User login
Cutaneous lesions? Consider C. diphtheriae in those with foreign travel
ATLANTA – Seven cases of imported Corynebacterium diphtheriae in Minnesota highlight the importance of maintaining suspicion that cutaneous lesions in individuals with recent travel to endemic countries might be associated with C. diphtheriae infection.
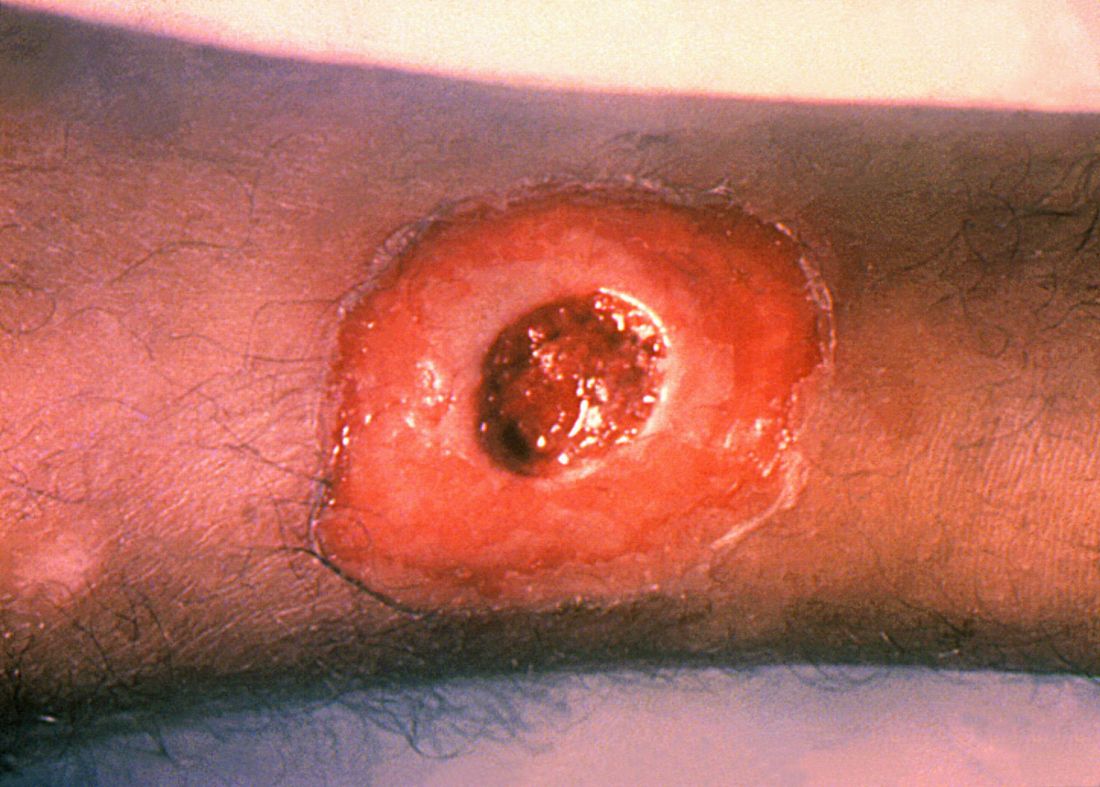
The cases also underscore the importance of referring C. diphtheriae isolates to state health departments for confirmatory testing, Jayne Griffith, of the Minnesota Department of Health, and her colleagues reported in a poster at the International Conference on Emerging Infectious Diseases.
“C. diphtheriae infections was not clinically suspected in any of these case-patients. All infections were initially identified solely by [matrix-assisted laser desorption/ionization time-of-flight spectrometry] testing performed at the clinical institutions,” the investigators wrote. “Confirmation and further toxigenicity testing allowed for prompt case investigation and public health response, preventing disease spread.”
Infections caused by toxigenic C. diphtheriae are rare in the United States because of widespread vaccination, but remain endemic in countries with suboptimal vaccine coverage. For this reason, infection is a concern for unvaccinated individuals traveling to diphtheria-endemic countries as well as for those who have contact with people from these areas. The investigators noted that “infections are primarily respiratory or cutaneous; respiratory infections can be life-threatening and cutaneous wounds may serve as a reservoir from which bacteria can be transmitted to susceptible contacts.”
The Minnesota cases involved patients who presented with cutaneous ulcers between 2014 and 2017. The Minnesota Department of Health confirmed C. diphtheriae status by culture after the initial identification at private institutions or providers using matrix-assisted laser desorption/ionization time-of-flight spectrometry. Isolates were sent to the Centers for Disease Control and Prevention Pertussis and Diphtheria Laboratory for biotyping and confirmation of toxigenicity.
The CDC confirmed that isolates from two patients were toxigenic C. diphtheriae biotype mitis. The remaining cases were nontoxigenic diphtheria, including C. diphtheriae mitis (three case-patients, including one who also had Staphylococcus aureus, and another who also had methicillin-resistant S. aureus) and two case-patients with C. diphtheriae belfanti.
The patients with toxigenic infection included a 35-year-old woman who developed an abdominal boil after spending months in Somalia, and a 48-year-old man who cut his leg while in Ethiopia.
The patients with nontoxigenic infection included four foreign-born individuals ranging in age from 7 to 66 years and one 24-year-old man from the United States. One of the foreign-born individuals had immigrated from a diphtheria-endemic country 3 months prior to his diagnosis, but none of the remaining four had traveled outside the United States in the 6 months prior to infection onset. One, however, lived in a home with family members who traveled frequently to eastern Africa. The vaccination status of these patients was unknown.
Contact tracing was conducted and prophylactic antibiotics were recommended as appropriate. Vaccination was recommended when a case-patient and/or contact was inadequately immunized.
Both patients with toxigenic infection experienced wound healing after appropriate antibiotic therapy.
Ms. Griffith reported having no disclosures.
ATLANTA – Seven cases of imported Corynebacterium diphtheriae in Minnesota highlight the importance of maintaining suspicion that cutaneous lesions in individuals with recent travel to endemic countries might be associated with C. diphtheriae infection.
The cases also underscore the importance of referring C. diphtheriae isolates to state health departments for confirmatory testing, Jayne Griffith, of the Minnesota Department of Health, and her colleagues reported in a poster at the International Conference on Emerging Infectious Diseases.
“C. diphtheriae infections was not clinically suspected in any of these case-patients. All infections were initially identified solely by [matrix-assisted laser desorption/ionization time-of-flight spectrometry] testing performed at the clinical institutions,” the investigators wrote. “Confirmation and further toxigenicity testing allowed for prompt case investigation and public health response, preventing disease spread.”
Infections caused by toxigenic C. diphtheriae are rare in the United States because of widespread vaccination, but remain endemic in countries with suboptimal vaccine coverage. For this reason, infection is a concern for unvaccinated individuals traveling to diphtheria-endemic countries as well as for those who have contact with people from these areas. The investigators noted that “infections are primarily respiratory or cutaneous; respiratory infections can be life-threatening and cutaneous wounds may serve as a reservoir from which bacteria can be transmitted to susceptible contacts.”
The Minnesota cases involved patients who presented with cutaneous ulcers between 2014 and 2017. The Minnesota Department of Health confirmed C. diphtheriae status by culture after the initial identification at private institutions or providers using matrix-assisted laser desorption/ionization time-of-flight spectrometry. Isolates were sent to the Centers for Disease Control and Prevention Pertussis and Diphtheria Laboratory for biotyping and confirmation of toxigenicity.
The CDC confirmed that isolates from two patients were toxigenic C. diphtheriae biotype mitis. The remaining cases were nontoxigenic diphtheria, including C. diphtheriae mitis (three case-patients, including one who also had Staphylococcus aureus, and another who also had methicillin-resistant S. aureus) and two case-patients with C. diphtheriae belfanti.
The patients with toxigenic infection included a 35-year-old woman who developed an abdominal boil after spending months in Somalia, and a 48-year-old man who cut his leg while in Ethiopia.
The patients with nontoxigenic infection included four foreign-born individuals ranging in age from 7 to 66 years and one 24-year-old man from the United States. One of the foreign-born individuals had immigrated from a diphtheria-endemic country 3 months prior to his diagnosis, but none of the remaining four had traveled outside the United States in the 6 months prior to infection onset. One, however, lived in a home with family members who traveled frequently to eastern Africa. The vaccination status of these patients was unknown.
Contact tracing was conducted and prophylactic antibiotics were recommended as appropriate. Vaccination was recommended when a case-patient and/or contact was inadequately immunized.
Both patients with toxigenic infection experienced wound healing after appropriate antibiotic therapy.
Ms. Griffith reported having no disclosures.
ATLANTA – Seven cases of imported Corynebacterium diphtheriae in Minnesota highlight the importance of maintaining suspicion that cutaneous lesions in individuals with recent travel to endemic countries might be associated with C. diphtheriae infection.
The cases also underscore the importance of referring C. diphtheriae isolates to state health departments for confirmatory testing, Jayne Griffith, of the Minnesota Department of Health, and her colleagues reported in a poster at the International Conference on Emerging Infectious Diseases.
“C. diphtheriae infections was not clinically suspected in any of these case-patients. All infections were initially identified solely by [matrix-assisted laser desorption/ionization time-of-flight spectrometry] testing performed at the clinical institutions,” the investigators wrote. “Confirmation and further toxigenicity testing allowed for prompt case investigation and public health response, preventing disease spread.”
Infections caused by toxigenic C. diphtheriae are rare in the United States because of widespread vaccination, but remain endemic in countries with suboptimal vaccine coverage. For this reason, infection is a concern for unvaccinated individuals traveling to diphtheria-endemic countries as well as for those who have contact with people from these areas. The investigators noted that “infections are primarily respiratory or cutaneous; respiratory infections can be life-threatening and cutaneous wounds may serve as a reservoir from which bacteria can be transmitted to susceptible contacts.”
The Minnesota cases involved patients who presented with cutaneous ulcers between 2014 and 2017. The Minnesota Department of Health confirmed C. diphtheriae status by culture after the initial identification at private institutions or providers using matrix-assisted laser desorption/ionization time-of-flight spectrometry. Isolates were sent to the Centers for Disease Control and Prevention Pertussis and Diphtheria Laboratory for biotyping and confirmation of toxigenicity.
The CDC confirmed that isolates from two patients were toxigenic C. diphtheriae biotype mitis. The remaining cases were nontoxigenic diphtheria, including C. diphtheriae mitis (three case-patients, including one who also had Staphylococcus aureus, and another who also had methicillin-resistant S. aureus) and two case-patients with C. diphtheriae belfanti.
The patients with toxigenic infection included a 35-year-old woman who developed an abdominal boil after spending months in Somalia, and a 48-year-old man who cut his leg while in Ethiopia.
The patients with nontoxigenic infection included four foreign-born individuals ranging in age from 7 to 66 years and one 24-year-old man from the United States. One of the foreign-born individuals had immigrated from a diphtheria-endemic country 3 months prior to his diagnosis, but none of the remaining four had traveled outside the United States in the 6 months prior to infection onset. One, however, lived in a home with family members who traveled frequently to eastern Africa. The vaccination status of these patients was unknown.
Contact tracing was conducted and prophylactic antibiotics were recommended as appropriate. Vaccination was recommended when a case-patient and/or contact was inadequately immunized.
Both patients with toxigenic infection experienced wound healing after appropriate antibiotic therapy.
Ms. Griffith reported having no disclosures.
REPORTING FROM ICEID 2018
Key clinical point: Corynebacterium diphtheriae should be considered in individuals who present with cutaneous lesions after traveling to diphtheria-endemic countries.
Major finding: Refer suspect C. diphtheriae isolates to state health departments.
Study details: A review of seven C. diphtheriae cases.
Disclosures: Ms. Griffith reported having no disclosures.
CDC supports Ebola response in DRC
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.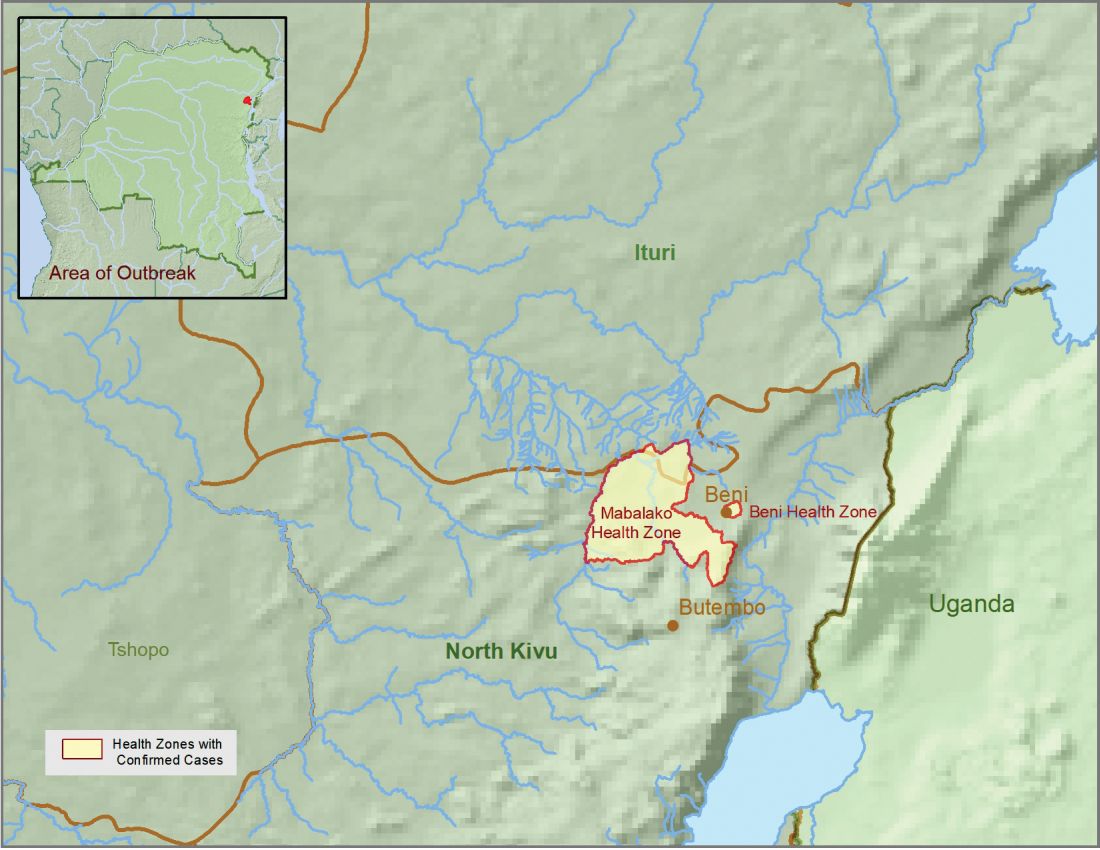
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
FDA: Krintafel approved as ‘radical cure’ for preventing malaria relapse
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.
The Food and Drug Administration has approved, under priority review, single-dose tafenoquine (to be marketed as Krintafel) for the prevention of relapse in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute Plasmodium vivax infection, according to an announcement by research partners GSK and the nonprofit Medicines for Malaria Venture.
Clinical efficacy and safety of the 300-mg single-dose tablet was provided by three randomized, double-blind studies: DETECTIVE Part 1 and Part 2 (TAF112582) and GATHER (TAF116564). The results of the two phase III studies were announced in June 2017.
Tafenoquine is referred to as a “radical cure,” because it targets the dormant liver forms of P. vivax and is coadministered with currently available antimalarials such as chloroquine or artemisinin-based combination therapies.
“The world has waited decades for a new medicine to counter P. vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance,” David Reddy, MD, CEO of MMV stated.
The FDA approval letter and the Krintafel label information are available online.
Female survivor transmits Ebola virus 1 year after outbreak
Molecular analysis showed that a Liberian woman who survived Ebola virus disease in 2014 had viral persistence or recurrent disease and transmitted the virus to other family members a year later, according to a study published online in The Lancet Infectious Diseases.

Although the original 2014-2015 Ebola virus disease epidemic in West Africa had been contained, subsequent clusters of infection continued to occur in the region, according to researchers. A particular cluster in Liberia in November 2015 was identified after a 15-year-old boy in Monrovia tested positive.
Based on serology and epidemiological and genomic data, the researchers concluded that this cluster was caused by a woman who survived Ebola virus disease in 2014 and transmitted the virus to three family members a year later.
Ebola transmission from persistently infected male survivors is well documented, but this is the first confirmed evidence for Ebola transmission from a persistently infected female survivor, according to Emily Kainne Dokubo, MD, of the Centers for Disease Control and Prevention and her colleagues.
“The findings from this and recent Ebola virus disease clusters highlight the risk of Ebola virus disease flare-ups even after an outbreak is declared over. Risk assessment and focused prevention efforts are needed for Ebola survivors and their close contacts,” Dr. Dokubo and her colleagues concluded.
The study was funded by the CDC, Defense Threat Reduction Agency, and WHO.
SOURCE: Dokubo EK et al. The Lancet Infectious Diseases, July 23, 2018.
Molecular analysis showed that a Liberian woman who survived Ebola virus disease in 2014 had viral persistence or recurrent disease and transmitted the virus to other family members a year later, according to a study published online in The Lancet Infectious Diseases.
Although the original 2014-2015 Ebola virus disease epidemic in West Africa had been contained, subsequent clusters of infection continued to occur in the region, according to researchers. A particular cluster in Liberia in November 2015 was identified after a 15-year-old boy in Monrovia tested positive.
Based on serology and epidemiological and genomic data, the researchers concluded that this cluster was caused by a woman who survived Ebola virus disease in 2014 and transmitted the virus to three family members a year later.
Ebola transmission from persistently infected male survivors is well documented, but this is the first confirmed evidence for Ebola transmission from a persistently infected female survivor, according to Emily Kainne Dokubo, MD, of the Centers for Disease Control and Prevention and her colleagues.
“The findings from this and recent Ebola virus disease clusters highlight the risk of Ebola virus disease flare-ups even after an outbreak is declared over. Risk assessment and focused prevention efforts are needed for Ebola survivors and their close contacts,” Dr. Dokubo and her colleagues concluded.
The study was funded by the CDC, Defense Threat Reduction Agency, and WHO.
SOURCE: Dokubo EK et al. The Lancet Infectious Diseases, July 23, 2018.
Molecular analysis showed that a Liberian woman who survived Ebola virus disease in 2014 had viral persistence or recurrent disease and transmitted the virus to other family members a year later, according to a study published online in The Lancet Infectious Diseases.
Although the original 2014-2015 Ebola virus disease epidemic in West Africa had been contained, subsequent clusters of infection continued to occur in the region, according to researchers. A particular cluster in Liberia in November 2015 was identified after a 15-year-old boy in Monrovia tested positive.
Based on serology and epidemiological and genomic data, the researchers concluded that this cluster was caused by a woman who survived Ebola virus disease in 2014 and transmitted the virus to three family members a year later.
Ebola transmission from persistently infected male survivors is well documented, but this is the first confirmed evidence for Ebola transmission from a persistently infected female survivor, according to Emily Kainne Dokubo, MD, of the Centers for Disease Control and Prevention and her colleagues.
“The findings from this and recent Ebola virus disease clusters highlight the risk of Ebola virus disease flare-ups even after an outbreak is declared over. Risk assessment and focused prevention efforts are needed for Ebola survivors and their close contacts,” Dr. Dokubo and her colleagues concluded.
The study was funded by the CDC, Defense Threat Reduction Agency, and WHO.
SOURCE: Dokubo EK et al. The Lancet Infectious Diseases, July 23, 2018.
FROM THE LANCET INFECTIOUS DISEASES
Mosaic HIV-1 vaccine stimulates broad antigenicity, enters phase 2b human efficacy trial
A mosaic adenovirus human immunodeficiency virus-1 (HIV-1) vaccine induced robust immune responses in humans and rhesus monkeys, and significantly protected the monkeys against repetitive simian/HIV (SHIV) mosaic challenges in rhesus monkeys. This vaccine concept is currently being evaluated in a phase 2b clinical efficacy study in sub-Saharan Africa, according to a report published online in The Lancet.
Because a mosaic combination of antigens can induce an immunogenic response to a broad geographic range of viral subtypes, such a vaccine can offer the theoretical possibility of developing a global HIV-1 vaccine, according to Dan H. Barouch, MD, of Harvard Medical School, Boston, and his colleagues.
The researchers conducted a multicenter, randomized, double-blind, placebo-controlled, phase 1/2a trial (APPROACH) with 393 healthy, HIV-1-uninfected participants (aged 18-50 years) who were considered at low risk for HIV-1 infection and were recruited from 12 clinics in East Africa, South Africa, Thailand, and the United States. Participants were primed at weeks 0 and 12 with Ad26.Mos.HIV (5 × 1010 viral particles per 0.5 mL) expressing mosaic HIV-1 envelope (Env)/Gag/Pol antigens and given boosters at weeks 24 and 48 with Ad26.Mos.HIV or modified vaccinia Ankara (MVA; 108 plaque-forming units per 0.5 mL) vectors with or without adjuvanted Env gp140 protein. The placebo group received 0.9% saline.
Eligible participants were randomly assigned to one of eight study groups: Ad26/Ad26 plus high-dose gp140; Ad26/Ad26 plus low-dose gp140; Ad26/Ad26; Ad26/MVA plus high-dose gp140; Ad26/MVA plus low-dose gp140; Ad26/MVA; Ad26/high-dose gp140; and placebo. “Overall, no substantial differences in safety or tolerability of any of the seven active vaccine groups were observed,” according to Dr. Barouch and his colleagues.
In addition, the researchers vaccinated 72 Indian-origin rhesus monkeys (Macaca mulatta) using a study design that was similar to that of the human APPROACH clinical study.
The mosaic HIV-1 vaccine “induced robust humoral and cellular immune responses in both humans and rhesus monkeys,” which were similar in both species in “magnitude, durability, and phenotype,” the investigators reported. In addition, the vaccine “provided 67% protection against acquisition of six intrarectal SHIV challenges in rhesus monkeys.”
Based on these results, the mosaic Ad26/Ad26 plus gp140 HIV-1 vaccine sufficiently met “pre-established safety and immunogenicity criteria to advance into a phase 2b clinical efficacy study in sub-Saharan Africa, which is now underway (NCT03060629),” according to the authors.
“Previous HIV-1 vaccine candidates have typically been limited to specific regions of the world. Optimized mosaic antigens offer the theoretical possibility of developing a global HIV-1 vaccine,” Dr. Barouch and his colleagues concluded.
This study was funded in part by Janssen Vaccines & Prevention BV and the National Institutes of Health. Dr. Barouch has received grant funding from Janssen Vaccines & Prevention BV and is a co-inventor on HIV-1 vaccine antigen patents that have been licensed to Janssen Vaccines & Prevention.
SOURCE: Barouch DH et al., The Lancet. 2018 July 6. doi: 10.1016/S0140-6736(18)31364-3.
A mosaic adenovirus human immunodeficiency virus-1 (HIV-1) vaccine induced robust immune responses in humans and rhesus monkeys, and significantly protected the monkeys against repetitive simian/HIV (SHIV) mosaic challenges in rhesus monkeys. This vaccine concept is currently being evaluated in a phase 2b clinical efficacy study in sub-Saharan Africa, according to a report published online in The Lancet.
Because a mosaic combination of antigens can induce an immunogenic response to a broad geographic range of viral subtypes, such a vaccine can offer the theoretical possibility of developing a global HIV-1 vaccine, according to Dan H. Barouch, MD, of Harvard Medical School, Boston, and his colleagues.
The researchers conducted a multicenter, randomized, double-blind, placebo-controlled, phase 1/2a trial (APPROACH) with 393 healthy, HIV-1-uninfected participants (aged 18-50 years) who were considered at low risk for HIV-1 infection and were recruited from 12 clinics in East Africa, South Africa, Thailand, and the United States. Participants were primed at weeks 0 and 12 with Ad26.Mos.HIV (5 × 1010 viral particles per 0.5 mL) expressing mosaic HIV-1 envelope (Env)/Gag/Pol antigens and given boosters at weeks 24 and 48 with Ad26.Mos.HIV or modified vaccinia Ankara (MVA; 108 plaque-forming units per 0.5 mL) vectors with or without adjuvanted Env gp140 protein. The placebo group received 0.9% saline.
Eligible participants were randomly assigned to one of eight study groups: Ad26/Ad26 plus high-dose gp140; Ad26/Ad26 plus low-dose gp140; Ad26/Ad26; Ad26/MVA plus high-dose gp140; Ad26/MVA plus low-dose gp140; Ad26/MVA; Ad26/high-dose gp140; and placebo. “Overall, no substantial differences in safety or tolerability of any of the seven active vaccine groups were observed,” according to Dr. Barouch and his colleagues.
In addition, the researchers vaccinated 72 Indian-origin rhesus monkeys (Macaca mulatta) using a study design that was similar to that of the human APPROACH clinical study.
The mosaic HIV-1 vaccine “induced robust humoral and cellular immune responses in both humans and rhesus monkeys,” which were similar in both species in “magnitude, durability, and phenotype,” the investigators reported. In addition, the vaccine “provided 67% protection against acquisition of six intrarectal SHIV challenges in rhesus monkeys.”
Based on these results, the mosaic Ad26/Ad26 plus gp140 HIV-1 vaccine sufficiently met “pre-established safety and immunogenicity criteria to advance into a phase 2b clinical efficacy study in sub-Saharan Africa, which is now underway (NCT03060629),” according to the authors.
“Previous HIV-1 vaccine candidates have typically been limited to specific regions of the world. Optimized mosaic antigens offer the theoretical possibility of developing a global HIV-1 vaccine,” Dr. Barouch and his colleagues concluded.
This study was funded in part by Janssen Vaccines & Prevention BV and the National Institutes of Health. Dr. Barouch has received grant funding from Janssen Vaccines & Prevention BV and is a co-inventor on HIV-1 vaccine antigen patents that have been licensed to Janssen Vaccines & Prevention.
SOURCE: Barouch DH et al., The Lancet. 2018 July 6. doi: 10.1016/S0140-6736(18)31364-3.
A mosaic adenovirus human immunodeficiency virus-1 (HIV-1) vaccine induced robust immune responses in humans and rhesus monkeys, and significantly protected the monkeys against repetitive simian/HIV (SHIV) mosaic challenges in rhesus monkeys. This vaccine concept is currently being evaluated in a phase 2b clinical efficacy study in sub-Saharan Africa, according to a report published online in The Lancet.
Because a mosaic combination of antigens can induce an immunogenic response to a broad geographic range of viral subtypes, such a vaccine can offer the theoretical possibility of developing a global HIV-1 vaccine, according to Dan H. Barouch, MD, of Harvard Medical School, Boston, and his colleagues.
The researchers conducted a multicenter, randomized, double-blind, placebo-controlled, phase 1/2a trial (APPROACH) with 393 healthy, HIV-1-uninfected participants (aged 18-50 years) who were considered at low risk for HIV-1 infection and were recruited from 12 clinics in East Africa, South Africa, Thailand, and the United States. Participants were primed at weeks 0 and 12 with Ad26.Mos.HIV (5 × 1010 viral particles per 0.5 mL) expressing mosaic HIV-1 envelope (Env)/Gag/Pol antigens and given boosters at weeks 24 and 48 with Ad26.Mos.HIV or modified vaccinia Ankara (MVA; 108 plaque-forming units per 0.5 mL) vectors with or without adjuvanted Env gp140 protein. The placebo group received 0.9% saline.
Eligible participants were randomly assigned to one of eight study groups: Ad26/Ad26 plus high-dose gp140; Ad26/Ad26 plus low-dose gp140; Ad26/Ad26; Ad26/MVA plus high-dose gp140; Ad26/MVA plus low-dose gp140; Ad26/MVA; Ad26/high-dose gp140; and placebo. “Overall, no substantial differences in safety or tolerability of any of the seven active vaccine groups were observed,” according to Dr. Barouch and his colleagues.
In addition, the researchers vaccinated 72 Indian-origin rhesus monkeys (Macaca mulatta) using a study design that was similar to that of the human APPROACH clinical study.
The mosaic HIV-1 vaccine “induced robust humoral and cellular immune responses in both humans and rhesus monkeys,” which were similar in both species in “magnitude, durability, and phenotype,” the investigators reported. In addition, the vaccine “provided 67% protection against acquisition of six intrarectal SHIV challenges in rhesus monkeys.”
Based on these results, the mosaic Ad26/Ad26 plus gp140 HIV-1 vaccine sufficiently met “pre-established safety and immunogenicity criteria to advance into a phase 2b clinical efficacy study in sub-Saharan Africa, which is now underway (NCT03060629),” according to the authors.
“Previous HIV-1 vaccine candidates have typically been limited to specific regions of the world. Optimized mosaic antigens offer the theoretical possibility of developing a global HIV-1 vaccine,” Dr. Barouch and his colleagues concluded.
This study was funded in part by Janssen Vaccines & Prevention BV and the National Institutes of Health. Dr. Barouch has received grant funding from Janssen Vaccines & Prevention BV and is a co-inventor on HIV-1 vaccine antigen patents that have been licensed to Janssen Vaccines & Prevention.
SOURCE: Barouch DH et al., The Lancet. 2018 July 6. doi: 10.1016/S0140-6736(18)31364-3.
FROM THE LANCET
Primary efficacy not met by new M. tuberculosis vaccine strategies
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.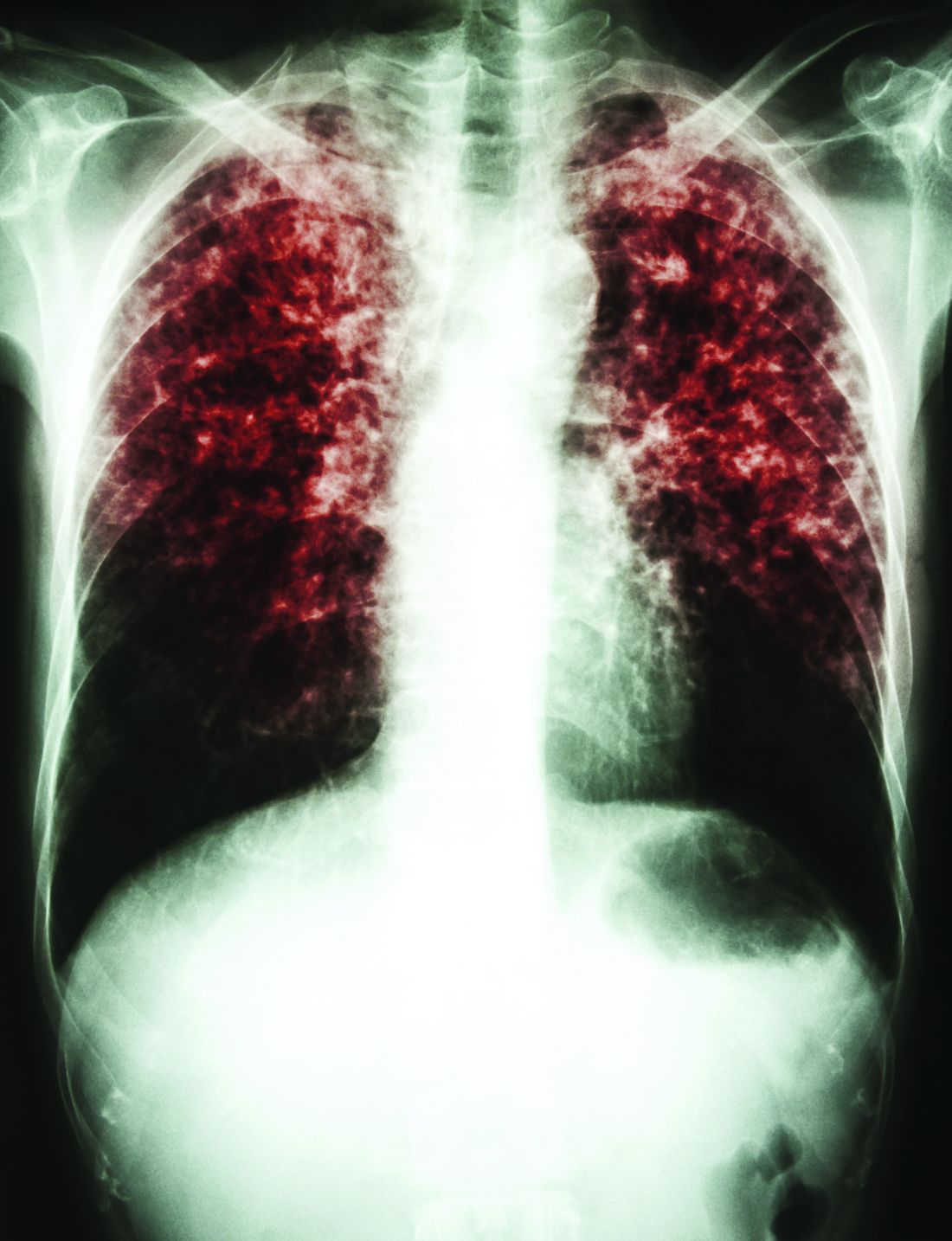
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint; however, both vaccines were immunogenic.
Major finding: For the secondary endpoint of sustained QuantiFERON-TB Gold In-Tube Assay (QFT) conversion, efficacy was 45.4% (P = .03) for BCG revaccination and 34.2% (P = .11) for H4:IC31, a candidate subunit vaccine.
Study details: A phase 2, randomized, placebo-controlled trial including 990 adolescents in South Africa who had received BCG vaccine in infancy.
Disclosures: The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
Source: Nemes E et al. N Engl J Med. 2018;379:138-49.
Cost is high for Japanese encephalitis vaccinations
Vaccination costs surrounding Japanese encephalitis is high, according to an economic analysis presented at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The analytic horizon for this research was 6 years, but productivity losses were evaluated over average life expectancy.
The researchers analyzed their data from two analytic perspectives: societal and traveler perspective. Factors analyzed included vaccine per dose, vaccine administration cost, and vaccine adverse events costs per vaccine.
The researchers assessed the risk to travelers based upon disease incidence among different groups. The highest-risk travelers (Group 1) – those who planned to spend a month or more in JE-endemic areas – had an incidence rate of 0.53/1 million travelers. Group 2 travelers – those who planned to stay less than a month and more than a fifth of their time outdoors – had an incidence rate of 0.25/1 million. Group 3 travelers, at lowest risk, had the lowest incidence rate at 0.04/1 million.
The calculated societal perspective cost per outcome averted was quite high for each risk group. For Group 1, the cost was $596 million per case averted. This cost rose to $1.3 billion for each case of long-term sequelae averted, and rose even higher to avert death, to $1.8 billion per death averted. These costs nearly doubled for Group 2, and in Group 3, the cost ballooned to $7.9 billion to avert one case of JE, $17 billion per prevention of long-term sequelae, and $23 billion per death averted.
The individual costs for JE vaccination are $292 per dose, with an administration fee of $46. Short-term treatment of JE costs nearly $30,000, and long-term treatment of JE also comes with a large bill of $8,437.
These costs are not simply monetary but are also felt in lost economic productivity. The cost of complete short-term recovery is nearly $60,000. Over an individual’s lifetime, this number rose to more than $1.5 million based on total loss of productivity.
Of the 67% of patients who survive JE, 32% recover completely while 68% deal with long-term sequelae. Of those, 28% experience mild symptoms while the remaining 72% have severe sequelae.
JE vaccine effectiveness does not appear to be an issue, with a 0.91 proportion of neutralizing antibodies seen after 1 year. With booster doses, vaccine effectiveness is 0.96.
There are limitations to this study, such as results being affected by the uncertainty of JE incidence. “The single most important variable is incidence,” stated Dr. Meltzer.
Vaccination costs surrounding Japanese encephalitis is high, according to an economic analysis presented at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The analytic horizon for this research was 6 years, but productivity losses were evaluated over average life expectancy.
The researchers analyzed their data from two analytic perspectives: societal and traveler perspective. Factors analyzed included vaccine per dose, vaccine administration cost, and vaccine adverse events costs per vaccine.
The researchers assessed the risk to travelers based upon disease incidence among different groups. The highest-risk travelers (Group 1) – those who planned to spend a month or more in JE-endemic areas – had an incidence rate of 0.53/1 million travelers. Group 2 travelers – those who planned to stay less than a month and more than a fifth of their time outdoors – had an incidence rate of 0.25/1 million. Group 3 travelers, at lowest risk, had the lowest incidence rate at 0.04/1 million.
The calculated societal perspective cost per outcome averted was quite high for each risk group. For Group 1, the cost was $596 million per case averted. This cost rose to $1.3 billion for each case of long-term sequelae averted, and rose even higher to avert death, to $1.8 billion per death averted. These costs nearly doubled for Group 2, and in Group 3, the cost ballooned to $7.9 billion to avert one case of JE, $17 billion per prevention of long-term sequelae, and $23 billion per death averted.
The individual costs for JE vaccination are $292 per dose, with an administration fee of $46. Short-term treatment of JE costs nearly $30,000, and long-term treatment of JE also comes with a large bill of $8,437.
These costs are not simply monetary but are also felt in lost economic productivity. The cost of complete short-term recovery is nearly $60,000. Over an individual’s lifetime, this number rose to more than $1.5 million based on total loss of productivity.
Of the 67% of patients who survive JE, 32% recover completely while 68% deal with long-term sequelae. Of those, 28% experience mild symptoms while the remaining 72% have severe sequelae.
JE vaccine effectiveness does not appear to be an issue, with a 0.91 proportion of neutralizing antibodies seen after 1 year. With booster doses, vaccine effectiveness is 0.96.
There are limitations to this study, such as results being affected by the uncertainty of JE incidence. “The single most important variable is incidence,” stated Dr. Meltzer.
Vaccination costs surrounding Japanese encephalitis is high, according to an economic analysis presented at a meeting of the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices.
The analytic horizon for this research was 6 years, but productivity losses were evaluated over average life expectancy.
The researchers analyzed their data from two analytic perspectives: societal and traveler perspective. Factors analyzed included vaccine per dose, vaccine administration cost, and vaccine adverse events costs per vaccine.
The researchers assessed the risk to travelers based upon disease incidence among different groups. The highest-risk travelers (Group 1) – those who planned to spend a month or more in JE-endemic areas – had an incidence rate of 0.53/1 million travelers. Group 2 travelers – those who planned to stay less than a month and more than a fifth of their time outdoors – had an incidence rate of 0.25/1 million. Group 3 travelers, at lowest risk, had the lowest incidence rate at 0.04/1 million.
The calculated societal perspective cost per outcome averted was quite high for each risk group. For Group 1, the cost was $596 million per case averted. This cost rose to $1.3 billion for each case of long-term sequelae averted, and rose even higher to avert death, to $1.8 billion per death averted. These costs nearly doubled for Group 2, and in Group 3, the cost ballooned to $7.9 billion to avert one case of JE, $17 billion per prevention of long-term sequelae, and $23 billion per death averted.
The individual costs for JE vaccination are $292 per dose, with an administration fee of $46. Short-term treatment of JE costs nearly $30,000, and long-term treatment of JE also comes with a large bill of $8,437.
These costs are not simply monetary but are also felt in lost economic productivity. The cost of complete short-term recovery is nearly $60,000. Over an individual’s lifetime, this number rose to more than $1.5 million based on total loss of productivity.
Of the 67% of patients who survive JE, 32% recover completely while 68% deal with long-term sequelae. Of those, 28% experience mild symptoms while the remaining 72% have severe sequelae.
JE vaccine effectiveness does not appear to be an issue, with a 0.91 proportion of neutralizing antibodies seen after 1 year. With booster doses, vaccine effectiveness is 0.96.
There are limitations to this study, such as results being affected by the uncertainty of JE incidence. “The single most important variable is incidence,” stated Dr. Meltzer.
REPORTING FROM AN ACIP MEETING
Oral tecovirimat for smallpox shows efficacy in animals, safety in humans
Tecovirimat, an oral antiviral agent, is being advanced as a treatment for smallpox based on recently reported studies showing efficacy data in animals and safety and pharmacokinetic data in humans.
“The aggregation of the results from these multiple studies involving animals and humans supports tecovirimat as potential smallpox antiviral drug,” Douglas W. Grosenbach, PhD, of SIGA Technologies and his coauthors reported in the New England Journal of Medicine.
Tecovirimat inhibits p37, a protein that is present in all orthopoxviruses, to prevent “formation and egress of enveloped virions, which are essential for virulence,” Dr. Grosenbach and his coauthors wrote in the report.
Smallpox, caused by the variola virus, was eradicated in 1980. However, the disease remains concerning because of the potential for intentional release of variola virus in an act of bioterrorism or biowarfare, according to the authors.
“A single case of smallpox anywhere in the world would be a global health emergency,” Dr. Grosenbach and his colleagues wrote.
Available vaccines are not used because of the risk of side effects and would not be used except in the case of known or suspected variola virus exposure, they added.
Since it would be unethical to intentionally expose humans to variola virus, tecovirimat is being developed in line with the Food and Drug Administration Animal Efficacy Rule, which gives the agency the authority to approve drugs based on animal data for diseases that have a low or nonexistent rate of natural occurrence.
The first treatment developed under the Animal Efficacy Rule, was raxibacumab, a biologic approved by the FDA in December 2012 for treatment of inhalation anthrax. Subsequently, several other agents for prophylaxis or treatment of anthrax and botulism have been developed based on the rule and approved.
Tecovirimat has demonstrated protective efficacy in pilot studies conducted in rabbits infected with rabbitpox virus and nonhuman primates infected with monkeypox virus, Dr. Grosenbach and his colleagues explained in the current report.
In nonhuman primate studies, doses of 3-10 mg/kilogram provided nearly full protection from death with a survival rate of approximately 95% versus 5% for placebo, along with reduced lesion counts and viral loads, according to investigators.
In a tecovirimat clinical trial, 452 volunteers were randomized to receive the antiviral agent twice daily at 600 mg or matching placebo for 14 days. Adverse events occurred in 37.3% of tecovirimat-treated and 33.3% of placebo-treated participants, with events of grade 3 or higher occurring in 1.1% of patients in both groups.
There was one death on the tecovirimat arm, which was caused by a pulmonary embolism in a participant who had a history of recurrent deep-vein thrombosis but was not receiving anticoagulant treatment, investigators said.
Dr. Grosenbach and colleagues also presented pharmacokinetic profiles and exposures for 48 volunteers in a fed state in their report in the New England Journal of Medicine, along with pharmacokinetic data from the animal studies.
The FDA has set a target date of Aug. 8, 2018, for final action on a new drug application submitted for oral tecovirimat for treatment of smallpox, SIGA Technologies said in a May 2018 news release.
Dr. Grosenbach and his coauthors reported a disclosure related to Biomedical Advanced Research and Development Authority (BARDA), grants from NIH during the conduct of the study, and personal fees from SIGA Technologies outside the submitted work.
SOURCE: Grosenbach DW et al. N Engl J Med. 2018 Jul 5;379(1):44-53.
Tecovirimat, an oral antiviral agent, is being advanced as a treatment for smallpox based on recently reported studies showing efficacy data in animals and safety and pharmacokinetic data in humans.
“The aggregation of the results from these multiple studies involving animals and humans supports tecovirimat as potential smallpox antiviral drug,” Douglas W. Grosenbach, PhD, of SIGA Technologies and his coauthors reported in the New England Journal of Medicine.
Tecovirimat inhibits p37, a protein that is present in all orthopoxviruses, to prevent “formation and egress of enveloped virions, which are essential for virulence,” Dr. Grosenbach and his coauthors wrote in the report.
Smallpox, caused by the variola virus, was eradicated in 1980. However, the disease remains concerning because of the potential for intentional release of variola virus in an act of bioterrorism or biowarfare, according to the authors.
“A single case of smallpox anywhere in the world would be a global health emergency,” Dr. Grosenbach and his colleagues wrote.
Available vaccines are not used because of the risk of side effects and would not be used except in the case of known or suspected variola virus exposure, they added.
Since it would be unethical to intentionally expose humans to variola virus, tecovirimat is being developed in line with the Food and Drug Administration Animal Efficacy Rule, which gives the agency the authority to approve drugs based on animal data for diseases that have a low or nonexistent rate of natural occurrence.
The first treatment developed under the Animal Efficacy Rule, was raxibacumab, a biologic approved by the FDA in December 2012 for treatment of inhalation anthrax. Subsequently, several other agents for prophylaxis or treatment of anthrax and botulism have been developed based on the rule and approved.
Tecovirimat has demonstrated protective efficacy in pilot studies conducted in rabbits infected with rabbitpox virus and nonhuman primates infected with monkeypox virus, Dr. Grosenbach and his colleagues explained in the current report.
In nonhuman primate studies, doses of 3-10 mg/kilogram provided nearly full protection from death with a survival rate of approximately 95% versus 5% for placebo, along with reduced lesion counts and viral loads, according to investigators.
In a tecovirimat clinical trial, 452 volunteers were randomized to receive the antiviral agent twice daily at 600 mg or matching placebo for 14 days. Adverse events occurred in 37.3% of tecovirimat-treated and 33.3% of placebo-treated participants, with events of grade 3 or higher occurring in 1.1% of patients in both groups.
There was one death on the tecovirimat arm, which was caused by a pulmonary embolism in a participant who had a history of recurrent deep-vein thrombosis but was not receiving anticoagulant treatment, investigators said.
Dr. Grosenbach and colleagues also presented pharmacokinetic profiles and exposures for 48 volunteers in a fed state in their report in the New England Journal of Medicine, along with pharmacokinetic data from the animal studies.
The FDA has set a target date of Aug. 8, 2018, for final action on a new drug application submitted for oral tecovirimat for treatment of smallpox, SIGA Technologies said in a May 2018 news release.
Dr. Grosenbach and his coauthors reported a disclosure related to Biomedical Advanced Research and Development Authority (BARDA), grants from NIH during the conduct of the study, and personal fees from SIGA Technologies outside the submitted work.
SOURCE: Grosenbach DW et al. N Engl J Med. 2018 Jul 5;379(1):44-53.
Tecovirimat, an oral antiviral agent, is being advanced as a treatment for smallpox based on recently reported studies showing efficacy data in animals and safety and pharmacokinetic data in humans.
“The aggregation of the results from these multiple studies involving animals and humans supports tecovirimat as potential smallpox antiviral drug,” Douglas W. Grosenbach, PhD, of SIGA Technologies and his coauthors reported in the New England Journal of Medicine.
Tecovirimat inhibits p37, a protein that is present in all orthopoxviruses, to prevent “formation and egress of enveloped virions, which are essential for virulence,” Dr. Grosenbach and his coauthors wrote in the report.
Smallpox, caused by the variola virus, was eradicated in 1980. However, the disease remains concerning because of the potential for intentional release of variola virus in an act of bioterrorism or biowarfare, according to the authors.
“A single case of smallpox anywhere in the world would be a global health emergency,” Dr. Grosenbach and his colleagues wrote.
Available vaccines are not used because of the risk of side effects and would not be used except in the case of known or suspected variola virus exposure, they added.
Since it would be unethical to intentionally expose humans to variola virus, tecovirimat is being developed in line with the Food and Drug Administration Animal Efficacy Rule, which gives the agency the authority to approve drugs based on animal data for diseases that have a low or nonexistent rate of natural occurrence.
The first treatment developed under the Animal Efficacy Rule, was raxibacumab, a biologic approved by the FDA in December 2012 for treatment of inhalation anthrax. Subsequently, several other agents for prophylaxis or treatment of anthrax and botulism have been developed based on the rule and approved.
Tecovirimat has demonstrated protective efficacy in pilot studies conducted in rabbits infected with rabbitpox virus and nonhuman primates infected with monkeypox virus, Dr. Grosenbach and his colleagues explained in the current report.
In nonhuman primate studies, doses of 3-10 mg/kilogram provided nearly full protection from death with a survival rate of approximately 95% versus 5% for placebo, along with reduced lesion counts and viral loads, according to investigators.
In a tecovirimat clinical trial, 452 volunteers were randomized to receive the antiviral agent twice daily at 600 mg or matching placebo for 14 days. Adverse events occurred in 37.3% of tecovirimat-treated and 33.3% of placebo-treated participants, with events of grade 3 or higher occurring in 1.1% of patients in both groups.
There was one death on the tecovirimat arm, which was caused by a pulmonary embolism in a participant who had a history of recurrent deep-vein thrombosis but was not receiving anticoagulant treatment, investigators said.
Dr. Grosenbach and colleagues also presented pharmacokinetic profiles and exposures for 48 volunteers in a fed state in their report in the New England Journal of Medicine, along with pharmacokinetic data from the animal studies.
The FDA has set a target date of Aug. 8, 2018, for final action on a new drug application submitted for oral tecovirimat for treatment of smallpox, SIGA Technologies said in a May 2018 news release.
Dr. Grosenbach and his coauthors reported a disclosure related to Biomedical Advanced Research and Development Authority (BARDA), grants from NIH during the conduct of the study, and personal fees from SIGA Technologies outside the submitted work.
SOURCE: Grosenbach DW et al. N Engl J Med. 2018 Jul 5;379(1):44-53.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Tecovirimat, an oral antiviral agent, is being advanced as a treatment for smallpox based on animal and human study data.
Major finding: The survival rate was approximately 95% in monkeypox-infected primates receiving doses of 3-10 mg/kilogram. In humans, the serious adverse event rate was 1.1% for both tecovirimat and placebo.
Study details: A report on multiple studies in small animals and nonhuman primates, along with a randomized safety trial involving 452 healthy human volunteers.
Disclosures: Study authors reported a disclosure related to Biomedical Advanced Research and Development Authority (BARDA), grants from NIH during the conduct of the study, and personal fees from SIGA Technologies outside the submitted work.
Source: Grosenbach DW et al. N Engl J Med. 2018 Jul 5;379(1):44-53.
FDA issues Ebola preparedness statement
In response to the Ebola outbreak in the Democratic Republic of Congo (DRC), the Food and Drug Administration has announced steps the agency is taking to make diagnostic and medical products available as part of critical response efforts.
In addition to providing scientific and regulatory advice to medical product developers, the FDA is using its authorities to ensure Merck’s investigational Ebola Zaire vaccine is made available appropriately to vaccinate high-risk populations in the DRC. Additionally, the FDA also is committed to facilitating the development of investigational drugs for the treatment of Ebola virus and supporting access to these products under appropriate regulatory pathways, FDA Commissioner Scott Gottlieb, MD, said in a statement.
Clinical trials that are adaptive to the circumstances of an outbreak are essential, he added. “During the 2014-2015 Ebola outbreak, the FDA recognized that some of the medical products that initially appeared to show great promise sometimes, when subjected to objective testing, were not effective or may have done more harm than good.”
Further, as there are no approved treatments or vaccines for Ebola, the agency will be monitoring for false product claims to protect consumers from fraudulent products claiming to prevent, treat, or cure the disease.
“The FDA knows that it takes a sustained, robust, and globally coordinated effort to best protect our nation from various infectious disease threats,” Dr. Gottlieb wrote. “We’re committed to supporting the people of the DRC and preventing a worsening circumstance during the current outbreak. And we remain highly engaged in the international response efforts.”
In response to the Ebola outbreak in the Democratic Republic of Congo (DRC), the Food and Drug Administration has announced steps the agency is taking to make diagnostic and medical products available as part of critical response efforts.
In addition to providing scientific and regulatory advice to medical product developers, the FDA is using its authorities to ensure Merck’s investigational Ebola Zaire vaccine is made available appropriately to vaccinate high-risk populations in the DRC. Additionally, the FDA also is committed to facilitating the development of investigational drugs for the treatment of Ebola virus and supporting access to these products under appropriate regulatory pathways, FDA Commissioner Scott Gottlieb, MD, said in a statement.
Clinical trials that are adaptive to the circumstances of an outbreak are essential, he added. “During the 2014-2015 Ebola outbreak, the FDA recognized that some of the medical products that initially appeared to show great promise sometimes, when subjected to objective testing, were not effective or may have done more harm than good.”
Further, as there are no approved treatments or vaccines for Ebola, the agency will be monitoring for false product claims to protect consumers from fraudulent products claiming to prevent, treat, or cure the disease.
“The FDA knows that it takes a sustained, robust, and globally coordinated effort to best protect our nation from various infectious disease threats,” Dr. Gottlieb wrote. “We’re committed to supporting the people of the DRC and preventing a worsening circumstance during the current outbreak. And we remain highly engaged in the international response efforts.”
In response to the Ebola outbreak in the Democratic Republic of Congo (DRC), the Food and Drug Administration has announced steps the agency is taking to make diagnostic and medical products available as part of critical response efforts.
In addition to providing scientific and regulatory advice to medical product developers, the FDA is using its authorities to ensure Merck’s investigational Ebola Zaire vaccine is made available appropriately to vaccinate high-risk populations in the DRC. Additionally, the FDA also is committed to facilitating the development of investigational drugs for the treatment of Ebola virus and supporting access to these products under appropriate regulatory pathways, FDA Commissioner Scott Gottlieb, MD, said in a statement.
Clinical trials that are adaptive to the circumstances of an outbreak are essential, he added. “During the 2014-2015 Ebola outbreak, the FDA recognized that some of the medical products that initially appeared to show great promise sometimes, when subjected to objective testing, were not effective or may have done more harm than good.”
Further, as there are no approved treatments or vaccines for Ebola, the agency will be monitoring for false product claims to protect consumers from fraudulent products claiming to prevent, treat, or cure the disease.
“The FDA knows that it takes a sustained, robust, and globally coordinated effort to best protect our nation from various infectious disease threats,” Dr. Gottlieb wrote. “We’re committed to supporting the people of the DRC and preventing a worsening circumstance during the current outbreak. And we remain highly engaged in the international response efforts.”
NIH launches early Ebola treatment trial
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
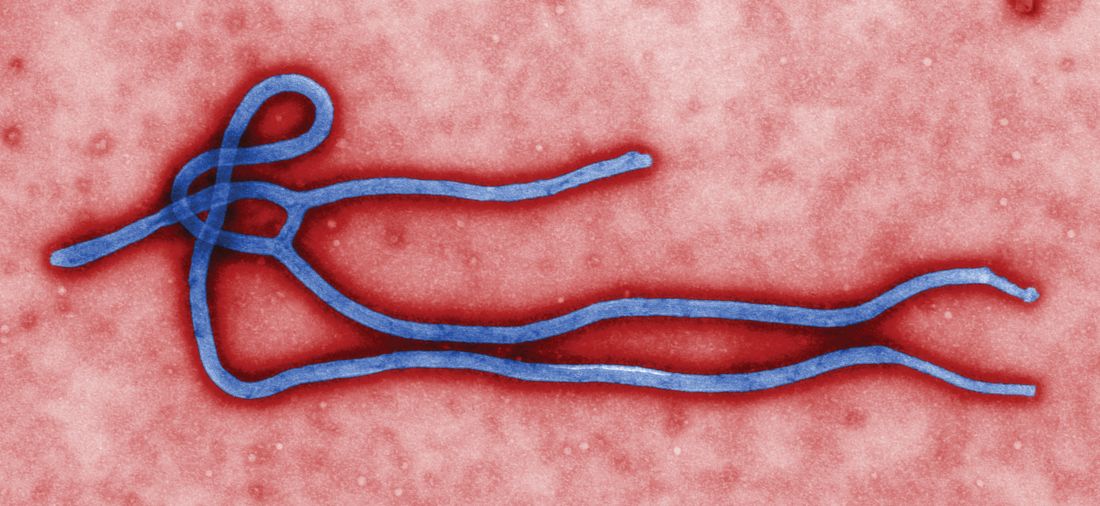
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.
A study of a potential new Ebola treatment has begun at the National Institutes of Health Clinical Center in Bethesda, Md. The small phase 1 clinical trial will examine the safety and tolerability of a single monoclonal antibody (mAb114), which was developed by scientists at the National Institute of Allergy and Infectious Diseases (NIAID) and their collaborators. Investigators plan to enroll between 18 and 30 healthy volunteers aged 18-60. The trial will not expose participants to Ebola virus, according to the NIH announcement.
MAb114 is a monoclonal antibody – a protein that binds to a single target on a pathogen — isolated from a human survivor of the 1995 Ebola outbreak in Kikwit, Democratic Republic of the Congo. Nancy Sullivan, PhD, chief of the Biodefense Research Section in NIAID’s Vaccine Research Center, and her team, in collaboration with researchers from the National Institute of Biomedical Research in the Democratic Republic of the Congo and the Institute for Biomedical Research in Switzerland, discovered that the survivor retained antibodies against Ebola 11 years after infection. They isolated and tested the antibodies and selected mAb114 as the most promising.
Although rVSV-ZEBOV, an experimental vaccine, is now available and in use in Africa during the current outbreak, specific treatment modalities are lacking.
More information can be found at www.clinicaltrials.gov, trial # NCT03478891.