User login
Nephrogenic Systemic Fibrosis in the Setting of Transient Renal Insufficiency
Nephrogenic systemic fibrosis (NSF) is a rare debilitating disorder characterized by dermal plaques, joint contractures, and fibrosis of the skin with possible involvement of muscles and internal organs.1-3 Originally identified in 1997 as nephrogenic fibrosing dermopathy to describe its characteristic cutaneous thickening and hardening, the name was changed to NSF to more accurately reflect the noncutaneous manifestations present in other organ tissues.2,4,5 Nephrogenic systemic fibrosis occurs in patients with a history of renal insufficiency and exposure to gadolinium-based contrast agents (GBCAs) used in magnetic resonance angiography and magnetic resonance imaging. There is no predilection for age, sex, or ethnicity.
Nephrogenic systemic fibrosis may develop over a period of days to several weeks. However, there have been cases of NSF developing 10 years after gadolinium exposure.2 In most cases, patients have a history of severe chronic renal disease requiring hemodialysis. There have been a few reported cases of NSF occurring in patients with resolved acute kidney injury or resolved acute on chronic renal disease.1,6-10 We present a case of NSF occurring in a patient with resolved transient renal insufficiency and no history of chronic renal disease.
Case Report
A 68-year-old woman presented with new dark, painless, pink plaques on the right thigh and calf. The patient stated the condition started and got worse after she was hospitalized 12 years prior for lower extremity cellulitis, sepsis, and acute renal failure. The patient developed complications during that hospital stay and underwent a renal biopsy and renal artery embolization requiring use of a GBCA. After the procedure, she noticed skin hardening in the extremities and decreased mobility in both legs while she was still in the hospital. It was thought that the lower leg changes were due to cellulitis. Therefore, when the renal issues resolved, she was discharged. Her skin and joint changes remained stable until 6 years later when she noticed new pink plaques appearing. Her medical history was positive for breast cancer, which was surgically and medically treated 16 years prior to presentation.
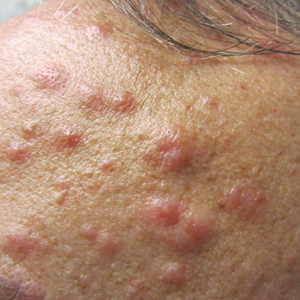
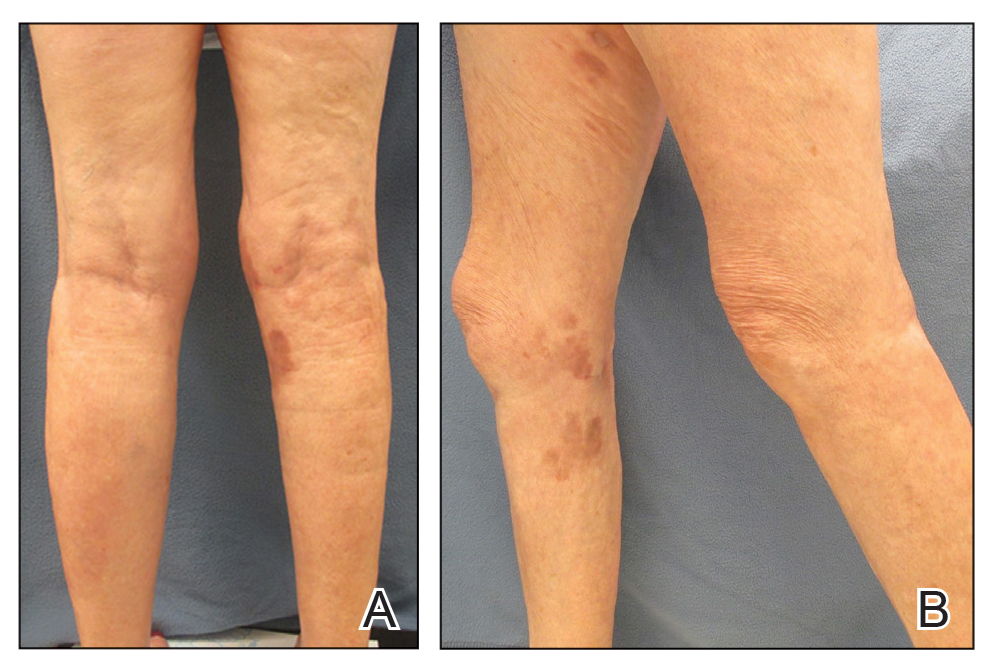
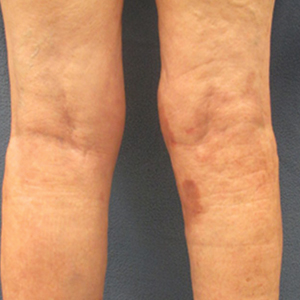
On presentation, physical examination revealed dark pink, hyperpigmented plaques on the right leg and a firm hypopigmented broad linear plaque on the right forearm. Palpation of the legs revealed thickened sclerotic plaques from the thighs down to the ankles (Figure 1). The plaques were not tender to palpation. She did have a decreased range of motion with eversion and inversion of the feet and ankles.
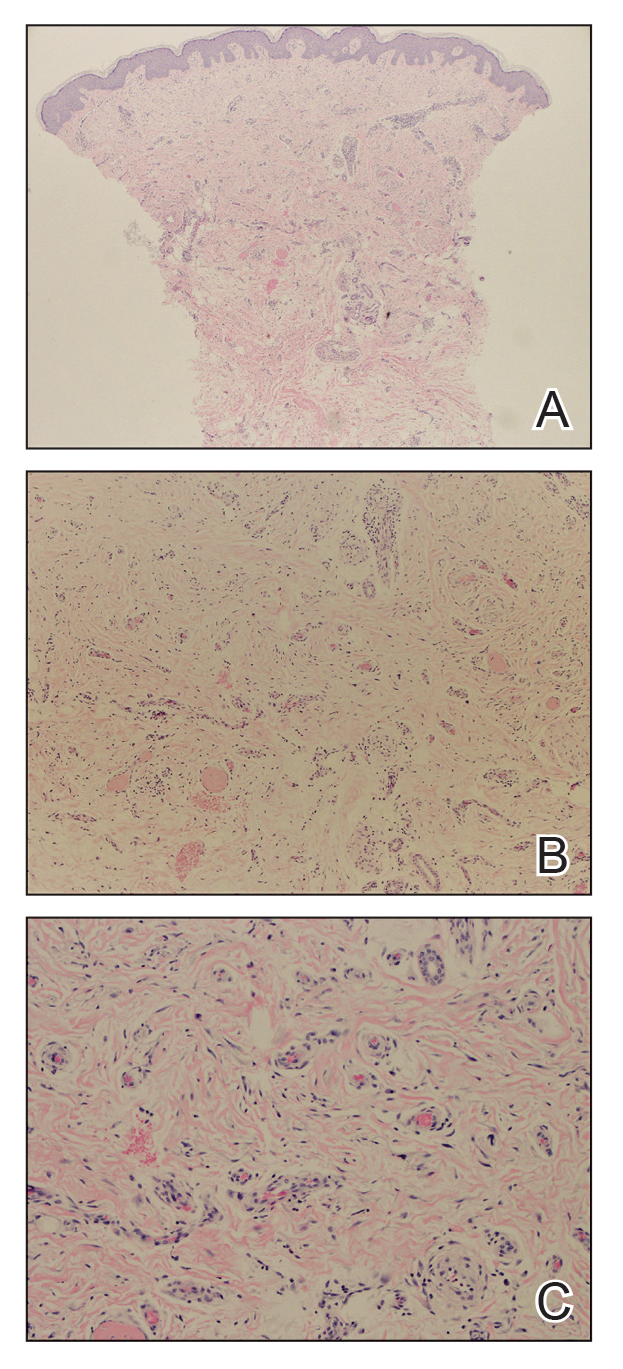
Biopsies from the right medial leg and right volar forearm showed increased bland dermal spindle cellularity associated with numerous round to ovoid osteoid aggregates encircling elastic fibers and surrounded by osteoblasts (Figure 2). CD34 immunohistochemistry showed general retention of staining within the dermal fibroblast population, and elastin stain showed general retention of elastic fiber bundles and thickening.
Laboratory workup included a complete blood cell count, comprehensive metabolic panel, thyroid-stimulating hormone level, and serum protein electrophoresis; results were all within reference range. The patient also had a urine element profile from an outside provider 1 month after presenting to our office that showed an elevated urine gadolinium level of 4.146 μg/g (reference range, 0–0.019 μg/g). The patient’s skin lesions have remained stable, and she is now working with physical therapy to help with her range of motion.
Comment
Gadolinium Causing Fibrosis—The incidence of NSF varies according to the severity of renal impairment, dosage level of GBCA used, and the history of GBCA use. In patients with normal renal function, gadolinium is excreted within 90 minutes. In patients with severe renal disease, the half-life can increase to up to 34.3 hours.11 Reduced renal clearance and increased half-life of gadolinium lead to prolonged excretion, causing the GBCA to become unstable and dissociate into its constituents, leading to tissue deposition of Gd3+ cations. This dissociation is thought to be due to differences in the stability of the various chelation complexes among the different formulations of GBCAs.12 The mechanism by which the dissociated gadolinium causes the fibrosis in the skin or other organs of the body is still unknown. Furthermore, even patients with normal renal function who undergo repeated administration of GBCA have been found to have higher levels of Gd3+ in their tissues, even in the absence of symptoms.13
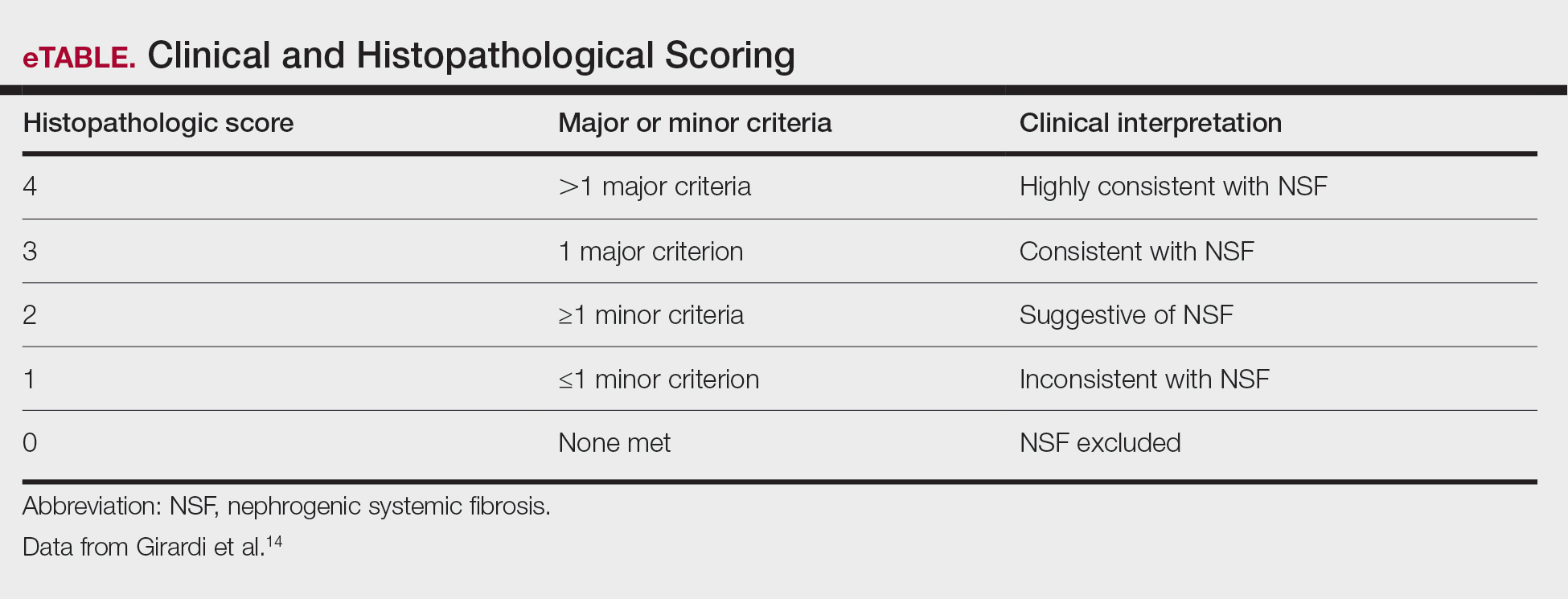
Diagnosing NSF—In 2011, Girardi et al14 created a clinical and histopathological scoring system to help diagnose NSF. Clinical findings can be broken down into major criteria and minor criteria. Major criteria consist of patterned plaques, joint contractures, cobblestoning, marked induration, or peau d’orange change. Minor criteria consist of puckering, linear banding, superficial plaques or patches, dermal papules, and scleral plaques. Histopathologic findings include increased dermal cellularity (score +1), CD34+ cells with tram tracking (score +1), thickened or thin collagen bundles (score +1), preserved elastic fibers (score −1), septal involvement (score +1), and osseous metaplasia (score +3)(eTable).14
Differential Diagnosis—The differential diagnosis of NSF includes scleromyxedema, scleroderma, eosinophilic fasciitis, eosinophilia-myalgia syndrome, lipodermatosclerosis, morphea, and chronic graft-vs-host disease. Histopathologic examination of scleromyxedema can look identical to NSF. Therefore, a review of the patient’s medical history, prior hospitalizations, and prior gadolinium exposure is important. Appropriate laboratory workups should be ordered to rule out the other differential diagnoses.
NSF and Kidney Injury—A PubMed search of articles indexed for MEDLINE using the terms NSF with kidney injury revealed 7 cases of NSF occurring in patients who either had resolved acute kidney injury or resolved acute on chronic kidney disease.1,6-10 Of those cases, 3 reported NSF occurring in patients with completely resolved acute kidney injury.6,7,10 One of those cases involved a 65-year-old man who developed acute renal failure due to acute tubular necrosis.7 He had no history of renal disease prior to hospitalization. His skin lesions continued to improve as his renal function normalized back to baseline after discharge.7 The second case involved a 42-year-old man who had repeated exposure to GBCAs during a brief period of acute kidney injury.6 Nephrogenic systemic fibrosis developed after his renal function normalized. The authors did not mention if there was clinical improvement.6 The third case involved a 22-year-old man who developed acute renal failure after ingestion of hair dye. He did not have a history of chronic renal disease, and as he recovered from the acute kidney injury, almost all of the skin lesions cleared after 1 year.10
Our patient did not have a history of chronic renal disease when she presented to the hospital for sepsis and acute tubular necrosis. Unlike 2 of the prior cases, she did not notice improvement of the skin lesions as the renal function returned to baseline. She continued to experience changes in the skin, even up to 5 years after, and then stabilized. Throughout that time, her renal function was normal. Interestingly, despite having a normal creatinine level, the patient had an elevated gadolinium level on the urine gadolinium test, which typically is not a standard test for NSF. However, the elevated value does shed light on the persistence of gadolinium in the patient despite her exposure having been more than 10 years earlier.
Treatment of NSF—There is no gold standard treatment of NSF, and reversing the fibrosis has proven to be difficult. Avoidance of GBCAs in acute kidney injury or chronic severe renal disease, as recommended by the US Food and Drug Administration, is key to preventing this debilitating disease.15 Restoration of renal function is essential for excreting the gadolinium and improvement in NSF.12 Physical and occupational therapy can improve joint mobility. Therapies such as extracorporeal photopheresis, sodium thiosulfate, pentoxifylline, glucocorticoids, plasmapheresis, intravenous immunoglobulin, cyclophosphamide, imatinib mesylate, intralesional interferon alfa, topical calcipotriene, corticosteroids, and UVA1 light therapy have been used with varying results.12 It has been suggested that renal transplantation can stop the progression of NSF. However, in the cases we reviewed, renal transplantation would not have benefited those patients because their renal function normalized.6,7,10 Additionally, even though our patient’s renal function normalized after discharge from the hospital, she continued to see more skin lesions developing, likely due to the accumulated gadolinium that was already in her tissue. The possibility of chelation therapy to remove the gadolinium has been proposed. In 1 case study involving deferoxamine injected intramuscularly in a patient with NSF, the urine excretion of gadolinium increased almost 2-fold, but there was no change in the serum concentration level of gadolinium or improvement in the patient’s clinical symptoms.16 We anticipate that our patient’s symptoms will slowly improve, as her body is still excreting the gadolinium. Our patient also was added to the International NSF Registry that was created by Dr. Shawn E. Cowper at the Yale School of Medicine (New Haven, Connecticut).
Conclusion
We report a rare case of NSF occurring in a patient with resolved acute kidney injury and no history of chronic renal disease. Our patient initially did not improve after her renal function normalized, as she continued to develop lesions 10 years after the exposure. Her elevated urine gadolinium excretion level also sheds light on the persistence of gadolinium in her body despite her normal renal function 10 years after her exposure. Although her clinical symptoms have stabilized, our case reiterates the complex pathology of this entity and challenge regarding treatment options. Physicians should be aware that NSF can still occur in healthy patients with no chronic renal disease who have had an episode of acute renal insufficiency along with exposure to a GBCA.
- Cowper SE, Su LD, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23:383-393.
- Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21:1104-1108.
- Larson KN, Gagnon AL, Darling MD, et al. Nephrogenic systemic fibrosis manifesting a decade after exposure to gadolinium. JAMA Dermatol. 2015;151:1117-1120.
- Mendoza FA, Artlett CM, Sandorfi N, et al. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35:238-249.
- Ting WW, Stone MS, Madison KC, et al. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol. 2003;139:903-906.
- Lu CF, Hsiao CH, Tjiu JW. Nephrogenic systemic fibrosis developed after recovery from acute renal failure: gadolinium as a possible aetiological factor. J Eur Acad Dermatol Venereol. 2009;23:339-340.
- Cassis TB, Jackson JM, Sonnier GB, et al. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45:56-59.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Mackay-Wiggan JM, Cohen DJ, Hardy MA, et al. Nephrogenic fibrosing dermopathy (scleromyxedema-like illness of renal disease). J Am Acad Dermatol. 2003;48:55-60.
- Reddy IS, Somani VK, Swarnalata G, et al. Nephrogenic systemic fibrosis following hair-dye ingestion induced acute renal failure. Indian J Dermatol Venereol Leprol. 2006;76:400-403.
- Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-2362.
- Cheong BYC, Muthupillai R. Nephrogenic systemic fibrosis: a concise review for cardiologists. Texas Heart Inst J. 2010;37:508-515.
- Rogosnitzky M, Branch S. Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. BioMetals. 2016;29:365-376.
- Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65:1095-1106.
- US Food and Drug Administration. FDA Drug Safety Communication: new warnings for using gadolinium-based contrast agents in patients with kidney dysfunction. Updated February 6, 2018. Accessed November 22, 2021. http://www.fda.gov/Drugs/DrugSafety/ucm223966.htm
- Leung N, Pittelkow MR, Lee CU, et al. Chelation of gadolinium with deferoxamine in a patient with nephrogenic systemic fibrosis. NDT Plus. 2009;2:309-311.
Nephrogenic systemic fibrosis (NSF) is a rare debilitating disorder characterized by dermal plaques, joint contractures, and fibrosis of the skin with possible involvement of muscles and internal organs.1-3 Originally identified in 1997 as nephrogenic fibrosing dermopathy to describe its characteristic cutaneous thickening and hardening, the name was changed to NSF to more accurately reflect the noncutaneous manifestations present in other organ tissues.2,4,5 Nephrogenic systemic fibrosis occurs in patients with a history of renal insufficiency and exposure to gadolinium-based contrast agents (GBCAs) used in magnetic resonance angiography and magnetic resonance imaging. There is no predilection for age, sex, or ethnicity.
Nephrogenic systemic fibrosis may develop over a period of days to several weeks. However, there have been cases of NSF developing 10 years after gadolinium exposure.2 In most cases, patients have a history of severe chronic renal disease requiring hemodialysis. There have been a few reported cases of NSF occurring in patients with resolved acute kidney injury or resolved acute on chronic renal disease.1,6-10 We present a case of NSF occurring in a patient with resolved transient renal insufficiency and no history of chronic renal disease.
Case Report
A 68-year-old woman presented with new dark, painless, pink plaques on the right thigh and calf. The patient stated the condition started and got worse after she was hospitalized 12 years prior for lower extremity cellulitis, sepsis, and acute renal failure. The patient developed complications during that hospital stay and underwent a renal biopsy and renal artery embolization requiring use of a GBCA. After the procedure, she noticed skin hardening in the extremities and decreased mobility in both legs while she was still in the hospital. It was thought that the lower leg changes were due to cellulitis. Therefore, when the renal issues resolved, she was discharged. Her skin and joint changes remained stable until 6 years later when she noticed new pink plaques appearing. Her medical history was positive for breast cancer, which was surgically and medically treated 16 years prior to presentation.
On presentation, physical examination revealed dark pink, hyperpigmented plaques on the right leg and a firm hypopigmented broad linear plaque on the right forearm. Palpation of the legs revealed thickened sclerotic plaques from the thighs down to the ankles (Figure 1). The plaques were not tender to palpation. She did have a decreased range of motion with eversion and inversion of the feet and ankles.
Biopsies from the right medial leg and right volar forearm showed increased bland dermal spindle cellularity associated with numerous round to ovoid osteoid aggregates encircling elastic fibers and surrounded by osteoblasts (Figure 2). CD34 immunohistochemistry showed general retention of staining within the dermal fibroblast population, and elastin stain showed general retention of elastic fiber bundles and thickening.
Laboratory workup included a complete blood cell count, comprehensive metabolic panel, thyroid-stimulating hormone level, and serum protein electrophoresis; results were all within reference range. The patient also had a urine element profile from an outside provider 1 month after presenting to our office that showed an elevated urine gadolinium level of 4.146 μg/g (reference range, 0–0.019 μg/g). The patient’s skin lesions have remained stable, and she is now working with physical therapy to help with her range of motion.
Comment
Gadolinium Causing Fibrosis—The incidence of NSF varies according to the severity of renal impairment, dosage level of GBCA used, and the history of GBCA use. In patients with normal renal function, gadolinium is excreted within 90 minutes. In patients with severe renal disease, the half-life can increase to up to 34.3 hours.11 Reduced renal clearance and increased half-life of gadolinium lead to prolonged excretion, causing the GBCA to become unstable and dissociate into its constituents, leading to tissue deposition of Gd3+ cations. This dissociation is thought to be due to differences in the stability of the various chelation complexes among the different formulations of GBCAs.12 The mechanism by which the dissociated gadolinium causes the fibrosis in the skin or other organs of the body is still unknown. Furthermore, even patients with normal renal function who undergo repeated administration of GBCA have been found to have higher levels of Gd3+ in their tissues, even in the absence of symptoms.13
Diagnosing NSF—In 2011, Girardi et al14 created a clinical and histopathological scoring system to help diagnose NSF. Clinical findings can be broken down into major criteria and minor criteria. Major criteria consist of patterned plaques, joint contractures, cobblestoning, marked induration, or peau d’orange change. Minor criteria consist of puckering, linear banding, superficial plaques or patches, dermal papules, and scleral plaques. Histopathologic findings include increased dermal cellularity (score +1), CD34+ cells with tram tracking (score +1), thickened or thin collagen bundles (score +1), preserved elastic fibers (score −1), septal involvement (score +1), and osseous metaplasia (score +3)(eTable).14
Differential Diagnosis—The differential diagnosis of NSF includes scleromyxedema, scleroderma, eosinophilic fasciitis, eosinophilia-myalgia syndrome, lipodermatosclerosis, morphea, and chronic graft-vs-host disease. Histopathologic examination of scleromyxedema can look identical to NSF. Therefore, a review of the patient’s medical history, prior hospitalizations, and prior gadolinium exposure is important. Appropriate laboratory workups should be ordered to rule out the other differential diagnoses.
NSF and Kidney Injury—A PubMed search of articles indexed for MEDLINE using the terms NSF with kidney injury revealed 7 cases of NSF occurring in patients who either had resolved acute kidney injury or resolved acute on chronic kidney disease.1,6-10 Of those cases, 3 reported NSF occurring in patients with completely resolved acute kidney injury.6,7,10 One of those cases involved a 65-year-old man who developed acute renal failure due to acute tubular necrosis.7 He had no history of renal disease prior to hospitalization. His skin lesions continued to improve as his renal function normalized back to baseline after discharge.7 The second case involved a 42-year-old man who had repeated exposure to GBCAs during a brief period of acute kidney injury.6 Nephrogenic systemic fibrosis developed after his renal function normalized. The authors did not mention if there was clinical improvement.6 The third case involved a 22-year-old man who developed acute renal failure after ingestion of hair dye. He did not have a history of chronic renal disease, and as he recovered from the acute kidney injury, almost all of the skin lesions cleared after 1 year.10
Our patient did not have a history of chronic renal disease when she presented to the hospital for sepsis and acute tubular necrosis. Unlike 2 of the prior cases, she did not notice improvement of the skin lesions as the renal function returned to baseline. She continued to experience changes in the skin, even up to 5 years after, and then stabilized. Throughout that time, her renal function was normal. Interestingly, despite having a normal creatinine level, the patient had an elevated gadolinium level on the urine gadolinium test, which typically is not a standard test for NSF. However, the elevated value does shed light on the persistence of gadolinium in the patient despite her exposure having been more than 10 years earlier.
Treatment of NSF—There is no gold standard treatment of NSF, and reversing the fibrosis has proven to be difficult. Avoidance of GBCAs in acute kidney injury or chronic severe renal disease, as recommended by the US Food and Drug Administration, is key to preventing this debilitating disease.15 Restoration of renal function is essential for excreting the gadolinium and improvement in NSF.12 Physical and occupational therapy can improve joint mobility. Therapies such as extracorporeal photopheresis, sodium thiosulfate, pentoxifylline, glucocorticoids, plasmapheresis, intravenous immunoglobulin, cyclophosphamide, imatinib mesylate, intralesional interferon alfa, topical calcipotriene, corticosteroids, and UVA1 light therapy have been used with varying results.12 It has been suggested that renal transplantation can stop the progression of NSF. However, in the cases we reviewed, renal transplantation would not have benefited those patients because their renal function normalized.6,7,10 Additionally, even though our patient’s renal function normalized after discharge from the hospital, she continued to see more skin lesions developing, likely due to the accumulated gadolinium that was already in her tissue. The possibility of chelation therapy to remove the gadolinium has been proposed. In 1 case study involving deferoxamine injected intramuscularly in a patient with NSF, the urine excretion of gadolinium increased almost 2-fold, but there was no change in the serum concentration level of gadolinium or improvement in the patient’s clinical symptoms.16 We anticipate that our patient’s symptoms will slowly improve, as her body is still excreting the gadolinium. Our patient also was added to the International NSF Registry that was created by Dr. Shawn E. Cowper at the Yale School of Medicine (New Haven, Connecticut).
Conclusion
We report a rare case of NSF occurring in a patient with resolved acute kidney injury and no history of chronic renal disease. Our patient initially did not improve after her renal function normalized, as she continued to develop lesions 10 years after the exposure. Her elevated urine gadolinium excretion level also sheds light on the persistence of gadolinium in her body despite her normal renal function 10 years after her exposure. Although her clinical symptoms have stabilized, our case reiterates the complex pathology of this entity and challenge regarding treatment options. Physicians should be aware that NSF can still occur in healthy patients with no chronic renal disease who have had an episode of acute renal insufficiency along with exposure to a GBCA.
Nephrogenic systemic fibrosis (NSF) is a rare debilitating disorder characterized by dermal plaques, joint contractures, and fibrosis of the skin with possible involvement of muscles and internal organs.1-3 Originally identified in 1997 as nephrogenic fibrosing dermopathy to describe its characteristic cutaneous thickening and hardening, the name was changed to NSF to more accurately reflect the noncutaneous manifestations present in other organ tissues.2,4,5 Nephrogenic systemic fibrosis occurs in patients with a history of renal insufficiency and exposure to gadolinium-based contrast agents (GBCAs) used in magnetic resonance angiography and magnetic resonance imaging. There is no predilection for age, sex, or ethnicity.
Nephrogenic systemic fibrosis may develop over a period of days to several weeks. However, there have been cases of NSF developing 10 years after gadolinium exposure.2 In most cases, patients have a history of severe chronic renal disease requiring hemodialysis. There have been a few reported cases of NSF occurring in patients with resolved acute kidney injury or resolved acute on chronic renal disease.1,6-10 We present a case of NSF occurring in a patient with resolved transient renal insufficiency and no history of chronic renal disease.
Case Report
A 68-year-old woman presented with new dark, painless, pink plaques on the right thigh and calf. The patient stated the condition started and got worse after she was hospitalized 12 years prior for lower extremity cellulitis, sepsis, and acute renal failure. The patient developed complications during that hospital stay and underwent a renal biopsy and renal artery embolization requiring use of a GBCA. After the procedure, she noticed skin hardening in the extremities and decreased mobility in both legs while she was still in the hospital. It was thought that the lower leg changes were due to cellulitis. Therefore, when the renal issues resolved, she was discharged. Her skin and joint changes remained stable until 6 years later when she noticed new pink plaques appearing. Her medical history was positive for breast cancer, which was surgically and medically treated 16 years prior to presentation.
On presentation, physical examination revealed dark pink, hyperpigmented plaques on the right leg and a firm hypopigmented broad linear plaque on the right forearm. Palpation of the legs revealed thickened sclerotic plaques from the thighs down to the ankles (Figure 1). The plaques were not tender to palpation. She did have a decreased range of motion with eversion and inversion of the feet and ankles.
Biopsies from the right medial leg and right volar forearm showed increased bland dermal spindle cellularity associated with numerous round to ovoid osteoid aggregates encircling elastic fibers and surrounded by osteoblasts (Figure 2). CD34 immunohistochemistry showed general retention of staining within the dermal fibroblast population, and elastin stain showed general retention of elastic fiber bundles and thickening.
Laboratory workup included a complete blood cell count, comprehensive metabolic panel, thyroid-stimulating hormone level, and serum protein electrophoresis; results were all within reference range. The patient also had a urine element profile from an outside provider 1 month after presenting to our office that showed an elevated urine gadolinium level of 4.146 μg/g (reference range, 0–0.019 μg/g). The patient’s skin lesions have remained stable, and she is now working with physical therapy to help with her range of motion.
Comment
Gadolinium Causing Fibrosis—The incidence of NSF varies according to the severity of renal impairment, dosage level of GBCA used, and the history of GBCA use. In patients with normal renal function, gadolinium is excreted within 90 minutes. In patients with severe renal disease, the half-life can increase to up to 34.3 hours.11 Reduced renal clearance and increased half-life of gadolinium lead to prolonged excretion, causing the GBCA to become unstable and dissociate into its constituents, leading to tissue deposition of Gd3+ cations. This dissociation is thought to be due to differences in the stability of the various chelation complexes among the different formulations of GBCAs.12 The mechanism by which the dissociated gadolinium causes the fibrosis in the skin or other organs of the body is still unknown. Furthermore, even patients with normal renal function who undergo repeated administration of GBCA have been found to have higher levels of Gd3+ in their tissues, even in the absence of symptoms.13
Diagnosing NSF—In 2011, Girardi et al14 created a clinical and histopathological scoring system to help diagnose NSF. Clinical findings can be broken down into major criteria and minor criteria. Major criteria consist of patterned plaques, joint contractures, cobblestoning, marked induration, or peau d’orange change. Minor criteria consist of puckering, linear banding, superficial plaques or patches, dermal papules, and scleral plaques. Histopathologic findings include increased dermal cellularity (score +1), CD34+ cells with tram tracking (score +1), thickened or thin collagen bundles (score +1), preserved elastic fibers (score −1), septal involvement (score +1), and osseous metaplasia (score +3)(eTable).14
Differential Diagnosis—The differential diagnosis of NSF includes scleromyxedema, scleroderma, eosinophilic fasciitis, eosinophilia-myalgia syndrome, lipodermatosclerosis, morphea, and chronic graft-vs-host disease. Histopathologic examination of scleromyxedema can look identical to NSF. Therefore, a review of the patient’s medical history, prior hospitalizations, and prior gadolinium exposure is important. Appropriate laboratory workups should be ordered to rule out the other differential diagnoses.
NSF and Kidney Injury—A PubMed search of articles indexed for MEDLINE using the terms NSF with kidney injury revealed 7 cases of NSF occurring in patients who either had resolved acute kidney injury or resolved acute on chronic kidney disease.1,6-10 Of those cases, 3 reported NSF occurring in patients with completely resolved acute kidney injury.6,7,10 One of those cases involved a 65-year-old man who developed acute renal failure due to acute tubular necrosis.7 He had no history of renal disease prior to hospitalization. His skin lesions continued to improve as his renal function normalized back to baseline after discharge.7 The second case involved a 42-year-old man who had repeated exposure to GBCAs during a brief period of acute kidney injury.6 Nephrogenic systemic fibrosis developed after his renal function normalized. The authors did not mention if there was clinical improvement.6 The third case involved a 22-year-old man who developed acute renal failure after ingestion of hair dye. He did not have a history of chronic renal disease, and as he recovered from the acute kidney injury, almost all of the skin lesions cleared after 1 year.10
Our patient did not have a history of chronic renal disease when she presented to the hospital for sepsis and acute tubular necrosis. Unlike 2 of the prior cases, she did not notice improvement of the skin lesions as the renal function returned to baseline. She continued to experience changes in the skin, even up to 5 years after, and then stabilized. Throughout that time, her renal function was normal. Interestingly, despite having a normal creatinine level, the patient had an elevated gadolinium level on the urine gadolinium test, which typically is not a standard test for NSF. However, the elevated value does shed light on the persistence of gadolinium in the patient despite her exposure having been more than 10 years earlier.
Treatment of NSF—There is no gold standard treatment of NSF, and reversing the fibrosis has proven to be difficult. Avoidance of GBCAs in acute kidney injury or chronic severe renal disease, as recommended by the US Food and Drug Administration, is key to preventing this debilitating disease.15 Restoration of renal function is essential for excreting the gadolinium and improvement in NSF.12 Physical and occupational therapy can improve joint mobility. Therapies such as extracorporeal photopheresis, sodium thiosulfate, pentoxifylline, glucocorticoids, plasmapheresis, intravenous immunoglobulin, cyclophosphamide, imatinib mesylate, intralesional interferon alfa, topical calcipotriene, corticosteroids, and UVA1 light therapy have been used with varying results.12 It has been suggested that renal transplantation can stop the progression of NSF. However, in the cases we reviewed, renal transplantation would not have benefited those patients because their renal function normalized.6,7,10 Additionally, even though our patient’s renal function normalized after discharge from the hospital, she continued to see more skin lesions developing, likely due to the accumulated gadolinium that was already in her tissue. The possibility of chelation therapy to remove the gadolinium has been proposed. In 1 case study involving deferoxamine injected intramuscularly in a patient with NSF, the urine excretion of gadolinium increased almost 2-fold, but there was no change in the serum concentration level of gadolinium or improvement in the patient’s clinical symptoms.16 We anticipate that our patient’s symptoms will slowly improve, as her body is still excreting the gadolinium. Our patient also was added to the International NSF Registry that was created by Dr. Shawn E. Cowper at the Yale School of Medicine (New Haven, Connecticut).
Conclusion
We report a rare case of NSF occurring in a patient with resolved acute kidney injury and no history of chronic renal disease. Our patient initially did not improve after her renal function normalized, as she continued to develop lesions 10 years after the exposure. Her elevated urine gadolinium excretion level also sheds light on the persistence of gadolinium in her body despite her normal renal function 10 years after her exposure. Although her clinical symptoms have stabilized, our case reiterates the complex pathology of this entity and challenge regarding treatment options. Physicians should be aware that NSF can still occur in healthy patients with no chronic renal disease who have had an episode of acute renal insufficiency along with exposure to a GBCA.
- Cowper SE, Su LD, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23:383-393.
- Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21:1104-1108.
- Larson KN, Gagnon AL, Darling MD, et al. Nephrogenic systemic fibrosis manifesting a decade after exposure to gadolinium. JAMA Dermatol. 2015;151:1117-1120.
- Mendoza FA, Artlett CM, Sandorfi N, et al. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35:238-249.
- Ting WW, Stone MS, Madison KC, et al. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol. 2003;139:903-906.
- Lu CF, Hsiao CH, Tjiu JW. Nephrogenic systemic fibrosis developed after recovery from acute renal failure: gadolinium as a possible aetiological factor. J Eur Acad Dermatol Venereol. 2009;23:339-340.
- Cassis TB, Jackson JM, Sonnier GB, et al. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45:56-59.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Mackay-Wiggan JM, Cohen DJ, Hardy MA, et al. Nephrogenic fibrosing dermopathy (scleromyxedema-like illness of renal disease). J Am Acad Dermatol. 2003;48:55-60.
- Reddy IS, Somani VK, Swarnalata G, et al. Nephrogenic systemic fibrosis following hair-dye ingestion induced acute renal failure. Indian J Dermatol Venereol Leprol. 2006;76:400-403.
- Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-2362.
- Cheong BYC, Muthupillai R. Nephrogenic systemic fibrosis: a concise review for cardiologists. Texas Heart Inst J. 2010;37:508-515.
- Rogosnitzky M, Branch S. Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. BioMetals. 2016;29:365-376.
- Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65:1095-1106.
- US Food and Drug Administration. FDA Drug Safety Communication: new warnings for using gadolinium-based contrast agents in patients with kidney dysfunction. Updated February 6, 2018. Accessed November 22, 2021. http://www.fda.gov/Drugs/DrugSafety/ucm223966.htm
- Leung N, Pittelkow MR, Lee CU, et al. Chelation of gadolinium with deferoxamine in a patient with nephrogenic systemic fibrosis. NDT Plus. 2009;2:309-311.
- Cowper SE, Su LD, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23:383-393.
- Grobner T. Gadolinium—a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21:1104-1108.
- Larson KN, Gagnon AL, Darling MD, et al. Nephrogenic systemic fibrosis manifesting a decade after exposure to gadolinium. JAMA Dermatol. 2015;151:1117-1120.
- Mendoza FA, Artlett CM, Sandorfi N, et al. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35:238-249.
- Ting WW, Stone MS, Madison KC, et al. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol. 2003;139:903-906.
- Lu CF, Hsiao CH, Tjiu JW. Nephrogenic systemic fibrosis developed after recovery from acute renal failure: gadolinium as a possible aetiological factor. J Eur Acad Dermatol Venereol. 2009;23:339-340.
- Cassis TB, Jackson JM, Sonnier GB, et al. Nephrogenic fibrosing dermopathy in a patient with acute renal failure never requiring dialysis. Int J Dermatol. 2006;45:56-59.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Mackay-Wiggan JM, Cohen DJ, Hardy MA, et al. Nephrogenic fibrosing dermopathy (scleromyxedema-like illness of renal disease). J Am Acad Dermatol. 2003;48:55-60.
- Reddy IS, Somani VK, Swarnalata G, et al. Nephrogenic systemic fibrosis following hair-dye ingestion induced acute renal failure. Indian J Dermatol Venereol Leprol. 2006;76:400-403.
- Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17:2359-2362.
- Cheong BYC, Muthupillai R. Nephrogenic systemic fibrosis: a concise review for cardiologists. Texas Heart Inst J. 2010;37:508-515.
- Rogosnitzky M, Branch S. Gadolinium-based contrast agent toxicity: a review of known and proposed mechanisms. BioMetals. 2016;29:365-376.
- Girardi M, Kay J, Elston DM, et al. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65:1095-1106.
- US Food and Drug Administration. FDA Drug Safety Communication: new warnings for using gadolinium-based contrast agents in patients with kidney dysfunction. Updated February 6, 2018. Accessed November 22, 2021. http://www.fda.gov/Drugs/DrugSafety/ucm223966.htm
- Leung N, Pittelkow MR, Lee CU, et al. Chelation of gadolinium with deferoxamine in a patient with nephrogenic systemic fibrosis. NDT Plus. 2009;2:309-311.
Practice Points
- Nephrogenic systemic fibrosis may occur in patients with a history of renal insufficiency and exposure to gadolinium-based contrast agents.
- Nephrogenic systemic fibrosis may develop over a period of days to several years after exposure.
- Symptoms of this rare disease can progress and get worse even after renal function normalizes.
Exuberant Lymphomatoid Papulosis of the Head and Upper Trunk
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
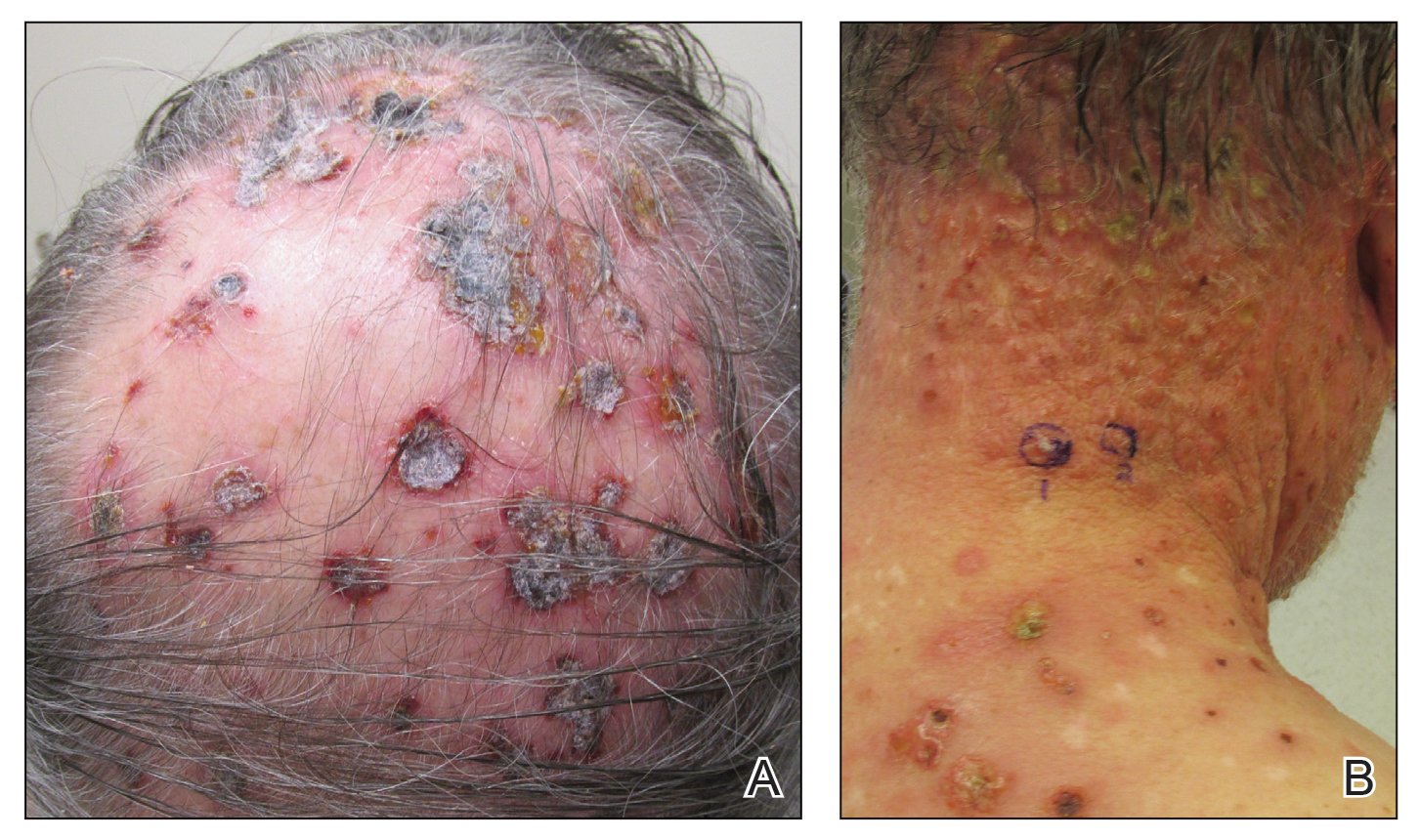
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
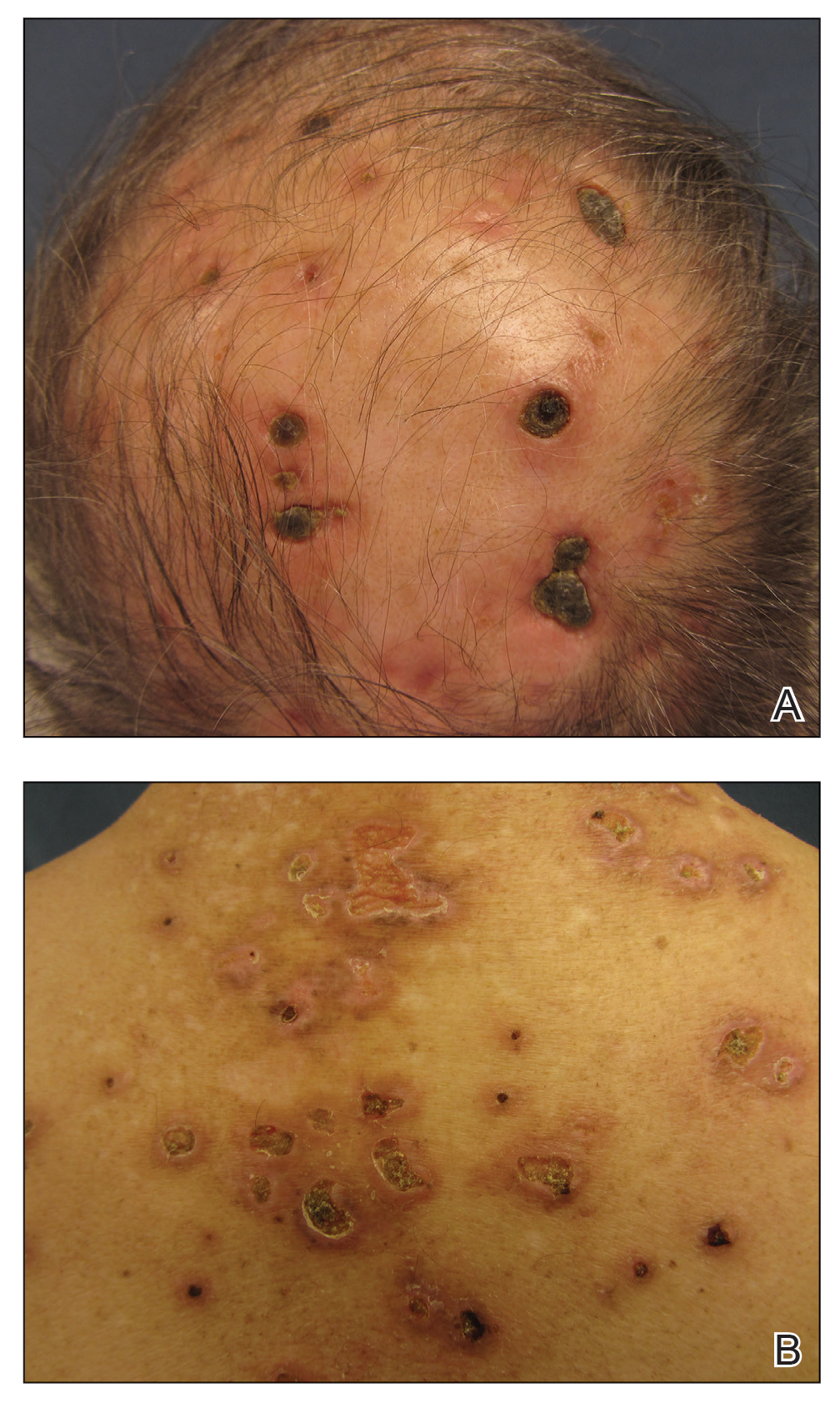
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
Practice Points
- Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder characterized by red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution.
- Histopathologically, LyP consists of a frequently CD30Mathematical Pi LT Std+ lymphocytic proliferation in multiple described patterns.
- Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy.
Scalp Arteriovenous Fistula With Intracranial Communication
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
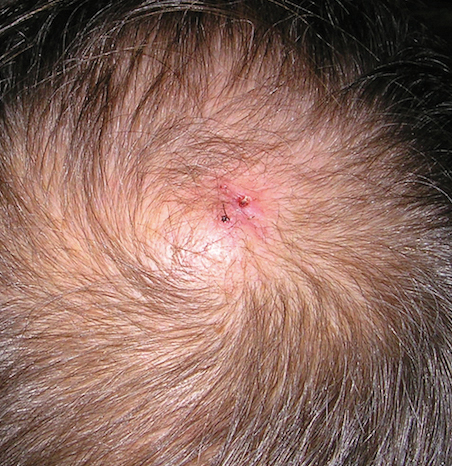
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
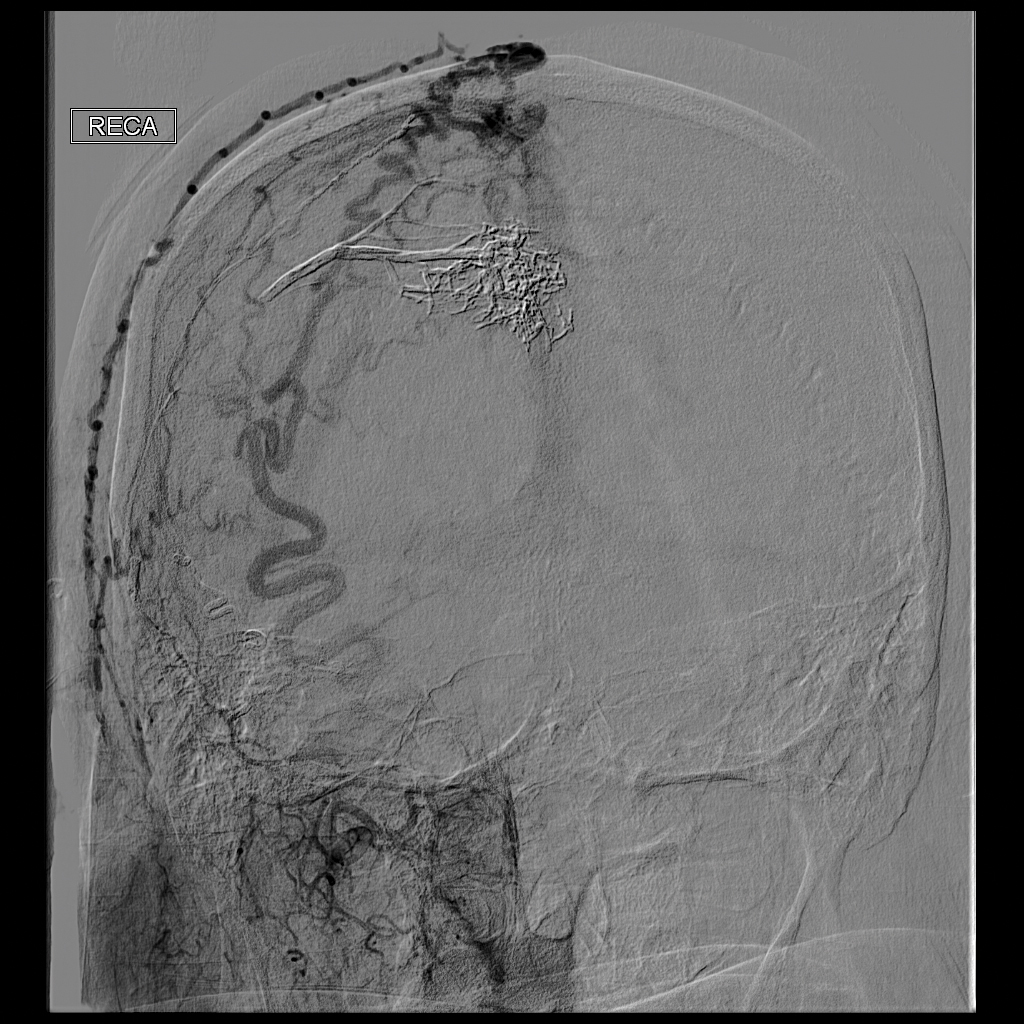
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
Practice Points
- Scalp arteriovenous fistulas may be traumatic or spontaneous and present as either an innocuous-looking scalp nodule or as a progressively enlarging pulsatile mass on the scalp.
- Clinical detection followed by appropriate imaging and referral to neurosurgery or interventional neuroradiology is vital to patient safety.
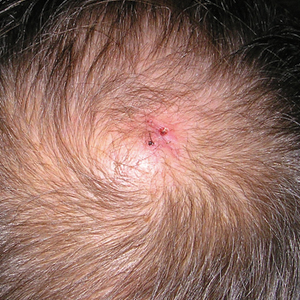
Field Cancerization With Multiple Keratoacanthomas Successfully Treated With Topical and Intralesional 5-Fluorouracil
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
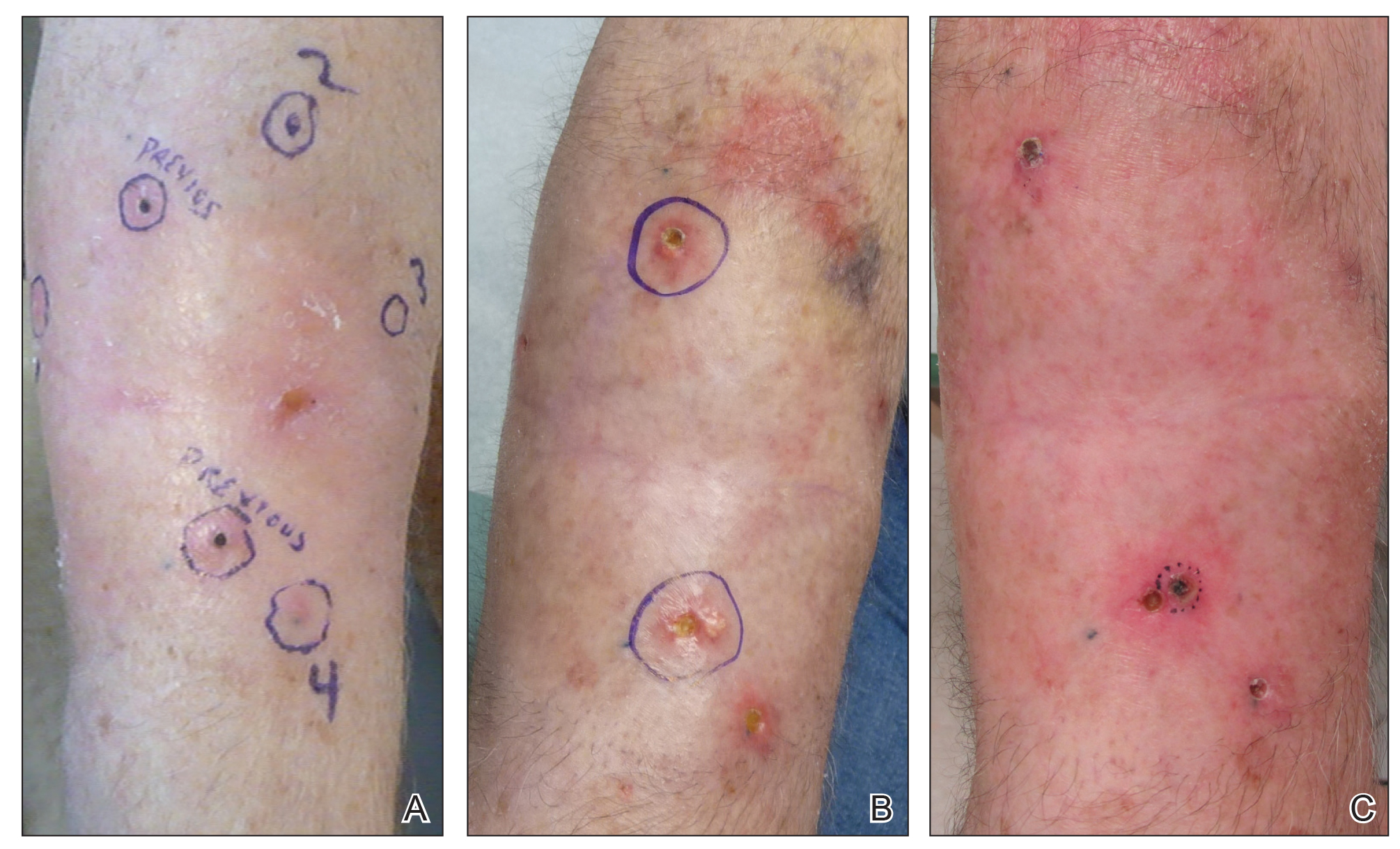
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
To the Editor:
The concept of field cancerization has been well described since its initial proposal by Slaughter et al1 in 1953. It describes a field of genetically altered cells where multiple clonally related neoplasms can develop.2,3 Treatment of patients with multiple neoplasms within an area of field cancerization can be especially challenging. We report a patient with field cancerization who had multiple squamous cell carcinomas (SCCs) and keratoacanthomas (KAs) that arose within the field.
A 78-year-old man initially presented with a papule on the right forearm of 3 months’ duration. He had a medical history of cutaneous SCC, myocardial infarction, type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, hypercholesterolemia, gout, and diverticulosis. He was not taking any chronic immunosuppressants that may have predisposed him to the development of nonmelanoma skin cancer. The papule was biopsied and diagnosed as a well-differentiated invasive SCC. A month later it was excised with clear margins.
Approximately 5 weeks after the excision, he returned with an enlarging lesion on the right forearm just medial to the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. Two months later the lesion was excised with clear margins. Four weeks later he returned with a new lesion adjacent to the medial aspect of the prior excision. The lesion was biopsied and diagnosed as a well-differentiated SCC. Four weeks later the lesion was excised with clear margins.
Another 4 weeks later the patient returned with a new lesion on the excision site. The lesion was biopsied and diagnosed as a well-differentiated SCC. The lesion was treated with radiotherapy, with a 5800-cGy course completed 2 months later. The next month, 2 papules just adjacent to the radiotherapy treatment field were biopsied and diagnosed as well-differentiated SCC, KA type. One week later, 2 additional new papules adjacent to the radiotherapy treatment field were biopsied and diagnosed as moderately differentiated SCC, KA type. At this time, the patient had 4 biopsy-proven KAs on the right forearm in the area of prior radiation (Figure, A). The radiation oncologist felt that further radiation was no longer indicated. A consultation was sought with surgical oncology, and wide excision of the field with sentinel lymph node biopsy and skin grafting was recommended. Computed tomography with contrast of the chest and right arm ordered by surgical oncology did not reveal metastatic disease.
After discussion of the risks, alternatives, and benefits of surgery, the patient elected to try nonsurgical treatment. He was treated with 5-fluorouracil (5-FU) cream 5% twice daily for 4 weeks. It was applied to the right arm from the elbow to the wrist and occluded under an elastic bandage. The patient stated that the biopsy sites became sore and inflamed during the treatment. After 4 weeks of treatment, all 4 KAs had healed without clinical evidence of tumor. During this time, however, the previously treated 2 sites had developed adjacent firm pink papules (Figure, B); these 2 lesions were then treated with intralesional 5-FU 50 mg/mL once weekly to resolution at 4 and 5 weeks, respectively. The proximal lesion was treated with 7.5 mg on week 1 and 5 mg on weeks 2, 3, and 4. The larger distal lesion was treated with 12.5 mg on week 1 and 5 mg on weeks 2, 3, 4, and 5. The volume injected was determined by ability to blanch and indurate the lesion and was decreased due to the shrinking size of the tumor. After 3 injections, both tumors had substantially decreased in size (Figure, C). The patient noted pain during injection but found the procedure tolerable and preferable to surgery. There were no other adverse events. At the end of treatment, both tumors had clinically resolved. No recurrence or development of new tumors was reported over 3 years of follow-up after the last injection.
Field cancerization was the outgrowth of the study of oral SCC in an effort to explain the development of multiple primary tumors and locally recurrent cancer.1,2 Histopathologically, the authors observed that oral cancer developed in multifocal areas of precancerous change, histologically abnormal hyperplastic tissue surrounded the tumors, oral cancer consisted of multiple independent areas that sometimes coalesced, and the persistence of abnormal tissue after surgery might explain local recurrences and the development of new lesions in a previously treated area.1,2 Since then, the concept has been applied to several other organ systems including the lungs, vulva, cervix, breasts, bladder, colon, and skin.2
In the skin, field cancerization involves clusters and contiguous patches of altered cells present in areas of chronic photodamage.2 Genetically altered fields form the foundation in which multiple clonally related neoplastic lesions can develop.2,3 These fields often remain after treatment of the primary tumor and may lead to new cancers that commonly are labeled as a second primary tumor or a local recurrence depending on the exact site and time interval.3 Brennan et al3 found clonal populations of infiltrating tumor cells harboring a p53 gene mutation in more than 50% of histopathologically negative surgical margins of patients with SCC of the head and neck. Furthermore, 40% of the patients with a margin positive for a p53 gene mutation had local recurrence vs none of the patients with negative margins.4 These findings were supported by several other studies where loss of heterozygosity, microsatellite alterations, chromosomal instability, or in situ hybridization was used to demonstrate genetically altered fields.2,4 Histopathologic patterns of epidermolytic hyperkeratosis, focal acantholytic dyskeratosis, and pronounced acantholysis as found in Hailey-Hailey disease may be a consequence of clonal expansion of mutated keratinocytes because of long-term exposure to mutagens such as UV light and human papillomavirus.5
The development of an expanding neoplastic field appears to play an important role in cutaneous carcinogenesis. It is necessary to consider the cutaneous field cancerization as a highly photodamaged area that contains clinical and subclinical lesions.2-4 The treatment of cutaneous neoplasms, SCC in particular, should focus not only on the tumor itself but also on the surrounding tissue. Adjunctive field-directed therapies should be considered after treatment of the primary tumor.4
Our patient continued to develop SCCs on the right forearm after multiple excisions with clear margins and subsequently was treated with radiation therapy. He then developed 4 KAs after radiation therapy to the right forearm. Topical 5-FU is a well-described treatment of field cancerization.2 In our patient, 5-FU cream 5% applied twice daily from the wrist to the elbow under occlusion for 4 weeks led to the involution of all 4 KAs. During this time, our patient developed 2 additional firm pink papules near the previously treated sites, which resolved with intralesional 5-FU weekly for 4 and 5 weeks, respectively.
Intralesional 5-FU has been described for the treatment of multiple and difficult-to-treat KAs. It is an antimetabolite and structural analog of uracil that disrupts DNA and RNA synthesis. It is contraindicated in liver disease, pregnancy or breastfeeding, and allergy to the medication.6 Intralesional 5-FU dosing recommendations for KAs include use of a 50-mg/mL solution and injecting 0.1 to 1 mL until the lesion blanches in color, which may be repeated every 1 to 4 weeks.7,8 The maximum recommended daily dose is 800 mg.6 Pretreatment with intralesional 1% lidocaine has been recommended by some authors due to pain with injection.8 Recommendations for laboratory monitoring include a complete blood cell count with differential at baseline and weekly. Side effects include local pain, erythema, crusting, ulceration, and necrosis. Systemic side effects include cytopenia and gastrointestinal tract upset.6 Intralesional 5-FU has been used successfully in a single dose of 10 mg per lesion in combination with systemic acitretin for the treatment of multiple KAs induced by vemurafenib.9 It also has been effective in the treatment of multiple recurrent reactive KAs developing in surgical margins.7 A review article reported that the use of intralesional 5-FU produced a 98% cure rate in 56 treated KAs.6 Alternative intralesional agents that may be considered for KAs include methotrexate, bleomycin, and interferon alfa-2b.6,7
Field cancerization may cause the development of multiple clonally related neoplasms within a field of genetically altered cells that may continue to develop after excision with clear margins or radiation therapy. Given the success of treatment in our patient, we recommend consideration for topical and intralesional 5-FU in patients who develop SCCs and KAs within an area of field cancerization.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
- Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Torezan LA, Festa-Neto C. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Brennan JA, Mao L, Hruban R, et al. Molecular assessment of histopathological staging in squamous-cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braakhuis, BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Carlson AJ, Scott D, Wharton J, et al. Incidental histopathologic patterns: possible evidence of “field cancerization” surrounding skin tumors. Am J Dermatopathol. 2001;23:494-496.
- Kirby J, Miller C. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63:689-702.
- Hadley J, Tristani-Firouzi P, Florell S, et al. Case series of multiple recurrent reactive keratoacanthomas developing at surgical margins. Dermatol Surg. 2009;35:2019-2024.
- Que S, Compton L, Schmults C. Eruptive squamous atypia (also known as eruptive keratoacanthoma): definition of the disease entity and successful management via intralesional 5-fluorouracil. J Am Acad Dermatol. 2019;81:111-122.
- LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas systemic acitretin and intralesional fluorouracil. JAMA Dermatol. 2013;149:279-281.
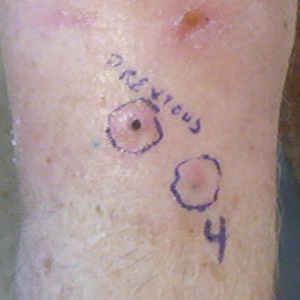
Darkening and Eruptive Nevi During Treatment With Erlotinib
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
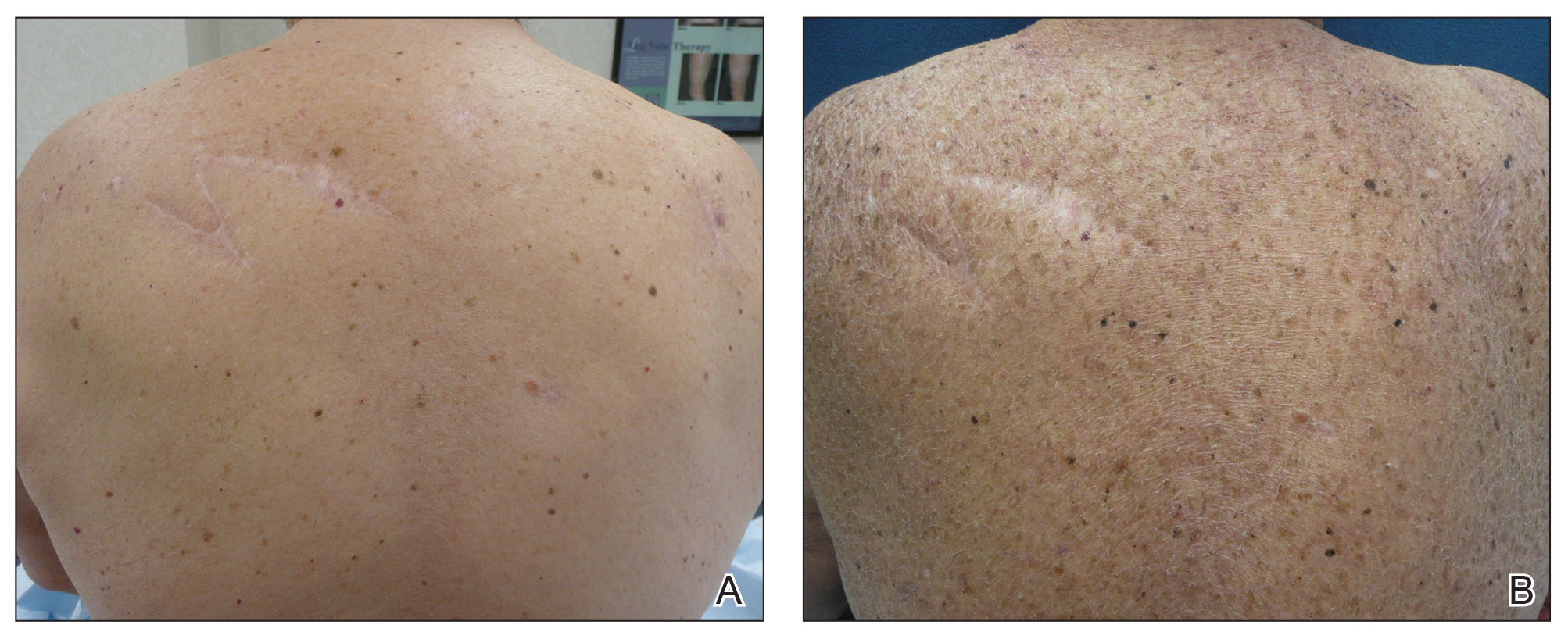
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
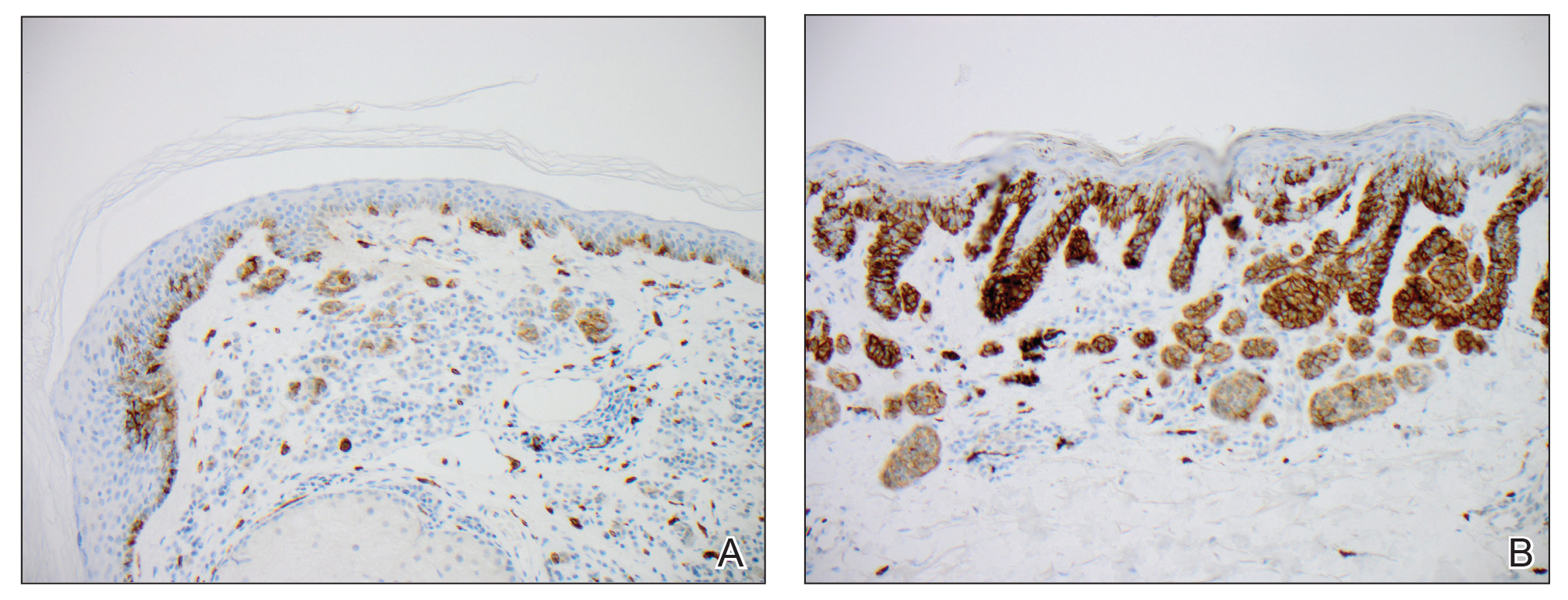
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
Practice Points
- Cutaneous side effects of erlotinib include acneform eruption, xerosis, paronychia, and pruritus.
- Clinicians should monitor patients for darkening and/or eruptive nevi as well as melanoma during treatment with erlotinib.
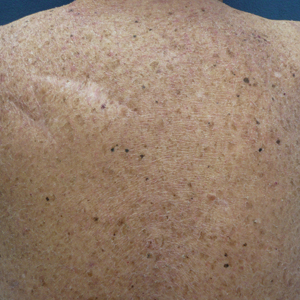
Acroangiodermatitis of Mali and Stewart-Bluefarb Syndrome
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
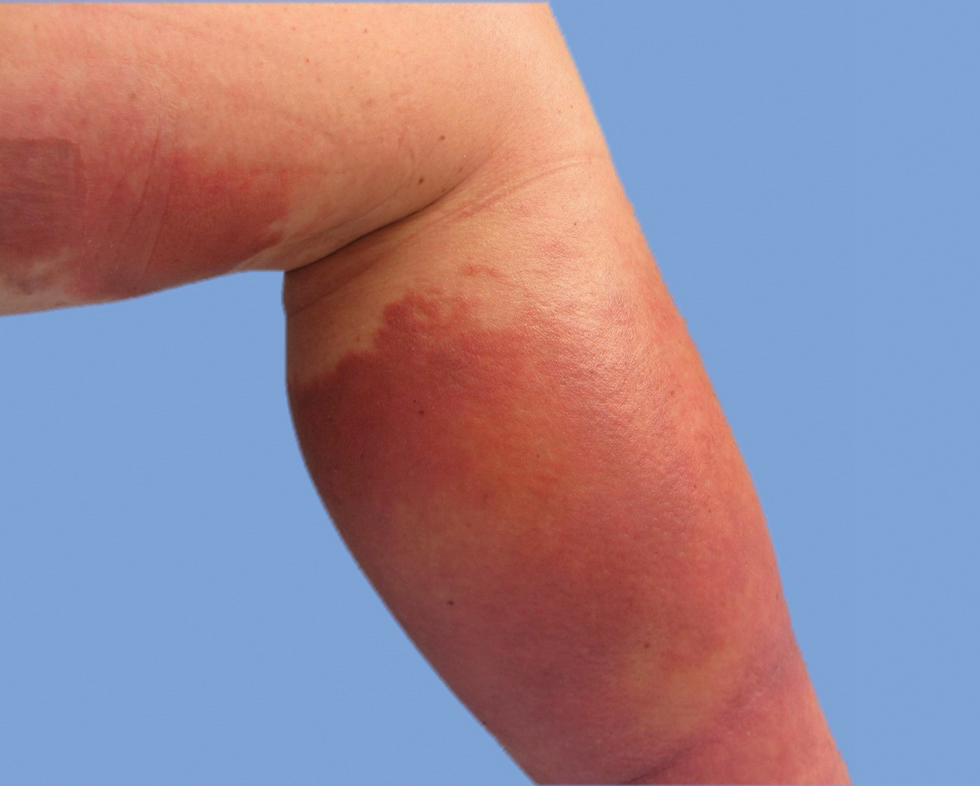
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
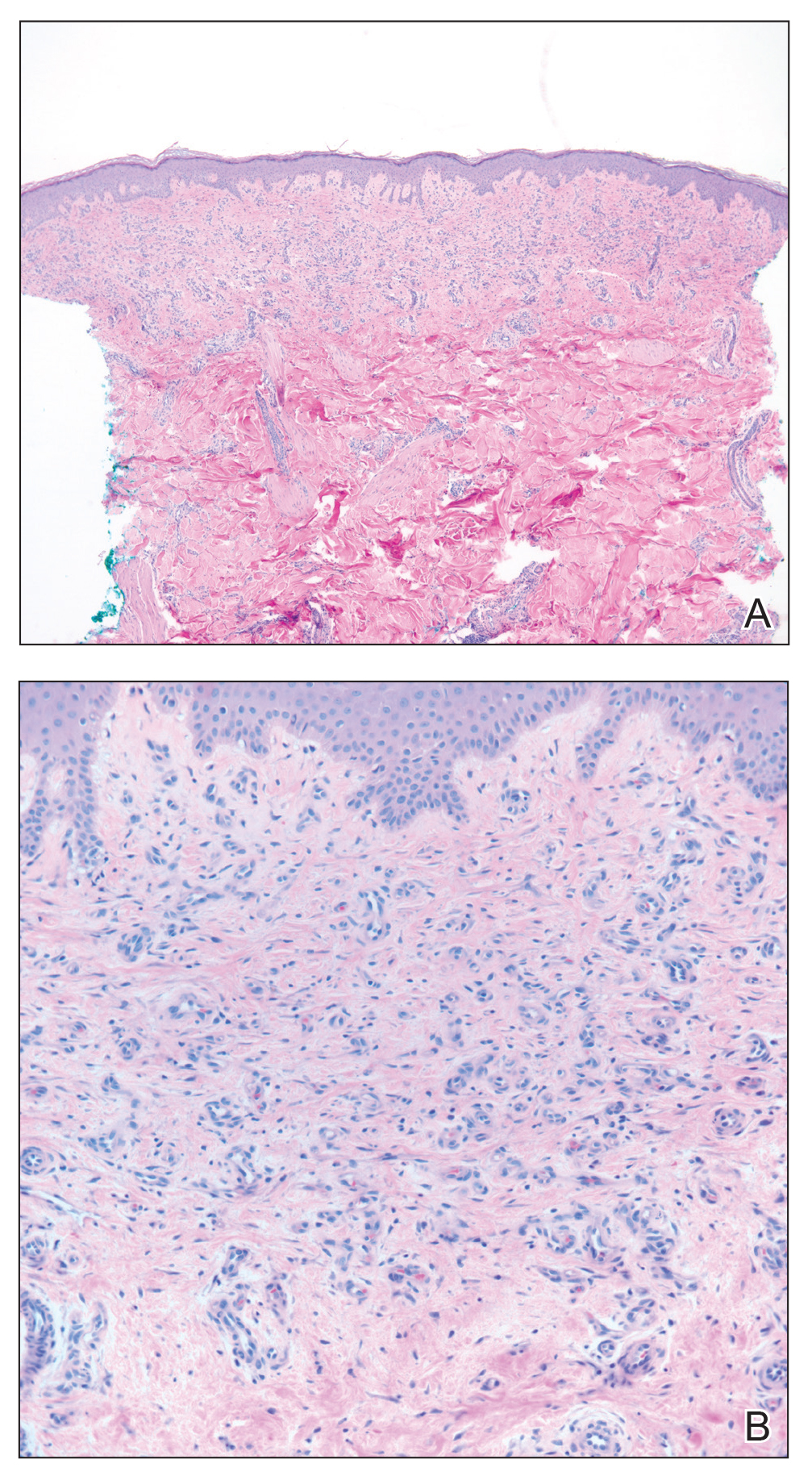
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
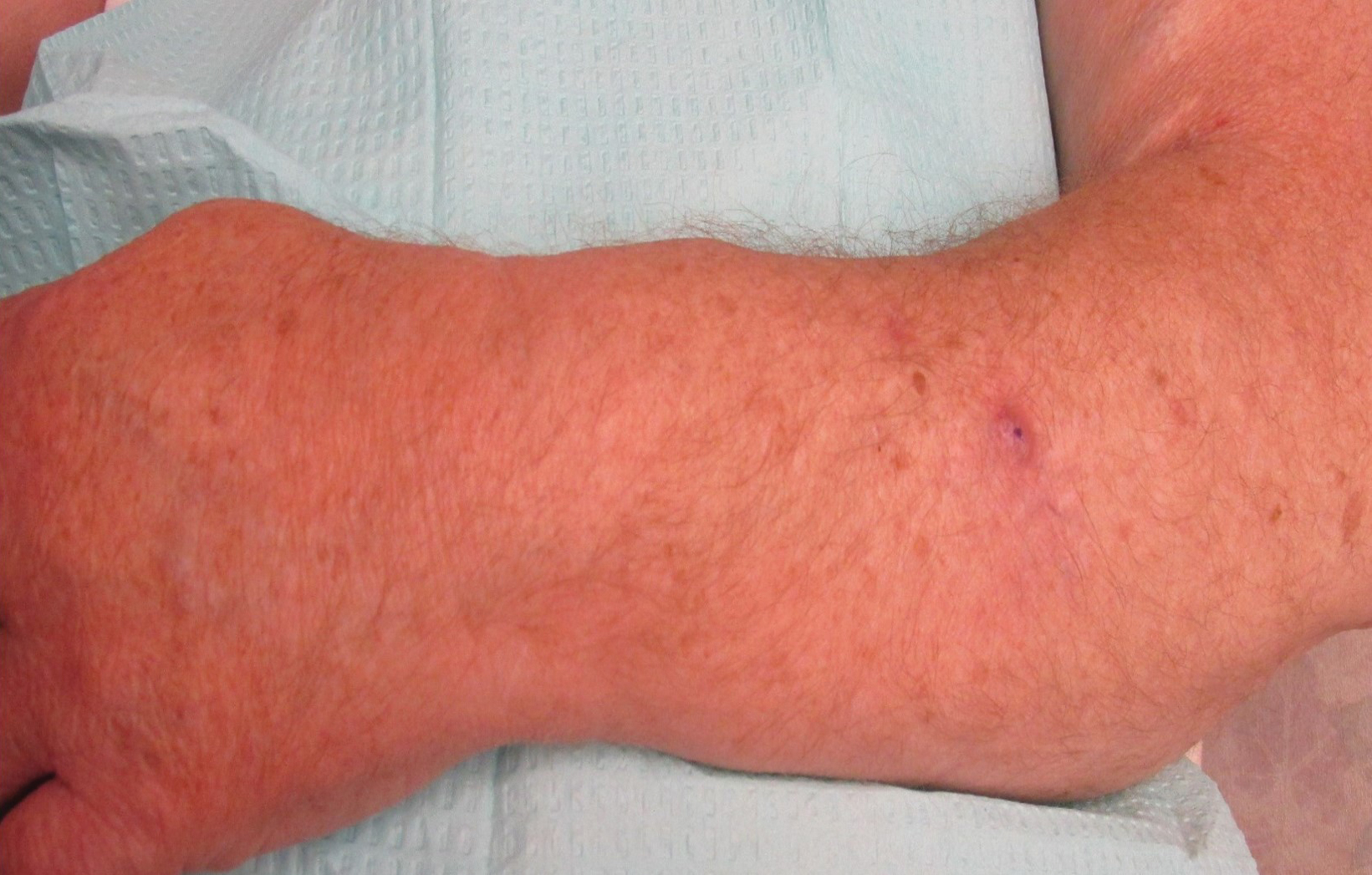
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
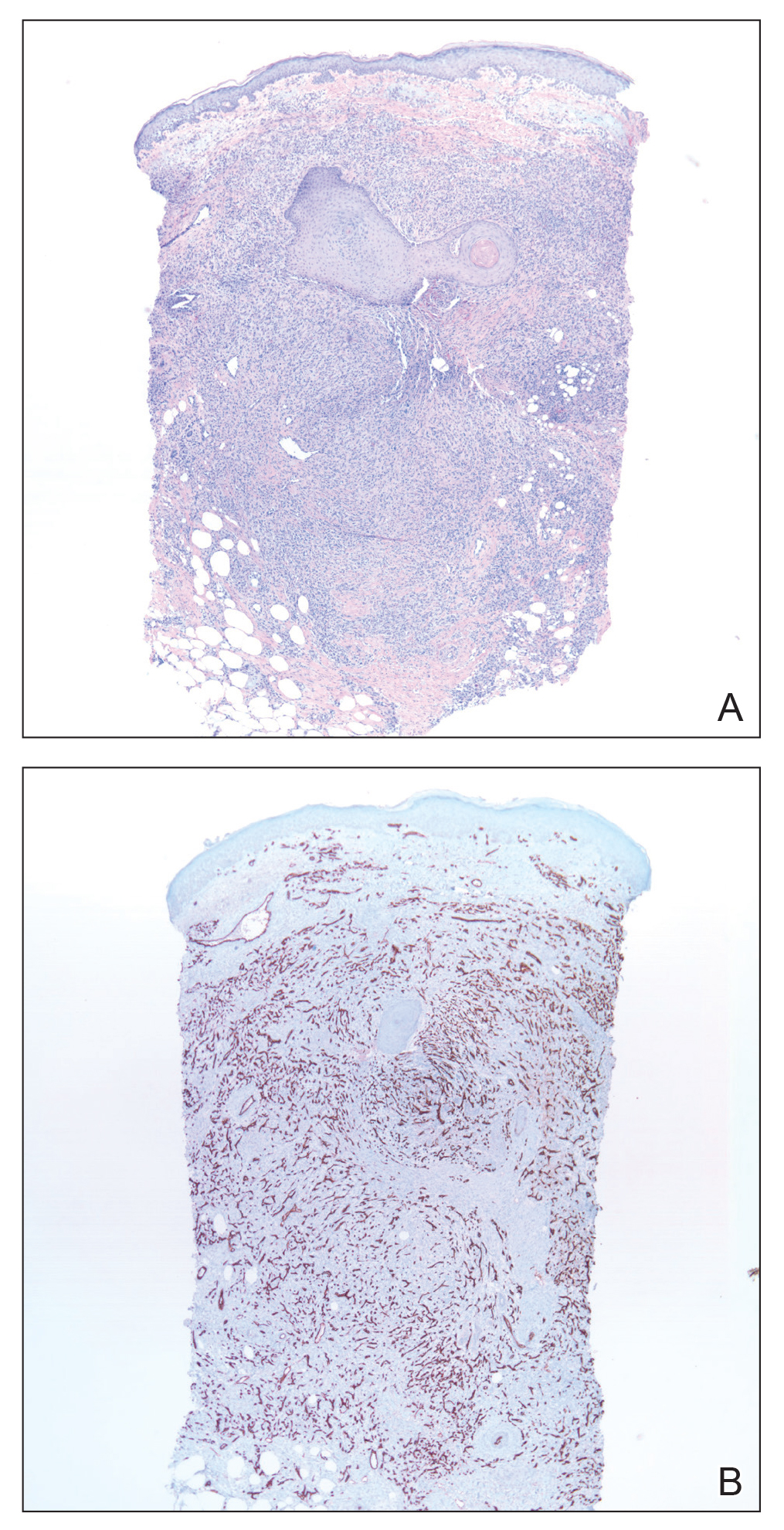
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
Case Reports
Patient 1
A 56-year-old white man with a history of hypertension, hyperlipidemia, sleep apnea, bilateral knee replacement, and cataract removal presented to the emergency department with a worsening rash on the left posterior medial leg of 6 months’ duration. He reported associated redness and tenderness with the plaques as well as increased swelling and firmness of the leg. He was admitted to the hospital where the infectious disease team treated him with cefazolin for presumed cellulitis. His condition did not improve, and another course of cefazolin was started in addition to oral fluconazole and clotrimazole–betamethasone dipropionate lotion for a possible fungal cause. Again, treatment provided no improvement.
He was then evaluated by dermatology. On physical examination, the patient had edema, warmth, and induration of the left lower leg. There also was an annular and serpiginous indurated plaque with minimal scale on the left lower leg (Figure 1). A firm, dark red to purple plaque on the left medial thigh with mild scale was present. There also was scaling of the right plantar foot.
Skin biopsy revealed a dermal capillary proliferation with a scattering of inflammatory cells including eosinophils as well as dermal fibrosis (Figure 2). Periodic acid–Schiff and human herpesvirus 8 (HHV-8) immunostains were negative. Considering the degree and depth of vascular proliferation, Mali-type acroangiodermatitis (AAD) was the favored diagnosis.
Patient 2
A 72-year-old white man presented with a firm asymptomatic growth on the left dorsal forearm of 3 months’ duration. It was located near the site of a prior squamous cell carcinoma that was excised 1 year prior to presentation. The patient had no treatment or biopsy of the presenting lesion. His medical and surgical history included polycystic kidney disease and renal transplantation 4 years prior to presentation. He also had an arteriovenous fistula of the left arm. His other chronic diseases included chronic obstructive lung disease, congestive heart failure, hypertension, type 2 diabetes mellitus, and obstructive sleep apnea.
On physical examination, the patient had a 1-cm violaceous nodule on the extensor surface of the left mid forearm. An arteriovenous fistula was present proximal to the lesion on the left arm (Figure 3).
Skin biopsy revealed a tightly packed proliferation of small vascular channels that tested negative for HHV-8, tumor protein p63, and cytokeratin 5/6. Erythrocytes were noted in the lumen of some of these vessels. Neutrophils were scattered and clustered throughout the specimen (Figure 4A). Blood vessels were highlighted with CD34 (Figure 4B). Grocott-Gomori methenamine-silver stain was negative for infectious agents. These findings favored AAD secondary to an arteriovenous malformation, consistent with Stewart-Bluefarb syndrome (SBS).
Comment
Presentation of AAD
Acroangiodermatitis is a rare chronic inflammatory skin process involving a reactive proliferation of capillaries and fibrosis of the skin that resembles Kaposi sarcoma both clinically and histopathologically. The condition has been reported in patients with chronic venous insufficiency,1 congenital arteriovenous malformation,2 acquired iatrogenic arteriovenous fistula,3 paralyzed extremity,4 suction socket lower limb prosthesis (amputees),5 and minor trauma.6-8 The lesions of AAD tend to be circumscribed, slowly evolving, red-violaceous (or brown or dusky) macules, papules, or plaques that may become verrucous or develop into painful ulcerations. They generally occur on the distal dorsal aspects of the lower legs and feet.110
Variants of AAD
Mali et al9 first reported cutaneous manifestations resembling Kaposi sarcoma in 18 patients with chronic venous insufficiency in 1965. Two years later, Bluefarb and Adams10 described kaposiform skin lesions in one patient with a congenital arteriovenous malformation without chronic venous insufficiency. It was not until 1974, however, that Earhart et al11 proposed the term pseudo-Kaposi sarcoma.10,11 Based on these findings, AAD is described as 2 variants: Mali type and SBS.
Mali-type AAD is more common and typically occurs in elderly men. It classically presents bilaterally on the lower extremities in association with severe chronic venous insufficiency.5 Skin lesions usually occur on the medial aspect of the lower legs (as in patient 1), dorsum of the heel, hallux, or second toe.12
The etiology of Mali-type AAD is poorly understood. The leading theory is that the condition involves reduced perfusion due to chronic edema, resulting in neovascularization, fibroblast proliferation, hypertrophy, and inflammatory skin changes. When AAD occurs in the setting of a suction socket prosthesis, the negative pressure of the stump-socket environment is thought to alter local circulation, leading to proliferation of small blood vessels.5,13
Stewart-Bluefarb syndrome usually involves a single extremity in young adults with congenital arteriovenous malformations, amputees, and individuals with hemiplegia or iatrogenic arteriovenous fistulae (as in patient 2).1 It was once thought to occur secondary to Klippel-Trenaunay-Weber syndrome; however, SBS rarely is accompanied by limb hypertrophy.9 Pathogenesis is thought to involve an angiogenic response to a high perfusion rate and high oxygen saturation, which leads to fibroblast proliferation and reactive endothelial hyperplasia.1,14
Diagnosis and Differential Diagnosis
Prompt identification of an underlying arteriovenous anomaly is critical, given the sequelae of high-flow shunts, which may result in skin ulceration, limb length discrepancy, cortical thinning of bone with regional osteoporosis, and congestive heart failure.1,5 Duplex ultrasonography is the first-line diagnostic modality because it is noninvasive and widely available. The key doppler feature of an arteriovenous malformation is low resistance and high diastolic pulsatile flow,1 which should be confirmed with magnetic resonance angiography or computed tomography angiography if present on ultrasonography.
The differential diagnosis of AAD includes Kaposi sarcoma, reactive angioendotheliomatosis, diffuse dermal angiomatosis, intravascular histiocytosis, glomeruloid angioendotheliomatosis, and angiopericytomatosis.15,16 These entities present as multiple erythematous, violaceous, purpuric patches and plaques generally on the extremities but can have a widely varied distribution. Some lesions evolve to necrosis or ulceration. Histopathologic analysis is useful to differentiate these entities.
Histopathology
The histopathologic features of AAD can be nonspecific; clinicopathologic correlation often is necessary to establish the diagnosis. Features include a proliferation of small thick-walled vessels, often in a lobular arrangement, in an edematous papillary dermis. Small thrombi may be observed. There may be increased fibroblasts; plump endothelial cells; a superficial mixed infiltrate comprised of lymphocytes, histiocytes, and eosinophils; and deposition of hemosiderin.2,5 These characteristics overlap with features of Kaposi sarcoma; AAD, however, lacks slitlike vascular spaces, perivascular CD34+ expression, and nuclear atypia. A negative HHV-8 stain will assist in ruling out Kaposi sarcoma.1,17
Management
Treatment reports are anecdotal. The goal is to correct underlying venous hypertension. Conservative measures with compression garments, intermittent pneumatic compression, and limb elevation are first line.18 Oral antibiotics and local wound care with topical emollients and corticosteroids have been shown to be effective treatments.19-21
Oral erythromycin 500 mg 4 times daily for 3 weeks and clobetasol propionate cream 0.05% healed a lower extremity ulcer in a patient with Mali-type AAD.21 In another patient, conservative treatment of Mali-type AAD failed, but rapid improvement of 2 lower extremity ulcers resulted after 3 weeks of oral dapsone 50 mg twice daily.22
Conclusion
Acroangiodermatitis is a rare entity that is characterized by erythematous violaceous papules and plaques of the extremities, commonly in the setting of chronic venous insufficiency or an arteriovenous shunt. Histopathologic analysis shows proliferation of capillaries with fibrosis, extravasation of erythrocytes, and deposition of hemosiderin without the spindle cells and slitlike vascular spaces characteristic of Kaposi sarcoma. Detection of an underlying arteriovenous malformation is essential, as the disease can have local and systemic consequences, such as skin ulceration and congestive heart failure.1 Treatment options are conservative, directed toward local wound care, compression, and management of complications, such as ulceration and infection, as well as obliterating any underlying arteriovenous malformation.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
- Parsi K, O’Connor AA, Bester L. Stewart-Bluefarb syndrome: report of five cases and a review of literature. Phlebology. 2015;30:505-514.
- Larralde M, Gonzalez V, Marietti R, et al. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol. 2001;18:325-327.
- Nakanishi G, Tachibana T, Soga H, et al. Pseudo-Kaposi’s sarcoma of the hand associated with acquired iatrogenic arteriovenous fistula. Indian J Dermatol. 2014;59:415-416.
- Landthaler M, Langehenke H, Holzmann H, et al. Mali’s acroangiodermatitis (pseudo-Kaposi) in paralyzed legs. Hautarzt. 1988;39:304-307.
- Trindade F, Requena L. Pseudo-Kaposi’s sarcoma because of suction socket lower limb prosthesis. J Cutan Pathol. 2009;36:482-485.
- Yu-Lu W, Tao Q, Hong-Zhong J, et al. Non-tender pedal plaques and nodules: pseudo-Kaposi’s sarcoma (Stewart-Bluefarb type) induced by trauma. J Dtsch Dermatol Ges. 2015;13:927-930.
- Del-Río E, Aguilar A, Ambrojo P, et al. Pseudo-Kaposi sarcoma induced by minor trauma in a patient with Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1993;18:151-153.
- Archie M, Khademi S, Aungst D, et al. A rare case of acroangiodermatitis associated with a congenital arteriovenous malformation (Stewart-Bluefarb Syndrome) in a young veteran: case report and review of the literature. Ann Vasc Surg. 2015;29:1448.e5-1448.e10.
- Mali JW, Kuiper JP, Hamers AA. Acro-angiodermatitis of the foot. Arch Dermatol. 1965;92:515-518.
- Bluefarb SM, Adams LA. Arteriovenous malformation with angiodermatitis. stasis dermatitis simulating Kaposi’s disease. Arch Dermatol. 1967;96:176-181.
- Earhart RN, Aeling JA, Nuss DD, et al. Pseudo-Kaposi sarcoma. A patient with arteriovenous malformation and skin lesions simulating Kaposi sarcoma. Arch Dermatol. 1974;110:907-910.
- Lugovic´ L, Pusic´ J, Situm M, et al. Acroangiodermatitis (pseudo-Kaposi sarcoma): three case reports. Acta Dermatovenerol Croat. 2007;15:152-157.
- Horiguchi Y, Takahashi K, Tanizaki H, et al. Case of bilateral acroangiodermatitis due to symmetrical arteriovenous fistulas of the soles. J Dermatol. 2015;42:989-991.
- Dog˘an S, Boztepe G, Karaduman A. Pseudo-Kaposi sarcoma: a challenging vascular phenomenon. Dermatol Online J. 2007;13:22.
- Mazloom SE, Stallings A, Kyei A. Differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol. 2017;39:33-39.
- Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
- Kanitakis J, Narvaez D, Claudy A. Expression of the CD34 antigen distinguishes Kaposi’s sarcoma from pseudo-Kaposi’s sarcoma (acroangiodermatitis). Br J Dermatol. 1996;134:44-46.
- Pires A, Depairon M, Ricci C, et al. Effect of compression therapy on a pseudo-Kaposi sarcoma. Dermatology. 1999;198:439-441.
- Hayek S, Atiyeh B, Zgheib E. Stewart-Bluefarb syndrome: review of the literature and case report of chronic ulcer treatment with heparan sulphate (Cacipliq20®). Int Wound J. 2015;12:169-172.
- Varyani N, Thukral A, Kumar N, et al. Nonhealing ulcer: acroangiodermatitis of Mali. Case Rep Dermatol Med. 2011;2011:909383.
- Mehta AA, Pereira RR, Nayak C, et al. Acroangiodermatitis of Mali: a rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76:553-556.
- Rashkovsky I, Gilead L, Schamroth J, et al. Acro-angiodermatitis: review of the literature and report of a case. Acta Derm Venereol. 1995;75:475-478.
Practice Points
- Acroangiodermatitis (AAD) may mimic Kaposi sarcoma clinically and histopathologically. A human herpesvirus 8 stain is helpful to differentiate these two entities.
- Diagnosis of AAD should prompt investigation of an underlying arteriovenous malformation, as the disease may have systemic consequences such as congestive heart failure.
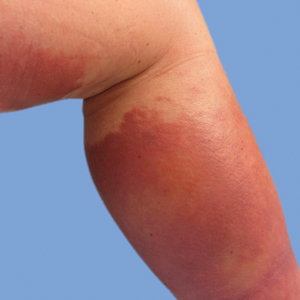
Relapsing Polychondritis in Human Immunodeficiency Virus
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
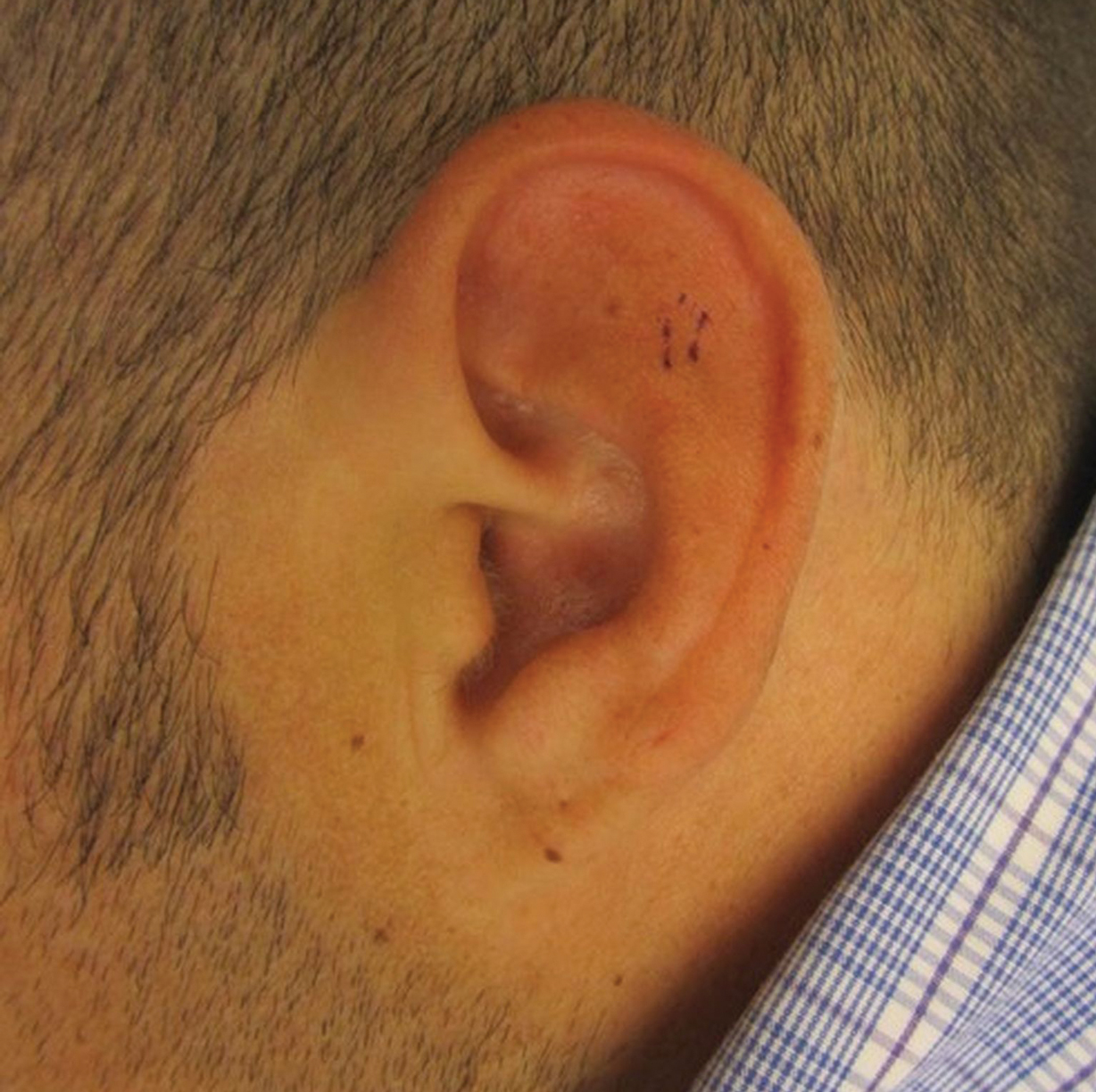
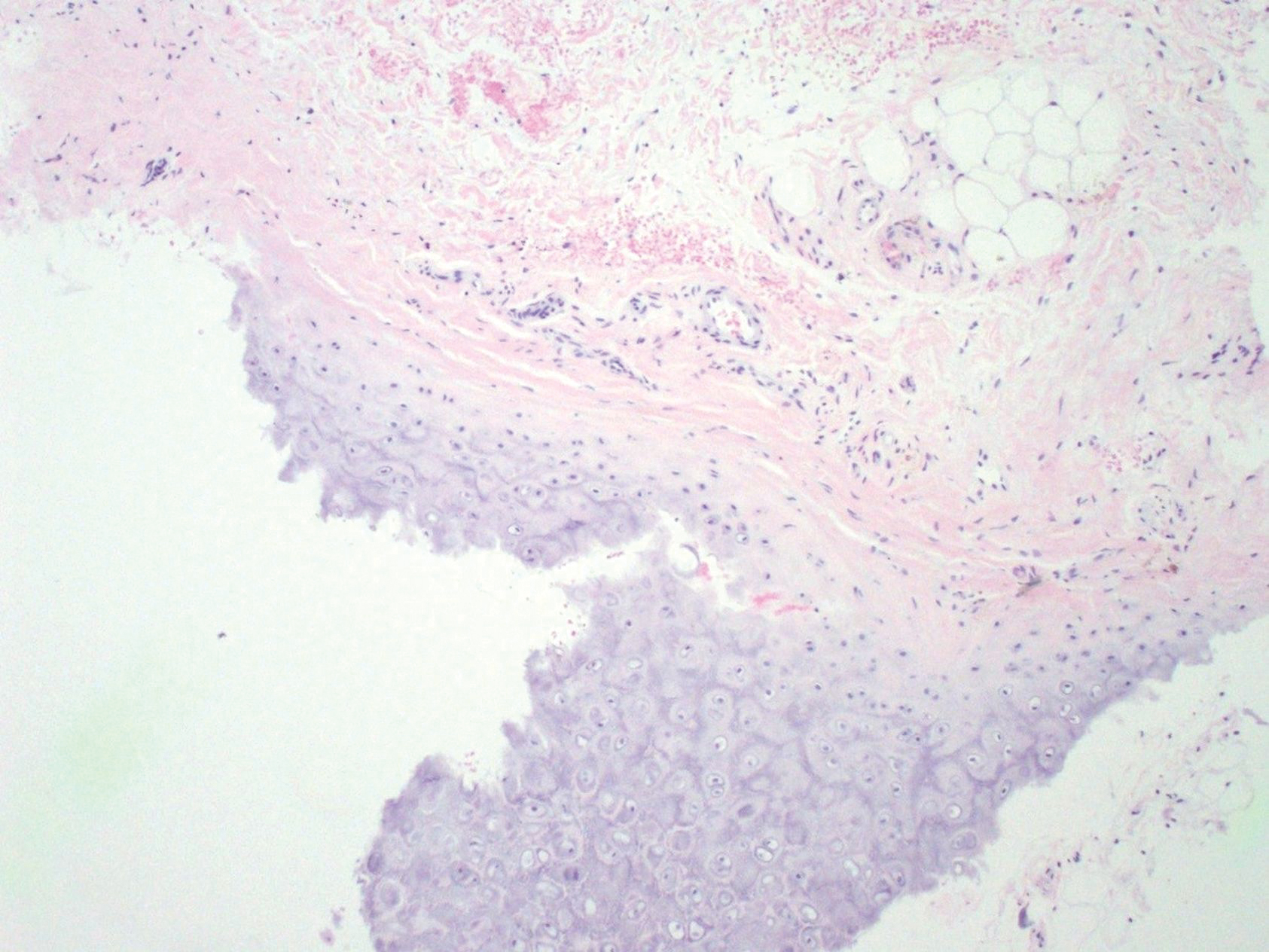
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Practice Points
- Relapsing polychondritis (RP) is characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures, most often manifesting as ear inflammation that involves the auricle but spares the lobe, nasal chondritis, and arthralgia.
- Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge.
- One-third of RP patients have coexisting autoimmune disease.
- Treatment of RP depends on severity of disease.
- Dermatologists must be aware of the potential for development of RP in the setting of human immunodeficiency virus infection; a missed diagnosis of this progressive disease has the potential to be life-threatening.
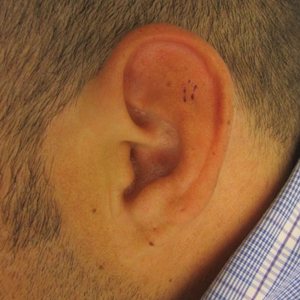
Necrobiosis Lipoidica With Superimposed Pyoderma Vegetans
Case Report
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.

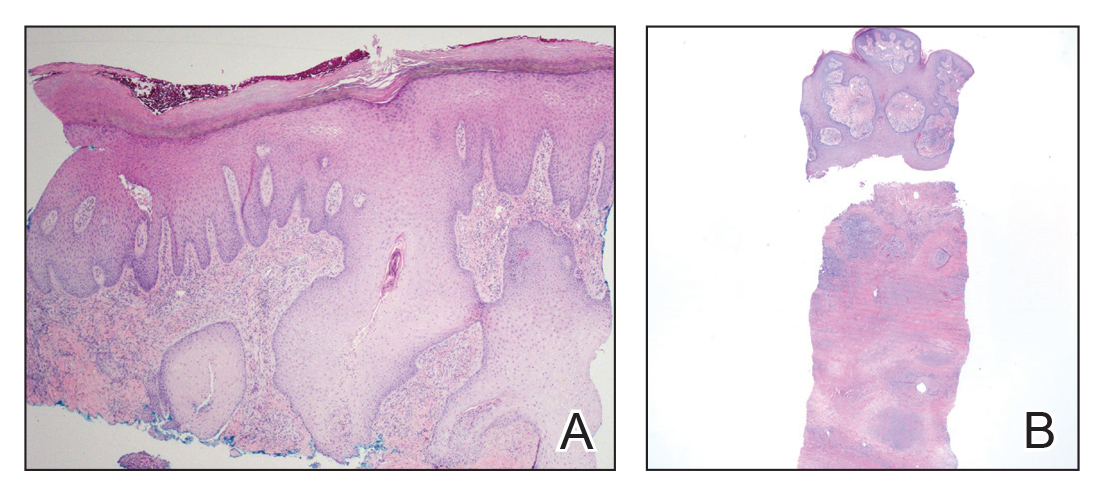
The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
Case Report
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.
The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
Case Report
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.
The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
Practice Points
- Necrobiosis lipoidica (NL), a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial-wall thickening, is most often seen in association with insulin-dependent diabetes mellitus (DM).
- Asymptomatic, well-circumscribed, violaceous papules and nodules coalesce into plaques on the lower extremities, face, or trunk in NL.
- Treatment mainstay is topical and intralesional corticosteroids at active borders of lesions. Other treatments used with some success include tumor necrosis factor 11α inhibitors, topical tretinoin, topical tacrolimus, and skin grafting. Control and management of DM can be helpful.
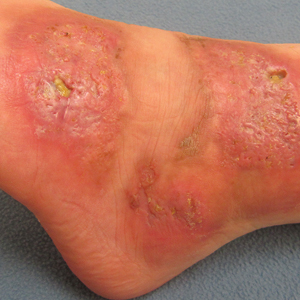
Inflammatory Linear Verrucous Epidermal Nevus Responsive to 308-nm Excimer Laser Treatment
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
 and after 18 treatment sessions with the 308-nm excimer laser (B)...")
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
Eosinophilic Pustular Folliculitis With Underlying Mantle Cell Lymphoma
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
, neck, and chest (B), as well as edematous pink papules and plaques on the forehead (C and D).")
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
(H&E, original magnifications ×10 and ×20).")
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Practice Points
- Recalcitrant folliculocentric papules and pustules involving the head, trunk, arms, and legs should raise suspicion of possible eosinophilic pustular folliculitis (EPF).
- Underlying hematopoietic malignancy may be associated with cases of EPF.