User login
Research and Reviews for the Practicing Oncologist
Intramedullary spinal cord and leptomeningeal metastases presenting as cauda equina syndrome in a patient with melanoma
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
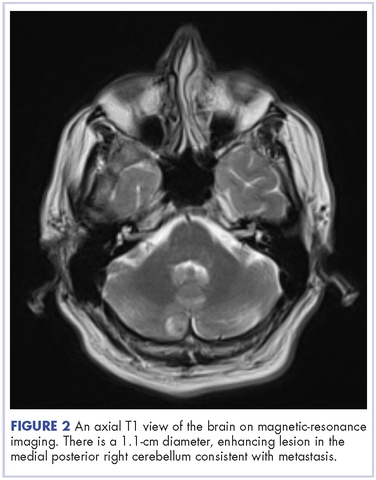
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).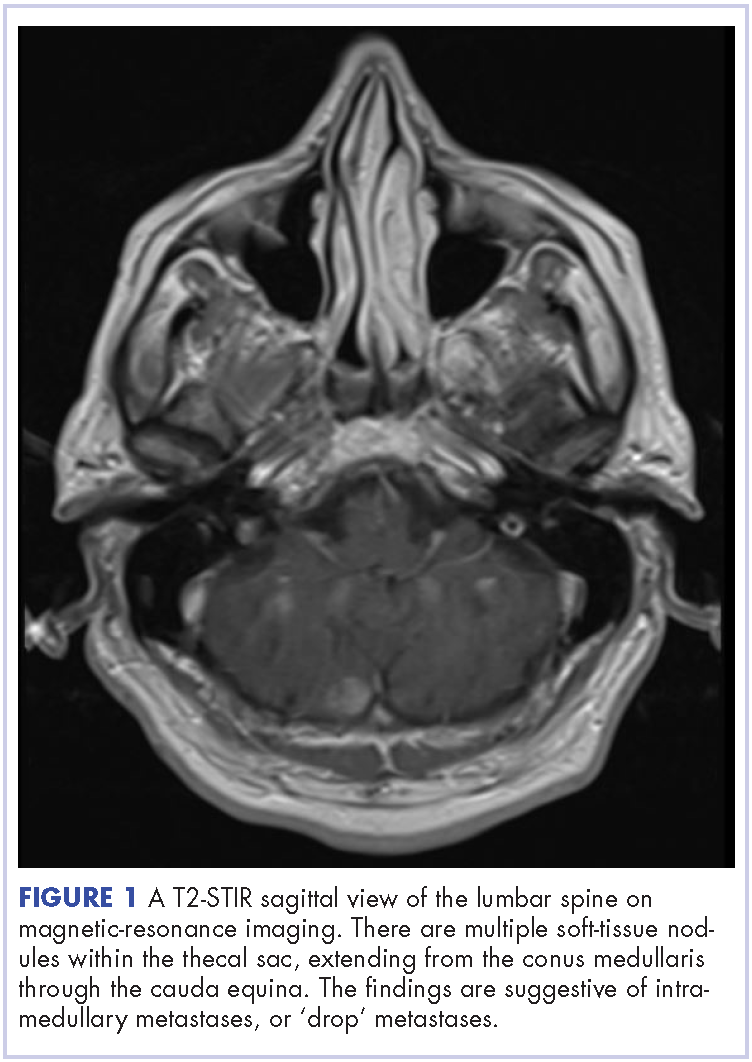

The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).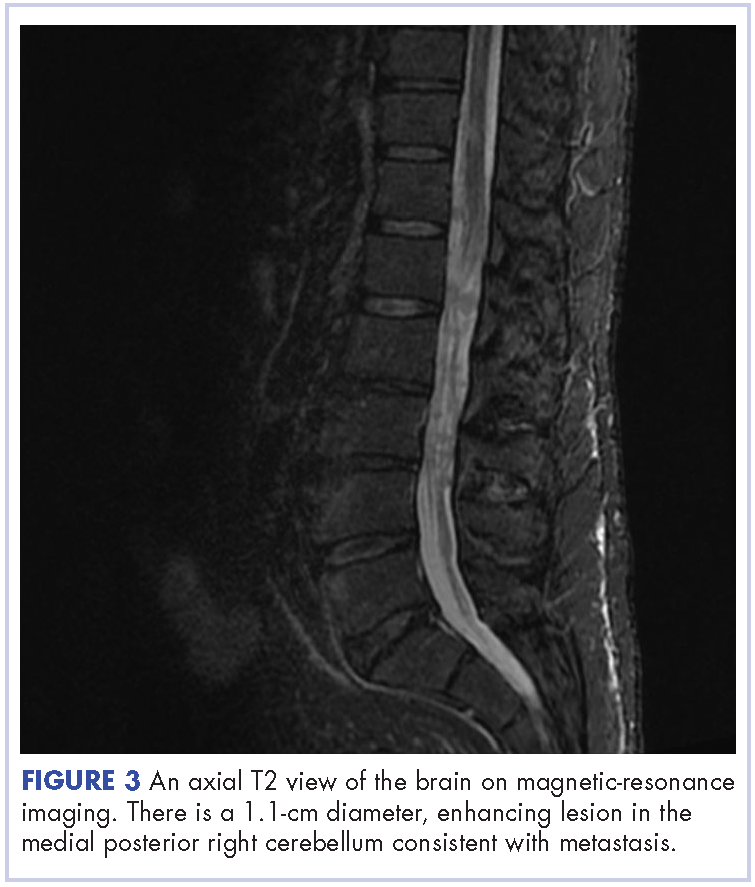
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).
The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
The incidence of malignant melanoma has been rising in the United States, especially among non-Hispanic white men and women. Death rates have increased for those aged 65 years or older, and incidence rates have increased for all age groups.1 It is a serious public health issue.
Given the unique biology of melanoma, metastatic disease can present in a variety of ways. In most cases, the lymph nodes and lungs are involved.2 The incidence of brain metastases is 10%-40%, however the percentage may be even higher based on reported incidence of autopsy reports.3 The most common forms of metastatic melanoma to the spine are vertebral and intramedullary.4 Specifically, leptomeningeal involvement can be found in 20% of patients in clinical studies and 44%-70% in autopsy series of patients with central nervous system (CNS) metastatic disease.5 Despite its incidence, leptomeningeal disease (LMD) from melanoma is rarely discussed in the literature and the diagnosis may be difficult. Even rarer is the documented presentation of intramedullary spinal cord metastases, or “drop metastases.”6 In our review of the literature, we found no published case reports to date of drop metastases from melanoma causing cauda equina syndrome.
The prognosis of patients with metastatic melanoma with brain metastases is very poor, with a median overall survival of about 4 months reported in several studies.7-9 Prognosis is even worse for patients with leptomeningeal involvement, and median survival without therapy is about 4-6 weeks.10 A combination of intrathecal and systemic chemotherapy can be used to treat LMD.11
Case presentation and summary
This is the case of a 56-year-old man with history of metastatic melanoma that had been initially diagnosed about 4 years before the current case presentation. Original sites of disease were a supraclavicular lymph node and solitary liver metastasis, both of which were resected. The patient then developed biopsy-proven lung involvement that required left and right wedge resections. Mutation testing for BRAF V600E and BRAF V600K was sent and not detected. Therefore the patient did not receive any BRAF-targeted therapies. Subsequently, recurrent metastatic disease to the brain with 2 dominant lesions in the cerebellum and the occiput as well as numerous small lesions at the gray-white matter junction was identified (Figure 1 and Figure 2).
The patient received whole-brain radiation (30 Gy in 10 fractions of 3 Gy each). There was no evidence of disease in his spine at that time. About 2 weeks after completing whole-brain radiation, the patient presented to the hospital with left lower extremity weakness, urinary retention, bowel incontinence, saddle anesthesia, and malaise. The symptoms had begun after he had finished whole-brain radiation and weakness progressed to the point at which he need a cane to be able to walk. A physical examination was significant for hyporreflexia, decreased strength and sensitivity on left lower extremity, saddle anesthesia, and lumbar spinal tenderness to palpation. The results of magnetic-resonance imaging (MRI) of the spine revealed multiple soft-tissue nodules extending from the conus medullaris throughout the cauda equina, consistent with intramedullary metastases, as well as concomitant leptomeningeal involvement (Figure 3).
The patient was started on steroids with minimal improvement in neurologic function. We consulted with our neurosurgery colleagues, but learned that no direct surgical intervention could be performed because of widespread involvement. We then proceeded with radiation, 30 Gy in 10 fractions to the lumbar spine. Intrathecal chemotherapy with methotrexate (12 mg twice a week) was also started, with a plan to complete 4 weeks. Shortly after starting radiation therapy and methotrexate, we observed clinical improvement in the patient, with mildly increased left lower extremity strength and increased ambulation with a physical therapist.
Cerebrospinal fluid studies (CSF) showed clearance of malignant cells after 2 treatments of intrathecal methotrexate as well as improvement in CSF chemistry parameters: the patient’s protein level decreased from 1,095 mg/dL to 42 mg/dL (15-45 mg/dL) and his glucose level increased from 3 mg/dL to 73 mg/dL (40-85 mg/dL) However, after completing 3 weeks of intrathecal chemotherapy, the hospital course was complicated by leukopenia, thrombocytopenia, and spontaneous intracranial hemorrhage. The cytopenias were thought to be secondary to systemic effect of intrathecal methotrexate in conjunction with the radiation treatments to the spine. Intrathecal chemotherapy was held.
The patient was not a candidate for systemic immunotherapy because of his decline in performance status. He continued to deteriorate neurologically, and the family decided to pursue inpatient hospice. He died a week after transfer to hospice and 5 weeks after the initial diagnosis of leptomeningeal and intramedullary metastases.
Conclusions
Although metastatic melanoma to the brain is not uncommon, leptomeningeal and intramedullary drop metastases are an infrequent presentation. Even more rare are intramedullary drop metastasis that are significant enough to cause cauda equina syndrome, as with our patient. The incidence of LMD has increased over the years and may continue to increase, likely because of the improved overall survival and a prolonged control of extracranial disease with newly approved systemic therapeutic drugs, such as molecularly targeted therapy and immunotherapy.12 Intramedullary metastases are extremely rare, but reported incidence has seemed to be increasing due to detection with MRI. Currently there are fewer than 100 case reports of intramedullary spinal cord metastasis.6 In one retrospective study, 40 patients with intramedullary metastatic disease secondary to systemic cancer were identified during 1980-1993.6 About half of those cases were from lung cancer, the second most common was breast cancer.
CNS involvement by melanoma can have debilitating complications and confers a poor prognosis. In another retrospective study, several patient characteristics were found to be associated with significantly shorter survival in patients with known brain metastases, including presence of neurologic symptoms and leptomeningeal involvement.3
Malignant cells can reach the CSF by several routes: direct extension, hematogenous, venous access, venous drainage from bone marrow and cranial and peripheral nerves. Once the tumor has reached the CSF, it can seed any portion of the nervous system that has contact with the CSF and become entangled among the cauda equine.13
Given the rarity of leptomeningeal and intramedullary involvement of melanoma, there are no standard treatment guidelines. Treatment for LMD usually consists of intrathecal and systemic chemotherapy. Commonly used intrathecal agents are methotrexate, liposomal cytarabine, and thiopeta.11 The goals of treatment are to improve or stabilize neurologic status of the patient and ideally prolong survival. The choice of agent for intrathecal chemotherapy is guided by the primary tumor, however, there is no strong evidence to choose one agent over the other.12,14 Methotrexate or cytarabine are generally recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Targeted therapy toward the primary tumor is occasionally used for treatment of LMD, for example rituximab can be given intrathecally for lymphoma,15 and trastuzumab has been given intrathecally for breast cancer.16 No intrathecal targeted agents are currently available for melanoma. Administration of intrathecal chemotherapy is given via lumbar puncture or Ommaya reservoir. Induction intrathecal chemotherapy is recommended by NCCN to be given for 4-6 weeks. The schedule of administration varies based on the agent used. Most systemic chemotherapy has poor CSF penetration, which is the basis behind using chemotherapy intrathecally in these patients.14 However, novel therapies for melanoma, such as ipilimumab, have shown activity in the CNS, and it is not known if intrathecal chemotherapy will continue to play role in the management of LMD.17
Systemic therapy for metastatic melanoma has changed with the development of novel agents, which have shown better efficacy than traditional chemotherapy. The recommendation for first-line systemic therapy of metastatic unresectable melanoma is based on several factors: BRAF mutation status, tempo of disease, and presence or absence of cancer-related symptoms. Immunotherapy for metastatic melanoma that is unresectable includes anti-programmed cell death protein-1 (PD-1) monotherapy (nivolumab or pembrolizumab) or combination therapy with nivolumab plus ipilimumab. Targeted therapy is preferred in cases with an identified BRAF mutation. Combination therapy with dabrafenib plus trametinib or with vemurafenib plus cobimetinib is recommended. Single-agent therapy may also be used with dabrafenib or vemurafenib.18
Ipilimumab is a monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 to potentiate an anti-tumor T-cell response that was approved in 2011 by the US Food and Drug Administration for the treatment of melanoma. A randomized, phase 3 clinical trial showed an increase in overall survival in patients with unresectable metastatic disease who had received previous treatment.19 Before that, no therapy had been shown to improve overall survival in patients with metastatic melanoma. Patients with CNS metastases were included in this study.19
The activity of ipilimumab specifically in patients with brain metastasis was further studied in a phase 2 trial that enrolled 72 patients, 1 cohort with symptomatic brain metastases and the other cohort with asymptomatic brain metastases.20 After 12 weeks of therapy, response was assessed by modified World Health Organization criteria for disease control (complete response plus partial response plus stable disease). In all, 18% of patients with asymptomatic brain metastasis achieved disease control, compared with 10% of patients with symptomatic brain metastases. When the brain alone was assessed, 24% of asymptomatic patients and 10% of symptomatic patients achieved disease control. No unexpected toxic effects occurred during the study. Anti-PD1 therapy such as nivolumab, which has shown durable responses in metastatic melanoma, has no published results specifically in patients with active brain metastases.
Of the BRAF-targeted therapy, dabrafenib and vemurafenib have also been studied in patients with brain metastases. For darafenib, 172 patients with BRAF-mutated metastatic melanoma were included in a phase 2 clinical trial that showed an intracranial response of 39% in previously untreated patients and 31% in patients whose brain metastases had progressed after previous local treatment.21 Vemurafenib has also shown intracranial response in a phase 2 clinical trial.22
The role of the aforementioned therapies in patients with metastatic melanoma with CNS disease should not be overlooked because these patients are typically excluded from clinical trials. As already noted, agents such as ipilimumab and the dabrafenib–vemurafenib combination have been studied in patients with brain metastases and have shown disease control, but more studies are needed to truly assess whether there is an improvement in overall survival and whether that will change treatment guidelines. Although patients with parenchymal brain metastases were included in these studies, it is not clear how patients with LMD and intramedullary spinal cord metastases, such as our patient, would be affected. It is also not clear whether intrathecal chemotherapy will continue to play a role in management of metastatic melanoma with LMD, especially if these newer agents have CNS activity in addition to controlling extracranial disease. Although rarely documented, leptomeningeal and intramedullary metastatic disease will likely become increasingly recognized as patients with cancer live longer and diagnostic studies improve. These initial studies showing intracranial disease control show compelling evidence to continue enrolling patients with active CNS disease in clinical trials.
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
1. Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S17.e1-S17.e11.
2. Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma: a study of 216 autopsy cases. Am J Surg. 1978;135(6):807-810.
3. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
4. Sun L, Song Y, Gong Q. Easily misdiagnosed delayed metastatic intraspinal extradural melanoma of the lumbar spine: a case report and review of the literature. Oncol Lett. 2013;5(6):1799-1802.
5. Moseley R, Davies A, Bourne S, et al. Neoplastic meningitis in malignant melanoma: diagnosis with monoclonal antibiodies. J Neurol Neurosurg Psychiatry. 1989;52:991-886.
6. Schiff D, O’Neill B. Intramedullary spinal cord metastases clinical features and treatment outcome. Neurology. 1996;47(4):906-912.
7. Fife KM, Colman MH, Stevens G, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293-1300.
8. Raizer J, Hwu W, Panageas K, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10(2):199-207.
9. Sampson JH, Carter JH Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
10. Abernethy AP. Central nervous system tumors. In: Loprinzi C, ed. ASCO-SEP: Medical Oncology Self-evaluation Program. 4th ed. Alexandria, VA: American Society of Clinical Oncology, 2015. Page 396. Print.
11. Pape E, Desmedt E, Zairi , et al. Leptomeningeal metastasis in melanoma: a prospective clinical study of nine patients. In Vivo. 2012;26(6):1079-1086.
12. Pavlidis N. The diagnostic and therapeutic management of leptomeningeal carcinomatosis. Ann Oncol. 2004;15(Suppl 4):iv285-291.
13. DeAngelis L, Posner JB. Neurologic complications of cancer. 2nd ed. New York, NY: Oxford University Press; 2008.
14. Chamberlain, M. Leptomeningeal metastasis. Curr Opin Oncol. 2010;22:627-635.
15. Chamberlain M, Johnston S, Van Horn A, Glantz MJ. Recurrent lymphomatous meningitis treated with intra-CSF rituximab and liposomal ara-C. J Neurooncol. 2009;91(3):271-277.
16. Zagouri F, Sergentanis T, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13-22.
17. Silk A, Bassetti M, West BT, Tsien C, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899-906.
18. [Behind paywall.] National Comprehensive Cancer Network. Melanoma (version 2.2016). http://www.nccn.org/professionals/physician_gls/pdf/melanoma.pdf. November 10, 2016. Accessed February 28, 2016
19. Hodi F, O’Day S, McDermott D, et al. Improved survival with ipilimumab in patients with metastatic melanoma. NEJM. 2010;363(8):711-723.
20. Margolin K, Ernstoff M, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459-465.
21. Long G, Trefzer U, Davies M, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
22. McArthur GA, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicenter study. Ann Oncol. 2017;
Evidence-based information for the entire oncology care team
The Journal of Community and Supportive Oncology is now available online only.
We publish a bimonthly digital edition, and accepted articles will be posted directly (and more speedily) to the web after going through the editing, production, and author-approval stages.
The look of the digital edition will be the same as the former print issues, and you will be able to print it out as a PDF.
In addition, we plan to expand our content to better serve the clinical and supportive practice needs of our readers and we'll continue to expand our reach by mailing twice-weekly electronic newsletters specially curated for the oncology care team.
Download the app for the digital edition at http://bit.ly/2nCEPIa -- it’s fast, easy, and there’s no charge!
Visit our website at www.jcso-online.com for regular updates on current practices in the community setting.
Submit a paper at www.editorialmanager.com/jso
Follow us on Twitter @jcs_onc and @davidhenrymd
Like us on Facebook
The Journal of Community and Supportive Oncology is now available online only.
We publish a bimonthly digital edition, and accepted articles will be posted directly (and more speedily) to the web after going through the editing, production, and author-approval stages.
The look of the digital edition will be the same as the former print issues, and you will be able to print it out as a PDF.
In addition, we plan to expand our content to better serve the clinical and supportive practice needs of our readers and we'll continue to expand our reach by mailing twice-weekly electronic newsletters specially curated for the oncology care team.
Download the app for the digital edition at http://bit.ly/2nCEPIa -- it’s fast, easy, and there’s no charge!
Visit our website at www.jcso-online.com for regular updates on current practices in the community setting.
Submit a paper at www.editorialmanager.com/jso
Follow us on Twitter @jcs_onc and @davidhenrymd
Like us on Facebook
The Journal of Community and Supportive Oncology is now available online only.
We publish a bimonthly digital edition, and accepted articles will be posted directly (and more speedily) to the web after going through the editing, production, and author-approval stages.
The look of the digital edition will be the same as the former print issues, and you will be able to print it out as a PDF.
In addition, we plan to expand our content to better serve the clinical and supportive practice needs of our readers and we'll continue to expand our reach by mailing twice-weekly electronic newsletters specially curated for the oncology care team.
Download the app for the digital edition at http://bit.ly/2nCEPIa -- it’s fast, easy, and there’s no charge!
Visit our website at www.jcso-online.com for regular updates on current practices in the community setting.
Submit a paper at www.editorialmanager.com/jso
Follow us on Twitter @jcs_onc and @davidhenrymd
Like us on Facebook
Ribociclib: another CDK inhibitor hits the mark in breast cancer
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
Perceived financial hardship among patients with advanced cancer
The American Cancer Society has identified a disparity in cancer death rates, noting that persons with lower socioeconomic status have higher rates of mortality.1 This is attributed to many factors, but it is largely owing to the higher burden of disease among lower-income individuals.1 A component of this disease burden is measured by assessing the patient-reported outcome of cancer-related distress. The National Comprehensive Cancer Network (NCCN) Distress Management Guidelines have defined distress as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social and/or spiritual nature that may interfere with the ability to cope with cancer, its physical symptoms and its treatment.”2
Financial hardship related to cancer diagnosis and treatment is increasingly being recognized as an important component of disease burden and distress. The advancements in costly cancer treatments have produced burdensome direct medical costs as well as numerous indirect costs that contribute to perceived financial hardship.3,4 These indirect costs include nonmedical expenses such as increased transportation needs or childcare, loss of earnings, or loss of household income due to caregiving needs.3 Moreover, indirect costs are often managed by patients and families through their use of savings, borrowing, reducing leisure activities, and selling possessions.3 Even though efforts to increase health coverage, such as the Affordable Care Act, have reduced the rates of individuals who are uninsured, persons with cancer who have insurance also face challenges because they cannot afford copays, monthly premiums, deductibles, and other high out-of-pocket expenses related to cancer treatment that are not covered by their insurance such as out-of-network services or providers.5-7
Thus, financial hardship may have an impact on several areas of a patient’s life and well-being, but the effects are commonly undetected.8-10 Research has established that financial strain can influence treatment choices and adherence to therapy.11 Furthermore, the effects of financial strain have been identified across the cancer care continuum, from diagnosis through survivorship, suggesting a bidirectional relationship between financial strain and well-being.11 Financial strain may reduce patient quality of life and worsen symptom burden because of the patient’s inability to access needed care, poor social supports, and/or increased stress.11-12 These worsening outcomes may also increase the use of financial reserves and affect their ability to work.7,11 Financial difficulties may also be associated with anxiety and depression, leading to worse quality of life and greater distress and symptom burden.12 Identifying groups at high risk for financial strain is crucial to ensure that resources are available to assist these populations.13 This burden can be even more pronounced in minority and underserved patients with cancer.7 Patients with advanced cancer are especially vulnerable to the burden of increased costs because of the use of expensive targeted therapies; their improved survival, which extends the time of expenditure; and increased use of financial reserves.9 Financial hardship in patients with advanced cancer is not well understood or characterized,9 which is why this study aimed to better quantify distress in advanced stage cancers by describing :
▪ A cohort of patients with advanced cancer and their levels of quality of life, symptom distress, cancer-related distress and perceived financial hardship;
▪ The relationship between perceived financial hardship, quality of life, symptom distress and overall cancer-related distress; and
▪ Quality of life, symptom distress, and overall cancer-related distress according to level of perceived financial hardship.
Methods
This study is a cross-sectional, descriptive, comparative study of distress, including perceived financial hardship, among patients with advanced cancer who were receiving palliative care treatment in two outpatient medical oncology clinics in Western Pennsylvania. The data were collected during May 2013-November 2014. The study protocol was approved by the Institutional Review Board at the University of Pittsburgh. Eligible participants had to be 18 years or older and have an advanced solid tumor of any kind, with a prognosis of 1 year or less confirmed by a physician or clinic nurse practitioner/physician assistant, and be able to read and understand English at the fourth-grade level. The sample was recruited from two clinics at the University of Pittsburgh Cancer Institute, a National Cancer Institute-designated Comprehensive Cancer Program.
Measurements
Sociodemographic factors. These were measured using an investigator-derived Sociodemographic Questionnaire, a 12-item form that includes variables such as age, race, marital status, cancer type, religion and spirituality, employment status, years of education, health insurance status, and income level.
Cancer-related distress. The NCCN Distress Thermometer is a self-report visual analog scale (0, no distress; 10, great distress) formed in the shape of a thermometer combined with a problem list that is often used in outpatient cancer settings for reporting of cancer-related distress.14-16 The sensitivity, specificity and convergent validity with the Brief Symptom Inventory and the Hospital Anxiety and Depression Scale have been established and appropriate cut-off score of the distress thermometer identified.14-16 A score of 4 or above indicates a clinically significant level of distress.14-16
Symptom distress. The McCorkle Symptom Distress Scale was developed in 1977 based on interviews that focused on the symptom experiences of patients. Psychometric testing among patients with cancer using the modified Symptom Distress Scale revealed high reliability (Cronbach alpha, 0.97).17 The instrument is a 13-item Likert scale (1-5) assessing the severity of distress experienced by a symptom. Total scores range from 13 to 65, where a higher score indicates greater distress. Moderate distress is indicated with a score of 25-33, and a score above 33 indicates severe distress, identifying the need for immediate intervention.17
Quality of life and spiritual well-being. The Functional Assessment of Cancer Therapy (FACT-G) is used to assess general cancer-related quality of life. It has four subscales: physical, emotional, social and family, and functional well-being, with a total score that ranges from 0-112, where higher scores show higher quality of life. The Spiritual Distress Well-Being questionnaire was used alongside the valid FACT-G assessment.18,19 The Spiritual Well-Being Short Form was developed with an ethnically diverse population and adds 12 items to the FACT-G. The items do not necessarily assume a faith in God, allowing a wide flexibility in application and tapping into issues such as faith, meaning, and finding peace and comfort despite advanced illness. Higher scores on the Spiritual Well-Being subscore (range, 0-48) are correlated with higher scores of quality of life. The possible scores for the combined FACT-G and Spiritual Well-Being assessment range from 0-160, with higher scores showing higher quality of life.
Economic hardship. Perceived financial hardship was measured using Barrera and colleagues’ Psychological Sense of Economic Hardship Scale. 20 The scale consists of 20-items broken down into 4 subscales: financial strain, inability to make ends meet, not enough money for necessities, and economic adjustments.20 Economic adjustments in the 3 months before administration of the questionnaire were assessed with 9 Yes or No items, such as added another job, received government assistance, or sold possessions to increase income. The subscale of not enough money for necessities was assessed with seven 5-point scale items in which respondents noted whether they felt they had enough money for housing, clothing, home furnishings, and a car over the previous 3 months. Inability to make ends meet included two 5-point scale items that assessed the difficulty in meeting financial demands in the previous 3 months. Financial strain consisted of two 5-point scale items concerned with expecting financial hardships in the coming 3 months. Scores can range from 20-73, with a higher score indicating worse economic hardship.
Data collection and analysis
In-person data collection occurred in the clinical waiting area before the clinician visit or in the treatment room with the patient using a consecutive, convenience sample. The nursing staff checked the clinic lists daily for possible patient participants. Patients with metastatic cancer were identified and then approached for consent. After we had received the patient’s consent, the administration of the instruments took about 20 minutes to complete. The data were then entered and verified in REDCap (Research Electronic Data Capture), which is hosted at the University of Pittsburgh.21The levels of symptom distress, quality of life, perceived financial hardship, and cancer-related distress were described through continuously measured variables. Descriptive statistics, measures of central tendency (mean and median), and dispersion (standard deviation and range), were obtained for the subscales and total scores. Correlation analysis was used to describe the relationship between perceived financial hardship and quality of life, symptom distress, and cancer-related distress. These primary outcome variables were further explored according to the level of dichotomized perceived financial hardship using mean score as the cut point. Independent sample t tests were used to compare patients experiencing high perceived financial hardship with those experiencing low perceived financial hardship.
Results
In all, 100 patients participated in the study. Any missing data points were replaced with the mean score for that variable, although this was minimal in this study. Most of the participants were women (67%), and the average age of the participants was 63.43 years (SD, 13.05; Table 1). Of the total number of participants, 73% were white, 26% were black, and 1% were Asian. Most of the participants were either retired and not working (39%) or disabled or unable to work (34%). Almost all of the participants had some form of insurance, with 99% having either private or public health insurance. A variety of cancer types were represented in this patient population, with higher percentages of breast (25%), gynecologic (10%), lung (19%), and colon/rectal cancer (15%). Of the total number of participants, 35% had annual household incomes below $20,000, and 50% had annual household incomes of more than $20,000. On average, participants had 13.48 years (SD, 2.78) of formal education.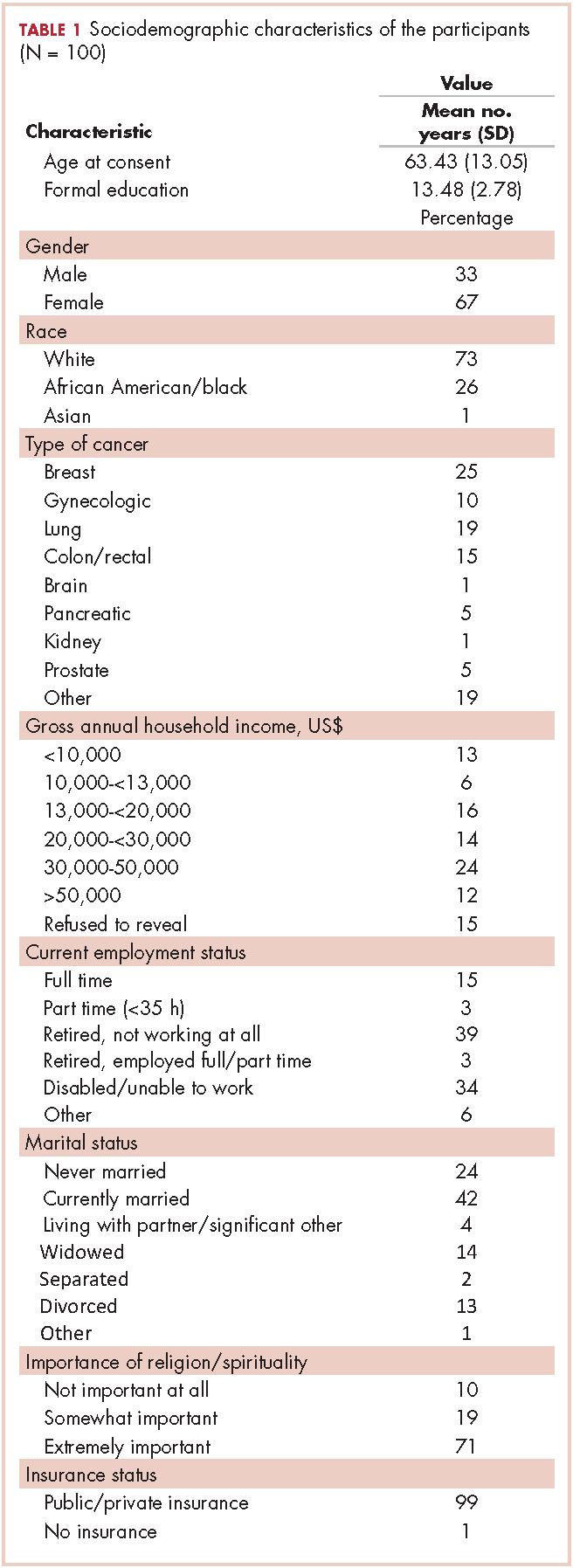
Descriptive statistics for the primary outcome variables can be found in Table 2. The average score for cancer-related distress based on the NCCN Distress Thermometer tool was 4.16 (SD, 3.26). The average score for the McCorkle Symptom Distress measurement was 25.45 (SD, 9.34). For quality of life, the average FACT-G total score was 73.77 (SD, 19.40). Of the FACT-G subscale average scores, physical well-being was 17.35 (SD, 7.50), social/family well-being 24.21 (SD, 5.25), emotional well-being 16.34 (SD, 5.42), and functional well-being 15.87 (SD, 6.78). Participants’ average score for the spiritual well-being measure was 35.20 (SD, 9.25) and the combined FACT-G and spiritual well-being average score was 108.97 (SD, 26.07). The total average score for perceived financial hardship was 35.70 (SD, 13.87), with subscale average scores of 3.44 (SD, 2.36) for financial strain, 5.73 (SD, 1.91) for inability to make ends meet, 16.43 (SD, 8.92) for not enough money for necessities, and 10.63 (SD, 2.70) for economic adjustments.
We conducted a bivariate correlation analysis to assess the relationship between perceived financial hardship and three other primary outcome variables (Table 3). These analyses showed significant low to moderate correlations with overall cancer-related distress (r, 0.439; P < .001), symptom distress (r, 0.409; P < .001) and overall quality of life scores (FACT-G and spiritual well-being combined score: r, -0.323; P < .001).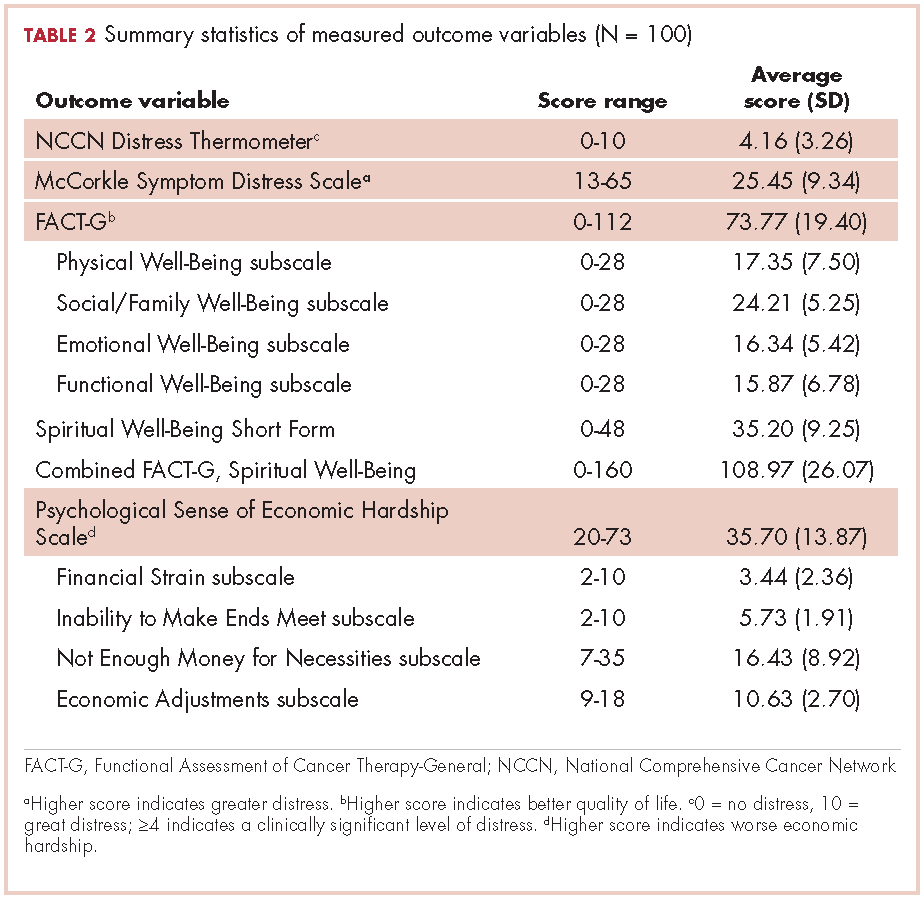
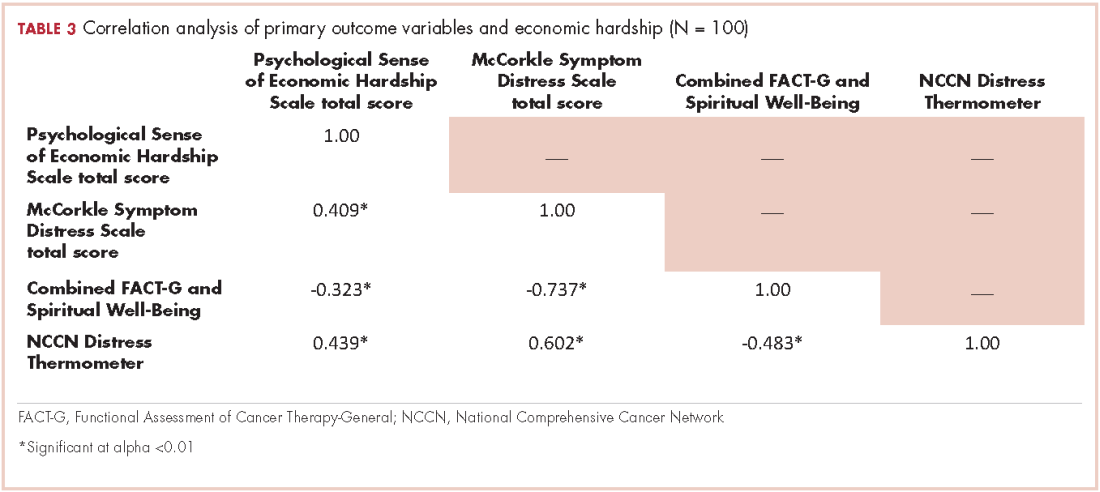
Forty-three participants reporting high perceived financial hardship experienced worse quality of life overall (FACT-G and spiritual well-being; P = .002), worse FACT-G total scores (P < .001), worse physical well-being (P < .001), worse social/family well-being (P = .029), worse emotional well-being, and no significant difference for functional (P = .082) or spiritual well-being (P = .453), compared with those with lower economic hardship. In overall cancer-related distress, participants with higher perceived financial hardship reported higher levels of cancer-related distress (P < .001) than those with lower perceived financial hardship. For those participants reporting higher perceived financial hardship there was also worse symptom distress (P < .001), compared with those with lower economic hardship (Table 4).
Discussion
Overall, this report provides data to illuminate our understanding of disparities in well-being that may be present in patients with advanced cancer. Our analysis found that patients with advanced cancer who have higher perceived financial hardship have significantly higher overall cancer-related distress, symptom distress, and poorer overall quality of life. In this study’s population of patients with advanced cancer, the most notable areas of economic hardship identified by participants were: not having enough money for necessities in the 3 months before the survey and the inability to make ends meet during the same time span, with difficulty paying bills and not having enough money left at the end of the month being most noteworthy among this study’s patient population. Financial strain and making economic adjustment were not as notable in the category of perceived financial hardship.
In regard to not having enough money, participants most commonly cited not being able to afford everyday necessities such as food, clothing, medical care, or a home, as well as leisure and recreational activities. These findings are further supported with the positive, moderate associations between perceived financial hardship and symptom distress and overall cancer-related distress found in this cohort of patients with advanced cancer and the negative, moderately associated relationship between perceived financial hardship and overall quality of life in this study’s sample.
Although these findings have been confirmed in the literature on cancer-related distress, our findings add to our knowledge on both economic and cancer-related distress exclusively in patients with advanced cancer.9,22 The broader cancer-related distress literature has also found an association between being younger and having a lower household income as risk factors for increased financial hardship; however, the perception of financial strain and magnitude was a more significant predictor of quality of life and perception of overall well-being.6,8-9,12,22-23 Furthermore, patients with cancer who noted having higher financial distress typically reported decreased satisfaction with cancer care which also influenced their adherence to treatment and quality of life.24
Our work now adds the important element of perceived financial hardship to the advanced cancer-related distress puzzle. We should consider integrating a financial distress assessment into routine cancer care, particularly with patients and families with advanced cancer, to proactively and routinely assess and intervene with available distress mitigating resources. Therefore, understanding the patients most likely to experience financial distress will help personalize supportive therapy.
This study’s results as well as the existing literature describing financial distress support the use of comprehensive screening instruments to capture elements of financial burden beyond out-of-pocket costs.8,25 This screening is particularly relevant because we are increasingly recognizing that gross annual household income does not always reflect financial hardship or distress. The instrument we used for this analysis, the Psychological Sense of Economic Hardship, provides a broad view of financial toxicity including the specific components of financial strain, the inability to make ends meet, not having enough money for necessities, and economic adjustments experienced by patients with advanced cancer.20 Another measure to evaluate financial toxicity among patients with cancer includes the Comprehensive Score for Financial Toxicity (COST), which is a widely used patient-reported outcome measure. It was developed with input from both patients and oncology experts.25 Use of a financial toxicity assessment tool adds to our understanding of the economic financial burden experienced by patients with cancer, specifically those with advanced cancer.
Tucker-Seeley and Yabroff have identified several areas in which the research agenda for financial toxicity should focus, including: documentation of the socioeconomic context among patients across all areas of the cancer care continuum, further identification and characterization of at risk populations to address health disparities, and the inclusion of cost discussions in the health care context.26 Furthermore, research is needed to identify key areas to target for interventions addressing financial toxicity, such as addressing lack of financial resources to cover the cost of cancer care, focusing on managing or preventing the distress that results from a lack of financial resources, or addressing coping behaviors used by families to manage the financial burden of cancer care.26 Although cost discussions between health care providers and patients have been identified as important in reducing the financial burden of cancer care, the content, timing, and goals of those discussions still need to be better articulated for different patient populations, including patients with advanced cancer.3,27-28 In addition, resources such as social workers, patient navigators, or financial counselors have been identified as effective in assisting patients with financial planning and accessing community resources to address financial burden and assistance.4
Design considerations
This study has limitations that need to be noted. Its cross-sectional design does not allow for the analysis of causal inferences. In addition, certain groups were underrepresented in this study’s sample, including uninsured patients, men, and some minority groups, which may have underestimated the amount of financial burden experienced by patients with advanced cancer. The lack of representativeness of uninsured individuals may be a result of the eligibility of persons with advanced cancer for Medicaid. However, a strength of this study is its ability to increase the representativeness of African American/black patients in the study of advanced cancer and financial hardship. In our study, just over a quarter of the participants (26 of 100; 26%) were black/African American, compared with the US Census Bureau’s national census level of 13.3% and 13.4% in Allegheny County, Pennsylvania .29
The lack of employed participants in this study could be because many were not able to work because of the advanced stage of their disease. The low level of partnered status is a limitation, although one study site was a low-income hospital where one generally tends to see higher levels of unpartnered status. This study did not control for demographic information such as gender or age, thus, the relationships between the primary outcome variables and financial hardship may be overestimated. Moreover, this analysis of financial distress is limited to the context of the United States due to our lack of universal health care and unique payment system. Although we included only patients who were in the palliative phase of cancer treatment, no medical record review was conducted to determine previous cancer history and treatments, which might have provided more insight into other financial loss or cost of cancer treatment. Furthermore, we note that it can be difficult to prognosticate with accuracy and identify that some patients with advanced cancer may have been excluded from the study due to the inclusion criteria of less than 1 year of survival.
Conclusion
Perceived financial hardship is an important assessment of the burden placed on patients due to the cost of disease; and is a good start in assessing indirect costs that patients take on when coping with advanced stages of cancer and can shed light on an aspect of distress experienced by this patient population that is not commonly addressed. Subjective measures of perceived financial hardship complement objective measures that are commonly indicative of economic resources and can further our understanding of the impact of financial distress experienced by patients with cancer. Further study of financial impacts of advanced cancer as well as predictors of financial distress are essential to the early identification of financial hardship and the development of interventions to support those at high risk or experiencing financial distress.
Acknowledgments
The authors acknowledge the patients and staff at the UPMC Mercy Cancer Center in Pittsburgh, Pennsylvania, who made this study possible, and Peggy Tate for her role in data collection. They also recognize the support of the Robert Wood Johnson Foundation through the Future Nursing Scholars program. They would also like to acknowledge that permission was granted for the use of the Psychological Sense of Economic Hardship study instrument.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf. Atlanta, GA: American Cancer Society. 2015. Accessed January 16, 2016.
2. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology distress management version 1.2014. http://williams.medicine.wisc.edu/distress.pdf. Updated May 2014. Accessed January 16, 2016.
3. de Souza JA, Wong Y-N. Financial distress in cancer patients. J Med Person. 2014;11(2):13-15.
4. Mcdougall JA, Ramsey SD, Hutchinson F, Shih Y-CT. Financial toxicity: a growing concern among cancer patients in the United States. ISPOR Connect. 2014;20(2):10-11.
5. Sharpe K, Shaw B, Seiler MB. Practical solutions when facing cost sharing: the American Cancer Society’s health insurance assistance service. Am J Manag Care. 2016;22(4):92-94.
6. Shankaran V, Jolly S, Blough D, Ramsey SD. Risk factors for financial hardship in patients receiving adjuvant chemotherapy for colon cancer : a population-based exploratory analysis. J Clin Oncol. 2012;30(14):1608-1614.
7. Meneses K, Azuero A, Hassey L, Mcnees P, Pisu M. Does economic burden influence quality of life in breast cancer survivors? Gynecol Oncol. 2012;124(3):437-443.
8. Catt S, Starkings R, Shilling V, Fallowfield L. Patient-reported outcome measures of the impact of cancer on patients’ everyday lives: a systematic review. J Cancer Surviv. 2017;11(2):211-232.
9. Delgado-Guay M, Ferrer J, Rieber AG, et al. Financial distress and its associations with physical and emotional symptoms and quality of life among advanced cancer patients. Oncologist. 2015;20:1092-1098.
10. Kale HP, Carroll N V. Self-reported financial burden of cancer care and its effect on physical and mental health-related quality of life among US cancer survivors. Cancer. 2016;122:1283-1289.
11. Lathan CS, Cronin A, Tucker-Seeley R, Zafar SY, Ayanian JZ, Schrag D. Association of financial strain with symptom burden and quality of life for patients with lung or colorectal cancer. J Clin Oncol. 2016;34(15):1732-1740.
12. Fenn KM, Evans SB, Mccorkle R, et al. Impact of financial burden of cancer on survivors’ quality of life. J Onocol Pract. 2014;10(5):332-339.
13. Azzani M, Roslani AC, Su TT. The perceived cancer-related financial hardship among patients and their families: a systematic review. Support Cancer Care. 2015;23:889-898.
14. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients a multicenter evaluation of the distress thermometer. Cancer. 2005;103:1494-1502.
15. Ransom S, Jacobsen PB, Booth-Jones M. Validation of the distress thermometer with bone marrow. Psychooncology. 2006;15:604-612.
16. Vodermaier A, Linden W, Siu C. Screening for emotional distress in cancer patients: a systematic review of assessment instruments. J Natl Cancer Inst. 2009;101:1464-1488.
17. McCorkle R, Quint-Benoliel J. Symptom distress, current concerns and mood disturbance after diagnosis of life-threatening disease. Soc Sci Med. 1983;17(7):431–8.
18. Cella DF, Tulsky DS, Gray G, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. J Clin Oncol. 1993;11:570-579.
19. Peterman AH, Fitchett G, Brady MJ, Hernandez L, Cella D. Measuring spiritual well-being in people with cancer: the functional assessment of chronic illness therapy – Spiritual Well-being Scale (FACIT-Sp). Ann Behav Med. 2002;24:49–58.
20. Barrera M, Caples H, Tein J. The psychological sense of economic hardship: measurement models, validity, and cross-ethnic equivalence for urban families. Am J Community Psychol. 2001;29:493-517.
21. Harris PA, Taylor R, Thielke R. Research electronic data capture (REDCap): A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
22. Zafar SY, McNeil RB, Thomas CM, Lathan CS, Ayanian JZ, Provenzale D. Population-based assessment of cancer survivors’ financial burden and quality of life: a prospective cohort study. J Oncol Pract. 2015;11(2):145-152.
23. Kent EE, Forsythe LP, Yabroff KR, et al. Are survivors who report cancer-related financial problems more likely to forgo or delay medical care? Cancer. 2013;119:3710-3717.
24. Chino F, Peppercorn J, Taylor Jr. DH, et al. Self-reported financial burden and satisfaction with care among patients with cancer. Oncologist. 2014;19:414-420.
25. De Souza JA, Yap BJ, Hlubocky FJ, et al. The development of a financial toxicity patient-reported outcome in cancer. Cancer. 2014;120:3245-3253.
26. Tucker-Seeley RD, Yabroff KR. Minimizing the “financial toxicity” associated with cancer care : advancing the research agenda. J Natl Cancer Inst. 2016;108(5):1-3.
27. Bestvina CM, Zullig LL, Rushing C, et al. Patient-oncologist cost communication, financial distress, and medication adherence. J Oncol Pract. 2014;10(3):162-168.
28. Irwin B, Kimmick G, Altomare I, et al. Patient experience and attitudes toward addressing the cost of breast cancer care. Oncologist. 2014;19:1135-1140.
29. US Census Bureau. United States. https://www.census.gov/quickfacts/. 2015.
The American Cancer Society has identified a disparity in cancer death rates, noting that persons with lower socioeconomic status have higher rates of mortality.1 This is attributed to many factors, but it is largely owing to the higher burden of disease among lower-income individuals.1 A component of this disease burden is measured by assessing the patient-reported outcome of cancer-related distress. The National Comprehensive Cancer Network (NCCN) Distress Management Guidelines have defined distress as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social and/or spiritual nature that may interfere with the ability to cope with cancer, its physical symptoms and its treatment.”2
Financial hardship related to cancer diagnosis and treatment is increasingly being recognized as an important component of disease burden and distress. The advancements in costly cancer treatments have produced burdensome direct medical costs as well as numerous indirect costs that contribute to perceived financial hardship.3,4 These indirect costs include nonmedical expenses such as increased transportation needs or childcare, loss of earnings, or loss of household income due to caregiving needs.3 Moreover, indirect costs are often managed by patients and families through their use of savings, borrowing, reducing leisure activities, and selling possessions.3 Even though efforts to increase health coverage, such as the Affordable Care Act, have reduced the rates of individuals who are uninsured, persons with cancer who have insurance also face challenges because they cannot afford copays, monthly premiums, deductibles, and other high out-of-pocket expenses related to cancer treatment that are not covered by their insurance such as out-of-network services or providers.5-7
Thus, financial hardship may have an impact on several areas of a patient’s life and well-being, but the effects are commonly undetected.8-10 Research has established that financial strain can influence treatment choices and adherence to therapy.11 Furthermore, the effects of financial strain have been identified across the cancer care continuum, from diagnosis through survivorship, suggesting a bidirectional relationship between financial strain and well-being.11 Financial strain may reduce patient quality of life and worsen symptom burden because of the patient’s inability to access needed care, poor social supports, and/or increased stress.11-12 These worsening outcomes may also increase the use of financial reserves and affect their ability to work.7,11 Financial difficulties may also be associated with anxiety and depression, leading to worse quality of life and greater distress and symptom burden.12 Identifying groups at high risk for financial strain is crucial to ensure that resources are available to assist these populations.13 This burden can be even more pronounced in minority and underserved patients with cancer.7 Patients with advanced cancer are especially vulnerable to the burden of increased costs because of the use of expensive targeted therapies; their improved survival, which extends the time of expenditure; and increased use of financial reserves.9 Financial hardship in patients with advanced cancer is not well understood or characterized,9 which is why this study aimed to better quantify distress in advanced stage cancers by describing :
▪ A cohort of patients with advanced cancer and their levels of quality of life, symptom distress, cancer-related distress and perceived financial hardship;
▪ The relationship between perceived financial hardship, quality of life, symptom distress and overall cancer-related distress; and
▪ Quality of life, symptom distress, and overall cancer-related distress according to level of perceived financial hardship.
Methods
This study is a cross-sectional, descriptive, comparative study of distress, including perceived financial hardship, among patients with advanced cancer who were receiving palliative care treatment in two outpatient medical oncology clinics in Western Pennsylvania. The data were collected during May 2013-November 2014. The study protocol was approved by the Institutional Review Board at the University of Pittsburgh. Eligible participants had to be 18 years or older and have an advanced solid tumor of any kind, with a prognosis of 1 year or less confirmed by a physician or clinic nurse practitioner/physician assistant, and be able to read and understand English at the fourth-grade level. The sample was recruited from two clinics at the University of Pittsburgh Cancer Institute, a National Cancer Institute-designated Comprehensive Cancer Program.
Measurements
Sociodemographic factors. These were measured using an investigator-derived Sociodemographic Questionnaire, a 12-item form that includes variables such as age, race, marital status, cancer type, religion and spirituality, employment status, years of education, health insurance status, and income level.
Cancer-related distress. The NCCN Distress Thermometer is a self-report visual analog scale (0, no distress; 10, great distress) formed in the shape of a thermometer combined with a problem list that is often used in outpatient cancer settings for reporting of cancer-related distress.14-16 The sensitivity, specificity and convergent validity with the Brief Symptom Inventory and the Hospital Anxiety and Depression Scale have been established and appropriate cut-off score of the distress thermometer identified.14-16 A score of 4 or above indicates a clinically significant level of distress.14-16
Symptom distress. The McCorkle Symptom Distress Scale was developed in 1977 based on interviews that focused on the symptom experiences of patients. Psychometric testing among patients with cancer using the modified Symptom Distress Scale revealed high reliability (Cronbach alpha, 0.97).17 The instrument is a 13-item Likert scale (1-5) assessing the severity of distress experienced by a symptom. Total scores range from 13 to 65, where a higher score indicates greater distress. Moderate distress is indicated with a score of 25-33, and a score above 33 indicates severe distress, identifying the need for immediate intervention.17
Quality of life and spiritual well-being. The Functional Assessment of Cancer Therapy (FACT-G) is used to assess general cancer-related quality of life. It has four subscales: physical, emotional, social and family, and functional well-being, with a total score that ranges from 0-112, where higher scores show higher quality of life. The Spiritual Distress Well-Being questionnaire was used alongside the valid FACT-G assessment.18,19 The Spiritual Well-Being Short Form was developed with an ethnically diverse population and adds 12 items to the FACT-G. The items do not necessarily assume a faith in God, allowing a wide flexibility in application and tapping into issues such as faith, meaning, and finding peace and comfort despite advanced illness. Higher scores on the Spiritual Well-Being subscore (range, 0-48) are correlated with higher scores of quality of life. The possible scores for the combined FACT-G and Spiritual Well-Being assessment range from 0-160, with higher scores showing higher quality of life.
Economic hardship. Perceived financial hardship was measured using Barrera and colleagues’ Psychological Sense of Economic Hardship Scale. 20 The scale consists of 20-items broken down into 4 subscales: financial strain, inability to make ends meet, not enough money for necessities, and economic adjustments.20 Economic adjustments in the 3 months before administration of the questionnaire were assessed with 9 Yes or No items, such as added another job, received government assistance, or sold possessions to increase income. The subscale of not enough money for necessities was assessed with seven 5-point scale items in which respondents noted whether they felt they had enough money for housing, clothing, home furnishings, and a car over the previous 3 months. Inability to make ends meet included two 5-point scale items that assessed the difficulty in meeting financial demands in the previous 3 months. Financial strain consisted of two 5-point scale items concerned with expecting financial hardships in the coming 3 months. Scores can range from 20-73, with a higher score indicating worse economic hardship.
Data collection and analysis
In-person data collection occurred in the clinical waiting area before the clinician visit or in the treatment room with the patient using a consecutive, convenience sample. The nursing staff checked the clinic lists daily for possible patient participants. Patients with metastatic cancer were identified and then approached for consent. After we had received the patient’s consent, the administration of the instruments took about 20 minutes to complete. The data were then entered and verified in REDCap (Research Electronic Data Capture), which is hosted at the University of Pittsburgh.21The levels of symptom distress, quality of life, perceived financial hardship, and cancer-related distress were described through continuously measured variables. Descriptive statistics, measures of central tendency (mean and median), and dispersion (standard deviation and range), were obtained for the subscales and total scores. Correlation analysis was used to describe the relationship between perceived financial hardship and quality of life, symptom distress, and cancer-related distress. These primary outcome variables were further explored according to the level of dichotomized perceived financial hardship using mean score as the cut point. Independent sample t tests were used to compare patients experiencing high perceived financial hardship with those experiencing low perceived financial hardship.
Results
In all, 100 patients participated in the study. Any missing data points were replaced with the mean score for that variable, although this was minimal in this study. Most of the participants were women (67%), and the average age of the participants was 63.43 years (SD, 13.05; Table 1). Of the total number of participants, 73% were white, 26% were black, and 1% were Asian. Most of the participants were either retired and not working (39%) or disabled or unable to work (34%). Almost all of the participants had some form of insurance, with 99% having either private or public health insurance. A variety of cancer types were represented in this patient population, with higher percentages of breast (25%), gynecologic (10%), lung (19%), and colon/rectal cancer (15%). Of the total number of participants, 35% had annual household incomes below $20,000, and 50% had annual household incomes of more than $20,000. On average, participants had 13.48 years (SD, 2.78) of formal education.
Descriptive statistics for the primary outcome variables can be found in Table 2. The average score for cancer-related distress based on the NCCN Distress Thermometer tool was 4.16 (SD, 3.26). The average score for the McCorkle Symptom Distress measurement was 25.45 (SD, 9.34). For quality of life, the average FACT-G total score was 73.77 (SD, 19.40). Of the FACT-G subscale average scores, physical well-being was 17.35 (SD, 7.50), social/family well-being 24.21 (SD, 5.25), emotional well-being 16.34 (SD, 5.42), and functional well-being 15.87 (SD, 6.78). Participants’ average score for the spiritual well-being measure was 35.20 (SD, 9.25) and the combined FACT-G and spiritual well-being average score was 108.97 (SD, 26.07). The total average score for perceived financial hardship was 35.70 (SD, 13.87), with subscale average scores of 3.44 (SD, 2.36) for financial strain, 5.73 (SD, 1.91) for inability to make ends meet, 16.43 (SD, 8.92) for not enough money for necessities, and 10.63 (SD, 2.70) for economic adjustments.
We conducted a bivariate correlation analysis to assess the relationship between perceived financial hardship and three other primary outcome variables (Table 3). These analyses showed significant low to moderate correlations with overall cancer-related distress (r, 0.439; P < .001), symptom distress (r, 0.409; P < .001) and overall quality of life scores (FACT-G and spiritual well-being combined score: r, -0.323; P < .001).
Forty-three participants reporting high perceived financial hardship experienced worse quality of life overall (FACT-G and spiritual well-being; P = .002), worse FACT-G total scores (P < .001), worse physical well-being (P < .001), worse social/family well-being (P = .029), worse emotional well-being, and no significant difference for functional (P = .082) or spiritual well-being (P = .453), compared with those with lower economic hardship. In overall cancer-related distress, participants with higher perceived financial hardship reported higher levels of cancer-related distress (P < .001) than those with lower perceived financial hardship. For those participants reporting higher perceived financial hardship there was also worse symptom distress (P < .001), compared with those with lower economic hardship (Table 4).
Discussion
Overall, this report provides data to illuminate our understanding of disparities in well-being that may be present in patients with advanced cancer. Our analysis found that patients with advanced cancer who have higher perceived financial hardship have significantly higher overall cancer-related distress, symptom distress, and poorer overall quality of life. In this study’s population of patients with advanced cancer, the most notable areas of economic hardship identified by participants were: not having enough money for necessities in the 3 months before the survey and the inability to make ends meet during the same time span, with difficulty paying bills and not having enough money left at the end of the month being most noteworthy among this study’s patient population. Financial strain and making economic adjustment were not as notable in the category of perceived financial hardship.
In regard to not having enough money, participants most commonly cited not being able to afford everyday necessities such as food, clothing, medical care, or a home, as well as leisure and recreational activities. These findings are further supported with the positive, moderate associations between perceived financial hardship and symptom distress and overall cancer-related distress found in this cohort of patients with advanced cancer and the negative, moderately associated relationship between perceived financial hardship and overall quality of life in this study’s sample.
Although these findings have been confirmed in the literature on cancer-related distress, our findings add to our knowledge on both economic and cancer-related distress exclusively in patients with advanced cancer.9,22 The broader cancer-related distress literature has also found an association between being younger and having a lower household income as risk factors for increased financial hardship; however, the perception of financial strain and magnitude was a more significant predictor of quality of life and perception of overall well-being.6,8-9,12,22-23 Furthermore, patients with cancer who noted having higher financial distress typically reported decreased satisfaction with cancer care which also influenced their adherence to treatment and quality of life.24
Our work now adds the important element of perceived financial hardship to the advanced cancer-related distress puzzle. We should consider integrating a financial distress assessment into routine cancer care, particularly with patients and families with advanced cancer, to proactively and routinely assess and intervene with available distress mitigating resources. Therefore, understanding the patients most likely to experience financial distress will help personalize supportive therapy.
This study’s results as well as the existing literature describing financial distress support the use of comprehensive screening instruments to capture elements of financial burden beyond out-of-pocket costs.8,25 This screening is particularly relevant because we are increasingly recognizing that gross annual household income does not always reflect financial hardship or distress. The instrument we used for this analysis, the Psychological Sense of Economic Hardship, provides a broad view of financial toxicity including the specific components of financial strain, the inability to make ends meet, not having enough money for necessities, and economic adjustments experienced by patients with advanced cancer.20 Another measure to evaluate financial toxicity among patients with cancer includes the Comprehensive Score for Financial Toxicity (COST), which is a widely used patient-reported outcome measure. It was developed with input from both patients and oncology experts.25 Use of a financial toxicity assessment tool adds to our understanding of the economic financial burden experienced by patients with cancer, specifically those with advanced cancer.
Tucker-Seeley and Yabroff have identified several areas in which the research agenda for financial toxicity should focus, including: documentation of the socioeconomic context among patients across all areas of the cancer care continuum, further identification and characterization of at risk populations to address health disparities, and the inclusion of cost discussions in the health care context.26 Furthermore, research is needed to identify key areas to target for interventions addressing financial toxicity, such as addressing lack of financial resources to cover the cost of cancer care, focusing on managing or preventing the distress that results from a lack of financial resources, or addressing coping behaviors used by families to manage the financial burden of cancer care.26 Although cost discussions between health care providers and patients have been identified as important in reducing the financial burden of cancer care, the content, timing, and goals of those discussions still need to be better articulated for different patient populations, including patients with advanced cancer.3,27-28 In addition, resources such as social workers, patient navigators, or financial counselors have been identified as effective in assisting patients with financial planning and accessing community resources to address financial burden and assistance.4
Design considerations
This study has limitations that need to be noted. Its cross-sectional design does not allow for the analysis of causal inferences. In addition, certain groups were underrepresented in this study’s sample, including uninsured patients, men, and some minority groups, which may have underestimated the amount of financial burden experienced by patients with advanced cancer. The lack of representativeness of uninsured individuals may be a result of the eligibility of persons with advanced cancer for Medicaid. However, a strength of this study is its ability to increase the representativeness of African American/black patients in the study of advanced cancer and financial hardship. In our study, just over a quarter of the participants (26 of 100; 26%) were black/African American, compared with the US Census Bureau’s national census level of 13.3% and 13.4% in Allegheny County, Pennsylvania .29
The lack of employed participants in this study could be because many were not able to work because of the advanced stage of their disease. The low level of partnered status is a limitation, although one study site was a low-income hospital where one generally tends to see higher levels of unpartnered status. This study did not control for demographic information such as gender or age, thus, the relationships between the primary outcome variables and financial hardship may be overestimated. Moreover, this analysis of financial distress is limited to the context of the United States due to our lack of universal health care and unique payment system. Although we included only patients who were in the palliative phase of cancer treatment, no medical record review was conducted to determine previous cancer history and treatments, which might have provided more insight into other financial loss or cost of cancer treatment. Furthermore, we note that it can be difficult to prognosticate with accuracy and identify that some patients with advanced cancer may have been excluded from the study due to the inclusion criteria of less than 1 year of survival.
Conclusion
Perceived financial hardship is an important assessment of the burden placed on patients due to the cost of disease; and is a good start in assessing indirect costs that patients take on when coping with advanced stages of cancer and can shed light on an aspect of distress experienced by this patient population that is not commonly addressed. Subjective measures of perceived financial hardship complement objective measures that are commonly indicative of economic resources and can further our understanding of the impact of financial distress experienced by patients with cancer. Further study of financial impacts of advanced cancer as well as predictors of financial distress are essential to the early identification of financial hardship and the development of interventions to support those at high risk or experiencing financial distress.
Acknowledgments
The authors acknowledge the patients and staff at the UPMC Mercy Cancer Center in Pittsburgh, Pennsylvania, who made this study possible, and Peggy Tate for her role in data collection. They also recognize the support of the Robert Wood Johnson Foundation through the Future Nursing Scholars program. They would also like to acknowledge that permission was granted for the use of the Psychological Sense of Economic Hardship study instrument.
The American Cancer Society has identified a disparity in cancer death rates, noting that persons with lower socioeconomic status have higher rates of mortality.1 This is attributed to many factors, but it is largely owing to the higher burden of disease among lower-income individuals.1 A component of this disease burden is measured by assessing the patient-reported outcome of cancer-related distress. The National Comprehensive Cancer Network (NCCN) Distress Management Guidelines have defined distress as “a multifactorial unpleasant emotional experience of a psychological (cognitive, behavioral, emotional), social and/or spiritual nature that may interfere with the ability to cope with cancer, its physical symptoms and its treatment.”2
Financial hardship related to cancer diagnosis and treatment is increasingly being recognized as an important component of disease burden and distress. The advancements in costly cancer treatments have produced burdensome direct medical costs as well as numerous indirect costs that contribute to perceived financial hardship.3,4 These indirect costs include nonmedical expenses such as increased transportation needs or childcare, loss of earnings, or loss of household income due to caregiving needs.3 Moreover, indirect costs are often managed by patients and families through their use of savings, borrowing, reducing leisure activities, and selling possessions.3 Even though efforts to increase health coverage, such as the Affordable Care Act, have reduced the rates of individuals who are uninsured, persons with cancer who have insurance also face challenges because they cannot afford copays, monthly premiums, deductibles, and other high out-of-pocket expenses related to cancer treatment that are not covered by their insurance such as out-of-network services or providers.5-7
Thus, financial hardship may have an impact on several areas of a patient’s life and well-being, but the effects are commonly undetected.8-10 Research has established that financial strain can influence treatment choices and adherence to therapy.11 Furthermore, the effects of financial strain have been identified across the cancer care continuum, from diagnosis through survivorship, suggesting a bidirectional relationship between financial strain and well-being.11 Financial strain may reduce patient quality of life and worsen symptom burden because of the patient’s inability to access needed care, poor social supports, and/or increased stress.11-12 These worsening outcomes may also increase the use of financial reserves and affect their ability to work.7,11 Financial difficulties may also be associated with anxiety and depression, leading to worse quality of life and greater distress and symptom burden.12 Identifying groups at high risk for financial strain is crucial to ensure that resources are available to assist these populations.13 This burden can be even more pronounced in minority and underserved patients with cancer.7 Patients with advanced cancer are especially vulnerable to the burden of increased costs because of the use of expensive targeted therapies; their improved survival, which extends the time of expenditure; and increased use of financial reserves.9 Financial hardship in patients with advanced cancer is not well understood or characterized,9 which is why this study aimed to better quantify distress in advanced stage cancers by describing :
▪ A cohort of patients with advanced cancer and their levels of quality of life, symptom distress, cancer-related distress and perceived financial hardship;
▪ The relationship between perceived financial hardship, quality of life, symptom distress and overall cancer-related distress; and
▪ Quality of life, symptom distress, and overall cancer-related distress according to level of perceived financial hardship.
Methods
This study is a cross-sectional, descriptive, comparative study of distress, including perceived financial hardship, among patients with advanced cancer who were receiving palliative care treatment in two outpatient medical oncology clinics in Western Pennsylvania. The data were collected during May 2013-November 2014. The study protocol was approved by the Institutional Review Board at the University of Pittsburgh. Eligible participants had to be 18 years or older and have an advanced solid tumor of any kind, with a prognosis of 1 year or less confirmed by a physician or clinic nurse practitioner/physician assistant, and be able to read and understand English at the fourth-grade level. The sample was recruited from two clinics at the University of Pittsburgh Cancer Institute, a National Cancer Institute-designated Comprehensive Cancer Program.
Measurements
Sociodemographic factors. These were measured using an investigator-derived Sociodemographic Questionnaire, a 12-item form that includes variables such as age, race, marital status, cancer type, religion and spirituality, employment status, years of education, health insurance status, and income level.
Cancer-related distress. The NCCN Distress Thermometer is a self-report visual analog scale (0, no distress; 10, great distress) formed in the shape of a thermometer combined with a problem list that is often used in outpatient cancer settings for reporting of cancer-related distress.14-16 The sensitivity, specificity and convergent validity with the Brief Symptom Inventory and the Hospital Anxiety and Depression Scale have been established and appropriate cut-off score of the distress thermometer identified.14-16 A score of 4 or above indicates a clinically significant level of distress.14-16
Symptom distress. The McCorkle Symptom Distress Scale was developed in 1977 based on interviews that focused on the symptom experiences of patients. Psychometric testing among patients with cancer using the modified Symptom Distress Scale revealed high reliability (Cronbach alpha, 0.97).17 The instrument is a 13-item Likert scale (1-5) assessing the severity of distress experienced by a symptom. Total scores range from 13 to 65, where a higher score indicates greater distress. Moderate distress is indicated with a score of 25-33, and a score above 33 indicates severe distress, identifying the need for immediate intervention.17
Quality of life and spiritual well-being. The Functional Assessment of Cancer Therapy (FACT-G) is used to assess general cancer-related quality of life. It has four subscales: physical, emotional, social and family, and functional well-being, with a total score that ranges from 0-112, where higher scores show higher quality of life. The Spiritual Distress Well-Being questionnaire was used alongside the valid FACT-G assessment.18,19 The Spiritual Well-Being Short Form was developed with an ethnically diverse population and adds 12 items to the FACT-G. The items do not necessarily assume a faith in God, allowing a wide flexibility in application and tapping into issues such as faith, meaning, and finding peace and comfort despite advanced illness. Higher scores on the Spiritual Well-Being subscore (range, 0-48) are correlated with higher scores of quality of life. The possible scores for the combined FACT-G and Spiritual Well-Being assessment range from 0-160, with higher scores showing higher quality of life.
Economic hardship. Perceived financial hardship was measured using Barrera and colleagues’ Psychological Sense of Economic Hardship Scale. 20 The scale consists of 20-items broken down into 4 subscales: financial strain, inability to make ends meet, not enough money for necessities, and economic adjustments.20 Economic adjustments in the 3 months before administration of the questionnaire were assessed with 9 Yes or No items, such as added another job, received government assistance, or sold possessions to increase income. The subscale of not enough money for necessities was assessed with seven 5-point scale items in which respondents noted whether they felt they had enough money for housing, clothing, home furnishings, and a car over the previous 3 months. Inability to make ends meet included two 5-point scale items that assessed the difficulty in meeting financial demands in the previous 3 months. Financial strain consisted of two 5-point scale items concerned with expecting financial hardships in the coming 3 months. Scores can range from 20-73, with a higher score indicating worse economic hardship.
Data collection and analysis
In-person data collection occurred in the clinical waiting area before the clinician visit or in the treatment room with the patient using a consecutive, convenience sample. The nursing staff checked the clinic lists daily for possible patient participants. Patients with metastatic cancer were identified and then approached for consent. After we had received the patient’s consent, the administration of the instruments took about 20 minutes to complete. The data were then entered and verified in REDCap (Research Electronic Data Capture), which is hosted at the University of Pittsburgh.21The levels of symptom distress, quality of life, perceived financial hardship, and cancer-related distress were described through continuously measured variables. Descriptive statistics, measures of central tendency (mean and median), and dispersion (standard deviation and range), were obtained for the subscales and total scores. Correlation analysis was used to describe the relationship between perceived financial hardship and quality of life, symptom distress, and cancer-related distress. These primary outcome variables were further explored according to the level of dichotomized perceived financial hardship using mean score as the cut point. Independent sample t tests were used to compare patients experiencing high perceived financial hardship with those experiencing low perceived financial hardship.
Results
In all, 100 patients participated in the study. Any missing data points were replaced with the mean score for that variable, although this was minimal in this study. Most of the participants were women (67%), and the average age of the participants was 63.43 years (SD, 13.05; Table 1). Of the total number of participants, 73% were white, 26% were black, and 1% were Asian. Most of the participants were either retired and not working (39%) or disabled or unable to work (34%). Almost all of the participants had some form of insurance, with 99% having either private or public health insurance. A variety of cancer types were represented in this patient population, with higher percentages of breast (25%), gynecologic (10%), lung (19%), and colon/rectal cancer (15%). Of the total number of participants, 35% had annual household incomes below $20,000, and 50% had annual household incomes of more than $20,000. On average, participants had 13.48 years (SD, 2.78) of formal education.
Descriptive statistics for the primary outcome variables can be found in Table 2. The average score for cancer-related distress based on the NCCN Distress Thermometer tool was 4.16 (SD, 3.26). The average score for the McCorkle Symptom Distress measurement was 25.45 (SD, 9.34). For quality of life, the average FACT-G total score was 73.77 (SD, 19.40). Of the FACT-G subscale average scores, physical well-being was 17.35 (SD, 7.50), social/family well-being 24.21 (SD, 5.25), emotional well-being 16.34 (SD, 5.42), and functional well-being 15.87 (SD, 6.78). Participants’ average score for the spiritual well-being measure was 35.20 (SD, 9.25) and the combined FACT-G and spiritual well-being average score was 108.97 (SD, 26.07). The total average score for perceived financial hardship was 35.70 (SD, 13.87), with subscale average scores of 3.44 (SD, 2.36) for financial strain, 5.73 (SD, 1.91) for inability to make ends meet, 16.43 (SD, 8.92) for not enough money for necessities, and 10.63 (SD, 2.70) for economic adjustments.
We conducted a bivariate correlation analysis to assess the relationship between perceived financial hardship and three other primary outcome variables (Table 3). These analyses showed significant low to moderate correlations with overall cancer-related distress (r, 0.439; P < .001), symptom distress (r, 0.409; P < .001) and overall quality of life scores (FACT-G and spiritual well-being combined score: r, -0.323; P < .001).
Forty-three participants reporting high perceived financial hardship experienced worse quality of life overall (FACT-G and spiritual well-being; P = .002), worse FACT-G total scores (P < .001), worse physical well-being (P < .001), worse social/family well-being (P = .029), worse emotional well-being, and no significant difference for functional (P = .082) or spiritual well-being (P = .453), compared with those with lower economic hardship. In overall cancer-related distress, participants with higher perceived financial hardship reported higher levels of cancer-related distress (P < .001) than those with lower perceived financial hardship. For those participants reporting higher perceived financial hardship there was also worse symptom distress (P < .001), compared with those with lower economic hardship (Table 4).
Discussion
Overall, this report provides data to illuminate our understanding of disparities in well-being that may be present in patients with advanced cancer. Our analysis found that patients with advanced cancer who have higher perceived financial hardship have significantly higher overall cancer-related distress, symptom distress, and poorer overall quality of life. In this study’s population of patients with advanced cancer, the most notable areas of economic hardship identified by participants were: not having enough money for necessities in the 3 months before the survey and the inability to make ends meet during the same time span, with difficulty paying bills and not having enough money left at the end of the month being most noteworthy among this study’s patient population. Financial strain and making economic adjustment were not as notable in the category of perceived financial hardship.
In regard to not having enough money, participants most commonly cited not being able to afford everyday necessities such as food, clothing, medical care, or a home, as well as leisure and recreational activities. These findings are further supported with the positive, moderate associations between perceived financial hardship and symptom distress and overall cancer-related distress found in this cohort of patients with advanced cancer and the negative, moderately associated relationship between perceived financial hardship and overall quality of life in this study’s sample.
Although these findings have been confirmed in the literature on cancer-related distress, our findings add to our knowledge on both economic and cancer-related distress exclusively in patients with advanced cancer.9,22 The broader cancer-related distress literature has also found an association between being younger and having a lower household income as risk factors for increased financial hardship; however, the perception of financial strain and magnitude was a more significant predictor of quality of life and perception of overall well-being.6,8-9,12,22-23 Furthermore, patients with cancer who noted having higher financial distress typically reported decreased satisfaction with cancer care which also influenced their adherence to treatment and quality of life.24
Our work now adds the important element of perceived financial hardship to the advanced cancer-related distress puzzle. We should consider integrating a financial distress assessment into routine cancer care, particularly with patients and families with advanced cancer, to proactively and routinely assess and intervene with available distress mitigating resources. Therefore, understanding the patients most likely to experience financial distress will help personalize supportive therapy.
This study’s results as well as the existing literature describing financial distress support the use of comprehensive screening instruments to capture elements of financial burden beyond out-of-pocket costs.8,25 This screening is particularly relevant because we are increasingly recognizing that gross annual household income does not always reflect financial hardship or distress. The instrument we used for this analysis, the Psychological Sense of Economic Hardship, provides a broad view of financial toxicity including the specific components of financial strain, the inability to make ends meet, not having enough money for necessities, and economic adjustments experienced by patients with advanced cancer.20 Another measure to evaluate financial toxicity among patients with cancer includes the Comprehensive Score for Financial Toxicity (COST), which is a widely used patient-reported outcome measure. It was developed with input from both patients and oncology experts.25 Use of a financial toxicity assessment tool adds to our understanding of the economic financial burden experienced by patients with cancer, specifically those with advanced cancer.
Tucker-Seeley and Yabroff have identified several areas in which the research agenda for financial toxicity should focus, including: documentation of the socioeconomic context among patients across all areas of the cancer care continuum, further identification and characterization of at risk populations to address health disparities, and the inclusion of cost discussions in the health care context.26 Furthermore, research is needed to identify key areas to target for interventions addressing financial toxicity, such as addressing lack of financial resources to cover the cost of cancer care, focusing on managing or preventing the distress that results from a lack of financial resources, or addressing coping behaviors used by families to manage the financial burden of cancer care.26 Although cost discussions between health care providers and patients have been identified as important in reducing the financial burden of cancer care, the content, timing, and goals of those discussions still need to be better articulated for different patient populations, including patients with advanced cancer.3,27-28 In addition, resources such as social workers, patient navigators, or financial counselors have been identified as effective in assisting patients with financial planning and accessing community resources to address financial burden and assistance.4
Design considerations
This study has limitations that need to be noted. Its cross-sectional design does not allow for the analysis of causal inferences. In addition, certain groups were underrepresented in this study’s sample, including uninsured patients, men, and some minority groups, which may have underestimated the amount of financial burden experienced by patients with advanced cancer. The lack of representativeness of uninsured individuals may be a result of the eligibility of persons with advanced cancer for Medicaid. However, a strength of this study is its ability to increase the representativeness of African American/black patients in the study of advanced cancer and financial hardship. In our study, just over a quarter of the participants (26 of 100; 26%) were black/African American, compared with the US Census Bureau’s national census level of 13.3% and 13.4% in Allegheny County, Pennsylvania .29
The lack of employed participants in this study could be because many were not able to work because of the advanced stage of their disease. The low level of partnered status is a limitation, although one study site was a low-income hospital where one generally tends to see higher levels of unpartnered status. This study did not control for demographic information such as gender or age, thus, the relationships between the primary outcome variables and financial hardship may be overestimated. Moreover, this analysis of financial distress is limited to the context of the United States due to our lack of universal health care and unique payment system. Although we included only patients who were in the palliative phase of cancer treatment, no medical record review was conducted to determine previous cancer history and treatments, which might have provided more insight into other financial loss or cost of cancer treatment. Furthermore, we note that it can be difficult to prognosticate with accuracy and identify that some patients with advanced cancer may have been excluded from the study due to the inclusion criteria of less than 1 year of survival.
Conclusion
Perceived financial hardship is an important assessment of the burden placed on patients due to the cost of disease; and is a good start in assessing indirect costs that patients take on when coping with advanced stages of cancer and can shed light on an aspect of distress experienced by this patient population that is not commonly addressed. Subjective measures of perceived financial hardship complement objective measures that are commonly indicative of economic resources and can further our understanding of the impact of financial distress experienced by patients with cancer. Further study of financial impacts of advanced cancer as well as predictors of financial distress are essential to the early identification of financial hardship and the development of interventions to support those at high risk or experiencing financial distress.
Acknowledgments
The authors acknowledge the patients and staff at the UPMC Mercy Cancer Center in Pittsburgh, Pennsylvania, who made this study possible, and Peggy Tate for her role in data collection. They also recognize the support of the Robert Wood Johnson Foundation through the Future Nursing Scholars program. They would also like to acknowledge that permission was granted for the use of the Psychological Sense of Economic Hardship study instrument.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf. Atlanta, GA: American Cancer Society. 2015. Accessed January 16, 2016.
2. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology distress management version 1.2014. http://williams.medicine.wisc.edu/distress.pdf. Updated May 2014. Accessed January 16, 2016.
3. de Souza JA, Wong Y-N. Financial distress in cancer patients. J Med Person. 2014;11(2):13-15.
4. Mcdougall JA, Ramsey SD, Hutchinson F, Shih Y-CT. Financial toxicity: a growing concern among cancer patients in the United States. ISPOR Connect. 2014;20(2):10-11.
5. Sharpe K, Shaw B, Seiler MB. Practical solutions when facing cost sharing: the American Cancer Society’s health insurance assistance service. Am J Manag Care. 2016;22(4):92-94.
6. Shankaran V, Jolly S, Blough D, Ramsey SD. Risk factors for financial hardship in patients receiving adjuvant chemotherapy for colon cancer : a population-based exploratory analysis. J Clin Oncol. 2012;30(14):1608-1614.
7. Meneses K, Azuero A, Hassey L, Mcnees P, Pisu M. Does economic burden influence quality of life in breast cancer survivors? Gynecol Oncol. 2012;124(3):437-443.
8. Catt S, Starkings R, Shilling V, Fallowfield L. Patient-reported outcome measures of the impact of cancer on patients’ everyday lives: a systematic review. J Cancer Surviv. 2017;11(2):211-232.
9. Delgado-Guay M, Ferrer J, Rieber AG, et al. Financial distress and its associations with physical and emotional symptoms and quality of life among advanced cancer patients. Oncologist. 2015;20:1092-1098.
10. Kale HP, Carroll N V. Self-reported financial burden of cancer care and its effect on physical and mental health-related quality of life among US cancer survivors. Cancer. 2016;122:1283-1289.
11. Lathan CS, Cronin A, Tucker-Seeley R, Zafar SY, Ayanian JZ, Schrag D. Association of financial strain with symptom burden and quality of life for patients with lung or colorectal cancer. J Clin Oncol. 2016;34(15):1732-1740.
12. Fenn KM, Evans SB, Mccorkle R, et al. Impact of financial burden of cancer on survivors’ quality of life. J Onocol Pract. 2014;10(5):332-339.
13. Azzani M, Roslani AC, Su TT. The perceived cancer-related financial hardship among patients and their families: a systematic review. Support Cancer Care. 2015;23:889-898.
14. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients a multicenter evaluation of the distress thermometer. Cancer. 2005;103:1494-1502.
15. Ransom S, Jacobsen PB, Booth-Jones M. Validation of the distress thermometer with bone marrow. Psychooncology. 2006;15:604-612.
16. Vodermaier A, Linden W, Siu C. Screening for emotional distress in cancer patients: a systematic review of assessment instruments. J Natl Cancer Inst. 2009;101:1464-1488.
17. McCorkle R, Quint-Benoliel J. Symptom distress, current concerns and mood disturbance after diagnosis of life-threatening disease. Soc Sci Med. 1983;17(7):431–8.
18. Cella DF, Tulsky DS, Gray G, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. J Clin Oncol. 1993;11:570-579.
19. Peterman AH, Fitchett G, Brady MJ, Hernandez L, Cella D. Measuring spiritual well-being in people with cancer: the functional assessment of chronic illness therapy – Spiritual Well-being Scale (FACIT-Sp). Ann Behav Med. 2002;24:49–58.
20. Barrera M, Caples H, Tein J. The psychological sense of economic hardship: measurement models, validity, and cross-ethnic equivalence for urban families. Am J Community Psychol. 2001;29:493-517.
21. Harris PA, Taylor R, Thielke R. Research electronic data capture (REDCap): A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
22. Zafar SY, McNeil RB, Thomas CM, Lathan CS, Ayanian JZ, Provenzale D. Population-based assessment of cancer survivors’ financial burden and quality of life: a prospective cohort study. J Oncol Pract. 2015;11(2):145-152.
23. Kent EE, Forsythe LP, Yabroff KR, et al. Are survivors who report cancer-related financial problems more likely to forgo or delay medical care? Cancer. 2013;119:3710-3717.
24. Chino F, Peppercorn J, Taylor Jr. DH, et al. Self-reported financial burden and satisfaction with care among patients with cancer. Oncologist. 2014;19:414-420.
25. De Souza JA, Yap BJ, Hlubocky FJ, et al. The development of a financial toxicity patient-reported outcome in cancer. Cancer. 2014;120:3245-3253.
26. Tucker-Seeley RD, Yabroff KR. Minimizing the “financial toxicity” associated with cancer care : advancing the research agenda. J Natl Cancer Inst. 2016;108(5):1-3.
27. Bestvina CM, Zullig LL, Rushing C, et al. Patient-oncologist cost communication, financial distress, and medication adherence. J Oncol Pract. 2014;10(3):162-168.
28. Irwin B, Kimmick G, Altomare I, et al. Patient experience and attitudes toward addressing the cost of breast cancer care. Oncologist. 2014;19:1135-1140.
29. US Census Bureau. United States. https://www.census.gov/quickfacts/. 2015.
1. American Cancer Society. Cancer Facts & Figures 2015. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2015/cancer-facts-and-figures-2015.pdf. Atlanta, GA: American Cancer Society. 2015. Accessed January 16, 2016.
2. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology distress management version 1.2014. http://williams.medicine.wisc.edu/distress.pdf. Updated May 2014. Accessed January 16, 2016.
3. de Souza JA, Wong Y-N. Financial distress in cancer patients. J Med Person. 2014;11(2):13-15.
4. Mcdougall JA, Ramsey SD, Hutchinson F, Shih Y-CT. Financial toxicity: a growing concern among cancer patients in the United States. ISPOR Connect. 2014;20(2):10-11.
5. Sharpe K, Shaw B, Seiler MB. Practical solutions when facing cost sharing: the American Cancer Society’s health insurance assistance service. Am J Manag Care. 2016;22(4):92-94.
6. Shankaran V, Jolly S, Blough D, Ramsey SD. Risk factors for financial hardship in patients receiving adjuvant chemotherapy for colon cancer : a population-based exploratory analysis. J Clin Oncol. 2012;30(14):1608-1614.
7. Meneses K, Azuero A, Hassey L, Mcnees P, Pisu M. Does economic burden influence quality of life in breast cancer survivors? Gynecol Oncol. 2012;124(3):437-443.
8. Catt S, Starkings R, Shilling V, Fallowfield L. Patient-reported outcome measures of the impact of cancer on patients’ everyday lives: a systematic review. J Cancer Surviv. 2017;11(2):211-232.
9. Delgado-Guay M, Ferrer J, Rieber AG, et al. Financial distress and its associations with physical and emotional symptoms and quality of life among advanced cancer patients. Oncologist. 2015;20:1092-1098.
10. Kale HP, Carroll N V. Self-reported financial burden of cancer care and its effect on physical and mental health-related quality of life among US cancer survivors. Cancer. 2016;122:1283-1289.
11. Lathan CS, Cronin A, Tucker-Seeley R, Zafar SY, Ayanian JZ, Schrag D. Association of financial strain with symptom burden and quality of life for patients with lung or colorectal cancer. J Clin Oncol. 2016;34(15):1732-1740.
12. Fenn KM, Evans SB, Mccorkle R, et al. Impact of financial burden of cancer on survivors’ quality of life. J Onocol Pract. 2014;10(5):332-339.
13. Azzani M, Roslani AC, Su TT. The perceived cancer-related financial hardship among patients and their families: a systematic review. Support Cancer Care. 2015;23:889-898.
14. Jacobsen PB, Donovan KA, Trask PC, et al. Screening for psychologic distress in ambulatory cancer patients a multicenter evaluation of the distress thermometer. Cancer. 2005;103:1494-1502.
15. Ransom S, Jacobsen PB, Booth-Jones M. Validation of the distress thermometer with bone marrow. Psychooncology. 2006;15:604-612.
16. Vodermaier A, Linden W, Siu C. Screening for emotional distress in cancer patients: a systematic review of assessment instruments. J Natl Cancer Inst. 2009;101:1464-1488.
17. McCorkle R, Quint-Benoliel J. Symptom distress, current concerns and mood disturbance after diagnosis of life-threatening disease. Soc Sci Med. 1983;17(7):431–8.
18. Cella DF, Tulsky DS, Gray G, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. J Clin Oncol. 1993;11:570-579.
19. Peterman AH, Fitchett G, Brady MJ, Hernandez L, Cella D. Measuring spiritual well-being in people with cancer: the functional assessment of chronic illness therapy – Spiritual Well-being Scale (FACIT-Sp). Ann Behav Med. 2002;24:49–58.
20. Barrera M, Caples H, Tein J. The psychological sense of economic hardship: measurement models, validity, and cross-ethnic equivalence for urban families. Am J Community Psychol. 2001;29:493-517.
21. Harris PA, Taylor R, Thielke R. Research electronic data capture (REDCap): A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377-381.
22. Zafar SY, McNeil RB, Thomas CM, Lathan CS, Ayanian JZ, Provenzale D. Population-based assessment of cancer survivors’ financial burden and quality of life: a prospective cohort study. J Oncol Pract. 2015;11(2):145-152.
23. Kent EE, Forsythe LP, Yabroff KR, et al. Are survivors who report cancer-related financial problems more likely to forgo or delay medical care? Cancer. 2013;119:3710-3717.
24. Chino F, Peppercorn J, Taylor Jr. DH, et al. Self-reported financial burden and satisfaction with care among patients with cancer. Oncologist. 2014;19:414-420.
25. De Souza JA, Yap BJ, Hlubocky FJ, et al. The development of a financial toxicity patient-reported outcome in cancer. Cancer. 2014;120:3245-3253.
26. Tucker-Seeley RD, Yabroff KR. Minimizing the “financial toxicity” associated with cancer care : advancing the research agenda. J Natl Cancer Inst. 2016;108(5):1-3.
27. Bestvina CM, Zullig LL, Rushing C, et al. Patient-oncologist cost communication, financial distress, and medication adherence. J Oncol Pract. 2014;10(3):162-168.
28. Irwin B, Kimmick G, Altomare I, et al. Patient experience and attitudes toward addressing the cost of breast cancer care. Oncologist. 2014;19:1135-1140.
29. US Census Bureau. United States. https://www.census.gov/quickfacts/. 2015.
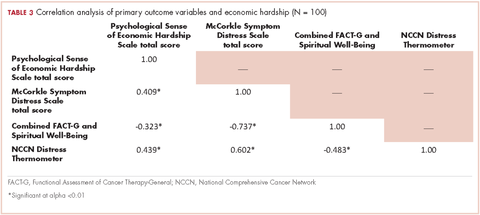
David Henry's JCSO podcast, July-August 2017
For the July-August issue of the Journal of Community and Supportive Oncology, the Editor-in-Chief, Dr David Henry, discusses a recap by Howard Burris, MD, of the top presentations at this year’s annual meeting of the American Society for Clinical Oncology, and a selection of articles on some of the key findings reported at the meeting. A number of articles, in keeping with the journal mission of delivering content that can inform or change daily practice in the community setting, provide ‘how-to’ clinical and supportive advice. They include an outline by Thomas J Smith of Johns Hopkins University of how to initiate goals-of-care conversations with patients and their family members; a review of managing polycythemia vera in the community oncology setting; a New Therapies feature on how immunotherapies are shaping the treatment of hematologic malignancies; and research articles on using Onodera’s Prognostic Nutritional Index to predict wound complications in patients with soft tissue sarcoma, and bone remodeling associated with CTLA-4 inhibition. A third research article assesses a multidisciplinary survivorship program in a group of predominantly Hispanic women with breast cancer. Also in the line-up for discussion are Case Reports, one on managing high-grade pleomorphic sarcoma in a patient with colon metastasis and another on intramedullary spinal cord and leptomeningeal metastases presenting as cauda equina syndrome in a patient with melanoma.
Listen to the podcast below.
For the July-August issue of the Journal of Community and Supportive Oncology, the Editor-in-Chief, Dr David Henry, discusses a recap by Howard Burris, MD, of the top presentations at this year’s annual meeting of the American Society for Clinical Oncology, and a selection of articles on some of the key findings reported at the meeting. A number of articles, in keeping with the journal mission of delivering content that can inform or change daily practice in the community setting, provide ‘how-to’ clinical and supportive advice. They include an outline by Thomas J Smith of Johns Hopkins University of how to initiate goals-of-care conversations with patients and their family members; a review of managing polycythemia vera in the community oncology setting; a New Therapies feature on how immunotherapies are shaping the treatment of hematologic malignancies; and research articles on using Onodera’s Prognostic Nutritional Index to predict wound complications in patients with soft tissue sarcoma, and bone remodeling associated with CTLA-4 inhibition. A third research article assesses a multidisciplinary survivorship program in a group of predominantly Hispanic women with breast cancer. Also in the line-up for discussion are Case Reports, one on managing high-grade pleomorphic sarcoma in a patient with colon metastasis and another on intramedullary spinal cord and leptomeningeal metastases presenting as cauda equina syndrome in a patient with melanoma.
Listen to the podcast below.
For the July-August issue of the Journal of Community and Supportive Oncology, the Editor-in-Chief, Dr David Henry, discusses a recap by Howard Burris, MD, of the top presentations at this year’s annual meeting of the American Society for Clinical Oncology, and a selection of articles on some of the key findings reported at the meeting. A number of articles, in keeping with the journal mission of delivering content that can inform or change daily practice in the community setting, provide ‘how-to’ clinical and supportive advice. They include an outline by Thomas J Smith of Johns Hopkins University of how to initiate goals-of-care conversations with patients and their family members; a review of managing polycythemia vera in the community oncology setting; a New Therapies feature on how immunotherapies are shaping the treatment of hematologic malignancies; and research articles on using Onodera’s Prognostic Nutritional Index to predict wound complications in patients with soft tissue sarcoma, and bone remodeling associated with CTLA-4 inhibition. A third research article assesses a multidisciplinary survivorship program in a group of predominantly Hispanic women with breast cancer. Also in the line-up for discussion are Case Reports, one on managing high-grade pleomorphic sarcoma in a patient with colon metastasis and another on intramedullary spinal cord and leptomeningeal metastases presenting as cauda equina syndrome in a patient with melanoma.
Listen to the podcast below.
Pancreatitis associated with newer classes of antineoplastic therapies
Patients with advanced malignancies may develop pancreatitis during therapy for their cancer. Acute pancreatitis is inflammation of the pancreas. Common symptoms include abdominal pain, nausea, vomiting, shortness of breath, dehydration. Laboratory evidence of acute pancreatitis includes elevations of the amylase and lipase. Mild pancreatitis occurs when there is no organ dysfunction, moderate pancreatitis is associated with one organ dysfunction, and severe pancreatitis is complicated by multiple organ dysfunction. Hypotension, hypocalcemia, or anemia suggest a more severe course of the pancreatitis. In some instances, the pancreatitis may be an adverse reaction to the therapy being given. However, other causes such as hypercalcemia, hypertriglyceridemia, cholelithiasis, and underlying malignancy must be ruled out before ascribing pancreatitis to a specific drug. To date, two classifications systems have been proposed by Trivedi1 and Badalov2 to evaluate the degree to which a drug is responsible for pancreatitis (Table 1). Furthermore, Naranjo and colleagues have proposed a more general method of assessing the causal relationship between drugs and adverse events.3 The Naranjo algorithm is not specific for pancreatitis. Jones and colleagues4 reported that 0.1%-2% of acute pancreatitis cases were owing to drugs. In 2015, they listed the older chemotherapy agents associated with pancreatitis. However, more recently, many new agents have been approved for the management of cancers. The newer classes of antineoplastic agents including proteasome inhibitors, immune-modulating agents, tyrosine kinase inhibitors, monoclonal antibodies against programmed cell death-1 (PD-1) and its ligand PD-L1 and antibody-toxin conjugates are now associated with acute pancreatitis.
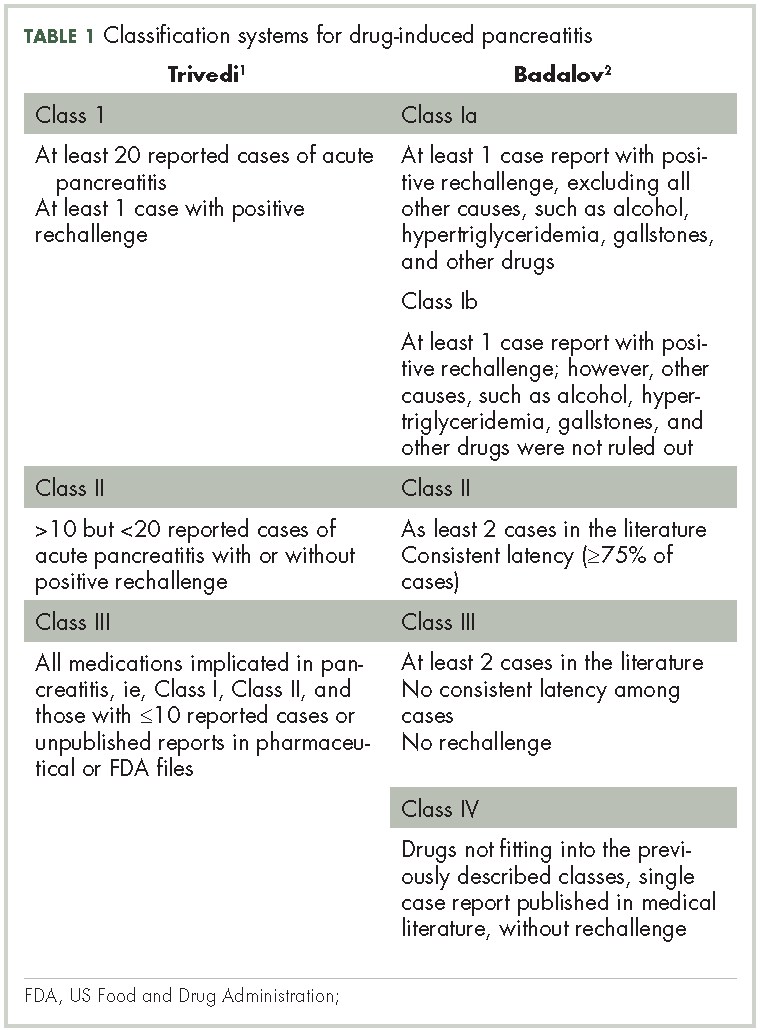
Methods
We conducted a search in PubMed, Google Scholar, and Micromedex for pancreatitis related to antineoplastic agents, including proteasome inhibitors, immune checkpoint inhibitors, monoclonal antibodies, immune-modulating agents, drug-induced pancreatitis. Terms used for the searches included each specific agent and pancreatitis, immunotherapy and pancreatitis, tyrosine kinase inhibitors and pancreatitis, auto immune pancreatitis, and toxicities of molecular target therapies. Reference lists from the identified manuscripts were reviewed for further studies of pancreatitis as a result of antineoplastic therapy. The most recent search date was February 15, 2017.
The degree to which each agent was associated with inducing pancreatitis was evaluated using the Badalov classification system2 in addition to the Naranjo Adverse Drug Reaction (ADR) Probability Scale.3 The Naranjo scale consists of 10 questions with values assigned to each answer. Total scores range from -4 to 13, where 13-9 indicates the reaction is considered definitely attributable to the drug; 8-5, probably attributable; 4-1, possibly attributable; and ≤0, doubtful if attributable.
A total of 67 manuscripts and abstracts were identified. Four manuscripts and 3 abstracts were excluded because they had insufficient information about possible pancreatitis or there was a presence of multiple other agents or conditions that might have caused pancreatitis. In total, 60 publications met inclusion criteria and were evaluated.
Results
Immune checkpoint inhibitors
In a review of toxicities of anti-programmed cell death-1 (PD-1) therapy, pancreatitis was reported to occur in about 1.8% of patients who received nivolumab or pembrolizumab.5 The 9 patients with pancreatitis attributed to an immune etiology were treated with corticosteroids. Pancreatitis was grade 2 in 3 patients (1.5-2 times upper limit of normal [ULN]), grade 3 in 4 patients (>2-5 ULN), and grade 4 ( >5 ULN) in 2 patients.
In asymptomatic individuals, pancreatitis has been detected on a positron-emission tomography–computed tomography (CT) scan after anti-PD-1 therapy.5 By contrast, there was a case report of a patient treated with nivolumab for lung cancer who developed anorexia, vomiting, and back pain on day 18 of therapy with an elevation of the amylase and lipase levels, but a negative CT.6 Later the patient developed a swollen pancreas on CT. Autoimmune pancreatitis comes in two forms. The most common relates to elevated levels of immunoglobulin G4 (IgG4; normal, 135 mg/dL ULN)7 The mechanism of immune pancreatitis associated with anti-PD-1 therapy is unknown.
Ipilimumab (an anti-CTLA4 antibody) has been approved by the US Food and Drug Administration (FDA) for the treatment of melanoma. Pancreatitis occurred in 1 patient in a phase 1 trial in pediatric patients.9 In a summary of 14 phase 1-3 trials of ipilimumab in advanced melanoma, pancreatitis was reported in fewer than 1% of the patients.10 In management guidelines for therapy with ipilimumab, pancreatitis may present as an asymptomatic increase in the levels of amylase and lipase, or with fevers, malaise, or abdominal pain. Oral prednisone or dexamethasone were given for the immune pancreatitis, but the decline in enzymes was slow, often taking months.11 In a preclinical model of autoimmune pancreatitis due to the blocking of CTLA4, there was suppression of regulatory T-cell function. The autoimmune pancreatitis responded to cyclosporin or rapamycin but there are no clinical data for these agents.12 The anti-PD-L1 agent atezolizumab has been associated with acute pancreatitis in 2 of 1,978 patients (0.1%).13 A review by Champiat and colleagues on dysimmune toxcities related to immune checkpoint inhibitors includes pancreatitis as an autoimmune complication of such therapies.14
Blinatumomab is an anti-CD19–directed CD3 T-cell engager that has been approved by the FDA for refractory B-cell acute lymphoblastic leukemia. In August 2016, the maker of the drug, Amgen, advised hematologists and oncologists that since February 2016, 10 patients out of more than 2,000 treated with blinatumomab had developed pancreatitis.15 Other medications the patients were receiving such as high-dose steroids might have caused or contributed to the pancreatitis. In one case, the pancreatitis improved with stopping blinatumomab but worsened with re-challenge. It is possible that the mechanism of the associated pancreatitis relates to a change in immune checkpoint inhibition. CD19-positive, CD24-high, CD27-positive regulatory B cells are decreased in autoimmune pancreatitis.16 Treatment with blinatumomab may decrease the CD19-positive cells.
Molecularly targeted agents, including TKIs
Molecularly targeted agents such as tyrosine kinase inhibitors (TKIs) or other kinase inhibitors have been associated with pancreatitis.17, 18 In a retrospective study by Tiruman and colleagues,19 the investigators found 91 patients with pancreatitis on imaging, of whom 15 were receiving molecularly target drugs. The pancreatitis was asymptomatic in 2 patients, but 13 had abdominal pain, many with nausea. Four of the patients also had gallstones, but the drug was deemed to be the cause of the pancreatitis. In 4 of the 9 patients in whom a rechallenge was done with the TKI, the pancreatitis relapsed. The pancreatitis resolved in 14 of the 15 patients; 1 patient died because of progressive cancer before the pancreatitis resolved. The pancreatitis was mild, 7 of the 15 patients had normal pancreatic enzymes and the pancreatitis was diagnosed by radiology.
Ghatlia and colleagues17 performed a meta-analysis of trials of TKI. They found 9 cases of pancreatitis in patients on sunitinib therapy. Of those, 4 patients were on sunitinib alone, and 5 were on other chemotherapy agents in combination with sunitinib. Eight cases of pancreatitis due to sorafenib were found. Three of the patients were on sorafenib alone, and 5 were on other chemotherapy including 1 on transcatheter embolization (TACE). Three cases of pancreatitis were associated with vandetanib; 2 of those patients had other concurrent chemotherapy. One case of axitinib induced pancreatitis was described.
Pancreatitis was reported in the phase 1 trials of sorafenib and sunitinib. In all, 3 of 69 patients treated with sorafenib had grade 3 pancreatitis and asymptomatic elevations of amylase and lipase levels were present in about 5% of patients receiving sunitinib.18,19
Other TKIs associated with pancreatitis include pazopanib,20,21 axitinib,22 and nilotinib.23 Pezzilli and coleagues24 described 5 patients with pancreatitis on sorafenib, 3 on sunitinib, 6 on nilotinib. It is possible that some of these cases appeared in other reviews. Ibrutinib, an inhibitor of Bruton’s tyrosine kinase, caused a single case of pancreatitis in 9 patients.25
Vemurafenib, a BRAF kinase inhibitor, was associated with pancreatitis in one case. In this case, the pancreatitis resolved on stopping the medication but recurred when vemurafenib rechallenge was attempted.26 There is a report of dabrafenib being associated with pancreatitis in 1 patient.27
Agents that inhibit the TKIs associated with BCR-ABL in chronic myelogenous leukemia are associated with acute pancreatitis. Imatinib-induced pancreatitis was reported in a small number of cases.28 Nilotinib has caused amylase/lipase elevations with and without symptomatic pancreatitis.29,30 Ponatinib, an inhibitor of BCR-ABL tyrosine kinase, is associated with pancreatitis.31 Pancreatitis occurred in 11 of 81 patients treated with ponatinib, and in 8 patients it was described as serious. Further elevation of amylase or lipase levels without clinical pancreatitis was noted in 7 other patients.
Proteosome inhibitors
In 2010, Elouni and colleagues32 reported a case of IV bortezomib-induced pancreatitis, which recurred on rechallenge with bortezomib. This same patient was also reported in an abstract in 2009.33 In 2009, there was an editorial comment which was added to the end of the abstract that the World Health Organization Adverse Drug Reaction database had 11 reports of bortezomib associated pancreatitis. Talamo and colleagues34 reported a case of bortezomib-induced pancreatitis due to bortezomib that had been administered subcutaneously. At that time, they also summarized 7 previous reports of bortezomib-associated pancreatitis. The mechanism of bortezomib-induced pancreatitis is not known.35-37
Fotoh and colleagues reported a patient with myeloma who had elevated triglyceride levels after bortezomib therapy.38 In one case of bortezomib-associated pancreatitis, the patient had an elevated triglyceride level, but it was not extremely high.39 Multiple myeloma itself may be associated with hyperlipidemia but only rarely.40 Gozetti and colleagues reported a patient who developed hyperlipidemia after two courses of bortezomib;41 stopping bisphosphonates may be associated with a rise in triglycerides. There was one case of carfilzomib causing pancreatitis during a phase 1 trial.42
Older chemotherapy agents
Reviews of drug-induced pancreatitis have listed many chemotherapy agents which may cause pancreatitis.1,43 The agent most frequently associated with acute pancreatitis has been asparaginase,44 with 2%-16% of patients undergoing asparaginase therapy developing pancreatitis. Asparaginase-related pancreatitis is grade 3 or 4 in 5%-10% of patients, and recurs in 63% of patients on rechallenge. Other chemotherapy agents associated with pancreatitis include: mercaptopurine, cytosine arabinoside, cisplatin, interferon alfa-2b, doxorubicin, tamoxifen, gefitinib, vinorelbine, oxaliplatin, levamisole, methotrexate, azathioprine, 5-fluorouracil, capecitabine, ifosfamide, paclitaxel, and all-trans retinoic acid.
Oxaliplatin carries a 0.1%-2% incidence of drug-induced pancreatitis. In one series of 6 patients, cessation of the agent allowed for resolution of symptoms and decrease in serum lipase and amylase levels.45 With capecitabine there are 2 case reports of pancreatitis.46 Cases of pancreatitis associated with trifluridine or tipiracil were not present in the literature.
Thalidomide caused severe pancreatitis in a patient when it was used to treat chronic graft-versus-host disease.47 This patient suffered recurrent pancreatitis on retreatment with the thalidomide. The authors further referenced two other suspected cases of thalidomide-induced, acute pancreatitis. However, in view of the extensive use of thalidomide for multiple myeloma before the development of lenalidomide, thalidomide-associated pancreatitis would be <1% of patients.
Agents that cause hypertriglyceridemia may cause pancreatitis. This mechanism has been reported as the cause of pancreatitis for everolimus48 and tamoxifen.49,50-52 Everolimus causes elevated triglycerides in 30%-50% of patients. There are case reports and a review of tamoxifen-associated pancreatitis caused by elevated triglycerides.52 There has also been a case of temsirolimus-associated pancreatitis,53 another agent that elevates triglycerides.
Pancreatitis associated with hepatic embolization or HIPEC
TACE leads to symptomatic acute pancreatitis in 0.4%-2% of patients, but nonselective TACE (into the hepatic artery and not just feeder vessels), may lead to elevated amylase levels in 15%-40% of patients.54-56 The risk of pancreatitis would depend on which chemotherapy drug is being infused into the liver. It would also be greater if the chemotherapy has to be infused into a larger part of the liver than into a small portion of the liver. In one patient, severe pancreatitis secondary to TACE occurred after two previous embolizations; prior embolization may have led to occlusion of the previously infused vessels.57 Radioembolization with 90Y microspheres was associated with one case of pancreatitis in 112 consecutive patients.58 The postembolization syndrome in the first 24 hours after the procedure may involve fever, abdominal pain, nausea, and vomiting due to acute pancreatitis in some instances.
Acute pancreatitis has also been described as a complication of hyperthermic intraperitoneal chemotherapy (HIPEC).59,60 Two of 13 patients receiving HIPEC for gastric cancer developed pancreatitis.59 In 25 patients with colon cancer who were treated with HIPEC, 1 patient had pancreatitis.60
Antibody-drug conjugates
Muzaffar and colleagues reported a patient with acute pancreatitis 3 days after starting therapy with ado-trastuzumab emtansine.61 Urru and colleagues62 reported a patient who developed acute pancreatitis after brentuximab vedotin therapy. Ghandi and colleagues63 identified 2 cases of fatal acute pancreatitis with brentuximab vedotin and 6 cases of nonfatal pancreatitis. Two of the nonfatal patients were rechallenged, and 1 developed recurrent pancreatitis. Because abdominal pain may occur in up to 18% of patients receiving brentuximab vedotin, the incidence of pancreatitis may be underestimated with this agent.64
In Table 2, ado-trastuzumab emtansine and brentuximab vedotin are listed with incidence and level of association given by the Baldavo2 and Naranjo.3 With greater awareness, the incidence of pancreatitis associated with these agents may rise or fall as more data is accumulated. In many instances, there are insufficient numbers of reported cases or insufficient information in single-case reports to complete the entire table.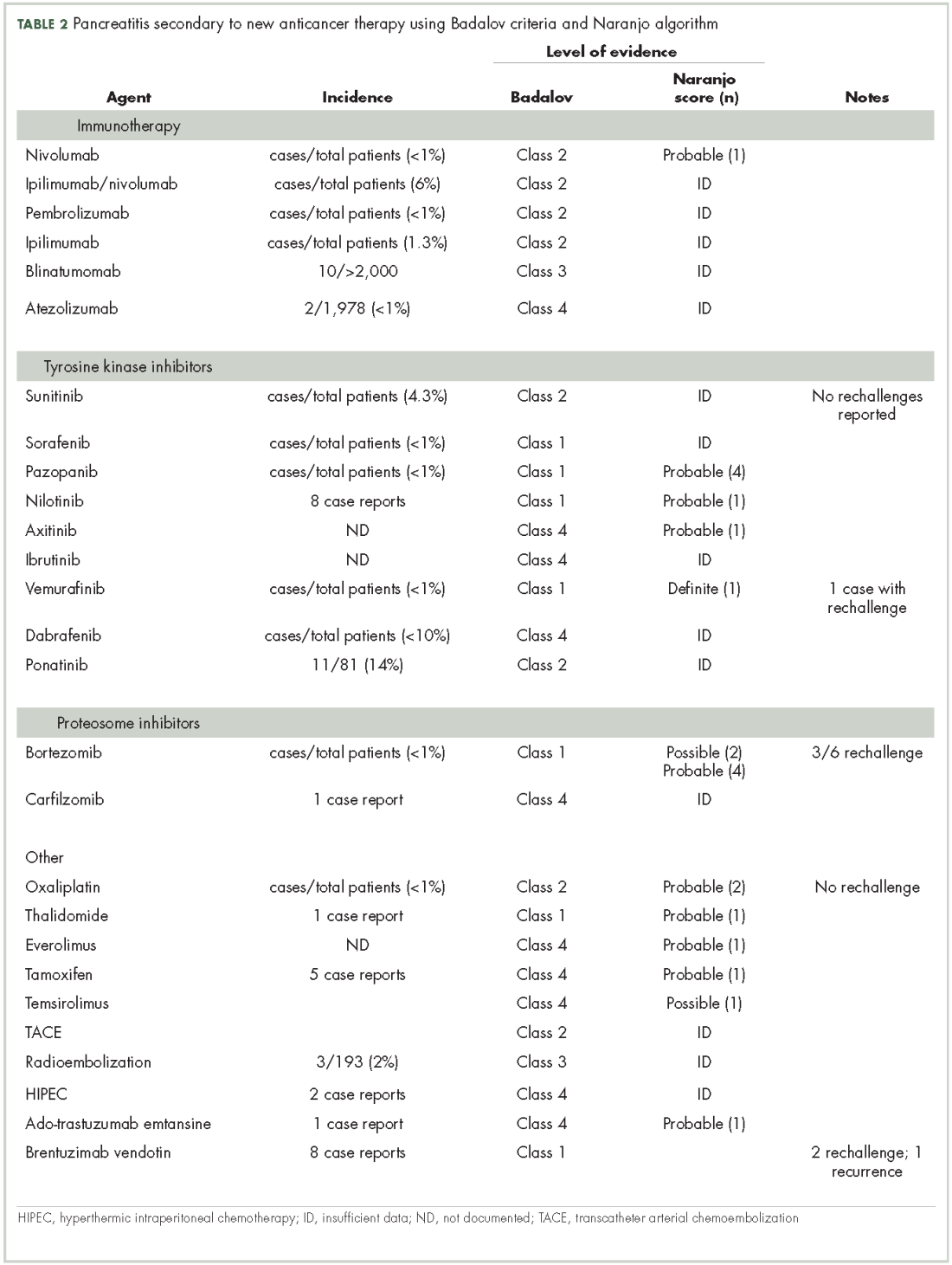
Discussion
Acute pancreatitis is an uncommon complication of tyrosine kinase inhibitors, other kinase inhibitors, proteasome inhibitors, monoclonal antibody-drug conjugates and anti-PD-1 immunotherapies. As nausea, abdominal pain, emesis are common in patients with cancer on antineoplastic therapy, some patients may have acute pancreatitis which is undiagnosed. It is not clear whether a patient with pancreatitis secondary to a TKI can be safely switched to a different TKI. As more molecularly targeted agents and more monoclonal antibodies targeting PD-L1 and PD-1 are under development, screening for amylase and lipase levels during phase 1/2 testing may prove helpful.
The natural history of cancer-drug–associated pancreatitis may depend on which agent is the cause. Although there are descriptions of the course of autoimmune pancreatitis, these studies have not included pancreatitis associated with anti-PD-L1 or -PD-1 therapies.65 It is possible that once an autoimmune pancreatitis has developed, simply stopping the inciting anti-PD-L1 or -PD-1 antibody may not lead to immediate resolution. Therapy with combined immune checkpoint blockade agents (eg, nivolumab and ipilimumab) may cause a higher incidence of pancreatitis than therapy with a single agent.66
In a report of 119 patients with melanoma who were treated with nivolumab and ipilimumab, there were 2 cases of acute pancreatitis, though 20% of patients had a grade 3 or higher amylase level, and just over 6% had a grade 3 or higher lipase.67 Stopping this type of immunotherapy early for grade 1,2, or 3 rises in pancreatic enzymes might prevent symptomatic pancreatitis from developing, but would stop potentially curative therapy for many patients who would have never developed clinically serious pancreatitis. Patients who suffer immune toxicities with anti-PD-1 therapies may be more apt to obtain some clinical benefit. The development of immune-related toxicities in patients treated with ipilimumab ( an anti CTLA4 antibody) seemed to correlate the tumor regression.68 This has also been suggested by the fact that the development of vitiligo correlates with clinical response in melanoma patients treated with nivolumab.69 Although clinically significant pancreatitis might be averted by stopping immune therapies earlier, stopping before it is deemed necessary might prevent cancer patients from receiving life-prolonging therapy.
Acute pancreatitis in general is severe in about 25% of cases and is associated with a significant risk of death. Scoring systems such as Ranson criteria and Apache 2 are used to assess the severity of pancreatitis although their utility is debated.70 Asparaginase is the chemotherapy agent most frequently associated with pancreatitis. It has been used to treat acute lymphoblastic leukemia for more than 30 years. This allowed for a study of 5,185 children and young adults who received asparaginase to determine what clinical factors and genomic factors were associated with the development of acute pancreatitis in 117 individuals.71 Further information gathered from programs such as the FDA and the adverse drug reaction program at Northwestern University in Chicago, coupled with the publication of other cases of pancreatitis associated with newer cancer agents may provide more insight into the mechanism causing pancreatitis due to a specific agent. With more cases being published, it may also become possible to determine if there are specific predisposing factors based on the clearance or metabolism of the offending agent or any genetic predisposition for drug-related pancreatitis.
1. Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol. 2005;29:709-716.
2. Badalov N, Baradarian R, Iswara K, et al. Drug-induced acute pancreatitis: an evidence-based review. Clin Gastroeneterol Hepatol. 2007;5:648-661.
3. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
4. Jones MR, Hall OM, Kaye AM, et al. Drug-induced acute pancreatitis: a review. Oschner J. 2015;15:45-51.
5. Hofmann L, Forschner A, Loquai C, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190-209.
6. Alabed YZ, Aghayev A, Sakellis C, et al. Pancreatitis secondary to anti-programmed death receptor 1 immunotherapy diagnosed by FDG PET/CT. Clin Nucl Med. 2015;40:e528-529.
7. Ikeuchi K, Okuma Y, Tabata T. Immune-related pancreatitis secondary to nivolumab in a patient with recurrent lung adenocarcinoma: a case report. Lung Cancer. 2016;90:148-150.
8. Webster GJ. Autoimmune pancreatitis – a riddle wrapped in an enigma. Dig Dis. 2016;34:532-539.
9. Merchant MS, Baird K, Wexler L, et al. Ipilimumab: first results of a phase I trial in pediatric patients with advanced solid tumors. J Clin Oncol. 2012;30:abstract 9545.
10. Ibrahim RA, Berman DM, Depril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma. J Clin Oncol. 2011;29:abstract 8583.
11. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30:2691-2697.
12. Mayerle J, van den Brandt C, Schwaiger T, et al. Blockage of CTLA-4 suggests that autoimmune pancreatitis is a T-cell mediated disease responsive to ciclosporin A and rapamycin . Pancreatology. 2012;12:579(abstract S8-3).
13. Tecentriq (package insert). South San Francisco, CA: Genentech Inc; 2016.
14. Champiat S, Lambotte E, Barreau E, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2015;27:559-574.
15. Amgen. New safety information for Blincyto (blinatumomab) Risk of pancreatitis. August 2016 and update to Micromedex 2016.
16. Sumimoto K, Uchida K, KusudaT, et al. The role of CD19+ CD24high CD38high and CD19+ CD24high, CD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis . Pancreatology. 2014;14:193-200.
17. Ghatalia P, Morgan CJ, Choueiri TK, et al. Pancreatitis with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol. 2015;94:136-145.
18. Sevin A, Chen A, Atkinson B. Tyrosine kinase inhibitor induced pancreatitis . J Oncol Pharm Pract. 2012;19:257-260.
19. Tirumani SH, Jagannathan JP, Shinagare AB, et al. Acute pancreatitis associated with molecular targeted therapies: a retrospective review of the clinico-radiological features, management and outcome. Pancreatology . 2013;13:461-467.
20. Russano M, Vincenzi B, Benditti O, et al. Pazopanib and pancreatic toxicity: a case report. BMC Res notes. 2015;8:196-198.
21. Kawakubo K, Hata H, Kawakami H, et al. Pazopanib induced severe acute pancreatitis. Case Rep Oncol. 2015;8:356-358.
22. Peron J, Khenifer S, Potier V, et al. Axitinib induced acute pancreatitis: a case report . Anticancer Drugs. 2014;25:478-479.
23. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis . Ann Pharmaco. 2013;37:33.
24. Pezzilli R, Corinaldesi R, Morselli-LabateAM. Tyrosine kinase inhibitors and acute pancreatitis. http://www.serena.unina.it/index.php/jop/article/view/3836/4278. Published May 5, 2010. Accessed May 22 , 2017.
25. Blum KA, Christian B, Flynn JM, et al. A phase I trial of the Bruton’s tyrosine kinase inhibitor, ibrutinib, in combination with rituximab and bendamustine in patients with relapsed/refractory non Hodgkin’s lymphoma. Blood. 2012;120:abstract 1643.
26. Muluneh B, Buie LW, Collichio F. Vemurafenib-associated pancreatitis: a case report. Pharmacotherapy. 2013;33:e43-e44.
27. Dabrafenib. In Life-Sciences-Europe.com from Tafinlar. EU Summary of Product Characteristics. 30 August 2013.
28. Varma MR, Mathew S, Krishnadas D, et al. Imatinib-induced pancreatitis. Indian J Pharmacol. 2010;42:50-52.
29. Palandri F, Castagnetti F, Soverinie S, et al. Pancreatic enzyme elevation in chronic myeloid leukemia patients treated with nilotinib after imatinib failure. Haematologica. 2009;94:1758-1761.
30. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis. Ann of Pharmacother. 2013;47:e.3
31. Cortesk JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadephia chromosome-positive leukemias. New Engl J Med. 2012;367:2075-2088.
32. Elouni B, Ben Salem C, Zamy M, et al. Bortezomib-induced acute pancreatitis [Letter]. J Pancreas. 2010;119:275-276.
33. Elouni B. Acute pancreatitis induced by Velcade ( bortezomib) with positive rechallenge. 9th Annual meeting of the International Society of Pharmacovigilance. Oct 2009 abstract 74.
34. Talamo G, Sikik J, Pandey MK, et al. Bortezomib-induced acute pancreatitis. Case report and review of the literature . J Oncol Pharm Prac. 2016;22:332-334.
35. SolakogluT, Akyol P, Guney T, et al. Acute pancreatitis caused by bortezomib. Pancreatology. 2013;13:189-190.
36. Mihaila RG. A possible rare complication of bortezomib treatment, acute pancreatitis. Acta Medica Transilvanica. 2013;2:269-171
37. Gupta H, Bansal R, Khanna S, et al. An unusual complication of bortezomib therapy: acute pancreatitis. Indian J Nephr. 2014;24:135-136.
38. Fotoh M, KitaharaT, Sakuta J, et al. Multiple lipoma with hyperlipidemia in a multiple myeloma patient treated with bortezomib/dexamethasone. Leuk Res. 2010;34:e120-121.
39. Wang HH, Tsui J, Wang XY, et al. Bortezomib induced acute pancreatitis in a patient with multiple myeloma. Leuk Lymphoma. 2014;55:1404-1405.
40. Misselwitz B, Goede JS, Pestalozzi BC, et al. Hyperlipidemic myeloma: review of 53 cases. Ann Hematol. 2010;89:569-577.
41. Gozzetti A, Fabbri A, Defina M, et al. Hyperlipidemia in a myeloma patient after bortezomib treatment. Leuk Research. 2010;34:e250.
42. Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013;121:893-897.
43. Runzi M, Layer P. Drug-associated pancreatitis: facts and fiction. Pancreas. 1996;13:100-109.
44. Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57:748-757.
45. Butt W, Saadati H, Wasif- Saif M. Oxaliplatin-induced pancreatitis: a case series. Anticancer Res. 2010;30:5113-5115.
46. Yucel H, Warmerdam LV. Capecitabine-induced pancreatitis. J Onc Pharm Pract. 2010;16:133-134.
47. Chung LW, Yeh S-P, Hsieh C-Y, et al. Life-threatening acute pancreatitis due to thalidomide therapy for chronic graft-versus-host disease. Ann Hematol. 2008;87:421-424.
48. Subramaniam S, Zell JA, Kunz PL. Everolimus causing severe hypertriglyceridemia and acute pancreatitis. J Natl Compr Canc Netw. 2013;11:5-9.
49. Wadood A, Chesner R, Mirza M, et al. Tamoxifen precipitation of familial hypertriglyceridaemia: a rare cause of acute pancreatitis. BMJ Case Rep. Published August 3, 2016. doi: 10.1136/bcr-2016-214837.
50. Sakhri J, BenSalem C, Fathallah H, et al. Severe pancreatitis due to tamoxifen induced hypertriglyceridemia with positive rechallenge. J Pancreas. 2010;11:382-384.
51. Elisaf MS, Nakou K, Liamis G, et al. Tamoxifen-induced severe hypertriglyceridemia and pancreatitis. Ann Oncol. 2000;11:1067-1069.
52. Artac M, Sari R, Altunbas J, et al. Asymptomatic acute pancreatitis due to tamoxifen-induced hypertriglyceridemia in a patient with diabetes mellitus and breast cancer. J Chemother. 2002;14:309-311.
53. [Author name not available]. Acute pancreatitis: 15 case reports. React Wkly. 2015;1546:29.
54. Ozcinar B, Guven K, Poylani A, et al. Necrotizing pancreatitis after transcatheter embolization for hepatocellular carcinoma. Diagn In
56. She WH, Chan ACY, Cheung TT, et al. Acute pancreatitis induced by transarterial chemoembolization: a single center experience of over 1500 cases. Hepatobiliary Pancreat Dis Int. 2016;15:93-98.
57. Bae SI, Yeon JE, Lee JM, et al. A case of necrotizing pancreatitis subsequent to transcatheter arterial chemoembolization in a patient with hepatocellular carcinoma. Clin Mol Hepatol. 2012;18:321-325.
58. Peterson JL, Vallow LA, Johnson DW, et al. Complications after 90Y microsphere radioembolization for unresectable hepatic tumors: an evaluation of 112 patients. Brachytherapy. 2013;12:573-579.
59. Piso P, Glockzin G, Schlitt HJ. Cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC) in patients with peritoneal carcinomatosis arising from gastric cancer. J Clin Oncol. 2011;29(suppl 4):abstract 132.
60. Sammartino P, Sibio S, Biacchi D, et al. Prevention of peritoneal metastases from colon cancer in high-risk patients: preliminary results of surgery plus prophylactic HIPEC. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3356888/?report=reader. Published 2012. Accessed May 23, 2017.
61. Muzaffar M, Jia J, Liles D, et al. Acute pancreatitis associated with ado-trastuzumab emtansine. Am J Ther. 2016;23:e572-574.
62. Urru SA, Mariotti E, Carta P, et al. Acute pancreatitis following brentuzimab vedotin therapy for refractory Hodgkin lymphoma: a case report. Drugs R D. 2014;14:9-11.
63. Gandhi MD, Evens AM, Fenske TS, et al. Pancreatitis in patients treated with brentuximab vedotin: a previously unrecognized serious adverse event. Blood. 2014;123:2895-2897.
64. Brentuximab vedotin in Micromedex solutions, Truven Health Analytics. 2016.
65. Okazaki K, Uchida K. Autoimmune pancreatitis: the past, present and future. Pancreas. 2015;44:1006-1016.
66. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab and ipilimumab in advanced melanoma. New Engl J Med. 2013;369:122-133.
67. Friedman CF, Clark V, Raikhel AV, et al. Thinking critically about classifying adverse events: incidence of pancreatitis in patients treated with nivolumab and ipilimumab. J Natl Cancer Inst. 2017;109:[page numbers not available].
68. Day D, Hansen AR. Immune-related adverse events associated with immune checkpoint inhibitors. BioDrugs. 2016;30:571-584.
69. Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
70 . Di MY, Liu H, Zu-Yao Y, et al. Prediction models of mortality in acute pancreatitis in adults. A systematic review. Ann Int Med. 2016;165:482-490.
71. Liu C, Yang W, Devidas M, et al. Clinical and genetic risk factors for acute pancreatitis in patients with acute lymphoblastic leukemia. J Clin Oncol. 2016;18:2133-2140.
Patients with advanced malignancies may develop pancreatitis during therapy for their cancer. Acute pancreatitis is inflammation of the pancreas. Common symptoms include abdominal pain, nausea, vomiting, shortness of breath, dehydration. Laboratory evidence of acute pancreatitis includes elevations of the amylase and lipase. Mild pancreatitis occurs when there is no organ dysfunction, moderate pancreatitis is associated with one organ dysfunction, and severe pancreatitis is complicated by multiple organ dysfunction. Hypotension, hypocalcemia, or anemia suggest a more severe course of the pancreatitis. In some instances, the pancreatitis may be an adverse reaction to the therapy being given. However, other causes such as hypercalcemia, hypertriglyceridemia, cholelithiasis, and underlying malignancy must be ruled out before ascribing pancreatitis to a specific drug. To date, two classifications systems have been proposed by Trivedi1 and Badalov2 to evaluate the degree to which a drug is responsible for pancreatitis (Table 1). Furthermore, Naranjo and colleagues have proposed a more general method of assessing the causal relationship between drugs and adverse events.3 The Naranjo algorithm is not specific for pancreatitis. Jones and colleagues4 reported that 0.1%-2% of acute pancreatitis cases were owing to drugs. In 2015, they listed the older chemotherapy agents associated with pancreatitis. However, more recently, many new agents have been approved for the management of cancers. The newer classes of antineoplastic agents including proteasome inhibitors, immune-modulating agents, tyrosine kinase inhibitors, monoclonal antibodies against programmed cell death-1 (PD-1) and its ligand PD-L1 and antibody-toxin conjugates are now associated with acute pancreatitis.
Methods
We conducted a search in PubMed, Google Scholar, and Micromedex for pancreatitis related to antineoplastic agents, including proteasome inhibitors, immune checkpoint inhibitors, monoclonal antibodies, immune-modulating agents, drug-induced pancreatitis. Terms used for the searches included each specific agent and pancreatitis, immunotherapy and pancreatitis, tyrosine kinase inhibitors and pancreatitis, auto immune pancreatitis, and toxicities of molecular target therapies. Reference lists from the identified manuscripts were reviewed for further studies of pancreatitis as a result of antineoplastic therapy. The most recent search date was February 15, 2017.
The degree to which each agent was associated with inducing pancreatitis was evaluated using the Badalov classification system2 in addition to the Naranjo Adverse Drug Reaction (ADR) Probability Scale.3 The Naranjo scale consists of 10 questions with values assigned to each answer. Total scores range from -4 to 13, where 13-9 indicates the reaction is considered definitely attributable to the drug; 8-5, probably attributable; 4-1, possibly attributable; and ≤0, doubtful if attributable.
A total of 67 manuscripts and abstracts were identified. Four manuscripts and 3 abstracts were excluded because they had insufficient information about possible pancreatitis or there was a presence of multiple other agents or conditions that might have caused pancreatitis. In total, 60 publications met inclusion criteria and were evaluated.
Results
Immune checkpoint inhibitors
In a review of toxicities of anti-programmed cell death-1 (PD-1) therapy, pancreatitis was reported to occur in about 1.8% of patients who received nivolumab or pembrolizumab.5 The 9 patients with pancreatitis attributed to an immune etiology were treated with corticosteroids. Pancreatitis was grade 2 in 3 patients (1.5-2 times upper limit of normal [ULN]), grade 3 in 4 patients (>2-5 ULN), and grade 4 ( >5 ULN) in 2 patients.
In asymptomatic individuals, pancreatitis has been detected on a positron-emission tomography–computed tomography (CT) scan after anti-PD-1 therapy.5 By contrast, there was a case report of a patient treated with nivolumab for lung cancer who developed anorexia, vomiting, and back pain on day 18 of therapy with an elevation of the amylase and lipase levels, but a negative CT.6 Later the patient developed a swollen pancreas on CT. Autoimmune pancreatitis comes in two forms. The most common relates to elevated levels of immunoglobulin G4 (IgG4; normal, 135 mg/dL ULN)7 The mechanism of immune pancreatitis associated with anti-PD-1 therapy is unknown.
Ipilimumab (an anti-CTLA4 antibody) has been approved by the US Food and Drug Administration (FDA) for the treatment of melanoma. Pancreatitis occurred in 1 patient in a phase 1 trial in pediatric patients.9 In a summary of 14 phase 1-3 trials of ipilimumab in advanced melanoma, pancreatitis was reported in fewer than 1% of the patients.10 In management guidelines for therapy with ipilimumab, pancreatitis may present as an asymptomatic increase in the levels of amylase and lipase, or with fevers, malaise, or abdominal pain. Oral prednisone or dexamethasone were given for the immune pancreatitis, but the decline in enzymes was slow, often taking months.11 In a preclinical model of autoimmune pancreatitis due to the blocking of CTLA4, there was suppression of regulatory T-cell function. The autoimmune pancreatitis responded to cyclosporin or rapamycin but there are no clinical data for these agents.12 The anti-PD-L1 agent atezolizumab has been associated with acute pancreatitis in 2 of 1,978 patients (0.1%).13 A review by Champiat and colleagues on dysimmune toxcities related to immune checkpoint inhibitors includes pancreatitis as an autoimmune complication of such therapies.14
Blinatumomab is an anti-CD19–directed CD3 T-cell engager that has been approved by the FDA for refractory B-cell acute lymphoblastic leukemia. In August 2016, the maker of the drug, Amgen, advised hematologists and oncologists that since February 2016, 10 patients out of more than 2,000 treated with blinatumomab had developed pancreatitis.15 Other medications the patients were receiving such as high-dose steroids might have caused or contributed to the pancreatitis. In one case, the pancreatitis improved with stopping blinatumomab but worsened with re-challenge. It is possible that the mechanism of the associated pancreatitis relates to a change in immune checkpoint inhibition. CD19-positive, CD24-high, CD27-positive regulatory B cells are decreased in autoimmune pancreatitis.16 Treatment with blinatumomab may decrease the CD19-positive cells.
Molecularly targeted agents, including TKIs
Molecularly targeted agents such as tyrosine kinase inhibitors (TKIs) or other kinase inhibitors have been associated with pancreatitis.17, 18 In a retrospective study by Tiruman and colleagues,19 the investigators found 91 patients with pancreatitis on imaging, of whom 15 were receiving molecularly target drugs. The pancreatitis was asymptomatic in 2 patients, but 13 had abdominal pain, many with nausea. Four of the patients also had gallstones, but the drug was deemed to be the cause of the pancreatitis. In 4 of the 9 patients in whom a rechallenge was done with the TKI, the pancreatitis relapsed. The pancreatitis resolved in 14 of the 15 patients; 1 patient died because of progressive cancer before the pancreatitis resolved. The pancreatitis was mild, 7 of the 15 patients had normal pancreatic enzymes and the pancreatitis was diagnosed by radiology.
Ghatlia and colleagues17 performed a meta-analysis of trials of TKI. They found 9 cases of pancreatitis in patients on sunitinib therapy. Of those, 4 patients were on sunitinib alone, and 5 were on other chemotherapy agents in combination with sunitinib. Eight cases of pancreatitis due to sorafenib were found. Three of the patients were on sorafenib alone, and 5 were on other chemotherapy including 1 on transcatheter embolization (TACE). Three cases of pancreatitis were associated with vandetanib; 2 of those patients had other concurrent chemotherapy. One case of axitinib induced pancreatitis was described.
Pancreatitis was reported in the phase 1 trials of sorafenib and sunitinib. In all, 3 of 69 patients treated with sorafenib had grade 3 pancreatitis and asymptomatic elevations of amylase and lipase levels were present in about 5% of patients receiving sunitinib.18,19
Other TKIs associated with pancreatitis include pazopanib,20,21 axitinib,22 and nilotinib.23 Pezzilli and coleagues24 described 5 patients with pancreatitis on sorafenib, 3 on sunitinib, 6 on nilotinib. It is possible that some of these cases appeared in other reviews. Ibrutinib, an inhibitor of Bruton’s tyrosine kinase, caused a single case of pancreatitis in 9 patients.25
Vemurafenib, a BRAF kinase inhibitor, was associated with pancreatitis in one case. In this case, the pancreatitis resolved on stopping the medication but recurred when vemurafenib rechallenge was attempted.26 There is a report of dabrafenib being associated with pancreatitis in 1 patient.27
Agents that inhibit the TKIs associated with BCR-ABL in chronic myelogenous leukemia are associated with acute pancreatitis. Imatinib-induced pancreatitis was reported in a small number of cases.28 Nilotinib has caused amylase/lipase elevations with and without symptomatic pancreatitis.29,30 Ponatinib, an inhibitor of BCR-ABL tyrosine kinase, is associated with pancreatitis.31 Pancreatitis occurred in 11 of 81 patients treated with ponatinib, and in 8 patients it was described as serious. Further elevation of amylase or lipase levels without clinical pancreatitis was noted in 7 other patients.
Proteosome inhibitors
In 2010, Elouni and colleagues32 reported a case of IV bortezomib-induced pancreatitis, which recurred on rechallenge with bortezomib. This same patient was also reported in an abstract in 2009.33 In 2009, there was an editorial comment which was added to the end of the abstract that the World Health Organization Adverse Drug Reaction database had 11 reports of bortezomib associated pancreatitis. Talamo and colleagues34 reported a case of bortezomib-induced pancreatitis due to bortezomib that had been administered subcutaneously. At that time, they also summarized 7 previous reports of bortezomib-associated pancreatitis. The mechanism of bortezomib-induced pancreatitis is not known.35-37
Fotoh and colleagues reported a patient with myeloma who had elevated triglyceride levels after bortezomib therapy.38 In one case of bortezomib-associated pancreatitis, the patient had an elevated triglyceride level, but it was not extremely high.39 Multiple myeloma itself may be associated with hyperlipidemia but only rarely.40 Gozetti and colleagues reported a patient who developed hyperlipidemia after two courses of bortezomib;41 stopping bisphosphonates may be associated with a rise in triglycerides. There was one case of carfilzomib causing pancreatitis during a phase 1 trial.42
Older chemotherapy agents
Reviews of drug-induced pancreatitis have listed many chemotherapy agents which may cause pancreatitis.1,43 The agent most frequently associated with acute pancreatitis has been asparaginase,44 with 2%-16% of patients undergoing asparaginase therapy developing pancreatitis. Asparaginase-related pancreatitis is grade 3 or 4 in 5%-10% of patients, and recurs in 63% of patients on rechallenge. Other chemotherapy agents associated with pancreatitis include: mercaptopurine, cytosine arabinoside, cisplatin, interferon alfa-2b, doxorubicin, tamoxifen, gefitinib, vinorelbine, oxaliplatin, levamisole, methotrexate, azathioprine, 5-fluorouracil, capecitabine, ifosfamide, paclitaxel, and all-trans retinoic acid.
Oxaliplatin carries a 0.1%-2% incidence of drug-induced pancreatitis. In one series of 6 patients, cessation of the agent allowed for resolution of symptoms and decrease in serum lipase and amylase levels.45 With capecitabine there are 2 case reports of pancreatitis.46 Cases of pancreatitis associated with trifluridine or tipiracil were not present in the literature.
Thalidomide caused severe pancreatitis in a patient when it was used to treat chronic graft-versus-host disease.47 This patient suffered recurrent pancreatitis on retreatment with the thalidomide. The authors further referenced two other suspected cases of thalidomide-induced, acute pancreatitis. However, in view of the extensive use of thalidomide for multiple myeloma before the development of lenalidomide, thalidomide-associated pancreatitis would be <1% of patients.
Agents that cause hypertriglyceridemia may cause pancreatitis. This mechanism has been reported as the cause of pancreatitis for everolimus48 and tamoxifen.49,50-52 Everolimus causes elevated triglycerides in 30%-50% of patients. There are case reports and a review of tamoxifen-associated pancreatitis caused by elevated triglycerides.52 There has also been a case of temsirolimus-associated pancreatitis,53 another agent that elevates triglycerides.
Pancreatitis associated with hepatic embolization or HIPEC
TACE leads to symptomatic acute pancreatitis in 0.4%-2% of patients, but nonselective TACE (into the hepatic artery and not just feeder vessels), may lead to elevated amylase levels in 15%-40% of patients.54-56 The risk of pancreatitis would depend on which chemotherapy drug is being infused into the liver. It would also be greater if the chemotherapy has to be infused into a larger part of the liver than into a small portion of the liver. In one patient, severe pancreatitis secondary to TACE occurred after two previous embolizations; prior embolization may have led to occlusion of the previously infused vessels.57 Radioembolization with 90Y microspheres was associated with one case of pancreatitis in 112 consecutive patients.58 The postembolization syndrome in the first 24 hours after the procedure may involve fever, abdominal pain, nausea, and vomiting due to acute pancreatitis in some instances.
Acute pancreatitis has also been described as a complication of hyperthermic intraperitoneal chemotherapy (HIPEC).59,60 Two of 13 patients receiving HIPEC for gastric cancer developed pancreatitis.59 In 25 patients with colon cancer who were treated with HIPEC, 1 patient had pancreatitis.60
Antibody-drug conjugates
Muzaffar and colleagues reported a patient with acute pancreatitis 3 days after starting therapy with ado-trastuzumab emtansine.61 Urru and colleagues62 reported a patient who developed acute pancreatitis after brentuximab vedotin therapy. Ghandi and colleagues63 identified 2 cases of fatal acute pancreatitis with brentuximab vedotin and 6 cases of nonfatal pancreatitis. Two of the nonfatal patients were rechallenged, and 1 developed recurrent pancreatitis. Because abdominal pain may occur in up to 18% of patients receiving brentuximab vedotin, the incidence of pancreatitis may be underestimated with this agent.64
In Table 2, ado-trastuzumab emtansine and brentuximab vedotin are listed with incidence and level of association given by the Baldavo2 and Naranjo.3 With greater awareness, the incidence of pancreatitis associated with these agents may rise or fall as more data is accumulated. In many instances, there are insufficient numbers of reported cases or insufficient information in single-case reports to complete the entire table.
Discussion
Acute pancreatitis is an uncommon complication of tyrosine kinase inhibitors, other kinase inhibitors, proteasome inhibitors, monoclonal antibody-drug conjugates and anti-PD-1 immunotherapies. As nausea, abdominal pain, emesis are common in patients with cancer on antineoplastic therapy, some patients may have acute pancreatitis which is undiagnosed. It is not clear whether a patient with pancreatitis secondary to a TKI can be safely switched to a different TKI. As more molecularly targeted agents and more monoclonal antibodies targeting PD-L1 and PD-1 are under development, screening for amylase and lipase levels during phase 1/2 testing may prove helpful.
The natural history of cancer-drug–associated pancreatitis may depend on which agent is the cause. Although there are descriptions of the course of autoimmune pancreatitis, these studies have not included pancreatitis associated with anti-PD-L1 or -PD-1 therapies.65 It is possible that once an autoimmune pancreatitis has developed, simply stopping the inciting anti-PD-L1 or -PD-1 antibody may not lead to immediate resolution. Therapy with combined immune checkpoint blockade agents (eg, nivolumab and ipilimumab) may cause a higher incidence of pancreatitis than therapy with a single agent.66
In a report of 119 patients with melanoma who were treated with nivolumab and ipilimumab, there were 2 cases of acute pancreatitis, though 20% of patients had a grade 3 or higher amylase level, and just over 6% had a grade 3 or higher lipase.67 Stopping this type of immunotherapy early for grade 1,2, or 3 rises in pancreatic enzymes might prevent symptomatic pancreatitis from developing, but would stop potentially curative therapy for many patients who would have never developed clinically serious pancreatitis. Patients who suffer immune toxicities with anti-PD-1 therapies may be more apt to obtain some clinical benefit. The development of immune-related toxicities in patients treated with ipilimumab ( an anti CTLA4 antibody) seemed to correlate the tumor regression.68 This has also been suggested by the fact that the development of vitiligo correlates with clinical response in melanoma patients treated with nivolumab.69 Although clinically significant pancreatitis might be averted by stopping immune therapies earlier, stopping before it is deemed necessary might prevent cancer patients from receiving life-prolonging therapy.
Acute pancreatitis in general is severe in about 25% of cases and is associated with a significant risk of death. Scoring systems such as Ranson criteria and Apache 2 are used to assess the severity of pancreatitis although their utility is debated.70 Asparaginase is the chemotherapy agent most frequently associated with pancreatitis. It has been used to treat acute lymphoblastic leukemia for more than 30 years. This allowed for a study of 5,185 children and young adults who received asparaginase to determine what clinical factors and genomic factors were associated with the development of acute pancreatitis in 117 individuals.71 Further information gathered from programs such as the FDA and the adverse drug reaction program at Northwestern University in Chicago, coupled with the publication of other cases of pancreatitis associated with newer cancer agents may provide more insight into the mechanism causing pancreatitis due to a specific agent. With more cases being published, it may also become possible to determine if there are specific predisposing factors based on the clearance or metabolism of the offending agent or any genetic predisposition for drug-related pancreatitis.
Patients with advanced malignancies may develop pancreatitis during therapy for their cancer. Acute pancreatitis is inflammation of the pancreas. Common symptoms include abdominal pain, nausea, vomiting, shortness of breath, dehydration. Laboratory evidence of acute pancreatitis includes elevations of the amylase and lipase. Mild pancreatitis occurs when there is no organ dysfunction, moderate pancreatitis is associated with one organ dysfunction, and severe pancreatitis is complicated by multiple organ dysfunction. Hypotension, hypocalcemia, or anemia suggest a more severe course of the pancreatitis. In some instances, the pancreatitis may be an adverse reaction to the therapy being given. However, other causes such as hypercalcemia, hypertriglyceridemia, cholelithiasis, and underlying malignancy must be ruled out before ascribing pancreatitis to a specific drug. To date, two classifications systems have been proposed by Trivedi1 and Badalov2 to evaluate the degree to which a drug is responsible for pancreatitis (Table 1). Furthermore, Naranjo and colleagues have proposed a more general method of assessing the causal relationship between drugs and adverse events.3 The Naranjo algorithm is not specific for pancreatitis. Jones and colleagues4 reported that 0.1%-2% of acute pancreatitis cases were owing to drugs. In 2015, they listed the older chemotherapy agents associated with pancreatitis. However, more recently, many new agents have been approved for the management of cancers. The newer classes of antineoplastic agents including proteasome inhibitors, immune-modulating agents, tyrosine kinase inhibitors, monoclonal antibodies against programmed cell death-1 (PD-1) and its ligand PD-L1 and antibody-toxin conjugates are now associated with acute pancreatitis.
Methods
We conducted a search in PubMed, Google Scholar, and Micromedex for pancreatitis related to antineoplastic agents, including proteasome inhibitors, immune checkpoint inhibitors, monoclonal antibodies, immune-modulating agents, drug-induced pancreatitis. Terms used for the searches included each specific agent and pancreatitis, immunotherapy and pancreatitis, tyrosine kinase inhibitors and pancreatitis, auto immune pancreatitis, and toxicities of molecular target therapies. Reference lists from the identified manuscripts were reviewed for further studies of pancreatitis as a result of antineoplastic therapy. The most recent search date was February 15, 2017.
The degree to which each agent was associated with inducing pancreatitis was evaluated using the Badalov classification system2 in addition to the Naranjo Adverse Drug Reaction (ADR) Probability Scale.3 The Naranjo scale consists of 10 questions with values assigned to each answer. Total scores range from -4 to 13, where 13-9 indicates the reaction is considered definitely attributable to the drug; 8-5, probably attributable; 4-1, possibly attributable; and ≤0, doubtful if attributable.
A total of 67 manuscripts and abstracts were identified. Four manuscripts and 3 abstracts were excluded because they had insufficient information about possible pancreatitis or there was a presence of multiple other agents or conditions that might have caused pancreatitis. In total, 60 publications met inclusion criteria and were evaluated.
Results
Immune checkpoint inhibitors
In a review of toxicities of anti-programmed cell death-1 (PD-1) therapy, pancreatitis was reported to occur in about 1.8% of patients who received nivolumab or pembrolizumab.5 The 9 patients with pancreatitis attributed to an immune etiology were treated with corticosteroids. Pancreatitis was grade 2 in 3 patients (1.5-2 times upper limit of normal [ULN]), grade 3 in 4 patients (>2-5 ULN), and grade 4 ( >5 ULN) in 2 patients.
In asymptomatic individuals, pancreatitis has been detected on a positron-emission tomography–computed tomography (CT) scan after anti-PD-1 therapy.5 By contrast, there was a case report of a patient treated with nivolumab for lung cancer who developed anorexia, vomiting, and back pain on day 18 of therapy with an elevation of the amylase and lipase levels, but a negative CT.6 Later the patient developed a swollen pancreas on CT. Autoimmune pancreatitis comes in two forms. The most common relates to elevated levels of immunoglobulin G4 (IgG4; normal, 135 mg/dL ULN)7 The mechanism of immune pancreatitis associated with anti-PD-1 therapy is unknown.
Ipilimumab (an anti-CTLA4 antibody) has been approved by the US Food and Drug Administration (FDA) for the treatment of melanoma. Pancreatitis occurred in 1 patient in a phase 1 trial in pediatric patients.9 In a summary of 14 phase 1-3 trials of ipilimumab in advanced melanoma, pancreatitis was reported in fewer than 1% of the patients.10 In management guidelines for therapy with ipilimumab, pancreatitis may present as an asymptomatic increase in the levels of amylase and lipase, or with fevers, malaise, or abdominal pain. Oral prednisone or dexamethasone were given for the immune pancreatitis, but the decline in enzymes was slow, often taking months.11 In a preclinical model of autoimmune pancreatitis due to the blocking of CTLA4, there was suppression of regulatory T-cell function. The autoimmune pancreatitis responded to cyclosporin or rapamycin but there are no clinical data for these agents.12 The anti-PD-L1 agent atezolizumab has been associated with acute pancreatitis in 2 of 1,978 patients (0.1%).13 A review by Champiat and colleagues on dysimmune toxcities related to immune checkpoint inhibitors includes pancreatitis as an autoimmune complication of such therapies.14
Blinatumomab is an anti-CD19–directed CD3 T-cell engager that has been approved by the FDA for refractory B-cell acute lymphoblastic leukemia. In August 2016, the maker of the drug, Amgen, advised hematologists and oncologists that since February 2016, 10 patients out of more than 2,000 treated with blinatumomab had developed pancreatitis.15 Other medications the patients were receiving such as high-dose steroids might have caused or contributed to the pancreatitis. In one case, the pancreatitis improved with stopping blinatumomab but worsened with re-challenge. It is possible that the mechanism of the associated pancreatitis relates to a change in immune checkpoint inhibition. CD19-positive, CD24-high, CD27-positive regulatory B cells are decreased in autoimmune pancreatitis.16 Treatment with blinatumomab may decrease the CD19-positive cells.
Molecularly targeted agents, including TKIs
Molecularly targeted agents such as tyrosine kinase inhibitors (TKIs) or other kinase inhibitors have been associated with pancreatitis.17, 18 In a retrospective study by Tiruman and colleagues,19 the investigators found 91 patients with pancreatitis on imaging, of whom 15 were receiving molecularly target drugs. The pancreatitis was asymptomatic in 2 patients, but 13 had abdominal pain, many with nausea. Four of the patients also had gallstones, but the drug was deemed to be the cause of the pancreatitis. In 4 of the 9 patients in whom a rechallenge was done with the TKI, the pancreatitis relapsed. The pancreatitis resolved in 14 of the 15 patients; 1 patient died because of progressive cancer before the pancreatitis resolved. The pancreatitis was mild, 7 of the 15 patients had normal pancreatic enzymes and the pancreatitis was diagnosed by radiology.
Ghatlia and colleagues17 performed a meta-analysis of trials of TKI. They found 9 cases of pancreatitis in patients on sunitinib therapy. Of those, 4 patients were on sunitinib alone, and 5 were on other chemotherapy agents in combination with sunitinib. Eight cases of pancreatitis due to sorafenib were found. Three of the patients were on sorafenib alone, and 5 were on other chemotherapy including 1 on transcatheter embolization (TACE). Three cases of pancreatitis were associated with vandetanib; 2 of those patients had other concurrent chemotherapy. One case of axitinib induced pancreatitis was described.
Pancreatitis was reported in the phase 1 trials of sorafenib and sunitinib. In all, 3 of 69 patients treated with sorafenib had grade 3 pancreatitis and asymptomatic elevations of amylase and lipase levels were present in about 5% of patients receiving sunitinib.18,19
Other TKIs associated with pancreatitis include pazopanib,20,21 axitinib,22 and nilotinib.23 Pezzilli and coleagues24 described 5 patients with pancreatitis on sorafenib, 3 on sunitinib, 6 on nilotinib. It is possible that some of these cases appeared in other reviews. Ibrutinib, an inhibitor of Bruton’s tyrosine kinase, caused a single case of pancreatitis in 9 patients.25
Vemurafenib, a BRAF kinase inhibitor, was associated with pancreatitis in one case. In this case, the pancreatitis resolved on stopping the medication but recurred when vemurafenib rechallenge was attempted.26 There is a report of dabrafenib being associated with pancreatitis in 1 patient.27
Agents that inhibit the TKIs associated with BCR-ABL in chronic myelogenous leukemia are associated with acute pancreatitis. Imatinib-induced pancreatitis was reported in a small number of cases.28 Nilotinib has caused amylase/lipase elevations with and without symptomatic pancreatitis.29,30 Ponatinib, an inhibitor of BCR-ABL tyrosine kinase, is associated with pancreatitis.31 Pancreatitis occurred in 11 of 81 patients treated with ponatinib, and in 8 patients it was described as serious. Further elevation of amylase or lipase levels without clinical pancreatitis was noted in 7 other patients.
Proteosome inhibitors
In 2010, Elouni and colleagues32 reported a case of IV bortezomib-induced pancreatitis, which recurred on rechallenge with bortezomib. This same patient was also reported in an abstract in 2009.33 In 2009, there was an editorial comment which was added to the end of the abstract that the World Health Organization Adverse Drug Reaction database had 11 reports of bortezomib associated pancreatitis. Talamo and colleagues34 reported a case of bortezomib-induced pancreatitis due to bortezomib that had been administered subcutaneously. At that time, they also summarized 7 previous reports of bortezomib-associated pancreatitis. The mechanism of bortezomib-induced pancreatitis is not known.35-37
Fotoh and colleagues reported a patient with myeloma who had elevated triglyceride levels after bortezomib therapy.38 In one case of bortezomib-associated pancreatitis, the patient had an elevated triglyceride level, but it was not extremely high.39 Multiple myeloma itself may be associated with hyperlipidemia but only rarely.40 Gozetti and colleagues reported a patient who developed hyperlipidemia after two courses of bortezomib;41 stopping bisphosphonates may be associated with a rise in triglycerides. There was one case of carfilzomib causing pancreatitis during a phase 1 trial.42
Older chemotherapy agents
Reviews of drug-induced pancreatitis have listed many chemotherapy agents which may cause pancreatitis.1,43 The agent most frequently associated with acute pancreatitis has been asparaginase,44 with 2%-16% of patients undergoing asparaginase therapy developing pancreatitis. Asparaginase-related pancreatitis is grade 3 or 4 in 5%-10% of patients, and recurs in 63% of patients on rechallenge. Other chemotherapy agents associated with pancreatitis include: mercaptopurine, cytosine arabinoside, cisplatin, interferon alfa-2b, doxorubicin, tamoxifen, gefitinib, vinorelbine, oxaliplatin, levamisole, methotrexate, azathioprine, 5-fluorouracil, capecitabine, ifosfamide, paclitaxel, and all-trans retinoic acid.
Oxaliplatin carries a 0.1%-2% incidence of drug-induced pancreatitis. In one series of 6 patients, cessation of the agent allowed for resolution of symptoms and decrease in serum lipase and amylase levels.45 With capecitabine there are 2 case reports of pancreatitis.46 Cases of pancreatitis associated with trifluridine or tipiracil were not present in the literature.
Thalidomide caused severe pancreatitis in a patient when it was used to treat chronic graft-versus-host disease.47 This patient suffered recurrent pancreatitis on retreatment with the thalidomide. The authors further referenced two other suspected cases of thalidomide-induced, acute pancreatitis. However, in view of the extensive use of thalidomide for multiple myeloma before the development of lenalidomide, thalidomide-associated pancreatitis would be <1% of patients.
Agents that cause hypertriglyceridemia may cause pancreatitis. This mechanism has been reported as the cause of pancreatitis for everolimus48 and tamoxifen.49,50-52 Everolimus causes elevated triglycerides in 30%-50% of patients. There are case reports and a review of tamoxifen-associated pancreatitis caused by elevated triglycerides.52 There has also been a case of temsirolimus-associated pancreatitis,53 another agent that elevates triglycerides.
Pancreatitis associated with hepatic embolization or HIPEC
TACE leads to symptomatic acute pancreatitis in 0.4%-2% of patients, but nonselective TACE (into the hepatic artery and not just feeder vessels), may lead to elevated amylase levels in 15%-40% of patients.54-56 The risk of pancreatitis would depend on which chemotherapy drug is being infused into the liver. It would also be greater if the chemotherapy has to be infused into a larger part of the liver than into a small portion of the liver. In one patient, severe pancreatitis secondary to TACE occurred after two previous embolizations; prior embolization may have led to occlusion of the previously infused vessels.57 Radioembolization with 90Y microspheres was associated with one case of pancreatitis in 112 consecutive patients.58 The postembolization syndrome in the first 24 hours after the procedure may involve fever, abdominal pain, nausea, and vomiting due to acute pancreatitis in some instances.
Acute pancreatitis has also been described as a complication of hyperthermic intraperitoneal chemotherapy (HIPEC).59,60 Two of 13 patients receiving HIPEC for gastric cancer developed pancreatitis.59 In 25 patients with colon cancer who were treated with HIPEC, 1 patient had pancreatitis.60
Antibody-drug conjugates
Muzaffar and colleagues reported a patient with acute pancreatitis 3 days after starting therapy with ado-trastuzumab emtansine.61 Urru and colleagues62 reported a patient who developed acute pancreatitis after brentuximab vedotin therapy. Ghandi and colleagues63 identified 2 cases of fatal acute pancreatitis with brentuximab vedotin and 6 cases of nonfatal pancreatitis. Two of the nonfatal patients were rechallenged, and 1 developed recurrent pancreatitis. Because abdominal pain may occur in up to 18% of patients receiving brentuximab vedotin, the incidence of pancreatitis may be underestimated with this agent.64
In Table 2, ado-trastuzumab emtansine and brentuximab vedotin are listed with incidence and level of association given by the Baldavo2 and Naranjo.3 With greater awareness, the incidence of pancreatitis associated with these agents may rise or fall as more data is accumulated. In many instances, there are insufficient numbers of reported cases or insufficient information in single-case reports to complete the entire table.
Discussion
Acute pancreatitis is an uncommon complication of tyrosine kinase inhibitors, other kinase inhibitors, proteasome inhibitors, monoclonal antibody-drug conjugates and anti-PD-1 immunotherapies. As nausea, abdominal pain, emesis are common in patients with cancer on antineoplastic therapy, some patients may have acute pancreatitis which is undiagnosed. It is not clear whether a patient with pancreatitis secondary to a TKI can be safely switched to a different TKI. As more molecularly targeted agents and more monoclonal antibodies targeting PD-L1 and PD-1 are under development, screening for amylase and lipase levels during phase 1/2 testing may prove helpful.
The natural history of cancer-drug–associated pancreatitis may depend on which agent is the cause. Although there are descriptions of the course of autoimmune pancreatitis, these studies have not included pancreatitis associated with anti-PD-L1 or -PD-1 therapies.65 It is possible that once an autoimmune pancreatitis has developed, simply stopping the inciting anti-PD-L1 or -PD-1 antibody may not lead to immediate resolution. Therapy with combined immune checkpoint blockade agents (eg, nivolumab and ipilimumab) may cause a higher incidence of pancreatitis than therapy with a single agent.66
In a report of 119 patients with melanoma who were treated with nivolumab and ipilimumab, there were 2 cases of acute pancreatitis, though 20% of patients had a grade 3 or higher amylase level, and just over 6% had a grade 3 or higher lipase.67 Stopping this type of immunotherapy early for grade 1,2, or 3 rises in pancreatic enzymes might prevent symptomatic pancreatitis from developing, but would stop potentially curative therapy for many patients who would have never developed clinically serious pancreatitis. Patients who suffer immune toxicities with anti-PD-1 therapies may be more apt to obtain some clinical benefit. The development of immune-related toxicities in patients treated with ipilimumab ( an anti CTLA4 antibody) seemed to correlate the tumor regression.68 This has also been suggested by the fact that the development of vitiligo correlates with clinical response in melanoma patients treated with nivolumab.69 Although clinically significant pancreatitis might be averted by stopping immune therapies earlier, stopping before it is deemed necessary might prevent cancer patients from receiving life-prolonging therapy.
Acute pancreatitis in general is severe in about 25% of cases and is associated with a significant risk of death. Scoring systems such as Ranson criteria and Apache 2 are used to assess the severity of pancreatitis although their utility is debated.70 Asparaginase is the chemotherapy agent most frequently associated with pancreatitis. It has been used to treat acute lymphoblastic leukemia for more than 30 years. This allowed for a study of 5,185 children and young adults who received asparaginase to determine what clinical factors and genomic factors were associated with the development of acute pancreatitis in 117 individuals.71 Further information gathered from programs such as the FDA and the adverse drug reaction program at Northwestern University in Chicago, coupled with the publication of other cases of pancreatitis associated with newer cancer agents may provide more insight into the mechanism causing pancreatitis due to a specific agent. With more cases being published, it may also become possible to determine if there are specific predisposing factors based on the clearance or metabolism of the offending agent or any genetic predisposition for drug-related pancreatitis.
1. Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol. 2005;29:709-716.
2. Badalov N, Baradarian R, Iswara K, et al. Drug-induced acute pancreatitis: an evidence-based review. Clin Gastroeneterol Hepatol. 2007;5:648-661.
3. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
4. Jones MR, Hall OM, Kaye AM, et al. Drug-induced acute pancreatitis: a review. Oschner J. 2015;15:45-51.
5. Hofmann L, Forschner A, Loquai C, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190-209.
6. Alabed YZ, Aghayev A, Sakellis C, et al. Pancreatitis secondary to anti-programmed death receptor 1 immunotherapy diagnosed by FDG PET/CT. Clin Nucl Med. 2015;40:e528-529.
7. Ikeuchi K, Okuma Y, Tabata T. Immune-related pancreatitis secondary to nivolumab in a patient with recurrent lung adenocarcinoma: a case report. Lung Cancer. 2016;90:148-150.
8. Webster GJ. Autoimmune pancreatitis – a riddle wrapped in an enigma. Dig Dis. 2016;34:532-539.
9. Merchant MS, Baird K, Wexler L, et al. Ipilimumab: first results of a phase I trial in pediatric patients with advanced solid tumors. J Clin Oncol. 2012;30:abstract 9545.
10. Ibrahim RA, Berman DM, Depril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma. J Clin Oncol. 2011;29:abstract 8583.
11. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30:2691-2697.
12. Mayerle J, van den Brandt C, Schwaiger T, et al. Blockage of CTLA-4 suggests that autoimmune pancreatitis is a T-cell mediated disease responsive to ciclosporin A and rapamycin . Pancreatology. 2012;12:579(abstract S8-3).
13. Tecentriq (package insert). South San Francisco, CA: Genentech Inc; 2016.
14. Champiat S, Lambotte E, Barreau E, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2015;27:559-574.
15. Amgen. New safety information for Blincyto (blinatumomab) Risk of pancreatitis. August 2016 and update to Micromedex 2016.
16. Sumimoto K, Uchida K, KusudaT, et al. The role of CD19+ CD24high CD38high and CD19+ CD24high, CD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis . Pancreatology. 2014;14:193-200.
17. Ghatalia P, Morgan CJ, Choueiri TK, et al. Pancreatitis with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol. 2015;94:136-145.
18. Sevin A, Chen A, Atkinson B. Tyrosine kinase inhibitor induced pancreatitis . J Oncol Pharm Pract. 2012;19:257-260.
19. Tirumani SH, Jagannathan JP, Shinagare AB, et al. Acute pancreatitis associated with molecular targeted therapies: a retrospective review of the clinico-radiological features, management and outcome. Pancreatology . 2013;13:461-467.
20. Russano M, Vincenzi B, Benditti O, et al. Pazopanib and pancreatic toxicity: a case report. BMC Res notes. 2015;8:196-198.
21. Kawakubo K, Hata H, Kawakami H, et al. Pazopanib induced severe acute pancreatitis. Case Rep Oncol. 2015;8:356-358.
22. Peron J, Khenifer S, Potier V, et al. Axitinib induced acute pancreatitis: a case report . Anticancer Drugs. 2014;25:478-479.
23. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis . Ann Pharmaco. 2013;37:33.
24. Pezzilli R, Corinaldesi R, Morselli-LabateAM. Tyrosine kinase inhibitors and acute pancreatitis. http://www.serena.unina.it/index.php/jop/article/view/3836/4278. Published May 5, 2010. Accessed May 22 , 2017.
25. Blum KA, Christian B, Flynn JM, et al. A phase I trial of the Bruton’s tyrosine kinase inhibitor, ibrutinib, in combination with rituximab and bendamustine in patients with relapsed/refractory non Hodgkin’s lymphoma. Blood. 2012;120:abstract 1643.
26. Muluneh B, Buie LW, Collichio F. Vemurafenib-associated pancreatitis: a case report. Pharmacotherapy. 2013;33:e43-e44.
27. Dabrafenib. In Life-Sciences-Europe.com from Tafinlar. EU Summary of Product Characteristics. 30 August 2013.
28. Varma MR, Mathew S, Krishnadas D, et al. Imatinib-induced pancreatitis. Indian J Pharmacol. 2010;42:50-52.
29. Palandri F, Castagnetti F, Soverinie S, et al. Pancreatic enzyme elevation in chronic myeloid leukemia patients treated with nilotinib after imatinib failure. Haematologica. 2009;94:1758-1761.
30. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis. Ann of Pharmacother. 2013;47:e.3
31. Cortesk JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadephia chromosome-positive leukemias. New Engl J Med. 2012;367:2075-2088.
32. Elouni B, Ben Salem C, Zamy M, et al. Bortezomib-induced acute pancreatitis [Letter]. J Pancreas. 2010;119:275-276.
33. Elouni B. Acute pancreatitis induced by Velcade ( bortezomib) with positive rechallenge. 9th Annual meeting of the International Society of Pharmacovigilance. Oct 2009 abstract 74.
34. Talamo G, Sikik J, Pandey MK, et al. Bortezomib-induced acute pancreatitis. Case report and review of the literature . J Oncol Pharm Prac. 2016;22:332-334.
35. SolakogluT, Akyol P, Guney T, et al. Acute pancreatitis caused by bortezomib. Pancreatology. 2013;13:189-190.
36. Mihaila RG. A possible rare complication of bortezomib treatment, acute pancreatitis. Acta Medica Transilvanica. 2013;2:269-171
37. Gupta H, Bansal R, Khanna S, et al. An unusual complication of bortezomib therapy: acute pancreatitis. Indian J Nephr. 2014;24:135-136.
38. Fotoh M, KitaharaT, Sakuta J, et al. Multiple lipoma with hyperlipidemia in a multiple myeloma patient treated with bortezomib/dexamethasone. Leuk Res. 2010;34:e120-121.
39. Wang HH, Tsui J, Wang XY, et al. Bortezomib induced acute pancreatitis in a patient with multiple myeloma. Leuk Lymphoma. 2014;55:1404-1405.
40. Misselwitz B, Goede JS, Pestalozzi BC, et al. Hyperlipidemic myeloma: review of 53 cases. Ann Hematol. 2010;89:569-577.
41. Gozzetti A, Fabbri A, Defina M, et al. Hyperlipidemia in a myeloma patient after bortezomib treatment. Leuk Research. 2010;34:e250.
42. Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013;121:893-897.
43. Runzi M, Layer P. Drug-associated pancreatitis: facts and fiction. Pancreas. 1996;13:100-109.
44. Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57:748-757.
45. Butt W, Saadati H, Wasif- Saif M. Oxaliplatin-induced pancreatitis: a case series. Anticancer Res. 2010;30:5113-5115.
46. Yucel H, Warmerdam LV. Capecitabine-induced pancreatitis. J Onc Pharm Pract. 2010;16:133-134.
47. Chung LW, Yeh S-P, Hsieh C-Y, et al. Life-threatening acute pancreatitis due to thalidomide therapy for chronic graft-versus-host disease. Ann Hematol. 2008;87:421-424.
48. Subramaniam S, Zell JA, Kunz PL. Everolimus causing severe hypertriglyceridemia and acute pancreatitis. J Natl Compr Canc Netw. 2013;11:5-9.
49. Wadood A, Chesner R, Mirza M, et al. Tamoxifen precipitation of familial hypertriglyceridaemia: a rare cause of acute pancreatitis. BMJ Case Rep. Published August 3, 2016. doi: 10.1136/bcr-2016-214837.
50. Sakhri J, BenSalem C, Fathallah H, et al. Severe pancreatitis due to tamoxifen induced hypertriglyceridemia with positive rechallenge. J Pancreas. 2010;11:382-384.
51. Elisaf MS, Nakou K, Liamis G, et al. Tamoxifen-induced severe hypertriglyceridemia and pancreatitis. Ann Oncol. 2000;11:1067-1069.
52. Artac M, Sari R, Altunbas J, et al. Asymptomatic acute pancreatitis due to tamoxifen-induced hypertriglyceridemia in a patient with diabetes mellitus and breast cancer. J Chemother. 2002;14:309-311.
53. [Author name not available]. Acute pancreatitis: 15 case reports. React Wkly. 2015;1546:29.
54. Ozcinar B, Guven K, Poylani A, et al. Necrotizing pancreatitis after transcatheter embolization for hepatocellular carcinoma. Diagn In
56. She WH, Chan ACY, Cheung TT, et al. Acute pancreatitis induced by transarterial chemoembolization: a single center experience of over 1500 cases. Hepatobiliary Pancreat Dis Int. 2016;15:93-98.
57. Bae SI, Yeon JE, Lee JM, et al. A case of necrotizing pancreatitis subsequent to transcatheter arterial chemoembolization in a patient with hepatocellular carcinoma. Clin Mol Hepatol. 2012;18:321-325.
58. Peterson JL, Vallow LA, Johnson DW, et al. Complications after 90Y microsphere radioembolization for unresectable hepatic tumors: an evaluation of 112 patients. Brachytherapy. 2013;12:573-579.
59. Piso P, Glockzin G, Schlitt HJ. Cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC) in patients with peritoneal carcinomatosis arising from gastric cancer. J Clin Oncol. 2011;29(suppl 4):abstract 132.
60. Sammartino P, Sibio S, Biacchi D, et al. Prevention of peritoneal metastases from colon cancer in high-risk patients: preliminary results of surgery plus prophylactic HIPEC. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3356888/?report=reader. Published 2012. Accessed May 23, 2017.
61. Muzaffar M, Jia J, Liles D, et al. Acute pancreatitis associated with ado-trastuzumab emtansine. Am J Ther. 2016;23:e572-574.
62. Urru SA, Mariotti E, Carta P, et al. Acute pancreatitis following brentuzimab vedotin therapy for refractory Hodgkin lymphoma: a case report. Drugs R D. 2014;14:9-11.
63. Gandhi MD, Evens AM, Fenske TS, et al. Pancreatitis in patients treated with brentuximab vedotin: a previously unrecognized serious adverse event. Blood. 2014;123:2895-2897.
64. Brentuximab vedotin in Micromedex solutions, Truven Health Analytics. 2016.
65. Okazaki K, Uchida K. Autoimmune pancreatitis: the past, present and future. Pancreas. 2015;44:1006-1016.
66. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab and ipilimumab in advanced melanoma. New Engl J Med. 2013;369:122-133.
67. Friedman CF, Clark V, Raikhel AV, et al. Thinking critically about classifying adverse events: incidence of pancreatitis in patients treated with nivolumab and ipilimumab. J Natl Cancer Inst. 2017;109:[page numbers not available].
68. Day D, Hansen AR. Immune-related adverse events associated with immune checkpoint inhibitors. BioDrugs. 2016;30:571-584.
69. Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
70 . Di MY, Liu H, Zu-Yao Y, et al. Prediction models of mortality in acute pancreatitis in adults. A systematic review. Ann Int Med. 2016;165:482-490.
71. Liu C, Yang W, Devidas M, et al. Clinical and genetic risk factors for acute pancreatitis in patients with acute lymphoblastic leukemia. J Clin Oncol. 2016;18:2133-2140.
1. Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol. 2005;29:709-716.
2. Badalov N, Baradarian R, Iswara K, et al. Drug-induced acute pancreatitis: an evidence-based review. Clin Gastroeneterol Hepatol. 2007;5:648-661.
3. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
4. Jones MR, Hall OM, Kaye AM, et al. Drug-induced acute pancreatitis: a review. Oschner J. 2015;15:45-51.
5. Hofmann L, Forschner A, Loquai C, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190-209.
6. Alabed YZ, Aghayev A, Sakellis C, et al. Pancreatitis secondary to anti-programmed death receptor 1 immunotherapy diagnosed by FDG PET/CT. Clin Nucl Med. 2015;40:e528-529.
7. Ikeuchi K, Okuma Y, Tabata T. Immune-related pancreatitis secondary to nivolumab in a patient with recurrent lung adenocarcinoma: a case report. Lung Cancer. 2016;90:148-150.
8. Webster GJ. Autoimmune pancreatitis – a riddle wrapped in an enigma. Dig Dis. 2016;34:532-539.
9. Merchant MS, Baird K, Wexler L, et al. Ipilimumab: first results of a phase I trial in pediatric patients with advanced solid tumors. J Clin Oncol. 2012;30:abstract 9545.
10. Ibrahim RA, Berman DM, Depril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma. J Clin Oncol. 2011;29:abstract 8583.
11. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30:2691-2697.
12. Mayerle J, van den Brandt C, Schwaiger T, et al. Blockage of CTLA-4 suggests that autoimmune pancreatitis is a T-cell mediated disease responsive to ciclosporin A and rapamycin . Pancreatology. 2012;12:579(abstract S8-3).
13. Tecentriq (package insert). South San Francisco, CA: Genentech Inc; 2016.
14. Champiat S, Lambotte E, Barreau E, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2015;27:559-574.
15. Amgen. New safety information for Blincyto (blinatumomab) Risk of pancreatitis. August 2016 and update to Micromedex 2016.
16. Sumimoto K, Uchida K, KusudaT, et al. The role of CD19+ CD24high CD38high and CD19+ CD24high, CD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis . Pancreatology. 2014;14:193-200.
17. Ghatalia P, Morgan CJ, Choueiri TK, et al. Pancreatitis with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol. 2015;94:136-145.
18. Sevin A, Chen A, Atkinson B. Tyrosine kinase inhibitor induced pancreatitis . J Oncol Pharm Pract. 2012;19:257-260.
19. Tirumani SH, Jagannathan JP, Shinagare AB, et al. Acute pancreatitis associated with molecular targeted therapies: a retrospective review of the clinico-radiological features, management and outcome. Pancreatology . 2013;13:461-467.
20. Russano M, Vincenzi B, Benditti O, et al. Pazopanib and pancreatic toxicity: a case report. BMC Res notes. 2015;8:196-198.
21. Kawakubo K, Hata H, Kawakami H, et al. Pazopanib induced severe acute pancreatitis. Case Rep Oncol. 2015;8:356-358.
22. Peron J, Khenifer S, Potier V, et al. Axitinib induced acute pancreatitis: a case report . Anticancer Drugs. 2014;25:478-479.
23. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis . Ann Pharmaco. 2013;37:33.
24. Pezzilli R, Corinaldesi R, Morselli-LabateAM. Tyrosine kinase inhibitors and acute pancreatitis. http://www.serena.unina.it/index.php/jop/article/view/3836/4278. Published May 5, 2010. Accessed May 22 , 2017.
25. Blum KA, Christian B, Flynn JM, et al. A phase I trial of the Bruton’s tyrosine kinase inhibitor, ibrutinib, in combination with rituximab and bendamustine in patients with relapsed/refractory non Hodgkin’s lymphoma. Blood. 2012;120:abstract 1643.
26. Muluneh B, Buie LW, Collichio F. Vemurafenib-associated pancreatitis: a case report. Pharmacotherapy. 2013;33:e43-e44.
27. Dabrafenib. In Life-Sciences-Europe.com from Tafinlar. EU Summary of Product Characteristics. 30 August 2013.
28. Varma MR, Mathew S, Krishnadas D, et al. Imatinib-induced pancreatitis. Indian J Pharmacol. 2010;42:50-52.
29. Palandri F, Castagnetti F, Soverinie S, et al. Pancreatic enzyme elevation in chronic myeloid leukemia patients treated with nilotinib after imatinib failure. Haematologica. 2009;94:1758-1761.
30. Engel T, Justo D, Amitai M, et al. Nilotinib-associated acute pancreatitis. Ann of Pharmacother. 2013;47:e.3
31. Cortesk JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadephia chromosome-positive leukemias. New Engl J Med. 2012;367:2075-2088.
32. Elouni B, Ben Salem C, Zamy M, et al. Bortezomib-induced acute pancreatitis [Letter]. J Pancreas. 2010;119:275-276.
33. Elouni B. Acute pancreatitis induced by Velcade ( bortezomib) with positive rechallenge. 9th Annual meeting of the International Society of Pharmacovigilance. Oct 2009 abstract 74.
34. Talamo G, Sikik J, Pandey MK, et al. Bortezomib-induced acute pancreatitis. Case report and review of the literature . J Oncol Pharm Prac. 2016;22:332-334.
35. SolakogluT, Akyol P, Guney T, et al. Acute pancreatitis caused by bortezomib. Pancreatology. 2013;13:189-190.
36. Mihaila RG. A possible rare complication of bortezomib treatment, acute pancreatitis. Acta Medica Transilvanica. 2013;2:269-171
37. Gupta H, Bansal R, Khanna S, et al. An unusual complication of bortezomib therapy: acute pancreatitis. Indian J Nephr. 2014;24:135-136.
38. Fotoh M, KitaharaT, Sakuta J, et al. Multiple lipoma with hyperlipidemia in a multiple myeloma patient treated with bortezomib/dexamethasone. Leuk Res. 2010;34:e120-121.
39. Wang HH, Tsui J, Wang XY, et al. Bortezomib induced acute pancreatitis in a patient with multiple myeloma. Leuk Lymphoma. 2014;55:1404-1405.
40. Misselwitz B, Goede JS, Pestalozzi BC, et al. Hyperlipidemic myeloma: review of 53 cases. Ann Hematol. 2010;89:569-577.
41. Gozzetti A, Fabbri A, Defina M, et al. Hyperlipidemia in a myeloma patient after bortezomib treatment. Leuk Research. 2010;34:e250.
42. Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013;121:893-897.
43. Runzi M, Layer P. Drug-associated pancreatitis: facts and fiction. Pancreas. 1996;13:100-109.
44. Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57:748-757.
45. Butt W, Saadati H, Wasif- Saif M. Oxaliplatin-induced pancreatitis: a case series. Anticancer Res. 2010;30:5113-5115.
46. Yucel H, Warmerdam LV. Capecitabine-induced pancreatitis. J Onc Pharm Pract. 2010;16:133-134.
47. Chung LW, Yeh S-P, Hsieh C-Y, et al. Life-threatening acute pancreatitis due to thalidomide therapy for chronic graft-versus-host disease. Ann Hematol. 2008;87:421-424.
48. Subramaniam S, Zell JA, Kunz PL. Everolimus causing severe hypertriglyceridemia and acute pancreatitis. J Natl Compr Canc Netw. 2013;11:5-9.
49. Wadood A, Chesner R, Mirza M, et al. Tamoxifen precipitation of familial hypertriglyceridaemia: a rare cause of acute pancreatitis. BMJ Case Rep. Published August 3, 2016. doi: 10.1136/bcr-2016-214837.
50. Sakhri J, BenSalem C, Fathallah H, et al. Severe pancreatitis due to tamoxifen induced hypertriglyceridemia with positive rechallenge. J Pancreas. 2010;11:382-384.
51. Elisaf MS, Nakou K, Liamis G, et al. Tamoxifen-induced severe hypertriglyceridemia and pancreatitis. Ann Oncol. 2000;11:1067-1069.
52. Artac M, Sari R, Altunbas J, et al. Asymptomatic acute pancreatitis due to tamoxifen-induced hypertriglyceridemia in a patient with diabetes mellitus and breast cancer. J Chemother. 2002;14:309-311.
53. [Author name not available]. Acute pancreatitis: 15 case reports. React Wkly. 2015;1546:29.
54. Ozcinar B, Guven K, Poylani A, et al. Necrotizing pancreatitis after transcatheter embolization for hepatocellular carcinoma. Diagn In
56. She WH, Chan ACY, Cheung TT, et al. Acute pancreatitis induced by transarterial chemoembolization: a single center experience of over 1500 cases. Hepatobiliary Pancreat Dis Int. 2016;15:93-98.
57. Bae SI, Yeon JE, Lee JM, et al. A case of necrotizing pancreatitis subsequent to transcatheter arterial chemoembolization in a patient with hepatocellular carcinoma. Clin Mol Hepatol. 2012;18:321-325.
58. Peterson JL, Vallow LA, Johnson DW, et al. Complications after 90Y microsphere radioembolization for unresectable hepatic tumors: an evaluation of 112 patients. Brachytherapy. 2013;12:573-579.
59. Piso P, Glockzin G, Schlitt HJ. Cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC) in patients with peritoneal carcinomatosis arising from gastric cancer. J Clin Oncol. 2011;29(suppl 4):abstract 132.
60. Sammartino P, Sibio S, Biacchi D, et al. Prevention of peritoneal metastases from colon cancer in high-risk patients: preliminary results of surgery plus prophylactic HIPEC. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3356888/?report=reader. Published 2012. Accessed May 23, 2017.
61. Muzaffar M, Jia J, Liles D, et al. Acute pancreatitis associated with ado-trastuzumab emtansine. Am J Ther. 2016;23:e572-574.
62. Urru SA, Mariotti E, Carta P, et al. Acute pancreatitis following brentuzimab vedotin therapy for refractory Hodgkin lymphoma: a case report. Drugs R D. 2014;14:9-11.
63. Gandhi MD, Evens AM, Fenske TS, et al. Pancreatitis in patients treated with brentuximab vedotin: a previously unrecognized serious adverse event. Blood. 2014;123:2895-2897.
64. Brentuximab vedotin in Micromedex solutions, Truven Health Analytics. 2016.
65. Okazaki K, Uchida K. Autoimmune pancreatitis: the past, present and future. Pancreas. 2015;44:1006-1016.
66. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab and ipilimumab in advanced melanoma. New Engl J Med. 2013;369:122-133.
67. Friedman CF, Clark V, Raikhel AV, et al. Thinking critically about classifying adverse events: incidence of pancreatitis in patients treated with nivolumab and ipilimumab. J Natl Cancer Inst. 2017;109:[page numbers not available].
68. Day D, Hansen AR. Immune-related adverse events associated with immune checkpoint inhibitors. BioDrugs. 2016;30:571-584.
69. Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
70 . Di MY, Liu H, Zu-Yao Y, et al. Prediction models of mortality in acute pancreatitis in adults. A systematic review. Ann Int Med. 2016;165:482-490.
71. Liu C, Yang W, Devidas M, et al. Clinical and genetic risk factors for acute pancreatitis in patients with acute lymphoblastic leukemia. J Clin Oncol. 2016;18:2133-2140.
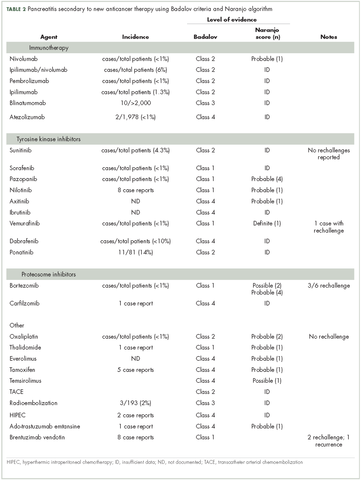
Prehabilitation for lymphedema in head and neck cancer patients at a community cancer center
Lymphedema is the swelling of tissue caused by the accumulation of interstitial fluid in any area of the body where lymphatic flow has been compromised.1 Secondary lymphedema is an acquired abnormality in lymph drainage1,2 and is the type commonly seen in cancer patients. Secondary lymphedema can be described as external or internal. Internal lymphedema, swelling of deep structures and tissues, is very difficult to quantify.
Lymphedema in patients with head and neck cancers
Lymphedema is a complicating morbidity frequently seen in head and neck cancer patients who have undergone treatment with surgery, radiation, and chemotherapy. However, although it is one of the most prevalent side effects of treatment, it is both under-recognized and under-treated.3
In head and neck cancer patients, internal swelling may develop in the soft tissues of the upper aero-digestive tract,4 affecting articulation and swallowing. Currently, there does not seem to be an effective practical and reliable tool with which to measure internal lymphedema. In addition, it is generally accepted that there is no effective way to treat internal lymphedema. By contrast, external lymphedema is more readily observed, but both subjective and objective assessments are difficult. External swelling may occur in the face, jaw, and neck. However, the subjective scales currently available are insufficient to capture very important characteristics of external lymphedema.5 The Edge Task Force on Head and Neck Cancer in 2015 was not able to recommend any outcome measures for objectively quantifying external edema.6 Furthermore, objective measurements of head and neck lymphedema can be expensive and time consuming.
Extent and risk
A combination of both internal and external swelling is seen in more than 50% of patients.7 Risk factors include “throat” tumors, multicancer treatment approaches, higher total radiation dose, a greater number of radiation procedures, and radiation at the surgical site.5 More than 500,000 survivors of head and neck cancer in the United States are at risk of lymphedema.5 Although recent advances in treatment have reduced the incidence of other morbidities, 50% of patients who are treated for head and neck cancer may still develop lymphedema.1,8 The reported incidence in some centers may be much higher, with up to 75% of patients developing lymphedema following treatment.9
Measurement modalities for clinical evaluation
There is little current research into lymphedema of the head and neck, despite the high prevalence of the condition.8 According to Deng and colleagues, measurement of head and neck lymphedema is a challenge, which has an impact on clinical assessment, diagnosis, and treatment of this under-recognized, under-reported and under-addressed problem in head and neck cancer patients.10 In a review of the literature, Deng and colleagues identified three measurement modalities available for clinical evaluation: patient-reported outcomes, clinician-reported outcomes, and technology.10 One major factor, though, in detecting lymphedema, is physician awareness: physicians, health care professionals, and even some lymphedema therapists are not well educated about this problem.8
Treatment
The effectiveness of traditional lymphedema treatment is not well defined.8 Currently, complete decongestive therapy (CDT), is considered the standard of care for lymphedema. The National Lymphedema Network has stated that modifications of CDT, especially manual lymphatic drainage and modified compressive garments for external lymphedema, have been shown to be beneficial for the treatment of lymphedema in head and neck cancer patients.11 Most findings in lymphedema research, mainly in breast cancer patients, have shown that early intervention is the best management and yields the best outcomes. As with other chronic conditions, early identification and timely, appropriate treatment of lymphedema is critical to improve clinical outcomes, to decrease symptom burden and functional impairment, and to improve overall quality of life in head and neck cancer patients.10
Improving recognition and treatment
Head and neck oncologic treatment is increasingly offered outside the network of specialist academic hospitals, at hospitals serving more localized communities where the neediest, sickest patient groups may be receiving less than optimal care.3 This challenges community hospitals to provide optimal treatment, similar to that being offered at nationally recognized institutions. In January 2012, we implemented a prehabilitation program in our community hospital cancer center to provide early intervention for our patients based on the understanding that proper and prompt treatment for patients with early signs of lymphedema should be a priority.12 In this article, we outline how we implemented the program and the describe improvements we observed before and after the implementation of the program.
The prehabilitation program
The role of the nurse navigator
Before the introduction of the prehabilitation program, our pattern of practice was to refer patients to oncology rehabilitation for lymphedema management after they had completed their medical treatment with surgery, radiation, and chemotherapy. In 2012, that was changed to a prehabilitation model of care that was overseen by a head and neck nurse navigator. This focus on prehabilitation begins with patients being referred to oncology rehabilitation at the time of cancer diagnosis for baseline assessment of head and neck swelling. In addition, there is assessment of the many possible other side effects associated with head and neck cancer and its treatment, namely loss of range of motion of the neck, jaw (trismus), and/or shoulders, postural deficits, functional loss, pain, balance dysfunction with fall risk, weakness, and fatigue. Therapeutic interventions are initiated as needed and appropriate. This process also raises awareness of a condition that has been described as under-recognized and under-treated.3
The nurse navigator sits in on each radiation oncology consultation and aids in “navigating” patients through their treatment. The nurse ensures that each patient is referred to different ancillary services from the outset, such as seeing a dietician, social worker, physical/occupational therapist and certified lymphedema therapist, speech pathologist, and financial assistance advisor, if necessary (Table 1).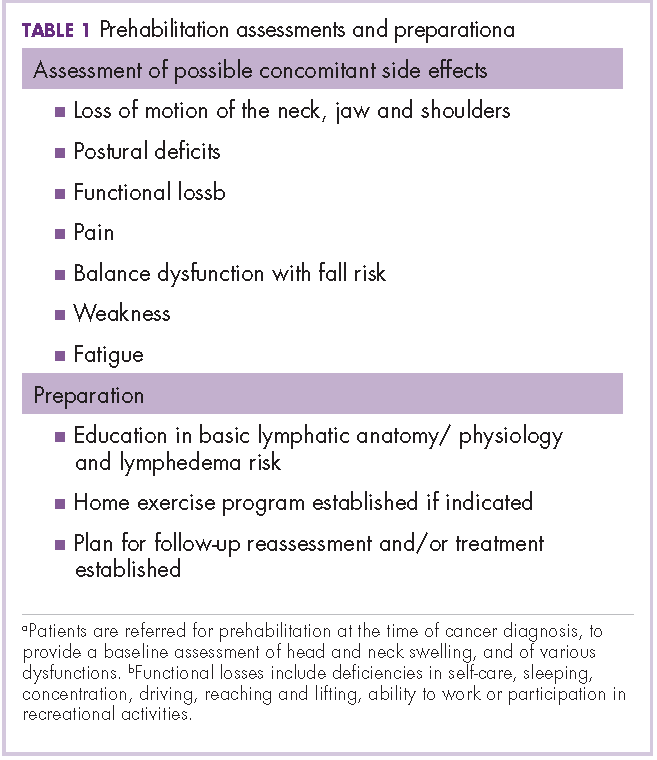
Assessment of lymphedema
Measurement of head and neck lymphedema is a challenge.10 In our program, the physical therapy assessment also includes the evaluation of several other morbidities associated with head and neck cancer and its treatment, such as range of motion, weakness, fatigue, radiation fibrosis, balance dysfunction, and risk of falling (Table 2).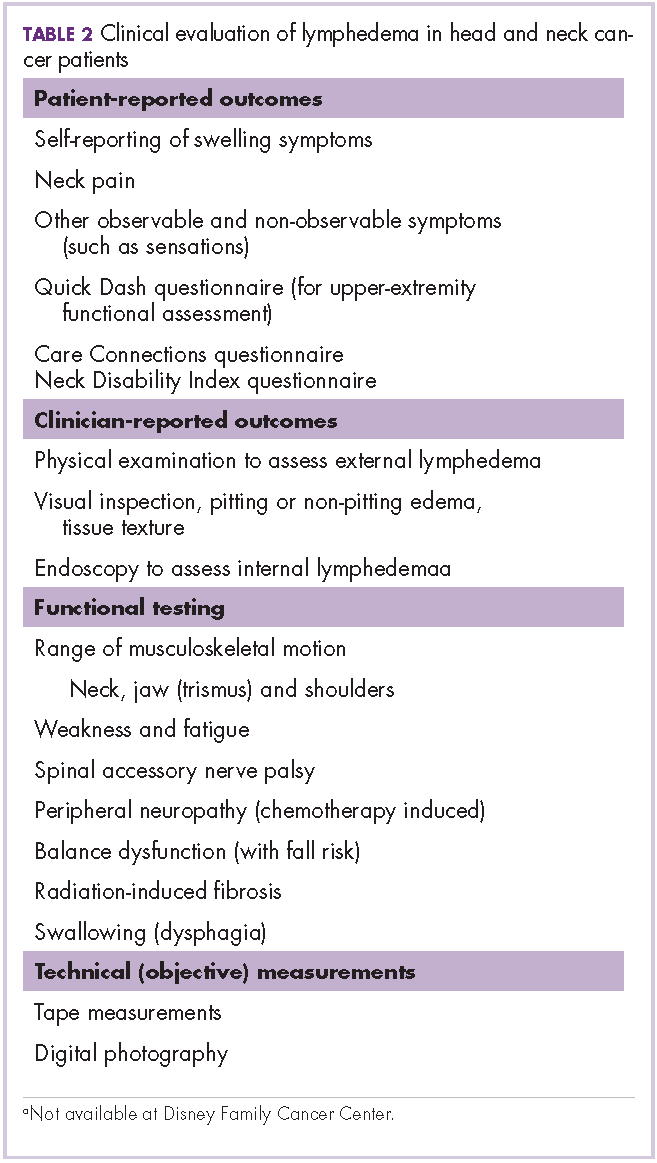
Patient-reported outcomes are essential to fully capture observable and unobservable symptoms (eg, sensations) as well as the functional impacts of lymphedema.10 In addition to lymphedema, there are many other morbidities that may be assessed on the basis of patient-reported outcome tools, such as upper extremity function with QuickDASH.13 At our clinic for head and neck cancer patients we use the Neck Disability Index (NDI)14 and Care Connections (CC)15 survey for the patient-reported outcomes. The Quick DASH, NDI, and CC tools all assess standard functional outcomes that are not specific to lymphedema, but are useful in documenting changes related to lymphedema. We initially used the CC survey and later transitioned to using the NDI. Neck pain is common with lymphedema in the head and neck region, and the NDI is a valid, reliable, responsive and internally consistent clinical tool to measure self-reported disability in patients with neck pain.16 These questionnaires were completed by the patients at their initial assessment, at reassessment, and at time of discharge.
Although objective criteria for external lymphedema have not been established, simple measurements such as using a tape measure to record neck circumference, allow a useful longitudinal assessment. Digital photography may be effective in the documentation and subjective evaluation of changes of external lymphedema.10,17 However, there are some limitations with photography because although external photographs (including digital photography and three-dimensional imaging) can capture some features, such as changes in contours, symmetry, and changes in skin quality and color, they do not detect changes in skin and soft tissue texture and compliance (Table 3).10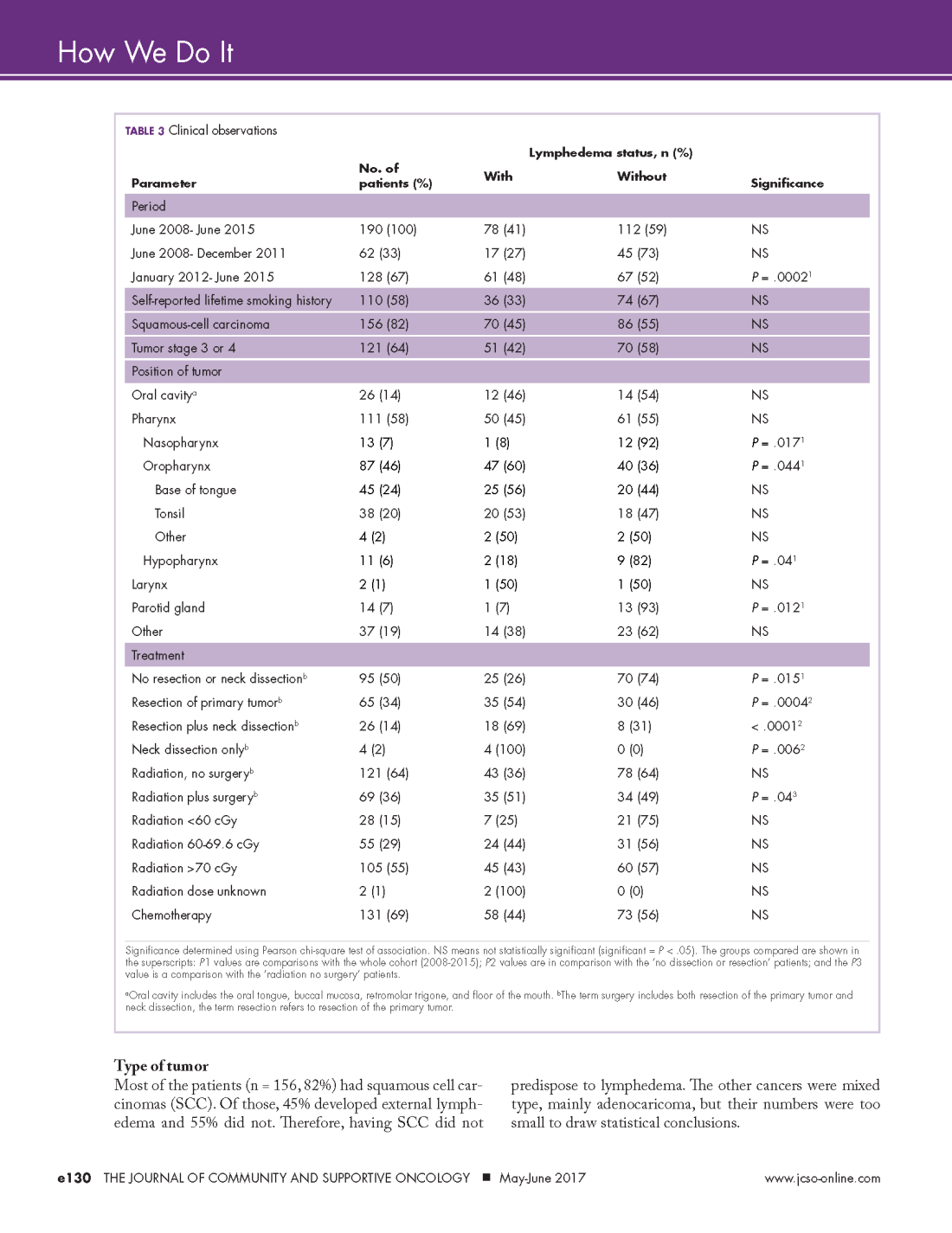
Impact on clinical outcomes
We retrospectively reviewed the medical records of 230 head and neck cancer patients who had been treated at our center between June 2008 and June 2015. Complete clinical data were available for 190 patients. The following information was extracted from each patient’s chart: whether they developed lymphedema, tumor stage, had surgery, radiation dose, type of chemotherapy given, their smoking history, if they had had a neck dissection and the primary site of the tumor (Table 3).
Incidence in different time periods. Of the 190 patients with complete records 78 (41%) were found to have lymphedema. These were all patients undergoing treatment for head and neck cancer during June 2008-June 2015. The prehabilitation program was initiated with the hiring of a nurse navigator for head and neck cancer, starting in January 2012. It is interesting to note that the incidence of lymphedema was 27% before the program was started, but after nurse navigator joined the team, the incidence increased significantly to 48% (P = .0002), in line with published expectations. This increase in recorded incidence may be attributable to the greater awareness of lymphedema intentionally fostered by the prehabilitation program.
Smoking history. Patients’ lifetime smoking history was retrieved from their medical records, based on their verbal admission of tobacco use. Most of the patients (n = 110) self-reported a history of smoking. Of those with a history of smoking, 36 (33%) developed external lymphedema after treatment for head and neck cancer, and 74 (67%) did not. However, this difference was not statistically significant. Hence, although smoking is a risk factor for head and neck cancer, it was not associated with the development of external lymphedema in our cohort of patients.
Type of tumor
Most of the patients (n = 156, 82%) had squamous cell carcinomas (SCC). Of those, 45% developed external lymphedema and 55% did not. Therefore, having SCC did not predispose to lymphedema. The other cancers were mixed type, mainly adenocaricoma, but their numbers were too small to draw statistical conclusions.
Stage of the tumor
About two thirds of the patients (n = 121, 64%) had stage 3 or 4 cancer. However, treatment of more advanced cancers was not associated with lymphedema development.
Site of the tumor
The literature suggests that patients with a primary tumor in the throat are at increased risk for lymphedema.5 The American Cancer Society has defined cancers of the oropharynx (throat) as including the base of the tongue (back third of the tongue), the soft palate, the tonsils, and the side and back walls of the throat.18 In our head and neck cancer cohort, patients with primary tumors of the oropharnyx were, perhaps, more susceptible to lymphedema (P = .044, Table 3). By contrast, in our cohort of patients, those with nasopharyngeal, hypopharyngeal, and parotid gland tumors were significantly less likely to develop lymphedema (Ps = .017, .04, .012, respectively).
No surgery
Half of our patients (n = 95) were not treated with surgery. In the patients who did not have surgery, 25 (26%) developed lymphedema, whereas 70 (74%) did not. Hence, although the incidence of lymphedema was significantly lower in patients who did not have surgery (P = .015), lymphedema did develop in patients who did not have a surgical procedure.
Resection of primary tumor without neck dissection
Of the 64 patients who had surgery, but without neck dissection, 35 (55%) developed external lymphedema. Compared with the no-surgery patients, the doubling of the incidence (from 26% to 55%) was highly significant (P = .0004). These findings are compatible with the literature reports that surgery increases the incidence of lymphedema, which is not surprising because surgery and subsequent scarring is known to compromise the lymphatic system.
Resection of primary tumor with neck dissection
The incidence of external lymphedema was increased to 69% when patients were subjected to both surgery and neck dissection. Compared with the June 2008-June 2015 cohort, there was a significant increase in the incidence of lymphedema in the neck dissection group (P = .007). Neck dissection involves the removal of lymph nodes and disruption of the lymphatic vessels, so it is not surprising that there is a higher incidence of external lymphedema. In our practice, neck dissections increased in frequency every year from June 2008 until December 2011, when 8 patients underwent neck dissections, 6 (75%) of whom developed lymphedema. Since January 2012, when the prehabilitation program was implemented, the number of neck dissections have declined, with more patients receiving chemoradiation and surgery being reserved for surgery. Hamoir and colleagues have reported that neck dissection is no longer justified unless there is clinically residual disease in the neck.19
Radiation
Lymphedema occurred in patients regardless of the dose of radiation received. Although the incidence of lymphedema seemed to be higher in patients who received more than 60 cGy, that difference was not statistically significant (Table 3). We had expected a relationship between radiation damage and greater lymphedema, but that was not evident in our patients.
Chemotherapy
The majority of patients (n = 131, 69%) received chemotherapy. The exposure to chemotherapy was not correlated with the risk of external lymphedema in our cohort of patients, with 58 of the 131 treated patients (44%) developing lymphedema, compared with 73 (56%) of treated patients who did not (Table 3).
Complete decongestive therapy
All patients with documented lymphedema were evaluated for complete decongestive therapy (CDT). Contraindications to CDT included congestive heart failure, renal failure, acute infection, peripheral artery disease, upper-quadrant deep vein thrombosis, and carotid artery stenosis. Eligible patients were referred to a certified lymphedema therapist for CDT. As the program evolved, patients at risk for lymphedema were referred for CDT early on, usually at the time of diagnosis, to improve early identification and surveillance of lymphedema.
CDT included manual lymph drainage, 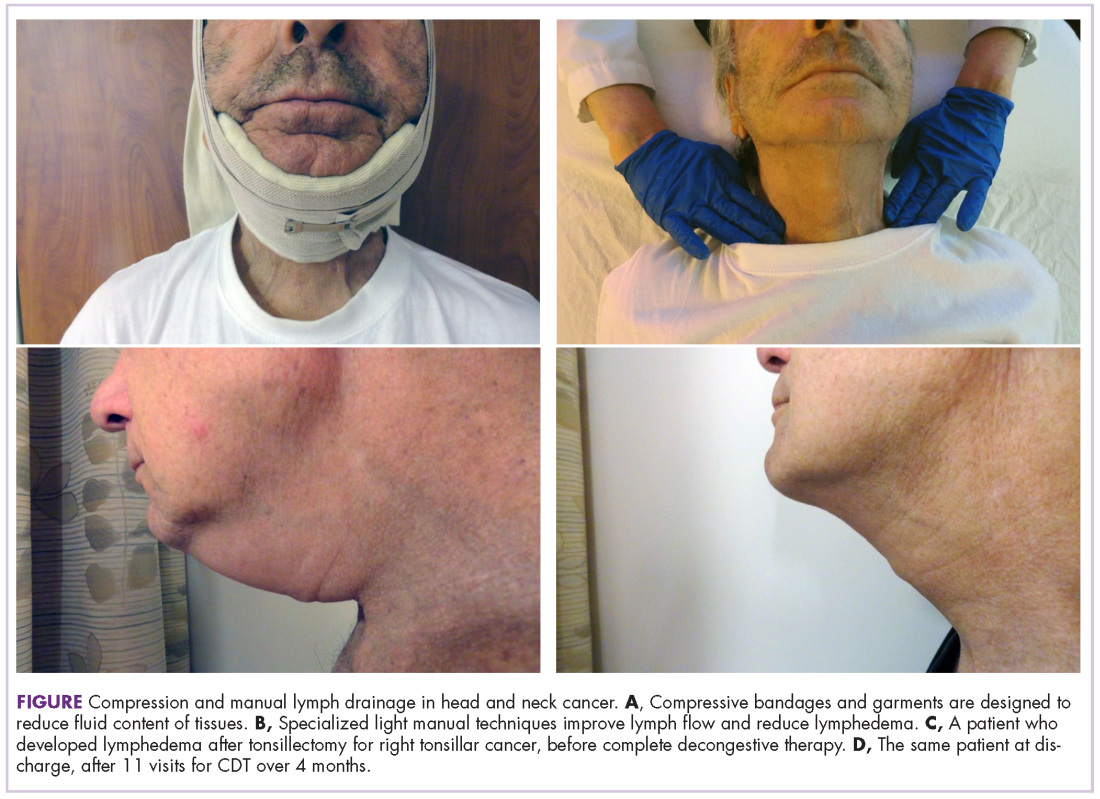
Patients’ responses to CDT were documented with digital photographs that were taken at each visit and, more recently, use of the NDI.
Communication and education
The head and neck cancer nurse navigator attends the cancer center’s multidisciplinary head and neck tumor board, which has representation from otolaryngology, diagnostic radiology, pathology, radiation oncology, medical oncology, reconstructive surgery, oncology rehabilitation (physical/occupational therapist), dietary services, speech pathology, social services and clinical research. This regular contact allows for earlier awareness about which patients are at greater risk for developing lymphedema, thus enabling early intervention (and patient education) in a timely manner.
Education of the patient, before cancer therapy, of the risks of lymphedema is very important. Before the implementation of the prehabilitation program, some patients did not fully comprehend what a painful and debilitating consequence of cancer treatment lymphedema could be.
Discussion
We introduced a prehabilitation program to detect and treat lymphedema in head and neck cancer patients in January 2012 part way through following an observation cohort from June2008 through June2015. Central to this, in our center, was the appointment of a nurse navigator whose primary focus was on head and neck cancer patients. We placed a high priority on the early detection and treatment of lymphedema because do so has been associated with better outcomes in other centers.
One immediate consequence of the inception of our program was the identification of more patients with external lymphedema. Our detected incidence rose significantly (P = .0002), from 27% in the period June 2008-December 20112010, before the program, to 48% during the January 2012-June 2015 period, after the inception of the program. This later incidence rate is in line with published incidence rates in most centers. However, it is still somewhat short of the 75% suggested in one center,9 which suggests we are either we are underdetecting lymphedema or there are differences in definition criteria or sensitivity levels for defining lymphedema.
There are currently no specific objective measures of lymphedema, so there is bound to be some variation in diagnosis rates. In our program, we rely heavily on the patient-reported outcome measures, the NDI instrument, and digital photography to detect and monitor lymphedema, starting with the pretreatment baseline values that are established for each patient.
The use of digital photography in our community hospital setting, which includes taking photographs before and after treatment and at each visit, motivates and encourages patients and provides a tool for clinical lymphedema therapists to visually document benefits of treatment. Patients’ motivation and compliance with their established home program for head and neck lymphedema self-management are essential. The elements of the home program may include self-manual lymph drainage, home-modified compression bandaging and garment wear, therapeutic exercises, and skin care. Patients with lymphedema who adhered closely with their therapy program were more than 8 times more likely to improve compared with noncompliant patients.17
Some groups of patients have a greater risk of developing lymphedema than others,5 so the development of an algorithm to predict lymphedema seemed possible. However, in our cohort of patients, only neck dissection, with its disruption of the lymphatic system of the neck, was strongly associated with external lymphedema (Table 3). It is important to note that some patients who did not undergo surgery developed lymphedema. In our patients, high doses of radiation alone did not seem to predispose to lymphedema. That suggests that no group of head and neck cancer patients should be ignored, which is why we did routine screening of all patients before, during, and after treatment.
Our protocol falls short in the detection of internal lymphedema. For example, information on swallowing gathered by our speech pathologists (in a different department) has not, so far, been included in our assessment. This is one opportunity to improve on our approach, especially because speech difficulties may be associated with internal lymphedema. In addition, we are not equipped for the requisite internal examinations. Unfortunately, there are no practical and successful treatments for patients suffering from internal swelling. This represents a challenge for the medical community to better meet this need. Therefore, although we are missing some assessments of internal lymphedema, this is of little therapeutic consequence at this time.
The increase in the detected incidence of external lymphedema points to a practice gap that has been resolved by the appointment of a dedicated nurse navigator who attends oncology reviews to share knowledge and information. Another educational effort has been made with the patients themselves to increase compliance and improve continuous care at home.
There is always room for improvement, however, either by feedback acquired from other institutions and hospitals or through the future introduction of more objective assessment techniques.
Conclusions
The introduction of the prehabilitation program at our center has coincided with a significantly improved detection rate for external lymphedema in head and neck cancer patients. It may be because the program emphasizes education about lymphedema that awareness of the condition has increased throughout the center. It is now widely recognized that all patients are at risk of lymphedema regardless of whether they fall into an acknowledged high-risk group. Our experience shows that there is no significant difference between treatment modalities apart from neck dissection. In our population, the use of this procedure is decreasing. External lymphedema can develop even in patients who do not have surgery. Therefore, there is no sound way to predict which patients are most likely to suffer from the accumulation of fluid in their head and neck after treatment for head and neck cancer. Thus, an assessment as described here, during and after treatment for all patients, is warranted. Patients are now being seen earlier as a part of the prehabilitation program, which facilitates access to complete decongestive treatment at an earlier stage, improves patient outcomes, and increases patient satisfaction with their treatment. Our prehabilitation program could serve as a model for other community hospital centers in achieving outcomes that are as good as those in academic centers.
Acknowledgments
The authors thank Irene Kadota and Heather Peters, from the Department of Radiation Oncology, and Julianne Courtenay, from the Department of Physical Therapy at the Disney Family Cancer Center, Burbank, California, for providing the original clinical data for analysis.
1. The National Lymphedema Medical Advisory Committee. The diagnosis and treatment of lymphedema. National Lymphedema Network. http://www.lymphnet.org/pdfDocs/nlntreatment.pdf. Updated February 2011. Accessed April 26, 2017.
2. McGarvey AC, Osmotherly PG, Hoffman GR, Chiarelli PE. Lymphedema following treatment for head and neck cancer: impact on patients, and beliefs of health professionals. Eur J Cancer Care (Engl). 2014;23(3):317-327.
3. Bhattacharyya N, Abemayor E. Patterns of hospital utilization for head and neck cancer care: changing demographics. JAMA Otolaryngol Head Neck Surg. 2015;141(4):307-312.
4. Deng J, Ridner SH, Dietrich MS, et al. Prevalence of secondary lymphedema in patients with head and neck cancer. J Pain Symptom Manage. 2012;43(2):244-252.
5. Deng J, Ridner SH, Dietrich MS, et al. Factors associated with external and internal lymphedema in patients with head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2012;84(3):e319-328.
6. Flores AM, Spinelli BA, Eden MM, Galantino ML. EDGE task force on head and neck cancer outcomes: a systematic review of outcomes measures for quantifying external lymphedema. Rehabil Oncol. 2015;33(2):15-23.
7. Ridner SH, Doersam J, Galford E. An update on lymphedema of the head and neck. http://www.lymphnet.org/pdfDocs/Vol_28-N2_Update_HN.pdf. Published April-June 2015. Accessed April 26, 2017.
8. Smith BG, Hutcheson KA, Little LG, et al. Lymphedema outcomes in patients with head and neck cancer. Otolaryngol Head Neck Surg. 2015;152(2);284-291.
9. Naqvi SHS, Karni RJ, Tan IC, et al. Int J Rad Oncol Biol Phys. 2016;4:927-928.
10. Deng J, Ridner SH, Aulino JM, Murphy BA. Assessment and measurement of head and neck lymphedema: state-of-the-science and future directions. Oral Oncol. 2015; 51(5):431-437.
11. Purcell A. Head and neck lymphedema management practices. J Lymphedema. 2013;8(2):8-15.
12. Paskett ED, Dean JA, Oliveri JM, Harrop JP. Cancer-related lymphedema risk factors, diagnosis treatment and impact: a review. J Clinl Oncol. 2012;30(30):3726-3733.
13. Quick DASH questionnaire. http://www.dash.iwh.on.ca/about-quickdash. [Last update not stated.] Accessed May 18, 2017.
14. Neck Disability Index questionnaire. www.aaos.org/uploadedFiles/NDI.pdf Accessed May 18, 2017.
15. Care Connections questionnaire. http://www.careconnections.com/. Accessed May 18, 2017.
16. Galantino ML, Eden MM, Spinelli BA, Flores AM. EDGE task force on head and neck cancer outcomes a systematic review of outcome measures for temporomandibular-related dysfunction. Rehabil Oncol. 2015;33(1):6-14.
17. Deng J, Ridner SH, Murphy BA. Lymphedema in patients with head and neck cancer. Oncol Nurs Forum. 2011;38(1):e1-e10.
18. What are oral cavity and oropharyngeal cancers? American Cancer Society. http://www.cancer.org/cancer/oralcavityandoropharyngealcancer/detailedguide/oral-cavity-and-oropharyngeal-cancer-what-is-oral-cavity-cancer. Last revised August 8, 2016. Accessed April 26, 2017.
19. Hamoir M, Schmitz S, Gregoire V. The role of neck dissection in squamous cell carcinoma of the head and neck. Curr Treat Options Oncol. 2014;15:611-624.
Lymphedema is the swelling of tissue caused by the accumulation of interstitial fluid in any area of the body where lymphatic flow has been compromised.1 Secondary lymphedema is an acquired abnormality in lymph drainage1,2 and is the type commonly seen in cancer patients. Secondary lymphedema can be described as external or internal. Internal lymphedema, swelling of deep structures and tissues, is very difficult to quantify.
Lymphedema in patients with head and neck cancers
Lymphedema is a complicating morbidity frequently seen in head and neck cancer patients who have undergone treatment with surgery, radiation, and chemotherapy. However, although it is one of the most prevalent side effects of treatment, it is both under-recognized and under-treated.3
In head and neck cancer patients, internal swelling may develop in the soft tissues of the upper aero-digestive tract,4 affecting articulation and swallowing. Currently, there does not seem to be an effective practical and reliable tool with which to measure internal lymphedema. In addition, it is generally accepted that there is no effective way to treat internal lymphedema. By contrast, external lymphedema is more readily observed, but both subjective and objective assessments are difficult. External swelling may occur in the face, jaw, and neck. However, the subjective scales currently available are insufficient to capture very important characteristics of external lymphedema.5 The Edge Task Force on Head and Neck Cancer in 2015 was not able to recommend any outcome measures for objectively quantifying external edema.6 Furthermore, objective measurements of head and neck lymphedema can be expensive and time consuming.
Extent and risk
A combination of both internal and external swelling is seen in more than 50% of patients.7 Risk factors include “throat” tumors, multicancer treatment approaches, higher total radiation dose, a greater number of radiation procedures, and radiation at the surgical site.5 More than 500,000 survivors of head and neck cancer in the United States are at risk of lymphedema.5 Although recent advances in treatment have reduced the incidence of other morbidities, 50% of patients who are treated for head and neck cancer may still develop lymphedema.1,8 The reported incidence in some centers may be much higher, with up to 75% of patients developing lymphedema following treatment.9
Measurement modalities for clinical evaluation
There is little current research into lymphedema of the head and neck, despite the high prevalence of the condition.8 According to Deng and colleagues, measurement of head and neck lymphedema is a challenge, which has an impact on clinical assessment, diagnosis, and treatment of this under-recognized, under-reported and under-addressed problem in head and neck cancer patients.10 In a review of the literature, Deng and colleagues identified three measurement modalities available for clinical evaluation: patient-reported outcomes, clinician-reported outcomes, and technology.10 One major factor, though, in detecting lymphedema, is physician awareness: physicians, health care professionals, and even some lymphedema therapists are not well educated about this problem.8
Treatment
The effectiveness of traditional lymphedema treatment is not well defined.8 Currently, complete decongestive therapy (CDT), is considered the standard of care for lymphedema. The National Lymphedema Network has stated that modifications of CDT, especially manual lymphatic drainage and modified compressive garments for external lymphedema, have been shown to be beneficial for the treatment of lymphedema in head and neck cancer patients.11 Most findings in lymphedema research, mainly in breast cancer patients, have shown that early intervention is the best management and yields the best outcomes. As with other chronic conditions, early identification and timely, appropriate treatment of lymphedema is critical to improve clinical outcomes, to decrease symptom burden and functional impairment, and to improve overall quality of life in head and neck cancer patients.10
Improving recognition and treatment
Head and neck oncologic treatment is increasingly offered outside the network of specialist academic hospitals, at hospitals serving more localized communities where the neediest, sickest patient groups may be receiving less than optimal care.3 This challenges community hospitals to provide optimal treatment, similar to that being offered at nationally recognized institutions. In January 2012, we implemented a prehabilitation program in our community hospital cancer center to provide early intervention for our patients based on the understanding that proper and prompt treatment for patients with early signs of lymphedema should be a priority.12 In this article, we outline how we implemented the program and the describe improvements we observed before and after the implementation of the program.
The prehabilitation program
The role of the nurse navigator
Before the introduction of the prehabilitation program, our pattern of practice was to refer patients to oncology rehabilitation for lymphedema management after they had completed their medical treatment with surgery, radiation, and chemotherapy. In 2012, that was changed to a prehabilitation model of care that was overseen by a head and neck nurse navigator. This focus on prehabilitation begins with patients being referred to oncology rehabilitation at the time of cancer diagnosis for baseline assessment of head and neck swelling. In addition, there is assessment of the many possible other side effects associated with head and neck cancer and its treatment, namely loss of range of motion of the neck, jaw (trismus), and/or shoulders, postural deficits, functional loss, pain, balance dysfunction with fall risk, weakness, and fatigue. Therapeutic interventions are initiated as needed and appropriate. This process also raises awareness of a condition that has been described as under-recognized and under-treated.3
The nurse navigator sits in on each radiation oncology consultation and aids in “navigating” patients through their treatment. The nurse ensures that each patient is referred to different ancillary services from the outset, such as seeing a dietician, social worker, physical/occupational therapist and certified lymphedema therapist, speech pathologist, and financial assistance advisor, if necessary (Table 1).
Assessment of lymphedema
Measurement of head and neck lymphedema is a challenge.10 In our program, the physical therapy assessment also includes the evaluation of several other morbidities associated with head and neck cancer and its treatment, such as range of motion, weakness, fatigue, radiation fibrosis, balance dysfunction, and risk of falling (Table 2).
Patient-reported outcomes are essential to fully capture observable and unobservable symptoms (eg, sensations) as well as the functional impacts of lymphedema.10 In addition to lymphedema, there are many other morbidities that may be assessed on the basis of patient-reported outcome tools, such as upper extremity function with QuickDASH.13 At our clinic for head and neck cancer patients we use the Neck Disability Index (NDI)14 and Care Connections (CC)15 survey for the patient-reported outcomes. The Quick DASH, NDI, and CC tools all assess standard functional outcomes that are not specific to lymphedema, but are useful in documenting changes related to lymphedema. We initially used the CC survey and later transitioned to using the NDI. Neck pain is common with lymphedema in the head and neck region, and the NDI is a valid, reliable, responsive and internally consistent clinical tool to measure self-reported disability in patients with neck pain.16 These questionnaires were completed by the patients at their initial assessment, at reassessment, and at time of discharge.
Although objective criteria for external lymphedema have not been established, simple measurements such as using a tape measure to record neck circumference, allow a useful longitudinal assessment. Digital photography may be effective in the documentation and subjective evaluation of changes of external lymphedema.10,17 However, there are some limitations with photography because although external photographs (including digital photography and three-dimensional imaging) can capture some features, such as changes in contours, symmetry, and changes in skin quality and color, they do not detect changes in skin and soft tissue texture and compliance (Table 3).10
Impact on clinical outcomes
We retrospectively reviewed the medical records of 230 head and neck cancer patients who had been treated at our center between June 2008 and June 2015. Complete clinical data were available for 190 patients. The following information was extracted from each patient’s chart: whether they developed lymphedema, tumor stage, had surgery, radiation dose, type of chemotherapy given, their smoking history, if they had had a neck dissection and the primary site of the tumor (Table 3).
Incidence in different time periods. Of the 190 patients with complete records 78 (41%) were found to have lymphedema. These were all patients undergoing treatment for head and neck cancer during June 2008-June 2015. The prehabilitation program was initiated with the hiring of a nurse navigator for head and neck cancer, starting in January 2012. It is interesting to note that the incidence of lymphedema was 27% before the program was started, but after nurse navigator joined the team, the incidence increased significantly to 48% (P = .0002), in line with published expectations. This increase in recorded incidence may be attributable to the greater awareness of lymphedema intentionally fostered by the prehabilitation program.
Smoking history. Patients’ lifetime smoking history was retrieved from their medical records, based on their verbal admission of tobacco use. Most of the patients (n = 110) self-reported a history of smoking. Of those with a history of smoking, 36 (33%) developed external lymphedema after treatment for head and neck cancer, and 74 (67%) did not. However, this difference was not statistically significant. Hence, although smoking is a risk factor for head and neck cancer, it was not associated with the development of external lymphedema in our cohort of patients.
Type of tumor
Most of the patients (n = 156, 82%) had squamous cell carcinomas (SCC). Of those, 45% developed external lymphedema and 55% did not. Therefore, having SCC did not predispose to lymphedema. The other cancers were mixed type, mainly adenocaricoma, but their numbers were too small to draw statistical conclusions.
Stage of the tumor
About two thirds of the patients (n = 121, 64%) had stage 3 or 4 cancer. However, treatment of more advanced cancers was not associated with lymphedema development.
Site of the tumor
The literature suggests that patients with a primary tumor in the throat are at increased risk for lymphedema.5 The American Cancer Society has defined cancers of the oropharynx (throat) as including the base of the tongue (back third of the tongue), the soft palate, the tonsils, and the side and back walls of the throat.18 In our head and neck cancer cohort, patients with primary tumors of the oropharnyx were, perhaps, more susceptible to lymphedema (P = .044, Table 3). By contrast, in our cohort of patients, those with nasopharyngeal, hypopharyngeal, and parotid gland tumors were significantly less likely to develop lymphedema (Ps = .017, .04, .012, respectively).
No surgery
Half of our patients (n = 95) were not treated with surgery. In the patients who did not have surgery, 25 (26%) developed lymphedema, whereas 70 (74%) did not. Hence, although the incidence of lymphedema was significantly lower in patients who did not have surgery (P = .015), lymphedema did develop in patients who did not have a surgical procedure.
Resection of primary tumor without neck dissection
Of the 64 patients who had surgery, but without neck dissection, 35 (55%) developed external lymphedema. Compared with the no-surgery patients, the doubling of the incidence (from 26% to 55%) was highly significant (P = .0004). These findings are compatible with the literature reports that surgery increases the incidence of lymphedema, which is not surprising because surgery and subsequent scarring is known to compromise the lymphatic system.
Resection of primary tumor with neck dissection
The incidence of external lymphedema was increased to 69% when patients were subjected to both surgery and neck dissection. Compared with the June 2008-June 2015 cohort, there was a significant increase in the incidence of lymphedema in the neck dissection group (P = .007). Neck dissection involves the removal of lymph nodes and disruption of the lymphatic vessels, so it is not surprising that there is a higher incidence of external lymphedema. In our practice, neck dissections increased in frequency every year from June 2008 until December 2011, when 8 patients underwent neck dissections, 6 (75%) of whom developed lymphedema. Since January 2012, when the prehabilitation program was implemented, the number of neck dissections have declined, with more patients receiving chemoradiation and surgery being reserved for surgery. Hamoir and colleagues have reported that neck dissection is no longer justified unless there is clinically residual disease in the neck.19
Radiation
Lymphedema occurred in patients regardless of the dose of radiation received. Although the incidence of lymphedema seemed to be higher in patients who received more than 60 cGy, that difference was not statistically significant (Table 3). We had expected a relationship between radiation damage and greater lymphedema, but that was not evident in our patients.
Chemotherapy
The majority of patients (n = 131, 69%) received chemotherapy. The exposure to chemotherapy was not correlated with the risk of external lymphedema in our cohort of patients, with 58 of the 131 treated patients (44%) developing lymphedema, compared with 73 (56%) of treated patients who did not (Table 3).
Complete decongestive therapy
All patients with documented lymphedema were evaluated for complete decongestive therapy (CDT). Contraindications to CDT included congestive heart failure, renal failure, acute infection, peripheral artery disease, upper-quadrant deep vein thrombosis, and carotid artery stenosis. Eligible patients were referred to a certified lymphedema therapist for CDT. As the program evolved, patients at risk for lymphedema were referred for CDT early on, usually at the time of diagnosis, to improve early identification and surveillance of lymphedema.
CDT included manual lymph drainage,
Patients’ responses to CDT were documented with digital photographs that were taken at each visit and, more recently, use of the NDI.
Communication and education
The head and neck cancer nurse navigator attends the cancer center’s multidisciplinary head and neck tumor board, which has representation from otolaryngology, diagnostic radiology, pathology, radiation oncology, medical oncology, reconstructive surgery, oncology rehabilitation (physical/occupational therapist), dietary services, speech pathology, social services and clinical research. This regular contact allows for earlier awareness about which patients are at greater risk for developing lymphedema, thus enabling early intervention (and patient education) in a timely manner.
Education of the patient, before cancer therapy, of the risks of lymphedema is very important. Before the implementation of the prehabilitation program, some patients did not fully comprehend what a painful and debilitating consequence of cancer treatment lymphedema could be.
Discussion
We introduced a prehabilitation program to detect and treat lymphedema in head and neck cancer patients in January 2012 part way through following an observation cohort from June2008 through June2015. Central to this, in our center, was the appointment of a nurse navigator whose primary focus was on head and neck cancer patients. We placed a high priority on the early detection and treatment of lymphedema because do so has been associated with better outcomes in other centers.
One immediate consequence of the inception of our program was the identification of more patients with external lymphedema. Our detected incidence rose significantly (P = .0002), from 27% in the period June 2008-December 20112010, before the program, to 48% during the January 2012-June 2015 period, after the inception of the program. This later incidence rate is in line with published incidence rates in most centers. However, it is still somewhat short of the 75% suggested in one center,9 which suggests we are either we are underdetecting lymphedema or there are differences in definition criteria or sensitivity levels for defining lymphedema.
There are currently no specific objective measures of lymphedema, so there is bound to be some variation in diagnosis rates. In our program, we rely heavily on the patient-reported outcome measures, the NDI instrument, and digital photography to detect and monitor lymphedema, starting with the pretreatment baseline values that are established for each patient.
The use of digital photography in our community hospital setting, which includes taking photographs before and after treatment and at each visit, motivates and encourages patients and provides a tool for clinical lymphedema therapists to visually document benefits of treatment. Patients’ motivation and compliance with their established home program for head and neck lymphedema self-management are essential. The elements of the home program may include self-manual lymph drainage, home-modified compression bandaging and garment wear, therapeutic exercises, and skin care. Patients with lymphedema who adhered closely with their therapy program were more than 8 times more likely to improve compared with noncompliant patients.17
Some groups of patients have a greater risk of developing lymphedema than others,5 so the development of an algorithm to predict lymphedema seemed possible. However, in our cohort of patients, only neck dissection, with its disruption of the lymphatic system of the neck, was strongly associated with external lymphedema (Table 3). It is important to note that some patients who did not undergo surgery developed lymphedema. In our patients, high doses of radiation alone did not seem to predispose to lymphedema. That suggests that no group of head and neck cancer patients should be ignored, which is why we did routine screening of all patients before, during, and after treatment.
Our protocol falls short in the detection of internal lymphedema. For example, information on swallowing gathered by our speech pathologists (in a different department) has not, so far, been included in our assessment. This is one opportunity to improve on our approach, especially because speech difficulties may be associated with internal lymphedema. In addition, we are not equipped for the requisite internal examinations. Unfortunately, there are no practical and successful treatments for patients suffering from internal swelling. This represents a challenge for the medical community to better meet this need. Therefore, although we are missing some assessments of internal lymphedema, this is of little therapeutic consequence at this time.
The increase in the detected incidence of external lymphedema points to a practice gap that has been resolved by the appointment of a dedicated nurse navigator who attends oncology reviews to share knowledge and information. Another educational effort has been made with the patients themselves to increase compliance and improve continuous care at home.
There is always room for improvement, however, either by feedback acquired from other institutions and hospitals or through the future introduction of more objective assessment techniques.
Conclusions
The introduction of the prehabilitation program at our center has coincided with a significantly improved detection rate for external lymphedema in head and neck cancer patients. It may be because the program emphasizes education about lymphedema that awareness of the condition has increased throughout the center. It is now widely recognized that all patients are at risk of lymphedema regardless of whether they fall into an acknowledged high-risk group. Our experience shows that there is no significant difference between treatment modalities apart from neck dissection. In our population, the use of this procedure is decreasing. External lymphedema can develop even in patients who do not have surgery. Therefore, there is no sound way to predict which patients are most likely to suffer from the accumulation of fluid in their head and neck after treatment for head and neck cancer. Thus, an assessment as described here, during and after treatment for all patients, is warranted. Patients are now being seen earlier as a part of the prehabilitation program, which facilitates access to complete decongestive treatment at an earlier stage, improves patient outcomes, and increases patient satisfaction with their treatment. Our prehabilitation program could serve as a model for other community hospital centers in achieving outcomes that are as good as those in academic centers.
Acknowledgments
The authors thank Irene Kadota and Heather Peters, from the Department of Radiation Oncology, and Julianne Courtenay, from the Department of Physical Therapy at the Disney Family Cancer Center, Burbank, California, for providing the original clinical data for analysis.
Lymphedema is the swelling of tissue caused by the accumulation of interstitial fluid in any area of the body where lymphatic flow has been compromised.1 Secondary lymphedema is an acquired abnormality in lymph drainage1,2 and is the type commonly seen in cancer patients. Secondary lymphedema can be described as external or internal. Internal lymphedema, swelling of deep structures and tissues, is very difficult to quantify.
Lymphedema in patients with head and neck cancers
Lymphedema is a complicating morbidity frequently seen in head and neck cancer patients who have undergone treatment with surgery, radiation, and chemotherapy. However, although it is one of the most prevalent side effects of treatment, it is both under-recognized and under-treated.3
In head and neck cancer patients, internal swelling may develop in the soft tissues of the upper aero-digestive tract,4 affecting articulation and swallowing. Currently, there does not seem to be an effective practical and reliable tool with which to measure internal lymphedema. In addition, it is generally accepted that there is no effective way to treat internal lymphedema. By contrast, external lymphedema is more readily observed, but both subjective and objective assessments are difficult. External swelling may occur in the face, jaw, and neck. However, the subjective scales currently available are insufficient to capture very important characteristics of external lymphedema.5 The Edge Task Force on Head and Neck Cancer in 2015 was not able to recommend any outcome measures for objectively quantifying external edema.6 Furthermore, objective measurements of head and neck lymphedema can be expensive and time consuming.
Extent and risk
A combination of both internal and external swelling is seen in more than 50% of patients.7 Risk factors include “throat” tumors, multicancer treatment approaches, higher total radiation dose, a greater number of radiation procedures, and radiation at the surgical site.5 More than 500,000 survivors of head and neck cancer in the United States are at risk of lymphedema.5 Although recent advances in treatment have reduced the incidence of other morbidities, 50% of patients who are treated for head and neck cancer may still develop lymphedema.1,8 The reported incidence in some centers may be much higher, with up to 75% of patients developing lymphedema following treatment.9
Measurement modalities for clinical evaluation
There is little current research into lymphedema of the head and neck, despite the high prevalence of the condition.8 According to Deng and colleagues, measurement of head and neck lymphedema is a challenge, which has an impact on clinical assessment, diagnosis, and treatment of this under-recognized, under-reported and under-addressed problem in head and neck cancer patients.10 In a review of the literature, Deng and colleagues identified three measurement modalities available for clinical evaluation: patient-reported outcomes, clinician-reported outcomes, and technology.10 One major factor, though, in detecting lymphedema, is physician awareness: physicians, health care professionals, and even some lymphedema therapists are not well educated about this problem.8
Treatment
The effectiveness of traditional lymphedema treatment is not well defined.8 Currently, complete decongestive therapy (CDT), is considered the standard of care for lymphedema. The National Lymphedema Network has stated that modifications of CDT, especially manual lymphatic drainage and modified compressive garments for external lymphedema, have been shown to be beneficial for the treatment of lymphedema in head and neck cancer patients.11 Most findings in lymphedema research, mainly in breast cancer patients, have shown that early intervention is the best management and yields the best outcomes. As with other chronic conditions, early identification and timely, appropriate treatment of lymphedema is critical to improve clinical outcomes, to decrease symptom burden and functional impairment, and to improve overall quality of life in head and neck cancer patients.10
Improving recognition and treatment
Head and neck oncologic treatment is increasingly offered outside the network of specialist academic hospitals, at hospitals serving more localized communities where the neediest, sickest patient groups may be receiving less than optimal care.3 This challenges community hospitals to provide optimal treatment, similar to that being offered at nationally recognized institutions. In January 2012, we implemented a prehabilitation program in our community hospital cancer center to provide early intervention for our patients based on the understanding that proper and prompt treatment for patients with early signs of lymphedema should be a priority.12 In this article, we outline how we implemented the program and the describe improvements we observed before and after the implementation of the program.
The prehabilitation program
The role of the nurse navigator
Before the introduction of the prehabilitation program, our pattern of practice was to refer patients to oncology rehabilitation for lymphedema management after they had completed their medical treatment with surgery, radiation, and chemotherapy. In 2012, that was changed to a prehabilitation model of care that was overseen by a head and neck nurse navigator. This focus on prehabilitation begins with patients being referred to oncology rehabilitation at the time of cancer diagnosis for baseline assessment of head and neck swelling. In addition, there is assessment of the many possible other side effects associated with head and neck cancer and its treatment, namely loss of range of motion of the neck, jaw (trismus), and/or shoulders, postural deficits, functional loss, pain, balance dysfunction with fall risk, weakness, and fatigue. Therapeutic interventions are initiated as needed and appropriate. This process also raises awareness of a condition that has been described as under-recognized and under-treated.3
The nurse navigator sits in on each radiation oncology consultation and aids in “navigating” patients through their treatment. The nurse ensures that each patient is referred to different ancillary services from the outset, such as seeing a dietician, social worker, physical/occupational therapist and certified lymphedema therapist, speech pathologist, and financial assistance advisor, if necessary (Table 1).
Assessment of lymphedema
Measurement of head and neck lymphedema is a challenge.10 In our program, the physical therapy assessment also includes the evaluation of several other morbidities associated with head and neck cancer and its treatment, such as range of motion, weakness, fatigue, radiation fibrosis, balance dysfunction, and risk of falling (Table 2).
Patient-reported outcomes are essential to fully capture observable and unobservable symptoms (eg, sensations) as well as the functional impacts of lymphedema.10 In addition to lymphedema, there are many other morbidities that may be assessed on the basis of patient-reported outcome tools, such as upper extremity function with QuickDASH.13 At our clinic for head and neck cancer patients we use the Neck Disability Index (NDI)14 and Care Connections (CC)15 survey for the patient-reported outcomes. The Quick DASH, NDI, and CC tools all assess standard functional outcomes that are not specific to lymphedema, but are useful in documenting changes related to lymphedema. We initially used the CC survey and later transitioned to using the NDI. Neck pain is common with lymphedema in the head and neck region, and the NDI is a valid, reliable, responsive and internally consistent clinical tool to measure self-reported disability in patients with neck pain.16 These questionnaires were completed by the patients at their initial assessment, at reassessment, and at time of discharge.
Although objective criteria for external lymphedema have not been established, simple measurements such as using a tape measure to record neck circumference, allow a useful longitudinal assessment. Digital photography may be effective in the documentation and subjective evaluation of changes of external lymphedema.10,17 However, there are some limitations with photography because although external photographs (including digital photography and three-dimensional imaging) can capture some features, such as changes in contours, symmetry, and changes in skin quality and color, they do not detect changes in skin and soft tissue texture and compliance (Table 3).10
Impact on clinical outcomes
We retrospectively reviewed the medical records of 230 head and neck cancer patients who had been treated at our center between June 2008 and June 2015. Complete clinical data were available for 190 patients. The following information was extracted from each patient’s chart: whether they developed lymphedema, tumor stage, had surgery, radiation dose, type of chemotherapy given, their smoking history, if they had had a neck dissection and the primary site of the tumor (Table 3).
Incidence in different time periods. Of the 190 patients with complete records 78 (41%) were found to have lymphedema. These were all patients undergoing treatment for head and neck cancer during June 2008-June 2015. The prehabilitation program was initiated with the hiring of a nurse navigator for head and neck cancer, starting in January 2012. It is interesting to note that the incidence of lymphedema was 27% before the program was started, but after nurse navigator joined the team, the incidence increased significantly to 48% (P = .0002), in line with published expectations. This increase in recorded incidence may be attributable to the greater awareness of lymphedema intentionally fostered by the prehabilitation program.
Smoking history. Patients’ lifetime smoking history was retrieved from their medical records, based on their verbal admission of tobacco use. Most of the patients (n = 110) self-reported a history of smoking. Of those with a history of smoking, 36 (33%) developed external lymphedema after treatment for head and neck cancer, and 74 (67%) did not. However, this difference was not statistically significant. Hence, although smoking is a risk factor for head and neck cancer, it was not associated with the development of external lymphedema in our cohort of patients.
Type of tumor
Most of the patients (n = 156, 82%) had squamous cell carcinomas (SCC). Of those, 45% developed external lymphedema and 55% did not. Therefore, having SCC did not predispose to lymphedema. The other cancers were mixed type, mainly adenocaricoma, but their numbers were too small to draw statistical conclusions.
Stage of the tumor
About two thirds of the patients (n = 121, 64%) had stage 3 or 4 cancer. However, treatment of more advanced cancers was not associated with lymphedema development.
Site of the tumor
The literature suggests that patients with a primary tumor in the throat are at increased risk for lymphedema.5 The American Cancer Society has defined cancers of the oropharynx (throat) as including the base of the tongue (back third of the tongue), the soft palate, the tonsils, and the side and back walls of the throat.18 In our head and neck cancer cohort, patients with primary tumors of the oropharnyx were, perhaps, more susceptible to lymphedema (P = .044, Table 3). By contrast, in our cohort of patients, those with nasopharyngeal, hypopharyngeal, and parotid gland tumors were significantly less likely to develop lymphedema (Ps = .017, .04, .012, respectively).
No surgery
Half of our patients (n = 95) were not treated with surgery. In the patients who did not have surgery, 25 (26%) developed lymphedema, whereas 70 (74%) did not. Hence, although the incidence of lymphedema was significantly lower in patients who did not have surgery (P = .015), lymphedema did develop in patients who did not have a surgical procedure.
Resection of primary tumor without neck dissection
Of the 64 patients who had surgery, but without neck dissection, 35 (55%) developed external lymphedema. Compared with the no-surgery patients, the doubling of the incidence (from 26% to 55%) was highly significant (P = .0004). These findings are compatible with the literature reports that surgery increases the incidence of lymphedema, which is not surprising because surgery and subsequent scarring is known to compromise the lymphatic system.
Resection of primary tumor with neck dissection
The incidence of external lymphedema was increased to 69% when patients were subjected to both surgery and neck dissection. Compared with the June 2008-June 2015 cohort, there was a significant increase in the incidence of lymphedema in the neck dissection group (P = .007). Neck dissection involves the removal of lymph nodes and disruption of the lymphatic vessels, so it is not surprising that there is a higher incidence of external lymphedema. In our practice, neck dissections increased in frequency every year from June 2008 until December 2011, when 8 patients underwent neck dissections, 6 (75%) of whom developed lymphedema. Since January 2012, when the prehabilitation program was implemented, the number of neck dissections have declined, with more patients receiving chemoradiation and surgery being reserved for surgery. Hamoir and colleagues have reported that neck dissection is no longer justified unless there is clinically residual disease in the neck.19
Radiation
Lymphedema occurred in patients regardless of the dose of radiation received. Although the incidence of lymphedema seemed to be higher in patients who received more than 60 cGy, that difference was not statistically significant (Table 3). We had expected a relationship between radiation damage and greater lymphedema, but that was not evident in our patients.
Chemotherapy
The majority of patients (n = 131, 69%) received chemotherapy. The exposure to chemotherapy was not correlated with the risk of external lymphedema in our cohort of patients, with 58 of the 131 treated patients (44%) developing lymphedema, compared with 73 (56%) of treated patients who did not (Table 3).
Complete decongestive therapy
All patients with documented lymphedema were evaluated for complete decongestive therapy (CDT). Contraindications to CDT included congestive heart failure, renal failure, acute infection, peripheral artery disease, upper-quadrant deep vein thrombosis, and carotid artery stenosis. Eligible patients were referred to a certified lymphedema therapist for CDT. As the program evolved, patients at risk for lymphedema were referred for CDT early on, usually at the time of diagnosis, to improve early identification and surveillance of lymphedema.
CDT included manual lymph drainage,
Patients’ responses to CDT were documented with digital photographs that were taken at each visit and, more recently, use of the NDI.
Communication and education
The head and neck cancer nurse navigator attends the cancer center’s multidisciplinary head and neck tumor board, which has representation from otolaryngology, diagnostic radiology, pathology, radiation oncology, medical oncology, reconstructive surgery, oncology rehabilitation (physical/occupational therapist), dietary services, speech pathology, social services and clinical research. This regular contact allows for earlier awareness about which patients are at greater risk for developing lymphedema, thus enabling early intervention (and patient education) in a timely manner.
Education of the patient, before cancer therapy, of the risks of lymphedema is very important. Before the implementation of the prehabilitation program, some patients did not fully comprehend what a painful and debilitating consequence of cancer treatment lymphedema could be.
Discussion
We introduced a prehabilitation program to detect and treat lymphedema in head and neck cancer patients in January 2012 part way through following an observation cohort from June2008 through June2015. Central to this, in our center, was the appointment of a nurse navigator whose primary focus was on head and neck cancer patients. We placed a high priority on the early detection and treatment of lymphedema because do so has been associated with better outcomes in other centers.
One immediate consequence of the inception of our program was the identification of more patients with external lymphedema. Our detected incidence rose significantly (P = .0002), from 27% in the period June 2008-December 20112010, before the program, to 48% during the January 2012-June 2015 period, after the inception of the program. This later incidence rate is in line with published incidence rates in most centers. However, it is still somewhat short of the 75% suggested in one center,9 which suggests we are either we are underdetecting lymphedema or there are differences in definition criteria or sensitivity levels for defining lymphedema.
There are currently no specific objective measures of lymphedema, so there is bound to be some variation in diagnosis rates. In our program, we rely heavily on the patient-reported outcome measures, the NDI instrument, and digital photography to detect and monitor lymphedema, starting with the pretreatment baseline values that are established for each patient.
The use of digital photography in our community hospital setting, which includes taking photographs before and after treatment and at each visit, motivates and encourages patients and provides a tool for clinical lymphedema therapists to visually document benefits of treatment. Patients’ motivation and compliance with their established home program for head and neck lymphedema self-management are essential. The elements of the home program may include self-manual lymph drainage, home-modified compression bandaging and garment wear, therapeutic exercises, and skin care. Patients with lymphedema who adhered closely with their therapy program were more than 8 times more likely to improve compared with noncompliant patients.17
Some groups of patients have a greater risk of developing lymphedema than others,5 so the development of an algorithm to predict lymphedema seemed possible. However, in our cohort of patients, only neck dissection, with its disruption of the lymphatic system of the neck, was strongly associated with external lymphedema (Table 3). It is important to note that some patients who did not undergo surgery developed lymphedema. In our patients, high doses of radiation alone did not seem to predispose to lymphedema. That suggests that no group of head and neck cancer patients should be ignored, which is why we did routine screening of all patients before, during, and after treatment.
Our protocol falls short in the detection of internal lymphedema. For example, information on swallowing gathered by our speech pathologists (in a different department) has not, so far, been included in our assessment. This is one opportunity to improve on our approach, especially because speech difficulties may be associated with internal lymphedema. In addition, we are not equipped for the requisite internal examinations. Unfortunately, there are no practical and successful treatments for patients suffering from internal swelling. This represents a challenge for the medical community to better meet this need. Therefore, although we are missing some assessments of internal lymphedema, this is of little therapeutic consequence at this time.
The increase in the detected incidence of external lymphedema points to a practice gap that has been resolved by the appointment of a dedicated nurse navigator who attends oncology reviews to share knowledge and information. Another educational effort has been made with the patients themselves to increase compliance and improve continuous care at home.
There is always room for improvement, however, either by feedback acquired from other institutions and hospitals or through the future introduction of more objective assessment techniques.
Conclusions
The introduction of the prehabilitation program at our center has coincided with a significantly improved detection rate for external lymphedema in head and neck cancer patients. It may be because the program emphasizes education about lymphedema that awareness of the condition has increased throughout the center. It is now widely recognized that all patients are at risk of lymphedema regardless of whether they fall into an acknowledged high-risk group. Our experience shows that there is no significant difference between treatment modalities apart from neck dissection. In our population, the use of this procedure is decreasing. External lymphedema can develop even in patients who do not have surgery. Therefore, there is no sound way to predict which patients are most likely to suffer from the accumulation of fluid in their head and neck after treatment for head and neck cancer. Thus, an assessment as described here, during and after treatment for all patients, is warranted. Patients are now being seen earlier as a part of the prehabilitation program, which facilitates access to complete decongestive treatment at an earlier stage, improves patient outcomes, and increases patient satisfaction with their treatment. Our prehabilitation program could serve as a model for other community hospital centers in achieving outcomes that are as good as those in academic centers.
Acknowledgments
The authors thank Irene Kadota and Heather Peters, from the Department of Radiation Oncology, and Julianne Courtenay, from the Department of Physical Therapy at the Disney Family Cancer Center, Burbank, California, for providing the original clinical data for analysis.
1. The National Lymphedema Medical Advisory Committee. The diagnosis and treatment of lymphedema. National Lymphedema Network. http://www.lymphnet.org/pdfDocs/nlntreatment.pdf. Updated February 2011. Accessed April 26, 2017.
2. McGarvey AC, Osmotherly PG, Hoffman GR, Chiarelli PE. Lymphedema following treatment for head and neck cancer: impact on patients, and beliefs of health professionals. Eur J Cancer Care (Engl). 2014;23(3):317-327.
3. Bhattacharyya N, Abemayor E. Patterns of hospital utilization for head and neck cancer care: changing demographics. JAMA Otolaryngol Head Neck Surg. 2015;141(4):307-312.
4. Deng J, Ridner SH, Dietrich MS, et al. Prevalence of secondary lymphedema in patients with head and neck cancer. J Pain Symptom Manage. 2012;43(2):244-252.
5. Deng J, Ridner SH, Dietrich MS, et al. Factors associated with external and internal lymphedema in patients with head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2012;84(3):e319-328.
6. Flores AM, Spinelli BA, Eden MM, Galantino ML. EDGE task force on head and neck cancer outcomes: a systematic review of outcomes measures for quantifying external lymphedema. Rehabil Oncol. 2015;33(2):15-23.
7. Ridner SH, Doersam J, Galford E. An update on lymphedema of the head and neck. http://www.lymphnet.org/pdfDocs/Vol_28-N2_Update_HN.pdf. Published April-June 2015. Accessed April 26, 2017.
8. Smith BG, Hutcheson KA, Little LG, et al. Lymphedema outcomes in patients with head and neck cancer. Otolaryngol Head Neck Surg. 2015;152(2);284-291.
9. Naqvi SHS, Karni RJ, Tan IC, et al. Int J Rad Oncol Biol Phys. 2016;4:927-928.
10. Deng J, Ridner SH, Aulino JM, Murphy BA. Assessment and measurement of head and neck lymphedema: state-of-the-science and future directions. Oral Oncol. 2015; 51(5):431-437.
11. Purcell A. Head and neck lymphedema management practices. J Lymphedema. 2013;8(2):8-15.
12. Paskett ED, Dean JA, Oliveri JM, Harrop JP. Cancer-related lymphedema risk factors, diagnosis treatment and impact: a review. J Clinl Oncol. 2012;30(30):3726-3733.
13. Quick DASH questionnaire. http://www.dash.iwh.on.ca/about-quickdash. [Last update not stated.] Accessed May 18, 2017.
14. Neck Disability Index questionnaire. www.aaos.org/uploadedFiles/NDI.pdf Accessed May 18, 2017.
15. Care Connections questionnaire. http://www.careconnections.com/. Accessed May 18, 2017.
16. Galantino ML, Eden MM, Spinelli BA, Flores AM. EDGE task force on head and neck cancer outcomes a systematic review of outcome measures for temporomandibular-related dysfunction. Rehabil Oncol. 2015;33(1):6-14.
17. Deng J, Ridner SH, Murphy BA. Lymphedema in patients with head and neck cancer. Oncol Nurs Forum. 2011;38(1):e1-e10.
18. What are oral cavity and oropharyngeal cancers? American Cancer Society. http://www.cancer.org/cancer/oralcavityandoropharyngealcancer/detailedguide/oral-cavity-and-oropharyngeal-cancer-what-is-oral-cavity-cancer. Last revised August 8, 2016. Accessed April 26, 2017.
19. Hamoir M, Schmitz S, Gregoire V. The role of neck dissection in squamous cell carcinoma of the head and neck. Curr Treat Options Oncol. 2014;15:611-624.
1. The National Lymphedema Medical Advisory Committee. The diagnosis and treatment of lymphedema. National Lymphedema Network. http://www.lymphnet.org/pdfDocs/nlntreatment.pdf. Updated February 2011. Accessed April 26, 2017.
2. McGarvey AC, Osmotherly PG, Hoffman GR, Chiarelli PE. Lymphedema following treatment for head and neck cancer: impact on patients, and beliefs of health professionals. Eur J Cancer Care (Engl). 2014;23(3):317-327.
3. Bhattacharyya N, Abemayor E. Patterns of hospital utilization for head and neck cancer care: changing demographics. JAMA Otolaryngol Head Neck Surg. 2015;141(4):307-312.
4. Deng J, Ridner SH, Dietrich MS, et al. Prevalence of secondary lymphedema in patients with head and neck cancer. J Pain Symptom Manage. 2012;43(2):244-252.
5. Deng J, Ridner SH, Dietrich MS, et al. Factors associated with external and internal lymphedema in patients with head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2012;84(3):e319-328.
6. Flores AM, Spinelli BA, Eden MM, Galantino ML. EDGE task force on head and neck cancer outcomes: a systematic review of outcomes measures for quantifying external lymphedema. Rehabil Oncol. 2015;33(2):15-23.
7. Ridner SH, Doersam J, Galford E. An update on lymphedema of the head and neck. http://www.lymphnet.org/pdfDocs/Vol_28-N2_Update_HN.pdf. Published April-June 2015. Accessed April 26, 2017.
8. Smith BG, Hutcheson KA, Little LG, et al. Lymphedema outcomes in patients with head and neck cancer. Otolaryngol Head Neck Surg. 2015;152(2);284-291.
9. Naqvi SHS, Karni RJ, Tan IC, et al. Int J Rad Oncol Biol Phys. 2016;4:927-928.
10. Deng J, Ridner SH, Aulino JM, Murphy BA. Assessment and measurement of head and neck lymphedema: state-of-the-science and future directions. Oral Oncol. 2015; 51(5):431-437.
11. Purcell A. Head and neck lymphedema management practices. J Lymphedema. 2013;8(2):8-15.
12. Paskett ED, Dean JA, Oliveri JM, Harrop JP. Cancer-related lymphedema risk factors, diagnosis treatment and impact: a review. J Clinl Oncol. 2012;30(30):3726-3733.
13. Quick DASH questionnaire. http://www.dash.iwh.on.ca/about-quickdash. [Last update not stated.] Accessed May 18, 2017.
14. Neck Disability Index questionnaire. www.aaos.org/uploadedFiles/NDI.pdf Accessed May 18, 2017.
15. Care Connections questionnaire. http://www.careconnections.com/. Accessed May 18, 2017.
16. Galantino ML, Eden MM, Spinelli BA, Flores AM. EDGE task force on head and neck cancer outcomes a systematic review of outcome measures for temporomandibular-related dysfunction. Rehabil Oncol. 2015;33(1):6-14.
17. Deng J, Ridner SH, Murphy BA. Lymphedema in patients with head and neck cancer. Oncol Nurs Forum. 2011;38(1):e1-e10.
18. What are oral cavity and oropharyngeal cancers? American Cancer Society. http://www.cancer.org/cancer/oralcavityandoropharyngealcancer/detailedguide/oral-cavity-and-oropharyngeal-cancer-what-is-oral-cavity-cancer. Last revised August 8, 2016. Accessed April 26, 2017.
19. Hamoir M, Schmitz S, Gregoire V. The role of neck dissection in squamous cell carcinoma of the head and neck. Curr Treat Options Oncol. 2014;15:611-624.
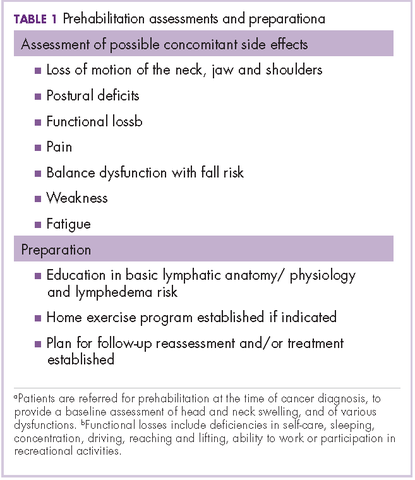
Oncology and the heart


A typical example is a patient who had Hodgkin disease in his teens and received mediastinal mantle radiation. Fifteen to 25 years later, the patient has a pacemaker for heart block, coronary artery disease that requires a stent, and most recently has two valves replaced—so aortic and mitral valve replacement because of late radiation effects. This scenario is typical for the “old” days. The 20-year cumulative incidence of radiation-induced cardiac toxicity is 15%-20% (Table, Figure).1 Sitting with a patient about to begin chest radiation, the absolute risks are unknown but presumed to be less as treatment is delivered according to the modern techniques that you described in the question.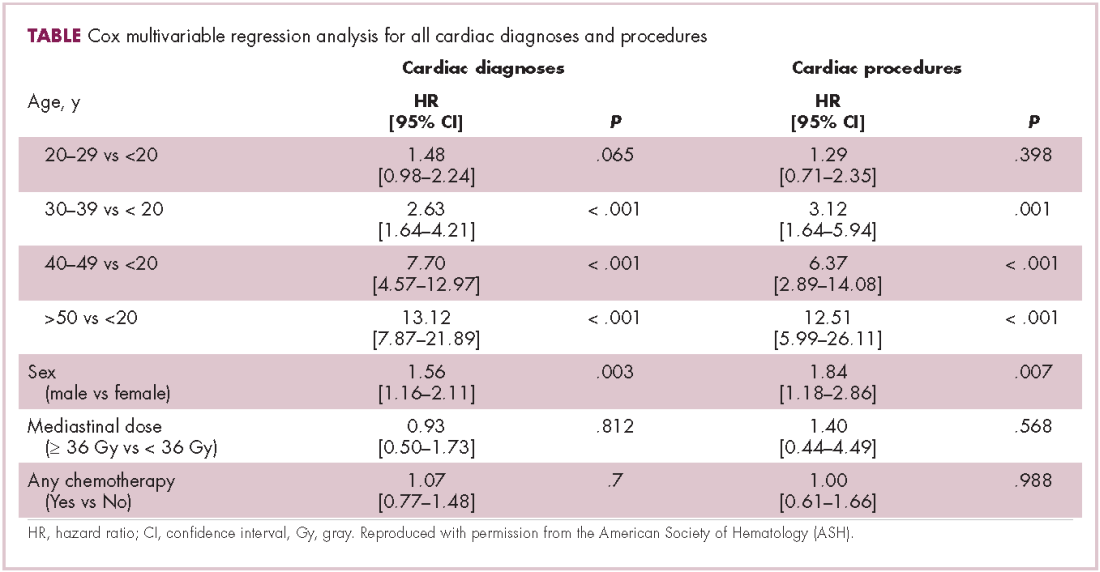
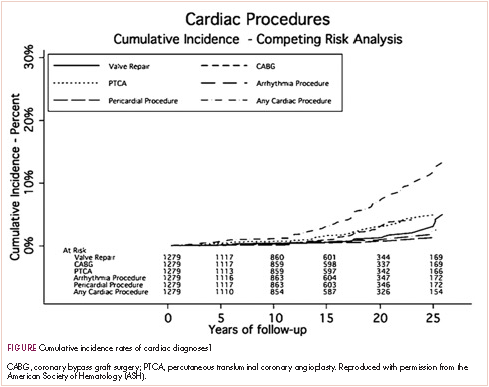
DH They’re so much better now, so this is less common.
JC With the shielding and breath-holding techniques and position changes, doing upright radiation rather than supine, and because the technology has improved both in the delivery of radiation and the technology in understanding where all the radiation is going, in today’s world, we can calculate pretty precisely how much radiation the heart actually receives. Ultimately, with the protective mechanisms that are in place going forward, the risks that I described for that survivor are probably exponentially less than what’s reported in the literature and what we see clinically. Radiation has become much, much safer. There is still probably some small risk of development of late changes, but I don’t think we know what that risk is today because the shielding and things we do to protect the heart have not yet been studied in the long term.
DH Of course, the patient is breathing and there’ll be some movement of the target. Some of the radiation techniques can follow the target despite the breathing?
JC Yes, definitely true. Radiation delivery is much more precise today. Not only has the delivery changed, but so has what we know about the location of potential arterial disease. For example, if you read any textbook, it says that for the coronaries, that it’s ostial and proximal disease of the left main, or the left anterior descending, or the right coronary artery. Today, somebody who gets chest/mediastinal radiation, for either breast cancer, lymphoma, or for a mediastinal tumor, the location of potential disease is more likely to mimic the location of classic coronary disease in the mid-portion of the left anterior descending artery rather than at the ostium. It’s going to be a different disease going forward.2,3
DH Let’s switch from radiation to chemotherapy. Of course, all of us worry about and are very familiar with the toxicity potential of doxorubicin and trastuzumab. I remember an American Society of Clinical Oncology meeting a few years ago, one of the speakers was a cardiologist and was advising us that perhaps the ejection fraction, albeit readily available and reproducible, was probably too simple and we should watch more closely with other techniques. My final question and then I’ll let you comment – I thought I recalled 5-fluorouracil (5-FU) infusions, which we do in some of our colorectal cancers, for example, can cause a vasospasm, Prinzmetal-type angina from time to time, and is that true in capecitabine? What are your thoughts on how to follow the doxorubicin, trastuzumab analogs, and anything about 5-FU and its analogs?
JC Okay, this is a giant question. I’ll take them in order. First, doxorubicin. Cumulative dose-related cardiotoxicity was first described by Von Hoff in 1979.4 That is, the more you get, the higher likelihood of developing cardiotoxicity. Up to a total of 400 mg/m2, the risk is <1%, with a sharp rise as the dose increases beyond this level.4 That being said, there is a clear large and individual variation: I’ve seen sarcoma patients who’ve gotten close to 1,000 mg/m2 without cardiac dysfunction, and some people with minimal exposure have full-blown cardiomyopathy. One of the protective strategies that we developed over the years is to give less of the drug, and with that get the same cancer treatment efficacy. There is definitely a risk for anthracyclines. Full-blown heart failure is probably in the 4%-8% range – and that’s cumulative lifetime – it’s not as high as we once thought it was. That doesn’t mean that it isn’t there, but, relatively speaking, from the standpoint of benefit of anthracyclines, the benefit certainly clearly outweighs the cardiac risk.
With administration of the anthracyclines, we try to do whatever protective things we can do. There are some people who believe that continuous infusion is safer for the heart than bolus injection. It’s pretty controversial. Dexrazoxane, which is a chelating agent, has been shown to reduce cardiotoxicity, and using a lipophilic anthracycline preparation may also have less cardiac toxicity.
DH I have a population in which a lot of liposomal doxorubicin is used and I’ve given a lot and rarely if ever get cardiac toxicity. You see that as well?
JC Yes. There’s a significant financial difference between doxorubicin and liposomal doxorubicin; the latter is more expensive. From the standpoint of safety, and from the standpoint of if I ever needed doxorubicin, I would probably jump on that and ask for the liposomal preparation and/or dexrazoxane.
DH For trastuzumab, we are getting echo- cardiograms every 9 weeks. That seems awfully simple, but there’s a whole algorithm we follow for particular change in ejection fraction and watch the drug or stop the drug. Are we doing that correctly?
JC The first statement I would make about that is that there are too many women who need trastuzumab whose therapy has been prematurely stopped because of just looking at the ejection fractions. So, there has to be more to decision-making other than just the number of the ejection fraction. We’re pretty aggressive and tend to try to get women to get the full dose and whatever dose-effective dose they need, especially with curative intent in the adjuvant setting that we make decisions based not only on the ejection fraction.
We also have, I would say, a handful of our medical breast oncologists who do not follow the package insert. We don’t get ejection fractions every 3 cycles. We have substituted a little bit by following biomarkers so that we use N-terminal pro b-type natriuretic peptide (NT-proBNP) to monitor people, either with each cycle or every third cycle. The benefit of BNP is its negative predictive value. If it’s normal, it’s hard to have any clinically significant myocardial dysfunction.
What we’re going to see over – I would hope – the next year or two is that the recommendations about getting echocardiograms frequently will go away.
DH That would be welcome because in our electronic medical records, it’s 9 weeks, stop, do this, etc. How about a comment on infusional 5-FU and possibly its cousins, such as capecitabine, and any coronary issues?
JC Let me come back, just one more thing about trastuzumab. For metastatic disease, we do whatever is necessary to continue effective cancer therapy and in the absence of any cardiac symptoms or abnormal physical findings, we continue cancer treatment without any serial echocardiographic monitoring.
DH You think the NT-proBNP might be useful? I know that’s excreted by the kidneys, so that might rise in renal failure, but we can adjust for that.
JC The negative predictive value of having a normal BNP is helpful. I think what I wanted to say was that screening echocardiograms and looking at ejection fraction in low-risk populations probably is clearly not cost-effective. It probably never alters decision making. If you have a 30-year-old person with no cardiac risk factors and no past cardiac history who develops B-cell lymphoma and is going to get anthracycline-based chemotherapy, the likelihood of finding a reason not to give that therapy based on an echocardiogram is quite small. I would even go further and say close to zero. We’ve begun to look at this. There is literature that supports the concept. Also, that in low-risk people – if you can define the low-risk population in an accurate way – for lymphoma patients or women with breast cancer getting either anthracyclines, trastuzumab, or the other human epidermal growth factor receptor-2 (HER2)-directed therapies, there’s probably little yield to even getting a baseline study.
DH Very interesting. I would agree with you.
JC We’re going to talk about 5-FU, of course. The 5-FU thing has become a passion of mine. Over the last two to two-and-a- half years we have gotten very aggressive with treating coronary spasm that’s induced by the fluoropyrimidines. That’s 5-FU and capecitabine, the oral version.
There is an incidence that the literature says is less than 1%. It probably is somewhere between 3% and 5%. It’s a little bit more common than has been reported. The reason is the way that it presents has classically been described in the literature as different than what occurs in real life. It is a phenomenon. It’s the most common cardiac side effect. Sometimes it is large epicardial coronary artery spasm. Sometimes it’s small vessel spasm. You can have chest pain with no electrocardiographic changes or ECG changes without chest pain (so-called silent ischemia). The description doesn’t always sound like classic angina but symptoms are temporally related to getting the drug.
So, we’ve developed a protocol to treat documented spasm as an outpatient to be able to continue those drugs to their logical conclusion from an oncologic standpoint. In fact, we just submitted a manuscript to the
DH Finally, it occurred to me that we cause problems with radiation. We cause problems with chemotherapy and other infusions. Are there particular cancers that you think of or you’re called in to see that you worry about cardiac involvement by their location? What comes to mind are cases I’ve had in which there is pericardial involvement and tamponade or restrictive pericarditis.
JC We see metastatic disease to the pericardium with breast cancer, lung cancer, and lymphoma. Renal cell has an interesting predilection to go to the pericardium. We’ve seen in the last probably 6 months 2 cases of bladder cancer with pericardial metastases. When we reviewed the literature, we were only able to find 9 or 10 case reports. It’s rare, but it occurs.
Fluid in the pericardium with and without tamponade is increasingly common, and because we do a better job in treating complicated cancer, people successively can receive cycles of sequential chemotherapeutic regimens – they are living longer, their cancer can get more complicated and/or resistant and with it, there’s more time for metastatic disease to occur. Tamponade is a common phenomenon. We always say that at 4 o’clock on Friday we always see somebody who has tamponade. We see a lot of pericardial disease.
Then, another area of a concern is the tyrosine kinase inhibitors that can cause hypertension, which is very common. We’ve become pretty aggressive. The oncologists recognize the importance of being able to follow and treat blood pressures to allow patients to get these treatments. I guess we couldn’t end without talking about checkpoint inhibitors and the recent lay press flurry about reporting myocarditis.
DH I haven’t personally experienced that. How common is that, and how do we watch for it?
JC Personally, I’ve seen probably four or five people who were referred because of heart failure on checkpoint inhibitors. For each of them, there was historically something as a preexisting problem before the checkpoint inhibitor. It was coincident that with either fluid changes or blood pressure changes associated with the treatment that they had a flare-up of heart failure.
We have not seen, fortunately, the dynamics that were reported in the
DH Well, certainly with the proliferation of the checkpoint inhibitors, and so many different tumors, and so much widespread use, it looks like there is a small safety signal there but still yet to be defined. How common is that, and what should we watch for?
JC Actually, it’s serendipitous that yesterday I was walking to the parking lot with one of the nurse practitioners who takes care of the melanoma population. She said to me, “Now, do you think that we should be getting BNP levels on everybody who is getting a checkpoint inhibitor?”
I don’t think that we’re there. Just the awareness to ask the right questions when you see a patient and before starting ask, is this somebody who, in the absence of a checkpoint inhibitor, could be at risk for myocardial disease? Recognize that and use the cardiology and oncology community to work together and try to make sure that you do whatever cardioprotective things you can do and to monitor them a little bit more closely. I’m not sure that everybody who is going to start a checkpoint inhibitor needs a cardiac evaluation, doesn’t need an echocardiogram, and doesn’t need baseline biomarkers to decide if there’s a potential cardiotoxicity problem.
DH Well certainly, you’ve raised my awareness. It was not something that I had been thinking of with checkpoint inhibitors. Now, I certainly would if the patient has some comorbid illness that involves the heart, maybe think about it, wait to see how these reports develop, and what you and the registry do.
JC You’ve seen people who get this sort of immunologic reaction that they require steroids for fluid accumulation, rash, or other things that are in this constellation. I wouldn’t be surprised if that group might have some subclinical myocarditis that just gets better when they get treated for the other things.
We have actually been trying to get a quick look at the left ventricle when patients on checkpoint inhibitors present with systemic, noncardiac symptoms to see if there is a cardiac signal we are missing. We have a handheld portable echocardiogram device called a Vscan (General Electric Company, Fairfield, CT). It’s not much bigger than the larger cellphones that are available. We’ve been going to the bedside when people have the reaction and sticking the transducer on to get a feeling of what the ventricle looks like. There’s a lot that we don’t know. It’s a fertile ground for investigation.
DH Well, I couldn’t ask you to end on a higher note than covering the checkpoint inhibitors, which are so popular and so interesting and used everywhere. We’re still managing that whole concept. I want to thank you very much.
JC It was a great pleasure. Thank you.
1. Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood. 2011;117(2):412-418.
2. Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013;368(11):987-998.
3. Specht L, Yahalom J, Illidge T, et al. Modern radiation therapy for Hodgkin lymphoma: field and dose guidelines from the International Lymphoma Radiation Oncology Group (ILROG). Int J Radiat Oncol Biol Phys. 2014;89(4):854-862.
4. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91(5):710-717.
5. Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749-1755.
A typical example is a patient who had Hodgkin disease in his teens and received mediastinal mantle radiation. Fifteen to 25 years later, the patient has a pacemaker for heart block, coronary artery disease that requires a stent, and most recently has two valves replaced—so aortic and mitral valve replacement because of late radiation effects. This scenario is typical for the “old” days. The 20-year cumulative incidence of radiation-induced cardiac toxicity is 15%-20% (Table, Figure).1 Sitting with a patient about to begin chest radiation, the absolute risks are unknown but presumed to be less as treatment is delivered according to the modern techniques that you described in the question.
DH They’re so much better now, so this is less common.
JC With the shielding and breath-holding techniques and position changes, doing upright radiation rather than supine, and because the technology has improved both in the delivery of radiation and the technology in understanding where all the radiation is going, in today’s world, we can calculate pretty precisely how much radiation the heart actually receives. Ultimately, with the protective mechanisms that are in place going forward, the risks that I described for that survivor are probably exponentially less than what’s reported in the literature and what we see clinically. Radiation has become much, much safer. There is still probably some small risk of development of late changes, but I don’t think we know what that risk is today because the shielding and things we do to protect the heart have not yet been studied in the long term.
DH Of course, the patient is breathing and there’ll be some movement of the target. Some of the radiation techniques can follow the target despite the breathing?
JC Yes, definitely true. Radiation delivery is much more precise today. Not only has the delivery changed, but so has what we know about the location of potential arterial disease. For example, if you read any textbook, it says that for the coronaries, that it’s ostial and proximal disease of the left main, or the left anterior descending, or the right coronary artery. Today, somebody who gets chest/mediastinal radiation, for either breast cancer, lymphoma, or for a mediastinal tumor, the location of potential disease is more likely to mimic the location of classic coronary disease in the mid-portion of the left anterior descending artery rather than at the ostium. It’s going to be a different disease going forward.2,3
DH Let’s switch from radiation to chemotherapy. Of course, all of us worry about and are very familiar with the toxicity potential of doxorubicin and trastuzumab. I remember an American Society of Clinical Oncology meeting a few years ago, one of the speakers was a cardiologist and was advising us that perhaps the ejection fraction, albeit readily available and reproducible, was probably too simple and we should watch more closely with other techniques. My final question and then I’ll let you comment – I thought I recalled 5-fluorouracil (5-FU) infusions, which we do in some of our colorectal cancers, for example, can cause a vasospasm, Prinzmetal-type angina from time to time, and is that true in capecitabine? What are your thoughts on how to follow the doxorubicin, trastuzumab analogs, and anything about 5-FU and its analogs?
JC Okay, this is a giant question. I’ll take them in order. First, doxorubicin. Cumulative dose-related cardiotoxicity was first described by Von Hoff in 1979.4 That is, the more you get, the higher likelihood of developing cardiotoxicity. Up to a total of 400 mg/m2, the risk is <1%, with a sharp rise as the dose increases beyond this level.4 That being said, there is a clear large and individual variation: I’ve seen sarcoma patients who’ve gotten close to 1,000 mg/m2 without cardiac dysfunction, and some people with minimal exposure have full-blown cardiomyopathy. One of the protective strategies that we developed over the years is to give less of the drug, and with that get the same cancer treatment efficacy. There is definitely a risk for anthracyclines. Full-blown heart failure is probably in the 4%-8% range – and that’s cumulative lifetime – it’s not as high as we once thought it was. That doesn’t mean that it isn’t there, but, relatively speaking, from the standpoint of benefit of anthracyclines, the benefit certainly clearly outweighs the cardiac risk.
With administration of the anthracyclines, we try to do whatever protective things we can do. There are some people who believe that continuous infusion is safer for the heart than bolus injection. It’s pretty controversial. Dexrazoxane, which is a chelating agent, has been shown to reduce cardiotoxicity, and using a lipophilic anthracycline preparation may also have less cardiac toxicity.
DH I have a population in which a lot of liposomal doxorubicin is used and I’ve given a lot and rarely if ever get cardiac toxicity. You see that as well?
JC Yes. There’s a significant financial difference between doxorubicin and liposomal doxorubicin; the latter is more expensive. From the standpoint of safety, and from the standpoint of if I ever needed doxorubicin, I would probably jump on that and ask for the liposomal preparation and/or dexrazoxane.
DH For trastuzumab, we are getting echo- cardiograms every 9 weeks. That seems awfully simple, but there’s a whole algorithm we follow for particular change in ejection fraction and watch the drug or stop the drug. Are we doing that correctly?
JC The first statement I would make about that is that there are too many women who need trastuzumab whose therapy has been prematurely stopped because of just looking at the ejection fractions. So, there has to be more to decision-making other than just the number of the ejection fraction. We’re pretty aggressive and tend to try to get women to get the full dose and whatever dose-effective dose they need, especially with curative intent in the adjuvant setting that we make decisions based not only on the ejection fraction.
We also have, I would say, a handful of our medical breast oncologists who do not follow the package insert. We don’t get ejection fractions every 3 cycles. We have substituted a little bit by following biomarkers so that we use N-terminal pro b-type natriuretic peptide (NT-proBNP) to monitor people, either with each cycle or every third cycle. The benefit of BNP is its negative predictive value. If it’s normal, it’s hard to have any clinically significant myocardial dysfunction.
What we’re going to see over – I would hope – the next year or two is that the recommendations about getting echocardiograms frequently will go away.
DH That would be welcome because in our electronic medical records, it’s 9 weeks, stop, do this, etc. How about a comment on infusional 5-FU and possibly its cousins, such as capecitabine, and any coronary issues?
JC Let me come back, just one more thing about trastuzumab. For metastatic disease, we do whatever is necessary to continue effective cancer therapy and in the absence of any cardiac symptoms or abnormal physical findings, we continue cancer treatment without any serial echocardiographic monitoring.
DH You think the NT-proBNP might be useful? I know that’s excreted by the kidneys, so that might rise in renal failure, but we can adjust for that.
JC The negative predictive value of having a normal BNP is helpful. I think what I wanted to say was that screening echocardiograms and looking at ejection fraction in low-risk populations probably is clearly not cost-effective. It probably never alters decision making. If you have a 30-year-old person with no cardiac risk factors and no past cardiac history who develops B-cell lymphoma and is going to get anthracycline-based chemotherapy, the likelihood of finding a reason not to give that therapy based on an echocardiogram is quite small. I would even go further and say close to zero. We’ve begun to look at this. There is literature that supports the concept. Also, that in low-risk people – if you can define the low-risk population in an accurate way – for lymphoma patients or women with breast cancer getting either anthracyclines, trastuzumab, or the other human epidermal growth factor receptor-2 (HER2)-directed therapies, there’s probably little yield to even getting a baseline study.
DH Very interesting. I would agree with you.
JC We’re going to talk about 5-FU, of course. The 5-FU thing has become a passion of mine. Over the last two to two-and-a- half years we have gotten very aggressive with treating coronary spasm that’s induced by the fluoropyrimidines. That’s 5-FU and capecitabine, the oral version.
There is an incidence that the literature says is less than 1%. It probably is somewhere between 3% and 5%. It’s a little bit more common than has been reported. The reason is the way that it presents has classically been described in the literature as different than what occurs in real life. It is a phenomenon. It’s the most common cardiac side effect. Sometimes it is large epicardial coronary artery spasm. Sometimes it’s small vessel spasm. You can have chest pain with no electrocardiographic changes or ECG changes without chest pain (so-called silent ischemia). The description doesn’t always sound like classic angina but symptoms are temporally related to getting the drug.
So, we’ve developed a protocol to treat documented spasm as an outpatient to be able to continue those drugs to their logical conclusion from an oncologic standpoint. In fact, we just submitted a manuscript to the
DH Finally, it occurred to me that we cause problems with radiation. We cause problems with chemotherapy and other infusions. Are there particular cancers that you think of or you’re called in to see that you worry about cardiac involvement by their location? What comes to mind are cases I’ve had in which there is pericardial involvement and tamponade or restrictive pericarditis.
JC We see metastatic disease to the pericardium with breast cancer, lung cancer, and lymphoma. Renal cell has an interesting predilection to go to the pericardium. We’ve seen in the last probably 6 months 2 cases of bladder cancer with pericardial metastases. When we reviewed the literature, we were only able to find 9 or 10 case reports. It’s rare, but it occurs.
Fluid in the pericardium with and without tamponade is increasingly common, and because we do a better job in treating complicated cancer, people successively can receive cycles of sequential chemotherapeutic regimens – they are living longer, their cancer can get more complicated and/or resistant and with it, there’s more time for metastatic disease to occur. Tamponade is a common phenomenon. We always say that at 4 o’clock on Friday we always see somebody who has tamponade. We see a lot of pericardial disease.
Then, another area of a concern is the tyrosine kinase inhibitors that can cause hypertension, which is very common. We’ve become pretty aggressive. The oncologists recognize the importance of being able to follow and treat blood pressures to allow patients to get these treatments. I guess we couldn’t end without talking about checkpoint inhibitors and the recent lay press flurry about reporting myocarditis.
DH I haven’t personally experienced that. How common is that, and how do we watch for it?
JC Personally, I’ve seen probably four or five people who were referred because of heart failure on checkpoint inhibitors. For each of them, there was historically something as a preexisting problem before the checkpoint inhibitor. It was coincident that with either fluid changes or blood pressure changes associated with the treatment that they had a flare-up of heart failure.
We have not seen, fortunately, the dynamics that were reported in the
DH Well, certainly with the proliferation of the checkpoint inhibitors, and so many different tumors, and so much widespread use, it looks like there is a small safety signal there but still yet to be defined. How common is that, and what should we watch for?
JC Actually, it’s serendipitous that yesterday I was walking to the parking lot with one of the nurse practitioners who takes care of the melanoma population. She said to me, “Now, do you think that we should be getting BNP levels on everybody who is getting a checkpoint inhibitor?”
I don’t think that we’re there. Just the awareness to ask the right questions when you see a patient and before starting ask, is this somebody who, in the absence of a checkpoint inhibitor, could be at risk for myocardial disease? Recognize that and use the cardiology and oncology community to work together and try to make sure that you do whatever cardioprotective things you can do and to monitor them a little bit more closely. I’m not sure that everybody who is going to start a checkpoint inhibitor needs a cardiac evaluation, doesn’t need an echocardiogram, and doesn’t need baseline biomarkers to decide if there’s a potential cardiotoxicity problem.
DH Well certainly, you’ve raised my awareness. It was not something that I had been thinking of with checkpoint inhibitors. Now, I certainly would if the patient has some comorbid illness that involves the heart, maybe think about it, wait to see how these reports develop, and what you and the registry do.
JC You’ve seen people who get this sort of immunologic reaction that they require steroids for fluid accumulation, rash, or other things that are in this constellation. I wouldn’t be surprised if that group might have some subclinical myocarditis that just gets better when they get treated for the other things.
We have actually been trying to get a quick look at the left ventricle when patients on checkpoint inhibitors present with systemic, noncardiac symptoms to see if there is a cardiac signal we are missing. We have a handheld portable echocardiogram device called a Vscan (General Electric Company, Fairfield, CT). It’s not much bigger than the larger cellphones that are available. We’ve been going to the bedside when people have the reaction and sticking the transducer on to get a feeling of what the ventricle looks like. There’s a lot that we don’t know. It’s a fertile ground for investigation.
DH Well, I couldn’t ask you to end on a higher note than covering the checkpoint inhibitors, which are so popular and so interesting and used everywhere. We’re still managing that whole concept. I want to thank you very much.
JC It was a great pleasure. Thank you.
A typical example is a patient who had Hodgkin disease in his teens and received mediastinal mantle radiation. Fifteen to 25 years later, the patient has a pacemaker for heart block, coronary artery disease that requires a stent, and most recently has two valves replaced—so aortic and mitral valve replacement because of late radiation effects. This scenario is typical for the “old” days. The 20-year cumulative incidence of radiation-induced cardiac toxicity is 15%-20% (Table, Figure).1 Sitting with a patient about to begin chest radiation, the absolute risks are unknown but presumed to be less as treatment is delivered according to the modern techniques that you described in the question.
DH They’re so much better now, so this is less common.
JC With the shielding and breath-holding techniques and position changes, doing upright radiation rather than supine, and because the technology has improved both in the delivery of radiation and the technology in understanding where all the radiation is going, in today’s world, we can calculate pretty precisely how much radiation the heart actually receives. Ultimately, with the protective mechanisms that are in place going forward, the risks that I described for that survivor are probably exponentially less than what’s reported in the literature and what we see clinically. Radiation has become much, much safer. There is still probably some small risk of development of late changes, but I don’t think we know what that risk is today because the shielding and things we do to protect the heart have not yet been studied in the long term.
DH Of course, the patient is breathing and there’ll be some movement of the target. Some of the radiation techniques can follow the target despite the breathing?
JC Yes, definitely true. Radiation delivery is much more precise today. Not only has the delivery changed, but so has what we know about the location of potential arterial disease. For example, if you read any textbook, it says that for the coronaries, that it’s ostial and proximal disease of the left main, or the left anterior descending, or the right coronary artery. Today, somebody who gets chest/mediastinal radiation, for either breast cancer, lymphoma, or for a mediastinal tumor, the location of potential disease is more likely to mimic the location of classic coronary disease in the mid-portion of the left anterior descending artery rather than at the ostium. It’s going to be a different disease going forward.2,3
DH Let’s switch from radiation to chemotherapy. Of course, all of us worry about and are very familiar with the toxicity potential of doxorubicin and trastuzumab. I remember an American Society of Clinical Oncology meeting a few years ago, one of the speakers was a cardiologist and was advising us that perhaps the ejection fraction, albeit readily available and reproducible, was probably too simple and we should watch more closely with other techniques. My final question and then I’ll let you comment – I thought I recalled 5-fluorouracil (5-FU) infusions, which we do in some of our colorectal cancers, for example, can cause a vasospasm, Prinzmetal-type angina from time to time, and is that true in capecitabine? What are your thoughts on how to follow the doxorubicin, trastuzumab analogs, and anything about 5-FU and its analogs?
JC Okay, this is a giant question. I’ll take them in order. First, doxorubicin. Cumulative dose-related cardiotoxicity was first described by Von Hoff in 1979.4 That is, the more you get, the higher likelihood of developing cardiotoxicity. Up to a total of 400 mg/m2, the risk is <1%, with a sharp rise as the dose increases beyond this level.4 That being said, there is a clear large and individual variation: I’ve seen sarcoma patients who’ve gotten close to 1,000 mg/m2 without cardiac dysfunction, and some people with minimal exposure have full-blown cardiomyopathy. One of the protective strategies that we developed over the years is to give less of the drug, and with that get the same cancer treatment efficacy. There is definitely a risk for anthracyclines. Full-blown heart failure is probably in the 4%-8% range – and that’s cumulative lifetime – it’s not as high as we once thought it was. That doesn’t mean that it isn’t there, but, relatively speaking, from the standpoint of benefit of anthracyclines, the benefit certainly clearly outweighs the cardiac risk.
With administration of the anthracyclines, we try to do whatever protective things we can do. There are some people who believe that continuous infusion is safer for the heart than bolus injection. It’s pretty controversial. Dexrazoxane, which is a chelating agent, has been shown to reduce cardiotoxicity, and using a lipophilic anthracycline preparation may also have less cardiac toxicity.
DH I have a population in which a lot of liposomal doxorubicin is used and I’ve given a lot and rarely if ever get cardiac toxicity. You see that as well?
JC Yes. There’s a significant financial difference between doxorubicin and liposomal doxorubicin; the latter is more expensive. From the standpoint of safety, and from the standpoint of if I ever needed doxorubicin, I would probably jump on that and ask for the liposomal preparation and/or dexrazoxane.
DH For trastuzumab, we are getting echo- cardiograms every 9 weeks. That seems awfully simple, but there’s a whole algorithm we follow for particular change in ejection fraction and watch the drug or stop the drug. Are we doing that correctly?
JC The first statement I would make about that is that there are too many women who need trastuzumab whose therapy has been prematurely stopped because of just looking at the ejection fractions. So, there has to be more to decision-making other than just the number of the ejection fraction. We’re pretty aggressive and tend to try to get women to get the full dose and whatever dose-effective dose they need, especially with curative intent in the adjuvant setting that we make decisions based not only on the ejection fraction.
We also have, I would say, a handful of our medical breast oncologists who do not follow the package insert. We don’t get ejection fractions every 3 cycles. We have substituted a little bit by following biomarkers so that we use N-terminal pro b-type natriuretic peptide (NT-proBNP) to monitor people, either with each cycle or every third cycle. The benefit of BNP is its negative predictive value. If it’s normal, it’s hard to have any clinically significant myocardial dysfunction.
What we’re going to see over – I would hope – the next year or two is that the recommendations about getting echocardiograms frequently will go away.
DH That would be welcome because in our electronic medical records, it’s 9 weeks, stop, do this, etc. How about a comment on infusional 5-FU and possibly its cousins, such as capecitabine, and any coronary issues?
JC Let me come back, just one more thing about trastuzumab. For metastatic disease, we do whatever is necessary to continue effective cancer therapy and in the absence of any cardiac symptoms or abnormal physical findings, we continue cancer treatment without any serial echocardiographic monitoring.
DH You think the NT-proBNP might be useful? I know that’s excreted by the kidneys, so that might rise in renal failure, but we can adjust for that.
JC The negative predictive value of having a normal BNP is helpful. I think what I wanted to say was that screening echocardiograms and looking at ejection fraction in low-risk populations probably is clearly not cost-effective. It probably never alters decision making. If you have a 30-year-old person with no cardiac risk factors and no past cardiac history who develops B-cell lymphoma and is going to get anthracycline-based chemotherapy, the likelihood of finding a reason not to give that therapy based on an echocardiogram is quite small. I would even go further and say close to zero. We’ve begun to look at this. There is literature that supports the concept. Also, that in low-risk people – if you can define the low-risk population in an accurate way – for lymphoma patients or women with breast cancer getting either anthracyclines, trastuzumab, or the other human epidermal growth factor receptor-2 (HER2)-directed therapies, there’s probably little yield to even getting a baseline study.
DH Very interesting. I would agree with you.
JC We’re going to talk about 5-FU, of course. The 5-FU thing has become a passion of mine. Over the last two to two-and-a- half years we have gotten very aggressive with treating coronary spasm that’s induced by the fluoropyrimidines. That’s 5-FU and capecitabine, the oral version.
There is an incidence that the literature says is less than 1%. It probably is somewhere between 3% and 5%. It’s a little bit more common than has been reported. The reason is the way that it presents has classically been described in the literature as different than what occurs in real life. It is a phenomenon. It’s the most common cardiac side effect. Sometimes it is large epicardial coronary artery spasm. Sometimes it’s small vessel spasm. You can have chest pain with no electrocardiographic changes or ECG changes without chest pain (so-called silent ischemia). The description doesn’t always sound like classic angina but symptoms are temporally related to getting the drug.
So, we’ve developed a protocol to treat documented spasm as an outpatient to be able to continue those drugs to their logical conclusion from an oncologic standpoint. In fact, we just submitted a manuscript to the
DH Finally, it occurred to me that we cause problems with radiation. We cause problems with chemotherapy and other infusions. Are there particular cancers that you think of or you’re called in to see that you worry about cardiac involvement by their location? What comes to mind are cases I’ve had in which there is pericardial involvement and tamponade or restrictive pericarditis.
JC We see metastatic disease to the pericardium with breast cancer, lung cancer, and lymphoma. Renal cell has an interesting predilection to go to the pericardium. We’ve seen in the last probably 6 months 2 cases of bladder cancer with pericardial metastases. When we reviewed the literature, we were only able to find 9 or 10 case reports. It’s rare, but it occurs.
Fluid in the pericardium with and without tamponade is increasingly common, and because we do a better job in treating complicated cancer, people successively can receive cycles of sequential chemotherapeutic regimens – they are living longer, their cancer can get more complicated and/or resistant and with it, there’s more time for metastatic disease to occur. Tamponade is a common phenomenon. We always say that at 4 o’clock on Friday we always see somebody who has tamponade. We see a lot of pericardial disease.
Then, another area of a concern is the tyrosine kinase inhibitors that can cause hypertension, which is very common. We’ve become pretty aggressive. The oncologists recognize the importance of being able to follow and treat blood pressures to allow patients to get these treatments. I guess we couldn’t end without talking about checkpoint inhibitors and the recent lay press flurry about reporting myocarditis.
DH I haven’t personally experienced that. How common is that, and how do we watch for it?
JC Personally, I’ve seen probably four or five people who were referred because of heart failure on checkpoint inhibitors. For each of them, there was historically something as a preexisting problem before the checkpoint inhibitor. It was coincident that with either fluid changes or blood pressure changes associated with the treatment that they had a flare-up of heart failure.
We have not seen, fortunately, the dynamics that were reported in the
DH Well, certainly with the proliferation of the checkpoint inhibitors, and so many different tumors, and so much widespread use, it looks like there is a small safety signal there but still yet to be defined. How common is that, and what should we watch for?
JC Actually, it’s serendipitous that yesterday I was walking to the parking lot with one of the nurse practitioners who takes care of the melanoma population. She said to me, “Now, do you think that we should be getting BNP levels on everybody who is getting a checkpoint inhibitor?”
I don’t think that we’re there. Just the awareness to ask the right questions when you see a patient and before starting ask, is this somebody who, in the absence of a checkpoint inhibitor, could be at risk for myocardial disease? Recognize that and use the cardiology and oncology community to work together and try to make sure that you do whatever cardioprotective things you can do and to monitor them a little bit more closely. I’m not sure that everybody who is going to start a checkpoint inhibitor needs a cardiac evaluation, doesn’t need an echocardiogram, and doesn’t need baseline biomarkers to decide if there’s a potential cardiotoxicity problem.
DH Well certainly, you’ve raised my awareness. It was not something that I had been thinking of with checkpoint inhibitors. Now, I certainly would if the patient has some comorbid illness that involves the heart, maybe think about it, wait to see how these reports develop, and what you and the registry do.
JC You’ve seen people who get this sort of immunologic reaction that they require steroids for fluid accumulation, rash, or other things that are in this constellation. I wouldn’t be surprised if that group might have some subclinical myocarditis that just gets better when they get treated for the other things.
We have actually been trying to get a quick look at the left ventricle when patients on checkpoint inhibitors present with systemic, noncardiac symptoms to see if there is a cardiac signal we are missing. We have a handheld portable echocardiogram device called a Vscan (General Electric Company, Fairfield, CT). It’s not much bigger than the larger cellphones that are available. We’ve been going to the bedside when people have the reaction and sticking the transducer on to get a feeling of what the ventricle looks like. There’s a lot that we don’t know. It’s a fertile ground for investigation.
DH Well, I couldn’t ask you to end on a higher note than covering the checkpoint inhibitors, which are so popular and so interesting and used everywhere. We’re still managing that whole concept. I want to thank you very much.
JC It was a great pleasure. Thank you.
1. Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood. 2011;117(2):412-418.
2. Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013;368(11):987-998.
3. Specht L, Yahalom J, Illidge T, et al. Modern radiation therapy for Hodgkin lymphoma: field and dose guidelines from the International Lymphoma Radiation Oncology Group (ILROG). Int J Radiat Oncol Biol Phys. 2014;89(4):854-862.
4. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91(5):710-717.
5. Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749-1755.
1. Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood. 2011;117(2):412-418.
2. Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013;368(11):987-998.
3. Specht L, Yahalom J, Illidge T, et al. Modern radiation therapy for Hodgkin lymphoma: field and dose guidelines from the International Lymphoma Radiation Oncology Group (ILROG). Int J Radiat Oncol Biol Phys. 2014;89(4):854-862.
4. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91(5):710-717.
5. Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749-1755.