User login
SGLT2 inhibitor use tied to fewer atrial arrhythmias
Patients with cardiac implantable electronic devices (CIEDs) who received treatment with an sodium-glucose cotransporter 2 inhibitor had significantly fewer atrial arrhythmia events, compared with those who never received such a drug, in a prospective analysis of nearly 14,000 patients with a device who were followed for an average of nearly 2 years.

The findings suggest that use of an agent from the class of SGLT2 inhibitors “is associated with a pronounced reduction in atrial arrhythmia burden and all-cause mortality in patients with a CIED in a real-world setting,” said Ilan Goldenberg, MD, at the American Heart Association scientific sessions. “These data indicate possible antiarrhythmic properties of SGLT2 inhibitors that are incremental to the beneficial effects of the drug on heart failure outcomes,” added Dr. Goldenberg, director of the Clinical Cardiovascular Research Center at the University of Rochester (N.Y.).
In a propensity score–matched analysis that included more than 5,000 of the enrolled patients with a CIED, treatment with an SGLT2 inhibitor was tied to a significant 23% relative reduction in atrial arrhythmia events and a 44% relative drop in all-cause death, he reported.
Effect mediated by reduced left atrial pressure?
“Other heart failure drugs have shown some decrease in the rate of sudden cardiac death, but this is the first [heart failure] drug to associate with a reduction in atrial arrhythmias,” Dr. Goldenberg noted. “We think that a reduction in left atrial pressure” produced by treatment with an SGLT2 inhibitor “may be linked to the reduction in atrial arrhythmias.”
The study did not show an association of SGLT2-inhibitor use and a change in ventricular arrhythmias, compared with patients with CIEDs who did not receive an agent from this class.
The findings suggest “expanding the possible indications for SGLT2 inhibitors,” commented Harriette G.C. Van Spall, MD, a cardiologist at McMaster University, Hamilton, Ont., who moderated the session where Dr. Goldenberg gave his report.
The study included 13,890 consecutive, prospectively enrolled patients who received a CIED during January 2015–April 2020 at any of five hospitals operated by either of two tertiary health care systems, one run by the University of Rochester and the second based at Sheba Medical Center in Tel HaShomer, Israel. The devices that made patients eligible for the study included permanent pacemakers, implantable cardioverter defibrillators, cardiac resynchronization therapy devices, and implantable cardiac monitors. A blinded adjudication committee composed of electrophysiologists identified the arrhythmic episodes.
At entry into the study (the time of device implantation), 12,992 patients were not receiving an SGLT2 inhibitor (94%) and 898 (6%) were receiving a drug from this class. Of those, 39% were on dapagliflozin (Farxiga), 35% were on empagliflozin (Jardiance), and 26% were on canagliflozin (Invokana).
Patients receiving an SGLT2 inhibitor at baseline were on average substantially younger than the patients not on this drug class (59 years vs. 69 years); they had a substantially higher prevalence of diabetes (78% vs. 25%), and ischemic cardiomyopathy (63% vs. 39%). Patients on an SGLT2 inhibitor at baseline also had more modestly higher prevalence rates of prior heart failure (38% vs. 31%), and hypertension (69% vs. 63%). Prevalence of a history of atrial fibrillation (AFib) was nearly the same in both groups: 31% in patients on an SGLT2 inhibitor and 35% in those not on these drugs.
The study’s primary endpoint was the total number of arrhythmia events during follow-up of 24,442 patient-years, during which patients exhibited 19,633 atrial arrhythmia events and 3,231 ventricular arrhythmia events.
1% absolute reduction in atrial arrhythmias
A multivariate analysis of the entire population – adjusted for baseline differences in age, diabetes, sex, and history of AFib – showed that treatment with an SGLT2 inhibitor at baseline was linked with a significant 24% relative reduction in incident atrial arrhythmia events, a significant 24% reduction in both atrial and ventricular arrhythmia events, and a 42% relative reduction in all-cause deaths, compared with no SGLT2-inhibitor treatment.
The only analyzed endpoint that showed no significant between-group difference was incidence of ventricular arrhythmias, which was a relative 7% lower in the SGLT2-inhibitor group.
On an absolute basis, treatment with an SGLT2 inhibitor was tied to about a 1% lower rate of atrial arrhythmia events per year, a reduction from a 2.5% rate in those not on an SGLT2 inhibitor to about a 1.5% rate in those taking this drug class.
A second, confirmatory analysis used propensity score matching to identify 5,323 patients not on an SGLT2 inhibitor at baseline who closely matched the 898 patients on an SGLT2 inhibitor. The multivariate modeling for this analysis also adjusted for age, diabetes, sex, and history of AFib.
The results of these analyses closely matched the calculations that used the entire study population. Relative to patients not on an SGLT2 inhibitor those on a drug from this class had 23% fewer atrial arrhythmias, 44% fewer total death, and 22% fewer atrial or ventricular arrhythmias, all significant differences. However, ventricular arrhythmias only reduced by a relative 5%, a nonsignificant difference.
In the propensity score–matched analysis, the absolute reduction in atrial arrhythmias in those on an SGLT2 inhibitor at baseline was roughly 1.3% fewer per year, compared with those not on this drug class.
The study was funded by an unrestricted grant to the University of Rochester from AstraZeneca, the company that markets the SGLT2 inhibitor dapagliflozin (Farxiga). Dr. Goldenberg and Dr. Van Spall had no disclosures.
Patients with cardiac implantable electronic devices (CIEDs) who received treatment with an sodium-glucose cotransporter 2 inhibitor had significantly fewer atrial arrhythmia events, compared with those who never received such a drug, in a prospective analysis of nearly 14,000 patients with a device who were followed for an average of nearly 2 years.
The findings suggest that use of an agent from the class of SGLT2 inhibitors “is associated with a pronounced reduction in atrial arrhythmia burden and all-cause mortality in patients with a CIED in a real-world setting,” said Ilan Goldenberg, MD, at the American Heart Association scientific sessions. “These data indicate possible antiarrhythmic properties of SGLT2 inhibitors that are incremental to the beneficial effects of the drug on heart failure outcomes,” added Dr. Goldenberg, director of the Clinical Cardiovascular Research Center at the University of Rochester (N.Y.).
In a propensity score–matched analysis that included more than 5,000 of the enrolled patients with a CIED, treatment with an SGLT2 inhibitor was tied to a significant 23% relative reduction in atrial arrhythmia events and a 44% relative drop in all-cause death, he reported.
Effect mediated by reduced left atrial pressure?
“Other heart failure drugs have shown some decrease in the rate of sudden cardiac death, but this is the first [heart failure] drug to associate with a reduction in atrial arrhythmias,” Dr. Goldenberg noted. “We think that a reduction in left atrial pressure” produced by treatment with an SGLT2 inhibitor “may be linked to the reduction in atrial arrhythmias.”
The study did not show an association of SGLT2-inhibitor use and a change in ventricular arrhythmias, compared with patients with CIEDs who did not receive an agent from this class.
The findings suggest “expanding the possible indications for SGLT2 inhibitors,” commented Harriette G.C. Van Spall, MD, a cardiologist at McMaster University, Hamilton, Ont., who moderated the session where Dr. Goldenberg gave his report.
The study included 13,890 consecutive, prospectively enrolled patients who received a CIED during January 2015–April 2020 at any of five hospitals operated by either of two tertiary health care systems, one run by the University of Rochester and the second based at Sheba Medical Center in Tel HaShomer, Israel. The devices that made patients eligible for the study included permanent pacemakers, implantable cardioverter defibrillators, cardiac resynchronization therapy devices, and implantable cardiac monitors. A blinded adjudication committee composed of electrophysiologists identified the arrhythmic episodes.
At entry into the study (the time of device implantation), 12,992 patients were not receiving an SGLT2 inhibitor (94%) and 898 (6%) were receiving a drug from this class. Of those, 39% were on dapagliflozin (Farxiga), 35% were on empagliflozin (Jardiance), and 26% were on canagliflozin (Invokana).
Patients receiving an SGLT2 inhibitor at baseline were on average substantially younger than the patients not on this drug class (59 years vs. 69 years); they had a substantially higher prevalence of diabetes (78% vs. 25%), and ischemic cardiomyopathy (63% vs. 39%). Patients on an SGLT2 inhibitor at baseline also had more modestly higher prevalence rates of prior heart failure (38% vs. 31%), and hypertension (69% vs. 63%). Prevalence of a history of atrial fibrillation (AFib) was nearly the same in both groups: 31% in patients on an SGLT2 inhibitor and 35% in those not on these drugs.
The study’s primary endpoint was the total number of arrhythmia events during follow-up of 24,442 patient-years, during which patients exhibited 19,633 atrial arrhythmia events and 3,231 ventricular arrhythmia events.
1% absolute reduction in atrial arrhythmias
A multivariate analysis of the entire population – adjusted for baseline differences in age, diabetes, sex, and history of AFib – showed that treatment with an SGLT2 inhibitor at baseline was linked with a significant 24% relative reduction in incident atrial arrhythmia events, a significant 24% reduction in both atrial and ventricular arrhythmia events, and a 42% relative reduction in all-cause deaths, compared with no SGLT2-inhibitor treatment.
The only analyzed endpoint that showed no significant between-group difference was incidence of ventricular arrhythmias, which was a relative 7% lower in the SGLT2-inhibitor group.
On an absolute basis, treatment with an SGLT2 inhibitor was tied to about a 1% lower rate of atrial arrhythmia events per year, a reduction from a 2.5% rate in those not on an SGLT2 inhibitor to about a 1.5% rate in those taking this drug class.
A second, confirmatory analysis used propensity score matching to identify 5,323 patients not on an SGLT2 inhibitor at baseline who closely matched the 898 patients on an SGLT2 inhibitor. The multivariate modeling for this analysis also adjusted for age, diabetes, sex, and history of AFib.
The results of these analyses closely matched the calculations that used the entire study population. Relative to patients not on an SGLT2 inhibitor those on a drug from this class had 23% fewer atrial arrhythmias, 44% fewer total death, and 22% fewer atrial or ventricular arrhythmias, all significant differences. However, ventricular arrhythmias only reduced by a relative 5%, a nonsignificant difference.
In the propensity score–matched analysis, the absolute reduction in atrial arrhythmias in those on an SGLT2 inhibitor at baseline was roughly 1.3% fewer per year, compared with those not on this drug class.
The study was funded by an unrestricted grant to the University of Rochester from AstraZeneca, the company that markets the SGLT2 inhibitor dapagliflozin (Farxiga). Dr. Goldenberg and Dr. Van Spall had no disclosures.
Patients with cardiac implantable electronic devices (CIEDs) who received treatment with an sodium-glucose cotransporter 2 inhibitor had significantly fewer atrial arrhythmia events, compared with those who never received such a drug, in a prospective analysis of nearly 14,000 patients with a device who were followed for an average of nearly 2 years.
The findings suggest that use of an agent from the class of SGLT2 inhibitors “is associated with a pronounced reduction in atrial arrhythmia burden and all-cause mortality in patients with a CIED in a real-world setting,” said Ilan Goldenberg, MD, at the American Heart Association scientific sessions. “These data indicate possible antiarrhythmic properties of SGLT2 inhibitors that are incremental to the beneficial effects of the drug on heart failure outcomes,” added Dr. Goldenberg, director of the Clinical Cardiovascular Research Center at the University of Rochester (N.Y.).
In a propensity score–matched analysis that included more than 5,000 of the enrolled patients with a CIED, treatment with an SGLT2 inhibitor was tied to a significant 23% relative reduction in atrial arrhythmia events and a 44% relative drop in all-cause death, he reported.
Effect mediated by reduced left atrial pressure?
“Other heart failure drugs have shown some decrease in the rate of sudden cardiac death, but this is the first [heart failure] drug to associate with a reduction in atrial arrhythmias,” Dr. Goldenberg noted. “We think that a reduction in left atrial pressure” produced by treatment with an SGLT2 inhibitor “may be linked to the reduction in atrial arrhythmias.”
The study did not show an association of SGLT2-inhibitor use and a change in ventricular arrhythmias, compared with patients with CIEDs who did not receive an agent from this class.
The findings suggest “expanding the possible indications for SGLT2 inhibitors,” commented Harriette G.C. Van Spall, MD, a cardiologist at McMaster University, Hamilton, Ont., who moderated the session where Dr. Goldenberg gave his report.
The study included 13,890 consecutive, prospectively enrolled patients who received a CIED during January 2015–April 2020 at any of five hospitals operated by either of two tertiary health care systems, one run by the University of Rochester and the second based at Sheba Medical Center in Tel HaShomer, Israel. The devices that made patients eligible for the study included permanent pacemakers, implantable cardioverter defibrillators, cardiac resynchronization therapy devices, and implantable cardiac monitors. A blinded adjudication committee composed of electrophysiologists identified the arrhythmic episodes.
At entry into the study (the time of device implantation), 12,992 patients were not receiving an SGLT2 inhibitor (94%) and 898 (6%) were receiving a drug from this class. Of those, 39% were on dapagliflozin (Farxiga), 35% were on empagliflozin (Jardiance), and 26% were on canagliflozin (Invokana).
Patients receiving an SGLT2 inhibitor at baseline were on average substantially younger than the patients not on this drug class (59 years vs. 69 years); they had a substantially higher prevalence of diabetes (78% vs. 25%), and ischemic cardiomyopathy (63% vs. 39%). Patients on an SGLT2 inhibitor at baseline also had more modestly higher prevalence rates of prior heart failure (38% vs. 31%), and hypertension (69% vs. 63%). Prevalence of a history of atrial fibrillation (AFib) was nearly the same in both groups: 31% in patients on an SGLT2 inhibitor and 35% in those not on these drugs.
The study’s primary endpoint was the total number of arrhythmia events during follow-up of 24,442 patient-years, during which patients exhibited 19,633 atrial arrhythmia events and 3,231 ventricular arrhythmia events.
1% absolute reduction in atrial arrhythmias
A multivariate analysis of the entire population – adjusted for baseline differences in age, diabetes, sex, and history of AFib – showed that treatment with an SGLT2 inhibitor at baseline was linked with a significant 24% relative reduction in incident atrial arrhythmia events, a significant 24% reduction in both atrial and ventricular arrhythmia events, and a 42% relative reduction in all-cause deaths, compared with no SGLT2-inhibitor treatment.
The only analyzed endpoint that showed no significant between-group difference was incidence of ventricular arrhythmias, which was a relative 7% lower in the SGLT2-inhibitor group.
On an absolute basis, treatment with an SGLT2 inhibitor was tied to about a 1% lower rate of atrial arrhythmia events per year, a reduction from a 2.5% rate in those not on an SGLT2 inhibitor to about a 1.5% rate in those taking this drug class.
A second, confirmatory analysis used propensity score matching to identify 5,323 patients not on an SGLT2 inhibitor at baseline who closely matched the 898 patients on an SGLT2 inhibitor. The multivariate modeling for this analysis also adjusted for age, diabetes, sex, and history of AFib.
The results of these analyses closely matched the calculations that used the entire study population. Relative to patients not on an SGLT2 inhibitor those on a drug from this class had 23% fewer atrial arrhythmias, 44% fewer total death, and 22% fewer atrial or ventricular arrhythmias, all significant differences. However, ventricular arrhythmias only reduced by a relative 5%, a nonsignificant difference.
In the propensity score–matched analysis, the absolute reduction in atrial arrhythmias in those on an SGLT2 inhibitor at baseline was roughly 1.3% fewer per year, compared with those not on this drug class.
The study was funded by an unrestricted grant to the University of Rochester from AstraZeneca, the company that markets the SGLT2 inhibitor dapagliflozin (Farxiga). Dr. Goldenberg and Dr. Van Spall had no disclosures.
FROM AHA 2021
Hospitalist movers and shakers – December 2021
Narine Sargsyan, MD, recently was named the 2021 Alton Memorial Hospital (Alton, Ill.) Chairman’s Award winner. Serving as BJC Medical Group’s hospitalist medical director and hospital department chief of medicine, Dr. Sargsyan won the award based on the nominations of her fellow physicians.

The Chairman’s Award goes to an Alton Memorial staff member acknowledged for contributions to the facility and the community, including promotion and execution of outstanding customer service. Dr. Sargsyan has been a point person for Alton’s treatment of patients during the COVID-19 pandemic, recruiting new hospitalists to treat hospital inpatients. She also served on a committee selecting the inaugural resident class for the Southern Illinois University School of Medicine’s Family Residency program.
Alice Tang, DO, recently was named chief medical officer at Sentara Northern Virginia Medical Center (Woodbridge, Va.). The former medical director at Sentara Lake Ridge Hospital also directed the stroke program at Sentara Northern Virginia Medical Center, so she is familiar with her new facility.
The hospital medicine veteran specialized in emergency medicine and earned her health care MBA from George Washington University. Dr. Tang said her goal as CMO is to enhance the care environment while simultaneously raising the level of the care given by Sentara providers.
Faisal Keen, MD, has been named 2021 Physician of the Year at Sarasota Memorial Hospital’s Sarasota (Fla.) campus. The award winner was selected by a panel of SMH physician leaders.

Dr. Keen has been a hospitalist at SMH Sarasota for the past 6 years.
In presenting Dr. Keen with the award, the staff paid particular compliment to the care he provided to the facility’s hundreds of COVID-19 patients throughout the pandemic. At one point during the surge, Dr. Keen worked 30 shifts during a single month. Among the praises he received during the award presentation were those for his efforts in hurricane preparedness and helping physicians at SMH utilize technology in their patient care.
Jeffrey Crowder, MSPA, PA-C, recently became the first physician assistant to be named chief of hospitalist service at Maine Veterans Affairs Medical Center (Augusta, Me.). He is the first PA to hold the position at any Maine VA hospital. Mr. Crowder held the role in an acting position for the previous year, helping Maine VA Augusta navigate the COVID-19 pandemic.
Mr. Crowder will oversee 13 physicians and 9 PAs in providing care to Maine’s veterans. Included in the facility are intensive care and medical surgery units. Mr. Crowder’s group is responsible for part-time coverage at the 60-bed Togus Community Living Center.
Southeast Iowa Regional Medical Center (West Burlington, Iowa) has expanded its hospitalist program, adding the service to its Fort Madison campus. The health system’s hospitalist program was initiated at SEIRMC’s West Burlington campus back in 2010. That facility now includes 12 full-time and five part-time hospitalist physicians.
OB Hospitalist Group (Greenville, S.C.) has been acquired by Kohlberg & Company LLC (Mount Kisco, N.Y.), giving the nation’s largest dedicated obstetric hospitalist provider a new stakeholder. OBHG hopes to expand its services, which already include 200 hospital partners across 34 states.
OBHG’s network of providers includes more than 1,100 clinicians, with sites normally featuring an OB emergency department with a practicing ob.gyn. on site around the clock. Kohlberg & Company was founded in 1987 and has organized nine private equity funds, raising $12 billion of equity capital.
Narine Sargsyan, MD, recently was named the 2021 Alton Memorial Hospital (Alton, Ill.) Chairman’s Award winner. Serving as BJC Medical Group’s hospitalist medical director and hospital department chief of medicine, Dr. Sargsyan won the award based on the nominations of her fellow physicians.
The Chairman’s Award goes to an Alton Memorial staff member acknowledged for contributions to the facility and the community, including promotion and execution of outstanding customer service. Dr. Sargsyan has been a point person for Alton’s treatment of patients during the COVID-19 pandemic, recruiting new hospitalists to treat hospital inpatients. She also served on a committee selecting the inaugural resident class for the Southern Illinois University School of Medicine’s Family Residency program.
Alice Tang, DO, recently was named chief medical officer at Sentara Northern Virginia Medical Center (Woodbridge, Va.). The former medical director at Sentara Lake Ridge Hospital also directed the stroke program at Sentara Northern Virginia Medical Center, so she is familiar with her new facility.
The hospital medicine veteran specialized in emergency medicine and earned her health care MBA from George Washington University. Dr. Tang said her goal as CMO is to enhance the care environment while simultaneously raising the level of the care given by Sentara providers.
Faisal Keen, MD, has been named 2021 Physician of the Year at Sarasota Memorial Hospital’s Sarasota (Fla.) campus. The award winner was selected by a panel of SMH physician leaders.
Dr. Keen has been a hospitalist at SMH Sarasota for the past 6 years.
In presenting Dr. Keen with the award, the staff paid particular compliment to the care he provided to the facility’s hundreds of COVID-19 patients throughout the pandemic. At one point during the surge, Dr. Keen worked 30 shifts during a single month. Among the praises he received during the award presentation were those for his efforts in hurricane preparedness and helping physicians at SMH utilize technology in their patient care.
Jeffrey Crowder, MSPA, PA-C, recently became the first physician assistant to be named chief of hospitalist service at Maine Veterans Affairs Medical Center (Augusta, Me.). He is the first PA to hold the position at any Maine VA hospital. Mr. Crowder held the role in an acting position for the previous year, helping Maine VA Augusta navigate the COVID-19 pandemic.
Mr. Crowder will oversee 13 physicians and 9 PAs in providing care to Maine’s veterans. Included in the facility are intensive care and medical surgery units. Mr. Crowder’s group is responsible for part-time coverage at the 60-bed Togus Community Living Center.
Southeast Iowa Regional Medical Center (West Burlington, Iowa) has expanded its hospitalist program, adding the service to its Fort Madison campus. The health system’s hospitalist program was initiated at SEIRMC’s West Burlington campus back in 2010. That facility now includes 12 full-time and five part-time hospitalist physicians.
OB Hospitalist Group (Greenville, S.C.) has been acquired by Kohlberg & Company LLC (Mount Kisco, N.Y.), giving the nation’s largest dedicated obstetric hospitalist provider a new stakeholder. OBHG hopes to expand its services, which already include 200 hospital partners across 34 states.
OBHG’s network of providers includes more than 1,100 clinicians, with sites normally featuring an OB emergency department with a practicing ob.gyn. on site around the clock. Kohlberg & Company was founded in 1987 and has organized nine private equity funds, raising $12 billion of equity capital.
Narine Sargsyan, MD, recently was named the 2021 Alton Memorial Hospital (Alton, Ill.) Chairman’s Award winner. Serving as BJC Medical Group’s hospitalist medical director and hospital department chief of medicine, Dr. Sargsyan won the award based on the nominations of her fellow physicians.
The Chairman’s Award goes to an Alton Memorial staff member acknowledged for contributions to the facility and the community, including promotion and execution of outstanding customer service. Dr. Sargsyan has been a point person for Alton’s treatment of patients during the COVID-19 pandemic, recruiting new hospitalists to treat hospital inpatients. She also served on a committee selecting the inaugural resident class for the Southern Illinois University School of Medicine’s Family Residency program.
Alice Tang, DO, recently was named chief medical officer at Sentara Northern Virginia Medical Center (Woodbridge, Va.). The former medical director at Sentara Lake Ridge Hospital also directed the stroke program at Sentara Northern Virginia Medical Center, so she is familiar with her new facility.
The hospital medicine veteran specialized in emergency medicine and earned her health care MBA from George Washington University. Dr. Tang said her goal as CMO is to enhance the care environment while simultaneously raising the level of the care given by Sentara providers.
Faisal Keen, MD, has been named 2021 Physician of the Year at Sarasota Memorial Hospital’s Sarasota (Fla.) campus. The award winner was selected by a panel of SMH physician leaders.
Dr. Keen has been a hospitalist at SMH Sarasota for the past 6 years.
In presenting Dr. Keen with the award, the staff paid particular compliment to the care he provided to the facility’s hundreds of COVID-19 patients throughout the pandemic. At one point during the surge, Dr. Keen worked 30 shifts during a single month. Among the praises he received during the award presentation were those for his efforts in hurricane preparedness and helping physicians at SMH utilize technology in their patient care.
Jeffrey Crowder, MSPA, PA-C, recently became the first physician assistant to be named chief of hospitalist service at Maine Veterans Affairs Medical Center (Augusta, Me.). He is the first PA to hold the position at any Maine VA hospital. Mr. Crowder held the role in an acting position for the previous year, helping Maine VA Augusta navigate the COVID-19 pandemic.
Mr. Crowder will oversee 13 physicians and 9 PAs in providing care to Maine’s veterans. Included in the facility are intensive care and medical surgery units. Mr. Crowder’s group is responsible for part-time coverage at the 60-bed Togus Community Living Center.
Southeast Iowa Regional Medical Center (West Burlington, Iowa) has expanded its hospitalist program, adding the service to its Fort Madison campus. The health system’s hospitalist program was initiated at SEIRMC’s West Burlington campus back in 2010. That facility now includes 12 full-time and five part-time hospitalist physicians.
OB Hospitalist Group (Greenville, S.C.) has been acquired by Kohlberg & Company LLC (Mount Kisco, N.Y.), giving the nation’s largest dedicated obstetric hospitalist provider a new stakeholder. OBHG hopes to expand its services, which already include 200 hospital partners across 34 states.
OBHG’s network of providers includes more than 1,100 clinicians, with sites normally featuring an OB emergency department with a practicing ob.gyn. on site around the clock. Kohlberg & Company was founded in 1987 and has organized nine private equity funds, raising $12 billion of equity capital.
First Omicron variant case identified in U.S.
He or she was fully vaccinated against COVID-19 and experienced only “mild symptoms that are improving,” officials with the Centers for Disease Control and Prevention said.
The patient, who was not named in the CDC’s announcement of the first U.S. case of the Omicron variant Dec. 1, is self-quarantining.
“All close contacts have been contacted and have tested negative,” officials said.
The announcement comes as no surprise to many as the Omicron variant, first identified in South Africa, has been reported in countries around the world in recent days. Hong Kong, the United Kingdom, and Germany each reported this variant, as have Italy and the Netherlands. Over the weekend, the first North American cases were identified in Canada.
Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, announced over the weekend that this newest variant was likely already in the United States, telling ABC’s This Week its appearance here was “inevitable.”
Similar to previous variants, this new strain likely started circulating in the United States before scientists could do genetic tests to confirm its presence.
The World Health Organization named Omicron a “variant of concern” on Nov. 26, even though much remains unknown about how well it spreads, how severe it can be, and how it may resist vaccines. In the meantime, the United States enacted travel bans from multiple South African countries.
It remains to be seen if Omicron will follow the pattern of the Delta variant, which was first identified in the United States in May and became the dominant strain by July. It’s also possible it will follow the path taken by the Mu variant. Mu emerged in March and April to much concern, only to fizzle out by September because it was unable to compete with the Delta variant.
A version of this article first appeared on WebMD.com.
He or she was fully vaccinated against COVID-19 and experienced only “mild symptoms that are improving,” officials with the Centers for Disease Control and Prevention said.
The patient, who was not named in the CDC’s announcement of the first U.S. case of the Omicron variant Dec. 1, is self-quarantining.
“All close contacts have been contacted and have tested negative,” officials said.
The announcement comes as no surprise to many as the Omicron variant, first identified in South Africa, has been reported in countries around the world in recent days. Hong Kong, the United Kingdom, and Germany each reported this variant, as have Italy and the Netherlands. Over the weekend, the first North American cases were identified in Canada.
Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, announced over the weekend that this newest variant was likely already in the United States, telling ABC’s This Week its appearance here was “inevitable.”
Similar to previous variants, this new strain likely started circulating in the United States before scientists could do genetic tests to confirm its presence.
The World Health Organization named Omicron a “variant of concern” on Nov. 26, even though much remains unknown about how well it spreads, how severe it can be, and how it may resist vaccines. In the meantime, the United States enacted travel bans from multiple South African countries.
It remains to be seen if Omicron will follow the pattern of the Delta variant, which was first identified in the United States in May and became the dominant strain by July. It’s also possible it will follow the path taken by the Mu variant. Mu emerged in March and April to much concern, only to fizzle out by September because it was unable to compete with the Delta variant.
A version of this article first appeared on WebMD.com.
He or she was fully vaccinated against COVID-19 and experienced only “mild symptoms that are improving,” officials with the Centers for Disease Control and Prevention said.
The patient, who was not named in the CDC’s announcement of the first U.S. case of the Omicron variant Dec. 1, is self-quarantining.
“All close contacts have been contacted and have tested negative,” officials said.
The announcement comes as no surprise to many as the Omicron variant, first identified in South Africa, has been reported in countries around the world in recent days. Hong Kong, the United Kingdom, and Germany each reported this variant, as have Italy and the Netherlands. Over the weekend, the first North American cases were identified in Canada.
Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, announced over the weekend that this newest variant was likely already in the United States, telling ABC’s This Week its appearance here was “inevitable.”
Similar to previous variants, this new strain likely started circulating in the United States before scientists could do genetic tests to confirm its presence.
The World Health Organization named Omicron a “variant of concern” on Nov. 26, even though much remains unknown about how well it spreads, how severe it can be, and how it may resist vaccines. In the meantime, the United States enacted travel bans from multiple South African countries.
It remains to be seen if Omicron will follow the pattern of the Delta variant, which was first identified in the United States in May and became the dominant strain by July. It’s also possible it will follow the path taken by the Mu variant. Mu emerged in March and April to much concern, only to fizzle out by September because it was unable to compete with the Delta variant.
A version of this article first appeared on WebMD.com.
Bedside frailty assessment can determine when CPR will be nonbeneficial
Background: Although average survival after in-hospital cardiac arrest is 17%-20%, many clinicians feel that survival is lower in older patients or patients with multiple comorbidities. The Clinical Frailty Scale (CFS) is a simple bedside visual tool that encapsulates patients’ mobility and functional status, with a score greater than 4 indicating frailty. How this measure of frailty correlates with outcomes after in-hospital cardiac arrest is unknown.

Study design: Retrospective review.
Setting: Tertiary referral center in England.
Synopsis: The study included patients over 60 years old who received CPR between May 2017 and December 2018. CFS was retroactively applied based on available chart data. The patients’ median age was 77 years old, and 71% were male. The initial cardiac rhythm was nonshockable in 82% of cases, and overall in-hospital mortality was 86%. Frailty was independently associated with increased mortality when controlling for age, comorbidities, and rhythm. No frail patients survived to hospital discharge, while 26% of patients with CFS greater than 4 survived. Although patients with a shockable rhythm had a better chance of survival overall, compared with those with a nonshockable rhythm (92% vs. 23%, P less than .001), 15% of frail patients had a shockable rhythm, and none survived to discharge. Limitations of the study include relatively small sample size and the possibility of confounding variables, such as comorbid conditions.
Bottom line: When adjusted for age and rhythm, no frail patients older than 60 who received CPR for cardiac arrest survived to hospital discharge. Clinicians should discuss the limited chance of survival and potential burdens of resuscitation with frail patients and their families to avoid inappropriate CPR at the end of life.
Citation: Ibitoye SE et al. Frailty status predicts futility of cardiopulmonary resuscitation in older adults. Age Ageing. 2020 Jun 5;[e-pub]. doi: doi.org/10.1093/ageing/afaa104.
Dr. Chokshi is a hospitalist in the Division of Hospital Medicine, Mount Sinai Health System, New York.
Background: Although average survival after in-hospital cardiac arrest is 17%-20%, many clinicians feel that survival is lower in older patients or patients with multiple comorbidities. The Clinical Frailty Scale (CFS) is a simple bedside visual tool that encapsulates patients’ mobility and functional status, with a score greater than 4 indicating frailty. How this measure of frailty correlates with outcomes after in-hospital cardiac arrest is unknown.
Study design: Retrospective review.
Setting: Tertiary referral center in England.
Synopsis: The study included patients over 60 years old who received CPR between May 2017 and December 2018. CFS was retroactively applied based on available chart data. The patients’ median age was 77 years old, and 71% were male. The initial cardiac rhythm was nonshockable in 82% of cases, and overall in-hospital mortality was 86%. Frailty was independently associated with increased mortality when controlling for age, comorbidities, and rhythm. No frail patients survived to hospital discharge, while 26% of patients with CFS greater than 4 survived. Although patients with a shockable rhythm had a better chance of survival overall, compared with those with a nonshockable rhythm (92% vs. 23%, P less than .001), 15% of frail patients had a shockable rhythm, and none survived to discharge. Limitations of the study include relatively small sample size and the possibility of confounding variables, such as comorbid conditions.
Bottom line: When adjusted for age and rhythm, no frail patients older than 60 who received CPR for cardiac arrest survived to hospital discharge. Clinicians should discuss the limited chance of survival and potential burdens of resuscitation with frail patients and their families to avoid inappropriate CPR at the end of life.
Citation: Ibitoye SE et al. Frailty status predicts futility of cardiopulmonary resuscitation in older adults. Age Ageing. 2020 Jun 5;[e-pub]. doi: doi.org/10.1093/ageing/afaa104.
Dr. Chokshi is a hospitalist in the Division of Hospital Medicine, Mount Sinai Health System, New York.
Background: Although average survival after in-hospital cardiac arrest is 17%-20%, many clinicians feel that survival is lower in older patients or patients with multiple comorbidities. The Clinical Frailty Scale (CFS) is a simple bedside visual tool that encapsulates patients’ mobility and functional status, with a score greater than 4 indicating frailty. How this measure of frailty correlates with outcomes after in-hospital cardiac arrest is unknown.
Study design: Retrospective review.
Setting: Tertiary referral center in England.
Synopsis: The study included patients over 60 years old who received CPR between May 2017 and December 2018. CFS was retroactively applied based on available chart data. The patients’ median age was 77 years old, and 71% were male. The initial cardiac rhythm was nonshockable in 82% of cases, and overall in-hospital mortality was 86%. Frailty was independently associated with increased mortality when controlling for age, comorbidities, and rhythm. No frail patients survived to hospital discharge, while 26% of patients with CFS greater than 4 survived. Although patients with a shockable rhythm had a better chance of survival overall, compared with those with a nonshockable rhythm (92% vs. 23%, P less than .001), 15% of frail patients had a shockable rhythm, and none survived to discharge. Limitations of the study include relatively small sample size and the possibility of confounding variables, such as comorbid conditions.
Bottom line: When adjusted for age and rhythm, no frail patients older than 60 who received CPR for cardiac arrest survived to hospital discharge. Clinicians should discuss the limited chance of survival and potential burdens of resuscitation with frail patients and their families to avoid inappropriate CPR at the end of life.
Citation: Ibitoye SE et al. Frailty status predicts futility of cardiopulmonary resuscitation in older adults. Age Ageing. 2020 Jun 5;[e-pub]. doi: doi.org/10.1093/ageing/afaa104.
Dr. Chokshi is a hospitalist in the Division of Hospital Medicine, Mount Sinai Health System, New York.
Moderna warns of material drop in vaccine efficacy against Omicron
“There is no world, I think, where [the effectiveness] is the same level … we had with Delta,” Stephane Bancel told the Financial Times .
“I think it’s going to be a material drop,” he said. “I just don’t know how much, because we need to wait for the data. But all the scientists I’ve talked to … are like, ‘This is not going to be good.’”
Vaccine companies are now studying whether the new Omicron variant could evade the current shots. Some data is expected in about 2 weeks.
Mr. Bancel said that if a new vaccine is needed, it could take several months to produce at scale. He estimated that Moderna could make billions of vaccine doses in 2022.
“[Moderna] and Pfizer cannot get a billion doses next week. The math doesn’t work,” he said. “But could we get the billion doses out by the summer? Sure.”
The news caused some panic on Nov. 30, prompting financial markets to fall sharply, according to Reuters. But the markets recovered after European officials gave a more reassuring outlook.
“Even if the new variant becomes more widespread, the vaccines we have will continue to provide protection,” Emer Cooke, executive director of the European Medicines Agency, told the European Parliament.
Mr. Cooke said the agency could approve new vaccines that target the Omicron variant within 3 to 4 months, if needed. Moderna and Pfizer have announced they are beginning to tailor a shot to address the Omicron variant in case the data shows they are necessary.
Also on Nov. 30, the European Centre for Disease Prevention and Control announced that 42 Omicron cases had been identified in 10 European Union countries, according to Reuters.
The cases were mild or had no symptoms, although they were found in younger people who may have mild or no symptoms anyway.
“For the assessment of whether [Omicron] escapes immunity, we still have to wait until investigations in the laboratories with [blood samples] from people who have recovered have been carried out,” Andrea Ammon, MD, chair of the agency, said during an online conference.
The University of Oxford, which developed a COVID-19 vaccine with AstraZeneca, said Nov. 30 that there’s no evidence that vaccines won’t prevent severe disease from the Omicron variant, according to Reuters.
“Despite the appearance of new variants over the past year, vaccines have continued to provide very high levels of protection against severe disease and there is no evidence so far that Omicron is any different,” the university said in a statement. “However, we have the necessary tools and processes in place for rapid development of an updated COVID-19 vaccine if it should be necessary.”
A version of this article first appeared on WebMD.com.
“There is no world, I think, where [the effectiveness] is the same level … we had with Delta,” Stephane Bancel told the Financial Times .
“I think it’s going to be a material drop,” he said. “I just don’t know how much, because we need to wait for the data. But all the scientists I’ve talked to … are like, ‘This is not going to be good.’”
Vaccine companies are now studying whether the new Omicron variant could evade the current shots. Some data is expected in about 2 weeks.
Mr. Bancel said that if a new vaccine is needed, it could take several months to produce at scale. He estimated that Moderna could make billions of vaccine doses in 2022.
“[Moderna] and Pfizer cannot get a billion doses next week. The math doesn’t work,” he said. “But could we get the billion doses out by the summer? Sure.”
The news caused some panic on Nov. 30, prompting financial markets to fall sharply, according to Reuters. But the markets recovered after European officials gave a more reassuring outlook.
“Even if the new variant becomes more widespread, the vaccines we have will continue to provide protection,” Emer Cooke, executive director of the European Medicines Agency, told the European Parliament.
Mr. Cooke said the agency could approve new vaccines that target the Omicron variant within 3 to 4 months, if needed. Moderna and Pfizer have announced they are beginning to tailor a shot to address the Omicron variant in case the data shows they are necessary.
Also on Nov. 30, the European Centre for Disease Prevention and Control announced that 42 Omicron cases had been identified in 10 European Union countries, according to Reuters.
The cases were mild or had no symptoms, although they were found in younger people who may have mild or no symptoms anyway.
“For the assessment of whether [Omicron] escapes immunity, we still have to wait until investigations in the laboratories with [blood samples] from people who have recovered have been carried out,” Andrea Ammon, MD, chair of the agency, said during an online conference.
The University of Oxford, which developed a COVID-19 vaccine with AstraZeneca, said Nov. 30 that there’s no evidence that vaccines won’t prevent severe disease from the Omicron variant, according to Reuters.
“Despite the appearance of new variants over the past year, vaccines have continued to provide very high levels of protection against severe disease and there is no evidence so far that Omicron is any different,” the university said in a statement. “However, we have the necessary tools and processes in place for rapid development of an updated COVID-19 vaccine if it should be necessary.”
A version of this article first appeared on WebMD.com.
“There is no world, I think, where [the effectiveness] is the same level … we had with Delta,” Stephane Bancel told the Financial Times .
“I think it’s going to be a material drop,” he said. “I just don’t know how much, because we need to wait for the data. But all the scientists I’ve talked to … are like, ‘This is not going to be good.’”
Vaccine companies are now studying whether the new Omicron variant could evade the current shots. Some data is expected in about 2 weeks.
Mr. Bancel said that if a new vaccine is needed, it could take several months to produce at scale. He estimated that Moderna could make billions of vaccine doses in 2022.
“[Moderna] and Pfizer cannot get a billion doses next week. The math doesn’t work,” he said. “But could we get the billion doses out by the summer? Sure.”
The news caused some panic on Nov. 30, prompting financial markets to fall sharply, according to Reuters. But the markets recovered after European officials gave a more reassuring outlook.
“Even if the new variant becomes more widespread, the vaccines we have will continue to provide protection,” Emer Cooke, executive director of the European Medicines Agency, told the European Parliament.
Mr. Cooke said the agency could approve new vaccines that target the Omicron variant within 3 to 4 months, if needed. Moderna and Pfizer have announced they are beginning to tailor a shot to address the Omicron variant in case the data shows they are necessary.
Also on Nov. 30, the European Centre for Disease Prevention and Control announced that 42 Omicron cases had been identified in 10 European Union countries, according to Reuters.
The cases were mild or had no symptoms, although they were found in younger people who may have mild or no symptoms anyway.
“For the assessment of whether [Omicron] escapes immunity, we still have to wait until investigations in the laboratories with [blood samples] from people who have recovered have been carried out,” Andrea Ammon, MD, chair of the agency, said during an online conference.
The University of Oxford, which developed a COVID-19 vaccine with AstraZeneca, said Nov. 30 that there’s no evidence that vaccines won’t prevent severe disease from the Omicron variant, according to Reuters.
“Despite the appearance of new variants over the past year, vaccines have continued to provide very high levels of protection against severe disease and there is no evidence so far that Omicron is any different,” the university said in a statement. “However, we have the necessary tools and processes in place for rapid development of an updated COVID-19 vaccine if it should be necessary.”
A version of this article first appeared on WebMD.com.
Children and COVID: New cases, vaccinations both decline
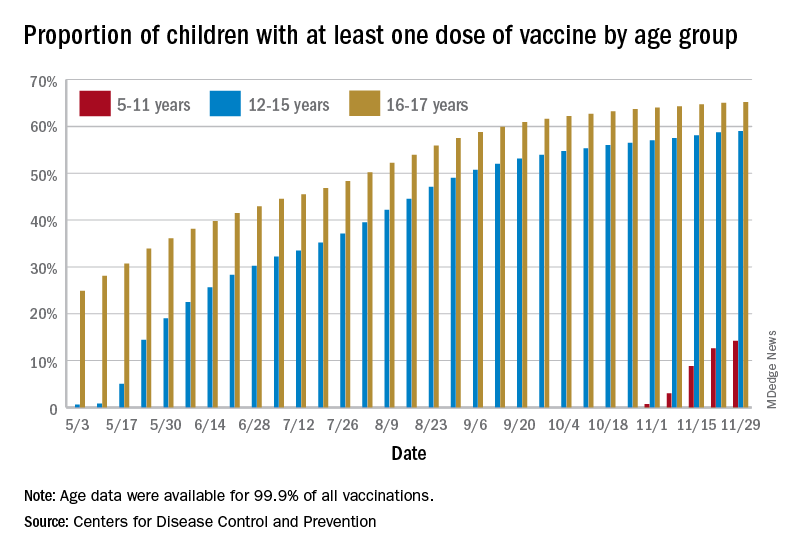
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
States reported 131,828 new pediatric cases for the week of Nov. 19-25, a decline of 7.1% over the previous week but still enough to surpass 100,000 for the 16th consecutive week. The weekly count had risen for 3 straight weeks since the last decrease in late October, the American Academy of Pediatrics and the Children’s Hospital Association said Nov. 30 in their weekly COVID report.
The AAP/CHA analysis, based on data from state and territorial health departments, puts the total number of cases in children at 6.9 million since the pandemic began, representing 17.0% of cases in Americans of all ages. The Centers for Disease Control and Prevention, which uses an age limit of 18 years to define a child, unlike some states, reports numbers of 6.1 million and 15.5%.
New vaccinations among the youngest eligible children, those aged 5-11 years, were down for the second week in a row after reaching almost 1.7 million during the first full week after approval on Nov. 2. Since then, the vaccination counts have been 1.2 million (Nov. 16-22) and 333,000 (Nov. 23-29), the CDC said on its COVID Data Tracker. A similar drop in the last week – from 127,000 to just 50,000 – also was seen for those aged 12-17 years.
Altogether, 14.2% of children aged 5-11, almost 4.1 million individuals, have received at least one dose of the vaccine, compared with 59.0% (10 million) of the 12- to 15-year-olds and 65.2% (5.5 million) of those aged 16-17. Just under 1% of the youngest group has been fully vaccinated, versus 49.0% and 55.8% for the older children, the CDC said.
It has been reported that Pfizer and BioNTech, which produce the only COVID vaccine approved for children, are planning to apply to the Food and Drug Administration during the first week of December for authorization for a booster dose for 16- and 17-year-olds.
Fauci: Omicron ‘very different from other variants’
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
The newly detected Omicron COVID-19 variant may be highly infectious and less responsive to available vaccines than other variants, but it is too early to know how it compares to the Delta variant, top infectious disease official Anthony S. Fauci, MD, said Nov. 30.
Dr. Fauci, speaking at a White House COVID-19 briefing, said there’s a “very unusual constellation of changes” across the COVID-19 genome that indicates it is unlike any variant we have seen so far.
“This mutational profile is very different from other variants of interest and concern, and although some mutations are also found in Delta, this is not Delta,” Dr. Fauci said. “These mutations have been associated with increased transmissibility and immune evasion.”
Omicron is the fifth designated COVID-19 variant of concern.
Detected first in South Africa, Omicron has been found in 20 countries so far. There are no known cases yet in the United States, but it has been detected in Canada.
Omicron has more than 30 mutations to the spike protein, the part of the virus that binds to human cells, Dr. Fauci said.
Cross-protection from boosters
Though the mutations suggest there is increased transmission of this variant, he said it is too soon to know how this compares to the Delta variant. And although the vaccines may not be as effective against Omicron, Dr. Fauci said there will likely be some protection.
“Remember, as with other variants, although partial immune escape may occur, vaccines, particularly boosters, give a level of antibodies that even with variants like Delta give you a degree of cross-protection, particularly against severe disease,” he said.
“When we say that although these mutations suggest a diminution of protection and a degree of immune evasion, we still, from experience with Delta, can make a reasonable conclusion that you would not eliminate all protection against this particular variant,” Dr. Fauci said.
So far, there is no reason to believe Omicron will cause more severe illness than other variants of concern.
“Although some preliminary information from South Africa suggests no unusual symptoms associated with variant, we do not know, and it is too early to tell,” Dr. Fauci said.
He recommended that people continue to wear masks, wash hands, and avoid crowded indoor venues. Most importantly, he recommended that everyone get their vaccines and boosters.
“One thing has become clear over the last 20 months: We can’t predict the future, but we can be prepared for it,” CDC Director Rochelle P. Walensky, MD, said at the briefing. “We have far more tools to fight the variant today than we did at this time last year.”
A version of this story first appeared on Medscape.com.
FDA panel backs first pill for COVID-19 by a small margin
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.

The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.
The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
, according to a panel of experts that advises the Food and Drug Administration on its regulatory decisions for these types of drugs.
The FDA’s Antimicrobial Drugs Advisory Committee narrowly voted to authorize the drug molnupiravir, voting 13 to 10 to support emergency use, which requires a medication to meet a lower standard of evidence than does full approval.
The FDA is not bound by the committee’s vote but typically follows its advice.
If authorized by the agency, molnupiravir would be the first antiviral agent available as a pill to treat COVID-19. Other therapies to treat the infection are available — monoclonal antibodies and the drug remdesivir — but they are given by infusion.
The United Kingdom has already authorized the use of Merck’s drug.
“This was clearly a difficult decision,” said committee member Michael Green, MD, a pediatric infectious disease expert at the University of Pittsburg School of Medicine.
Green said he voted yes, and that the drug’s ability to prevent deaths in the study weighed heavily on his decision. He said given uncertainties around the drug both the company and FDA should keep a close eye on patients taking the drug going forward.
“Should an alternative oral agent become available that had a better safety profile and equal or better efficacy profile, the agency might reconsider its authorization,” he said.
Others didn’t agree that the drug should be allowed onto the market.
“I voted no,” said Jennifer Le, PharmD, a professor of clinical pharmacy at the University of California. Dr. Le said the modest benefit of the medication didn’t outweigh all the potential safety issues. “I think I just need more efficacy and safety data,” she said.
Initial results from the first half of people enrolled in the clinical trial found the pill cut the risk of hospitalization or death by 50% in patients at higher risk of severe outcomes from COVID-19.
But later results, released just days before the meeting, showed that the drug’s effectiveness had dropped to about 30%.
In the updated analysis, 48 patients out of the 709 who were taking the drug were hospitalized or died within 29 days compared to 68 out of 699 who randomly got the placebo. There was one death in the group that got molnupiravir compared to nine in the placebo group. Nearly all those deaths occurred during the first phase of the study.
On Nov. 30 Merck explained that the drug’s efficacy appeared to fall, in part, because the placebo group had experienced fewer hospitalizations and deaths than expected during the second half of the study, making the drug look less beneficial by comparison.
The company said it wasn’t sure why patients in the placebo group had fared so much better in later trial enrollments.
“The efficacy of this product is not overwhelmingly good,” said committee member David Hardy, MD, an infectious disease expert at Charles Drew University School of Medicine in Los Angeles. “And I think that makes all of us a little uncomfortable about whether this is an advanced therapeutic because it’s an oral medication rather than an intravenous medication,” he said during the panel’s deliberations.
“I think we have to be very careful about how we’re going to allow people to use this,” Dr. Hardy said.
Many who voted for authorization thought use of the drug should be restricted to unvaccinated people who were at high risk of severe COVID-19 outcomes, the same population enrolled in the clinical trial. People in the trial were considered at higher risk if they were over age 60, had cancer, chronic kidney disease, chronic obstructive pulmonary disease, were obese, or had heart disease or diabetes.
There are some significant limitations of the study that may affect how the drug is used. Vaccinated people couldn’t enroll in the study, so it’s not known if the medication would have any benefit for them. Nearly two-thirds of the U.S. population is fully vaccinated. The study found no additional benefit of the medication compared to the placebo in people who had detectable antibodies, presumably from a prior infection.
Animal studies found that the drug — which kills the virus by forcing it to make errors as it copies its genetic material inside cells — could disrupt bone formation. For that reason, the manufacturer and the FDA agreed that it should not be used in anyone younger than age 18.
Animal studies also indicated that the drug could cause birth defects. For that reason, the company said the drug shouldn’t be given to women who are pregnant or breastfeeding and said doctors should make sure women of childbearing age aren’t pregnant before taking the medication.
Some members of the panel felt that pregnant women and their doctors should be given the choice of whether or not to use the drug, given that pregnant women are at high risk for severe COVID-19 outcomes and infused therapies may not be available in all settings.
Other members of the committee said they were uncomfortable authorizing the drug given its potential to mutate the virus.
The drug, which forces the virus to mutate as it copies its RNA, eventually causes the virus to make so many errors in its genetic material that it can no longer make more of itself and the immune system clears it out of the body.
But it takes a few days to work — the drug is designed to be taken for 5 consecutive days -- and studies of the viral loads of patients taking the drug show that through the first 2 days, viral loads remain detectable as these mutations occur.
Studies by the FDA show some of those mutations in the spike protein are the same ones that have helped the virus become more transmissible and escape the protection of vaccines.
So the question is whether someone taking the medication could develop a dangerous mutation and then infect someone else, sparking the spread of a new variant.
Nicholas Kartsonis, MD, a vice president at Merck, said that the company was still analyzing data.
“Even if the probability is very low — 1 in 10,000 or 1 in 100,000 -- that this drug would induce an escape mutant for which the vaccines we have would not cover, that would be catastrophic for the whole world, actually,” said committee member James Hildreth, MD, an immunologist and president of Meharry Medical College, Nashville. “Do you have sufficient data on the likelihood of that happening?” he asked Dr. Kartsonis of Merck.
“So we don’t,” Dr. Kartsonis said.
He said, in theory, the risk of mutation with molnupiravir is the same as seen with the use of vaccines or monoclonal antibody therapies. Dr. Hildreth wasn’t satisfied with that answer.
“With all respect, the mechanism of your drug is to drive [genetic mutations], so it’s not the same as the vaccine. It’s not the same as monoclonal antibodies,” he said.
Dr. Hildreth later said he didn’t feel comfortable voting for authorization given the uncertainties around escape mutants. He voted no.
“It was an easy vote for me,” he said.
A version of this article first appeared on Medscape.com.
Optimizing perioperative cardiac risk assessment and management for noncardiac surgery
Background: There are extensive publications regarding preoperative risk assessment and optimization of risk management. This article is a review of current aggregate data from various meta-analyses and observational studies. It explores a systematic approach to preoperative risk assessment.

Study design: Literature review of meta-analyses and observational studies.
Setting: A review of the current literature available in the MEDLINE database and Cochrane Library from 1949 to January 2020, favoring meta-analyses and clinical practice guidelines.
Synopsis: A total of 92 publications were included in this review, which found history, physical exam, and functional capacity to be the best assessments of cardiac risk and should guide further preoperative management. Cardiovascular testing is rarely indicated except in those with clinical signs and symptoms of active cardiac conditions or with poor functional status undergoing high-risk surgery. Cardiac consultation should be considered for those with prior stents; high-risk conditions, including acute coronary syndrome, severe valvular disease, or active heart failure, among other conditions; or high-risk findings on cardiovascular testing. Preoperative medications should be individualized to patient-specific conditions. This study is limited by current available evidence and expert opinion, and the systematic approach suggested here has not been prospectively tested.
Bottom line: Preoperative risk assessment and management should be largely based on individualized history, physical exam, and functional status. Cardiovascular work-up should be pursued only if it would influence surgical decision-making and perioperative care.
Citation: Smilowitz NR, Berger JS. Perioperative cardiovascular risk assessment and management for noncardiac surgery: A review. JAMA. 2020 Jul 21;324:279-90. doi:
Dr. Young is a hospitalist at Northwestern Memorial Hospital and instructor of medicine, Feinberg School of Medicine, both in Chicago.
Background: There are extensive publications regarding preoperative risk assessment and optimization of risk management. This article is a review of current aggregate data from various meta-analyses and observational studies. It explores a systematic approach to preoperative risk assessment.
Study design: Literature review of meta-analyses and observational studies.
Setting: A review of the current literature available in the MEDLINE database and Cochrane Library from 1949 to January 2020, favoring meta-analyses and clinical practice guidelines.
Synopsis: A total of 92 publications were included in this review, which found history, physical exam, and functional capacity to be the best assessments of cardiac risk and should guide further preoperative management. Cardiovascular testing is rarely indicated except in those with clinical signs and symptoms of active cardiac conditions or with poor functional status undergoing high-risk surgery. Cardiac consultation should be considered for those with prior stents; high-risk conditions, including acute coronary syndrome, severe valvular disease, or active heart failure, among other conditions; or high-risk findings on cardiovascular testing. Preoperative medications should be individualized to patient-specific conditions. This study is limited by current available evidence and expert opinion, and the systematic approach suggested here has not been prospectively tested.
Bottom line: Preoperative risk assessment and management should be largely based on individualized history, physical exam, and functional status. Cardiovascular work-up should be pursued only if it would influence surgical decision-making and perioperative care.
Citation: Smilowitz NR, Berger JS. Perioperative cardiovascular risk assessment and management for noncardiac surgery: A review. JAMA. 2020 Jul 21;324:279-90. doi:
Dr. Young is a hospitalist at Northwestern Memorial Hospital and instructor of medicine, Feinberg School of Medicine, both in Chicago.
Background: There are extensive publications regarding preoperative risk assessment and optimization of risk management. This article is a review of current aggregate data from various meta-analyses and observational studies. It explores a systematic approach to preoperative risk assessment.
Study design: Literature review of meta-analyses and observational studies.
Setting: A review of the current literature available in the MEDLINE database and Cochrane Library from 1949 to January 2020, favoring meta-analyses and clinical practice guidelines.
Synopsis: A total of 92 publications were included in this review, which found history, physical exam, and functional capacity to be the best assessments of cardiac risk and should guide further preoperative management. Cardiovascular testing is rarely indicated except in those with clinical signs and symptoms of active cardiac conditions or with poor functional status undergoing high-risk surgery. Cardiac consultation should be considered for those with prior stents; high-risk conditions, including acute coronary syndrome, severe valvular disease, or active heart failure, among other conditions; or high-risk findings on cardiovascular testing. Preoperative medications should be individualized to patient-specific conditions. This study is limited by current available evidence and expert opinion, and the systematic approach suggested here has not been prospectively tested.
Bottom line: Preoperative risk assessment and management should be largely based on individualized history, physical exam, and functional status. Cardiovascular work-up should be pursued only if it would influence surgical decision-making and perioperative care.
Citation: Smilowitz NR, Berger JS. Perioperative cardiovascular risk assessment and management for noncardiac surgery: A review. JAMA. 2020 Jul 21;324:279-90. doi:
Dr. Young is a hospitalist at Northwestern Memorial Hospital and instructor of medicine, Feinberg School of Medicine, both in Chicago.
Merck’s COVID-19 pill may be less effective than first hoped
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.
According to an analysis by scientists at the Food and Drug Administration, the experimental pill cut the risk of hospitalization or death from COVID-19 by about 30%, compared to a placebo, and the pill showed no benefit for people with antibodies against COVID-19 from prior infection.
The updated analysis showed 48 hospitalizations or deaths among study participants who were randomly assigned to take the antiviral drug, compared to 68 among those who took a placebo.
Those results come from the full set of 1,433 patients who were randomized in the clinical trial, which just became available last week.
Initial results from the first 775 patients enrolled in the clinical trial, which were issued in a company news release in October, had said the drug cut the risk of hospitalization or death for patients at high risk of severe disease by about 50%.
Merck has been producing millions of doses of molnupiravir, which is the first antiviral pill to treat COVID-19 infections. The United Kingdom’s drug regulator authorized use of the medication in early November. The company said it expected to distribute the medication globally by the end of 2021.
In October, two Indian drug companies halted late-stage clinical trials of a generic version of molnupiravir after the studies failed to find any benefit to patients with moderate COVID-19. Trials in patients with milder symptoms are still ongoing.
On Nov. 27, the New England Journal of Medicine postponed its planned early release of the molnupiravir study results, citing “new information.”
The medication is designed to be given as four pills taken every 12 hours for 5 days. It’s most effective when taken within the first few days of new symptoms, something that requires convenient and affordable testing.
The new results seem to put molnupiravir far below the effectiveness of existing treatments.
The infused monoclonal antibody cocktail REGEN-COV, which the FDA has already authorized for emergency use, is about 85% effective at preventing hospitalization or death in patients who are at risk for severe COVID-19 outcomes, and it appears to be just as effective in people who already have antibodies against COVID-19, which is why it is being given to both vaccinated and unvaccinated patients, the FDA said.
In early November, Pfizer said its experimental antiviral pill Paxlovid cut the risk of hospitalization or death by 89%.
In briefing documents posted ahead of an advisory committee meeting Nov. 30, the FDA highlights other potential safety issues with the Merck drug, which works by causing the virus to make mistakes as it copies itself, eventually causing the virus to mutate itself to death.
The agency has asked the advisory committee to weigh in on the right patient population for the drug: Should pregnant women get it? Could the drug harm a developing fetus?
Should vaccinated people with breakthrough infections get it? Would it work for them? People with reduced immune function are more likely to get a breakthrough infection. They’re also more likely to shed virus for a longer period of time, making them perfect incubators for variants. What could happen if we give this type of patient a drug that increases mutations?
And what about mutations caused by the medication? Could they increase the potential for more variants? The agency concluded the risk of this happening was low.
In animal studies, the drug impacted bone formation. For this reason, the agency has agreed with the drug company that molnupiravir should not be given to anyone under the age of 18.
Aside from these concerns, the FDA says there were no major safety issues among people who took part in the clinical trial, though they acknowledge that number is small.
A version of this article first appeared on WebMD.com.