User login
Extreme Athlete, 18, With Worsening Cough
IN THIS ARTICLE
- Adverse effects of ciprofloxacin
- Symptoms of common tick-borne diseases
- Symptoms of phase 1 and late-phase disease
- Additional resources
Jane, an 18-year-old college student, presents in early November with a three-week history of worsening cough and sinus congestion. Recently, the cough has been interrupting her sleep and yellow-green nasal drainage and sinus pressure have increased. Ordinarily very fit and athletic, she reports that since she arrived at college two months ago, her body has become “more fragile.”
Further questioning reveals that, over the past two months, the patient’s symptoms have included extreme fatigue, severe unremitting headache, blurred vision, shortness of breath, and a racing heart rate on exertion. Her symptoms make it impossible for her to maintain her demanding exercise routine, a development that compounds her frustration and sadness. She has also been forced to limit her participation in school activities, with significant academic decline as a result.
Aside from depression (well controlled with bupropion HCl extended release, 300 mg/d), Jane’s medical history is unremarkable. She reports having “excellent health” until she arrived at her mid-Atlantic urban college.
A complicated history
Born and raised in Connecticut, Jane is an avid runner who competes in extreme sports. This past summer, she trained for and participated in two “mud run” events (ie, endurance races of several miles with numerous challenges and obstacles) in Connecticut and New York. Training included endurance runs and sprints, as well as crawling through mud-laden fields and woods.
She also did a three-week summer internship on an oyster farm. There, she was required to shuck oysters and stand in brackish water for six-hour shifts to examine oyster beds. In the process, she sustained numerous cuts and bruises on her hands, arms, and legs.
A week or so after returning to college in late August, Jane developed blisters on both heels, which progressed to infected ulcerations. She was evaluated at the university hospital emergency department (ED) and treated with a 21-day course of ciprofloxacin. When left-sided unilateral knee swelling developed about two weeks later, she underwent arthrocentesis at the university health center, but joint aspirate was not sent for analysis. A two-week course of antibiotic therapy was initiated.
From October to her presentation in early November, Jane has experienced intermittent fevers and chills, with a temperature as high as 101°F. In addition, she complains of fasciculations and weakness in her lower limbs; dyspnea, tachycardia, and dizziness during or after any exertion; unremitting posterior neck pain; and a constant, severe headache located primarily in the bitemporal region. She developed bilateral conjunctivitis, which resolved spontaneously in about one week; persistent blurred vision; a transient petechial chest rash; recurring episodes of syncope; pyelonephritis; a persistent vaginal yeast infection; decreased appetite; and a 7-lb weight loss (5% of her total body weight).
Jane’s academic and athletic performance has been severely impaired. Once a long-distance runner, she can no longer walk any distance without frequent rest. In the four months since the mud runs, the patient reports, she has been seen in the student health center four times and in the ED twice. Additionally, she has undergone thorough examinations by clinicians specializing in infectious disease, pulmonology, neurology, and neuro-ophthalmology. She has undergone lab work, including
• Complete blood cell count with differential
• Comprehensive metabolic panel
• Urinalysis and urine culture
• Lyme antibody and blood polymerase chain reaction (PCR)
• HIV testing
• Rheumatoid factor
• Erythrocyte sedimentation rate (ESR)
• C-reactive protein (CRP)
• Epstein-Barr virus IgM
• Cytomegalovirus (CMV) IgM
• Human granulocytic ehrlichiosis (HGE) antibody and human anaplasma phagocytophilum (HGA)
• HGA PCR
• Rickettsia antibody panel
• Babesia microti antibodies
• Pregnancy testing
• Chest x-ray
• Lumbar puncture
All lab results were within normal range. In light of this, several clinicians have told Jane that her illness is “all in her head.”
Continue for the patient investigates >>
The patient investigates
In mid-December, after she has returned home from college, Jane’s symptoms abruptly worsen. She complains of feeling “shakier,” with weakness in her legs and what she calls “brain fog.” Her headache, blurred vision, and dizziness have worsened. Frightened and concerned, she returns to the ED. Results of a thorough evaluation, including lumbar puncture, reveal no abnormality.
Jane has become extremely frail. She is losing weight, her hair has lost its luster, and her nails are cracking and bleeding. She is unable to walk without concern for falling and cannot climb the 20 steps to her bedroom. Once a healthy and vibrant 18-year-old, she now spends most of her time in a lethargic state on a first-floor living room couch.
Frustrated by her unexplained declining health, she begins to research illnesses associated with extreme sports and prolonged marine exposure. She returns to ask about three possible explanations for her condition:
1. Adverse effects of ciprofloxacin use, which include fever or chills, dizziness, racing heartbeat, headache, and nausea.1
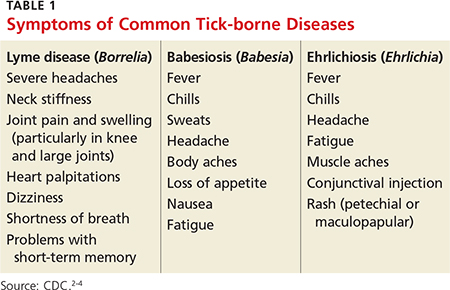
2. A tick-borne disease, possibly contracted during her practice runs in the Connecticut woods (see Table 1).2-4 Each year, she recalls, she has found and removed four or five embedded ticks. In the northeastern United States, the most common tick-borne diseases are borreliosis, babesiosis, and ehrlichiosis.5-7
3. Leptospirosis, contracted through the patient’s exposure to mud and brackish water during her summer activities. According to her research, more than 25 outbreaks and 600 cases of leptospirosis (between 1931 and 1998) have been associated with fresh pond, creek, or river water.8
Based on Jane’s symptoms and history, and in accord with her research, early-phase leptospirosis is identified as a diagnosis of exclusion (with a possible comorbid tick-borne zoonosis).
Continue for discussion >>
DISCUSSION
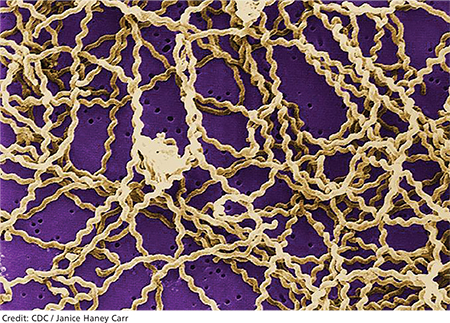
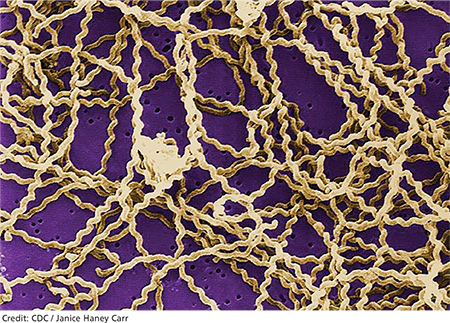
Leptospirosis develops when humans come into contact with animal urine infected by leptospires—that is, pathogenic spirochetes excreted via the renal tubules of infected host animals.9,10 While host animals include dogs, pigs, cattle, reptiles, and amphibians, the animal most commonly associated with human infection is the brown rat (Rattus norvegicus).11-15
Leptospires enter the human host through mucous membranes, cuts, or abrasions in the skin. Individuals at increased risk for infection include those whose work or other activities expose them “to animal reservoirs or contaminated environments”—including participants in water sports and similar recreation.11-14 As Mwachui et al explain, “recreational exposure to [Leptospira-]contaminated water has become more important for sport enthusiasts, swimmers and travellers from industrialized countries,” whereas flooding is usually involved in infection in undeveloped countries.16
The largest outbreak of leptospirosis reported in the US to date occurred in 1998, when heavy rains preceded a triathlon in Springfield, Illinois. When many participants became ill after the event, researchers from the National Center for Infectious Diseases were able to contact and test 834 of the 876 competing athletes; of these, 98 (12%) reported being ill and 52 (11%) tested positive for leptospirosis. Additionally, 14 of the 248 community residents who were sickened (6%) tested positive.17 According to CDC estimates, between 100 and 200 cases of leptospirosis develop annually in the US, with about half occurring in Hawaii.9
Onset of symptoms, which are described as protean and nonspecific, occurs two days to four weeks after exposure, making leptospirosis difficult to diagnosewithout a high degree of suspicion; zoonotic exposure (as with freshwater or mud sports) or a history of travel to Hawaii, Tahiti, Thailand, Indonesia, the Caribbean, and/or Costa Rica may raise suspicion.12-14,18 In early-phase leptospirosis, symptoms can mimic those of influenza, meningitis, malaria, dengue fever, scrub typhus, rickettsial disease, and typhoid fever (see Table 2).10 Thus, when a patient presents with these symptoms, it is imperative that the clinician consider leptospirosis.19Of note: Flu-like symptoms with conjunctival suffusion are considered pathognomonic for leptospirosis.18

About 10% of patients with early-phase leptospirosis will develop late-phase disease (ie, Weil’s disease), with severe symptoms that include jaundice, meningitis, pulmonary hemorrhage, and acute kidney injury (see Table 3 for a more detailed list).20 The case patient’s history and symptoms were consistent with a diagnosis of early-phase leptospirosis.

Epidemiology
In 2015, leptospirosis was estimated to affect more than 1 million persons worldwide, with 58,900 deaths attributed to the disease each year—making leptospirosis the leading cause of death attributable to zoonotic illness.11 Historically, leptospirosis-associated morbidity and mortality have been greatest in resource-poor countries with tropical climates (eg, southern and Southeast Asia, Central America and tropical Latin America, and East Sub-Saharan Africa).11,12
However, illness resulting from recreational exposures to contaminated water has been linked to increasing travel to exotic destinations, participation in adventure travel, and the growing popularity of extreme sports involving fresh water.9 Recreational mud run events, for example, involve swimming in potentially contaminated waters and crawling through flooded farm fields where animal urine can be present—an ideal environment for Leptospira to thrive and for participants to contract the disease.14,15
Continue for laboratory work-up >>
Laboratory work-up
Diagnosis of leptospirosis is challenging.21 Laboratory tests vary, depending on the timing and stage of infection, and are mostly unavailable in resource-poor countries. Test results for the patient with early-phase leptospirosis may demonstrate renal or hepatic abnormalities.18 However, laboratory confirmation of leptospirosis requires22
• A fourfold increase in antibody titer between acute and convalescent serum samples, as detected by microscopic agglutination testing (MAT) or
• A high MAT titer (> 1:400 to 1:800), in single or paired samples or
• Isolation of pathogenic Leptospira species from a normally sterile site or
• Detection of DNA from pathogenic Leptospira species by PCR
A positive laboratory result is, of course, confirmatory. However, negative laboratory findings must be viewed with healthy skepticism.12 A false-negative result may merely indicate the shortcoming of the testing method to accurately assess the presence of Leptospira.
Treatment options
The high mortality rate associated with severe leptospirosis makes early diagnosis and treatment essential.23 The World Health Organization warns that antibiotic treatment for leptospirosis must be instituted within five days of symptom onset.10
Treatment options for an ambulatory patient with mild symptoms and no organ involvement include oral doxycycline (100 mg bid for 5-7 d) or oral azithromycin (500 mg/d for 5-7 d). For patients with organ involvement, IV penicillin (1.5 million U every 6 h for 7 d), ceftriaxone (1 g/d for 7 d), or cefotaxime (1 g every 6 h for 7 d) may be considered.12,20
OUTCOME FOR THE CASE PATIENT
With leptospirosis as the diagnosis of exclusion, Jane was treated successfully with a 21-day course of oral doxycycline (100 mg bid). She has been symptom free since completing the regimen. After undergoing physical therapy and athletic training, she has been able to resume her full exercise regimen, and her recovery is considered complete.
CONCLUSION
The growing popularity of adventure travel and “extreme sports” events, particularly triathlons and mud runs, may precipitate an increase in associated infections with Leptospira and other zoonotic pathogens. For patients with flulike symptoms who routinely engage in such sports—especially those who present with conjunctival suffusion—leptospirosis should be considered in the differential diagnosis.

REFERENCES
1. Owens RC Jr, Ambrose PG. Antimicrobial safety: focus on fluoroquinolones. Clin Infect Dis. 2005;41(suppl 2):S144-S157.
2. CDC. Signs and symptoms of untreated Lyme disease (2015). www.cdc.gov/lyme/signs_symptoms/index.html. Accessed June 7, 2016.
3. CDC. Parasites: babesiosis (2014). www.cdc.gov/parasites/babesiosis/disease.html. Accessed June 7, 2016.
4. CDC. Ehrlichiosis: symptoms, diagnosis, and treatment (2013). www.cdc.gov/Ehrlichiosis/symptoms/index.html. Accessed June 7, 2016.
5. Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016 Feb 5. [Epub ahead of print]
6. Choi E, Pyzocha NJ, Maurer DM. Tick-borne illnesses. Curr Sports Med Rep. 2016;15(2):98-104.
7. Chomel B. Lyme disease. Rev Sci Tech. 2015;34(2):569-576.
8. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14(2):296-326.
9. CDC. Leptospirosis: signs and symptoms (2016). www.cdc.gov/leptospirosis/symptoms/index.html. Accessed June 7, 2016.
10. World Health Organization, International Leptospirosis Society. Human Leptospirosis: Guidance for Diagnosis, Surveillance, and Control (2003). http://apps.who.int/iris/bitstream/10665/42667/1/WHO_CDS_CSR_EPH_2002.23.pdf. Accessed June 7, 2016.
11. Costa F, Hagan JE, Calcagno J, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003898.
12. Haake DA, Levett PN. Leptospirosis in humans. Curr Top Microbiol Immunol. 2015;387:65-97.
13. Picardeau M. Diagnosis and epidemiology of leptospirosis. Médecine et Maladies Infectieuses. 2013;43(1):1-9.
14. Picardeau M. Leptospirosis: updating the global picture of an emerging neglected disease. PLoS Negl Trop Dis. 2015;9(9):e0004039.
15. Zavitsanou A, Babatsikou F. Leptospirosis: epidemiology and preventive measures. Health Sci J. 2008;2(2):75-82.
16. Mwachui MA, Crump L, Hartskeerl R, et al. Environmental and behavioural determinants of leptospirosis transmission: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003843.
17. Morgan J, Bornstein SL, Karpati AM, et al. Outbreak of leptospirosis among triathlon participants and community residents in Springfield, Illinois, 1998. Clin Infect Dis. 2002;34(12):1593-1599.
18. Katz AR, Ansdell VE, Effler PV, et al. Assessment of the clinical presentation and treatment of 353 cases of laboratory-confirmed leptospirosis in Hawaii, 1974-1998. Clin Infect Dis. 2001;33(11):1834-1841.
19. Yaakob Y, Rodrigues KF, John DV. Leptospirosis: recent incidents and available diagnostics—a review. Med J Malaysia. 2015;70(6):351-355.
20. Seguro AC, Andrade L. Pathophysiology of leptospirosis. Shock. 2013;39(suppl 1):17-23.
21. Musso D, La Scola B. Laboratory diagnosis of leptospirosis: a challenge. J Microbiol Immunol Infect. 2013;46(4):245-252.
22. Waggoner JJ, Balassiano I, Mohamed-Hadley A, et al. Reverse-transcriptase PCR detection of Leptospira: absence of agreement with single-specimen microscopic agglutination testing. PLoS One. 2015;10(7):e0132988.
23. Iwasaki H, Chagan-Yasutan H, Leano PS, et al. Combined antibody and DNA detection for early diagnosis of leptospirosis after a disaster. Diagn Microbiol Infect Dis. 2016;84(4):287-291
IN THIS ARTICLE
- Adverse effects of ciprofloxacin
- Symptoms of common tick-borne diseases
- Symptoms of phase 1 and late-phase disease
- Additional resources
Jane, an 18-year-old college student, presents in early November with a three-week history of worsening cough and sinus congestion. Recently, the cough has been interrupting her sleep and yellow-green nasal drainage and sinus pressure have increased. Ordinarily very fit and athletic, she reports that since she arrived at college two months ago, her body has become “more fragile.”
Further questioning reveals that, over the past two months, the patient’s symptoms have included extreme fatigue, severe unremitting headache, blurred vision, shortness of breath, and a racing heart rate on exertion. Her symptoms make it impossible for her to maintain her demanding exercise routine, a development that compounds her frustration and sadness. She has also been forced to limit her participation in school activities, with significant academic decline as a result.
Aside from depression (well controlled with bupropion HCl extended release, 300 mg/d), Jane’s medical history is unremarkable. She reports having “excellent health” until she arrived at her mid-Atlantic urban college.
A complicated history
Born and raised in Connecticut, Jane is an avid runner who competes in extreme sports. This past summer, she trained for and participated in two “mud run” events (ie, endurance races of several miles with numerous challenges and obstacles) in Connecticut and New York. Training included endurance runs and sprints, as well as crawling through mud-laden fields and woods.
She also did a three-week summer internship on an oyster farm. There, she was required to shuck oysters and stand in brackish water for six-hour shifts to examine oyster beds. In the process, she sustained numerous cuts and bruises on her hands, arms, and legs.
A week or so after returning to college in late August, Jane developed blisters on both heels, which progressed to infected ulcerations. She was evaluated at the university hospital emergency department (ED) and treated with a 21-day course of ciprofloxacin. When left-sided unilateral knee swelling developed about two weeks later, she underwent arthrocentesis at the university health center, but joint aspirate was not sent for analysis. A two-week course of antibiotic therapy was initiated.
From October to her presentation in early November, Jane has experienced intermittent fevers and chills, with a temperature as high as 101°F. In addition, she complains of fasciculations and weakness in her lower limbs; dyspnea, tachycardia, and dizziness during or after any exertion; unremitting posterior neck pain; and a constant, severe headache located primarily in the bitemporal region. She developed bilateral conjunctivitis, which resolved spontaneously in about one week; persistent blurred vision; a transient petechial chest rash; recurring episodes of syncope; pyelonephritis; a persistent vaginal yeast infection; decreased appetite; and a 7-lb weight loss (5% of her total body weight).
Jane’s academic and athletic performance has been severely impaired. Once a long-distance runner, she can no longer walk any distance without frequent rest. In the four months since the mud runs, the patient reports, she has been seen in the student health center four times and in the ED twice. Additionally, she has undergone thorough examinations by clinicians specializing in infectious disease, pulmonology, neurology, and neuro-ophthalmology. She has undergone lab work, including
• Complete blood cell count with differential
• Comprehensive metabolic panel
• Urinalysis and urine culture
• Lyme antibody and blood polymerase chain reaction (PCR)
• HIV testing
• Rheumatoid factor
• Erythrocyte sedimentation rate (ESR)
• C-reactive protein (CRP)
• Epstein-Barr virus IgM
• Cytomegalovirus (CMV) IgM
• Human granulocytic ehrlichiosis (HGE) antibody and human anaplasma phagocytophilum (HGA)
• HGA PCR
• Rickettsia antibody panel
• Babesia microti antibodies
• Pregnancy testing
• Chest x-ray
• Lumbar puncture
All lab results were within normal range. In light of this, several clinicians have told Jane that her illness is “all in her head.”
Continue for the patient investigates >>
The patient investigates
In mid-December, after she has returned home from college, Jane’s symptoms abruptly worsen. She complains of feeling “shakier,” with weakness in her legs and what she calls “brain fog.” Her headache, blurred vision, and dizziness have worsened. Frightened and concerned, she returns to the ED. Results of a thorough evaluation, including lumbar puncture, reveal no abnormality.
Jane has become extremely frail. She is losing weight, her hair has lost its luster, and her nails are cracking and bleeding. She is unable to walk without concern for falling and cannot climb the 20 steps to her bedroom. Once a healthy and vibrant 18-year-old, she now spends most of her time in a lethargic state on a first-floor living room couch.
Frustrated by her unexplained declining health, she begins to research illnesses associated with extreme sports and prolonged marine exposure. She returns to ask about three possible explanations for her condition:
1. Adverse effects of ciprofloxacin use, which include fever or chills, dizziness, racing heartbeat, headache, and nausea.1
2. A tick-borne disease, possibly contracted during her practice runs in the Connecticut woods (see Table 1).2-4 Each year, she recalls, she has found and removed four or five embedded ticks. In the northeastern United States, the most common tick-borne diseases are borreliosis, babesiosis, and ehrlichiosis.5-7
3. Leptospirosis, contracted through the patient’s exposure to mud and brackish water during her summer activities. According to her research, more than 25 outbreaks and 600 cases of leptospirosis (between 1931 and 1998) have been associated with fresh pond, creek, or river water.8
Based on Jane’s symptoms and history, and in accord with her research, early-phase leptospirosis is identified as a diagnosis of exclusion (with a possible comorbid tick-borne zoonosis).
Continue for discussion >>
DISCUSSION
Leptospirosis develops when humans come into contact with animal urine infected by leptospires—that is, pathogenic spirochetes excreted via the renal tubules of infected host animals.9,10 While host animals include dogs, pigs, cattle, reptiles, and amphibians, the animal most commonly associated with human infection is the brown rat (Rattus norvegicus).11-15
Leptospires enter the human host through mucous membranes, cuts, or abrasions in the skin. Individuals at increased risk for infection include those whose work or other activities expose them “to animal reservoirs or contaminated environments”—including participants in water sports and similar recreation.11-14 As Mwachui et al explain, “recreational exposure to [Leptospira-]contaminated water has become more important for sport enthusiasts, swimmers and travellers from industrialized countries,” whereas flooding is usually involved in infection in undeveloped countries.16
The largest outbreak of leptospirosis reported in the US to date occurred in 1998, when heavy rains preceded a triathlon in Springfield, Illinois. When many participants became ill after the event, researchers from the National Center for Infectious Diseases were able to contact and test 834 of the 876 competing athletes; of these, 98 (12%) reported being ill and 52 (11%) tested positive for leptospirosis. Additionally, 14 of the 248 community residents who were sickened (6%) tested positive.17 According to CDC estimates, between 100 and 200 cases of leptospirosis develop annually in the US, with about half occurring in Hawaii.9
Onset of symptoms, which are described as protean and nonspecific, occurs two days to four weeks after exposure, making leptospirosis difficult to diagnosewithout a high degree of suspicion; zoonotic exposure (as with freshwater or mud sports) or a history of travel to Hawaii, Tahiti, Thailand, Indonesia, the Caribbean, and/or Costa Rica may raise suspicion.12-14,18 In early-phase leptospirosis, symptoms can mimic those of influenza, meningitis, malaria, dengue fever, scrub typhus, rickettsial disease, and typhoid fever (see Table 2).10 Thus, when a patient presents with these symptoms, it is imperative that the clinician consider leptospirosis.19Of note: Flu-like symptoms with conjunctival suffusion are considered pathognomonic for leptospirosis.18
About 10% of patients with early-phase leptospirosis will develop late-phase disease (ie, Weil’s disease), with severe symptoms that include jaundice, meningitis, pulmonary hemorrhage, and acute kidney injury (see Table 3 for a more detailed list).20 The case patient’s history and symptoms were consistent with a diagnosis of early-phase leptospirosis.
Epidemiology
In 2015, leptospirosis was estimated to affect more than 1 million persons worldwide, with 58,900 deaths attributed to the disease each year—making leptospirosis the leading cause of death attributable to zoonotic illness.11 Historically, leptospirosis-associated morbidity and mortality have been greatest in resource-poor countries with tropical climates (eg, southern and Southeast Asia, Central America and tropical Latin America, and East Sub-Saharan Africa).11,12
However, illness resulting from recreational exposures to contaminated water has been linked to increasing travel to exotic destinations, participation in adventure travel, and the growing popularity of extreme sports involving fresh water.9 Recreational mud run events, for example, involve swimming in potentially contaminated waters and crawling through flooded farm fields where animal urine can be present—an ideal environment for Leptospira to thrive and for participants to contract the disease.14,15
Continue for laboratory work-up >>
Laboratory work-up
Diagnosis of leptospirosis is challenging.21 Laboratory tests vary, depending on the timing and stage of infection, and are mostly unavailable in resource-poor countries. Test results for the patient with early-phase leptospirosis may demonstrate renal or hepatic abnormalities.18 However, laboratory confirmation of leptospirosis requires22
• A fourfold increase in antibody titer between acute and convalescent serum samples, as detected by microscopic agglutination testing (MAT) or
• A high MAT titer (> 1:400 to 1:800), in single or paired samples or
• Isolation of pathogenic Leptospira species from a normally sterile site or
• Detection of DNA from pathogenic Leptospira species by PCR
A positive laboratory result is, of course, confirmatory. However, negative laboratory findings must be viewed with healthy skepticism.12 A false-negative result may merely indicate the shortcoming of the testing method to accurately assess the presence of Leptospira.
Treatment options
The high mortality rate associated with severe leptospirosis makes early diagnosis and treatment essential.23 The World Health Organization warns that antibiotic treatment for leptospirosis must be instituted within five days of symptom onset.10
Treatment options for an ambulatory patient with mild symptoms and no organ involvement include oral doxycycline (100 mg bid for 5-7 d) or oral azithromycin (500 mg/d for 5-7 d). For patients with organ involvement, IV penicillin (1.5 million U every 6 h for 7 d), ceftriaxone (1 g/d for 7 d), or cefotaxime (1 g every 6 h for 7 d) may be considered.12,20
OUTCOME FOR THE CASE PATIENT
With leptospirosis as the diagnosis of exclusion, Jane was treated successfully with a 21-day course of oral doxycycline (100 mg bid). She has been symptom free since completing the regimen. After undergoing physical therapy and athletic training, she has been able to resume her full exercise regimen, and her recovery is considered complete.
CONCLUSION
The growing popularity of adventure travel and “extreme sports” events, particularly triathlons and mud runs, may precipitate an increase in associated infections with Leptospira and other zoonotic pathogens. For patients with flulike symptoms who routinely engage in such sports—especially those who present with conjunctival suffusion—leptospirosis should be considered in the differential diagnosis.
REFERENCES
1. Owens RC Jr, Ambrose PG. Antimicrobial safety: focus on fluoroquinolones. Clin Infect Dis. 2005;41(suppl 2):S144-S157.
2. CDC. Signs and symptoms of untreated Lyme disease (2015). www.cdc.gov/lyme/signs_symptoms/index.html. Accessed June 7, 2016.
3. CDC. Parasites: babesiosis (2014). www.cdc.gov/parasites/babesiosis/disease.html. Accessed June 7, 2016.
4. CDC. Ehrlichiosis: symptoms, diagnosis, and treatment (2013). www.cdc.gov/Ehrlichiosis/symptoms/index.html. Accessed June 7, 2016.
5. Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016 Feb 5. [Epub ahead of print]
6. Choi E, Pyzocha NJ, Maurer DM. Tick-borne illnesses. Curr Sports Med Rep. 2016;15(2):98-104.
7. Chomel B. Lyme disease. Rev Sci Tech. 2015;34(2):569-576.
8. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14(2):296-326.
9. CDC. Leptospirosis: signs and symptoms (2016). www.cdc.gov/leptospirosis/symptoms/index.html. Accessed June 7, 2016.
10. World Health Organization, International Leptospirosis Society. Human Leptospirosis: Guidance for Diagnosis, Surveillance, and Control (2003). http://apps.who.int/iris/bitstream/10665/42667/1/WHO_CDS_CSR_EPH_2002.23.pdf. Accessed June 7, 2016.
11. Costa F, Hagan JE, Calcagno J, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003898.
12. Haake DA, Levett PN. Leptospirosis in humans. Curr Top Microbiol Immunol. 2015;387:65-97.
13. Picardeau M. Diagnosis and epidemiology of leptospirosis. Médecine et Maladies Infectieuses. 2013;43(1):1-9.
14. Picardeau M. Leptospirosis: updating the global picture of an emerging neglected disease. PLoS Negl Trop Dis. 2015;9(9):e0004039.
15. Zavitsanou A, Babatsikou F. Leptospirosis: epidemiology and preventive measures. Health Sci J. 2008;2(2):75-82.
16. Mwachui MA, Crump L, Hartskeerl R, et al. Environmental and behavioural determinants of leptospirosis transmission: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003843.
17. Morgan J, Bornstein SL, Karpati AM, et al. Outbreak of leptospirosis among triathlon participants and community residents in Springfield, Illinois, 1998. Clin Infect Dis. 2002;34(12):1593-1599.
18. Katz AR, Ansdell VE, Effler PV, et al. Assessment of the clinical presentation and treatment of 353 cases of laboratory-confirmed leptospirosis in Hawaii, 1974-1998. Clin Infect Dis. 2001;33(11):1834-1841.
19. Yaakob Y, Rodrigues KF, John DV. Leptospirosis: recent incidents and available diagnostics—a review. Med J Malaysia. 2015;70(6):351-355.
20. Seguro AC, Andrade L. Pathophysiology of leptospirosis. Shock. 2013;39(suppl 1):17-23.
21. Musso D, La Scola B. Laboratory diagnosis of leptospirosis: a challenge. J Microbiol Immunol Infect. 2013;46(4):245-252.
22. Waggoner JJ, Balassiano I, Mohamed-Hadley A, et al. Reverse-transcriptase PCR detection of Leptospira: absence of agreement with single-specimen microscopic agglutination testing. PLoS One. 2015;10(7):e0132988.
23. Iwasaki H, Chagan-Yasutan H, Leano PS, et al. Combined antibody and DNA detection for early diagnosis of leptospirosis after a disaster. Diagn Microbiol Infect Dis. 2016;84(4):287-291
IN THIS ARTICLE
- Adverse effects of ciprofloxacin
- Symptoms of common tick-borne diseases
- Symptoms of phase 1 and late-phase disease
- Additional resources
Jane, an 18-year-old college student, presents in early November with a three-week history of worsening cough and sinus congestion. Recently, the cough has been interrupting her sleep and yellow-green nasal drainage and sinus pressure have increased. Ordinarily very fit and athletic, she reports that since she arrived at college two months ago, her body has become “more fragile.”
Further questioning reveals that, over the past two months, the patient’s symptoms have included extreme fatigue, severe unremitting headache, blurred vision, shortness of breath, and a racing heart rate on exertion. Her symptoms make it impossible for her to maintain her demanding exercise routine, a development that compounds her frustration and sadness. She has also been forced to limit her participation in school activities, with significant academic decline as a result.
Aside from depression (well controlled with bupropion HCl extended release, 300 mg/d), Jane’s medical history is unremarkable. She reports having “excellent health” until she arrived at her mid-Atlantic urban college.
A complicated history
Born and raised in Connecticut, Jane is an avid runner who competes in extreme sports. This past summer, she trained for and participated in two “mud run” events (ie, endurance races of several miles with numerous challenges and obstacles) in Connecticut and New York. Training included endurance runs and sprints, as well as crawling through mud-laden fields and woods.
She also did a three-week summer internship on an oyster farm. There, she was required to shuck oysters and stand in brackish water for six-hour shifts to examine oyster beds. In the process, she sustained numerous cuts and bruises on her hands, arms, and legs.
A week or so after returning to college in late August, Jane developed blisters on both heels, which progressed to infected ulcerations. She was evaluated at the university hospital emergency department (ED) and treated with a 21-day course of ciprofloxacin. When left-sided unilateral knee swelling developed about two weeks later, she underwent arthrocentesis at the university health center, but joint aspirate was not sent for analysis. A two-week course of antibiotic therapy was initiated.
From October to her presentation in early November, Jane has experienced intermittent fevers and chills, with a temperature as high as 101°F. In addition, she complains of fasciculations and weakness in her lower limbs; dyspnea, tachycardia, and dizziness during or after any exertion; unremitting posterior neck pain; and a constant, severe headache located primarily in the bitemporal region. She developed bilateral conjunctivitis, which resolved spontaneously in about one week; persistent blurred vision; a transient petechial chest rash; recurring episodes of syncope; pyelonephritis; a persistent vaginal yeast infection; decreased appetite; and a 7-lb weight loss (5% of her total body weight).
Jane’s academic and athletic performance has been severely impaired. Once a long-distance runner, she can no longer walk any distance without frequent rest. In the four months since the mud runs, the patient reports, she has been seen in the student health center four times and in the ED twice. Additionally, she has undergone thorough examinations by clinicians specializing in infectious disease, pulmonology, neurology, and neuro-ophthalmology. She has undergone lab work, including
• Complete blood cell count with differential
• Comprehensive metabolic panel
• Urinalysis and urine culture
• Lyme antibody and blood polymerase chain reaction (PCR)
• HIV testing
• Rheumatoid factor
• Erythrocyte sedimentation rate (ESR)
• C-reactive protein (CRP)
• Epstein-Barr virus IgM
• Cytomegalovirus (CMV) IgM
• Human granulocytic ehrlichiosis (HGE) antibody and human anaplasma phagocytophilum (HGA)
• HGA PCR
• Rickettsia antibody panel
• Babesia microti antibodies
• Pregnancy testing
• Chest x-ray
• Lumbar puncture
All lab results were within normal range. In light of this, several clinicians have told Jane that her illness is “all in her head.”
Continue for the patient investigates >>
The patient investigates
In mid-December, after she has returned home from college, Jane’s symptoms abruptly worsen. She complains of feeling “shakier,” with weakness in her legs and what she calls “brain fog.” Her headache, blurred vision, and dizziness have worsened. Frightened and concerned, she returns to the ED. Results of a thorough evaluation, including lumbar puncture, reveal no abnormality.
Jane has become extremely frail. She is losing weight, her hair has lost its luster, and her nails are cracking and bleeding. She is unable to walk without concern for falling and cannot climb the 20 steps to her bedroom. Once a healthy and vibrant 18-year-old, she now spends most of her time in a lethargic state on a first-floor living room couch.
Frustrated by her unexplained declining health, she begins to research illnesses associated with extreme sports and prolonged marine exposure. She returns to ask about three possible explanations for her condition:
1. Adverse effects of ciprofloxacin use, which include fever or chills, dizziness, racing heartbeat, headache, and nausea.1
2. A tick-borne disease, possibly contracted during her practice runs in the Connecticut woods (see Table 1).2-4 Each year, she recalls, she has found and removed four or five embedded ticks. In the northeastern United States, the most common tick-borne diseases are borreliosis, babesiosis, and ehrlichiosis.5-7
3. Leptospirosis, contracted through the patient’s exposure to mud and brackish water during her summer activities. According to her research, more than 25 outbreaks and 600 cases of leptospirosis (between 1931 and 1998) have been associated with fresh pond, creek, or river water.8
Based on Jane’s symptoms and history, and in accord with her research, early-phase leptospirosis is identified as a diagnosis of exclusion (with a possible comorbid tick-borne zoonosis).
Continue for discussion >>
DISCUSSION
Leptospirosis develops when humans come into contact with animal urine infected by leptospires—that is, pathogenic spirochetes excreted via the renal tubules of infected host animals.9,10 While host animals include dogs, pigs, cattle, reptiles, and amphibians, the animal most commonly associated with human infection is the brown rat (Rattus norvegicus).11-15
Leptospires enter the human host through mucous membranes, cuts, or abrasions in the skin. Individuals at increased risk for infection include those whose work or other activities expose them “to animal reservoirs or contaminated environments”—including participants in water sports and similar recreation.11-14 As Mwachui et al explain, “recreational exposure to [Leptospira-]contaminated water has become more important for sport enthusiasts, swimmers and travellers from industrialized countries,” whereas flooding is usually involved in infection in undeveloped countries.16
The largest outbreak of leptospirosis reported in the US to date occurred in 1998, when heavy rains preceded a triathlon in Springfield, Illinois. When many participants became ill after the event, researchers from the National Center for Infectious Diseases were able to contact and test 834 of the 876 competing athletes; of these, 98 (12%) reported being ill and 52 (11%) tested positive for leptospirosis. Additionally, 14 of the 248 community residents who were sickened (6%) tested positive.17 According to CDC estimates, between 100 and 200 cases of leptospirosis develop annually in the US, with about half occurring in Hawaii.9
Onset of symptoms, which are described as protean and nonspecific, occurs two days to four weeks after exposure, making leptospirosis difficult to diagnosewithout a high degree of suspicion; zoonotic exposure (as with freshwater or mud sports) or a history of travel to Hawaii, Tahiti, Thailand, Indonesia, the Caribbean, and/or Costa Rica may raise suspicion.12-14,18 In early-phase leptospirosis, symptoms can mimic those of influenza, meningitis, malaria, dengue fever, scrub typhus, rickettsial disease, and typhoid fever (see Table 2).10 Thus, when a patient presents with these symptoms, it is imperative that the clinician consider leptospirosis.19Of note: Flu-like symptoms with conjunctival suffusion are considered pathognomonic for leptospirosis.18
About 10% of patients with early-phase leptospirosis will develop late-phase disease (ie, Weil’s disease), with severe symptoms that include jaundice, meningitis, pulmonary hemorrhage, and acute kidney injury (see Table 3 for a more detailed list).20 The case patient’s history and symptoms were consistent with a diagnosis of early-phase leptospirosis.
Epidemiology
In 2015, leptospirosis was estimated to affect more than 1 million persons worldwide, with 58,900 deaths attributed to the disease each year—making leptospirosis the leading cause of death attributable to zoonotic illness.11 Historically, leptospirosis-associated morbidity and mortality have been greatest in resource-poor countries with tropical climates (eg, southern and Southeast Asia, Central America and tropical Latin America, and East Sub-Saharan Africa).11,12
However, illness resulting from recreational exposures to contaminated water has been linked to increasing travel to exotic destinations, participation in adventure travel, and the growing popularity of extreme sports involving fresh water.9 Recreational mud run events, for example, involve swimming in potentially contaminated waters and crawling through flooded farm fields where animal urine can be present—an ideal environment for Leptospira to thrive and for participants to contract the disease.14,15
Continue for laboratory work-up >>
Laboratory work-up
Diagnosis of leptospirosis is challenging.21 Laboratory tests vary, depending on the timing and stage of infection, and are mostly unavailable in resource-poor countries. Test results for the patient with early-phase leptospirosis may demonstrate renal or hepatic abnormalities.18 However, laboratory confirmation of leptospirosis requires22
• A fourfold increase in antibody titer between acute and convalescent serum samples, as detected by microscopic agglutination testing (MAT) or
• A high MAT titer (> 1:400 to 1:800), in single or paired samples or
• Isolation of pathogenic Leptospira species from a normally sterile site or
• Detection of DNA from pathogenic Leptospira species by PCR
A positive laboratory result is, of course, confirmatory. However, negative laboratory findings must be viewed with healthy skepticism.12 A false-negative result may merely indicate the shortcoming of the testing method to accurately assess the presence of Leptospira.
Treatment options
The high mortality rate associated with severe leptospirosis makes early diagnosis and treatment essential.23 The World Health Organization warns that antibiotic treatment for leptospirosis must be instituted within five days of symptom onset.10
Treatment options for an ambulatory patient with mild symptoms and no organ involvement include oral doxycycline (100 mg bid for 5-7 d) or oral azithromycin (500 mg/d for 5-7 d). For patients with organ involvement, IV penicillin (1.5 million U every 6 h for 7 d), ceftriaxone (1 g/d for 7 d), or cefotaxime (1 g every 6 h for 7 d) may be considered.12,20
OUTCOME FOR THE CASE PATIENT
With leptospirosis as the diagnosis of exclusion, Jane was treated successfully with a 21-day course of oral doxycycline (100 mg bid). She has been symptom free since completing the regimen. After undergoing physical therapy and athletic training, she has been able to resume her full exercise regimen, and her recovery is considered complete.
CONCLUSION
The growing popularity of adventure travel and “extreme sports” events, particularly triathlons and mud runs, may precipitate an increase in associated infections with Leptospira and other zoonotic pathogens. For patients with flulike symptoms who routinely engage in such sports—especially those who present with conjunctival suffusion—leptospirosis should be considered in the differential diagnosis.
REFERENCES
1. Owens RC Jr, Ambrose PG. Antimicrobial safety: focus on fluoroquinolones. Clin Infect Dis. 2005;41(suppl 2):S144-S157.
2. CDC. Signs and symptoms of untreated Lyme disease (2015). www.cdc.gov/lyme/signs_symptoms/index.html. Accessed June 7, 2016.
3. CDC. Parasites: babesiosis (2014). www.cdc.gov/parasites/babesiosis/disease.html. Accessed June 7, 2016.
4. CDC. Ehrlichiosis: symptoms, diagnosis, and treatment (2013). www.cdc.gov/Ehrlichiosis/symptoms/index.html. Accessed June 7, 2016.
5. Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016 Feb 5. [Epub ahead of print]
6. Choi E, Pyzocha NJ, Maurer DM. Tick-borne illnesses. Curr Sports Med Rep. 2016;15(2):98-104.
7. Chomel B. Lyme disease. Rev Sci Tech. 2015;34(2):569-576.
8. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14(2):296-326.
9. CDC. Leptospirosis: signs and symptoms (2016). www.cdc.gov/leptospirosis/symptoms/index.html. Accessed June 7, 2016.
10. World Health Organization, International Leptospirosis Society. Human Leptospirosis: Guidance for Diagnosis, Surveillance, and Control (2003). http://apps.who.int/iris/bitstream/10665/42667/1/WHO_CDS_CSR_EPH_2002.23.pdf. Accessed June 7, 2016.
11. Costa F, Hagan JE, Calcagno J, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003898.
12. Haake DA, Levett PN. Leptospirosis in humans. Curr Top Microbiol Immunol. 2015;387:65-97.
13. Picardeau M. Diagnosis and epidemiology of leptospirosis. Médecine et Maladies Infectieuses. 2013;43(1):1-9.
14. Picardeau M. Leptospirosis: updating the global picture of an emerging neglected disease. PLoS Negl Trop Dis. 2015;9(9):e0004039.
15. Zavitsanou A, Babatsikou F. Leptospirosis: epidemiology and preventive measures. Health Sci J. 2008;2(2):75-82.
16. Mwachui MA, Crump L, Hartskeerl R, et al. Environmental and behavioural determinants of leptospirosis transmission: a systematic review. PLoS Negl Trop Dis. 2015;9(9):e0003843.
17. Morgan J, Bornstein SL, Karpati AM, et al. Outbreak of leptospirosis among triathlon participants and community residents in Springfield, Illinois, 1998. Clin Infect Dis. 2002;34(12):1593-1599.
18. Katz AR, Ansdell VE, Effler PV, et al. Assessment of the clinical presentation and treatment of 353 cases of laboratory-confirmed leptospirosis in Hawaii, 1974-1998. Clin Infect Dis. 2001;33(11):1834-1841.
19. Yaakob Y, Rodrigues KF, John DV. Leptospirosis: recent incidents and available diagnostics—a review. Med J Malaysia. 2015;70(6):351-355.
20. Seguro AC, Andrade L. Pathophysiology of leptospirosis. Shock. 2013;39(suppl 1):17-23.
21. Musso D, La Scola B. Laboratory diagnosis of leptospirosis: a challenge. J Microbiol Immunol Infect. 2013;46(4):245-252.
22. Waggoner JJ, Balassiano I, Mohamed-Hadley A, et al. Reverse-transcriptase PCR detection of Leptospira: absence of agreement with single-specimen microscopic agglutination testing. PLoS One. 2015;10(7):e0132988.
23. Iwasaki H, Chagan-Yasutan H, Leano PS, et al. Combined antibody and DNA detection for early diagnosis of leptospirosis after a disaster. Diagn Microbiol Infect Dis. 2016;84(4):287-291
Febrile, Immunocompromised Man With Rash
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
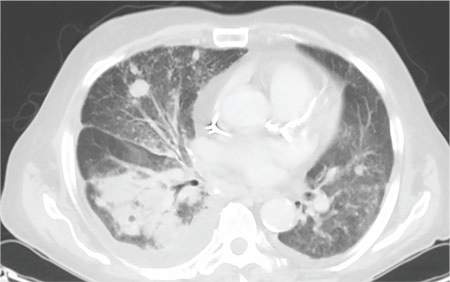
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
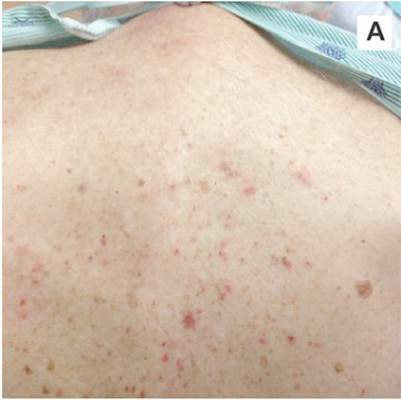 | 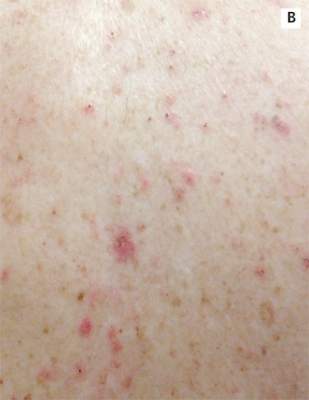 |
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
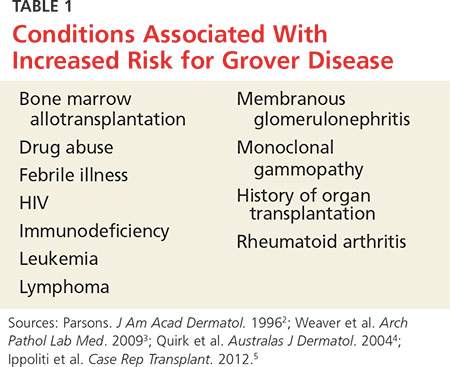
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.

Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
| |
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.
Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.
IN THIS ARTICLE
- Conditions associated with increased risk for case disease
- Outcome for the case patient
- Differential diagnosis
A 78-year-old white man with chronic lymphocytic leukemia is admitted to the hospital with worsening cough, shortness of breath, and fever. His medical history is significant for pneumonia caused by Pneumocystis jirovecii in the past year. In the weeks preceding hospital admission, the patient developed an erythematous rash over his trunk (see photographs).
During the man’s hospital stay, this eruption becomes increasingly pruritic and spreads to his proximal extremities. His pulmonary symptoms improve slightly following the initiation of broad-spectrum antibiotic therapy (piperacillin/tazobactam and vancomycin), but CT performed one week after admission reveals worsening pulmonary disease (see image). The radiologist’s differential diagnosis includes neoplasm, fungal infection, Kaposi sarcoma, and autoimmune disease.
| |
| A. The patient's back shows a distribution of lesions, with areas of excoriation caused by scratching. | B. A close-up reveals erythematous papules and keratotic papules. |
Suspecting that the progressive rash is related to the systemic process, the provider orders a punch biopsy in an effort to reach a diagnosis with minimally invasive studies. When the patient’s clinical status further declines, he undergoes video-assisted thoracoscopic surgery to obtain an excisional biopsy of one of the pulmonary nodules. Subsequent analysis reveals fungal organisms consistent with histoplasmosis. Interestingly, in the histologic review of the skin biopsy, focal acantholytic dyskeratosis—suggestive of Grover disease—is identified.
Continue for discussion >>
DISCUSSION
Grover disease (GD), also known as transient acantholytic dermatosis, is a skin condition of uncertain pathophysiology. Its clinical presentation can be difficult to distinguish from other dermopathies.1,2
Incidence
GD most commonly appears in fair-skinned persons of late middle age, with men affected at two to three times the rate seen in women.1,2 Although GD has been documented in patients ranging in age from 4 to 100, this dermopathy is rare in younger patients.1-3 Persons with a prior history of atopic dermatitis, contact dermatitis, or xerosis cutis are at increased risk for GD—likely due to an increased dermatologic sensitivity to irritants resulting from the aforementioned disorders.1,4 Risk for GD is also elevated in patients with chronic medical conditions, immunodeficiency, febrile illnesses, or malignancies (see Table 1).2-5
The true incidence of GD is not known; biopsy-proven GD is uncommon, and specific data on the incidence and prevalence of the condition are lacking. Swiss researchers who reviewed more than 30,000 skin biopsies in the late 1990s noted only 24 diagnosed cases of GD, and similar findings have been reported in the United States.1,6 However, the variable presentation and often mild nature of GD may result in cases of misdiagnosis, lack of diagnosis, or empiric treatment in the absence of a formal diagnosis.7
Causative factors
Although the pathophysiology of GD is uncertain, the most likely cause is an occlusion of the eccrine glands.3 This is followed by acantholysis, or separation of keratinocytes within the epidermis, which in turn leads to the development of vesicular lesions.
Though diagnosed most often in the winter, GD has also been associated with exposure to sunlight, heat, xerosis, and diaphoresis.1,3 Hospitalized or bedridden patients are at risk for occlusion of the eccrine glands and thus for GD. Use of certain therapies, including sulfadoxine/pyrimethamine (an antimalarial treatment), ionizing radiation, and interleukin-4, may also be precursors for the condition.2
Other exacerbating factors have been suggested, but reports are largely limited to case studies and other anecdotal publications.2 Concrete data regarding the etiology and pathophysiology of GD are still relatively scarce.
Clinical presentation
Patients with GD present with pruritic dermatitis on the trunk and proximal extremities, most classically on the anterior chest and mid back.2,3 The severity of the rash does not necessarily correlate to the degree of pruritus. Some patients report only mild pruritus, while others experience debilitating discomfort and pain. In most cases, erythematous and violaceous papules and vesicles appear first, followed by keratotic erosions.3
GD is a self-limited disorder that often resolves within a few weeks, although some cases will persist for several months.3,5 Severity and duration of symptoms appear to be correlated with increasing age; elderly patients experience worse pruritus for longer periods than do younger patients.2
Although the condition is sometimes referred to as transient acantholytic dermatosis, there are three typical presentations of GD: transient eruptive, persistent pruritic, and chronic asymptomatic.4 Transient eruptive GD presents suddenly, with intense pruritus, and tends to subside over several weeks. Persistent pruritic disease generally causes a milder pruritus, with symptoms that last for several months and are not well controlled by medication. Chronic asymptomatic GD can be difficult to treat medically, yet this form of the disease typically causes little to no irritation and requires minimal therapeutic intervention.4
Systemic symptoms of GD have not been observed. Pruritus and rash are the main features in most affected patients. However, pruritic papulovesicular eruptions are commonly seen in other conditions with similar characteristics (see Table 2,3,4), and GD is comparatively rare. While clinical appearance alone may suggest a diagnosis of GD, further testing may be needed to eliminate other conditions from the differential.
Treatment and prognosis
In the absence of randomized therapeutic trials for GD, there are no strict guidelines for treatment. When irritation, inflammation, and pruritus become bothersome, several interventions may be considered. The first step may consist of efforts to modify aggravating factors, such as dry skin, occlusion, excess heat, and rapid temperature changes. Indeed, for mild cases of GD, this may be all that is required.
The firstline pharmacotherapy for GD is medium- to high-potency topical corticosteroids, which reduce inflammation and pruritus in approximately half of affected patients.3,6,8 Topical emollients and oral antihistamines can also provide symptom relief. Vitamin D analogues are considered secondline therapy, and retinoids (both topical and systemic) have also been shown to reduce GD severity.3,4,8
Severe, refractory cases may require more aggressive systemic therapy with corticosteroids or retinoids. For pruritic relief, several weeks of oral corticosteroids may be necessary—and GD may rebound after treatment ceases.3,4 Therefore, oral corticosteroids should only be considered for severe or persistent cases, since the systemic adverse effects (eg, immunosuppression, weight gain, dysglycemia) of these drugs may outweigh the benefits in patients with GD. Other interventions, including phototherapy and immunosuppressive drugs (eg, etanercept) have also demonstrated benefit in select patients.4,9,10
The self-limited nature of GD, along with its lack of systemic symptoms, is associated with a generally benign course of disease and no long-term sequelae.3,5
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
This case involved an immunocompromised patient with systemic symptoms, vasculitic cutaneous lesions, and significant pulmonary disease. The differential diagnosis was extensive, and diagnosis based on clinical grounds alone was extremely challenging. In these circumstances, diagnostic testing was essential to reach a final diagnosis.
In this case, the skin biopsy yielded a diagnosis of GD, and the rash was found to be unrelated to the patient’s systemic and pulmonary symptoms. The providers were then able to focus on the diagnosis of histoplasmosis, with only minimal intervention for the patient’s GD (ie, oral diphenhydramine prn for pruritus).
CONCLUSION
In many cases of GD, skin biopsy can guide providers when the history and physical examination do not yield a clear diagnosis. The histopathology of affected tissue can provide invaluable information about an underlying disease process, particularly in complex cases such as this patient’s. Skin biopsy provides a minimally invasive opportunity to obtain a diagnosis in patients with a condition that affects multiple organ systems, and its use should be considered in disease processes with cutaneous manifestations.
REFERENCES
1. Scheinfeld N, Mones J. Seasonal variation of transient acantholytic dyskeratosis (Grover’s disease). J Am Acad Dermatol. 2006;55(2): 263-268.
2. Parsons JM. Transient acantholytic dermatosis (Grover’s disease): a global perspective. J Am Acad Dermatol. 1996;35(5 part 1):653-666.
3. Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133(9):1490-1494.
4. Quirk CJ, Heenan PJ. Grover’s disease: 34 years on. Australas J Dermatol. 2004;45(2):83-86.
5. Ippoliti G, Paulli M, Lucioni M, et al. Grover’s disease after heart transplantation: a case report. Case Rep Transplant. 2012;2012:126592.
6. Streit M, Paredes BE, Braathen LR, Brand CU. Transitory acantholytic dermatosis (Grover’s disease): an analysis of the clinical spectrum based on 21 histologically assessed cases [in German]. Hautarzt. 2000;51:244-249.
7. Joshi R, Taneja A. Grover’s disease with acrosyringeal acantholysis: a rare histological presentation of an uncommon disease. Indian J Dermatol. 2014;59(6):621-623.
8. Riemann H, High WA. Grover’s disease (transient and persistent acantholytic dermatosis). UpToDate. 2015. www.uptodate.com/contents/grovers-disease-transient-and-persistent-acantholytic-dermatosis. Accessed June 4, 2016.
9. Breuckmann F, Appelhans C, Altmeyer P, Kreuter A. Medium-dose ultraviolet A1 phototherapy in transient acantholytic dermatosis (Grover’s disease). J Am Acad Dermatol. 2005;52(1):169-170.
10. Norman R, Chau V. Use of etanercept in treating pruritus and preventing new lesions in Grover disease. J Am Acad Dermatol. 2011;64(4):796-798.
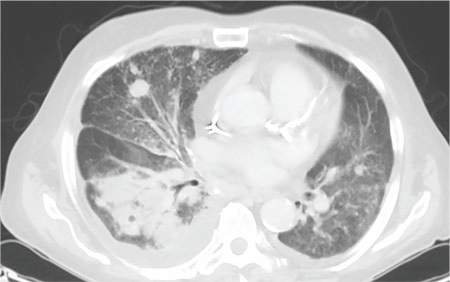
Woman, 35, With Jaundice and Altered Mental Status
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
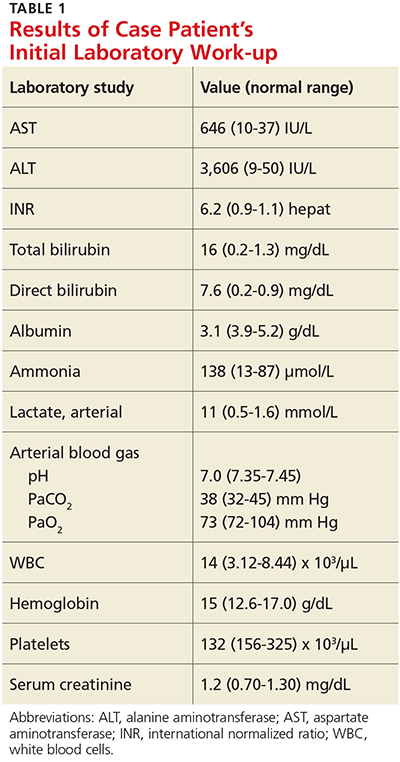
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5

In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5
In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5
In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
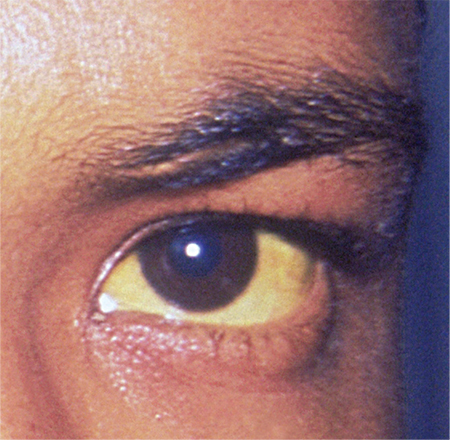
Girl, 5, With Fever and Hip Pain
A 5-year-old Filipino girl was brought to a pediatric clinic for follow up of an unresolved fever and for new-onset right hip pain, which occurred intermittently for the past week and was associated with a right-sided limp. She had been experiencing nightly fevers ranging from 101°F to 105°F for the past two weeks, for which her parents had been giving ibuprofen with mixed results; she remained afebrile during daytime hours.
Using the Wong-Baker FACES pain scale, the patient rated the pain as a 4/10 in severity (“Hurts a Little More” face).1 Standing and walking aggravated the pain but did not limit activity. Although ibuprofen decreased the fever, it did not alleviate the hip pain. Other symptoms included vomiting one to two times daily, without hematemesis, and four to five episodes of diarrhea daily, without abdominal pain, hematochezia, or melena. She also experienced decreased appetite, but her parents reported no change in her dietary or fluid intake. The patient and her parents denied additional symptoms.
Further investigation revealed that the patient had been seen a week earlier by two other clinicians in the office for complaints of fever, rash, nausea, hematemesis, and diarrhea. She had been diagnosed with a herpes simplex viral (HSV) lesion of the nose, epistaxis, and viral gastroenteritis. Her treatment plan consisted of acyclovir ointment for the HSV lesion and symptomatic support for the gastroenteritis associated diarrhea. The complaint of hematemesis was attributed to postnasal drip from the epistaxis, and reassurance was provided to the patient and family. In addition, six weeks earlier, the patient had been treated for otitis media with a full course of amoxicillin.
Medical history was negative for surgeries, trauma, injuries, and chronic medical conditions. She took no medications or supplements on a regular basis. Her parents denied any known drug allergies and stated that her immunizations were up to date.
The patient lived at home with her biological parents and two brothers, all of whom were healthy, without any recent infections or illnesses. Of significance, the family had travelled to the Philippines for vacation about four months earlier. Results of a tuberculin skin test done six weeks earlier (because the patient presented with respiratory symptoms shortly after traveling to the Philippines) were negative.
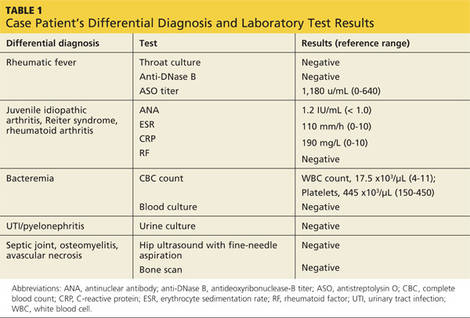
Physical examination revealed a well-developed, well-nourished 40-lb girl, in no acute distress, who was active and playful with her brother while in the exam room. Vital signs were significant for a fever of 101.9°F (last dose of ibuprofen was approximately six hours earlier) but were otherwise stable. Skin exam revealed that the prior HSV lesion of the nose had resolved. HEENT, cardiovascular, and pulmonary exam findings were noncontributory. Urine dipstick was negative.
Abdominal exam revealed normoactive bowel sounds in all four quadrants, and on palpation, the abdomen was soft, nontender, and without organomegaly. Specialized abdominal exams to assess for peritonitis, including those to elicit Rovsing, rebound tenderness, obturator, and psoas signs, were all negative. Bilateral extremity exams of the hips, knees, and ankles revealed full range of motion (active and passive), with normal muscle strength throughout. The only significant finding on the physical exam was mild pain of the right anterior hip at 15° of flexion, appreciated while the patient was supine on the exam table. The patient was also observed pushing off her right lower extremity when climbing onto the exam table, and she skipped down the hall when leaving the exam.
With fever of unknown origin (FUO) and a largely negative history and physical, the working list of differential diagnoses included
• Avascular necrosis
• Bacteremia
• Juvenile idiopathic arthritis
• Osteomyelitis
• Pyelonephritis
• Reiter syndrome
• Rheumatic fever
• Rheumatoid arthritis
• Septic joint
• Urinary tract infection
To begin the diagnostic process, a number of laboratory tests and imaging procedures were ordered. Table 1 presents the results of these studies. A tuberculin skin test was not repeated. While awaiting test results, the patient was started on naproxen oral suspension (125 mg/5 mL; 4 mL bid) for fever and pain control.
Based on findings consistent with an inflammatory pattern, the history of otitis media (of possible streptococcal origin) six weeks prior to this visit, and the elevated ASO titer, the patient was started on penicillin V (250 mg bid) and instructed to return for follow up in two days.
At the follow-up visit, no improvement was noted; the patient continued to experience nightly fevers and hip pain. Rovsing, rebound tenderness, obturator, and psoas signs continued to be negative. Physical examination did, however, reveal a mild abdominal tenderness in the right lower quadrant.
Due to this new finding, an abdominal ultrasound was ordered to screen for appendicitis. Despite the parents’ appropriate concern for the child, misunderstanding about the urgent need to obtain the abdominal ultrasound led to a two-day delay in scheduling the exam. Results of ultrasonography revealed psoas abscess, and the patient was promptly admitted to the pediatric floor of the local hospital.
Continue for discussion >>
DISCUSSION
Psoas abscess is a collection of pus in the iliopsoas compartment, an extraperitoneal space containing the psoas and iliacus muscles.2 It can be life-threatening if the infection progresses to septic shock. Historically, psoas abscesses were a frequent complication of tuberculosis (TB) of the spine; but with modern TB treatment, these abscesses have become rare.2 Paradoxically, increased utilization of CT to evaluate sepsis of unknown etiology has led to a recent increase in the frequency of psoas abscess diagnosis.3
Psoas abscesses are categorized as either primary or secondary, with primary infections originating in the psoas muscle and secondary infections spreading from adjacent organs.2 In 42% to 88% of cases (depending on the study), primary psoas abscesses are caused by the hematogenous spread of Staphylococcus aureus from distant infection sites.2,4,5 The psoas muscle is particularly susceptible to this mode of infection because of its rich vascular supply.6 Children, immunosuppressed adults (ie, patients with diabetes, HIV/AIDS, or renal failure), IV drug users, and patients with a history of trauma to the muscle are most susceptible to developing a primary psoas abscess.2,5
Secondary psoas abscesses are caused by infections involving adjacent structures of the gastrointestinal, urinary, and skeletal systems. They are most frequently associated with intra-abdominal inflammatory processes, with the most common etiology being Crohn disease.5 Secondary psoas abscesses, though more diverse in their bacterial flora, tend to follow certain microbiologic patterns based on the inoculating source; Escherichia coli is the most common pathogen in secondary abscesses caused by gastrointestinal (42%) and urinary (61%) sources, and S aureus the most common (35%) from skeletal origins (ie, osteomyelitis).4,5Mycobacterium tuberculosis is the more frequently found cause in developing countries but should be considered if the patient has recently travelled outside the United States.
Review of the literature suggests that the incidence of methicillin-resistant S aureus (MRSA) as the causative agent of psoas abscesses may be increasing. However, there is a wide variance in the incidence reported, ranging from 1.1% to 12% of confirmed microbial infections.5,7,8
The classic historical presentation of psoas abscess has been described as the triad of back pain, fever, and limp5,6; however, this triad has only been described in approximately 30% of cases.5 The typical presentation consists of flank or lower limb pain (91%), fever (75%), anorexia (46%), and/or weakness (43%).4 Laboratory abnormalities include leukocytosis (67%) and elevated markers of inflammation (eg, erythrocyte sedimentation rate, seen in 73% of cases).4
Imaging via abdominal ultrasound may be helpful to screen for psoas abscess; however, its utility is limited by a low diagnostic yield of 60% or less.2,4 Direct visualization of the retroperitoneal structures, for example, can be problematic due to the presence of bowel gas.9 Abdominal CT is considered the gold standard for the definitive diagnosis of psoas abscess due to its high sensitivity (100%) and specificity (77%); it can also be used simultaneously to guide percutaneous drainage to treat the abscess if needed.7 However, some clinicians prefer abdominal MRI because of its ability to enhance soft-tissue visualization without requiring use of IV contrast.2,4
The approach to treating psoas abscess varies from a strictly antibiotic regimen to percutaneous drainage, and in rare circumstances, open surgical drainage. Antibiotic therapy without drainage or surgical intervention is a sufficient starting point for treatment of abscesses less than 3 cm in size.3
The antibiotic regimen choice depends on the suspected pathogen. In cases of suspected S aureus, empiric antistaphylococcal antibiotics should be initiated while culture results are pending.2,4 Secondary psoas abscesses thought to be derived from a urinary or gastrointestinal source should prompt use of a broader spectrum antibiotic due to the higher probability of gram-negative, anaerobic, or polymicrobial involvement.2,4
Once final culture and sensitivity results are obtained, antibiotic therapy should be modified to target the isolated pathogen(s). Treatment duration is typically six weeks but may vary, depending on serial culture results and the inoculating source.4 Review of the literature reveals that abscesses resulting from skeletal sources have traditionally been treated longer, usually with antibiotics alone, than those from urinary or gastrointestinal sources, which are often treated with the combination of antibiotics and percutaneous drainage.4
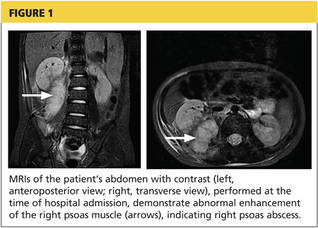
In cases of psoas abscesses larger than 3 cm, management should include both appropriate antibiotics and percutaneous drainage of the abscess.2 Percutaneous drainage is preferred to open surgical drainage because outcomes are similar, it is less invasive, and there is less risk of spreading abscess contents.2-4 In a retrospective analysis by Dietrich et al, 50% of patients treated with antibiotics and percutaneous drainage responded after one drainage, but the success rate increased to 100% after a second drainage.7 In addition, percutaneous drainage was associated with a lower mortality rate and a shorter hospital stay when compared to open surgical drainage.7
Open surgical drainage is rarely performed and usually only considered if the patient is not responding to a combination of focused antibiotic treatment and percutaneous drainage or has associated comorbidities, such as Crohn ileocolitis.2-4 In a retrospective analysis by Tabrizian et al, percutaneous drainage served as a bridge to open surgical drainage in nearly all patients with a gastrointestinal origin, such as Crohn disease, diverticulitis, appendicitis, and/or pancreatitis.6
Treatment of psoas abscesses has an overall failure rate of 15.8%, with an associated mortality rate of less than 7%.4 Overall prognosis is good, but outcomes can be negatively affected by such factors as advanced age, delay in diagnosis, bacteremia, and other comorbidities.4
Next page: Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
The patient required an 11-day hospitalization; her day-by-day course is described briefly below.
Day 1. Upon admission, abdominal MRI was ordered (see Figure 1) and empiric piperacillin/tazobactam IV was initiated. C-reactive protein (CRP) level and white blood cell (WBC) counts were elevated (see Table 2). Infectious disease, surgery, and urology consults were obtained.
Day 2. Fine-needle aspiration of the abscess was performed for cultures, and 40 mL of purulent fluid was drained. Piperacillin/tazobactam administration was continued, but the patient experienced ongoing fever and vomiting.
Day 3. Preliminary aspirate culture results revealed S aureus infection. Piperacillin/tazobactam was discontinued, and vancomycin IV was started. CRP levels and WBC counts decreased, as did fever and vomiting.
Day 4. Final aspirate culture results identified MRSA infection, sensitive to clindamycin. Vancomycin was discontinued, and clindamycin IV was started. Although the patient’s condition improved somewhat, fever and vomiting persisted.
Day 5. Both CRP levels and WBC counts increased from day 3. A surgical consult was sought.
Day 6. Repeat abdominal MRI revealed a decrease in the size of the abscess (see Figure 2, page 30. CRP levels and WBC counts remained high, with persistent fever and vomiting.
Day 7. The clinical team, in consultation with the parents, determined that placement of a peripherally inserted central catheter (PICC) line for drainage of the abscess was necessary.
Day 8. A 10-French pigtail catheter was inserted into the abscess, 20 mL of purulent fluid was drained, and a PICC line was inserted. Clindamycin IV was continued and, eight hours after the catheter was placed, fever and vomiting resolved.
Day 9. Both CRP levels and WBC counts dropped by half (WBC count was normal), while 10 mL of clear fluid drained from the catheter. The patient remained afebrile, without nausea or vomiting, on clindamycin IV.
Day 10. After 36 hours of clear drainage, the catheter was removed. CRP level further decreased. Clindamycin IV was discontinued, and the patient, now asymptomatic, was started on oral clindamycin.
Day 11. The patient was discharged on a regimen of oral clindamycin for six weeks, with weekly abdominal ultrasounds. She completed her entire course of antibiotics and fully recovered from the infection.
Next page: Conclusion >>
CONCLUSION
Since children generally compensate well during times of increased stress on the body, it is vital that persistent FUOs continue to be evaluated until a definitive source is identified, especially in this population. Early diagnosis and treatment of psoas abscess is essential for better outcomes, since delay is associated with a greater risk for sepsis.
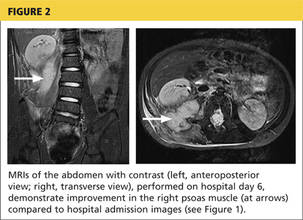
While the likelihood of developing psoas abscess is low, it is worth keeping the diagnosis in mind for cases of unexplained lower abdominal pain, flank pain, or hip pain when more common etiologies have been excluded. This is especially important in the setting of recent travel to a developing country due to the fact that a psoas abscess can be a complication of TB of the spine.
The authors would like to thank Jeff Brand, MD, for his assistance in the preparation of this manuscript.
REFERENCES
1. Wong-Baker Faces Corporation. Wong-Baker FACES Pain Rating Scale. www.wongbakerfaces.org. Accessed May 19, 2015.
2. Mallick IH, Thoufreeq MH, Rajendren TP. Iliopsoas abscesses. Postgrad Med J. 2004;80(946):459-462.
3. Yacoub WN, Sohn HJ, Chan S, et al. Psoas abscess rarely requires surgical intervention. Am J Surg. 2008;196(2):223-227.
4. Lopez VN, Ramos JM, Meseguer V, et al; The Infectious Diseases Study Group of the Spanish Society of Internal Medicine. Microbiology and outcome of iliopsoas abscess in 124 patients. Medicine. 2009;88(2):120-130.
5. Shields D, Robinson P, Crowley TP. Iliopsoas abscess—a review and update on the literature. Int J Surg. 2012;10(9):466-469.
6. Tabrizian P, Nguyen SQ, Greenstein A, et al. Management and treatment of iliopsoas abscess. Arch Surg. 2009;144(10):946-949.
7. Dietrich A, Vaccarezza H, Vaccaro CA. Iliopsoas abscess: presentation, management, and outcomes. Surg Laparosc Endosc Percutan Tech. 2013;23(1):45-48.
8. Wong OF, Ho PL, Lam SK. Retrospective review of clinical presentations, microbiology, and outcomes of patients with psoas abscess. Hong Kong Med J. 2013;19(5):416-423.
9. Woo MY. Psoas abscess. J Emerg Med. 2014;47(5):e129-e130.
A 5-year-old Filipino girl was brought to a pediatric clinic for follow up of an unresolved fever and for new-onset right hip pain, which occurred intermittently for the past week and was associated with a right-sided limp. She had been experiencing nightly fevers ranging from 101°F to 105°F for the past two weeks, for which her parents had been giving ibuprofen with mixed results; she remained afebrile during daytime hours.
Using the Wong-Baker FACES pain scale, the patient rated the pain as a 4/10 in severity (“Hurts a Little More” face).1 Standing and walking aggravated the pain but did not limit activity. Although ibuprofen decreased the fever, it did not alleviate the hip pain. Other symptoms included vomiting one to two times daily, without hematemesis, and four to five episodes of diarrhea daily, without abdominal pain, hematochezia, or melena. She also experienced decreased appetite, but her parents reported no change in her dietary or fluid intake. The patient and her parents denied additional symptoms.
Further investigation revealed that the patient had been seen a week earlier by two other clinicians in the office for complaints of fever, rash, nausea, hematemesis, and diarrhea. She had been diagnosed with a herpes simplex viral (HSV) lesion of the nose, epistaxis, and viral gastroenteritis. Her treatment plan consisted of acyclovir ointment for the HSV lesion and symptomatic support for the gastroenteritis associated diarrhea. The complaint of hematemesis was attributed to postnasal drip from the epistaxis, and reassurance was provided to the patient and family. In addition, six weeks earlier, the patient had been treated for otitis media with a full course of amoxicillin.
Medical history was negative for surgeries, trauma, injuries, and chronic medical conditions. She took no medications or supplements on a regular basis. Her parents denied any known drug allergies and stated that her immunizations were up to date.
The patient lived at home with her biological parents and two brothers, all of whom were healthy, without any recent infections or illnesses. Of significance, the family had travelled to the Philippines for vacation about four months earlier. Results of a tuberculin skin test done six weeks earlier (because the patient presented with respiratory symptoms shortly after traveling to the Philippines) were negative.
Physical examination revealed a well-developed, well-nourished 40-lb girl, in no acute distress, who was active and playful with her brother while in the exam room. Vital signs were significant for a fever of 101.9°F (last dose of ibuprofen was approximately six hours earlier) but were otherwise stable. Skin exam revealed that the prior HSV lesion of the nose had resolved. HEENT, cardiovascular, and pulmonary exam findings were noncontributory. Urine dipstick was negative.
Abdominal exam revealed normoactive bowel sounds in all four quadrants, and on palpation, the abdomen was soft, nontender, and without organomegaly. Specialized abdominal exams to assess for peritonitis, including those to elicit Rovsing, rebound tenderness, obturator, and psoas signs, were all negative. Bilateral extremity exams of the hips, knees, and ankles revealed full range of motion (active and passive), with normal muscle strength throughout. The only significant finding on the physical exam was mild pain of the right anterior hip at 15° of flexion, appreciated while the patient was supine on the exam table. The patient was also observed pushing off her right lower extremity when climbing onto the exam table, and she skipped down the hall when leaving the exam.
With fever of unknown origin (FUO) and a largely negative history and physical, the working list of differential diagnoses included
• Avascular necrosis
• Bacteremia
• Juvenile idiopathic arthritis
• Osteomyelitis
• Pyelonephritis
• Reiter syndrome
• Rheumatic fever
• Rheumatoid arthritis
• Septic joint
• Urinary tract infection
To begin the diagnostic process, a number of laboratory tests and imaging procedures were ordered. Table 1 presents the results of these studies. A tuberculin skin test was not repeated. While awaiting test results, the patient was started on naproxen oral suspension (125 mg/5 mL; 4 mL bid) for fever and pain control.
Based on findings consistent with an inflammatory pattern, the history of otitis media (of possible streptococcal origin) six weeks prior to this visit, and the elevated ASO titer, the patient was started on penicillin V (250 mg bid) and instructed to return for follow up in two days.
At the follow-up visit, no improvement was noted; the patient continued to experience nightly fevers and hip pain. Rovsing, rebound tenderness, obturator, and psoas signs continued to be negative. Physical examination did, however, reveal a mild abdominal tenderness in the right lower quadrant.
Due to this new finding, an abdominal ultrasound was ordered to screen for appendicitis. Despite the parents’ appropriate concern for the child, misunderstanding about the urgent need to obtain the abdominal ultrasound led to a two-day delay in scheduling the exam. Results of ultrasonography revealed psoas abscess, and the patient was promptly admitted to the pediatric floor of the local hospital.
Continue for discussion >>
DISCUSSION
Psoas abscess is a collection of pus in the iliopsoas compartment, an extraperitoneal space containing the psoas and iliacus muscles.2 It can be life-threatening if the infection progresses to septic shock. Historically, psoas abscesses were a frequent complication of tuberculosis (TB) of the spine; but with modern TB treatment, these abscesses have become rare.2 Paradoxically, increased utilization of CT to evaluate sepsis of unknown etiology has led to a recent increase in the frequency of psoas abscess diagnosis.3
Psoas abscesses are categorized as either primary or secondary, with primary infections originating in the psoas muscle and secondary infections spreading from adjacent organs.2 In 42% to 88% of cases (depending on the study), primary psoas abscesses are caused by the hematogenous spread of Staphylococcus aureus from distant infection sites.2,4,5 The psoas muscle is particularly susceptible to this mode of infection because of its rich vascular supply.6 Children, immunosuppressed adults (ie, patients with diabetes, HIV/AIDS, or renal failure), IV drug users, and patients with a history of trauma to the muscle are most susceptible to developing a primary psoas abscess.2,5
Secondary psoas abscesses are caused by infections involving adjacent structures of the gastrointestinal, urinary, and skeletal systems. They are most frequently associated with intra-abdominal inflammatory processes, with the most common etiology being Crohn disease.5 Secondary psoas abscesses, though more diverse in their bacterial flora, tend to follow certain microbiologic patterns based on the inoculating source; Escherichia coli is the most common pathogen in secondary abscesses caused by gastrointestinal (42%) and urinary (61%) sources, and S aureus the most common (35%) from skeletal origins (ie, osteomyelitis).4,5Mycobacterium tuberculosis is the more frequently found cause in developing countries but should be considered if the patient has recently travelled outside the United States.
Review of the literature suggests that the incidence of methicillin-resistant S aureus (MRSA) as the causative agent of psoas abscesses may be increasing. However, there is a wide variance in the incidence reported, ranging from 1.1% to 12% of confirmed microbial infections.5,7,8
The classic historical presentation of psoas abscess has been described as the triad of back pain, fever, and limp5,6; however, this triad has only been described in approximately 30% of cases.5 The typical presentation consists of flank or lower limb pain (91%), fever (75%), anorexia (46%), and/or weakness (43%).4 Laboratory abnormalities include leukocytosis (67%) and elevated markers of inflammation (eg, erythrocyte sedimentation rate, seen in 73% of cases).4
Imaging via abdominal ultrasound may be helpful to screen for psoas abscess; however, its utility is limited by a low diagnostic yield of 60% or less.2,4 Direct visualization of the retroperitoneal structures, for example, can be problematic due to the presence of bowel gas.9 Abdominal CT is considered the gold standard for the definitive diagnosis of psoas abscess due to its high sensitivity (100%) and specificity (77%); it can also be used simultaneously to guide percutaneous drainage to treat the abscess if needed.7 However, some clinicians prefer abdominal MRI because of its ability to enhance soft-tissue visualization without requiring use of IV contrast.2,4
The approach to treating psoas abscess varies from a strictly antibiotic regimen to percutaneous drainage, and in rare circumstances, open surgical drainage. Antibiotic therapy without drainage or surgical intervention is a sufficient starting point for treatment of abscesses less than 3 cm in size.3
The antibiotic regimen choice depends on the suspected pathogen. In cases of suspected S aureus, empiric antistaphylococcal antibiotics should be initiated while culture results are pending.2,4 Secondary psoas abscesses thought to be derived from a urinary or gastrointestinal source should prompt use of a broader spectrum antibiotic due to the higher probability of gram-negative, anaerobic, or polymicrobial involvement.2,4
Once final culture and sensitivity results are obtained, antibiotic therapy should be modified to target the isolated pathogen(s). Treatment duration is typically six weeks but may vary, depending on serial culture results and the inoculating source.4 Review of the literature reveals that abscesses resulting from skeletal sources have traditionally been treated longer, usually with antibiotics alone, than those from urinary or gastrointestinal sources, which are often treated with the combination of antibiotics and percutaneous drainage.4
In cases of psoas abscesses larger than 3 cm, management should include both appropriate antibiotics and percutaneous drainage of the abscess.2 Percutaneous drainage is preferred to open surgical drainage because outcomes are similar, it is less invasive, and there is less risk of spreading abscess contents.2-4 In a retrospective analysis by Dietrich et al, 50% of patients treated with antibiotics and percutaneous drainage responded after one drainage, but the success rate increased to 100% after a second drainage.7 In addition, percutaneous drainage was associated with a lower mortality rate and a shorter hospital stay when compared to open surgical drainage.7
Open surgical drainage is rarely performed and usually only considered if the patient is not responding to a combination of focused antibiotic treatment and percutaneous drainage or has associated comorbidities, such as Crohn ileocolitis.2-4 In a retrospective analysis by Tabrizian et al, percutaneous drainage served as a bridge to open surgical drainage in nearly all patients with a gastrointestinal origin, such as Crohn disease, diverticulitis, appendicitis, and/or pancreatitis.6
Treatment of psoas abscesses has an overall failure rate of 15.8%, with an associated mortality rate of less than 7%.4 Overall prognosis is good, but outcomes can be negatively affected by such factors as advanced age, delay in diagnosis, bacteremia, and other comorbidities.4
Next page: Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
The patient required an 11-day hospitalization; her day-by-day course is described briefly below.
Day 1. Upon admission, abdominal MRI was ordered (see Figure 1) and empiric piperacillin/tazobactam IV was initiated. C-reactive protein (CRP) level and white blood cell (WBC) counts were elevated (see Table 2). Infectious disease, surgery, and urology consults were obtained.
Day 2. Fine-needle aspiration of the abscess was performed for cultures, and 40 mL of purulent fluid was drained. Piperacillin/tazobactam administration was continued, but the patient experienced ongoing fever and vomiting.
Day 3. Preliminary aspirate culture results revealed S aureus infection. Piperacillin/tazobactam was discontinued, and vancomycin IV was started. CRP levels and WBC counts decreased, as did fever and vomiting.
Day 4. Final aspirate culture results identified MRSA infection, sensitive to clindamycin. Vancomycin was discontinued, and clindamycin IV was started. Although the patient’s condition improved somewhat, fever and vomiting persisted.
Day 5. Both CRP levels and WBC counts increased from day 3. A surgical consult was sought.
Day 6. Repeat abdominal MRI revealed a decrease in the size of the abscess (see Figure 2, page 30. CRP levels and WBC counts remained high, with persistent fever and vomiting.
Day 7. The clinical team, in consultation with the parents, determined that placement of a peripherally inserted central catheter (PICC) line for drainage of the abscess was necessary.
Day 8. A 10-French pigtail catheter was inserted into the abscess, 20 mL of purulent fluid was drained, and a PICC line was inserted. Clindamycin IV was continued and, eight hours after the catheter was placed, fever and vomiting resolved.
Day 9. Both CRP levels and WBC counts dropped by half (WBC count was normal), while 10 mL of clear fluid drained from the catheter. The patient remained afebrile, without nausea or vomiting, on clindamycin IV.
Day 10. After 36 hours of clear drainage, the catheter was removed. CRP level further decreased. Clindamycin IV was discontinued, and the patient, now asymptomatic, was started on oral clindamycin.
Day 11. The patient was discharged on a regimen of oral clindamycin for six weeks, with weekly abdominal ultrasounds. She completed her entire course of antibiotics and fully recovered from the infection.
Next page: Conclusion >>
CONCLUSION
Since children generally compensate well during times of increased stress on the body, it is vital that persistent FUOs continue to be evaluated until a definitive source is identified, especially in this population. Early diagnosis and treatment of psoas abscess is essential for better outcomes, since delay is associated with a greater risk for sepsis.
While the likelihood of developing psoas abscess is low, it is worth keeping the diagnosis in mind for cases of unexplained lower abdominal pain, flank pain, or hip pain when more common etiologies have been excluded. This is especially important in the setting of recent travel to a developing country due to the fact that a psoas abscess can be a complication of TB of the spine.
The authors would like to thank Jeff Brand, MD, for his assistance in the preparation of this manuscript.
REFERENCES
1. Wong-Baker Faces Corporation. Wong-Baker FACES Pain Rating Scale. www.wongbakerfaces.org. Accessed May 19, 2015.
2. Mallick IH, Thoufreeq MH, Rajendren TP. Iliopsoas abscesses. Postgrad Med J. 2004;80(946):459-462.
3. Yacoub WN, Sohn HJ, Chan S, et al. Psoas abscess rarely requires surgical intervention. Am J Surg. 2008;196(2):223-227.
4. Lopez VN, Ramos JM, Meseguer V, et al; The Infectious Diseases Study Group of the Spanish Society of Internal Medicine. Microbiology and outcome of iliopsoas abscess in 124 patients. Medicine. 2009;88(2):120-130.
5. Shields D, Robinson P, Crowley TP. Iliopsoas abscess—a review and update on the literature. Int J Surg. 2012;10(9):466-469.
6. Tabrizian P, Nguyen SQ, Greenstein A, et al. Management and treatment of iliopsoas abscess. Arch Surg. 2009;144(10):946-949.
7. Dietrich A, Vaccarezza H, Vaccaro CA. Iliopsoas abscess: presentation, management, and outcomes. Surg Laparosc Endosc Percutan Tech. 2013;23(1):45-48.
8. Wong OF, Ho PL, Lam SK. Retrospective review of clinical presentations, microbiology, and outcomes of patients with psoas abscess. Hong Kong Med J. 2013;19(5):416-423.
9. Woo MY. Psoas abscess. J Emerg Med. 2014;47(5):e129-e130.
A 5-year-old Filipino girl was brought to a pediatric clinic for follow up of an unresolved fever and for new-onset right hip pain, which occurred intermittently for the past week and was associated with a right-sided limp. She had been experiencing nightly fevers ranging from 101°F to 105°F for the past two weeks, for which her parents had been giving ibuprofen with mixed results; she remained afebrile during daytime hours.
Using the Wong-Baker FACES pain scale, the patient rated the pain as a 4/10 in severity (“Hurts a Little More” face).1 Standing and walking aggravated the pain but did not limit activity. Although ibuprofen decreased the fever, it did not alleviate the hip pain. Other symptoms included vomiting one to two times daily, without hematemesis, and four to five episodes of diarrhea daily, without abdominal pain, hematochezia, or melena. She also experienced decreased appetite, but her parents reported no change in her dietary or fluid intake. The patient and her parents denied additional symptoms.
Further investigation revealed that the patient had been seen a week earlier by two other clinicians in the office for complaints of fever, rash, nausea, hematemesis, and diarrhea. She had been diagnosed with a herpes simplex viral (HSV) lesion of the nose, epistaxis, and viral gastroenteritis. Her treatment plan consisted of acyclovir ointment for the HSV lesion and symptomatic support for the gastroenteritis associated diarrhea. The complaint of hematemesis was attributed to postnasal drip from the epistaxis, and reassurance was provided to the patient and family. In addition, six weeks earlier, the patient had been treated for otitis media with a full course of amoxicillin.
Medical history was negative for surgeries, trauma, injuries, and chronic medical conditions. She took no medications or supplements on a regular basis. Her parents denied any known drug allergies and stated that her immunizations were up to date.
The patient lived at home with her biological parents and two brothers, all of whom were healthy, without any recent infections or illnesses. Of significance, the family had travelled to the Philippines for vacation about four months earlier. Results of a tuberculin skin test done six weeks earlier (because the patient presented with respiratory symptoms shortly after traveling to the Philippines) were negative.
Physical examination revealed a well-developed, well-nourished 40-lb girl, in no acute distress, who was active and playful with her brother while in the exam room. Vital signs were significant for a fever of 101.9°F (last dose of ibuprofen was approximately six hours earlier) but were otherwise stable. Skin exam revealed that the prior HSV lesion of the nose had resolved. HEENT, cardiovascular, and pulmonary exam findings were noncontributory. Urine dipstick was negative.
Abdominal exam revealed normoactive bowel sounds in all four quadrants, and on palpation, the abdomen was soft, nontender, and without organomegaly. Specialized abdominal exams to assess for peritonitis, including those to elicit Rovsing, rebound tenderness, obturator, and psoas signs, were all negative. Bilateral extremity exams of the hips, knees, and ankles revealed full range of motion (active and passive), with normal muscle strength throughout. The only significant finding on the physical exam was mild pain of the right anterior hip at 15° of flexion, appreciated while the patient was supine on the exam table. The patient was also observed pushing off her right lower extremity when climbing onto the exam table, and she skipped down the hall when leaving the exam.
With fever of unknown origin (FUO) and a largely negative history and physical, the working list of differential diagnoses included
• Avascular necrosis
• Bacteremia
• Juvenile idiopathic arthritis
• Osteomyelitis
• Pyelonephritis
• Reiter syndrome
• Rheumatic fever
• Rheumatoid arthritis
• Septic joint
• Urinary tract infection
To begin the diagnostic process, a number of laboratory tests and imaging procedures were ordered. Table 1 presents the results of these studies. A tuberculin skin test was not repeated. While awaiting test results, the patient was started on naproxen oral suspension (125 mg/5 mL; 4 mL bid) for fever and pain control.
Based on findings consistent with an inflammatory pattern, the history of otitis media (of possible streptococcal origin) six weeks prior to this visit, and the elevated ASO titer, the patient was started on penicillin V (250 mg bid) and instructed to return for follow up in two days.
At the follow-up visit, no improvement was noted; the patient continued to experience nightly fevers and hip pain. Rovsing, rebound tenderness, obturator, and psoas signs continued to be negative. Physical examination did, however, reveal a mild abdominal tenderness in the right lower quadrant.
Due to this new finding, an abdominal ultrasound was ordered to screen for appendicitis. Despite the parents’ appropriate concern for the child, misunderstanding about the urgent need to obtain the abdominal ultrasound led to a two-day delay in scheduling the exam. Results of ultrasonography revealed psoas abscess, and the patient was promptly admitted to the pediatric floor of the local hospital.
Continue for discussion >>
DISCUSSION
Psoas abscess is a collection of pus in the iliopsoas compartment, an extraperitoneal space containing the psoas and iliacus muscles.2 It can be life-threatening if the infection progresses to septic shock. Historically, psoas abscesses were a frequent complication of tuberculosis (TB) of the spine; but with modern TB treatment, these abscesses have become rare.2 Paradoxically, increased utilization of CT to evaluate sepsis of unknown etiology has led to a recent increase in the frequency of psoas abscess diagnosis.3
Psoas abscesses are categorized as either primary or secondary, with primary infections originating in the psoas muscle and secondary infections spreading from adjacent organs.2 In 42% to 88% of cases (depending on the study), primary psoas abscesses are caused by the hematogenous spread of Staphylococcus aureus from distant infection sites.2,4,5 The psoas muscle is particularly susceptible to this mode of infection because of its rich vascular supply.6 Children, immunosuppressed adults (ie, patients with diabetes, HIV/AIDS, or renal failure), IV drug users, and patients with a history of trauma to the muscle are most susceptible to developing a primary psoas abscess.2,5
Secondary psoas abscesses are caused by infections involving adjacent structures of the gastrointestinal, urinary, and skeletal systems. They are most frequently associated with intra-abdominal inflammatory processes, with the most common etiology being Crohn disease.5 Secondary psoas abscesses, though more diverse in their bacterial flora, tend to follow certain microbiologic patterns based on the inoculating source; Escherichia coli is the most common pathogen in secondary abscesses caused by gastrointestinal (42%) and urinary (61%) sources, and S aureus the most common (35%) from skeletal origins (ie, osteomyelitis).4,5Mycobacterium tuberculosis is the more frequently found cause in developing countries but should be considered if the patient has recently travelled outside the United States.
Review of the literature suggests that the incidence of methicillin-resistant S aureus (MRSA) as the causative agent of psoas abscesses may be increasing. However, there is a wide variance in the incidence reported, ranging from 1.1% to 12% of confirmed microbial infections.5,7,8
The classic historical presentation of psoas abscess has been described as the triad of back pain, fever, and limp5,6; however, this triad has only been described in approximately 30% of cases.5 The typical presentation consists of flank or lower limb pain (91%), fever (75%), anorexia (46%), and/or weakness (43%).4 Laboratory abnormalities include leukocytosis (67%) and elevated markers of inflammation (eg, erythrocyte sedimentation rate, seen in 73% of cases).4
Imaging via abdominal ultrasound may be helpful to screen for psoas abscess; however, its utility is limited by a low diagnostic yield of 60% or less.2,4 Direct visualization of the retroperitoneal structures, for example, can be problematic due to the presence of bowel gas.9 Abdominal CT is considered the gold standard for the definitive diagnosis of psoas abscess due to its high sensitivity (100%) and specificity (77%); it can also be used simultaneously to guide percutaneous drainage to treat the abscess if needed.7 However, some clinicians prefer abdominal MRI because of its ability to enhance soft-tissue visualization without requiring use of IV contrast.2,4
The approach to treating psoas abscess varies from a strictly antibiotic regimen to percutaneous drainage, and in rare circumstances, open surgical drainage. Antibiotic therapy without drainage or surgical intervention is a sufficient starting point for treatment of abscesses less than 3 cm in size.3
The antibiotic regimen choice depends on the suspected pathogen. In cases of suspected S aureus, empiric antistaphylococcal antibiotics should be initiated while culture results are pending.2,4 Secondary psoas abscesses thought to be derived from a urinary or gastrointestinal source should prompt use of a broader spectrum antibiotic due to the higher probability of gram-negative, anaerobic, or polymicrobial involvement.2,4
Once final culture and sensitivity results are obtained, antibiotic therapy should be modified to target the isolated pathogen(s). Treatment duration is typically six weeks but may vary, depending on serial culture results and the inoculating source.4 Review of the literature reveals that abscesses resulting from skeletal sources have traditionally been treated longer, usually with antibiotics alone, than those from urinary or gastrointestinal sources, which are often treated with the combination of antibiotics and percutaneous drainage.4
In cases of psoas abscesses larger than 3 cm, management should include both appropriate antibiotics and percutaneous drainage of the abscess.2 Percutaneous drainage is preferred to open surgical drainage because outcomes are similar, it is less invasive, and there is less risk of spreading abscess contents.2-4 In a retrospective analysis by Dietrich et al, 50% of patients treated with antibiotics and percutaneous drainage responded after one drainage, but the success rate increased to 100% after a second drainage.7 In addition, percutaneous drainage was associated with a lower mortality rate and a shorter hospital stay when compared to open surgical drainage.7
Open surgical drainage is rarely performed and usually only considered if the patient is not responding to a combination of focused antibiotic treatment and percutaneous drainage or has associated comorbidities, such as Crohn ileocolitis.2-4 In a retrospective analysis by Tabrizian et al, percutaneous drainage served as a bridge to open surgical drainage in nearly all patients with a gastrointestinal origin, such as Crohn disease, diverticulitis, appendicitis, and/or pancreatitis.6
Treatment of psoas abscesses has an overall failure rate of 15.8%, with an associated mortality rate of less than 7%.4 Overall prognosis is good, but outcomes can be negatively affected by such factors as advanced age, delay in diagnosis, bacteremia, and other comorbidities.4
Next page: Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
The patient required an 11-day hospitalization; her day-by-day course is described briefly below.
Day 1. Upon admission, abdominal MRI was ordered (see Figure 1) and empiric piperacillin/tazobactam IV was initiated. C-reactive protein (CRP) level and white blood cell (WBC) counts were elevated (see Table 2). Infectious disease, surgery, and urology consults were obtained.
Day 2. Fine-needle aspiration of the abscess was performed for cultures, and 40 mL of purulent fluid was drained. Piperacillin/tazobactam administration was continued, but the patient experienced ongoing fever and vomiting.
Day 3. Preliminary aspirate culture results revealed S aureus infection. Piperacillin/tazobactam was discontinued, and vancomycin IV was started. CRP levels and WBC counts decreased, as did fever and vomiting.
Day 4. Final aspirate culture results identified MRSA infection, sensitive to clindamycin. Vancomycin was discontinued, and clindamycin IV was started. Although the patient’s condition improved somewhat, fever and vomiting persisted.
Day 5. Both CRP levels and WBC counts increased from day 3. A surgical consult was sought.
Day 6. Repeat abdominal MRI revealed a decrease in the size of the abscess (see Figure 2, page 30. CRP levels and WBC counts remained high, with persistent fever and vomiting.
Day 7. The clinical team, in consultation with the parents, determined that placement of a peripherally inserted central catheter (PICC) line for drainage of the abscess was necessary.
Day 8. A 10-French pigtail catheter was inserted into the abscess, 20 mL of purulent fluid was drained, and a PICC line was inserted. Clindamycin IV was continued and, eight hours after the catheter was placed, fever and vomiting resolved.
Day 9. Both CRP levels and WBC counts dropped by half (WBC count was normal), while 10 mL of clear fluid drained from the catheter. The patient remained afebrile, without nausea or vomiting, on clindamycin IV.
Day 10. After 36 hours of clear drainage, the catheter was removed. CRP level further decreased. Clindamycin IV was discontinued, and the patient, now asymptomatic, was started on oral clindamycin.
Day 11. The patient was discharged on a regimen of oral clindamycin for six weeks, with weekly abdominal ultrasounds. She completed her entire course of antibiotics and fully recovered from the infection.
Next page: Conclusion >>
CONCLUSION
Since children generally compensate well during times of increased stress on the body, it is vital that persistent FUOs continue to be evaluated until a definitive source is identified, especially in this population. Early diagnosis and treatment of psoas abscess is essential for better outcomes, since delay is associated with a greater risk for sepsis.
While the likelihood of developing psoas abscess is low, it is worth keeping the diagnosis in mind for cases of unexplained lower abdominal pain, flank pain, or hip pain when more common etiologies have been excluded. This is especially important in the setting of recent travel to a developing country due to the fact that a psoas abscess can be a complication of TB of the spine.
The authors would like to thank Jeff Brand, MD, for his assistance in the preparation of this manuscript.
REFERENCES
1. Wong-Baker Faces Corporation. Wong-Baker FACES Pain Rating Scale. www.wongbakerfaces.org. Accessed May 19, 2015.
2. Mallick IH, Thoufreeq MH, Rajendren TP. Iliopsoas abscesses. Postgrad Med J. 2004;80(946):459-462.
3. Yacoub WN, Sohn HJ, Chan S, et al. Psoas abscess rarely requires surgical intervention. Am J Surg. 2008;196(2):223-227.
4. Lopez VN, Ramos JM, Meseguer V, et al; The Infectious Diseases Study Group of the Spanish Society of Internal Medicine. Microbiology and outcome of iliopsoas abscess in 124 patients. Medicine. 2009;88(2):120-130.
5. Shields D, Robinson P, Crowley TP. Iliopsoas abscess—a review and update on the literature. Int J Surg. 2012;10(9):466-469.
6. Tabrizian P, Nguyen SQ, Greenstein A, et al. Management and treatment of iliopsoas abscess. Arch Surg. 2009;144(10):946-949.
7. Dietrich A, Vaccarezza H, Vaccaro CA. Iliopsoas abscess: presentation, management, and outcomes. Surg Laparosc Endosc Percutan Tech. 2013;23(1):45-48.
8. Wong OF, Ho PL, Lam SK. Retrospective review of clinical presentations, microbiology, and outcomes of patients with psoas abscess. Hong Kong Med J. 2013;19(5):416-423.
9. Woo MY. Psoas abscess. J Emerg Med. 2014;47(5):e129-e130.
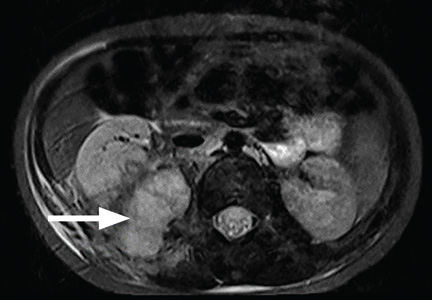
Woman, 64, With Eye Pain, Swelling, and Tearing
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
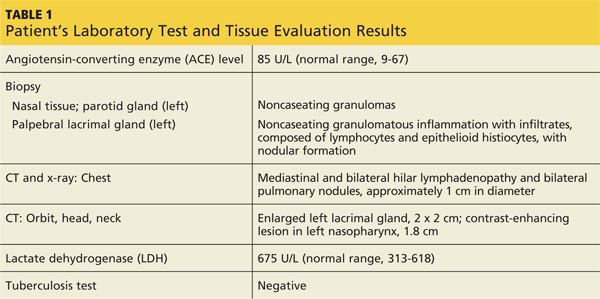
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
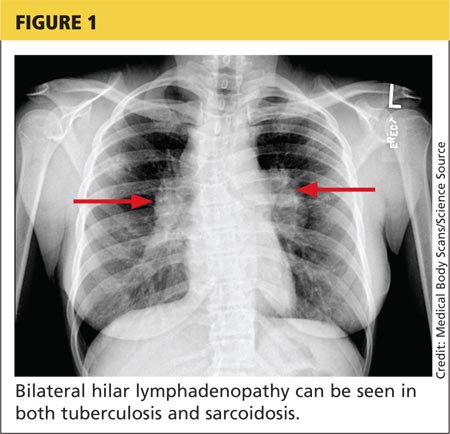
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
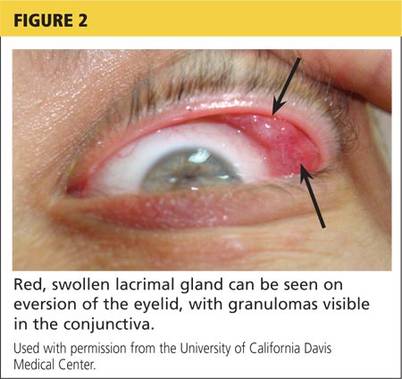
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
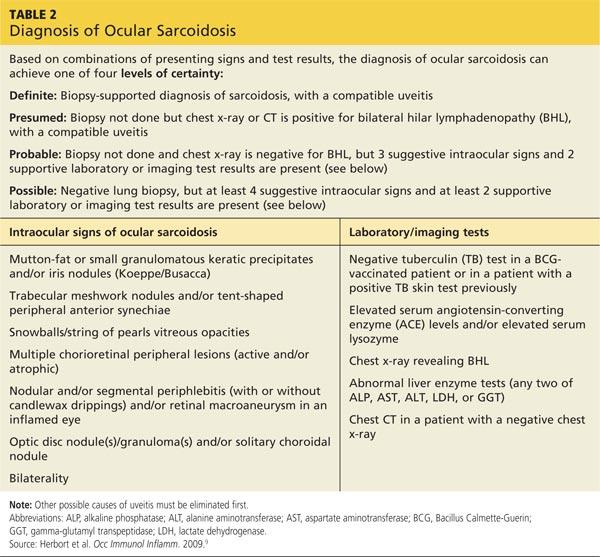
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
A 64-year-old woman presented to the clinic with a two-to-three-week history of significant pain, swelling, and excessive tearing of the left eye. The patient had a persistent cough but denied wheezing or shortness of breath.
Medical history was remarkable for uveitis, severe recurrent sinusitis, and allergic rhinitis. The patient reported that she had been exposed to benzene and burning paint fumes about 10 years ago but had no known symptoms or problems at the time.
Vital signs included a temperature of 97.0°F; respiratory rate, 18 breaths/min; pulse, 100 beats/min; and blood pressure, 144/80 mm Hg. Her height was 65 in; weight, 122 lb; and O2 saturation, 100% on room air.
Physical examination revealed a left palpebral lacrimal mass with an enlarged lacrimal gland. The left lacrimal gland and conjunctiva were mildly erythematous, with a cobblestone appearance. The right eye was stable, with no significant inflammation. Pupils were equal, round, and reactive to light and accommodation. Extraocular movements were intact. Nasal turbinates were swollen and mildly erythematous. Oropharynx was stable and tonsils absent. Left parotid gland was slightly swollen and tender.
The neck was supple with no jugular venous distension. Palpable cervical and supraclavicular lymphadenopathy, measuring approximately 1.5 x 1.5 cm bilaterally, was present. The lungs were clear to auscultation and percussion. The heart rate and rhythm were regular, with normal S1 and S2 sounds. The abdomen was soft, nontender, and without hepatosplenomegaly. Extremities were stable, with no rashes, lesions, or cutaneous skin nodules.
The patient was referred to a specialist for a complete ophthalmologic examination and further work-up. This included a complete blood count, comprehensive metabolic panel, tissue biopsies of the affected lacrimal gland and parotid gland, CT, and x-rays; results are shown in Table 1. In addition, the patient’s persistent nasosinus congestion was determined, by otolaryngologic consultation, to be the result of a deviated septum, for which she underwent endoscopic nasal septal repair with tissue biopsy.
The lacrimal gland biopsy led to a diagnosis of chronic noncaseating granulomatous dacryoadenitis, with an extensive area of necrosis. Significant findings included histiocytes and discrete nodules in the gland. Biopsies of the parotid gland and nasal tissue also identified noncaseating granulomas.
The patient’s test results suggested several possible diagnoses, including
• Granulomatosis with polyangiitis
• Tuberculosis (TB) or similar pulmonary infectious disease
• Sarcoidosis (ocular and/or pulmonary)
Continue for differential diagnosis >>
DISCUSSION
Differential diagnosis
Granulomatosis with polyangiitis. GPA, also known as Wegener granulomatosis, is characterized by necrotizing granulomatous inflammation with necrotizing vasculitis, usually of small and medium vessels; ocular involvement is frequent.1 Ocular granulomas of GPA can be mistaken for those caused by other diseases, such as mycobacterial or syphilitic infection or idiopathic uveitis.2
Tuberculosis. Common symptoms of TB include fever, cough, dyspnea, weight loss, malaise, and fatigue. Granulomas are typically necrotizing but are occasionally nonnecrotizing.3 TB can manifest with hilar and diffuse lymphadenopathy,4 which the patient’s chest imaging revealed (see Figure 1). Granulomas produced by Mycobacterium tuberculosis and atypical mycobacteria are similar histopathologically to sarcoidosis granulomas, complicating the diagnostic process.5
Next page: Sarcoidosis >>
Sarcoidosis. Sarcoidosis is a multisystem inflammatory disease characterized by noncaseating epithelioid granulomas in affected organs.6 More than 90% of patients with sarcoidosis present with pulmonary symptoms, including shortness of breath, cough, and pleuritic chest pain.6-8 Ocular manifestations, such as uveitis, iritis, or conjunctivitis, are less common, developing in 30% to 60% of patients.2,9,10 In addition, rashes, lesions, or cutaneous skin nodules, including erythema nodosum and lupus pernio, are seen in 25% to 35% of patients.2,6
In up to two-thirds of patients, sarcoidosis resolves spontaneously2; in others, it may become chronic and progressive.4 Patients may have few or no symptoms; some require no treatment, while others may be severely affected by the disease.
Ocular involvement in sarcoidosis generally manifests as uveitis, most commonly in the anterior chamber. Uveitis is a potentially vision-threatening inflammatory disease involving both the uveal tract and adjacent structures.11 In a review of records for 2,619 patients with uveitis, 59.9% had anterior disease, of whom 2.1% were diagnosed with sarcoidosis.11
While the etiology of sarcoidosis continues to be studied,7 the prevailing theory is that, in genetically predisposed individuals, sarcoidosis is a cell-mediated immune response to as-yet unknown antigen triggers that leads to granuloma formation.3,6,7
CD4+ activated T-cells stimulate the immune reaction against an antigen, producing cytokines that activate immune cells (eg, B cells, macrophages, monocytes, and neutrophils).2 Immune cells accumulate and aggregate at antigen sites in an exaggerated response, resulting in the formation of granulomas (see Figure 2).7,12,13
Infectious agents have long been investigated as possible causative agents in sarcoidosis, with Mycobacterium species most frequently identified.5 Additional possibilities include Propionibacterium acnes (found predominantly in skin lesions) and herpesviruses, although viruses are not known to cause epitheliod granulomas.14
Environmental triggers have also been explored. One large study found a possible association between exposure to insecticides, agricultural environments, and microbial bioaerosols and sarcoidosis.15
The difficulty of pinpointing a single etiology for sarcoidosis—with its varying clinical manifestations, severity, and disease course—suggests that sarcoidosis may be a spectrum of disorders caused by the interaction of genetic, immunologic, infectious, and environmental factors.14
Next page: Diagnosis of sarcoidosis >>
Diagnosis
The diagnosis of sarcoidosis is based on clinical and radiologic features, histologic evidence of noncaseating granulomas, and exclusion of other possible causes of granulomas.2,12 In addition, when ocular sarcoidosis is suspected, other possible causes of uveitis must be excluded.
In an effort to address these challenges, the International Workshop on Ocular Sarcoidosis (IWOS) developed a standardized approach to diagnosis.9 The group first identified seven intraocular signs of ocular sarcoidosis and then five laboratory or imaging tests that are of value in making the diagnosis in patients with these signs. Last, they established four levels of certainty for the diagnosis of ocular sarcoidosis, based on these signs, tests, and biopsy results, if available (see Table 2).
Treatment
Anterior uveitis in sarcoidosis is usually treated initially with a topical corticosteroid (eg, prednisolone or difluprednate drops), particularly if the patient’s symptoms are mild. In more severe cases (eg, posterior or bilateral uveitis) or when topical corticosteroids are ineffective, systemic (oral) corticosteroids (eg, prednisone) may be initiated. Topical therapy can also be added to an oral regimen as a means of decreasing the oral dosage and thereby reducing the adverse effects of systemic corticosteroids. When the patient’s disease is refractory to corticosteroids or there are concerns about long-term adverse effects, chronic cases may be treated with immunosuppressive agents (eg, methotrexate, azathioprine, mycophenolate mofetil). Finally, refractory cases of ocular sarcoidosis may be treated with anti–tumor necrosis factor α (TNF-α) biologic agents such as infliximab and adalimumab.10,17
Continue for case patient outcome >>
OUTCOME FOR THE CASE PATIENT
Histologic evaluation of tissue from the lacrimal gland, parotid gland, and sinus cavity revealed inflammatory noncaseating granulomas, strongly suggestive of sarcoidosis. Diagnosis of ocular sarcoidosis was based on the noncaseating granulomas in the lacrimal gland.9,16 Pulmonary sarcoidosis was also diagnosed, based on the presence of hilar and mediastinal lymphadenopathy.7
The mass in the patient’s lacrimal gland was surgically removed. She was treated with a combination of topical and oral corticosteroids tapered over two weeks, which induced remission of her ocular disease. The patient will be seen annually by an ophthalmologic specialist and was advised to contact her clinician immediately if acute ocular symptoms recurred.10,17
The patient’s persistent cough was determined to be secondary to acute bronchitis, rather than to her pulmonary sarcoidosis, which required no treatment. She received a short course of antibiotics and antitussives for her bronchitis. Systemic corticosteroid treatment of her ocular sarcoidosis also had the benefit of decreasing the size of her pulmonary nodules. She will be followed with annual CT and chest x-rays to monitor the status of her hilar and mediastinal lymphadenopathy and the nodules.3 Periodic pulmonary function testing will also be performed.7
Continue for conclusion >>
CONCLUSION
The elusive nature of the diagnosis of sarcoidosis is well documented in the medical literature. In this case, histologic evaluation of biopsied tissue, correlated with clinical symptoms and radiographic findings, were essential in making the diagnosis.
Primary care providers may be the first to evaluate patients with ocular sarcoidosis and will oversee long-term management. Patients who present with symptoms of eye pain, visual disturbances, abnormal inflammatory ocular features, or swollen lacrimal glands should be referred to an ophthalmologic specialist for further evaluation.
REFERENCES
1. Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
2. Culver DA. Sarcoidosis. Immunol Allergy Clin North Am. 2012;32(4):487-511.
3. Spagnolo P, Luppi F, Roversi P, et al. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012;125(2):118-125.
4. Dempsey OJ, Peterson EW, Kerr KM, Denison AR. Sarcoidosis. BMJ. 2009;339:620-625.
5. Brownell I, Ramirez-Valle F, Sanchez M, Prystowsky S. Evidence for mycobacteria in sarcoidosis. Am J Respir Cell Mol Biol. 2011;45(5):899-905.
6. Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399.
7. Baughman MD, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2011;183(5):573-581.
8. Koyama T, Ueda H, Togashi K, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24(1):87-104.
9. Herbort CP, Rao NA, Mochizuki M; for the Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009; 17(3):160-169.
10. Jamilloux Y, Kodjikian L, Broussolle C, Seve P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13(8):840-849.
11. Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis. 2012;7:57.
12. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. New Engl J Med. 2007;357(21):2153-2165.
13. Fontenot A, King T. Pathogenesis of sarcoidosis. www.uptodate.com/contents/pathogenesis-of-sarcoidosis?source=search_result&search=Pathogenesis+of+sarcoidosis&selectedTitle=1%7E150. Accessed February 17, 2015.
14. Saidha S, Sotirchos ES, Eckstein C. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med. 2012;85(1):133-141.
15. Newman LS, Rose CS, Bresnitz EA, et al; for the ACCESS Research Group. A case control etiologic study of sarcoidosis. Environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
16. Kawaguchi T, Hanada A, Horie S, et al. Evaluation of characteristic ocular signs and systemic investigations in ocular sarcoidosis patients. Jpn J Opthalmol. 2007;51(2):121-126.
17. Bodaghi B, Touitou V, Fardeau C, et al. Ocular sarcoidosis. Presse Med. 2012;41(6 Pt 2):e349-e354.
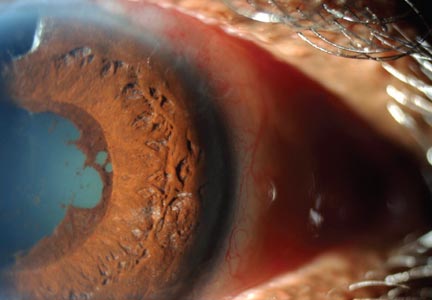
75-Year-Old Woman With Elevated Liver Enzymes
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
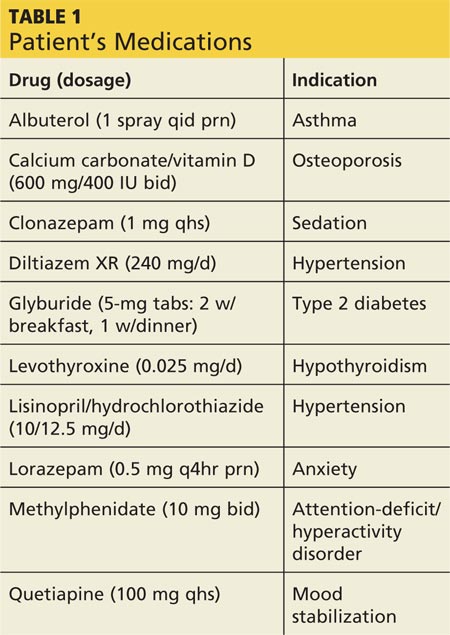
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
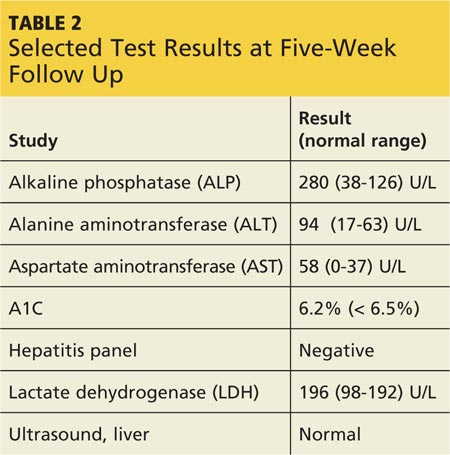
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
Woman, 32, With Crusty Red Blisters
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.

Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
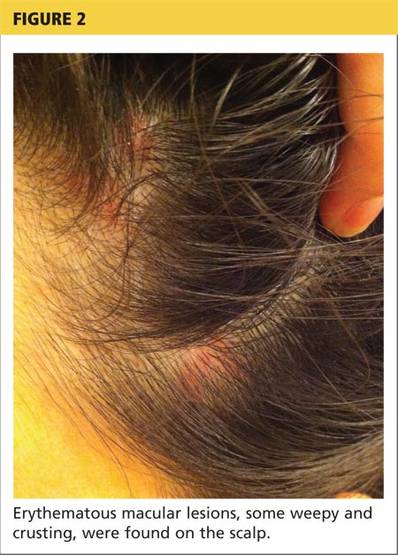
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
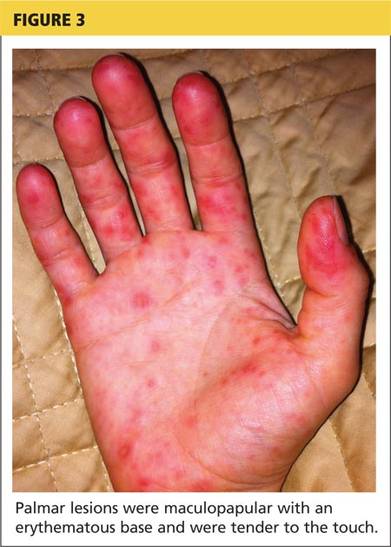
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
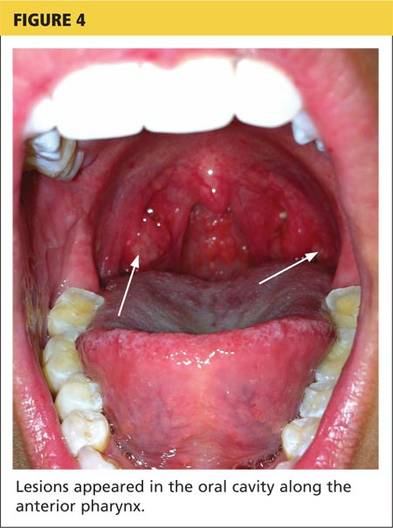
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
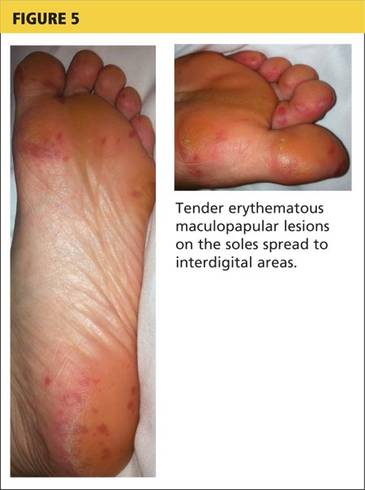
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
A 32-year-old Korean woman presented with a rash on her scalp, face, palms, soles, and genital region and with sores in the oral cavity. The blisters were red and flat with some crusting, particularly on the scalp and face. The patient described the blisters as very painful, adding that it hurt to walk, grasp objects, and drink fluids. Associated symptoms included painful urination, sore throat, malaise, and fever of up to 103°F. She was taking acetaminophen and ibuprofen to alleviate the fever and pain.
Medical history was unremarkable. Social history was negative for recent changes in sexual partner or travel to foreign countries.
Physical examination revealed numerous flat, erythematous lesions. Lesions on the face and scalp had developed a weeping, honey-colored crust (see Figure 1 and Figure 2). The lesions were tender to the touch, particularly on the palms and soles.
Further questioning revealed that the patient’s 18-month-old son had exhibited similar symptoms two to three days prior to her illness.
Continue for differential diagnosis >>
DIFFERENTIAL DIAGNOSIS
Because multiple bacterial and viral diseases manifest in this fashion, the differential diagnosis included the following disorders:
Erythema multiforme. This skin condition may result from an allergic or hypersensitivity reaction to certain drugs or from infections. Infections that can cause erythema multiforme include herpes simplex virus and mycoplasma. Patients present with lesions on the palms (see Figure 3), soles, extremities, face, or trunk. The lesions can appear as a nodule, papule, macule, or vesicle. Initially, in the mild form, the lesions may appear as hives or target-shaped rashes, occurring on the face and acral surfaces. A severe form of erythema multiforme, known as Stevens-Johnson syndrome, is characterized by rash, mucosal involvement, and systemic symptoms.1
Herpes zoster. This viral infection is caused by the varicella-zoster virus, which also causes chicken pox. The virus lies dormant within a single sensory ganglion and may reappear as shingles along the dermatome of that nerve. Patients may experience burning or shooting pain with tingling or itching before the rash appears; vesicular lesions with erythematous bases appear days later. The rash occurs unilaterally on the body or face and does not cross the midline. A viral culture may be obtained for identification.2
Herpetic gingivostomatitis. This infection is most commonly caused by herpes simplex virus type 1, the same virus that causes cold sores. Patients may present with ulcerations along the buccal mucosa and gums. The infection manifests with systemic symptoms, including malaise, fever, irritability, and cervical adenopathy. A viral culture will identify the etiology.3
Impetigo. This skin infection is typically caused by bacteria, predominantly Staphylococcus aureus, Streptococcus pyogenes, or a combination. Infections generally occur after a break in the skin surface. The most common presentation is a rash that spreads to different parts of the body after scratching. Skin lesions can occur on the face, lips, or extremities. Initially vesicular, the lesions generally form a honey-colored crust after fluid discharge. The clinician should take a skin or fluid sample from the lesion to culture, which may identify the pathogen.4
Syphilis. This sexually transmitted infection is caused by the spirochete Treponema pallidum. In primary syphilis, patients can develop a painless sore, or chancre, on the genitals, rectal area, or mouth. If left untreated, the disease can progress to secondary syphilis, manifesting as a pale pink or reddish maculopapular rash on the palms and soles. The rash can be associated with fever, sore throat, myalgia, and fatigue. It is important to rule out syphilis because, left untreated, it can lead to cardiac and neurologic complications. Screening tests include VDRL and the rapid plasma reagin (RPR) test, both of which assess for antibodies to the organism, and dark field microscopy of ulcerations to identify the organism.5
Also included in the differential diagnosis for the patient’s symptoms was hand-foot-mouth disease (HFMD), discussed below.
Next page: Discussion >>
DISCUSSION
HFMD is an acute viral illness most often affecting children younger than 5 and occurring in summer to early fall months. HFMD manifests with fever and papulovesicular eruptions; lesions often appear in the oral cavity first, spreading to the palms, soles, and buttocks. Route of transmission is usually fecal-oral or through respiratory droplets, oral secretions, or direct contact with fluid-filled vesicles.6-8 The highly contagious nature of the virus causes it to spread to close contacts and family members and leads to outbreaks in schools and daycare centers.9,10
In the United States, the most common etiology of HFMD is coxsackievirus A16.7 Another causative agent, enterovirus 71 (EV71), has been found responsible for HFMD epidemics in southeast Asia and Australia.11,12 Recently, the coxsackievirus A6 (CV A6) strain has been linked to outbreaks of HFMD.7,9 This strain may produce an atypical manifestation of skin lesions on the face, trunk, and extremities. The lesions may also appear larger than usual and have a vesiculobullous rather than the more typical papulovesicular appearance. The course of the illness differs in severity depending on the strain of the virus causing HFMD.9
CLINICAL PRESENTATION
The acute phase of HFMD typically begins with prodromal symptoms such as fever, malaise, and sore throat. Erythematous ulcerations usually appear in the oral cavity first (see Figure 4) and often cause symptoms such as sore throat, dysphagia, or dryness. As the disease progresses, cutaneous lesions spread to the face, extremities, interdigital areas (see Figure 5), trunk, and perianal area (which may cause dysuria). The lesions may initially appear as erythematous macules or papules, transforming to vesicles as the disease progresses. The mucocutaneous lesions are usually asymptomatic but can be tender to touch or pressure and may leak fluid.7,10
In only a few cases—caused by CV A6—have lesions in the scalp been reported; the mechanism of action is unknown.10 Instances of lesions invading the nails have been reported, causing desquamation and shedding. This condition is known as onychomadesis.9,11
Continue for diagnosis >>
DIAGNOSIS
Serologic testing and viral cultures can identify the exact strain of virus causing HFMD and are particularly useful in unusual presentations. Polymerase chain reaction (PCR) testing yields a high sensitivity and specificity for the causative agent. Histologic examination of skin biopsies may show lymphocytic infiltrates and areas of degeneration along the epidermis. However, most cases are diagnosed based on clinical presentation alone.6,12
TREATMENT AND MANAGEMENT
Management of HFMD is primarily symptomatic, consisting of supportive care that includes use of antipyretics, NSAIDs, and adequate fluid intake to prevent dehydration. The disease is usually self-limited, resolving within seven to 10 days without sequelae.6,9 Aseptic meningitis and other severe complications (especially pulmonary and neurologic), most often associated with EV71 infection, can occur in vulnerable populations, including elderly, pregnant, and immunocompromised patients.9,11 Because the virus is excreted directly from palmar lesions onto the hands, proper hygiene and handwashing techniques offer an exceptionally strong protective effect, preventing transmission and reducing morbidity.8
PATIENT OUTCOME
Based on clinical findings and patient history, the patient was diagnosed with HFMD, which she contracted from her son. Laboratory testing and viral cultures were deemed unnecessary in this case. Treatment was symptomatic, and her skin lesions resolved in one to two weeks.
At follow-up, the patient stated her skin lesions resolved completely without leaving any scars. She also indicated that her nails peeled and shed approximately four weeks after diagnosis, but began to regrow normally four months after diagnosis.
CONCLUSION
Clinicians need to recognize that, although it is uncommon outside the pediatric population, HFMD may occur in adults with intact immune systems. The presentation of HFMD in adults may be atypical, including cutaneous lesions in the scalp and shedding of the nails several weeks after diagnosis. Depending on the viral strain involved, adult patients may have more severe illness and may take longer to recover. Therefore, early diagnosis is important to help prevent the spread of infection and reduce the severity of complications.
REFERENCES
1. Patel NN, Patel DN. Erythema multiforme syndrome. Am J Med. 2009;122(7):623-625.
2. Bader MS. Herpes zoster: diagnostic, therapeutic, and preventive approaches. Postgrad Med. 2013;125(5):78-91.
3. Avci O, Ertam I. Viral infections of the face. Clin Derm. 2014;32:715-733.
4. Hartman-Adams H, Banvard C, Juckett G. Impetigo: diagnosis and treatment. Am Fam Physician. 2014;90(4):229-235.
5. Markle W, Conti T, Kad M. Sexually transmitted diseases. Prim Care Clin Office Pract. 2013;40:557-587.
6. Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22(2):216-218.
7. CDC. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6 - Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61(12):213-214.
8. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127(4):e898-e904.
9. Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth Disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5(2):203-209.
10. Lønnberg AS, Elberling J, Fischer TK, Skov L. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93(4):467-468.
11. Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerging Infect Dis. 2009;15(9):1485-1488.
12. Shea YF, Chan CY, Hung IFN, Chan KH. Hand, foot and mouth disease in an immunocompetent adult due to Coxsackievirus A6. Hong Kong Med J. 2013;19(3):262-264.
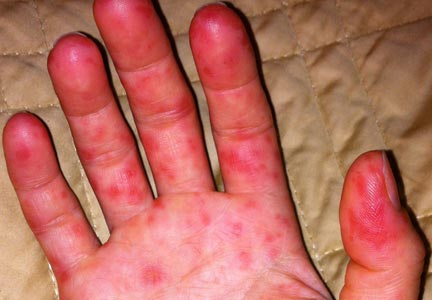
Woman, 66, With Persistent Abdominal and Back Pain
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.

Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
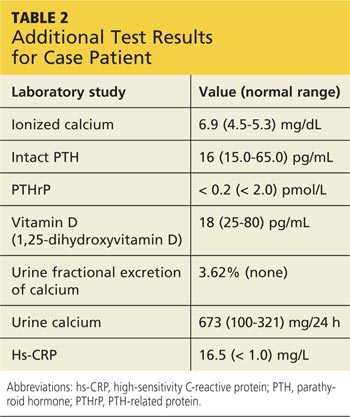
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
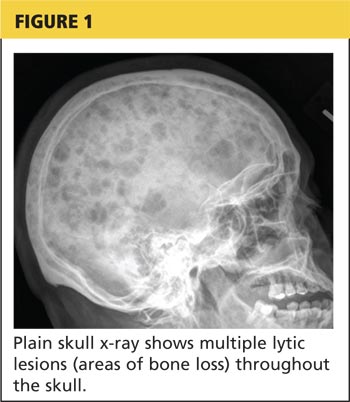
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
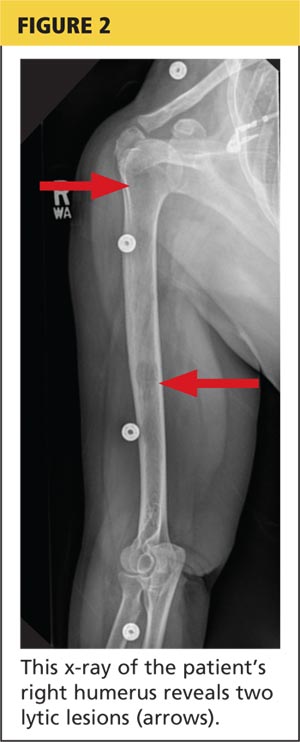
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
A 66-year-old Latin American woman presented to the emergency department (ED) with persistent abdominal and back pain of about one month’s duration. She had visited another ED eight days earlier for similar symptoms and was discharged home with a mild opioid pain medication and a proton pump inhibitor. However, she said that she had received neither a diagnosis nor an explanation for her symptoms.
Medical history, obtained with the assistance of an interpreter because the patient was not fluent in English, included hypertension, coronary artery disease, and hyperlipidemia; these had gone untreated for at least two years. She denied any personal or family history of cancer or endocrine disorders. Surgical history included a cholecystectomy and a percutaneous coronary intervention for an unknown coronary artery lesion.
She had a 14-pack-year history of cigarette smoking. Her medications included only ibuprofen and hydrocodone, and she had no known drug allergies. The patient denied use of herbal preparations or vitamin supplements and unusual dietary practices.
Review of systems revealed occasional dizziness, constipation, decreased appetite, and some mild confusion noted by family members, but no fever, chills, palpitations, chest pain, shortness of breath, muscle spasm, or weakness. Vital signs were normal. Physical examination was remarkable for tenderness of the upper quadrants of the abdomen with deep palpation, without guarding or rebound. Bony tenderness at the right anterior costal margin of the rib cage was also noted.
Laboratory work-up revealed marked hypercalcemia (15.4 mg/dL), electrolyte abnormalities, anemia, impaired renal function, and elevated alkaline phosphatase and globulin levels (see Table 1). In addition, a plain abdominal x-ray series was negative for acute findings, but x-rays of the right ribs revealed a fracture of the sixth rib and osteopenia.
Continued >>
The patient was admitted to the hospital for treatment of hypercalcemia and hypokalemia and for work-up of elevated alkaline phosphatase and abdominal pain. Upon admission, serum ionized calcium measurement confirmed true hypercalcemia. Additional diagnostic tests were then ordered to help differentiate between parathyroid hormone (PTH)–mediated and non-PTH–mediated causes for the hypercalcemia (see Table 2).
The patient’s PTH level was normal and the urine fractional excretion of calcium level was high, ruling out familial hypocalciuric hypercalcemia (FHH), in which urine calcium level is low. A measurement of PTH-related protein (PTHrP), secreted by some cancers, was normal, suggesting exclusion of solid tumor malignancy. Vitamin D toxicity was ruled out because the patient’s 1,25-dihydroxyvitamin D level was low.
The patient continued to experience vague abdominal, back, and rib pain that seemed to migrate daily and worsened with movement. A skeletal x-ray was performed and revealed numerous lytic lesions of the skull (Figure 1), midright humerus (Figure 2), and distal left radius.
Continue for discussion >>
DISCUSSION
Hypercalcemia is a relatively common presentation in primary care. The most frequent causes are primary hyperparathyroidism and malignancy.1 One in 500 patients will be diagnosed incidentally with asymptomatic hypercalcemia caused by underlying hyperparathyroidism.1
Clinical manifestations of hypercalcemia can range from no symptoms to multisystem disease. Fatigue, nausea, vomiting, constipation, bone pain, osteoporosis, nephrolithiasis, mental status changes, hypertension, anemia, elevated creatinine, and cardiac arrhythmias are among the more common clinical conditions associated with hypercalcemia (hence the mnemonic “stones, bones, abdominal moans, and psychic groans” for its signs and symptoms).1
Diagnostic overview
Causes of hypercalcemia are numerous and can be broken down into two categories: PTH-mediated and non-PTH–mediated. PTH-mediated causes include primary and secondary hyperparathyroidism and FHH. Non-PTH–mediated causes include vitamin D toxicity, solid tumor malignancy with or without metastasis, multiple myeloma (MM) and other plasma cell dyscrasias, granulomatous disease such as sarcoid, and some medications.1
The differential diagnosis for hypercalcemia begins with measurement of the patient’s intact PTH level. An elevated or high-normal result indicates a PTH-mediated cause, so 24-hour measurement of excretion of urinary calcium is the next step. A low or low-normal PTH level (< 20 pg/mL), however, suggests the cause is non-PTH-mediated.2 The diagnostic approach in this situation is more challenging because testing to exclude or confirm various potential causes can be expensive and time-consuming. The degree of hypercalcemia, however, can aid in the diagnosis: Primary hyperparathyroidism is often associated with borderline or mild hypercalcemia, while calcium values > 13 mg/dL are more common in patients with malignancies.2
PTH elevates calcium levels in the blood when ionized (free) calcium levels are low by increasing gastrointestinal absorption, decreasing urinary excretion, and increasing bone resorption.3 With malignant tumors such as lung, breast, and renal cell, osteolytic metastases can destroy the bone, resulting in release of calcium. In other cases, solid-tumor cancers produce PTHrP, which increases serum calcium. This latter situation is referred to as humoral hypercalcemia of malignancy.3 Lymphoma and granulomatous disease, such as sarcoid, can be associated with excess production of 1,25-dihydroxyvitamin D.2 If vitamin D is elevated but PTH and PTHrP are normal, a chest x-ray should be obtained to evaluate the patient for sarcoid or lymphoma.
Hypercalcemia work-up
The work-up for suspected hypercalcemia begins with measurement of the patient’s calcium level. Because calcium is bound to albumin in the blood, the standard serum calcium test may not reflect the true calcium level. (If albumin is high, the calcium level will be high, and vice versa.)1 The true (serum ionized) calcium level (also known as corrected calcium level) should always be calculated to confirm true hypercalcemia. A formula commonly used to calculate the corrected calcium level is
Corrected calcium (mg/dL) = (measured calcium [mg/dL]) + 0.8 (4.0 – serum albumin [mg/dL])
Direct measurement of the serum ionized calcium level is not affected by the albumin level and can also confirm true hypercalcemia.4
Once hypercalcemia is confirmed, the next step is to measure the patient’s intact PTH level.
If intact PTH is elevated or high normal, consider primary hyperparathyroidism or FHH and confirm by obtaining the urine calcium level.
• If urine calcium level is high (> 200 mg/24 h), the diagnosis is primary hyperparathyroidism.
• If urine calcium level is low (< 100 mg/24 h), the diagnosis is FHH.
If intact PTH is low, consider non–PTH-mediated causes and confirm by obtaining PTHrP and vitamin D levels.
• If PTHrP level is elevated, scan for malignancy.
• If 1,25-dihydroxyvitamin D level is elevated, order a chest x-ray to rule out sarcoid or lymphoma.
• If both PTHrP and vitamin D levels are normal, order both serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP) with immunofixation to rule out MM.
• If vitamin D level is elevated, check vitamin and herbal supplement use for excessive vitamin D intake.
Treatment of symptomatic hypercalcemia
The goals of treatment of symptomatic hypercalcemia are to reduce the serum calcium level to the normal range and to treat the underlying cause.1 Mild hypercalcemia (calcium level, 10-12 mg/dL) is typically asymptomatic and does not need to be treated. Moderate hypercalcemia (calcium level, 12-14 mg/dL) may not require treatment unless the patient is symptomatic and/or has had an acute rise in calcium level.1 In mild to moderate hypercalcemia, the serum calcium level should be monitored to establish a trend.
Treatment for symptomatic moderate and severe hypercalcemia (calcium level, > 14 mg/dL) typically involves a similar regimen:
• Volume expansion with isotonic saline at an initial rate of 2 to 4 L/d, which is then adjusted to achieve 200 mL/h of continuous urine output. IV furosemide can be used with caution (10-20 mg IV as needed) to promote diuresis if volume overload is a concern (furosemide promotes renal excretion of calcium).
• Administration of subcutaneous calcitonin (4-8 IU/kg, repeated every 6 h for 24 h). Calcitonin works rapidly to lower calcium levels in 4 to 6 h.
• Concurrent administration of IV bisphosphonate (zoledronic acid [4 mg over 15 min] or pamidronate [60-90 mg over 4 h]). Pamidronate is superior for reversal of malignancy-related hypercalcemia.1
Hypercalcemia and multiple myeloma
MM is a malignant neoplasm of plasma cells that accounts for approximately 1% of all cancers and about 10% of hematologic malignancies in the United States, with a median patient age of 70 at diagnosis.5-7 In MM, myeloma cells induce the secretion of cytokines and growth factors that alter plasma cells, activate osteoclasts, suppress osteoblasts, cause abnormal interactions between plasma cells and bone marrow, and stimulate aberrant angiogenesis.8 Osteoclastic bone resorption produces hypercalcemia as well as the lytic lesions seen on x-ray.7
Approximately 74% of patients present with typical MM symptoms of calcium elevation in the blood, renal insufficiency, anemia, and bone lesions, known as CRAB symptoms, but other myeloma-related manifestations may be present.9
Diagnostic criteria for MM include the following (all three must be present):10
• Monoclonal bone marrow plasma cells ≥ 10% and/or a biopsy-proven plasmacytoma
• Monoclonal protein in the serum and/or urine (if none is detected, disease is nonsecretory and diagnosis requires ≥ 30% bone marrow plasma cells and/or biopsy-proven plasmacytoma)
• Myeloma-related organ dysfunction, indicated by at least one of the CRAB symptoms.
In the absence of CRAB symptoms, an asymptomatic patient may have an MM precursor syndrome: monoclonal gammopathy of undetermined significance or smoldering (or indolent) MM.10
Treatment of multiple myeloma
In recent years, the use of induction therapy followed by autologous stem cell transplantation and the development of novel therapeutic agents have extended overall survival for patients with MM. These agents include proteasome inhibitors (bortezomib and the second-generation carfilzomib)11 and immunomodulators (thalidomide and the second-generation lenalidomide). Early diagnosis and treatment can improve progression-free survival as well as overall survival, including recovery of renal function for patients with renal failure.12 With survival ranging from one year or less—with aggressive disease—to 10 years or more for patients with responsive disease,7 there remains no cure for MM.
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Further work-up included SPEP and UPEP with immunofixation, which revealed marked IgG free λ light chains with an M (monoclonal) component, making MM a very likely diagnosis. Confirmation by means of bone marrow biopsy was indicated, but the patient refused the procedure.
The patient’s hypercalcemia was treated by IV administration of calcitonin with isotonic saline. This reduced the serum calcium level from 15.4 mg/dL to 10.6 mg/dL within 48 hours. One dose of ergocalciferol (vitamin D2) was then administered to promote intestinal absorption of calcium and support bone mineralization, further lowering the patient’s serum calcium level to a normal 8.9 mg/dL. Hypokalemia was treated with oral potassium supplementation.
The patient, now stable, was referred to the hematology/oncology and bone mineral metabolism clinics and was discharged from the hospital. She did not keep those appointments and was lost to follow-up.
CONCLUSION
The most common causes of hypercalcemia are hyperparathyroidism and malignancy. Most cases do not require treatment unless the calcium level is >14 mg/dL and/or the patient is symptomatic. Red flag symptoms include weakness, abdominal pain, mental status changes, and coma.4 Primary care clinicians should suspect MM in older patients with laboratory findings of hypercalcemia, anemia, and renal dysfunction, with lytic lesions on x-ray.
REFERENCES
1. Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician. 2003;67(9):1959-1966.
2. Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45(12):
954-963.
3. Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35(2):215-237.
4. Sharma B, Misicko NE. How should you evaluate elevated calcium in an asymptomatic patient? J Fam Pract. 2008;57(4):267-269.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
6. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33.
7. Shaheen SP, Talwalkar SS, Medeiros LJ. Multiple myeloma and immunosecretory disorders: an update. Adv Anat Pathol. 2008;15(4):196-210.
8. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11): 1046-1060.
9. Talamo G, Farooq U, Zangari M, et al. Beyond the CRAB symptoms: a study of presenting clinical manifestations of multiple myeloma. Clin Lymphoma Myeloma Leuk. 2010;10(6):464-468.
10. Palumbo A, Sezer O, Kyle R, et al. International Myeloma Working Group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transportation. Leukemia. 2009;23(10):1716-1730.
11. Kyprolis [package insert]. South San Francisco, CA: Onyx Pharmaceuticals, Inc; 2012.
12. Suyani E, Sucak GT, Erten Y, et al. Evaluation of multiple myeloma patients presenting with renal failure in a university hospital in the year 2010. Ren Fail. 2012;34(2):257-262.
Woman With Blue-Gray Palate and Nail Beds
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
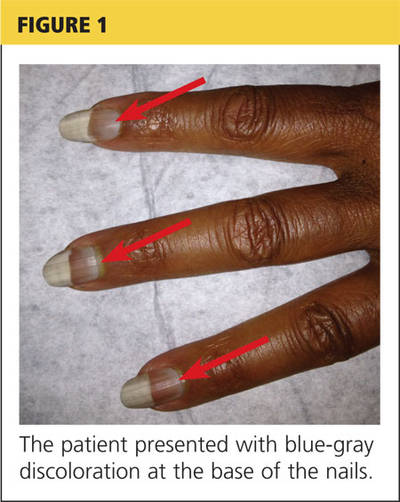
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
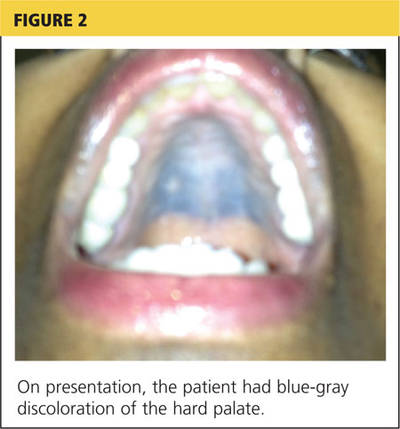
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
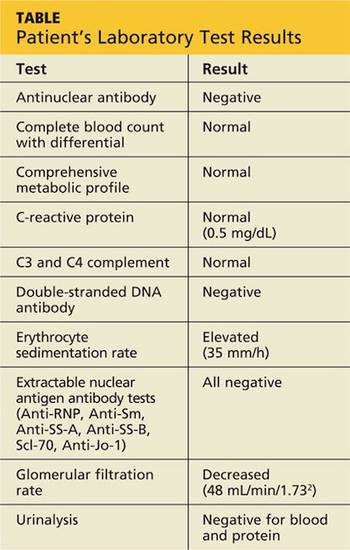
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
A 62-year-old African-American woman presented for evaluation of a bluish discoloration of the hard palate and nail beds, noticeable for several months. In addition, she had complaints of fatigue and arthralgia. She reported that she had been taking hydroxychloroquine 400 mg/d and quinacrine 100 mg/d for several years for the treatment of systemic lupus erythematosus (SLE). Her medical history was also significant for dry mouth syndrome treated with pilocarpine.
The patient’s vital signs included a temperature of 97°F;
respiratory rate, 15 breaths/min; pulse, 72 beats/min; and blood pressure, 130/80 mm Hg. Height was 62 in, weight was 189 lb, and BMI was 34.56. A bluish gray color was noted in the subungual areas of her nails (see Figure 1). There were several circumferential areas of skin hyperpigmentation resulting from healed lupus skin lesions on her arms. Nailfold capillaroscopy revealed several dilated blood vessels. The sclerae appeared dry, but no erythema or inflammation was noted.
Examination of the mouth revealed a bluish discoloration of the hard palate (see Figure 2) and decreased salivary pool. Respiratory, cardiovascular, and abdominal examination findings were normal. Musculoskeletal examination was unremarkable for acute joint tenderness or synovitis. Crepitation and bony changes were noted in the left knee, without effusion or decreased range of motion.
Laboratory studies were ordered, and the results are listed in the table.
DISCUSSION
Hyperpigmentation of the oral mucosa can be associated with a number of conditions, including adrenal insufficiency, Peutz-Jeghers syndrome, hemochromatosis, polyostotic fibrous dysplasia, hyperparathyroidism, neurofibromatosis, and bronchogenic malignancy.1,2 Other causes of oral hyperpigmentation include physiologic pigmentary or postinflammatory changes, oral melanoacanthosis, blue nevus, and melanoma.2,3 While these diagnoses should be considered when encountering a mucosal lesion, they were unlikely in this patient because of the color changes in her nail beds.
Systemic skin and mucous membrane discoloration can also occur with the use of certain drugs and other substances, including chemotherapeutic agents, benzodiazepines, hormones, carotenoids, phenolphthalein, heavy metal salts, and several antimicrobial agents.1 In dark-skinned individuals, hyperpigmentation of the oral mucosa can be caused by a physiologic deposition of melanin.4
Pigmentary Changes
The use of antimalarial drugs, such as quinacrine, chloroquine, and hydroxychloroquine, has long been associated with pigmentary changes to the palatal mucosa and subungual areas.1,3 These drugs can stimulate melanin production and cause hemosiderin deposition, resulting in pigmentary changes.5 Skin discoloration is believed to be the result of the formation of a melanin-drug complex in areas with an elevated affinity for melanin.1 Besides malaria, these drugs are commonly used to treat SLE and discoid lupus erythematosus, rheumatoid arthritis, and other rheumatologic conditions.5
The diagnosis of drug-induced hyperpigmentation is generally clinical, supported by the patient’s history—which often includes the use of antimalarial drugs—and presentation.1 If a clear cause cannot be determined by clinical evaluation, then a biopsy to confirm a drug-induced cause may be necessary.2 A classic study by Tuffanelli et al reported that the onset of hyperpigmentation related to antimalarial drug therapy may not occur until 4 to 70 months after initiation of treatment.6 Once the offending drug is discontinued, pigmentation changes slowly fade but often do not completely resolve,7 and patients should be advised of this.
Ocular Retinopathy
While pigmentary changes associated with antimalarial drugs are benign,3 a rare but serious adverse effect of antimalarials is retinal toxicity. Ocular retinopathy related to chloroquine and hydroxychloroquine therapy has been well documented and may result in irreversible vision loss.8,9 The most recent recommendations from the American Academy of Ophthalmology suggest a baseline eye examination at initiation of antimalarial treatment and annual examinations starting after five years of therapy because the risk for toxicity relates to the cumulative dose.8 More frequent ophthalmologic evaluations are recommended for individuals at higher risk, such as those with preexisting retinal or macular disease.9
Outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
A biopsy of the roof of the patient’s mouth confirmed that the palatal hyperpigmentation was caused by her antimalarial medications. Since the patient displayed no evidence of active lupus skin lesions and laboratory results indicated that her SLE was inactive, one of the drugs, quinacrine, was discontinued.
The patient was referred for an ophthalmologic evaluation. No evidence of retinal toxicity was found.
Follow-up evaluations at two months and six months revealed no significant improvement in the discoloration of the patient’s oral mucosa or nail beds. At the six-month visit, her dosage of hydroxychloroquine was reevaluated.
The patient’s hydroxychloroquine dosage was determined based on 7.3 mg/kg/d. In the case of an overweight patient, especially one of shorter-than-average stature, hydroxychloroquine dosing should be based on ideal body weight to minimize the risk for overdosage; in general, a maximum dosage of 6.5 mg/kg/d is recommended.8,9 As a result, the patient’s dosage was decreased to 300 mg/d.
At her nine-month follow-up evaluation, the discoloration to the patient’s oral mucosa had faded but had not resolved completely (see Figure 3). No significant change was noted in the subungual discoloration. The patient had experienced no exacerbations of lupus-related symptoms since her medication adjustments.
CONCLUSION
Although this patient’s hyperpigmentation was benign, staying alert to this potential adverse effect of antimalarial drugs is important in making a diagnosis. As with many skin lesions, if the clinical evaluation does not provide a clear cause, a biopsy may be needed. For anyone taking antimalarial drugs, regular ophthalmologic evaluations are recommended to facilitate early detection of the rare adverse effect of retinal toxicity. Nevertheless, with careful monitoring, antimalarial drugs are safe and effective for the treatment of inflammatory conditions such as SLE and rheumatoid arthritis.
REFERENCES
1. Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):189-194.
2. Gondak R-O, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Pathol Oral Cir Bucal. 2012;17(6):e919-e924.
3. Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;
107:8-12.
4. Kalampalikis A, Goetze S, Elsner P. Isolated hyperpigmentation of the oral mucosa due to hydroxychloroquine. J Dtsch Dermatol Ges. 2012; 10(12):921-922.
5. de Andrade BA, Fonseca FP, Pires FR, et al. Hard palate hyperpigmentation secondary to chronic chloroquine therapy: report of five cases.
J Cutan Pathol. 2013;40(9):833-838.
6. Tuffanelli D, Abraham RK, Dubois EI. Pigmentation from antimalarial therapy: its possible relationship to the ocular lesions. Arch Derm. 1963; 88:419-426.
7. Melikoglu MA, Melikoglu M, Gurbuz U, et al. Hydroxychloroquine-induced hyperpigmentation: a case report. J Clin Pharm Ther. 2008; 33(6):699-701.
8. Marmor MF, Kellner U, Lai YY, et al; American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):
415-422.
9. Screening for hydroxychloroquine retinopathy. Position statement, American College of Rheumatology. www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed July 17, 2014.
Man, 26, With Sudden-Onset Right Lower Quadrant Pain
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
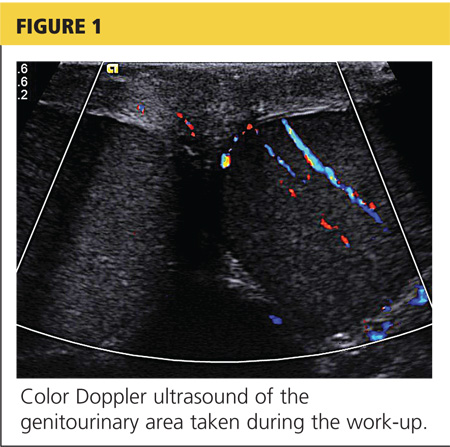
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
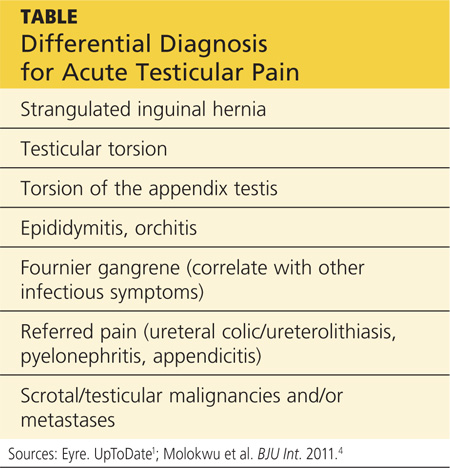
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
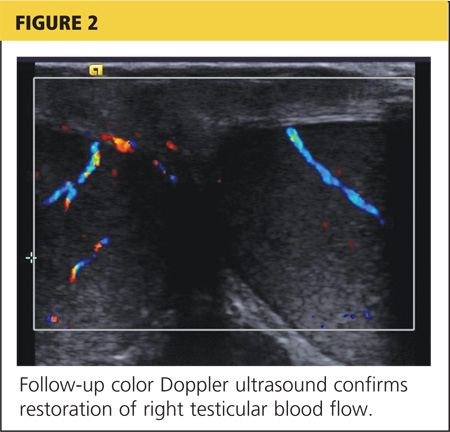
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.
A 26-year-old man presented to the emergency department (ED) with a chief complaint of abdominal pain. After triage was complete, he was transported to an examination room, where the clinician obtained the history of presenting illness. The onset of pain was approximately 90 minutes prior to arrival at the ED and woke the patient from a “sound sleep.” He stated that the pain initially started as a “3 out of 10” but had progressed to a “12 out of 10,” and he described it as being in the right lower quadrant of his abdomen, with radiation to his right testicle. However, he was unsure where the pain started or if it was worse in either location. Nausea was the primary associated symptom, but he denied vomiting, diarrhea, fever, dysuria, or hematuria. Last, the patient denied history of trauma.
Medical history was noncontributory: He denied previous gastrointestinal diseases, and there was no history of renal stones, urinary tract infection, or any other genitourinary disease. He had no surgical history. The patient smoked less than a pack of cigarettes per day but denied alcohol or drug use.
Physical examination revealed a young man in moderate discomfort. Despite describing his pain as a “12 out of 10,” he had a blood pressure of 121/72 mm Hg; pulse, 59 beats/min; respiratory rate, 20 breaths/min; and temperature, 96.8°F. HEENT and cardiovascular, respiratory, musculoskeletal, and neurologic exam results were all within normal limits. Abdominal examination revealed a mildly tender right lower quadrant with deep palpation, but no rebound or guarding. Murphy sign was negative.
Because of the complaint of pain radiating to the testicles, a genitourinary examination was performed. The penis appeared unremarkable, with no lesions or discharge. There was no inguinal lymphadenopathy. The scrotum appeared appropriate in size and was also grossly unremarkable. The left testicle was nontender. However, palpation of the right testicle elicited moderate to severe pain. There was no visible swelling, and there were no palpable hernias or other masses. Cremasteric reflex was assessed bilaterally and deemed to be absent on the right side.
A workup was initiated that included a complete blood count, comprehensive metabolic panel, and urinalysis; the results of these tests were unremarkable. A differential diagnosis was formed, with emphasis on appendicitis and testicular torsion. Because of the specific nature and location of the pain, both ultrasound and CT of the abdomen/pelvis were considered. It was decided to order the ultrasound, with a plan to perform CT only if ultrasound was unremarkable. The patient was medicated for his pain and the ultrasound commenced. Halfway through the imaging, the clinician and attending physician were summoned to the examination room to review the image seen in Figure 1.
On the next page: Discussion and diagnosis >>
DISCUSSION
Testicular torsion may occur if the testicle twists or rotates on the spermatic cord. The twisting causes arterial ischemia and venous outflow obstruction, cutting off the testicle’s blood supply.1,2 Torsion may be extravaginal or intravaginal, depending on the extent of involvement of the surrounding structures.2
Extravaginal torsion is most commonly seen in neonates and occurs because the entire testicle may freely rotate prior to fixation to the scrotal wall via the tunica vaginalis.2Intravaginal torsion is more common in adolescents and often occurs as a result of a condition known as bell clapper deformity. This congenital abnormality enables the testicle to rotate within the tunica vaginalis and rest transversely in the scrotum instead of in a more vertical orientation.2,3 Torsion occurs if the testicle rotates 90° to 180°, with complete torsion occurring at 360° (torsion may extend to as much as 720°).2 Torsion may also occur as a result of trauma.1
Peak incidence of testicular torsion occurs at ages 13 to 14, but it can occur at any age; torsion affects approximately 1 in 4,000 males younger than 25.2-5 Ninety-five percent of all torsions are intravaginal.2 Torsion is the most common pathology for males who undergo surgical exploration for scrotal pain.3
The main goal in the diagnosis and treatment of torsion is testicular salvage. Torsion is considered a urologic emergency, making early diagnosis and treatment critical to prevent testicular loss. In fact, a review of the relevant literature reveals that the rate of testicular salvage is much higher if the diagnosis is made within 6 to 12 hours.1,2,5 Potential sequelae from delayed treatment include testicular infarction, loss of testicle, infertility problems, infections, cosmetic deformity, and increased risk for testicular malignancy.2
Because many men hesitate to seek medical attention for symptoms of testicular pain and swelling, the primary care clinician should openly discuss testicular disorders, especially with preadolescent males, during testicular examinations.6
Diagnosis
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. The cremasteric reflex should be assessed because it can help differentiate among the causes of testicular pain.7 It is performed by gently stroking the upper inner thigh and observing for contraction of the ipsilateral testicle. One study found that, in cases of torsion, the absence of a cremasteric reflex had a sensitivity of 96% and a specificity of 88%.7 See the Table for the differential diagnosis for acute testicular pain.
While it is often possible to make the diagnosis of testicular torsion clinically, ultrasound with color Doppler is the diagnostic test of choice in cases for which the cause of acute scrotal pain is unclear.8 Ultrasound provides anatomic detail of the scrotum and its contents, and perfusion is assessed by adding the color Doppler images.8 It is important to note that, while the absence of blood flow is considered diagnostic for testicular torsion, the presence of flow does not necessarily exclude it.4
On the next page: Treatment >>
Treatment
Surgical exploration with intraoperative detorsion and orchiopexy (fixation of the testicle to the scrotal wall) is the mainstay of treatment for testicular torsion.1 Orchiopexy is often performed bilaterally in order to prevent future torsion of the unaffected testicle. In about 40% of males with the bell clapper deformity, the condition is present on both sides.2 Orchiectomy, the complete removal of the testicle, is necessary when the degree of torsion and subsequent ischemia have caused irreversible damage to the testicle.6 In one study in which 2,248 cases of torsion were reviewed, approximately 34% of males required orchiectomy.6
If surgery may be delayed, the clinician may attempt manual detorsion at the bedside. Despite the “open book” method described in many texts—which instructs the practitioner to rotate the testicle laterally—a review of the literature reveals that torsion takes place medially only 70% of the time.1,5 The clinician should always consider this when any attempts at manual detorsion are made and correlate his or her technique with physical examination and the patient’s response.5
Relief of pain and return of the testicle to its natural longitudinal lie are considered indicators of successful detorsion.1 Color Doppler ultrasound should be used to confirm the return of circulation. However, in one case review of pediatric patients who underwent surgical exploration after manual detorsion, some degree of residual torsion remained in 32%.5 Because of this risk, surgery is still indicated even in cases of successful bedside detorsion.5
On the next page: Case continuation >>
CASE CONTINUATION
The decision to perform bedside ultrasound was made because the diagnosis of testicular torsion is a surgical emergency, and the window of time to prevent complications can be extremely narrow. If the ultrasound had been normal, then a CT scan may have provided additional data on which to base the diagnosis.
The patient was given adequate parenteral pain medication. After color Doppler ultrasound confirmed the torsion, the testicle was laterally rotated approximately 360°. The patient reported alleviation of his symptoms. Color Doppler was again performed to confirm the return of hyperemic blood flow to the affected testicle (Figure 2). The urologist arrived shortly thereafter and the patient was taken to the operating room, where he underwent scrotal exploration and bilateral orchiopexy.
On the next page: Conclusion >>
CONCLUSION
A testicular examination should be performed on any male presenting with a chief complaint of lower abdominal pain, back/flank pain, or any pain that radiates to the groin. Testicular torsion is most commonly seen in infants and adolescents but can occur at any age. The condition is a surgical emergency and the goal is testicular salvage, which is most likely to occur before 12 hours have elapsed since the onset of symptoms. An important component of the physical examination is attempting to elicit the cremasteric reflex, which is likely to be absent in the presence of torsion.
The primary care provider’s goal is to rapidly diagnose testicular torsion, then refer the patient immediately to a urologist or ED. The skilled clinician may attempt manual detorsion, based on his/her expertise and comfort level; however, this procedure should never delay prompt surgical intervention.
REFERENCES
1. Eyre RC. Evaluation of the acute scrotum in adults. www.uptodate.com/contents/evaluation-of-the-acute-scrotum-in-adults. Accessed May 16, 2014.
2. Ogunyemi OI, Weiker M, Abel EJ. Testicular torsion. http://emedicine.medscape.com/article/2036003-overview. Accessed May 16, 2014.
3. Khan F, Muoka O, Watson GM. Bell clapper testis, torsion, and detorsion: a case report. Case Rep Urol. 2011;2011:631970.
4. Molokwu CN, Somani BK, Goodman CM. Outcomes of scrotal exploration for acute scrotal pain suspicious of testicular torsion: a consecutive case series of 173 patients. BJU Int. 2011;107(6):990-993.
5. Sessions AE, Rabinowitz R, Hulbert WC, et al. Testicular torsion: direction, degree, duration and disinformation. J Urol. 2003;169(2):663-665.
6. Mansbach JM, Forbes P, Peters C. Testicular torsion and risk factors for orchiectomy. Arch Pediatr Adolesc Med. 2005;159:1167-1171.
7. Schmitz D, Safranek S. How useful is a physical exam in diagnosing testicular torsion? J Fam Pract. 2009;58(8):433-434.
8. D’Andrea A, Coppolino F, Cesarano E, et al. US in the assessment of acute scrotum. Crit Ultrasound J. 2013;5(suppl 1):S8. www.criticalultrasound journal.com/content/5/S1/S8/. Accessed May 16, 2014.