User login
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.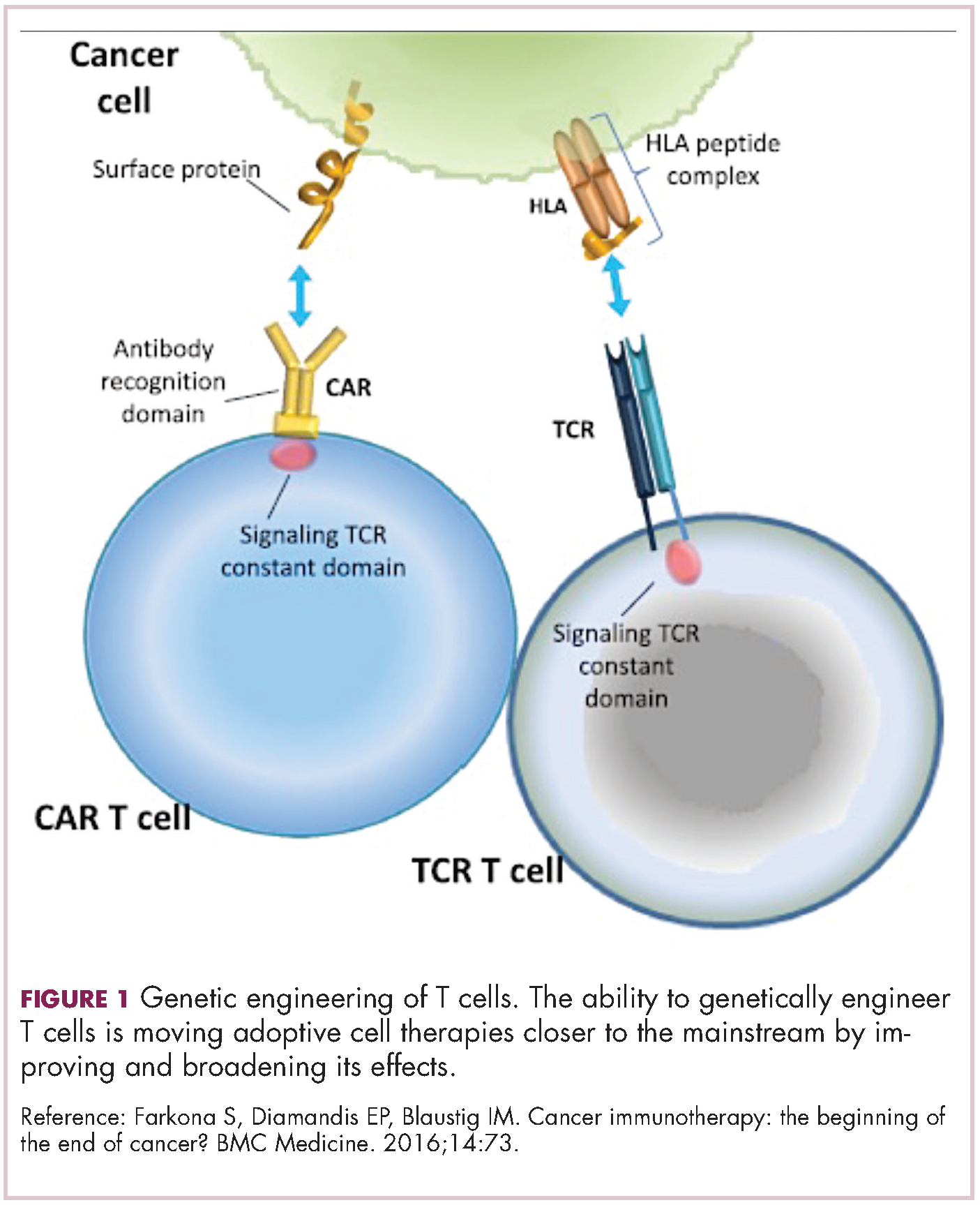
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17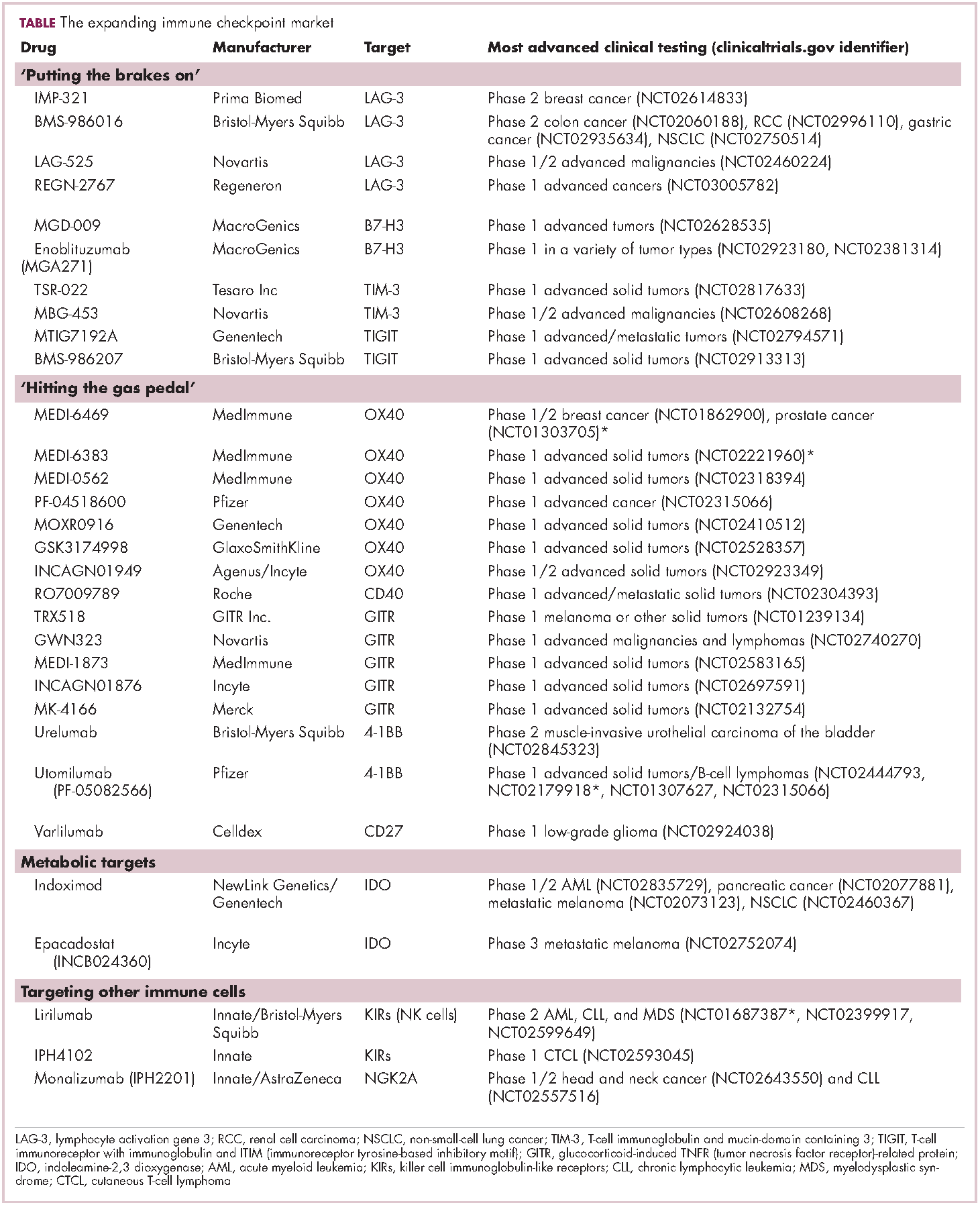
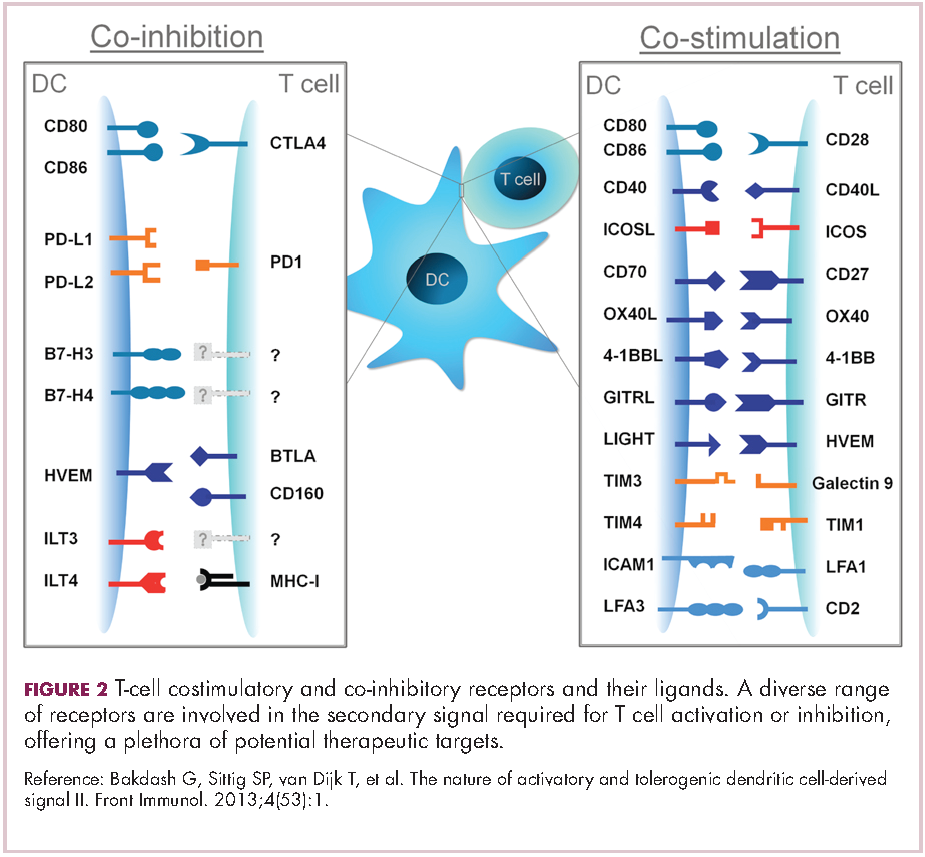
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).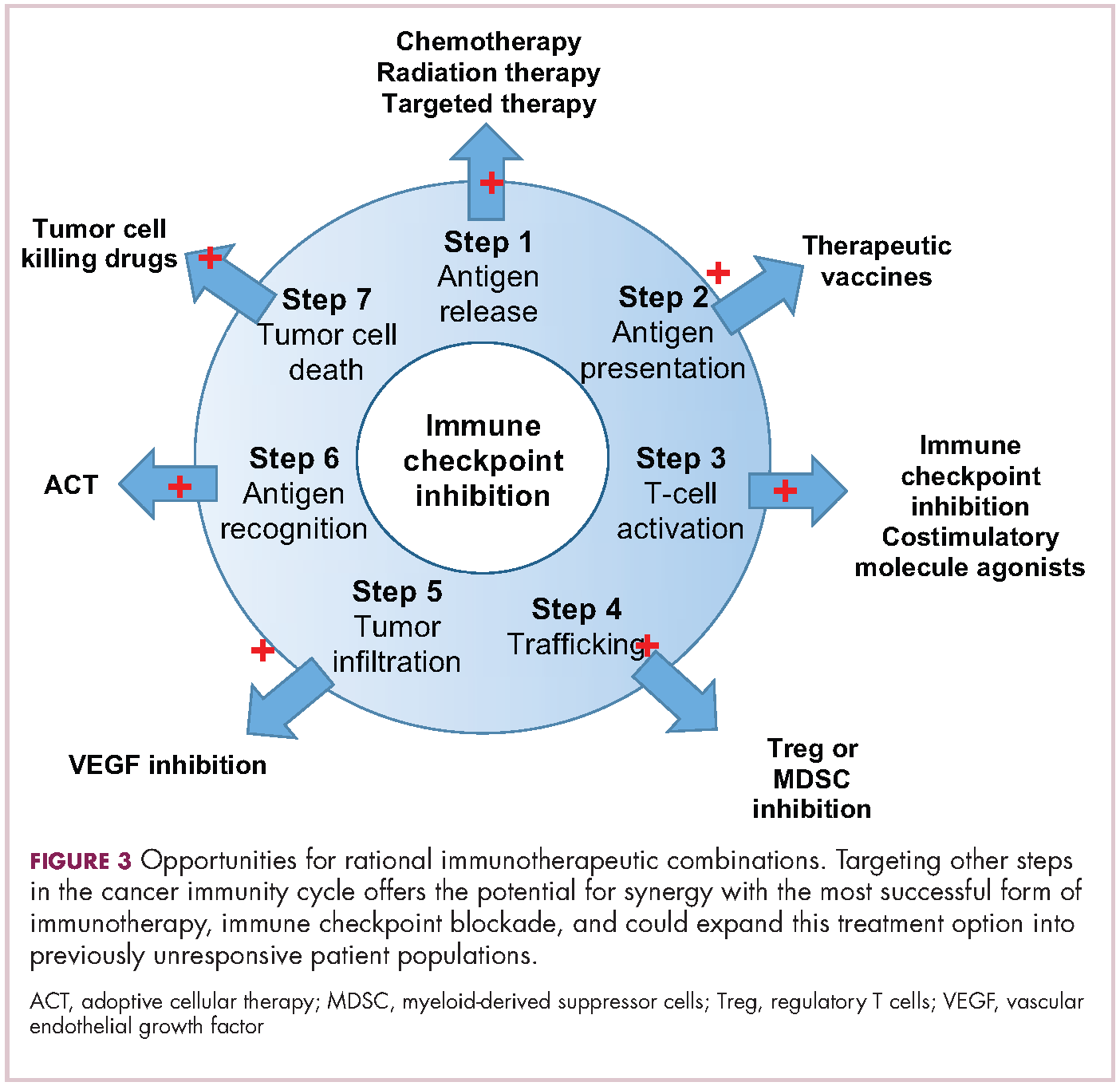
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Expanded approval for daratumumab in multiple myeloma
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.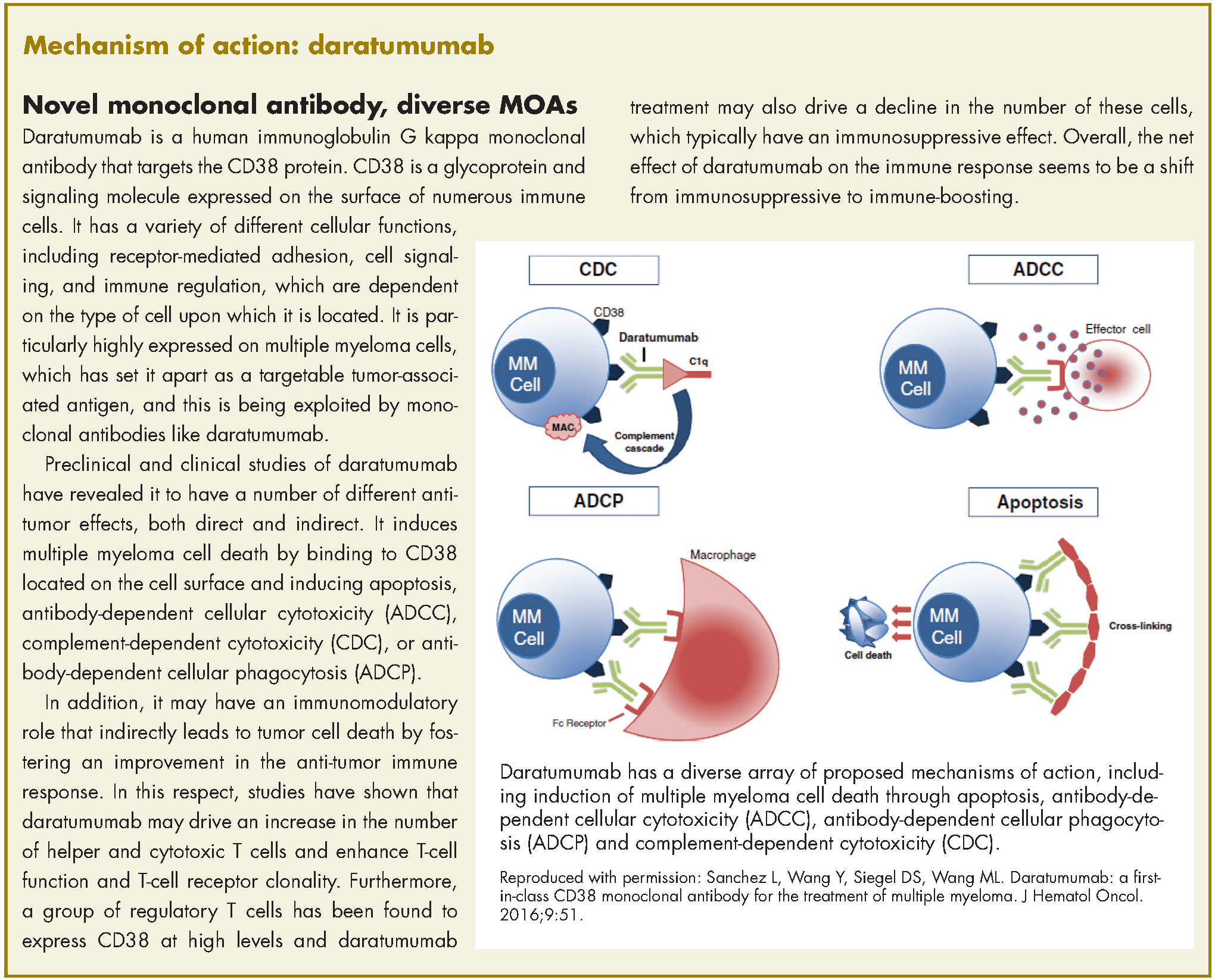
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
Neuromodulation for Treatment-Refractory PTSD (FULL)
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
Journal of Hospital Medicine – Dec. 2017
BACKGROUND: Identifying hospitals that are both early and consistent adopters of high-value care can help shed light on the culture and practices at those institutions that are necessary to promote high-value care nationwide. The use of troponin testing to diagnose acute myocardial infarction (AMI), and not testing for myoglobin or creatine kinase-MB (CK-MB), is a high-value recommendation of the Choosing Wisely® campaign.
OBJECTIVE: To examine the variation in cardiac biomarker testing and the effect of the Choosing Wisely® troponin-only testing recommendation for the diagnosis of AMI.
DESIGN: A retrospective, observational study using administrative ordering data from Vizient’s Clinical Database/Resource Manager.

PATIENTS: Hospitalized patients with a principal discharge diagnosis of AMI.
INTERVENTION: The Choosing Wisely® recommendation to order troponin-only testing to diagnose AMI was released during the first quarter of 2015.
RESULTS: In 19 hospitals, troponin-only testing was consistently ordered to diagnose AMI before the Choosing Wisely® recommendation and throughout the study period. In 34 hospitals, both troponin testing and myoglobin/CK-MB testing were ordered to diagnose AMI even after the Choosing Wisely® recommendation. In 26 hospitals with low rates of troponin-only testing before the Choosing Wisely® recommendation, the release of the recommendation was associated with a statistically significant increase in the rate of troponin-only testing to diagnose AMI.
CONCLUSION: In institutions with low rates of troponin-only testing prior to the Choosing Wisely® recommendation, the recommendation was associated with a significant increase in the rate of troponin-only testing.
Read the entire article in the Dec. 2017 issue of the Journal of Hospital Medicine.
Also in JHM this month
Hospital perceptions of Medicare’s Sepsis Quality Reporting Initiative
AUTHORS: Ian J. Barbash, MD, MS; Kimberly J. Rak, PhD; Courtney C. Kuza, MPH; and Jeremy M. Kahn, MD, MS
Health literacy and hospital length of stay: An inpatient cohort study
AUTHORS: Ethan G. Jaffee, MD; Vineet M. Arora, MD, MAPP; Madeleine I. Matthiesen, MD; David O. Meltzer, MD, PhD, MHM; and Valerie G. Press, MD, FAAP, FACP, MPH
How exemplary teaching physicians interact with hospitalized patients
AUTHORS: Sanjay Saint, MD, MPH, FHM; Molly Harrod, PhD; Karen E. Fowler, MPH; and Nathan Houchens, MD, FACP, FHM
A randomized cohort controlled trial to compare intern sign-out training interventions
AUTHORS: Soo-Hoon Lee, PhD; Christopher Terndrup, MD; Phillip H. Phan, PhD; Sandra E. Zaeh, MD; Kwame Atsina, MD; Nicole Minkove, MD; Alexander Billioux, MD; DPhil, Souvik Chatterjee, MD; Idoreyin Montague, MD; Bennett Clark, MD; Andrew Hughes, MD; and Sanjay V. Desai, MD
BACKGROUND: Identifying hospitals that are both early and consistent adopters of high-value care can help shed light on the culture and practices at those institutions that are necessary to promote high-value care nationwide. The use of troponin testing to diagnose acute myocardial infarction (AMI), and not testing for myoglobin or creatine kinase-MB (CK-MB), is a high-value recommendation of the Choosing Wisely® campaign.
OBJECTIVE: To examine the variation in cardiac biomarker testing and the effect of the Choosing Wisely® troponin-only testing recommendation for the diagnosis of AMI.
DESIGN: A retrospective, observational study using administrative ordering data from Vizient’s Clinical Database/Resource Manager.
PATIENTS: Hospitalized patients with a principal discharge diagnosis of AMI.
INTERVENTION: The Choosing Wisely® recommendation to order troponin-only testing to diagnose AMI was released during the first quarter of 2015.
RESULTS: In 19 hospitals, troponin-only testing was consistently ordered to diagnose AMI before the Choosing Wisely® recommendation and throughout the study period. In 34 hospitals, both troponin testing and myoglobin/CK-MB testing were ordered to diagnose AMI even after the Choosing Wisely® recommendation. In 26 hospitals with low rates of troponin-only testing before the Choosing Wisely® recommendation, the release of the recommendation was associated with a statistically significant increase in the rate of troponin-only testing to diagnose AMI.
CONCLUSION: In institutions with low rates of troponin-only testing prior to the Choosing Wisely® recommendation, the recommendation was associated with a significant increase in the rate of troponin-only testing.
Read the entire article in the Dec. 2017 issue of the Journal of Hospital Medicine.
Also in JHM this month
Hospital perceptions of Medicare’s Sepsis Quality Reporting Initiative
AUTHORS: Ian J. Barbash, MD, MS; Kimberly J. Rak, PhD; Courtney C. Kuza, MPH; and Jeremy M. Kahn, MD, MS
Health literacy and hospital length of stay: An inpatient cohort study
AUTHORS: Ethan G. Jaffee, MD; Vineet M. Arora, MD, MAPP; Madeleine I. Matthiesen, MD; David O. Meltzer, MD, PhD, MHM; and Valerie G. Press, MD, FAAP, FACP, MPH
How exemplary teaching physicians interact with hospitalized patients
AUTHORS: Sanjay Saint, MD, MPH, FHM; Molly Harrod, PhD; Karen E. Fowler, MPH; and Nathan Houchens, MD, FACP, FHM
A randomized cohort controlled trial to compare intern sign-out training interventions
AUTHORS: Soo-Hoon Lee, PhD; Christopher Terndrup, MD; Phillip H. Phan, PhD; Sandra E. Zaeh, MD; Kwame Atsina, MD; Nicole Minkove, MD; Alexander Billioux, MD; DPhil, Souvik Chatterjee, MD; Idoreyin Montague, MD; Bennett Clark, MD; Andrew Hughes, MD; and Sanjay V. Desai, MD
BACKGROUND: Identifying hospitals that are both early and consistent adopters of high-value care can help shed light on the culture and practices at those institutions that are necessary to promote high-value care nationwide. The use of troponin testing to diagnose acute myocardial infarction (AMI), and not testing for myoglobin or creatine kinase-MB (CK-MB), is a high-value recommendation of the Choosing Wisely® campaign.
OBJECTIVE: To examine the variation in cardiac biomarker testing and the effect of the Choosing Wisely® troponin-only testing recommendation for the diagnosis of AMI.
DESIGN: A retrospective, observational study using administrative ordering data from Vizient’s Clinical Database/Resource Manager.
PATIENTS: Hospitalized patients with a principal discharge diagnosis of AMI.
INTERVENTION: The Choosing Wisely® recommendation to order troponin-only testing to diagnose AMI was released during the first quarter of 2015.
RESULTS: In 19 hospitals, troponin-only testing was consistently ordered to diagnose AMI before the Choosing Wisely® recommendation and throughout the study period. In 34 hospitals, both troponin testing and myoglobin/CK-MB testing were ordered to diagnose AMI even after the Choosing Wisely® recommendation. In 26 hospitals with low rates of troponin-only testing before the Choosing Wisely® recommendation, the release of the recommendation was associated with a statistically significant increase in the rate of troponin-only testing to diagnose AMI.
CONCLUSION: In institutions with low rates of troponin-only testing prior to the Choosing Wisely® recommendation, the recommendation was associated with a significant increase in the rate of troponin-only testing.
Read the entire article in the Dec. 2017 issue of the Journal of Hospital Medicine.
Also in JHM this month
Hospital perceptions of Medicare’s Sepsis Quality Reporting Initiative
AUTHORS: Ian J. Barbash, MD, MS; Kimberly J. Rak, PhD; Courtney C. Kuza, MPH; and Jeremy M. Kahn, MD, MS
Health literacy and hospital length of stay: An inpatient cohort study
AUTHORS: Ethan G. Jaffee, MD; Vineet M. Arora, MD, MAPP; Madeleine I. Matthiesen, MD; David O. Meltzer, MD, PhD, MHM; and Valerie G. Press, MD, FAAP, FACP, MPH
How exemplary teaching physicians interact with hospitalized patients
AUTHORS: Sanjay Saint, MD, MPH, FHM; Molly Harrod, PhD; Karen E. Fowler, MPH; and Nathan Houchens, MD, FACP, FHM
A randomized cohort controlled trial to compare intern sign-out training interventions
AUTHORS: Soo-Hoon Lee, PhD; Christopher Terndrup, MD; Phillip H. Phan, PhD; Sandra E. Zaeh, MD; Kwame Atsina, MD; Nicole Minkove, MD; Alexander Billioux, MD; DPhil, Souvik Chatterjee, MD; Idoreyin Montague, MD; Bennett Clark, MD; Andrew Hughes, MD; and Sanjay V. Desai, MD
Ribociclib: another CDK inhibitor hits the mark in breast cancer
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
Rucaparib – second PARP inhibitor hits the market for ovarian cancer
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
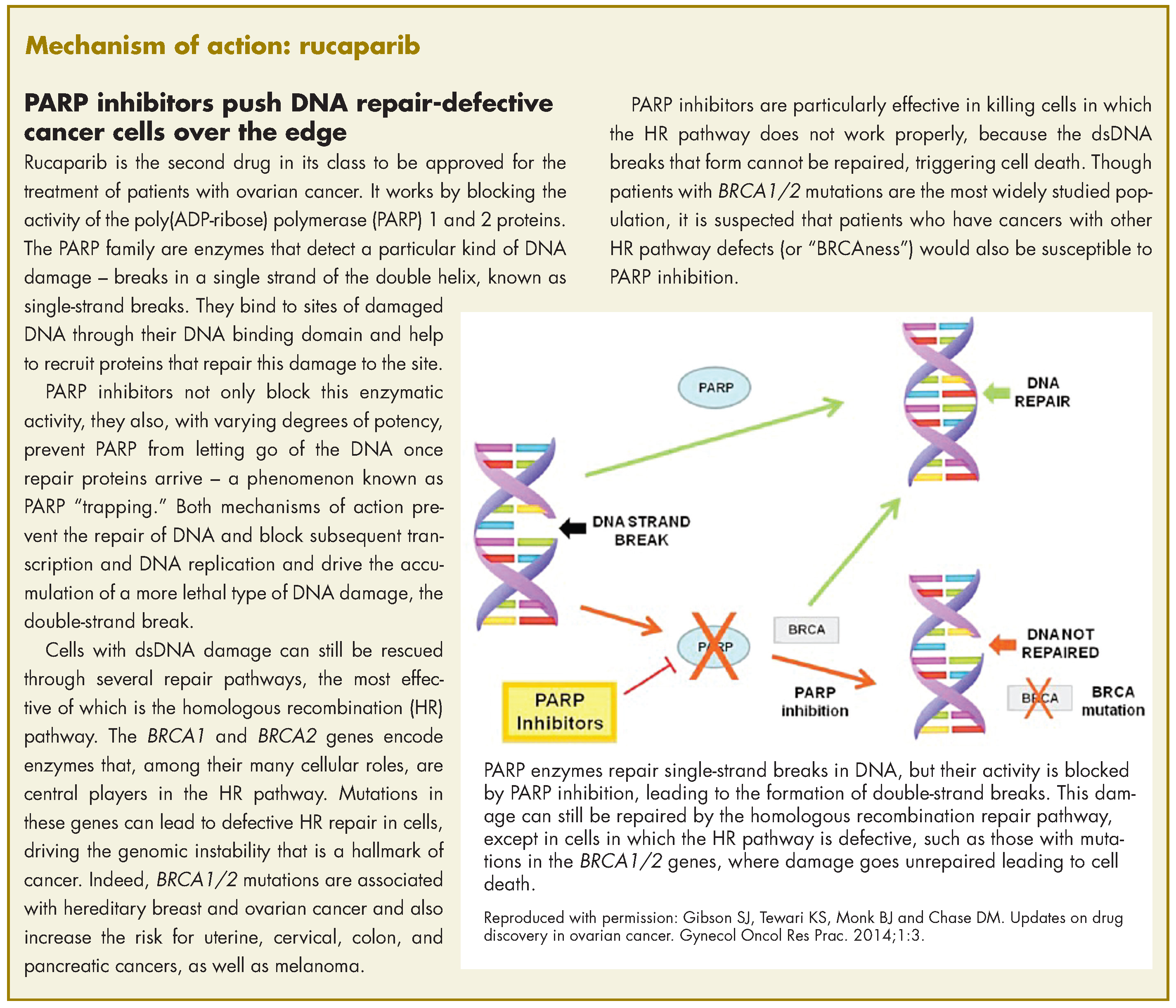
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
Lenvatinib expands its reach into renal cell carcinoma
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
Liquid gold: blood-based biopsies make headway
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10
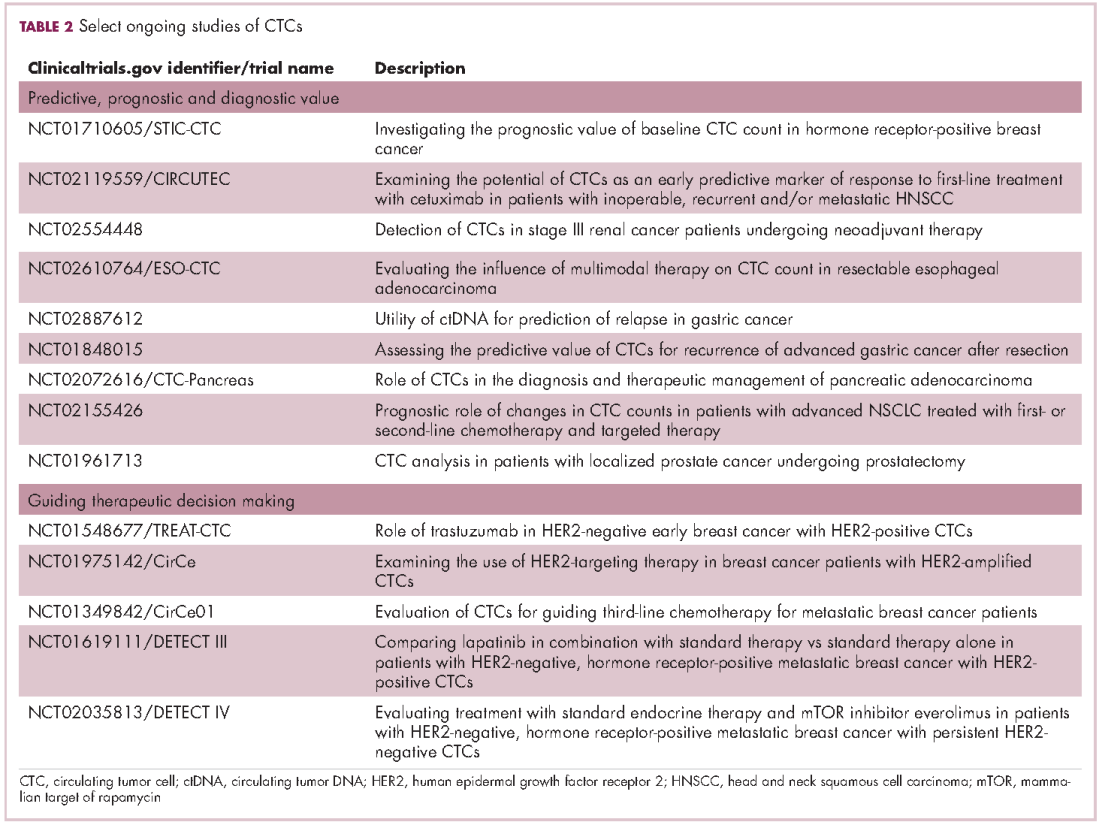
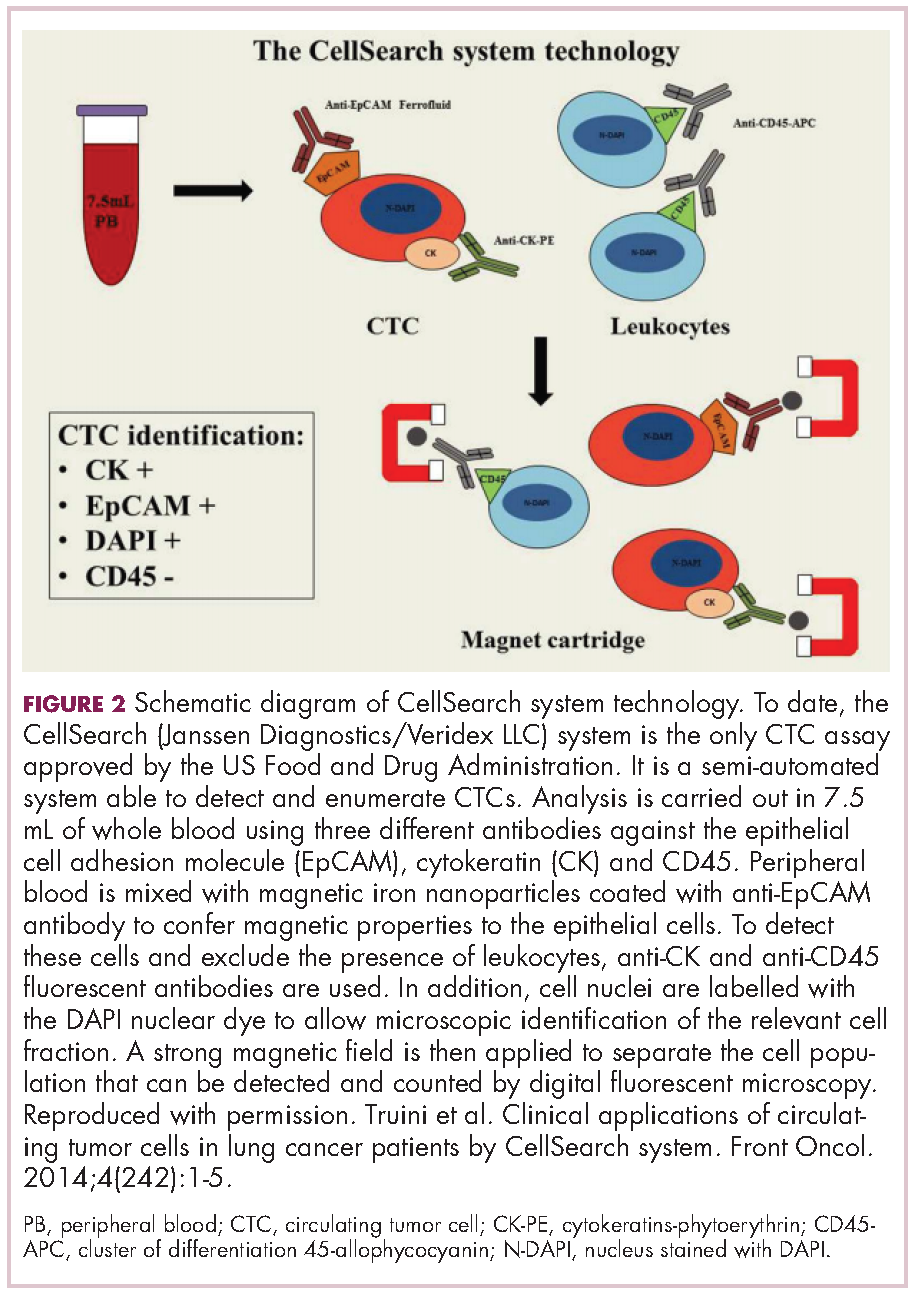
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
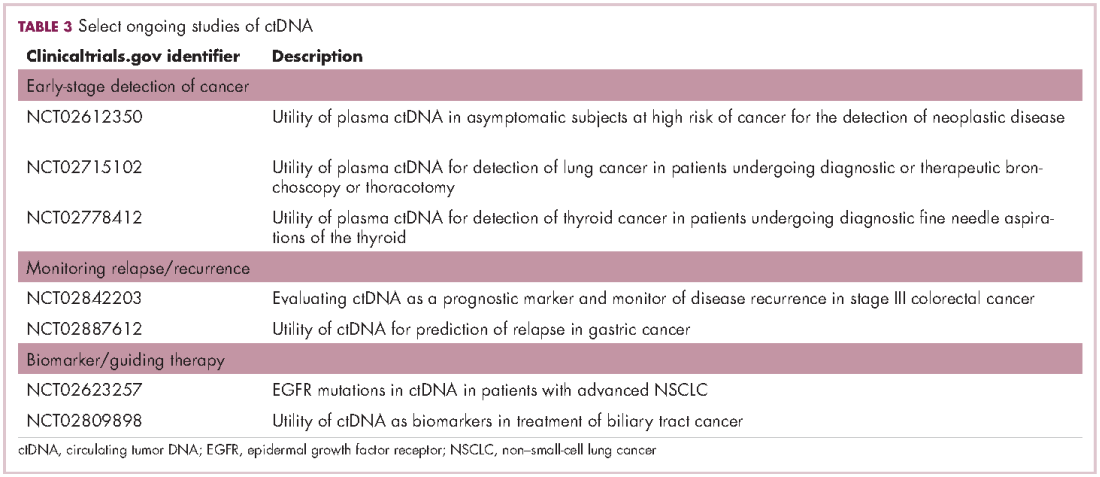
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
Complexity at Hospital Discharge
Patient complexity is associated with greater hospital readmission rates,1,2 poorer quality of care,3 and lower patient satisfaction.4 Improving outcomes for complex patients is a global priority,5 and local initiatives such as Ontario’s Health Links are being developed, yet evidence to inform care is lacking.6-8
The prevalence of patients living with multiple comorbidities is increasing as advances in medicine enable people to live and manage chronic diseases.9-11 However, these medical gains have resulted in an increased burden on both patients and healthcare systems. Socioeconomic status and co-occurring psychosocial challenges further complicate health and healthcare in marginalized populations.12,13
Human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) is one example of a disease that medicine has transformed. Individuals living with HIV today, on antiretroviral medications, may be able to manage their chronic illness for decades.14,15 However, in addition to social determinants of health that influence ongoing adherence and engagement in care, these medications do not completely eradicate the impact of HIV and, as a result, HIV-positive individuals are at a greater risk of developing additional comorbidities.15 People living with HIV may, therefore, represent an important patient population in which healthcare interventions and system improvements for complex patients should be explored.
Improving health systems and better supporting complex patients requires a broader understanding of the patient experience and the challenges encountered, especially during high-risk periods such as hospital discharge. Qualitative research approaches are designed to help us understand social phenomena in their “natural” settings,16 and thus suited to achieve this goal, providing critical insight to inform healthcare systems and policies.17,18 This study sought to answer the question, “What are the obstacles and challenges faced by complex patients during hospital discharge and post-discharge transition?” We approached patient complexity holistically, using a unified Complexity Framework6 that connects 5 health dimensions—social capital, mental health, demographics, health and social experiences, and physical health—identified as important to understanding complex patients and their interaction with healthcare. A longitudinal case study approach was used, with multiple sources of data, to understand the clinical context and discharge plans in relation to the lived experience of patients over time, exploring potential misalignment and areas for improvement.
METHODS
This community-based research study was conducted at Casey House, a 13-bed subacute care hospital in Toronto, Canada that provides in-patient and community programs to a complex patient group. All patients are HIV-positive. Inpatient hospital care is provided by an interdisciplinary team, including physicians, social workers, nurses, and healthcare aides. A harm reduction approach is taken to substance use. Twelve beds are for general admission. Patients may be transferred from acute-care hospitals or referred by community-based providers. One bed is reserved for scheduled 2-week respite stays.
The primary research team for this community-based project consisted of clinicians and community and academic researchers. The study was conducted in collaboration with housing, healthcare, and HIV service providers and was advised by 2 individuals with lived experience of discharge from Casey House. Community members with lived experience attended team meetings, provided feedback on all stages of the project (ie, interview guides, recruitment, analysis and dissemination), and helped facilitate community engagement sessions with other patients at the start and the end of the project.
Standard practice for discharge planning involves clinicians determining a tentative discharge date and identifying strategies to support the patient. Planning is informed by knowledge gathered by the interdisciplinary team throughout the admission, including social determinants of health (ie, housing, social support, food security). Patients are encouraged to invite an individual from their social support network to attend a discharge meeting, where the care team reviews goals for admission, course of treatment, referrals, and important follow-up dates.
We used a multi-case study approach to explore the discharge process and post-discharge period. A case was defined as the discharge and transition of a patient from hospital to community. Data were collected through serial interviews with patients (n = 4), medical chart abstraction, and review of discharge summaries. Serial interviews, although not frequently used in clinical research, have been proposed as a strong approach for exploring complex processes and to build trust between researcher and participant,19 both of which were relevant in this study. Patient interviews were conducted by the Master’s trained research coordinator (SM) using tailored semi-structured interview guides for 4 time points: before the discharge meeting (I1); after the discharge meeting but before discharge (I2); within a week of discharge (I3); and approximately 30 days after discharge (I4). Interviews were audio recorded and transcribed verbatim.
Cases were eligible if the patient had a general admission and a planned discharge to the community, and was able to communicate in English and direct his/her own care. Patient-initiated discharges and discharges to another healthcare facility were excluded. Casey House clinical staff approached consecutive potentially eligible patients for their willingness to speak with the researcher coordinator. The research coordinator met with patients to assess eligibility and obtain informed consent to participate. All participants provided informed written consent. The study was approved by the University of Toronto HIV Research Ethics Board.
Interview data, managed with MAXQDA software (VERBI GmbH, Berlin, Germany), were analyzed using a framework analysis approach.20,21 At least 3 authors read each transcript in its entirety. Priority questions/topics identified a priori by stakeholders as important to inform change in care and practices were used as the first draft of the coding framework. The framework was modified through team discussion in the analysis phase to integrate emerging themes. Participant demographic and clinical data were extracted using a structured data collection form.
Preliminary data analysis was completed for the separate data sources including inter- and intra-case comparisons: exploring how experiences and perceptions changed over time and themes that emerged across cases at the same time point. Data sources were combined to strengthen the understanding of the cases and identify relationships and discrepancies across sources.22 Audit trails, reflexive journaling, group coding and analysis meetings and member-checking, were used to enhance analytical rigor.
RESULTS
The results focus on the patient experience of the “discharge plan” and are presented in terms of 3 pre-identified categories: 1) social support; 2) discharge process and transition experience; and 3) post-discharge follow-up and referrals; and 1 emergent theme, patient priorities.

Participants experienced complex medical and psychosocial challenges (Table 1, participant characteristics). All participants were living with HIV plus a mean of 5 additional comorbidities, the most common being hepatitis C (n = 3), chronic obstructive pulmonary disease (n = 2), herpes (n = 2) and opportunistic infections (n = 2). Eight of 9 participants had a history of an Axis 1 diagnosis, most commonly mood disorder (n = 4). Substance use was identified in all participants. An overview of each case is presented in Table 2.

Three patients declined to be considered for the study. Informed consent was obtained for 10 cases. One participant withdrew after interview 1. Data are presented here for 9 cases, including 32 interviews, between October 2013 and June 2014. Interviews 1 (I1) and 2 (I2) were combined for 3 participants. Two participants were lost to follow-up for interview 4.
Social Support
For the purposes of this paper, we define “social support” as the emotional or instrumental assistance an individual perceives and experiences from people in his/her self-identified network (ie, family, friends). Participants’ discharge-related experience of social support did not align, in most cases, with the information from their medical charts or their expectations. At admission, 8 of 9 participants identified at least 1 person in their social support network, yet only 1 participant had someone attend the discharge meeting. One participant said she had expected “my daughter, my mother, my brother, somebody. At least somebody. But they never show up.” (P5, I2).
The complexity of her relationship with her family and her unmet needs for support continued after discharge:
I try and be as independent as possible. I don’t have to call them for nothing. Because, even the other day, I called my mom and I asked her, I said, “Mom, I’m going to give you $400 [to pay back a personal loan] and I’m going to give you an extra $100, you could buy me some food.” And she goes “Okay.” But, I didn’t give it to her yet. I don’t know, she seems money hungry right now, so I’m like no, I’ll wait. (P5, I4)
In the hospital, participants frequently spoke about discharge and transition planning that was inclusive of their social support networks. However, a sense of isolation and loneliness was common post-discharge. Often, friends and family members did not provide the support that participants anticipated, but instead were sources of anxiety and stress. One participant conveyed his experience with a friend he listed as a social support:
I gave him some money to get me some groceries, to make sure I had some food in the house when I got home. He didn’t do that. All of a sudden he was called away to [another city]. He told me his father had a heart attack. He told [others] his father had a slip. I still have yet to receive my money. (P7, I4)
Discharge Process and Transition Experience
While some participants were excited about the thought of freedom of being home, others were anxious about the burdens of returning to life outside of the hospital.
I kind of feel like, yeah, I want to go home, but then I think to myself what am I going to do when I get home. Am I just going to go back to what I’ve been doing? Am I going to really change? Am I going to forget to take my pill one day because I’m home and stuff like that. (P4, I1)
The discharge process was often perceived by participants to be rushed. Some participants found the discharge meetings helpful, while others did not feel the process empowered them to engage in a meaningful conversation with hospital staff.
There was no one there with me to even help me with my brain, to think. But it’s afterwards I’m like why didn’t I say that, like that’s what I meant to say. The brain just doesn’t function that way. (P8, I2).
This participant struggled with the transition. One week after discharge when she was asked how her health was she replied:
Terrible. I’ve got no energy. I haven’t eaten for 3 days. I haven’t drank for 3 days. I’ve got diarrhea galore […] Just no appetite whatsoever. I can’t even make it up the stairs without losing my breath. If I make it up the stairs, I have to sit for 15 or 20 minutes… (P8, I3)
The weight of maintaining activities of daily living was prominent in all post-discharge interviews, in many cases accentuated by declining health. The transition to home was more challenging than participants expected; the experience was strongly influenced by the stability of their health, their environment, and the complexity of their lives.
Follow-up and Referrals
Discharge summaries included a mean of 7 referrals. All participants were referred to a case coordinator, nurse, and family physician. Other referrals included pharmacist (n = 8); personal support worker (n = 6); housing (n = 5); and food-support programs (n = 5).
Several factors led to challenges accessing and receiving services. Participants identified: difficulty with requisite paperwork; mobility and financial constraints; personal and logistical challenges with home-care providers; and competing priorities, such as caring for family. These experiences were frequently accompanied by frustration and anxiety.
Because, if I’m in [city where girlfriend lives], I will not get the support that I get when I’m home. Like my nurse comes. [She] was supposed to come and see me twice and I missed that. I missed like 4 [appointments]. You understand? Certain things I’ve been missing. (P6, I4)
When one participant was asked if she had followed up with the food support program she had been referred to, she responded:
Oh, baby, no. I’ve been so confused. I’ve had ODSP [referring to Ontario Disability Support Program, a government disability program] on my case. I’ve got all the files all mixed up. My worker’s a real bitch. She hates me, big time. I was supposed to go bring in papers today, but I couldn’t get out of bed. I don’t know how much trouble I’m going to be in with ODSP now. (P8, I3)
Despite comprehensive discharge plans and referrals, all participants experienced delays and difficulties in accessing and receiving services. In most cases, there was no single contributing factor to these challenges; the unique experiences were a result of the complex interplay of multiple factors for each individual.
Patient Priorities
In the hospital, participants primarily identified goals of improving physical health and medication adherence. However, these goals often shifted to meeting basic living necessities and supporting others upon discharge. Barriers to adequate food and mobility were prominent themes.
One participant spoke about the challenges of supporting her son while struggling with her own health after discharge:
Well, I’ve been dying, I can’t even walk, and yet I’m the one that still has to go to WalMart, to grab milk and bread for my kid. It’s not like I need any of that stuff, because I don’t even eat. (P8, I3)
Participants were admitted on a mean of 6 medications and discharged with a mean of 14 (Table 1). In the hospital, medications are dispensed directly to patients; however, maintaining optimal adherence at home was complex. When 1 participant was asked about her medications after being home for a week, she said:
My meds, you know I have the cream that I’m supposed to put … and I can’t find it. I lost it yesterday. I used it yesterday morning and all day yesterday I’m looking, like, did it fall behind there? But, obviously, I can’t look over there [because of mobility challenges] … I don’t think I can get it covered [by insurance to replace it]. (P5, I3)
Participants found it difficult to follow a specific dosing schedule, ensure food intake corresponded to medication guidelines, and navigate the impact of substance use. Substance use for some was associated with nonadherence. A participant, explaining his quickly declining health, spoke about the impact of using crack cocaine:
Yeah, when I use I don’t think about medicating, taking my pills or anything like that. That’s not even on your mind. It doesn’t come across your mind. […] I guess, that’s part of the addictive personality. It wants to grab hold of you and say “no, focus on me, focus on me.” (P7, I4)
Others used marijuana as an appetite stimulant and a critical piece of their medication adherence routine.
DISCUSSION
This study followed complex patients through hospital discharge and transition back into the community. In the hospital, participants focused on medical goals, but following discharge basic living needs became the priority. Despite a comprehensive plan to provide support upon discharge, participants found executing and following up with referrals, services, and medication adherence was often overwhelming and not achieved in the month post-hospitalization.
Our study provides depth and context to support and understand the findings of reviews evaluating interventions to improve transitions in care.23,24 A systematic review of interventions to decrease 30-day readmission rates concluded that comprehensive support interventions (with many components) contributed to the greatest reduction in risk of readmission.16 Components that showed the greatest impact were those that were designed to improve patients’ capacity for self-care (including their ability to access and follow through with post-discharge care plans) and those that involved more individuals in the delivery of care.23
Our results also support and expand on other qualitative findings of complex patients. Kangovi et al.25 interviewed patients with low socioeconomic status at a single time point post-discharge to identify common experiences. They summarized their findings in 6 themes: powerlessness during hospitalization; incongruence of patient and clinical team goals; competing issues influencing prominence of health behaviors; socioeconomic constraints on patients’ ability to perform recommended behaviors; sense of abandonment after discharge; and loss of self-efficacy resulting from the “failure” to follow the discharge plan. Our findings tell a very similar story but provide the additional context and understanding of the lived experience over time. We found that the transition experience was most challenging when the home environment was unstable, resulting in a shift in priorities from those set during hospitalization.
While increased support may improve outcomes, there is a need to improve awareness, integration, and support for building capacity within complex patients.26 Capacity is defined here as the sum of resources and abilities that a patient can draw on, and includes physical and mental as well as social, financial, personal, and environmental capabilities and resources.27 This includes understanding the potential negative impact of developing a clinical plan which, in order to operationalize, requires resources in excess of the patient’s capacity at that time.27 Minimally disruptive medicine, a promising theoretical approach for improving the care of complex clients, embodies the awareness of capacity in achieving patient-centered care while “imposing the smallest possible treatment burden on patients’ lives.”28
This study, although not without its limitations, provides an in-depth exploration of the experiences of a small number of patients living with HIV, recruited from a single facility in Toronto, Canada after relatively long hospital stays. There are specific context issues related to HIV, such as stigma and severe consequences for suboptimal medication adherence. Furthermore, this study took place where many urban health resources exist; complex patients in rural settings or in environments less tailored to the needs associated with complex medical, psychiatric, and social conditions may experience greater barriers in the transition process. Although this study captured data from medical charts and documents relevant to the cases, further exploration of the clinician decision-making process in creating the discharge plans and additional sources of data on health outcomes post-discharge would be beneficial.
Despite its limitations, this study provides detail and depth to understand some of the most complex patients who suffer from significant challenges in the health system and who are amongst the highest-cost healthcare users. The case study approach, with serial interviews, is an important strength of this study, enabling meaningful insight into hospital discharge processes and challenges experienced by complex patients that can inform individual-level care practice and the development of new programs and interventions.
This study builds on recent research with complex patients in calling for a new approach to clinical care.6,29,30 In order to support complex patients through discharge, clinical goals and referrals must be made in light of a patient’s capacity in the community. Structural changes may be made to improve coordination and access to services, decreasing the burden and improving the healthcare experience. Albreht et al.31 highlight a number of promising programs across Europe (such as the Clinic for Multimorbidity and Polypharmacy in Denmark) designed to improve the health and healthcare for individuals living with multiple chronic conditions. Small-scale changes are also important such as increasing conversations about the capacity and limitations of individuals listed as social supports, and making appropriate and realistic referrals based on an understanding of a patient’s capacity and motivation for follow-up. Shippee et al.32 identify a list of approaches in line with minimally disruptive medicine that can be integrated into existing systems as part of a developing “toolkit” (eg, elicitation of transcendent patient goals, and integration of patient-reported outcome tracking of challenges and burdens associated with health and daily living). The findings of this study suggest that the elements of the toolkit may provide a foundation for future interventions and research to improve hospital care and discharge outcomes for complex patients.
Disclosures
This project was funded by a Canadian Institutes of Health Research (CIHR) HIV/AIDS Community-based Research Catalyst Grant (#126669). Dr. Brennan’s research is supported by an Ontario HIV Treatment Network (OHTN) Applied HIV Research Chair. Dr. Chan Carusone reports grants from Canadian Institutes of Health Research during the conduct of the study.
1. Allaudeen N, Vidyarthi A, Masselli J, Auerback A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6:54-60. PubMed
2. Hu J, Gonsahn MD, Nerenz DR. Socioeconomic status and readmissions: evidence from an urban teaching hospital. Health Aff (Millwood). 2014;33:778-785. PubMed
3. Panagioti M, Stokes J, Esmail A, et al. Multimorbidity and patient safety incidents in primary care: a systematic review and meta-analysis. PLoS One. 2015;10:e0135947. PubMed
4. Paddison CA, Saunders CL, Abel GA, Payne RA, Campbell JL, Roland M. Why do patients with multimorbidity in England report worse experiences in primary care? Evidence from the General Practice Patient Survey. BMJ Open. 2015;5:e006172. PubMed
5. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
6. Schaink AK, Kuluski K, Lyons RF, et al. A scoping review and thematic classification of patient complexity: offering a unifying framework. J Comorbidity. 2012;2:1-9.
7. Roland M, Paddison C. Better management of patients with multimorbidity. BMJ. 2013;346:f2510. PubMed
8. Smith SM, Soubhi H, Fortin M, Hudon C, O’Dowd T. Managing patients with multimorbidity: a systematic review of interventions in primary care and community settings. BMJ. 2012;345:e5205. PubMed
9. Afshar S, Roderick PJ, Kowal P, Dimitrov BD, Hill AG. Multimorbidity and the inequalities of global ageing: a cross-sectional study of 28 countries using the World Health Surveys. BMC Public Health. 2015;15:776. PubMed
10. Pefoyo AJK, Bronskill SE, Gruneir A, et al. The increasing burden and complexity of multimorbidity. BMC Public Health. 2015;15:415. PubMed
11. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis. 2013;10:E65. PubMed
12. World Health Organization. Commission on Social Determinants of Health Final Report: Closing the Gap in a Generation: Health Equity through Action on Social Determinants of Health. Geneva, Switzerland: World Health Organization, 2008.
13. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B.. Epidemiology of multimorbidity and implications for health care, research and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
14. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8:e81355. PubMed
15. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525-1533. PubMed
16. Mays N, Pope C. Qualitative research: rigour and qualitative research. BMJ. 1995;311:109-112. PubMed
17. Gilson L, Hanson K, Sheikh K, Agyepong IA, Ssengooba F, Bennett S. Building the field of health policy and systems research: social science matters. PLoS Med. 2011;8:e1001079. PubMed
18. Stoto MA, Nelson CD, Klaiman T. Getting from what to why: using qualitative research to conduct public health systems research. AcademyHealth; August 2013. http://www.academyhealth.org/files/publications/qmforph.pdf. Accessed May 24, 2016.
19. Murray SA, Kendall M, Carduff E, et al. Use of serial qualitative interviews to understand patients’ evolving experiences and needs. BMJ. 2009;339:b3702. PubMed
20. Pope C, Ziebland S, Mays N. Qualitative research in health care. Analysing qualitative data. BMJ. 2000;320:114-116. PubMed
21. Dixon-Woods M. Using framework-based synthesis for conducting reviews of qualitative studies. BMC Med. 2011;9:39. PubMed
22. Yin RK. Case Study Research: Design and Methods. 5th ed. Thousand Oaks, CA: Sage Publications, Inc.; 2014.
23. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174:1095-1107. PubMed
24. Kansagara D, Chiovaro JC, Kagen D, et al. So many options, where do we start? An overview of the care transitions literature. J Hosp Med. 2016;11:221-230. PubMed
25. Kangovi S, Barg FK, Carter T, et al. Challenges faced by patients with low socioeconomic status during the post-hospital transition. J Gen Intern Med. 2013;29:283-289. PubMed
26. Gill A, Kuluski K, Jaakimainen L, Naganathan G, Upshur R, Wodchis WP. “Where do we go from here?” Health system frustrations expressed by patients with multimorbidity, their caregivers and family physicians. Healthc Policy. 2014;9:73-89. PubMed
27. Shippee ND, Shah ND, May CR, Mair FS, Montori VM. Cumulative complexity: a functional, patient-centered model of patient complexity can improve research and practice. J Clin Epidemiol. 2012;65:1041-1051. PubMed
28. Leppin AL, Montori VM, Gionfriddo MR. Minimally disruptive medicine: a pragmatically comprehensive model for delivering care to patients with multiple chronic conditions. Healthcare (Basel). 2015;3:50-63. PubMed
29. Salisbury C. Multimorbidity: redesigning health care for people who use it. Lancet. 2012;380:7-9. PubMed
30. Upshur R, Tracy S. Chronicity and complexity: is what’s good for the diseases always good for the patients? Can Fam Physician. 2008;54:1655-1658. PubMed
31. Albreht A, Dyakova M, Schellevis FG, Van den Broucke S. Many diseases, one model of care? J Comorbidity. 2016;6:12-20.
32. Shippee ND, Allen SV, Leppin AL, May CR, Montori VM. Attaining minimally disruptive medicine: context, challenges and a roadmap for implementation. J R Coll Physicians Edinb. 2015;45:118-122. PubMed
Patient complexity is associated with greater hospital readmission rates,1,2 poorer quality of care,3 and lower patient satisfaction.4 Improving outcomes for complex patients is a global priority,5 and local initiatives such as Ontario’s Health Links are being developed, yet evidence to inform care is lacking.6-8
The prevalence of patients living with multiple comorbidities is increasing as advances in medicine enable people to live and manage chronic diseases.9-11 However, these medical gains have resulted in an increased burden on both patients and healthcare systems. Socioeconomic status and co-occurring psychosocial challenges further complicate health and healthcare in marginalized populations.12,13
Human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) is one example of a disease that medicine has transformed. Individuals living with HIV today, on antiretroviral medications, may be able to manage their chronic illness for decades.14,15 However, in addition to social determinants of health that influence ongoing adherence and engagement in care, these medications do not completely eradicate the impact of HIV and, as a result, HIV-positive individuals are at a greater risk of developing additional comorbidities.15 People living with HIV may, therefore, represent an important patient population in which healthcare interventions and system improvements for complex patients should be explored.
Improving health systems and better supporting complex patients requires a broader understanding of the patient experience and the challenges encountered, especially during high-risk periods such as hospital discharge. Qualitative research approaches are designed to help us understand social phenomena in their “natural” settings,16 and thus suited to achieve this goal, providing critical insight to inform healthcare systems and policies.17,18 This study sought to answer the question, “What are the obstacles and challenges faced by complex patients during hospital discharge and post-discharge transition?” We approached patient complexity holistically, using a unified Complexity Framework6 that connects 5 health dimensions—social capital, mental health, demographics, health and social experiences, and physical health—identified as important to understanding complex patients and their interaction with healthcare. A longitudinal case study approach was used, with multiple sources of data, to understand the clinical context and discharge plans in relation to the lived experience of patients over time, exploring potential misalignment and areas for improvement.
METHODS
This community-based research study was conducted at Casey House, a 13-bed subacute care hospital in Toronto, Canada that provides in-patient and community programs to a complex patient group. All patients are HIV-positive. Inpatient hospital care is provided by an interdisciplinary team, including physicians, social workers, nurses, and healthcare aides. A harm reduction approach is taken to substance use. Twelve beds are for general admission. Patients may be transferred from acute-care hospitals or referred by community-based providers. One bed is reserved for scheduled 2-week respite stays.
The primary research team for this community-based project consisted of clinicians and community and academic researchers. The study was conducted in collaboration with housing, healthcare, and HIV service providers and was advised by 2 individuals with lived experience of discharge from Casey House. Community members with lived experience attended team meetings, provided feedback on all stages of the project (ie, interview guides, recruitment, analysis and dissemination), and helped facilitate community engagement sessions with other patients at the start and the end of the project.
Standard practice for discharge planning involves clinicians determining a tentative discharge date and identifying strategies to support the patient. Planning is informed by knowledge gathered by the interdisciplinary team throughout the admission, including social determinants of health (ie, housing, social support, food security). Patients are encouraged to invite an individual from their social support network to attend a discharge meeting, where the care team reviews goals for admission, course of treatment, referrals, and important follow-up dates.
We used a multi-case study approach to explore the discharge process and post-discharge period. A case was defined as the discharge and transition of a patient from hospital to community. Data were collected through serial interviews with patients (n = 4), medical chart abstraction, and review of discharge summaries. Serial interviews, although not frequently used in clinical research, have been proposed as a strong approach for exploring complex processes and to build trust between researcher and participant,19 both of which were relevant in this study. Patient interviews were conducted by the Master’s trained research coordinator (SM) using tailored semi-structured interview guides for 4 time points: before the discharge meeting (I1); after the discharge meeting but before discharge (I2); within a week of discharge (I3); and approximately 30 days after discharge (I4). Interviews were audio recorded and transcribed verbatim.
Cases were eligible if the patient had a general admission and a planned discharge to the community, and was able to communicate in English and direct his/her own care. Patient-initiated discharges and discharges to another healthcare facility were excluded. Casey House clinical staff approached consecutive potentially eligible patients for their willingness to speak with the researcher coordinator. The research coordinator met with patients to assess eligibility and obtain informed consent to participate. All participants provided informed written consent. The study was approved by the University of Toronto HIV Research Ethics Board.
Interview data, managed with MAXQDA software (VERBI GmbH, Berlin, Germany), were analyzed using a framework analysis approach.20,21 At least 3 authors read each transcript in its entirety. Priority questions/topics identified a priori by stakeholders as important to inform change in care and practices were used as the first draft of the coding framework. The framework was modified through team discussion in the analysis phase to integrate emerging themes. Participant demographic and clinical data were extracted using a structured data collection form.
Preliminary data analysis was completed for the separate data sources including inter- and intra-case comparisons: exploring how experiences and perceptions changed over time and themes that emerged across cases at the same time point. Data sources were combined to strengthen the understanding of the cases and identify relationships and discrepancies across sources.22 Audit trails, reflexive journaling, group coding and analysis meetings and member-checking, were used to enhance analytical rigor.
RESULTS
The results focus on the patient experience of the “discharge plan” and are presented in terms of 3 pre-identified categories: 1) social support; 2) discharge process and transition experience; and 3) post-discharge follow-up and referrals; and 1 emergent theme, patient priorities.
Participants experienced complex medical and psychosocial challenges (Table 1, participant characteristics). All participants were living with HIV plus a mean of 5 additional comorbidities, the most common being hepatitis C (n = 3), chronic obstructive pulmonary disease (n = 2), herpes (n = 2) and opportunistic infections (n = 2). Eight of 9 participants had a history of an Axis 1 diagnosis, most commonly mood disorder (n = 4). Substance use was identified in all participants. An overview of each case is presented in Table 2.
Three patients declined to be considered for the study. Informed consent was obtained for 10 cases. One participant withdrew after interview 1. Data are presented here for 9 cases, including 32 interviews, between October 2013 and June 2014. Interviews 1 (I1) and 2 (I2) were combined for 3 participants. Two participants were lost to follow-up for interview 4.
Social Support
For the purposes of this paper, we define “social support” as the emotional or instrumental assistance an individual perceives and experiences from people in his/her self-identified network (ie, family, friends). Participants’ discharge-related experience of social support did not align, in most cases, with the information from their medical charts or their expectations. At admission, 8 of 9 participants identified at least 1 person in their social support network, yet only 1 participant had someone attend the discharge meeting. One participant said she had expected “my daughter, my mother, my brother, somebody. At least somebody. But they never show up.” (P5, I2).
The complexity of her relationship with her family and her unmet needs for support continued after discharge:
I try and be as independent as possible. I don’t have to call them for nothing. Because, even the other day, I called my mom and I asked her, I said, “Mom, I’m going to give you $400 [to pay back a personal loan] and I’m going to give you an extra $100, you could buy me some food.” And she goes “Okay.” But, I didn’t give it to her yet. I don’t know, she seems money hungry right now, so I’m like no, I’ll wait. (P5, I4)
In the hospital, participants frequently spoke about discharge and transition planning that was inclusive of their social support networks. However, a sense of isolation and loneliness was common post-discharge. Often, friends and family members did not provide the support that participants anticipated, but instead were sources of anxiety and stress. One participant conveyed his experience with a friend he listed as a social support:
I gave him some money to get me some groceries, to make sure I had some food in the house when I got home. He didn’t do that. All of a sudden he was called away to [another city]. He told me his father had a heart attack. He told [others] his father had a slip. I still have yet to receive my money. (P7, I4)
Discharge Process and Transition Experience
While some participants were excited about the thought of freedom of being home, others were anxious about the burdens of returning to life outside of the hospital.
I kind of feel like, yeah, I want to go home, but then I think to myself what am I going to do when I get home. Am I just going to go back to what I’ve been doing? Am I going to really change? Am I going to forget to take my pill one day because I’m home and stuff like that. (P4, I1)
The discharge process was often perceived by participants to be rushed. Some participants found the discharge meetings helpful, while others did not feel the process empowered them to engage in a meaningful conversation with hospital staff.
There was no one there with me to even help me with my brain, to think. But it’s afterwards I’m like why didn’t I say that, like that’s what I meant to say. The brain just doesn’t function that way. (P8, I2).
This participant struggled with the transition. One week after discharge when she was asked how her health was she replied:
Terrible. I’ve got no energy. I haven’t eaten for 3 days. I haven’t drank for 3 days. I’ve got diarrhea galore […] Just no appetite whatsoever. I can’t even make it up the stairs without losing my breath. If I make it up the stairs, I have to sit for 15 or 20 minutes… (P8, I3)
The weight of maintaining activities of daily living was prominent in all post-discharge interviews, in many cases accentuated by declining health. The transition to home was more challenging than participants expected; the experience was strongly influenced by the stability of their health, their environment, and the complexity of their lives.
Follow-up and Referrals
Discharge summaries included a mean of 7 referrals. All participants were referred to a case coordinator, nurse, and family physician. Other referrals included pharmacist (n = 8); personal support worker (n = 6); housing (n = 5); and food-support programs (n = 5).
Several factors led to challenges accessing and receiving services. Participants identified: difficulty with requisite paperwork; mobility and financial constraints; personal and logistical challenges with home-care providers; and competing priorities, such as caring for family. These experiences were frequently accompanied by frustration and anxiety.
Because, if I’m in [city where girlfriend lives], I will not get the support that I get when I’m home. Like my nurse comes. [She] was supposed to come and see me twice and I missed that. I missed like 4 [appointments]. You understand? Certain things I’ve been missing. (P6, I4)
When one participant was asked if she had followed up with the food support program she had been referred to, she responded:
Oh, baby, no. I’ve been so confused. I’ve had ODSP [referring to Ontario Disability Support Program, a government disability program] on my case. I’ve got all the files all mixed up. My worker’s a real bitch. She hates me, big time. I was supposed to go bring in papers today, but I couldn’t get out of bed. I don’t know how much trouble I’m going to be in with ODSP now. (P8, I3)
Despite comprehensive discharge plans and referrals, all participants experienced delays and difficulties in accessing and receiving services. In most cases, there was no single contributing factor to these challenges; the unique experiences were a result of the complex interplay of multiple factors for each individual.
Patient Priorities
In the hospital, participants primarily identified goals of improving physical health and medication adherence. However, these goals often shifted to meeting basic living necessities and supporting others upon discharge. Barriers to adequate food and mobility were prominent themes.
One participant spoke about the challenges of supporting her son while struggling with her own health after discharge:
Well, I’ve been dying, I can’t even walk, and yet I’m the one that still has to go to WalMart, to grab milk and bread for my kid. It’s not like I need any of that stuff, because I don’t even eat. (P8, I3)
Participants were admitted on a mean of 6 medications and discharged with a mean of 14 (Table 1). In the hospital, medications are dispensed directly to patients; however, maintaining optimal adherence at home was complex. When 1 participant was asked about her medications after being home for a week, she said:
My meds, you know I have the cream that I’m supposed to put … and I can’t find it. I lost it yesterday. I used it yesterday morning and all day yesterday I’m looking, like, did it fall behind there? But, obviously, I can’t look over there [because of mobility challenges] … I don’t think I can get it covered [by insurance to replace it]. (P5, I3)
Participants found it difficult to follow a specific dosing schedule, ensure food intake corresponded to medication guidelines, and navigate the impact of substance use. Substance use for some was associated with nonadherence. A participant, explaining his quickly declining health, spoke about the impact of using crack cocaine:
Yeah, when I use I don’t think about medicating, taking my pills or anything like that. That’s not even on your mind. It doesn’t come across your mind. […] I guess, that’s part of the addictive personality. It wants to grab hold of you and say “no, focus on me, focus on me.” (P7, I4)
Others used marijuana as an appetite stimulant and a critical piece of their medication adherence routine.
DISCUSSION
This study followed complex patients through hospital discharge and transition back into the community. In the hospital, participants focused on medical goals, but following discharge basic living needs became the priority. Despite a comprehensive plan to provide support upon discharge, participants found executing and following up with referrals, services, and medication adherence was often overwhelming and not achieved in the month post-hospitalization.
Our study provides depth and context to support and understand the findings of reviews evaluating interventions to improve transitions in care.23,24 A systematic review of interventions to decrease 30-day readmission rates concluded that comprehensive support interventions (with many components) contributed to the greatest reduction in risk of readmission.16 Components that showed the greatest impact were those that were designed to improve patients’ capacity for self-care (including their ability to access and follow through with post-discharge care plans) and those that involved more individuals in the delivery of care.23
Our results also support and expand on other qualitative findings of complex patients. Kangovi et al.25 interviewed patients with low socioeconomic status at a single time point post-discharge to identify common experiences. They summarized their findings in 6 themes: powerlessness during hospitalization; incongruence of patient and clinical team goals; competing issues influencing prominence of health behaviors; socioeconomic constraints on patients’ ability to perform recommended behaviors; sense of abandonment after discharge; and loss of self-efficacy resulting from the “failure” to follow the discharge plan. Our findings tell a very similar story but provide the additional context and understanding of the lived experience over time. We found that the transition experience was most challenging when the home environment was unstable, resulting in a shift in priorities from those set during hospitalization.
While increased support may improve outcomes, there is a need to improve awareness, integration, and support for building capacity within complex patients.26 Capacity is defined here as the sum of resources and abilities that a patient can draw on, and includes physical and mental as well as social, financial, personal, and environmental capabilities and resources.27 This includes understanding the potential negative impact of developing a clinical plan which, in order to operationalize, requires resources in excess of the patient’s capacity at that time.27 Minimally disruptive medicine, a promising theoretical approach for improving the care of complex clients, embodies the awareness of capacity in achieving patient-centered care while “imposing the smallest possible treatment burden on patients’ lives.”28
This study, although not without its limitations, provides an in-depth exploration of the experiences of a small number of patients living with HIV, recruited from a single facility in Toronto, Canada after relatively long hospital stays. There are specific context issues related to HIV, such as stigma and severe consequences for suboptimal medication adherence. Furthermore, this study took place where many urban health resources exist; complex patients in rural settings or in environments less tailored to the needs associated with complex medical, psychiatric, and social conditions may experience greater barriers in the transition process. Although this study captured data from medical charts and documents relevant to the cases, further exploration of the clinician decision-making process in creating the discharge plans and additional sources of data on health outcomes post-discharge would be beneficial.
Despite its limitations, this study provides detail and depth to understand some of the most complex patients who suffer from significant challenges in the health system and who are amongst the highest-cost healthcare users. The case study approach, with serial interviews, is an important strength of this study, enabling meaningful insight into hospital discharge processes and challenges experienced by complex patients that can inform individual-level care practice and the development of new programs and interventions.
This study builds on recent research with complex patients in calling for a new approach to clinical care.6,29,30 In order to support complex patients through discharge, clinical goals and referrals must be made in light of a patient’s capacity in the community. Structural changes may be made to improve coordination and access to services, decreasing the burden and improving the healthcare experience. Albreht et al.31 highlight a number of promising programs across Europe (such as the Clinic for Multimorbidity and Polypharmacy in Denmark) designed to improve the health and healthcare for individuals living with multiple chronic conditions. Small-scale changes are also important such as increasing conversations about the capacity and limitations of individuals listed as social supports, and making appropriate and realistic referrals based on an understanding of a patient’s capacity and motivation for follow-up. Shippee et al.32 identify a list of approaches in line with minimally disruptive medicine that can be integrated into existing systems as part of a developing “toolkit” (eg, elicitation of transcendent patient goals, and integration of patient-reported outcome tracking of challenges and burdens associated with health and daily living). The findings of this study suggest that the elements of the toolkit may provide a foundation for future interventions and research to improve hospital care and discharge outcomes for complex patients.
Disclosures
This project was funded by a Canadian Institutes of Health Research (CIHR) HIV/AIDS Community-based Research Catalyst Grant (#126669). Dr. Brennan’s research is supported by an Ontario HIV Treatment Network (OHTN) Applied HIV Research Chair. Dr. Chan Carusone reports grants from Canadian Institutes of Health Research during the conduct of the study.
Patient complexity is associated with greater hospital readmission rates,1,2 poorer quality of care,3 and lower patient satisfaction.4 Improving outcomes for complex patients is a global priority,5 and local initiatives such as Ontario’s Health Links are being developed, yet evidence to inform care is lacking.6-8
The prevalence of patients living with multiple comorbidities is increasing as advances in medicine enable people to live and manage chronic diseases.9-11 However, these medical gains have resulted in an increased burden on both patients and healthcare systems. Socioeconomic status and co-occurring psychosocial challenges further complicate health and healthcare in marginalized populations.12,13
Human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) is one example of a disease that medicine has transformed. Individuals living with HIV today, on antiretroviral medications, may be able to manage their chronic illness for decades.14,15 However, in addition to social determinants of health that influence ongoing adherence and engagement in care, these medications do not completely eradicate the impact of HIV and, as a result, HIV-positive individuals are at a greater risk of developing additional comorbidities.15 People living with HIV may, therefore, represent an important patient population in which healthcare interventions and system improvements for complex patients should be explored.
Improving health systems and better supporting complex patients requires a broader understanding of the patient experience and the challenges encountered, especially during high-risk periods such as hospital discharge. Qualitative research approaches are designed to help us understand social phenomena in their “natural” settings,16 and thus suited to achieve this goal, providing critical insight to inform healthcare systems and policies.17,18 This study sought to answer the question, “What are the obstacles and challenges faced by complex patients during hospital discharge and post-discharge transition?” We approached patient complexity holistically, using a unified Complexity Framework6 that connects 5 health dimensions—social capital, mental health, demographics, health and social experiences, and physical health—identified as important to understanding complex patients and their interaction with healthcare. A longitudinal case study approach was used, with multiple sources of data, to understand the clinical context and discharge plans in relation to the lived experience of patients over time, exploring potential misalignment and areas for improvement.
METHODS
This community-based research study was conducted at Casey House, a 13-bed subacute care hospital in Toronto, Canada that provides in-patient and community programs to a complex patient group. All patients are HIV-positive. Inpatient hospital care is provided by an interdisciplinary team, including physicians, social workers, nurses, and healthcare aides. A harm reduction approach is taken to substance use. Twelve beds are for general admission. Patients may be transferred from acute-care hospitals or referred by community-based providers. One bed is reserved for scheduled 2-week respite stays.
The primary research team for this community-based project consisted of clinicians and community and academic researchers. The study was conducted in collaboration with housing, healthcare, and HIV service providers and was advised by 2 individuals with lived experience of discharge from Casey House. Community members with lived experience attended team meetings, provided feedback on all stages of the project (ie, interview guides, recruitment, analysis and dissemination), and helped facilitate community engagement sessions with other patients at the start and the end of the project.
Standard practice for discharge planning involves clinicians determining a tentative discharge date and identifying strategies to support the patient. Planning is informed by knowledge gathered by the interdisciplinary team throughout the admission, including social determinants of health (ie, housing, social support, food security). Patients are encouraged to invite an individual from their social support network to attend a discharge meeting, where the care team reviews goals for admission, course of treatment, referrals, and important follow-up dates.
We used a multi-case study approach to explore the discharge process and post-discharge period. A case was defined as the discharge and transition of a patient from hospital to community. Data were collected through serial interviews with patients (n = 4), medical chart abstraction, and review of discharge summaries. Serial interviews, although not frequently used in clinical research, have been proposed as a strong approach for exploring complex processes and to build trust between researcher and participant,19 both of which were relevant in this study. Patient interviews were conducted by the Master’s trained research coordinator (SM) using tailored semi-structured interview guides for 4 time points: before the discharge meeting (I1); after the discharge meeting but before discharge (I2); within a week of discharge (I3); and approximately 30 days after discharge (I4). Interviews were audio recorded and transcribed verbatim.
Cases were eligible if the patient had a general admission and a planned discharge to the community, and was able to communicate in English and direct his/her own care. Patient-initiated discharges and discharges to another healthcare facility were excluded. Casey House clinical staff approached consecutive potentially eligible patients for their willingness to speak with the researcher coordinator. The research coordinator met with patients to assess eligibility and obtain informed consent to participate. All participants provided informed written consent. The study was approved by the University of Toronto HIV Research Ethics Board.
Interview data, managed with MAXQDA software (VERBI GmbH, Berlin, Germany), were analyzed using a framework analysis approach.20,21 At least 3 authors read each transcript in its entirety. Priority questions/topics identified a priori by stakeholders as important to inform change in care and practices were used as the first draft of the coding framework. The framework was modified through team discussion in the analysis phase to integrate emerging themes. Participant demographic and clinical data were extracted using a structured data collection form.
Preliminary data analysis was completed for the separate data sources including inter- and intra-case comparisons: exploring how experiences and perceptions changed over time and themes that emerged across cases at the same time point. Data sources were combined to strengthen the understanding of the cases and identify relationships and discrepancies across sources.22 Audit trails, reflexive journaling, group coding and analysis meetings and member-checking, were used to enhance analytical rigor.
RESULTS
The results focus on the patient experience of the “discharge plan” and are presented in terms of 3 pre-identified categories: 1) social support; 2) discharge process and transition experience; and 3) post-discharge follow-up and referrals; and 1 emergent theme, patient priorities.
Participants experienced complex medical and psychosocial challenges (Table 1, participant characteristics). All participants were living with HIV plus a mean of 5 additional comorbidities, the most common being hepatitis C (n = 3), chronic obstructive pulmonary disease (n = 2), herpes (n = 2) and opportunistic infections (n = 2). Eight of 9 participants had a history of an Axis 1 diagnosis, most commonly mood disorder (n = 4). Substance use was identified in all participants. An overview of each case is presented in Table 2.
Three patients declined to be considered for the study. Informed consent was obtained for 10 cases. One participant withdrew after interview 1. Data are presented here for 9 cases, including 32 interviews, between October 2013 and June 2014. Interviews 1 (I1) and 2 (I2) were combined for 3 participants. Two participants were lost to follow-up for interview 4.
Social Support
For the purposes of this paper, we define “social support” as the emotional or instrumental assistance an individual perceives and experiences from people in his/her self-identified network (ie, family, friends). Participants’ discharge-related experience of social support did not align, in most cases, with the information from their medical charts or their expectations. At admission, 8 of 9 participants identified at least 1 person in their social support network, yet only 1 participant had someone attend the discharge meeting. One participant said she had expected “my daughter, my mother, my brother, somebody. At least somebody. But they never show up.” (P5, I2).
The complexity of her relationship with her family and her unmet needs for support continued after discharge:
I try and be as independent as possible. I don’t have to call them for nothing. Because, even the other day, I called my mom and I asked her, I said, “Mom, I’m going to give you $400 [to pay back a personal loan] and I’m going to give you an extra $100, you could buy me some food.” And she goes “Okay.” But, I didn’t give it to her yet. I don’t know, she seems money hungry right now, so I’m like no, I’ll wait. (P5, I4)
In the hospital, participants frequently spoke about discharge and transition planning that was inclusive of their social support networks. However, a sense of isolation and loneliness was common post-discharge. Often, friends and family members did not provide the support that participants anticipated, but instead were sources of anxiety and stress. One participant conveyed his experience with a friend he listed as a social support:
I gave him some money to get me some groceries, to make sure I had some food in the house when I got home. He didn’t do that. All of a sudden he was called away to [another city]. He told me his father had a heart attack. He told [others] his father had a slip. I still have yet to receive my money. (P7, I4)
Discharge Process and Transition Experience
While some participants were excited about the thought of freedom of being home, others were anxious about the burdens of returning to life outside of the hospital.
I kind of feel like, yeah, I want to go home, but then I think to myself what am I going to do when I get home. Am I just going to go back to what I’ve been doing? Am I going to really change? Am I going to forget to take my pill one day because I’m home and stuff like that. (P4, I1)
The discharge process was often perceived by participants to be rushed. Some participants found the discharge meetings helpful, while others did not feel the process empowered them to engage in a meaningful conversation with hospital staff.
There was no one there with me to even help me with my brain, to think. But it’s afterwards I’m like why didn’t I say that, like that’s what I meant to say. The brain just doesn’t function that way. (P8, I2).
This participant struggled with the transition. One week after discharge when she was asked how her health was she replied:
Terrible. I’ve got no energy. I haven’t eaten for 3 days. I haven’t drank for 3 days. I’ve got diarrhea galore […] Just no appetite whatsoever. I can’t even make it up the stairs without losing my breath. If I make it up the stairs, I have to sit for 15 or 20 minutes… (P8, I3)
The weight of maintaining activities of daily living was prominent in all post-discharge interviews, in many cases accentuated by declining health. The transition to home was more challenging than participants expected; the experience was strongly influenced by the stability of their health, their environment, and the complexity of their lives.
Follow-up and Referrals
Discharge summaries included a mean of 7 referrals. All participants were referred to a case coordinator, nurse, and family physician. Other referrals included pharmacist (n = 8); personal support worker (n = 6); housing (n = 5); and food-support programs (n = 5).
Several factors led to challenges accessing and receiving services. Participants identified: difficulty with requisite paperwork; mobility and financial constraints; personal and logistical challenges with home-care providers; and competing priorities, such as caring for family. These experiences were frequently accompanied by frustration and anxiety.
Because, if I’m in [city where girlfriend lives], I will not get the support that I get when I’m home. Like my nurse comes. [She] was supposed to come and see me twice and I missed that. I missed like 4 [appointments]. You understand? Certain things I’ve been missing. (P6, I4)
When one participant was asked if she had followed up with the food support program she had been referred to, she responded:
Oh, baby, no. I’ve been so confused. I’ve had ODSP [referring to Ontario Disability Support Program, a government disability program] on my case. I’ve got all the files all mixed up. My worker’s a real bitch. She hates me, big time. I was supposed to go bring in papers today, but I couldn’t get out of bed. I don’t know how much trouble I’m going to be in with ODSP now. (P8, I3)
Despite comprehensive discharge plans and referrals, all participants experienced delays and difficulties in accessing and receiving services. In most cases, there was no single contributing factor to these challenges; the unique experiences were a result of the complex interplay of multiple factors for each individual.
Patient Priorities
In the hospital, participants primarily identified goals of improving physical health and medication adherence. However, these goals often shifted to meeting basic living necessities and supporting others upon discharge. Barriers to adequate food and mobility were prominent themes.
One participant spoke about the challenges of supporting her son while struggling with her own health after discharge:
Well, I’ve been dying, I can’t even walk, and yet I’m the one that still has to go to WalMart, to grab milk and bread for my kid. It’s not like I need any of that stuff, because I don’t even eat. (P8, I3)
Participants were admitted on a mean of 6 medications and discharged with a mean of 14 (Table 1). In the hospital, medications are dispensed directly to patients; however, maintaining optimal adherence at home was complex. When 1 participant was asked about her medications after being home for a week, she said:
My meds, you know I have the cream that I’m supposed to put … and I can’t find it. I lost it yesterday. I used it yesterday morning and all day yesterday I’m looking, like, did it fall behind there? But, obviously, I can’t look over there [because of mobility challenges] … I don’t think I can get it covered [by insurance to replace it]. (P5, I3)
Participants found it difficult to follow a specific dosing schedule, ensure food intake corresponded to medication guidelines, and navigate the impact of substance use. Substance use for some was associated with nonadherence. A participant, explaining his quickly declining health, spoke about the impact of using crack cocaine:
Yeah, when I use I don’t think about medicating, taking my pills or anything like that. That’s not even on your mind. It doesn’t come across your mind. […] I guess, that’s part of the addictive personality. It wants to grab hold of you and say “no, focus on me, focus on me.” (P7, I4)
Others used marijuana as an appetite stimulant and a critical piece of their medication adherence routine.
DISCUSSION
This study followed complex patients through hospital discharge and transition back into the community. In the hospital, participants focused on medical goals, but following discharge basic living needs became the priority. Despite a comprehensive plan to provide support upon discharge, participants found executing and following up with referrals, services, and medication adherence was often overwhelming and not achieved in the month post-hospitalization.
Our study provides depth and context to support and understand the findings of reviews evaluating interventions to improve transitions in care.23,24 A systematic review of interventions to decrease 30-day readmission rates concluded that comprehensive support interventions (with many components) contributed to the greatest reduction in risk of readmission.16 Components that showed the greatest impact were those that were designed to improve patients’ capacity for self-care (including their ability to access and follow through with post-discharge care plans) and those that involved more individuals in the delivery of care.23
Our results also support and expand on other qualitative findings of complex patients. Kangovi et al.25 interviewed patients with low socioeconomic status at a single time point post-discharge to identify common experiences. They summarized their findings in 6 themes: powerlessness during hospitalization; incongruence of patient and clinical team goals; competing issues influencing prominence of health behaviors; socioeconomic constraints on patients’ ability to perform recommended behaviors; sense of abandonment after discharge; and loss of self-efficacy resulting from the “failure” to follow the discharge plan. Our findings tell a very similar story but provide the additional context and understanding of the lived experience over time. We found that the transition experience was most challenging when the home environment was unstable, resulting in a shift in priorities from those set during hospitalization.
While increased support may improve outcomes, there is a need to improve awareness, integration, and support for building capacity within complex patients.26 Capacity is defined here as the sum of resources and abilities that a patient can draw on, and includes physical and mental as well as social, financial, personal, and environmental capabilities and resources.27 This includes understanding the potential negative impact of developing a clinical plan which, in order to operationalize, requires resources in excess of the patient’s capacity at that time.27 Minimally disruptive medicine, a promising theoretical approach for improving the care of complex clients, embodies the awareness of capacity in achieving patient-centered care while “imposing the smallest possible treatment burden on patients’ lives.”28
This study, although not without its limitations, provides an in-depth exploration of the experiences of a small number of patients living with HIV, recruited from a single facility in Toronto, Canada after relatively long hospital stays. There are specific context issues related to HIV, such as stigma and severe consequences for suboptimal medication adherence. Furthermore, this study took place where many urban health resources exist; complex patients in rural settings or in environments less tailored to the needs associated with complex medical, psychiatric, and social conditions may experience greater barriers in the transition process. Although this study captured data from medical charts and documents relevant to the cases, further exploration of the clinician decision-making process in creating the discharge plans and additional sources of data on health outcomes post-discharge would be beneficial.
Despite its limitations, this study provides detail and depth to understand some of the most complex patients who suffer from significant challenges in the health system and who are amongst the highest-cost healthcare users. The case study approach, with serial interviews, is an important strength of this study, enabling meaningful insight into hospital discharge processes and challenges experienced by complex patients that can inform individual-level care practice and the development of new programs and interventions.
This study builds on recent research with complex patients in calling for a new approach to clinical care.6,29,30 In order to support complex patients through discharge, clinical goals and referrals must be made in light of a patient’s capacity in the community. Structural changes may be made to improve coordination and access to services, decreasing the burden and improving the healthcare experience. Albreht et al.31 highlight a number of promising programs across Europe (such as the Clinic for Multimorbidity and Polypharmacy in Denmark) designed to improve the health and healthcare for individuals living with multiple chronic conditions. Small-scale changes are also important such as increasing conversations about the capacity and limitations of individuals listed as social supports, and making appropriate and realistic referrals based on an understanding of a patient’s capacity and motivation for follow-up. Shippee et al.32 identify a list of approaches in line with minimally disruptive medicine that can be integrated into existing systems as part of a developing “toolkit” (eg, elicitation of transcendent patient goals, and integration of patient-reported outcome tracking of challenges and burdens associated with health and daily living). The findings of this study suggest that the elements of the toolkit may provide a foundation for future interventions and research to improve hospital care and discharge outcomes for complex patients.
Disclosures
This project was funded by a Canadian Institutes of Health Research (CIHR) HIV/AIDS Community-based Research Catalyst Grant (#126669). Dr. Brennan’s research is supported by an Ontario HIV Treatment Network (OHTN) Applied HIV Research Chair. Dr. Chan Carusone reports grants from Canadian Institutes of Health Research during the conduct of the study.
1. Allaudeen N, Vidyarthi A, Masselli J, Auerback A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6:54-60. PubMed
2. Hu J, Gonsahn MD, Nerenz DR. Socioeconomic status and readmissions: evidence from an urban teaching hospital. Health Aff (Millwood). 2014;33:778-785. PubMed
3. Panagioti M, Stokes J, Esmail A, et al. Multimorbidity and patient safety incidents in primary care: a systematic review and meta-analysis. PLoS One. 2015;10:e0135947. PubMed
4. Paddison CA, Saunders CL, Abel GA, Payne RA, Campbell JL, Roland M. Why do patients with multimorbidity in England report worse experiences in primary care? Evidence from the General Practice Patient Survey. BMJ Open. 2015;5:e006172. PubMed
5. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
6. Schaink AK, Kuluski K, Lyons RF, et al. A scoping review and thematic classification of patient complexity: offering a unifying framework. J Comorbidity. 2012;2:1-9.
7. Roland M, Paddison C. Better management of patients with multimorbidity. BMJ. 2013;346:f2510. PubMed
8. Smith SM, Soubhi H, Fortin M, Hudon C, O’Dowd T. Managing patients with multimorbidity: a systematic review of interventions in primary care and community settings. BMJ. 2012;345:e5205. PubMed
9. Afshar S, Roderick PJ, Kowal P, Dimitrov BD, Hill AG. Multimorbidity and the inequalities of global ageing: a cross-sectional study of 28 countries using the World Health Surveys. BMC Public Health. 2015;15:776. PubMed
10. Pefoyo AJK, Bronskill SE, Gruneir A, et al. The increasing burden and complexity of multimorbidity. BMC Public Health. 2015;15:415. PubMed
11. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis. 2013;10:E65. PubMed
12. World Health Organization. Commission on Social Determinants of Health Final Report: Closing the Gap in a Generation: Health Equity through Action on Social Determinants of Health. Geneva, Switzerland: World Health Organization, 2008.
13. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B.. Epidemiology of multimorbidity and implications for health care, research and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
14. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8:e81355. PubMed
15. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525-1533. PubMed
16. Mays N, Pope C. Qualitative research: rigour and qualitative research. BMJ. 1995;311:109-112. PubMed
17. Gilson L, Hanson K, Sheikh K, Agyepong IA, Ssengooba F, Bennett S. Building the field of health policy and systems research: social science matters. PLoS Med. 2011;8:e1001079. PubMed
18. Stoto MA, Nelson CD, Klaiman T. Getting from what to why: using qualitative research to conduct public health systems research. AcademyHealth; August 2013. http://www.academyhealth.org/files/publications/qmforph.pdf. Accessed May 24, 2016.
19. Murray SA, Kendall M, Carduff E, et al. Use of serial qualitative interviews to understand patients’ evolving experiences and needs. BMJ. 2009;339:b3702. PubMed
20. Pope C, Ziebland S, Mays N. Qualitative research in health care. Analysing qualitative data. BMJ. 2000;320:114-116. PubMed
21. Dixon-Woods M. Using framework-based synthesis for conducting reviews of qualitative studies. BMC Med. 2011;9:39. PubMed
22. Yin RK. Case Study Research: Design and Methods. 5th ed. Thousand Oaks, CA: Sage Publications, Inc.; 2014.
23. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174:1095-1107. PubMed
24. Kansagara D, Chiovaro JC, Kagen D, et al. So many options, where do we start? An overview of the care transitions literature. J Hosp Med. 2016;11:221-230. PubMed
25. Kangovi S, Barg FK, Carter T, et al. Challenges faced by patients with low socioeconomic status during the post-hospital transition. J Gen Intern Med. 2013;29:283-289. PubMed
26. Gill A, Kuluski K, Jaakimainen L, Naganathan G, Upshur R, Wodchis WP. “Where do we go from here?” Health system frustrations expressed by patients with multimorbidity, their caregivers and family physicians. Healthc Policy. 2014;9:73-89. PubMed
27. Shippee ND, Shah ND, May CR, Mair FS, Montori VM. Cumulative complexity: a functional, patient-centered model of patient complexity can improve research and practice. J Clin Epidemiol. 2012;65:1041-1051. PubMed
28. Leppin AL, Montori VM, Gionfriddo MR. Minimally disruptive medicine: a pragmatically comprehensive model for delivering care to patients with multiple chronic conditions. Healthcare (Basel). 2015;3:50-63. PubMed
29. Salisbury C. Multimorbidity: redesigning health care for people who use it. Lancet. 2012;380:7-9. PubMed
30. Upshur R, Tracy S. Chronicity and complexity: is what’s good for the diseases always good for the patients? Can Fam Physician. 2008;54:1655-1658. PubMed
31. Albreht A, Dyakova M, Schellevis FG, Van den Broucke S. Many diseases, one model of care? J Comorbidity. 2016;6:12-20.
32. Shippee ND, Allen SV, Leppin AL, May CR, Montori VM. Attaining minimally disruptive medicine: context, challenges and a roadmap for implementation. J R Coll Physicians Edinb. 2015;45:118-122. PubMed
1. Allaudeen N, Vidyarthi A, Masselli J, Auerback A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6:54-60. PubMed
2. Hu J, Gonsahn MD, Nerenz DR. Socioeconomic status and readmissions: evidence from an urban teaching hospital. Health Aff (Millwood). 2014;33:778-785. PubMed
3. Panagioti M, Stokes J, Esmail A, et al. Multimorbidity and patient safety incidents in primary care: a systematic review and meta-analysis. PLoS One. 2015;10:e0135947. PubMed
4. Paddison CA, Saunders CL, Abel GA, Payne RA, Campbell JL, Roland M. Why do patients with multimorbidity in England report worse experiences in primary care? Evidence from the General Practice Patient Survey. BMJ Open. 2015;5:e006172. PubMed
5. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
6. Schaink AK, Kuluski K, Lyons RF, et al. A scoping review and thematic classification of patient complexity: offering a unifying framework. J Comorbidity. 2012;2:1-9.
7. Roland M, Paddison C. Better management of patients with multimorbidity. BMJ. 2013;346:f2510. PubMed
8. Smith SM, Soubhi H, Fortin M, Hudon C, O’Dowd T. Managing patients with multimorbidity: a systematic review of interventions in primary care and community settings. BMJ. 2012;345:e5205. PubMed
9. Afshar S, Roderick PJ, Kowal P, Dimitrov BD, Hill AG. Multimorbidity and the inequalities of global ageing: a cross-sectional study of 28 countries using the World Health Surveys. BMC Public Health. 2015;15:776. PubMed
10. Pefoyo AJK, Bronskill SE, Gruneir A, et al. The increasing burden and complexity of multimorbidity. BMC Public Health. 2015;15:415. PubMed
11. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis. 2013;10:E65. PubMed
12. World Health Organization. Commission on Social Determinants of Health Final Report: Closing the Gap in a Generation: Health Equity through Action on Social Determinants of Health. Geneva, Switzerland: World Health Organization, 2008.
13. Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B.. Epidemiology of multimorbidity and implications for health care, research and medical education: a cross-sectional study. Lancet. 2012;380:37-43. PubMed
14. Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8:e81355. PubMed
15. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525-1533. PubMed
16. Mays N, Pope C. Qualitative research: rigour and qualitative research. BMJ. 1995;311:109-112. PubMed
17. Gilson L, Hanson K, Sheikh K, Agyepong IA, Ssengooba F, Bennett S. Building the field of health policy and systems research: social science matters. PLoS Med. 2011;8:e1001079. PubMed
18. Stoto MA, Nelson CD, Klaiman T. Getting from what to why: using qualitative research to conduct public health systems research. AcademyHealth; August 2013. http://www.academyhealth.org/files/publications/qmforph.pdf. Accessed May 24, 2016.
19. Murray SA, Kendall M, Carduff E, et al. Use of serial qualitative interviews to understand patients’ evolving experiences and needs. BMJ. 2009;339:b3702. PubMed
20. Pope C, Ziebland S, Mays N. Qualitative research in health care. Analysing qualitative data. BMJ. 2000;320:114-116. PubMed
21. Dixon-Woods M. Using framework-based synthesis for conducting reviews of qualitative studies. BMC Med. 2011;9:39. PubMed
22. Yin RK. Case Study Research: Design and Methods. 5th ed. Thousand Oaks, CA: Sage Publications, Inc.; 2014.
23. Leppin AL, Gionfriddo MR, Kessler M, et al. Preventing 30-day hospital readmissions: a systematic review and meta-analysis of randomized trials. JAMA Intern Med. 2014;174:1095-1107. PubMed
24. Kansagara D, Chiovaro JC, Kagen D, et al. So many options, where do we start? An overview of the care transitions literature. J Hosp Med. 2016;11:221-230. PubMed
25. Kangovi S, Barg FK, Carter T, et al. Challenges faced by patients with low socioeconomic status during the post-hospital transition. J Gen Intern Med. 2013;29:283-289. PubMed
26. Gill A, Kuluski K, Jaakimainen L, Naganathan G, Upshur R, Wodchis WP. “Where do we go from here?” Health system frustrations expressed by patients with multimorbidity, their caregivers and family physicians. Healthc Policy. 2014;9:73-89. PubMed
27. Shippee ND, Shah ND, May CR, Mair FS, Montori VM. Cumulative complexity: a functional, patient-centered model of patient complexity can improve research and practice. J Clin Epidemiol. 2012;65:1041-1051. PubMed
28. Leppin AL, Montori VM, Gionfriddo MR. Minimally disruptive medicine: a pragmatically comprehensive model for delivering care to patients with multiple chronic conditions. Healthcare (Basel). 2015;3:50-63. PubMed
29. Salisbury C. Multimorbidity: redesigning health care for people who use it. Lancet. 2012;380:7-9. PubMed
30. Upshur R, Tracy S. Chronicity and complexity: is what’s good for the diseases always good for the patients? Can Fam Physician. 2008;54:1655-1658. PubMed
31. Albreht A, Dyakova M, Schellevis FG, Van den Broucke S. Many diseases, one model of care? J Comorbidity. 2016;6:12-20.
32. Shippee ND, Allen SV, Leppin AL, May CR, Montori VM. Attaining minimally disruptive medicine: context, challenges and a roadmap for implementation. J R Coll Physicians Edinb. 2015;45:118-122. PubMed
© 2017 Society of Hospital Medicine