User login
Itchy, vesicular rash
Pemphigoid gestationis
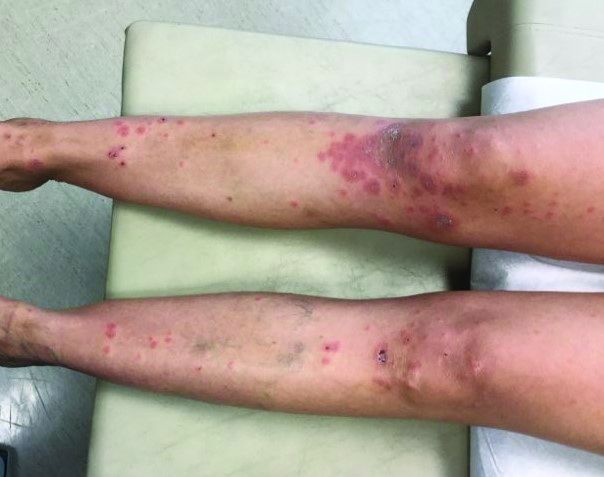
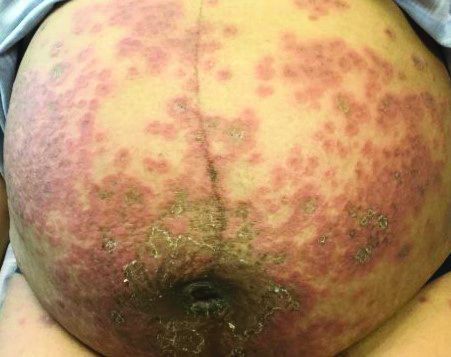
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.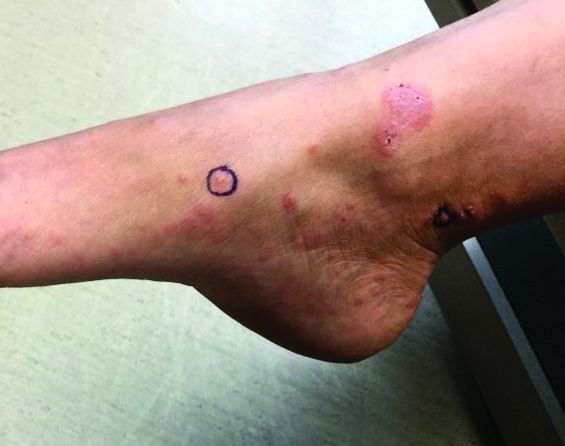
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Pemphigoid gestationis
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Pemphigoid gestationis
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
What's your diagnosis?
A punch biopsy of one of the lesions showed a superficial and deep mixed inflammatory cell infiltrate, including neutrophils and eosinophils. There was also vasculitis, karyorrhexis and extravasated red blood cells. The findings are those of leukocytoclastic vasculitis, suggestive of acute hemorrhagic edema of infancy. Direct immunofluorescence was positive for IgM, C3, and fibrinogen, but negative for IgA.
Acute hemorrhagic edema of infancy (AHEI), also known as Finkelstein disease, is form of leukocytoclastic vasculitis that occurs in infants and toddlers aged between4 months and 3 years.
The lesions start as petechiae or edematous, erythematous to violaceous nodules that later coalesce and form “cockade”-like plaques with a central clearing on the face and extremities. Gastrointestinal, renal, and joint involvement are rare.1 AHEI follows a benign course with resolution of the lesions and symptoms within days to weeks. The etiology of this condition is not known but infection triggers have been reported including coronavirus infections, coxsackie virus infections, Escherichia coli urinary tract infections, herpes simplex virus stomatitis, and pneumococcal bacteremia.2,3 Our patient had a prior history of pneumococcal pneumonia and metapneumovirus infection. MMR vaccine also has been reported as a possible trigger, as well as some medications.
Laboratory results are usually normal, but some patients may have elevated inflammatory markers (C-reactive protein and erythrocyte sedimentation rate), as noted in our patient, and leukocytosis, thrombocytosis, and eosinophilia. Microscopic analysis demonstrates leukocytoclastic vasculitis of small vessels with associated karyorrhexis and extravasated red blood cells.
The differential diagnosis includes other vasculitic conditions, primarily Henoch-Schönlein purpura (HSP). Patients with HSP tend to be older in age and the lesions described as palpable purpura commonly affect the lower extremities and buttocks. These patients can present with abdominal pain and arthritis; renal compromise also can occur. Direct immunofluorescence can commonly be positive for IgA, which was negative in our patient.
AHEI and HSP are considered different entities, but both present with leukocytoclastic vasculitis.1 Another condition to consider in patients with fever, rash, and edema is Kawasaki disease, also a form of vasculitis, that affects small- and medium-size muscular vessels with predilection for the coronary arteries. Patients with Kawasaki disease present with fever (usually longer than 5 days), facial and extremity edema (similar to AHEI), skin lesions (which may have multiple presentations, the most common being macular, papular and erythematous, and urticarial eruptions), but also lymphadenopathy and conjunctivitis. These patients appear sicker than children with AHEI. Their laboratory results show leukocytosis, thrombocytosis or thrombocytopenia, elevated inflammatory markers, and sterile pyuria.4
Patients with erythema nodosum present with tender erythematous nodules, which can look like early AHEI lesions. The most common location is the lower extremities, but in children erythema nodosum can occur on the face, trunk, and arms. The lesions can occur secondary to infections such as streptococcus, mycoplasma, tuberculosis, coccidioidomycosis, and sarcoidosis, as well as to malignancy or medications. These patients do not appear sick, are not febrile, and are rarely seen under 2 years of age.5
Acute febrile neutrophilic dermatosis – Sweets’ syndrome – also should be considered in a patient with tender nodules, fever, and leukocytosis. The skin lesions in Sweets’ syndrome, compared with those in AHEI, are painful and can present as papules, nodules, and bullae on the face and extremities. A prior history of an upper respiratory infection is commonly described in children with Sweets’ syndrome. These patients present with fever, which may start days to weeks prior to the lesions starting. Children with Sweets’ syndrome also can have conjunctivitis, myalgias, polyarthritis, and in severe cases septic shock and multiorgan dysfunction. Sweets’ syndrome can be seen in patients with inflammatory bowel disease, systemic lupus erythematosus, chronic multifocal osteomyelitis, and malignancy; it also may be induced by certain medications.6
As mentioned above, the course of AHEI is benign, and the condition resolves within days to weeks. Treatment is supportive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. F1000Res. 2019;8:1771.
2. Pediatr Dermatol. 2006 Jul-Aug;23(4):361-4.
3. Pediatr Dermatol. 2015 Nov-Dec;32(6):e309-11.
4. Clin Dermatol. 2017 Nov-Dec;35(6):530-40.
5. Yonsei Med J. 2019 Mar;60(3):312-4.
6. Pediatr Dermatol. 2015 Jul-Aug;32(4):437-46.
A punch biopsy of one of the lesions showed a superficial and deep mixed inflammatory cell infiltrate, including neutrophils and eosinophils. There was also vasculitis, karyorrhexis and extravasated red blood cells. The findings are those of leukocytoclastic vasculitis, suggestive of acute hemorrhagic edema of infancy. Direct immunofluorescence was positive for IgM, C3, and fibrinogen, but negative for IgA.
Acute hemorrhagic edema of infancy (AHEI), also known as Finkelstein disease, is form of leukocytoclastic vasculitis that occurs in infants and toddlers aged between4 months and 3 years.
The lesions start as petechiae or edematous, erythematous to violaceous nodules that later coalesce and form “cockade”-like plaques with a central clearing on the face and extremities. Gastrointestinal, renal, and joint involvement are rare.1 AHEI follows a benign course with resolution of the lesions and symptoms within days to weeks. The etiology of this condition is not known but infection triggers have been reported including coronavirus infections, coxsackie virus infections, Escherichia coli urinary tract infections, herpes simplex virus stomatitis, and pneumococcal bacteremia.2,3 Our patient had a prior history of pneumococcal pneumonia and metapneumovirus infection. MMR vaccine also has been reported as a possible trigger, as well as some medications.
Laboratory results are usually normal, but some patients may have elevated inflammatory markers (C-reactive protein and erythrocyte sedimentation rate), as noted in our patient, and leukocytosis, thrombocytosis, and eosinophilia. Microscopic analysis demonstrates leukocytoclastic vasculitis of small vessels with associated karyorrhexis and extravasated red blood cells.
The differential diagnosis includes other vasculitic conditions, primarily Henoch-Schönlein purpura (HSP). Patients with HSP tend to be older in age and the lesions described as palpable purpura commonly affect the lower extremities and buttocks. These patients can present with abdominal pain and arthritis; renal compromise also can occur. Direct immunofluorescence can commonly be positive for IgA, which was negative in our patient.
AHEI and HSP are considered different entities, but both present with leukocytoclastic vasculitis.1 Another condition to consider in patients with fever, rash, and edema is Kawasaki disease, also a form of vasculitis, that affects small- and medium-size muscular vessels with predilection for the coronary arteries. Patients with Kawasaki disease present with fever (usually longer than 5 days), facial and extremity edema (similar to AHEI), skin lesions (which may have multiple presentations, the most common being macular, papular and erythematous, and urticarial eruptions), but also lymphadenopathy and conjunctivitis. These patients appear sicker than children with AHEI. Their laboratory results show leukocytosis, thrombocytosis or thrombocytopenia, elevated inflammatory markers, and sterile pyuria.4
Patients with erythema nodosum present with tender erythematous nodules, which can look like early AHEI lesions. The most common location is the lower extremities, but in children erythema nodosum can occur on the face, trunk, and arms. The lesions can occur secondary to infections such as streptococcus, mycoplasma, tuberculosis, coccidioidomycosis, and sarcoidosis, as well as to malignancy or medications. These patients do not appear sick, are not febrile, and are rarely seen under 2 years of age.5
Acute febrile neutrophilic dermatosis – Sweets’ syndrome – also should be considered in a patient with tender nodules, fever, and leukocytosis. The skin lesions in Sweets’ syndrome, compared with those in AHEI, are painful and can present as papules, nodules, and bullae on the face and extremities. A prior history of an upper respiratory infection is commonly described in children with Sweets’ syndrome. These patients present with fever, which may start days to weeks prior to the lesions starting. Children with Sweets’ syndrome also can have conjunctivitis, myalgias, polyarthritis, and in severe cases septic shock and multiorgan dysfunction. Sweets’ syndrome can be seen in patients with inflammatory bowel disease, systemic lupus erythematosus, chronic multifocal osteomyelitis, and malignancy; it also may be induced by certain medications.6
As mentioned above, the course of AHEI is benign, and the condition resolves within days to weeks. Treatment is supportive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. F1000Res. 2019;8:1771.
2. Pediatr Dermatol. 2006 Jul-Aug;23(4):361-4.
3. Pediatr Dermatol. 2015 Nov-Dec;32(6):e309-11.
4. Clin Dermatol. 2017 Nov-Dec;35(6):530-40.
5. Yonsei Med J. 2019 Mar;60(3):312-4.
6. Pediatr Dermatol. 2015 Jul-Aug;32(4):437-46.
A punch biopsy of one of the lesions showed a superficial and deep mixed inflammatory cell infiltrate, including neutrophils and eosinophils. There was also vasculitis, karyorrhexis and extravasated red blood cells. The findings are those of leukocytoclastic vasculitis, suggestive of acute hemorrhagic edema of infancy. Direct immunofluorescence was positive for IgM, C3, and fibrinogen, but negative for IgA.
Acute hemorrhagic edema of infancy (AHEI), also known as Finkelstein disease, is form of leukocytoclastic vasculitis that occurs in infants and toddlers aged between4 months and 3 years.
The lesions start as petechiae or edematous, erythematous to violaceous nodules that later coalesce and form “cockade”-like plaques with a central clearing on the face and extremities. Gastrointestinal, renal, and joint involvement are rare.1 AHEI follows a benign course with resolution of the lesions and symptoms within days to weeks. The etiology of this condition is not known but infection triggers have been reported including coronavirus infections, coxsackie virus infections, Escherichia coli urinary tract infections, herpes simplex virus stomatitis, and pneumococcal bacteremia.2,3 Our patient had a prior history of pneumococcal pneumonia and metapneumovirus infection. MMR vaccine also has been reported as a possible trigger, as well as some medications.
Laboratory results are usually normal, but some patients may have elevated inflammatory markers (C-reactive protein and erythrocyte sedimentation rate), as noted in our patient, and leukocytosis, thrombocytosis, and eosinophilia. Microscopic analysis demonstrates leukocytoclastic vasculitis of small vessels with associated karyorrhexis and extravasated red blood cells.
The differential diagnosis includes other vasculitic conditions, primarily Henoch-Schönlein purpura (HSP). Patients with HSP tend to be older in age and the lesions described as palpable purpura commonly affect the lower extremities and buttocks. These patients can present with abdominal pain and arthritis; renal compromise also can occur. Direct immunofluorescence can commonly be positive for IgA, which was negative in our patient.
AHEI and HSP are considered different entities, but both present with leukocytoclastic vasculitis.1 Another condition to consider in patients with fever, rash, and edema is Kawasaki disease, also a form of vasculitis, that affects small- and medium-size muscular vessels with predilection for the coronary arteries. Patients with Kawasaki disease present with fever (usually longer than 5 days), facial and extremity edema (similar to AHEI), skin lesions (which may have multiple presentations, the most common being macular, papular and erythematous, and urticarial eruptions), but also lymphadenopathy and conjunctivitis. These patients appear sicker than children with AHEI. Their laboratory results show leukocytosis, thrombocytosis or thrombocytopenia, elevated inflammatory markers, and sterile pyuria.4
Patients with erythema nodosum present with tender erythematous nodules, which can look like early AHEI lesions. The most common location is the lower extremities, but in children erythema nodosum can occur on the face, trunk, and arms. The lesions can occur secondary to infections such as streptococcus, mycoplasma, tuberculosis, coccidioidomycosis, and sarcoidosis, as well as to malignancy or medications. These patients do not appear sick, are not febrile, and are rarely seen under 2 years of age.5
Acute febrile neutrophilic dermatosis – Sweets’ syndrome – also should be considered in a patient with tender nodules, fever, and leukocytosis. The skin lesions in Sweets’ syndrome, compared with those in AHEI, are painful and can present as papules, nodules, and bullae on the face and extremities. A prior history of an upper respiratory infection is commonly described in children with Sweets’ syndrome. These patients present with fever, which may start days to weeks prior to the lesions starting. Children with Sweets’ syndrome also can have conjunctivitis, myalgias, polyarthritis, and in severe cases septic shock and multiorgan dysfunction. Sweets’ syndrome can be seen in patients with inflammatory bowel disease, systemic lupus erythematosus, chronic multifocal osteomyelitis, and malignancy; it also may be induced by certain medications.6
As mentioned above, the course of AHEI is benign, and the condition resolves within days to weeks. Treatment is supportive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. F1000Res. 2019;8:1771.
2. Pediatr Dermatol. 2006 Jul-Aug;23(4):361-4.
3. Pediatr Dermatol. 2015 Nov-Dec;32(6):e309-11.
4. Clin Dermatol. 2017 Nov-Dec;35(6):530-40.
5. Yonsei Med J. 2019 Mar;60(3):312-4.
6. Pediatr Dermatol. 2015 Jul-Aug;32(4):437-46.
At 3 a.m., you receive a call from the ED for a baby with a new rash on the arms, legs, and face. Some of the lesions appear to be tender. He has a mild fever of 38.4° C (101.1° F) and is not in acute distress. He is drinking, but not eating much.
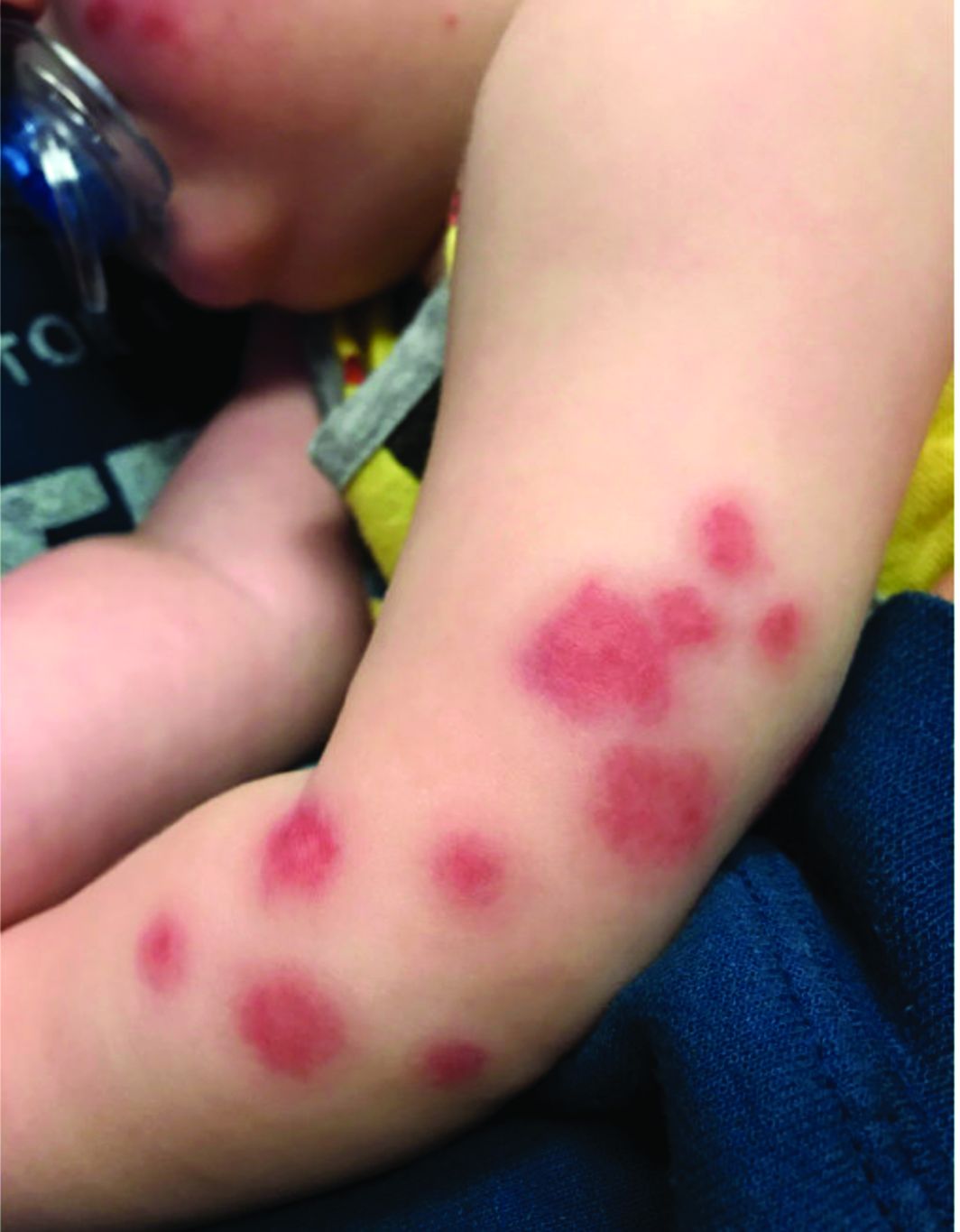
The parents also have noted some swelling on the hands and the feet. He has no upper respiratory or gastrointestinal symptoms. He is not walking yet.
He was admitted to the hospital 3 weeks prior for streptococcal pneumonia and metapneumovirus infection. He was treated with ceftriaxone, supportive respiratory care, and an albuterol inhaler. Influenza and respiratory syncytial virus tests were negative.
On physical exam, the child is tired and sleeping in his mom's arms. He has red and some purpuric papules on the face. On the arms and legs, he has purpuric papules and nodules. There is some edema on the face, hands, and feet. His conjunctiva is normal, and he has no oral lesions. He has no lymphadenopathy or hepatosplenomegaly.
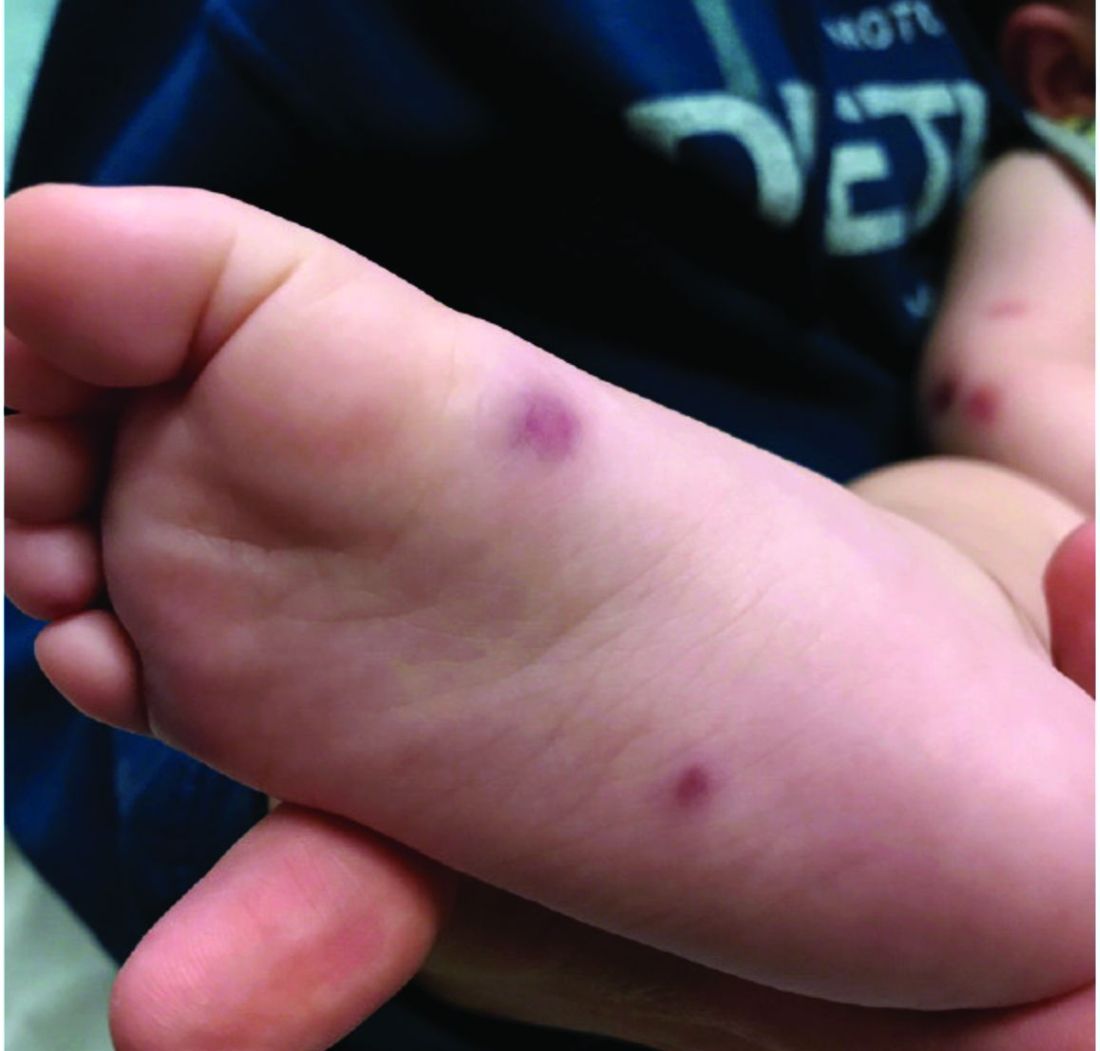
Blood work shows normal complete blood count, coagulation tests, comprehensive metabolic panel, and urinalysis, but he has an elevated C-reactive protein of 114 mg/L and an elevated erythrocyte sedimentation rate of 71 mm/hour.
April 2020
Shiitake mushroom flagellate dermatitis
that resemble whiplash marks. The lesions may be extremely pruritic, and petechiae may be present in the streaks. The trunk is most commonly affected, although lesions can occur on the limbs. Mucosa is not affected. Sun exposure may exacerbate the condition. The dermatitis has been described in all ages and races, and males seem to be more affected than females.
Shiitake mushroom flagellate dermatitis typically occurs following the ingestion of raw or undercooked shiitake mushrooms (Lentinula edodes). The mushrooms contain a polysaccharide called lentinan. Ingestion of lentinan activates interleukin-1 (IL-1), resulting in vasodilation and the subsequent dermatitis that can occur within a few hours and up to 5 days post ingestion. Associated gastrointestinal symptoms, fever, and localized swelling have been reported. The rash will resolve spontaneously over a few days to weeks.
Flagellate erythema has been described with bleomycin treatment. Other reported associations include peplomycin (a bleomycin derivative) and docetaxel. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anywhere from 1 day to several months after exposure. Over time, the erythema will develop into postinflammatory hyperpigmentation.
Dermatomyositis may present with flagellate erythema. Other symptoms include muscle weakness and an inflammatory myopathy. A heliotrope rash on the eyelids, Gottron’s papules on the hands, ragged cuticles with prominent vessels on nail folds may be seen. Blood work may reveal elevated antinuclear antibodies (ANA), anti–Mi-2 and anti–Jo-1. Adult-onset Still disease is characterized by fever, arthritis, and salmon-colored patches.
Our patient’s dermatitis resolved spontaneously without treatment.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Shiitake mushroom flagellate dermatitis
that resemble whiplash marks. The lesions may be extremely pruritic, and petechiae may be present in the streaks. The trunk is most commonly affected, although lesions can occur on the limbs. Mucosa is not affected. Sun exposure may exacerbate the condition. The dermatitis has been described in all ages and races, and males seem to be more affected than females.
Shiitake mushroom flagellate dermatitis typically occurs following the ingestion of raw or undercooked shiitake mushrooms (Lentinula edodes). The mushrooms contain a polysaccharide called lentinan. Ingestion of lentinan activates interleukin-1 (IL-1), resulting in vasodilation and the subsequent dermatitis that can occur within a few hours and up to 5 days post ingestion. Associated gastrointestinal symptoms, fever, and localized swelling have been reported. The rash will resolve spontaneously over a few days to weeks.
Flagellate erythema has been described with bleomycin treatment. Other reported associations include peplomycin (a bleomycin derivative) and docetaxel. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anywhere from 1 day to several months after exposure. Over time, the erythema will develop into postinflammatory hyperpigmentation.
Dermatomyositis may present with flagellate erythema. Other symptoms include muscle weakness and an inflammatory myopathy. A heliotrope rash on the eyelids, Gottron’s papules on the hands, ragged cuticles with prominent vessels on nail folds may be seen. Blood work may reveal elevated antinuclear antibodies (ANA), anti–Mi-2 and anti–Jo-1. Adult-onset Still disease is characterized by fever, arthritis, and salmon-colored patches.
Our patient’s dermatitis resolved spontaneously without treatment.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Shiitake mushroom flagellate dermatitis
that resemble whiplash marks. The lesions may be extremely pruritic, and petechiae may be present in the streaks. The trunk is most commonly affected, although lesions can occur on the limbs. Mucosa is not affected. Sun exposure may exacerbate the condition. The dermatitis has been described in all ages and races, and males seem to be more affected than females.
Shiitake mushroom flagellate dermatitis typically occurs following the ingestion of raw or undercooked shiitake mushrooms (Lentinula edodes). The mushrooms contain a polysaccharide called lentinan. Ingestion of lentinan activates interleukin-1 (IL-1), resulting in vasodilation and the subsequent dermatitis that can occur within a few hours and up to 5 days post ingestion. Associated gastrointestinal symptoms, fever, and localized swelling have been reported. The rash will resolve spontaneously over a few days to weeks.
Flagellate erythema has been described with bleomycin treatment. Other reported associations include peplomycin (a bleomycin derivative) and docetaxel. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anywhere from 1 day to several months after exposure. Over time, the erythema will develop into postinflammatory hyperpigmentation.
Dermatomyositis may present with flagellate erythema. Other symptoms include muscle weakness and an inflammatory myopathy. A heliotrope rash on the eyelids, Gottron’s papules on the hands, ragged cuticles with prominent vessels on nail folds may be seen. Blood work may reveal elevated antinuclear antibodies (ANA), anti–Mi-2 and anti–Jo-1. Adult-onset Still disease is characterized by fever, arthritis, and salmon-colored patches.
Our patient’s dermatitis resolved spontaneously without treatment.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.

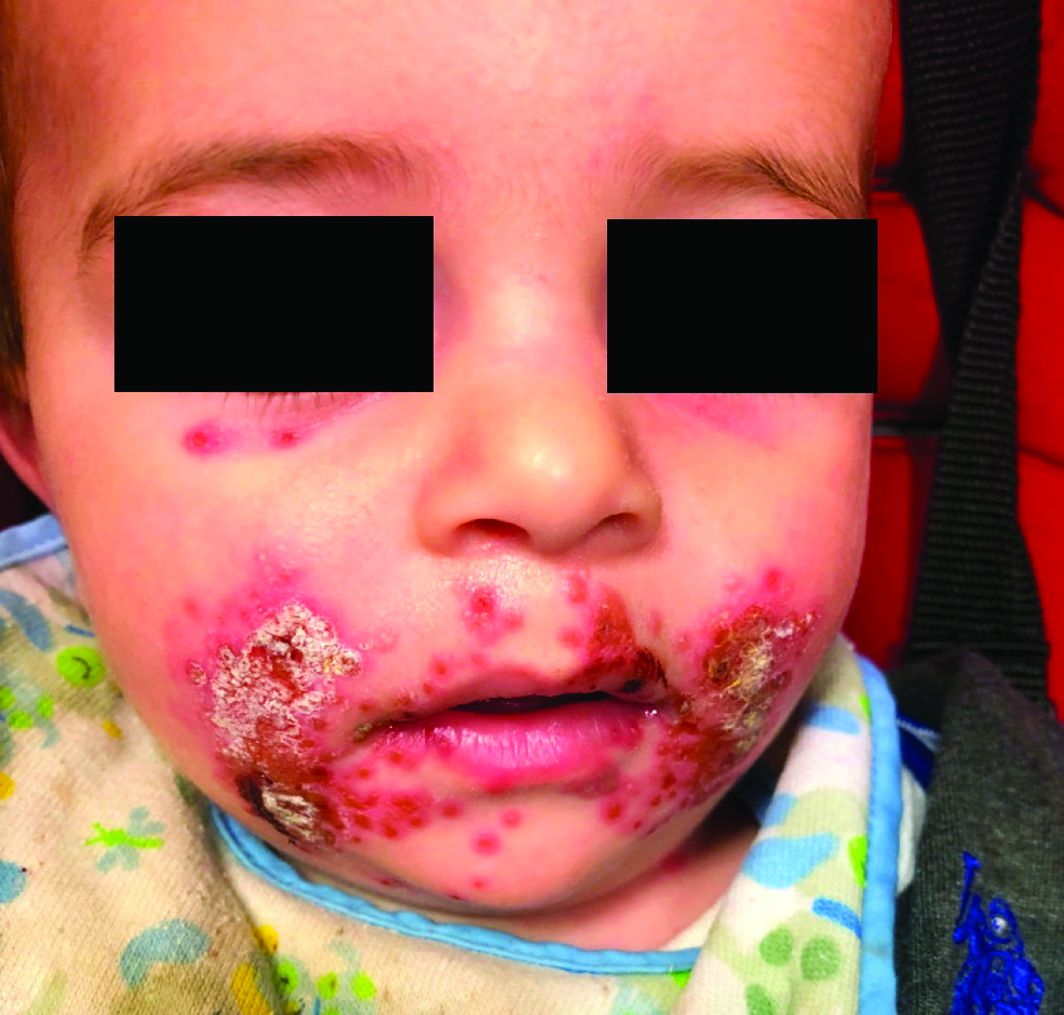
A 7-month-old male presents with perioral rash and fever
Patients with atopic dermatitis are at risk for developing the herpes simplex virus (HSV)–related skin complication “eczema herpeticum,” also known as Kaposi’s varicelliform eruption. Eczema herpeticum is characterized by cutaneous pain and vesicular skin lesions, most commonly secondary to infection with HSV-1. The condition may affect individuals with atopic dermatitis or other inflammatory skin disorders. Eczema herpeticum develops when the virus infects large areas of skin, rather than being confined to a small area as in the common cold sore. Eczema herpeticum often appears on the face and neck, although it can appear anywhere on the body. In some cases, the rash may be difficult to distinguish from a patient’s baseline eczema if the latter is poorly controlled. Skin symptoms of eczema herpeticum include clusters of small blisters that are itchy and painful; vesicles that appear red, purple, or black; purulent blisters; or crusting. Classically, the morphology of vesicles or crusted lesions shows a “cluster of grapes” appearance. Eczema herpeticum may present with a high fever, chills, and swollen lymph glands.
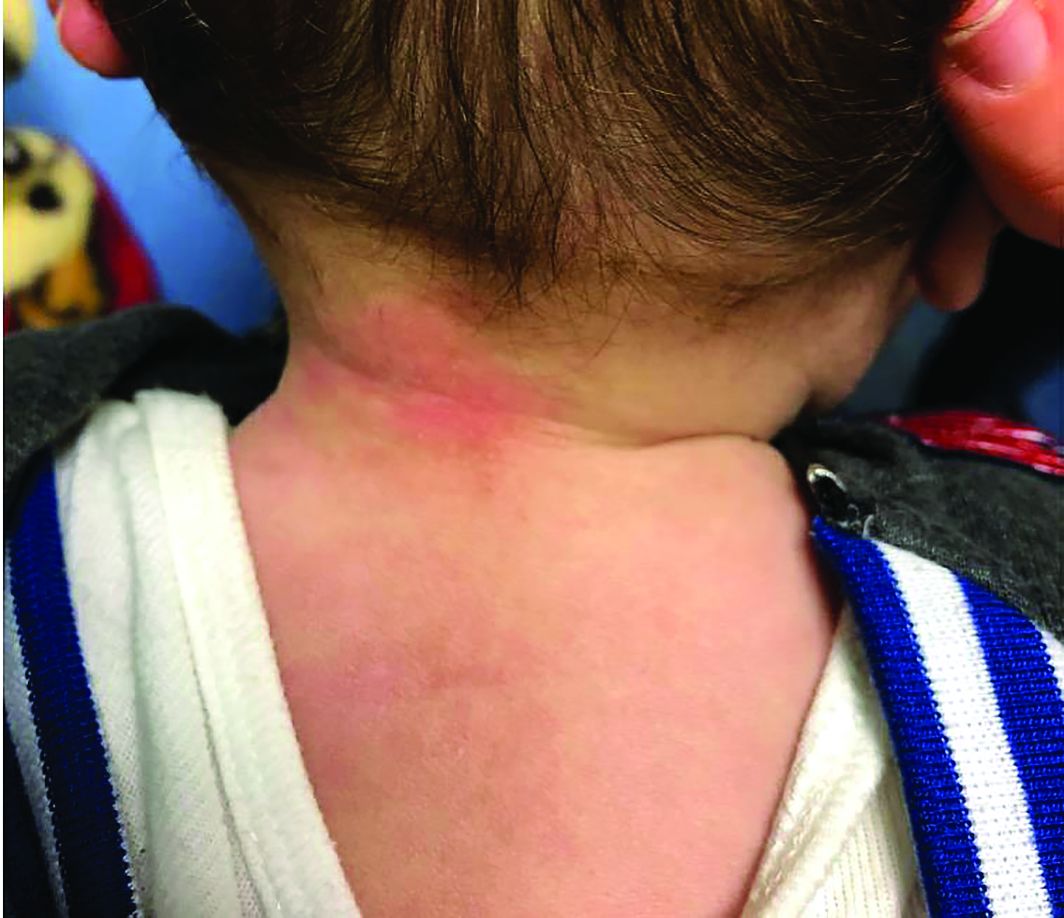
While a clinical diagnosis based on the history, physical findings, and morphologic appearance of the rash is reasonable, testing may confirm the diagnosis. The most sensitive and specific tests are polymerase chain reaction sequencing for HSV, direct fluorescent antibody stain, and/or viral culture, while Tzanck smear may show characteristic histologic changes. Treatment is with oral antiviral therapy and treatment of the eczema.
Hand, foot, and mouth disease (HFMD) is a common viral illness usually affecting infants and children. The infection often involves the hands, feet, mouth, and sometimes, the genitals and buttocks. The viral exanthem is most commonly caused by the coxsackievirus, of the enterovirus family. Coxsackievirus A16 and enterovirus A71 are the serotypes that are most commonly implicated as the causative agents. HFMD initially presents with a low-grade fever, reduced appetite, and general malaise. About 1-2 days later, the child may develop painful mouth sores with an exanthem that involves the dorsum of the hands, soles of the feet, buttocks, legs, and arms. The exanthem consists of vesicles surrounded by a thin halo of erythema, eventually rupturing and forming superficial ulcers with a gray-yellow base and erythematous rim. The exanthem is itchy, and can be macular, papular, or vesicular. The lesions are nonpruritic, and typically not painful. The diagnosis of HFMD usually is made clinically, although a physician can swab the mouth or get a stool sample for polymerase chain reaction, which will show the virus; treatment is supportive. In children with atopic dermatitis, lesions also can tend to concentrate in areas previously or currently affected by the dermatitis, similar to eczema herpeticum, and the terms eczema coxsackium or atypical HFMD are applicable. In young adults, the disease may present with erythematous papulovesicular lesions on the face, oral mucosa, extensor surfaces of the upper and lower extremities, and palms and soles; confluent, hemorrhagic, and crusted lesions also can be seen on the extremities. Systemic symptoms usually subside in a few days; the skin lesions resolve without scarring in days to weeks.
Secondary bacterial infection is not uncommon in eczema herpeticum patients, reflecting common Staphylococcus aureus infection in atopic dermatitis patients. Streptococcus also may be seen as a concurrent infection. Treatment of secondary bacterial infection may be considered based on clinic context and culture.
Impetiginized eczema also is in the differential diagnosis of eczema herpeticum. S. aureus and Streptococci are the most important causative organisms. Lesions can manifest as a single red papule or macule that quickly becomes vesicular or eroded. Subsequently, the content dries, forming honey-colored crusts. Impetigo may resolve spontaneously, although in the context of infected eczema both topical anti-inflammatory agents (e.g. topical corticosteroids) along with systemic antibiotics may be a reasonable treatment option. Although our patient had honey-colored crusting, the wound culture showed normal bacterial flora.
Primary varicella infection causes acute fever and rash, with an initial exanthem of disseminated pruritic erythematous macules that progress beyond the papular stage, forming clear, fluid-filled vesicles (like dewdrops on a rose petal). In children, the rash presents on the stomach, back, and face, and then spreads to other parts of the body. Blisters also can arise inside the mouth.
In this patient, perioral HSV PCR 1 was positive, and wound culture showed normal oral flora with no organisms or white blood cells seen. The patient responded well to oral acyclovir, and treatment of his underlying atopic dermatitis with low-potency topical corticosteroids.
Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians had relevant financial disclosures. Email them at pdnews@mdedge.com.
Sources
Can Fam Physician. 2012 Dec;58(12):1358-61.
William L Weston, MD., William Howe, MD. UpToDate. Treatment of atopic dermatitis (eczema).
Christine Johnson, MD, Anna Wald, MD, MPH. UpToDate. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection.
Robert Sidbury, MD, MPH. UpToDate. Atypical exanthems in children.
National Eczema Association. Eczema herpeticum.
Centers for Disease Control and Prevention. Symptoms and diagnosis of hand, foot, and mouth disease (HFMD).
Patients with atopic dermatitis are at risk for developing the herpes simplex virus (HSV)–related skin complication “eczema herpeticum,” also known as Kaposi’s varicelliform eruption. Eczema herpeticum is characterized by cutaneous pain and vesicular skin lesions, most commonly secondary to infection with HSV-1. The condition may affect individuals with atopic dermatitis or other inflammatory skin disorders. Eczema herpeticum develops when the virus infects large areas of skin, rather than being confined to a small area as in the common cold sore. Eczema herpeticum often appears on the face and neck, although it can appear anywhere on the body. In some cases, the rash may be difficult to distinguish from a patient’s baseline eczema if the latter is poorly controlled. Skin symptoms of eczema herpeticum include clusters of small blisters that are itchy and painful; vesicles that appear red, purple, or black; purulent blisters; or crusting. Classically, the morphology of vesicles or crusted lesions shows a “cluster of grapes” appearance. Eczema herpeticum may present with a high fever, chills, and swollen lymph glands.
While a clinical diagnosis based on the history, physical findings, and morphologic appearance of the rash is reasonable, testing may confirm the diagnosis. The most sensitive and specific tests are polymerase chain reaction sequencing for HSV, direct fluorescent antibody stain, and/or viral culture, while Tzanck smear may show characteristic histologic changes. Treatment is with oral antiviral therapy and treatment of the eczema.
Hand, foot, and mouth disease (HFMD) is a common viral illness usually affecting infants and children. The infection often involves the hands, feet, mouth, and sometimes, the genitals and buttocks. The viral exanthem is most commonly caused by the coxsackievirus, of the enterovirus family. Coxsackievirus A16 and enterovirus A71 are the serotypes that are most commonly implicated as the causative agents. HFMD initially presents with a low-grade fever, reduced appetite, and general malaise. About 1-2 days later, the child may develop painful mouth sores with an exanthem that involves the dorsum of the hands, soles of the feet, buttocks, legs, and arms. The exanthem consists of vesicles surrounded by a thin halo of erythema, eventually rupturing and forming superficial ulcers with a gray-yellow base and erythematous rim. The exanthem is itchy, and can be macular, papular, or vesicular. The lesions are nonpruritic, and typically not painful. The diagnosis of HFMD usually is made clinically, although a physician can swab the mouth or get a stool sample for polymerase chain reaction, which will show the virus; treatment is supportive. In children with atopic dermatitis, lesions also can tend to concentrate in areas previously or currently affected by the dermatitis, similar to eczema herpeticum, and the terms eczema coxsackium or atypical HFMD are applicable. In young adults, the disease may present with erythematous papulovesicular lesions on the face, oral mucosa, extensor surfaces of the upper and lower extremities, and palms and soles; confluent, hemorrhagic, and crusted lesions also can be seen on the extremities. Systemic symptoms usually subside in a few days; the skin lesions resolve without scarring in days to weeks.
Secondary bacterial infection is not uncommon in eczema herpeticum patients, reflecting common Staphylococcus aureus infection in atopic dermatitis patients. Streptococcus also may be seen as a concurrent infection. Treatment of secondary bacterial infection may be considered based on clinic context and culture.
Impetiginized eczema also is in the differential diagnosis of eczema herpeticum. S. aureus and Streptococci are the most important causative organisms. Lesions can manifest as a single red papule or macule that quickly becomes vesicular or eroded. Subsequently, the content dries, forming honey-colored crusts. Impetigo may resolve spontaneously, although in the context of infected eczema both topical anti-inflammatory agents (e.g. topical corticosteroids) along with systemic antibiotics may be a reasonable treatment option. Although our patient had honey-colored crusting, the wound culture showed normal bacterial flora.
Primary varicella infection causes acute fever and rash, with an initial exanthem of disseminated pruritic erythematous macules that progress beyond the papular stage, forming clear, fluid-filled vesicles (like dewdrops on a rose petal). In children, the rash presents on the stomach, back, and face, and then spreads to other parts of the body. Blisters also can arise inside the mouth.
In this patient, perioral HSV PCR 1 was positive, and wound culture showed normal oral flora with no organisms or white blood cells seen. The patient responded well to oral acyclovir, and treatment of his underlying atopic dermatitis with low-potency topical corticosteroids.
Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians had relevant financial disclosures. Email them at pdnews@mdedge.com.
Sources
Can Fam Physician. 2012 Dec;58(12):1358-61.
William L Weston, MD., William Howe, MD. UpToDate. Treatment of atopic dermatitis (eczema).
Christine Johnson, MD, Anna Wald, MD, MPH. UpToDate. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection.
Robert Sidbury, MD, MPH. UpToDate. Atypical exanthems in children.
National Eczema Association. Eczema herpeticum.
Centers for Disease Control and Prevention. Symptoms and diagnosis of hand, foot, and mouth disease (HFMD).
Patients with atopic dermatitis are at risk for developing the herpes simplex virus (HSV)–related skin complication “eczema herpeticum,” also known as Kaposi’s varicelliform eruption. Eczema herpeticum is characterized by cutaneous pain and vesicular skin lesions, most commonly secondary to infection with HSV-1. The condition may affect individuals with atopic dermatitis or other inflammatory skin disorders. Eczema herpeticum develops when the virus infects large areas of skin, rather than being confined to a small area as in the common cold sore. Eczema herpeticum often appears on the face and neck, although it can appear anywhere on the body. In some cases, the rash may be difficult to distinguish from a patient’s baseline eczema if the latter is poorly controlled. Skin symptoms of eczema herpeticum include clusters of small blisters that are itchy and painful; vesicles that appear red, purple, or black; purulent blisters; or crusting. Classically, the morphology of vesicles or crusted lesions shows a “cluster of grapes” appearance. Eczema herpeticum may present with a high fever, chills, and swollen lymph glands.
While a clinical diagnosis based on the history, physical findings, and morphologic appearance of the rash is reasonable, testing may confirm the diagnosis. The most sensitive and specific tests are polymerase chain reaction sequencing for HSV, direct fluorescent antibody stain, and/or viral culture, while Tzanck smear may show characteristic histologic changes. Treatment is with oral antiviral therapy and treatment of the eczema.
Hand, foot, and mouth disease (HFMD) is a common viral illness usually affecting infants and children. The infection often involves the hands, feet, mouth, and sometimes, the genitals and buttocks. The viral exanthem is most commonly caused by the coxsackievirus, of the enterovirus family. Coxsackievirus A16 and enterovirus A71 are the serotypes that are most commonly implicated as the causative agents. HFMD initially presents with a low-grade fever, reduced appetite, and general malaise. About 1-2 days later, the child may develop painful mouth sores with an exanthem that involves the dorsum of the hands, soles of the feet, buttocks, legs, and arms. The exanthem consists of vesicles surrounded by a thin halo of erythema, eventually rupturing and forming superficial ulcers with a gray-yellow base and erythematous rim. The exanthem is itchy, and can be macular, papular, or vesicular. The lesions are nonpruritic, and typically not painful. The diagnosis of HFMD usually is made clinically, although a physician can swab the mouth or get a stool sample for polymerase chain reaction, which will show the virus; treatment is supportive. In children with atopic dermatitis, lesions also can tend to concentrate in areas previously or currently affected by the dermatitis, similar to eczema herpeticum, and the terms eczema coxsackium or atypical HFMD are applicable. In young adults, the disease may present with erythematous papulovesicular lesions on the face, oral mucosa, extensor surfaces of the upper and lower extremities, and palms and soles; confluent, hemorrhagic, and crusted lesions also can be seen on the extremities. Systemic symptoms usually subside in a few days; the skin lesions resolve without scarring in days to weeks.
Secondary bacterial infection is not uncommon in eczema herpeticum patients, reflecting common Staphylococcus aureus infection in atopic dermatitis patients. Streptococcus also may be seen as a concurrent infection. Treatment of secondary bacterial infection may be considered based on clinic context and culture.
Impetiginized eczema also is in the differential diagnosis of eczema herpeticum. S. aureus and Streptococci are the most important causative organisms. Lesions can manifest as a single red papule or macule that quickly becomes vesicular or eroded. Subsequently, the content dries, forming honey-colored crusts. Impetigo may resolve spontaneously, although in the context of infected eczema both topical anti-inflammatory agents (e.g. topical corticosteroids) along with systemic antibiotics may be a reasonable treatment option. Although our patient had honey-colored crusting, the wound culture showed normal bacterial flora.
Primary varicella infection causes acute fever and rash, with an initial exanthem of disseminated pruritic erythematous macules that progress beyond the papular stage, forming clear, fluid-filled vesicles (like dewdrops on a rose petal). In children, the rash presents on the stomach, back, and face, and then spreads to other parts of the body. Blisters also can arise inside the mouth.
In this patient, perioral HSV PCR 1 was positive, and wound culture showed normal oral flora with no organisms or white blood cells seen. The patient responded well to oral acyclovir, and treatment of his underlying atopic dermatitis with low-potency topical corticosteroids.
Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither of the physicians had relevant financial disclosures. Email them at pdnews@mdedge.com.
Sources
Can Fam Physician. 2012 Dec;58(12):1358-61.
William L Weston, MD., William Howe, MD. UpToDate. Treatment of atopic dermatitis (eczema).
Christine Johnson, MD, Anna Wald, MD, MPH. UpToDate. Epidemiology, clinical manifestations, and diagnosis of herpes simplex virus type 1 infection.
Robert Sidbury, MD, MPH. UpToDate. Atypical exanthems in children.
National Eczema Association. Eczema herpeticum.
Centers for Disease Control and Prevention. Symptoms and diagnosis of hand, foot, and mouth disease (HFMD).
Nail dystrophy and nail plate thinning
At a follow-up visit, a biopsy of the skin on the fingertips was performed, which showed lichenoid lymphocytic inflammatory infiltrate with associated hyperkeratosis, hypergranulosis, and acanthosis.
No fungal elements were seen. The findings were consistent with lichen planus.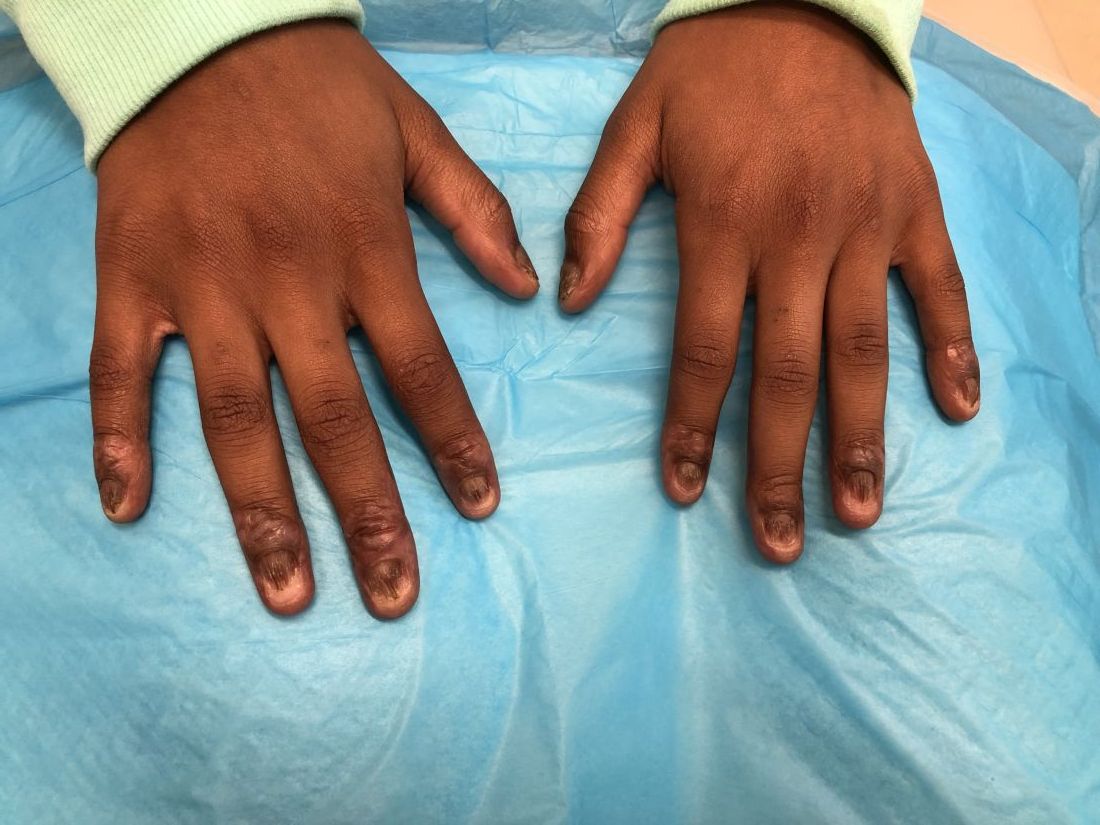
The patient was started on hydroxychloroquine. It was recommended she start a 6-week course of oral prednisone, but the mother was opposed to systemic treatment because of potential side effects.
She continued topical betamethasone without much change. Topical tacrolimus later was recommended to use on off days of betamethasone, which led to no improvement. Narrow-band UVB also was started with minimal improvement. Unfortunately,
Nail lichen planus (NLP) in children is not a common condition.1 In a recent series from Chiheb et al., NLP was reported in 90 patients, of which 40% were children; a quarter of the patients reported having extracutaneous involvement as well.2 In another childhood LP series,14 % of the children presented with nail disease.3 It can be a severe disease that, if not treated aggressively, may lead to destruction of the nail bed. This condition seems to be more prevalent in boys than girls and more prevalent in African American children.3 Unfortunately, in this patient’s case, the mother was hesitant to use systemic therapy and aggressive treatment was delayed.
Possible but not clear associations with autoimmune conditions such as vitiligo, autoimmune thyroiditis, myasthenia gravis, alopecia areata, thymoma, autoimmune polyendocrinopathy, atopic dermatitis, and lichen nitidus have been described in children with LP.
The clinical characteristics of NLP include nail plate thinning with longitudinal ridging and fissuring, with or without pterygium; trachyonychia; and erythema of the lunula when the nail matrix is involved. When the nail bed is affected, the patient can present with onycholysis with or without subungual hyperkeratosis and violaceous hue of the nail bed.4 NLP can have three different clinical presentations described by Tosti et al., which include typical NLP, 20‐nail dystrophy (trachyonychia), and idiopathic nail atrophy. Idiopathic nail atrophy is described solely in children as an acute and rapid progression that leads to destruction of the nail within months, which appears to be the clinical presentation in our patient.
The differential diagnosis of nail dystrophy in children includes infectious processes such as onychomycosis, especially when children present with onycholysis and subungual hyperkeratosis. Because of this, it is recommended to perform a nail culture or submit a sample of nail clippings for microscopic evaluation to confirm the diagnosis of onychomycosis prior to starting systemic therapy in children. Fingernail involvement without toenail involvement is an unusual presentation of onychomycosis.
Twenty-nail dystrophy – also known as trachyonychia – can be caused by several inflammatory skin conditions such as lichen planus, psoriasis, eczema, pemphigus vulgaris, and alopecia areata. Clinically, there is uniformly monomorphic thinning of the nail plate with longitudinal ridging without splitting or pterygium.1 This is a benign condition and should not cause scarring. About 10% of the cases of 20-nail dystrophy are caused by lichen planus.
Nail psoriasis is characterized by nail pitting, oil spots on the nail plate, leukonychia, subungual hyperkeratosis, and onycholysis, as well as nail crumbling, which were not seen in our patient. Although her initial presentation was of 20-nail dystrophy, which also can be a presentation of nail psoriasis, its rapid evolution with associated nail atrophy and pterygium make it unlikely to be psoriasis in this particular patient.
Patients with pachyonychia congenita – which is a genetic disorder or keratinization caused by mutations on several genes encoding keratin such as K6a, K16, K17, K6b, and possibly K6c – present with nail thickening (pachyonychia) and discoloration of the nails, as well as pincer nails. These patients also present with oral leukokeratosis and focal palmoplantar keratoderma.
The main treatment of lichen planus is potent topical corticosteroids.
For nail disease, topical treatment may not be effective and systemic treatment may be necessary. Systemic corticosteroids have been used in several pediatric series varying from a short course given at a dose of 1- 2 mg/kg per day for 2 weeks to a longer 3-month course followed by tapering.3 There are several protocols of intramuscular triamcinolone at a dose of 0.5 mg/kg in children in once a month injections for about 3 months that have been reported successful with minimal side effects.1 Other medications reported useful in patients with NLP include dapsone and acitretin. Other treatment options include narrow-band UVB and PUVA.3
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Dermatol. 2001 Aug;137(8):1027-32.
2. Ann Dermatol Venereol. 2015 Jan;142(1):21-5.
3. Pediatr Dermatol. 2014 Jan-Feb;31(1):59-67.
4. Dermatological diseases, in “Nails: Diagnosis, Therapy, and Surgery,” 3rd ed. (Oxford: Elsevier Saunders, 2005, p. 105).
At a follow-up visit, a biopsy of the skin on the fingertips was performed, which showed lichenoid lymphocytic inflammatory infiltrate with associated hyperkeratosis, hypergranulosis, and acanthosis.
No fungal elements were seen. The findings were consistent with lichen planus.
The patient was started on hydroxychloroquine. It was recommended she start a 6-week course of oral prednisone, but the mother was opposed to systemic treatment because of potential side effects.
She continued topical betamethasone without much change. Topical tacrolimus later was recommended to use on off days of betamethasone, which led to no improvement. Narrow-band UVB also was started with minimal improvement. Unfortunately,
Nail lichen planus (NLP) in children is not a common condition.1 In a recent series from Chiheb et al., NLP was reported in 90 patients, of which 40% were children; a quarter of the patients reported having extracutaneous involvement as well.2 In another childhood LP series,14 % of the children presented with nail disease.3 It can be a severe disease that, if not treated aggressively, may lead to destruction of the nail bed. This condition seems to be more prevalent in boys than girls and more prevalent in African American children.3 Unfortunately, in this patient’s case, the mother was hesitant to use systemic therapy and aggressive treatment was delayed.
Possible but not clear associations with autoimmune conditions such as vitiligo, autoimmune thyroiditis, myasthenia gravis, alopecia areata, thymoma, autoimmune polyendocrinopathy, atopic dermatitis, and lichen nitidus have been described in children with LP.
The clinical characteristics of NLP include nail plate thinning with longitudinal ridging and fissuring, with or without pterygium; trachyonychia; and erythema of the lunula when the nail matrix is involved. When the nail bed is affected, the patient can present with onycholysis with or without subungual hyperkeratosis and violaceous hue of the nail bed.4 NLP can have three different clinical presentations described by Tosti et al., which include typical NLP, 20‐nail dystrophy (trachyonychia), and idiopathic nail atrophy. Idiopathic nail atrophy is described solely in children as an acute and rapid progression that leads to destruction of the nail within months, which appears to be the clinical presentation in our patient.
The differential diagnosis of nail dystrophy in children includes infectious processes such as onychomycosis, especially when children present with onycholysis and subungual hyperkeratosis. Because of this, it is recommended to perform a nail culture or submit a sample of nail clippings for microscopic evaluation to confirm the diagnosis of onychomycosis prior to starting systemic therapy in children. Fingernail involvement without toenail involvement is an unusual presentation of onychomycosis.
Twenty-nail dystrophy – also known as trachyonychia – can be caused by several inflammatory skin conditions such as lichen planus, psoriasis, eczema, pemphigus vulgaris, and alopecia areata. Clinically, there is uniformly monomorphic thinning of the nail plate with longitudinal ridging without splitting or pterygium.1 This is a benign condition and should not cause scarring. About 10% of the cases of 20-nail dystrophy are caused by lichen planus.
Nail psoriasis is characterized by nail pitting, oil spots on the nail plate, leukonychia, subungual hyperkeratosis, and onycholysis, as well as nail crumbling, which were not seen in our patient. Although her initial presentation was of 20-nail dystrophy, which also can be a presentation of nail psoriasis, its rapid evolution with associated nail atrophy and pterygium make it unlikely to be psoriasis in this particular patient.
Patients with pachyonychia congenita – which is a genetic disorder or keratinization caused by mutations on several genes encoding keratin such as K6a, K16, K17, K6b, and possibly K6c – present with nail thickening (pachyonychia) and discoloration of the nails, as well as pincer nails. These patients also present with oral leukokeratosis and focal palmoplantar keratoderma.
The main treatment of lichen planus is potent topical corticosteroids.
For nail disease, topical treatment may not be effective and systemic treatment may be necessary. Systemic corticosteroids have been used in several pediatric series varying from a short course given at a dose of 1- 2 mg/kg per day for 2 weeks to a longer 3-month course followed by tapering.3 There are several protocols of intramuscular triamcinolone at a dose of 0.5 mg/kg in children in once a month injections for about 3 months that have been reported successful with minimal side effects.1 Other medications reported useful in patients with NLP include dapsone and acitretin. Other treatment options include narrow-band UVB and PUVA.3
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Dermatol. 2001 Aug;137(8):1027-32.
2. Ann Dermatol Venereol. 2015 Jan;142(1):21-5.
3. Pediatr Dermatol. 2014 Jan-Feb;31(1):59-67.
4. Dermatological diseases, in “Nails: Diagnosis, Therapy, and Surgery,” 3rd ed. (Oxford: Elsevier Saunders, 2005, p. 105).
At a follow-up visit, a biopsy of the skin on the fingertips was performed, which showed lichenoid lymphocytic inflammatory infiltrate with associated hyperkeratosis, hypergranulosis, and acanthosis.
No fungal elements were seen. The findings were consistent with lichen planus.
The patient was started on hydroxychloroquine. It was recommended she start a 6-week course of oral prednisone, but the mother was opposed to systemic treatment because of potential side effects.
She continued topical betamethasone without much change. Topical tacrolimus later was recommended to use on off days of betamethasone, which led to no improvement. Narrow-band UVB also was started with minimal improvement. Unfortunately,
Nail lichen planus (NLP) in children is not a common condition.1 In a recent series from Chiheb et al., NLP was reported in 90 patients, of which 40% were children; a quarter of the patients reported having extracutaneous involvement as well.2 In another childhood LP series,14 % of the children presented with nail disease.3 It can be a severe disease that, if not treated aggressively, may lead to destruction of the nail bed. This condition seems to be more prevalent in boys than girls and more prevalent in African American children.3 Unfortunately, in this patient’s case, the mother was hesitant to use systemic therapy and aggressive treatment was delayed.
Possible but not clear associations with autoimmune conditions such as vitiligo, autoimmune thyroiditis, myasthenia gravis, alopecia areata, thymoma, autoimmune polyendocrinopathy, atopic dermatitis, and lichen nitidus have been described in children with LP.
The clinical characteristics of NLP include nail plate thinning with longitudinal ridging and fissuring, with or without pterygium; trachyonychia; and erythema of the lunula when the nail matrix is involved. When the nail bed is affected, the patient can present with onycholysis with or without subungual hyperkeratosis and violaceous hue of the nail bed.4 NLP can have three different clinical presentations described by Tosti et al., which include typical NLP, 20‐nail dystrophy (trachyonychia), and idiopathic nail atrophy. Idiopathic nail atrophy is described solely in children as an acute and rapid progression that leads to destruction of the nail within months, which appears to be the clinical presentation in our patient.
The differential diagnosis of nail dystrophy in children includes infectious processes such as onychomycosis, especially when children present with onycholysis and subungual hyperkeratosis. Because of this, it is recommended to perform a nail culture or submit a sample of nail clippings for microscopic evaluation to confirm the diagnosis of onychomycosis prior to starting systemic therapy in children. Fingernail involvement without toenail involvement is an unusual presentation of onychomycosis.
Twenty-nail dystrophy – also known as trachyonychia – can be caused by several inflammatory skin conditions such as lichen planus, psoriasis, eczema, pemphigus vulgaris, and alopecia areata. Clinically, there is uniformly monomorphic thinning of the nail plate with longitudinal ridging without splitting or pterygium.1 This is a benign condition and should not cause scarring. About 10% of the cases of 20-nail dystrophy are caused by lichen planus.
Nail psoriasis is characterized by nail pitting, oil spots on the nail plate, leukonychia, subungual hyperkeratosis, and onycholysis, as well as nail crumbling, which were not seen in our patient. Although her initial presentation was of 20-nail dystrophy, which also can be a presentation of nail psoriasis, its rapid evolution with associated nail atrophy and pterygium make it unlikely to be psoriasis in this particular patient.
Patients with pachyonychia congenita – which is a genetic disorder or keratinization caused by mutations on several genes encoding keratin such as K6a, K16, K17, K6b, and possibly K6c – present with nail thickening (pachyonychia) and discoloration of the nails, as well as pincer nails. These patients also present with oral leukokeratosis and focal palmoplantar keratoderma.
The main treatment of lichen planus is potent topical corticosteroids.
For nail disease, topical treatment may not be effective and systemic treatment may be necessary. Systemic corticosteroids have been used in several pediatric series varying from a short course given at a dose of 1- 2 mg/kg per day for 2 weeks to a longer 3-month course followed by tapering.3 There are several protocols of intramuscular triamcinolone at a dose of 0.5 mg/kg in children in once a month injections for about 3 months that have been reported successful with minimal side effects.1 Other medications reported useful in patients with NLP include dapsone and acitretin. Other treatment options include narrow-band UVB and PUVA.3
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Dermatol. 2001 Aug;137(8):1027-32.
2. Ann Dermatol Venereol. 2015 Jan;142(1):21-5.
3. Pediatr Dermatol. 2014 Jan-Feb;31(1):59-67.
4. Dermatological diseases, in “Nails: Diagnosis, Therapy, and Surgery,” 3rd ed. (Oxford: Elsevier Saunders, 2005, p. 105).
An 8-year-old female child comes to our pediatric dermatology clinic for evaluation of onychomycosis on her fingernails. The mother stated the child started developing funny-looking nails 1 year prior to the visit. It started with only two fingernails affected and now has spread to all her fingernails. Her toenails are not involved.
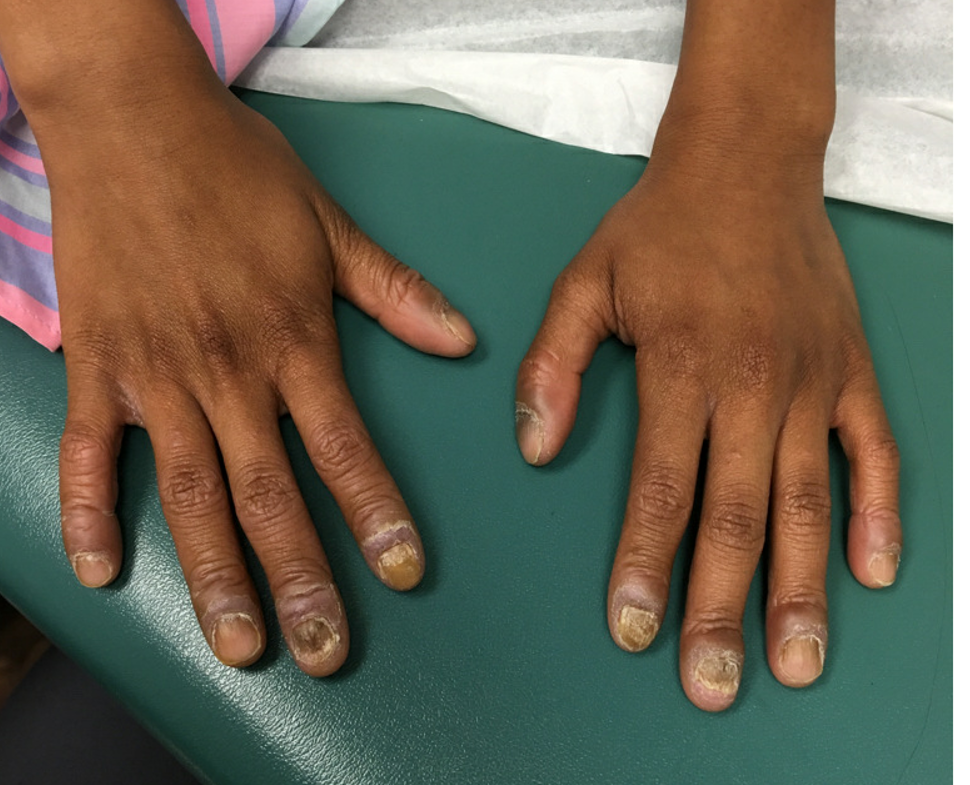
She denied any pain or itching. She initially was treated with topical antifungal medications as well as tea tree oil, apple cider vinegar, and a 6-week course of oral griseofulvin without any improvement. Her nails progressively have gotten much worse. She has no history of atopic dermatitis or any other skin conditions. She denied any joint pain, sun sensitivity, hair loss, or any other symptoms. The mother denied any family history of nail fungus, ringworm, psoriasis, or eczema.
She likes to play basketball and enjoys arts and crafts. She has a cat and a dog; neither of them have any skin problems.
On physical examination, there is nail dystrophy with nail plate thinning and longitudinal fissuring of all fingernails but not of the toenails. She also has hyperpigmented violaceous plaques on the surrounding periungual skin. There are no other skin lesions, and there are no oral or genital lesions. There is no scalp involvement or hair loss. At follow-up several months later, she had complete destruction of the nail plate with scar formation.
A fungal culture was performed, as well as microscopic analysis of the nail with periodic acid fast and giemsa stains, which showed no fungal organisms.
She initially was treated with topical betamethasone twice a day for 6 weeks and then 2 weeks on and 2 weeks off without much change.
Make the Diagnosis - March 2020
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.
Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.
Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
The patient’s biopsy showed sparse and grouped and slightly enlarged atypical stained mononuclear cells in mostly perifollicular areas with focal epidermotropism. CD30 staining was positive. She responded to potent topical steroids.
The etiology of LyP is unknown. It is unclear whether the proliferation of T-cells is a benign and chronic disorder, or an indolent T-cell malignancy.
In addition, 10% of LyP cases are associated with anaplastic large-cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin lymphoma. Borderline cases are those that overlap LyP and lymphoma.
Patients typically present with crops of asymptomatic erythematous to brown papules that may become pustular, vesicular, or necrotic. Lesions tend to resolve within 2-8 weeks with or without scarring. The trunk and extremities are commonly affected. The condition tends to be chronic over months to years. The waxing and waning course is characteristic of LyP. Constitutional symptoms are generally absent in cases not associated with systemic disease.
Histopathologic examination reveals a dense wedge-shaped dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils. Epidermotropism may be present and lymphocytes stain positive for CD30+. Vessels in the dermis may exhibit fibrin deposition and red blood cell extravasation. Histologically, LyP can be classified as Type A to E. These subtypes are determined by the size and type of atypical cells, location and amount of infiltrate, and staining of CD30 and CD8.
The differential diagnosis of LyP includes pityriasis lichenoides, anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, arthropod assault, Langerhans cell histiocytosis, and leukemia cutis. Treatment is symptomatic. Mild forms of LyP can many times be managed with superpotent topical corticosteroids. Bexarotene gel has been used for early lesions. For more widespread or persistent disease, intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate have been reported to be effective. Refractory cases may respond to interferon alpha or oral bexarotene. Routine evaluations are recommended as patients may be at increased risk for the development of lymphoma.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
February 2020
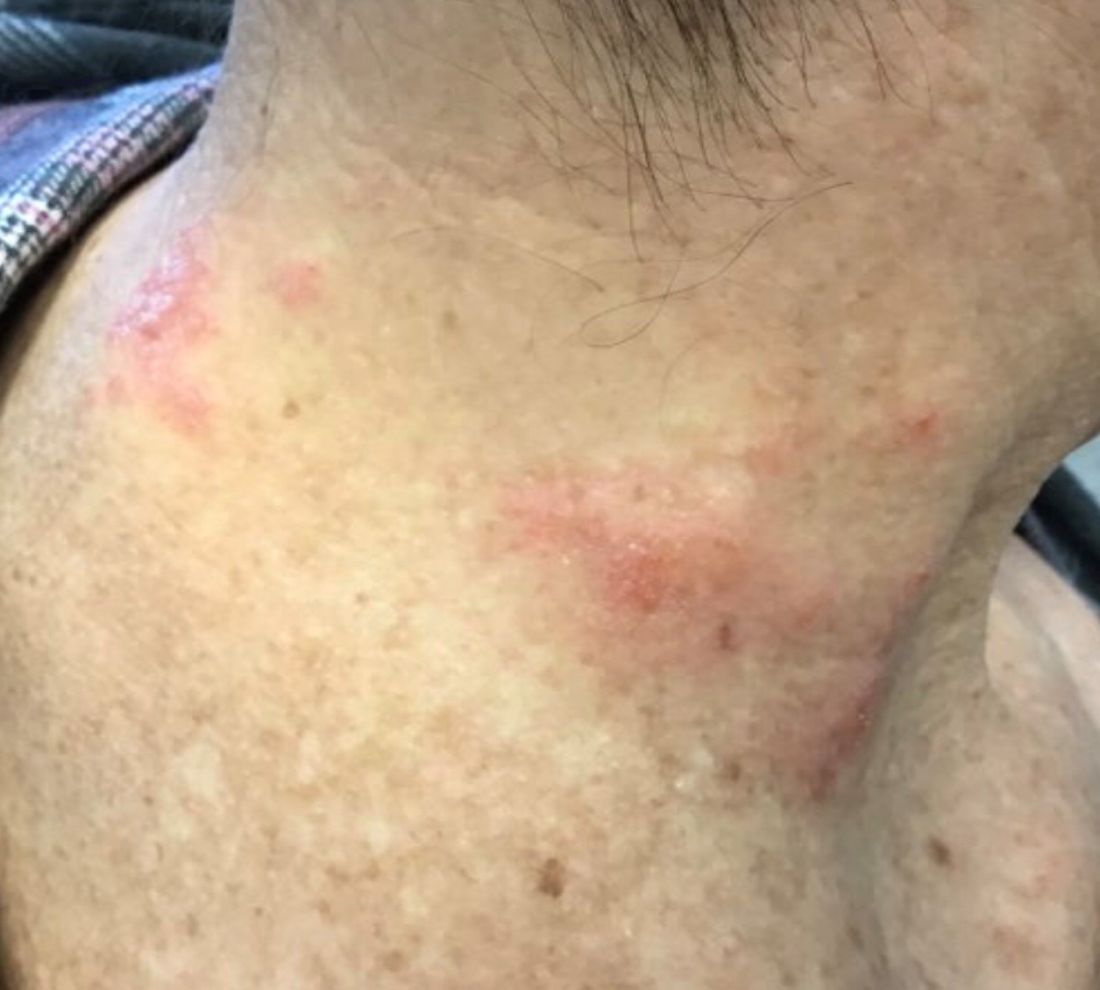
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Subacute cutaneous lupus erythematosus
Subacute cutaneous lupus erythematosus (SCLE) is a type of cutaneous lupus erythematosus that may occur independently of or in combination with systemic lupus erythematosus. About 10%-15% of patients with SCLE will develop systemic lupus erythematosus. White females are more typically affected.
SCLE lesions often present as scaly, annular, or polycyclic scaly patches and plaques with central clearing. They may appear psoriasiform. They heal without atrophy or scarring but may leave dyspigmentation. Follicular plugging is absent. Lesions generally occur on sun exposed areas such as the neck, V of the chest, and upper extremities. Up to 75% of patients may exhibit associated symptoms such as photosensitivity, oral ulcers, and arthritis. Less than 20% of patients will develop internal disease, including nephritis and pulmonary disease. Symptoms of Sjögren’s syndrome and SCLE may overlap in some patients, and will portend higher risk for internal disease.
The differential diagnosis includes eczema, psoriasis, dermatophytosis, granuloma annulare, and erythema annulare centrifugum. Histology reveals epidermal atrophy and keratinocyte apoptosis, with a superficial and perivascular lymphohistiocytic infiltrate in the upper dermis. Interface changes at the dermal-epidermal junction can be seen. Direct immunofluorescence of lesional skin is positive in one-third of cases, often revealing granular deposits of IgG and IgM at the dermal-epidermal junction and around hair follicles (called the lupus-band test). Serology in SCLE may reveal a positive antinuclear antigen test, as well as positive Ro/SSA antigen. Other lupus serologies such as La/SSB, dsDNA, antihistone, and Sm antibodies may be positive, but are less commonly seen.
Several drugs may cause SCLE, such as hydrochlorothiazide, terbinafine, ACE inhibitors, NSAIDs, calcium-channel blockers, interferons, anticonvulsants, griseofulvin, penicillamine, spironolactone, tumor necrosis factor–alpha inhibitors, and statins. Discontinuing the offending medications may clear the lesions, but not always.
Treatment includes sunscreen and avoidance of sun exposure. Potent topical corticosteroids are helpful. If systemic treatment is indicated, antimalarials are first line.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
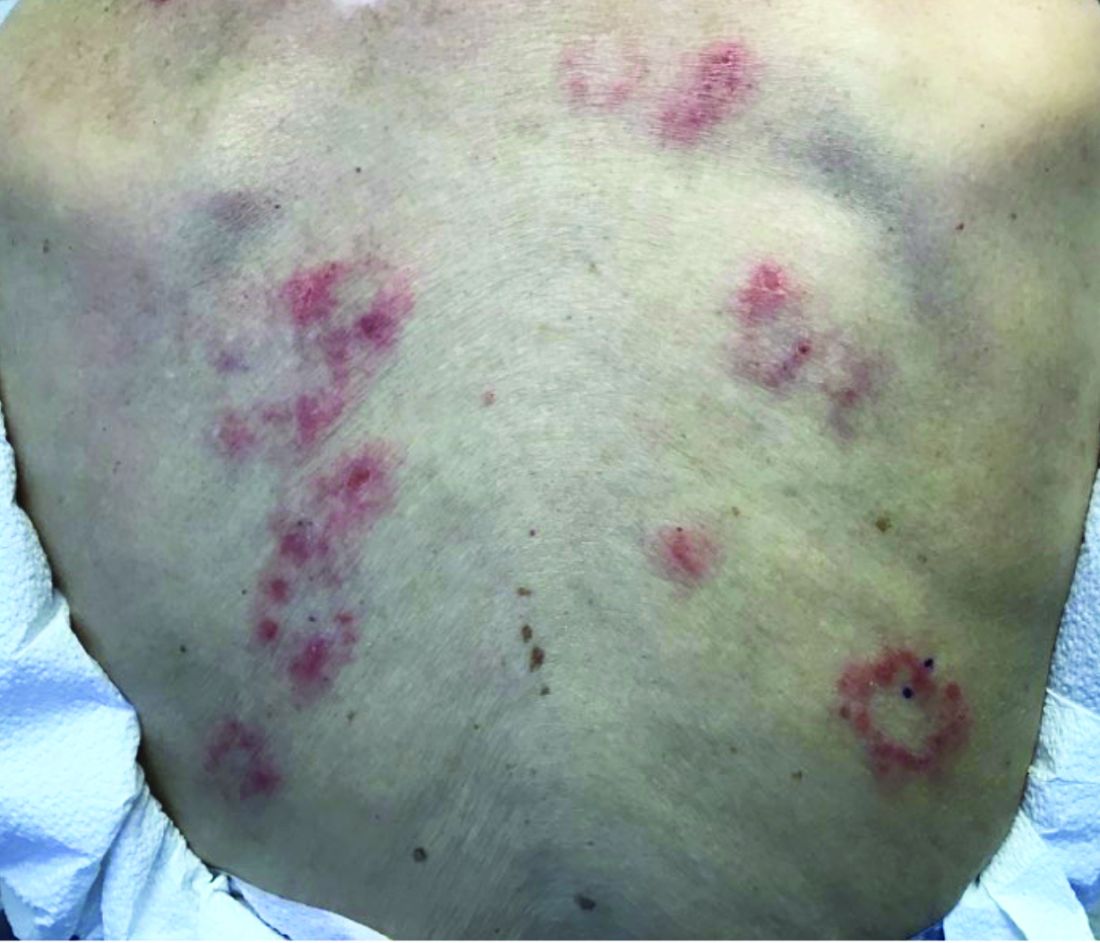
Rash on hands and feet
Lichenoid dermatoses are a heterogeneous group of diseases with varying clinical presentations. The term “lichenoid” refers to the popular lesions of certain skin disorders of which lichen planus (LP) is the prototype. The papules are shiny, flat topped, polygonal, of different sizes, and occur in clusters creating a pattern that resembles lichen growing on a rock. Lichenoid eruptions are quite common in children and can result from many different origins. In most instances the precise mechanism of disease is not known, although it is usually believed to be immunologic in nature. Certain disorders are common in children, whereas others more often affect the adult population.
Lichen striatus, lichen nitidus (LN), and lichen spinulosus are lichenoid lesions that are more common in children than adults.
LN – as seen in the patient described here – is an uncommon benign inflammatory skin disease, primarily of children. Individual lesions are sharply demarcated, pinpoint to pinhead sized, round or polygonal, and strikingly monomorphous in nature. The papules are usually flesh colored, however, the color varies from yellow and brown to violet hues depending on the background color of the patient’s skin. This variation in color is in contrast with LP which is characteristically violaceous. The surfaces of the papules are flat, shiny, and slightly elevated. They may have a fine scale or a hyperkeratotic plug. The lesions tend to occur in groups, primarily on the abdomen, chest, glans penis, and upper extremities. The Koebner phenomenon is observed and is a hallmark for the disorder. LN is generally asymptomatic, unlike LP, which is exceedingly pruritic.
The cause of LN is unknown; however, it has been proposed that LN, in particular generalized LN, may be associated with immune alterations in the patient. The course of LN is slowly progressive with a tendency toward remission. The lesions can remain stationary for years; however, they sometimes disappear spontaneously and completely.
The differential diagnosis of LN beyond the entities discussed above includes frictional lichenoid eruption, lichenoid drug eruption, LP, and keratosis pilaris.
LP is the classic lichenoid eruption. It is rare in children and occurs most frequently in individuals aged 30-60 years. LP usually manifests as an extremely pruritic eruption of flat-topped polygonal and violaceous papules that often have fine linear white scales known as Wickham striae. The distribution is usually bilateral and symmetric with most of the papules and plaques located on the legs, flexor wrists, neck, and genitalia. The lesions may exhibit the Koebner phenomenon, appearing in a linear pattern along the site of a scratch. Generally, in childhood cases there is reported itching, and oral and nail lesions are less common.
Frictional lichenoid eruption occurs in childhood. The lesions consist of lichenoid papules with regular borders 1-2 mm in diameter that generally are asymptomatic, although they may be mildly pruritic. The papules are found in a very characteristic distribution with almost exclusive involvement of the backs of the hands, fingers, elbows, and knees with occasional involvement of the extensor forearms and cheeks. This disorder occurs in predisposed children who have been exposed to significant frictional force during play, and typically resolves spontaneously after removal of the stimulus.
Keratosis pilaris is a rash that usually is found on the outer areas of the upper arms, upper thighs, buttocks, and cheeks. It consists of small bumps that are flesh colored to red. The bumps generally don’t hurt or itch.
The lack of symptoms and spontaneous healing have rendered treatment unnecessary in most cases. LN generally is self-limiting, thus treatment may not be necessary. However, topical treatment with mid- to high-potency corticosteroids has hastened resolution of lesions in some children, as have topical dinitrochlorobenzene and systemic treatment with psoralens, astemizole, etretinate, and psoralen-UVA.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Neither Dr. Eichenfield nor Dr. Bhatti has any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
Pickert A. Cutis. 2012 Sep;90(3):E1-3. https://mdedge-files-live.s3.us-east-2.amazonaws.com/files/s3fs-public/Document/September-2017/0900300E1.pdf Tziotzios C et al. J Am Acad Dermatol. 2018 Nov;79(5):789-804. Tilly JJ et al. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
Lichenoid dermatoses are a heterogeneous group of diseases with varying clinical presentations. The term “lichenoid” refers to the popular lesions of certain skin disorders of which lichen planus (LP) is the prototype. The papules are shiny, flat topped, polygonal, of different sizes, and occur in clusters creating a pattern that resembles lichen growing on a rock. Lichenoid eruptions are quite common in children and can result from many different origins. In most instances the precise mechanism of disease is not known, although it is usually believed to be immunologic in nature. Certain disorders are common in children, whereas others more often affect the adult population.
Lichen striatus, lichen nitidus (LN), and lichen spinulosus are lichenoid lesions that are more common in children than adults.
LN – as seen in the patient described here – is an uncommon benign inflammatory skin disease, primarily of children. Individual lesions are sharply demarcated, pinpoint to pinhead sized, round or polygonal, and strikingly monomorphous in nature. The papules are usually flesh colored, however, the color varies from yellow and brown to violet hues depending on the background color of the patient’s skin. This variation in color is in contrast with LP which is characteristically violaceous. The surfaces of the papules are flat, shiny, and slightly elevated. They may have a fine scale or a hyperkeratotic plug. The lesions tend to occur in groups, primarily on the abdomen, chest, glans penis, and upper extremities. The Koebner phenomenon is observed and is a hallmark for the disorder. LN is generally asymptomatic, unlike LP, which is exceedingly pruritic.
The cause of LN is unknown; however, it has been proposed that LN, in particular generalized LN, may be associated with immune alterations in the patient. The course of LN is slowly progressive with a tendency toward remission. The lesions can remain stationary for years; however, they sometimes disappear spontaneously and completely.
The differential diagnosis of LN beyond the entities discussed above includes frictional lichenoid eruption, lichenoid drug eruption, LP, and keratosis pilaris.
LP is the classic lichenoid eruption. It is rare in children and occurs most frequently in individuals aged 30-60 years. LP usually manifests as an extremely pruritic eruption of flat-topped polygonal and violaceous papules that often have fine linear white scales known as Wickham striae. The distribution is usually bilateral and symmetric with most of the papules and plaques located on the legs, flexor wrists, neck, and genitalia. The lesions may exhibit the Koebner phenomenon, appearing in a linear pattern along the site of a scratch. Generally, in childhood cases there is reported itching, and oral and nail lesions are less common.
Frictional lichenoid eruption occurs in childhood. The lesions consist of lichenoid papules with regular borders 1-2 mm in diameter that generally are asymptomatic, although they may be mildly pruritic. The papules are found in a very characteristic distribution with almost exclusive involvement of the backs of the hands, fingers, elbows, and knees with occasional involvement of the extensor forearms and cheeks. This disorder occurs in predisposed children who have been exposed to significant frictional force during play, and typically resolves spontaneously after removal of the stimulus.
Keratosis pilaris is a rash that usually is found on the outer areas of the upper arms, upper thighs, buttocks, and cheeks. It consists of small bumps that are flesh colored to red. The bumps generally don’t hurt or itch.
The lack of symptoms and spontaneous healing have rendered treatment unnecessary in most cases. LN generally is self-limiting, thus treatment may not be necessary. However, topical treatment with mid- to high-potency corticosteroids has hastened resolution of lesions in some children, as have topical dinitrochlorobenzene and systemic treatment with psoralens, astemizole, etretinate, and psoralen-UVA.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Neither Dr. Eichenfield nor Dr. Bhatti has any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
Pickert A. Cutis. 2012 Sep;90(3):E1-3. https://mdedge-files-live.s3.us-east-2.amazonaws.com/files/s3fs-public/Document/September-2017/0900300E1.pdf Tziotzios C et al. J Am Acad Dermatol. 2018 Nov;79(5):789-804. Tilly JJ et al. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
Lichenoid dermatoses are a heterogeneous group of diseases with varying clinical presentations. The term “lichenoid” refers to the popular lesions of certain skin disorders of which lichen planus (LP) is the prototype. The papules are shiny, flat topped, polygonal, of different sizes, and occur in clusters creating a pattern that resembles lichen growing on a rock. Lichenoid eruptions are quite common in children and can result from many different origins. In most instances the precise mechanism of disease is not known, although it is usually believed to be immunologic in nature. Certain disorders are common in children, whereas others more often affect the adult population.
Lichen striatus, lichen nitidus (LN), and lichen spinulosus are lichenoid lesions that are more common in children than adults.
LN – as seen in the patient described here – is an uncommon benign inflammatory skin disease, primarily of children. Individual lesions are sharply demarcated, pinpoint to pinhead sized, round or polygonal, and strikingly monomorphous in nature. The papules are usually flesh colored, however, the color varies from yellow and brown to violet hues depending on the background color of the patient’s skin. This variation in color is in contrast with LP which is characteristically violaceous. The surfaces of the papules are flat, shiny, and slightly elevated. They may have a fine scale or a hyperkeratotic plug. The lesions tend to occur in groups, primarily on the abdomen, chest, glans penis, and upper extremities. The Koebner phenomenon is observed and is a hallmark for the disorder. LN is generally asymptomatic, unlike LP, which is exceedingly pruritic.
The cause of LN is unknown; however, it has been proposed that LN, in particular generalized LN, may be associated with immune alterations in the patient. The course of LN is slowly progressive with a tendency toward remission. The lesions can remain stationary for years; however, they sometimes disappear spontaneously and completely.
The differential diagnosis of LN beyond the entities discussed above includes frictional lichenoid eruption, lichenoid drug eruption, LP, and keratosis pilaris.
LP is the classic lichenoid eruption. It is rare in children and occurs most frequently in individuals aged 30-60 years. LP usually manifests as an extremely pruritic eruption of flat-topped polygonal and violaceous papules that often have fine linear white scales known as Wickham striae. The distribution is usually bilateral and symmetric with most of the papules and plaques located on the legs, flexor wrists, neck, and genitalia. The lesions may exhibit the Koebner phenomenon, appearing in a linear pattern along the site of a scratch. Generally, in childhood cases there is reported itching, and oral and nail lesions are less common.
Frictional lichenoid eruption occurs in childhood. The lesions consist of lichenoid papules with regular borders 1-2 mm in diameter that generally are asymptomatic, although they may be mildly pruritic. The papules are found in a very characteristic distribution with almost exclusive involvement of the backs of the hands, fingers, elbows, and knees with occasional involvement of the extensor forearms and cheeks. This disorder occurs in predisposed children who have been exposed to significant frictional force during play, and typically resolves spontaneously after removal of the stimulus.
Keratosis pilaris is a rash that usually is found on the outer areas of the upper arms, upper thighs, buttocks, and cheeks. It consists of small bumps that are flesh colored to red. The bumps generally don’t hurt or itch.
The lack of symptoms and spontaneous healing have rendered treatment unnecessary in most cases. LN generally is self-limiting, thus treatment may not be necessary. However, topical treatment with mid- to high-potency corticosteroids has hastened resolution of lesions in some children, as have topical dinitrochlorobenzene and systemic treatment with psoralens, astemizole, etretinate, and psoralen-UVA.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Bhatti is a research fellow in pediatric dermatology at Rady Children’s Hospital and the University of California, San Diego. Neither Dr. Eichenfield nor Dr. Bhatti has any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
Pickert A. Cutis. 2012 Sep;90(3):E1-3. https://mdedge-files-live.s3.us-east-2.amazonaws.com/files/s3fs-public/Document/September-2017/0900300E1.pdf Tziotzios C et al. J Am Acad Dermatol. 2018 Nov;79(5):789-804. Tilly JJ et al. J Am Acad Dermatol. 2004 Oct;51(4):606-24.
A 9-year-old healthy Kuwaiti male with no significant past medical history presents with a rash on his hands and feet that has been present for 3 years.
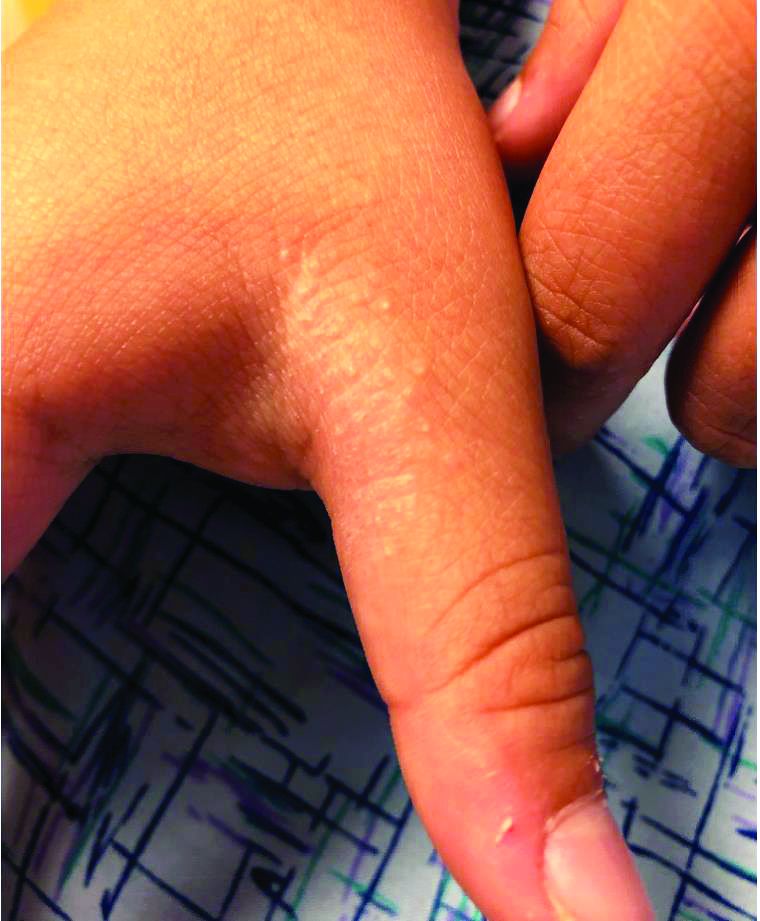
His mother reports that he has been seen by dermatologists in various countries and was last seen by a dermatologist in Kuwait 3 years ago. At that time, he was told that it was dryness and advised to not shower daily. Since then he has been taking showers three times weekly and using Cetaphil once weekly without improvement. He was seen by his pediatrician 6 months ago, diagnosed with xerosis, and was given hydrocortisone 2.5% to use twice daily, again without any improvement.
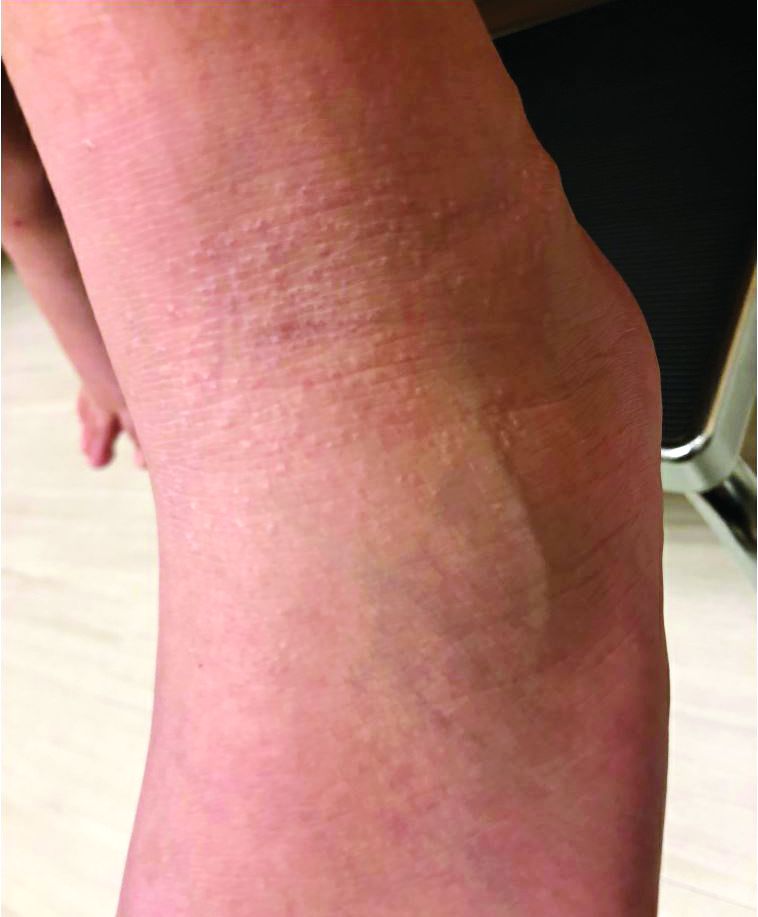
The rash is not itchy, and he has no oral lesions or nail involvement. Exam revealed lichenoid papules on bilateral dorsal hands and feet, bilateral upper arms, bilateral axilla, lower abdomen, and left upper chest.
Make the Diagnosis
A skin biopsy of one of the lesions showed granulomatous inflammation composed of lymphocytes, macrophages, and giant cells around hair follicles with negative mycobacterium stains and fungal stains, consistent with granulomatous periorificial dermatitis. Tissue cultures from a skin biopsy for aerobic bacteria, mycobacteria, and fungus all were negative.
The patient initially was treated with erythromycin, but after 2 weeks, he reported abdominal pain and nausea and was unable to tolerate the medication. He was switched to clarithromycin, which he took for 6 weeks with clearance of the lesions.
A year later, some of the lesions recurred. He was treated again with clarithromycin and the lesions resolved.
Childhood granulomatous periorificial dermatitis (CGPD) is a benign skin eruption that occurs in prepubertal children. It also has been called facial Afro-Caribbean childhood eruption (FACE), and it tends to occur most commonly in children of darker skin types.1 but there are some cases reported of extra facial involvement.2 The lesions usually are not symptomatic, and they are more common in boys. The cause of this condition is not known, but possible triggers could include prior exposure to topical and systemic corticosteroids, as well as exposure to certain allergens such as formaldehyde.1
In histopathology, the lesions are characterized by granulomatous infiltrates around the hair follicles and the upper dermis. The granulomas are formed of macrophages, lymphocytes, and giant cell, as were seen in our patient.3
Several conditions can look very similar to CGPD; these include sarcoidosis, lupus miliaris disseminatus faciei (LMDF), and granulomatous rosacea.
Sarcoidosis is a rare condition in children, and the lesions can be similar to the ones seen in our patient. Patients with sarcoidosis usually present with other systemic symptoms including fever, weight loss, respiratory symptoms, and fatigue; none of these were seen in our patient. Under the microscope, the lesions are characterized by “naked granulomas” instead of the inflammatory granulomas seen on our patient.
Lupus miliaris disseminatus faciei is a rare inflammatory skin condition commonly seen in young adults and is thought to be a variant of rosacea. It is characterized by skin-color to pink to yellow dome-shaped papules on the central face. Histologically, the lesions present as dermal epithelioid cell granulomas with central necrosis and surrounding lymphocytic infiltrate with multinucleate giant cells.4
Granulomatous rosacea and CGPD are considered two separate entities. Granulomatous rosacea tends to have a more chronic course, is not that common in children, and clinically presents with pustules, papules, and cysts around the eyes and cheeks.
Infectious processes like tuberculosis and fungal infections were ruled out in our patient with cultures and histopathology. Allergic contact dermatitis on the face can present with skin-color to pink papules, but they usually are very pruritic and improve with topical corticosteroids, while these medications can worsen CGPD.
CGPD can be a self-limiting condition. When mild, it can be treated with topical metronidazole, topical erythromycin, topical clindamycin solution, or pimecrolimus. Our patient failed treatment with pimecrolimus. For severe presentations, oral tetracyclines, erythromycin, and other macrolides, metronidazole, and oral isotretinoin can help clear the lesions.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Ann Dermatol. 2011 Aug;23(3):386-8.
2. Int J Dermatol. 2007 Feb;46(2):143-5.
3. J Cutan Med Surg. 2009 Feb 28;13(2):115-8.
4. An Bras Dermatol. 2017 Nov-Dec;92(6):851-3.
5. Indian Dermatol Online J. 2018 Jan-Feb; 9(1):68-70.
A skin biopsy of one of the lesions showed granulomatous inflammation composed of lymphocytes, macrophages, and giant cells around hair follicles with negative mycobacterium stains and fungal stains, consistent with granulomatous periorificial dermatitis. Tissue cultures from a skin biopsy for aerobic bacteria, mycobacteria, and fungus all were negative.
The patient initially was treated with erythromycin, but after 2 weeks, he reported abdominal pain and nausea and was unable to tolerate the medication. He was switched to clarithromycin, which he took for 6 weeks with clearance of the lesions.
A year later, some of the lesions recurred. He was treated again with clarithromycin and the lesions resolved.
Childhood granulomatous periorificial dermatitis (CGPD) is a benign skin eruption that occurs in prepubertal children. It also has been called facial Afro-Caribbean childhood eruption (FACE), and it tends to occur most commonly in children of darker skin types.1 but there are some cases reported of extra facial involvement.2 The lesions usually are not symptomatic, and they are more common in boys. The cause of this condition is not known, but possible triggers could include prior exposure to topical and systemic corticosteroids, as well as exposure to certain allergens such as formaldehyde.1
In histopathology, the lesions are characterized by granulomatous infiltrates around the hair follicles and the upper dermis. The granulomas are formed of macrophages, lymphocytes, and giant cell, as were seen in our patient.3
Several conditions can look very similar to CGPD; these include sarcoidosis, lupus miliaris disseminatus faciei (LMDF), and granulomatous rosacea.
Sarcoidosis is a rare condition in children, and the lesions can be similar to the ones seen in our patient. Patients with sarcoidosis usually present with other systemic symptoms including fever, weight loss, respiratory symptoms, and fatigue; none of these were seen in our patient. Under the microscope, the lesions are characterized by “naked granulomas” instead of the inflammatory granulomas seen on our patient.
Lupus miliaris disseminatus faciei is a rare inflammatory skin condition commonly seen in young adults and is thought to be a variant of rosacea. It is characterized by skin-color to pink to yellow dome-shaped papules on the central face. Histologically, the lesions present as dermal epithelioid cell granulomas with central necrosis and surrounding lymphocytic infiltrate with multinucleate giant cells.4
Granulomatous rosacea and CGPD are considered two separate entities. Granulomatous rosacea tends to have a more chronic course, is not that common in children, and clinically presents with pustules, papules, and cysts around the eyes and cheeks.
Infectious processes like tuberculosis and fungal infections were ruled out in our patient with cultures and histopathology. Allergic contact dermatitis on the face can present with skin-color to pink papules, but they usually are very pruritic and improve with topical corticosteroids, while these medications can worsen CGPD.
CGPD can be a self-limiting condition. When mild, it can be treated with topical metronidazole, topical erythromycin, topical clindamycin solution, or pimecrolimus. Our patient failed treatment with pimecrolimus. For severe presentations, oral tetracyclines, erythromycin, and other macrolides, metronidazole, and oral isotretinoin can help clear the lesions.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Ann Dermatol. 2011 Aug;23(3):386-8.
2. Int J Dermatol. 2007 Feb;46(2):143-5.
3. J Cutan Med Surg. 2009 Feb 28;13(2):115-8.
4. An Bras Dermatol. 2017 Nov-Dec;92(6):851-3.
5. Indian Dermatol Online J. 2018 Jan-Feb; 9(1):68-70.
A skin biopsy of one of the lesions showed granulomatous inflammation composed of lymphocytes, macrophages, and giant cells around hair follicles with negative mycobacterium stains and fungal stains, consistent with granulomatous periorificial dermatitis. Tissue cultures from a skin biopsy for aerobic bacteria, mycobacteria, and fungus all were negative.
The patient initially was treated with erythromycin, but after 2 weeks, he reported abdominal pain and nausea and was unable to tolerate the medication. He was switched to clarithromycin, which he took for 6 weeks with clearance of the lesions.
A year later, some of the lesions recurred. He was treated again with clarithromycin and the lesions resolved.
Childhood granulomatous periorificial dermatitis (CGPD) is a benign skin eruption that occurs in prepubertal children. It also has been called facial Afro-Caribbean childhood eruption (FACE), and it tends to occur most commonly in children of darker skin types.1 but there are some cases reported of extra facial involvement.2 The lesions usually are not symptomatic, and they are more common in boys. The cause of this condition is not known, but possible triggers could include prior exposure to topical and systemic corticosteroids, as well as exposure to certain allergens such as formaldehyde.1
In histopathology, the lesions are characterized by granulomatous infiltrates around the hair follicles and the upper dermis. The granulomas are formed of macrophages, lymphocytes, and giant cell, as were seen in our patient.3
Several conditions can look very similar to CGPD; these include sarcoidosis, lupus miliaris disseminatus faciei (LMDF), and granulomatous rosacea.
Sarcoidosis is a rare condition in children, and the lesions can be similar to the ones seen in our patient. Patients with sarcoidosis usually present with other systemic symptoms including fever, weight loss, respiratory symptoms, and fatigue; none of these were seen in our patient. Under the microscope, the lesions are characterized by “naked granulomas” instead of the inflammatory granulomas seen on our patient.
Lupus miliaris disseminatus faciei is a rare inflammatory skin condition commonly seen in young adults and is thought to be a variant of rosacea. It is characterized by skin-color to pink to yellow dome-shaped papules on the central face. Histologically, the lesions present as dermal epithelioid cell granulomas with central necrosis and surrounding lymphocytic infiltrate with multinucleate giant cells.4
Granulomatous rosacea and CGPD are considered two separate entities. Granulomatous rosacea tends to have a more chronic course, is not that common in children, and clinically presents with pustules, papules, and cysts around the eyes and cheeks.
Infectious processes like tuberculosis and fungal infections were ruled out in our patient with cultures and histopathology. Allergic contact dermatitis on the face can present with skin-color to pink papules, but they usually are very pruritic and improve with topical corticosteroids, while these medications can worsen CGPD.
CGPD can be a self-limiting condition. When mild, it can be treated with topical metronidazole, topical erythromycin, topical clindamycin solution, or pimecrolimus. Our patient failed treatment with pimecrolimus. For severe presentations, oral tetracyclines, erythromycin, and other macrolides, metronidazole, and oral isotretinoin can help clear the lesions.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Ann Dermatol. 2011 Aug;23(3):386-8.
2. Int J Dermatol. 2007 Feb;46(2):143-5.
3. J Cutan Med Surg. 2009 Feb 28;13(2):115-8.
4. An Bras Dermatol. 2017 Nov-Dec;92(6):851-3.
5. Indian Dermatol Online J. 2018 Jan-Feb; 9(1):68-70.
An 8-year-old African American male presented to our pediatric dermatology clinic for evaluation of a 3-month history of flesh-colored bumps on the face. According to the patient's mother, the lesions started with small pimple-like lesions around the nose and then spread to the whole face. Some lesions were crusty and somewhat itchy. He was treated with cephalexin and pimecrolimus with no improvement. The mother was very concerned because the lesions were close to the eyes and spreading.
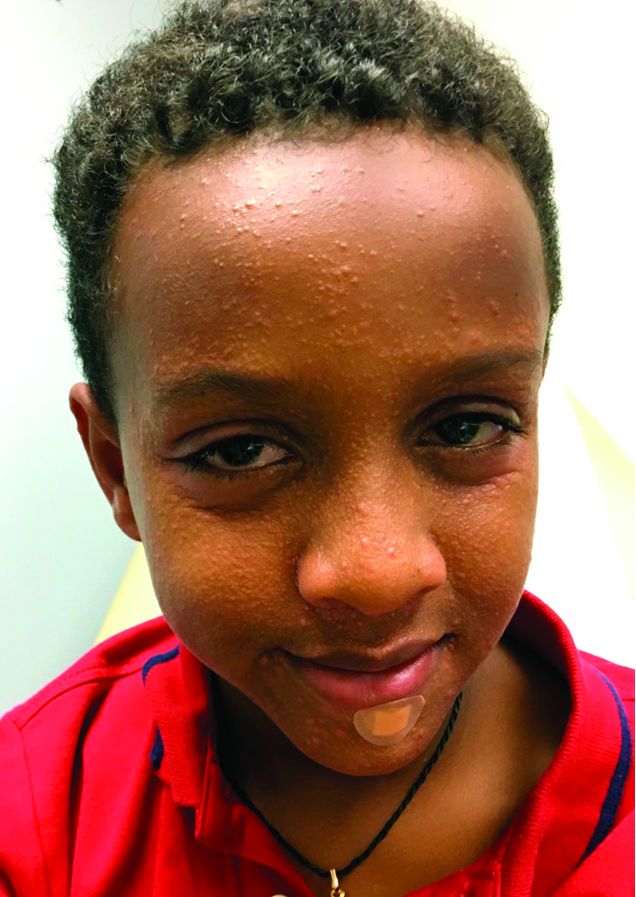
He had no fevers, arthritis, or upper respiratory or gastrointestinal symptoms. He recently came back from a trip to Africa to visit his family. No other family members were affected. He used some new soaps, sunscreens, and moisturizers while he was in Africa.
On physical examination, the boy was in no acute distress. He had multiple flesh-colored papules on the face, especially around the eyes, nose, and mouth, where some lesions appeared crusted. There were no other skin lesions elsewhere on his body. There was no lymphadenopathy or hepatosplenomegaly.
Progressive, pruritic eruption of firm, skin-colored papules
Scleromyxedema, or generalized lichen myxedematosus, is a primary cutaneous mucinosis with unknown pathogenesis characterized by generalized firm, skin-colored papules and is commonly associated with an underlying monoclonal gammopathy (usually Ig-gamma paraproteinemia).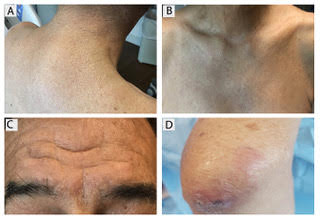
Scleromyxedema may have associated internal involvement, including neurologic, gastrointestinal, pulmonary, renal, cardiovascular, ophthalmological, or musculoskeletal. Histopathology demonstrates mucin in the dermis seen with Alcian blue staining, proliferation of fibroblasts, and increased collagen deposition.
The condition is chronic and progressive. Intravenous immunoglobulin is considered first-line treatment. Thalidomide and corticosteroids have been reported to also be efficacious.
It is associated with hematologic disorders, including IgA monoclonal gammopathy, as well as myeloproliferative disorders, leukemia, infections, and inflammatory bowel disease. Although its pathophysiology is not well understood, vascular immune complex deposition, repetitive inflammation, and subsequent fibrosis may play a role. On histology, there is leukocytoclastic vasculitis with polymorphonuclear cell infiltrate and fibrin deposition in the superficial and mid-dermis and onion-skin fibrosis.
EED often self-resolves within 5-10 years, although it can become chronic and recurrent. Dapsone, niacinamide, antimalarials, NSAIDs, tetracyclines, corticosteroids, colchicine, and plasmapheresis are reported treatments. This patient’s EED was recalcitrant to prednisone and responded to colchicine.
Scleromyxedema and EED are both rare, distinct cutaneous entities associated with different underlying paraproteinemias and to the best of our knowledge, have not been previously reported to coexist in a single patient.
This case and the photos were submitted by Rachel Fayne, BA; Yumeng Li, MD, MS; Fabrizio Galimberti, MD, PhD; and Brian Morrison, MD, of the department of dermatology and cutaneous surgery at the University of Miami.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Scleromyxedema, or generalized lichen myxedematosus, is a primary cutaneous mucinosis with unknown pathogenesis characterized by generalized firm, skin-colored papules and is commonly associated with an underlying monoclonal gammopathy (usually Ig-gamma paraproteinemia).
Scleromyxedema may have associated internal involvement, including neurologic, gastrointestinal, pulmonary, renal, cardiovascular, ophthalmological, or musculoskeletal. Histopathology demonstrates mucin in the dermis seen with Alcian blue staining, proliferation of fibroblasts, and increased collagen deposition.
The condition is chronic and progressive. Intravenous immunoglobulin is considered first-line treatment. Thalidomide and corticosteroids have been reported to also be efficacious.
It is associated with hematologic disorders, including IgA monoclonal gammopathy, as well as myeloproliferative disorders, leukemia, infections, and inflammatory bowel disease. Although its pathophysiology is not well understood, vascular immune complex deposition, repetitive inflammation, and subsequent fibrosis may play a role. On histology, there is leukocytoclastic vasculitis with polymorphonuclear cell infiltrate and fibrin deposition in the superficial and mid-dermis and onion-skin fibrosis.
EED often self-resolves within 5-10 years, although it can become chronic and recurrent. Dapsone, niacinamide, antimalarials, NSAIDs, tetracyclines, corticosteroids, colchicine, and plasmapheresis are reported treatments. This patient’s EED was recalcitrant to prednisone and responded to colchicine.
Scleromyxedema and EED are both rare, distinct cutaneous entities associated with different underlying paraproteinemias and to the best of our knowledge, have not been previously reported to coexist in a single patient.
This case and the photos were submitted by Rachel Fayne, BA; Yumeng Li, MD, MS; Fabrizio Galimberti, MD, PhD; and Brian Morrison, MD, of the department of dermatology and cutaneous surgery at the University of Miami.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Scleromyxedema, or generalized lichen myxedematosus, is a primary cutaneous mucinosis with unknown pathogenesis characterized by generalized firm, skin-colored papules and is commonly associated with an underlying monoclonal gammopathy (usually Ig-gamma paraproteinemia).
Scleromyxedema may have associated internal involvement, including neurologic, gastrointestinal, pulmonary, renal, cardiovascular, ophthalmological, or musculoskeletal. Histopathology demonstrates mucin in the dermis seen with Alcian blue staining, proliferation of fibroblasts, and increased collagen deposition.
The condition is chronic and progressive. Intravenous immunoglobulin is considered first-line treatment. Thalidomide and corticosteroids have been reported to also be efficacious.
It is associated with hematologic disorders, including IgA monoclonal gammopathy, as well as myeloproliferative disorders, leukemia, infections, and inflammatory bowel disease. Although its pathophysiology is not well understood, vascular immune complex deposition, repetitive inflammation, and subsequent fibrosis may play a role. On histology, there is leukocytoclastic vasculitis with polymorphonuclear cell infiltrate and fibrin deposition in the superficial and mid-dermis and onion-skin fibrosis.
EED often self-resolves within 5-10 years, although it can become chronic and recurrent. Dapsone, niacinamide, antimalarials, NSAIDs, tetracyclines, corticosteroids, colchicine, and plasmapheresis are reported treatments. This patient’s EED was recalcitrant to prednisone and responded to colchicine.
Scleromyxedema and EED are both rare, distinct cutaneous entities associated with different underlying paraproteinemias and to the best of our knowledge, have not been previously reported to coexist in a single patient.
This case and the photos were submitted by Rachel Fayne, BA; Yumeng Li, MD, MS; Fabrizio Galimberti, MD, PhD; and Brian Morrison, MD, of the department of dermatology and cutaneous surgery at the University of Miami.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
