User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
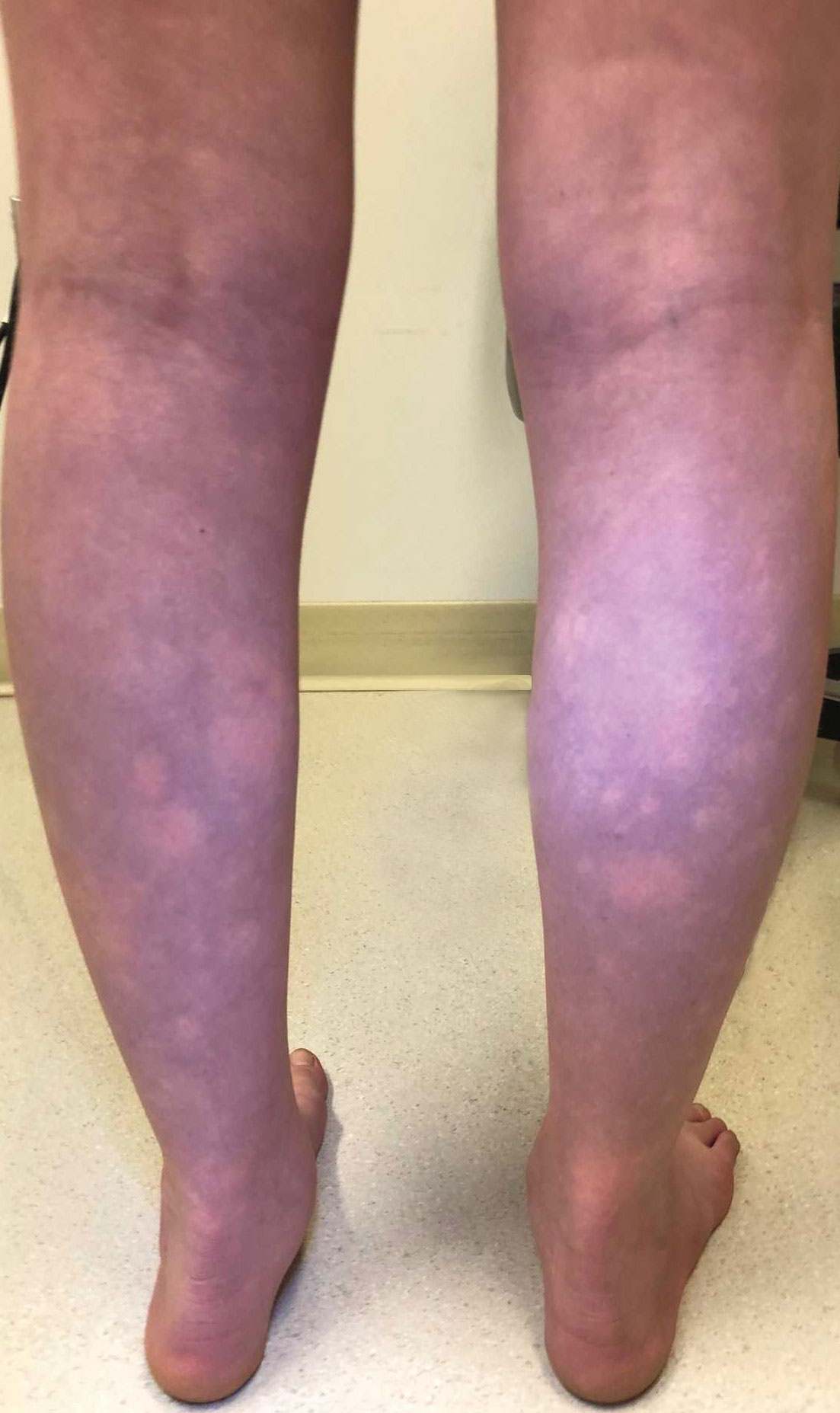
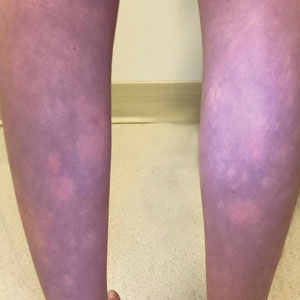
Transient Symmetric Blanching Macules on a Background of Reticulate Erythema
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
An 11-year-old girl was referred to the dermatology clinic for evaluation of a rash on the legs and feet of 1 year’s duration. The rash appeared every time she was standing for longer than 10 to 15 minutes and resolved when sitting or laying down. After the initial onset, the rash did not spread to other body areas but became more prominent in appearance. The patient endorsed intense pruritus associated with the rash. A review of systems was negative for fever, headaches, history of blood clots, and joint pain. She did not have any known medical conditions or take any medications. The patient’s mother reported that the patient experienced episodes of leg numbness while sitting in vehicles from 6 to 10 years of age. There was no family history of rheumatologic, hematologic, or cardiac conditions. The patient’s mother had experienced 2 miscarriages but denied any other obstetric complications. The patient had 1 sibling who was unaffected. Physical examination revealed reticulate erythema on the calves with scattered regions of blanching and evanescent pink macules as well as dermatographism.
One month prior to presenting to dermatology, the patient was evaluated by rheumatology, endocrinology, and hematology. Laboratory workup completed at age 3 years included antinuclear antibody, anticardiolipin antibody, and antithrombin III activity; factor V Leiden; cryoglobulins; quantitation (human chorionic gonadotropin); proteins S and C activity; antineutrophil cytoplasmic antibody screen; thyroid studies; prothrombin time; and partial thromboplastin time. All laboratory results were within reference range.
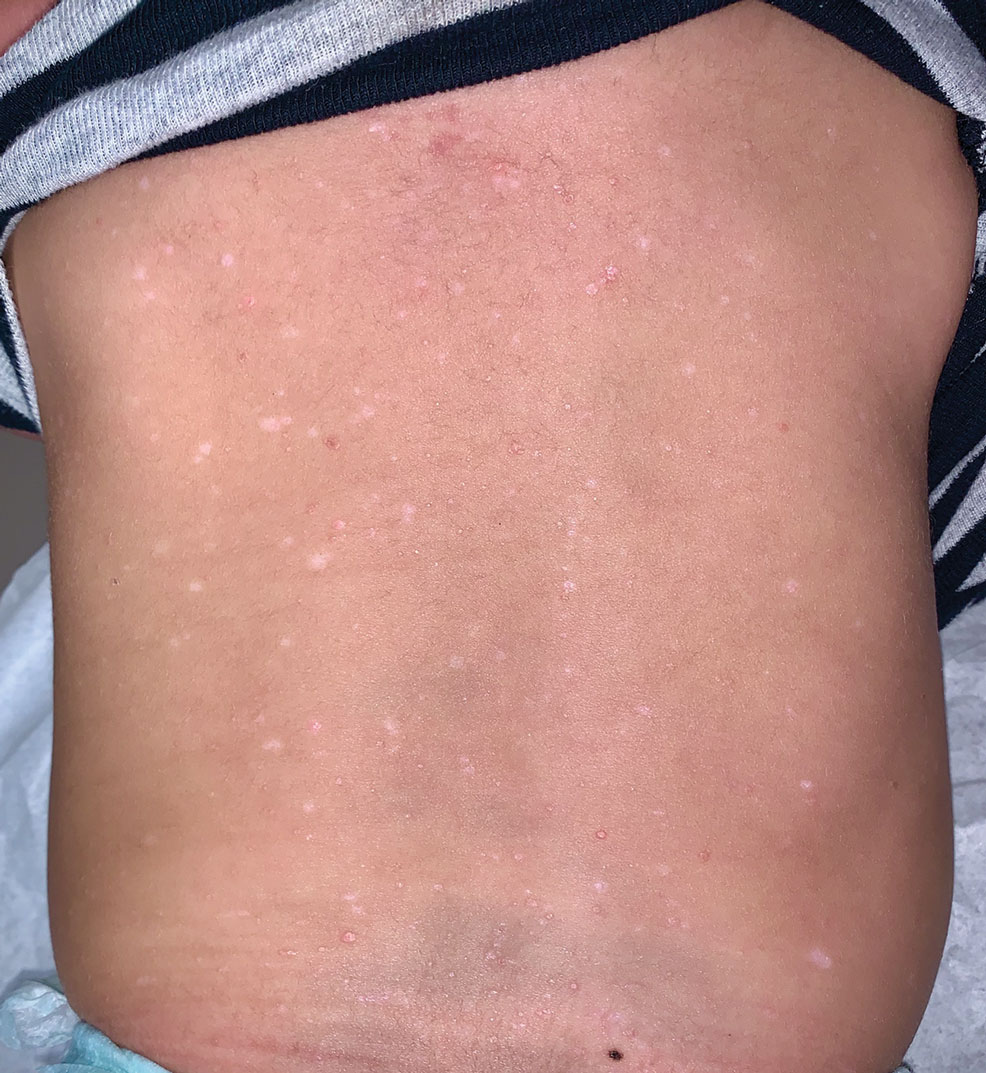
Hypopigmented Cutaneous Langerhans Cell Histiocytosis in a Hispanic Infant
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
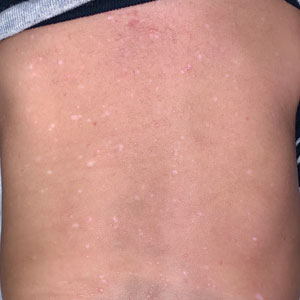
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
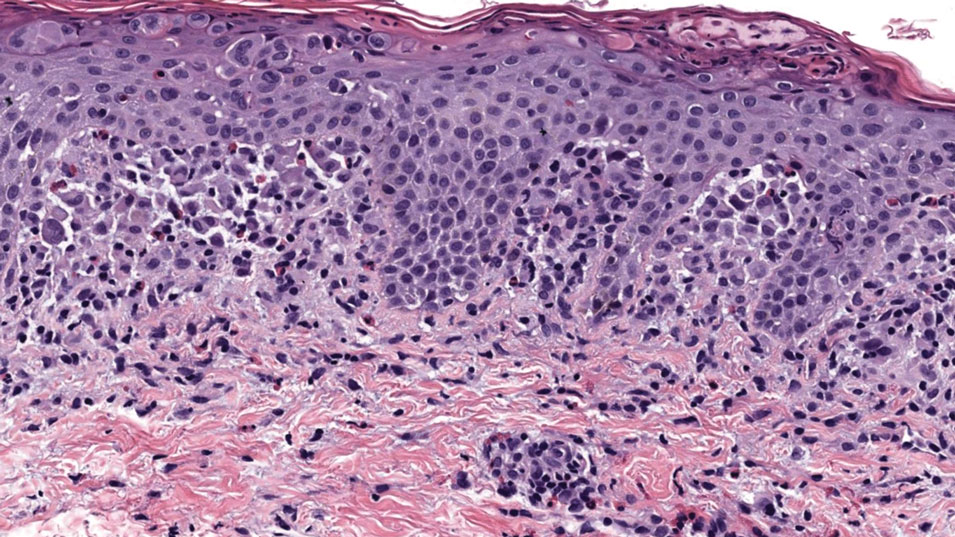
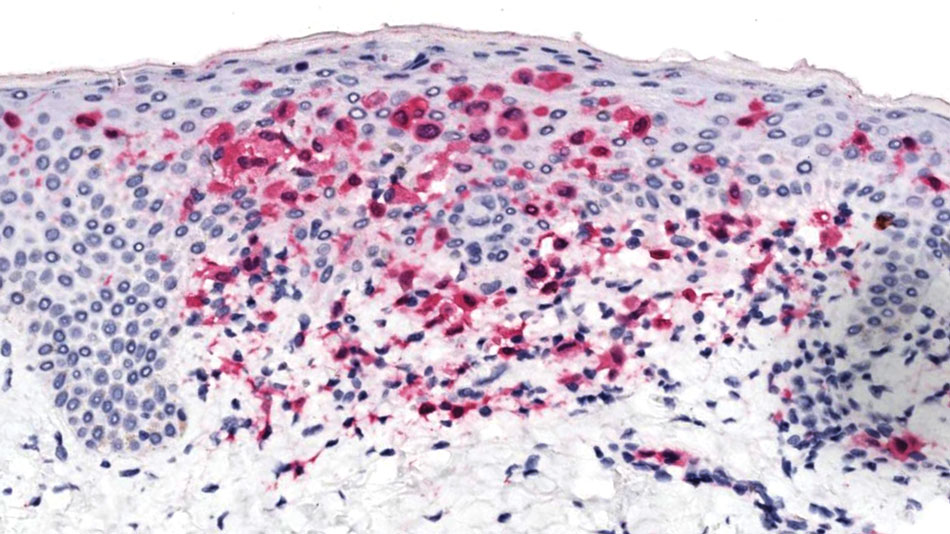
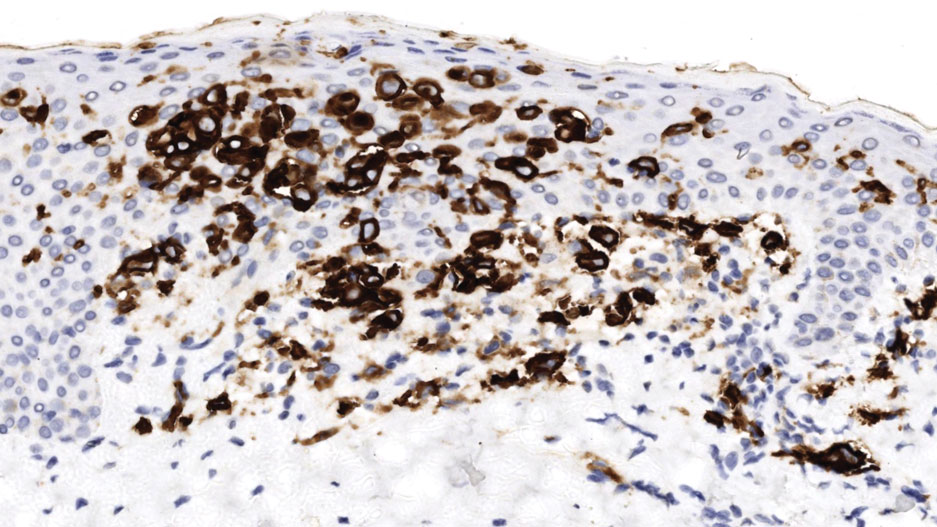
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
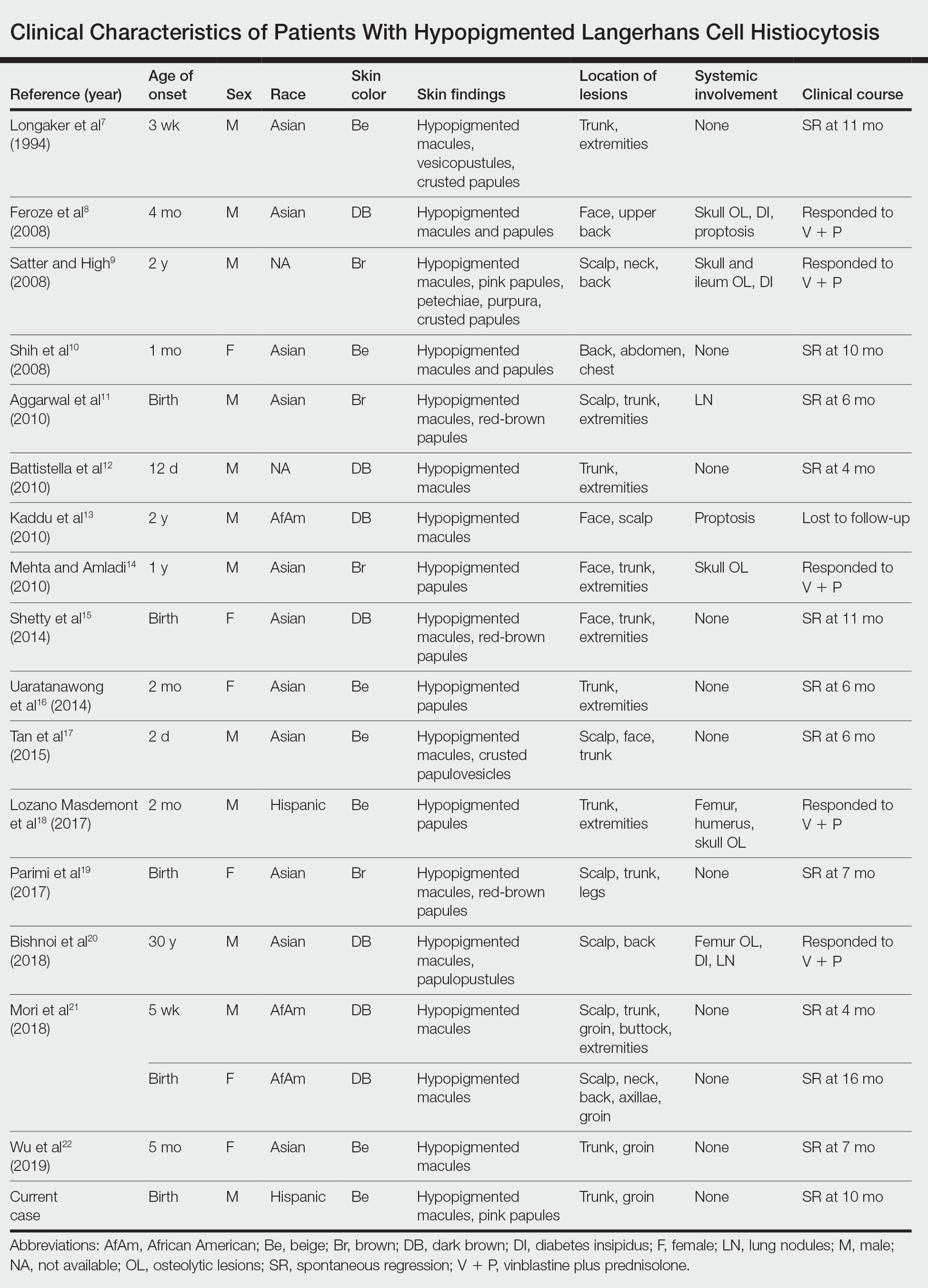
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
Practice Points
- Dermatologists should be aware of the hypopigmented variant of cutaneous Langerhans cell histiocytosis (LCH), which has been reported exclusively in patients with skin of color.
- Langerhans cell histiocytosis should be included in the differential diagnosis of hypopigmented macules, which may be the only cutaneous manifestation or may coincide with typical lesions of LCH.
- Hypopigmented cutaneous LCH may be more common in newborns and associated with a better prognosis.
Exploring Skin Pigmentation Adaptation: A Systematic Review on the Vitamin D Adaptation Hypothesis
The risk for developing skin cancer can be somewhat attributed to variations in skin pigmentation. Historically, lighter skin pigmentation has been observed in populations living in higher latitudes and darker pigmentation in populations near the equator. Although skin pigmentation is a conglomeration of genetic and environmental factors, anthropologic studies have demonstrated an association of human skin lightening with historic human migratory patterns.1 It is postulated that migration to latitudes with less UVB light penetration has resulted in a compensatory natural selection of lighter skin types. Furthermore, the driving force behind this migration-associated skin lightening has remained unclear.1
The need for folate metabolism, vitamin D synthesis, and barrier protection, as well as cultural practices, has been postulated as driving factors for skin pigmentation variation. Synthesis of vitamin D is a UV radiation (UVR)–dependent process and has remained a prominent theoretical driver for the basis of evolutionary skin lightening. Vitamin D can be acquired both exogenously or endogenously via dietary supplementation or sunlight; however, historically it has been obtained through UVB exposure primarily. Once UVB is absorbed by the skin, it catalyzes conversion of 7-dehydrocholesterol to previtamin D3, which is converted to vitamin D in the kidneys.2,3 It is suggested that lighter skin tones have an advantage over darker skin tones in synthesizing vitamin D at higher latitudes where there is less UVB, thus leading to the adaptation process.1 In this systematic review, we analyzed the evolutionary vitamin D adaptation hypothesis and assessed the validity of evidence supporting this theory in the literature.
Methods
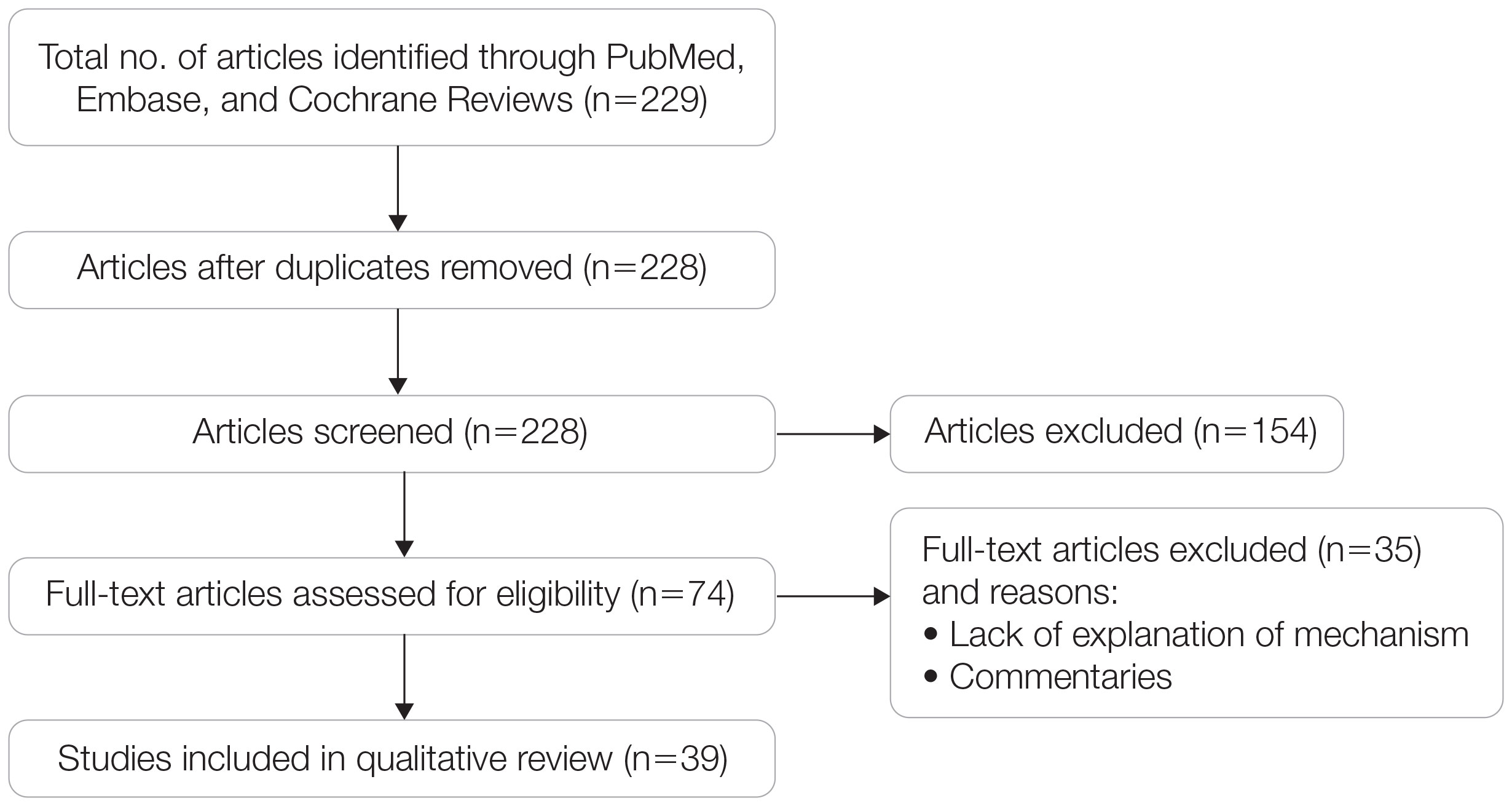
A search of PubMed, Embase, and the Cochrane Reviews database was conducted using the terms evolution, vitamin D, and skin to generate articles published from 2010 to 2022 that evaluated the influence of UVR-dependent production of vitamin D on skin pigmentation through historical migration patterns (Figure). Studies were excluded during an initial screening of abstracts followed by full-text assessment if they only had abstracts and if articles were inaccessible for review or in the form of case reports and commentaries.
The following data were extracted from each included study: reference citation, affiliated institutions of authors, author specialties, journal name, year of publication, study period, type of article, type of study, mechanism of adaptation, data concluding or supporting vitamin D as the driver, and data concluding or suggesting against vitamin D as the driver. Data concluding or supporting vitamin D as the driver were recorded from statistically significant results, study conclusions, and direct quotations. Data concluding or suggesting against vitamin D as the driver also were recorded from significant results, study conclusions, and direct quotes. The mechanism of adaptation was based on vitamin D synthesis modulation, melanin upregulation, genetic selections, genetic drift, mating patterns, increased vitamin D sensitivity, interbreeding, and diet.
Studies included in the analysis were placed into 1 of 3 categories: supporting, neutral, and against. Strength of Recommendation Taxonomy (SORT) criteria were used to classify the level of evidence of each article.4 Each article’s level of evidence was then graded (Table 1). The SORT grading levels were based on quality and evidence type: level 1 signified good-quality, patient-oriented evidence; level 2 signified limited-quality, patient-oriented evidence; and level 3 signified other evidence.4
Results
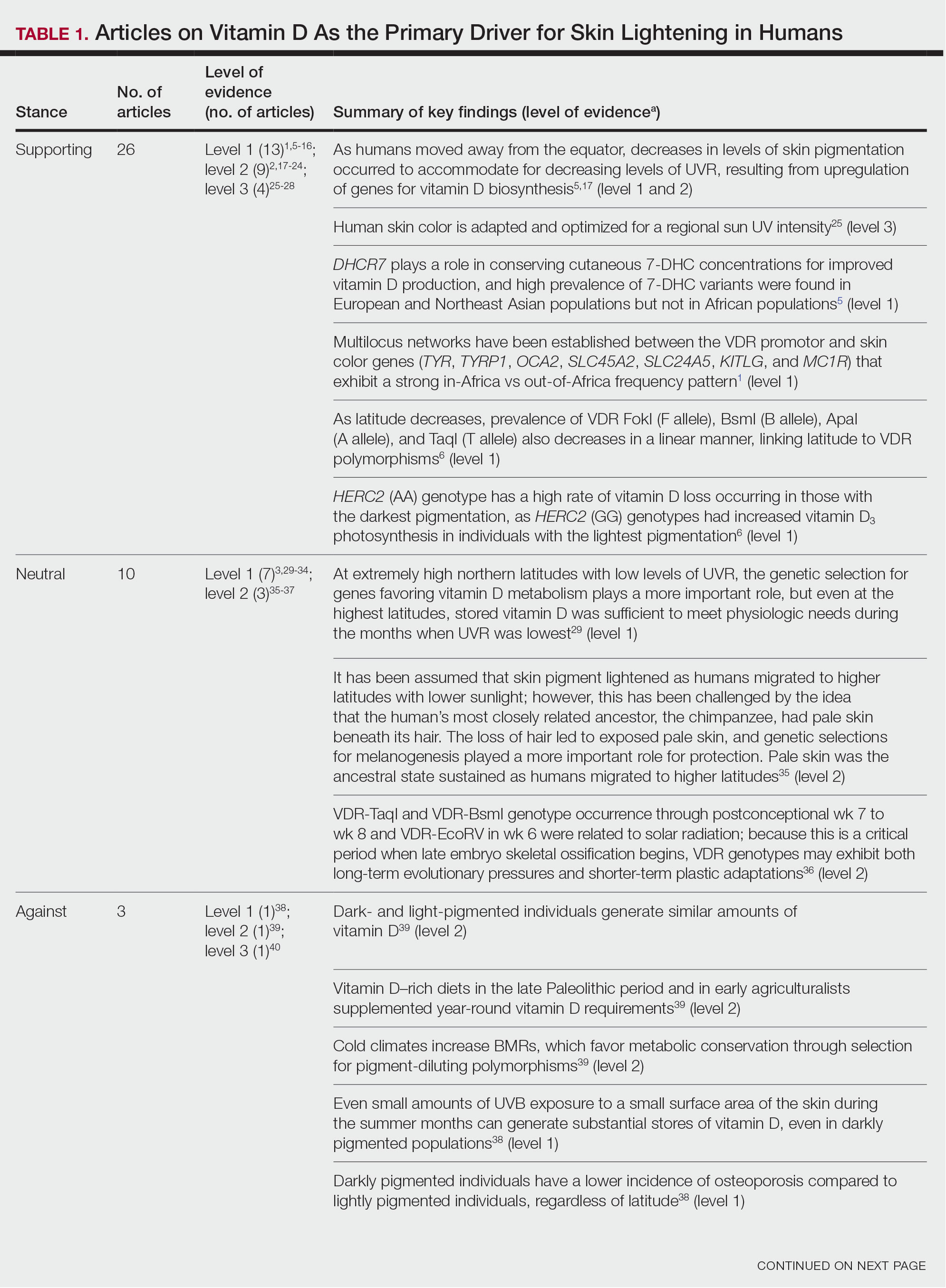
Article Selection—A total of 229 articles were identified for screening, and 39 studies met inclusion criteria.1-3,5-40 Systematic and retrospective reviews were the most common types of studies. Genomic analysis/sequencing/genome-wide association studies (GWAS) were the most common methods of analysis. Of these 39 articles, 26 were classified as supporting the evolutionary vitamin D adaptation hypothesis, 10 were classified as neutral, and 3 were classified as against (Table 1).
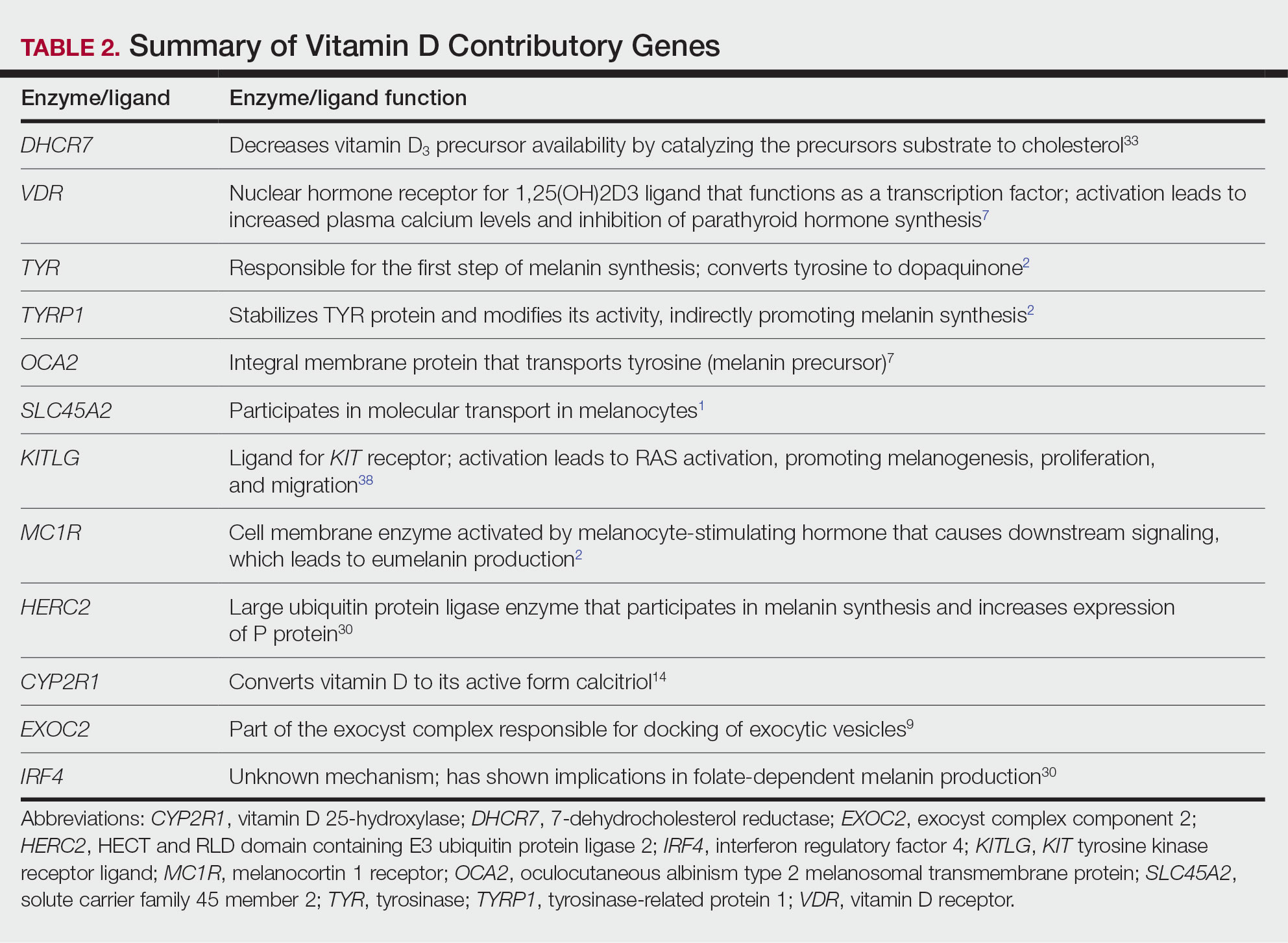
Of the articles classified as supporting the vitamin D hypothesis, 13 articles were level 1 evidence, 9 were level 2, and 4 were level 3. Key findings supporting the vitamin D hypothesis included genetic natural selection favoring vitamin D synthesis genes at higher latitudes with lower UVR and the skin lightening that occurred to protect against vitamin D deficiency (Table 1). Specific genes supporting these findings included 7-dehydrocholesterol reductase (DHCR7), vitamin D receptor (VDR), tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), oculocutaneous albinism type 2 melanosomal transmembrane protein (OCA2), solute carrier family 45 member 2 (SLC45A2), solute carrier family 4 member 5 (SLC24A5), Kit ligand (KITLG), melanocortin 1 receptor (MC1R), and HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2)(Table 2).
Of the articles classified as being against the vitamin D hypothesis, 1 article was level 1 evidence, 1 was level 2, and 1 was level 3. Key findings refuting the vitamin D hypothesis included similar amounts of vitamin D synthesis in contemporary dark- and light-pigmented individuals, vitamin D–rich diets in the late Paleolithic period and in early agriculturalists, and metabolic conservation being the primary driver (Table 1).
Of the articles classified as neutral to the hypothesis, 7 articles were level 1 evidence and 3 were level 2. Key findings of these articles included genetic selection favoring vitamin D synthesis only for populations at extremely northern latitudes, skin lightening that was sustained in northern latitudes from the neighboring human ancestor the chimpanzee, and evidence for long-term evolutionary pressures and short-term plastic adaptations in vitamin D genes (Table 1).
Comment
The importance of appropriate vitamin D levels is hypothesized as a potent driver in skin lightening because the vitamin is essential for many biochemical processes within the human body. Proper calcification of bones requires activated vitamin D to prevent rickets in childhood. Pelvic deformation in women with rickets can obstruct childbirth in primitive medical environments.15 This direct reproductive impairment suggests a strong selective pressure for skin lightening in populations that migrated northward to enhance vitamin D synthesis.
Of the 39 articles that we reviewed, the majority (n=26 [66.7%]) supported the hypothesis that vitamin D synthesis was the main driver behind skin lightening, whereas 3 (7.7%) did not support the hypothesis and 10 (25.6%) were neutral. Other leading theories explaining skin lightening included the idea that enhanced melanogenesis protected against folate degradation; genetic selection for light-skin alleles due to genetic drift; skin lightening being the result of sexual selection; and a combination of factors, including dietary choices, clothing preferences, and skin permeability barriers.
Articles With Supporting Evidence for the Vitamin D Theory—As Homo sapiens migrated out of Africa, migration patterns demonstrated the correlation between distance from the equator and skin pigmentation from natural selection. Individuals with darker skin pigment required higher levels of UVR to synthesize vitamin D. According to Beleza et al,1 as humans migrated to areas of higher latitudes with lower levels of UVR, natural selection favored the development of lighter skin to maximize vitamin D production. Vitamin D is linked to calcium metabolism, and its deficiency can lead to bone malformations and poor immune function.35 Several genes affecting melanogenesis and skin pigment have been found to have geospatial patterns that map to different geographic locations of various populations, indicating how human migration patterns out of Africa created this natural selection for skin lightening. The gene KITLG—associated with lighter skin pigmentation—has been found in high frequencies in both European and East Asian populations and is proposed to have increased in frequency after the migration out of Africa. However, the genes TYRP1, SLC24A5, and SLC45A2 were found at high frequencies only in European populations, and this selection occurred 11,000 to 19,000 years ago during the Last Glacial Maximum (15,000–20,000 years ago), demonstrating the selection for European over East Asian characteristics. During this period, seasonal changes increased the risk for vitamin D deficiency and provided an urgency for selection to a lighter skin pigment.1
The migration of H sapiens to northern latitudes prompted the selection of alleles that would increasevitamin D synthesis to counteract the reduced UV exposure. Genetic analysis studies have found key associations between genes encoding for the metabolism of vitamin D and pigmentation. Among this complex network are the essential downstream enzymes in the melanocortin receptor 1 pathway, including TYR and TYRP1. Forty-six of 960 single-nucleotide polymorphisms located in 29 different genes involved in skin pigmentation that were analyzed in a cohort of 2970 individuals were significantly associated with serum vitamin D levels (P<.05). The exocyst complex component 2 (EXOC2), TYR, and TYRP1 gene variants were shown to have the greatest influence on vitamin D status.9 These data reveal how pigment genotypes are predictive of vitamin D levels and the epistatic potential among many genes in this complex network.
Gene variation plays an important role in vitamin D status when comparing genetic polymorphisms in populations in northern latitudes to African populations. Vitamin D3 precursor availability is decreased by 7-DHCR catalyzing the precursors substrate to cholesterol. In a study using GWAS, it was found that “variations in DHCR7 may aid vitamin D production by conserving cutaneous 7-DHC levels. A high prevalence of DHCR7 variants were found in European and Northeast Asian populations but not in African populations, suggesting that selection occurred for these DHCR7 mutations in populations who migrated to more northern latitudes.5 Multilocus networks have been established between the VDR promotor and skin color genes (Table 2) that exhibit a strong in-Africa vs out-of-Africa frequency pattern. It also has been shown that genetic variation (suggesting a long-term evolutionary inclination) and epigenetic modification (indicative of short-term exposure) of VDR lends support to the vitamin D hypothesis. As latitude decreases, prevalence of VDR FokI (F allele), BsmI (B allele), ApaI (A allele), and TaqI (T allele) also decreases in a linear manner, linking latitude to VDR polymorphisms. Plasma vitamin D levels and photoperiod of conception—UV exposure during the periconceptional period—also were extrapolative of VDR methylation in a study involving 80 participants, where these 2 factors accounted for 17% of variance in methylation.6
Other noteworthy genes included HERC2, which has implications in the expression of OCA2 (melanocyte-specific transporter protein), and IRF4, which encodes for an important enzyme in folate-dependent melanin production. In an Australian cross-sectional study that analyzed vitamin D and pigmentation gene polymorphisms in conjunction with plasma vitamin D levels, the most notable rate of vitamin D loss occurred in individuals with the darkest pigmentation HERC2 (AA) genotype.31 In contrast, the lightest pigmentation HERC2 (GG) genotypes had increased vitamin D3 photosynthesis. Interestingly, the lightest interferon regulatory factor 4 (IRF4) TT genotype and the darkest HERC2 AA genotype, rendering the greatest folate loss and largest synthesis of vitamin D3, were not seen in combination in any of the participants.30 In addition to HERC2, derived alleles from pigment-associated genes SLC24A5*A and SLC45A2*G demonstrated greater frequencies in Europeans (>90%) compared to Africans and East Asians, where the allelic frequencies were either rare or absent.1 This evidence delineates not only the complexity but also the strong relationship between skin pigmentation, latitude, and vitamin D status. The GWAS also have supported this concept. In comparing European populations to African populations, there was a 4-fold increase in the frequencies of “derived alleles of the vitamin D transport protein (GC, rs3755967), the 25(OH)D3 synthesizing enzyme (CYP2R1, rs10741657), VDR (rs2228570 (commonly known as FokI polymorphism), rs1544410 (Bsm1), and rs731236 (Taq1) and the VDR target genes CYP24A1 (rs17216707), CD14 (rs2569190), and CARD9 (rs4077515).”32
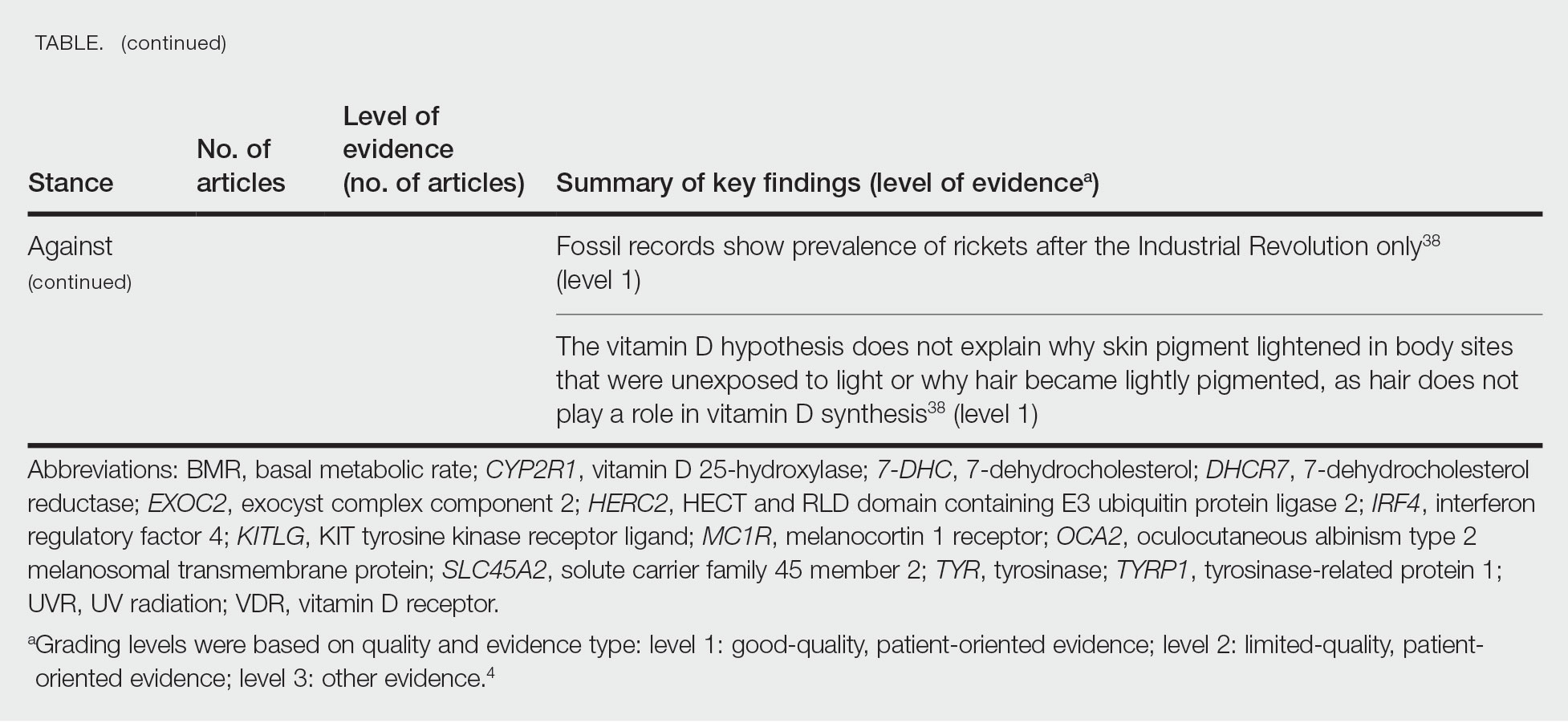
Articles With Evidence Against the Vitamin D Theory—This review analyzed the level of support for the theory that vitamin D was the main driver for skin lightening. Although most articles supported this theory, there were articles that listed other plausible counterarguments. Jablonski and Chaplin3 suggested that humans living in higher latitudes compensated for increased demand of vitamin D by placing cultural importance on a diet of vitamin D–rich foods and thus would not have experienced decreased vitamin D levels, which we hypothesize were the driver for skin lightening. Elias et al39 argued that initial pigment dilution may have instead served to improve metabolic conservation, as the authors found no evidence of rickets—the sequelae of vitamin D deficiency—in pre–industrial age human fossils. Elias and Williams38 proposed that differences in skin pigment are due to a more intact skin permeability barrier as “a requirement for life in a desiccating terrestrial environment,” which is seen in darker skin tones compared to lighter skin tones and thus can survive better in warmer climates with less risk of infections or dehydration.
Articles With Neutral Evidence for the Vitamin D Theory—Greaves41 argued against the idea that skin evolved to become lighter to protect against vitamin D deficiency. They proposed that the chimpanzee, which is the human’s most closely related species, had light skin covered by hair, and the loss of this hair led to exposed pale skin that created a need for increased melanin production for protection from UVR. Greaves41 stated that the MC1R gene (associated with darker pigmentation) was selected for in African populations, and those with pale skin retained their original pigment as they migrated to higher latitudes. Further research has demonstrated that the genetic natural selection for skin pigment is a complex process that involves multiple gene variants found throughout cultures across the globe.
Conclusion
Skin pigmentation has continuously evolved alongside humans. Genetic selection for lighter skin coincides with a favorable selection for genes involved in vitamin D synthesis as humans migrated to northern latitudes, which enabled humans to produce adequate levels of exogenous vitamin D in low-UVR areas and in turn promoted survival. Early humans without access to supplementation or foods rich in vitamin D acquired vitamin D primarily through sunlight. In comparison to modern society, where vitamin D supplementation is accessible and human lifespans are prolonged, lighter skin tone is now a risk factor for malignant cancers of the skin rather than being a protective adaptation. Current sun behavior recommendations conclude that the body’s need for vitamin D is satisfied by UV exposure to the arms, legs, hands, and/or face for only 5 to 30 minutes between 10
The hypothesis that skin lightening primarily was driven by the need for vitamin D can only be partially supported by our review. Studies have shown that there is a corresponding complex network of genes that determines skin pigmentation as well as vitamin D synthesis and conservation. However, there is sufficient evidence that skin lightening is multifactorial in nature, and vitamin D alone may not be the sole driver. The information in this review can be used by health care providers to educate patients on sun protection, given the lesser threat of severe vitamin D deficiency in developed communities today that have access to adequate nutrition and supplementation.
Skin lightening and its coinciding evolutionary drivers are a rather neglected area of research. Due to heterogeneous cohorts and conservative data analysis, GWAS studies run the risk of type II error, yielding a limitation in our data analysis.9 Furthermore, the data regarding specific time frames in evolutionary skin lightening as well as the intensity of gene polymorphisms are limited.1 Further studies are needed to determine the interconnectedness of the current skin-lightening theories to identify other important factors that may play a role in the process. Determining the key event can help us better understand skin-adaptation mechanisms and create a framework for understanding the vital process involved in adaptation, survival, and disease manifestation in different patient populations.
- Beleza S, Santos AM, McEvoy B, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013;30:24-35. doi:10.1093/molbev/mss207
- Carlberg C. Nutrigenomics of vitamin D. Nutrients. 2019;11:676. doi:10.3390/nu11030676
- Jablonski NG, Chaplin G. The roles of vitamin D and cutaneous vitamin D production in human evolution and health. Int J Paleopathol. 2018;23:54-59. doi:10.1016/j.ijpp.2018.01.005
- Weiss BD. SORT: strength of recommendation taxonomy. Fam Med. 2004;36:141-143.
- Wolf ST, Kenney WL. The vitamin D–folate hypothesis in human vascular health. Am J Physiol Regul Integr Comp Physiology. 2019;317:R491-R501. doi:10.1152/ajpregu.00136.2019
- Lucock M, Jones P, Martin C, et al. Photobiology of vitamins. Nutr Rev. 2018;76:512-525. doi:10.1093/nutrit/nuy013
- Hochberg Z, Hochberg I. Evolutionary perspective in rickets and vitamin D. Front Endocrinol (Lausanne). 2019;10:306. doi:10.3389/fendo.2019.00306
- Rossberg W, Saternus R, Wagenpfeil S, et al. Human pigmentation, cutaneous vitamin D synthesis and evolution: variants of genes (SNPs) involved in skin pigmentation are associated with 25(OH)D serum concentration. Anticancer Res. 2016;36:1429-1437.
- Saternus R, Pilz S, Gräber S, et al. A closer look at evolution: variants (SNPs) of genes involved in skin pigmentation, including EXOC2, TYR, TYRP1, and DCT, are associated with 25(OH)D serum concentration. Endocrinology. 2015;156:39-47. doi:10.1210/en.2014-1238
- López S, García Ó, Yurrebaso I, et al. The interplay between natural selection and susceptibility to melanoma on allele 374F of SLC45A2 gene in a south European population. PloS One. 2014;9:E104367. doi:1371/journal.pone.0104367
- Lucock M, Yates Z, Martin C, et al. Vitamin D, folate, and potential early lifecycle environmental origin of significant adult phenotypes. Evol Med Public Health. 2014;2014:69-91. doi:10.1093/emph/eou013
- Hudjashov G, Villems R, Kivisild T. Global patterns of diversity and selection in human tyrosinase gene. PloS One. 2013;8:E74307. doi:10.1371/journal.pone.0074307
- Khan R, Khan BSR. Diet, disease and pigment variation in humans. Med Hypotheses. 2010;75:363-367. doi:10.1016/j.mehy.2010.03.033
- Kuan V, Martineau AR, Griffiths CJ, et al. DHCR7 mutations linked to higher vitamin D status allowed early human migration to northern latitudes. BMC Evol Biol. 2013;13:144. doi:10.1186/1471-2148-13-144
- Omenn GS. Evolution and public health. Proc National Acad Sci. 2010;107(suppl 1):1702-1709. doi:10.1073/pnas.0906198106
- Yuen AWC, Jablonski NG. Vitamin D: in the evolution of human skin colour. Med Hypotheses. 2010;74:39-44. doi:10.1016/j.mehy.2009.08.007
- Vieth R. Weaker bones and white skin as adaptions to improve anthropological “fitness” for northern environments. Osteoporosis Int. 2020;31:617-624. doi:10.1007/s00198-019-05167-4
- Carlberg C. Vitamin D: a micronutrient regulating genes. Curr Pharm Des. 2019;25:1740-1746. doi:10.2174/1381612825666190705193227
- Haddadeen C, Lai C, Cho SY, et al. Variants of the melanocortin‐1 receptor: do they matter clinically? Exp Dermatol. 2015;1:5-9. doi:10.1111/exd.12540
- Yao S, Ambrosone CB. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J Steroid Biochem Mol Biol. 2013;136:337-341. doi:10.1016/j.jsbmb.2012.09.010
- Jablonski N. The evolution of human skin colouration and its relevance to health in the modern world. J Royal Coll Physicians Edinb. 2012;42:58-63. doi:10.4997/jrcpe.2012.114
- Jablonski NG, Chaplin G. Human skin pigmentation as an adaptation to UV radiation. Proc National Acad Sci. 2010;107(suppl 2):8962-8968. doi:10.1073/pnas.0914628107
- Hochberg Z, Templeton AR. Evolutionary perspective in skin color, vitamin D and its receptor. Hormones. 2010;9:307-311. doi:10.14310/horm.2002.1281
- Jones P, Lucock M, Veysey M, et al. The vitamin D–folate hypothesis as an evolutionary model for skin pigmentation: an update and integration of current ideas. Nutrients. 2018;10:554. doi:10.3390/nu10050554
- Lindqvist PG, Epstein E, Landin-Olsson M, et al. Women with fair phenotypes seem to confer a survival advantage in a low UV milieu. a nested matched case control study. PloS One. 2020;15:E0228582. doi:10.1371/journal.pone.0228582
- Holick MF. Shedding new light on the role of the sunshine vitamin D for skin health: the lncRNA–skin cancer connection. Exp Dermatol. 2014;23:391-392. doi:10.1111/exd.12386
- Jablonski NG, Chaplin G. Epidermal pigmentation in the human lineage is an adaptation to ultraviolet radiation. J Hum Evol. 2013;65:671-675. doi:10.1016/j.jhevol.2013.06.004
- Jablonski NG, Chaplin G. The evolution of skin pigmentation and hair texture in people of African ancestry. Dermatol Clin. 2014;32:113-121. doi:10.1016/j.det.2013.11.003
- Jablonski NG. The evolution of human skin pigmentation involved the interactions of genetic, environmental, and cultural variables. Pigment Cell Melanoma Res. 2021;34:707-7 doi:10.1111/pcmr.12976
- Lucock MD, Jones PR, Veysey M, et al. Biophysical evidence to support and extend the vitamin D‐folate hypothesis as a paradigm for the evolution of human skin pigmentation. Am J Hum Biol. 2022;34:E23667. doi:10.1002/ajhb.23667
- Missaggia BO, Reales G, Cybis GB, et al. Adaptation and co‐adaptation of skin pigmentation and vitamin D genes in native Americans. Am J Med Genet C Semin Med Genet. 2020;184:1060-1077. doi:10.1002/ajmg.c.31873
- Hanel A, Carlberg C. Skin colour and vitamin D: an update. Exp Dermatol. 2020;29:864-875. doi:10.1111/exd.14142
- Hanel A, Carlberg C. Vitamin D and evolution: pharmacologic implications. Biochem Pharmacol. 2020;173:113595. doi:10.1016/j.bcp.2019.07.024
- Flegr J, Sýkorová K, Fiala V, et al. Increased 25(OH)D3 level in redheaded people: could redheadedness be an adaptation to temperate climate? Exp Dermatol. 2020;29:598-609. doi:10.1111/exd.14119
- James WPT, Johnson RJ, Speakman JR, et al. Nutrition and its role in human evolution. J Intern Med. 2019;285:533-549. doi:10.1111/joim.12878
- Lucock M, Jones P, Martin C, et al. Vitamin D: beyond metabolism. J Evid Based Complementary Altern Med. 2015;20:310-322. doi:10.1177/2156587215580491
- Jarrett P, Scragg R. Evolution, prehistory and vitamin D. Int J Environ Res Public Health. 2020;17:646. doi:10.3390/ijerph17020646
- Elias PM, Williams ML. Re-appraisal of current theories for thedevelopment and loss of epidermal pigmentation in hominins and modern humans. J Hum Evol. 2013;64:687-692. doi:10.1016/j.jhevol.2013.02.003
- Elias PM, Williams ML. Basis for the gain and subsequent dilution of epidermal pigmentation during human evolution: the barrier and metabolic conservation hypotheses revisited. Am J Phys Anthropol. 2016;161:189-207. doi:10.1002/ajpa.23030
- Williams JD, Jacobson EL, Kim H, et al. Water soluble vitamins, clinical research and future application. Subcell Biochem. 2011;56:181-197. doi:10.1007/978-94-007-2199-9_10
- Greaves M. Was skin cancer a selective force for black pigmentation in early hominin evolution [published online February 26, 2014]? Proc Biol Sci. 2014;281:20132955. doi:10.1098/rspb.2013.2955
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281. doi:10.1056/nejmra070553
- Bouillon R. Comparative analysis of nutritional guidelines for vitamin D. Nat Rev Endocrinol. 2017;13:466-479. doi:10.1038/nrendo.2017.31
- US Department of Health and Human Services. The Surgeon General’s Call to Action to Prevent Skin Cancer. US Dept of Health and Human Services, Office of the Surgeon General; 2014. Accessed April 29, 2024. https://www.hhs.gov/sites/default/files/call-to-action-prevent-skin-cancer.pdf
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press; 2011. https://www.ncbi.nlm.nih.gov/books/NBK56070/
The risk for developing skin cancer can be somewhat attributed to variations in skin pigmentation. Historically, lighter skin pigmentation has been observed in populations living in higher latitudes and darker pigmentation in populations near the equator. Although skin pigmentation is a conglomeration of genetic and environmental factors, anthropologic studies have demonstrated an association of human skin lightening with historic human migratory patterns.1 It is postulated that migration to latitudes with less UVB light penetration has resulted in a compensatory natural selection of lighter skin types. Furthermore, the driving force behind this migration-associated skin lightening has remained unclear.1
The need for folate metabolism, vitamin D synthesis, and barrier protection, as well as cultural practices, has been postulated as driving factors for skin pigmentation variation. Synthesis of vitamin D is a UV radiation (UVR)–dependent process and has remained a prominent theoretical driver for the basis of evolutionary skin lightening. Vitamin D can be acquired both exogenously or endogenously via dietary supplementation or sunlight; however, historically it has been obtained through UVB exposure primarily. Once UVB is absorbed by the skin, it catalyzes conversion of 7-dehydrocholesterol to previtamin D3, which is converted to vitamin D in the kidneys.2,3 It is suggested that lighter skin tones have an advantage over darker skin tones in synthesizing vitamin D at higher latitudes where there is less UVB, thus leading to the adaptation process.1 In this systematic review, we analyzed the evolutionary vitamin D adaptation hypothesis and assessed the validity of evidence supporting this theory in the literature.
Methods
A search of PubMed, Embase, and the Cochrane Reviews database was conducted using the terms evolution, vitamin D, and skin to generate articles published from 2010 to 2022 that evaluated the influence of UVR-dependent production of vitamin D on skin pigmentation through historical migration patterns (Figure). Studies were excluded during an initial screening of abstracts followed by full-text assessment if they only had abstracts and if articles were inaccessible for review or in the form of case reports and commentaries.
The following data were extracted from each included study: reference citation, affiliated institutions of authors, author specialties, journal name, year of publication, study period, type of article, type of study, mechanism of adaptation, data concluding or supporting vitamin D as the driver, and data concluding or suggesting against vitamin D as the driver. Data concluding or supporting vitamin D as the driver were recorded from statistically significant results, study conclusions, and direct quotations. Data concluding or suggesting against vitamin D as the driver also were recorded from significant results, study conclusions, and direct quotes. The mechanism of adaptation was based on vitamin D synthesis modulation, melanin upregulation, genetic selections, genetic drift, mating patterns, increased vitamin D sensitivity, interbreeding, and diet.
Studies included in the analysis were placed into 1 of 3 categories: supporting, neutral, and against. Strength of Recommendation Taxonomy (SORT) criteria were used to classify the level of evidence of each article.4 Each article’s level of evidence was then graded (Table 1). The SORT grading levels were based on quality and evidence type: level 1 signified good-quality, patient-oriented evidence; level 2 signified limited-quality, patient-oriented evidence; and level 3 signified other evidence.4
Results
Article Selection—A total of 229 articles were identified for screening, and 39 studies met inclusion criteria.1-3,5-40 Systematic and retrospective reviews were the most common types of studies. Genomic analysis/sequencing/genome-wide association studies (GWAS) were the most common methods of analysis. Of these 39 articles, 26 were classified as supporting the evolutionary vitamin D adaptation hypothesis, 10 were classified as neutral, and 3 were classified as against (Table 1).
Of the articles classified as supporting the vitamin D hypothesis, 13 articles were level 1 evidence, 9 were level 2, and 4 were level 3. Key findings supporting the vitamin D hypothesis included genetic natural selection favoring vitamin D synthesis genes at higher latitudes with lower UVR and the skin lightening that occurred to protect against vitamin D deficiency (Table 1). Specific genes supporting these findings included 7-dehydrocholesterol reductase (DHCR7), vitamin D receptor (VDR), tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), oculocutaneous albinism type 2 melanosomal transmembrane protein (OCA2), solute carrier family 45 member 2 (SLC45A2), solute carrier family 4 member 5 (SLC24A5), Kit ligand (KITLG), melanocortin 1 receptor (MC1R), and HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2)(Table 2).
Of the articles classified as being against the vitamin D hypothesis, 1 article was level 1 evidence, 1 was level 2, and 1 was level 3. Key findings refuting the vitamin D hypothesis included similar amounts of vitamin D synthesis in contemporary dark- and light-pigmented individuals, vitamin D–rich diets in the late Paleolithic period and in early agriculturalists, and metabolic conservation being the primary driver (Table 1).
Of the articles classified as neutral to the hypothesis, 7 articles were level 1 evidence and 3 were level 2. Key findings of these articles included genetic selection favoring vitamin D synthesis only for populations at extremely northern latitudes, skin lightening that was sustained in northern latitudes from the neighboring human ancestor the chimpanzee, and evidence for long-term evolutionary pressures and short-term plastic adaptations in vitamin D genes (Table 1).
Comment
The importance of appropriate vitamin D levels is hypothesized as a potent driver in skin lightening because the vitamin is essential for many biochemical processes within the human body. Proper calcification of bones requires activated vitamin D to prevent rickets in childhood. Pelvic deformation in women with rickets can obstruct childbirth in primitive medical environments.15 This direct reproductive impairment suggests a strong selective pressure for skin lightening in populations that migrated northward to enhance vitamin D synthesis.
Of the 39 articles that we reviewed, the majority (n=26 [66.7%]) supported the hypothesis that vitamin D synthesis was the main driver behind skin lightening, whereas 3 (7.7%) did not support the hypothesis and 10 (25.6%) were neutral. Other leading theories explaining skin lightening included the idea that enhanced melanogenesis protected against folate degradation; genetic selection for light-skin alleles due to genetic drift; skin lightening being the result of sexual selection; and a combination of factors, including dietary choices, clothing preferences, and skin permeability barriers.
Articles With Supporting Evidence for the Vitamin D Theory—As Homo sapiens migrated out of Africa, migration patterns demonstrated the correlation between distance from the equator and skin pigmentation from natural selection. Individuals with darker skin pigment required higher levels of UVR to synthesize vitamin D. According to Beleza et al,1 as humans migrated to areas of higher latitudes with lower levels of UVR, natural selection favored the development of lighter skin to maximize vitamin D production. Vitamin D is linked to calcium metabolism, and its deficiency can lead to bone malformations and poor immune function.35 Several genes affecting melanogenesis and skin pigment have been found to have geospatial patterns that map to different geographic locations of various populations, indicating how human migration patterns out of Africa created this natural selection for skin lightening. The gene KITLG—associated with lighter skin pigmentation—has been found in high frequencies in both European and East Asian populations and is proposed to have increased in frequency after the migration out of Africa. However, the genes TYRP1, SLC24A5, and SLC45A2 were found at high frequencies only in European populations, and this selection occurred 11,000 to 19,000 years ago during the Last Glacial Maximum (15,000–20,000 years ago), demonstrating the selection for European over East Asian characteristics. During this period, seasonal changes increased the risk for vitamin D deficiency and provided an urgency for selection to a lighter skin pigment.1
The migration of H sapiens to northern latitudes prompted the selection of alleles that would increasevitamin D synthesis to counteract the reduced UV exposure. Genetic analysis studies have found key associations between genes encoding for the metabolism of vitamin D and pigmentation. Among this complex network are the essential downstream enzymes in the melanocortin receptor 1 pathway, including TYR and TYRP1. Forty-six of 960 single-nucleotide polymorphisms located in 29 different genes involved in skin pigmentation that were analyzed in a cohort of 2970 individuals were significantly associated with serum vitamin D levels (P<.05). The exocyst complex component 2 (EXOC2), TYR, and TYRP1 gene variants were shown to have the greatest influence on vitamin D status.9 These data reveal how pigment genotypes are predictive of vitamin D levels and the epistatic potential among many genes in this complex network.
Gene variation plays an important role in vitamin D status when comparing genetic polymorphisms in populations in northern latitudes to African populations. Vitamin D3 precursor availability is decreased by 7-DHCR catalyzing the precursors substrate to cholesterol. In a study using GWAS, it was found that “variations in DHCR7 may aid vitamin D production by conserving cutaneous 7-DHC levels. A high prevalence of DHCR7 variants were found in European and Northeast Asian populations but not in African populations, suggesting that selection occurred for these DHCR7 mutations in populations who migrated to more northern latitudes.5 Multilocus networks have been established between the VDR promotor and skin color genes (Table 2) that exhibit a strong in-Africa vs out-of-Africa frequency pattern. It also has been shown that genetic variation (suggesting a long-term evolutionary inclination) and epigenetic modification (indicative of short-term exposure) of VDR lends support to the vitamin D hypothesis. As latitude decreases, prevalence of VDR FokI (F allele), BsmI (B allele), ApaI (A allele), and TaqI (T allele) also decreases in a linear manner, linking latitude to VDR polymorphisms. Plasma vitamin D levels and photoperiod of conception—UV exposure during the periconceptional period—also were extrapolative of VDR methylation in a study involving 80 participants, where these 2 factors accounted for 17% of variance in methylation.6
Other noteworthy genes included HERC2, which has implications in the expression of OCA2 (melanocyte-specific transporter protein), and IRF4, which encodes for an important enzyme in folate-dependent melanin production. In an Australian cross-sectional study that analyzed vitamin D and pigmentation gene polymorphisms in conjunction with plasma vitamin D levels, the most notable rate of vitamin D loss occurred in individuals with the darkest pigmentation HERC2 (AA) genotype.31 In contrast, the lightest pigmentation HERC2 (GG) genotypes had increased vitamin D3 photosynthesis. Interestingly, the lightest interferon regulatory factor 4 (IRF4) TT genotype and the darkest HERC2 AA genotype, rendering the greatest folate loss and largest synthesis of vitamin D3, were not seen in combination in any of the participants.30 In addition to HERC2, derived alleles from pigment-associated genes SLC24A5*A and SLC45A2*G demonstrated greater frequencies in Europeans (>90%) compared to Africans and East Asians, where the allelic frequencies were either rare or absent.1 This evidence delineates not only the complexity but also the strong relationship between skin pigmentation, latitude, and vitamin D status. The GWAS also have supported this concept. In comparing European populations to African populations, there was a 4-fold increase in the frequencies of “derived alleles of the vitamin D transport protein (GC, rs3755967), the 25(OH)D3 synthesizing enzyme (CYP2R1, rs10741657), VDR (rs2228570 (commonly known as FokI polymorphism), rs1544410 (Bsm1), and rs731236 (Taq1) and the VDR target genes CYP24A1 (rs17216707), CD14 (rs2569190), and CARD9 (rs4077515).”32
Articles With Evidence Against the Vitamin D Theory—This review analyzed the level of support for the theory that vitamin D was the main driver for skin lightening. Although most articles supported this theory, there were articles that listed other plausible counterarguments. Jablonski and Chaplin3 suggested that humans living in higher latitudes compensated for increased demand of vitamin D by placing cultural importance on a diet of vitamin D–rich foods and thus would not have experienced decreased vitamin D levels, which we hypothesize were the driver for skin lightening. Elias et al39 argued that initial pigment dilution may have instead served to improve metabolic conservation, as the authors found no evidence of rickets—the sequelae of vitamin D deficiency—in pre–industrial age human fossils. Elias and Williams38 proposed that differences in skin pigment are due to a more intact skin permeability barrier as “a requirement for life in a desiccating terrestrial environment,” which is seen in darker skin tones compared to lighter skin tones and thus can survive better in warmer climates with less risk of infections or dehydration.
Articles With Neutral Evidence for the Vitamin D Theory—Greaves41 argued against the idea that skin evolved to become lighter to protect against vitamin D deficiency. They proposed that the chimpanzee, which is the human’s most closely related species, had light skin covered by hair, and the loss of this hair led to exposed pale skin that created a need for increased melanin production for protection from UVR. Greaves41 stated that the MC1R gene (associated with darker pigmentation) was selected for in African populations, and those with pale skin retained their original pigment as they migrated to higher latitudes. Further research has demonstrated that the genetic natural selection for skin pigment is a complex process that involves multiple gene variants found throughout cultures across the globe.
Conclusion
Skin pigmentation has continuously evolved alongside humans. Genetic selection for lighter skin coincides with a favorable selection for genes involved in vitamin D synthesis as humans migrated to northern latitudes, which enabled humans to produce adequate levels of exogenous vitamin D in low-UVR areas and in turn promoted survival. Early humans without access to supplementation or foods rich in vitamin D acquired vitamin D primarily through sunlight. In comparison to modern society, where vitamin D supplementation is accessible and human lifespans are prolonged, lighter skin tone is now a risk factor for malignant cancers of the skin rather than being a protective adaptation. Current sun behavior recommendations conclude that the body’s need for vitamin D is satisfied by UV exposure to the arms, legs, hands, and/or face for only 5 to 30 minutes between 10
The hypothesis that skin lightening primarily was driven by the need for vitamin D can only be partially supported by our review. Studies have shown that there is a corresponding complex network of genes that determines skin pigmentation as well as vitamin D synthesis and conservation. However, there is sufficient evidence that skin lightening is multifactorial in nature, and vitamin D alone may not be the sole driver. The information in this review can be used by health care providers to educate patients on sun protection, given the lesser threat of severe vitamin D deficiency in developed communities today that have access to adequate nutrition and supplementation.
Skin lightening and its coinciding evolutionary drivers are a rather neglected area of research. Due to heterogeneous cohorts and conservative data analysis, GWAS studies run the risk of type II error, yielding a limitation in our data analysis.9 Furthermore, the data regarding specific time frames in evolutionary skin lightening as well as the intensity of gene polymorphisms are limited.1 Further studies are needed to determine the interconnectedness of the current skin-lightening theories to identify other important factors that may play a role in the process. Determining the key event can help us better understand skin-adaptation mechanisms and create a framework for understanding the vital process involved in adaptation, survival, and disease manifestation in different patient populations.
The risk for developing skin cancer can be somewhat attributed to variations in skin pigmentation. Historically, lighter skin pigmentation has been observed in populations living in higher latitudes and darker pigmentation in populations near the equator. Although skin pigmentation is a conglomeration of genetic and environmental factors, anthropologic studies have demonstrated an association of human skin lightening with historic human migratory patterns.1 It is postulated that migration to latitudes with less UVB light penetration has resulted in a compensatory natural selection of lighter skin types. Furthermore, the driving force behind this migration-associated skin lightening has remained unclear.1
The need for folate metabolism, vitamin D synthesis, and barrier protection, as well as cultural practices, has been postulated as driving factors for skin pigmentation variation. Synthesis of vitamin D is a UV radiation (UVR)–dependent process and has remained a prominent theoretical driver for the basis of evolutionary skin lightening. Vitamin D can be acquired both exogenously or endogenously via dietary supplementation or sunlight; however, historically it has been obtained through UVB exposure primarily. Once UVB is absorbed by the skin, it catalyzes conversion of 7-dehydrocholesterol to previtamin D3, which is converted to vitamin D in the kidneys.2,3 It is suggested that lighter skin tones have an advantage over darker skin tones in synthesizing vitamin D at higher latitudes where there is less UVB, thus leading to the adaptation process.1 In this systematic review, we analyzed the evolutionary vitamin D adaptation hypothesis and assessed the validity of evidence supporting this theory in the literature.
Methods
A search of PubMed, Embase, and the Cochrane Reviews database was conducted using the terms evolution, vitamin D, and skin to generate articles published from 2010 to 2022 that evaluated the influence of UVR-dependent production of vitamin D on skin pigmentation through historical migration patterns (Figure). Studies were excluded during an initial screening of abstracts followed by full-text assessment if they only had abstracts and if articles were inaccessible for review or in the form of case reports and commentaries.
The following data were extracted from each included study: reference citation, affiliated institutions of authors, author specialties, journal name, year of publication, study period, type of article, type of study, mechanism of adaptation, data concluding or supporting vitamin D as the driver, and data concluding or suggesting against vitamin D as the driver. Data concluding or supporting vitamin D as the driver were recorded from statistically significant results, study conclusions, and direct quotations. Data concluding or suggesting against vitamin D as the driver also were recorded from significant results, study conclusions, and direct quotes. The mechanism of adaptation was based on vitamin D synthesis modulation, melanin upregulation, genetic selections, genetic drift, mating patterns, increased vitamin D sensitivity, interbreeding, and diet.
Studies included in the analysis were placed into 1 of 3 categories: supporting, neutral, and against. Strength of Recommendation Taxonomy (SORT) criteria were used to classify the level of evidence of each article.4 Each article’s level of evidence was then graded (Table 1). The SORT grading levels were based on quality and evidence type: level 1 signified good-quality, patient-oriented evidence; level 2 signified limited-quality, patient-oriented evidence; and level 3 signified other evidence.4
Results
Article Selection—A total of 229 articles were identified for screening, and 39 studies met inclusion criteria.1-3,5-40 Systematic and retrospective reviews were the most common types of studies. Genomic analysis/sequencing/genome-wide association studies (GWAS) were the most common methods of analysis. Of these 39 articles, 26 were classified as supporting the evolutionary vitamin D adaptation hypothesis, 10 were classified as neutral, and 3 were classified as against (Table 1).
Of the articles classified as supporting the vitamin D hypothesis, 13 articles were level 1 evidence, 9 were level 2, and 4 were level 3. Key findings supporting the vitamin D hypothesis included genetic natural selection favoring vitamin D synthesis genes at higher latitudes with lower UVR and the skin lightening that occurred to protect against vitamin D deficiency (Table 1). Specific genes supporting these findings included 7-dehydrocholesterol reductase (DHCR7), vitamin D receptor (VDR), tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), oculocutaneous albinism type 2 melanosomal transmembrane protein (OCA2), solute carrier family 45 member 2 (SLC45A2), solute carrier family 4 member 5 (SLC24A5), Kit ligand (KITLG), melanocortin 1 receptor (MC1R), and HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2)(Table 2).
Of the articles classified as being against the vitamin D hypothesis, 1 article was level 1 evidence, 1 was level 2, and 1 was level 3. Key findings refuting the vitamin D hypothesis included similar amounts of vitamin D synthesis in contemporary dark- and light-pigmented individuals, vitamin D–rich diets in the late Paleolithic period and in early agriculturalists, and metabolic conservation being the primary driver (Table 1).
Of the articles classified as neutral to the hypothesis, 7 articles were level 1 evidence and 3 were level 2. Key findings of these articles included genetic selection favoring vitamin D synthesis only for populations at extremely northern latitudes, skin lightening that was sustained in northern latitudes from the neighboring human ancestor the chimpanzee, and evidence for long-term evolutionary pressures and short-term plastic adaptations in vitamin D genes (Table 1).
Comment
The importance of appropriate vitamin D levels is hypothesized as a potent driver in skin lightening because the vitamin is essential for many biochemical processes within the human body. Proper calcification of bones requires activated vitamin D to prevent rickets in childhood. Pelvic deformation in women with rickets can obstruct childbirth in primitive medical environments.15 This direct reproductive impairment suggests a strong selective pressure for skin lightening in populations that migrated northward to enhance vitamin D synthesis.
Of the 39 articles that we reviewed, the majority (n=26 [66.7%]) supported the hypothesis that vitamin D synthesis was the main driver behind skin lightening, whereas 3 (7.7%) did not support the hypothesis and 10 (25.6%) were neutral. Other leading theories explaining skin lightening included the idea that enhanced melanogenesis protected against folate degradation; genetic selection for light-skin alleles due to genetic drift; skin lightening being the result of sexual selection; and a combination of factors, including dietary choices, clothing preferences, and skin permeability barriers.
Articles With Supporting Evidence for the Vitamin D Theory—As Homo sapiens migrated out of Africa, migration patterns demonstrated the correlation between distance from the equator and skin pigmentation from natural selection. Individuals with darker skin pigment required higher levels of UVR to synthesize vitamin D. According to Beleza et al,1 as humans migrated to areas of higher latitudes with lower levels of UVR, natural selection favored the development of lighter skin to maximize vitamin D production. Vitamin D is linked to calcium metabolism, and its deficiency can lead to bone malformations and poor immune function.35 Several genes affecting melanogenesis and skin pigment have been found to have geospatial patterns that map to different geographic locations of various populations, indicating how human migration patterns out of Africa created this natural selection for skin lightening. The gene KITLG—associated with lighter skin pigmentation—has been found in high frequencies in both European and East Asian populations and is proposed to have increased in frequency after the migration out of Africa. However, the genes TYRP1, SLC24A5, and SLC45A2 were found at high frequencies only in European populations, and this selection occurred 11,000 to 19,000 years ago during the Last Glacial Maximum (15,000–20,000 years ago), demonstrating the selection for European over East Asian characteristics. During this period, seasonal changes increased the risk for vitamin D deficiency and provided an urgency for selection to a lighter skin pigment.1
The migration of H sapiens to northern latitudes prompted the selection of alleles that would increasevitamin D synthesis to counteract the reduced UV exposure. Genetic analysis studies have found key associations between genes encoding for the metabolism of vitamin D and pigmentation. Among this complex network are the essential downstream enzymes in the melanocortin receptor 1 pathway, including TYR and TYRP1. Forty-six of 960 single-nucleotide polymorphisms located in 29 different genes involved in skin pigmentation that were analyzed in a cohort of 2970 individuals were significantly associated with serum vitamin D levels (P<.05). The exocyst complex component 2 (EXOC2), TYR, and TYRP1 gene variants were shown to have the greatest influence on vitamin D status.9 These data reveal how pigment genotypes are predictive of vitamin D levels and the epistatic potential among many genes in this complex network.
Gene variation plays an important role in vitamin D status when comparing genetic polymorphisms in populations in northern latitudes to African populations. Vitamin D3 precursor availability is decreased by 7-DHCR catalyzing the precursors substrate to cholesterol. In a study using GWAS, it was found that “variations in DHCR7 may aid vitamin D production by conserving cutaneous 7-DHC levels. A high prevalence of DHCR7 variants were found in European and Northeast Asian populations but not in African populations, suggesting that selection occurred for these DHCR7 mutations in populations who migrated to more northern latitudes.5 Multilocus networks have been established between the VDR promotor and skin color genes (Table 2) that exhibit a strong in-Africa vs out-of-Africa frequency pattern. It also has been shown that genetic variation (suggesting a long-term evolutionary inclination) and epigenetic modification (indicative of short-term exposure) of VDR lends support to the vitamin D hypothesis. As latitude decreases, prevalence of VDR FokI (F allele), BsmI (B allele), ApaI (A allele), and TaqI (T allele) also decreases in a linear manner, linking latitude to VDR polymorphisms. Plasma vitamin D levels and photoperiod of conception—UV exposure during the periconceptional period—also were extrapolative of VDR methylation in a study involving 80 participants, where these 2 factors accounted for 17% of variance in methylation.6
Other noteworthy genes included HERC2, which has implications in the expression of OCA2 (melanocyte-specific transporter protein), and IRF4, which encodes for an important enzyme in folate-dependent melanin production. In an Australian cross-sectional study that analyzed vitamin D and pigmentation gene polymorphisms in conjunction with plasma vitamin D levels, the most notable rate of vitamin D loss occurred in individuals with the darkest pigmentation HERC2 (AA) genotype.31 In contrast, the lightest pigmentation HERC2 (GG) genotypes had increased vitamin D3 photosynthesis. Interestingly, the lightest interferon regulatory factor 4 (IRF4) TT genotype and the darkest HERC2 AA genotype, rendering the greatest folate loss and largest synthesis of vitamin D3, were not seen in combination in any of the participants.30 In addition to HERC2, derived alleles from pigment-associated genes SLC24A5*A and SLC45A2*G demonstrated greater frequencies in Europeans (>90%) compared to Africans and East Asians, where the allelic frequencies were either rare or absent.1 This evidence delineates not only the complexity but also the strong relationship between skin pigmentation, latitude, and vitamin D status. The GWAS also have supported this concept. In comparing European populations to African populations, there was a 4-fold increase in the frequencies of “derived alleles of the vitamin D transport protein (GC, rs3755967), the 25(OH)D3 synthesizing enzyme (CYP2R1, rs10741657), VDR (rs2228570 (commonly known as FokI polymorphism), rs1544410 (Bsm1), and rs731236 (Taq1) and the VDR target genes CYP24A1 (rs17216707), CD14 (rs2569190), and CARD9 (rs4077515).”32
Articles With Evidence Against the Vitamin D Theory—This review analyzed the level of support for the theory that vitamin D was the main driver for skin lightening. Although most articles supported this theory, there were articles that listed other plausible counterarguments. Jablonski and Chaplin3 suggested that humans living in higher latitudes compensated for increased demand of vitamin D by placing cultural importance on a diet of vitamin D–rich foods and thus would not have experienced decreased vitamin D levels, which we hypothesize were the driver for skin lightening. Elias et al39 argued that initial pigment dilution may have instead served to improve metabolic conservation, as the authors found no evidence of rickets—the sequelae of vitamin D deficiency—in pre–industrial age human fossils. Elias and Williams38 proposed that differences in skin pigment are due to a more intact skin permeability barrier as “a requirement for life in a desiccating terrestrial environment,” which is seen in darker skin tones compared to lighter skin tones and thus can survive better in warmer climates with less risk of infections or dehydration.
Articles With Neutral Evidence for the Vitamin D Theory—Greaves41 argued against the idea that skin evolved to become lighter to protect against vitamin D deficiency. They proposed that the chimpanzee, which is the human’s most closely related species, had light skin covered by hair, and the loss of this hair led to exposed pale skin that created a need for increased melanin production for protection from UVR. Greaves41 stated that the MC1R gene (associated with darker pigmentation) was selected for in African populations, and those with pale skin retained their original pigment as they migrated to higher latitudes. Further research has demonstrated that the genetic natural selection for skin pigment is a complex process that involves multiple gene variants found throughout cultures across the globe.
Conclusion
Skin pigmentation has continuously evolved alongside humans. Genetic selection for lighter skin coincides with a favorable selection for genes involved in vitamin D synthesis as humans migrated to northern latitudes, which enabled humans to produce adequate levels of exogenous vitamin D in low-UVR areas and in turn promoted survival. Early humans without access to supplementation or foods rich in vitamin D acquired vitamin D primarily through sunlight. In comparison to modern society, where vitamin D supplementation is accessible and human lifespans are prolonged, lighter skin tone is now a risk factor for malignant cancers of the skin rather than being a protective adaptation. Current sun behavior recommendations conclude that the body’s need for vitamin D is satisfied by UV exposure to the arms, legs, hands, and/or face for only 5 to 30 minutes between 10
The hypothesis that skin lightening primarily was driven by the need for vitamin D can only be partially supported by our review. Studies have shown that there is a corresponding complex network of genes that determines skin pigmentation as well as vitamin D synthesis and conservation. However, there is sufficient evidence that skin lightening is multifactorial in nature, and vitamin D alone may not be the sole driver. The information in this review can be used by health care providers to educate patients on sun protection, given the lesser threat of severe vitamin D deficiency in developed communities today that have access to adequate nutrition and supplementation.
Skin lightening and its coinciding evolutionary drivers are a rather neglected area of research. Due to heterogeneous cohorts and conservative data analysis, GWAS studies run the risk of type II error, yielding a limitation in our data analysis.9 Furthermore, the data regarding specific time frames in evolutionary skin lightening as well as the intensity of gene polymorphisms are limited.1 Further studies are needed to determine the interconnectedness of the current skin-lightening theories to identify other important factors that may play a role in the process. Determining the key event can help us better understand skin-adaptation mechanisms and create a framework for understanding the vital process involved in adaptation, survival, and disease manifestation in different patient populations.
- Beleza S, Santos AM, McEvoy B, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013;30:24-35. doi:10.1093/molbev/mss207
- Carlberg C. Nutrigenomics of vitamin D. Nutrients. 2019;11:676. doi:10.3390/nu11030676
- Jablonski NG, Chaplin G. The roles of vitamin D and cutaneous vitamin D production in human evolution and health. Int J Paleopathol. 2018;23:54-59. doi:10.1016/j.ijpp.2018.01.005
- Weiss BD. SORT: strength of recommendation taxonomy. Fam Med. 2004;36:141-143.
- Wolf ST, Kenney WL. The vitamin D–folate hypothesis in human vascular health. Am J Physiol Regul Integr Comp Physiology. 2019;317:R491-R501. doi:10.1152/ajpregu.00136.2019
- Lucock M, Jones P, Martin C, et al. Photobiology of vitamins. Nutr Rev. 2018;76:512-525. doi:10.1093/nutrit/nuy013
- Hochberg Z, Hochberg I. Evolutionary perspective in rickets and vitamin D. Front Endocrinol (Lausanne). 2019;10:306. doi:10.3389/fendo.2019.00306
- Rossberg W, Saternus R, Wagenpfeil S, et al. Human pigmentation, cutaneous vitamin D synthesis and evolution: variants of genes (SNPs) involved in skin pigmentation are associated with 25(OH)D serum concentration. Anticancer Res. 2016;36:1429-1437.
- Saternus R, Pilz S, Gräber S, et al. A closer look at evolution: variants (SNPs) of genes involved in skin pigmentation, including EXOC2, TYR, TYRP1, and DCT, are associated with 25(OH)D serum concentration. Endocrinology. 2015;156:39-47. doi:10.1210/en.2014-1238
- López S, García Ó, Yurrebaso I, et al. The interplay between natural selection and susceptibility to melanoma on allele 374F of SLC45A2 gene in a south European population. PloS One. 2014;9:E104367. doi:1371/journal.pone.0104367
- Lucock M, Yates Z, Martin C, et al. Vitamin D, folate, and potential early lifecycle environmental origin of significant adult phenotypes. Evol Med Public Health. 2014;2014:69-91. doi:10.1093/emph/eou013
- Hudjashov G, Villems R, Kivisild T. Global patterns of diversity and selection in human tyrosinase gene. PloS One. 2013;8:E74307. doi:10.1371/journal.pone.0074307
- Khan R, Khan BSR. Diet, disease and pigment variation in humans. Med Hypotheses. 2010;75:363-367. doi:10.1016/j.mehy.2010.03.033
- Kuan V, Martineau AR, Griffiths CJ, et al. DHCR7 mutations linked to higher vitamin D status allowed early human migration to northern latitudes. BMC Evol Biol. 2013;13:144. doi:10.1186/1471-2148-13-144
- Omenn GS. Evolution and public health. Proc National Acad Sci. 2010;107(suppl 1):1702-1709. doi:10.1073/pnas.0906198106
- Yuen AWC, Jablonski NG. Vitamin D: in the evolution of human skin colour. Med Hypotheses. 2010;74:39-44. doi:10.1016/j.mehy.2009.08.007
- Vieth R. Weaker bones and white skin as adaptions to improve anthropological “fitness” for northern environments. Osteoporosis Int. 2020;31:617-624. doi:10.1007/s00198-019-05167-4
- Carlberg C. Vitamin D: a micronutrient regulating genes. Curr Pharm Des. 2019;25:1740-1746. doi:10.2174/1381612825666190705193227
- Haddadeen C, Lai C, Cho SY, et al. Variants of the melanocortin‐1 receptor: do they matter clinically? Exp Dermatol. 2015;1:5-9. doi:10.1111/exd.12540
- Yao S, Ambrosone CB. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J Steroid Biochem Mol Biol. 2013;136:337-341. doi:10.1016/j.jsbmb.2012.09.010
- Jablonski N. The evolution of human skin colouration and its relevance to health in the modern world. J Royal Coll Physicians Edinb. 2012;42:58-63. doi:10.4997/jrcpe.2012.114
- Jablonski NG, Chaplin G. Human skin pigmentation as an adaptation to UV radiation. Proc National Acad Sci. 2010;107(suppl 2):8962-8968. doi:10.1073/pnas.0914628107
- Hochberg Z, Templeton AR. Evolutionary perspective in skin color, vitamin D and its receptor. Hormones. 2010;9:307-311. doi:10.14310/horm.2002.1281
- Jones P, Lucock M, Veysey M, et al. The vitamin D–folate hypothesis as an evolutionary model for skin pigmentation: an update and integration of current ideas. Nutrients. 2018;10:554. doi:10.3390/nu10050554
- Lindqvist PG, Epstein E, Landin-Olsson M, et al. Women with fair phenotypes seem to confer a survival advantage in a low UV milieu. a nested matched case control study. PloS One. 2020;15:E0228582. doi:10.1371/journal.pone.0228582
- Holick MF. Shedding new light on the role of the sunshine vitamin D for skin health: the lncRNA–skin cancer connection. Exp Dermatol. 2014;23:391-392. doi:10.1111/exd.12386
- Jablonski NG, Chaplin G. Epidermal pigmentation in the human lineage is an adaptation to ultraviolet radiation. J Hum Evol. 2013;65:671-675. doi:10.1016/j.jhevol.2013.06.004
- Jablonski NG, Chaplin G. The evolution of skin pigmentation and hair texture in people of African ancestry. Dermatol Clin. 2014;32:113-121. doi:10.1016/j.det.2013.11.003
- Jablonski NG. The evolution of human skin pigmentation involved the interactions of genetic, environmental, and cultural variables. Pigment Cell Melanoma Res. 2021;34:707-7 doi:10.1111/pcmr.12976
- Lucock MD, Jones PR, Veysey M, et al. Biophysical evidence to support and extend the vitamin D‐folate hypothesis as a paradigm for the evolution of human skin pigmentation. Am J Hum Biol. 2022;34:E23667. doi:10.1002/ajhb.23667
- Missaggia BO, Reales G, Cybis GB, et al. Adaptation and co‐adaptation of skin pigmentation and vitamin D genes in native Americans. Am J Med Genet C Semin Med Genet. 2020;184:1060-1077. doi:10.1002/ajmg.c.31873
- Hanel A, Carlberg C. Skin colour and vitamin D: an update. Exp Dermatol. 2020;29:864-875. doi:10.1111/exd.14142
- Hanel A, Carlberg C. Vitamin D and evolution: pharmacologic implications. Biochem Pharmacol. 2020;173:113595. doi:10.1016/j.bcp.2019.07.024
- Flegr J, Sýkorová K, Fiala V, et al. Increased 25(OH)D3 level in redheaded people: could redheadedness be an adaptation to temperate climate? Exp Dermatol. 2020;29:598-609. doi:10.1111/exd.14119
- James WPT, Johnson RJ, Speakman JR, et al. Nutrition and its role in human evolution. J Intern Med. 2019;285:533-549. doi:10.1111/joim.12878
- Lucock M, Jones P, Martin C, et al. Vitamin D: beyond metabolism. J Evid Based Complementary Altern Med. 2015;20:310-322. doi:10.1177/2156587215580491
- Jarrett P, Scragg R. Evolution, prehistory and vitamin D. Int J Environ Res Public Health. 2020;17:646. doi:10.3390/ijerph17020646
- Elias PM, Williams ML. Re-appraisal of current theories for thedevelopment and loss of epidermal pigmentation in hominins and modern humans. J Hum Evol. 2013;64:687-692. doi:10.1016/j.jhevol.2013.02.003
- Elias PM, Williams ML. Basis for the gain and subsequent dilution of epidermal pigmentation during human evolution: the barrier and metabolic conservation hypotheses revisited. Am J Phys Anthropol. 2016;161:189-207. doi:10.1002/ajpa.23030
- Williams JD, Jacobson EL, Kim H, et al. Water soluble vitamins, clinical research and future application. Subcell Biochem. 2011;56:181-197. doi:10.1007/978-94-007-2199-9_10
- Greaves M. Was skin cancer a selective force for black pigmentation in early hominin evolution [published online February 26, 2014]? Proc Biol Sci. 2014;281:20132955. doi:10.1098/rspb.2013.2955
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281. doi:10.1056/nejmra070553
- Bouillon R. Comparative analysis of nutritional guidelines for vitamin D. Nat Rev Endocrinol. 2017;13:466-479. doi:10.1038/nrendo.2017.31
- US Department of Health and Human Services. The Surgeon General’s Call to Action to Prevent Skin Cancer. US Dept of Health and Human Services, Office of the Surgeon General; 2014. Accessed April 29, 2024. https://www.hhs.gov/sites/default/files/call-to-action-prevent-skin-cancer.pdf
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press; 2011. https://www.ncbi.nlm.nih.gov/books/NBK56070/
- Beleza S, Santos AM, McEvoy B, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013;30:24-35. doi:10.1093/molbev/mss207
- Carlberg C. Nutrigenomics of vitamin D. Nutrients. 2019;11:676. doi:10.3390/nu11030676
- Jablonski NG, Chaplin G. The roles of vitamin D and cutaneous vitamin D production in human evolution and health. Int J Paleopathol. 2018;23:54-59. doi:10.1016/j.ijpp.2018.01.005
- Weiss BD. SORT: strength of recommendation taxonomy. Fam Med. 2004;36:141-143.
- Wolf ST, Kenney WL. The vitamin D–folate hypothesis in human vascular health. Am J Physiol Regul Integr Comp Physiology. 2019;317:R491-R501. doi:10.1152/ajpregu.00136.2019
- Lucock M, Jones P, Martin C, et al. Photobiology of vitamins. Nutr Rev. 2018;76:512-525. doi:10.1093/nutrit/nuy013
- Hochberg Z, Hochberg I. Evolutionary perspective in rickets and vitamin D. Front Endocrinol (Lausanne). 2019;10:306. doi:10.3389/fendo.2019.00306
- Rossberg W, Saternus R, Wagenpfeil S, et al. Human pigmentation, cutaneous vitamin D synthesis and evolution: variants of genes (SNPs) involved in skin pigmentation are associated with 25(OH)D serum concentration. Anticancer Res. 2016;36:1429-1437.
- Saternus R, Pilz S, Gräber S, et al. A closer look at evolution: variants (SNPs) of genes involved in skin pigmentation, including EXOC2, TYR, TYRP1, and DCT, are associated with 25(OH)D serum concentration. Endocrinology. 2015;156:39-47. doi:10.1210/en.2014-1238
- López S, García Ó, Yurrebaso I, et al. The interplay between natural selection and susceptibility to melanoma on allele 374F of SLC45A2 gene in a south European population. PloS One. 2014;9:E104367. doi:1371/journal.pone.0104367
- Lucock M, Yates Z, Martin C, et al. Vitamin D, folate, and potential early lifecycle environmental origin of significant adult phenotypes. Evol Med Public Health. 2014;2014:69-91. doi:10.1093/emph/eou013
- Hudjashov G, Villems R, Kivisild T. Global patterns of diversity and selection in human tyrosinase gene. PloS One. 2013;8:E74307. doi:10.1371/journal.pone.0074307
- Khan R, Khan BSR. Diet, disease and pigment variation in humans. Med Hypotheses. 2010;75:363-367. doi:10.1016/j.mehy.2010.03.033
- Kuan V, Martineau AR, Griffiths CJ, et al. DHCR7 mutations linked to higher vitamin D status allowed early human migration to northern latitudes. BMC Evol Biol. 2013;13:144. doi:10.1186/1471-2148-13-144
- Omenn GS. Evolution and public health. Proc National Acad Sci. 2010;107(suppl 1):1702-1709. doi:10.1073/pnas.0906198106
- Yuen AWC, Jablonski NG. Vitamin D: in the evolution of human skin colour. Med Hypotheses. 2010;74:39-44. doi:10.1016/j.mehy.2009.08.007
- Vieth R. Weaker bones and white skin as adaptions to improve anthropological “fitness” for northern environments. Osteoporosis Int. 2020;31:617-624. doi:10.1007/s00198-019-05167-4
- Carlberg C. Vitamin D: a micronutrient regulating genes. Curr Pharm Des. 2019;25:1740-1746. doi:10.2174/1381612825666190705193227
- Haddadeen C, Lai C, Cho SY, et al. Variants of the melanocortin‐1 receptor: do they matter clinically? Exp Dermatol. 2015;1:5-9. doi:10.1111/exd.12540
- Yao S, Ambrosone CB. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J Steroid Biochem Mol Biol. 2013;136:337-341. doi:10.1016/j.jsbmb.2012.09.010
- Jablonski N. The evolution of human skin colouration and its relevance to health in the modern world. J Royal Coll Physicians Edinb. 2012;42:58-63. doi:10.4997/jrcpe.2012.114
- Jablonski NG, Chaplin G. Human skin pigmentation as an adaptation to UV radiation. Proc National Acad Sci. 2010;107(suppl 2):8962-8968. doi:10.1073/pnas.0914628107
- Hochberg Z, Templeton AR. Evolutionary perspective in skin color, vitamin D and its receptor. Hormones. 2010;9:307-311. doi:10.14310/horm.2002.1281
- Jones P, Lucock M, Veysey M, et al. The vitamin D–folate hypothesis as an evolutionary model for skin pigmentation: an update and integration of current ideas. Nutrients. 2018;10:554. doi:10.3390/nu10050554
- Lindqvist PG, Epstein E, Landin-Olsson M, et al. Women with fair phenotypes seem to confer a survival advantage in a low UV milieu. a nested matched case control study. PloS One. 2020;15:E0228582. doi:10.1371/journal.pone.0228582
- Holick MF. Shedding new light on the role of the sunshine vitamin D for skin health: the lncRNA–skin cancer connection. Exp Dermatol. 2014;23:391-392. doi:10.1111/exd.12386
- Jablonski NG, Chaplin G. Epidermal pigmentation in the human lineage is an adaptation to ultraviolet radiation. J Hum Evol. 2013;65:671-675. doi:10.1016/j.jhevol.2013.06.004
- Jablonski NG, Chaplin G. The evolution of skin pigmentation and hair texture in people of African ancestry. Dermatol Clin. 2014;32:113-121. doi:10.1016/j.det.2013.11.003
- Jablonski NG. The evolution of human skin pigmentation involved the interactions of genetic, environmental, and cultural variables. Pigment Cell Melanoma Res. 2021;34:707-7 doi:10.1111/pcmr.12976
- Lucock MD, Jones PR, Veysey M, et al. Biophysical evidence to support and extend the vitamin D‐folate hypothesis as a paradigm for the evolution of human skin pigmentation. Am J Hum Biol. 2022;34:E23667. doi:10.1002/ajhb.23667
- Missaggia BO, Reales G, Cybis GB, et al. Adaptation and co‐adaptation of skin pigmentation and vitamin D genes in native Americans. Am J Med Genet C Semin Med Genet. 2020;184:1060-1077. doi:10.1002/ajmg.c.31873
- Hanel A, Carlberg C. Skin colour and vitamin D: an update. Exp Dermatol. 2020;29:864-875. doi:10.1111/exd.14142
- Hanel A, Carlberg C. Vitamin D and evolution: pharmacologic implications. Biochem Pharmacol. 2020;173:113595. doi:10.1016/j.bcp.2019.07.024
- Flegr J, Sýkorová K, Fiala V, et al. Increased 25(OH)D3 level in redheaded people: could redheadedness be an adaptation to temperate climate? Exp Dermatol. 2020;29:598-609. doi:10.1111/exd.14119
- James WPT, Johnson RJ, Speakman JR, et al. Nutrition and its role in human evolution. J Intern Med. 2019;285:533-549. doi:10.1111/joim.12878
- Lucock M, Jones P, Martin C, et al. Vitamin D: beyond metabolism. J Evid Based Complementary Altern Med. 2015;20:310-322. doi:10.1177/2156587215580491
- Jarrett P, Scragg R. Evolution, prehistory and vitamin D. Int J Environ Res Public Health. 2020;17:646. doi:10.3390/ijerph17020646
- Elias PM, Williams ML. Re-appraisal of current theories for thedevelopment and loss of epidermal pigmentation in hominins and modern humans. J Hum Evol. 2013;64:687-692. doi:10.1016/j.jhevol.2013.02.003
- Elias PM, Williams ML. Basis for the gain and subsequent dilution of epidermal pigmentation during human evolution: the barrier and metabolic conservation hypotheses revisited. Am J Phys Anthropol. 2016;161:189-207. doi:10.1002/ajpa.23030
- Williams JD, Jacobson EL, Kim H, et al. Water soluble vitamins, clinical research and future application. Subcell Biochem. 2011;56:181-197. doi:10.1007/978-94-007-2199-9_10
- Greaves M. Was skin cancer a selective force for black pigmentation in early hominin evolution [published online February 26, 2014]? Proc Biol Sci. 2014;281:20132955. doi:10.1098/rspb.2013.2955
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281. doi:10.1056/nejmra070553
- Bouillon R. Comparative analysis of nutritional guidelines for vitamin D. Nat Rev Endocrinol. 2017;13:466-479. doi:10.1038/nrendo.2017.31
- US Department of Health and Human Services. The Surgeon General’s Call to Action to Prevent Skin Cancer. US Dept of Health and Human Services, Office of the Surgeon General; 2014. Accessed April 29, 2024. https://www.hhs.gov/sites/default/files/call-to-action-prevent-skin-cancer.pdf
- Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Ross AC, Taylor CL, Yaktine AL, et al, eds. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press; 2011. https://www.ncbi.nlm.nih.gov/books/NBK56070/
Practice Points
- Sufficient UV radiation exposure is required to synthesize vitamin D, but excess exposure increases skin cancer risk.
- Genes associated with vitamin D production and melanin synthesis form an interconnected network that explains skin tone polymorphisms and their influence on healthy sun behaviors.
- Adaptations in genetics of skin pigmentation and vitamin D metabolism due to anthropologic patterns of migration to northern latitudes may help explain predisposition to dermatologic diseases such as skin cancer.
Reactive Granulomatous Dermatitis: Variability of the Predominant Inflammatory Cell Type
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
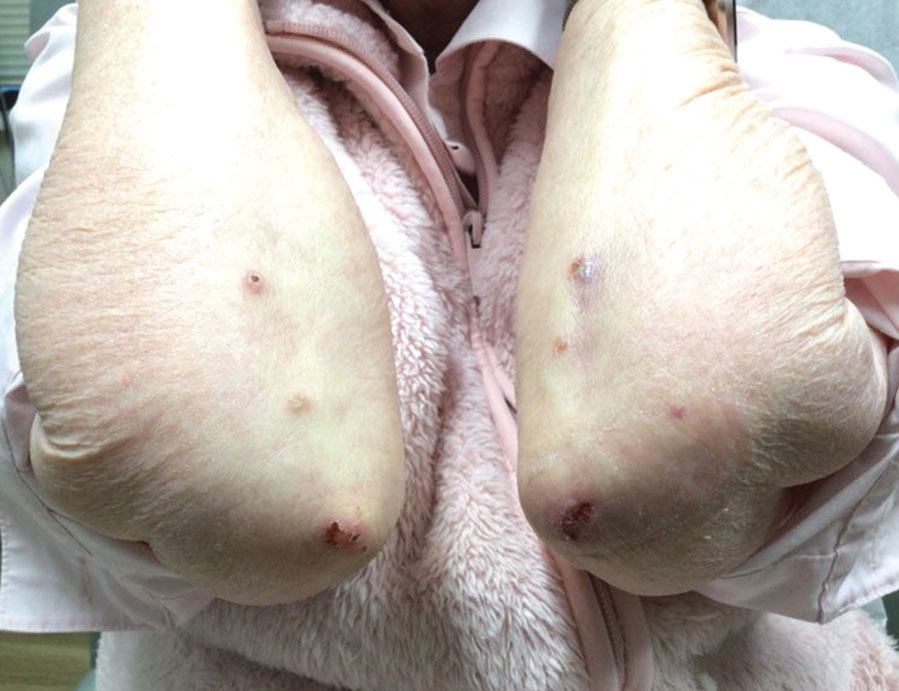
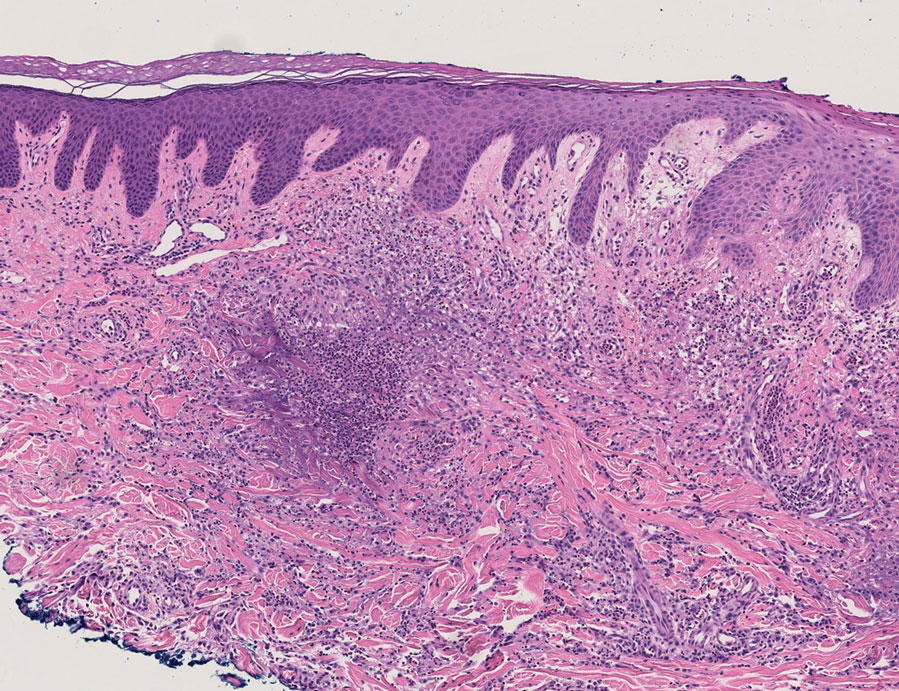
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
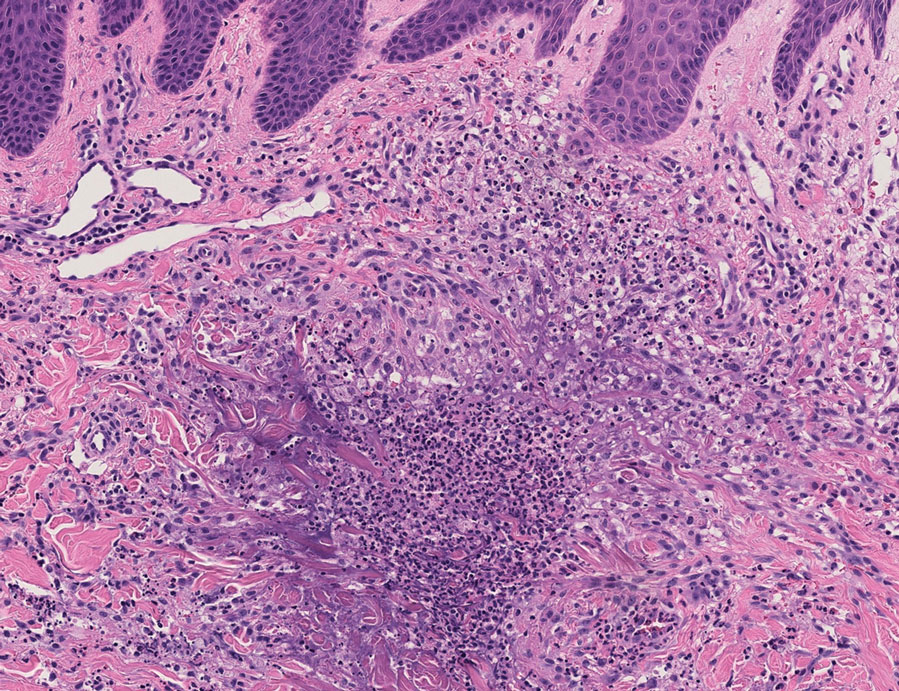
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
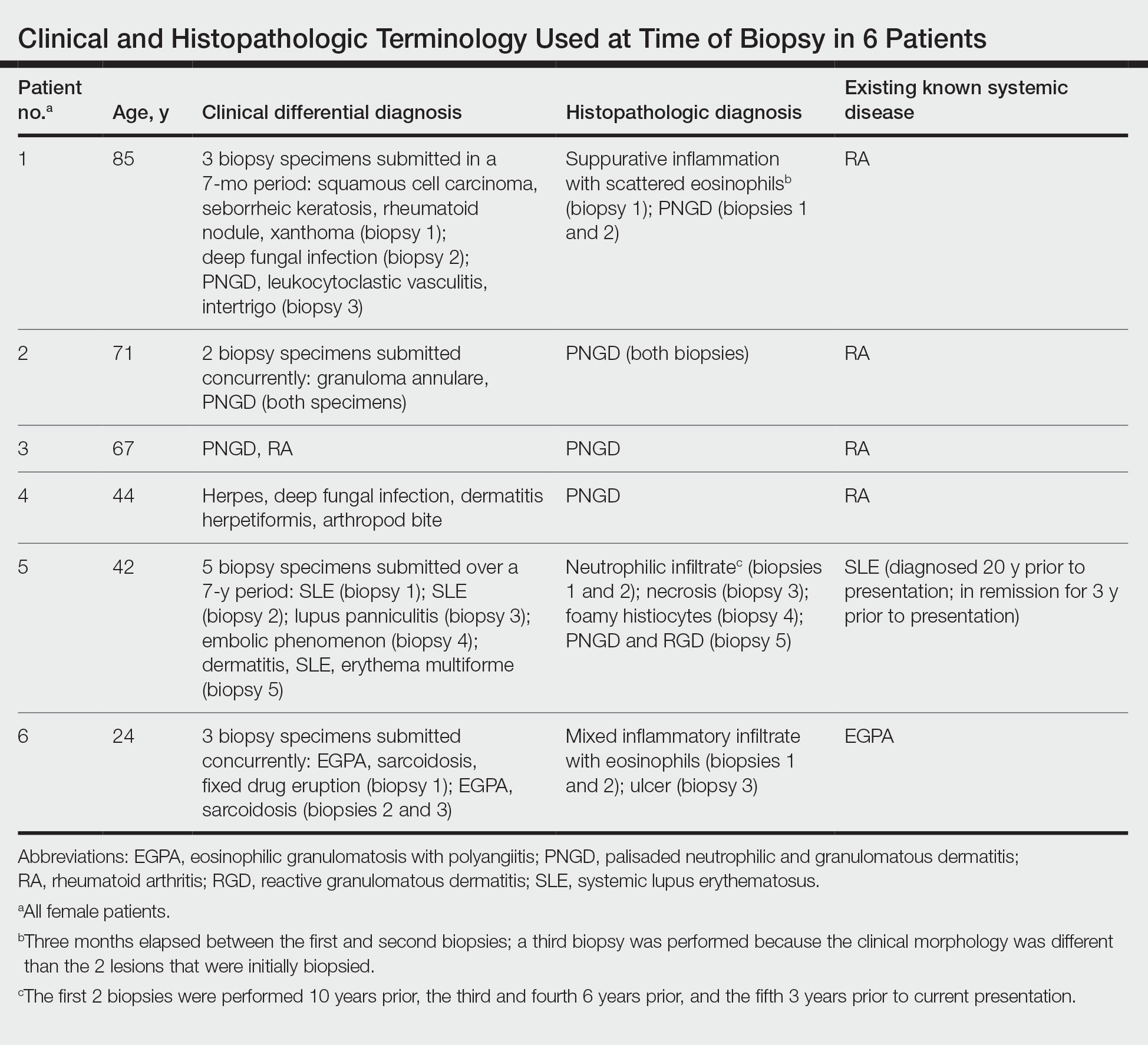
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
Practice Points
- The term reactive granulomatous dermatitis (RGD) provides a unifying rubric for palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction.
- Reactive granulomatous dermatitis can have a variable infiltrate that includes neutrophils, histiocytes, and/or eosinophils.
- Awareness of the variability in inflammatory cell type is important for the diagnosis of RGD.
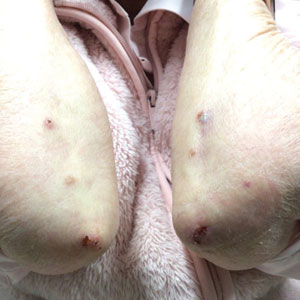
Growing Periumbilical Plaque: A Case of Perforating Calcific Elastosis
To the Editor:
Pseudoxanthoma elasticum (PXE) is a genetic perforating dermatosis characterized by fragmentation and calcification of elastic fibers that most commonly manifests on the skin, eyes, gastrointestinal tract, or cardiovascular system.1 Classic skin findings include multiple symmetric yellowish papules favoring the flexural surfaces of the body and neck as well as the periumbilical and inguinal regions.1,2 Many life-threatening complications from this disease can occur due to calcification of elastic fibers in other parts of the body, such as the internal elastic lamina of arteries, which can cause gastrointestinal tract bleeding and accelerated cardiovascular disease including valvular disease.2,3 If PXE is localized to the skin only without systemic involvement or a family history, a diagnosis of perforating calcific elastosis (PCE) can be made. We report a case of PCE in a patient with a growing umbilical lesion.

A 49-year-old multiparous (gravida 3, para 3) woman presented for evaluation of an evolving periumbilical lesion of 4 months’ duration. She denied pain, bleeding, or drainage from the area, as well as any systemic symptoms. The patient had a surgical history of a laparoscopic hysterectomy 7 years prior to the current presentation due to uterine fibroids, which resulted in a periumbilical scar. At the current presentation, physical examination revealed 2 hyperpigmented to violaceous periumbilical papules coalescing into a plaque with overlying hyperkeratosis and crusting (Figure 1). A punch biopsy was performed and histopathology showed diffuse dermal collections of degenerated eosinophilic distorted elastic fibers with calcification (Figure 2). Further sections showed a transepidermal channel in which the elastic fibers extruded from the dermis through the epidermis (Figure 3). The diagnosis of acquired PCE was made based on the clinical presentation, relevant medical history, and lack of underlying medical conditions or family history of PXE. No further workup was needed, and the patient reported no further progression and rather some improvement (decrease in size) of the lesion at 3-month follow-up.

Perforating calcific elastosis (also known as periumbilical perforating PXE) is a rare acquired condition that is seen predominantly in multiparous middle-aged women.4-6 This diagnosis consists of degenerated calcified elastic fibers that may perforate the skin of the abdominal or periumbilical region. It clinically manifests as multiple painless hyperkeratotic papules surrounding the periumbilical region.4-6

The etiology and pathogenesis of PCE have not been defined but have been attributed to recurrent stressing of elastic fibers due to repeat traumas,1 which is proposed to lead to degeneration of elastic fibers and calcification of damaged tissue.4-7 As a result, PCE most commonly manifests in multiparous, obese, middle-aged women and patients with multiple abdominal surgeries or ascites.1 It also has been reported in patients with renal failure due to deposition of abnormal calcium phosphate products onto elastic fibers.4 In our patient, the development of PCE was related to both multiparity and trauma from prior surgery.
The histopathologic findings of PCE and PXE are similar, warranting differentiation via thorough clinical examination as well as further investigation of the patient’s medical and family history. Both show degenerated, fragmented, curly elastic fibers with calcium deposition throughout the dermis and a transepidermal channel extruding these elastic fibers.7,8 The biopsies stain positive for elastic fibers and calcium deposition. Calcium staining can help to differentiate these entities from elastosis perforans serpiginosa, which lacks the presence of calcium staining.7
There are no definitive treatments for PCE. A single case report of a patient with PCE and renal failure showed regression with hemodialysis.9 In a study evaluating patients with inherited PXE, notable improvement was seen in skin lesions treated with bisphosphonates, possibly suggesting that regulating serum calcium may contribute to improvement of the disease.3 Most cases spontaneously resolve with atrophic plaques. Our patient required no additional treatment with no further progression and reported improvement of the lesion with spontaneous decrease in size.
- Jha AK, Zheeshan MD, Sinha BK, et al. Periumbilical perforating pseudoxanthoma elasticum: a rare case report. Dermatol Pract Concept. 2018;8:75-77. doi:10.5826/dpc.0802a02
- Ko JH, Shih YC, Huang YC, et al. Pseudoxanthoma elasticum. Lancet. 2013;381:565.
- Sherer DW, Singer G, Uribarri J, et al. Oral phosphate binders in the treatment of pseudoxanthoma elasticum. J Am Acad Dermatol. 2005;53:610-615.
- Lal NR, Bandyopadhyay D, Verma R, et al. Perforating calcific elastosis: revisiting a rare entity. Indian J Dermatol. 2018;63:186-188. doi:10.4103/ijd.IJD_111_17
- Kocatürk E, Kavala M, Zindanci I, et al. Periumbilical perforating pseudoxanthoma elasticum. Indian J Dermatol Venereol Leprol. 2009;75:329.
- Bressan AL, Vasconcelos BN, Silva RDS, et al. Periumbilical and periareolar perforating pseudoxanthoma elasticum. An Bras Dermatol. 2010;85:705-707. doi:10.1590/s0365-05962010000500018
- Hosen MJ, Lamoen A, De Paepe A, et al. Histopathology of pseudoxanthoma elasticum and related disorders: histological hallmarks and diagnostic clues. Scientifica (Cairo). 2012;2012:598262.
- Bathina M, Hedge SP, Shanavaz AA, et al. Pruritic periumbilical plaque as a presentation of rare perforating dermatosis. Indian Dermatol Online J. 2020;11:68-71. doi:10.4103/idoj.IDOJ_95_19
- Sapadin AN, Lebwohl MG, Teich SA, et al. Periumbilical pseudoxanthoma elasticum associated with chronic renal failure and angioid streaks—apparent regression with hemodialysis. J Am Acad Dermatol. 1998;39:338-344.
To the Editor:
Pseudoxanthoma elasticum (PXE) is a genetic perforating dermatosis characterized by fragmentation and calcification of elastic fibers that most commonly manifests on the skin, eyes, gastrointestinal tract, or cardiovascular system.1 Classic skin findings include multiple symmetric yellowish papules favoring the flexural surfaces of the body and neck as well as the periumbilical and inguinal regions.1,2 Many life-threatening complications from this disease can occur due to calcification of elastic fibers in other parts of the body, such as the internal elastic lamina of arteries, which can cause gastrointestinal tract bleeding and accelerated cardiovascular disease including valvular disease.2,3 If PXE is localized to the skin only without systemic involvement or a family history, a diagnosis of perforating calcific elastosis (PCE) can be made. We report a case of PCE in a patient with a growing umbilical lesion.
A 49-year-old multiparous (gravida 3, para 3) woman presented for evaluation of an evolving periumbilical lesion of 4 months’ duration. She denied pain, bleeding, or drainage from the area, as well as any systemic symptoms. The patient had a surgical history of a laparoscopic hysterectomy 7 years prior to the current presentation due to uterine fibroids, which resulted in a periumbilical scar. At the current presentation, physical examination revealed 2 hyperpigmented to violaceous periumbilical papules coalescing into a plaque with overlying hyperkeratosis and crusting (Figure 1). A punch biopsy was performed and histopathology showed diffuse dermal collections of degenerated eosinophilic distorted elastic fibers with calcification (Figure 2). Further sections showed a transepidermal channel in which the elastic fibers extruded from the dermis through the epidermis (Figure 3). The diagnosis of acquired PCE was made based on the clinical presentation, relevant medical history, and lack of underlying medical conditions or family history of PXE. No further workup was needed, and the patient reported no further progression and rather some improvement (decrease in size) of the lesion at 3-month follow-up.
Perforating calcific elastosis (also known as periumbilical perforating PXE) is a rare acquired condition that is seen predominantly in multiparous middle-aged women.4-6 This diagnosis consists of degenerated calcified elastic fibers that may perforate the skin of the abdominal or periumbilical region. It clinically manifests as multiple painless hyperkeratotic papules surrounding the periumbilical region.4-6
The etiology and pathogenesis of PCE have not been defined but have been attributed to recurrent stressing of elastic fibers due to repeat traumas,1 which is proposed to lead to degeneration of elastic fibers and calcification of damaged tissue.4-7 As a result, PCE most commonly manifests in multiparous, obese, middle-aged women and patients with multiple abdominal surgeries or ascites.1 It also has been reported in patients with renal failure due to deposition of abnormal calcium phosphate products onto elastic fibers.4 In our patient, the development of PCE was related to both multiparity and trauma from prior surgery.
The histopathologic findings of PCE and PXE are similar, warranting differentiation via thorough clinical examination as well as further investigation of the patient’s medical and family history. Both show degenerated, fragmented, curly elastic fibers with calcium deposition throughout the dermis and a transepidermal channel extruding these elastic fibers.7,8 The biopsies stain positive for elastic fibers and calcium deposition. Calcium staining can help to differentiate these entities from elastosis perforans serpiginosa, which lacks the presence of calcium staining.7
There are no definitive treatments for PCE. A single case report of a patient with PCE and renal failure showed regression with hemodialysis.9 In a study evaluating patients with inherited PXE, notable improvement was seen in skin lesions treated with bisphosphonates, possibly suggesting that regulating serum calcium may contribute to improvement of the disease.3 Most cases spontaneously resolve with atrophic plaques. Our patient required no additional treatment with no further progression and reported improvement of the lesion with spontaneous decrease in size.
To the Editor:
Pseudoxanthoma elasticum (PXE) is a genetic perforating dermatosis characterized by fragmentation and calcification of elastic fibers that most commonly manifests on the skin, eyes, gastrointestinal tract, or cardiovascular system.1 Classic skin findings include multiple symmetric yellowish papules favoring the flexural surfaces of the body and neck as well as the periumbilical and inguinal regions.1,2 Many life-threatening complications from this disease can occur due to calcification of elastic fibers in other parts of the body, such as the internal elastic lamina of arteries, which can cause gastrointestinal tract bleeding and accelerated cardiovascular disease including valvular disease.2,3 If PXE is localized to the skin only without systemic involvement or a family history, a diagnosis of perforating calcific elastosis (PCE) can be made. We report a case of PCE in a patient with a growing umbilical lesion.
A 49-year-old multiparous (gravida 3, para 3) woman presented for evaluation of an evolving periumbilical lesion of 4 months’ duration. She denied pain, bleeding, or drainage from the area, as well as any systemic symptoms. The patient had a surgical history of a laparoscopic hysterectomy 7 years prior to the current presentation due to uterine fibroids, which resulted in a periumbilical scar. At the current presentation, physical examination revealed 2 hyperpigmented to violaceous periumbilical papules coalescing into a plaque with overlying hyperkeratosis and crusting (Figure 1). A punch biopsy was performed and histopathology showed diffuse dermal collections of degenerated eosinophilic distorted elastic fibers with calcification (Figure 2). Further sections showed a transepidermal channel in which the elastic fibers extruded from the dermis through the epidermis (Figure 3). The diagnosis of acquired PCE was made based on the clinical presentation, relevant medical history, and lack of underlying medical conditions or family history of PXE. No further workup was needed, and the patient reported no further progression and rather some improvement (decrease in size) of the lesion at 3-month follow-up.
Perforating calcific elastosis (also known as periumbilical perforating PXE) is a rare acquired condition that is seen predominantly in multiparous middle-aged women.4-6 This diagnosis consists of degenerated calcified elastic fibers that may perforate the skin of the abdominal or periumbilical region. It clinically manifests as multiple painless hyperkeratotic papules surrounding the periumbilical region.4-6
The etiology and pathogenesis of PCE have not been defined but have been attributed to recurrent stressing of elastic fibers due to repeat traumas,1 which is proposed to lead to degeneration of elastic fibers and calcification of damaged tissue.4-7 As a result, PCE most commonly manifests in multiparous, obese, middle-aged women and patients with multiple abdominal surgeries or ascites.1 It also has been reported in patients with renal failure due to deposition of abnormal calcium phosphate products onto elastic fibers.4 In our patient, the development of PCE was related to both multiparity and trauma from prior surgery.
The histopathologic findings of PCE and PXE are similar, warranting differentiation via thorough clinical examination as well as further investigation of the patient’s medical and family history. Both show degenerated, fragmented, curly elastic fibers with calcium deposition throughout the dermis and a transepidermal channel extruding these elastic fibers.7,8 The biopsies stain positive for elastic fibers and calcium deposition. Calcium staining can help to differentiate these entities from elastosis perforans serpiginosa, which lacks the presence of calcium staining.7
There are no definitive treatments for PCE. A single case report of a patient with PCE and renal failure showed regression with hemodialysis.9 In a study evaluating patients with inherited PXE, notable improvement was seen in skin lesions treated with bisphosphonates, possibly suggesting that regulating serum calcium may contribute to improvement of the disease.3 Most cases spontaneously resolve with atrophic plaques. Our patient required no additional treatment with no further progression and reported improvement of the lesion with spontaneous decrease in size.
- Jha AK, Zheeshan MD, Sinha BK, et al. Periumbilical perforating pseudoxanthoma elasticum: a rare case report. Dermatol Pract Concept. 2018;8:75-77. doi:10.5826/dpc.0802a02
- Ko JH, Shih YC, Huang YC, et al. Pseudoxanthoma elasticum. Lancet. 2013;381:565.
- Sherer DW, Singer G, Uribarri J, et al. Oral phosphate binders in the treatment of pseudoxanthoma elasticum. J Am Acad Dermatol. 2005;53:610-615.
- Lal NR, Bandyopadhyay D, Verma R, et al. Perforating calcific elastosis: revisiting a rare entity. Indian J Dermatol. 2018;63:186-188. doi:10.4103/ijd.IJD_111_17
- Kocatürk E, Kavala M, Zindanci I, et al. Periumbilical perforating pseudoxanthoma elasticum. Indian J Dermatol Venereol Leprol. 2009;75:329.
- Bressan AL, Vasconcelos BN, Silva RDS, et al. Periumbilical and periareolar perforating pseudoxanthoma elasticum. An Bras Dermatol. 2010;85:705-707. doi:10.1590/s0365-05962010000500018
- Hosen MJ, Lamoen A, De Paepe A, et al. Histopathology of pseudoxanthoma elasticum and related disorders: histological hallmarks and diagnostic clues. Scientifica (Cairo). 2012;2012:598262.
- Bathina M, Hedge SP, Shanavaz AA, et al. Pruritic periumbilical plaque as a presentation of rare perforating dermatosis. Indian Dermatol Online J. 2020;11:68-71. doi:10.4103/idoj.IDOJ_95_19
- Sapadin AN, Lebwohl MG, Teich SA, et al. Periumbilical pseudoxanthoma elasticum associated with chronic renal failure and angioid streaks—apparent regression with hemodialysis. J Am Acad Dermatol. 1998;39:338-344.
- Jha AK, Zheeshan MD, Sinha BK, et al. Periumbilical perforating pseudoxanthoma elasticum: a rare case report. Dermatol Pract Concept. 2018;8:75-77. doi:10.5826/dpc.0802a02
- Ko JH, Shih YC, Huang YC, et al. Pseudoxanthoma elasticum. Lancet. 2013;381:565.
- Sherer DW, Singer G, Uribarri J, et al. Oral phosphate binders in the treatment of pseudoxanthoma elasticum. J Am Acad Dermatol. 2005;53:610-615.
- Lal NR, Bandyopadhyay D, Verma R, et al. Perforating calcific elastosis: revisiting a rare entity. Indian J Dermatol. 2018;63:186-188. doi:10.4103/ijd.IJD_111_17
- Kocatürk E, Kavala M, Zindanci I, et al. Periumbilical perforating pseudoxanthoma elasticum. Indian J Dermatol Venereol Leprol. 2009;75:329.
- Bressan AL, Vasconcelos BN, Silva RDS, et al. Periumbilical and periareolar perforating pseudoxanthoma elasticum. An Bras Dermatol. 2010;85:705-707. doi:10.1590/s0365-05962010000500018
- Hosen MJ, Lamoen A, De Paepe A, et al. Histopathology of pseudoxanthoma elasticum and related disorders: histological hallmarks and diagnostic clues. Scientifica (Cairo). 2012;2012:598262.
- Bathina M, Hedge SP, Shanavaz AA, et al. Pruritic periumbilical plaque as a presentation of rare perforating dermatosis. Indian Dermatol Online J. 2020;11:68-71. doi:10.4103/idoj.IDOJ_95_19
- Sapadin AN, Lebwohl MG, Teich SA, et al. Periumbilical pseudoxanthoma elasticum associated with chronic renal failure and angioid streaks—apparent regression with hemodialysis. J Am Acad Dermatol. 1998;39:338-344.
PRACTICE POINTS
- Perforating calcific elastosis (PCE) is a rare, localized, acquired variant of the inherited connective tissue disorder pseudoxanthoma elasticum (PXE).
- Histopathologic findings are identical for PCE and PXE, warranting differentiation via thorough clinical examination as well as further investigation of the patient’s medical and family history.
- Although there are no definitive treatments, most cases of PCE resolve spontaneously.
- Dermatologists should be aware of the importance of clinically differentiating PCE from PXE to prevent extensive workup, which can lead to unnecessary testing and increased morbidity in patients.

Early Treatment of Lyme Disease Prompted by Histopathologic Analysis of the Abdomen of an Engorged Tick
To the Editor:
Lyme disease is caused by spirochetes of the Borrelia burgdorferi sensu lato species complex and transmitted to humans by the bite of the Ixodes scapularis tick. It was first classified as a nationally notifiable disease in 1991, and the incidence has risen remarkably since then.1 More than 63,000 cases are reported annually to the Centers for Disease Control and Prevention; however, this number reflects severe underreporting, as the true incidence of the disease is projected to be closer to 476,000 cases per year.2 Additionally, 95% of US cases occur in the Northeast and upper Midwest.3 Given the pervasiveness of Lyme disease, early and reliable diagnostic methodology is critical, especially in cases in which the timeline of inoculation is unclear. We present a case of Lyme disease that was discovered during a routine dermatologic visit.
A 77-year-old White man with no relevant medical history presented to a dermatology clinic in west-central Virginia for a routine skin check. Physical examination revealed a well-appearing patient without overt skin abnormalities. However, on closer evaluation, a 0.2×0.1-cm engorged black I scapularis tick was visualized on the left lateral upper back. There was a surrounding zone of erythema that measured less than the 5-cm-diameter criterion for erythema migrans.1
Upon questioning, the patient reported that he was unaware of the tick and could not provide a timeline for inoculation. To ensure proper treatment, the tick was removed in the office and a specimen was sent for histopathology. The arthropod was formalin fixed and paraffin embedded, and it was examined using hematoxylin and eosin and Warthin-Starry stains. Histopathology of the specimen revealed a blood-engorged arthropod. Warthin-Starry stain of the abdomen of the tick highlighted tiny strandlike spirochetes within the gut that were compatible with B burgdorferi (Figure). This finding prompted treatment with a 3-week course of doxycycline. Following treatment, erythema resolved. The patient experienced no sequelae.

Lyme disease can cause a range of serious complications if left untreated, including arthritis, neurologic deficits, and heart block. During the early stages of disease, the sensitivity and specificity of diagnostic methods such as serologic testing are limited.4 The gold standard for the diagnosis of Lyme disease comprises culture and subsequent confirmation by polymerase chain reaction.1 However, cultivation of B burgdorferi is challenging.5 The Centers for Disease Control and Prevention recommends 2-tiered serologic antibody analysis, which has 27% sensitivity during the first week of cutaneous symptoms, and involves an enzyme-linked immunoassay followed by reflexive immunoblotting for positive or indeterminate cases.2,6 The precision of this method is limited by several variables; for example, seroconversion fails to occur in approximately 40% of cases, even after proven exposure to the spirochete.7 Furthermore, the sensitivity of the test is particularly low during the first 4 to 6 weeks of infection—before the body mounts a proper immune response; fewer than 50% of patients exhibit a positive response to the test at initial presentation.3
Clinical diagnosis of Lyme disease is possible, though the pathognomonic erythema migrans rash can be delayed for as long as 30 days and remains absent in 20% to 30% of patients.1 Prophylactic treatment can be offered to individuals who reside in a hyperendemic area and have a rash or have had an engorged Ixodes tick attached for longer than 36 hours.8
More definitive techniques for early diagnosis are needed to enable selective and accurate treatment. The standard of care for Lyme disease includes a 10-day course of doxycycline or a 14-day course of cefuroxime axetil or amoxicillin.9 Many patients tolerate treatment and achieve resolution of disease, but antibiotics are not benign, as some patients experience drug-related adverse effects such as photosensitivity, urticaria, diarrhea, nausea, vomiting, esophagitis, hepatotoxicity, and the Jarisch-Herxheimer reaction (fever, chills, rigors, nausea and vomiting, headache, tachycardia, hypotension, hyperventilation, flushing, myalgia, and exacerbation of lesions).10,11 In a group of 123 patients with Lyme disease, 30% treated with cefuroxime axetil and 32% treated with doxycycline had 1 or more drug-related adverse events.10 Additionally, avoidable antibiotic use is associated with increasing antibiotic resistance.12 Improved diagnostic accuracy would prevent unnecessary treatment. Galan and colleagues7 reported that Warthin-Starry staining of prepared sections of the abdomen of a tick allowed for detection of B burgdorferi with a sensitivity of 71% and specificity of 83%. This technique did not delay the final biopsy report and may be a promising adjunct to the diagnosis of early Lyme disease.7
Anecdotally, many patients who present with an attached and engorged tick are unaware of the timeline of their exposure. Histologic analysis of a removed tick could aid in early clinical decision-making—ie, when the diagnosis is unclear and treatment guidelines vary by region and circumstance. Improved sensitivity and specificity of diagnosis can prevent unnecessary antibiotic treatment, which is associated with adverse effects and escalation of antibiotic resistance.
- Borchers AT, Keen CL, Huntley AC, et al. Lyme disease: a rigorous review of diagnostic criteria and treatment. J Autoimmun. 2015;57:82-115. doi:10.1016/j.jaut.2014.09.004
- Centers for Disease Control and Prevention. Lyme disease: data and surveillance. February 14, 2024. Accessed March 5, 2024. https://www.cdc.gov/lyme/datasurveillance/index.html
- Marques AR. Laboratory diagnosis of Lyme disease. Infect Dis Clin North Am. 2015;29:295-307. doi:10.1016/j.idc.2015.02.005
- Bratton RL, Whiteside JW, Hovan MJ, et al. Diagnosis and treatment of Lyme disease. Mayo Clin Proc. 2008;83:566-571. doi:10.4065/83.5.566
- Berger B, Johnson R, Kodner C. Cultivation of Borrelia burgdorferi from human tick bite sites: a guide to the risk of infection. J Am Acad Dermatol. 1995;32(2 pt 1):184-187. doi:10.1016/0190-9622(95)90123-x
- Branda JA, Linskey K, Kim YA, et al. Two-tiered antibody testing for Lyme disease with use of 2 enzyme immunoassays, a whole-cell sonicate enzyme immunoassay followed by a VlsE C6 peptide enzyme immunoassay. Clin Infect Dis. 2011;53:541-547. doi:10.1093/cid/cir464
- Galan A, Kupernik P, Cowper SE. Detection of Borrelia in Ixodes scapularis ticks by silver stain, immunohistochemical and direct immunofluorescent methods. J Cutan Pathol. 2018;45:473-477. doi:10.1111/cup.13143
- Nadelman RB, Nowakowski J, Fish D, et al; Prophylaxis with single-dose doxycycline for the prevention of Lyme disease after an Ixodes scapularis tick bite. N Engl J Med. 2001;345:79-84. doi:10.1056/NEJM200107123450201
- Lantos PM, Rumbaugh J, Bockenstedt LK, et al. Clinical practice guidelines by the Infectious Diseases Society of America (IDSA), American Academy of Neurology (AAN), and American College of Rheumatology (ACR): 2020 guidelines for the prevention, diagnosis, and treatment of Lyme disease. Arthritis Rheumatol. 2021;73:12-20. doi:10.1002/art.41562
- Nadelman RB, Luger SW, Frank E, et al. Comparison of cefuroxime axetil and doxycycline in the treatment of early Lyme disease. Ann Intern Med. 1992;117:273-280. doi:10.7326/0003-4819-117-4-273
- Gresser U. Amoxicillin–clavulanic acid therapy may be associated with severe side effects—review of the literature. Eur J Med Res. 2001;6:139-149.
- Nathan C, Cars O. Antibiotic resistance—problems, progress, and prospects. N Engl J Med. 2014;371:1761-1763. doi:10.1056/NEJMp1408040
To the Editor:
Lyme disease is caused by spirochetes of the Borrelia burgdorferi sensu lato species complex and transmitted to humans by the bite of the Ixodes scapularis tick. It was first classified as a nationally notifiable disease in 1991, and the incidence has risen remarkably since then.1 More than 63,000 cases are reported annually to the Centers for Disease Control and Prevention; however, this number reflects severe underreporting, as the true incidence of the disease is projected to be closer to 476,000 cases per year.2 Additionally, 95% of US cases occur in the Northeast and upper Midwest.3 Given the pervasiveness of Lyme disease, early and reliable diagnostic methodology is critical, especially in cases in which the timeline of inoculation is unclear. We present a case of Lyme disease that was discovered during a routine dermatologic visit.
A 77-year-old White man with no relevant medical history presented to a dermatology clinic in west-central Virginia for a routine skin check. Physical examination revealed a well-appearing patient without overt skin abnormalities. However, on closer evaluation, a 0.2×0.1-cm engorged black I scapularis tick was visualized on the left lateral upper back. There was a surrounding zone of erythema that measured less than the 5-cm-diameter criterion for erythema migrans.1
Upon questioning, the patient reported that he was unaware of the tick and could not provide a timeline for inoculation. To ensure proper treatment, the tick was removed in the office and a specimen was sent for histopathology. The arthropod was formalin fixed and paraffin embedded, and it was examined using hematoxylin and eosin and Warthin-Starry stains. Histopathology of the specimen revealed a blood-engorged arthropod. Warthin-Starry stain of the abdomen of the tick highlighted tiny strandlike spirochetes within the gut that were compatible with B burgdorferi (Figure). This finding prompted treatment with a 3-week course of doxycycline. Following treatment, erythema resolved. The patient experienced no sequelae.
Lyme disease can cause a range of serious complications if left untreated, including arthritis, neurologic deficits, and heart block. During the early stages of disease, the sensitivity and specificity of diagnostic methods such as serologic testing are limited.4 The gold standard for the diagnosis of Lyme disease comprises culture and subsequent confirmation by polymerase chain reaction.1 However, cultivation of B burgdorferi is challenging.5 The Centers for Disease Control and Prevention recommends 2-tiered serologic antibody analysis, which has 27% sensitivity during the first week of cutaneous symptoms, and involves an enzyme-linked immunoassay followed by reflexive immunoblotting for positive or indeterminate cases.2,6 The precision of this method is limited by several variables; for example, seroconversion fails to occur in approximately 40% of cases, even after proven exposure to the spirochete.7 Furthermore, the sensitivity of the test is particularly low during the first 4 to 6 weeks of infection—before the body mounts a proper immune response; fewer than 50% of patients exhibit a positive response to the test at initial presentation.3
Clinical diagnosis of Lyme disease is possible, though the pathognomonic erythema migrans rash can be delayed for as long as 30 days and remains absent in 20% to 30% of patients.1 Prophylactic treatment can be offered to individuals who reside in a hyperendemic area and have a rash or have had an engorged Ixodes tick attached for longer than 36 hours.8
More definitive techniques for early diagnosis are needed to enable selective and accurate treatment. The standard of care for Lyme disease includes a 10-day course of doxycycline or a 14-day course of cefuroxime axetil or amoxicillin.9 Many patients tolerate treatment and achieve resolution of disease, but antibiotics are not benign, as some patients experience drug-related adverse effects such as photosensitivity, urticaria, diarrhea, nausea, vomiting, esophagitis, hepatotoxicity, and the Jarisch-Herxheimer reaction (fever, chills, rigors, nausea and vomiting, headache, tachycardia, hypotension, hyperventilation, flushing, myalgia, and exacerbation of lesions).10,11 In a group of 123 patients with Lyme disease, 30% treated with cefuroxime axetil and 32% treated with doxycycline had 1 or more drug-related adverse events.10 Additionally, avoidable antibiotic use is associated with increasing antibiotic resistance.12 Improved diagnostic accuracy would prevent unnecessary treatment. Galan and colleagues7 reported that Warthin-Starry staining of prepared sections of the abdomen of a tick allowed for detection of B burgdorferi with a sensitivity of 71% and specificity of 83%. This technique did not delay the final biopsy report and may be a promising adjunct to the diagnosis of early Lyme disease.7
Anecdotally, many patients who present with an attached and engorged tick are unaware of the timeline of their exposure. Histologic analysis of a removed tick could aid in early clinical decision-making—ie, when the diagnosis is unclear and treatment guidelines vary by region and circumstance. Improved sensitivity and specificity of diagnosis can prevent unnecessary antibiotic treatment, which is associated with adverse effects and escalation of antibiotic resistance.
To the Editor:
Lyme disease is caused by spirochetes of the Borrelia burgdorferi sensu lato species complex and transmitted to humans by the bite of the Ixodes scapularis tick. It was first classified as a nationally notifiable disease in 1991, and the incidence has risen remarkably since then.1 More than 63,000 cases are reported annually to the Centers for Disease Control and Prevention; however, this number reflects severe underreporting, as the true incidence of the disease is projected to be closer to 476,000 cases per year.2 Additionally, 95% of US cases occur in the Northeast and upper Midwest.3 Given the pervasiveness of Lyme disease, early and reliable diagnostic methodology is critical, especially in cases in which the timeline of inoculation is unclear. We present a case of Lyme disease that was discovered during a routine dermatologic visit.
A 77-year-old White man with no relevant medical history presented to a dermatology clinic in west-central Virginia for a routine skin check. Physical examination revealed a well-appearing patient without overt skin abnormalities. However, on closer evaluation, a 0.2×0.1-cm engorged black I scapularis tick was visualized on the left lateral upper back. There was a surrounding zone of erythema that measured less than the 5-cm-diameter criterion for erythema migrans.1
Upon questioning, the patient reported that he was unaware of the tick and could not provide a timeline for inoculation. To ensure proper treatment, the tick was removed in the office and a specimen was sent for histopathology. The arthropod was formalin fixed and paraffin embedded, and it was examined using hematoxylin and eosin and Warthin-Starry stains. Histopathology of the specimen revealed a blood-engorged arthropod. Warthin-Starry stain of the abdomen of the tick highlighted tiny strandlike spirochetes within the gut that were compatible with B burgdorferi (Figure). This finding prompted treatment with a 3-week course of doxycycline. Following treatment, erythema resolved. The patient experienced no sequelae.
Lyme disease can cause a range of serious complications if left untreated, including arthritis, neurologic deficits, and heart block. During the early stages of disease, the sensitivity and specificity of diagnostic methods such as serologic testing are limited.4 The gold standard for the diagnosis of Lyme disease comprises culture and subsequent confirmation by polymerase chain reaction.1 However, cultivation of B burgdorferi is challenging.5 The Centers for Disease Control and Prevention recommends 2-tiered serologic antibody analysis, which has 27% sensitivity during the first week of cutaneous symptoms, and involves an enzyme-linked immunoassay followed by reflexive immunoblotting for positive or indeterminate cases.2,6 The precision of this method is limited by several variables; for example, seroconversion fails to occur in approximately 40% of cases, even after proven exposure to the spirochete.7 Furthermore, the sensitivity of the test is particularly low during the first 4 to 6 weeks of infection—before the body mounts a proper immune response; fewer than 50% of patients exhibit a positive response to the test at initial presentation.3
Clinical diagnosis of Lyme disease is possible, though the pathognomonic erythema migrans rash can be delayed for as long as 30 days and remains absent in 20% to 30% of patients.1 Prophylactic treatment can be offered to individuals who reside in a hyperendemic area and have a rash or have had an engorged Ixodes tick attached for longer than 36 hours.8
More definitive techniques for early diagnosis are needed to enable selective and accurate treatment. The standard of care for Lyme disease includes a 10-day course of doxycycline or a 14-day course of cefuroxime axetil or amoxicillin.9 Many patients tolerate treatment and achieve resolution of disease, but antibiotics are not benign, as some patients experience drug-related adverse effects such as photosensitivity, urticaria, diarrhea, nausea, vomiting, esophagitis, hepatotoxicity, and the Jarisch-Herxheimer reaction (fever, chills, rigors, nausea and vomiting, headache, tachycardia, hypotension, hyperventilation, flushing, myalgia, and exacerbation of lesions).10,11 In a group of 123 patients with Lyme disease, 30% treated with cefuroxime axetil and 32% treated with doxycycline had 1 or more drug-related adverse events.10 Additionally, avoidable antibiotic use is associated with increasing antibiotic resistance.12 Improved diagnostic accuracy would prevent unnecessary treatment. Galan and colleagues7 reported that Warthin-Starry staining of prepared sections of the abdomen of a tick allowed for detection of B burgdorferi with a sensitivity of 71% and specificity of 83%. This technique did not delay the final biopsy report and may be a promising adjunct to the diagnosis of early Lyme disease.7
Anecdotally, many patients who present with an attached and engorged tick are unaware of the timeline of their exposure. Histologic analysis of a removed tick could aid in early clinical decision-making—ie, when the diagnosis is unclear and treatment guidelines vary by region and circumstance. Improved sensitivity and specificity of diagnosis can prevent unnecessary antibiotic treatment, which is associated with adverse effects and escalation of antibiotic resistance.
- Borchers AT, Keen CL, Huntley AC, et al. Lyme disease: a rigorous review of diagnostic criteria and treatment. J Autoimmun. 2015;57:82-115. doi:10.1016/j.jaut.2014.09.004
- Centers for Disease Control and Prevention. Lyme disease: data and surveillance. February 14, 2024. Accessed March 5, 2024. https://www.cdc.gov/lyme/datasurveillance/index.html
- Marques AR. Laboratory diagnosis of Lyme disease. Infect Dis Clin North Am. 2015;29:295-307. doi:10.1016/j.idc.2015.02.005
- Bratton RL, Whiteside JW, Hovan MJ, et al. Diagnosis and treatment of Lyme disease. Mayo Clin Proc. 2008;83:566-571. doi:10.4065/83.5.566
- Berger B, Johnson R, Kodner C. Cultivation of Borrelia burgdorferi from human tick bite sites: a guide to the risk of infection. J Am Acad Dermatol. 1995;32(2 pt 1):184-187. doi:10.1016/0190-9622(95)90123-x
- Branda JA, Linskey K, Kim YA, et al. Two-tiered antibody testing for Lyme disease with use of 2 enzyme immunoassays, a whole-cell sonicate enzyme immunoassay followed by a VlsE C6 peptide enzyme immunoassay. Clin Infect Dis. 2011;53:541-547. doi:10.1093/cid/cir464
- Galan A, Kupernik P, Cowper SE. Detection of Borrelia in Ixodes scapularis ticks by silver stain, immunohistochemical and direct immunofluorescent methods. J Cutan Pathol. 2018;45:473-477. doi:10.1111/cup.13143
- Nadelman RB, Nowakowski J, Fish D, et al; Prophylaxis with single-dose doxycycline for the prevention of Lyme disease after an Ixodes scapularis tick bite. N Engl J Med. 2001;345:79-84. doi:10.1056/NEJM200107123450201
- Lantos PM, Rumbaugh J, Bockenstedt LK, et al. Clinical practice guidelines by the Infectious Diseases Society of America (IDSA), American Academy of Neurology (AAN), and American College of Rheumatology (ACR): 2020 guidelines for the prevention, diagnosis, and treatment of Lyme disease. Arthritis Rheumatol. 2021;73:12-20. doi:10.1002/art.41562
- Nadelman RB, Luger SW, Frank E, et al. Comparison of cefuroxime axetil and doxycycline in the treatment of early Lyme disease. Ann Intern Med. 1992;117:273-280. doi:10.7326/0003-4819-117-4-273
- Gresser U. Amoxicillin–clavulanic acid therapy may be associated with severe side effects—review of the literature. Eur J Med Res. 2001;6:139-149.
- Nathan C, Cars O. Antibiotic resistance—problems, progress, and prospects. N Engl J Med. 2014;371:1761-1763. doi:10.1056/NEJMp1408040
- Borchers AT, Keen CL, Huntley AC, et al. Lyme disease: a rigorous review of diagnostic criteria and treatment. J Autoimmun. 2015;57:82-115. doi:10.1016/j.jaut.2014.09.004
- Centers for Disease Control and Prevention. Lyme disease: data and surveillance. February 14, 2024. Accessed March 5, 2024. https://www.cdc.gov/lyme/datasurveillance/index.html
- Marques AR. Laboratory diagnosis of Lyme disease. Infect Dis Clin North Am. 2015;29:295-307. doi:10.1016/j.idc.2015.02.005
- Bratton RL, Whiteside JW, Hovan MJ, et al. Diagnosis and treatment of Lyme disease. Mayo Clin Proc. 2008;83:566-571. doi:10.4065/83.5.566
- Berger B, Johnson R, Kodner C. Cultivation of Borrelia burgdorferi from human tick bite sites: a guide to the risk of infection. J Am Acad Dermatol. 1995;32(2 pt 1):184-187. doi:10.1016/0190-9622(95)90123-x
- Branda JA, Linskey K, Kim YA, et al. Two-tiered antibody testing for Lyme disease with use of 2 enzyme immunoassays, a whole-cell sonicate enzyme immunoassay followed by a VlsE C6 peptide enzyme immunoassay. Clin Infect Dis. 2011;53:541-547. doi:10.1093/cid/cir464
- Galan A, Kupernik P, Cowper SE. Detection of Borrelia in Ixodes scapularis ticks by silver stain, immunohistochemical and direct immunofluorescent methods. J Cutan Pathol. 2018;45:473-477. doi:10.1111/cup.13143
- Nadelman RB, Nowakowski J, Fish D, et al; Prophylaxis with single-dose doxycycline for the prevention of Lyme disease after an Ixodes scapularis tick bite. N Engl J Med. 2001;345:79-84. doi:10.1056/NEJM200107123450201
- Lantos PM, Rumbaugh J, Bockenstedt LK, et al. Clinical practice guidelines by the Infectious Diseases Society of America (IDSA), American Academy of Neurology (AAN), and American College of Rheumatology (ACR): 2020 guidelines for the prevention, diagnosis, and treatment of Lyme disease. Arthritis Rheumatol. 2021;73:12-20. doi:10.1002/art.41562
- Nadelman RB, Luger SW, Frank E, et al. Comparison of cefuroxime axetil and doxycycline in the treatment of early Lyme disease. Ann Intern Med. 1992;117:273-280. doi:10.7326/0003-4819-117-4-273
- Gresser U. Amoxicillin–clavulanic acid therapy may be associated with severe side effects—review of the literature. Eur J Med Res. 2001;6:139-149.
- Nathan C, Cars O. Antibiotic resistance—problems, progress, and prospects. N Engl J Med. 2014;371:1761-1763. doi:10.1056/NEJMp1408040
PRACTICE POINTS
- Lyme disease is increasingly common in the United States.
- Lyme disease can cause debilitating sequelae if left untreated, including arthritis, neurologic deficits, and heart block.
- Diagnostic methods for identifying early Lyme disease have limited sensitivity and specificity, necessitating alternative strategies for making an accurate diagnosis and initiating treatment.

Purpuric Eruption in a Patient With Hairy Cell Leukemia
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
A 68-year-old woman presented to the emergency department with neutropenic fever and a rash over the body after receiving 2 doses of cladribine therapy for hairy cell leukemia. Physical examination demonstrated marked facial (top), lip, and tongue swelling, as well as a diffuse dusky nonpalpable purpuric rash on the abdomen (bottom) and back involving 90% of the body surface area. Bilateral ear edema was appreciated with accentuation of the earlobe crease. The patient exhibited subconjunctival hemorrhage, ectropion, and scleral injection. A punch biopsy of the thigh was performed.
Multiple Asymptomatic Dome-Shaped Papules on the Scalp
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).

Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5

Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7

Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.

- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).
Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5
Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7
Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.
The Diagnosis: Spiradenocylindroma
Shave biopsies of our patient’s lesions showed wellcircumscribed dermal nodules resembling a spiradenoma with 3 cell populations: those with lighter nuclei, darker nuclei, and scattered lymphocytes. However, the conspicuous globules of basement membrane material were reminiscent of a cylindroma. These overlapping features and the patient’s history of cylindroma were suggestive of a diagnosis of spiradenocylindroma.
Spiradenocylindroma is an uncommon dermal tumor with features that overlap with spiradenoma and cylindroma.1 It may manifest as a solitary lesion or multiple lesions and can occur sporadically or in the context of a family history. Histologically, it must be distinguished from other intradermal basaloid neoplasms including conventional cylindroma and spiradenoma, dermal duct tumor, hidradenoma, and trichoblastoma.
When patients present with multiple cylindromas, spiradenomas, or spiradenocylindromas, physicians should consider genetic testing and review of the family history to assess for cylindromatosis gene mutations or Brooke-Spiegler syndrome. Biopsy and histologic examination are important because malignant tumors can evolve from pre-existing spiradenocylindromas, cylindromas, and spiradenomas,2 with an increased risk in patients with Brooke-Spiegler syndrome.1 Our patient declined further genetic workup but continues to follow up with dermatology for monitoring of lesions.
Dermal duct tumors are morphologic variants of poromas that are derived from sweat gland lineage and usually manifest as solitary dome-shaped papules, plaques, or nodules most often seen on acral surfaces as well as the head and neck.3 Clinically, they may be indistinguishable from spiradenocylindromas and require biopsy for histologic evaluation. They can be distinguished from spiradenocylindroma by the presence of small dermal nodules composed of cuboidal cells with ample pink cytoplasm and cuticle-lined ducts (Figure 1).
Trichoblastomas typically are deep-seated basaloid follicular neoplasms on the scalp with papillary mesenchyme resembling the normal fibrous sheath of the hair follicle, often replete with papillary mesenchymal bodies (Figure 2). There generally are no retraction spaces between its basaloid nests and the surrounding stroma, which is unlikely to contain mucin relative to basal cell carcinoma (BCC).4,5
Adenoid cystic carcinoma is a rare salivary gland tumor that can metastasize to the skin and rarely arises as a primary skin adnexal tumor. It manifests as a slowgrowing mass that can be tender to palpation.6 Histologic examination shows dermal islands with cribriform blue and pink spaces. Compared to BCC, adenoid cystic carcinoma cells are enlarged and epithelioid with relatively scarce cytoplasm (Figure 3).6,7 Adenoid cystic carcinoma can show variable growth patterns including infiltrative nests and trabeculae. Perineural invasion is common, and there is a high risk for local recurrence.7 First-line therapy usually is surgical, and postoperative radiotherapy may be required.6,7
Nodular BCC commonly manifests as an enlarging nonhealing lesion on sun-exposed skin and has many subtypes, typically with arborizing telangiectases on dermoscopy. Histopathologic examination of nodular BCC reveals a nest of basaloid follicular germinative cells in the dermis with peripheral palisading and a fibromyxoid stroma (Figure 4).8 Patients with Brooke-Spiegler syndrome are at increased risk for nodular BCC, which may be clinically indistinguishable from spiradenoma, cylindroma, and spiradenocylindroma, necessitating histologic assessment.
- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
- Facchini V, Colangeli W, Bozza F, et al. A rare histopathological spiradenocylindroma: a case report. Clin Ter. 2022;173:292-294. doi:10.7417/ CT.2022.2433
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update [published online March 14, 2016]. Head Neck Pathol. 2016;10:125-30. doi:10.1007/s12105-016-0705-x
- Miller AC, Adjei S, Temiz LA, et al. Dermal duct tumor: a diagnostic dilemma. Dermatopathology (Basel). 2022;9:36-47. doi:10.3390/dermatopathology9010007
- Elston DM. Pilar and sebaceous neoplasms. In: Elston DM, Ferringer T, Ko C, et al. Dermatopathology. 3rd ed. Elsevier; 2018:71-85.
- McCalmont TH, Pincus LB. Adnexal neoplasms. In: Bolognia J, Schaffer J, Cerroni, L. Dermatology. 4th ed. Elsevier; 2017:1930-1953.
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update [published online May 2, 2015]. Oral Oncol. 2015;51:652-661. doi:10.1016/j.oraloncology.2015.04.005
- Tonev ID, Pirgova YS, Conev NV. Primary adenoid cystic carcinoma of the skin with multiple local recurrences. Case Rep Oncol. 2015;8:251- 255. doi:10.1159/000431082
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations [published online May 18, 2018]. J Am Acad Dermatol. 2019;80:303-317. doi:10.1016/j.jaad.2018.03.060
A 62-year-old man with a history of cylindromas presented to our clinic with multiple asymptomatic, 3- to 4-mm, nonmobile, dome-shaped, telangiectatic, pink papules over the parietal and vertex scalp that had been present for more than 10 years without change. Several family members had similar lesions that had not been evaluated by a physician, and there had been no genetic evaluation. Shave biopsies of several lesions were performed.


Impact of the COVID-19 Pandemic on Care for Patients With Skin Cancer
To the Editor:
The most common malignancy in the United States is skin cancer, with melanoma accounting for the majority of skin cancer deaths.1 Despite the lack of established guidelines for routine total-body skin examinations, many patients regularly visit their dermatologist for assessment of pigmented skin lesions.2 During the COVID-19 pandemic, many patients were unable to attend in-person dermatology visits, which resulted in many high-risk individuals not receiving care or alternatively seeking virtual care for cutaneous lesions.3 There has been a lack of research in the United States exploring the utilization of teledermatology during the pandemic and its overall impact on the care of patients with a history of skin cancer. We explored the impact of the COVID-19 pandemic on care for patients with skin cancer in a large US population.
 With and Without a History of Skin Cancera in 2020-2021 NHIS (N=46,679)")
 With and Without a History of Skin Cancera in 2020-2021 NHIS (N=46,679)")
Using anonymous survey data from the 2020-2021 National Health Interview Survey,4 we conducted a population-based, cross-sectional study to evaluate access to care during the COVID-19 pandemic for patients with a self-reported history of skin cancer—melanoma, nonmelanoma skin cancer, or unknown skin cancer. The 3 outcome variables included having a virtual medical appointment in the past 12 months (yes/no), delaying medical care due to the COVID-19 pandemic (yes/no), and not receiving care due to the COVID-19 pandemic (yes/no). Multivariable logistic regression models evaluating the relationship between a history of skin cancer and access to care were constructed using Stata/MP 17.0 (StataCorp LLC). We controlled for patient age; education; race/ethnicity; received public assistance or welfare payments; sex; region; US citizenship status; health insurance status; comorbidities including history of hypertension, diabetes, and hypercholesterolemia; and birthplace in the United States in the logistic regression models.

Our analysis included 46,679 patients aged 18 years or older, of whom 3.4% (weighted)(n=2204) reported a history of skin cancer (eTable 1). The weighted percentage was calculated using National Health Interview Survey design parameters (accounting for the multistage sampling design) to represent the general US population. Compared with those with no history of skin cancer, patients with a history of skin cancer were significantly more likely to delay medical care (adjusted odds ratio [AOR], 1.37; 95% CI, 1.21-1.54; P<.001) or not receive care (AOR, 1.35; 95% CI, 1.16-1.57; P<.001) due to the pandemic and were more likely to have had a virtual medical visit in the past 12 months (AOR, 1.12; 95% CI, 1.00-1.26; P=.05). Additionally, subgroup analysis revealed that females were more likely than males to forego medical care (eTable 2). β Coefficients for independent and dependent variables were further analyzed using logistic regression (eTable 3).

After adjusting for various potential confounders including comorbidities, our results revealed that patients with a history of skin cancer reported that they were less likely to receive in-person medical care due to the COVID-19 pandemic, as high-risk individuals with a history of skin cancer may have stopped receiving total-body skin examinations and dermatology care during the pandemic. Our findings showed that patients with a history of skin cancer were more likely than those without skin cancer to delay or forego care due to the pandemic, which may contribute to a higher incidence of advanced-stage melanomas postpandemic. Trepanowski et al5 reported an increased incidence of patients presenting with more advanced melanomas during the pandemic. Telemedicine was more commonly utilized by patients with a history of skin cancer during the pandemic.
In the future, virtual care may help limit advanced stages of skin cancer by serving as a viable alternative to in-person care.6 It has been reported that telemedicine can serve as a useful triage service reducing patient wait times.7 Teledermatology should not replace in-person care, as there is no evidence of the diagnostic accuracy of this service and many patients still will need to be seen in-person for confirmation of their diagnosis and potential biopsy. Further studies are needed to assess for missed skin cancer diagnoses due to the utilization of telemedicine.
Limitations of this study included a self-reported history of skin cancer, β coefficients that may suggest a high degree of collinearity, and lack of specific survey questions regarding dermatologic care during the COVID-19 pandemic. Further long-term studies exploring the clinical applicability and diagnostic accuracy of virtual medicine visits for cutaneous malignancies are vital, as teledermatology may play an essential role in curbing rising skin cancer rates even beyond the pandemic.
- Guy GP Jr, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections—United States, 1982-2030. MMWR Morb Mortal Wkly Rep. 2015;64:591-596.
- Whiteman DC, Olsen CM, MacGregor S, et al; QSkin Study. The effect of screening on melanoma incidence and biopsy rates. Br J Dermatol. 2022;187:515-522. doi:10.1111/bjd.21649
- Jobbágy A, Kiss N, Meznerics FA, et al. Emergency use and efficacy of an asynchronous teledermatology system as a novel tool for early diagnosis of skin cancer during the first wave of COVID-19 pandemic. Int J Environ Res Public Health. 2022;19:2699. doi:10.3390/ijerph19052699
- National Center for Health Statistics. NHIS Data, Questionnaires and Related Documentation. Centers for Disease Control and Prevention website. Accessed April 19, 2023. https://www.cdc.gov/nchs/nhis/data-questionnaires-documentation.htm
- Trepanowski N, Chang MS, Zhou G, et al. Delays in melanoma presentation during the COVID-19 pandemic: a nationwide multi-institutional cohort study. J Am Acad Dermatol. 2022;87:1217-1219. doi:10.1016/j.jaad.2022.06.031
- Chiru MR, Hindocha S, Burova E, et al. Management of the two-week wait pathway for skin cancer patients, before and during the pandemic: is virtual consultation an option? J Pers Med. 2022;12:1258. doi:10.3390/jpm12081258
- Finnane A Dallest K Janda M et al. Teledermatology for the diagnosis and management of skin cancer: a systematic review. JAMA Dermatol. 2017;153:319-327. doi:10.1001/jamadermatol.2016.4361
To the Editor:
The most common malignancy in the United States is skin cancer, with melanoma accounting for the majority of skin cancer deaths.1 Despite the lack of established guidelines for routine total-body skin examinations, many patients regularly visit their dermatologist for assessment of pigmented skin lesions.2 During the COVID-19 pandemic, many patients were unable to attend in-person dermatology visits, which resulted in many high-risk individuals not receiving care or alternatively seeking virtual care for cutaneous lesions.3 There has been a lack of research in the United States exploring the utilization of teledermatology during the pandemic and its overall impact on the care of patients with a history of skin cancer. We explored the impact of the COVID-19 pandemic on care for patients with skin cancer in a large US population.
Using anonymous survey data from the 2020-2021 National Health Interview Survey,4 we conducted a population-based, cross-sectional study to evaluate access to care during the COVID-19 pandemic for patients with a self-reported history of skin cancer—melanoma, nonmelanoma skin cancer, or unknown skin cancer. The 3 outcome variables included having a virtual medical appointment in the past 12 months (yes/no), delaying medical care due to the COVID-19 pandemic (yes/no), and not receiving care due to the COVID-19 pandemic (yes/no). Multivariable logistic regression models evaluating the relationship between a history of skin cancer and access to care were constructed using Stata/MP 17.0 (StataCorp LLC). We controlled for patient age; education; race/ethnicity; received public assistance or welfare payments; sex; region; US citizenship status; health insurance status; comorbidities including history of hypertension, diabetes, and hypercholesterolemia; and birthplace in the United States in the logistic regression models.
Our analysis included 46,679 patients aged 18 years or older, of whom 3.4% (weighted)(n=2204) reported a history of skin cancer (eTable 1). The weighted percentage was calculated using National Health Interview Survey design parameters (accounting for the multistage sampling design) to represent the general US population. Compared with those with no history of skin cancer, patients with a history of skin cancer were significantly more likely to delay medical care (adjusted odds ratio [AOR], 1.37; 95% CI, 1.21-1.54; P<.001) or not receive care (AOR, 1.35; 95% CI, 1.16-1.57; P<.001) due to the pandemic and were more likely to have had a virtual medical visit in the past 12 months (AOR, 1.12; 95% CI, 1.00-1.26; P=.05). Additionally, subgroup analysis revealed that females were more likely than males to forego medical care (eTable 2). β Coefficients for independent and dependent variables were further analyzed using logistic regression (eTable 3).
After adjusting for various potential confounders including comorbidities, our results revealed that patients with a history of skin cancer reported that they were less likely to receive in-person medical care due to the COVID-19 pandemic, as high-risk individuals with a history of skin cancer may have stopped receiving total-body skin examinations and dermatology care during the pandemic. Our findings showed that patients with a history of skin cancer were more likely than those without skin cancer to delay or forego care due to the pandemic, which may contribute to a higher incidence of advanced-stage melanomas postpandemic. Trepanowski et al5 reported an increased incidence of patients presenting with more advanced melanomas during the pandemic. Telemedicine was more commonly utilized by patients with a history of skin cancer during the pandemic.
In the future, virtual care may help limit advanced stages of skin cancer by serving as a viable alternative to in-person care.6 It has been reported that telemedicine can serve as a useful triage service reducing patient wait times.7 Teledermatology should not replace in-person care, as there is no evidence of the diagnostic accuracy of this service and many patients still will need to be seen in-person for confirmation of their diagnosis and potential biopsy. Further studies are needed to assess for missed skin cancer diagnoses due to the utilization of telemedicine.
Limitations of this study included a self-reported history of skin cancer, β coefficients that may suggest a high degree of collinearity, and lack of specific survey questions regarding dermatologic care during the COVID-19 pandemic. Further long-term studies exploring the clinical applicability and diagnostic accuracy of virtual medicine visits for cutaneous malignancies are vital, as teledermatology may play an essential role in curbing rising skin cancer rates even beyond the pandemic.
To the Editor:
The most common malignancy in the United States is skin cancer, with melanoma accounting for the majority of skin cancer deaths.1 Despite the lack of established guidelines for routine total-body skin examinations, many patients regularly visit their dermatologist for assessment of pigmented skin lesions.2 During the COVID-19 pandemic, many patients were unable to attend in-person dermatology visits, which resulted in many high-risk individuals not receiving care or alternatively seeking virtual care for cutaneous lesions.3 There has been a lack of research in the United States exploring the utilization of teledermatology during the pandemic and its overall impact on the care of patients with a history of skin cancer. We explored the impact of the COVID-19 pandemic on care for patients with skin cancer in a large US population.
Using anonymous survey data from the 2020-2021 National Health Interview Survey,4 we conducted a population-based, cross-sectional study to evaluate access to care during the COVID-19 pandemic for patients with a self-reported history of skin cancer—melanoma, nonmelanoma skin cancer, or unknown skin cancer. The 3 outcome variables included having a virtual medical appointment in the past 12 months (yes/no), delaying medical care due to the COVID-19 pandemic (yes/no), and not receiving care due to the COVID-19 pandemic (yes/no). Multivariable logistic regression models evaluating the relationship between a history of skin cancer and access to care were constructed using Stata/MP 17.0 (StataCorp LLC). We controlled for patient age; education; race/ethnicity; received public assistance or welfare payments; sex; region; US citizenship status; health insurance status; comorbidities including history of hypertension, diabetes, and hypercholesterolemia; and birthplace in the United States in the logistic regression models.
Our analysis included 46,679 patients aged 18 years or older, of whom 3.4% (weighted)(n=2204) reported a history of skin cancer (eTable 1). The weighted percentage was calculated using National Health Interview Survey design parameters (accounting for the multistage sampling design) to represent the general US population. Compared with those with no history of skin cancer, patients with a history of skin cancer were significantly more likely to delay medical care (adjusted odds ratio [AOR], 1.37; 95% CI, 1.21-1.54; P<.001) or not receive care (AOR, 1.35; 95% CI, 1.16-1.57; P<.001) due to the pandemic and were more likely to have had a virtual medical visit in the past 12 months (AOR, 1.12; 95% CI, 1.00-1.26; P=.05). Additionally, subgroup analysis revealed that females were more likely than males to forego medical care (eTable 2). β Coefficients for independent and dependent variables were further analyzed using logistic regression (eTable 3).
After adjusting for various potential confounders including comorbidities, our results revealed that patients with a history of skin cancer reported that they were less likely to receive in-person medical care due to the COVID-19 pandemic, as high-risk individuals with a history of skin cancer may have stopped receiving total-body skin examinations and dermatology care during the pandemic. Our findings showed that patients with a history of skin cancer were more likely than those without skin cancer to delay or forego care due to the pandemic, which may contribute to a higher incidence of advanced-stage melanomas postpandemic. Trepanowski et al5 reported an increased incidence of patients presenting with more advanced melanomas during the pandemic. Telemedicine was more commonly utilized by patients with a history of skin cancer during the pandemic.
In the future, virtual care may help limit advanced stages of skin cancer by serving as a viable alternative to in-person care.6 It has been reported that telemedicine can serve as a useful triage service reducing patient wait times.7 Teledermatology should not replace in-person care, as there is no evidence of the diagnostic accuracy of this service and many patients still will need to be seen in-person for confirmation of their diagnosis and potential biopsy. Further studies are needed to assess for missed skin cancer diagnoses due to the utilization of telemedicine.
Limitations of this study included a self-reported history of skin cancer, β coefficients that may suggest a high degree of collinearity, and lack of specific survey questions regarding dermatologic care during the COVID-19 pandemic. Further long-term studies exploring the clinical applicability and diagnostic accuracy of virtual medicine visits for cutaneous malignancies are vital, as teledermatology may play an essential role in curbing rising skin cancer rates even beyond the pandemic.
- Guy GP Jr, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections—United States, 1982-2030. MMWR Morb Mortal Wkly Rep. 2015;64:591-596.
- Whiteman DC, Olsen CM, MacGregor S, et al; QSkin Study. The effect of screening on melanoma incidence and biopsy rates. Br J Dermatol. 2022;187:515-522. doi:10.1111/bjd.21649
- Jobbágy A, Kiss N, Meznerics FA, et al. Emergency use and efficacy of an asynchronous teledermatology system as a novel tool for early diagnosis of skin cancer during the first wave of COVID-19 pandemic. Int J Environ Res Public Health. 2022;19:2699. doi:10.3390/ijerph19052699
- National Center for Health Statistics. NHIS Data, Questionnaires and Related Documentation. Centers for Disease Control and Prevention website. Accessed April 19, 2023. https://www.cdc.gov/nchs/nhis/data-questionnaires-documentation.htm
- Trepanowski N, Chang MS, Zhou G, et al. Delays in melanoma presentation during the COVID-19 pandemic: a nationwide multi-institutional cohort study. J Am Acad Dermatol. 2022;87:1217-1219. doi:10.1016/j.jaad.2022.06.031
- Chiru MR, Hindocha S, Burova E, et al. Management of the two-week wait pathway for skin cancer patients, before and during the pandemic: is virtual consultation an option? J Pers Med. 2022;12:1258. doi:10.3390/jpm12081258
- Finnane A Dallest K Janda M et al. Teledermatology for the diagnosis and management of skin cancer: a systematic review. JAMA Dermatol. 2017;153:319-327. doi:10.1001/jamadermatol.2016.4361
- Guy GP Jr, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections—United States, 1982-2030. MMWR Morb Mortal Wkly Rep. 2015;64:591-596.
- Whiteman DC, Olsen CM, MacGregor S, et al; QSkin Study. The effect of screening on melanoma incidence and biopsy rates. Br J Dermatol. 2022;187:515-522. doi:10.1111/bjd.21649
- Jobbágy A, Kiss N, Meznerics FA, et al. Emergency use and efficacy of an asynchronous teledermatology system as a novel tool for early diagnosis of skin cancer during the first wave of COVID-19 pandemic. Int J Environ Res Public Health. 2022;19:2699. doi:10.3390/ijerph19052699
- National Center for Health Statistics. NHIS Data, Questionnaires and Related Documentation. Centers for Disease Control and Prevention website. Accessed April 19, 2023. https://www.cdc.gov/nchs/nhis/data-questionnaires-documentation.htm
- Trepanowski N, Chang MS, Zhou G, et al. Delays in melanoma presentation during the COVID-19 pandemic: a nationwide multi-institutional cohort study. J Am Acad Dermatol. 2022;87:1217-1219. doi:10.1016/j.jaad.2022.06.031
- Chiru MR, Hindocha S, Burova E, et al. Management of the two-week wait pathway for skin cancer patients, before and during the pandemic: is virtual consultation an option? J Pers Med. 2022;12:1258. doi:10.3390/jpm12081258
- Finnane A Dallest K Janda M et al. Teledermatology for the diagnosis and management of skin cancer: a systematic review. JAMA Dermatol. 2017;153:319-327. doi:10.1001/jamadermatol.2016.4361
PRACTICE POINTS
- The COVID-19 pandemic has altered the landscape of medicine, as many individuals are now utilizing telemedicine to receive care.
- Many individuals will continue to receive telemedicine moving forward, making it crucial to understand access to care.
Comment on “Skin Cancer Screening: The Paradox of Melanoma and Improved All-Cause Mortality”
To the Editor:
I was unsurprised and gratified by the information presented in the Viewpoint on skin cancer screening by Ngo1 (Cutis. 2024;113:94-96). In my 30 years as a community dermatologist, I have observed that patients who opt to have periodic full-body skin examinations usually are more health literate, more likely to have a primary care physician (PCP) who has encouraged them to do so (ie, a conscientious practitioner directing their preventive care), more likely to have a strong will to live, and less likely to have multiple stressors that preclude self-care (eg, may be less likely to have a spouse for whom they are a caregiver) compared to those who do not get screened.
Findings on a full-body skin examination may impact patients in many ways, not only by the detection of skin cancers. I have discovered the following:
- evidence of diabetes/insulin resistance in the form of acanthosis nigricans, tinea corporis, erythrasma;
- evidence of rosacea associated with excessive alcohol intake;
- evidence of smoking-related issues such as psoriasis or hidradenitis suppurativa;
- cutaneous evidence of other systemic diseases (eg, autoimmune disease, cancer);
- elucidation of other chronic health problems (eg, psoriasis of the skin as a clue for undiagnosed psoriatic arthritis); and
- detection of parasites on the skin (eg, ticks) or signs of infection that may have notable ramifications (eg, interdigital maceration of a diabetic patient with tinea pedis).
I even saw a patient who had been sent for magnetic resonance imaging for back pain by her internist without any physical examination when she actually had an erosion over the sacrum from a rug burn!
When conducting full-body skin examinations, dermatologists should not underestimate these principles:
- The “magic” of using a relatively noninvasive and sensitive screening tool—comfort and stress reduction for the patient from a thorough visual, tactile, olfactory, and auditory examination.
- Human interaction—especially when the patient is seen annually or even more frequently over a period of years or decades, and especially when an excellent patient-physician rapport has been established.
- The impact of improving a patient’s appearance on their overall sense of well-being (eg, by controlling rosacea).
- The opportunity to introduce concepts (ie, educate patients) such as alcohol avoidance, smoking cessation, weight reduction, hygiene, diet, and exercise in a more tangential way than a PCP, as well as to consider with patients the idea that lifestyle modification may be an adjunct, if not a replacement, for prescription treatments.
- The stress reduction that ensues when a variety of self-identified health issues are addressed, for which the only treatment may be reassurance.
I would add to Dr. Ngo’s argument that stratifying patients into skin cancer risk categories may be a useful measure if the only goal of periodic dermatologic evaluation is skin cancer detection. One size rarely fits all when it comes to health recommendations.
In sum, I believe that periodic full-body skin examination is absolutely beneficial to patient care, and I am not at all surprised that all-cause mortality was lower in patients who have those examinations. Furthermore, when I offer my healthy, low-risk patients the option to return in 2 years rather than 1, the vast majority insist on 1 year. My mother used to say, “It’s better to be looked over than to be overlooked,” and I tell my patients that, too—but it seems they already know that instinctively.
- Ngo BT. Skin cancer screening: the paradox of melanoma and improved all-cause mortality. Cutis. 2024;113:94-96. doi:10.12788/cutis.0948
To the Editor:
I was unsurprised and gratified by the information presented in the Viewpoint on skin cancer screening by Ngo1 (Cutis. 2024;113:94-96). In my 30 years as a community dermatologist, I have observed that patients who opt to have periodic full-body skin examinations usually are more health literate, more likely to have a primary care physician (PCP) who has encouraged them to do so (ie, a conscientious practitioner directing their preventive care), more likely to have a strong will to live, and less likely to have multiple stressors that preclude self-care (eg, may be less likely to have a spouse for whom they are a caregiver) compared to those who do not get screened.
Findings on a full-body skin examination may impact patients in many ways, not only by the detection of skin cancers. I have discovered the following:
- evidence of diabetes/insulin resistance in the form of acanthosis nigricans, tinea corporis, erythrasma;
- evidence of rosacea associated with excessive alcohol intake;
- evidence of smoking-related issues such as psoriasis or hidradenitis suppurativa;
- cutaneous evidence of other systemic diseases (eg, autoimmune disease, cancer);
- elucidation of other chronic health problems (eg, psoriasis of the skin as a clue for undiagnosed psoriatic arthritis); and
- detection of parasites on the skin (eg, ticks) or signs of infection that may have notable ramifications (eg, interdigital maceration of a diabetic patient with tinea pedis).
I even saw a patient who had been sent for magnetic resonance imaging for back pain by her internist without any physical examination when she actually had an erosion over the sacrum from a rug burn!
When conducting full-body skin examinations, dermatologists should not underestimate these principles:
- The “magic” of using a relatively noninvasive and sensitive screening tool—comfort and stress reduction for the patient from a thorough visual, tactile, olfactory, and auditory examination.
- Human interaction—especially when the patient is seen annually or even more frequently over a period of years or decades, and especially when an excellent patient-physician rapport has been established.
- The impact of improving a patient’s appearance on their overall sense of well-being (eg, by controlling rosacea).
- The opportunity to introduce concepts (ie, educate patients) such as alcohol avoidance, smoking cessation, weight reduction, hygiene, diet, and exercise in a more tangential way than a PCP, as well as to consider with patients the idea that lifestyle modification may be an adjunct, if not a replacement, for prescription treatments.
- The stress reduction that ensues when a variety of self-identified health issues are addressed, for which the only treatment may be reassurance.
I would add to Dr. Ngo’s argument that stratifying patients into skin cancer risk categories may be a useful measure if the only goal of periodic dermatologic evaluation is skin cancer detection. One size rarely fits all when it comes to health recommendations.
In sum, I believe that periodic full-body skin examination is absolutely beneficial to patient care, and I am not at all surprised that all-cause mortality was lower in patients who have those examinations. Furthermore, when I offer my healthy, low-risk patients the option to return in 2 years rather than 1, the vast majority insist on 1 year. My mother used to say, “It’s better to be looked over than to be overlooked,” and I tell my patients that, too—but it seems they already know that instinctively.
To the Editor:
I was unsurprised and gratified by the information presented in the Viewpoint on skin cancer screening by Ngo1 (Cutis. 2024;113:94-96). In my 30 years as a community dermatologist, I have observed that patients who opt to have periodic full-body skin examinations usually are more health literate, more likely to have a primary care physician (PCP) who has encouraged them to do so (ie, a conscientious practitioner directing their preventive care), more likely to have a strong will to live, and less likely to have multiple stressors that preclude self-care (eg, may be less likely to have a spouse for whom they are a caregiver) compared to those who do not get screened.
Findings on a full-body skin examination may impact patients in many ways, not only by the detection of skin cancers. I have discovered the following:
- evidence of diabetes/insulin resistance in the form of acanthosis nigricans, tinea corporis, erythrasma;
- evidence of rosacea associated with excessive alcohol intake;
- evidence of smoking-related issues such as psoriasis or hidradenitis suppurativa;
- cutaneous evidence of other systemic diseases (eg, autoimmune disease, cancer);
- elucidation of other chronic health problems (eg, psoriasis of the skin as a clue for undiagnosed psoriatic arthritis); and
- detection of parasites on the skin (eg, ticks) or signs of infection that may have notable ramifications (eg, interdigital maceration of a diabetic patient with tinea pedis).
I even saw a patient who had been sent for magnetic resonance imaging for back pain by her internist without any physical examination when she actually had an erosion over the sacrum from a rug burn!
When conducting full-body skin examinations, dermatologists should not underestimate these principles:
- The “magic” of using a relatively noninvasive and sensitive screening tool—comfort and stress reduction for the patient from a thorough visual, tactile, olfactory, and auditory examination.
- Human interaction—especially when the patient is seen annually or even more frequently over a period of years or decades, and especially when an excellent patient-physician rapport has been established.
- The impact of improving a patient’s appearance on their overall sense of well-being (eg, by controlling rosacea).
- The opportunity to introduce concepts (ie, educate patients) such as alcohol avoidance, smoking cessation, weight reduction, hygiene, diet, and exercise in a more tangential way than a PCP, as well as to consider with patients the idea that lifestyle modification may be an adjunct, if not a replacement, for prescription treatments.
- The stress reduction that ensues when a variety of self-identified health issues are addressed, for which the only treatment may be reassurance.
I would add to Dr. Ngo’s argument that stratifying patients into skin cancer risk categories may be a useful measure if the only goal of periodic dermatologic evaluation is skin cancer detection. One size rarely fits all when it comes to health recommendations.
In sum, I believe that periodic full-body skin examination is absolutely beneficial to patient care, and I am not at all surprised that all-cause mortality was lower in patients who have those examinations. Furthermore, when I offer my healthy, low-risk patients the option to return in 2 years rather than 1, the vast majority insist on 1 year. My mother used to say, “It’s better to be looked over than to be overlooked,” and I tell my patients that, too—but it seems they already know that instinctively.
- Ngo BT. Skin cancer screening: the paradox of melanoma and improved all-cause mortality. Cutis. 2024;113:94-96. doi:10.12788/cutis.0948
- Ngo BT. Skin cancer screening: the paradox of melanoma and improved all-cause mortality. Cutis. 2024;113:94-96. doi:10.12788/cutis.0948