User login
Laser vaginal rejuvenation procedures
VIENNA – Vaginal rejuvenation is a major practice growth opportunity for dermatologists who have expertise with lasers, Peter Bjerring, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
The rejuvenation procedures involve heating the connective tissue of the vaginal wall to 40-42 C in order to stimulate tissue remodeling with formation of new collagen and elastic fibers. The evidence base for vaginal rejuvenation using a variety of noninvasive energy-based devices developed for vaginal use isn’t nearly as extensive as it is for skin rejuvenation using lasers. Research is beginning to increase for this indication, but current results are primarily limited to small single-arm studies based on self-reported improvements. Further, studies don’t compare outcomes vs another treatment, such as estrogen cream.
Despite the minimal evidence base for laser procedures, “feminine rejuvenation is becoming very popular. These are (women) who might present to a dermatologist or to a gynecologist,” observed Dr. Bjerring, medical director and head of the Skin and Laser Center at Malholm (Denmark) Hospital.
Minimally ablative or nonablative fractional laser therapy for vaginal rejuvenation requires no anesthesia and no downtime. The lasers being used for this purpose are similar to those already used most often for skin resurfacing and rejuvenation of the face and neck: fractional CO2 lasers at the 10,600-nm wavelength, such as the MonaLisa Touch or FemTouch, and 2,940-nm nonablative erbium:yttrium-aluminum-garnet (Er:YAG) lasers such as the IntimaLase. A course of treatment with these devices typically consists of three, 15-minute sessions at 4- to 6-week intervals.
Dr. Bjerring noted that this mechanism of benefit has been demonstrated by Italian investigators who conducted a histologic study of the effects of microablative fractional CO2 laser therapy on ex vivo specimens of atrophic vaginal tissue obtained from women with vulvovaginal atrophy who underwent major surgery for pelvic organ prolapse.
The investigators treated one side of the atrophic vaginal wall specimen with the microablative fractional CO2 laser and left the contralateral area untreated as a control. They documented that laser therapy restored the vaginal squamous stratified epithelium, with enhanced storage of glycogen in the epithelial cells and shedding of glycogen-rich cells at the epithelial surface. In the connective tissue, activated fibroblasts synthesized new collagen-laden extracellular matrix. All this was accomplished without damage to adjacent untreated tissue (Menopause 2015 Aug;22(8):845-9).
In a single-arm study performed by many of the same Italian investigators, 77 postmenopausal women underwent a course of fractional microablative CO2 laser therapy because they experienced painful sexual intercourse due to vulvovaginal atrophy. At 12 weeks of followup, the group reported significant improvement in sexual function and satisfaction with their sexual life as measured by the Short Form-12 and the Female Sexual Function Index. Self-rated scores of vaginal burning, dryness, and itching improved significantly, as did complaints of pain during intercourse or urination. At baseline, 20 of the 77 women were not sexually active due to the severity of their vulvovaginal atrophy; at followup, 17 of the 20 had reestablished sexual activity (Climacteric. 2015 Apr;18(2):219-25).
Dr. Bjerring also highlighted an American study, again single-arm, in which 27 women with genitourinary syndrome of menopause were examined at baseline and again 3 months after their third and final treatment with a fractional CO2 laser. At follow up, 26 of the 27 pronounced themselves satisfied or extremely satisfied with the results, with significant improvement in the same outcome measures used in the Italian study. At follow up, 25 of the women had an increase in comfortable dilator size (Menopause 2016 Oct;23(10):1102-7).
Dr. Bjerring reported having no financial conflicts of interest regarding his presentation.
VIENNA – Vaginal rejuvenation is a major practice growth opportunity for dermatologists who have expertise with lasers, Peter Bjerring, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
The rejuvenation procedures involve heating the connective tissue of the vaginal wall to 40-42 C in order to stimulate tissue remodeling with formation of new collagen and elastic fibers. The evidence base for vaginal rejuvenation using a variety of noninvasive energy-based devices developed for vaginal use isn’t nearly as extensive as it is for skin rejuvenation using lasers. Research is beginning to increase for this indication, but current results are primarily limited to small single-arm studies based on self-reported improvements. Further, studies don’t compare outcomes vs another treatment, such as estrogen cream.
Despite the minimal evidence base for laser procedures, “feminine rejuvenation is becoming very popular. These are (women) who might present to a dermatologist or to a gynecologist,” observed Dr. Bjerring, medical director and head of the Skin and Laser Center at Malholm (Denmark) Hospital.
Minimally ablative or nonablative fractional laser therapy for vaginal rejuvenation requires no anesthesia and no downtime. The lasers being used for this purpose are similar to those already used most often for skin resurfacing and rejuvenation of the face and neck: fractional CO2 lasers at the 10,600-nm wavelength, such as the MonaLisa Touch or FemTouch, and 2,940-nm nonablative erbium:yttrium-aluminum-garnet (Er:YAG) lasers such as the IntimaLase. A course of treatment with these devices typically consists of three, 15-minute sessions at 4- to 6-week intervals.
Dr. Bjerring noted that this mechanism of benefit has been demonstrated by Italian investigators who conducted a histologic study of the effects of microablative fractional CO2 laser therapy on ex vivo specimens of atrophic vaginal tissue obtained from women with vulvovaginal atrophy who underwent major surgery for pelvic organ prolapse.
The investigators treated one side of the atrophic vaginal wall specimen with the microablative fractional CO2 laser and left the contralateral area untreated as a control. They documented that laser therapy restored the vaginal squamous stratified epithelium, with enhanced storage of glycogen in the epithelial cells and shedding of glycogen-rich cells at the epithelial surface. In the connective tissue, activated fibroblasts synthesized new collagen-laden extracellular matrix. All this was accomplished without damage to adjacent untreated tissue (Menopause 2015 Aug;22(8):845-9).
In a single-arm study performed by many of the same Italian investigators, 77 postmenopausal women underwent a course of fractional microablative CO2 laser therapy because they experienced painful sexual intercourse due to vulvovaginal atrophy. At 12 weeks of followup, the group reported significant improvement in sexual function and satisfaction with their sexual life as measured by the Short Form-12 and the Female Sexual Function Index. Self-rated scores of vaginal burning, dryness, and itching improved significantly, as did complaints of pain during intercourse or urination. At baseline, 20 of the 77 women were not sexually active due to the severity of their vulvovaginal atrophy; at followup, 17 of the 20 had reestablished sexual activity (Climacteric. 2015 Apr;18(2):219-25).
Dr. Bjerring also highlighted an American study, again single-arm, in which 27 women with genitourinary syndrome of menopause were examined at baseline and again 3 months after their third and final treatment with a fractional CO2 laser. At follow up, 26 of the 27 pronounced themselves satisfied or extremely satisfied with the results, with significant improvement in the same outcome measures used in the Italian study. At follow up, 25 of the women had an increase in comfortable dilator size (Menopause 2016 Oct;23(10):1102-7).
Dr. Bjerring reported having no financial conflicts of interest regarding his presentation.
VIENNA – Vaginal rejuvenation is a major practice growth opportunity for dermatologists who have expertise with lasers, Peter Bjerring, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
The rejuvenation procedures involve heating the connective tissue of the vaginal wall to 40-42 C in order to stimulate tissue remodeling with formation of new collagen and elastic fibers. The evidence base for vaginal rejuvenation using a variety of noninvasive energy-based devices developed for vaginal use isn’t nearly as extensive as it is for skin rejuvenation using lasers. Research is beginning to increase for this indication, but current results are primarily limited to small single-arm studies based on self-reported improvements. Further, studies don’t compare outcomes vs another treatment, such as estrogen cream.
Despite the minimal evidence base for laser procedures, “feminine rejuvenation is becoming very popular. These are (women) who might present to a dermatologist or to a gynecologist,” observed Dr. Bjerring, medical director and head of the Skin and Laser Center at Malholm (Denmark) Hospital.
Minimally ablative or nonablative fractional laser therapy for vaginal rejuvenation requires no anesthesia and no downtime. The lasers being used for this purpose are similar to those already used most often for skin resurfacing and rejuvenation of the face and neck: fractional CO2 lasers at the 10,600-nm wavelength, such as the MonaLisa Touch or FemTouch, and 2,940-nm nonablative erbium:yttrium-aluminum-garnet (Er:YAG) lasers such as the IntimaLase. A course of treatment with these devices typically consists of three, 15-minute sessions at 4- to 6-week intervals.
Dr. Bjerring noted that this mechanism of benefit has been demonstrated by Italian investigators who conducted a histologic study of the effects of microablative fractional CO2 laser therapy on ex vivo specimens of atrophic vaginal tissue obtained from women with vulvovaginal atrophy who underwent major surgery for pelvic organ prolapse.
The investigators treated one side of the atrophic vaginal wall specimen with the microablative fractional CO2 laser and left the contralateral area untreated as a control. They documented that laser therapy restored the vaginal squamous stratified epithelium, with enhanced storage of glycogen in the epithelial cells and shedding of glycogen-rich cells at the epithelial surface. In the connective tissue, activated fibroblasts synthesized new collagen-laden extracellular matrix. All this was accomplished without damage to adjacent untreated tissue (Menopause 2015 Aug;22(8):845-9).
In a single-arm study performed by many of the same Italian investigators, 77 postmenopausal women underwent a course of fractional microablative CO2 laser therapy because they experienced painful sexual intercourse due to vulvovaginal atrophy. At 12 weeks of followup, the group reported significant improvement in sexual function and satisfaction with their sexual life as measured by the Short Form-12 and the Female Sexual Function Index. Self-rated scores of vaginal burning, dryness, and itching improved significantly, as did complaints of pain during intercourse or urination. At baseline, 20 of the 77 women were not sexually active due to the severity of their vulvovaginal atrophy; at followup, 17 of the 20 had reestablished sexual activity (Climacteric. 2015 Apr;18(2):219-25).
Dr. Bjerring also highlighted an American study, again single-arm, in which 27 women with genitourinary syndrome of menopause were examined at baseline and again 3 months after their third and final treatment with a fractional CO2 laser. At follow up, 26 of the 27 pronounced themselves satisfied or extremely satisfied with the results, with significant improvement in the same outcome measures used in the Italian study. At follow up, 25 of the women had an increase in comfortable dilator size (Menopause 2016 Oct;23(10):1102-7).
Dr. Bjerring reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
A sweet new solution for rosacea
VIENNA – A medical-grade topical honey product proved safe and effective for the treatment of rosacea in a randomized, placebo-controlled clinical trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The product, known as Honevo, is a cream consisting of 90% New Zealand kanuka honey and 10% glycerine. It is applied twice daily as a face mask. Honevo is designed to wash off easily and is less sticky than honey.
The primary outcome in the trial was at least a 2-point improvement from baseline on the 7-point Investigator Global Assessment of Rosacea Severity Score (IGA-RSS) as assessed by blinded investigators. This outcome, which reflects a clinical improvement from severe to moderate or from moderate to mild, was achieved in 34% of the Honevo group, compared with 17% of controls. Significant improvement in the honey group was seen after 2 weeks.
Rosacea resolved in 13% of the Honevo group and in 3% of controls, based on a week 8 IGA-RSS of zero, noted Dr. Dreno, professor and chair of the department of dermatology at the University of Nantes (France).
The investigators, from the Medical Research Institute of New Zealand and the University of Otago in Wellington, observed that the study outcomes look at least as good as the results of placebo-controlled studies of topical metronidazole or azelaic cream. They plan to conduct randomized, head-to-head comparative trials of those prescription drugs versus Honevo, which is an OTC product.
The mechanism of action of kanuka honey in treating rosacea is believed to involve its previously reported antibacterial and anti-inflammatory effects, according to the investigators (BMJ Open. 2015 Jun 24;5[6]:e007651. doi: 10.1136/bmjopen-2015-007651).
The researchers noted that many rosacea patients aren’t interested in long-term antibiotic therapy. They want a natural product that doesn’t contribute to the global antibiotic resistance problem and is available OTC. And Honevo is one of the few natural or complementary medicine therapies backed by data from a rigorous clinical trial, in this case one registered in the Australian New Zealand Clinical Trials Registry (ACTRN12614000004662).
Dr. Dreno wasn’t involved in the study but included it in a talk in which she examined the strengths and weaknesses of current rosacea therapies. She is waiting for a confirmatory study before she incorporates Honevo in her own treatment algorithm. She said that she also would like to see studies examining whether combining the topical honey product with prescription drugs for rosacea provides synergistic efficacy.
HoneyLab, which funded the clinical trial and markets Honevo, ships the product to customers worldwide from its New Zealand headquarters.
VIENNA – A medical-grade topical honey product proved safe and effective for the treatment of rosacea in a randomized, placebo-controlled clinical trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The product, known as Honevo, is a cream consisting of 90% New Zealand kanuka honey and 10% glycerine. It is applied twice daily as a face mask. Honevo is designed to wash off easily and is less sticky than honey.
The primary outcome in the trial was at least a 2-point improvement from baseline on the 7-point Investigator Global Assessment of Rosacea Severity Score (IGA-RSS) as assessed by blinded investigators. This outcome, which reflects a clinical improvement from severe to moderate or from moderate to mild, was achieved in 34% of the Honevo group, compared with 17% of controls. Significant improvement in the honey group was seen after 2 weeks.
Rosacea resolved in 13% of the Honevo group and in 3% of controls, based on a week 8 IGA-RSS of zero, noted Dr. Dreno, professor and chair of the department of dermatology at the University of Nantes (France).
The investigators, from the Medical Research Institute of New Zealand and the University of Otago in Wellington, observed that the study outcomes look at least as good as the results of placebo-controlled studies of topical metronidazole or azelaic cream. They plan to conduct randomized, head-to-head comparative trials of those prescription drugs versus Honevo, which is an OTC product.
The mechanism of action of kanuka honey in treating rosacea is believed to involve its previously reported antibacterial and anti-inflammatory effects, according to the investigators (BMJ Open. 2015 Jun 24;5[6]:e007651. doi: 10.1136/bmjopen-2015-007651).
The researchers noted that many rosacea patients aren’t interested in long-term antibiotic therapy. They want a natural product that doesn’t contribute to the global antibiotic resistance problem and is available OTC. And Honevo is one of the few natural or complementary medicine therapies backed by data from a rigorous clinical trial, in this case one registered in the Australian New Zealand Clinical Trials Registry (ACTRN12614000004662).
Dr. Dreno wasn’t involved in the study but included it in a talk in which she examined the strengths and weaknesses of current rosacea therapies. She is waiting for a confirmatory study before she incorporates Honevo in her own treatment algorithm. She said that she also would like to see studies examining whether combining the topical honey product with prescription drugs for rosacea provides synergistic efficacy.
HoneyLab, which funded the clinical trial and markets Honevo, ships the product to customers worldwide from its New Zealand headquarters.
VIENNA – A medical-grade topical honey product proved safe and effective for the treatment of rosacea in a randomized, placebo-controlled clinical trial, Brigitte Dreno, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The product, known as Honevo, is a cream consisting of 90% New Zealand kanuka honey and 10% glycerine. It is applied twice daily as a face mask. Honevo is designed to wash off easily and is less sticky than honey.
The primary outcome in the trial was at least a 2-point improvement from baseline on the 7-point Investigator Global Assessment of Rosacea Severity Score (IGA-RSS) as assessed by blinded investigators. This outcome, which reflects a clinical improvement from severe to moderate or from moderate to mild, was achieved in 34% of the Honevo group, compared with 17% of controls. Significant improvement in the honey group was seen after 2 weeks.
Rosacea resolved in 13% of the Honevo group and in 3% of controls, based on a week 8 IGA-RSS of zero, noted Dr. Dreno, professor and chair of the department of dermatology at the University of Nantes (France).
The investigators, from the Medical Research Institute of New Zealand and the University of Otago in Wellington, observed that the study outcomes look at least as good as the results of placebo-controlled studies of topical metronidazole or azelaic cream. They plan to conduct randomized, head-to-head comparative trials of those prescription drugs versus Honevo, which is an OTC product.
The mechanism of action of kanuka honey in treating rosacea is believed to involve its previously reported antibacterial and anti-inflammatory effects, according to the investigators (BMJ Open. 2015 Jun 24;5[6]:e007651. doi: 10.1136/bmjopen-2015-007651).
The researchers noted that many rosacea patients aren’t interested in long-term antibiotic therapy. They want a natural product that doesn’t contribute to the global antibiotic resistance problem and is available OTC. And Honevo is one of the few natural or complementary medicine therapies backed by data from a rigorous clinical trial, in this case one registered in the Australian New Zealand Clinical Trials Registry (ACTRN12614000004662).
Dr. Dreno wasn’t involved in the study but included it in a talk in which she examined the strengths and weaknesses of current rosacea therapies. She is waiting for a confirmatory study before she incorporates Honevo in her own treatment algorithm. She said that she also would like to see studies examining whether combining the topical honey product with prescription drugs for rosacea provides synergistic efficacy.
HoneyLab, which funded the clinical trial and markets Honevo, ships the product to customers worldwide from its New Zealand headquarters.
AT THE EADV CONGRESS
Key clinical point:
Major finding: 34% of rosacea patients experienced clinically meaningful improvement in response to twice-daily kanuka honey face masks, a rate twice that in controls.
Data source: This was a randomized, placebo-controlled, single-blind, 8-week clinical trial involving 138 adults with rosacea.
Disclosures: The presenter reported having no financial conflicts of interest regarding the study.
Promising pipeline for chronic pruritus
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
VIENNA – Help is on the way for physicians stymied in their efforts to treat patients with severe chronic itch, Sonja Ständer, MD, declared at the annual congress of the European Academy of Dermatology and Venereology.
Now working their way through the developmental pipeline are two promising novel classes of drugs designed to target the physiologic mechanisms underlying this common and challenging problem: neurokinin 1 (NK1) receptor antagonists and selective opioid receptor agonists, explained Dr. Ständer, professor of dermatology and head of the Center for Chronic Pruritus at the University of Münster (Germany).
She led a landmark study that outlined the previously unappreciated dimensions of chronic pruritus as a clinical issue. This was a cross-sectional study of nearly 12,000 German employees in various industries that demonstrated the prevalence of chronic pruritus was 16.8%. One-quarter of affected individuals had chronic pruritus for longer than 5 years. The prevalence climbed with age, from 12% in workers up to age 30 years to 20% in 61- to 70-year-olds (Dermatology. 2010;221[3]:229-35).
One in three patients who present to dermatologists’ offices have chronic pruritus, she added.
Chronic pruritus can have a multitude of different causes: not only chronic skin diseases, but incurable renal and liver diseases, psychiatric conditions, neurologic disorders, drug side effects, and various forms of cancer, with hematologic malignancies figuring prominently. The quality of life impact is huge. And even though a plethora of topical and systemic therapies are available for itchy skin, they often are ineffective in patients with severe chronic pruritus. Thus, there is a significant unmet need for new and effective therapies, Dr. Ständer continued.
NK1 receptor antagonists: Substance P is a neuropeptide which plays a major role in the induction and maintenance of pruritus. The NK1 receptor, which is abundantly expressed in the skin and CNS, is substance P’s receptor – and therefore a logical target for novel anti-itch therapy. Bind that receptor and a key signaling pathway in pruritus is disrupted.
Aprepitant is an oral NK1 receptor antagonist approved as Emend more than a decade ago for prevention of chemotherapy-induced nausea and vomiting. Dr. Ständer was lead author of an early single-arm pilot study showing aprepitant is also dramatically effective for severe refractory chronic pruritus (PLoS One. 2010 Jun 4;5[6]:e10968).
Aprepitant is approved only for 3 days of use for its licensed indication. However, Dr. Ständer said she and other dermatologists have used it off-label for as long as 4 weeks in patients with chronic pruritus and found it to be safe, with mild nonlimiting side effects and relief that is often long lasting after treatment discontinuation.
Oral aprepitant is under study in several ongoing phase II clinical trials for treatment of severe pruritus resulting from targeted biologic therapies for various cancers. In addition, Leo Pharma is developing a topical gel formulation of aprepitant for chronic pruritus which is now in phase II studies.
Serlopitant is a once-daily oral NK1 receptor antagonist under development specifically for treatment of severe chronic pruritus. In a recent as-yet unpublished multicenter, double-blind, placebo-controlled phase II clinical trial involving 257 patients with severe refractory chronic pruritus of various etiologies, 6 weeks of serlopitant at 1 or 5 mg/day was markedly more effective than placebo in reducing itch intensity. Based upon these favorable results, Menlo Therapeutics has begun two new phase II randomized, placebo-controlled studies of the drug: ATOMIK, a roughly 450-patient, 40 U.S.-site study in patients with atopic dermatitis; and AUBURN, involving roughly 150 burn patients with chronic pruritus at 20 centers.
Selective opioid receptor agonists: These agents target kappa- and/or mu-opioid receptors on peripheral pain-sensing neurons in order to inhibit itch without activating other opioid receptors linked to classic opioid side effects such as respiratory depression, constipation, and addiction. These selective agents are essentially designed to be nonnarcotic opioids.
One such agent is nalfurafine, an oral kappa-opioid receptor agonist marketed in Japan for treatment of uremic pruritus in patients with chronic kidney disease undergoing hemodialysis and for refractory pruritus in chronic liver disease. The drug is in phase II studies in the United States.
Nalbuphine, a dual kappa-opiod agonist and partial mu-opioid antagonist, is Food and Drug Administration–approved as an injectable agent, known as Nubain, for moderate to severe pain. An investigational extended-release tablet formulation has successfully completed a U.S. multicenter, double-blind, placebo-controlled phase II/III clinical trial in hemodialysis patients with severe chronic uremic pruritus and a phase II study in patients with prurigo nodularis. Because the drug was effective in two conditions having very different sources of itch, it is likely to be of benefit in many forms of severe chronic pruritus, Dr. Ständer said.
She reported having no financial conflicts of interest.
EXPERT ANALYSIS FROM THE EADV
Initial suboptimal responders to secukinumab usually bloom later
VIENNA – When the occasional patient on secukinumab for moderate to severe psoriasis fails to achieve a PASI 75 response initially, don’t despair: Continuing treatment with the biologic usually gets them over that bar, Christopher E. Griffiths, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A new secondary pooled analysis of four phase III, 52-week, pivotal clinical trials of secukinumab (Cosentyx) indicates that more than three-quarters of initial suboptimal responders will go on to achieve a PASI 75 response. Moreover, more than one-third will have a PASI 90 response by week 16, which is sustained through week 52. And almost one in five slow responders will have a PASI 100 response – clear skin – at week 52, according to Dr. Griffiths, professor of dermatology at the University of Manchester, England.
He presented a secondary analysis of four phase III studies: ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis), FEATURE (First Study of Secukinumab in Pre-filled Syringes in Subjects With Chronic Plaque-type Psoriasis: Response at 12 Weeks), FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis), and JUNCTURE (Judging the Efficacy of Secukinumab in Patients With Psoriasis Using AutoiNjector: a Clinical Trial Evaluating Treatment Results). The analysis was conducted to provide additional perspective on the product labeling statement that treatment discontinuation should be considered in patients who haven’t responded to secukinumab by week 16.
The four studies featured a total of 2,405 patients with moderate to severe psoriasis on secukinumab at the approved dosing schedule.
The key findings: At week 12 – the primary endpoint in the four trials – only 5.2% of patients on secukinumab had not achieved a PASI 75 response. Yet just 4 weeks later, at week 16, 56% of this group had managed to get there. Seventy-seven percent of early non- or partial responders achieved a PASI 75 response at some point during weeks 13-52, and 55% had a PASI 75 response at 52 weeks.
Thirty-five percent of early poor responders achieved PASI 90 at 16 weeks and 37% at 52 weeks. Twelve percent of patients who didn’t get to PASI 75 at 12 weeks had a PASI 100 response by 16 weeks, and nearly 18% did by week 52.
This analysis was supported by secukinumab manufacturer Novartis. Dr. Griffiths reported receiving research funds from and serving as a consultant to Novartis and numerous other pharmaceutical companies.
VIENNA – When the occasional patient on secukinumab for moderate to severe psoriasis fails to achieve a PASI 75 response initially, don’t despair: Continuing treatment with the biologic usually gets them over that bar, Christopher E. Griffiths, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A new secondary pooled analysis of four phase III, 52-week, pivotal clinical trials of secukinumab (Cosentyx) indicates that more than three-quarters of initial suboptimal responders will go on to achieve a PASI 75 response. Moreover, more than one-third will have a PASI 90 response by week 16, which is sustained through week 52. And almost one in five slow responders will have a PASI 100 response – clear skin – at week 52, according to Dr. Griffiths, professor of dermatology at the University of Manchester, England.
He presented a secondary analysis of four phase III studies: ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis), FEATURE (First Study of Secukinumab in Pre-filled Syringes in Subjects With Chronic Plaque-type Psoriasis: Response at 12 Weeks), FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis), and JUNCTURE (Judging the Efficacy of Secukinumab in Patients With Psoriasis Using AutoiNjector: a Clinical Trial Evaluating Treatment Results). The analysis was conducted to provide additional perspective on the product labeling statement that treatment discontinuation should be considered in patients who haven’t responded to secukinumab by week 16.
The four studies featured a total of 2,405 patients with moderate to severe psoriasis on secukinumab at the approved dosing schedule.
The key findings: At week 12 – the primary endpoint in the four trials – only 5.2% of patients on secukinumab had not achieved a PASI 75 response. Yet just 4 weeks later, at week 16, 56% of this group had managed to get there. Seventy-seven percent of early non- or partial responders achieved a PASI 75 response at some point during weeks 13-52, and 55% had a PASI 75 response at 52 weeks.
Thirty-five percent of early poor responders achieved PASI 90 at 16 weeks and 37% at 52 weeks. Twelve percent of patients who didn’t get to PASI 75 at 12 weeks had a PASI 100 response by 16 weeks, and nearly 18% did by week 52.
This analysis was supported by secukinumab manufacturer Novartis. Dr. Griffiths reported receiving research funds from and serving as a consultant to Novartis and numerous other pharmaceutical companies.
VIENNA – When the occasional patient on secukinumab for moderate to severe psoriasis fails to achieve a PASI 75 response initially, don’t despair: Continuing treatment with the biologic usually gets them over that bar, Christopher E. Griffiths, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A new secondary pooled analysis of four phase III, 52-week, pivotal clinical trials of secukinumab (Cosentyx) indicates that more than three-quarters of initial suboptimal responders will go on to achieve a PASI 75 response. Moreover, more than one-third will have a PASI 90 response by week 16, which is sustained through week 52. And almost one in five slow responders will have a PASI 100 response – clear skin – at week 52, according to Dr. Griffiths, professor of dermatology at the University of Manchester, England.
He presented a secondary analysis of four phase III studies: ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis), FEATURE (First Study of Secukinumab in Pre-filled Syringes in Subjects With Chronic Plaque-type Psoriasis: Response at 12 Weeks), FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis), and JUNCTURE (Judging the Efficacy of Secukinumab in Patients With Psoriasis Using AutoiNjector: a Clinical Trial Evaluating Treatment Results). The analysis was conducted to provide additional perspective on the product labeling statement that treatment discontinuation should be considered in patients who haven’t responded to secukinumab by week 16.
The four studies featured a total of 2,405 patients with moderate to severe psoriasis on secukinumab at the approved dosing schedule.
The key findings: At week 12 – the primary endpoint in the four trials – only 5.2% of patients on secukinumab had not achieved a PASI 75 response. Yet just 4 weeks later, at week 16, 56% of this group had managed to get there. Seventy-seven percent of early non- or partial responders achieved a PASI 75 response at some point during weeks 13-52, and 55% had a PASI 75 response at 52 weeks.
Thirty-five percent of early poor responders achieved PASI 90 at 16 weeks and 37% at 52 weeks. Twelve percent of patients who didn’t get to PASI 75 at 12 weeks had a PASI 100 response by 16 weeks, and nearly 18% did by week 52.
This analysis was supported by secukinumab manufacturer Novartis. Dr. Griffiths reported receiving research funds from and serving as a consultant to Novartis and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Most psoriasis patients who don’t achieve a PASI 75 response by week 12 on secukinumab will do so by week 16 and will maintain that response through week 52.
Data source: A pooled secondary analysis of PASI response rates in four phase III randomized clinical trials of secukinumab featuring 2,405 patients with moderate to severe psoriasis who were on the biologic for 52 weeks, including the 119 who did not achieve a PASI 75 response by week 12.
Disclosures: This analysis of four phase III clinical trials was sponsored by Novartis, as were the trials. The presenter reported receiving research funding from and serving as a consultant to Novartis and other pharmaceutical companies.
Secukinumab tames severe scalp psoriasis
VIENNA – Secukinumab proved highly effective specifically for the treatment of moderate to severe scalp psoriasis in a phase IIIb clinical trial, Mark G. Lebwohl, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The scalp is one of the areas most commonly affected by psoriasis, yet few treatment trials have focused on patients with primarily moderate to severe scalp psoriasis. This phase IIIb study was designed to do just that. The 102 participants had psoriasis over a mean of 60% of their scalp for at least 6 months at baseline despite various forms of therapy; 40% had 70% or greater scalp involvement. The study population’s mean baseline Psoriasis Scalp Severity Index score was 34 out of a possible 72, noted Dr. Lebwohl, professor and chairman of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
“The mean involved body surface area was only 11.2%, and the PASI was 8.4. That is below the entry score required for most biologic studies, yet scalp involvement was substantial,” he observed.
Participants in the double-blind trial were randomized to either subcutaneous secukinumab (Cosentyx) at 300 mg on the approved treatment schedule for psoriasis or to placebo, with the primary endpoint being at least a 90% improvement in Psoriasis Area and Severity Index scores (PASI 90 response) at 12 weeks.
“The results were striking. Quite stunning,” Dr. Lebwohl said.
A PASI 90 response was achieved in 53% of secukinumab-treated patients, compared with 2% of controls. Already by week 3 a significant difference was apparent between the two study arms: At that early point, 12% of the secukinumab group, but none of the controls, had a PASI 90 response.
The secondary endpoint was change in the Investigator’s Global Assessment of scalp disease. At baseline, roughly 80% of patients had an IGA of 3 out of a possible 4 and the rest were at 4. At 3 weeks, 26% of the secukinumab group had a score of 0 or 1, signifying a clear or almost clear scalp, compared with 6% of controls. At 12 weeks, 57% of patients on secukinumab had an IGA of 0 or 1, as did 6% of those on placebo.
Side effects of secukinumab in the 12-week study were minimal. There were no serious adverse events. One case of candidiasis occurred in each study arm. Both responded readily to standard therapy.
Secukinumab is a fully human monoclonal antibody that inhibits interleukin-17A. It’s approved for treatment of moderate-to-severe psoriasis, psoriatic arthritis, and ankylosing spondylitis.
This phase IIIb clinical trial was sponsored by Novartis. Dr. Lebwohl reported that his department receives research funding from Novartis and roughly a dozen other pharmaceutical companies.
VIENNA – Secukinumab proved highly effective specifically for the treatment of moderate to severe scalp psoriasis in a phase IIIb clinical trial, Mark G. Lebwohl, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The scalp is one of the areas most commonly affected by psoriasis, yet few treatment trials have focused on patients with primarily moderate to severe scalp psoriasis. This phase IIIb study was designed to do just that. The 102 participants had psoriasis over a mean of 60% of their scalp for at least 6 months at baseline despite various forms of therapy; 40% had 70% or greater scalp involvement. The study population’s mean baseline Psoriasis Scalp Severity Index score was 34 out of a possible 72, noted Dr. Lebwohl, professor and chairman of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
“The mean involved body surface area was only 11.2%, and the PASI was 8.4. That is below the entry score required for most biologic studies, yet scalp involvement was substantial,” he observed.
Participants in the double-blind trial were randomized to either subcutaneous secukinumab (Cosentyx) at 300 mg on the approved treatment schedule for psoriasis or to placebo, with the primary endpoint being at least a 90% improvement in Psoriasis Area and Severity Index scores (PASI 90 response) at 12 weeks.
“The results were striking. Quite stunning,” Dr. Lebwohl said.
A PASI 90 response was achieved in 53% of secukinumab-treated patients, compared with 2% of controls. Already by week 3 a significant difference was apparent between the two study arms: At that early point, 12% of the secukinumab group, but none of the controls, had a PASI 90 response.
The secondary endpoint was change in the Investigator’s Global Assessment of scalp disease. At baseline, roughly 80% of patients had an IGA of 3 out of a possible 4 and the rest were at 4. At 3 weeks, 26% of the secukinumab group had a score of 0 or 1, signifying a clear or almost clear scalp, compared with 6% of controls. At 12 weeks, 57% of patients on secukinumab had an IGA of 0 or 1, as did 6% of those on placebo.
Side effects of secukinumab in the 12-week study were minimal. There were no serious adverse events. One case of candidiasis occurred in each study arm. Both responded readily to standard therapy.
Secukinumab is a fully human monoclonal antibody that inhibits interleukin-17A. It’s approved for treatment of moderate-to-severe psoriasis, psoriatic arthritis, and ankylosing spondylitis.
This phase IIIb clinical trial was sponsored by Novartis. Dr. Lebwohl reported that his department receives research funding from Novartis and roughly a dozen other pharmaceutical companies.
VIENNA – Secukinumab proved highly effective specifically for the treatment of moderate to severe scalp psoriasis in a phase IIIb clinical trial, Mark G. Lebwohl, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The scalp is one of the areas most commonly affected by psoriasis, yet few treatment trials have focused on patients with primarily moderate to severe scalp psoriasis. This phase IIIb study was designed to do just that. The 102 participants had psoriasis over a mean of 60% of their scalp for at least 6 months at baseline despite various forms of therapy; 40% had 70% or greater scalp involvement. The study population’s mean baseline Psoriasis Scalp Severity Index score was 34 out of a possible 72, noted Dr. Lebwohl, professor and chairman of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
“The mean involved body surface area was only 11.2%, and the PASI was 8.4. That is below the entry score required for most biologic studies, yet scalp involvement was substantial,” he observed.
Participants in the double-blind trial were randomized to either subcutaneous secukinumab (Cosentyx) at 300 mg on the approved treatment schedule for psoriasis or to placebo, with the primary endpoint being at least a 90% improvement in Psoriasis Area and Severity Index scores (PASI 90 response) at 12 weeks.
“The results were striking. Quite stunning,” Dr. Lebwohl said.
A PASI 90 response was achieved in 53% of secukinumab-treated patients, compared with 2% of controls. Already by week 3 a significant difference was apparent between the two study arms: At that early point, 12% of the secukinumab group, but none of the controls, had a PASI 90 response.
The secondary endpoint was change in the Investigator’s Global Assessment of scalp disease. At baseline, roughly 80% of patients had an IGA of 3 out of a possible 4 and the rest were at 4. At 3 weeks, 26% of the secukinumab group had a score of 0 or 1, signifying a clear or almost clear scalp, compared with 6% of controls. At 12 weeks, 57% of patients on secukinumab had an IGA of 0 or 1, as did 6% of those on placebo.
Side effects of secukinumab in the 12-week study were minimal. There were no serious adverse events. One case of candidiasis occurred in each study arm. Both responded readily to standard therapy.
Secukinumab is a fully human monoclonal antibody that inhibits interleukin-17A. It’s approved for treatment of moderate-to-severe psoriasis, psoriatic arthritis, and ankylosing spondylitis.
This phase IIIb clinical trial was sponsored by Novartis. Dr. Lebwohl reported that his department receives research funding from Novartis and roughly a dozen other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: 53% of patients with chronic moderate to severe scalp psoriasis experienced at least a 90% improvement after 12 weeks on secukinumab, compared with 2% of controls.
Data source: This prospective, double-blind, phase IIIb clinical trial randomized 102 patients with moderate to severe scalp psoriasis to secukinumab or placebo.
Disclosures: The study was sponsored by Novartis. The presenter reported that his academic department receives research funding from Novartis and roughly a dozen other pharmaceutical companies.
Ixekizumab proves highly effective for palmoplantar, scalp psoriasis
VIENNA – Ixekizumab proved markedly more effective than etanercept for treatment of palmoplantar psoriasis in a head-to-head contest in the landmark phase III UNCOVER-3 trial, Alan Menter, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Significant improvement in palmoplantar disease was seen as early as week 2 in patients randomized to ixekizumab (Taltz), a humanized monoclonal antibody directed against interleukin-17A. Moreover, the early improvement was maintained out to week 60 with administration of 80 mg of ixekizumab by subcutaneous injection every 4 weeks in the open-label extension phase of UNCOVER-3. This pivotal trial, including 1,346 patients with moderate to severe psoriasis, helped win regulatory approval for ixekizumab for treatment of chronic plaque psoriasis.
The primary results of UNCOVER-3 have been published. At week 60, at least 80% of patients on maintenance therapy with ixekizumab had a PASI 75 response and at least 73% were rated clear or almost clear (N Engl J Med. 2016 Jul 28;375[4]:345-56).
Dr. Menter and Dr. Reich presented new subgroup analyses focused specifically on palmoplantar and scalp psoriasis because these two expressions of the disease are very important in clinical practice, albeit for different reasons.
Scalp psoriasis is extremely common in patients with plaque psoriasis. In fact, nearly 91% of subjects in UNCOVER-3 had scalp involvement.
“That’s a higher percentage than we’re accustomed to seeing in daily practice. It suggests scalp psoriasis may be more common than previously thought in patients with moderate or severe psoriasis,” said Dr. Reich, professor of dermatology at Georg-August-University in Gottingen, Germany, and a partner at the Dermatologikum Hamburg.
At week 60, more than 77% of patients on ixekizumab achieved a Psoriasis Scalp Severity Index 100 response (PSSI 100), meaning they had complete resolution of their scalp psoriasis. More than 80% achieved a PSSI 90 response indicative of complete or near complete resolution of their scalp involvement, the dermatologist reported.
“I often tell my patients that it’s because of palmoplantar psoriasis that I have so many white hairs. It’s certainly a disease that none of us cope with well topically, phototherapy-wise, PUVA-wise, or with systemic therapy. All of the studies done to date with our systemic therapies show significantly lower effect on palmoplantar psoriasis than for psoriasis at other sites. When I did the REVEAL study for Humira [adalimumab], we published a week 16 PASI 75 rate of 71%. When we did the palmoplantar psoriasis cohort, it was less than 40%,” recalled Dr. Menter, who is chair of dermatology at Baylor University Medical Center, Dallas.
“Even though palmoplantar disease affects less than 5% of the body surface area, the quality of life impact for patients with significant palmoplantar pustular or plaque psoriasis is very significant,” Dr. Menter continued. “We’ve worked with our hand surgeons and our foot surgeons to show that the impairment equals that seen in patients with severe rheumatoid arthritis or osteoarthritis of the hands and feet. So it is a huge issue.”
He reported on the 115 UNCOVER-3 participants with palmoplantar involvement. Within 2 weeks after the first 80-mg dose of ixekizumab, recipients had a 60% improvement in their Palmoplantar Psoriasis Area and Severity Index (PPSI) scores.
“It was very dramatic. These are figures that we haven’t seen with methotrexate, with retinoids, or with TNF-alpha blockers,” according to Dr. Menter.
At week 12 in UNCOVER-3, patients randomized to ixekizumab at 80 mg every 2 weeks showed an 85% improvement from baseline in PPSI scores. Those on ixekizumab at 80 mg every 4 weeks had a 78% improvement from baseline, while patients on etanercept at 50 mg twice weekly showed a 52% improvement.
At 60 weeks, PPSI 100 response rates – that is, clear palms and soles – were 60%-70% in the various ixekizumab-treated groups.
“To me, the big issue now is what about palmoplantar pustulosis, a totally different disease, and a disease with equally serious issues for our patients. I’m looking forward to studies in that population. I sincerely hope these new agents such as ixekizumab will have a significant role to play,” he said.
Dr. Menter and Dr. Reich reported receiving research support from and serving as consultants to Eli Lilly, which sponsored the UNCOVER-3 trial and markets ixekizumab, as well as numerous other pharmaceutical companies.
VIENNA – Ixekizumab proved markedly more effective than etanercept for treatment of palmoplantar psoriasis in a head-to-head contest in the landmark phase III UNCOVER-3 trial, Alan Menter, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Significant improvement in palmoplantar disease was seen as early as week 2 in patients randomized to ixekizumab (Taltz), a humanized monoclonal antibody directed against interleukin-17A. Moreover, the early improvement was maintained out to week 60 with administration of 80 mg of ixekizumab by subcutaneous injection every 4 weeks in the open-label extension phase of UNCOVER-3. This pivotal trial, including 1,346 patients with moderate to severe psoriasis, helped win regulatory approval for ixekizumab for treatment of chronic plaque psoriasis.
The primary results of UNCOVER-3 have been published. At week 60, at least 80% of patients on maintenance therapy with ixekizumab had a PASI 75 response and at least 73% were rated clear or almost clear (N Engl J Med. 2016 Jul 28;375[4]:345-56).
Dr. Menter and Dr. Reich presented new subgroup analyses focused specifically on palmoplantar and scalp psoriasis because these two expressions of the disease are very important in clinical practice, albeit for different reasons.
Scalp psoriasis is extremely common in patients with plaque psoriasis. In fact, nearly 91% of subjects in UNCOVER-3 had scalp involvement.
“That’s a higher percentage than we’re accustomed to seeing in daily practice. It suggests scalp psoriasis may be more common than previously thought in patients with moderate or severe psoriasis,” said Dr. Reich, professor of dermatology at Georg-August-University in Gottingen, Germany, and a partner at the Dermatologikum Hamburg.
At week 60, more than 77% of patients on ixekizumab achieved a Psoriasis Scalp Severity Index 100 response (PSSI 100), meaning they had complete resolution of their scalp psoriasis. More than 80% achieved a PSSI 90 response indicative of complete or near complete resolution of their scalp involvement, the dermatologist reported.
“I often tell my patients that it’s because of palmoplantar psoriasis that I have so many white hairs. It’s certainly a disease that none of us cope with well topically, phototherapy-wise, PUVA-wise, or with systemic therapy. All of the studies done to date with our systemic therapies show significantly lower effect on palmoplantar psoriasis than for psoriasis at other sites. When I did the REVEAL study for Humira [adalimumab], we published a week 16 PASI 75 rate of 71%. When we did the palmoplantar psoriasis cohort, it was less than 40%,” recalled Dr. Menter, who is chair of dermatology at Baylor University Medical Center, Dallas.
“Even though palmoplantar disease affects less than 5% of the body surface area, the quality of life impact for patients with significant palmoplantar pustular or plaque psoriasis is very significant,” Dr. Menter continued. “We’ve worked with our hand surgeons and our foot surgeons to show that the impairment equals that seen in patients with severe rheumatoid arthritis or osteoarthritis of the hands and feet. So it is a huge issue.”
He reported on the 115 UNCOVER-3 participants with palmoplantar involvement. Within 2 weeks after the first 80-mg dose of ixekizumab, recipients had a 60% improvement in their Palmoplantar Psoriasis Area and Severity Index (PPSI) scores.
“It was very dramatic. These are figures that we haven’t seen with methotrexate, with retinoids, or with TNF-alpha blockers,” according to Dr. Menter.
At week 12 in UNCOVER-3, patients randomized to ixekizumab at 80 mg every 2 weeks showed an 85% improvement from baseline in PPSI scores. Those on ixekizumab at 80 mg every 4 weeks had a 78% improvement from baseline, while patients on etanercept at 50 mg twice weekly showed a 52% improvement.
At 60 weeks, PPSI 100 response rates – that is, clear palms and soles – were 60%-70% in the various ixekizumab-treated groups.
“To me, the big issue now is what about palmoplantar pustulosis, a totally different disease, and a disease with equally serious issues for our patients. I’m looking forward to studies in that population. I sincerely hope these new agents such as ixekizumab will have a significant role to play,” he said.
Dr. Menter and Dr. Reich reported receiving research support from and serving as consultants to Eli Lilly, which sponsored the UNCOVER-3 trial and markets ixekizumab, as well as numerous other pharmaceutical companies.
VIENNA – Ixekizumab proved markedly more effective than etanercept for treatment of palmoplantar psoriasis in a head-to-head contest in the landmark phase III UNCOVER-3 trial, Alan Menter, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Significant improvement in palmoplantar disease was seen as early as week 2 in patients randomized to ixekizumab (Taltz), a humanized monoclonal antibody directed against interleukin-17A. Moreover, the early improvement was maintained out to week 60 with administration of 80 mg of ixekizumab by subcutaneous injection every 4 weeks in the open-label extension phase of UNCOVER-3. This pivotal trial, including 1,346 patients with moderate to severe psoriasis, helped win regulatory approval for ixekizumab for treatment of chronic plaque psoriasis.
The primary results of UNCOVER-3 have been published. At week 60, at least 80% of patients on maintenance therapy with ixekizumab had a PASI 75 response and at least 73% were rated clear or almost clear (N Engl J Med. 2016 Jul 28;375[4]:345-56).
Dr. Menter and Dr. Reich presented new subgroup analyses focused specifically on palmoplantar and scalp psoriasis because these two expressions of the disease are very important in clinical practice, albeit for different reasons.
Scalp psoriasis is extremely common in patients with plaque psoriasis. In fact, nearly 91% of subjects in UNCOVER-3 had scalp involvement.
“That’s a higher percentage than we’re accustomed to seeing in daily practice. It suggests scalp psoriasis may be more common than previously thought in patients with moderate or severe psoriasis,” said Dr. Reich, professor of dermatology at Georg-August-University in Gottingen, Germany, and a partner at the Dermatologikum Hamburg.
At week 60, more than 77% of patients on ixekizumab achieved a Psoriasis Scalp Severity Index 100 response (PSSI 100), meaning they had complete resolution of their scalp psoriasis. More than 80% achieved a PSSI 90 response indicative of complete or near complete resolution of their scalp involvement, the dermatologist reported.
“I often tell my patients that it’s because of palmoplantar psoriasis that I have so many white hairs. It’s certainly a disease that none of us cope with well topically, phototherapy-wise, PUVA-wise, or with systemic therapy. All of the studies done to date with our systemic therapies show significantly lower effect on palmoplantar psoriasis than for psoriasis at other sites. When I did the REVEAL study for Humira [adalimumab], we published a week 16 PASI 75 rate of 71%. When we did the palmoplantar psoriasis cohort, it was less than 40%,” recalled Dr. Menter, who is chair of dermatology at Baylor University Medical Center, Dallas.
“Even though palmoplantar disease affects less than 5% of the body surface area, the quality of life impact for patients with significant palmoplantar pustular or plaque psoriasis is very significant,” Dr. Menter continued. “We’ve worked with our hand surgeons and our foot surgeons to show that the impairment equals that seen in patients with severe rheumatoid arthritis or osteoarthritis of the hands and feet. So it is a huge issue.”
He reported on the 115 UNCOVER-3 participants with palmoplantar involvement. Within 2 weeks after the first 80-mg dose of ixekizumab, recipients had a 60% improvement in their Palmoplantar Psoriasis Area and Severity Index (PPSI) scores.
“It was very dramatic. These are figures that we haven’t seen with methotrexate, with retinoids, or with TNF-alpha blockers,” according to Dr. Menter.
At week 12 in UNCOVER-3, patients randomized to ixekizumab at 80 mg every 2 weeks showed an 85% improvement from baseline in PPSI scores. Those on ixekizumab at 80 mg every 4 weeks had a 78% improvement from baseline, while patients on etanercept at 50 mg twice weekly showed a 52% improvement.
At 60 weeks, PPSI 100 response rates – that is, clear palms and soles – were 60%-70% in the various ixekizumab-treated groups.
“To me, the big issue now is what about palmoplantar pustulosis, a totally different disease, and a disease with equally serious issues for our patients. I’m looking forward to studies in that population. I sincerely hope these new agents such as ixekizumab will have a significant role to play,” he said.
Dr. Menter and Dr. Reich reported receiving research support from and serving as consultants to Eli Lilly, which sponsored the UNCOVER-3 trial and markets ixekizumab, as well as numerous other pharmaceutical companies.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Survey shines new light on weighty comorbidity burden in adult atopic dermatitis
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.
Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.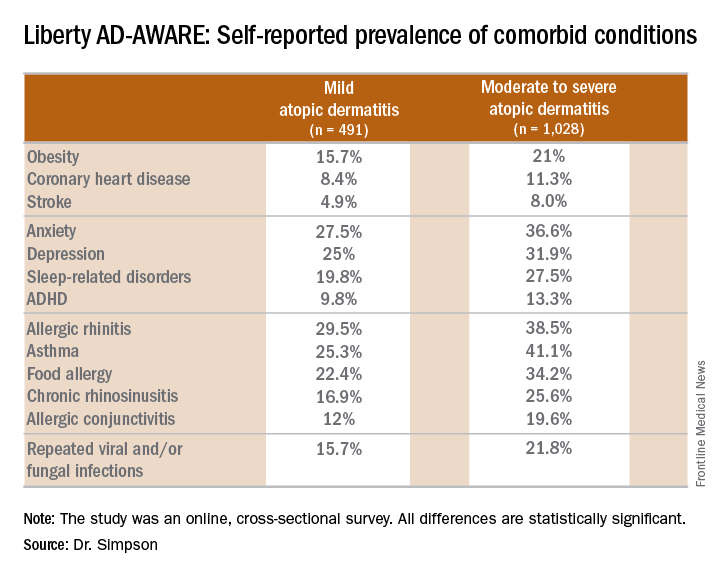
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.
Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.
Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Latest ixekizumab safety data called ‘very reassuring’
VIENNA – Updated longer-term safety data for ixekizumab in patients with moderate to severe plaque psoriasis continue to show no new safety signals, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The safety database now includes 4,213 psoriasis patients on ixekizumab (Taltz) for a total of 7,843 patient-years of regular ongoing follow-up in seven different phase I-III clinical trials. And with a large group of patients now having been on the novel humanized monoclonal antibody targeting interleukin-17A for 2 years and smaller numbers out to 5 years, there have been no surprises, according to Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
The key trends in the safety analysis are that the number of patients with an adverse event resulting in ixekizumab discontinuation is very low, yet adverse event rates are declining over time.
“This is pretty common in clinical trials,” according to the dermatologist. “In those first 12 weeks of a study you are seeing the patients very frequently, asking them very detailed questions, and we often pick up adverse events more frequently as a result. Upper respiratory infections are a good example: In the first month a patient will remember what happened last week. But if you haven’t seen a patient in 3 months they might not remember that 10 weeks ago they had a little cold. That’s why we tend to see URI rates go down over time. Now, if you see adverse events go up over time – especially for things like malignancy – then there is certainly cause for concern that there’s a cumulative problem with toxicity. That is clearly not a problem with this drug.”
Turning to selected adverse events of interest, Dr. Kimball noted that 2.1% of patients have experienced oral candidiasis while on ixekizumab.
“Oral Candida infection is one of the known side effects with this drug. It doesn’t happen very frequently, and to date, the infections have been very manageable, but it is something you want to have in your mind because it does happen,” she noted.
Serious infections have occurred in 105 patients, 2.5% of those on ixekizumab. Major adverse cardiovascular events have occurred in 1.0%, nonmelanoma skin cancers in 0.7%, and other cancers in 1.1%. Of note, only 5 patients (0.1%) have developed Crohn’s disease, 10 have been diagnosed with ulcerative colitis, and there have been no completed suicides.
The safety follow-up is ongoing.
The safety registry is supported by Eli Lilly, which markets ixekizumab. Dr. Kimball reported receiving research funding from and serving as a consultant to Eli Lilly and numerous other pharmaceutical companies.
VIENNA – Updated longer-term safety data for ixekizumab in patients with moderate to severe plaque psoriasis continue to show no new safety signals, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The safety database now includes 4,213 psoriasis patients on ixekizumab (Taltz) for a total of 7,843 patient-years of regular ongoing follow-up in seven different phase I-III clinical trials. And with a large group of patients now having been on the novel humanized monoclonal antibody targeting interleukin-17A for 2 years and smaller numbers out to 5 years, there have been no surprises, according to Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
The key trends in the safety analysis are that the number of patients with an adverse event resulting in ixekizumab discontinuation is very low, yet adverse event rates are declining over time.
“This is pretty common in clinical trials,” according to the dermatologist. “In those first 12 weeks of a study you are seeing the patients very frequently, asking them very detailed questions, and we often pick up adverse events more frequently as a result. Upper respiratory infections are a good example: In the first month a patient will remember what happened last week. But if you haven’t seen a patient in 3 months they might not remember that 10 weeks ago they had a little cold. That’s why we tend to see URI rates go down over time. Now, if you see adverse events go up over time – especially for things like malignancy – then there is certainly cause for concern that there’s a cumulative problem with toxicity. That is clearly not a problem with this drug.”
Turning to selected adverse events of interest, Dr. Kimball noted that 2.1% of patients have experienced oral candidiasis while on ixekizumab.
“Oral Candida infection is one of the known side effects with this drug. It doesn’t happen very frequently, and to date, the infections have been very manageable, but it is something you want to have in your mind because it does happen,” she noted.
Serious infections have occurred in 105 patients, 2.5% of those on ixekizumab. Major adverse cardiovascular events have occurred in 1.0%, nonmelanoma skin cancers in 0.7%, and other cancers in 1.1%. Of note, only 5 patients (0.1%) have developed Crohn’s disease, 10 have been diagnosed with ulcerative colitis, and there have been no completed suicides.
The safety follow-up is ongoing.
The safety registry is supported by Eli Lilly, which markets ixekizumab. Dr. Kimball reported receiving research funding from and serving as a consultant to Eli Lilly and numerous other pharmaceutical companies.
VIENNA – Updated longer-term safety data for ixekizumab in patients with moderate to severe plaque psoriasis continue to show no new safety signals, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
The safety database now includes 4,213 psoriasis patients on ixekizumab (Taltz) for a total of 7,843 patient-years of regular ongoing follow-up in seven different phase I-III clinical trials. And with a large group of patients now having been on the novel humanized monoclonal antibody targeting interleukin-17A for 2 years and smaller numbers out to 5 years, there have been no surprises, according to Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
The key trends in the safety analysis are that the number of patients with an adverse event resulting in ixekizumab discontinuation is very low, yet adverse event rates are declining over time.
“This is pretty common in clinical trials,” according to the dermatologist. “In those first 12 weeks of a study you are seeing the patients very frequently, asking them very detailed questions, and we often pick up adverse events more frequently as a result. Upper respiratory infections are a good example: In the first month a patient will remember what happened last week. But if you haven’t seen a patient in 3 months they might not remember that 10 weeks ago they had a little cold. That’s why we tend to see URI rates go down over time. Now, if you see adverse events go up over time – especially for things like malignancy – then there is certainly cause for concern that there’s a cumulative problem with toxicity. That is clearly not a problem with this drug.”
Turning to selected adverse events of interest, Dr. Kimball noted that 2.1% of patients have experienced oral candidiasis while on ixekizumab.
“Oral Candida infection is one of the known side effects with this drug. It doesn’t happen very frequently, and to date, the infections have been very manageable, but it is something you want to have in your mind because it does happen,” she noted.
Serious infections have occurred in 105 patients, 2.5% of those on ixekizumab. Major adverse cardiovascular events have occurred in 1.0%, nonmelanoma skin cancers in 0.7%, and other cancers in 1.1%. Of note, only 5 patients (0.1%) have developed Crohn’s disease, 10 have been diagnosed with ulcerative colitis, and there have been no completed suicides.
The safety follow-up is ongoing.
The safety registry is supported by Eli Lilly, which markets ixekizumab. Dr. Kimball reported receiving research funding from and serving as a consultant to Eli Lilly and numerous other pharmaceutical companies.
THE EADV CONGRESS
Pharyngeal reservoir drives gonorrhea epidemic in gay men
VIENNA – , Colm O’Mahony, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We think pharyngeal gonorrhea is now the most important factor in this continuing epidemic,” according to Dr. O’Mahony, professor of medicine and a venereologist at the University of Chester (England).
Recent studies have shown that men can carry Neisseria gonorrhoeae in their throats for weeks or months without symptoms and can readily spread the infection through unprotected oral sex.
Also, surveys of men who have sex with men (MSM) indicate that saliva is commonly used as a lubricant for anal sex. Australian investigators have estimated that about half of rectal gonorrhea in MSM would be eliminated if they stopped using their partner’s saliva for this purpose (Sex Trans Infect. 2016 Mar 3. pii:sextrans-2015-052502. doi: 10.1136/sextrans-2015-052502).
And then there is French kissing.
“Men don’t go out to a nightclub and have indiscriminate anal sex anymore. It’s not like that,” according to Dr. O’Mahony. “But they do kiss quite a lot of other men over the course of an evening, and it’s deep kissing. They may French kiss 15-20 other young men. And we think there’s actually a significant risk of transmission of gonorrhea from this simple deep kissing.”
Indeed, Australian investigators are now conducting a study examining whether having young gay men take a bottle of mouthwash with them when they go clubbing so they can take a good swish in between kissing will protect against N. gonorrhoeae infection.
“Apparently gonorrhea is quite sensitive to mouthwashes like Listerine. So we await those study results with interest,” he continued.
Dr. O’Mahony warned that the problem of multidrug-resistant gonorrhea is further along than most noninfectious disease experts realize. That’s a frightening prospect, given that an estimated 800,000 cases of gonorrhea occur annually in the United States alone. Because of well-documented treatment failures with cefixime and other oral cephalosporins, the Centers for Disease Control and Prevention now recommends only one regimen for the treatment of gonorrhea: dual treatment with a single intramuscular dose of 250 mg of ceftriaxone (Rocephin) plus 1 g of azithromycin in a single dose.
“There have already been some cases of ceftriaxone-resistant gonorrhea reported in Japan, Spain, and other parts of Europe. And we’re now seeing azithromycin-resistant gonorrhea throughout the U.K., which is a problem. So we are really worried that we will end up with untreatable gonorrhea within a couple of years,” Dr. O’Mahony said.
The evolving antimicrobial resistance scenario is reminiscent of the quinolone experience, he added.
“In 1992, I published the first reported case of quinolone-resistant gonorrhea in Liverpool. Five years later we had to stop using quinolones because more than 10% of gonorrhea was resistant,” the venereologist said.
In mid-2016 the CDC published the first-ever comprehensive data from its Gonococcal Isolate Surveillance Project. An analysis of more than 5,000 N. gonorrhoeae isolates obtained from men with gonococcal urethritis presenting at U.S. STD clinics showed that 25.3% of samples were resistant to tetracycline, 19.2% to ciprofloxacin, and 16.2% to penicillin. The prevalence of reduced azithromycin susceptibility jumped from 0.6% in 2013 to 2.5% in 2014. Reduced ceftriaxone susceptibility doubled from 0.4% in 2013 to 0.8% the following year. Antimicrobial susceptibility patterns varied by geographic region, with the highest rates of reduced susceptibility being seen in the Midwest and among MSM (MMWR Surveill Summ. 2016;65[No. SS-7]:1–19. doi: 10.15585/mmwr.ss6507a1).
Dr. O’Mahony reported having no relevant financial conflicts.
VIENNA – , Colm O’Mahony, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We think pharyngeal gonorrhea is now the most important factor in this continuing epidemic,” according to Dr. O’Mahony, professor of medicine and a venereologist at the University of Chester (England).
Recent studies have shown that men can carry Neisseria gonorrhoeae in their throats for weeks or months without symptoms and can readily spread the infection through unprotected oral sex.
Also, surveys of men who have sex with men (MSM) indicate that saliva is commonly used as a lubricant for anal sex. Australian investigators have estimated that about half of rectal gonorrhea in MSM would be eliminated if they stopped using their partner’s saliva for this purpose (Sex Trans Infect. 2016 Mar 3. pii:sextrans-2015-052502. doi: 10.1136/sextrans-2015-052502).
And then there is French kissing.
“Men don’t go out to a nightclub and have indiscriminate anal sex anymore. It’s not like that,” according to Dr. O’Mahony. “But they do kiss quite a lot of other men over the course of an evening, and it’s deep kissing. They may French kiss 15-20 other young men. And we think there’s actually a significant risk of transmission of gonorrhea from this simple deep kissing.”
Indeed, Australian investigators are now conducting a study examining whether having young gay men take a bottle of mouthwash with them when they go clubbing so they can take a good swish in between kissing will protect against N. gonorrhoeae infection.
“Apparently gonorrhea is quite sensitive to mouthwashes like Listerine. So we await those study results with interest,” he continued.
Dr. O’Mahony warned that the problem of multidrug-resistant gonorrhea is further along than most noninfectious disease experts realize. That’s a frightening prospect, given that an estimated 800,000 cases of gonorrhea occur annually in the United States alone. Because of well-documented treatment failures with cefixime and other oral cephalosporins, the Centers for Disease Control and Prevention now recommends only one regimen for the treatment of gonorrhea: dual treatment with a single intramuscular dose of 250 mg of ceftriaxone (Rocephin) plus 1 g of azithromycin in a single dose.
“There have already been some cases of ceftriaxone-resistant gonorrhea reported in Japan, Spain, and other parts of Europe. And we’re now seeing azithromycin-resistant gonorrhea throughout the U.K., which is a problem. So we are really worried that we will end up with untreatable gonorrhea within a couple of years,” Dr. O’Mahony said.
The evolving antimicrobial resistance scenario is reminiscent of the quinolone experience, he added.
“In 1992, I published the first reported case of quinolone-resistant gonorrhea in Liverpool. Five years later we had to stop using quinolones because more than 10% of gonorrhea was resistant,” the venereologist said.
In mid-2016 the CDC published the first-ever comprehensive data from its Gonococcal Isolate Surveillance Project. An analysis of more than 5,000 N. gonorrhoeae isolates obtained from men with gonococcal urethritis presenting at U.S. STD clinics showed that 25.3% of samples were resistant to tetracycline, 19.2% to ciprofloxacin, and 16.2% to penicillin. The prevalence of reduced azithromycin susceptibility jumped from 0.6% in 2013 to 2.5% in 2014. Reduced ceftriaxone susceptibility doubled from 0.4% in 2013 to 0.8% the following year. Antimicrobial susceptibility patterns varied by geographic region, with the highest rates of reduced susceptibility being seen in the Midwest and among MSM (MMWR Surveill Summ. 2016;65[No. SS-7]:1–19. doi: 10.15585/mmwr.ss6507a1).
Dr. O’Mahony reported having no relevant financial conflicts.
VIENNA – , Colm O’Mahony, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“We think pharyngeal gonorrhea is now the most important factor in this continuing epidemic,” according to Dr. O’Mahony, professor of medicine and a venereologist at the University of Chester (England).
Recent studies have shown that men can carry Neisseria gonorrhoeae in their throats for weeks or months without symptoms and can readily spread the infection through unprotected oral sex.
Also, surveys of men who have sex with men (MSM) indicate that saliva is commonly used as a lubricant for anal sex. Australian investigators have estimated that about half of rectal gonorrhea in MSM would be eliminated if they stopped using their partner’s saliva for this purpose (Sex Trans Infect. 2016 Mar 3. pii:sextrans-2015-052502. doi: 10.1136/sextrans-2015-052502).
And then there is French kissing.
“Men don’t go out to a nightclub and have indiscriminate anal sex anymore. It’s not like that,” according to Dr. O’Mahony. “But they do kiss quite a lot of other men over the course of an evening, and it’s deep kissing. They may French kiss 15-20 other young men. And we think there’s actually a significant risk of transmission of gonorrhea from this simple deep kissing.”
Indeed, Australian investigators are now conducting a study examining whether having young gay men take a bottle of mouthwash with them when they go clubbing so they can take a good swish in between kissing will protect against N. gonorrhoeae infection.
“Apparently gonorrhea is quite sensitive to mouthwashes like Listerine. So we await those study results with interest,” he continued.
Dr. O’Mahony warned that the problem of multidrug-resistant gonorrhea is further along than most noninfectious disease experts realize. That’s a frightening prospect, given that an estimated 800,000 cases of gonorrhea occur annually in the United States alone. Because of well-documented treatment failures with cefixime and other oral cephalosporins, the Centers for Disease Control and Prevention now recommends only one regimen for the treatment of gonorrhea: dual treatment with a single intramuscular dose of 250 mg of ceftriaxone (Rocephin) plus 1 g of azithromycin in a single dose.
“There have already been some cases of ceftriaxone-resistant gonorrhea reported in Japan, Spain, and other parts of Europe. And we’re now seeing azithromycin-resistant gonorrhea throughout the U.K., which is a problem. So we are really worried that we will end up with untreatable gonorrhea within a couple of years,” Dr. O’Mahony said.
The evolving antimicrobial resistance scenario is reminiscent of the quinolone experience, he added.
“In 1992, I published the first reported case of quinolone-resistant gonorrhea in Liverpool. Five years later we had to stop using quinolones because more than 10% of gonorrhea was resistant,” the venereologist said.
In mid-2016 the CDC published the first-ever comprehensive data from its Gonococcal Isolate Surveillance Project. An analysis of more than 5,000 N. gonorrhoeae isolates obtained from men with gonococcal urethritis presenting at U.S. STD clinics showed that 25.3% of samples were resistant to tetracycline, 19.2% to ciprofloxacin, and 16.2% to penicillin. The prevalence of reduced azithromycin susceptibility jumped from 0.6% in 2013 to 2.5% in 2014. Reduced ceftriaxone susceptibility doubled from 0.4% in 2013 to 0.8% the following year. Antimicrobial susceptibility patterns varied by geographic region, with the highest rates of reduced susceptibility being seen in the Midwest and among MSM (MMWR Surveill Summ. 2016;65[No. SS-7]:1–19. doi: 10.15585/mmwr.ss6507a1).
Dr. O’Mahony reported having no relevant financial conflicts.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Novel oral antifungal headed to phase III for onychomycosis
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.
He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks. 
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.
He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks.
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
VIENNA – A once-weekly oral antifungal drug known as VT-1161 will move into phase III clinical testing in 2017 based on its impressive performance in an interim analysis of a phase IIb study, Amir Tavakkol, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We saw robust evidence of clinical and mycologic efficacy in patients with moderate to severe onychomycosis. This was true even in patients with nails considered very difficult to treat because of dermatophytomas and spikes, which are usually poor prognostic elements,” said Dr. Tavakkol, chief development officer at Viamet Pharmaceuticals in Durham, N.C., which is developing VT-1161.
He reported on 107 patients with toenail onychomycosis who enrolled in the phase IIb, double-blind, placebo-controlled RENOVATE (Restoring Nail: An Oral VT-1161 Tablet Evaluation) study. At enrollment they averaged 47% disease involvement of the big toenail, the target nail for the trial. Participants in the five study arms had an average of 4.2-5.0 affected toenails. Both the percentage of nail involvement and the number of diseased toenails were roughly twice as great as is typical in studies of topical antifungals, underscoring that participants in the VT-1161 trial had fairly severe onychomycosis.
Patients were assigned to one of five study arms: 300 mg of VT-1161 once weekly for 12 weeks, then 12 weeks of placebo; 600 mg of VT-1161 once weekly for 12 weeks, followed by 12 weeks of placebo; either 300 or 600 mg of the antifungal agent once weekly for the full 24 weeks; or 24 weeks of once-per-week placebo. Immediately prior to the formal start of the study, however, everyone received 14 days of a once-daily loading dose of VT-1161 or placebo at the dose they would subsequently take once weekly during the trial.
The primary outcome in the ongoing study is the percentage of patients with a complete cure, both mycologic and clinical, at 48 weeks. Those data aren’t in yet, but Dr. Tavakkol presented the results of the prespecified interim analysis at 24 weeks.
“Please keep in mind that this is only at most 24 weeks of therapy. Given the rate of nail growth at 1 mm per month when it is healthy, these are remarkable data. Ten percent or less nail involvement is basically 1-2 mm at the distal end. I believe a substantial percentage of these patients will reach clinical cure by 48 weeks,” he said.
VT-1161, a molecule with high selectivity for fungal CYP51, blocks the production of ergosterol, a key component of the fungal cell membrane, according to the company. It has no known drug interactions. At all doses studied in the trial, it was safe and well tolerated, with no abnormal liver function tests, no effect on bilirubin, and no change in QTc interval. Only 8 of the 107 patients reported adverse events deemed possibly related to the study drug by blinded investigators. No one dropped out of the study.
“VT-1161 is also being developed for recurrent vulvovaginal candidiasis. The results there are outstanding, too,” Dr. Tavakkol said.
The trial was funded by Viamet, where he is employed.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Among onychomycosis patients with an average 47% target toenail involvement at baseline, 35% had improved to no more than 10% nail involvement at 24 weeks after 12 weeks of once-weekly VT-1161 followed by 12 weeks of placebo.
Data source: A double-blind, randomized, phase IIb clinical trial involving 107 patients with toenail onychomycosis.
Disclosures: The trial was funded by Viamet Pharmaceuticals, where the study presenter is employed.