User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Recurrent Aphthous Stomatitis: Clinical Experience From a University Hospital in Brazil
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide,usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide,usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
To the Editor:
Recurrent aphthous stomatitis (RAS) is a mucocutaneous condition characterized by single or multiple, painful,1,2 round ulcerations of variable sizes with a tendency for recurrence, most commonly located in nonkeratinized areas of the oral mucosa. Pathergy commonly is observed.3 Although many authors consider the terms RAS andaphtha to be synonymous,4,5 differentiating the clinical lesion (aphthous ulceration) from the disease (aphtha or RAS) can be useful, as several other diseases can at times manifest with similar ulcers (called aphthoid lesions), such as pemphigus vulgaris, mucous membrane pemphigoid, and erythema multiforme.6
It is estimated that approximately 20% of individuals worldwide have at least one episode of aphtha during their lifetime,7 and it is considered the most common disease of the oral mucosa.8,9 However, only patients presenting with severe acute outbreaks or frequent relapses typically seek medical treatment. Clinically, aphthous ulcers are classified as aphtha minor (small number of small lesions), aphtha major (large deep lesions that also can affect the minor salivary glands with intense necrosis, difficulty in healing, and mucosal scarring), and aphtha herpetiformis (innumerous tiny lesions that reappear in recurring outbreaks).1-3 The term complex aphthosis was introduced in 198510 and is defined as recurrent oral and genital aphthous ulcerations or recurring multiple oral aphthous ulcers in the absence of systemic manifestations or Behçet disease11,12; however, complex aphthosis also has been reported as frequent episodes of ulcerations that may be associated with systemic diseases including Behçet disease.13,14
Currently, RAS is considered an immunologically mediated alteration in cutaneous mucosal reactivity with a multifactorial systemic cause. Underlying conditions such as Behçet disease, inflammatory bowel disease (IBD), iatrogenic immunosuppression (eg, following solid organ transplantation), AIDS, and cyclic neutropenia may or may not be detected.11-13
Our retrospective study explored the systemic nature of RAS. We reviewed patient records to evaluate underlying systemic conditions associated with the diagnosis of RAS and the use of oral medications in managing the disease. Medical records from the Department of Dermatology of the University of São Paulo, Brazil, from 2003 to 2017 were reviewed to identify patients with a diagnosis of RAS. Clinical classification of RAS—minor, major, or herpetiform—as well as the presence of aphthous lesions in other locations and the presence of other associated inflammatory cutaneous manifestations also were noted. Associated systemic diseases and treatments for RAS were recorded. Patients for whom the diagnosis of RAS was changed during follow-up were excluded. Because this was a retrospective analysis of medical records and without any patient risk, informed consent was not needed.
Medical records for 125 patients were reviewed; 63 were male (50.4%), and 62 were female (49.6%). The age at onset of symptoms, which ranged from a few months after birth to 74 years, was reported in only 92 (73.6%) patient medical records. Of these, 30 (32.6%) reported onset before 20 years of age, 39 (42.4%) between 20 and 39 years, 17 (18.5%) between 40 and 59 years, and 6 (6.5%) at 60 years or older. Morphologically, 72 (57.6%) had minor, 42 (33.6%) had major, and 11 (8.8%) had herpetiform aphthous ulcers. None of the patients presented with sporadic lesions; the disease was long-standing and persistent in all cases (complex aphthosis).
Regarding the location of the ulcers, 92 (73.6%) patients had lesions on the oral mucosa only. Some patients had lesions in more than one site in addition to the oral mucosa: 32 (25.6%) had aphthae in the genital/groin region and 4 (3.2%) presented with perianal/anal aphthae. Nineteen patients (19.2%) presented other cutaneous manifestations in addition to aphthae: 11 (45.8%) had folliculitis/pseudofolliculitis, and 8 (33.3%) had erythema nodosum (EN). Eight patients (33.3%) presented with uveitis, and 6 (25%) presented with concomitant arthralgia/arthritis. Fifty-four patients (43.2%) had confirmed or suspected associated disease: Behçet disease (21 [38.9%]), IBD (10 [18.5%]), solid organ transplantation (7 [13.0%])(kidney, 4 [57.1%]; heart, 2 [28.6%]; liver, 1 [14.3%]), HIV infection (6 [11.1%]), lymphoma (1 [1.9%]), aplastic anemia (1 [1.9%]), or myelodysplastic syndrome (1 [1.9%]). Ten patients (18.5%) presented with other diseases under investigation (eg, unidentified rheumatologic disease, unexplained neutropenia, undiagnosed immunodeficiencies, autoinflammatory syndromes, possible cyclic neutropenia).
Biopsies of the oral mucosa were performed in 31 patients. Histopathologic findings will be discussed in a future publication (unpublished data).
Five patients (4.0%) were lost to follow-up and did not receive treatment; 10 (8.0%) received only topical treatment (analgesics and/or corticosteroids). All 9 (7.2%) patients undergoing intralesional corticosteroid injections also were on a systemic treatment. One hundred ten (88.0%) patients were treated systemically—with colchicine (84/110 [76.4%]), thalidomide (43/110 [39.1%]), small pulses of oral corticosteroids (26 [23.6%]), dapsone (12/110 [10.9%]), or pentoxifylline (3 [2.7%]). Furthermore, in patients with associated diseases, treatment of the underlying condition was conducted when available, and follow-up was carried out in conjunction with the appropriate specialists. For treatment of the associated disease, patients received other medications such as methotrexate, azathioprine, cyclophosphamide, intravenous corticosteroid pulse, and immunobiologics.
The prevalence of RAS between sexes in our study population was similar (50.4% male; 49.6% female). Results from prior studies have been mixed; some reported a higher prevalence in females,15-18 while others found no predilection for sex among patients diagnosed with RAS.19,20 In our analysis, 75% of patients experienced symptoms of RAS before 40 years of age; in prior studies, up to 56% of patients experienced symptoms between the ages of 20 and 40 years.21,22
In our study, 26.4% of patients had extraoral aphthae. Genital lesions have been described as infrequent,23 and lesions manifesting in other mucous membranes or on the skin are rare.24 A study reported genital involvement in 8% to 13% of patients with oral aphtha.25 We observed genital involvement in 25.6% of patients. Likewise, this higher value may be due to our study population of patients referred to our university hospital. In our study, 19.2% of patients presented with other inflammatory manifestations in addition to aphthous ulcerations (eg, folliculitis, EN, uveitis, arthritis). As dermatologists in a tertiary reference hospital, we actively look for such associations in every aphtha patient, which may not be the case in many nondermatologic oral care services.
In our study population, 43.2% of patients were diagnosed with or were under investigation for systemic diseases known to be associated with RAS. We found associations with Behçet disease most frequently, followed by IBD,26 solid organ transplantation, and HIV. In this group of patients, the respective systemic disease was active or poorly controlled. In transplant recipients, aphtha major was the most common type, similar to other studies.27 We observed no notable difference in the clinical picture of the oral ulcers in patients with a well-established systemic disease vs those without.
Most of our cases did not present findings other than aphtha, indicating that the intrinsic defect that predisposes to RAS is always systemic. Even mild and sporadic cases may be attributable to a systemic disorder of cutaneous-mucosal reactivity. The predisposition to RAS never originates in the oral cavity, hence the confusion caused and the uselessness of studies that relate aphthae to factors such as local food allergies, pH changes, or local infection with microorganisms.5,28 The disease course (reducing the frequency of lesion appearance and accelerating the healing of extensive lesions) is only modified with systemic treatment, with local measures proving to be only moderately useful to relieve pain. We believe that RAS can in many ways be compared to EN and pyoderma gangrenosum (PG): some systemic conditions that predispose patients to EN and PG also may predispose them to RAS (eg, IBD, hematologic disorders). Similar to RAS, many cases of EN and PG are idiopathic. In addition, pathergy also occurs in PG.11,13
We were unable to observe or establish any predictive clinical element that could indicate a better or worse response to the prescribed treatments, which also has been noted by other authors.3,4 Treatment of RAS is empiric, generally starting with drugs that are easier to prescribe and with fewer adverse effects, then progressing to more complex drugs when a good response is not obtained. Colchicine was the most commonly prescribed medication (76.4% [84/110]). It has been proposed by several authors3,4 as a first-line systemic medication for the treatment of recurrent aphthae, as it has been shown to be effective and safe. The dosage ranged from 0.5 mg twice daily to 0.5 mg 4 times daily. Dapsone is an established drug for aphtha29,30 and was used in 12 of our patients. The dosage used in our patients ranged from 50 to 100 mg/d. Adverse effects such as hemolytic anemia frequently are seen, and one of the patients in our study developed DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome in response to dapsone. In 7 cases, colchicine and dapsone were used together, which is believed to potentiate the therapeutic effects. This combination may be useful in patients for whom thalidomide cannot be used or those who have not improved with monotherapy.29 Thalidomide is considered one of the most effective drugs for RAS.30,31 Forty-three patients in our analysis were treated with thalidomide,usually as a first choice. The dosage ranged from 100 to 200 mg/d. It was mainly chosen in disabling pediatric cases, adult men with aphthous major, and women with no risk for pregnancy. Due to its potential adverse effects, thalidomide has been recommended when there is no response with other medications that are dose dependent; severe adverse effects such as thromboembolism and peripheral neuropathy are rare.31 Oral corticosteroids were used in 26 patients, aiming at rapid improvement in very symptomatic cases; however, due to the potential for long-term adverse effects, in all cases they were prescribed in combination with another medication that was maintained after the corticosteroid was discontinued.
We highlight the systemic nature of RAS as well as its frequent association with systemic diseases and other correlated manifestations (pustules, EN, arthralgia). We also emphasize the importance of using oral medications to adequately control the disease and do not recommend topical medications aimed at treating local causes. Dermatologists should be consulted in managing severe cases of RAS.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
- Buño IJ, Huff JC, Weston WL, et al. Elevated levels of interferon gamma, tumor necrosis factor alpha, interleukins 2, 4, and 5, but not interleukin 10, are present in recurrent aphthous stomatitis. Arch Dermatol. 1998;134:827-831.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728- 732.
- Natah SS, Konttinen YT, Enattah NS, et al. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg. 2004;33:221-234.
- Zunt SL. Recurrent aphthous stomatitis. Dermatol Clin. 2003;21:33-39.
- Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. number VI. recurrent aphthous stomatitis. Oral Dis. 2006;12:1-21.
- Chams-Davatchi C, Shizarpour M, Davatchi F, et al. Comparison of oral aphthae in Behçet’s disease and idiopathic recurrent aphthous stomatitis. Adv Exp Med Biol. 2003;528:317-320.
- Schemel-Suárez M, López-López J, Chimenos-Küstner E. Oral ulcers: differential diagnosis and treatment [in Spanish]. Med Clin (Barc). 2015;145:499-503.
- S´lebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205-215.
- Edgar NR, Saleh D, Miller RA. Recurrent aphthous stomatitis: a review. J Clin Aesthet Dermatol. 2017;10:26-36.
- Jorizzo JL, Taylor RS, Schmalstieg FC, et al. Complex aphthosis: a forme fruste of Behçet’s syndrome? J Am Acad Dermatol. 1985;13:80-84.
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet’s disease. Dermatol Clin. 2003;21:41-48.
- Bulur I, Melrem O. Behçet disease: new aspects. Clin Dermatol. 2017;35:421-434.
- Cui RZ, Rogers RS 3rd. Recurrent aphthous stomatitis. Clin Dermatol. 2016;34:475-481.
- Femiano F, Lanza A, Buonaiuto C, et al. Guidelines for diagnosis and management of aphthous stomatitis. Pediatr Infect Dis J. 2007;26:728-732.
- Ship II. Epidemiologic aspects of recurrent aphthous ulcerations. Oral Surg Oral Med Oral Pathol. 1972;33:400-406.
- Ship JA. Recurrent aphthous stomatitis. an update. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:141-147.
- Wilhelmsen NS, Weber R, Monteiro F, et al. Correlation between histocompatibility antigens and recurrent aphthous stomatitis in the Brazilian population. Braz J Otorhinolaryngol. 2009;75:426-431.
- S´lebioda Z, Dorocka-Bobkowska B. Systemic and environmental risk factors for recurrent aphthous stomatitis in a Polish cohort of patients. Postepy Dermatol Alergol. 2019;36:196-201.
- Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int. 2000;31:95-112.
- Brocklehurst P, Tickle M, Glenny AM, et al. Systemic interventions for recurrent aphthous stomatitis (mouth ulcers). Cochrane Database Syst Rev. 2012;12:CD005411.
- Belenguer-Guallar I, Jiménez-Soriano Y, Ariadna Claramunt-Lozano A. Treatment of recurrent aphthous stomatitis. a literature review. J Clin Exp Dent. 2014;6:E168-E174.
- Bagán JV, Sanchis JM, Milián MA, et al. Recurrent aphthous stomatitis. a study of the clinical characteristics of lesions in 93 cases. J Oral Pathol Med. 1991;20:395-397.
- Huppert JS, Gerber MA, Deitch HR, et al. Vulvar ulcers in young females: a manifestation of aphthosis. J Pediatr Adolesc Gynecol. 2006;19:195-204.
- Scully C, Porter S. Recurrent aphthous stomatitis: current concepts of etiology, pathogenesis and management. J Oral Pathol Med. 1989;18:21-27
- Chapel TA. Origins of penile ulcerations. Arch Androl. 1979; 3: 351-357.
- Lourenço SV, Hussein TP, Bologna SB, et al. Oral manifestations of inflammatory bowel disease: a review based on the observation of six cases. J Eur Acad Dermatol Venereol. 2010;24:204-207.
- Nico MM, Brito AE, Martins LE, et al. Oral ulcers in an immunosuppressed 5-year-old boy. Clin Exp Dermatol. 2008;33:367-368.
- Trakji B, Baroudi K, Kharma Y. The effect of dietary habits on the development of the recurrent aphthous stomatitis. Niger Med J. 2012;53:9-11.
- Lynde CB, Bruce AJ, Rogers RS 3rd. Successful treatment of complex aphthosis with colchicine and dapsone. Arch Dermatol. 2009;145:273-276.
- Letsinger JA, McCarty MA, Jorizzo JL. Complex aphthosis: a large case series with evaluation algorithm and therapeutic ladder from topicals to thalidomide. J Am Acad Dermatol. 2005(3 pt 1);52:500-508.
- Hello M, Barbarot S, Bastuji-Garin S, et al. Use of thalidomide for severe recurrent aphthous stomatitis: a multicenter cohort analysis. Medicine (Baltimore). 2010;89:176-182.
Practice Points
- The process that leads to the formation of aphthous ulcerations is always systemic, not local, even in the absence of a diagnosable systemic disease.
- Relapsing cases of aphthae should be treated with systemic medication.
Advancements in Targeted Therapies for Vitiligo: Prioritizing Equity in Drug Development
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).

Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).
Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
Vitiligo is a common acquired autoimmune disease that causes depigmented patches to develop throughout the skin , with descriptions dating back more than 3000 years to the earliest known Indian and Egyptian texts. Approximately 1.4% of the worldwide population has vitiligo,1 and onset follows a bimodal age distribution with an early-onset population (mean age at onset, 10.3 years) as well as an adult-onset population (mean age at onset, 34 years).2 Vitiligo manifests as well-defined, irregular, depigmented macules and patches surrounded by normal skin. The patches can vary in size from a few millimeters to several centimeters. There may be signs of inflammation, and the lesions can be itchy, but in most cases vitiligo is asymptomatic. In nonsegmental vitiligo, the depigmented patches are ymmetrical, can appear in any area of the body, and commonly progress slowly. In segmental vitiligo, the patches are unilateral, rarely cross the midline of the body, and are localized to one area. Segmental vitiligo commonly appears in childhood and progresses rapidly but stops abruptly within 6 to 12 months and remains stable, usually for life.3 Although the condition may be more apparent in patients with skin of color, vitiligo manifests at a similar rate in individuals of all races and ethnicities.4
Similar to most autoimmune diseases, vitiligo has a strong genetic predisposition. Although the overall prevalence of vitiligo is less than 2%, having a family history of vitiligo (ie, a first-degree relative with vitiligo) increases an individual’s risk to 6%, while concordance in identical twins is 23%.5 Beyond genetic predisposition, there is strong evidence that environmental exposures, such as hair dyes, contribute to risk for disease.6 Interestingly, vitiligo is associated with polyautoimmunity—the presence of multiple autoimmune diseases in a single patient,7 such as type 1 diabetes mellitus, rheumatoid arthritis, autoimmune thyroid disease, pernicious anemia, and Addison disease. Similar to vitiligo itself, polyautoimmunity likely is driven by a combination of genetic and environmental factors.5
We provide a brief overview of clinical trial results of Janus kinase (JAK) inhibitors for treating vitiligo and discuss the trial cohorts, with an emphasis on the impact of cohort demographic composition for individuals with skin of color. We recommend factors that investigators should consider to ensure equitable representation of individuals with skin of color in future clinical trials.
Autoimmune Pathogenesis and Treatment With JAK Inhibitors
Vitiligo is driven by autoreactive CD8+ T cells that target melanocytes and secrete IFN-g. Signaling of IFN-g occurs through the JAK–signal transducer and activator of transcription (JAK-STAT) pathway, leading to transcriptional changes that activate proinflammatory genes such as the chemokine CXCL10, which is required for the directed accumulation of melanocyte-specific CD8+ T cells at the epidermis where melanocytes reside.8 Once vitiligo has been initiated, the disease persists due to the presence of resident memory T cells that remain in the skin and destroy new melanocytes.9,10
Given the central role of IFN-g signaling in the pathogenesis of vitiligo, drugs that inhibit JAK signaling are appealing to treat the disease. These JAK inhibitors bind to the kinase domain of JAK to prevent its activation, thus preventing downstream signaling events including STAT phosphorylation and its translocation to the nucleus, which ultimately stops the upregulation of inflammatory gene transcription. This process attenuates the autoimmune response in the skin and results in repigmentation of vitiligo lesions. In 2022, the US Food and Drug Administration approved the topical JAK inhibitor ruxolitinib for the treatment of vitiligo. Additional clinical trials have been initiated to test oral JAK inhibitors—ritlecitinib (ClinicalTrials.gov identifiers NCT06163326, NCT06072183, NCT05583526), povorcitinib (NCT04818346, NCT06113445, NCT06113471), and upadacitinib (NCT04927975, NCT06118411)—with strong results reported so far.11
The effects of JAK inhibitors can be striking, as shown in the Figure. A patient of one of the authors (J.E.H.) used topical ruxolitinib on only the left arm for approximately 36 weeks and results were as expected—strong repigmentation of only the treated area, which is possible with JAK inhibitors. Indeed, 2 phase 3 studies—Topical Ruxolitinib Evaluation in Vitiligo (TRuE-V1 and TRuE-V2)—showed that approximately 30% of participants in TRuE-V1 (N=330) and 30.9% of participants in TRuE-V2 (N=344) achieved at least 75% improvement over baseline in the facial vitiligo area scoring index (VASI).12 In the oral ritlecitinib phase 2b study, 12.1% of the 187 participants on the highest tested dose of ritlecitinib (loading dose of 200 mg/d for 28 days, followed by 50 mg/d maintenance dose) achieved at least 75% improvement over baseline in the VASI at 24 weeks.11 Although this rate is lower than for topical ruxolitinib, this trial required all participants to have active disease (unlike the TRuE-V trials of ruxolitinib), which likely created a higher bar for repigmentation and thus resulted in fewer participants achieving the primary outcome at the early 6-month end point. Extension of treatment through 48 weeks demonstrated continued improvement over baseline without any evidence of plateau.11 Although treatment with JAK inhibitors can result in dramatic repigmentation of vitiligo patches, it falls short of providing a permanent cure, as stopping treatment results in relapse (ie, the return of depigmented lesions).
Racial Disparities in Clinical Trials
Even though vitiligo affects all skin types and races/ethnicities with similar prevalence and severity, the proportion of individuals with darker skin types enrolled in these clinical trials fails to match their representation in the population as a whole. A study examining the prevalence of vitiligo in the United States reported that Black or African American individuals represented 15.8% of vitiligo diagnoses in the United States4 even though they are only 12.7% of the total US population. However, Black or African American individuals comprised only 5% of the combined participants in the TRuE-V clinical trials for topical ruxolitinib12 and 2.7% of the participants in the phase 2b study of oral ritlecitinib.11 This lack of appropriate representation is not unique to JAK inhibitors or other vitiligo trials. Indeed, the US Food and Drug Administration reported that Black or African American individuals comprised only 8% of participants for all clinical trials in 2020.13
Efficacy Metrics Beyond Repigmentation
Disparities in quality-of-life (QOL) metrics in diseases affecting individuals with skin of color also exist. In vitiligo, the contrast between affected and unaffected skin is greater in patients with skin of color, which means that for a given VASI score, the visibility of depigmentation as well as repigmentation may be variable among patients. Additionally, there is evidence that QOL concerns vary between patients with skin of color and those with lighter skin types. Ezzedine et al14 found that QOL concerns in vitiligo patients with darker skin focused more on appearance, while concerns in vitiligo patients with lighter skin focused more on skin cancer risk. In addition to QOL differences among individuals with different skin types, there also are well-documented differences in attitudes to vitiligo among certain ethnic or cultural groups.15 For example, the Rigveda (an ancient Hindu text) indicates that individuals with vitiligo and their progeny are disqualified from marriage. Although the JAK inhibitor clinical trials for vitiligo did not appear to show differences in the degree of repigmentation among different skin types or races/ethnicities, QOL measures were not collected as a secondary end point in these studies—despite the fact that at least 1 study had documented that QOL measures were not uniform across patients when stratified by age and extent of disease.1,11,12 This same study also presented limited data suggestive of lower QOL in patients with the darkest skin phototype.1
Considerations for Future Clinical Trials
It is logical to assume that every clinical trialist in dermatology seeks equitable representation among a diverse set of races, ethnicities, and skin types, but achieving this goal remains elusive. Two recent publications16,17 outlined the challenges and examined solutions to address enrollment disparities, including several barriers to diversity among clinical trial participants: awareness of the clinical trials among minority populations; easy access to clinical trial sites; reluctance to participate because of prior experiences of discrimination, even if unrelated to clinical trials; and a lack of workforce diversity among the clinical trialist teams. To overcome these barriers, a multifaceted approach is needed that requires action at the level of the patient, provider, community, and institution. Once diverse representation is achieved, investigators should consider the need for QOL metrics as a secondary outcome in their trials, which will ensure that the intended clinical effect is matched by patient expectations across different races and ethnicities based on the potential differential impact that diseases such as vitiligo can have on patients with skin of color.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
- Bibeau K, Pandya AG, Ezzedine K, et al. Vitiligo prevalence and quality of life among adults in Europe, Japan and the USA. J Eur Acad Dermatol Venereol. 2022;36:1831-1844.
- Jin Y, Roberts GHL, Ferrara TM, et al. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat Commun. 2019;10:391.
- Rodrigues M, Ezzedine K, Hamzavi I, et al; Vitiligo Working Group. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13.
- Gandhi K, Ezzedine K, Anastassopoulos KP, et al. Prevalence of vitiligo among adults in the United States. JAMA Dermatol. 2022;158:43-50.
- Spritz RA, Santorico SA. The genetic basis of vitiligo. J Invest Dermatol. 2021;141:265-73.
- Harris JE. Chemical-induced vitiligo. Dermatol Clin. 2017;35:151-161.
- Ahmed F, Moseley I, Ragi SD, et al. Vitiligo in underrepresented communities: an all of us database analysis. J Am Acad Dermatol. 2023;88:945-948.
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621-648.
- Richmond JM, Strassner JP, Zapata L Jr, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:eaam7710.
- Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139:769-778.
- Ezzedine K, Peeva E, Yamaguchi Y, et al. Efficacy and safety of oral ritlecitinib for the treatment of active nonsegmental vitiligo: a randomized phase 2b clinical trial. J Am Acad Dermatol. 2023;88:395-403.
- Rosmarin D, Passeron T, Pandya AG, et al. Two phase 3, randomized, controlled trials of ruxolitinib cream for vitiligo. N Engl J Med. 2022;387:1445-1455.
- Cavazzoni P, Anagnostiadis E, Lolic M. Drug trials snapshots summary report. US Food and Drug Administration website. Accessed March 19, 2024. https://www.fda.gov/media/145718/download
- Ezzedine K, Grimes PE, Meurant JM, et al. Living with vitiligo: results from a national survey indicate differences between skin phototypes. Br J Dermatol. 2015;173:607-609.
- Elbuluk N, Ezzedine K. Quality of life, burden of disease, co-morbidities, and systemic effects in vitiligo patients. Dermatol Clin. 2017;35:117-128.
- Kahn JM, Gray DM 2nd, Oliveri JM, et al. Strategies to improve diversity, equity, and inclusion in clinical trials. Cancer. 2022;128:216-221.
- Nolan TS, McKoy A, Gray DM 2nd, et al. Virtual community engagement for retention of black men in clinical research. Am J Mens Health. 2023;17:15579883221147767.
Practice Points
- Vitiligo is an autoimmune disease of the skin that affects all skin types but can be particularly disfiguring in those with skin of color.
- Ruxolitinib, a topical Janus kinase (JAK) inhibitor, is the only US Food and Drug Administration–approved treatment to repigment the skin in vitiligo and has shown efficacy for individuals with all skin phototypes.
- Individuals with skin of color are underrepresented in patient cohorts for JAK inhibitor clinical trials for vitiligo, mirroring a phenomenon seen in the majority of clinical trials. Ensuring diverse participant enrollment and measuring quality-of-life metrics will strengthen future clinical trials for treatment of vitiligo and other skin diseases impacting patients with skin of color.

Best Practices for Clinical Image Collection and Utilization in Patients With Skin of Color
Clinical images are integral to dermatologic care, research, and education. Studies have highlighted the underrepresentation of images of skin of color (SOC) in educational materials,1 clinical trials,2 and research publications.3 Recognition of this disparity has ignited a call to action by dermatologists and dermatologic organizations to address the gap by improving the collection and use of SOC images.4 It is critical to remind dermatologists of the importance of properly obtaining informed consent and ensuring images are not used without a patient’s permission, as images in journal articles, conference presentations, and educational materials can be widely distributed and shared. Herein, we summarize current practices of clinical image storage and make general recommendations on how dermatologists can better protect patient privacy. Certain cultural and social factors in patients with SOC should be considered when obtaining informed consent and collecting images.
Clinical Image Acquisition
Consenting procedures are crucial components of proper image usage. However, current consenting practices are inconsistent across various platforms, including academic journals, websites, printed text, social media, and educational presentations.5
Current regulations for use of patient health information in the United States are governed by the Health Insurance Portability and Accountability Act (HIPAA)of 1996. Although this act explicitly prohibits use of “full face photographic images and any comparable images” without consent from the patient or the patient’s representative, there is less restriction regarding the use of deidentified images.6 Some clinicians or researchers may consider using a black bar or a masking technique over the eyes or face, but this is not always a sufficient method of anonymizing an image.
One study investigating the different requirements listed by the top 20 dermatology journals (as determined by the Google Scholar h5-index) found that while 95% (19/20) of journals stated that written or signed consent or permission was a requirement for use of patient images, only 20% (4/20) instructed authors to inform the patient or the patient’s representative that images may become available on the internet.5 Once an article is accepted for publication by a medical journal, it eventually may be accessible online; however, patients may not be aware of this factor, which is particularly concerning for those with SOC due to the increased demand for diverse dermatologic resources and images as well as the highly digitalized manner in which we access and share media.
Furthermore, cultural and social factors exist that present challenges to informed decision-making during the consenting process for certain SOC populations such as a lack of trust in the medical and scientific research community, inadequate comprehension of the consent material, health illiteracy, language barriers, or use of complex terminology in consent documentation.7,8 Studies also have shown that patients in ethnic minority groups have greater barriers to health literacy compared to other patient groups, and patients with limited health literacy are less likely to ask questions during their medical visits.9,10 Therefore, when obtaining informed consent for images, it is important that measures are taken to ensure that the patient has full knowledge and understanding of what the consent covers, including the extent to which the images will be used and/or shared and whether the patient’s confidentiality and/or anonymity are at risk.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Encourage influential dermatology organizations such as the American Academy of Dermatology to establish standardized consenting procedures for image acquisition and use, including requirements to provide (a) written consent for all patient images and (b) specific details as to where and how the image may be used and/or shared.
2. Ensure that consent terminology is presented at a sixth-grade reading level or below, minimize the use of medical jargon and complex terms, and provide consent documentation in the patient’s preferred language.
3. Allow patients to take the consent document home so they can have additional time to comprehensively review the material or have it reviewed by family or friends.
4. Employ strategies such as teach-back methods and encourage questions to maximize the level of understanding during the consent process.
Clinical Image Storage
Clinical image storage procedures can have an impact on a patient’s health information remaining anonymous and confidential. In a survey evaluating medical photography use among 153 US board-certified dermatologists, 69.1% of respondents reported emailing or texting images between patients and colleagues. Additionally, 30.3% (46/152) reported having patient photographs stored on their personal phone at the time of the survey, and 39.1% (18/46) of those individuals had images that showed identifiable features, such as the patient’s face or a tattoo.11
Although most providers state that their devices are password protected, it cannot be guaranteed that the device and consequently the images remain secure and inaccessible to unauthorized individuals. As sharing and viewing images continue to play an essential role in assessing disease state, progression, treatment response, and inclusion in research, we must establish and encourage clear guidelines for the storage and retention of such images.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Store clinical images exclusively on password-protected devices and in password-protected files.
2. Use work-related cameras or electronic devices rather than personal devices, unless the personal device is being used to upload directly into the patient’s medical record. In such cases, use a HIPAA-compliant electronic medical record mobile application that does not store images on the application or the device itself.
3. Avoid using text-messaging systems or unencrypted email to share identifying images without clear patient consent.
Clinical Image Use
Once a thorough consenting process has been completed, it is crucial that the use and distribution of the clinical image are in accordance with the terms specified in the original consent. With the current state of technologic advancement, widespread social media usage, and constant sharing of information, adherence to these terms can be challenging. For example, an image initially intended for use in an educational presentation at a professional conference can be shared on social media if an audience member captures a photo of it. In another example, a patient may consent to their image being shown on a dermatologic website but that image can be duplicated and shared on other unauthorized sites and locations. This situation can be particularly distressing to patients whose image may include all or most of their face, an intimate area, or other physical features that they did not wish to share widely.
Individuals identifying as Black/African American, Latino/Hispanic, or Asian have been shown to express less comfort with providing permission for images of a nonidentifiable sensitive area to be taken (or obtained) or for use for teaching irrespective of identifiability compared to their White counterparts,12 which may be due to the aforementioned lack of trust in medical providers and the health care system in general, both of which may contribute to concerns with how a clinical image is used and/or shared. Although consent from a patient or the patient’s representative can be granted, we must ensure that the use of these images adheres to the patient’s initial agreement. Ultimately, medical providers, researchers, and other parties involved in acquiring or sharing patient images have both an ethical and legal responsibility to ensure that anonymity, privacy, and confidentiality are preserved to the greatest extent possible.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Display a message on websites containing patient images stating that the sharing of the images outside the established guidelines and intended use is prohibited.
2. Place a watermark on images to discourage unauthorized duplication.
3. Issue explicit instructions to audiences prohibiting the copying or reproducing of any patient images during teaching events or presentations.
Final Thoughts
The use of clinical images is an essential component of dermatologic care, education, and research. Due to the higher demand for diverse and representative images and the dearth of images in the medical literature, many SOC images have been widely disseminated and utilized by dermatologists, raising concerns of the adequacy of informed consent for the storage and use of such material. Therefore, dermatologists should implement streamlined guidelines and consent procedures to ensure a patient’s informed consent is provided with full knowledge of how and where their images might be used and shared. Additional efforts should be made to protect patients’ privacy and unauthorized use of their images. Furthermore, we encourage our leading dermatology organizations to develop expert consensus on best practices for appropriate clinical image consent, storage, and use.
- Alvarado SM, Feng H. Representation of dark skin images of common dermatologic conditions in educational resources: a cross-sectional analysis [published online June 18, 2020]. J Am Acad Dermatol. 2021;84:1427-1431. doi:10.1016/j.jaad.2020.06.041
- Charrow A, Xia FD, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198. doi:10.1001/jamadermatol.2016.4129
- Marroquin NA, Carboni A, Zueger M, et al. Skin of color representation trends in JAAD case reports 2015-2021: content analysis. JMIR Dermatol. 2023;6:e40816. doi:10.2196/40816
- Kim Y, Miller JJ, Hollins LC. Skin of color matters: a call to action. J Am Acad Dermatol. 2021;84:E273-E274. doi:10.1016/j.jaad.2020.11.026
- Nanda JK, Marchetti MA. Consent and deidentification of patient images in dermatology journals: observational study. JMIR Dermatol. 2022;5:E37398. doi:10.2196/37398
- US Department of Health and Human Services. Summary of the HIPAA privacy rule. Updated October 19, 2022. Accessed March 15, 2024. https://www.hhs.gov/hipaa/for-professionals/privacy/laws-regulations/index.html
- Quinn SC, Garza MA, Butler J, et al. Improving informed consent with minority participants: results from researcher and community surveys. J Empir Res Hum Res Ethics. 2012;7:44-55. doi:10.1525/jer.2012.7.5.44
- Hadden KB, Prince LY, Moore TD, et al. Improving readability of informed consents for research at an academic medical institution. J Clin Transl Sci. 2017;1:361-365. doi:10.1017/cts.2017.312
- Muvuka B, Combs RM, Ayangeakaa SD, et al. Health literacy in African-American communities: barriers and strategies. Health Lit Res Pract. 2020;4:E138-E143. doi:10.3928/24748307-20200617-01
- Menendez ME, van Hoorn BT, Mackert M, et al. Patients with limited health literacy ask fewer questions during office visits with hand surgeons. Clin Orthop Relat Res. 2017;475:1291-1297. doi:10.1007/s11999-016-5140-5
- Milam EC, Leger MC. Use of medical photography among dermatologists: a nationwide online survey study. J Eur Acad Dermatol Venereol. 2018;32:1804-1809. doi:10.1111/jdv.14839
- Leger MC, Wu T, Haimovic A, et al. Patient perspectives on medical photography in dermatology. Dermatol Surg. 2014;40:1028-1037. doi:10.1097/01.DSS.0000452632.22081.79
Clinical images are integral to dermatologic care, research, and education. Studies have highlighted the underrepresentation of images of skin of color (SOC) in educational materials,1 clinical trials,2 and research publications.3 Recognition of this disparity has ignited a call to action by dermatologists and dermatologic organizations to address the gap by improving the collection and use of SOC images.4 It is critical to remind dermatologists of the importance of properly obtaining informed consent and ensuring images are not used without a patient’s permission, as images in journal articles, conference presentations, and educational materials can be widely distributed and shared. Herein, we summarize current practices of clinical image storage and make general recommendations on how dermatologists can better protect patient privacy. Certain cultural and social factors in patients with SOC should be considered when obtaining informed consent and collecting images.
Clinical Image Acquisition
Consenting procedures are crucial components of proper image usage. However, current consenting practices are inconsistent across various platforms, including academic journals, websites, printed text, social media, and educational presentations.5
Current regulations for use of patient health information in the United States are governed by the Health Insurance Portability and Accountability Act (HIPAA)of 1996. Although this act explicitly prohibits use of “full face photographic images and any comparable images” without consent from the patient or the patient’s representative, there is less restriction regarding the use of deidentified images.6 Some clinicians or researchers may consider using a black bar or a masking technique over the eyes or face, but this is not always a sufficient method of anonymizing an image.
One study investigating the different requirements listed by the top 20 dermatology journals (as determined by the Google Scholar h5-index) found that while 95% (19/20) of journals stated that written or signed consent or permission was a requirement for use of patient images, only 20% (4/20) instructed authors to inform the patient or the patient’s representative that images may become available on the internet.5 Once an article is accepted for publication by a medical journal, it eventually may be accessible online; however, patients may not be aware of this factor, which is particularly concerning for those with SOC due to the increased demand for diverse dermatologic resources and images as well as the highly digitalized manner in which we access and share media.
Furthermore, cultural and social factors exist that present challenges to informed decision-making during the consenting process for certain SOC populations such as a lack of trust in the medical and scientific research community, inadequate comprehension of the consent material, health illiteracy, language barriers, or use of complex terminology in consent documentation.7,8 Studies also have shown that patients in ethnic minority groups have greater barriers to health literacy compared to other patient groups, and patients with limited health literacy are less likely to ask questions during their medical visits.9,10 Therefore, when obtaining informed consent for images, it is important that measures are taken to ensure that the patient has full knowledge and understanding of what the consent covers, including the extent to which the images will be used and/or shared and whether the patient’s confidentiality and/or anonymity are at risk.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Encourage influential dermatology organizations such as the American Academy of Dermatology to establish standardized consenting procedures for image acquisition and use, including requirements to provide (a) written consent for all patient images and (b) specific details as to where and how the image may be used and/or shared.
2. Ensure that consent terminology is presented at a sixth-grade reading level or below, minimize the use of medical jargon and complex terms, and provide consent documentation in the patient’s preferred language.
3. Allow patients to take the consent document home so they can have additional time to comprehensively review the material or have it reviewed by family or friends.
4. Employ strategies such as teach-back methods and encourage questions to maximize the level of understanding during the consent process.
Clinical Image Storage
Clinical image storage procedures can have an impact on a patient’s health information remaining anonymous and confidential. In a survey evaluating medical photography use among 153 US board-certified dermatologists, 69.1% of respondents reported emailing or texting images between patients and colleagues. Additionally, 30.3% (46/152) reported having patient photographs stored on their personal phone at the time of the survey, and 39.1% (18/46) of those individuals had images that showed identifiable features, such as the patient’s face or a tattoo.11
Although most providers state that their devices are password protected, it cannot be guaranteed that the device and consequently the images remain secure and inaccessible to unauthorized individuals. As sharing and viewing images continue to play an essential role in assessing disease state, progression, treatment response, and inclusion in research, we must establish and encourage clear guidelines for the storage and retention of such images.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Store clinical images exclusively on password-protected devices and in password-protected files.
2. Use work-related cameras or electronic devices rather than personal devices, unless the personal device is being used to upload directly into the patient’s medical record. In such cases, use a HIPAA-compliant electronic medical record mobile application that does not store images on the application or the device itself.
3. Avoid using text-messaging systems or unencrypted email to share identifying images without clear patient consent.
Clinical Image Use
Once a thorough consenting process has been completed, it is crucial that the use and distribution of the clinical image are in accordance with the terms specified in the original consent. With the current state of technologic advancement, widespread social media usage, and constant sharing of information, adherence to these terms can be challenging. For example, an image initially intended for use in an educational presentation at a professional conference can be shared on social media if an audience member captures a photo of it. In another example, a patient may consent to their image being shown on a dermatologic website but that image can be duplicated and shared on other unauthorized sites and locations. This situation can be particularly distressing to patients whose image may include all or most of their face, an intimate area, or other physical features that they did not wish to share widely.
Individuals identifying as Black/African American, Latino/Hispanic, or Asian have been shown to express less comfort with providing permission for images of a nonidentifiable sensitive area to be taken (or obtained) or for use for teaching irrespective of identifiability compared to their White counterparts,12 which may be due to the aforementioned lack of trust in medical providers and the health care system in general, both of which may contribute to concerns with how a clinical image is used and/or shared. Although consent from a patient or the patient’s representative can be granted, we must ensure that the use of these images adheres to the patient’s initial agreement. Ultimately, medical providers, researchers, and other parties involved in acquiring or sharing patient images have both an ethical and legal responsibility to ensure that anonymity, privacy, and confidentiality are preserved to the greatest extent possible.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Display a message on websites containing patient images stating that the sharing of the images outside the established guidelines and intended use is prohibited.
2. Place a watermark on images to discourage unauthorized duplication.
3. Issue explicit instructions to audiences prohibiting the copying or reproducing of any patient images during teaching events or presentations.
Final Thoughts
The use of clinical images is an essential component of dermatologic care, education, and research. Due to the higher demand for diverse and representative images and the dearth of images in the medical literature, many SOC images have been widely disseminated and utilized by dermatologists, raising concerns of the adequacy of informed consent for the storage and use of such material. Therefore, dermatologists should implement streamlined guidelines and consent procedures to ensure a patient’s informed consent is provided with full knowledge of how and where their images might be used and shared. Additional efforts should be made to protect patients’ privacy and unauthorized use of their images. Furthermore, we encourage our leading dermatology organizations to develop expert consensus on best practices for appropriate clinical image consent, storage, and use.
Clinical images are integral to dermatologic care, research, and education. Studies have highlighted the underrepresentation of images of skin of color (SOC) in educational materials,1 clinical trials,2 and research publications.3 Recognition of this disparity has ignited a call to action by dermatologists and dermatologic organizations to address the gap by improving the collection and use of SOC images.4 It is critical to remind dermatologists of the importance of properly obtaining informed consent and ensuring images are not used without a patient’s permission, as images in journal articles, conference presentations, and educational materials can be widely distributed and shared. Herein, we summarize current practices of clinical image storage and make general recommendations on how dermatologists can better protect patient privacy. Certain cultural and social factors in patients with SOC should be considered when obtaining informed consent and collecting images.
Clinical Image Acquisition
Consenting procedures are crucial components of proper image usage. However, current consenting practices are inconsistent across various platforms, including academic journals, websites, printed text, social media, and educational presentations.5
Current regulations for use of patient health information in the United States are governed by the Health Insurance Portability and Accountability Act (HIPAA)of 1996. Although this act explicitly prohibits use of “full face photographic images and any comparable images” without consent from the patient or the patient’s representative, there is less restriction regarding the use of deidentified images.6 Some clinicians or researchers may consider using a black bar or a masking technique over the eyes or face, but this is not always a sufficient method of anonymizing an image.
One study investigating the different requirements listed by the top 20 dermatology journals (as determined by the Google Scholar h5-index) found that while 95% (19/20) of journals stated that written or signed consent or permission was a requirement for use of patient images, only 20% (4/20) instructed authors to inform the patient or the patient’s representative that images may become available on the internet.5 Once an article is accepted for publication by a medical journal, it eventually may be accessible online; however, patients may not be aware of this factor, which is particularly concerning for those with SOC due to the increased demand for diverse dermatologic resources and images as well as the highly digitalized manner in which we access and share media.
Furthermore, cultural and social factors exist that present challenges to informed decision-making during the consenting process for certain SOC populations such as a lack of trust in the medical and scientific research community, inadequate comprehension of the consent material, health illiteracy, language barriers, or use of complex terminology in consent documentation.7,8 Studies also have shown that patients in ethnic minority groups have greater barriers to health literacy compared to other patient groups, and patients with limited health literacy are less likely to ask questions during their medical visits.9,10 Therefore, when obtaining informed consent for images, it is important that measures are taken to ensure that the patient has full knowledge and understanding of what the consent covers, including the extent to which the images will be used and/or shared and whether the patient’s confidentiality and/or anonymity are at risk.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Encourage influential dermatology organizations such as the American Academy of Dermatology to establish standardized consenting procedures for image acquisition and use, including requirements to provide (a) written consent for all patient images and (b) specific details as to where and how the image may be used and/or shared.
2. Ensure that consent terminology is presented at a sixth-grade reading level or below, minimize the use of medical jargon and complex terms, and provide consent documentation in the patient’s preferred language.
3. Allow patients to take the consent document home so they can have additional time to comprehensively review the material or have it reviewed by family or friends.
4. Employ strategies such as teach-back methods and encourage questions to maximize the level of understanding during the consent process.
Clinical Image Storage
Clinical image storage procedures can have an impact on a patient’s health information remaining anonymous and confidential. In a survey evaluating medical photography use among 153 US board-certified dermatologists, 69.1% of respondents reported emailing or texting images between patients and colleagues. Additionally, 30.3% (46/152) reported having patient photographs stored on their personal phone at the time of the survey, and 39.1% (18/46) of those individuals had images that showed identifiable features, such as the patient’s face or a tattoo.11
Although most providers state that their devices are password protected, it cannot be guaranteed that the device and consequently the images remain secure and inaccessible to unauthorized individuals. As sharing and viewing images continue to play an essential role in assessing disease state, progression, treatment response, and inclusion in research, we must establish and encourage clear guidelines for the storage and retention of such images.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Store clinical images exclusively on password-protected devices and in password-protected files.
2. Use work-related cameras or electronic devices rather than personal devices, unless the personal device is being used to upload directly into the patient’s medical record. In such cases, use a HIPAA-compliant electronic medical record mobile application that does not store images on the application or the device itself.
3. Avoid using text-messaging systems or unencrypted email to share identifying images without clear patient consent.
Clinical Image Use
Once a thorough consenting process has been completed, it is crucial that the use and distribution of the clinical image are in accordance with the terms specified in the original consent. With the current state of technologic advancement, widespread social media usage, and constant sharing of information, adherence to these terms can be challenging. For example, an image initially intended for use in an educational presentation at a professional conference can be shared on social media if an audience member captures a photo of it. In another example, a patient may consent to their image being shown on a dermatologic website but that image can be duplicated and shared on other unauthorized sites and locations. This situation can be particularly distressing to patients whose image may include all or most of their face, an intimate area, or other physical features that they did not wish to share widely.
Individuals identifying as Black/African American, Latino/Hispanic, or Asian have been shown to express less comfort with providing permission for images of a nonidentifiable sensitive area to be taken (or obtained) or for use for teaching irrespective of identifiability compared to their White counterparts,12 which may be due to the aforementioned lack of trust in medical providers and the health care system in general, both of which may contribute to concerns with how a clinical image is used and/or shared. Although consent from a patient or the patient’s representative can be granted, we must ensure that the use of these images adheres to the patient’s initial agreement. Ultimately, medical providers, researchers, and other parties involved in acquiring or sharing patient images have both an ethical and legal responsibility to ensure that anonymity, privacy, and confidentiality are preserved to the greatest extent possible.
Recommendations—We propose that dermatologists should follow these recommendations:
1. Display a message on websites containing patient images stating that the sharing of the images outside the established guidelines and intended use is prohibited.
2. Place a watermark on images to discourage unauthorized duplication.
3. Issue explicit instructions to audiences prohibiting the copying or reproducing of any patient images during teaching events or presentations.
Final Thoughts
The use of clinical images is an essential component of dermatologic care, education, and research. Due to the higher demand for diverse and representative images and the dearth of images in the medical literature, many SOC images have been widely disseminated and utilized by dermatologists, raising concerns of the adequacy of informed consent for the storage and use of such material. Therefore, dermatologists should implement streamlined guidelines and consent procedures to ensure a patient’s informed consent is provided with full knowledge of how and where their images might be used and shared. Additional efforts should be made to protect patients’ privacy and unauthorized use of their images. Furthermore, we encourage our leading dermatology organizations to develop expert consensus on best practices for appropriate clinical image consent, storage, and use.
- Alvarado SM, Feng H. Representation of dark skin images of common dermatologic conditions in educational resources: a cross-sectional analysis [published online June 18, 2020]. J Am Acad Dermatol. 2021;84:1427-1431. doi:10.1016/j.jaad.2020.06.041
- Charrow A, Xia FD, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198. doi:10.1001/jamadermatol.2016.4129
- Marroquin NA, Carboni A, Zueger M, et al. Skin of color representation trends in JAAD case reports 2015-2021: content analysis. JMIR Dermatol. 2023;6:e40816. doi:10.2196/40816
- Kim Y, Miller JJ, Hollins LC. Skin of color matters: a call to action. J Am Acad Dermatol. 2021;84:E273-E274. doi:10.1016/j.jaad.2020.11.026
- Nanda JK, Marchetti MA. Consent and deidentification of patient images in dermatology journals: observational study. JMIR Dermatol. 2022;5:E37398. doi:10.2196/37398
- US Department of Health and Human Services. Summary of the HIPAA privacy rule. Updated October 19, 2022. Accessed March 15, 2024. https://www.hhs.gov/hipaa/for-professionals/privacy/laws-regulations/index.html
- Quinn SC, Garza MA, Butler J, et al. Improving informed consent with minority participants: results from researcher and community surveys. J Empir Res Hum Res Ethics. 2012;7:44-55. doi:10.1525/jer.2012.7.5.44
- Hadden KB, Prince LY, Moore TD, et al. Improving readability of informed consents for research at an academic medical institution. J Clin Transl Sci. 2017;1:361-365. doi:10.1017/cts.2017.312
- Muvuka B, Combs RM, Ayangeakaa SD, et al. Health literacy in African-American communities: barriers and strategies. Health Lit Res Pract. 2020;4:E138-E143. doi:10.3928/24748307-20200617-01
- Menendez ME, van Hoorn BT, Mackert M, et al. Patients with limited health literacy ask fewer questions during office visits with hand surgeons. Clin Orthop Relat Res. 2017;475:1291-1297. doi:10.1007/s11999-016-5140-5
- Milam EC, Leger MC. Use of medical photography among dermatologists: a nationwide online survey study. J Eur Acad Dermatol Venereol. 2018;32:1804-1809. doi:10.1111/jdv.14839
- Leger MC, Wu T, Haimovic A, et al. Patient perspectives on medical photography in dermatology. Dermatol Surg. 2014;40:1028-1037. doi:10.1097/01.DSS.0000452632.22081.79
- Alvarado SM, Feng H. Representation of dark skin images of common dermatologic conditions in educational resources: a cross-sectional analysis [published online June 18, 2020]. J Am Acad Dermatol. 2021;84:1427-1431. doi:10.1016/j.jaad.2020.06.041
- Charrow A, Xia FD, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198. doi:10.1001/jamadermatol.2016.4129
- Marroquin NA, Carboni A, Zueger M, et al. Skin of color representation trends in JAAD case reports 2015-2021: content analysis. JMIR Dermatol. 2023;6:e40816. doi:10.2196/40816
- Kim Y, Miller JJ, Hollins LC. Skin of color matters: a call to action. J Am Acad Dermatol. 2021;84:E273-E274. doi:10.1016/j.jaad.2020.11.026
- Nanda JK, Marchetti MA. Consent and deidentification of patient images in dermatology journals: observational study. JMIR Dermatol. 2022;5:E37398. doi:10.2196/37398
- US Department of Health and Human Services. Summary of the HIPAA privacy rule. Updated October 19, 2022. Accessed March 15, 2024. https://www.hhs.gov/hipaa/for-professionals/privacy/laws-regulations/index.html
- Quinn SC, Garza MA, Butler J, et al. Improving informed consent with minority participants: results from researcher and community surveys. J Empir Res Hum Res Ethics. 2012;7:44-55. doi:10.1525/jer.2012.7.5.44
- Hadden KB, Prince LY, Moore TD, et al. Improving readability of informed consents for research at an academic medical institution. J Clin Transl Sci. 2017;1:361-365. doi:10.1017/cts.2017.312
- Muvuka B, Combs RM, Ayangeakaa SD, et al. Health literacy in African-American communities: barriers and strategies. Health Lit Res Pract. 2020;4:E138-E143. doi:10.3928/24748307-20200617-01
- Menendez ME, van Hoorn BT, Mackert M, et al. Patients with limited health literacy ask fewer questions during office visits with hand surgeons. Clin Orthop Relat Res. 2017;475:1291-1297. doi:10.1007/s11999-016-5140-5
- Milam EC, Leger MC. Use of medical photography among dermatologists: a nationwide online survey study. J Eur Acad Dermatol Venereol. 2018;32:1804-1809. doi:10.1111/jdv.14839
- Leger MC, Wu T, Haimovic A, et al. Patient perspectives on medical photography in dermatology. Dermatol Surg. 2014;40:1028-1037. doi:10.1097/01.DSS.0000452632.22081.79

Enhancing Cosmetic and Functional Improvement of Recalcitrant Nail Lichen Planus With Resin Nail
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
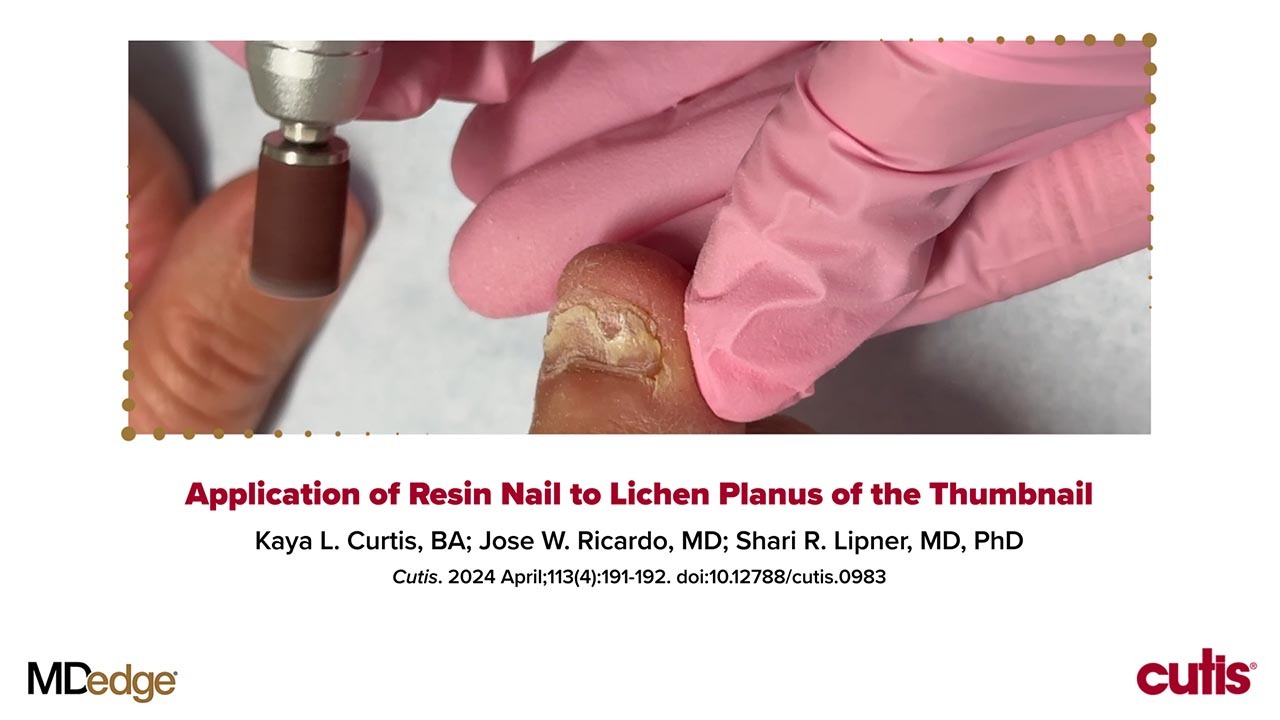
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.

Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.
Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.
Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056

Evaluating the Cost Burden of Alopecia Areata Treatment: A Comprehensive Review for Dermatologists
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
 treatment.")
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29


Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29
Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29
Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
Practice Points
- Hair loss treatments and concealment techniques cost the average patient thousands of dollars. Much of this cost burden comes from items not covered by insurance.
- Providers should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective option.
- Self-administering diphencyprone at home is more cost- and time-effective than in-office diphencyprone administration and does not decrease efficacy.

The Impact of Primary Tumor Site on Survival in Mycosis Fungoides
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.


Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
 for overall survival and disease-specific survival.")

For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).


Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).


Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.
Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).
Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).
Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.
Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).
Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).
Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
Practice Points
- Mycosis fungoides (MF) is the most common cutaneous T-cell lymphoma.
- Because MF is associated with diagnostic challenges due to its indolent course, data regarding primary tumor site as a prognostic factor are limited.
- Although MF originating from the head and neck region did not appear to influence survival, it was found that patients who were older or who had a larger tumor size at diagnosis, a higher T stage, lymph node involvement, or presence of metastasis had poorer survival overall and may benefit from additional counseling regarding their prognosis.
Centrifugally Spreading Lymphocutaneous Sporotrichosis: A Rare Cutaneous Manifestation
To the Editor:
Sporotrichosis refers to a subacute to chronic fungal infection that usually involves the cutaneous and subcutaneous tissues and is caused by the introduction of Sporothrix, a dimorphic fungus, through the skin. We present a case of chronic atypical lymphocutaneous sporotrichosis.
A 46-year-old man presented to the outpatient dermatology clinic for follow-up for a rash on the right leg that spread to the thigh and became painful and pruritic. It initially developed 8 years prior to the current presentation after he sustained trauma to the leg from an electroshock weapon. One year prior to the current presentation, he had presented to the emergency department and was prescribed doxycycline 100 mg twice daily for 7 days as well as bacitracin ointment. He also was instructed to follow up with dermatology, but a lack of health insurance and other socioeconomic barriers prevented him from seeking dermatologic care. Nine months later, he again presented to the emergency department due to a motor vehicle accident. Computed tomography (CT) of the right leg revealed exophytic dermal masses, inflammatory stranding of the subcutaneous tissue, and right inguinal lymph nodes measuring up to 1.4 cm; there was no osteoarticular involvement. At that time, the patient was applying gentian violet to the skin lesions and taking hydroxyzine 50 mg 3 times daily as needed for pruritus with minimal relief. Financial support was provided for follow-up with dermatology, which occurred almost 5 months later.
At the current presentation, physical examination revealed a large annular plaque with verrucous, scaly, erythematous borders and a hypopigmented atrophic center extending from the medial aspect of the right leg to the posterior thigh. Numerous pink, scaly, crusted nodules were scattered primarily along the periphery, with some evidence of draining sinus tracts. In addition, a fibrotic pink linear plaque extended from the medial right leg to the popliteal fossa, consistent with a keloid. Violet staining along the periphery of the lesion also was appreciated secondary to the application of topical gentian violet (Figure 1).

Based on the chronic history and morphology, a diagnosis of a chronic fungal or atypical mycobacterial infection was favored. In particular, chromoblastomycosis, cutaneous tuberculosis (eg, scrofuloderma, lupus vulgaris, tuberculosis verrucosa cutis), and atypical mycobacterial infection were highest on the differential, as these conditions often exhibit annular, nodular, verrucous, and/or atrophic lesions. The nodularity, crusting, and draining sinus tracts also raised the possibility of mycetoma. Given the extension of the lesion from the lower to upper leg, a sporotrichoid infection also was considered but was thought to be less likely based on the annular configuration.
Two 4-mm punch biopsies were taken from a peripheral nodule—one for routine histology and another for bacterial, fungal, and mycobacterial cultures. An interferon-gamma release assay also was ordered to evaluate for immune responses indicative of prior Mycobacterium tuberculosis infection, but the patient did not obtain this for unknown reasons. Histology demonstrated pseudoepitheliomatous hyperplasia and necrotizing granulomas, which suggested an infectious etiology, but no organisms were identified on tissue staining and all cultures were negative for growth at 6 weeks. The patient was asked to return at that point, and 4 additional scouting biopsies were performed and sent for routine histology, M tuberculosis nucleic acid amplification testing, and microbiologic cultures (ie, bacterial, mycobacterial, fungal, nocardia, actinomycetes). Within 1 week, a filamentous organism with pigmentation visible on the front and back of a Sabouraud dextrose agar plate was identified on fungal culture (Figure 2). Microscopic evaluation of this mold with lactophenol blue stain revealed thin septate hyphae with conidiophores arising at right angles that bore clusters of microconidia (Figure 3). Sequencing analysis ultimately identified this organism as Sporothrix schenckii. Routine histology demonstrated pseudoepitheliomatous hyperplasia with scattered intraepidermal collections of neutrophils (Figure 4). The dermis showed a dense, superficial, and deep infiltrate composed of lymphocytes, histiocytes, and plasma cells with occasional neutrophils and eosinophils. A Grocott-Gomori methenamine-silver stain revealed a cluster of ovoid yeast forms within the stratum corneum (Figure 5). The patient was referred to infectious disease for follow-up and treatment.

The patient later visited a community clinic providing dermatologic care for patients without insurance. He was started on itraconazole 200 mg daily for a total of 6 months until dermatologic clearance of the cutaneous lesions was observed. He was followed by the clinic with laboratory tests including a liver function test. At follow-up 8 months later, a repeat biopsy was performed to ensure histologic clearance of the sporotrichosis, which revealed a dermal scar and no evidence of residual infection.
.")
Sporothrix schenckii was first isolated in 1898 by Benjamin Schenck, a student at Johns Hopkins Medicine (Baltimore, Maryland), and identified by a mycologist as sporotricha.1 Species within the genus Sporothrix are unique in that the fungi are both dimorphic (growing as a mold at 25 °C but as a yeast at 37 °C) and dematiaceous (dark pigmentation from melanin is visible on inspection of the anterior and reverse sides of culture plates). Infection usually occurs when cutaneous or subcutaneous tissues are exposed to the fungus via microabrasions; activities thought to contribute to exposure include gardening, agricultural work, animal husbandry, and feline scratches.2 Although skin trauma frequently is considered the primary route of infection, patient recall is variable, with one study noting that only 37.7% of patients recalled trauma and another study similarly demonstrating a patient recall rate of 25%.3,4

Lymphocutaneous sporotrichosis is the most common presentation of the fungal infection,5 and clinical cases may be classified into 1 of 4 categories: (1) lymphangitic lesions—papules at the site of inoculation with spread along the lymphatic channels; (2) localized (fixed) cutaneous lesions—1 or 2 lesions at the inoculation site; (3) disseminated (multifocal) cutaneous lesions; and (4) extracutaneous lesions.6 Extracutaneous manifestations of this infection most notably have been reported as pulmonary disease through inhalation of conidia or through dissemination in immunocompromised hosts.7 Our patient’s infection was categorized as lymphangitic lesions due to spread from the lower to upper leg, albeit in a highly atypical, annular fashion. A review of systems was otherwise negative, and CT ruled out osteoarticular involvement.
.")
In addition to socioeconomic barriers, several factors contributed to a delayed diagnosis in this patient including the annular presentation with central hypopigmentation and atrophy, negative initial microbiological cultures and lack of visualization of organisms on histopathology, and the consequent need for repeat biopsies. For lymphocutaneous sporotrichosis, the typical presentation consists of a papule or ulcerated nodule at the site of inoculation with subsequent linear spread along lymphatic channels. This classic sporotrichoid pattern is a key diagnostic clue for identifying sporotrichosis but was absent at the time our patient presented for medical care. Rather, the sporotrichoid spread seemed to have occurred in a centrifugal fashion up the leg. Few case reports have documented an annular presentation of lymphocutaneous sporotrichosis,8-13 and one report described central atrophy and hypopigmentation.10 Pain and pruritus, which were present in our patient, rarely are documented.9 Finally, the diagnosis of cutaneous fungal infections may require multiple biopsies due to the variable abundance of viable organisms in tissue specimens as well as the fastidious growth characteristics of these organisms. Furthermore, sensitivity often is low for both fungal and mycobacterial cultures, and cultures may take days to weeks to yield growth.14,15 For these reasons, empiric therapy and repeat biopsies often are pursued if clinical suspicion is high enough.16 Our patient returned for multiple scouting biopsies after the initial tissue culture was negative and was even considered for empiric treatment against Mycobacterium prior to positive fungal cultures.
Another unique aspect of our case was the presence of a keloid. It is difficult to know if this keloid was secondary to the trauma the patient sustained in the inciting incident or formed from the fungal infection. Interestingly, it has been hypothesized that fungal infections may contribute to keloid and hypertrophic scar formation.17 In a case series of 3 patients with either keloids or hypertrophic scars and concomitant tinea infection, there was notable improvement in the appearance of the scars 2 weeks after beginning itraconazole therapy.17 However, it is not yet known if a fungal infection can contribute to the pathogenesis of keloid formation.
As with other aspects of this case, the length of time the patient went without diagnosis and treatment was unusual and may help explain the atypical presentation. Although the incubation period for S schenckii can vary, most reports identify patients as seeking medical attention within 1 year of rash onset.18-20 In our case, the patient was not diagnosed until 8 years after his symptoms began, requiring multiple referrals, multiple health system touchpoints, and an institution-specific financial aid program. As such, this case also highlights the potential need for a multidisciplinary team approach when caring for patients with poor access to health care.
In conclusion, this case illustrates a unique presentation of lymphocutaneous sporotrichosis that may mimic other chronic infections and result in delayed diagnosis. Although lymphangitic sporotrichosis generally is recognized as having a linear distribution, mounting evidence from this report and others suggests an annular presentation also is possible. Pruritus or pain is rare but should not preclude a diagnosis of sporotrichosis if present. For patients with limited access to health care resources, it is especially important to involve multiple members of the health care team, including social workers and specialists, to prevent a protracted and severe course of disease.
- Schenck BR. On refractory subcutaneous abscesses caused by a fungus possibly related to the sporotricha. Bulletin of the Johns Hopkins Hospital. 1898;93:286-290.
- de Lima Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654. doi:10.1128/CMR.00007-11
- Crevasse L, Ellner PD. An outbreak of sporotrichosis in florida. J Am Med Assoc. 1960;173:29-33. doi:10.1001/jama.1960.03020190031006
- Mayorga R, Cáceres A, Toriello C, et al. An endemic area of sporotrichosis in Guatemala [in French]. Sabouraudia. 1978;16:185-198.
- Morris-Jones R. Sporotrichosis. Clin Exp Dermatol. 2002;27:427-431. doi:10.1046/j.1365-2230.2002.01087.x
- Sampaio SA, Da Lacaz CS. Clinical and statistical studies on sporotrichosis in Sao Paulo (Brazil). Article in German. Hautarzt. 1959;10:490-493.
- Ramos-e-Silva M, Vasconcelos C, Carneiro S, et al. Sporotrichosis. Clin Dermatol. 2007;25:181-187. doi:10.1016/j.clindermatol.2006.05.006
- Williams BA, Jennings TA, Rushing EC, et al. Sporotrichosis on the face of a 7-year-old boy following a bicycle accident. Pediatr Dermatol. 2013;30:E246-E247. doi:10.1111/j.1525-1470.2011.01696.x
- Vaishampayan SS, Borde P. An unusual presentation of sporotrichosis. Indian J Dermatol. 2013;58:409. doi:10.4103/0019-5154.117350
- Qin J, Zhang J. Sporotrichosis. N Engl J Med. 2019;380:771. doi:10.1056/NEJMicm1809179
- Patel A, Mudenda V, Lakhi S, et al. A 27-year-old severely immunosuppressed female with misleading clinical features of disseminated cutaneous sporotrichosis. Case Rep Dermatol Med. 2016;2016:1-4. doi:10.1155/2016/9403690
- de Oliveira-Esteves ICMR, Almeida Rosa da Silva G, Eyer-Silva WA, et al. Rapidly progressive disseminated sporotrichosis as the first presentation of HIV infection in a patient with a very low CD4 cell count. Case Rep Infect Dis. 2017;2017:4713140. doi:10.1155/2017/4713140
- Singh S, Bachaspatimayum R, Meetei U, et al. Terbinafine in fixed cutaneous sporotrichosis: a case series. J Clin Diagnostic Res. 2018;12:FR01-FR03. doi:10.7860/JCDR/2018/25315.12223
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Peters F, Batinica M, Plum G, et al. Bug or no bug: challenges in diagnosing cutaneous mycobacterial infections. J Ger Soc Dermatol. 2016;14:1227-1236. doi:10.1111/ddg.13001
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973. doi:10.1155/2018/7201973
- Okada E, Maruyama Y. Are keloids and hypertrophic scars caused by fungal infection? . Plast Reconstr Surg. 2007;120:814-815. doi:10.1097/01.prs.0000278813.23244.3f
- Pappas PG, Tellez I, Deep AE, et al. Sporotrichosis in Peru: description of an area of hyperendemicity. Clin Infect Dis. 2000;30:65-70. doi:10.1086/313607
- McGuinness SL, Boyd R, Kidd S, et al. Epidemiological investigation of an outbreak of cutaneous sporotrichosis, Northern Territory, Australia. BMC Infect Dis. 2016;16:1-7. doi:10.1186/s12879-016-1338-0
- Rojas FD, Fernández MS, Lucchelli JM, et al. Cavitary pulmonary sporotrichosis: case report and literature review. Mycopathologia. 2017;182:1119-1123. doi:10.1007/s11046-017-0197-6
To the Editor:
Sporotrichosis refers to a subacute to chronic fungal infection that usually involves the cutaneous and subcutaneous tissues and is caused by the introduction of Sporothrix, a dimorphic fungus, through the skin. We present a case of chronic atypical lymphocutaneous sporotrichosis.
A 46-year-old man presented to the outpatient dermatology clinic for follow-up for a rash on the right leg that spread to the thigh and became painful and pruritic. It initially developed 8 years prior to the current presentation after he sustained trauma to the leg from an electroshock weapon. One year prior to the current presentation, he had presented to the emergency department and was prescribed doxycycline 100 mg twice daily for 7 days as well as bacitracin ointment. He also was instructed to follow up with dermatology, but a lack of health insurance and other socioeconomic barriers prevented him from seeking dermatologic care. Nine months later, he again presented to the emergency department due to a motor vehicle accident. Computed tomography (CT) of the right leg revealed exophytic dermal masses, inflammatory stranding of the subcutaneous tissue, and right inguinal lymph nodes measuring up to 1.4 cm; there was no osteoarticular involvement. At that time, the patient was applying gentian violet to the skin lesions and taking hydroxyzine 50 mg 3 times daily as needed for pruritus with minimal relief. Financial support was provided for follow-up with dermatology, which occurred almost 5 months later.
At the current presentation, physical examination revealed a large annular plaque with verrucous, scaly, erythematous borders and a hypopigmented atrophic center extending from the medial aspect of the right leg to the posterior thigh. Numerous pink, scaly, crusted nodules were scattered primarily along the periphery, with some evidence of draining sinus tracts. In addition, a fibrotic pink linear plaque extended from the medial right leg to the popliteal fossa, consistent with a keloid. Violet staining along the periphery of the lesion also was appreciated secondary to the application of topical gentian violet (Figure 1).
Based on the chronic history and morphology, a diagnosis of a chronic fungal or atypical mycobacterial infection was favored. In particular, chromoblastomycosis, cutaneous tuberculosis (eg, scrofuloderma, lupus vulgaris, tuberculosis verrucosa cutis), and atypical mycobacterial infection were highest on the differential, as these conditions often exhibit annular, nodular, verrucous, and/or atrophic lesions. The nodularity, crusting, and draining sinus tracts also raised the possibility of mycetoma. Given the extension of the lesion from the lower to upper leg, a sporotrichoid infection also was considered but was thought to be less likely based on the annular configuration.
Two 4-mm punch biopsies were taken from a peripheral nodule—one for routine histology and another for bacterial, fungal, and mycobacterial cultures. An interferon-gamma release assay also was ordered to evaluate for immune responses indicative of prior Mycobacterium tuberculosis infection, but the patient did not obtain this for unknown reasons. Histology demonstrated pseudoepitheliomatous hyperplasia and necrotizing granulomas, which suggested an infectious etiology, but no organisms were identified on tissue staining and all cultures were negative for growth at 6 weeks. The patient was asked to return at that point, and 4 additional scouting biopsies were performed and sent for routine histology, M tuberculosis nucleic acid amplification testing, and microbiologic cultures (ie, bacterial, mycobacterial, fungal, nocardia, actinomycetes). Within 1 week, a filamentous organism with pigmentation visible on the front and back of a Sabouraud dextrose agar plate was identified on fungal culture (Figure 2). Microscopic evaluation of this mold with lactophenol blue stain revealed thin septate hyphae with conidiophores arising at right angles that bore clusters of microconidia (Figure 3). Sequencing analysis ultimately identified this organism as Sporothrix schenckii. Routine histology demonstrated pseudoepitheliomatous hyperplasia with scattered intraepidermal collections of neutrophils (Figure 4). The dermis showed a dense, superficial, and deep infiltrate composed of lymphocytes, histiocytes, and plasma cells with occasional neutrophils and eosinophils. A Grocott-Gomori methenamine-silver stain revealed a cluster of ovoid yeast forms within the stratum corneum (Figure 5). The patient was referred to infectious disease for follow-up and treatment.
The patient later visited a community clinic providing dermatologic care for patients without insurance. He was started on itraconazole 200 mg daily for a total of 6 months until dermatologic clearance of the cutaneous lesions was observed. He was followed by the clinic with laboratory tests including a liver function test. At follow-up 8 months later, a repeat biopsy was performed to ensure histologic clearance of the sporotrichosis, which revealed a dermal scar and no evidence of residual infection.
Sporothrix schenckii was first isolated in 1898 by Benjamin Schenck, a student at Johns Hopkins Medicine (Baltimore, Maryland), and identified by a mycologist as sporotricha.1 Species within the genus Sporothrix are unique in that the fungi are both dimorphic (growing as a mold at 25 °C but as a yeast at 37 °C) and dematiaceous (dark pigmentation from melanin is visible on inspection of the anterior and reverse sides of culture plates). Infection usually occurs when cutaneous or subcutaneous tissues are exposed to the fungus via microabrasions; activities thought to contribute to exposure include gardening, agricultural work, animal husbandry, and feline scratches.2 Although skin trauma frequently is considered the primary route of infection, patient recall is variable, with one study noting that only 37.7% of patients recalled trauma and another study similarly demonstrating a patient recall rate of 25%.3,4
Lymphocutaneous sporotrichosis is the most common presentation of the fungal infection,5 and clinical cases may be classified into 1 of 4 categories: (1) lymphangitic lesions—papules at the site of inoculation with spread along the lymphatic channels; (2) localized (fixed) cutaneous lesions—1 or 2 lesions at the inoculation site; (3) disseminated (multifocal) cutaneous lesions; and (4) extracutaneous lesions.6 Extracutaneous manifestations of this infection most notably have been reported as pulmonary disease through inhalation of conidia or through dissemination in immunocompromised hosts.7 Our patient’s infection was categorized as lymphangitic lesions due to spread from the lower to upper leg, albeit in a highly atypical, annular fashion. A review of systems was otherwise negative, and CT ruled out osteoarticular involvement.
In addition to socioeconomic barriers, several factors contributed to a delayed diagnosis in this patient including the annular presentation with central hypopigmentation and atrophy, negative initial microbiological cultures and lack of visualization of organisms on histopathology, and the consequent need for repeat biopsies. For lymphocutaneous sporotrichosis, the typical presentation consists of a papule or ulcerated nodule at the site of inoculation with subsequent linear spread along lymphatic channels. This classic sporotrichoid pattern is a key diagnostic clue for identifying sporotrichosis but was absent at the time our patient presented for medical care. Rather, the sporotrichoid spread seemed to have occurred in a centrifugal fashion up the leg. Few case reports have documented an annular presentation of lymphocutaneous sporotrichosis,8-13 and one report described central atrophy and hypopigmentation.10 Pain and pruritus, which were present in our patient, rarely are documented.9 Finally, the diagnosis of cutaneous fungal infections may require multiple biopsies due to the variable abundance of viable organisms in tissue specimens as well as the fastidious growth characteristics of these organisms. Furthermore, sensitivity often is low for both fungal and mycobacterial cultures, and cultures may take days to weeks to yield growth.14,15 For these reasons, empiric therapy and repeat biopsies often are pursued if clinical suspicion is high enough.16 Our patient returned for multiple scouting biopsies after the initial tissue culture was negative and was even considered for empiric treatment against Mycobacterium prior to positive fungal cultures.
Another unique aspect of our case was the presence of a keloid. It is difficult to know if this keloid was secondary to the trauma the patient sustained in the inciting incident or formed from the fungal infection. Interestingly, it has been hypothesized that fungal infections may contribute to keloid and hypertrophic scar formation.17 In a case series of 3 patients with either keloids or hypertrophic scars and concomitant tinea infection, there was notable improvement in the appearance of the scars 2 weeks after beginning itraconazole therapy.17 However, it is not yet known if a fungal infection can contribute to the pathogenesis of keloid formation.
As with other aspects of this case, the length of time the patient went without diagnosis and treatment was unusual and may help explain the atypical presentation. Although the incubation period for S schenckii can vary, most reports identify patients as seeking medical attention within 1 year of rash onset.18-20 In our case, the patient was not diagnosed until 8 years after his symptoms began, requiring multiple referrals, multiple health system touchpoints, and an institution-specific financial aid program. As such, this case also highlights the potential need for a multidisciplinary team approach when caring for patients with poor access to health care.
In conclusion, this case illustrates a unique presentation of lymphocutaneous sporotrichosis that may mimic other chronic infections and result in delayed diagnosis. Although lymphangitic sporotrichosis generally is recognized as having a linear distribution, mounting evidence from this report and others suggests an annular presentation also is possible. Pruritus or pain is rare but should not preclude a diagnosis of sporotrichosis if present. For patients with limited access to health care resources, it is especially important to involve multiple members of the health care team, including social workers and specialists, to prevent a protracted and severe course of disease.
To the Editor:
Sporotrichosis refers to a subacute to chronic fungal infection that usually involves the cutaneous and subcutaneous tissues and is caused by the introduction of Sporothrix, a dimorphic fungus, through the skin. We present a case of chronic atypical lymphocutaneous sporotrichosis.
A 46-year-old man presented to the outpatient dermatology clinic for follow-up for a rash on the right leg that spread to the thigh and became painful and pruritic. It initially developed 8 years prior to the current presentation after he sustained trauma to the leg from an electroshock weapon. One year prior to the current presentation, he had presented to the emergency department and was prescribed doxycycline 100 mg twice daily for 7 days as well as bacitracin ointment. He also was instructed to follow up with dermatology, but a lack of health insurance and other socioeconomic barriers prevented him from seeking dermatologic care. Nine months later, he again presented to the emergency department due to a motor vehicle accident. Computed tomography (CT) of the right leg revealed exophytic dermal masses, inflammatory stranding of the subcutaneous tissue, and right inguinal lymph nodes measuring up to 1.4 cm; there was no osteoarticular involvement. At that time, the patient was applying gentian violet to the skin lesions and taking hydroxyzine 50 mg 3 times daily as needed for pruritus with minimal relief. Financial support was provided for follow-up with dermatology, which occurred almost 5 months later.
At the current presentation, physical examination revealed a large annular plaque with verrucous, scaly, erythematous borders and a hypopigmented atrophic center extending from the medial aspect of the right leg to the posterior thigh. Numerous pink, scaly, crusted nodules were scattered primarily along the periphery, with some evidence of draining sinus tracts. In addition, a fibrotic pink linear plaque extended from the medial right leg to the popliteal fossa, consistent with a keloid. Violet staining along the periphery of the lesion also was appreciated secondary to the application of topical gentian violet (Figure 1).
Based on the chronic history and morphology, a diagnosis of a chronic fungal or atypical mycobacterial infection was favored. In particular, chromoblastomycosis, cutaneous tuberculosis (eg, scrofuloderma, lupus vulgaris, tuberculosis verrucosa cutis), and atypical mycobacterial infection were highest on the differential, as these conditions often exhibit annular, nodular, verrucous, and/or atrophic lesions. The nodularity, crusting, and draining sinus tracts also raised the possibility of mycetoma. Given the extension of the lesion from the lower to upper leg, a sporotrichoid infection also was considered but was thought to be less likely based on the annular configuration.
Two 4-mm punch biopsies were taken from a peripheral nodule—one for routine histology and another for bacterial, fungal, and mycobacterial cultures. An interferon-gamma release assay also was ordered to evaluate for immune responses indicative of prior Mycobacterium tuberculosis infection, but the patient did not obtain this for unknown reasons. Histology demonstrated pseudoepitheliomatous hyperplasia and necrotizing granulomas, which suggested an infectious etiology, but no organisms were identified on tissue staining and all cultures were negative for growth at 6 weeks. The patient was asked to return at that point, and 4 additional scouting biopsies were performed and sent for routine histology, M tuberculosis nucleic acid amplification testing, and microbiologic cultures (ie, bacterial, mycobacterial, fungal, nocardia, actinomycetes). Within 1 week, a filamentous organism with pigmentation visible on the front and back of a Sabouraud dextrose agar plate was identified on fungal culture (Figure 2). Microscopic evaluation of this mold with lactophenol blue stain revealed thin septate hyphae with conidiophores arising at right angles that bore clusters of microconidia (Figure 3). Sequencing analysis ultimately identified this organism as Sporothrix schenckii. Routine histology demonstrated pseudoepitheliomatous hyperplasia with scattered intraepidermal collections of neutrophils (Figure 4). The dermis showed a dense, superficial, and deep infiltrate composed of lymphocytes, histiocytes, and plasma cells with occasional neutrophils and eosinophils. A Grocott-Gomori methenamine-silver stain revealed a cluster of ovoid yeast forms within the stratum corneum (Figure 5). The patient was referred to infectious disease for follow-up and treatment.
The patient later visited a community clinic providing dermatologic care for patients without insurance. He was started on itraconazole 200 mg daily for a total of 6 months until dermatologic clearance of the cutaneous lesions was observed. He was followed by the clinic with laboratory tests including a liver function test. At follow-up 8 months later, a repeat biopsy was performed to ensure histologic clearance of the sporotrichosis, which revealed a dermal scar and no evidence of residual infection.
Sporothrix schenckii was first isolated in 1898 by Benjamin Schenck, a student at Johns Hopkins Medicine (Baltimore, Maryland), and identified by a mycologist as sporotricha.1 Species within the genus Sporothrix are unique in that the fungi are both dimorphic (growing as a mold at 25 °C but as a yeast at 37 °C) and dematiaceous (dark pigmentation from melanin is visible on inspection of the anterior and reverse sides of culture plates). Infection usually occurs when cutaneous or subcutaneous tissues are exposed to the fungus via microabrasions; activities thought to contribute to exposure include gardening, agricultural work, animal husbandry, and feline scratches.2 Although skin trauma frequently is considered the primary route of infection, patient recall is variable, with one study noting that only 37.7% of patients recalled trauma and another study similarly demonstrating a patient recall rate of 25%.3,4
Lymphocutaneous sporotrichosis is the most common presentation of the fungal infection,5 and clinical cases may be classified into 1 of 4 categories: (1) lymphangitic lesions—papules at the site of inoculation with spread along the lymphatic channels; (2) localized (fixed) cutaneous lesions—1 or 2 lesions at the inoculation site; (3) disseminated (multifocal) cutaneous lesions; and (4) extracutaneous lesions.6 Extracutaneous manifestations of this infection most notably have been reported as pulmonary disease through inhalation of conidia or through dissemination in immunocompromised hosts.7 Our patient’s infection was categorized as lymphangitic lesions due to spread from the lower to upper leg, albeit in a highly atypical, annular fashion. A review of systems was otherwise negative, and CT ruled out osteoarticular involvement.
In addition to socioeconomic barriers, several factors contributed to a delayed diagnosis in this patient including the annular presentation with central hypopigmentation and atrophy, negative initial microbiological cultures and lack of visualization of organisms on histopathology, and the consequent need for repeat biopsies. For lymphocutaneous sporotrichosis, the typical presentation consists of a papule or ulcerated nodule at the site of inoculation with subsequent linear spread along lymphatic channels. This classic sporotrichoid pattern is a key diagnostic clue for identifying sporotrichosis but was absent at the time our patient presented for medical care. Rather, the sporotrichoid spread seemed to have occurred in a centrifugal fashion up the leg. Few case reports have documented an annular presentation of lymphocutaneous sporotrichosis,8-13 and one report described central atrophy and hypopigmentation.10 Pain and pruritus, which were present in our patient, rarely are documented.9 Finally, the diagnosis of cutaneous fungal infections may require multiple biopsies due to the variable abundance of viable organisms in tissue specimens as well as the fastidious growth characteristics of these organisms. Furthermore, sensitivity often is low for both fungal and mycobacterial cultures, and cultures may take days to weeks to yield growth.14,15 For these reasons, empiric therapy and repeat biopsies often are pursued if clinical suspicion is high enough.16 Our patient returned for multiple scouting biopsies after the initial tissue culture was negative and was even considered for empiric treatment against Mycobacterium prior to positive fungal cultures.
Another unique aspect of our case was the presence of a keloid. It is difficult to know if this keloid was secondary to the trauma the patient sustained in the inciting incident or formed from the fungal infection. Interestingly, it has been hypothesized that fungal infections may contribute to keloid and hypertrophic scar formation.17 In a case series of 3 patients with either keloids or hypertrophic scars and concomitant tinea infection, there was notable improvement in the appearance of the scars 2 weeks after beginning itraconazole therapy.17 However, it is not yet known if a fungal infection can contribute to the pathogenesis of keloid formation.
As with other aspects of this case, the length of time the patient went without diagnosis and treatment was unusual and may help explain the atypical presentation. Although the incubation period for S schenckii can vary, most reports identify patients as seeking medical attention within 1 year of rash onset.18-20 In our case, the patient was not diagnosed until 8 years after his symptoms began, requiring multiple referrals, multiple health system touchpoints, and an institution-specific financial aid program. As such, this case also highlights the potential need for a multidisciplinary team approach when caring for patients with poor access to health care.
In conclusion, this case illustrates a unique presentation of lymphocutaneous sporotrichosis that may mimic other chronic infections and result in delayed diagnosis. Although lymphangitic sporotrichosis generally is recognized as having a linear distribution, mounting evidence from this report and others suggests an annular presentation also is possible. Pruritus or pain is rare but should not preclude a diagnosis of sporotrichosis if present. For patients with limited access to health care resources, it is especially important to involve multiple members of the health care team, including social workers and specialists, to prevent a protracted and severe course of disease.
- Schenck BR. On refractory subcutaneous abscesses caused by a fungus possibly related to the sporotricha. Bulletin of the Johns Hopkins Hospital. 1898;93:286-290.
- de Lima Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654. doi:10.1128/CMR.00007-11
- Crevasse L, Ellner PD. An outbreak of sporotrichosis in florida. J Am Med Assoc. 1960;173:29-33. doi:10.1001/jama.1960.03020190031006
- Mayorga R, Cáceres A, Toriello C, et al. An endemic area of sporotrichosis in Guatemala [in French]. Sabouraudia. 1978;16:185-198.
- Morris-Jones R. Sporotrichosis. Clin Exp Dermatol. 2002;27:427-431. doi:10.1046/j.1365-2230.2002.01087.x
- Sampaio SA, Da Lacaz CS. Clinical and statistical studies on sporotrichosis in Sao Paulo (Brazil). Article in German. Hautarzt. 1959;10:490-493.
- Ramos-e-Silva M, Vasconcelos C, Carneiro S, et al. Sporotrichosis. Clin Dermatol. 2007;25:181-187. doi:10.1016/j.clindermatol.2006.05.006
- Williams BA, Jennings TA, Rushing EC, et al. Sporotrichosis on the face of a 7-year-old boy following a bicycle accident. Pediatr Dermatol. 2013;30:E246-E247. doi:10.1111/j.1525-1470.2011.01696.x
- Vaishampayan SS, Borde P. An unusual presentation of sporotrichosis. Indian J Dermatol. 2013;58:409. doi:10.4103/0019-5154.117350
- Qin J, Zhang J. Sporotrichosis. N Engl J Med. 2019;380:771. doi:10.1056/NEJMicm1809179
- Patel A, Mudenda V, Lakhi S, et al. A 27-year-old severely immunosuppressed female with misleading clinical features of disseminated cutaneous sporotrichosis. Case Rep Dermatol Med. 2016;2016:1-4. doi:10.1155/2016/9403690
- de Oliveira-Esteves ICMR, Almeida Rosa da Silva G, Eyer-Silva WA, et al. Rapidly progressive disseminated sporotrichosis as the first presentation of HIV infection in a patient with a very low CD4 cell count. Case Rep Infect Dis. 2017;2017:4713140. doi:10.1155/2017/4713140
- Singh S, Bachaspatimayum R, Meetei U, et al. Terbinafine in fixed cutaneous sporotrichosis: a case series. J Clin Diagnostic Res. 2018;12:FR01-FR03. doi:10.7860/JCDR/2018/25315.12223
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Peters F, Batinica M, Plum G, et al. Bug or no bug: challenges in diagnosing cutaneous mycobacterial infections. J Ger Soc Dermatol. 2016;14:1227-1236. doi:10.1111/ddg.13001
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973. doi:10.1155/2018/7201973
- Okada E, Maruyama Y. Are keloids and hypertrophic scars caused by fungal infection? . Plast Reconstr Surg. 2007;120:814-815. doi:10.1097/01.prs.0000278813.23244.3f
- Pappas PG, Tellez I, Deep AE, et al. Sporotrichosis in Peru: description of an area of hyperendemicity. Clin Infect Dis. 2000;30:65-70. doi:10.1086/313607
- McGuinness SL, Boyd R, Kidd S, et al. Epidemiological investigation of an outbreak of cutaneous sporotrichosis, Northern Territory, Australia. BMC Infect Dis. 2016;16:1-7. doi:10.1186/s12879-016-1338-0
- Rojas FD, Fernández MS, Lucchelli JM, et al. Cavitary pulmonary sporotrichosis: case report and literature review. Mycopathologia. 2017;182:1119-1123. doi:10.1007/s11046-017-0197-6
- Schenck BR. On refractory subcutaneous abscesses caused by a fungus possibly related to the sporotricha. Bulletin of the Johns Hopkins Hospital. 1898;93:286-290.
- de Lima Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654. doi:10.1128/CMR.00007-11
- Crevasse L, Ellner PD. An outbreak of sporotrichosis in florida. J Am Med Assoc. 1960;173:29-33. doi:10.1001/jama.1960.03020190031006
- Mayorga R, Cáceres A, Toriello C, et al. An endemic area of sporotrichosis in Guatemala [in French]. Sabouraudia. 1978;16:185-198.
- Morris-Jones R. Sporotrichosis. Clin Exp Dermatol. 2002;27:427-431. doi:10.1046/j.1365-2230.2002.01087.x
- Sampaio SA, Da Lacaz CS. Clinical and statistical studies on sporotrichosis in Sao Paulo (Brazil). Article in German. Hautarzt. 1959;10:490-493.
- Ramos-e-Silva M, Vasconcelos C, Carneiro S, et al. Sporotrichosis. Clin Dermatol. 2007;25:181-187. doi:10.1016/j.clindermatol.2006.05.006
- Williams BA, Jennings TA, Rushing EC, et al. Sporotrichosis on the face of a 7-year-old boy following a bicycle accident. Pediatr Dermatol. 2013;30:E246-E247. doi:10.1111/j.1525-1470.2011.01696.x
- Vaishampayan SS, Borde P. An unusual presentation of sporotrichosis. Indian J Dermatol. 2013;58:409. doi:10.4103/0019-5154.117350
- Qin J, Zhang J. Sporotrichosis. N Engl J Med. 2019;380:771. doi:10.1056/NEJMicm1809179
- Patel A, Mudenda V, Lakhi S, et al. A 27-year-old severely immunosuppressed female with misleading clinical features of disseminated cutaneous sporotrichosis. Case Rep Dermatol Med. 2016;2016:1-4. doi:10.1155/2016/9403690
- de Oliveira-Esteves ICMR, Almeida Rosa da Silva G, Eyer-Silva WA, et al. Rapidly progressive disseminated sporotrichosis as the first presentation of HIV infection in a patient with a very low CD4 cell count. Case Rep Infect Dis. 2017;2017:4713140. doi:10.1155/2017/4713140
- Singh S, Bachaspatimayum R, Meetei U, et al. Terbinafine in fixed cutaneous sporotrichosis: a case series. J Clin Diagnostic Res. 2018;12:FR01-FR03. doi:10.7860/JCDR/2018/25315.12223
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280. doi:10.1128/CMR.00053-10
- Peters F, Batinica M, Plum G, et al. Bug or no bug: challenges in diagnosing cutaneous mycobacterial infections. J Ger Soc Dermatol. 2016;14:1227-1236. doi:10.1111/ddg.13001
- Khadka P, Koirala S, Thapaliya J. Cutaneous tuberculosis: clinicopathologic arrays and diagnostic challenges. Dermatol Res Pract. 2018;2018:7201973. doi:10.1155/2018/7201973
- Okada E, Maruyama Y. Are keloids and hypertrophic scars caused by fungal infection? . Plast Reconstr Surg. 2007;120:814-815. doi:10.1097/01.prs.0000278813.23244.3f
- Pappas PG, Tellez I, Deep AE, et al. Sporotrichosis in Peru: description of an area of hyperendemicity. Clin Infect Dis. 2000;30:65-70. doi:10.1086/313607
- McGuinness SL, Boyd R, Kidd S, et al. Epidemiological investigation of an outbreak of cutaneous sporotrichosis, Northern Territory, Australia. BMC Infect Dis. 2016;16:1-7. doi:10.1186/s12879-016-1338-0
- Rojas FD, Fernández MS, Lucchelli JM, et al. Cavitary pulmonary sporotrichosis: case report and literature review. Mycopathologia. 2017;182:1119-1123. doi:10.1007/s11046-017-0197-6
Practice Points
- An atypical presentation of lymphocutaneous sporotrichosis may pose challenges to timely diagnosis and treatment.
- Although lymphocutaneous sporotrichosis spreads most commonly in a linear fashion along lymphatic channels, an annular configuration is possible.
- Initial tissue cultures and histopathology of lymphocutaneous sporotrichosis may not yield a diagnosis, necessitating repeat biopsies when clinical suspicion is high.

Eosinophilic Pustular Folliculitis in the Setting of Untreated Chronic Lymphocytic Leukemia
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.

A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.

Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.
A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.
Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.
A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.
Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
Practice Points
- Eosinophilic pustular folliculitis (EPF) is associated with an immunosuppressed state, as in patients with underlying hematologic malignancy.
- Topical corticosteroids remain the first-line treatment for EPF; however, antimicrobial agents have been used with moderate success when topical therapies have failed.

Papulosquamous Dermatophytid Reaction in a Child With Tinea Capitis
To the Editor:
Tinea capitis is a common childhood infection seen worldwide and is more prevalent in children of African descent.1 Treatment can be effective; however, the diagnosis may be delayed due to variability in presentation, camouflage of scalp scale with ointment, and the diagnostic experience of the provider. A common complication of tinea capitis is the dermatophytid (id) reaction, which commonly manifests as multiple 1- to 2-mm monomorphic papules. We report a case of a papulosquamous variant of an id reaction secondary to tinea capitis.
An 8-year-old African American child presented with annular hyperpigmented patches on the face and trunk of several months’ duration. There was no preceding fever, illness, scalp pruritus, or alopecia according to the patient’s mother. The hyperpigmented patches persisted despite use of hydrocortisone and antifungal creams prescribed by a primary care provider. A fungal culture of a scalp specimen was negative. Physical examination during the initial dermatology visit revealed multiple annular hyperpigmented patches on the trunk and extremities. No plaques were evident; however, the mother reported that when the lesions first developed, they were raised and mildly pruritic. The patient was prescribed triamcinolone ointment 0.1% twice daily as needed for itching, and sun protection was emphasized.
At the follow-up visit weeks later, the patient’s mother reported that the ointment had helped the lesions resolve faster, but new lesions continued to appear. Physical examination at this visit was notable for scattered hyperpigmented patches, annular hyperpigmented plaques, and erythematous plaques on the trunk, arms, and legs, in addition to papulosquamous plaques and hyperpigmented patches on the forehead (Figure 1). Suspicion for tinea capitis was discussed, a repeat scalp fungal culture was performed, and oral terbinafine 250 mg once daily was started empirically. The culture was positive for Trichophyton tonsurans supporting the diagnosis of concomitant tinea capitis. The rash resolved with terbinafine, and annular patches of postinflammatory hyperpigmentation remained.

Dermatophytid reactions are immunologically mediated, disseminated, eczematous eruptions occurring after cutaneous infections or inflammatory skin conditions. Reactions occur days to weeks after exposure to antigens of dermatophytes causing tinea pedis or capitis.2
Common culprits include Microsporum canis and T tonsurans.3 Dermatophytid reactions with tinea capitis exhibit morphologic variability including a symmetric distribution of grouped or diffuse,4 pruritic, erythematous or flesh-colored, follicular papules on the trunk, with or without progression to the face, torso, upper extremities, and/or lower extremities.3 Other reported manifestations include erythema multiforme, erythema nodosum,3 or lupuslike lesions, and crops of dyshidrotic vesicles on the hands in the setting of Trichophyton mentagrophytes–induced tinea pedis.5
The papulosquamous variant id reaction should be considered in a wider differential that includes psoriasis, nummular eczema, and pityriasis rosea. Unlike psoriasis, the id reaction is not chronic and responds to systemic antifungal therapy. Nummular eczema can be ruled out, though not entirely, by a lack of personal or family history of atopy. The characteristic cleavage lines of pityriasis rosea on the trunk are absent in patients with an id reaction, and there would be no preceding illness or herald patches seen in the id reaction.
Tinea capitis may cause a variety of id manifestations, including the papulosquamous phenotype. This case addresses practice gaps that may lead to delayed diagnosis. It also highlights the importance of recognizing uncommon morphologies, performing repeat cultures of the scalp after a negative fungal culture, and lowering the threshold of suspicion for tinea capitis in the appropriate age group and demographic, specifically pediatric patients of African descent.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
- Mayser P. Dermatophyte: current situation [in German]. Hautarzt. 2017;68:316-323.
- Nowicki R. Allergic phenomena in the course of dermatomycoses [in Polish]. Pol Merkur Lekarski. 2003;14:532-534.
5. Boralevi F, Léauté-Labrèze C, Roul S, et al. Lupus-erythematosus-like eruption induced by Trichophyton mentagrophytes infection. Dermatology. 2003;206:303-306.
To the Editor:
Tinea capitis is a common childhood infection seen worldwide and is more prevalent in children of African descent.1 Treatment can be effective; however, the diagnosis may be delayed due to variability in presentation, camouflage of scalp scale with ointment, and the diagnostic experience of the provider. A common complication of tinea capitis is the dermatophytid (id) reaction, which commonly manifests as multiple 1- to 2-mm monomorphic papules. We report a case of a papulosquamous variant of an id reaction secondary to tinea capitis.
An 8-year-old African American child presented with annular hyperpigmented patches on the face and trunk of several months’ duration. There was no preceding fever, illness, scalp pruritus, or alopecia according to the patient’s mother. The hyperpigmented patches persisted despite use of hydrocortisone and antifungal creams prescribed by a primary care provider. A fungal culture of a scalp specimen was negative. Physical examination during the initial dermatology visit revealed multiple annular hyperpigmented patches on the trunk and extremities. No plaques were evident; however, the mother reported that when the lesions first developed, they were raised and mildly pruritic. The patient was prescribed triamcinolone ointment 0.1% twice daily as needed for itching, and sun protection was emphasized.
At the follow-up visit weeks later, the patient’s mother reported that the ointment had helped the lesions resolve faster, but new lesions continued to appear. Physical examination at this visit was notable for scattered hyperpigmented patches, annular hyperpigmented plaques, and erythematous plaques on the trunk, arms, and legs, in addition to papulosquamous plaques and hyperpigmented patches on the forehead (Figure 1). Suspicion for tinea capitis was discussed, a repeat scalp fungal culture was performed, and oral terbinafine 250 mg once daily was started empirically. The culture was positive for Trichophyton tonsurans supporting the diagnosis of concomitant tinea capitis. The rash resolved with terbinafine, and annular patches of postinflammatory hyperpigmentation remained.
Dermatophytid reactions are immunologically mediated, disseminated, eczematous eruptions occurring after cutaneous infections or inflammatory skin conditions. Reactions occur days to weeks after exposure to antigens of dermatophytes causing tinea pedis or capitis.2
Common culprits include Microsporum canis and T tonsurans.3 Dermatophytid reactions with tinea capitis exhibit morphologic variability including a symmetric distribution of grouped or diffuse,4 pruritic, erythematous or flesh-colored, follicular papules on the trunk, with or without progression to the face, torso, upper extremities, and/or lower extremities.3 Other reported manifestations include erythema multiforme, erythema nodosum,3 or lupuslike lesions, and crops of dyshidrotic vesicles on the hands in the setting of Trichophyton mentagrophytes–induced tinea pedis.5
The papulosquamous variant id reaction should be considered in a wider differential that includes psoriasis, nummular eczema, and pityriasis rosea. Unlike psoriasis, the id reaction is not chronic and responds to systemic antifungal therapy. Nummular eczema can be ruled out, though not entirely, by a lack of personal or family history of atopy. The characteristic cleavage lines of pityriasis rosea on the trunk are absent in patients with an id reaction, and there would be no preceding illness or herald patches seen in the id reaction.
Tinea capitis may cause a variety of id manifestations, including the papulosquamous phenotype. This case addresses practice gaps that may lead to delayed diagnosis. It also highlights the importance of recognizing uncommon morphologies, performing repeat cultures of the scalp after a negative fungal culture, and lowering the threshold of suspicion for tinea capitis in the appropriate age group and demographic, specifically pediatric patients of African descent.
To the Editor:
Tinea capitis is a common childhood infection seen worldwide and is more prevalent in children of African descent.1 Treatment can be effective; however, the diagnosis may be delayed due to variability in presentation, camouflage of scalp scale with ointment, and the diagnostic experience of the provider. A common complication of tinea capitis is the dermatophytid (id) reaction, which commonly manifests as multiple 1- to 2-mm monomorphic papules. We report a case of a papulosquamous variant of an id reaction secondary to tinea capitis.
An 8-year-old African American child presented with annular hyperpigmented patches on the face and trunk of several months’ duration. There was no preceding fever, illness, scalp pruritus, or alopecia according to the patient’s mother. The hyperpigmented patches persisted despite use of hydrocortisone and antifungal creams prescribed by a primary care provider. A fungal culture of a scalp specimen was negative. Physical examination during the initial dermatology visit revealed multiple annular hyperpigmented patches on the trunk and extremities. No plaques were evident; however, the mother reported that when the lesions first developed, they were raised and mildly pruritic. The patient was prescribed triamcinolone ointment 0.1% twice daily as needed for itching, and sun protection was emphasized.
At the follow-up visit weeks later, the patient’s mother reported that the ointment had helped the lesions resolve faster, but new lesions continued to appear. Physical examination at this visit was notable for scattered hyperpigmented patches, annular hyperpigmented plaques, and erythematous plaques on the trunk, arms, and legs, in addition to papulosquamous plaques and hyperpigmented patches on the forehead (Figure 1). Suspicion for tinea capitis was discussed, a repeat scalp fungal culture was performed, and oral terbinafine 250 mg once daily was started empirically. The culture was positive for Trichophyton tonsurans supporting the diagnosis of concomitant tinea capitis. The rash resolved with terbinafine, and annular patches of postinflammatory hyperpigmentation remained.
Dermatophytid reactions are immunologically mediated, disseminated, eczematous eruptions occurring after cutaneous infections or inflammatory skin conditions. Reactions occur days to weeks after exposure to antigens of dermatophytes causing tinea pedis or capitis.2
Common culprits include Microsporum canis and T tonsurans.3 Dermatophytid reactions with tinea capitis exhibit morphologic variability including a symmetric distribution of grouped or diffuse,4 pruritic, erythematous or flesh-colored, follicular papules on the trunk, with or without progression to the face, torso, upper extremities, and/or lower extremities.3 Other reported manifestations include erythema multiforme, erythema nodosum,3 or lupuslike lesions, and crops of dyshidrotic vesicles on the hands in the setting of Trichophyton mentagrophytes–induced tinea pedis.5
The papulosquamous variant id reaction should be considered in a wider differential that includes psoriasis, nummular eczema, and pityriasis rosea. Unlike psoriasis, the id reaction is not chronic and responds to systemic antifungal therapy. Nummular eczema can be ruled out, though not entirely, by a lack of personal or family history of atopy. The characteristic cleavage lines of pityriasis rosea on the trunk are absent in patients with an id reaction, and there would be no preceding illness or herald patches seen in the id reaction.
Tinea capitis may cause a variety of id manifestations, including the papulosquamous phenotype. This case addresses practice gaps that may lead to delayed diagnosis. It also highlights the importance of recognizing uncommon morphologies, performing repeat cultures of the scalp after a negative fungal culture, and lowering the threshold of suspicion for tinea capitis in the appropriate age group and demographic, specifically pediatric patients of African descent.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
- Mayser P. Dermatophyte: current situation [in German]. Hautarzt. 2017;68:316-323.
- Nowicki R. Allergic phenomena in the course of dermatomycoses [in Polish]. Pol Merkur Lekarski. 2003;14:532-534.
5. Boralevi F, Léauté-Labrèze C, Roul S, et al. Lupus-erythematosus-like eruption induced by Trichophyton mentagrophytes infection. Dermatology. 2003;206:303-306.
- Sharma V, Silverberg NB, Howard R, et al. Do hair care practices affect the acquisition of tinea capitis? a case-control study. Arch Pediatr Adolesc Med. 2001;155:818-821.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
- Mayser P. Dermatophyte: current situation [in German]. Hautarzt. 2017;68:316-323.
- Nowicki R. Allergic phenomena in the course of dermatomycoses [in Polish]. Pol Merkur Lekarski. 2003;14:532-534.
5. Boralevi F, Léauté-Labrèze C, Roul S, et al. Lupus-erythematosus-like eruption induced by Trichophyton mentagrophytes infection. Dermatology. 2003;206:303-306.
Practice Points
- Dermatophytid (id) reactions can manifest as papulosquamous eruptions after cutaneous infections or inflammatory skin conditions.
- High clinical suspicion for id reaction in patients of the appropriate age group and demographic—pediatric patients of African descent—is imperative for reaching the correct diagnosis.
- Repeat cultures of the scalp may be indicated in patients with high clinical probability for an id reaction despite a negative fungal culture or empiric systemic treatment.
