User login
Interview with Andrew Solomon, MD, on diagnosing multiple sclerosis

Andrew Solomon, MD, is a neurologist and Associate Professor in the Department of Neurological Sciences and Division Chief of Multiple Sclerosis at The University of Vermont. We sat down to talk with Dr. Solomon about his experience with multiple sclerosis (MS) misdiagnosis and what can be done to improve MS diagnosis going forward.
How prevalent is the misdiagnosis of MS and what are the effects that it has on patients?
The first thing to clarify is what we mean by MS misdiagnosis. In this case, we’re talking about patients who are incorrectly assigned a diagnosis of MS. MS is hard to diagnose. Sometimes we take too long to diagnose people who have MS, sometimes we incorrectly diagnose MS in people who don’t have it.
We don’t have very good data in terms of how frequent misdiagnosis is, but we have some. The earliest data we have is from the 1980s. In 1988 there was a study published involving approximately 500 patients who had been diagnosed with MS in life and subsequently died between the 1960s and the 1980s and had a postmortem exam. 6% of those people who had a diagnosis of MS during their lifetime didn’t actually have MS.1
In 2012, we did a survey of 122 MS specialists.2 We asked them if they had encountered patients incorrectly assigned a diagnosis of MS in the past year, and 95% of them had seen such a patient. This was not the most scientific study because it was just a survey and subject to recall bias. Still, many of these MS providers recalled having seen three to ten such patients in the last year where they strongly felt that a pre-existing MS diagnosis made by another provider was incorrect.
Another study was recently published by Dr. Kaisey.3 She looked at referrals to two academic MS centers on the West Coast, and specifically new patient referrals over 12 months to those two MS centers. She found that out of 241 patients referred to the two MS centers, almost one in five who was referred with a pre-existing diagnosis of MS who came to these MS centers was subsequently found not have MS. This included 17% of patients at Cedars Sinai and 19% at UCLA. That’s an alarmingly high number of patients. And that’s the best data we have right now.
We don’t know how representative that number is of other MS centers and clinical practice nationally, but the bottom line is that misdiagnosis of MS is, unfortunately, fairly common and there’s a lot of risks to patients associated with misdiagnosis, as well as costs to our health care system.
Can you elaborate on the risks to patients?
We published a study where we looked at records from 110 patients who had been incorrectly assigned a diagnosis of MS. Twenty-three MS specialists from 4 different MS centers participated in this study that was published in Neurology in 2016.4
70% of these patients had been receiving disease modifying therapy for MS, which certainly has risks and side effects associated with it. 24% received disease modifying therapy with a known risk of PML, which is a frequently fatal brain infection associated with these therapies. Approximately 30% of these patients who did not have MS were on disease modifying therapy for in 3 to 9 years and 30% were on disease modifying therapy for 10 years or more.
Dr. Kaisey’s study also supports our findings. She found approximately 110 patient-years of exposure to unnecessary disease modifying therapy in the misdiagnosed patients in her study.3 Patients suffer side effects in addition to unnecessary risk related to these therapies.
It’s also important to emphasize that many of these patients also received inadequate treatment for their correct diagnoses.
How did the 2017 revision to the McDonald criteria address the challenge of MS diagnosis?
The problem of MS misdiagnosis is prominently discussed in the 2017 McDonald criteria.5 Unfortunately, one of the likely causes of misdiagnosis is that many clinicians may not read the full manuscript of the McDonald criteria itself. They instead rely on summary cards or reproductions—condensed versions of how to diagnose MS—which often don’t really provide the full context that would allow physicians to think critically and avoid a misdiagnosis.
MS is still a clinical diagnosis, which means we’re reliant on physician decision-making to confirm a diagnosis of MS. There are multiple steps to it.
First, we must determine if somebody has a syndrome typical for MS and they have objective evidence of a CNS lesion. After that the McDonald criteria can be applied to determine if they meet dissemination in time, dissemination in space, and have no other explanation for this syndrome before making a diagnosis of MS. Each one of those steps is reliant on thoughtful clinical decision-making and may be prone to potential error.
Knowing the ins and outs and the details of each of those steps is important. Reading the diagnostic criteria is probably the first step in becoming skilled at MS diagnosis. It’s important to know the criteria were not developed as a screening tool. The neurologist is essentially the screening tool. The diagnostic criteria can’t be used until a MS-typical syndrome with objective evidence of a CNS lesion is confirmed.
In what way was the previous criteria flawed?
I wouldn’t say any of the MS diagnostic criteria were flawed; they have evolved along with data in our field that has helped us make diagnosis of MS earlier in many patients. When using the criteria, it’s important to understand the types of patients in the cohorts used to validate our diagnostic criteria. They were primarily younger than 50, and usually Caucasian. They had only syndromes typical for MS with objective evidence of CNS damage corroborating these syndromes. Using the criteria more broadly in patients who do not fit this profile can reduce its specificity and lead to misdiagnosis.
For determination of MRI dissemination in time and dissemination space, there are also some misconceptions that frequently lead to misdiagnosis. Knowing which areas are required for dissemination in space in crucial. For example, the optic nerve currently is not an area that can be used to fulfill dissemination in space, which is a mistake people frequently make. Knowing that the terms “juxtacortical” and “periventricular” means touching the ventricle and touching the cortex is very important. This is a mistake that’s often made as well, and many disorders present with MRI lesions near but not touching the cortex or ventricle. Knowing each element of our diagnostic criteria and what those terms specifically mean is important. In the 2017 McDonald criteria there’s an excellent glossary that helps clinicians understand these terms and how to use them appropriately.5
What more needs to be done to prevent MS misdiagnosis?
First, we need to figure out how to better educate clinicians on how to use our diagnostic criteria appropriately.
We recently completed a study that suggests that residents in training, and even MS specialists, have trouble using the diagnostic criteria. This study was presented at the American Academy of Neurology Annual meeting but has not been published yet. Education on how to use the diagnostic criteria, and in which patients to use the criteria (and in which patients the criteria do not apply) is important, particularly when new revisions to the diagnostic criteria are published.
We published a paper recently that provided guidance on how to avoid misdiagnosis using the 2017 McDonald criteria, and how to approach patients where the diagnostic criteria didn’t apply.6 Sometimes additional clinical, laboratory, or MRI evaluation and monitoring is required in such patients to either confirm a diagnosis of MS, or determine that a patient does not have MS.
We also desperately need biomarkers that may help us diagnose MS more accurately in patients who have neurological symptoms and an abnormal MRI and are seeing a neurologist for the first time. There’s research going on now to find relevant biomarkers in the form of blood tests, as well as MRI assessments. In particular, ongoing research focused on the MRI finding we have termed the “central vein sign” suggests this approach may be helpful as a MRI-specific biomarker for MS.7,8 We need multicenter studies evaluating this and other biomarkers in patients who come to our clinics for a new evaluation for MS, to confirm that they are accurate. We need more researchers working on how to improve diagnosis of MS.
References:
1. Engell T. A clinico-pathoanatomical study of multiple sclerosis diagnosis. Acta Neurol Scand. 1988;78(1):39-44.
2. Solomon AJ, Klein EP, Bourdette D. "Undiagnosing" multiple sclerosis: the challenge of misdiagnosis in MS. Neurology. 2012;78(24):1986-1991.
3. Kaisey M, Solomon AJ, Luu M, Giesser BS, Sicotte NL. Incidence of multiple sclerosis misdiagnosis in referrals to two academic centers. Mult Scler Relat Disord. 2019;30:51-56.
4. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
5. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173..
6. Solomon AJ, Naismith RT, Cross AH. Misdiagnosis of multiple sclerosis: impact of the 2017 McDonald criteria on clinical practice. Neurology. 2019;92(1):26-33.
7. Sati P, Oh J, Constable RT, et al; NAIMS Cooperative. The central vein sign and its clinical evaluation for the diagnosis of multiple sclerosis: a consensus statement from the North American Imaging in Multiple Sclerosis Cooperative. Nat Rev Neurol. 2016;12(12):714-722.
8. Sinnecker T, Clarke MA, Meier D, et al. Evaluation of the central vein sign as a diagnostic imaging biomarker in multiple sclerosis [published online ahead of print August 19, 2019]. JAMA Neurology. 2019: doi: 10.1001/jamaneurol.2019.2478.
Andrew Solomon, MD, is a neurologist and Associate Professor in the Department of Neurological Sciences and Division Chief of Multiple Sclerosis at The University of Vermont. We sat down to talk with Dr. Solomon about his experience with multiple sclerosis (MS) misdiagnosis and what can be done to improve MS diagnosis going forward.
How prevalent is the misdiagnosis of MS and what are the effects that it has on patients?
The first thing to clarify is what we mean by MS misdiagnosis. In this case, we’re talking about patients who are incorrectly assigned a diagnosis of MS. MS is hard to diagnose. Sometimes we take too long to diagnose people who have MS, sometimes we incorrectly diagnose MS in people who don’t have it.
We don’t have very good data in terms of how frequent misdiagnosis is, but we have some. The earliest data we have is from the 1980s. In 1988 there was a study published involving approximately 500 patients who had been diagnosed with MS in life and subsequently died between the 1960s and the 1980s and had a postmortem exam. 6% of those people who had a diagnosis of MS during their lifetime didn’t actually have MS.1
In 2012, we did a survey of 122 MS specialists.2 We asked them if they had encountered patients incorrectly assigned a diagnosis of MS in the past year, and 95% of them had seen such a patient. This was not the most scientific study because it was just a survey and subject to recall bias. Still, many of these MS providers recalled having seen three to ten such patients in the last year where they strongly felt that a pre-existing MS diagnosis made by another provider was incorrect.
Another study was recently published by Dr. Kaisey.3 She looked at referrals to two academic MS centers on the West Coast, and specifically new patient referrals over 12 months to those two MS centers. She found that out of 241 patients referred to the two MS centers, almost one in five who was referred with a pre-existing diagnosis of MS who came to these MS centers was subsequently found not have MS. This included 17% of patients at Cedars Sinai and 19% at UCLA. That’s an alarmingly high number of patients. And that’s the best data we have right now.
We don’t know how representative that number is of other MS centers and clinical practice nationally, but the bottom line is that misdiagnosis of MS is, unfortunately, fairly common and there’s a lot of risks to patients associated with misdiagnosis, as well as costs to our health care system.
Can you elaborate on the risks to patients?
We published a study where we looked at records from 110 patients who had been incorrectly assigned a diagnosis of MS. Twenty-three MS specialists from 4 different MS centers participated in this study that was published in Neurology in 2016.4
70% of these patients had been receiving disease modifying therapy for MS, which certainly has risks and side effects associated with it. 24% received disease modifying therapy with a known risk of PML, which is a frequently fatal brain infection associated with these therapies. Approximately 30% of these patients who did not have MS were on disease modifying therapy for in 3 to 9 years and 30% were on disease modifying therapy for 10 years or more.
Dr. Kaisey’s study also supports our findings. She found approximately 110 patient-years of exposure to unnecessary disease modifying therapy in the misdiagnosed patients in her study.3 Patients suffer side effects in addition to unnecessary risk related to these therapies.
It’s also important to emphasize that many of these patients also received inadequate treatment for their correct diagnoses.
How did the 2017 revision to the McDonald criteria address the challenge of MS diagnosis?
The problem of MS misdiagnosis is prominently discussed in the 2017 McDonald criteria.5 Unfortunately, one of the likely causes of misdiagnosis is that many clinicians may not read the full manuscript of the McDonald criteria itself. They instead rely on summary cards or reproductions—condensed versions of how to diagnose MS—which often don’t really provide the full context that would allow physicians to think critically and avoid a misdiagnosis.
MS is still a clinical diagnosis, which means we’re reliant on physician decision-making to confirm a diagnosis of MS. There are multiple steps to it.
First, we must determine if somebody has a syndrome typical for MS and they have objective evidence of a CNS lesion. After that the McDonald criteria can be applied to determine if they meet dissemination in time, dissemination in space, and have no other explanation for this syndrome before making a diagnosis of MS. Each one of those steps is reliant on thoughtful clinical decision-making and may be prone to potential error.
Knowing the ins and outs and the details of each of those steps is important. Reading the diagnostic criteria is probably the first step in becoming skilled at MS diagnosis. It’s important to know the criteria were not developed as a screening tool. The neurologist is essentially the screening tool. The diagnostic criteria can’t be used until a MS-typical syndrome with objective evidence of a CNS lesion is confirmed.
In what way was the previous criteria flawed?
I wouldn’t say any of the MS diagnostic criteria were flawed; they have evolved along with data in our field that has helped us make diagnosis of MS earlier in many patients. When using the criteria, it’s important to understand the types of patients in the cohorts used to validate our diagnostic criteria. They were primarily younger than 50, and usually Caucasian. They had only syndromes typical for MS with objective evidence of CNS damage corroborating these syndromes. Using the criteria more broadly in patients who do not fit this profile can reduce its specificity and lead to misdiagnosis.
For determination of MRI dissemination in time and dissemination space, there are also some misconceptions that frequently lead to misdiagnosis. Knowing which areas are required for dissemination in space in crucial. For example, the optic nerve currently is not an area that can be used to fulfill dissemination in space, which is a mistake people frequently make. Knowing that the terms “juxtacortical” and “periventricular” means touching the ventricle and touching the cortex is very important. This is a mistake that’s often made as well, and many disorders present with MRI lesions near but not touching the cortex or ventricle. Knowing each element of our diagnostic criteria and what those terms specifically mean is important. In the 2017 McDonald criteria there’s an excellent glossary that helps clinicians understand these terms and how to use them appropriately.5
What more needs to be done to prevent MS misdiagnosis?
First, we need to figure out how to better educate clinicians on how to use our diagnostic criteria appropriately.
We recently completed a study that suggests that residents in training, and even MS specialists, have trouble using the diagnostic criteria. This study was presented at the American Academy of Neurology Annual meeting but has not been published yet. Education on how to use the diagnostic criteria, and in which patients to use the criteria (and in which patients the criteria do not apply) is important, particularly when new revisions to the diagnostic criteria are published.
We published a paper recently that provided guidance on how to avoid misdiagnosis using the 2017 McDonald criteria, and how to approach patients where the diagnostic criteria didn’t apply.6 Sometimes additional clinical, laboratory, or MRI evaluation and monitoring is required in such patients to either confirm a diagnosis of MS, or determine that a patient does not have MS.
We also desperately need biomarkers that may help us diagnose MS more accurately in patients who have neurological symptoms and an abnormal MRI and are seeing a neurologist for the first time. There’s research going on now to find relevant biomarkers in the form of blood tests, as well as MRI assessments. In particular, ongoing research focused on the MRI finding we have termed the “central vein sign” suggests this approach may be helpful as a MRI-specific biomarker for MS.7,8 We need multicenter studies evaluating this and other biomarkers in patients who come to our clinics for a new evaluation for MS, to confirm that they are accurate. We need more researchers working on how to improve diagnosis of MS.
References:
1. Engell T. A clinico-pathoanatomical study of multiple sclerosis diagnosis. Acta Neurol Scand. 1988;78(1):39-44.
2. Solomon AJ, Klein EP, Bourdette D. "Undiagnosing" multiple sclerosis: the challenge of misdiagnosis in MS. Neurology. 2012;78(24):1986-1991.
3. Kaisey M, Solomon AJ, Luu M, Giesser BS, Sicotte NL. Incidence of multiple sclerosis misdiagnosis in referrals to two academic centers. Mult Scler Relat Disord. 2019;30:51-56.
4. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
5. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173..
6. Solomon AJ, Naismith RT, Cross AH. Misdiagnosis of multiple sclerosis: impact of the 2017 McDonald criteria on clinical practice. Neurology. 2019;92(1):26-33.
7. Sati P, Oh J, Constable RT, et al; NAIMS Cooperative. The central vein sign and its clinical evaluation for the diagnosis of multiple sclerosis: a consensus statement from the North American Imaging in Multiple Sclerosis Cooperative. Nat Rev Neurol. 2016;12(12):714-722.
8. Sinnecker T, Clarke MA, Meier D, et al. Evaluation of the central vein sign as a diagnostic imaging biomarker in multiple sclerosis [published online ahead of print August 19, 2019]. JAMA Neurology. 2019: doi: 10.1001/jamaneurol.2019.2478.
Andrew Solomon, MD, is a neurologist and Associate Professor in the Department of Neurological Sciences and Division Chief of Multiple Sclerosis at The University of Vermont. We sat down to talk with Dr. Solomon about his experience with multiple sclerosis (MS) misdiagnosis and what can be done to improve MS diagnosis going forward.
How prevalent is the misdiagnosis of MS and what are the effects that it has on patients?
The first thing to clarify is what we mean by MS misdiagnosis. In this case, we’re talking about patients who are incorrectly assigned a diagnosis of MS. MS is hard to diagnose. Sometimes we take too long to diagnose people who have MS, sometimes we incorrectly diagnose MS in people who don’t have it.
We don’t have very good data in terms of how frequent misdiagnosis is, but we have some. The earliest data we have is from the 1980s. In 1988 there was a study published involving approximately 500 patients who had been diagnosed with MS in life and subsequently died between the 1960s and the 1980s and had a postmortem exam. 6% of those people who had a diagnosis of MS during their lifetime didn’t actually have MS.1
In 2012, we did a survey of 122 MS specialists.2 We asked them if they had encountered patients incorrectly assigned a diagnosis of MS in the past year, and 95% of them had seen such a patient. This was not the most scientific study because it was just a survey and subject to recall bias. Still, many of these MS providers recalled having seen three to ten such patients in the last year where they strongly felt that a pre-existing MS diagnosis made by another provider was incorrect.
Another study was recently published by Dr. Kaisey.3 She looked at referrals to two academic MS centers on the West Coast, and specifically new patient referrals over 12 months to those two MS centers. She found that out of 241 patients referred to the two MS centers, almost one in five who was referred with a pre-existing diagnosis of MS who came to these MS centers was subsequently found not have MS. This included 17% of patients at Cedars Sinai and 19% at UCLA. That’s an alarmingly high number of patients. And that’s the best data we have right now.
We don’t know how representative that number is of other MS centers and clinical practice nationally, but the bottom line is that misdiagnosis of MS is, unfortunately, fairly common and there’s a lot of risks to patients associated with misdiagnosis, as well as costs to our health care system.
Can you elaborate on the risks to patients?
We published a study where we looked at records from 110 patients who had been incorrectly assigned a diagnosis of MS. Twenty-three MS specialists from 4 different MS centers participated in this study that was published in Neurology in 2016.4
70% of these patients had been receiving disease modifying therapy for MS, which certainly has risks and side effects associated with it. 24% received disease modifying therapy with a known risk of PML, which is a frequently fatal brain infection associated with these therapies. Approximately 30% of these patients who did not have MS were on disease modifying therapy for in 3 to 9 years and 30% were on disease modifying therapy for 10 years or more.
Dr. Kaisey’s study also supports our findings. She found approximately 110 patient-years of exposure to unnecessary disease modifying therapy in the misdiagnosed patients in her study.3 Patients suffer side effects in addition to unnecessary risk related to these therapies.
It’s also important to emphasize that many of these patients also received inadequate treatment for their correct diagnoses.
How did the 2017 revision to the McDonald criteria address the challenge of MS diagnosis?
The problem of MS misdiagnosis is prominently discussed in the 2017 McDonald criteria.5 Unfortunately, one of the likely causes of misdiagnosis is that many clinicians may not read the full manuscript of the McDonald criteria itself. They instead rely on summary cards or reproductions—condensed versions of how to diagnose MS—which often don’t really provide the full context that would allow physicians to think critically and avoid a misdiagnosis.
MS is still a clinical diagnosis, which means we’re reliant on physician decision-making to confirm a diagnosis of MS. There are multiple steps to it.
First, we must determine if somebody has a syndrome typical for MS and they have objective evidence of a CNS lesion. After that the McDonald criteria can be applied to determine if they meet dissemination in time, dissemination in space, and have no other explanation for this syndrome before making a diagnosis of MS. Each one of those steps is reliant on thoughtful clinical decision-making and may be prone to potential error.
Knowing the ins and outs and the details of each of those steps is important. Reading the diagnostic criteria is probably the first step in becoming skilled at MS diagnosis. It’s important to know the criteria were not developed as a screening tool. The neurologist is essentially the screening tool. The diagnostic criteria can’t be used until a MS-typical syndrome with objective evidence of a CNS lesion is confirmed.
In what way was the previous criteria flawed?
I wouldn’t say any of the MS diagnostic criteria were flawed; they have evolved along with data in our field that has helped us make diagnosis of MS earlier in many patients. When using the criteria, it’s important to understand the types of patients in the cohorts used to validate our diagnostic criteria. They were primarily younger than 50, and usually Caucasian. They had only syndromes typical for MS with objective evidence of CNS damage corroborating these syndromes. Using the criteria more broadly in patients who do not fit this profile can reduce its specificity and lead to misdiagnosis.
For determination of MRI dissemination in time and dissemination space, there are also some misconceptions that frequently lead to misdiagnosis. Knowing which areas are required for dissemination in space in crucial. For example, the optic nerve currently is not an area that can be used to fulfill dissemination in space, which is a mistake people frequently make. Knowing that the terms “juxtacortical” and “periventricular” means touching the ventricle and touching the cortex is very important. This is a mistake that’s often made as well, and many disorders present with MRI lesions near but not touching the cortex or ventricle. Knowing each element of our diagnostic criteria and what those terms specifically mean is important. In the 2017 McDonald criteria there’s an excellent glossary that helps clinicians understand these terms and how to use them appropriately.5
What more needs to be done to prevent MS misdiagnosis?
First, we need to figure out how to better educate clinicians on how to use our diagnostic criteria appropriately.
We recently completed a study that suggests that residents in training, and even MS specialists, have trouble using the diagnostic criteria. This study was presented at the American Academy of Neurology Annual meeting but has not been published yet. Education on how to use the diagnostic criteria, and in which patients to use the criteria (and in which patients the criteria do not apply) is important, particularly when new revisions to the diagnostic criteria are published.
We published a paper recently that provided guidance on how to avoid misdiagnosis using the 2017 McDonald criteria, and how to approach patients where the diagnostic criteria didn’t apply.6 Sometimes additional clinical, laboratory, or MRI evaluation and monitoring is required in such patients to either confirm a diagnosis of MS, or determine that a patient does not have MS.
We also desperately need biomarkers that may help us diagnose MS more accurately in patients who have neurological symptoms and an abnormal MRI and are seeing a neurologist for the first time. There’s research going on now to find relevant biomarkers in the form of blood tests, as well as MRI assessments. In particular, ongoing research focused on the MRI finding we have termed the “central vein sign” suggests this approach may be helpful as a MRI-specific biomarker for MS.7,8 We need multicenter studies evaluating this and other biomarkers in patients who come to our clinics for a new evaluation for MS, to confirm that they are accurate. We need more researchers working on how to improve diagnosis of MS.
References:
1. Engell T. A clinico-pathoanatomical study of multiple sclerosis diagnosis. Acta Neurol Scand. 1988;78(1):39-44.
2. Solomon AJ, Klein EP, Bourdette D. "Undiagnosing" multiple sclerosis: the challenge of misdiagnosis in MS. Neurology. 2012;78(24):1986-1991.
3. Kaisey M, Solomon AJ, Luu M, Giesser BS, Sicotte NL. Incidence of multiple sclerosis misdiagnosis in referrals to two academic centers. Mult Scler Relat Disord. 2019;30:51-56.
4. Solomon AJ, Bourdette DN, Cross AH, et al. The contemporary spectrum of multiple sclerosis misdiagnosis: a multicenter study. Neurology. 2016;87(13):1393-1399.
5. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173..
6. Solomon AJ, Naismith RT, Cross AH. Misdiagnosis of multiple sclerosis: impact of the 2017 McDonald criteria on clinical practice. Neurology. 2019;92(1):26-33.
7. Sati P, Oh J, Constable RT, et al; NAIMS Cooperative. The central vein sign and its clinical evaluation for the diagnosis of multiple sclerosis: a consensus statement from the North American Imaging in Multiple Sclerosis Cooperative. Nat Rev Neurol. 2016;12(12):714-722.
8. Sinnecker T, Clarke MA, Meier D, et al. Evaluation of the central vein sign as a diagnostic imaging biomarker in multiple sclerosis [published online ahead of print August 19, 2019]. JAMA Neurology. 2019: doi: 10.1001/jamaneurol.2019.2478.

Postinflammatory Hyperpigmentation Following Treatment of Hyperkeratosis Lenticularis Perstans With Tazarotene Cream 0.1%
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.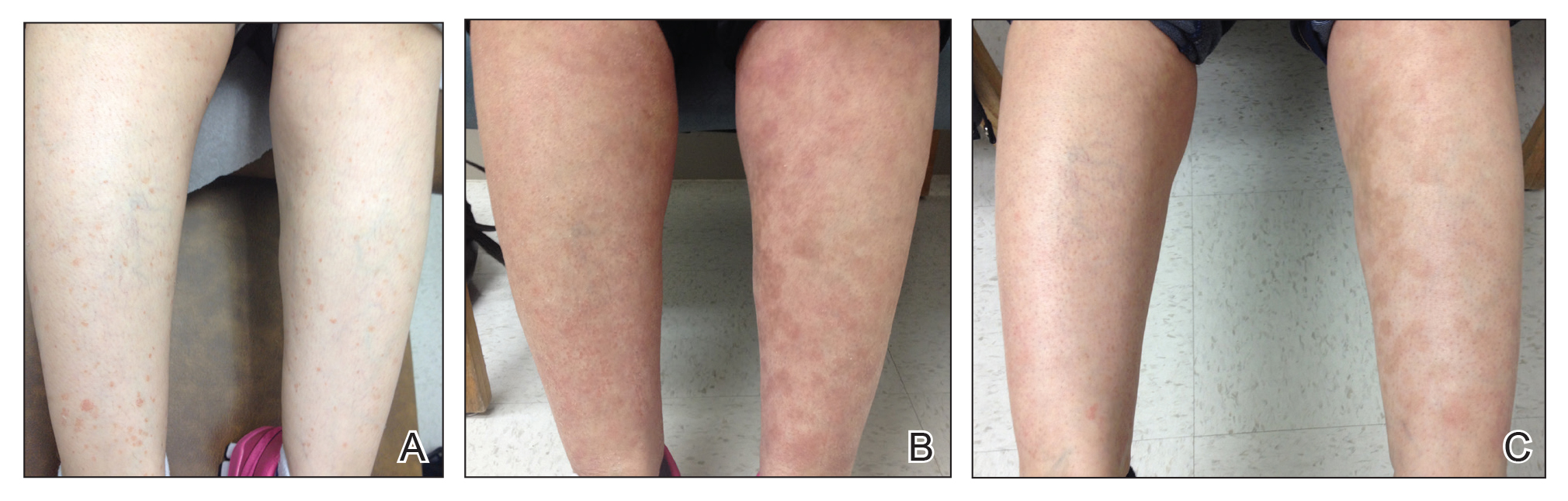
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
To the Editor:
Hyperkeratosis lenticularis perstans (HLP), or Flegel disease, is a rare keratinization disorder characterized by asymptomatic, red-brown, 1- to 5-mm papules with irregular horny scales commonly seen on the dorsal feet and lower legs.1 Hyperkeratosis lenticularis perstans is notorious for being difficult to treat. Various treatment options, including 5-fluorouracil, topical and oral retinoids, vitamin D3 derivatives, psoralen plus UVA therapy, and dermabrasion, have been explored but none have proven to be consistently effective.
A woman in her 50s presented with an asymptomatic eruption on the legs and thighs that had been present for the last 20 years. She had been misdiagnosed by multiple outside providers with atopic dermatitis and was treated with topical steroids without considerable improvement. Upon initial presentation to our clinic , physical examination revealed a woman with Fitzpatrick skin type II with multiple hyperpigmented, red-brown, 2- to 6-mm papules on the extensor surfaces of the lower legs and upper thighs (Figure, A). A 3-mm punch biopsy of a lesion on the right upper thigh revealed hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate. The clinical and histopathologic findings were consistent with HLP.
The patient was started on treatment with 5-fluorouracil cream on the right leg and tazarotene cream 0.1% on the left leg to determine which agent would work best. After 9 weeks of treatment, slight improvement was observed on both legs, but the lesions were still erythematous (Figure, B). Treatment was continued, and after 14 weeks complete resolution of the lesions was noted on both legs; however, postinflammatory hyperpigmentation (PIH) was observed on the left leg, which had been treated with tazarotene (Figure, C). The patient was lost to follow-up prior to treatment of the PIH.
Postinflammatory hyperpigmentation is an acquired excess of pigment due to a prior disease process such as an infection, allergic reaction, trauma, inflammatory disease, or drug reaction. In our patient, this finding was unusual because tazarotene has been shown to be an effective treatment of PIH.2,3
In PIH, there is either abnormal production or distribution of melanin pigment in the epidermis and/or dermis. Several mechanisms for PIH have been suggested. One potential mechanism is disruption of the basal cell layer due to dermal lymphocytic inflammation, causing melanin to be released and trapped by macrophages present in the dermal papillae. Another possible mechanism is epidermal hypermelanosis, in which the release and oxidation of arachidonic acid to prostaglandins and leukotrienes alters immune cells and melanocytes, causing an increase in melanin and increased transfer of melanin to keratinocytes in the surrounding epidermis.4
Treatment of PIH can be a difficult and prolonged process, especially when a dermal rather than epidermal melanosis is observed. Topical retinoids, topical hydroquinone, azelaic acid, corticosteroids, tretinoin cream, glycolic acid, and trichloroacetic acid have been shown to be effective in treating epidermal PIH. Tazarotene is a synthetic retinoid that has been proven to be an effective treatment of PIH3; however, in our patient the PIH progressed with treatment. One plausible explanation is that irritation caused by the medication led to further PIH.2,5
It is uncommon for tazarotene to cause PIH. Hyperpigmentation is listed as an adverse effect observed during the postmarketing experience according to one manufacturer6 and the US Food and Drug Administration; however, details about prior incidents of hyperpigmentation have not been reported in the literature. Our case is unique because both treatments showed considerable improvement in HLP, but more PIH was observed on the tazarotene-treated leg.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
- Bean SF. Hyperkeratosis lenticularis perstans. a clinical, histopathologic, and genetic study. Arch Dermatol. 1969;99:705-709.
- Callender V, St. Surin-Lord S, Davis E, et al. Postinflammatory hyperpigmentation: etiologic and therapeutic considerations. Am J Clin Dermatol. 2011;12:87-99.
- McEvoy G. Tazarotene (topical). In: AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, Inc; 2014:84-92.
- Lacz N, Vafaie J, Kihiczak N, et al. Postinflammatory hyperpigmentation: a common but troubling condition. Int J Dermatol. 2004;43:362-365.
- Tazorac (tazarotene) cream [package insert]. Irvine, CA: Allergan, Inc; 2013.
- Tazorac (tazarotene) gel [package insert]. Irvine, CA: Allergan, Inc; 2014.
Practice Points
- Hyperkeratosis lenticularis perstans is a rare keratinization disorder that presents with asymptomatic red-brown papules with irregular horny scales on the lower extremities.
- Hyperkeratosis lenticularis perstans can be difficult to diagnose and treat. Hematoxylin and eosin staining generally will show hyperkeratosis and parakeratosis with basal layer degeneration and a perivascular lymphocytic infiltrate.
- Tazarotene cream 0.1% is a synthetic retinoid sometimes used for treatment of hyperpigmentation, but it also can cause postinflammatory hyperpigmentation.
Fox Chase faculty receive grants for cancer research, education
Faculty members at Fox Chase Cancer Center have received grants to promote education about liver cancer, study pancreatic and breast cancer, and examine burnout among physician assistants (PAs).

Eric D. Tetzlaff, a PA at Fox Chase in Philadelphia, has received a 3-year grant from the Association of Physician Assistants in Oncology. With this $15,000 grant, Mr. Tetzlaff plans to conduct a longitudinal study that will explore burnout among PAs working in oncology.
The goals of his study are to “understand the impact of the attitudes of oncology PAs regarding teamwork, expectations for their professional role, type of collaborative practice, organizational context of the job environment, and moral distress, on burnout and career satisfaction,” according to Fox Chase.
Jaye Gardiner, PhD, a postdoctoral researcher in the Edna Cukierman laboratory at Fox Chase, has received a $163,500 grant from the American Cancer Society. With this grant, Dr. Gardiner will investigate the role of tumor stroma in pancreatic cancer.
Dr. Gardiner plans to explore how cancer-associated fibroblasts in the pancreatic stroma “communicate with one another and how this communication is altered in tumor-promoting versus tumor-restricting conditions,” according to Fox Chase.
Dietmar J. Kappes, PhD, a professor of blood cell development and cancer and director of the Transgenic Mouse Facility at Fox Chase, has received a 5-year grant from the National Institutes of Health. With this $626,072 grant, Dr. Kappes will investigate the role of the transcription factor ThPOK in breast cancer.
Dr. Kappes and colleagues previously found a link between high cytoplasmic levels of ThPOK and poor outcomes in breast cancer. Now, Dr. Kappes plans to “further elucidate the role of ThPOK in breast cancer by combining novel animal models and molecular approaches,” according to Fox Chase.
Evelyn González, senior director of the Fox Chase’s Office of Community Outreach, and Shannon Lynch, PhD, who is with the Cancer Prevention and Control program, have received a 2-year grant from the Pennsylvania Department of Human Services.
The pair will use this $125,000 grant to provide liver cancer and hepatitis education to communities in the Philadelphia area with the greatest burden of liver cancer and related risk factors. Dr. Lynch will find these at-risk communities, and the Office of Community Outreach will work with partner groups in those areas to provide bilingual education about hepatitis and how it relates to liver cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
Faculty members at Fox Chase Cancer Center have received grants to promote education about liver cancer, study pancreatic and breast cancer, and examine burnout among physician assistants (PAs).
Eric D. Tetzlaff, a PA at Fox Chase in Philadelphia, has received a 3-year grant from the Association of Physician Assistants in Oncology. With this $15,000 grant, Mr. Tetzlaff plans to conduct a longitudinal study that will explore burnout among PAs working in oncology.
The goals of his study are to “understand the impact of the attitudes of oncology PAs regarding teamwork, expectations for their professional role, type of collaborative practice, organizational context of the job environment, and moral distress, on burnout and career satisfaction,” according to Fox Chase.
Jaye Gardiner, PhD, a postdoctoral researcher in the Edna Cukierman laboratory at Fox Chase, has received a $163,500 grant from the American Cancer Society. With this grant, Dr. Gardiner will investigate the role of tumor stroma in pancreatic cancer.
Dr. Gardiner plans to explore how cancer-associated fibroblasts in the pancreatic stroma “communicate with one another and how this communication is altered in tumor-promoting versus tumor-restricting conditions,” according to Fox Chase.
Dietmar J. Kappes, PhD, a professor of blood cell development and cancer and director of the Transgenic Mouse Facility at Fox Chase, has received a 5-year grant from the National Institutes of Health. With this $626,072 grant, Dr. Kappes will investigate the role of the transcription factor ThPOK in breast cancer.
Dr. Kappes and colleagues previously found a link between high cytoplasmic levels of ThPOK and poor outcomes in breast cancer. Now, Dr. Kappes plans to “further elucidate the role of ThPOK in breast cancer by combining novel animal models and molecular approaches,” according to Fox Chase.
Evelyn González, senior director of the Fox Chase’s Office of Community Outreach, and Shannon Lynch, PhD, who is with the Cancer Prevention and Control program, have received a 2-year grant from the Pennsylvania Department of Human Services.
The pair will use this $125,000 grant to provide liver cancer and hepatitis education to communities in the Philadelphia area with the greatest burden of liver cancer and related risk factors. Dr. Lynch will find these at-risk communities, and the Office of Community Outreach will work with partner groups in those areas to provide bilingual education about hepatitis and how it relates to liver cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
Faculty members at Fox Chase Cancer Center have received grants to promote education about liver cancer, study pancreatic and breast cancer, and examine burnout among physician assistants (PAs).
Eric D. Tetzlaff, a PA at Fox Chase in Philadelphia, has received a 3-year grant from the Association of Physician Assistants in Oncology. With this $15,000 grant, Mr. Tetzlaff plans to conduct a longitudinal study that will explore burnout among PAs working in oncology.
The goals of his study are to “understand the impact of the attitudes of oncology PAs regarding teamwork, expectations for their professional role, type of collaborative practice, organizational context of the job environment, and moral distress, on burnout and career satisfaction,” according to Fox Chase.
Jaye Gardiner, PhD, a postdoctoral researcher in the Edna Cukierman laboratory at Fox Chase, has received a $163,500 grant from the American Cancer Society. With this grant, Dr. Gardiner will investigate the role of tumor stroma in pancreatic cancer.
Dr. Gardiner plans to explore how cancer-associated fibroblasts in the pancreatic stroma “communicate with one another and how this communication is altered in tumor-promoting versus tumor-restricting conditions,” according to Fox Chase.
Dietmar J. Kappes, PhD, a professor of blood cell development and cancer and director of the Transgenic Mouse Facility at Fox Chase, has received a 5-year grant from the National Institutes of Health. With this $626,072 grant, Dr. Kappes will investigate the role of the transcription factor ThPOK in breast cancer.
Dr. Kappes and colleagues previously found a link between high cytoplasmic levels of ThPOK and poor outcomes in breast cancer. Now, Dr. Kappes plans to “further elucidate the role of ThPOK in breast cancer by combining novel animal models and molecular approaches,” according to Fox Chase.
Evelyn González, senior director of the Fox Chase’s Office of Community Outreach, and Shannon Lynch, PhD, who is with the Cancer Prevention and Control program, have received a 2-year grant from the Pennsylvania Department of Human Services.
The pair will use this $125,000 grant to provide liver cancer and hepatitis education to communities in the Philadelphia area with the greatest burden of liver cancer and related risk factors. Dr. Lynch will find these at-risk communities, and the Office of Community Outreach will work with partner groups in those areas to provide bilingual education about hepatitis and how it relates to liver cancer.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at hematologynews@mdedge.com, and you could be featured in Movers in Medicine.
Chronic hypertension in pregnancy increased 13-fold since 1970
The rate of chronic hypertension during pregnancy has increased significantly in the United States since 1970 and is more common in older women and in black women, according to a population-based, cross-sectional analysis.

Researchers analyzed data from more than 151 million women with delivery-related hospitalizations in the United States between 1970 and 2010 and found that the rate of chronic hypertension in pregnancy increased steadily over time from 1970 to 1990, plateaued from 1990 to 2000, then increased again to 2010.
The analysis revealed an average annual increase of 6% – which was higher among white women than among black women – and an overall 13-fold increase from 1970 to 2010. These increases appeared to be independent of rates of obesity and smoking. The findings were published in Hypertension.
The rates of chronic hypertension also increased with maternal age, among both black and white women.
“The strong association between age and rates of chronic hypertension underscores the potential for both biological and social determinants of health to influence risk,” wrote Cande V. Ananth, PhD, from the Rutgers University, New Brunswick, N.J., and coauthors. “The period effect in chronic hypertension in pregnancy is thus largely a product of the age effect and the increasing mean age at first birth in the U.S.”
The overall prevalence of chronic hypertension in pregnancy was 0.63%, but was twofold higher in black women, compared with white women (1.24% vs. 0.53%). The authors noted that black women experienced disproportionally higher rates of ischemic placental disease, pregestational and gestational diabetes, preterm delivery and perinatal mortality, which may be a consequences of higher rates of obesity, social disadvantage, smoking, and less access to care.
“This disparity may also be related to the higher tendency of black women to develop vascular disease at an earlier age than white women, which may also explain why the age-associated increase in chronic hypertension among black women is relatively smaller than white women,” they wrote. “The persistent race disparity in chronic hypertension is also a cause for continued concern and underscores the role of complex population dynamics that shape risks.”
This was the largest study to evaluate changes in the prevalence of chronic hypertension in pregnancy over time and particularly how the prevalence is influenced by age, period, and birth cohort.
In regard to the 13-fold increase from 1970 to 2010, the researchers suggested that changing diagnostic criteria for hypertension, as well as earlier access to prenatal care, may have played a part. For example, the American College of Cardiology recently modified their guidelines to include patients with systolic and diastolic blood pressures of 130-139 mm Hg and 80-89 mm Hg as stage 1 hypertension, which they noted would increase the prevalence rates of chronic hypertension during pregnancy.
The researchers reported having no outside funding and no conflicts of interest.
SOURCE: Ananth CV et al. Hypertension. 2019 Sept 9. doi: 10.1161/HYPERTENSIONAHA.119.12968.
The rate of chronic hypertension during pregnancy has increased significantly in the United States since 1970 and is more common in older women and in black women, according to a population-based, cross-sectional analysis.
Researchers analyzed data from more than 151 million women with delivery-related hospitalizations in the United States between 1970 and 2010 and found that the rate of chronic hypertension in pregnancy increased steadily over time from 1970 to 1990, plateaued from 1990 to 2000, then increased again to 2010.
The analysis revealed an average annual increase of 6% – which was higher among white women than among black women – and an overall 13-fold increase from 1970 to 2010. These increases appeared to be independent of rates of obesity and smoking. The findings were published in Hypertension.
The rates of chronic hypertension also increased with maternal age, among both black and white women.
“The strong association between age and rates of chronic hypertension underscores the potential for both biological and social determinants of health to influence risk,” wrote Cande V. Ananth, PhD, from the Rutgers University, New Brunswick, N.J., and coauthors. “The period effect in chronic hypertension in pregnancy is thus largely a product of the age effect and the increasing mean age at first birth in the U.S.”
The overall prevalence of chronic hypertension in pregnancy was 0.63%, but was twofold higher in black women, compared with white women (1.24% vs. 0.53%). The authors noted that black women experienced disproportionally higher rates of ischemic placental disease, pregestational and gestational diabetes, preterm delivery and perinatal mortality, which may be a consequences of higher rates of obesity, social disadvantage, smoking, and less access to care.
“This disparity may also be related to the higher tendency of black women to develop vascular disease at an earlier age than white women, which may also explain why the age-associated increase in chronic hypertension among black women is relatively smaller than white women,” they wrote. “The persistent race disparity in chronic hypertension is also a cause for continued concern and underscores the role of complex population dynamics that shape risks.”
This was the largest study to evaluate changes in the prevalence of chronic hypertension in pregnancy over time and particularly how the prevalence is influenced by age, period, and birth cohort.
In regard to the 13-fold increase from 1970 to 2010, the researchers suggested that changing diagnostic criteria for hypertension, as well as earlier access to prenatal care, may have played a part. For example, the American College of Cardiology recently modified their guidelines to include patients with systolic and diastolic blood pressures of 130-139 mm Hg and 80-89 mm Hg as stage 1 hypertension, which they noted would increase the prevalence rates of chronic hypertension during pregnancy.
The researchers reported having no outside funding and no conflicts of interest.
SOURCE: Ananth CV et al. Hypertension. 2019 Sept 9. doi: 10.1161/HYPERTENSIONAHA.119.12968.
The rate of chronic hypertension during pregnancy has increased significantly in the United States since 1970 and is more common in older women and in black women, according to a population-based, cross-sectional analysis.
Researchers analyzed data from more than 151 million women with delivery-related hospitalizations in the United States between 1970 and 2010 and found that the rate of chronic hypertension in pregnancy increased steadily over time from 1970 to 1990, plateaued from 1990 to 2000, then increased again to 2010.
The analysis revealed an average annual increase of 6% – which was higher among white women than among black women – and an overall 13-fold increase from 1970 to 2010. These increases appeared to be independent of rates of obesity and smoking. The findings were published in Hypertension.
The rates of chronic hypertension also increased with maternal age, among both black and white women.
“The strong association between age and rates of chronic hypertension underscores the potential for both biological and social determinants of health to influence risk,” wrote Cande V. Ananth, PhD, from the Rutgers University, New Brunswick, N.J., and coauthors. “The period effect in chronic hypertension in pregnancy is thus largely a product of the age effect and the increasing mean age at first birth in the U.S.”
The overall prevalence of chronic hypertension in pregnancy was 0.63%, but was twofold higher in black women, compared with white women (1.24% vs. 0.53%). The authors noted that black women experienced disproportionally higher rates of ischemic placental disease, pregestational and gestational diabetes, preterm delivery and perinatal mortality, which may be a consequences of higher rates of obesity, social disadvantage, smoking, and less access to care.
“This disparity may also be related to the higher tendency of black women to develop vascular disease at an earlier age than white women, which may also explain why the age-associated increase in chronic hypertension among black women is relatively smaller than white women,” they wrote. “The persistent race disparity in chronic hypertension is also a cause for continued concern and underscores the role of complex population dynamics that shape risks.”
This was the largest study to evaluate changes in the prevalence of chronic hypertension in pregnancy over time and particularly how the prevalence is influenced by age, period, and birth cohort.
In regard to the 13-fold increase from 1970 to 2010, the researchers suggested that changing diagnostic criteria for hypertension, as well as earlier access to prenatal care, may have played a part. For example, the American College of Cardiology recently modified their guidelines to include patients with systolic and diastolic blood pressures of 130-139 mm Hg and 80-89 mm Hg as stage 1 hypertension, which they noted would increase the prevalence rates of chronic hypertension during pregnancy.
The researchers reported having no outside funding and no conflicts of interest.
SOURCE: Ananth CV et al. Hypertension. 2019 Sept 9. doi: 10.1161/HYPERTENSIONAHA.119.12968.
FROM HYPERTENSION

Abstracts Presented at the 2019 AVAHO Annual Meeting (Digital Edition)


Can we eradicate malaria by 2050?
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
A new report by members of the Lancet Commission on Malaria Eradication has called for ending malaria in Africa within a generation, specifically aiming at the year 2050.
The Lancet Commission on Malaria Eradication is a joint endeavor between The Lancet and the University of California, San Francisco, and was convened in 2017 to consider the feasibility and affordability of malaria eradication, as well as to identify priority actions for the achievement of the goal. Eradication was considered “a necessary one given the never-ending struggle against drug and insecticide resistance and the social and economic costs associated with a failure to eradicate.”
Between 2000 and 2017, the worldwide annual incidence of malaria declined by 36%, and the annual death rate declined by 60%, according to the report. In 2007, Bill and Melinda Gates proposed that controlling malaria was not enough and complete eradication was the only scientifically and ethically defensible objective. This goal was adopted by the World Health Organization and other interested parties, and by 2015, global strategies and a potential timeline for eradication were developed.
“Global progress has stalled since 2015 and the malaria community is now at a critical moment, faced with a decision to either temper its ambitions as it did in 1969 or recommit to an eradication goal,” according to the report.
In the report, the authors used new modeling analysis to estimate plausible scenarios for the distribution and intensity of malaria in 2030 and 2050. Socioeconomic and environmental trends, together with enhanced access to high-quality diagnosis, treatment, and vector control, could lead to a “world largely free of malaria” by 2050, but with pockets of low-level transmission persisting across a belt of Africa.
Current statistics lend weight to the promise of eventual eradication, according to the report.
Between 2000 and 2017, 20 countries – constituting about one-fifth of the 106 malaria-endemic countries in 2000 – eliminated malaria transmission within their borders, reporting zero indigenous malaria cases for at least 1 year. However, this was counterbalanced by the fact that between 2015 and 2017, 55 countries had an increase in cases, and 38 countries had an increase in deaths.
“The good news is that 38 countries had incidences of fewer than ten cases per 1,000 population in 2017, with 25 countries reporting fewer than one case per 1,000 population. The same 38 countries reported just 5% of total malaria deaths. Nearly all of these low-burden countries are actively working towards national and regional elimination goals of 2030 or earlier,” according to the report.
The analysis undertaken for the report consisted of the following four steps:
1. Development of a machine-learning model to capture associations between malaria endemicity data and a wide range of socioeconomic and environmental geospatial covariates.
2. Mapping of covariate estimates to the years 2030 and 2050 on the basis of projected global trends.
3. Application of the associations learned in the first step to projected covariates generated in the second step to estimate the possible future global landscape of malaria endemicity.
4. Use of a mathematical transmission model to explore the potential effect of differing levels of malaria interventions.
The report indicates that an annual spending of $6 billion or more is required, while the current global expenditure is approximately $4.3 billion. An additional investment of $2 billion per year is necessary, with a quarter of the funds coming from increased development assistance from external donors and the rest from government health spending in malaria-endemic countries, according to the report.
However, other areas of concern remain, including the current lack of effective and widely deployable outdoor biting technologies, though these are expected to be available within the next decade, according to the report.
In terms of the modeling used in the report, the authors noted that past performance does not “capture the effect of mass drug administration or mass chemoprevention because these interventions are either relatively new or have yet to be applied widely. These underestimates might be counteracted by the absence of drug or insecticide resistance from our projections,which result in overly optimistic estimates for the continued efficacy of current tools.”
The commission was launched in October 2017 by the Global Health Group at the University of California, San Francisco. The commission built on the 2010 Lancet Malaria Elimination Series, “which evaluated the operational, technical, and financial requirements for malaria elimination and helped shape and build early support for the eradication agenda,” according to the report.
SOURCE: Feachem RGA et al. Lancet. 2019 Sept 8. doi: 10.1016/S0140-6736(19)31139-0.
FROM THE LANCET
Chemotherapy may raise CVD risk in pediatric cancer survivors
Pediatric cancer survivors have a higher likelihood of experiencing a cardiac event, developing diabetes, or having hypertension at a median 10-year follow-up, according to results from a recent research letter published in Circulation.
Ashna Khanna of the University of Toronto and colleagues identified 7,289 pediatric patients in the Pediatric Oncology Group of Ontario Networked Information System who were diagnosed with cancer at median age of 7 years old, who were treated between 1987 and 2010, and who were cancer survivors for 5 years. Each patient was matched to five cancer-free control subjects who were a median of 24 years old at the 10-year follow-up (36,205 cancer-free individuals). The researchers studied whether pediatric cancer survivors experienced cardiac events, such as heart failure, arrhythmia, pericardial disease, valvular disease, or coronary artery disease. They also evaluated the incidence of diabetes and hypertension in each group.
Of the children who survived cancer, 2.8% (n = 203) experienced at least one cardiac event versus 0.9% of controls (P less than .001). The cancer survivors experienced 3.2 cardiac events per 1,000 person-years (95% confidence interval, 2.8-3.6), compared with the control group in which there was a rate of 0.9 cardiac events per 1,000 person-years (95% CI, 0.9-1.9).
With regard to cardiovascular disease (CVD) risk, associated factors included cancer relapse or subsequent cancer (hazard ratio, 1.7; 95% CI, 1.1-2.7) and a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy, compared with a dose of less than 250 mg/m2 or no anthracycline chemotherapy (HR, 2.0; 95% CI, 1.4-2.9). Patients who developed diabetes mellitus before a CVD diagnosis were also at higher risk of CVD (HR, 3.0; 95% CI, 1.6-5.8).
Heart failure risk was also statistically significant in patients with relapse and subsequent childhood cancer (HR, 2.0; 95% CI, 1.1-3.7), a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy (HR, 8.6; 95% CI, 4.5-16.6), diabetes (HR, 4.3; 95% CI, 1.8-10.7), and hypertension (HR, 3.1; 95% CI, 1.3-7.9).
“While anthracycline chemotherapy may induce heart disease, many patients require this cancer treatment to survive,” Paul Nathan, MD, of the Hospital for Sick Children in Canada and a study coauthor said in a statement. “Doctors should address heart disease risk factors – such as diabetes and hypertension – that can be modified.”
This study was funded in part from a grant by the Canadian Institutes for Health Research. Several authors reported support from noncommercial sources. The other authors reported having no relevant conflicts of interest.
SOURCE: Khanna A et al. Circulation. 2019 Aug 26. doi: 10.1161/CIRCULATIONAHA.119.041403.
Pediatric cancer survivors have a higher likelihood of experiencing a cardiac event, developing diabetes, or having hypertension at a median 10-year follow-up, according to results from a recent research letter published in Circulation.
Ashna Khanna of the University of Toronto and colleagues identified 7,289 pediatric patients in the Pediatric Oncology Group of Ontario Networked Information System who were diagnosed with cancer at median age of 7 years old, who were treated between 1987 and 2010, and who were cancer survivors for 5 years. Each patient was matched to five cancer-free control subjects who were a median of 24 years old at the 10-year follow-up (36,205 cancer-free individuals). The researchers studied whether pediatric cancer survivors experienced cardiac events, such as heart failure, arrhythmia, pericardial disease, valvular disease, or coronary artery disease. They also evaluated the incidence of diabetes and hypertension in each group.
Of the children who survived cancer, 2.8% (n = 203) experienced at least one cardiac event versus 0.9% of controls (P less than .001). The cancer survivors experienced 3.2 cardiac events per 1,000 person-years (95% confidence interval, 2.8-3.6), compared with the control group in which there was a rate of 0.9 cardiac events per 1,000 person-years (95% CI, 0.9-1.9).
With regard to cardiovascular disease (CVD) risk, associated factors included cancer relapse or subsequent cancer (hazard ratio, 1.7; 95% CI, 1.1-2.7) and a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy, compared with a dose of less than 250 mg/m2 or no anthracycline chemotherapy (HR, 2.0; 95% CI, 1.4-2.9). Patients who developed diabetes mellitus before a CVD diagnosis were also at higher risk of CVD (HR, 3.0; 95% CI, 1.6-5.8).
Heart failure risk was also statistically significant in patients with relapse and subsequent childhood cancer (HR, 2.0; 95% CI, 1.1-3.7), a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy (HR, 8.6; 95% CI, 4.5-16.6), diabetes (HR, 4.3; 95% CI, 1.8-10.7), and hypertension (HR, 3.1; 95% CI, 1.3-7.9).
“While anthracycline chemotherapy may induce heart disease, many patients require this cancer treatment to survive,” Paul Nathan, MD, of the Hospital for Sick Children in Canada and a study coauthor said in a statement. “Doctors should address heart disease risk factors – such as diabetes and hypertension – that can be modified.”
This study was funded in part from a grant by the Canadian Institutes for Health Research. Several authors reported support from noncommercial sources. The other authors reported having no relevant conflicts of interest.
SOURCE: Khanna A et al. Circulation. 2019 Aug 26. doi: 10.1161/CIRCULATIONAHA.119.041403.
Pediatric cancer survivors have a higher likelihood of experiencing a cardiac event, developing diabetes, or having hypertension at a median 10-year follow-up, according to results from a recent research letter published in Circulation.
Ashna Khanna of the University of Toronto and colleagues identified 7,289 pediatric patients in the Pediatric Oncology Group of Ontario Networked Information System who were diagnosed with cancer at median age of 7 years old, who were treated between 1987 and 2010, and who were cancer survivors for 5 years. Each patient was matched to five cancer-free control subjects who were a median of 24 years old at the 10-year follow-up (36,205 cancer-free individuals). The researchers studied whether pediatric cancer survivors experienced cardiac events, such as heart failure, arrhythmia, pericardial disease, valvular disease, or coronary artery disease. They also evaluated the incidence of diabetes and hypertension in each group.
Of the children who survived cancer, 2.8% (n = 203) experienced at least one cardiac event versus 0.9% of controls (P less than .001). The cancer survivors experienced 3.2 cardiac events per 1,000 person-years (95% confidence interval, 2.8-3.6), compared with the control group in which there was a rate of 0.9 cardiac events per 1,000 person-years (95% CI, 0.9-1.9).
With regard to cardiovascular disease (CVD) risk, associated factors included cancer relapse or subsequent cancer (hazard ratio, 1.7; 95% CI, 1.1-2.7) and a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy, compared with a dose of less than 250 mg/m2 or no anthracycline chemotherapy (HR, 2.0; 95% CI, 1.4-2.9). Patients who developed diabetes mellitus before a CVD diagnosis were also at higher risk of CVD (HR, 3.0; 95% CI, 1.6-5.8).
Heart failure risk was also statistically significant in patients with relapse and subsequent childhood cancer (HR, 2.0; 95% CI, 1.1-3.7), a 250-mg/m2 or greater dose of doxorubicin-equivalent anthracycline chemotherapy (HR, 8.6; 95% CI, 4.5-16.6), diabetes (HR, 4.3; 95% CI, 1.8-10.7), and hypertension (HR, 3.1; 95% CI, 1.3-7.9).
“While anthracycline chemotherapy may induce heart disease, many patients require this cancer treatment to survive,” Paul Nathan, MD, of the Hospital for Sick Children in Canada and a study coauthor said in a statement. “Doctors should address heart disease risk factors – such as diabetes and hypertension – that can be modified.”
This study was funded in part from a grant by the Canadian Institutes for Health Research. Several authors reported support from noncommercial sources. The other authors reported having no relevant conflicts of interest.
SOURCE: Khanna A et al. Circulation. 2019 Aug 26. doi: 10.1161/CIRCULATIONAHA.119.041403.
FROM CIRCULATION
U.S. and African programs aim to improve understanding, treatment of sickle cell disease
Researchers are leading several programs designed to serve the sickle cell community in the United States and sub-Saharan Africa, officials at the National Heart, Lung, and Blood Institute (NHLBI) said during a recent webinar.

One program based in the United States is focused on building a registry for patients with sickle cell disease (SCD) and conducting studies designed to improve SCD care. Another program involves building “an information-sharing network and patient-powered registry” in the United States.
The programs in sub-Saharan Africa were designed to establish a database of SCD patients, optimize the use of hydroxyurea in children with SCD, and aid genomic studies of SCD.
W. Keith Hoots, MD, director of the Division of Blood Diseases and Resources at NHLBI, began the webinar with an overview of the programs in sub-Saharan Africa. He described four programs with sites in nine countries (Angola, Cameroon, Democratic Republic of Congo, Ghana, Kenya, Nigeria, South Africa, Tanzania, and Uganda).
SPARCO and SADaCC
Dr. Hoots outlined the scope the Sickle Pan-African Research Consortium (SPARCO) and the Sickle Africa Data Coordinating Center (SADaCC), both part of the Sickle In Africa consortium.
A major goal of SPARCO and SADaCC is to create a Research Electronic Data Capture database that encompasses SCD patients in sub-Saharan Africa. As of April 2019, the database included 6,578 patients. The target is 13,000 patients.
Other goals of SPARCO and SADaCC are to “harmonize” SCD phenotype definitions and ontologies, create clinical guidelines for SCD management in sub-Saharan Africa, plan future cohort studies, and develop programs for newborn screening, infection prevention, and increased use of hydroxyurea.
“So far, they’re well along in establishing a registry and a database system,” Dr. Hoots said. “They’ve agreed on the database elements, phenotype definitions, and ontologies, they’ve developed some regionally appropriate clinical management guidelines, and they’ve begun skills development on the ground at all respective sites.”
REACH
Another program Dr. Hoots discussed is Realizing Effectiveness Across Continents With Hydroxyurea (REACH), a phase 1/2 pilot study of hydroxyurea in children (aged 1-10 years) with SCD in sub-Saharan Africa.
The goals of REACH are to determine the optimal dose of hydroxyurea in this population; teach African physicians how to administer hydroxyurea; assess the safety, feasibility, and benefits of hydroxyurea; study variability in response to hydroxyurea; gather data for the Research Electronic Data Capture database; and establish a research infrastructure for future collaborations.
Results from more than 600 children enrolled in REACH were presented at the 2018 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2019 Jan 10; 380[2]:121-31).
SickleGenAfrica
SickleGenAfrica is part of the H3Africa consortium and aims to “build capacity for genomic research in Africa,” Dr. Hoots said.
Under this program, researchers will conduct three studies to test the hypothesis that genetic variation affects the defense against hemolysis and organ damage in patients with SCD. The researchers will study existing cohorts of SCD patients including children and adults.
Other goals of SickleGenAfrica are to establish a molecular hematology and sickle cell mouse core, an SCD biorepository core, a bioinformatics core, and an administrative core for the coordination of activities. The program will also be used to train “future science leaders” in SCD research, Dr. Hoots said.
SCDIC
Cheryl Anne Boyce, PhD, chief of the Implementation Science Branch at the Center for Translation Research and Implementation Science at NHLBI, discussed the United States–based Sickle Cell Disease Implementation Consortium (SCDIC).
“The goals of the consortium are to develop a registry in collaboration with other centers and the NHLBI, as well as a needs-based community assessment of the barriers to care for subjects with sickle cell disease,” Dr. Boyce said. “We also wanted to design implementation research studies that address the identified barriers to care.”
Dr. Boyce said the SCDIC’s registry is open to patients aged 15-45 years who have a confirmed SCD diagnosis, speak English, and are able to consent to and complete a survey. The registry has enrolled almost 2,400 patients from eight centers over 18 months.
The SCDIC has also performed a needs assessment that prompted the development of three implementation research studies. The first study involves using mobile health interventions to, ideally, increase patient adherence to hydroxyurea and improve provider knowledge of hydroxyurea.
With the second study, researchers aim to improve the care of SCD patients in the emergency department by using an inpatient portal. The goals of the third study are to establish a standard definition for unaffiliated patients, conduct a needs assessment for this group, and develop an intervention that can provide these patients with guideline-based SCD care.
Get Connected
Kim Smith-Whitley, MD, director of the Comprehensive Sickle Cell Center at the Children’s Hospital of Philadelphia and a board member of the Sickle Cell Disease Association of America (SCDAA), described Get Connected, “an information-sharing network and patient-powered registry” created by SCDAA.
Dr. Smith-Whitley said one purpose of Get Connected is to provide a network that facilitates “the distribution of information related to clinical care, research, health services, health policy, and advocacy.”
The network is open to families living with SCD and sickle cell trait, SCDAA member organizations, health care providers, clinical researchers, and community-based organizations.
Get Connected also includes a registry for SCD patients that stores information on their diagnosis and treatment, as well as online communities that can be used to share information and provide psychosocial support.
Thus far, Get Connected has enrolled 6,329 individuals. This includes 5,100 children and adults with SCD, 652 children and adults with sickle cell trait, and 577 nonpatients.
The webinar presenters did not disclose any conflicts of interest.
Researchers are leading several programs designed to serve the sickle cell community in the United States and sub-Saharan Africa, officials at the National Heart, Lung, and Blood Institute (NHLBI) said during a recent webinar.
One program based in the United States is focused on building a registry for patients with sickle cell disease (SCD) and conducting studies designed to improve SCD care. Another program involves building “an information-sharing network and patient-powered registry” in the United States.
The programs in sub-Saharan Africa were designed to establish a database of SCD patients, optimize the use of hydroxyurea in children with SCD, and aid genomic studies of SCD.
W. Keith Hoots, MD, director of the Division of Blood Diseases and Resources at NHLBI, began the webinar with an overview of the programs in sub-Saharan Africa. He described four programs with sites in nine countries (Angola, Cameroon, Democratic Republic of Congo, Ghana, Kenya, Nigeria, South Africa, Tanzania, and Uganda).
SPARCO and SADaCC
Dr. Hoots outlined the scope the Sickle Pan-African Research Consortium (SPARCO) and the Sickle Africa Data Coordinating Center (SADaCC), both part of the Sickle In Africa consortium.
A major goal of SPARCO and SADaCC is to create a Research Electronic Data Capture database that encompasses SCD patients in sub-Saharan Africa. As of April 2019, the database included 6,578 patients. The target is 13,000 patients.
Other goals of SPARCO and SADaCC are to “harmonize” SCD phenotype definitions and ontologies, create clinical guidelines for SCD management in sub-Saharan Africa, plan future cohort studies, and develop programs for newborn screening, infection prevention, and increased use of hydroxyurea.
“So far, they’re well along in establishing a registry and a database system,” Dr. Hoots said. “They’ve agreed on the database elements, phenotype definitions, and ontologies, they’ve developed some regionally appropriate clinical management guidelines, and they’ve begun skills development on the ground at all respective sites.”
REACH
Another program Dr. Hoots discussed is Realizing Effectiveness Across Continents With Hydroxyurea (REACH), a phase 1/2 pilot study of hydroxyurea in children (aged 1-10 years) with SCD in sub-Saharan Africa.
The goals of REACH are to determine the optimal dose of hydroxyurea in this population; teach African physicians how to administer hydroxyurea; assess the safety, feasibility, and benefits of hydroxyurea; study variability in response to hydroxyurea; gather data for the Research Electronic Data Capture database; and establish a research infrastructure for future collaborations.
Results from more than 600 children enrolled in REACH were presented at the 2018 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2019 Jan 10; 380[2]:121-31).
SickleGenAfrica
SickleGenAfrica is part of the H3Africa consortium and aims to “build capacity for genomic research in Africa,” Dr. Hoots said.
Under this program, researchers will conduct three studies to test the hypothesis that genetic variation affects the defense against hemolysis and organ damage in patients with SCD. The researchers will study existing cohorts of SCD patients including children and adults.
Other goals of SickleGenAfrica are to establish a molecular hematology and sickle cell mouse core, an SCD biorepository core, a bioinformatics core, and an administrative core for the coordination of activities. The program will also be used to train “future science leaders” in SCD research, Dr. Hoots said.
SCDIC
Cheryl Anne Boyce, PhD, chief of the Implementation Science Branch at the Center for Translation Research and Implementation Science at NHLBI, discussed the United States–based Sickle Cell Disease Implementation Consortium (SCDIC).
“The goals of the consortium are to develop a registry in collaboration with other centers and the NHLBI, as well as a needs-based community assessment of the barriers to care for subjects with sickle cell disease,” Dr. Boyce said. “We also wanted to design implementation research studies that address the identified barriers to care.”
Dr. Boyce said the SCDIC’s registry is open to patients aged 15-45 years who have a confirmed SCD diagnosis, speak English, and are able to consent to and complete a survey. The registry has enrolled almost 2,400 patients from eight centers over 18 months.
The SCDIC has also performed a needs assessment that prompted the development of three implementation research studies. The first study involves using mobile health interventions to, ideally, increase patient adherence to hydroxyurea and improve provider knowledge of hydroxyurea.
With the second study, researchers aim to improve the care of SCD patients in the emergency department by using an inpatient portal. The goals of the third study are to establish a standard definition for unaffiliated patients, conduct a needs assessment for this group, and develop an intervention that can provide these patients with guideline-based SCD care.
Get Connected
Kim Smith-Whitley, MD, director of the Comprehensive Sickle Cell Center at the Children’s Hospital of Philadelphia and a board member of the Sickle Cell Disease Association of America (SCDAA), described Get Connected, “an information-sharing network and patient-powered registry” created by SCDAA.
Dr. Smith-Whitley said one purpose of Get Connected is to provide a network that facilitates “the distribution of information related to clinical care, research, health services, health policy, and advocacy.”
The network is open to families living with SCD and sickle cell trait, SCDAA member organizations, health care providers, clinical researchers, and community-based organizations.
Get Connected also includes a registry for SCD patients that stores information on their diagnosis and treatment, as well as online communities that can be used to share information and provide psychosocial support.
Thus far, Get Connected has enrolled 6,329 individuals. This includes 5,100 children and adults with SCD, 652 children and adults with sickle cell trait, and 577 nonpatients.
The webinar presenters did not disclose any conflicts of interest.
Researchers are leading several programs designed to serve the sickle cell community in the United States and sub-Saharan Africa, officials at the National Heart, Lung, and Blood Institute (NHLBI) said during a recent webinar.
One program based in the United States is focused on building a registry for patients with sickle cell disease (SCD) and conducting studies designed to improve SCD care. Another program involves building “an information-sharing network and patient-powered registry” in the United States.
The programs in sub-Saharan Africa were designed to establish a database of SCD patients, optimize the use of hydroxyurea in children with SCD, and aid genomic studies of SCD.
W. Keith Hoots, MD, director of the Division of Blood Diseases and Resources at NHLBI, began the webinar with an overview of the programs in sub-Saharan Africa. He described four programs with sites in nine countries (Angola, Cameroon, Democratic Republic of Congo, Ghana, Kenya, Nigeria, South Africa, Tanzania, and Uganda).
SPARCO and SADaCC
Dr. Hoots outlined the scope the Sickle Pan-African Research Consortium (SPARCO) and the Sickle Africa Data Coordinating Center (SADaCC), both part of the Sickle In Africa consortium.
A major goal of SPARCO and SADaCC is to create a Research Electronic Data Capture database that encompasses SCD patients in sub-Saharan Africa. As of April 2019, the database included 6,578 patients. The target is 13,000 patients.
Other goals of SPARCO and SADaCC are to “harmonize” SCD phenotype definitions and ontologies, create clinical guidelines for SCD management in sub-Saharan Africa, plan future cohort studies, and develop programs for newborn screening, infection prevention, and increased use of hydroxyurea.
“So far, they’re well along in establishing a registry and a database system,” Dr. Hoots said. “They’ve agreed on the database elements, phenotype definitions, and ontologies, they’ve developed some regionally appropriate clinical management guidelines, and they’ve begun skills development on the ground at all respective sites.”
REACH
Another program Dr. Hoots discussed is Realizing Effectiveness Across Continents With Hydroxyurea (REACH), a phase 1/2 pilot study of hydroxyurea in children (aged 1-10 years) with SCD in sub-Saharan Africa.
The goals of REACH are to determine the optimal dose of hydroxyurea in this population; teach African physicians how to administer hydroxyurea; assess the safety, feasibility, and benefits of hydroxyurea; study variability in response to hydroxyurea; gather data for the Research Electronic Data Capture database; and establish a research infrastructure for future collaborations.
Results from more than 600 children enrolled in REACH were presented at the 2018 annual meeting of the American Society of Hematology and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2019 Jan 10; 380[2]:121-31).
SickleGenAfrica
SickleGenAfrica is part of the H3Africa consortium and aims to “build capacity for genomic research in Africa,” Dr. Hoots said.
Under this program, researchers will conduct three studies to test the hypothesis that genetic variation affects the defense against hemolysis and organ damage in patients with SCD. The researchers will study existing cohorts of SCD patients including children and adults.
Other goals of SickleGenAfrica are to establish a molecular hematology and sickle cell mouse core, an SCD biorepository core, a bioinformatics core, and an administrative core for the coordination of activities. The program will also be used to train “future science leaders” in SCD research, Dr. Hoots said.
SCDIC
Cheryl Anne Boyce, PhD, chief of the Implementation Science Branch at the Center for Translation Research and Implementation Science at NHLBI, discussed the United States–based Sickle Cell Disease Implementation Consortium (SCDIC).
“The goals of the consortium are to develop a registry in collaboration with other centers and the NHLBI, as well as a needs-based community assessment of the barriers to care for subjects with sickle cell disease,” Dr. Boyce said. “We also wanted to design implementation research studies that address the identified barriers to care.”
Dr. Boyce said the SCDIC’s registry is open to patients aged 15-45 years who have a confirmed SCD diagnosis, speak English, and are able to consent to and complete a survey. The registry has enrolled almost 2,400 patients from eight centers over 18 months.
The SCDIC has also performed a needs assessment that prompted the development of three implementation research studies. The first study involves using mobile health interventions to, ideally, increase patient adherence to hydroxyurea and improve provider knowledge of hydroxyurea.
With the second study, researchers aim to improve the care of SCD patients in the emergency department by using an inpatient portal. The goals of the third study are to establish a standard definition for unaffiliated patients, conduct a needs assessment for this group, and develop an intervention that can provide these patients with guideline-based SCD care.
Get Connected
Kim Smith-Whitley, MD, director of the Comprehensive Sickle Cell Center at the Children’s Hospital of Philadelphia and a board member of the Sickle Cell Disease Association of America (SCDAA), described Get Connected, “an information-sharing network and patient-powered registry” created by SCDAA.
Dr. Smith-Whitley said one purpose of Get Connected is to provide a network that facilitates “the distribution of information related to clinical care, research, health services, health policy, and advocacy.”
The network is open to families living with SCD and sickle cell trait, SCDAA member organizations, health care providers, clinical researchers, and community-based organizations.
Get Connected also includes a registry for SCD patients that stores information on their diagnosis and treatment, as well as online communities that can be used to share information and provide psychosocial support.
Thus far, Get Connected has enrolled 6,329 individuals. This includes 5,100 children and adults with SCD, 652 children and adults with sickle cell trait, and 577 nonpatients.
The webinar presenters did not disclose any conflicts of interest.
Pediatric HSCT recipients still risking sunburn
Young people who have received allogeneic hematopoietic stem cell transplants (HSCTs) are more likely to wear hats, sunscreen and other sun protection, but still intentionally tan and experience sunburn at the same rate as their peers, new research suggests.

In a survey‐based, cross‐sectional cohort study, researchers compared sun-protection behaviors and sun exposure in 85 children aged 21 years and younger who had undergone HSCT and 85 age-, sex-, and skin type–matched controls. The findings were published in Pediatric Dermatology.
HSCT recipients have a higher risk of long-term complications such as skin cancer, for which sun exposure is a major modifiable environmental risk factor.
“Therefore, consistent sun avoidance and protection as well as regular dermatologic evaluations are important for HSCT recipients,” wrote Edward B. Li, PhD, from Harvard Medical School, Boston, and coauthors.
The survey found no significant difference between the transplant and control group in the amount of intentional sun exposure, such as the amount of time spent outside on weekdays and weekends during the peak sun intensity hours of 10 a.m. and 4 p.m. More than one in five transplant recipients (21.2%) reported spending at least 3 hours a day outside between 10 a.m. and 4 p.m. on weekdays, as did 36.5% of transplant recipients on weekends.
There were also no significant differences between the two groups in terms of time spent tanning, either in the sun or in a tanning bed. Additionally, a similar number of transplant recipients and controls experienced one or more red or painful sunburns in the past year (25.9% vs. 27.1%).
However, transplant patients did practice better sun protection behaviors than did the control group, with 60% reporting that they always wore sunscreen, compared with 29.4% of controls. The transplant recipients were also significantly more likely to wear sunglasses and a hat and to stay in the shade or use an umbrella.
“While these data may reflect that HSCT patients are not practicing adequate sun avoidance, it may also suggest that these long‐term survivors are able to enjoy being outdoors as much as their peers and have a similar desire to have a tanned appearance,” the researchers wrote. “While a healthy and active lifestyle should be encouraged for all children, our results emphasize the need for pediatric HSCT survivors to be educated on their increased risk for UV‐related skin cancers, counseled on avoidance of intentional tanning, and advised on the importance of sun protection behaviors in an effort to improve long-term outcomes.”
The researchers noted that transplant recipients were significantly more likely to have had a full body skin exam from a health care professional than were individuals in the control group (61.2% vs. 4.7%) and were more likely to have done a self-check or been checked by a partner in the previous year.
The study was supported by the Society for Pediatric Dermatology, the Dermatology Foundation, the National Institutes of Health, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. One author declared a financial interest in a company developing a dermatological product. No other conflicts of interest were declared.
SOURCE: Li EB et al. Pediatr Dermatol. 2019 Aug 13. doi: 10.1111/pde.13984.
Young people who have received allogeneic hematopoietic stem cell transplants (HSCTs) are more likely to wear hats, sunscreen and other sun protection, but still intentionally tan and experience sunburn at the same rate as their peers, new research suggests.
In a survey‐based, cross‐sectional cohort study, researchers compared sun-protection behaviors and sun exposure in 85 children aged 21 years and younger who had undergone HSCT and 85 age-, sex-, and skin type–matched controls. The findings were published in Pediatric Dermatology.
HSCT recipients have a higher risk of long-term complications such as skin cancer, for which sun exposure is a major modifiable environmental risk factor.
“Therefore, consistent sun avoidance and protection as well as regular dermatologic evaluations are important for HSCT recipients,” wrote Edward B. Li, PhD, from Harvard Medical School, Boston, and coauthors.
The survey found no significant difference between the transplant and control group in the amount of intentional sun exposure, such as the amount of time spent outside on weekdays and weekends during the peak sun intensity hours of 10 a.m. and 4 p.m. More than one in five transplant recipients (21.2%) reported spending at least 3 hours a day outside between 10 a.m. and 4 p.m. on weekdays, as did 36.5% of transplant recipients on weekends.
There were also no significant differences between the two groups in terms of time spent tanning, either in the sun or in a tanning bed. Additionally, a similar number of transplant recipients and controls experienced one or more red or painful sunburns in the past year (25.9% vs. 27.1%).
However, transplant patients did practice better sun protection behaviors than did the control group, with 60% reporting that they always wore sunscreen, compared with 29.4% of controls. The transplant recipients were also significantly more likely to wear sunglasses and a hat and to stay in the shade or use an umbrella.
“While these data may reflect that HSCT patients are not practicing adequate sun avoidance, it may also suggest that these long‐term survivors are able to enjoy being outdoors as much as their peers and have a similar desire to have a tanned appearance,” the researchers wrote. “While a healthy and active lifestyle should be encouraged for all children, our results emphasize the need for pediatric HSCT survivors to be educated on their increased risk for UV‐related skin cancers, counseled on avoidance of intentional tanning, and advised on the importance of sun protection behaviors in an effort to improve long-term outcomes.”
The researchers noted that transplant recipients were significantly more likely to have had a full body skin exam from a health care professional than were individuals in the control group (61.2% vs. 4.7%) and were more likely to have done a self-check or been checked by a partner in the previous year.
The study was supported by the Society for Pediatric Dermatology, the Dermatology Foundation, the National Institutes of Health, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. One author declared a financial interest in a company developing a dermatological product. No other conflicts of interest were declared.
SOURCE: Li EB et al. Pediatr Dermatol. 2019 Aug 13. doi: 10.1111/pde.13984.
Young people who have received allogeneic hematopoietic stem cell transplants (HSCTs) are more likely to wear hats, sunscreen and other sun protection, but still intentionally tan and experience sunburn at the same rate as their peers, new research suggests.
In a survey‐based, cross‐sectional cohort study, researchers compared sun-protection behaviors and sun exposure in 85 children aged 21 years and younger who had undergone HSCT and 85 age-, sex-, and skin type–matched controls. The findings were published in Pediatric Dermatology.
HSCT recipients have a higher risk of long-term complications such as skin cancer, for which sun exposure is a major modifiable environmental risk factor.
“Therefore, consistent sun avoidance and protection as well as regular dermatologic evaluations are important for HSCT recipients,” wrote Edward B. Li, PhD, from Harvard Medical School, Boston, and coauthors.
The survey found no significant difference between the transplant and control group in the amount of intentional sun exposure, such as the amount of time spent outside on weekdays and weekends during the peak sun intensity hours of 10 a.m. and 4 p.m. More than one in five transplant recipients (21.2%) reported spending at least 3 hours a day outside between 10 a.m. and 4 p.m. on weekdays, as did 36.5% of transplant recipients on weekends.
There were also no significant differences between the two groups in terms of time spent tanning, either in the sun or in a tanning bed. Additionally, a similar number of transplant recipients and controls experienced one or more red or painful sunburns in the past year (25.9% vs. 27.1%).
However, transplant patients did practice better sun protection behaviors than did the control group, with 60% reporting that they always wore sunscreen, compared with 29.4% of controls. The transplant recipients were also significantly more likely to wear sunglasses and a hat and to stay in the shade or use an umbrella.
“While these data may reflect that HSCT patients are not practicing adequate sun avoidance, it may also suggest that these long‐term survivors are able to enjoy being outdoors as much as their peers and have a similar desire to have a tanned appearance,” the researchers wrote. “While a healthy and active lifestyle should be encouraged for all children, our results emphasize the need for pediatric HSCT survivors to be educated on their increased risk for UV‐related skin cancers, counseled on avoidance of intentional tanning, and advised on the importance of sun protection behaviors in an effort to improve long-term outcomes.”
The researchers noted that transplant recipients were significantly more likely to have had a full body skin exam from a health care professional than were individuals in the control group (61.2% vs. 4.7%) and were more likely to have done a self-check or been checked by a partner in the previous year.
The study was supported by the Society for Pediatric Dermatology, the Dermatology Foundation, the National Institutes of Health, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. One author declared a financial interest in a company developing a dermatological product. No other conflicts of interest were declared.
SOURCE: Li EB et al. Pediatr Dermatol. 2019 Aug 13. doi: 10.1111/pde.13984.
FROM PEDIATRIC DERMATOLOGY
IASLC survey: Ongoing and intensified molecular testing education needed globally
BARCELONA – Molecular testing to guide treatment in patients with lung cancer remains underused, and awareness of related evidence-based guidelines is suboptimal, results of an international survey suggest.

Overall, 61% of the 2,537 respondents from 102 countries and across multiple relevant medical specialties reported that molecular testing rates in their country were less than 50%, with the lowest rates reported in Latin America. And 33% of those requesting molecular testing said they were unaware of the most updated guidelines supporting the use of such testing in lung cancer, Matthew Smeltzer, PhD, of the University of Memphis (Tenn.) reported during a press conference at the World Conference on Lung Cancer.
The findings from the International Association for the Study of Lung Cancer (IASLC) Global Survey on Molecular Testing in Lung Cancer also showed that 41% of respondents who perform or interpret molecular testing assays report being dissatisfied with the conditions of molecular testing in their country, 17% said they feel that patients are not satisfied, and 35% said they aren’t sure about the state of testing in their country.
Specific concerns reported by respondents included trouble understanding results, the time it takes to receive the results, and the reliability of samples.
The top five barriers to molecular testing included cost, quality, access, awareness, and time, Dr. Smeltzer said at the meeting which is sponsored by the IASLC.
“These five were the same five top barriers in each region of the world,” he said, noting that the ordering of the barriers differed somewhat among regions.
The survey included a seven-question introduction, with 32 additional questions for respondents who request tests and treat patients, 45 questions on performing and interpreting assays, and 24 questions on tissue acquisition. Additionally, all respondents were asked to list barriers that impede their country’s ability to offer molecular testing.
“I’d say we got a pretty good geographic distribution of responses; 56% of these responses were from developing countries, 44% from developed countries,” he said, noting that medical oncologists constituted the highest percentage of respondents, followed by pulmonologists, thoracic surgeons, pathologists, and other scientists.
When asked specifically what would prompt molecular testing, respondents most often listed adenocarcinoma, never-smoker status, female gender, and young age, Dr. Smeltzer said.
“Overall, we’re still finding that many in the lung cancer community are not satisfied with the current state of molecular testing. We’ve got suboptimal awareness of the evidence-based guidelines. We have barriers that remain to molecular testing, which we’ve identified, and [we’re] recommending continuous education around molecular testing, and that should be intensified on a national and international level to ensure that patients receive optimal therapy,” he concluded.
The IASLC survey was funded by AstraZeneca. Dr. Smeltzer reported receiving research support from the Bristol Myers Squibb Foundation.
BARCELONA – Molecular testing to guide treatment in patients with lung cancer remains underused, and awareness of related evidence-based guidelines is suboptimal, results of an international survey suggest.
Overall, 61% of the 2,537 respondents from 102 countries and across multiple relevant medical specialties reported that molecular testing rates in their country were less than 50%, with the lowest rates reported in Latin America. And 33% of those requesting molecular testing said they were unaware of the most updated guidelines supporting the use of such testing in lung cancer, Matthew Smeltzer, PhD, of the University of Memphis (Tenn.) reported during a press conference at the World Conference on Lung Cancer.
The findings from the International Association for the Study of Lung Cancer (IASLC) Global Survey on Molecular Testing in Lung Cancer also showed that 41% of respondents who perform or interpret molecular testing assays report being dissatisfied with the conditions of molecular testing in their country, 17% said they feel that patients are not satisfied, and 35% said they aren’t sure about the state of testing in their country.
Specific concerns reported by respondents included trouble understanding results, the time it takes to receive the results, and the reliability of samples.
The top five barriers to molecular testing included cost, quality, access, awareness, and time, Dr. Smeltzer said at the meeting which is sponsored by the IASLC.
“These five were the same five top barriers in each region of the world,” he said, noting that the ordering of the barriers differed somewhat among regions.
The survey included a seven-question introduction, with 32 additional questions for respondents who request tests and treat patients, 45 questions on performing and interpreting assays, and 24 questions on tissue acquisition. Additionally, all respondents were asked to list barriers that impede their country’s ability to offer molecular testing.
“I’d say we got a pretty good geographic distribution of responses; 56% of these responses were from developing countries, 44% from developed countries,” he said, noting that medical oncologists constituted the highest percentage of respondents, followed by pulmonologists, thoracic surgeons, pathologists, and other scientists.
When asked specifically what would prompt molecular testing, respondents most often listed adenocarcinoma, never-smoker status, female gender, and young age, Dr. Smeltzer said.
“Overall, we’re still finding that many in the lung cancer community are not satisfied with the current state of molecular testing. We’ve got suboptimal awareness of the evidence-based guidelines. We have barriers that remain to molecular testing, which we’ve identified, and [we’re] recommending continuous education around molecular testing, and that should be intensified on a national and international level to ensure that patients receive optimal therapy,” he concluded.
The IASLC survey was funded by AstraZeneca. Dr. Smeltzer reported receiving research support from the Bristol Myers Squibb Foundation.
BARCELONA – Molecular testing to guide treatment in patients with lung cancer remains underused, and awareness of related evidence-based guidelines is suboptimal, results of an international survey suggest.
Overall, 61% of the 2,537 respondents from 102 countries and across multiple relevant medical specialties reported that molecular testing rates in their country were less than 50%, with the lowest rates reported in Latin America. And 33% of those requesting molecular testing said they were unaware of the most updated guidelines supporting the use of such testing in lung cancer, Matthew Smeltzer, PhD, of the University of Memphis (Tenn.) reported during a press conference at the World Conference on Lung Cancer.
The findings from the International Association for the Study of Lung Cancer (IASLC) Global Survey on Molecular Testing in Lung Cancer also showed that 41% of respondents who perform or interpret molecular testing assays report being dissatisfied with the conditions of molecular testing in their country, 17% said they feel that patients are not satisfied, and 35% said they aren’t sure about the state of testing in their country.
Specific concerns reported by respondents included trouble understanding results, the time it takes to receive the results, and the reliability of samples.
The top five barriers to molecular testing included cost, quality, access, awareness, and time, Dr. Smeltzer said at the meeting which is sponsored by the IASLC.
“These five were the same five top barriers in each region of the world,” he said, noting that the ordering of the barriers differed somewhat among regions.
The survey included a seven-question introduction, with 32 additional questions for respondents who request tests and treat patients, 45 questions on performing and interpreting assays, and 24 questions on tissue acquisition. Additionally, all respondents were asked to list barriers that impede their country’s ability to offer molecular testing.
“I’d say we got a pretty good geographic distribution of responses; 56% of these responses were from developing countries, 44% from developed countries,” he said, noting that medical oncologists constituted the highest percentage of respondents, followed by pulmonologists, thoracic surgeons, pathologists, and other scientists.
When asked specifically what would prompt molecular testing, respondents most often listed adenocarcinoma, never-smoker status, female gender, and young age, Dr. Smeltzer said.
“Overall, we’re still finding that many in the lung cancer community are not satisfied with the current state of molecular testing. We’ve got suboptimal awareness of the evidence-based guidelines. We have barriers that remain to molecular testing, which we’ve identified, and [we’re] recommending continuous education around molecular testing, and that should be intensified on a national and international level to ensure that patients receive optimal therapy,” he concluded.
The IASLC survey was funded by AstraZeneca. Dr. Smeltzer reported receiving research support from the Bristol Myers Squibb Foundation.
REPORTING FROM WCLC 2019