User login
Two probiotic products don’t prevent gastroenteritis in children, studies show
Two probiotic products containing strains of Lactobacillus rhamnosus failed to prevent moderate-to-severe gastroenteritis in children, according to the results of large, randomized trials published in the New England Journal of Medicine.
Neither probiotic formulation significantly reduced duration of diarrhea or vomiting, or improved endpoints such as daycare absenteeism in the double-blind, placebo-controlled trials, which together included 1,857 infants or children with acute infectious gastroenteritis treated in the United States or Canada.
In one of the two trials, conducted at 10 U.S. pediatric emergency departments, a 5-day course of L. rhamnosus GG did not improve outcomes, versus placebo, according to investigators, led by David Schnadower, MD, of Cincinnati (Ohio) Children’s Hospital Medical Center.
Results of the trial, which comprised 971 children aged 3 months to 4 years, sharply contrast with results of previous studies and meta-analyses suggesting probiotics do improve outcomes in children with acute gastroenteritis.
However, those studies were hampered by small sample sizes, lack of probiotic quality control, and endpoints “of questionable relevance,” among other limitations, according to Dr. Schnadower and his coauthors.
“The rigor of our research design calls into question recommendations to use L. rhamnosus GG in the treatment of children with acute gastroenteritis,” the authors said in their published report.
Moderate to severe gastroenteritis within 14 days of enrollment, the trial’s primary endpoint, occurred in 11.8% of children who received the probiotic, and in 12.6% of those who received placebo (P = .83).
Diarrhea duration was similar, at 49.7 hours and 50.9 hours in the probiotic and placebo groups, respectively (P = .26). Likewise, there were no significant differences in duration of vomiting, daycare absenteeism, or rate of household transmission between the study arms, investigators reported.
In the Canadian trial, which was similar to the U.S. trial but conducted independently, a probiotic product containing L. rhamnosus R0011 and L. helveticus R0052 also showed no significant benefit over placebo in reducing incidence of moderate to severe gastroenteritis within 14 days of enrollment.
That endpoint occurred in 26.1% of children assigned to probiotics, and 24.7% assigned to placebo (P = .72). The trial comprised 886 children 3-48 months presenting to one of six pediatric emergency departments in Canada.
As in the U.S. trial, investigators said there were no significant differences in diarrhea duration, at 52.5 and 55.5 hours in the probiotic and placebo groups, respectively (P = .31). And there were no significant differences in duration of vomiting, unscheduled health care provider visits, or adverse events.
Both trials used a modified Vesikari scale symptom score of 9 or higher (range, 0-20) to define moderate to severe gastroenteritis.
Rather than focusing on a single symptom such as diarrhea, the modified Vesikari scale score shows a “constellation of symptoms” associated with gastroenteritis, according to the Canadian investigators, led by Stephen B. Freedman, MDCM, of the department of pediatrics at Alberta Children’s Hospital and Research Institute, University of Calgary.
Although the use of composite measures has been questioned, the modified Vesikari scale is externally validated and produced consistent findings for individual symptoms, according to the authors. “Analysis of all individual score elements supported the conclusions based on our primary outcome,” they wrote.
Despite the findings, the conclusions about the particular probiotic product evaluated in the trial cannot be generalized to others in the market, according to Dr. Freedman and his colleagues. Other “large, well conducted trials have aroused similar concerns regarding the effectiveness of probiotics for other conditions,” they added. “Nonetheless, there may be specific indications and populations that will benefit from alternative probiotic agents.”
The U.S. study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, among other sources. Dr. Schnadower reported that he received grants from the NICHD and nonfinancial support from iHealth.
The Canadian study was supported by the Canadian Institutes of Health Research, among other sources. Dr. Freedman reported that he received nonfinancial support from Calgary Laboratory Services, Copan Italia, Lallemand Health Solutions, Luminex, and ProvLab Alberta, along with grants from the Canadian Institutes of Health Research and Alberta Children’s Hospital Foundation.
SOURCES: Schnadower D et al. N Engl J Med. 2018 Nov 22;379(21):2002-14; Freedman SB et al. N Engl J Med. 2018 Nov 22;379(21);2015-26.
These two studies, which are large and well conducted, do not support use of probiotics that contain Lactobacillus rhamnosus for moderate to severe gastroenteritis in children, according to J. Thomas LaMont, MD.
“These negative trial data will be valuable to clinicians and professional bodies in making decisions regarding the use of either of these probiotic formulations in children with diarrhea,” Dr. LaMont said in an editorial.
Recommendations to use probiotics to treat acute gastroenteritis, as published by some professional societies, rely largely on studies that were underpowered or had issues related to study design or choice of endpoint, Dr. LaMont cautioned.
That said, there are many other probiotic formulations beyond the two evaluated in these trials, he added. Other probiotic agents have different mechanisms of action and ability to colonize the bowel, compared with L. rhamnosus, and thus could be effective against infectious diarrhea in children.
A probiotic formula including L. plantarum significantly reduced the sepsis rate in healthy newborns in one recent placebo-controlled trial in India, he added. That probiotic strain can colonize the intestinal tract for extended periods, compared with other probiotics.
“With their low cost and minimal toxic effects, probiotics have potential for the treatment of a variety of gastrointestinal and other diseases, but rigorous trials such as those described in this [study] are required to determine any potential efficacy or effectiveness,” Dr. LaMont concluded.
Dr. LaMont is with the division of gastroenterology, Beth Israel Deaconess Medical Center, Boston. He had no disclosures related to his editorial ( N Engl J Med. 2018 Nov 22;379[21]:2076-7 ).
These two studies, which are large and well conducted, do not support use of probiotics that contain Lactobacillus rhamnosus for moderate to severe gastroenteritis in children, according to J. Thomas LaMont, MD.
“These negative trial data will be valuable to clinicians and professional bodies in making decisions regarding the use of either of these probiotic formulations in children with diarrhea,” Dr. LaMont said in an editorial.
Recommendations to use probiotics to treat acute gastroenteritis, as published by some professional societies, rely largely on studies that were underpowered or had issues related to study design or choice of endpoint, Dr. LaMont cautioned.
That said, there are many other probiotic formulations beyond the two evaluated in these trials, he added. Other probiotic agents have different mechanisms of action and ability to colonize the bowel, compared with L. rhamnosus, and thus could be effective against infectious diarrhea in children.
A probiotic formula including L. plantarum significantly reduced the sepsis rate in healthy newborns in one recent placebo-controlled trial in India, he added. That probiotic strain can colonize the intestinal tract for extended periods, compared with other probiotics.
“With their low cost and minimal toxic effects, probiotics have potential for the treatment of a variety of gastrointestinal and other diseases, but rigorous trials such as those described in this [study] are required to determine any potential efficacy or effectiveness,” Dr. LaMont concluded.
Dr. LaMont is with the division of gastroenterology, Beth Israel Deaconess Medical Center, Boston. He had no disclosures related to his editorial ( N Engl J Med. 2018 Nov 22;379[21]:2076-7 ).
These two studies, which are large and well conducted, do not support use of probiotics that contain Lactobacillus rhamnosus for moderate to severe gastroenteritis in children, according to J. Thomas LaMont, MD.
“These negative trial data will be valuable to clinicians and professional bodies in making decisions regarding the use of either of these probiotic formulations in children with diarrhea,” Dr. LaMont said in an editorial.
Recommendations to use probiotics to treat acute gastroenteritis, as published by some professional societies, rely largely on studies that were underpowered or had issues related to study design or choice of endpoint, Dr. LaMont cautioned.
That said, there are many other probiotic formulations beyond the two evaluated in these trials, he added. Other probiotic agents have different mechanisms of action and ability to colonize the bowel, compared with L. rhamnosus, and thus could be effective against infectious diarrhea in children.
A probiotic formula including L. plantarum significantly reduced the sepsis rate in healthy newborns in one recent placebo-controlled trial in India, he added. That probiotic strain can colonize the intestinal tract for extended periods, compared with other probiotics.
“With their low cost and minimal toxic effects, probiotics have potential for the treatment of a variety of gastrointestinal and other diseases, but rigorous trials such as those described in this [study] are required to determine any potential efficacy or effectiveness,” Dr. LaMont concluded.
Dr. LaMont is with the division of gastroenterology, Beth Israel Deaconess Medical Center, Boston. He had no disclosures related to his editorial ( N Engl J Med. 2018 Nov 22;379[21]:2076-7 ).
Two probiotic products containing strains of Lactobacillus rhamnosus failed to prevent moderate-to-severe gastroenteritis in children, according to the results of large, randomized trials published in the New England Journal of Medicine.
Neither probiotic formulation significantly reduced duration of diarrhea or vomiting, or improved endpoints such as daycare absenteeism in the double-blind, placebo-controlled trials, which together included 1,857 infants or children with acute infectious gastroenteritis treated in the United States or Canada.
In one of the two trials, conducted at 10 U.S. pediatric emergency departments, a 5-day course of L. rhamnosus GG did not improve outcomes, versus placebo, according to investigators, led by David Schnadower, MD, of Cincinnati (Ohio) Children’s Hospital Medical Center.
Results of the trial, which comprised 971 children aged 3 months to 4 years, sharply contrast with results of previous studies and meta-analyses suggesting probiotics do improve outcomes in children with acute gastroenteritis.
However, those studies were hampered by small sample sizes, lack of probiotic quality control, and endpoints “of questionable relevance,” among other limitations, according to Dr. Schnadower and his coauthors.
“The rigor of our research design calls into question recommendations to use L. rhamnosus GG in the treatment of children with acute gastroenteritis,” the authors said in their published report.
Moderate to severe gastroenteritis within 14 days of enrollment, the trial’s primary endpoint, occurred in 11.8% of children who received the probiotic, and in 12.6% of those who received placebo (P = .83).
Diarrhea duration was similar, at 49.7 hours and 50.9 hours in the probiotic and placebo groups, respectively (P = .26). Likewise, there were no significant differences in duration of vomiting, daycare absenteeism, or rate of household transmission between the study arms, investigators reported.
In the Canadian trial, which was similar to the U.S. trial but conducted independently, a probiotic product containing L. rhamnosus R0011 and L. helveticus R0052 also showed no significant benefit over placebo in reducing incidence of moderate to severe gastroenteritis within 14 days of enrollment.
That endpoint occurred in 26.1% of children assigned to probiotics, and 24.7% assigned to placebo (P = .72). The trial comprised 886 children 3-48 months presenting to one of six pediatric emergency departments in Canada.
As in the U.S. trial, investigators said there were no significant differences in diarrhea duration, at 52.5 and 55.5 hours in the probiotic and placebo groups, respectively (P = .31). And there were no significant differences in duration of vomiting, unscheduled health care provider visits, or adverse events.
Both trials used a modified Vesikari scale symptom score of 9 or higher (range, 0-20) to define moderate to severe gastroenteritis.
Rather than focusing on a single symptom such as diarrhea, the modified Vesikari scale score shows a “constellation of symptoms” associated with gastroenteritis, according to the Canadian investigators, led by Stephen B. Freedman, MDCM, of the department of pediatrics at Alberta Children’s Hospital and Research Institute, University of Calgary.
Although the use of composite measures has been questioned, the modified Vesikari scale is externally validated and produced consistent findings for individual symptoms, according to the authors. “Analysis of all individual score elements supported the conclusions based on our primary outcome,” they wrote.
Despite the findings, the conclusions about the particular probiotic product evaluated in the trial cannot be generalized to others in the market, according to Dr. Freedman and his colleagues. Other “large, well conducted trials have aroused similar concerns regarding the effectiveness of probiotics for other conditions,” they added. “Nonetheless, there may be specific indications and populations that will benefit from alternative probiotic agents.”
The U.S. study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, among other sources. Dr. Schnadower reported that he received grants from the NICHD and nonfinancial support from iHealth.
The Canadian study was supported by the Canadian Institutes of Health Research, among other sources. Dr. Freedman reported that he received nonfinancial support from Calgary Laboratory Services, Copan Italia, Lallemand Health Solutions, Luminex, and ProvLab Alberta, along with grants from the Canadian Institutes of Health Research and Alberta Children’s Hospital Foundation.
SOURCES: Schnadower D et al. N Engl J Med. 2018 Nov 22;379(21):2002-14; Freedman SB et al. N Engl J Med. 2018 Nov 22;379(21);2015-26.
Two probiotic products containing strains of Lactobacillus rhamnosus failed to prevent moderate-to-severe gastroenteritis in children, according to the results of large, randomized trials published in the New England Journal of Medicine.
Neither probiotic formulation significantly reduced duration of diarrhea or vomiting, or improved endpoints such as daycare absenteeism in the double-blind, placebo-controlled trials, which together included 1,857 infants or children with acute infectious gastroenteritis treated in the United States or Canada.
In one of the two trials, conducted at 10 U.S. pediatric emergency departments, a 5-day course of L. rhamnosus GG did not improve outcomes, versus placebo, according to investigators, led by David Schnadower, MD, of Cincinnati (Ohio) Children’s Hospital Medical Center.
Results of the trial, which comprised 971 children aged 3 months to 4 years, sharply contrast with results of previous studies and meta-analyses suggesting probiotics do improve outcomes in children with acute gastroenteritis.
However, those studies were hampered by small sample sizes, lack of probiotic quality control, and endpoints “of questionable relevance,” among other limitations, according to Dr. Schnadower and his coauthors.
“The rigor of our research design calls into question recommendations to use L. rhamnosus GG in the treatment of children with acute gastroenteritis,” the authors said in their published report.
Moderate to severe gastroenteritis within 14 days of enrollment, the trial’s primary endpoint, occurred in 11.8% of children who received the probiotic, and in 12.6% of those who received placebo (P = .83).
Diarrhea duration was similar, at 49.7 hours and 50.9 hours in the probiotic and placebo groups, respectively (P = .26). Likewise, there were no significant differences in duration of vomiting, daycare absenteeism, or rate of household transmission between the study arms, investigators reported.
In the Canadian trial, which was similar to the U.S. trial but conducted independently, a probiotic product containing L. rhamnosus R0011 and L. helveticus R0052 also showed no significant benefit over placebo in reducing incidence of moderate to severe gastroenteritis within 14 days of enrollment.
That endpoint occurred in 26.1% of children assigned to probiotics, and 24.7% assigned to placebo (P = .72). The trial comprised 886 children 3-48 months presenting to one of six pediatric emergency departments in Canada.
As in the U.S. trial, investigators said there were no significant differences in diarrhea duration, at 52.5 and 55.5 hours in the probiotic and placebo groups, respectively (P = .31). And there were no significant differences in duration of vomiting, unscheduled health care provider visits, or adverse events.
Both trials used a modified Vesikari scale symptom score of 9 or higher (range, 0-20) to define moderate to severe gastroenteritis.
Rather than focusing on a single symptom such as diarrhea, the modified Vesikari scale score shows a “constellation of symptoms” associated with gastroenteritis, according to the Canadian investigators, led by Stephen B. Freedman, MDCM, of the department of pediatrics at Alberta Children’s Hospital and Research Institute, University of Calgary.
Although the use of composite measures has been questioned, the modified Vesikari scale is externally validated and produced consistent findings for individual symptoms, according to the authors. “Analysis of all individual score elements supported the conclusions based on our primary outcome,” they wrote.
Despite the findings, the conclusions about the particular probiotic product evaluated in the trial cannot be generalized to others in the market, according to Dr. Freedman and his colleagues. Other “large, well conducted trials have aroused similar concerns regarding the effectiveness of probiotics for other conditions,” they added. “Nonetheless, there may be specific indications and populations that will benefit from alternative probiotic agents.”
The U.S. study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, among other sources. Dr. Schnadower reported that he received grants from the NICHD and nonfinancial support from iHealth.
The Canadian study was supported by the Canadian Institutes of Health Research, among other sources. Dr. Freedman reported that he received nonfinancial support from Calgary Laboratory Services, Copan Italia, Lallemand Health Solutions, Luminex, and ProvLab Alberta, along with grants from the Canadian Institutes of Health Research and Alberta Children’s Hospital Foundation.
SOURCES: Schnadower D et al. N Engl J Med. 2018 Nov 22;379(21):2002-14; Freedman SB et al. N Engl J Med. 2018 Nov 22;379(21);2015-26.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Two probiotic products containing strains of Lactobacillus rhamnosus did not prevent moderate to severe gastroenteritis in children.
Major finding: Neither probiotic formulation significantly reduced duration of diarrhea or vomiting, or improved endpoints such as daycare absenteeism.
Study details: Two randomized, controlled trials, comprising 1,857 infants or children with acute infectious gastroenteritis.
Disclosures: The U.S. study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, among other sources. Dr. Schnadower reported that he received grants from the NICHD and nonfinancial support from iHealth. The Canadian study was supported by the Canadian Institutes of Health Research, among other sources. Dr. Freedman reported that he received nonfinancial support from Calgary Laboratory Services, Copan Italia, Lallemand Health Solutions, Luminex, and ProvLab Alberta, along with grants from the Canadian Institutes of Health Research and Alberta Children’s Hospital Foundation.
Sources: Schnadower D et al. N Engl J Med. 2018 Nov 22;379(21):2002-14; Freedman SB et al. N Engl J Med. 2018 Nov 22;379(21);2015-26.
FDA approves glasdegib for AML
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.

The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration has approved the hedgehog pathway inhibitor glasdegib (Daurismo) for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed acute myeloid leukemia who are aged 75 years and older or who are ineligible for intensive chemotherapy.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038). This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions. The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n = 84) or LDAC alone (n = 41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib-LDAC arm and 2.6% (1/38) in the LDAC-only arm. The median overall survival was 8.3 months in the glasdegib-LDAC arm and 4.3 months in the LDAC-only arm (hazard ratio, 0.46; P = .0002).
The most common adverse events in the first 90 days of treatment, occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively, were anemia (43% and 42%), fatigue (36% and 32%), hemorrhage (36% and 42%), febrile neutropenia (31% and 22%), musculoskeletal pain (30% and 17%), edema (30% and 20%), and thrombocytopenia (30% and 27%).
The incidence of serious adverse events was 79% in the glasdegib arm, and the most common events were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%) and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib.
Glasdegib is a product of Pfizer.
FDA approves venetoclax for AML

The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval to venetoclax (Venclexta®) for use in acute myeloid leukemia (AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit.
Therefore, continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies—the phase 1 b M14-358 trial (NCT02203773) and the phase 1/2 M14-387 trial (NCT02287233).
M14-358 trial
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively.
The most common adverse events (AEs)—occurring in at least 30% of patients in both arms—were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia.
The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome.
The incidence of fatal AEs was 1.5% within 30 days of treatment initiation.
M14-387 trial
The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea.
The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection.
The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche. It is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the United States and by AbbVie elsewhere.
FDA approves glasdegib for AML

The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
The U.S. Food and Drug Administration (FDA) has approved the hedgehog pathway inhibitor glasdegib (Daurismo™) to treat certain patients with acute myeloid leukemia (AML).
Glasdegib is approved for use in combination with low-dose cytarabine (LDAC) to treat adults with newly diagnosed AML who are age 75 and older or who are ineligible for intensive chemotherapy.
Glasdegib was approved under priority review and also received orphan drug designation from the FDA.
The prescribing information for glasdegib includes a boxed warning detailing the risk of embryo-fetal death or severe birth defects associated with the drug.
The FDA’s approval of glasdegib is based on results from the phase 2 BRIGHT AML 1003 trial (NCT01546038).
This trial included 111 adults with newly diagnosed AML and 14 patients with other conditions.
The patients were randomized to receive glasdegib (at 100 mg daily) in combination with LDAC (n=84) or LDAC alone (n=41).
The complete response rate among the AML patients was 18.2% (14/77) in the glasdegib arm and 2.6% (1/38) in the LDAC arm.
The median overall survival was 8.3 months in the glasdegib arm and 4.3 months in the LDAC arm (hazard ratio=0.46; P=0.0002).
The most common adverse events (AEs) in the first 90 days of treatment—occurring in at least 30% of patients in either arm (glasdegib-LDAC and LDAC alone, respectively)—were:
- Anemia (43% and 42%)
- Fatigue (36% and 32%)
- Hemorrhage (36% and 42%)
- Febrile neutropenia (31% and 22%)
- Musculoskeletal pain (30% and 17%)
- Edema (30% and 20%)
- Thrombocytopenia (30% and 27%).
The incidence of serious AEs was 79% in the glasdegib arm. The most common serious AEs occurring in at least 5% of patients in this arm were febrile neutropenia (29%), pneumonia (23%), hemorrhage (12%), anemia (7%), and sepsis (7%).
Additional data from this trial are included in the prescribing information for glasdegib. Glasdegib is a product of Pfizer.
Venetoclax gets accelerated approval in older AML patients
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
ASH 2018 coming attractions look at the big picture
In the closest thing the medical world has to movie trailers, the American Society of Hematology held a press conference offering
Shorter R-CHOP regimen for DLBCL
Under the heading “Big Trials, Big Results” will be data from the FLYER trial, a phase 3, randomized, deescalation trial in 592 patients aged 18-60 years with favorable-prognosis diffuse large B-cell lymphoma. The investigators report that both progression-free survival and overall survival with four cycles of R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone) were noninferior to those for patients treated with six cycles of R-CHOP (abstract 781).

Ibrutinib mastery in CLL
Also on the program are results of a study showing that ibrutinib (Imbruvica), either alone or in combination with rituximab, is associated with superior progression-free survival than bendamustine and rituximab in older patients with chronic lymphocytic leukemia (CLL).
The trial, the Alliance North American Intergroup Study A041202 (abstract 6) is the first major trial to pit ibrutinib against the modern standard of immunochemotherapy rather than the older standard of chlorambucil, Dr. Brodsky noted.
Anemia support in beta-thalassemia, MDS
In nonmalignant disease, investigators in the randomized, phase 3 BELIEVE trial are reporting results of their study showing that the first-in-class erythroid maturation agent luspatercept was associated with significant reductions in the need for RBC transfusion in adults with transfusion-dependent beta-thalassemia.
The investigators report that the experimental agent was “generally well tolerated” (abstract 163).
“Beyond a proof of principle, [this is] certainly a very exciting advancement in this group of patients who otherwise had very few treatment options,” said Alexis A. Thompson, MD, associate director of equity and minority health at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, and the current ASH president.
Dr. Thompson also highlighted the MEDALIST trial (abstract 1), a phase 3, randomized study showing that luspatercept significantly reduced transfusion burden, compared with placebo, in patients with anemia caused by very low–, low-, or intermediate-risk myelodysplastic syndrome with ring sideroblasts who require RBC transfusions.
“This group of patients were individuals who were refractory or were not responders or did not tolerate erythropoietic stimulating agents and therefore were requiring regular transfusion,” Dr. Thompson said.
Worth the wait
The late-breaking abstract program was stretched from the usual six abstracts to seven this year because of the unusually high quality of the science, Dr. Brodsky said.
Among these star attractions are results of a phase 3, randomized study of daratumumab (Darzalex) plus lenalidomide and dexamethasone versus lenalidomide-dexamethasone alone for patients with newly diagnosed multiple myeloma who are ineligible for transplant.
The investigators found that adding daratumumab reduced the risk of disease progression or death by close to 50%, supporting the combination as a new standard of care in these patients, according to Thierry Facon, MD, from the Hospital Claude Huriez in Lille, France, and colleagues (abstract LBA-2).
Two other late-breakers deal with CLL. The first, a randomized, phase 3 study of ibrutinib-based therapy versus standard fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy in younger patients with untreated CLL, found that ibrutinib and rituximab provided significantly better progression-free survival and overall survival (abstract LBA-4).
“These findings have immediate practice-changing implications and establish ibrutinib-based therapy as the most efficacious first-line therapy for patients with CLL,” wrote Tait D. Shanafelt, MD, from Stanford (Calif.) University, and colleagues.
On a less positive note, Australian researchers report their discovery of a recurrent mutation in BCL2 that confers resistance to venetoclax (Venclexta) in patients with progressive CLL (abstract LBA-7).
“This mutation provides new insights into the pathobiology of venetoclax resistance and provides a potential biomarker of impending clinical relapse,” wrote Piers Blombery, MBBS, from the University of Melbourne, and colleagues.
Finally, investigators from children’s hospitals in the United States and Europe report promising findings on the safety and efficacy of emapalumab for the treatment of patients with the rare genetic disorder primary hemophagocytic lymphohistiocytosis (HLH).
The drug, newly approved by the Food and Drug Administration, was able to control HLH’s hyperinflammatory activity, and allowed a substantial proportion of patients to survive to hematopoietic stem cell transplantation, the investigators said (abstract LBA-6).
In the closest thing the medical world has to movie trailers, the American Society of Hematology held a press conference offering
Shorter R-CHOP regimen for DLBCL
Under the heading “Big Trials, Big Results” will be data from the FLYER trial, a phase 3, randomized, deescalation trial in 592 patients aged 18-60 years with favorable-prognosis diffuse large B-cell lymphoma. The investigators report that both progression-free survival and overall survival with four cycles of R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone) were noninferior to those for patients treated with six cycles of R-CHOP (abstract 781).
Ibrutinib mastery in CLL
Also on the program are results of a study showing that ibrutinib (Imbruvica), either alone or in combination with rituximab, is associated with superior progression-free survival than bendamustine and rituximab in older patients with chronic lymphocytic leukemia (CLL).
The trial, the Alliance North American Intergroup Study A041202 (abstract 6) is the first major trial to pit ibrutinib against the modern standard of immunochemotherapy rather than the older standard of chlorambucil, Dr. Brodsky noted.
Anemia support in beta-thalassemia, MDS
In nonmalignant disease, investigators in the randomized, phase 3 BELIEVE trial are reporting results of their study showing that the first-in-class erythroid maturation agent luspatercept was associated with significant reductions in the need for RBC transfusion in adults with transfusion-dependent beta-thalassemia.
The investigators report that the experimental agent was “generally well tolerated” (abstract 163).
“Beyond a proof of principle, [this is] certainly a very exciting advancement in this group of patients who otherwise had very few treatment options,” said Alexis A. Thompson, MD, associate director of equity and minority health at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, and the current ASH president.
Dr. Thompson also highlighted the MEDALIST trial (abstract 1), a phase 3, randomized study showing that luspatercept significantly reduced transfusion burden, compared with placebo, in patients with anemia caused by very low–, low-, or intermediate-risk myelodysplastic syndrome with ring sideroblasts who require RBC transfusions.
“This group of patients were individuals who were refractory or were not responders or did not tolerate erythropoietic stimulating agents and therefore were requiring regular transfusion,” Dr. Thompson said.
Worth the wait
The late-breaking abstract program was stretched from the usual six abstracts to seven this year because of the unusually high quality of the science, Dr. Brodsky said.
Among these star attractions are results of a phase 3, randomized study of daratumumab (Darzalex) plus lenalidomide and dexamethasone versus lenalidomide-dexamethasone alone for patients with newly diagnosed multiple myeloma who are ineligible for transplant.
The investigators found that adding daratumumab reduced the risk of disease progression or death by close to 50%, supporting the combination as a new standard of care in these patients, according to Thierry Facon, MD, from the Hospital Claude Huriez in Lille, France, and colleagues (abstract LBA-2).
Two other late-breakers deal with CLL. The first, a randomized, phase 3 study of ibrutinib-based therapy versus standard fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy in younger patients with untreated CLL, found that ibrutinib and rituximab provided significantly better progression-free survival and overall survival (abstract LBA-4).
“These findings have immediate practice-changing implications and establish ibrutinib-based therapy as the most efficacious first-line therapy for patients with CLL,” wrote Tait D. Shanafelt, MD, from Stanford (Calif.) University, and colleagues.
On a less positive note, Australian researchers report their discovery of a recurrent mutation in BCL2 that confers resistance to venetoclax (Venclexta) in patients with progressive CLL (abstract LBA-7).
“This mutation provides new insights into the pathobiology of venetoclax resistance and provides a potential biomarker of impending clinical relapse,” wrote Piers Blombery, MBBS, from the University of Melbourne, and colleagues.
Finally, investigators from children’s hospitals in the United States and Europe report promising findings on the safety and efficacy of emapalumab for the treatment of patients with the rare genetic disorder primary hemophagocytic lymphohistiocytosis (HLH).
The drug, newly approved by the Food and Drug Administration, was able to control HLH’s hyperinflammatory activity, and allowed a substantial proportion of patients to survive to hematopoietic stem cell transplantation, the investigators said (abstract LBA-6).
In the closest thing the medical world has to movie trailers, the American Society of Hematology held a press conference offering
Shorter R-CHOP regimen for DLBCL
Under the heading “Big Trials, Big Results” will be data from the FLYER trial, a phase 3, randomized, deescalation trial in 592 patients aged 18-60 years with favorable-prognosis diffuse large B-cell lymphoma. The investigators report that both progression-free survival and overall survival with four cycles of R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone) were noninferior to those for patients treated with six cycles of R-CHOP (abstract 781).
Ibrutinib mastery in CLL
Also on the program are results of a study showing that ibrutinib (Imbruvica), either alone or in combination with rituximab, is associated with superior progression-free survival than bendamustine and rituximab in older patients with chronic lymphocytic leukemia (CLL).
The trial, the Alliance North American Intergroup Study A041202 (abstract 6) is the first major trial to pit ibrutinib against the modern standard of immunochemotherapy rather than the older standard of chlorambucil, Dr. Brodsky noted.
Anemia support in beta-thalassemia, MDS
In nonmalignant disease, investigators in the randomized, phase 3 BELIEVE trial are reporting results of their study showing that the first-in-class erythroid maturation agent luspatercept was associated with significant reductions in the need for RBC transfusion in adults with transfusion-dependent beta-thalassemia.
The investigators report that the experimental agent was “generally well tolerated” (abstract 163).
“Beyond a proof of principle, [this is] certainly a very exciting advancement in this group of patients who otherwise had very few treatment options,” said Alexis A. Thompson, MD, associate director of equity and minority health at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, and the current ASH president.
Dr. Thompson also highlighted the MEDALIST trial (abstract 1), a phase 3, randomized study showing that luspatercept significantly reduced transfusion burden, compared with placebo, in patients with anemia caused by very low–, low-, or intermediate-risk myelodysplastic syndrome with ring sideroblasts who require RBC transfusions.
“This group of patients were individuals who were refractory or were not responders or did not tolerate erythropoietic stimulating agents and therefore were requiring regular transfusion,” Dr. Thompson said.
Worth the wait
The late-breaking abstract program was stretched from the usual six abstracts to seven this year because of the unusually high quality of the science, Dr. Brodsky said.
Among these star attractions are results of a phase 3, randomized study of daratumumab (Darzalex) plus lenalidomide and dexamethasone versus lenalidomide-dexamethasone alone for patients with newly diagnosed multiple myeloma who are ineligible for transplant.
The investigators found that adding daratumumab reduced the risk of disease progression or death by close to 50%, supporting the combination as a new standard of care in these patients, according to Thierry Facon, MD, from the Hospital Claude Huriez in Lille, France, and colleagues (abstract LBA-2).
Two other late-breakers deal with CLL. The first, a randomized, phase 3 study of ibrutinib-based therapy versus standard fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy in younger patients with untreated CLL, found that ibrutinib and rituximab provided significantly better progression-free survival and overall survival (abstract LBA-4).
“These findings have immediate practice-changing implications and establish ibrutinib-based therapy as the most efficacious first-line therapy for patients with CLL,” wrote Tait D. Shanafelt, MD, from Stanford (Calif.) University, and colleagues.
On a less positive note, Australian researchers report their discovery of a recurrent mutation in BCL2 that confers resistance to venetoclax (Venclexta) in patients with progressive CLL (abstract LBA-7).
“This mutation provides new insights into the pathobiology of venetoclax resistance and provides a potential biomarker of impending clinical relapse,” wrote Piers Blombery, MBBS, from the University of Melbourne, and colleagues.
Finally, investigators from children’s hospitals in the United States and Europe report promising findings on the safety and efficacy of emapalumab for the treatment of patients with the rare genetic disorder primary hemophagocytic lymphohistiocytosis (HLH).
The drug, newly approved by the Food and Drug Administration, was able to control HLH’s hyperinflammatory activity, and allowed a substantial proportion of patients to survive to hematopoietic stem cell transplantation, the investigators said (abstract LBA-6).
Abortion measures continue to fall
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.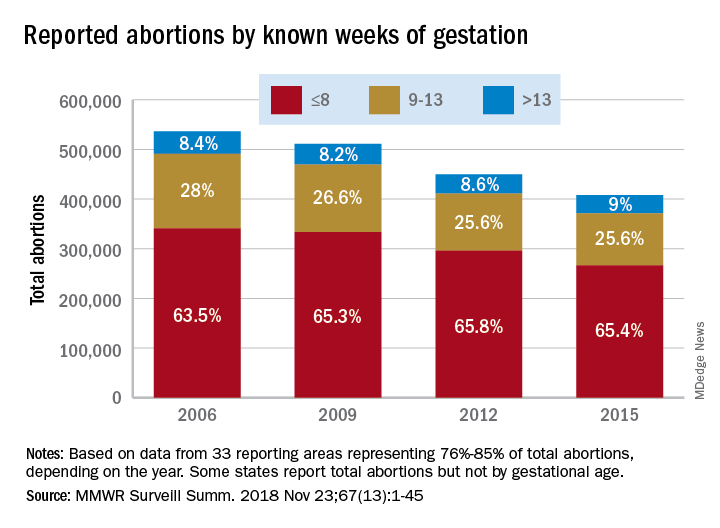
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
Three important national measures of abortion dropped by at least 19% from 2006 to 2015, according to the Centers for Disease Control and Prevention.
Surgical and medical abortions reported to the CDC dropped by 24%, going from almost 843,000 in 2006 to 638,000 in 2015, with declines occurring every year, Tara C. Jatlaoui, MD, and her associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, reported in Morbidity and Mortality Weekly Report Surveillance Summaries.
Over that same time period, the abortion rate fell from 15.9 per 1,000 women aged 15-44 years to 11.8 per 1,000 – a decline of 26%. Abortion ratio – the number of abortions per 1,000 live births within a given population – declined by 19%, dropping from 233 abortions per 1,000 live births in 2006 to 188 abortions per 1,000 live births in 2015, the investigators reported. The findings were based on data from 49 areas that continuously reported over the study period (excludes California, Maryland, and New Hampshire but includes the District of Columbia and New York City).
Abortion rates were highest for women aged 20-29 years for the study period, and this age group accounted for the largest share among the 44 reporting areas that provided data by maternal age each year. Those under age 15 years had the largest drops by age in total number of abortions (40%) and abortion rate (58%) but had the highest, by far, abortion ratio for each year of the study (700 per 1,000 live births in that age group in 2015. The abortion ratio for 15- to 19-year-olds was 289 per 1,000 live births).
The percentage of abortions performed before 14 weeks’ gestation changed little, going from 91.5% in 2006 to 91% in 2015, but “a shift occurred toward earlier gestational ages,” they noted. The percentage of abortions performed before or at 8 weeks increased 3% and those done at 9-13 weeks dropped almost 9% among the 33 areas that reported gestational age every year. Abortions done after 13 weeks gestation represented 9% of all abortions during the study period, with an increase of 7% occurring from 2006 to 2015, Dr. Jatlaoui and her associates said.
Removing barriers such as cost, “insufficient provider reimbursement and training, inadequate client-centered counseling, lack of youth-friendly services, and low client awareness ... might help improve contraceptive use, potentially reducing the number of unintended pregnancies and the number of abortions performed in the United States,” the researchers wrote.
SOURCE: Jatlaoui TC et al. MMWR Surveill Summ. 2018 Nov 23(13):1-45.
FROM MMWR SURVEILLANCE SUMMARIES
Mylan issues voluntary recall of certain valsartan-containing products
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.

The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.
The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
Mylan has announced that it is voluntarily recalling 15 lots of products containing valsartan because of the detection of trace amounts of N-nitrosodiethylamine within the active ingredient, according to a company announcement posted on the website of the Food and Drug Administration.
The affected products include six lots of amlodipine/valsartan tablets (5-mg/160-mg, 10-mg/160-mg, and 10-mg/320-mg strengths), seven lots of valsartan tablets (40-mg, 80-mg, 160-mg, and 320-mg strengths), and two lots of valsartan/hydrochlorothiazide tablets (320-mg/25-mg strength). All products were distributed between March 2017 and November 2018.
“Patients should contact their pharmacist or physician who can advise them about an alternative treatment prior to returning their medication. Patients who are on valsartan should continue taking their medication, as the risk of harm to the patient’s health may be higher if the treatment is stopped immediately without any alternative treatment,” the company statement said.
N-nitrosodiethylamine is a naturally occurring substance that has been identified as a human carcinogen by the International Agency for Research on Cancer.
The lot and NDC (National Drug Code) numbers of the affected products can be found in the full press release on the FDA website.
Mylan is notifying its distributors and customers by letter and is arranging for return of all recalled products. Wholesalers, retailers, and consumers in possession of recalled product should contact Stericycle at 1-888-406-9305 for the return of the recalled product. Normal business hours are Monday through Friday, 8 a.m. to 5 p.m. EST, according to the statement.
Bariatric surgery ups risk of suicide, self-harm
NASHVILLE – , according to findings from a meta-analysis presented at the meeting.

Overall, the odds ratio was 1.69 for self-harm or suicide after bariatric surgery (95% confidence interval, 1.62-1.76; P less than .001), “indicating a nearly 70% increase in risk for self-harm or suicide following bariatric surgery,” wrote Dawn Roberts, PhD, of Bradley University, Peoria, Ill., and her coauthor, Nicole Pearl of Washington University, St. Louis, in the poster accompanying the presentation.
Further, as elapsed time from surgery grew, the suicide rate dropped (effect size covariance, r = –0.25). “Thus, the greatest risk for self-harm or suicide appears to emerge in the years immediately following surgery,” Dr. Roberts said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
The investigators had a primary objective of characterizing the association between bariatric surgery and suicide or self-harm. The secondary purpose of the meta-analysis was to find moderators of the association that could explain some of the variability that had previously been seen in studies looking at mental health outcomes after bariatric surgery.
Some of the potential moderators, explained the investigators, included the surgery type. With Roux-en-Y gastric bypass (RYGB), more tissue is removed, potentially causing “more extensive disruption of neural pathways,” the investigators wrote. With greater loss of small-intestine surface area might come more disruption of the gut-brain axis, along with unknown effects on metabolism and pharmacokinetics of psychiatric medication. Additionally, alcohol use disorder might have a more profound effect after gastric bypass surgery.
It had previously been shown that more than two-thirds of suicides happen within the first 3 years after bariatric surgery. With the initial weight loss comes renegotiation of personal relationships, and the potential for more mobility and perhaps expanded career choices; stress accompanies even positive changes in these major life domains. Some patients will also experience weight regain within the first 3 years, after an initial nadir. This also can cause deterioration in quality of life, the investigators explained.
Dr. Roberts and her coinvestigator acknowledge that some of the potential moderators may have been missed in the initial data reporting. For example, the “presurgical sample may be at higher risk for suicide or self-injury but withhold psychiatric history during evaluation for fear of being rejected for surgery.”
From an initial 2,676 studies identified for consideration, investigators in the end extracted data from 28 studies from the United States, Canada, Sweden, and Brazil. The studies had considerable heterogeneity in methods; some included presurgery/postsurgery analyses of the same patients, some had a comparator nonsurgical group, and some used a single postsurgical assessment.
In studies where no nonbariatric comparison sample was available, the investigators assigned interpolated comparison. To arrive at this measure, they drew on the World Health Organization–reported base rate of suicides in the study country at the approximate year of assessment. Suicide was the only interpolated outcome.
Various measures of suicide and self-harm were captured, including completed and probable suicides, suicide attempts, and self-harm events. In some studies, information was drawn from a suicide-specific questionnaire, or from a suicide item on another type of questionnaire.
There was significant variability in the odds ratios for suicide or self-harm across studies, Dr. Roberts said.
The researchers plan to continue analyzing additional measures captured in the meta-analysis, such as gender, age, initial body mass index, surgery type, and the percent of excess weight lost at the time of assessment for suicide or self-harm risk. They reported no outside sources of funding, and no conflicts of interest.
SOURCE: Roberts, D et al. Obesity Week 2018, Poster A433.
NASHVILLE – , according to findings from a meta-analysis presented at the meeting.
Overall, the odds ratio was 1.69 for self-harm or suicide after bariatric surgery (95% confidence interval, 1.62-1.76; P less than .001), “indicating a nearly 70% increase in risk for self-harm or suicide following bariatric surgery,” wrote Dawn Roberts, PhD, of Bradley University, Peoria, Ill., and her coauthor, Nicole Pearl of Washington University, St. Louis, in the poster accompanying the presentation.
Further, as elapsed time from surgery grew, the suicide rate dropped (effect size covariance, r = –0.25). “Thus, the greatest risk for self-harm or suicide appears to emerge in the years immediately following surgery,” Dr. Roberts said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
The investigators had a primary objective of characterizing the association between bariatric surgery and suicide or self-harm. The secondary purpose of the meta-analysis was to find moderators of the association that could explain some of the variability that had previously been seen in studies looking at mental health outcomes after bariatric surgery.
Some of the potential moderators, explained the investigators, included the surgery type. With Roux-en-Y gastric bypass (RYGB), more tissue is removed, potentially causing “more extensive disruption of neural pathways,” the investigators wrote. With greater loss of small-intestine surface area might come more disruption of the gut-brain axis, along with unknown effects on metabolism and pharmacokinetics of psychiatric medication. Additionally, alcohol use disorder might have a more profound effect after gastric bypass surgery.
It had previously been shown that more than two-thirds of suicides happen within the first 3 years after bariatric surgery. With the initial weight loss comes renegotiation of personal relationships, and the potential for more mobility and perhaps expanded career choices; stress accompanies even positive changes in these major life domains. Some patients will also experience weight regain within the first 3 years, after an initial nadir. This also can cause deterioration in quality of life, the investigators explained.
Dr. Roberts and her coinvestigator acknowledge that some of the potential moderators may have been missed in the initial data reporting. For example, the “presurgical sample may be at higher risk for suicide or self-injury but withhold psychiatric history during evaluation for fear of being rejected for surgery.”
From an initial 2,676 studies identified for consideration, investigators in the end extracted data from 28 studies from the United States, Canada, Sweden, and Brazil. The studies had considerable heterogeneity in methods; some included presurgery/postsurgery analyses of the same patients, some had a comparator nonsurgical group, and some used a single postsurgical assessment.
In studies where no nonbariatric comparison sample was available, the investigators assigned interpolated comparison. To arrive at this measure, they drew on the World Health Organization–reported base rate of suicides in the study country at the approximate year of assessment. Suicide was the only interpolated outcome.
Various measures of suicide and self-harm were captured, including completed and probable suicides, suicide attempts, and self-harm events. In some studies, information was drawn from a suicide-specific questionnaire, or from a suicide item on another type of questionnaire.
There was significant variability in the odds ratios for suicide or self-harm across studies, Dr. Roberts said.
The researchers plan to continue analyzing additional measures captured in the meta-analysis, such as gender, age, initial body mass index, surgery type, and the percent of excess weight lost at the time of assessment for suicide or self-harm risk. They reported no outside sources of funding, and no conflicts of interest.
SOURCE: Roberts, D et al. Obesity Week 2018, Poster A433.
NASHVILLE – , according to findings from a meta-analysis presented at the meeting.
Overall, the odds ratio was 1.69 for self-harm or suicide after bariatric surgery (95% confidence interval, 1.62-1.76; P less than .001), “indicating a nearly 70% increase in risk for self-harm or suicide following bariatric surgery,” wrote Dawn Roberts, PhD, of Bradley University, Peoria, Ill., and her coauthor, Nicole Pearl of Washington University, St. Louis, in the poster accompanying the presentation.
Further, as elapsed time from surgery grew, the suicide rate dropped (effect size covariance, r = –0.25). “Thus, the greatest risk for self-harm or suicide appears to emerge in the years immediately following surgery,” Dr. Roberts said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
The investigators had a primary objective of characterizing the association between bariatric surgery and suicide or self-harm. The secondary purpose of the meta-analysis was to find moderators of the association that could explain some of the variability that had previously been seen in studies looking at mental health outcomes after bariatric surgery.
Some of the potential moderators, explained the investigators, included the surgery type. With Roux-en-Y gastric bypass (RYGB), more tissue is removed, potentially causing “more extensive disruption of neural pathways,” the investigators wrote. With greater loss of small-intestine surface area might come more disruption of the gut-brain axis, along with unknown effects on metabolism and pharmacokinetics of psychiatric medication. Additionally, alcohol use disorder might have a more profound effect after gastric bypass surgery.
It had previously been shown that more than two-thirds of suicides happen within the first 3 years after bariatric surgery. With the initial weight loss comes renegotiation of personal relationships, and the potential for more mobility and perhaps expanded career choices; stress accompanies even positive changes in these major life domains. Some patients will also experience weight regain within the first 3 years, after an initial nadir. This also can cause deterioration in quality of life, the investigators explained.
Dr. Roberts and her coinvestigator acknowledge that some of the potential moderators may have been missed in the initial data reporting. For example, the “presurgical sample may be at higher risk for suicide or self-injury but withhold psychiatric history during evaluation for fear of being rejected for surgery.”
From an initial 2,676 studies identified for consideration, investigators in the end extracted data from 28 studies from the United States, Canada, Sweden, and Brazil. The studies had considerable heterogeneity in methods; some included presurgery/postsurgery analyses of the same patients, some had a comparator nonsurgical group, and some used a single postsurgical assessment.
In studies where no nonbariatric comparison sample was available, the investigators assigned interpolated comparison. To arrive at this measure, they drew on the World Health Organization–reported base rate of suicides in the study country at the approximate year of assessment. Suicide was the only interpolated outcome.
Various measures of suicide and self-harm were captured, including completed and probable suicides, suicide attempts, and self-harm events. In some studies, information was drawn from a suicide-specific questionnaire, or from a suicide item on another type of questionnaire.
There was significant variability in the odds ratios for suicide or self-harm across studies, Dr. Roberts said.
The researchers plan to continue analyzing additional measures captured in the meta-analysis, such as gender, age, initial body mass index, surgery type, and the percent of excess weight lost at the time of assessment for suicide or self-harm risk. They reported no outside sources of funding, and no conflicts of interest.
SOURCE: Roberts, D et al. Obesity Week 2018, Poster A433.
REPORTING FROM OBESITY WEEK 2018
Key clinical point: Bariatric surgery patients have an elevated risk for suicide or self-harm.
Major finding: The odds ratio for suicide or self-harm was 1.69 after bariatric surgery.
Study details: Meta-analysis of 28 studies of bariatric surgery patients.
Disclosures: The authors reported no conflicts of interest and no outside sources of funding.
Source: Roberts D et al. Obesity Week 2018, Poster A433.

Probative pee, Pilgrim obesity, and med school baked bribes
He tweets, he (doesn’t) score!
We all know that less sleep equals poor job performance. Up way too late on a Sunday night means you might fall face first into your keyboard the next morning and accidentally send an email that ends with “hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh.” So, yeah, sleep is important.

It’s especially important when you are a professional athlete and your entire livelihood depends on you being in tip-top shape. Researchers from the State University of New York at Stony Brook studied the performance of NBA players in relation to their late-night Twitter binges. Unsurprisingly, tweeting in the wee hours correlated with fewer points and fewer rebounds in the next day’s game. We’re sure coaches are just thrilled to hear about their players spending precious night hours @-ing random trolls on Twitter. Does this mean less tweeting and more sleep means anyone can be the next LeBron? Or, um, the next @KingJames? Probably not … but give it a try.
Poppy seeds and probative pee
As you tuck into your Thanksgiving leftovers, give a thought to a valiant physician who ate and drank – and peed – for science. Not just once, but twice.

At the recent Pain Care for Primary Care symposium in San Diego, Mount Sinai Beth Israel addiction specialist Edwin Salsitz, MD, gave a presentation about drug screening and mentioned his own homegrown investigation into two reputed sources of false positives.
A few years ago, a patient tested positive for opiates and, like many before him, blamed his fondness for poppy-seed bagels. Dr. Salsitz asked the patient to buy him a poppy bagel from his usual source, then the doctor went home and ate it on a Sunday prior to collecting his own pee. The doctor’s subsequent urine test was positive for opiates, and the patient was off the hook. (For more about the poppy-seed menace to accurate opiate testing, check this clinical update from the Aegis testing company.)
Later, it was time to check another possible urban legend. Dr. Salsitz got some mate de coca tea from a friend who’d returned from South America. Again, he took time out of a Sunday, this time to enjoy a hot beverage, mate de coca style, and collect his own pee. The urine test was positive this time, too – for cocaine.
The moral of the story? If you have a drug test looming, be safe and just stick to a croissant and coffee.
Psst … want a cookie?
In medical school, the best and brightest sacrifice their bodies and social lives to absorb knowledge like human sponges. Or maybe it’s where they absorb cookies in exchange for positive end-of-course evaluations.

Investigators from the University of Münster (Germany) decided to give 118 of the school’s third-year medical students a little test. During a course on emergency medicine, some groups were given access to free chocolate cookies (Discus deliciosum spp.) in their sessions, and some groups were not. When it came time to fill out their “student evaluations of teaching” at the end of the semester, the “cookie group” was more generous in its ratings of the course material and gave significantly higher scores to the teachers and to the course overall, compared with the control group (Med Educ. 2018 Oct;52[10]:1064-72).
This all seemed a little suspicious, so we did a little digging. Turns out that the cookie group – the one that provided all that warm, chocolatey positive reinforcement – was chock full of the usual suspects: Ernie the elf, Mrs. Fields, Famous Amos, and Mr. Big himself, Cookie Monster.
Why Myles Standish wasn’t fat
In the autumn of 1621, obesity didn’t dine with the 53 Pilgrims who gave culinary thanks for surviving their first disappointing Boston Bruins season. Er, for their first harvest after a brutal New England winter. Why was that first Thanksgiving such a svelte affair, free of the high-BMI epidemic that afflicts so many Bruins faithful nearly 4 centuries later? Was it the free-range turkey? The lean venison? The Wampanoag guests’ demands for a DASH-diet dinner?

A modern study may help reveal the historical truth: 17th century Plymouth Plantation wasn’t yet bisected by the 21st century Cape Cod traffic snarling the Pilgrims Highway, a.k.a. Massachusetts Route 3.
It was Spanish researchers, not English Puritans, who unbuckled the portly puzzle’s Pilgrim hat. Investigators with the Barcelona Institute for Global Health examined the link between traffic noise exposure and obesity markers among a group of Swiss adults. The verdict? Those exposed to the highest levels of traffic noise ran the greatest risk of becoming obese. Specifically, every 10-decibel rise in road noise packed on another 17% increase in obesity. Seems tractor-trailer downshifts and honking horns may disturb sleep, gridlocking glucose metabolism and diverting everyone to the nearest drive-thru.
Next on the Spaniards’ research to-do list: Can your New England uncle’s annual Turkey Day tales of Red Sox triumphs trigger psychosis among familial Yankees fans?
He tweets, he (doesn’t) score!
We all know that less sleep equals poor job performance. Up way too late on a Sunday night means you might fall face first into your keyboard the next morning and accidentally send an email that ends with “hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh.” So, yeah, sleep is important.
It’s especially important when you are a professional athlete and your entire livelihood depends on you being in tip-top shape. Researchers from the State University of New York at Stony Brook studied the performance of NBA players in relation to their late-night Twitter binges. Unsurprisingly, tweeting in the wee hours correlated with fewer points and fewer rebounds in the next day’s game. We’re sure coaches are just thrilled to hear about their players spending precious night hours @-ing random trolls on Twitter. Does this mean less tweeting and more sleep means anyone can be the next LeBron? Or, um, the next @KingJames? Probably not … but give it a try.
Poppy seeds and probative pee
As you tuck into your Thanksgiving leftovers, give a thought to a valiant physician who ate and drank – and peed – for science. Not just once, but twice.
At the recent Pain Care for Primary Care symposium in San Diego, Mount Sinai Beth Israel addiction specialist Edwin Salsitz, MD, gave a presentation about drug screening and mentioned his own homegrown investigation into two reputed sources of false positives.
A few years ago, a patient tested positive for opiates and, like many before him, blamed his fondness for poppy-seed bagels. Dr. Salsitz asked the patient to buy him a poppy bagel from his usual source, then the doctor went home and ate it on a Sunday prior to collecting his own pee. The doctor’s subsequent urine test was positive for opiates, and the patient was off the hook. (For more about the poppy-seed menace to accurate opiate testing, check this clinical update from the Aegis testing company.)
Later, it was time to check another possible urban legend. Dr. Salsitz got some mate de coca tea from a friend who’d returned from South America. Again, he took time out of a Sunday, this time to enjoy a hot beverage, mate de coca style, and collect his own pee. The urine test was positive this time, too – for cocaine.
The moral of the story? If you have a drug test looming, be safe and just stick to a croissant and coffee.
Psst … want a cookie?
In medical school, the best and brightest sacrifice their bodies and social lives to absorb knowledge like human sponges. Or maybe it’s where they absorb cookies in exchange for positive end-of-course evaluations.
Investigators from the University of Münster (Germany) decided to give 118 of the school’s third-year medical students a little test. During a course on emergency medicine, some groups were given access to free chocolate cookies (Discus deliciosum spp.) in their sessions, and some groups were not. When it came time to fill out their “student evaluations of teaching” at the end of the semester, the “cookie group” was more generous in its ratings of the course material and gave significantly higher scores to the teachers and to the course overall, compared with the control group (Med Educ. 2018 Oct;52[10]:1064-72).
This all seemed a little suspicious, so we did a little digging. Turns out that the cookie group – the one that provided all that warm, chocolatey positive reinforcement – was chock full of the usual suspects: Ernie the elf, Mrs. Fields, Famous Amos, and Mr. Big himself, Cookie Monster.
Why Myles Standish wasn’t fat
In the autumn of 1621, obesity didn’t dine with the 53 Pilgrims who gave culinary thanks for surviving their first disappointing Boston Bruins season. Er, for their first harvest after a brutal New England winter. Why was that first Thanksgiving such a svelte affair, free of the high-BMI epidemic that afflicts so many Bruins faithful nearly 4 centuries later? Was it the free-range turkey? The lean venison? The Wampanoag guests’ demands for a DASH-diet dinner?
A modern study may help reveal the historical truth: 17th century Plymouth Plantation wasn’t yet bisected by the 21st century Cape Cod traffic snarling the Pilgrims Highway, a.k.a. Massachusetts Route 3.
It was Spanish researchers, not English Puritans, who unbuckled the portly puzzle’s Pilgrim hat. Investigators with the Barcelona Institute for Global Health examined the link between traffic noise exposure and obesity markers among a group of Swiss adults. The verdict? Those exposed to the highest levels of traffic noise ran the greatest risk of becoming obese. Specifically, every 10-decibel rise in road noise packed on another 17% increase in obesity. Seems tractor-trailer downshifts and honking horns may disturb sleep, gridlocking glucose metabolism and diverting everyone to the nearest drive-thru.
Next on the Spaniards’ research to-do list: Can your New England uncle’s annual Turkey Day tales of Red Sox triumphs trigger psychosis among familial Yankees fans?
He tweets, he (doesn’t) score!
We all know that less sleep equals poor job performance. Up way too late on a Sunday night means you might fall face first into your keyboard the next morning and accidentally send an email that ends with “hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhh.” So, yeah, sleep is important.
It’s especially important when you are a professional athlete and your entire livelihood depends on you being in tip-top shape. Researchers from the State University of New York at Stony Brook studied the performance of NBA players in relation to their late-night Twitter binges. Unsurprisingly, tweeting in the wee hours correlated with fewer points and fewer rebounds in the next day’s game. We’re sure coaches are just thrilled to hear about their players spending precious night hours @-ing random trolls on Twitter. Does this mean less tweeting and more sleep means anyone can be the next LeBron? Or, um, the next @KingJames? Probably not … but give it a try.
Poppy seeds and probative pee
As you tuck into your Thanksgiving leftovers, give a thought to a valiant physician who ate and drank – and peed – for science. Not just once, but twice.
At the recent Pain Care for Primary Care symposium in San Diego, Mount Sinai Beth Israel addiction specialist Edwin Salsitz, MD, gave a presentation about drug screening and mentioned his own homegrown investigation into two reputed sources of false positives.
A few years ago, a patient tested positive for opiates and, like many before him, blamed his fondness for poppy-seed bagels. Dr. Salsitz asked the patient to buy him a poppy bagel from his usual source, then the doctor went home and ate it on a Sunday prior to collecting his own pee. The doctor’s subsequent urine test was positive for opiates, and the patient was off the hook. (For more about the poppy-seed menace to accurate opiate testing, check this clinical update from the Aegis testing company.)
Later, it was time to check another possible urban legend. Dr. Salsitz got some mate de coca tea from a friend who’d returned from South America. Again, he took time out of a Sunday, this time to enjoy a hot beverage, mate de coca style, and collect his own pee. The urine test was positive this time, too – for cocaine.
The moral of the story? If you have a drug test looming, be safe and just stick to a croissant and coffee.
Psst … want a cookie?
In medical school, the best and brightest sacrifice their bodies and social lives to absorb knowledge like human sponges. Or maybe it’s where they absorb cookies in exchange for positive end-of-course evaluations.
Investigators from the University of Münster (Germany) decided to give 118 of the school’s third-year medical students a little test. During a course on emergency medicine, some groups were given access to free chocolate cookies (Discus deliciosum spp.) in their sessions, and some groups were not. When it came time to fill out their “student evaluations of teaching” at the end of the semester, the “cookie group” was more generous in its ratings of the course material and gave significantly higher scores to the teachers and to the course overall, compared with the control group (Med Educ. 2018 Oct;52[10]:1064-72).
This all seemed a little suspicious, so we did a little digging. Turns out that the cookie group – the one that provided all that warm, chocolatey positive reinforcement – was chock full of the usual suspects: Ernie the elf, Mrs. Fields, Famous Amos, and Mr. Big himself, Cookie Monster.
Why Myles Standish wasn’t fat
In the autumn of 1621, obesity didn’t dine with the 53 Pilgrims who gave culinary thanks for surviving their first disappointing Boston Bruins season. Er, for their first harvest after a brutal New England winter. Why was that first Thanksgiving such a svelte affair, free of the high-BMI epidemic that afflicts so many Bruins faithful nearly 4 centuries later? Was it the free-range turkey? The lean venison? The Wampanoag guests’ demands for a DASH-diet dinner?
A modern study may help reveal the historical truth: 17th century Plymouth Plantation wasn’t yet bisected by the 21st century Cape Cod traffic snarling the Pilgrims Highway, a.k.a. Massachusetts Route 3.
It was Spanish researchers, not English Puritans, who unbuckled the portly puzzle’s Pilgrim hat. Investigators with the Barcelona Institute for Global Health examined the link between traffic noise exposure and obesity markers among a group of Swiss adults. The verdict? Those exposed to the highest levels of traffic noise ran the greatest risk of becoming obese. Specifically, every 10-decibel rise in road noise packed on another 17% increase in obesity. Seems tractor-trailer downshifts and honking horns may disturb sleep, gridlocking glucose metabolism and diverting everyone to the nearest drive-thru.
Next on the Spaniards’ research to-do list: Can your New England uncle’s annual Turkey Day tales of Red Sox triumphs trigger psychosis among familial Yankees fans?