and treats TFFD; it involves rubbing the affected area with a 70% isopropyl alcohol pad.1
TFFD typically affects the face, neck, trunk, or ankles, but the scalp, axilla, back, and pubis also can be affected.1 Histopathology will show negligible amounts of dermal inflammation, hyperkeratosis with mild acanthosis, and hyperkeratosis and papillomatosis.1 Most patients diagnosed with TFFD report that the hyperpigmentation does not improve despite washing with soap and water.2
Hygiene is not a factor
In 2015, Greywal and Cohen followed the case presentations of 10 Caucasian patients with TFFD who presented with “brown and/or black plaques or papules or both.”2 Many of the individuals followed in this case series reported “[practicing] good hygiene and showered a minimum of every other day or daily.”2 The same was reported by the patient in this case. This suggests that TFFD is not a consequence of poor hygiene but perhaps a result of “sticky” sebum that produces a buildup of keratin debris, sebum, and bacteria on the skin.3 This produces the hyperpigmentation seen clinically.
Differential includes post-inflammatory hyperpigmentation
Several other hyperpigmentation disorders were considered on the initial differential diagnosis for this case, including melasma and post-inflammatory hyperpigmentation. However, these 2 conditions are macular, whereas this hyperpigmented condition had a rough, mildly papular texture. Additionally, melasma flares up in the summer with UV exposure, and post-inflammatory hyperpigmentation presents with pruritus and/or a pre-existing rash.4 This patient reported that the condition did not itch nor change with increased sunlight, thus making melasma and post-inflammatory hyperpigmentation unlikely diagnoses.
Acanthosis nigricans also was considered because it presents with a velvety brown pigmentation similar to what was seen with this patient. Acanthosis nigricans, however, primarily affects flexural areas, not the face, making it improbable.
Continue to: Our patient
Our patient. A “wipe test” was performed on the patient. This removed the brown flaky scaling and revealed the underlying normal skin. We instructed the patient to wash daily with a soapy wash cloth and scrub with 70% isopropyl alcohol should the hyperpigmentation recur. The patient did not return.
CORRESPONDENCE
Robert T. Brodell, MD, Department of Dermatology, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; rbrodell@umc.edu
A 59-year-old woman presented to a dermatology clinic with an asymptomatic brown facial hyperpigmentation that had developed several years earlier, and had persisted, despite regular face washing. Physicians who previously treated this patient interpreted this as melasma and advised her to wear sunscreen. The condition was not aggravated by sun exposure. The patient reported that she was otherwise healthy.
Physical examination revealed a brown discoloration with a slightly rough texture. Upon rubbing the affected area with a 70% isopropyl alcohol pad, normal skin was revealed (FIGURE 1A) and brown flakes were apparent on the gauze (FIGURE 1B).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Dx: Terra firma-forme dermatosis
The physician diagnosed terra firma-forme dermatosis (TFFD) in this patient, noting the “dirty brown coloration” and distribution that did not suggest post-inflammatory hyperpigmentation or melasma. TFFD is a rare and benign form of acquired hyperpigmentation characterized by “velvety, pigmented patches or plaques.”1 A simple bedside test, known as an “alcohol wipe test,” both confirms and treats TFFD; it involves rubbing the affected area with a 70% isopropyl alcohol pad.1
TFFD typically affects the face, neck, trunk, or ankles, but the scalp, axilla, back, and pubis also can be affected.1 Histopathology will show negligible amounts of dermal inflammation, hyperkeratosis with mild acanthosis, and hyperkeratosis and papillomatosis.1 Most patients diagnosed with TFFD report that the hyperpigmentation does not improve despite washing with soap and water.2
Hygiene is not a factor
In 2015, Greywal and Cohen followed the case presentations of 10 Caucasian patients with TFFD who presented with “brown and/or black plaques or papules or both.”2 Many of the individuals followed in this case series reported “[practicing] good hygiene and showered a minimum of every other day or daily.”2 The same was reported by the patient in this case. This suggests that TFFD is not a consequence of poor hygiene but perhaps a result of “sticky” sebum that produces a buildup of keratin debris, sebum, and bacteria on the skin.3 This produces the hyperpigmentation seen clinically.
Differential includes post-inflammatory hyperpigmentation
Several other hyperpigmentation disorders were considered on the initial differential diagnosis for this case, including melasma and post-inflammatory hyperpigmentation. However, these 2 conditions are macular, whereas this hyperpigmented condition had a rough, mildly papular texture. Additionally, melasma flares up in the summer with UV exposure, and post-inflammatory hyperpigmentation presents with pruritus and/or a pre-existing rash.4 This patient reported that the condition did not itch nor change with increased sunlight, thus making melasma and post-inflammatory hyperpigmentation unlikely diagnoses.
Acanthosis nigricans also was considered because it presents with a velvety brown pigmentation similar to what was seen with this patient. Acanthosis nigricans, however, primarily affects flexural areas, not the face, making it improbable.
Continue to: Our patient
Our patient. A “wipe test” was performed on the patient. This removed the brown flaky scaling and revealed the underlying normal skin. We instructed the patient to wash daily with a soapy wash cloth and scrub with 70% isopropyl alcohol should the hyperpigmentation recur. The patient did not return.
CORRESPONDENCE
Robert T. Brodell, MD, Department of Dermatology, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; rbrodell@umc.edu
1. Lunge S, Supraja C. Terra firma-forme dermatosis—a dirty dermatosis: report of two cases. Our Dermatol Online. 2016;7:338-340.
2. Greywal T, Cohen PR. Terra firma-forme dermatosis: A report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
3. Alonso-Usero V, Gavrilova M, et al. Dermatosis neglecta or terra firma-forme dermatosis. Actas Dermosifiliogr. 2012;103:932-934.
4. Lucas J, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J. 2006;12:5.
1. Lunge S, Supraja C. Terra firma-forme dermatosis—a dirty dermatosis: report of two cases. Our Dermatol Online. 2016;7:338-340.
2. Greywal T, Cohen PR. Terra firma-forme dermatosis: A report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
3. Alonso-Usero V, Gavrilova M, et al. Dermatosis neglecta or terra firma-forme dermatosis. Actas Dermosifiliogr. 2012;103:932-934.
4. Lucas J, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J. 2006;12:5.

Tinea Incognito in a Tattoo
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.

Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
Practice Points
- Health care providers should have a low threshold to perform a potassium hydroxide preparation when the possibility of a superficial fungal infection is considered.
- Tinea incognito occurs when a superficial fungal infection has unusual clinical features in the setting of local immune suppression.

Idiopathic Livedo Racemosa Presenting With Splenomegaly and Diffuse Lymphadenopathy
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
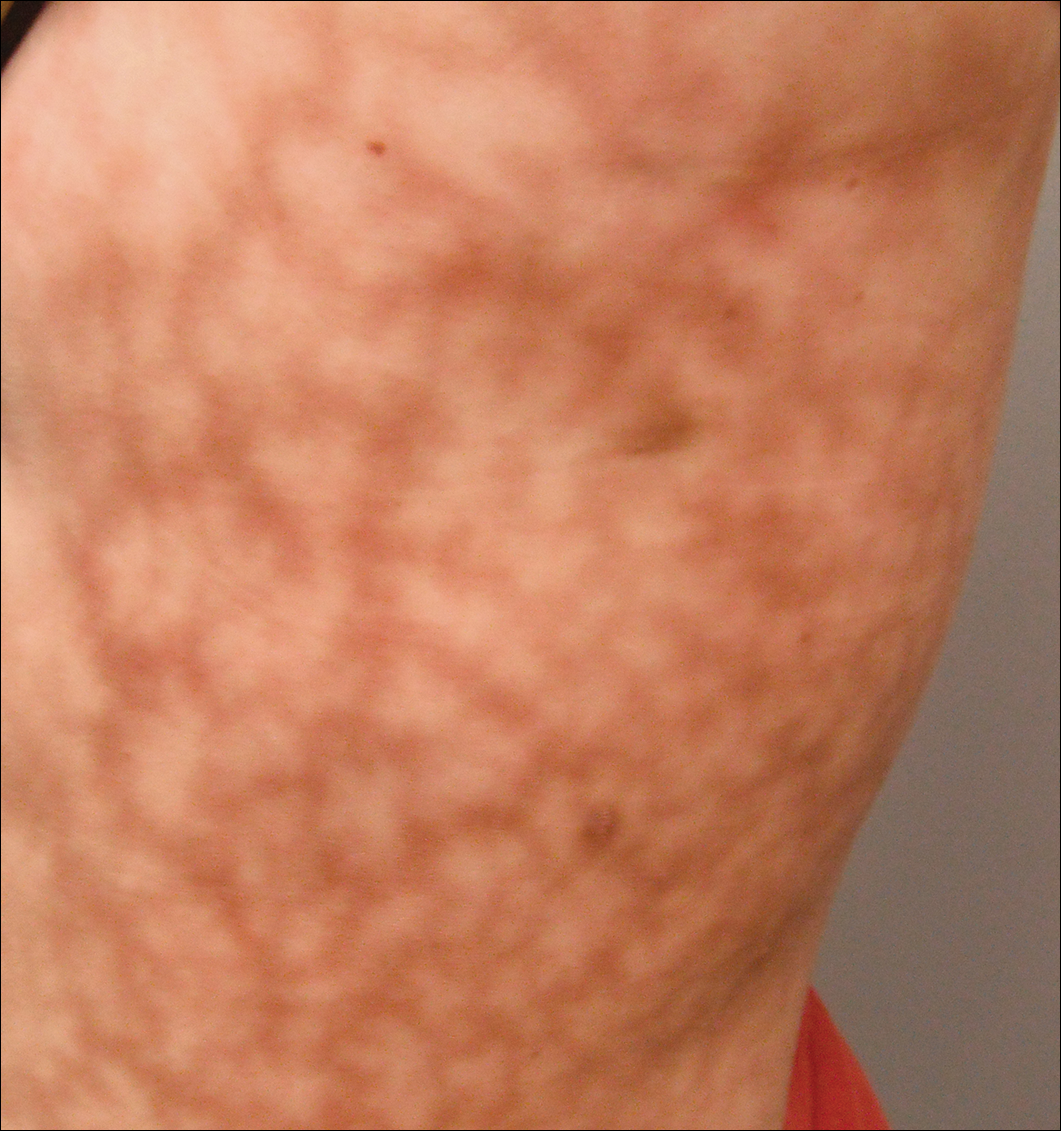
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
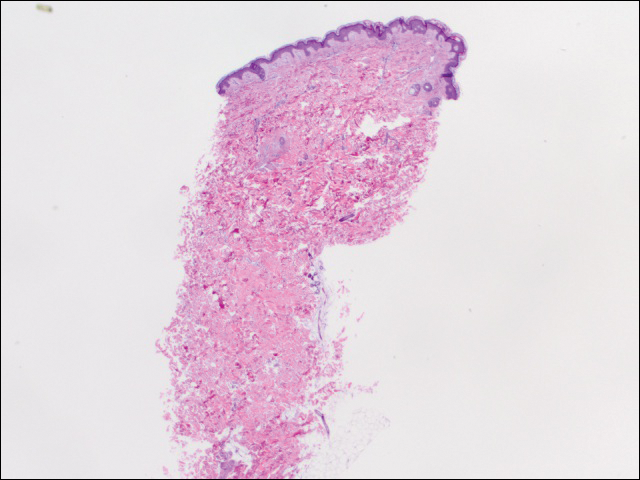
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
Sneddon syndrome (SS) was first described in 1965 in patients with persistent livedo racemosa and neurological events.1 Because the other manifestations of SS are nonspecific (eg, hypertension, cardiac valvulopathy, arterial and venous occlusion), the diagnosis often is delayed. Many patients who experience prodromal neurologic symptoms such as headaches, depression, anxiety, dizziness, and neuropathy often present to a physician prior to developing ischemic brain manifestations2 but seldom receive the correct diagnosis. Onset of cerebral occlusive events typically occurs in patients younger than 45 years and may present as a transient ischemic attack, stroke, or intracranial hemorrhage.3 The disease is more prevalent in females than males (2:1 ratio). The exact pathogenesis of SS is still unknown, and although it has been thought of as a separate entity from systemic lupus erythematosus and other antiphospholipid disorders, it has been postulated that an immunological dysfunction damages vessel walls leading to thrombosis.
Cutaneous findings associated with SS involve small- to medium-sized dermal-subdermal arteries. Histopathology in some patients demonstrates proliferation of the endothelium and fibrin deposits with subsequent obliteration of involved arteries.4 In many patients including our patient, histopathologic examination of involved skin fails to show specific abnormalities.1 Zelger et al5 reported the sequence of histopathologic skin events in a series of antiphospholipid-negative SS patients. The authors reported that only small arteries at the dermis-subcutis junction were involved and a progression of endothelial dysfunction was observed. The authors believed there were several nonspecific stages prior to fibrin occlusion of involved arteries.5 Stage I involved loosening of endothelial cells with nonspecific perivascular lymphocytic infiltration with perivascular inflammation and lymphocytic infiltration representing the prime mover of the disease.5,6 This stage is thought to be short lived, thus the reason why it has gone undetected for many years in SS patients. Stages II to IV progress through fibrin deposition and occlusion.5 Histological features of stages I to II have not been reported because of late diagnosis of SS. Stage I patients typically present with an average duration of symptoms of 6 months with few neurologic symptoms, the most common being paresthesia of the legs.5
Case Report
A 37-year-old woman with epigastric tenderness on the left side and splenomegaly seen on computed tomography was referred by a hematologist for evaluation of a reticular rash on the left side of the flank of 9 months’ duration with a presumed diagnosis of focal melanoderma. Her medical history was remarkable for a congenital ventricular septal defect and coarctation of the aorta, as well as endometriosis, myalgia, and joint stiffness that had all developed over the last year. Her medical history also was remarkable for nephrolithiasis, irritable bowel syndrome, and chronic sinusitis, as well as psychiatric depression and anxiety disorders. She recently had been diagnosed with moderate hypertension and had experienced difficulty getting pregnant for the last several years with 3 consecutive miscarriages in the first trimester. Neurologic symptoms included neuropathy involving the feet, intermittent paresthesia of the legs, and a history of chronic migraine headaches for several months.
Dermatologic examination revealed a slightly overweight woman with a 25×30-cm dusky, erythematous, irregular, netlike pattern on the left side of the upper and lower trunk (Figure 1). Extensive livedo racemosa was not altered by changes in temperature and had been unchanged for more than 9 months. There were no signs of pruritus or ulcerations, and areas of livedo racemosa were slightly tender to palpation.
We performed 2 sets of three 4-mm biopsies. The first set targeted areas within the violaceous pattern, while the second set targeted areas of normal tissue between the mottled areas. All 6 specimens demonstrated superficial perivascular lymphocytic infiltrate with no evidence of vasculitis or connective tissue disease. The vessels showed no microthrombi or surrounding fibrosis. No eosinophils were identified within the epidermis. There was no evidence of increased dermal mucin. Both the superficial and deep vascular plexuses were unremarkable and showed no evidence of damage to the walls (Figure 2).
To rule out other possible causes of livedo racemosa, complete blood cell count, comprehensive metabolic panel, coagulation profile, lipase test, urinalysis, serologic testing, and immunologic workup were performed. Lipase was within reference range. The complete blood cell count revealed mild anemia, while the rest of the values were within reference range. An immunologic workup included Sjögren syndrome antigen A, Sjögren syndrome antigen B, anticardiolipin antibodies, and antinuclear antibody, which were all negative. Family history was remarkable for first-degree relatives with systemic lupus erythematosus and Crohn disease.
Computed tomography revealed enlargement of the spleen, as well as periaortic, portacaval, and porta hepatis lymphadenopathy. Based on the laboratory findings and clinical presentation as well as the patient’s medical history, the diagnosis of exclusion was idiopathic livedo racemosa with unknown progression to full-blown SS. The patient did not meet the current diagnostic criteria for SS, and her immunologic studies failed to confirm any present antibodies, but involvement of the reticuloendothelial system pointed to production of antibodies that were not yet detectable on laboratory testing.
Comment
More than 50 years after the first case of SS was diagnosed, better laboratory workup is available and more information is known about the pathophysiology. Sneddon syndrome is a rare disorder, affecting only approximately 4 patients per million each year worldwide. Seronegative antiphospholipid antibody syndrome (SNAPS) describes patients with clinical presentations of antiphospholipid syndrome (APS) without detectable serological markers.7 Antiphospholipid-negative SS, which was seen in our patient, would be categorized under SNAPS. A PubMed search of articles indexed for MEDLINE using the terms livedo racemosa, Sneddon syndrome, and SNAPS and splenomegaly revealed there currently are no known cases of SNAPS that have been reported with splenomegaly and lymphadenopathy. Our patient presented with the following clinical features of SS: livedo racemosa, history of miscarriage, psychiatric disturbances, and hypertension. Surprisingly, biopsies from affected skin did not show any fibrin deposition or microthrombi but did reveal perivascular lymphocytic infiltrations. Magnetic resonance imaging did not show any pathological lesions or vascular changes.
Sneddon syndrome and APS share a common pathway to occlusive arteriolopathy for which 4 stages have been described by Zelger et al.5 Stage I involves a nonspecific Langerhans cell infiltrate with polymorphonuclear leukocytes. The tunica media and elastic lamina usually are unaltered at this early stage, while the surrounding connective tissue may appear edematous.5 This early stage of histopathology has not been evaluated in SS patients, primarily because of delay of diagnosis. Late stages III and IV will show fibrin deposition and shrinkage of affected vessels.7
A PubMed search using the terms Sneddon syndrome, lymphadenopathy and livedo racemosa, and Sneddon syndrome and lymphadenopathy revealed that splenomegaly and lymphadenopathy have not been reported in patients with SS. In patients with antiphospholipid-negative SS, one can assume that antibodies to other phospholipids not tested must exist because of striking similarities between APS and antiphospholipid-negative SS.8 Although our patient did not test positive for any of these antibodies, she did present with lymphadenopathy and splenic enlargement, leading us to believe that involvement of the reticuloendothelial system may be a feature of SS that has not been previously reported. Further studies are required to name specific antigens responsible for clinical manifestations in SS.
Currently, no single diagnostic test for SS exists, thus delaying both diagnosis and initiation of treatment. Histopathologic examination may be helpful, but in many cases it is nonspecific, as are serologic markers. Neuroradiological confirmation of involvement usually is the confirmatory feature in many patients with late-stage diagnosis.2 A diagnostic schematic for SS, which was first described by Daoud et al,2 illustrates classification of symptoms and aids in diagnosis. A working diagnosis of idiopathic livedo racemosa is made after ruling out other causes of SS in a patient with nonspecific biopsy findings and negative magnetic resonance imaging results with prodromal symptoms. The prognosis for such patients progressing to full SS is unknown with or without management using anticoagulant therapy.
Conclusion
Early diagnosis of livedo racemosa and SS is essential, as prevention of cerebrovascular accidents, myocardial infarction, and other thromboembolic diseases can be minimized by attacking risk factors such as smoking, taking oral contraceptive pills, becoming pregnant,9 and by initiating either antiplatelet or anticoagulation treatments. These treatments have been shown to delay the development of neurovascular damage and early-onset dementia. We present this case to demonstrate the variability of early-presenting symptoms in idiopathic livedo racemosa. Recognizing some of the early manifestations can lead to early diagnosis and initiation of treatment.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
- Sneddon IB. Cerebro-vascular lesions and livedo reticularis. Br J Dermatol. 1965;77:180-185.
- Daoud MS, Wilmoth GJ, Su WP, et al. Sneddon syndrome. Semin Dermatol. 1995;14:166-172.
- Besnier R, Francès C, Ankri A, et al. Factor V Leiden mutation in Sneddon syndrome. Lupus. 2003;12:406-408.
- K aragülle AT, Karadağ D, Erden A, et al. Sneddon’s syndrome: MR imaging findings. Eur Radiol. 2002;12:144-146.
- Zelg er B, Sepp N, Schmid KW, et al. Life-history of cutaneous vascular-lesions in Sneddon’s syndrome. Hum Pathol. 1992;23:668-675.
- Ayoub N, Esposito G, Barete S, et al. Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004;35:1329-1332.
- Duva l A, Darnige L, Glowacki F, et al. Livedo, dementia, thrombocytopenia, and endotheliitis without antiphospholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009;61:1076-1078.
- Kala shnikova LA, Nasonov EL, Kushekbaeva AE, et al. Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990;40:464-467.
- Wohl rab J, Fischer M, Wolter M, et al. Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. a report of 15 cases. Br J Dermatol. 2001;145:285-288.
Practice Points
- The classic physical diagnostic finding of Sneddon syndrome (SS) is livedo racemosa.
- Early identification and treatment of SS can prevent serious morbidity due to stroke, myocardial infarction, and other thrombotic events.
- Preventive care in SS should include antiplatelet therapy or anticoagulants and smoking cessation along with avoidance of birth control pills.