User login
Download the SVSConnect Mobile App
If you’re looking to have the best remote user experience on SVSConnect, download the Member Centric app. Don’t limit your time on the community to only when you have a desktop or laptop computer available. The app gives you easy access to the Open Forum, member directory and shared resources directly from your mobile device. You're able to download this from either Google Play or the App Store. Read how to download the app here.
As always, use your SVS credentials to sign in and reach out to communications@Vascularsociety.org with questions.
If you’re looking to have the best remote user experience on SVSConnect, download the Member Centric app. Don’t limit your time on the community to only when you have a desktop or laptop computer available. The app gives you easy access to the Open Forum, member directory and shared resources directly from your mobile device. You're able to download this from either Google Play or the App Store. Read how to download the app here.
As always, use your SVS credentials to sign in and reach out to communications@Vascularsociety.org with questions.
If you’re looking to have the best remote user experience on SVSConnect, download the Member Centric app. Don’t limit your time on the community to only when you have a desktop or laptop computer available. The app gives you easy access to the Open Forum, member directory and shared resources directly from your mobile device. You're able to download this from either Google Play or the App Store. Read how to download the app here.
As always, use your SVS credentials to sign in and reach out to communications@Vascularsociety.org with questions.
Apply for SVS Membership
The third membership deadline of 2019 is a few short days away on Sept. 1. SVS members enjoy many benefits such as SVSConnect, discounted pricing on publications and meeting registration, practice management resources and much more. Visit the membership page and start your application today. If you have any questions, please contact the SVS membership department at membership@vascularsociety.org.
The third membership deadline of 2019 is a few short days away on Sept. 1. SVS members enjoy many benefits such as SVSConnect, discounted pricing on publications and meeting registration, practice management resources and much more. Visit the membership page and start your application today. If you have any questions, please contact the SVS membership department at membership@vascularsociety.org.
The third membership deadline of 2019 is a few short days away on Sept. 1. SVS members enjoy many benefits such as SVSConnect, discounted pricing on publications and meeting registration, practice management resources and much more. Visit the membership page and start your application today. If you have any questions, please contact the SVS membership department at membership@vascularsociety.org.
Last call for VAM 2020 Proposals
The Society for Vascular Surgery seeks proposals for invited sessions for the 2020 Vascular Annual Meeting, to be held June 17 to 20 in Toronto, Ontario, Canada. Scientific sessions will be June 18 to 20 and exhibits will be open June 18 to 19. Proposals should include the session's educational benefit, a short outline of the program topic, session goals and target audience, among other information. Obtain the information/submission form here and send in before tomorrow, Aug. 28. Email completed forms to achurilla@vascularsociety.org.
The Society for Vascular Surgery seeks proposals for invited sessions for the 2020 Vascular Annual Meeting, to be held June 17 to 20 in Toronto, Ontario, Canada. Scientific sessions will be June 18 to 20 and exhibits will be open June 18 to 19. Proposals should include the session's educational benefit, a short outline of the program topic, session goals and target audience, among other information. Obtain the information/submission form here and send in before tomorrow, Aug. 28. Email completed forms to achurilla@vascularsociety.org.
The Society for Vascular Surgery seeks proposals for invited sessions for the 2020 Vascular Annual Meeting, to be held June 17 to 20 in Toronto, Ontario, Canada. Scientific sessions will be June 18 to 20 and exhibits will be open June 18 to 19. Proposals should include the session's educational benefit, a short outline of the program topic, session goals and target audience, among other information. Obtain the information/submission form here and send in before tomorrow, Aug. 28. Email completed forms to achurilla@vascularsociety.org.
Tell us what you think of our Patient Education Materials
Last year, the SVS Foundation updated and rewrote its popular patient education fliers. These are designed to fulfill the Foundation’s mission to support patient education, vascular health and community awareness of vascular disease. SVS members are encouraged to download and print the fliers to use in their offices. If you’ve taken advantage of these fliers, we’d like to hear how you’ve benefited from them. Please take this short survey and provide your feedback. If you have yet to see the fliers, view them here.
Last year, the SVS Foundation updated and rewrote its popular patient education fliers. These are designed to fulfill the Foundation’s mission to support patient education, vascular health and community awareness of vascular disease. SVS members are encouraged to download and print the fliers to use in their offices. If you’ve taken advantage of these fliers, we’d like to hear how you’ve benefited from them. Please take this short survey and provide your feedback. If you have yet to see the fliers, view them here.
Last year, the SVS Foundation updated and rewrote its popular patient education fliers. These are designed to fulfill the Foundation’s mission to support patient education, vascular health and community awareness of vascular disease. SVS members are encouraged to download and print the fliers to use in their offices. If you’ve taken advantage of these fliers, we’d like to hear how you’ve benefited from them. Please take this short survey and provide your feedback. If you have yet to see the fliers, view them here.
Insurance networks: Is it time to abandon them?
Bob has been coming to therapy for a few months now. Initially, we met every week, but as his depression lifted, he asked to space the sessions out to twice a month, even as he continues to struggle with many challenges in his life. The cost, he says, is prohibitive, and while Bob believed he had good commercial insurance coverage, he’s learned a few things about insurance and mental health care.

Bob was referred to me by his internist. He knew I did not participate in his insurance network, but his policy covers out-of-network care. He’d had a number of imaging studies and then knee surgery earlier in the year, so he believed he’d met his deductible. He learned that, while he’d met his in-network deductible, he’d had no out-of-network expenses and there was a separate, much higher deductible – one he was not likely to meet with outpatient psychiatric care. In fact, the full cost of his treatment was not being subtracted from the deductible he needed to meet, but rather he was getting credit for lower usual and customary evaluation and management and psychotherapy fees for each session. It became clear that it would be many months – if ever – before Bob could expect any reimbursement for his out-of-network visits.
What Bob didn’t know was that, had he decided to switch to an in-network psychiatrist, he might well have trouble finding one, since half of psychiatrists don’t participate with any health insurance plans. And if he did see an in-network psychiatrist, he would likely need to find a separate in-network social worker or psychologist for psychotherapy, because most in-network psychiatrists see patients for short medication-management appointments. While the insurance companies would give Bob a list of providers, those lists are not kept up to date and include psychiatrists who have died, moved, aren’t taking new patients, or who have retired. The insurance company clearly states on its voicemail that verification of services does not guarantee payment, and Bob was told that the only way he could be certain of the reimbursement would be to submit the claims and wait. He went into treatment fully understanding that he might get no help with the cost from his health insurance.
When I first started in private practice in the 1990s, I joined only one panel. An older colleague told me I was foolish to hesitate, and that soon the panels would fill and it would be too late; psychiatrists wouldn’t be able to get on to the panels and would be unable to attract patients. A few years later, that same psychiatrist withdrew from all the insurance panels he was on; working on their terms was not rewarding. This division of in-network and out-of-network care is crucial to managed care: They must attract panels of doctors who will work for lower rates or with stipulations on how the doctor practices in order to save money.
But managed care came with a price: An entire administration was created to oversee the regulation of treatment. With time, some aspects of care management have vanished; it has been years since I have been asked to fill out a treatment plan to justify a need for outpatient psychotherapy. In Bob’s case, it’s clear how they save money; since he will not reach his high deductible, he will bear the full cost of his psychotherapy. Other patients who cannot afford to go out of network may give up searching and decide to go without treatment – this is not always an easy service to negotiate when one is distressed and compromised. Half of people with serious psychiatric disorders are not in treatment, and barriers to getting care are certainly one reason why.
Many psychiatrists have discovered that they can maintain a practice without being on insurance panels, as managed care only works if there are enough players willing to toss the ball. As psychiatrists have shied away from these panels, insurers have raised their reimbursement rates, and in Maryland, Medicaid also has had to raise their rates. The struggle has become one of how to get enough mental health professionals, and psychiatrists in particular, to join insurance panels in what is a shortage field.
Perhaps there are better ways to spend health care dollars than on the administration and management that come with limiting which doctors patients can see. The logistics of in-network versus out-of-network care are an expensive one, and create unconscionable scenarios in other fields. For example, such scenarios include ones in which a patient is brought in for emergency care to a facility where the doctors are not in network, or a patient has a procedure with an in-network surgeon but is unaware that the anesthesiologist or other members of the care team are not on the panel.
What if insurers controlled costs by setting a reasonable fee they would pay for services and paid any licensed physician for these services? Would market forces sort this out? Would fees then set to one which insurers would be willing to pay and physicians would be willing to accept? Or would those who are ill and impoverished be blocked from getting any care? What if we tried a whole new paradigm for psychiatric care?
Richard G. Frank, PhD, is a professor of health economics at Harvard. He specializes in mental health economics and is coauthor of the book “Better But Not Well: Mental Health Policy in the United States Since 1950” (Baltimore: Johns Hopkins University Press, 2006). Dr. Frank is a proponent of insurance panels.
“In the world we live in, we need to have panels; they are essential to controlling cost. They create a balance in terms of cost and utilization control in ways that protect patients, and they create a way for the insurance companies to bargain,” he said.
Dr. Frank noted that the concept that insurers should pay a set reasonable fee to any physician a patient wants to see has been tried. “It’s called ‘reference pricing,’ and when they did that with hip replacement surgery in California, it just hasn’t worked out. Patients ended up getting larger bills than they anticipated.
“The issues with psychiatry are different,” he continued. “There is a lot of bad behavior on the part of insurers and it’s an issue of parity. We shouldn’t let insurers differentially pay psychiatrists less. They have had an incentive to reduce the availability of mental health care and it’s bad for patients. It’s about lower payments to psychiatrists and the way those payments are currently structured drives patients out of care and defeats the purpose of insurance.”
Steven Sharfstein, MD, is the former CEO of Sheppard Pratt Health Systems, a past president of the American Psychiatric Association, and coauthor of several books on the economics of psychiatry. He refers to the current practice of credentialing network psychiatrists as a means of “rationing by supply.”
“The networks create a barrier to accessing care. I don’t think it’s an efficient way to take on the high cost of care, and it creates a tiered system.” Like Dr. Frank, Dr. Sharfstein believes parity is a large part of the problem. When asked about the idea of ending networks and establishing uniform deductibles and reimbursement rates, Dr. Sharfstein replied: “It really depends. We would need fees to hit a sweet spot that supports care while controlling costs.”
One thing is clear: With the current paradigm, it is often difficult to access treatment, and the well-insured patients may bear a significant and disproportionate cost (if not the entire cost) for getting care. Many who need care do not get it, regardless of their insurance status. Our status quo for psychiatric care falls short and I don’t predict that psychiatrists will rush to join networks so long as the demand for psychiatrists is greater than the supply.
Might another financial model work better? We know the system is lacking and one option, as Dr. Frank suggests, is to find solutions within the current model. But perhaps the question should not be one of how to get more psychiatrists to join networks, but of how to rework the system without the assumption that networks are the only way. While Bob is pleased that his symptoms are getting better and he’s managed a way to tackle his own bills, it’s certainly time to explore new ways of delivering and reimbursing psychiatric care.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle of Inpatient Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and has a private practice in Baltimore.
Bob has been coming to therapy for a few months now. Initially, we met every week, but as his depression lifted, he asked to space the sessions out to twice a month, even as he continues to struggle with many challenges in his life. The cost, he says, is prohibitive, and while Bob believed he had good commercial insurance coverage, he’s learned a few things about insurance and mental health care.
Bob was referred to me by his internist. He knew I did not participate in his insurance network, but his policy covers out-of-network care. He’d had a number of imaging studies and then knee surgery earlier in the year, so he believed he’d met his deductible. He learned that, while he’d met his in-network deductible, he’d had no out-of-network expenses and there was a separate, much higher deductible – one he was not likely to meet with outpatient psychiatric care. In fact, the full cost of his treatment was not being subtracted from the deductible he needed to meet, but rather he was getting credit for lower usual and customary evaluation and management and psychotherapy fees for each session. It became clear that it would be many months – if ever – before Bob could expect any reimbursement for his out-of-network visits.
What Bob didn’t know was that, had he decided to switch to an in-network psychiatrist, he might well have trouble finding one, since half of psychiatrists don’t participate with any health insurance plans. And if he did see an in-network psychiatrist, he would likely need to find a separate in-network social worker or psychologist for psychotherapy, because most in-network psychiatrists see patients for short medication-management appointments. While the insurance companies would give Bob a list of providers, those lists are not kept up to date and include psychiatrists who have died, moved, aren’t taking new patients, or who have retired. The insurance company clearly states on its voicemail that verification of services does not guarantee payment, and Bob was told that the only way he could be certain of the reimbursement would be to submit the claims and wait. He went into treatment fully understanding that he might get no help with the cost from his health insurance.
When I first started in private practice in the 1990s, I joined only one panel. An older colleague told me I was foolish to hesitate, and that soon the panels would fill and it would be too late; psychiatrists wouldn’t be able to get on to the panels and would be unable to attract patients. A few years later, that same psychiatrist withdrew from all the insurance panels he was on; working on their terms was not rewarding. This division of in-network and out-of-network care is crucial to managed care: They must attract panels of doctors who will work for lower rates or with stipulations on how the doctor practices in order to save money.
But managed care came with a price: An entire administration was created to oversee the regulation of treatment. With time, some aspects of care management have vanished; it has been years since I have been asked to fill out a treatment plan to justify a need for outpatient psychotherapy. In Bob’s case, it’s clear how they save money; since he will not reach his high deductible, he will bear the full cost of his psychotherapy. Other patients who cannot afford to go out of network may give up searching and decide to go without treatment – this is not always an easy service to negotiate when one is distressed and compromised. Half of people with serious psychiatric disorders are not in treatment, and barriers to getting care are certainly one reason why.
Many psychiatrists have discovered that they can maintain a practice without being on insurance panels, as managed care only works if there are enough players willing to toss the ball. As psychiatrists have shied away from these panels, insurers have raised their reimbursement rates, and in Maryland, Medicaid also has had to raise their rates. The struggle has become one of how to get enough mental health professionals, and psychiatrists in particular, to join insurance panels in what is a shortage field.
Perhaps there are better ways to spend health care dollars than on the administration and management that come with limiting which doctors patients can see. The logistics of in-network versus out-of-network care are an expensive one, and create unconscionable scenarios in other fields. For example, such scenarios include ones in which a patient is brought in for emergency care to a facility where the doctors are not in network, or a patient has a procedure with an in-network surgeon but is unaware that the anesthesiologist or other members of the care team are not on the panel.
What if insurers controlled costs by setting a reasonable fee they would pay for services and paid any licensed physician for these services? Would market forces sort this out? Would fees then set to one which insurers would be willing to pay and physicians would be willing to accept? Or would those who are ill and impoverished be blocked from getting any care? What if we tried a whole new paradigm for psychiatric care?
Richard G. Frank, PhD, is a professor of health economics at Harvard. He specializes in mental health economics and is coauthor of the book “Better But Not Well: Mental Health Policy in the United States Since 1950” (Baltimore: Johns Hopkins University Press, 2006). Dr. Frank is a proponent of insurance panels.
“In the world we live in, we need to have panels; they are essential to controlling cost. They create a balance in terms of cost and utilization control in ways that protect patients, and they create a way for the insurance companies to bargain,” he said.
Dr. Frank noted that the concept that insurers should pay a set reasonable fee to any physician a patient wants to see has been tried. “It’s called ‘reference pricing,’ and when they did that with hip replacement surgery in California, it just hasn’t worked out. Patients ended up getting larger bills than they anticipated.
“The issues with psychiatry are different,” he continued. “There is a lot of bad behavior on the part of insurers and it’s an issue of parity. We shouldn’t let insurers differentially pay psychiatrists less. They have had an incentive to reduce the availability of mental health care and it’s bad for patients. It’s about lower payments to psychiatrists and the way those payments are currently structured drives patients out of care and defeats the purpose of insurance.”
Steven Sharfstein, MD, is the former CEO of Sheppard Pratt Health Systems, a past president of the American Psychiatric Association, and coauthor of several books on the economics of psychiatry. He refers to the current practice of credentialing network psychiatrists as a means of “rationing by supply.”
“The networks create a barrier to accessing care. I don’t think it’s an efficient way to take on the high cost of care, and it creates a tiered system.” Like Dr. Frank, Dr. Sharfstein believes parity is a large part of the problem. When asked about the idea of ending networks and establishing uniform deductibles and reimbursement rates, Dr. Sharfstein replied: “It really depends. We would need fees to hit a sweet spot that supports care while controlling costs.”
One thing is clear: With the current paradigm, it is often difficult to access treatment, and the well-insured patients may bear a significant and disproportionate cost (if not the entire cost) for getting care. Many who need care do not get it, regardless of their insurance status. Our status quo for psychiatric care falls short and I don’t predict that psychiatrists will rush to join networks so long as the demand for psychiatrists is greater than the supply.
Might another financial model work better? We know the system is lacking and one option, as Dr. Frank suggests, is to find solutions within the current model. But perhaps the question should not be one of how to get more psychiatrists to join networks, but of how to rework the system without the assumption that networks are the only way. While Bob is pleased that his symptoms are getting better and he’s managed a way to tackle his own bills, it’s certainly time to explore new ways of delivering and reimbursing psychiatric care.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle of Inpatient Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and has a private practice in Baltimore.
Bob has been coming to therapy for a few months now. Initially, we met every week, but as his depression lifted, he asked to space the sessions out to twice a month, even as he continues to struggle with many challenges in his life. The cost, he says, is prohibitive, and while Bob believed he had good commercial insurance coverage, he’s learned a few things about insurance and mental health care.
Bob was referred to me by his internist. He knew I did not participate in his insurance network, but his policy covers out-of-network care. He’d had a number of imaging studies and then knee surgery earlier in the year, so he believed he’d met his deductible. He learned that, while he’d met his in-network deductible, he’d had no out-of-network expenses and there was a separate, much higher deductible – one he was not likely to meet with outpatient psychiatric care. In fact, the full cost of his treatment was not being subtracted from the deductible he needed to meet, but rather he was getting credit for lower usual and customary evaluation and management and psychotherapy fees for each session. It became clear that it would be many months – if ever – before Bob could expect any reimbursement for his out-of-network visits.
What Bob didn’t know was that, had he decided to switch to an in-network psychiatrist, he might well have trouble finding one, since half of psychiatrists don’t participate with any health insurance plans. And if he did see an in-network psychiatrist, he would likely need to find a separate in-network social worker or psychologist for psychotherapy, because most in-network psychiatrists see patients for short medication-management appointments. While the insurance companies would give Bob a list of providers, those lists are not kept up to date and include psychiatrists who have died, moved, aren’t taking new patients, or who have retired. The insurance company clearly states on its voicemail that verification of services does not guarantee payment, and Bob was told that the only way he could be certain of the reimbursement would be to submit the claims and wait. He went into treatment fully understanding that he might get no help with the cost from his health insurance.
When I first started in private practice in the 1990s, I joined only one panel. An older colleague told me I was foolish to hesitate, and that soon the panels would fill and it would be too late; psychiatrists wouldn’t be able to get on to the panels and would be unable to attract patients. A few years later, that same psychiatrist withdrew from all the insurance panels he was on; working on their terms was not rewarding. This division of in-network and out-of-network care is crucial to managed care: They must attract panels of doctors who will work for lower rates or with stipulations on how the doctor practices in order to save money.
But managed care came with a price: An entire administration was created to oversee the regulation of treatment. With time, some aspects of care management have vanished; it has been years since I have been asked to fill out a treatment plan to justify a need for outpatient psychotherapy. In Bob’s case, it’s clear how they save money; since he will not reach his high deductible, he will bear the full cost of his psychotherapy. Other patients who cannot afford to go out of network may give up searching and decide to go without treatment – this is not always an easy service to negotiate when one is distressed and compromised. Half of people with serious psychiatric disorders are not in treatment, and barriers to getting care are certainly one reason why.
Many psychiatrists have discovered that they can maintain a practice without being on insurance panels, as managed care only works if there are enough players willing to toss the ball. As psychiatrists have shied away from these panels, insurers have raised their reimbursement rates, and in Maryland, Medicaid also has had to raise their rates. The struggle has become one of how to get enough mental health professionals, and psychiatrists in particular, to join insurance panels in what is a shortage field.
Perhaps there are better ways to spend health care dollars than on the administration and management that come with limiting which doctors patients can see. The logistics of in-network versus out-of-network care are an expensive one, and create unconscionable scenarios in other fields. For example, such scenarios include ones in which a patient is brought in for emergency care to a facility where the doctors are not in network, or a patient has a procedure with an in-network surgeon but is unaware that the anesthesiologist or other members of the care team are not on the panel.
What if insurers controlled costs by setting a reasonable fee they would pay for services and paid any licensed physician for these services? Would market forces sort this out? Would fees then set to one which insurers would be willing to pay and physicians would be willing to accept? Or would those who are ill and impoverished be blocked from getting any care? What if we tried a whole new paradigm for psychiatric care?
Richard G. Frank, PhD, is a professor of health economics at Harvard. He specializes in mental health economics and is coauthor of the book “Better But Not Well: Mental Health Policy in the United States Since 1950” (Baltimore: Johns Hopkins University Press, 2006). Dr. Frank is a proponent of insurance panels.
“In the world we live in, we need to have panels; they are essential to controlling cost. They create a balance in terms of cost and utilization control in ways that protect patients, and they create a way for the insurance companies to bargain,” he said.
Dr. Frank noted that the concept that insurers should pay a set reasonable fee to any physician a patient wants to see has been tried. “It’s called ‘reference pricing,’ and when they did that with hip replacement surgery in California, it just hasn’t worked out. Patients ended up getting larger bills than they anticipated.
“The issues with psychiatry are different,” he continued. “There is a lot of bad behavior on the part of insurers and it’s an issue of parity. We shouldn’t let insurers differentially pay psychiatrists less. They have had an incentive to reduce the availability of mental health care and it’s bad for patients. It’s about lower payments to psychiatrists and the way those payments are currently structured drives patients out of care and defeats the purpose of insurance.”
Steven Sharfstein, MD, is the former CEO of Sheppard Pratt Health Systems, a past president of the American Psychiatric Association, and coauthor of several books on the economics of psychiatry. He refers to the current practice of credentialing network psychiatrists as a means of “rationing by supply.”
“The networks create a barrier to accessing care. I don’t think it’s an efficient way to take on the high cost of care, and it creates a tiered system.” Like Dr. Frank, Dr. Sharfstein believes parity is a large part of the problem. When asked about the idea of ending networks and establishing uniform deductibles and reimbursement rates, Dr. Sharfstein replied: “It really depends. We would need fees to hit a sweet spot that supports care while controlling costs.”
One thing is clear: With the current paradigm, it is often difficult to access treatment, and the well-insured patients may bear a significant and disproportionate cost (if not the entire cost) for getting care. Many who need care do not get it, regardless of their insurance status. Our status quo for psychiatric care falls short and I don’t predict that psychiatrists will rush to join networks so long as the demand for psychiatrists is greater than the supply.
Might another financial model work better? We know the system is lacking and one option, as Dr. Frank suggests, is to find solutions within the current model. But perhaps the question should not be one of how to get more psychiatrists to join networks, but of how to rework the system without the assumption that networks are the only way. While Bob is pleased that his symptoms are getting better and he’s managed a way to tackle his own bills, it’s certainly time to explore new ways of delivering and reimbursing psychiatric care.
Dr. Miller is coauthor with Annette Hanson, MD, of “Committed: The Battle of Inpatient Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and has a private practice in Baltimore.
Pretreatment CT data may help predict immunotherapy benefit in ovarian cancer
Pretreatment CT data may help identify responders to immunotherapy in ovarian cancer, according to a new study.
Specifically, fewer sites of disease and lower intratumor heterogeneity on contrast-enhanced CT may indicate a higher likelihood of durable response to immune checkpoint inhibitors, according to results of the retrospective study, recently published in JCO Precision Oncology.
“Our results suggest that quantitative analysis of baseline contrast-enhanced CT may facilitate the delivery of precision medicine to patients with ovarian cancer by identifying patients who may benefit from immunotherapy,” wrote Yuki Himoto, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues.
The study leverages findings from the emerging field of radiomics, which the investigators note allows for “virtual sampling” of tumor heterogeneity within a single lesion and between lesions.
“This information may complement molecular profiling in personalizing medical decisions,” Dr. Himoto and coauthors explained.
The study cohort included 75 patients with recurrent ovarian cancer who were enrolled in ongoing, prospective trials of immunotherapy, according to the researchers. Of that group, just under one in five derived a durable clinical benefit, defined as progression-free survival lasting at least 24 weeks.
In univariable analysis, they found a number of contrast-enhanced CT variables were linked to durable clinical benefit, including fewer disease sites, lower cluster-site entropy and dissimilarity, which they wrote were an indicator of lower intertumor heterogeneity, and higher energy in the largest-volume lesion, which they described as an indicator of lower intratumor heterogeneity.
However, in multivariable analysis, the only variables that were still associated with durable clinical benefit were fewer disease sites (odds ratio, 1.64; 95% confidence interval, 1.19-2.27; P = .012) and higher energy in the largest lesion (odds ratio, 1.41; 95% CI, 1.11-1.81; P = .006), according to the report.
Those two factors combined were a composite indicator of durable clinical benefit (C-index, 0.821).
These findings could represent a step forward in the provision of immunotherapy in ovarian cancer, which exhibits poor response to immune checkpoint inhibitors, compared with some other cancer types, the investigators wrote.
More insights are needed, however, to help personalize the selection of immunotherapy in ovarian cancer, including a better understanding of cancer immune reactions and retooling of immune response criteria, they added.
“Composite multimodal multifaceted biomarkers that noninvasively capture spatiotemporal tumor heterogeneity will likely be necessary to comprehensively assess immune the tumor microenvironment and serve as clinical decision support for prognosis inference and prediction of response,” Dr. Himoto and associates wrote.
The study was supported by the National Cancer Institute, among other sources. Study authors reported disclosures related to Merck, Bristol-Myers Squibb, Genentech, Celgene, AstraZeneca, Y-mAbs Therapeutics, and others.
SOURCE: Himoto Y et al. JCO Precis Oncol. 2019 Aug 13. doi: 10.1200/PO.19.00038.
Pretreatment CT data may help identify responders to immunotherapy in ovarian cancer, according to a new study.
Specifically, fewer sites of disease and lower intratumor heterogeneity on contrast-enhanced CT may indicate a higher likelihood of durable response to immune checkpoint inhibitors, according to results of the retrospective study, recently published in JCO Precision Oncology.
“Our results suggest that quantitative analysis of baseline contrast-enhanced CT may facilitate the delivery of precision medicine to patients with ovarian cancer by identifying patients who may benefit from immunotherapy,” wrote Yuki Himoto, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues.
The study leverages findings from the emerging field of radiomics, which the investigators note allows for “virtual sampling” of tumor heterogeneity within a single lesion and between lesions.
“This information may complement molecular profiling in personalizing medical decisions,” Dr. Himoto and coauthors explained.
The study cohort included 75 patients with recurrent ovarian cancer who were enrolled in ongoing, prospective trials of immunotherapy, according to the researchers. Of that group, just under one in five derived a durable clinical benefit, defined as progression-free survival lasting at least 24 weeks.
In univariable analysis, they found a number of contrast-enhanced CT variables were linked to durable clinical benefit, including fewer disease sites, lower cluster-site entropy and dissimilarity, which they wrote were an indicator of lower intertumor heterogeneity, and higher energy in the largest-volume lesion, which they described as an indicator of lower intratumor heterogeneity.
However, in multivariable analysis, the only variables that were still associated with durable clinical benefit were fewer disease sites (odds ratio, 1.64; 95% confidence interval, 1.19-2.27; P = .012) and higher energy in the largest lesion (odds ratio, 1.41; 95% CI, 1.11-1.81; P = .006), according to the report.
Those two factors combined were a composite indicator of durable clinical benefit (C-index, 0.821).
These findings could represent a step forward in the provision of immunotherapy in ovarian cancer, which exhibits poor response to immune checkpoint inhibitors, compared with some other cancer types, the investigators wrote.
More insights are needed, however, to help personalize the selection of immunotherapy in ovarian cancer, including a better understanding of cancer immune reactions and retooling of immune response criteria, they added.
“Composite multimodal multifaceted biomarkers that noninvasively capture spatiotemporal tumor heterogeneity will likely be necessary to comprehensively assess immune the tumor microenvironment and serve as clinical decision support for prognosis inference and prediction of response,” Dr. Himoto and associates wrote.
The study was supported by the National Cancer Institute, among other sources. Study authors reported disclosures related to Merck, Bristol-Myers Squibb, Genentech, Celgene, AstraZeneca, Y-mAbs Therapeutics, and others.
SOURCE: Himoto Y et al. JCO Precis Oncol. 2019 Aug 13. doi: 10.1200/PO.19.00038.
Pretreatment CT data may help identify responders to immunotherapy in ovarian cancer, according to a new study.
Specifically, fewer sites of disease and lower intratumor heterogeneity on contrast-enhanced CT may indicate a higher likelihood of durable response to immune checkpoint inhibitors, according to results of the retrospective study, recently published in JCO Precision Oncology.
“Our results suggest that quantitative analysis of baseline contrast-enhanced CT may facilitate the delivery of precision medicine to patients with ovarian cancer by identifying patients who may benefit from immunotherapy,” wrote Yuki Himoto, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, and colleagues.
The study leverages findings from the emerging field of radiomics, which the investigators note allows for “virtual sampling” of tumor heterogeneity within a single lesion and between lesions.
“This information may complement molecular profiling in personalizing medical decisions,” Dr. Himoto and coauthors explained.
The study cohort included 75 patients with recurrent ovarian cancer who were enrolled in ongoing, prospective trials of immunotherapy, according to the researchers. Of that group, just under one in five derived a durable clinical benefit, defined as progression-free survival lasting at least 24 weeks.
In univariable analysis, they found a number of contrast-enhanced CT variables were linked to durable clinical benefit, including fewer disease sites, lower cluster-site entropy and dissimilarity, which they wrote were an indicator of lower intertumor heterogeneity, and higher energy in the largest-volume lesion, which they described as an indicator of lower intratumor heterogeneity.
However, in multivariable analysis, the only variables that were still associated with durable clinical benefit were fewer disease sites (odds ratio, 1.64; 95% confidence interval, 1.19-2.27; P = .012) and higher energy in the largest lesion (odds ratio, 1.41; 95% CI, 1.11-1.81; P = .006), according to the report.
Those two factors combined were a composite indicator of durable clinical benefit (C-index, 0.821).
These findings could represent a step forward in the provision of immunotherapy in ovarian cancer, which exhibits poor response to immune checkpoint inhibitors, compared with some other cancer types, the investigators wrote.
More insights are needed, however, to help personalize the selection of immunotherapy in ovarian cancer, including a better understanding of cancer immune reactions and retooling of immune response criteria, they added.
“Composite multimodal multifaceted biomarkers that noninvasively capture spatiotemporal tumor heterogeneity will likely be necessary to comprehensively assess immune the tumor microenvironment and serve as clinical decision support for prognosis inference and prediction of response,” Dr. Himoto and associates wrote.
The study was supported by the National Cancer Institute, among other sources. Study authors reported disclosures related to Merck, Bristol-Myers Squibb, Genentech, Celgene, AstraZeneca, Y-mAbs Therapeutics, and others.
SOURCE: Himoto Y et al. JCO Precis Oncol. 2019 Aug 13. doi: 10.1200/PO.19.00038.
FROM JCO PRECISION ONCOLOGY
Researchers win grants to study real-world cancer data
Three researchers have won CancerLinQ Discovery Research Support Grants from the American Society of Clinical Oncology, and two researchers have received a National Institutes of Health R21 grant.

Igor Astsaturov, MD, PhD, and Edna Cukierman, PhD, both of Fox Chase Cancer Center in Philadelphia, won the R21 grant. The pair will receive $432,410 over 2 years for their research on pancreatic cancer.
With their work, Dr. Astsaturov and Dr. Cukierman are “hoping to describe the structural and functional nature of cell-cell contact, or oncogenic synapses, associated with cancer-associated fibroblasts and pancreatic cells,” according to Fox Chase.
Three other researchers have won ASCO’s CancerLinQ Discovery Research Support Grants. The recipients will conduct projects using CancerLinQ, which collects and analyzes real-world data from cancer patients at practices across the United States.
Each 1-year grant covers the cost of a CancerLinQ Discovery data set and a meeting at ASCO headquarters. The grants also contribute to personnel and/or research expenses. The grants are funded by the ASCO Foundation’s Mission Endowment of Conquer Cancer.

With his CancerLinQ Discovery grant, Sadiq Rehmani, MD, of Icahn School of Medicine at Mount Sinai in New York, will study immunotherapy in older lung cancer patients with comorbidities.
Grant recipient Yasmin Karimi, MD, of Stanford (Calif.) University, will study how osteoclast inhibitors affect skeletal-related events and mortality in “real-world” patients with metastatic breast cancer and bone metastasis.

Grant recipient Vinayak Muralidhar, MD, of Brigham and Women’s Hospital and the radiation oncology program at Harvard Medical School, Boston, will study the use of androgen-deprivation therapy and hypofractionation in prostate cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
Three researchers have won CancerLinQ Discovery Research Support Grants from the American Society of Clinical Oncology, and two researchers have received a National Institutes of Health R21 grant.
Igor Astsaturov, MD, PhD, and Edna Cukierman, PhD, both of Fox Chase Cancer Center in Philadelphia, won the R21 grant. The pair will receive $432,410 over 2 years for their research on pancreatic cancer.
With their work, Dr. Astsaturov and Dr. Cukierman are “hoping to describe the structural and functional nature of cell-cell contact, or oncogenic synapses, associated with cancer-associated fibroblasts and pancreatic cells,” according to Fox Chase.
Three other researchers have won ASCO’s CancerLinQ Discovery Research Support Grants. The recipients will conduct projects using CancerLinQ, which collects and analyzes real-world data from cancer patients at practices across the United States.
Each 1-year grant covers the cost of a CancerLinQ Discovery data set and a meeting at ASCO headquarters. The grants also contribute to personnel and/or research expenses. The grants are funded by the ASCO Foundation’s Mission Endowment of Conquer Cancer.
With his CancerLinQ Discovery grant, Sadiq Rehmani, MD, of Icahn School of Medicine at Mount Sinai in New York, will study immunotherapy in older lung cancer patients with comorbidities.
Grant recipient Yasmin Karimi, MD, of Stanford (Calif.) University, will study how osteoclast inhibitors affect skeletal-related events and mortality in “real-world” patients with metastatic breast cancer and bone metastasis.
Grant recipient Vinayak Muralidhar, MD, of Brigham and Women’s Hospital and the radiation oncology program at Harvard Medical School, Boston, will study the use of androgen-deprivation therapy and hypofractionation in prostate cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
Three researchers have won CancerLinQ Discovery Research Support Grants from the American Society of Clinical Oncology, and two researchers have received a National Institutes of Health R21 grant.
Igor Astsaturov, MD, PhD, and Edna Cukierman, PhD, both of Fox Chase Cancer Center in Philadelphia, won the R21 grant. The pair will receive $432,410 over 2 years for their research on pancreatic cancer.
With their work, Dr. Astsaturov and Dr. Cukierman are “hoping to describe the structural and functional nature of cell-cell contact, or oncogenic synapses, associated with cancer-associated fibroblasts and pancreatic cells,” according to Fox Chase.
Three other researchers have won ASCO’s CancerLinQ Discovery Research Support Grants. The recipients will conduct projects using CancerLinQ, which collects and analyzes real-world data from cancer patients at practices across the United States.
Each 1-year grant covers the cost of a CancerLinQ Discovery data set and a meeting at ASCO headquarters. The grants also contribute to personnel and/or research expenses. The grants are funded by the ASCO Foundation’s Mission Endowment of Conquer Cancer.
With his CancerLinQ Discovery grant, Sadiq Rehmani, MD, of Icahn School of Medicine at Mount Sinai in New York, will study immunotherapy in older lung cancer patients with comorbidities.
Grant recipient Yasmin Karimi, MD, of Stanford (Calif.) University, will study how osteoclast inhibitors affect skeletal-related events and mortality in “real-world” patients with metastatic breast cancer and bone metastasis.
Grant recipient Vinayak Muralidhar, MD, of Brigham and Women’s Hospital and the radiation oncology program at Harvard Medical School, Boston, will study the use of androgen-deprivation therapy and hypofractionation in prostate cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
What is your diagnosis? - September 2019
Erosive protein-losing enteropathy secondary to disseminated histoplasmosis
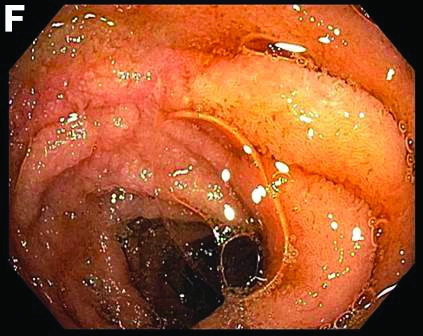
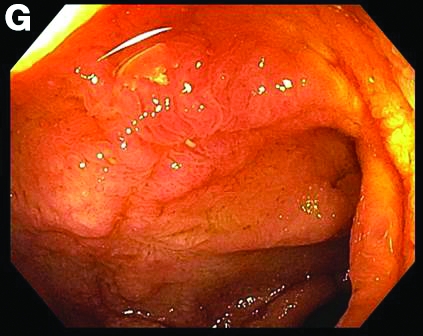
This patient was treated with amphotericin B and transitioned to oral itraconazole with frequent blood level monitoring to ensure absorption. His symptoms improved gradually. Small-bowel enteroscopy 3 weeks after presentation showed a normal duodenum and healing, superficial ulcers in the proximal jejunum (Figure F, G). Blood albumin levels had recovered to 3.1 g/dL (normal, 3.5–5.0 g/dL).
Protein-losing enteropathy (PLE) is a rare syndrome characterized by loss of serum proteins in the gastrointestinal (GI) tract, resulting in significant hypoproteinemia and consequent edema.1 PLE can also result in ascites, pleural and pericardial effusions, and, in prolonged cases, malnutrition. There are a variety of causes of PLE that can be broadly grouped into erosive GI disorders, disorders of increased GI mucosal permeability, and disorders of increased interstitial pressure. The clinical presentation depends on the underlying etiology, but commonly includes generalized edema owing to hypoproteinemia and resulting reduced oncotic pressure. GI symptoms are not frequently observed. The initial step in evaluating a patient with symptoms concerning for PLE is to rule out more common causes of hypoproteinemia, such as renal or hepatic disease, and malnutrition. To confirm enteric protein loss, alpha 1-antitrypsin clearance with a 24-hour stool collection is commonly and reliably used. Treatment of PLE is centered on treating the underlying cause while monitoring and treating malnutrition, including micronutrient deficiencies.
Fungal infections are a rare cause of PLE, but important to recognize as a potential complication of tumor necrosis factor–therapy, because these medications are commonly used for a variety of autoimmune diseases.2 Although histoplasmosis is an uncommon cause of GI inflammation, disseminated histoplasmosis causing PLE has been previously reported.3 In our patient, Histoplasma capsulatum infection caused diffuse GI ulcers, which allowed protein loss in the GI tract (erosive PLE). Antifungal treatment resulted in healing of intestinal ulcers and correction of hypoalbuminemia, thereby confirming the diagnosis of PLE and obviating the need for a confirmatory alpha 1-antitrypsin clearance study.
References
1. Umar SB, DiBaise JK. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105:43-9.
2. Tsiodras S, Samonis G, Boumpas DT. et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-94.
3. Kok J, Chen SC, Anderson L, et al. Protein-losing enteropathy and hypogammaglobulinaemia as first manifestations of disseminated histoplasmosis coincident with Nocardia infection. J Med Microbiol. 2010;59:610-3.
Erosive protein-losing enteropathy secondary to disseminated histoplasmosis
This patient was treated with amphotericin B and transitioned to oral itraconazole with frequent blood level monitoring to ensure absorption. His symptoms improved gradually. Small-bowel enteroscopy 3 weeks after presentation showed a normal duodenum and healing, superficial ulcers in the proximal jejunum (Figure F, G). Blood albumin levels had recovered to 3.1 g/dL (normal, 3.5–5.0 g/dL).
Protein-losing enteropathy (PLE) is a rare syndrome characterized by loss of serum proteins in the gastrointestinal (GI) tract, resulting in significant hypoproteinemia and consequent edema.1 PLE can also result in ascites, pleural and pericardial effusions, and, in prolonged cases, malnutrition. There are a variety of causes of PLE that can be broadly grouped into erosive GI disorders, disorders of increased GI mucosal permeability, and disorders of increased interstitial pressure. The clinical presentation depends on the underlying etiology, but commonly includes generalized edema owing to hypoproteinemia and resulting reduced oncotic pressure. GI symptoms are not frequently observed. The initial step in evaluating a patient with symptoms concerning for PLE is to rule out more common causes of hypoproteinemia, such as renal or hepatic disease, and malnutrition. To confirm enteric protein loss, alpha 1-antitrypsin clearance with a 24-hour stool collection is commonly and reliably used. Treatment of PLE is centered on treating the underlying cause while monitoring and treating malnutrition, including micronutrient deficiencies.
Fungal infections are a rare cause of PLE, but important to recognize as a potential complication of tumor necrosis factor–therapy, because these medications are commonly used for a variety of autoimmune diseases.2 Although histoplasmosis is an uncommon cause of GI inflammation, disseminated histoplasmosis causing PLE has been previously reported.3 In our patient, Histoplasma capsulatum infection caused diffuse GI ulcers, which allowed protein loss in the GI tract (erosive PLE). Antifungal treatment resulted in healing of intestinal ulcers and correction of hypoalbuminemia, thereby confirming the diagnosis of PLE and obviating the need for a confirmatory alpha 1-antitrypsin clearance study.
References
1. Umar SB, DiBaise JK. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105:43-9.
2. Tsiodras S, Samonis G, Boumpas DT. et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-94.
3. Kok J, Chen SC, Anderson L, et al. Protein-losing enteropathy and hypogammaglobulinaemia as first manifestations of disseminated histoplasmosis coincident with Nocardia infection. J Med Microbiol. 2010;59:610-3.
Erosive protein-losing enteropathy secondary to disseminated histoplasmosis
This patient was treated with amphotericin B and transitioned to oral itraconazole with frequent blood level monitoring to ensure absorption. His symptoms improved gradually. Small-bowel enteroscopy 3 weeks after presentation showed a normal duodenum and healing, superficial ulcers in the proximal jejunum (Figure F, G). Blood albumin levels had recovered to 3.1 g/dL (normal, 3.5–5.0 g/dL).
Protein-losing enteropathy (PLE) is a rare syndrome characterized by loss of serum proteins in the gastrointestinal (GI) tract, resulting in significant hypoproteinemia and consequent edema.1 PLE can also result in ascites, pleural and pericardial effusions, and, in prolonged cases, malnutrition. There are a variety of causes of PLE that can be broadly grouped into erosive GI disorders, disorders of increased GI mucosal permeability, and disorders of increased interstitial pressure. The clinical presentation depends on the underlying etiology, but commonly includes generalized edema owing to hypoproteinemia and resulting reduced oncotic pressure. GI symptoms are not frequently observed. The initial step in evaluating a patient with symptoms concerning for PLE is to rule out more common causes of hypoproteinemia, such as renal or hepatic disease, and malnutrition. To confirm enteric protein loss, alpha 1-antitrypsin clearance with a 24-hour stool collection is commonly and reliably used. Treatment of PLE is centered on treating the underlying cause while monitoring and treating malnutrition, including micronutrient deficiencies.
Fungal infections are a rare cause of PLE, but important to recognize as a potential complication of tumor necrosis factor–therapy, because these medications are commonly used for a variety of autoimmune diseases.2 Although histoplasmosis is an uncommon cause of GI inflammation, disseminated histoplasmosis causing PLE has been previously reported.3 In our patient, Histoplasma capsulatum infection caused diffuse GI ulcers, which allowed protein loss in the GI tract (erosive PLE). Antifungal treatment resulted in healing of intestinal ulcers and correction of hypoalbuminemia, thereby confirming the diagnosis of PLE and obviating the need for a confirmatory alpha 1-antitrypsin clearance study.
References
1. Umar SB, DiBaise JK. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105:43-9.
2. Tsiodras S, Samonis G, Boumpas DT. et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-94.
3. Kok J, Chen SC, Anderson L, et al. Protein-losing enteropathy and hypogammaglobulinaemia as first manifestations of disseminated histoplasmosis coincident with Nocardia infection. J Med Microbiol. 2010;59:610-3.
A 34-year-old man with a medical history of psoriasis, on adalimumab, presented with a 2-week history of progressively worsening abdominal pain, nausea, vomiting, melenic diarrhea, subjective fevers, and generalized weakness. One week into the illness, he developed progressive bilateral extremity and scrotal swelling.
His vital signs included a temperature of 36.8°C, heart rate of 104 beats per minute, respiratory rate of 18 breaths per minute, and a blood pressure of 114/71 mm Hg. The physical examination was notable for a well-nourished appearance, diffuse abdominal tenderness to palpation without distension, organomegaly, or rigidity, and pitting lower extremity edema.
Laboratory evaluation showed hemoglobin 10.3 g/dL (normal, 13.5–17.5 g/dL), leukocytes 10 × 109/L (normal, 3.5–10.5 × 109/L), platelets 212 × 109/L (normal, 150–450 × 109/L), sodium 131 mmol/L (normal, 135–145 mmol/L), creatinine 1 mg/dL (normal, 0.8–1.3 mg/dL), albumin 1.8 g/dL (normal, 3.5–5.0 g/dL), and C-reactive protein 53 mg/L (normal, less than 8 mg/L). Liver chemistries were all normal.
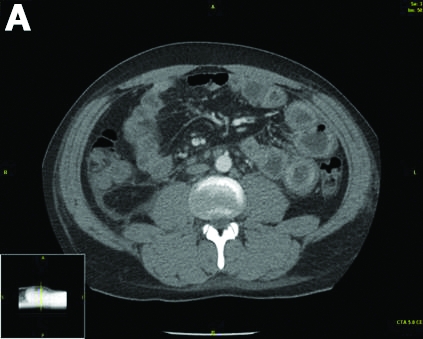
Urinalysis was unremarkable with normal urine protein levels. The enteric pathogen panel by polymerase chain reaction was negative. Computed tomography (CT) of the abdomen and pelvis showed marked circumferential wall thickening with mural enhancement of multiple loops of jejunum (Figure A).
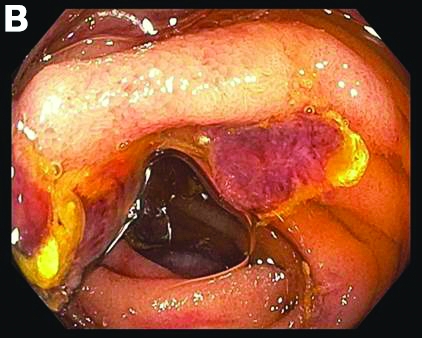
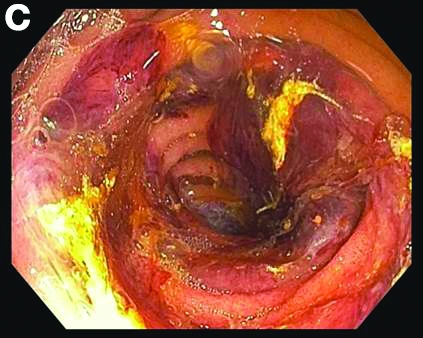
Small-bowel enteroscopy showed diffuse erosions in the entire duodenum and many oozing superficial ulcers with edematous and erythematous mucosa in the proximal jejunum (Figures B, C).
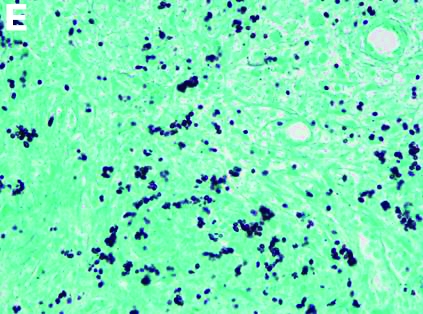
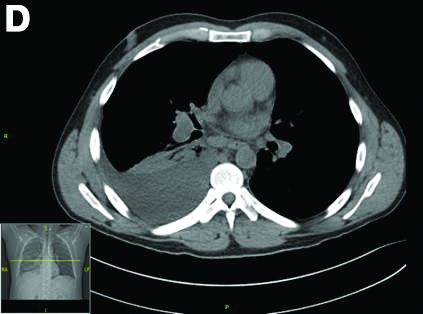
CT scan of the chest showed right lower lobe consolidation associated with a large right pleural effusion, and mediastinal, bilateral, hilar and abdominal lymphadenopathy (Figure D). Endobronchial ultrasound-guided transbronchial biopsy of lymph nodes was positive for oval-shaped organisms exhibiting narrow-based budding on GMS stain (Figure E).
Based on the clinical scenario and images, what is the most likely diagnosis?
Granuloma Annulare Presenting as Firm Nodules on the Forehead and Scalp in a Child
To the Editor:
A 3.5-year-old boy presented with asymptomatic subcutaneous nodules on the left side of the forehead and frontal scalp of approximately 6 months’ duration. There was no history of trauma or preceding rash. His medical history was remarkable only for allergic rhinitis. A review of systems was otherwise negative. Computed tomography ordered by the patient’s pediatrician prior to referral to dermatology showed soft tissue masses on the forehead and frontal scalp with no associated bone or brain parenchymal signal abnormalities.
At presentation to dermatology, physical examination revealed a 6×7-cm, flesh-colored cluster of firm, nontender, fixed, subcutaneous nodules on the left superior forehead and anterior part of the left frontal scalp (Figure 1A), as well as 2×1.5-cm, firm, fixed, flesh-colored nodule inferior to the larger cluster of lesions (Figure 1B). The remainder of the skin examination was unremarkable.
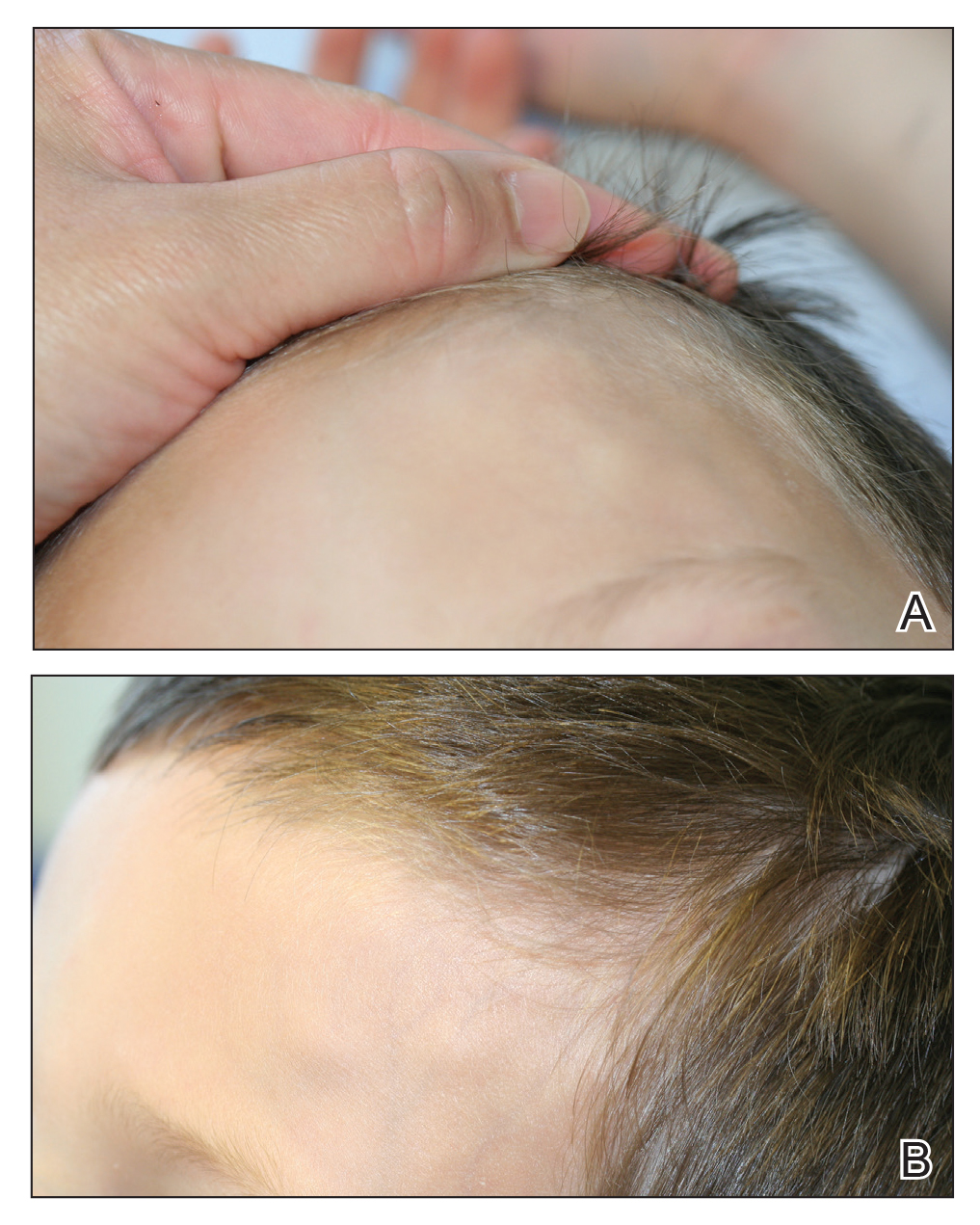
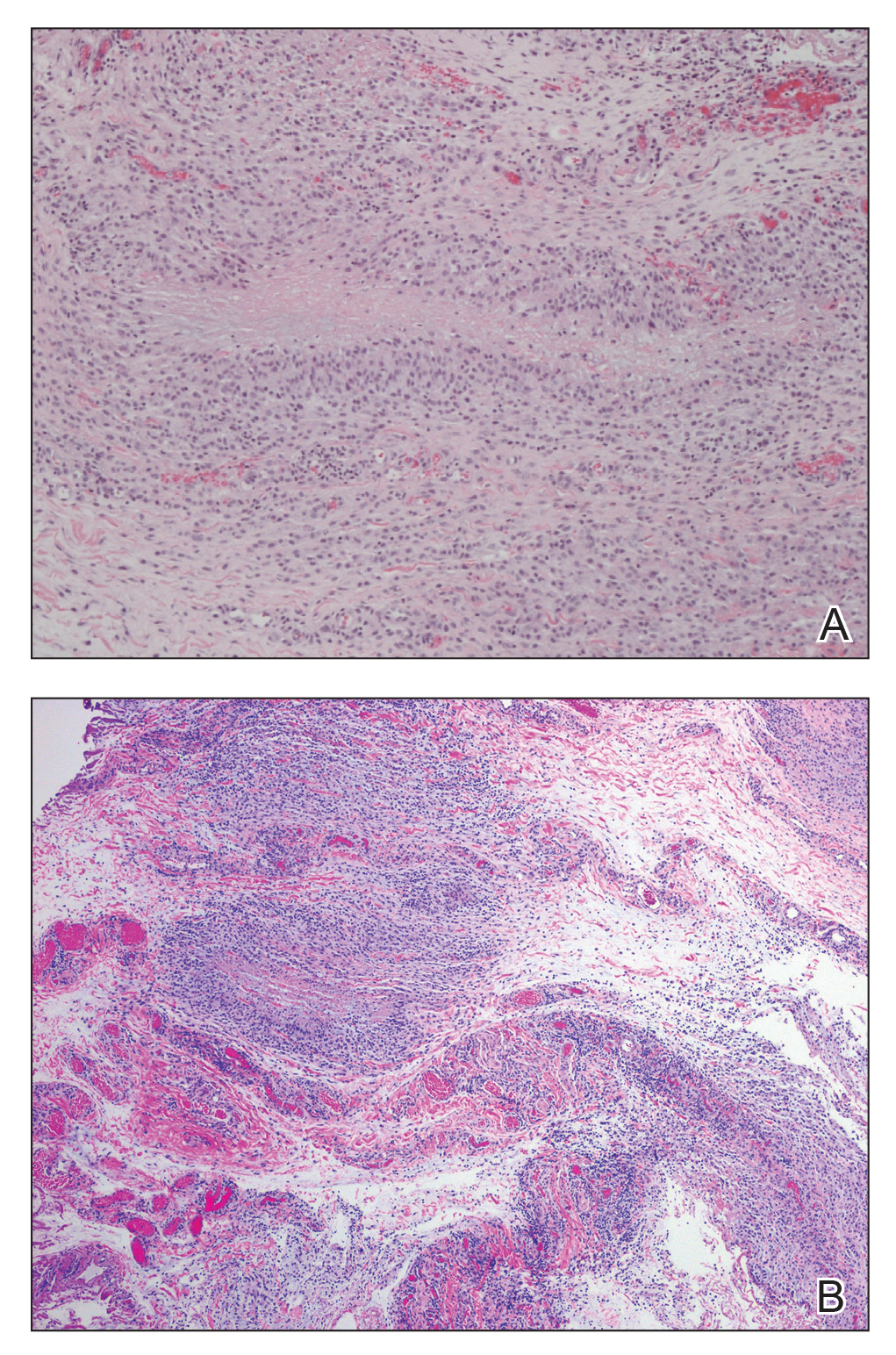
The patient was referred to plastic surgery for an incisional biopsy. The histopathologic findings demonstrated a marked mixed inflammatory infiltrate composed of lymphocytes and histiocytes with rare eosinophils and neutrophils in the subcutaneous tissue. The histiocytes were arranged in a palisading pattern surrounding central areas of necrosis (Figure 2). These features were consistent with a diagnosis of subcutaneous granuloma annulare (GA).
After the histologic diagnosis was elucidated, the patient’s family was provided reassurance regarding the benign nature and self-resolving course of GA in most children. No treatment was initiated, and no further laboratory studies or imaging were performed. At 9-month follow-up, the nodules were considerably smaller, and the patient remained asymptomatic.
Granuloma annulare is a benign dermatosis that presents in various forms, including localized, generalized, perforating, and subcutaneous subtypes. Subcutaneous GA (SGA) occurs most commonly in young children. The typical presentation of SGA is single or multiple flesh-colored to pink subcutaneous nodules on the arms, legs, or scalp.1 On the scalp, SGA has a predilection for the parietal and occipital regions. In some cases, there may be a history of preceding trauma to the area. Typically, lesions on the arms and legs are mobile whereas lesions on the scalp may be fixed due to their close proximity to the periosteum. Patients often are asymptomatic, and in the majority of cases, lesions resolve spontaneously over several months to years. Approximately 50% of cases resolve within 2 years of onset.2
Histologically, SGA appears as a nodule of fibrinoid or necrotic degeneration surrounded by palisaded histiocytes and lymphocytes in the deep dermis or subcutaneous tissue. Subcutaneous granuloma annulare is an accurate term for our case due to the subcutaneous location of the granulomatous change; however, some practitioners may prefer to use the term deep GA when the histologic findings are in the deep dermis vs the subcutis. Often, prominent deposition of mucin may be found. Histologically, SGA can closely resemble a rheumatoid nodule or necrobiosis lipoidica.1
Subcutaneous GA presenting on the scalp and forehead, such as in our case, can present a diagnostic challenge due to the extensive differential diagnoses that must be considered, including trauma, infection, bone or skin disease, and inflammatory or autoimmune disease.2,3 Additionally, the firm fixed characteristics of some lesions may raise additional concerns for possible malignant diagnoses such as epithelial sarcoma, peripheral nerve sheath tumor, rhabdomyosarcoma, or Langerhans cell histiocytosis, as highlighted by Agrawal et al.4 For these reasons, imaging and biopsy often are necessary for histologic diagnosis.
There is no consensus on the etiology of SGA, and no specific disease associations have been proven. Some case reports and case series have proposed a link between SGA and type 1 diabetes mellitus.1,4 In one retrospective case series, Grogg and Nascimento1 found that 2 of 34 (5.9%) pediatric patients with SGA had known or subsequently diagnosed diabetes mellitus; however, a definitive association between the 2 entities has not been elucidated. Underlying type 1 diabetes mellitus should be considered in patients with isolated SGA, but laboratory screening for diabetes is not necessary in patients with a negative review of systems.
Treatment of SGA typically is not required, as it is a self-limited condition. Often, once a histologic diagnosis is established, no further evaluation or treatment is warranted. Multiple treatment modalities have been reported, including intralesional and topical corticosteroids, laser therapy, cryotherapy, and systemic agents such as isotretinoin or corticosteroids; however, no treatment has been shown to be consistently efficacious.5 Excision of the lesions may be performed, but the risk for recurrence often precludes it as a viable option. In some case series, there have been recurrence rates as high as 40% to 75% in the months to years following surgical excision/biopsy.1,6
Patients presenting with presumed SGA should undergo a thorough history, review of systems, and full-body skin examination. In some cases, imaging and biopsy may be necessary to make a definitive diagnosis and exclude a more serious condition.
- Grogg KL, Nascimento AG. Subcutaneous granuloma annulare in childhood: clinicopathologic features in 34 cases. Pediatrics. 2001;107:E42.
- Sabuncuoglu H, Oge K, Soylemezoglu F, et al. Subcutaneous granuloma annulare of the scalp in childhood: a case report and review of the literature. Turk Neurosurg. 2007;17:19-22.
- Whelan JP, Zembowicz A. A 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Agrawal AK, Kammen BF, Guo H, et al. An unusual presentation of subcutaneous granuloma annulare in association with juvenile-onset diabetes: case report and literature review. Pediatr Dermatol. 2012;29:202-205.
- Cronquist SD, Stashower ME, Benson PM. Deep dermal granuloma annulare presenting as an eyelid tumor in a child, with review of pediatric eyelid lesions. Pediatr Dermatol. 1999;16:377-380.
- Jankowski PP, Krishna PH, Rutledge JC, et al. Surgical management and outcome of scalp subcutaneous granuloma annulare in children: case report. Neurosurgery. 2008;63:E1002, discussion E1002.
To the Editor:
A 3.5-year-old boy presented with asymptomatic subcutaneous nodules on the left side of the forehead and frontal scalp of approximately 6 months’ duration. There was no history of trauma or preceding rash. His medical history was remarkable only for allergic rhinitis. A review of systems was otherwise negative. Computed tomography ordered by the patient’s pediatrician prior to referral to dermatology showed soft tissue masses on the forehead and frontal scalp with no associated bone or brain parenchymal signal abnormalities.
At presentation to dermatology, physical examination revealed a 6×7-cm, flesh-colored cluster of firm, nontender, fixed, subcutaneous nodules on the left superior forehead and anterior part of the left frontal scalp (Figure 1A), as well as 2×1.5-cm, firm, fixed, flesh-colored nodule inferior to the larger cluster of lesions (Figure 1B). The remainder of the skin examination was unremarkable.
The patient was referred to plastic surgery for an incisional biopsy. The histopathologic findings demonstrated a marked mixed inflammatory infiltrate composed of lymphocytes and histiocytes with rare eosinophils and neutrophils in the subcutaneous tissue. The histiocytes were arranged in a palisading pattern surrounding central areas of necrosis (Figure 2). These features were consistent with a diagnosis of subcutaneous granuloma annulare (GA).
After the histologic diagnosis was elucidated, the patient’s family was provided reassurance regarding the benign nature and self-resolving course of GA in most children. No treatment was initiated, and no further laboratory studies or imaging were performed. At 9-month follow-up, the nodules were considerably smaller, and the patient remained asymptomatic.
Granuloma annulare is a benign dermatosis that presents in various forms, including localized, generalized, perforating, and subcutaneous subtypes. Subcutaneous GA (SGA) occurs most commonly in young children. The typical presentation of SGA is single or multiple flesh-colored to pink subcutaneous nodules on the arms, legs, or scalp.1 On the scalp, SGA has a predilection for the parietal and occipital regions. In some cases, there may be a history of preceding trauma to the area. Typically, lesions on the arms and legs are mobile whereas lesions on the scalp may be fixed due to their close proximity to the periosteum. Patients often are asymptomatic, and in the majority of cases, lesions resolve spontaneously over several months to years. Approximately 50% of cases resolve within 2 years of onset.2
Histologically, SGA appears as a nodule of fibrinoid or necrotic degeneration surrounded by palisaded histiocytes and lymphocytes in the deep dermis or subcutaneous tissue. Subcutaneous granuloma annulare is an accurate term for our case due to the subcutaneous location of the granulomatous change; however, some practitioners may prefer to use the term deep GA when the histologic findings are in the deep dermis vs the subcutis. Often, prominent deposition of mucin may be found. Histologically, SGA can closely resemble a rheumatoid nodule or necrobiosis lipoidica.1
Subcutaneous GA presenting on the scalp and forehead, such as in our case, can present a diagnostic challenge due to the extensive differential diagnoses that must be considered, including trauma, infection, bone or skin disease, and inflammatory or autoimmune disease.2,3 Additionally, the firm fixed characteristics of some lesions may raise additional concerns for possible malignant diagnoses such as epithelial sarcoma, peripheral nerve sheath tumor, rhabdomyosarcoma, or Langerhans cell histiocytosis, as highlighted by Agrawal et al.4 For these reasons, imaging and biopsy often are necessary for histologic diagnosis.
There is no consensus on the etiology of SGA, and no specific disease associations have been proven. Some case reports and case series have proposed a link between SGA and type 1 diabetes mellitus.1,4 In one retrospective case series, Grogg and Nascimento1 found that 2 of 34 (5.9%) pediatric patients with SGA had known or subsequently diagnosed diabetes mellitus; however, a definitive association between the 2 entities has not been elucidated. Underlying type 1 diabetes mellitus should be considered in patients with isolated SGA, but laboratory screening for diabetes is not necessary in patients with a negative review of systems.
Treatment of SGA typically is not required, as it is a self-limited condition. Often, once a histologic diagnosis is established, no further evaluation or treatment is warranted. Multiple treatment modalities have been reported, including intralesional and topical corticosteroids, laser therapy, cryotherapy, and systemic agents such as isotretinoin or corticosteroids; however, no treatment has been shown to be consistently efficacious.5 Excision of the lesions may be performed, but the risk for recurrence often precludes it as a viable option. In some case series, there have been recurrence rates as high as 40% to 75% in the months to years following surgical excision/biopsy.1,6
Patients presenting with presumed SGA should undergo a thorough history, review of systems, and full-body skin examination. In some cases, imaging and biopsy may be necessary to make a definitive diagnosis and exclude a more serious condition.
To the Editor:
A 3.5-year-old boy presented with asymptomatic subcutaneous nodules on the left side of the forehead and frontal scalp of approximately 6 months’ duration. There was no history of trauma or preceding rash. His medical history was remarkable only for allergic rhinitis. A review of systems was otherwise negative. Computed tomography ordered by the patient’s pediatrician prior to referral to dermatology showed soft tissue masses on the forehead and frontal scalp with no associated bone or brain parenchymal signal abnormalities.
At presentation to dermatology, physical examination revealed a 6×7-cm, flesh-colored cluster of firm, nontender, fixed, subcutaneous nodules on the left superior forehead and anterior part of the left frontal scalp (Figure 1A), as well as 2×1.5-cm, firm, fixed, flesh-colored nodule inferior to the larger cluster of lesions (Figure 1B). The remainder of the skin examination was unremarkable.
The patient was referred to plastic surgery for an incisional biopsy. The histopathologic findings demonstrated a marked mixed inflammatory infiltrate composed of lymphocytes and histiocytes with rare eosinophils and neutrophils in the subcutaneous tissue. The histiocytes were arranged in a palisading pattern surrounding central areas of necrosis (Figure 2). These features were consistent with a diagnosis of subcutaneous granuloma annulare (GA).
After the histologic diagnosis was elucidated, the patient’s family was provided reassurance regarding the benign nature and self-resolving course of GA in most children. No treatment was initiated, and no further laboratory studies or imaging were performed. At 9-month follow-up, the nodules were considerably smaller, and the patient remained asymptomatic.
Granuloma annulare is a benign dermatosis that presents in various forms, including localized, generalized, perforating, and subcutaneous subtypes. Subcutaneous GA (SGA) occurs most commonly in young children. The typical presentation of SGA is single or multiple flesh-colored to pink subcutaneous nodules on the arms, legs, or scalp.1 On the scalp, SGA has a predilection for the parietal and occipital regions. In some cases, there may be a history of preceding trauma to the area. Typically, lesions on the arms and legs are mobile whereas lesions on the scalp may be fixed due to their close proximity to the periosteum. Patients often are asymptomatic, and in the majority of cases, lesions resolve spontaneously over several months to years. Approximately 50% of cases resolve within 2 years of onset.2
Histologically, SGA appears as a nodule of fibrinoid or necrotic degeneration surrounded by palisaded histiocytes and lymphocytes in the deep dermis or subcutaneous tissue. Subcutaneous granuloma annulare is an accurate term for our case due to the subcutaneous location of the granulomatous change; however, some practitioners may prefer to use the term deep GA when the histologic findings are in the deep dermis vs the subcutis. Often, prominent deposition of mucin may be found. Histologically, SGA can closely resemble a rheumatoid nodule or necrobiosis lipoidica.1
Subcutaneous GA presenting on the scalp and forehead, such as in our case, can present a diagnostic challenge due to the extensive differential diagnoses that must be considered, including trauma, infection, bone or skin disease, and inflammatory or autoimmune disease.2,3 Additionally, the firm fixed characteristics of some lesions may raise additional concerns for possible malignant diagnoses such as epithelial sarcoma, peripheral nerve sheath tumor, rhabdomyosarcoma, or Langerhans cell histiocytosis, as highlighted by Agrawal et al.4 For these reasons, imaging and biopsy often are necessary for histologic diagnosis.
There is no consensus on the etiology of SGA, and no specific disease associations have been proven. Some case reports and case series have proposed a link between SGA and type 1 diabetes mellitus.1,4 In one retrospective case series, Grogg and Nascimento1 found that 2 of 34 (5.9%) pediatric patients with SGA had known or subsequently diagnosed diabetes mellitus; however, a definitive association between the 2 entities has not been elucidated. Underlying type 1 diabetes mellitus should be considered in patients with isolated SGA, but laboratory screening for diabetes is not necessary in patients with a negative review of systems.
Treatment of SGA typically is not required, as it is a self-limited condition. Often, once a histologic diagnosis is established, no further evaluation or treatment is warranted. Multiple treatment modalities have been reported, including intralesional and topical corticosteroids, laser therapy, cryotherapy, and systemic agents such as isotretinoin or corticosteroids; however, no treatment has been shown to be consistently efficacious.5 Excision of the lesions may be performed, but the risk for recurrence often precludes it as a viable option. In some case series, there have been recurrence rates as high as 40% to 75% in the months to years following surgical excision/biopsy.1,6
Patients presenting with presumed SGA should undergo a thorough history, review of systems, and full-body skin examination. In some cases, imaging and biopsy may be necessary to make a definitive diagnosis and exclude a more serious condition.
- Grogg KL, Nascimento AG. Subcutaneous granuloma annulare in childhood: clinicopathologic features in 34 cases. Pediatrics. 2001;107:E42.
- Sabuncuoglu H, Oge K, Soylemezoglu F, et al. Subcutaneous granuloma annulare of the scalp in childhood: a case report and review of the literature. Turk Neurosurg. 2007;17:19-22.
- Whelan JP, Zembowicz A. A 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Agrawal AK, Kammen BF, Guo H, et al. An unusual presentation of subcutaneous granuloma annulare in association with juvenile-onset diabetes: case report and literature review. Pediatr Dermatol. 2012;29:202-205.
- Cronquist SD, Stashower ME, Benson PM. Deep dermal granuloma annulare presenting as an eyelid tumor in a child, with review of pediatric eyelid lesions. Pediatr Dermatol. 1999;16:377-380.
- Jankowski PP, Krishna PH, Rutledge JC, et al. Surgical management and outcome of scalp subcutaneous granuloma annulare in children: case report. Neurosurgery. 2008;63:E1002, discussion E1002.
- Grogg KL, Nascimento AG. Subcutaneous granuloma annulare in childhood: clinicopathologic features in 34 cases. Pediatrics. 2001;107:E42.
- Sabuncuoglu H, Oge K, Soylemezoglu F, et al. Subcutaneous granuloma annulare of the scalp in childhood: a case report and review of the literature. Turk Neurosurg. 2007;17:19-22.
- Whelan JP, Zembowicz A. A 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Agrawal AK, Kammen BF, Guo H, et al. An unusual presentation of subcutaneous granuloma annulare in association with juvenile-onset diabetes: case report and literature review. Pediatr Dermatol. 2012;29:202-205.
- Cronquist SD, Stashower ME, Benson PM. Deep dermal granuloma annulare presenting as an eyelid tumor in a child, with review of pediatric eyelid lesions. Pediatr Dermatol. 1999;16:377-380.
- Jankowski PP, Krishna PH, Rutledge JC, et al. Surgical management and outcome of scalp subcutaneous granuloma annulare in children: case report. Neurosurgery. 2008;63:E1002, discussion E1002.
Practice Points
- Subcutaneous granuloma annulare (GA) is an important diagnosis to consider in the differential of subcutaneous nodules in children.
- The subcutaneous variant of GA can present without any typical GA lesions.
- Subcutaneous GA typically has a self-resolving course in most children.
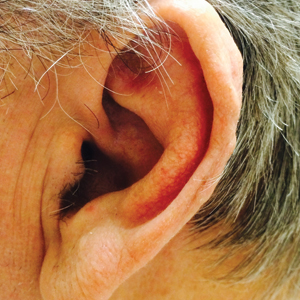
Idiopathic Bilateral Auricular Ossificans
To the Editor:
A 60-year-old man with a history of basal cell carcinoma, rosacea, and seborrheic dermatitis presented for a routine skin examination. The patient mentioned incidentally that both of his ears were “rock hard” and had been so for the last 10 to 20 years. He was not experiencing hearing abnormalities and denied any history of external ear trauma, frostbite injury of the ears, or history of endocrinopathy. Physical examination revealed normal-appearing skin on the bilateral ears (Figure 1), and palpation confirmed a bonelike consistency of the auricles with sparing of the earlobes. The differential diagnosis included osteoma cutis, ectopic calcification, tophaceous gout, localized scleroderma, and relapsing polychondritis. Incisional biopsy of the left posterior auricle to the level of cartilage revealed a histologically normal epidermis and dermis, with small fragments of cartilage calcification and lacunar bone formation (Figure 2). A radiograph of the skull showed faint calcification of the auricular cartilage, but no other osseous abnormalities were observed (Figure 3). Further laboratory workup including a comprehensive metabolic panel; complete blood cell count; and thyroid stimulating hormone, parathyroid hormone, and cortisol levels were normal. Based on these findings, a diagnosis of bilateral idiopathic auricular ossificans was made. Therapy was not pursued because the patient was asymptomatic, but referral to otolaryngology would have been considered if hearing impairment had occurred.

with auricular ossificans. Palpation of the auricle revealed a
bonelike consistency.
Auricular ossificans, which is characterized by the replacement of external ear cartilage by bone, is a rare condition with as few as 22 histologically proven cases documented in the literature.1,2 One case was reported in 2012 with consistent clinical and radiographic findings, but the patient declined biopsy.3 Similar to our patient, many pathologically documented cases have been determined to be idiopathic after workup, with identifiable triggering factors including cold injury, trauma, perichondritis, Addison disease, diabetes, and postpartum hypopituitarism.1 The male-to-female ratio is 18 to 5, the average age at diagnosis is 57 years, and as many as 70% of cases have demonstrated bilateral involvement.1,2
The majority of cases of auricular ossificans are asymptomatic at presentation with an insidious onset of the disease process over several years. Physical examination of the ear generally reveals a petrified auricle with sparing of the lobule and an otherwise normal clinical appearance. Radiographs demonstrate calcification, sometimes exactly mimicking the pattern seen in normal bone. Biopsy and histologic analysis show not only calcified cartilage but also actual lamellar bone formation.4 Depending on the precipitating factors, laboratory workup may uncover underlying metabolic abnormalities but often is unremarkable. Expert opinion generally recommends against extensive workup, which should be guided by the clinical presentation and the physician’s judgement.5 An insufficient number of patients with auricular ossificans have been definitively identified to provide clearer evidence-based recommendations.
Most patients present without pain or hearing abnormalities and do not require treatment. In one case, involvement of both the auricle and external ear canal resulted in intractable cerumen impaction that caused conductive hearing loss, eventually requiring resection of the ossified external ear cartilage and tragus.6 The most common reason for treatment has been discomfort impairing sleep, with surgical intervention including conchal reduction or wedge resection.5 A combined paucity of cases and poor understanding of the pathophysiologic mechanisms behind auricular ossificans limits current therapeutic options. Fortunately, this process appears to be benign in the majority of patients and generally represents a phenomenon that is much more interesting to the clinician than it is vexing to the patient.
- Calderon-Komaromy A, Cordoba S, Tardio JC, et al. Bilateral ossification of the auricular cartilage [published online November 28, 2014]. Actas Dermosifiliogr. 2015;106:433-435.
- Chang KH, Kim DK, Kim JH, et al. Idiopathic acquired ectopic auricular ossification: a case report and review of the literature. Ear Nose Throat J. 2011;90:424-427.
- Buikema KE, Adams EG. A rare case of petrified ear [published online October 15, 2012]. Case Rep Dermatol Med. 2012;2012:410601.
- Mastronikolis NS, Zampakis P, Kalogeropoulou C, et al. Bilateral ossification of the auricles: an unusual entity and review of the literature. Head Face Med. 2009;5:17.
- High WA, Larson MJ, Hoang MP. Idiopathic bilateral auricular ossificans: a case report and review of the literature. Arch Pathol Lab Med. 2004;128:1432-1434.
- Manni JJ, Berénos-Riley LC. Ossification of the external ear: a case report and review of the literature [published online June 18, 2005]. Eur Arch Otorhinolaryngol. 2005;262:961-964.
To the Editor:
A 60-year-old man with a history of basal cell carcinoma, rosacea, and seborrheic dermatitis presented for a routine skin examination. The patient mentioned incidentally that both of his ears were “rock hard” and had been so for the last 10 to 20 years. He was not experiencing hearing abnormalities and denied any history of external ear trauma, frostbite injury of the ears, or history of endocrinopathy. Physical examination revealed normal-appearing skin on the bilateral ears (Figure 1), and palpation confirmed a bonelike consistency of the auricles with sparing of the earlobes. The differential diagnosis included osteoma cutis, ectopic calcification, tophaceous gout, localized scleroderma, and relapsing polychondritis. Incisional biopsy of the left posterior auricle to the level of cartilage revealed a histologically normal epidermis and dermis, with small fragments of cartilage calcification and lacunar bone formation (Figure 2). A radiograph of the skull showed faint calcification of the auricular cartilage, but no other osseous abnormalities were observed (Figure 3). Further laboratory workup including a comprehensive metabolic panel; complete blood cell count; and thyroid stimulating hormone, parathyroid hormone, and cortisol levels were normal. Based on these findings, a diagnosis of bilateral idiopathic auricular ossificans was made. Therapy was not pursued because the patient was asymptomatic, but referral to otolaryngology would have been considered if hearing impairment had occurred.
with auricular ossificans. Palpation of the auricle revealed a
bonelike consistency.
Auricular ossificans, which is characterized by the replacement of external ear cartilage by bone, is a rare condition with as few as 22 histologically proven cases documented in the literature.1,2 One case was reported in 2012 with consistent clinical and radiographic findings, but the patient declined biopsy.3 Similar to our patient, many pathologically documented cases have been determined to be idiopathic after workup, with identifiable triggering factors including cold injury, trauma, perichondritis, Addison disease, diabetes, and postpartum hypopituitarism.1 The male-to-female ratio is 18 to 5, the average age at diagnosis is 57 years, and as many as 70% of cases have demonstrated bilateral involvement.1,2
The majority of cases of auricular ossificans are asymptomatic at presentation with an insidious onset of the disease process over several years. Physical examination of the ear generally reveals a petrified auricle with sparing of the lobule and an otherwise normal clinical appearance. Radiographs demonstrate calcification, sometimes exactly mimicking the pattern seen in normal bone. Biopsy and histologic analysis show not only calcified cartilage but also actual lamellar bone formation.4 Depending on the precipitating factors, laboratory workup may uncover underlying metabolic abnormalities but often is unremarkable. Expert opinion generally recommends against extensive workup, which should be guided by the clinical presentation and the physician’s judgement.5 An insufficient number of patients with auricular ossificans have been definitively identified to provide clearer evidence-based recommendations.
Most patients present without pain or hearing abnormalities and do not require treatment. In one case, involvement of both the auricle and external ear canal resulted in intractable cerumen impaction that caused conductive hearing loss, eventually requiring resection of the ossified external ear cartilage and tragus.6 The most common reason for treatment has been discomfort impairing sleep, with surgical intervention including conchal reduction or wedge resection.5 A combined paucity of cases and poor understanding of the pathophysiologic mechanisms behind auricular ossificans limits current therapeutic options. Fortunately, this process appears to be benign in the majority of patients and generally represents a phenomenon that is much more interesting to the clinician than it is vexing to the patient.
To the Editor:
A 60-year-old man with a history of basal cell carcinoma, rosacea, and seborrheic dermatitis presented for a routine skin examination. The patient mentioned incidentally that both of his ears were “rock hard” and had been so for the last 10 to 20 years. He was not experiencing hearing abnormalities and denied any history of external ear trauma, frostbite injury of the ears, or history of endocrinopathy. Physical examination revealed normal-appearing skin on the bilateral ears (Figure 1), and palpation confirmed a bonelike consistency of the auricles with sparing of the earlobes. The differential diagnosis included osteoma cutis, ectopic calcification, tophaceous gout, localized scleroderma, and relapsing polychondritis. Incisional biopsy of the left posterior auricle to the level of cartilage revealed a histologically normal epidermis and dermis, with small fragments of cartilage calcification and lacunar bone formation (Figure 2). A radiograph of the skull showed faint calcification of the auricular cartilage, but no other osseous abnormalities were observed (Figure 3). Further laboratory workup including a comprehensive metabolic panel; complete blood cell count; and thyroid stimulating hormone, parathyroid hormone, and cortisol levels were normal. Based on these findings, a diagnosis of bilateral idiopathic auricular ossificans was made. Therapy was not pursued because the patient was asymptomatic, but referral to otolaryngology would have been considered if hearing impairment had occurred.
with auricular ossificans. Palpation of the auricle revealed a
bonelike consistency.
Auricular ossificans, which is characterized by the replacement of external ear cartilage by bone, is a rare condition with as few as 22 histologically proven cases documented in the literature.1,2 One case was reported in 2012 with consistent clinical and radiographic findings, but the patient declined biopsy.3 Similar to our patient, many pathologically documented cases have been determined to be idiopathic after workup, with identifiable triggering factors including cold injury, trauma, perichondritis, Addison disease, diabetes, and postpartum hypopituitarism.1 The male-to-female ratio is 18 to 5, the average age at diagnosis is 57 years, and as many as 70% of cases have demonstrated bilateral involvement.1,2
The majority of cases of auricular ossificans are asymptomatic at presentation with an insidious onset of the disease process over several years. Physical examination of the ear generally reveals a petrified auricle with sparing of the lobule and an otherwise normal clinical appearance. Radiographs demonstrate calcification, sometimes exactly mimicking the pattern seen in normal bone. Biopsy and histologic analysis show not only calcified cartilage but also actual lamellar bone formation.4 Depending on the precipitating factors, laboratory workup may uncover underlying metabolic abnormalities but often is unremarkable. Expert opinion generally recommends against extensive workup, which should be guided by the clinical presentation and the physician’s judgement.5 An insufficient number of patients with auricular ossificans have been definitively identified to provide clearer evidence-based recommendations.
Most patients present without pain or hearing abnormalities and do not require treatment. In one case, involvement of both the auricle and external ear canal resulted in intractable cerumen impaction that caused conductive hearing loss, eventually requiring resection of the ossified external ear cartilage and tragus.6 The most common reason for treatment has been discomfort impairing sleep, with surgical intervention including conchal reduction or wedge resection.5 A combined paucity of cases and poor understanding of the pathophysiologic mechanisms behind auricular ossificans limits current therapeutic options. Fortunately, this process appears to be benign in the majority of patients and generally represents a phenomenon that is much more interesting to the clinician than it is vexing to the patient.
- Calderon-Komaromy A, Cordoba S, Tardio JC, et al. Bilateral ossification of the auricular cartilage [published online November 28, 2014]. Actas Dermosifiliogr. 2015;106:433-435.
- Chang KH, Kim DK, Kim JH, et al. Idiopathic acquired ectopic auricular ossification: a case report and review of the literature. Ear Nose Throat J. 2011;90:424-427.
- Buikema KE, Adams EG. A rare case of petrified ear [published online October 15, 2012]. Case Rep Dermatol Med. 2012;2012:410601.
- Mastronikolis NS, Zampakis P, Kalogeropoulou C, et al. Bilateral ossification of the auricles: an unusual entity and review of the literature. Head Face Med. 2009;5:17.
- High WA, Larson MJ, Hoang MP. Idiopathic bilateral auricular ossificans: a case report and review of the literature. Arch Pathol Lab Med. 2004;128:1432-1434.
- Manni JJ, Berénos-Riley LC. Ossification of the external ear: a case report and review of the literature [published online June 18, 2005]. Eur Arch Otorhinolaryngol. 2005;262:961-964.
- Calderon-Komaromy A, Cordoba S, Tardio JC, et al. Bilateral ossification of the auricular cartilage [published online November 28, 2014]. Actas Dermosifiliogr. 2015;106:433-435.
- Chang KH, Kim DK, Kim JH, et al. Idiopathic acquired ectopic auricular ossification: a case report and review of the literature. Ear Nose Throat J. 2011;90:424-427.
- Buikema KE, Adams EG. A rare case of petrified ear [published online October 15, 2012]. Case Rep Dermatol Med. 2012;2012:410601.
- Mastronikolis NS, Zampakis P, Kalogeropoulou C, et al. Bilateral ossification of the auricles: an unusual entity and review of the literature. Head Face Med. 2009;5:17.
- High WA, Larson MJ, Hoang MP. Idiopathic bilateral auricular ossificans: a case report and review of the literature. Arch Pathol Lab Med. 2004;128:1432-1434.
- Manni JJ, Berénos-Riley LC. Ossification of the external ear: a case report and review of the literature [published online June 18, 2005]. Eur Arch Otorhinolaryngol. 2005;262:961-964.
Practice Points
- Auricular ossificans is a rare condition characterized by the replacement of external ear cartilage by bone.
- The majority of cases are asymptomatic at presentation with an insidious onset of the disease process over several years.
- Most patients present without pain or hearing abnormalities and do not require treatment.