User login
GLP-1 RA dulaglutide yields cardiac gains, even in non–at-risk patients
SAN FRANCISCO – A large, long-term study is linking yet another glucagon-like peptide–1 receptor agonist diabetes drug to positive cardiovascular outcomes: Patients with type 2 diabetes and heart disease risk factors who took dulaglutide for about 5 years during the REWIND study had a 12% lower risk of major adverse cardiovascular events, compared with those who took placebo.
These new findings on cardiac risk are unusual compared with other newer-generation diabetes drugs, because a high percentage of the participants did not have existing cardiovascular disease. In addition, the study population had a higher percentage of women, compared with previous studies.
“We feel very strongly that the participants were similar to the ... ambulatory patients with type 2 diabetes with cardiovascular risk who are routinely seen in clinical practice,” study coauthor Jeffrey L. Probstfield, MD, of the University of Washington, Seattle, saidin a presentation at the annual scientific sessions of the American Diabetes Association. The findings were published simultaneously in The Lancet (2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
Dulaglutide’s serious adverse-effect profile was similar to that of placebo, the study authors noted, and the drug also showed benefits in renal outcomes, as reported in a separate study (Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31150-3.
The Food and Drug Administration has mandated that six glucagon-like peptide–1 receptor agonists (GLP-1 RAs) – albiglutide (Tanzeum), exenatide (Byetta), liraglutide (Victoza), lixisenatide (Adlyxin), semaglutide (Ozempic) and dulaglutide (Trulicity) – undergo testing of cardiovascular outcomes. Dulaglutide is the fourth, following albiglutide, liraglutide, and semaglutide, to show consistent, statistically significant reduction in major adverse cardiovascular events (MACE).
For the double-blind, randomized, placebo-controlled REWIND study, researchers recruited 9,901 participants who were at least 50 years old with type 2 diabetes, a hemoglobin A1c (HbA1c) level of 9.5% or less, and a body mass index of more than 23 kg/m2. The participants came from 371 sites in 24 countries, including the United States and Canada. More than 80% were taking an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker, and other blood pressure drugs were also common.
The average mean age was 66 years; 46% of the participants were women, three-quarters were white, and 31% had previous cardiovascular disease. Previous GLP-1 RA studies of this type had markedly lower percentages of women – the other studies comprised 30%-39% women – and included higher percentages of participants with previous cardiovascular disease (73%-100%).
Of the participants in the current study, 4,949 were assigned to receive dulaglutide and 4,952 to receive placebo. They were followed for a median 5.4 years. About 57% never stopped using the drug, and 11% of those in the drug group and 7.5% in the placebo group stopped use because of adverse effects.
In regard to diabetes outcomes, HbA1c levels fell in the drug group by a mean –0.61% (95% confidence interval, –0.65 to –0.58; P less than .0001), compared with placebo. Their weight decreased by a mean –1.5 kg (95% CI, -1.7 to -1.3; P less than .0001) Systolic blood pressure and LDL cholesterol levels were slightly lower in the drug group, but heart rate was higher.
On the heart front, MACE fell by 12% in the drug group, compared with placebo (hazard ratio, 0.88; 95% CI, 0.79-0.99; P = .026). “The effect of the intervention begins [within] the first year and continued in a progressive, proportional fashion throughout the follow-up period,” said study lead author Hertzel C. Gerstein, MD, of McMaster University and Hamilton Health Sciences, Hamilton, Ont.

There was an especially large decline in the number of nonfatal stroke cases in the drug group, compared with placebo (135 vs. 175, respectively; HR, 0.76; 95% CI, 0.61-0.95; P = .017). The drug did not have a statistically significant effect on cardiovascular death.
The researchers found no difference in the drug’s effects on MACE in subgroups including age, gender, ethnicity, duration of diabetes, and history of cardiovascular disease.
They also reported a decline in a renal composite outcome (first macroalbuminuria, sustained decline in estimated glomerular filtration rate of 30% or more, chronic renal replacement) in the drug group (HR, 0.85; 95% CI, 0.77-0.93; P = .0004).
Rates of serious adverse effects were similar in the drug and placebo groups. Gastrointestinal adverse effects – including nausea, constipation, and diarrhea – were as expected, Dr. Gerstein said.
“The addition of dulaglutide could be considered for both primary and secondary cardiovascular prevention in middle-aged patients with type 2 diabetes and cardiovascular risk factors,” Dr. Gerstein said.
In an independent commentary at the meeting presentation, Sophia Zoungas, MBBS (Hons), FRCP, PhD, of Monash University, Melbourne, praised the study and applauded the findings.

However, she called attention to the results that pinpointed higher levels of microvascular-related eye outcomes (HR, 1.24; 95% CI, 0.92-1.68) and fatal myocardial infarction (HR, 1.29; 95% CI, 0.72-2.30) in the dulaglutide group. Both of those outcomes were rare – 171 eye outcomes and 46 fatal myocardial infarctions overall. She also questioned whether the adherence rates would be as high in a real-world setting.
Eli Lilly funded the study. Three of the authors were employees of Eli Lilly, eight reported financial relationships with the company, five reported financial relationships with other firms, and the remaining authors reported no competing interests.
SOURCE: Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
SAN FRANCISCO – A large, long-term study is linking yet another glucagon-like peptide–1 receptor agonist diabetes drug to positive cardiovascular outcomes: Patients with type 2 diabetes and heart disease risk factors who took dulaglutide for about 5 years during the REWIND study had a 12% lower risk of major adverse cardiovascular events, compared with those who took placebo.
These new findings on cardiac risk are unusual compared with other newer-generation diabetes drugs, because a high percentage of the participants did not have existing cardiovascular disease. In addition, the study population had a higher percentage of women, compared with previous studies.
“We feel very strongly that the participants were similar to the ... ambulatory patients with type 2 diabetes with cardiovascular risk who are routinely seen in clinical practice,” study coauthor Jeffrey L. Probstfield, MD, of the University of Washington, Seattle, saidin a presentation at the annual scientific sessions of the American Diabetes Association. The findings were published simultaneously in The Lancet (2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
Dulaglutide’s serious adverse-effect profile was similar to that of placebo, the study authors noted, and the drug also showed benefits in renal outcomes, as reported in a separate study (Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31150-3.
The Food and Drug Administration has mandated that six glucagon-like peptide–1 receptor agonists (GLP-1 RAs) – albiglutide (Tanzeum), exenatide (Byetta), liraglutide (Victoza), lixisenatide (Adlyxin), semaglutide (Ozempic) and dulaglutide (Trulicity) – undergo testing of cardiovascular outcomes. Dulaglutide is the fourth, following albiglutide, liraglutide, and semaglutide, to show consistent, statistically significant reduction in major adverse cardiovascular events (MACE).
For the double-blind, randomized, placebo-controlled REWIND study, researchers recruited 9,901 participants who were at least 50 years old with type 2 diabetes, a hemoglobin A1c (HbA1c) level of 9.5% or less, and a body mass index of more than 23 kg/m2. The participants came from 371 sites in 24 countries, including the United States and Canada. More than 80% were taking an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker, and other blood pressure drugs were also common.
The average mean age was 66 years; 46% of the participants were women, three-quarters were white, and 31% had previous cardiovascular disease. Previous GLP-1 RA studies of this type had markedly lower percentages of women – the other studies comprised 30%-39% women – and included higher percentages of participants with previous cardiovascular disease (73%-100%).
Of the participants in the current study, 4,949 were assigned to receive dulaglutide and 4,952 to receive placebo. They were followed for a median 5.4 years. About 57% never stopped using the drug, and 11% of those in the drug group and 7.5% in the placebo group stopped use because of adverse effects.
In regard to diabetes outcomes, HbA1c levels fell in the drug group by a mean –0.61% (95% confidence interval, –0.65 to –0.58; P less than .0001), compared with placebo. Their weight decreased by a mean –1.5 kg (95% CI, -1.7 to -1.3; P less than .0001) Systolic blood pressure and LDL cholesterol levels were slightly lower in the drug group, but heart rate was higher.
On the heart front, MACE fell by 12% in the drug group, compared with placebo (hazard ratio, 0.88; 95% CI, 0.79-0.99; P = .026). “The effect of the intervention begins [within] the first year and continued in a progressive, proportional fashion throughout the follow-up period,” said study lead author Hertzel C. Gerstein, MD, of McMaster University and Hamilton Health Sciences, Hamilton, Ont.
There was an especially large decline in the number of nonfatal stroke cases in the drug group, compared with placebo (135 vs. 175, respectively; HR, 0.76; 95% CI, 0.61-0.95; P = .017). The drug did not have a statistically significant effect on cardiovascular death.
The researchers found no difference in the drug’s effects on MACE in subgroups including age, gender, ethnicity, duration of diabetes, and history of cardiovascular disease.
They also reported a decline in a renal composite outcome (first macroalbuminuria, sustained decline in estimated glomerular filtration rate of 30% or more, chronic renal replacement) in the drug group (HR, 0.85; 95% CI, 0.77-0.93; P = .0004).
Rates of serious adverse effects were similar in the drug and placebo groups. Gastrointestinal adverse effects – including nausea, constipation, and diarrhea – were as expected, Dr. Gerstein said.
“The addition of dulaglutide could be considered for both primary and secondary cardiovascular prevention in middle-aged patients with type 2 diabetes and cardiovascular risk factors,” Dr. Gerstein said.
In an independent commentary at the meeting presentation, Sophia Zoungas, MBBS (Hons), FRCP, PhD, of Monash University, Melbourne, praised the study and applauded the findings.
However, she called attention to the results that pinpointed higher levels of microvascular-related eye outcomes (HR, 1.24; 95% CI, 0.92-1.68) and fatal myocardial infarction (HR, 1.29; 95% CI, 0.72-2.30) in the dulaglutide group. Both of those outcomes were rare – 171 eye outcomes and 46 fatal myocardial infarctions overall. She also questioned whether the adherence rates would be as high in a real-world setting.
Eli Lilly funded the study. Three of the authors were employees of Eli Lilly, eight reported financial relationships with the company, five reported financial relationships with other firms, and the remaining authors reported no competing interests.
SOURCE: Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
SAN FRANCISCO – A large, long-term study is linking yet another glucagon-like peptide–1 receptor agonist diabetes drug to positive cardiovascular outcomes: Patients with type 2 diabetes and heart disease risk factors who took dulaglutide for about 5 years during the REWIND study had a 12% lower risk of major adverse cardiovascular events, compared with those who took placebo.
These new findings on cardiac risk are unusual compared with other newer-generation diabetes drugs, because a high percentage of the participants did not have existing cardiovascular disease. In addition, the study population had a higher percentage of women, compared with previous studies.
“We feel very strongly that the participants were similar to the ... ambulatory patients with type 2 diabetes with cardiovascular risk who are routinely seen in clinical practice,” study coauthor Jeffrey L. Probstfield, MD, of the University of Washington, Seattle, saidin a presentation at the annual scientific sessions of the American Diabetes Association. The findings were published simultaneously in The Lancet (2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
Dulaglutide’s serious adverse-effect profile was similar to that of placebo, the study authors noted, and the drug also showed benefits in renal outcomes, as reported in a separate study (Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31150-3.
The Food and Drug Administration has mandated that six glucagon-like peptide–1 receptor agonists (GLP-1 RAs) – albiglutide (Tanzeum), exenatide (Byetta), liraglutide (Victoza), lixisenatide (Adlyxin), semaglutide (Ozempic) and dulaglutide (Trulicity) – undergo testing of cardiovascular outcomes. Dulaglutide is the fourth, following albiglutide, liraglutide, and semaglutide, to show consistent, statistically significant reduction in major adverse cardiovascular events (MACE).
For the double-blind, randomized, placebo-controlled REWIND study, researchers recruited 9,901 participants who were at least 50 years old with type 2 diabetes, a hemoglobin A1c (HbA1c) level of 9.5% or less, and a body mass index of more than 23 kg/m2. The participants came from 371 sites in 24 countries, including the United States and Canada. More than 80% were taking an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker, and other blood pressure drugs were also common.
The average mean age was 66 years; 46% of the participants were women, three-quarters were white, and 31% had previous cardiovascular disease. Previous GLP-1 RA studies of this type had markedly lower percentages of women – the other studies comprised 30%-39% women – and included higher percentages of participants with previous cardiovascular disease (73%-100%).
Of the participants in the current study, 4,949 were assigned to receive dulaglutide and 4,952 to receive placebo. They were followed for a median 5.4 years. About 57% never stopped using the drug, and 11% of those in the drug group and 7.5% in the placebo group stopped use because of adverse effects.
In regard to diabetes outcomes, HbA1c levels fell in the drug group by a mean –0.61% (95% confidence interval, –0.65 to –0.58; P less than .0001), compared with placebo. Their weight decreased by a mean –1.5 kg (95% CI, -1.7 to -1.3; P less than .0001) Systolic blood pressure and LDL cholesterol levels were slightly lower in the drug group, but heart rate was higher.
On the heart front, MACE fell by 12% in the drug group, compared with placebo (hazard ratio, 0.88; 95% CI, 0.79-0.99; P = .026). “The effect of the intervention begins [within] the first year and continued in a progressive, proportional fashion throughout the follow-up period,” said study lead author Hertzel C. Gerstein, MD, of McMaster University and Hamilton Health Sciences, Hamilton, Ont.
There was an especially large decline in the number of nonfatal stroke cases in the drug group, compared with placebo (135 vs. 175, respectively; HR, 0.76; 95% CI, 0.61-0.95; P = .017). The drug did not have a statistically significant effect on cardiovascular death.
The researchers found no difference in the drug’s effects on MACE in subgroups including age, gender, ethnicity, duration of diabetes, and history of cardiovascular disease.
They also reported a decline in a renal composite outcome (first macroalbuminuria, sustained decline in estimated glomerular filtration rate of 30% or more, chronic renal replacement) in the drug group (HR, 0.85; 95% CI, 0.77-0.93; P = .0004).
Rates of serious adverse effects were similar in the drug and placebo groups. Gastrointestinal adverse effects – including nausea, constipation, and diarrhea – were as expected, Dr. Gerstein said.
“The addition of dulaglutide could be considered for both primary and secondary cardiovascular prevention in middle-aged patients with type 2 diabetes and cardiovascular risk factors,” Dr. Gerstein said.
In an independent commentary at the meeting presentation, Sophia Zoungas, MBBS (Hons), FRCP, PhD, of Monash University, Melbourne, praised the study and applauded the findings.
However, she called attention to the results that pinpointed higher levels of microvascular-related eye outcomes (HR, 1.24; 95% CI, 0.92-1.68) and fatal myocardial infarction (HR, 1.29; 95% CI, 0.72-2.30) in the dulaglutide group. Both of those outcomes were rare – 171 eye outcomes and 46 fatal myocardial infarctions overall. She also questioned whether the adherence rates would be as high in a real-world setting.
Eli Lilly funded the study. Three of the authors were employees of Eli Lilly, eight reported financial relationships with the company, five reported financial relationships with other firms, and the remaining authors reported no competing interests.
SOURCE: Gerstein HC et al. Lancet. 2019 Jun 10. doi: 10.1016/S0140-6736(19)31149-3.
REPORTING FROM ADA 2019
Bullous Systemic Lupus Erythematosus Successfully Treated With Rituximab
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
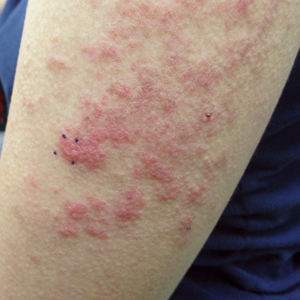
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
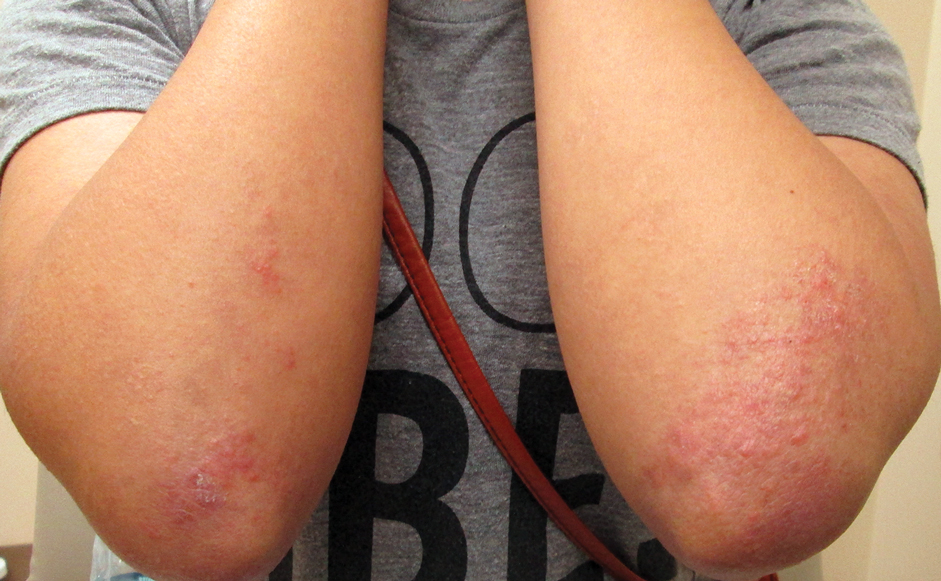
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
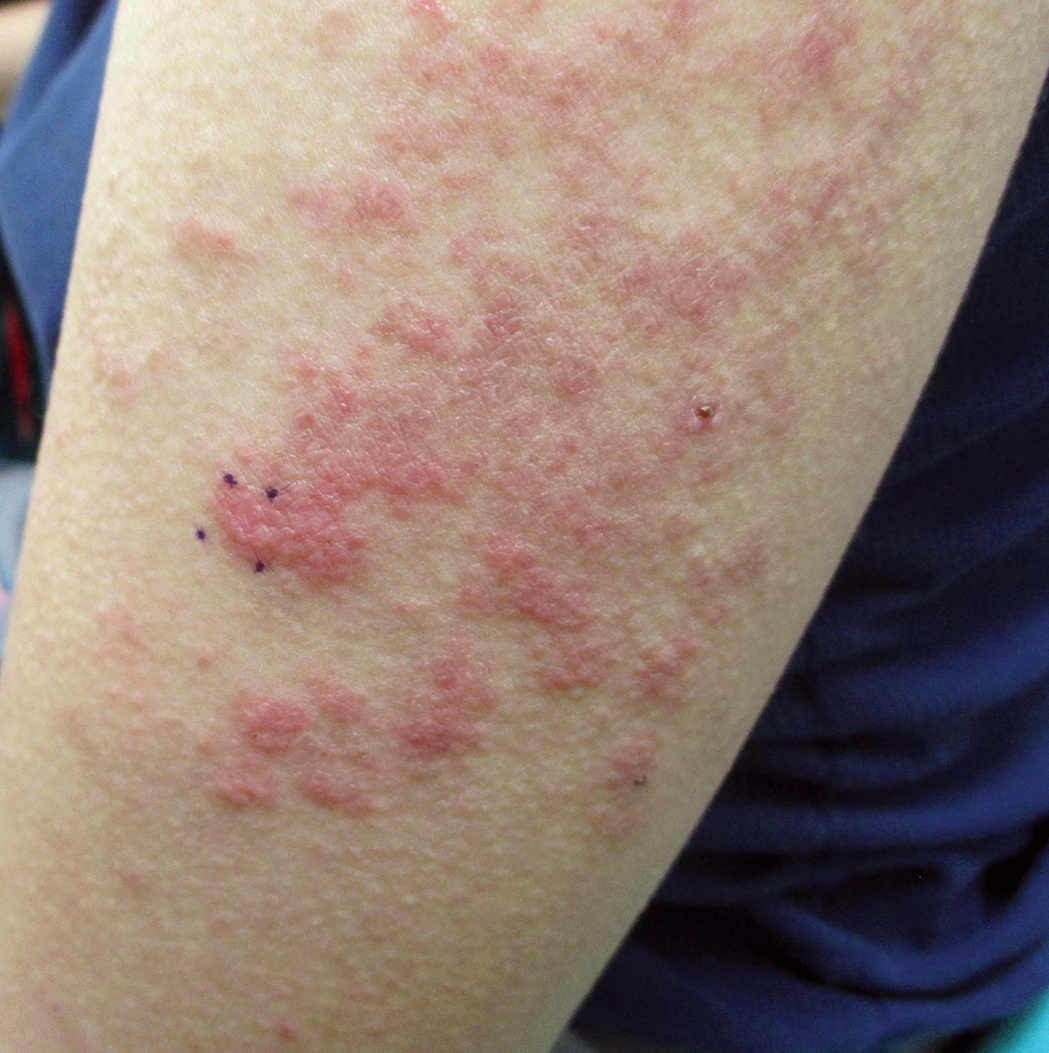
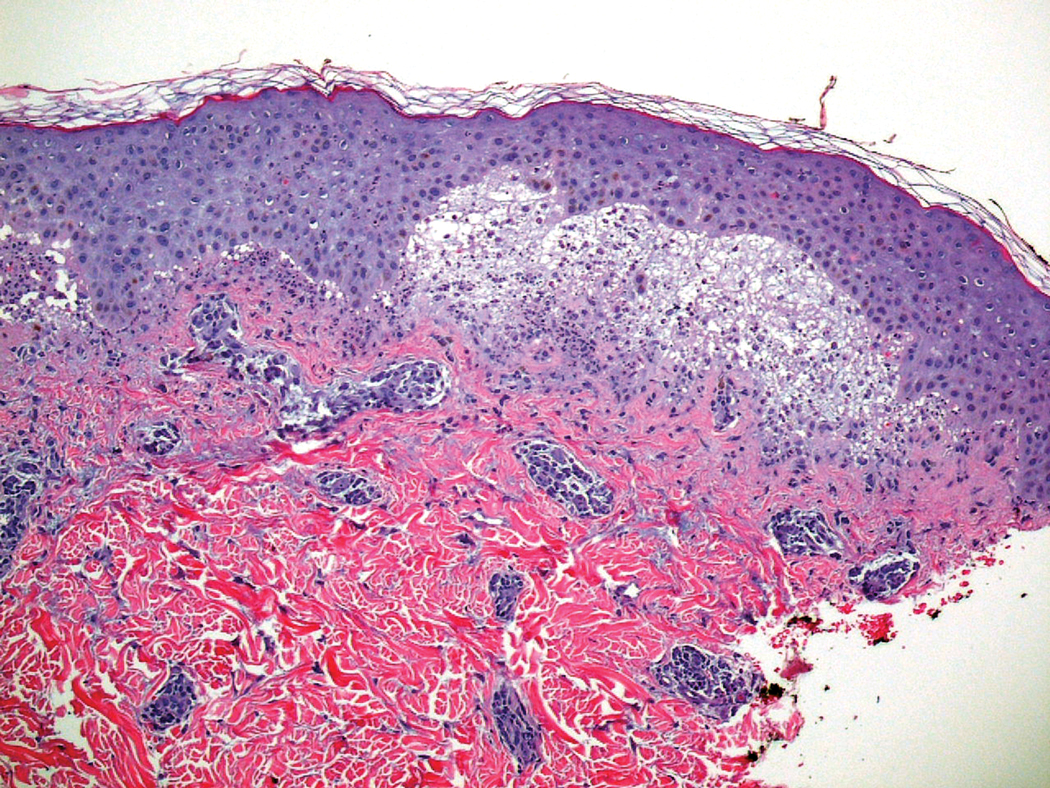
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Practice Points
- Bullous systemic lupus erythematosus (BSLE) can present with a waxing and waning course punctuated by flares.
- Different clinical presentations can occur over the disease course.
- Rituximab is a viable treatment option in BSLE.
Acute Graft-vs-host Disease Following Liver Transplantation
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
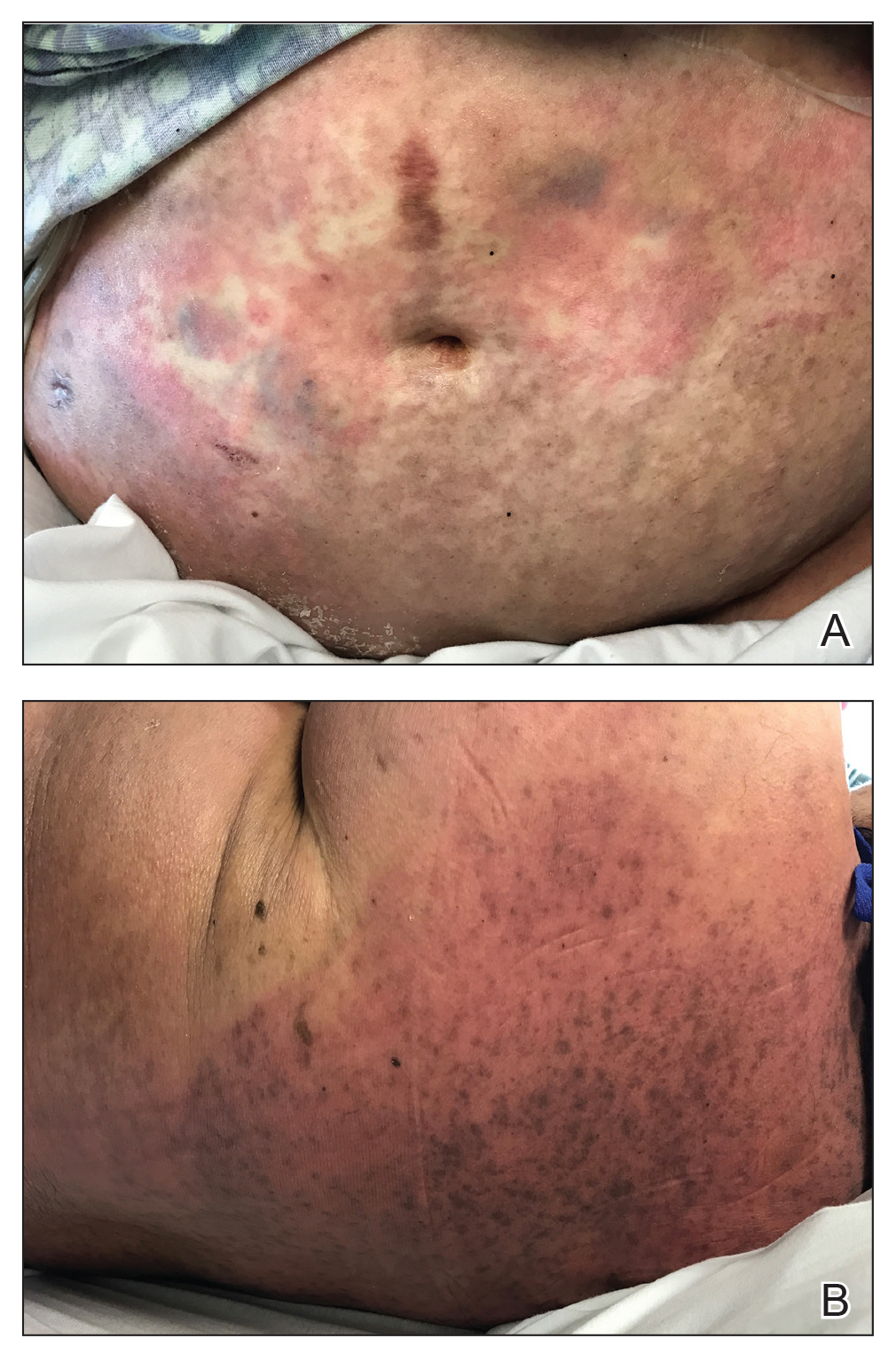
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
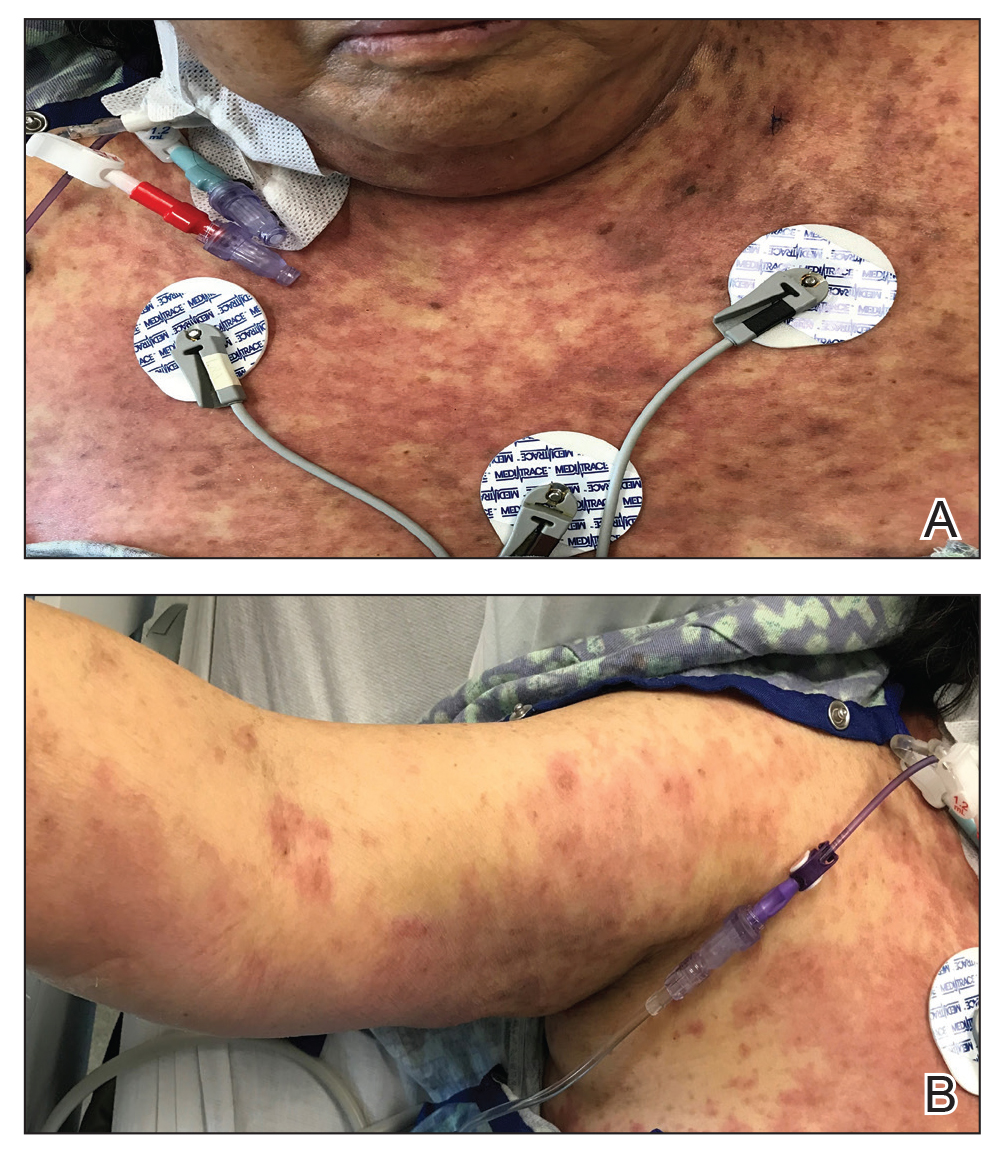
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
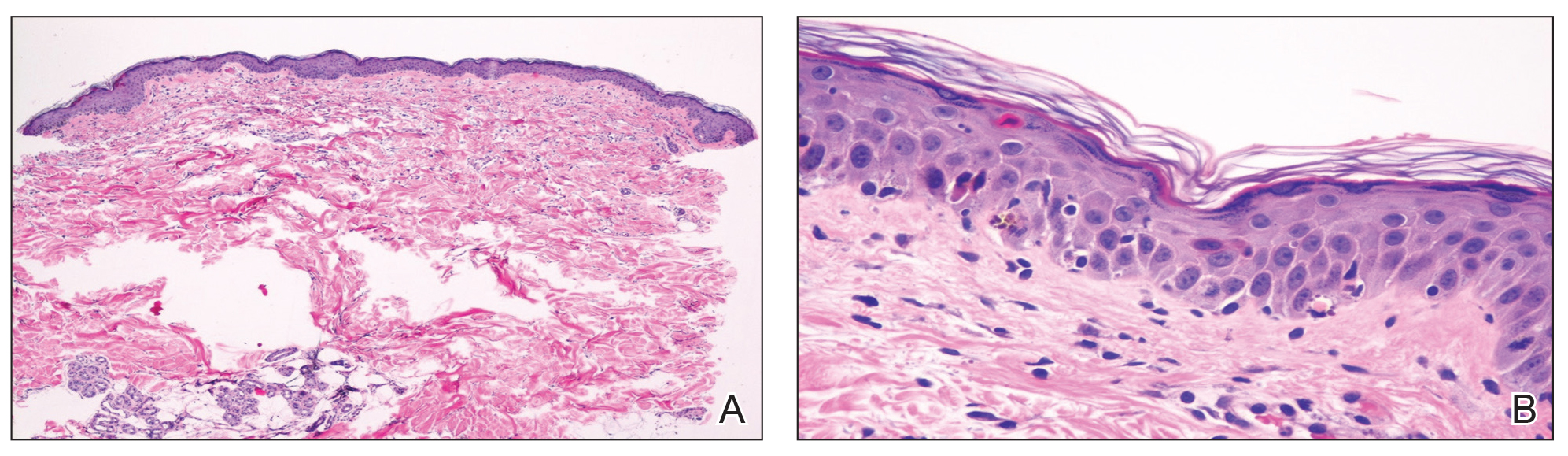
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
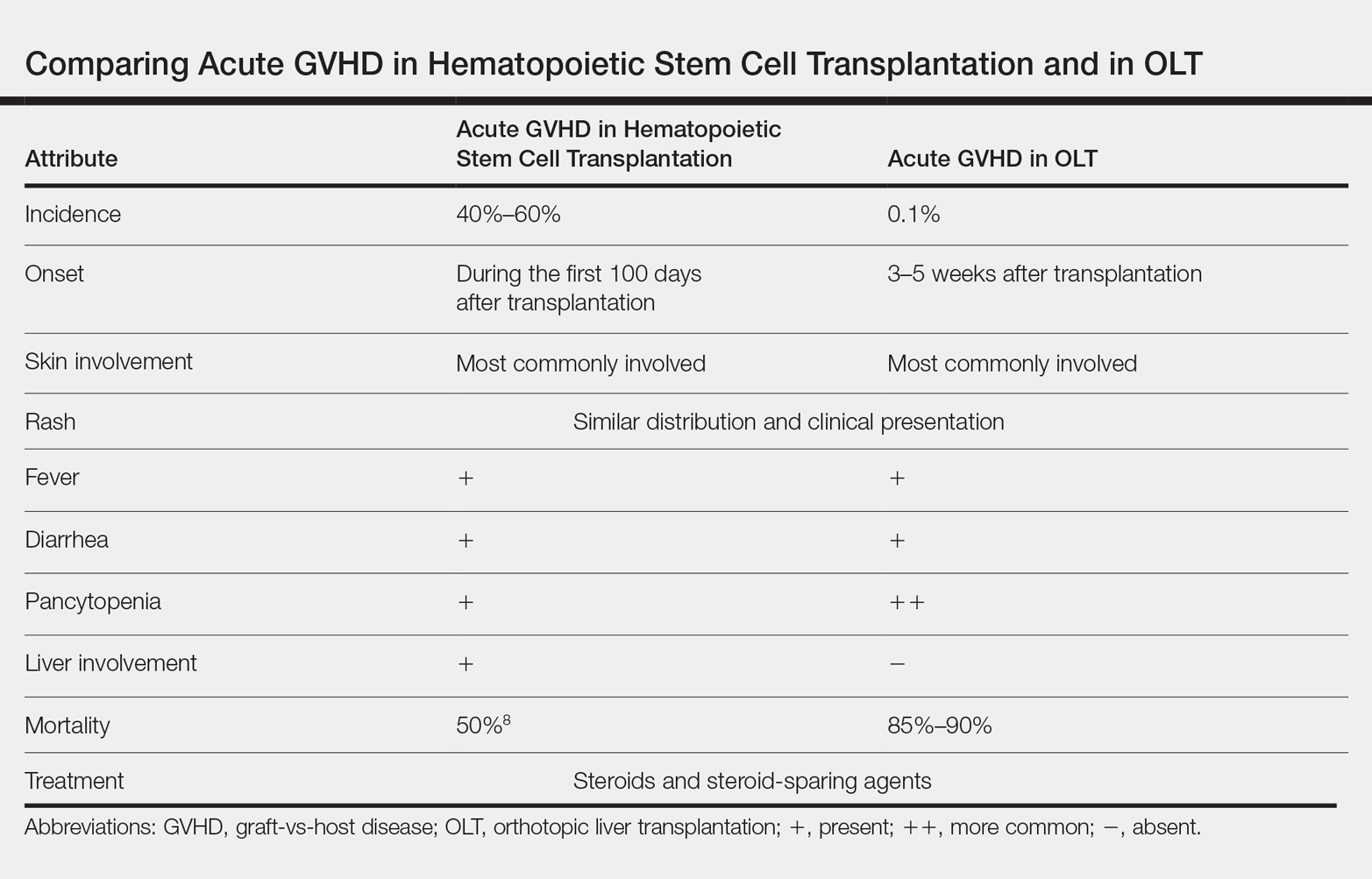
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Practice Points
- Acute graft-vs-host disease (GVHD) is a T cell–mediated reaction in which donor T lymphocytes attack host tissue in the setting of immunosuppression.
- Acute GVHD is more common in allogeneic stem cell transplantation but can occur in the setting of solid organ transplantation.
- Symptoms of acute GVHD include rash with or without pruritus, fever, pancytopenia, and diarrhea.
- Early recognition and treatment with systemic steroids can improve mortality.
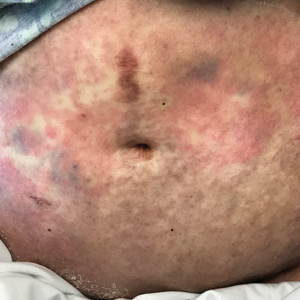
Adding ipilimumab to nivolumab provides no benefit in SCC trial
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.

However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.
The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.
The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.
The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
REPORTING FROM ASCO 2019
Key clinical point: Ipilimumab plus nivolumab appears no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
Major finding: The median progression-free survival was 3.8 months in the ipilimumab plus nivolumab arm and 2.9 months in the nivolumab arm (P = .19). The median overall survival was 10.0 months and 11.0 months, respectively (P = .82).
Study details: A phase 3 trial of 275 previously treated patients with stage IV or recurrent squamous cell lung cancer.
Disclosures: This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research. The researchers reported relationships with a range of companies. Source: Bazhenova L et al. ASCO 2019, Abstract 9014.
FDA overlooked red flags in esketamine testing
For some patients, it also has dwelled in the shadows of conventional medicine as a depression treatment – prescribed by their doctors, but not approved for that purpose by the federal agency responsible for determining which treatments are “safe and effective.”
That effectively changed in March, when the Food and Drug Administration approved a ketamine cousin called esketamine, taken as a nasal spray, for patients with intractable depression. With that, the esketamine nasal spray, under the brand name Spravato, was introduced as a miracle drug – announced in press releases, celebrated on the evening news, and embraced by major health care providers like the Department of Veterans Affairs.
The problem, critics say, is that the drug’s manufacturer, Janssen, provided the FDA with at best modest evidence it worked and then only in limited trials. It presented no information about the safety of Spravato for long-term use beyond 60 weeks. And three patients who received the drug died by suicide during clinical trials, compared with none in the control group, which raised red flags Janssen and the FDA dismissed.
The FDA, under political pressure to rapidly green-light drugs that treat life-threatening conditions, approved it anyway. And, though Spravato’s appearance on the market was greeted with public applause, some deep misgivings were expressed at its day-long review meeting and in the agency’s own briefing materials, according to public recordings, documents, and interviews with participants, KHN found.
Jess Fiedorowicz, MD, director of the Mood Disorders Center at the University of Iowa, Iowa City, and a member of the FDA advisory committee that reviewed the drug, described its benefit as “almost certainly exaggerated” after hearing the evidence.
Dr. Fiedorowicz said he expected at least a split decision by the committee. “And then it went strongly in favor, which surprised me,” he said in an interview.
Esketamine’s trajectory to approval shows – step by step – how drugmakers can take advantage of shortcuts in the FDA process with the agency’s blessing and maneuver through safety and efficacy reviews to bring a lucrative drug to market.
Step 1: In late 2013, Janssen got the FDA to designate esketamine a “breakthrough therapy” because it showed the potential to reverse depression rapidly — a holy grail for suicidal patients, such as those in an emergency room. That potential was based on a 2-day study during which 30 patients were given esketamine intravenously.
“Breakthrough therapy” status puts drugs on a fast track to approval, with more frequent input from the FDA.
Step 2: But discussions between regulators and drug manufacturers can affect the amount and quality of evidence required by the agency. In the case of Spravato, they involved questions like “How many drugs must fail before a patient’s depression is considered intractable or ‘treatment resistant’?” and “How many successful clinical trials are necessary for FDA approval?”
Step 3: Any prior agreements can leave the FDA’s expert advisory committees hamstrung in reaching a verdict. Dr. Fiedorowicz abstained on Spravato because, though he considered Janssen’s study design flawed, the FDA had approved it.
The expert panel cleared the drug according to the evidence that the agency and Janssen had determined was sufficient. Matthew Rudorfer, MD, an associate director at the National Institute of Mental Health, concluded that the “benefits outweighed the risks.” Explaining his “yes” vote, he said, “I think we’re all agreeing on the very important, and sometimes life-or-death, risk of inadequately treated depression that factored into my equation.”
But others who also voted “yes” were more explicit in their qualms. “I don’t think that we really understand what happens when you take this week after week for weeks and months and years,” said Steven Meisel, PharmD, system director of medication safety for Fairview Health Services based in Minneapolis.
A Nasal Spray Offers A Path To A Patent
Spravato is available only under supervision at a certified facility where patients must be monitored for at least two hours after taking the drug to watch for side effects like dizziness, detachment from reality, and increased blood pressure, as well as to reduce the risk of abuse. Patients must take it with an oral antidepressant.
Despite those requirements, Janssen, part of Johnson & Johnson, defended its new offering. “Until the recent FDA approval of Spravato, health care providers haven’t had any new medication options,” Kristina Chang, a Janssen spokeswoman, wrote in an emailed statement.
Esketamine is the first new type of drug approved to treat severe depression in about three decades.
Although ketamine has been used off-label for years to treat depression and posttraumatic stress disorder, drugmakers saw little profit in doing the studies to prove to the FDA that it worked for that purpose. But a nasal spray of esketamine, which is derived from ketamine and is (in some studies) more potent, could be patented as a new drug.
Although Spravato costs more than $4,700 for the first month of treatment (not including the cost of monitoring or the oral antidepressant), insurers are more likely to reimburse for Spravato than for ketamine, since the latter is not approved for depression.
Shortly before the committee began voting, a study participant identifying herself only as “Patient 20015525” said, “I am offering real-world proof of efficacy, and that is I am both alive and here today.”
The drug did not work “for the majority of people who took it,” Dr. Meisel, the medication safety expert, said in an interview. “But for a subset of those for whom it did work, it was dramatic.”
Concerns About Testing Precedents
Those considerations apparently helped outweigh several scientific red flags that committee members called out during the hearing.
Although the drug had gotten breakthrough status because of its potential for results within 24 hours, the trials were not persuasive enough for the FDA to label it “rapid acting.”
The FDA typically requires that applicants provide at least two clinical trials demonstrating the drug’s efficacy, “each convincing on its own.” Janssen provided just one successful short-term, double-blind trial of esketamine. Two other trials it ran to test efficacy fell short.
To reach the two-trial threshold, the FDA broke its precedent for psychiatric drugs and allowed the company to count a trial conducted to study a different topic: relapse and remission trends. But, by definition, every patient in the trial had already taken and seen improvement from esketamine.
What’s more, that single positive efficacy trial showed just a 4-point improvement in depression symptoms, compared with the placebo treatment, on a 60-point scale some clinicians use to measure depression severity. Some committee members noted the trial wasn’t really blind since participants could recognize they were getting the drug from side effects like a temporary out-of-body sensation.
Finally, the FDA lowered the bar for “treatment-resistant depression.” Initially, for inclusion, trial participants would have had to have failed two classes of oral antidepressants.
Less than 2 years later, the FDA loosened that definition, saying a patient needed only to have taken two different pills, no matter the class.
Forty-nine of the 227 people who participated in Janssen’s only successful efficacy trial had failed just one class of oral antidepressants. “They weeded out the true treatment-resistant patients,” said Erick Turner, MD, a former FDA reviewer who serves on the committee but did not attend the meeting.
Six participants died during the studies, three by suicide. Janssen and the FDA dismissed the deaths as unrelated to the drug, noting the low number and lack of a pattern among hundreds of participants. They also pointed out that suicidal behavior is associated with severe depression – even though those who had suicidal ideation with some intent to act in the previous 6 months, or a history of suicidal behavior in the previous year, were excluded from the studies.
In a recent commentary in the American Journal of Psychiatry, Alan Schatzberg, MD, a Stanford (Calif.) University researcher who has studied ketamine, suggested there might be a link caused by “a protracted withdrawal reaction, as has been reported with opioids,” since ketamine appears to interact with the brain’s opioid receptors (Am J Psych. 2019. doi: 10.1176/appi.ajp.2019.19040423).
Kim Witczak, the committee’s consumer representative, found Janssen’s conclusion about the suicides unsatisfying. “I just feel like it was kind of a quick brush-over,” Ms. Witczak said in an interview. She voted against the drug.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
For some patients, it also has dwelled in the shadows of conventional medicine as a depression treatment – prescribed by their doctors, but not approved for that purpose by the federal agency responsible for determining which treatments are “safe and effective.”
That effectively changed in March, when the Food and Drug Administration approved a ketamine cousin called esketamine, taken as a nasal spray, for patients with intractable depression. With that, the esketamine nasal spray, under the brand name Spravato, was introduced as a miracle drug – announced in press releases, celebrated on the evening news, and embraced by major health care providers like the Department of Veterans Affairs.
The problem, critics say, is that the drug’s manufacturer, Janssen, provided the FDA with at best modest evidence it worked and then only in limited trials. It presented no information about the safety of Spravato for long-term use beyond 60 weeks. And three patients who received the drug died by suicide during clinical trials, compared with none in the control group, which raised red flags Janssen and the FDA dismissed.
The FDA, under political pressure to rapidly green-light drugs that treat life-threatening conditions, approved it anyway. And, though Spravato’s appearance on the market was greeted with public applause, some deep misgivings were expressed at its day-long review meeting and in the agency’s own briefing materials, according to public recordings, documents, and interviews with participants, KHN found.
Jess Fiedorowicz, MD, director of the Mood Disorders Center at the University of Iowa, Iowa City, and a member of the FDA advisory committee that reviewed the drug, described its benefit as “almost certainly exaggerated” after hearing the evidence.
Dr. Fiedorowicz said he expected at least a split decision by the committee. “And then it went strongly in favor, which surprised me,” he said in an interview.
Esketamine’s trajectory to approval shows – step by step – how drugmakers can take advantage of shortcuts in the FDA process with the agency’s blessing and maneuver through safety and efficacy reviews to bring a lucrative drug to market.
Step 1: In late 2013, Janssen got the FDA to designate esketamine a “breakthrough therapy” because it showed the potential to reverse depression rapidly — a holy grail for suicidal patients, such as those in an emergency room. That potential was based on a 2-day study during which 30 patients were given esketamine intravenously.
“Breakthrough therapy” status puts drugs on a fast track to approval, with more frequent input from the FDA.
Step 2: But discussions between regulators and drug manufacturers can affect the amount and quality of evidence required by the agency. In the case of Spravato, they involved questions like “How many drugs must fail before a patient’s depression is considered intractable or ‘treatment resistant’?” and “How many successful clinical trials are necessary for FDA approval?”
Step 3: Any prior agreements can leave the FDA’s expert advisory committees hamstrung in reaching a verdict. Dr. Fiedorowicz abstained on Spravato because, though he considered Janssen’s study design flawed, the FDA had approved it.
The expert panel cleared the drug according to the evidence that the agency and Janssen had determined was sufficient. Matthew Rudorfer, MD, an associate director at the National Institute of Mental Health, concluded that the “benefits outweighed the risks.” Explaining his “yes” vote, he said, “I think we’re all agreeing on the very important, and sometimes life-or-death, risk of inadequately treated depression that factored into my equation.”
But others who also voted “yes” were more explicit in their qualms. “I don’t think that we really understand what happens when you take this week after week for weeks and months and years,” said Steven Meisel, PharmD, system director of medication safety for Fairview Health Services based in Minneapolis.
A Nasal Spray Offers A Path To A Patent
Spravato is available only under supervision at a certified facility where patients must be monitored for at least two hours after taking the drug to watch for side effects like dizziness, detachment from reality, and increased blood pressure, as well as to reduce the risk of abuse. Patients must take it with an oral antidepressant.
Despite those requirements, Janssen, part of Johnson & Johnson, defended its new offering. “Until the recent FDA approval of Spravato, health care providers haven’t had any new medication options,” Kristina Chang, a Janssen spokeswoman, wrote in an emailed statement.
Esketamine is the first new type of drug approved to treat severe depression in about three decades.
Although ketamine has been used off-label for years to treat depression and posttraumatic stress disorder, drugmakers saw little profit in doing the studies to prove to the FDA that it worked for that purpose. But a nasal spray of esketamine, which is derived from ketamine and is (in some studies) more potent, could be patented as a new drug.
Although Spravato costs more than $4,700 for the first month of treatment (not including the cost of monitoring or the oral antidepressant), insurers are more likely to reimburse for Spravato than for ketamine, since the latter is not approved for depression.
Shortly before the committee began voting, a study participant identifying herself only as “Patient 20015525” said, “I am offering real-world proof of efficacy, and that is I am both alive and here today.”
The drug did not work “for the majority of people who took it,” Dr. Meisel, the medication safety expert, said in an interview. “But for a subset of those for whom it did work, it was dramatic.”
Concerns About Testing Precedents
Those considerations apparently helped outweigh several scientific red flags that committee members called out during the hearing.
Although the drug had gotten breakthrough status because of its potential for results within 24 hours, the trials were not persuasive enough for the FDA to label it “rapid acting.”
The FDA typically requires that applicants provide at least two clinical trials demonstrating the drug’s efficacy, “each convincing on its own.” Janssen provided just one successful short-term, double-blind trial of esketamine. Two other trials it ran to test efficacy fell short.
To reach the two-trial threshold, the FDA broke its precedent for psychiatric drugs and allowed the company to count a trial conducted to study a different topic: relapse and remission trends. But, by definition, every patient in the trial had already taken and seen improvement from esketamine.
What’s more, that single positive efficacy trial showed just a 4-point improvement in depression symptoms, compared with the placebo treatment, on a 60-point scale some clinicians use to measure depression severity. Some committee members noted the trial wasn’t really blind since participants could recognize they were getting the drug from side effects like a temporary out-of-body sensation.
Finally, the FDA lowered the bar for “treatment-resistant depression.” Initially, for inclusion, trial participants would have had to have failed two classes of oral antidepressants.
Less than 2 years later, the FDA loosened that definition, saying a patient needed only to have taken two different pills, no matter the class.
Forty-nine of the 227 people who participated in Janssen’s only successful efficacy trial had failed just one class of oral antidepressants. “They weeded out the true treatment-resistant patients,” said Erick Turner, MD, a former FDA reviewer who serves on the committee but did not attend the meeting.
Six participants died during the studies, three by suicide. Janssen and the FDA dismissed the deaths as unrelated to the drug, noting the low number and lack of a pattern among hundreds of participants. They also pointed out that suicidal behavior is associated with severe depression – even though those who had suicidal ideation with some intent to act in the previous 6 months, or a history of suicidal behavior in the previous year, were excluded from the studies.
In a recent commentary in the American Journal of Psychiatry, Alan Schatzberg, MD, a Stanford (Calif.) University researcher who has studied ketamine, suggested there might be a link caused by “a protracted withdrawal reaction, as has been reported with opioids,” since ketamine appears to interact with the brain’s opioid receptors (Am J Psych. 2019. doi: 10.1176/appi.ajp.2019.19040423).
Kim Witczak, the committee’s consumer representative, found Janssen’s conclusion about the suicides unsatisfying. “I just feel like it was kind of a quick brush-over,” Ms. Witczak said in an interview. She voted against the drug.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
For some patients, it also has dwelled in the shadows of conventional medicine as a depression treatment – prescribed by their doctors, but not approved for that purpose by the federal agency responsible for determining which treatments are “safe and effective.”
That effectively changed in March, when the Food and Drug Administration approved a ketamine cousin called esketamine, taken as a nasal spray, for patients with intractable depression. With that, the esketamine nasal spray, under the brand name Spravato, was introduced as a miracle drug – announced in press releases, celebrated on the evening news, and embraced by major health care providers like the Department of Veterans Affairs.
The problem, critics say, is that the drug’s manufacturer, Janssen, provided the FDA with at best modest evidence it worked and then only in limited trials. It presented no information about the safety of Spravato for long-term use beyond 60 weeks. And three patients who received the drug died by suicide during clinical trials, compared with none in the control group, which raised red flags Janssen and the FDA dismissed.
The FDA, under political pressure to rapidly green-light drugs that treat life-threatening conditions, approved it anyway. And, though Spravato’s appearance on the market was greeted with public applause, some deep misgivings were expressed at its day-long review meeting and in the agency’s own briefing materials, according to public recordings, documents, and interviews with participants, KHN found.
Jess Fiedorowicz, MD, director of the Mood Disorders Center at the University of Iowa, Iowa City, and a member of the FDA advisory committee that reviewed the drug, described its benefit as “almost certainly exaggerated” after hearing the evidence.
Dr. Fiedorowicz said he expected at least a split decision by the committee. “And then it went strongly in favor, which surprised me,” he said in an interview.
Esketamine’s trajectory to approval shows – step by step – how drugmakers can take advantage of shortcuts in the FDA process with the agency’s blessing and maneuver through safety and efficacy reviews to bring a lucrative drug to market.
Step 1: In late 2013, Janssen got the FDA to designate esketamine a “breakthrough therapy” because it showed the potential to reverse depression rapidly — a holy grail for suicidal patients, such as those in an emergency room. That potential was based on a 2-day study during which 30 patients were given esketamine intravenously.
“Breakthrough therapy” status puts drugs on a fast track to approval, with more frequent input from the FDA.
Step 2: But discussions between regulators and drug manufacturers can affect the amount and quality of evidence required by the agency. In the case of Spravato, they involved questions like “How many drugs must fail before a patient’s depression is considered intractable or ‘treatment resistant’?” and “How many successful clinical trials are necessary for FDA approval?”
Step 3: Any prior agreements can leave the FDA’s expert advisory committees hamstrung in reaching a verdict. Dr. Fiedorowicz abstained on Spravato because, though he considered Janssen’s study design flawed, the FDA had approved it.
The expert panel cleared the drug according to the evidence that the agency and Janssen had determined was sufficient. Matthew Rudorfer, MD, an associate director at the National Institute of Mental Health, concluded that the “benefits outweighed the risks.” Explaining his “yes” vote, he said, “I think we’re all agreeing on the very important, and sometimes life-or-death, risk of inadequately treated depression that factored into my equation.”
But others who also voted “yes” were more explicit in their qualms. “I don’t think that we really understand what happens when you take this week after week for weeks and months and years,” said Steven Meisel, PharmD, system director of medication safety for Fairview Health Services based in Minneapolis.
A Nasal Spray Offers A Path To A Patent
Spravato is available only under supervision at a certified facility where patients must be monitored for at least two hours after taking the drug to watch for side effects like dizziness, detachment from reality, and increased blood pressure, as well as to reduce the risk of abuse. Patients must take it with an oral antidepressant.
Despite those requirements, Janssen, part of Johnson & Johnson, defended its new offering. “Until the recent FDA approval of Spravato, health care providers haven’t had any new medication options,” Kristina Chang, a Janssen spokeswoman, wrote in an emailed statement.
Esketamine is the first new type of drug approved to treat severe depression in about three decades.
Although ketamine has been used off-label for years to treat depression and posttraumatic stress disorder, drugmakers saw little profit in doing the studies to prove to the FDA that it worked for that purpose. But a nasal spray of esketamine, which is derived from ketamine and is (in some studies) more potent, could be patented as a new drug.
Although Spravato costs more than $4,700 for the first month of treatment (not including the cost of monitoring or the oral antidepressant), insurers are more likely to reimburse for Spravato than for ketamine, since the latter is not approved for depression.
Shortly before the committee began voting, a study participant identifying herself only as “Patient 20015525” said, “I am offering real-world proof of efficacy, and that is I am both alive and here today.”
The drug did not work “for the majority of people who took it,” Dr. Meisel, the medication safety expert, said in an interview. “But for a subset of those for whom it did work, it was dramatic.”
Concerns About Testing Precedents
Those considerations apparently helped outweigh several scientific red flags that committee members called out during the hearing.
Although the drug had gotten breakthrough status because of its potential for results within 24 hours, the trials were not persuasive enough for the FDA to label it “rapid acting.”
The FDA typically requires that applicants provide at least two clinical trials demonstrating the drug’s efficacy, “each convincing on its own.” Janssen provided just one successful short-term, double-blind trial of esketamine. Two other trials it ran to test efficacy fell short.
To reach the two-trial threshold, the FDA broke its precedent for psychiatric drugs and allowed the company to count a trial conducted to study a different topic: relapse and remission trends. But, by definition, every patient in the trial had already taken and seen improvement from esketamine.
What’s more, that single positive efficacy trial showed just a 4-point improvement in depression symptoms, compared with the placebo treatment, on a 60-point scale some clinicians use to measure depression severity. Some committee members noted the trial wasn’t really blind since participants could recognize they were getting the drug from side effects like a temporary out-of-body sensation.
Finally, the FDA lowered the bar for “treatment-resistant depression.” Initially, for inclusion, trial participants would have had to have failed two classes of oral antidepressants.
Less than 2 years later, the FDA loosened that definition, saying a patient needed only to have taken two different pills, no matter the class.
Forty-nine of the 227 people who participated in Janssen’s only successful efficacy trial had failed just one class of oral antidepressants. “They weeded out the true treatment-resistant patients,” said Erick Turner, MD, a former FDA reviewer who serves on the committee but did not attend the meeting.
Six participants died during the studies, three by suicide. Janssen and the FDA dismissed the deaths as unrelated to the drug, noting the low number and lack of a pattern among hundreds of participants. They also pointed out that suicidal behavior is associated with severe depression – even though those who had suicidal ideation with some intent to act in the previous 6 months, or a history of suicidal behavior in the previous year, were excluded from the studies.
In a recent commentary in the American Journal of Psychiatry, Alan Schatzberg, MD, a Stanford (Calif.) University researcher who has studied ketamine, suggested there might be a link caused by “a protracted withdrawal reaction, as has been reported with opioids,” since ketamine appears to interact with the brain’s opioid receptors (Am J Psych. 2019. doi: 10.1176/appi.ajp.2019.19040423).
Kim Witczak, the committee’s consumer representative, found Janssen’s conclusion about the suicides unsatisfying. “I just feel like it was kind of a quick brush-over,” Ms. Witczak said in an interview. She voted against the drug.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Intranasal esketamine plus antidepressant deflects relapse
Esketamine nasal spray used with an oral antidepressant was significantly more effective at delaying a relapse of depression compared with placebo, based on data from 297 adults in remission.
Patients with treatment-resistant depression are more likely to relapse, wrote Ella J. Daly, MD, of Janssen Research and Development, Titusville, N.J., and colleagues.
In the SUSTAIN-1 study, published in JAMA Psychiatry, the researchers randomized 297 adults in the maintenance phase of depression treatment to esketamine hydrochloride or placebo. The average age of the patients was 46 years, and 66% were female.
among the 176 patients who achieved stable remission. In addition, relapse occurred in 25.8% in the esketamine and antidepressant group and 57.6 in the antidepressant and placebo among the patients who achieved stable response.
The median duration esketamine use during the maintenance phase was 17.7 weeks among patients who achieved stable remission and 19.4 among those who achieved stable response.
The study was designed to include a 4-week screening and prospective observation phase, a 4-week open-label induction phase, a 12-week optimization phase, a maintenance phase, and a 2-week posttreatment follow-up phase.
The most common adverse events reported by esketamine patients were transient dysgeusia, vertigo, dissociation, somnolence, and dizziness. Most of these effects were moderate; no cases of respiratory depression, interstitial cystitis, or death were reported.
The results were limited by several factors, including the transient effects of esketamine that made blinding difficult, the researchers noted. However, the findings support the safety of the spray and “significant, clinically meaningful superiority” of the spray, compared with placebo, for relapse prevention in patients with treatment-resistant depression, they concluded.
Janssen Research and Development funded the study. Dr. Daly and several coauthors are employees of the company.
SOURCE: Daly E et al. JAMA Psychiatry. 2019 Jun 5. doi: 10.1001/jamapsychiatry.2019.1189.
Esketamine nasal spray used with an oral antidepressant was significantly more effective at delaying a relapse of depression compared with placebo, based on data from 297 adults in remission.
Patients with treatment-resistant depression are more likely to relapse, wrote Ella J. Daly, MD, of Janssen Research and Development, Titusville, N.J., and colleagues.
In the SUSTAIN-1 study, published in JAMA Psychiatry, the researchers randomized 297 adults in the maintenance phase of depression treatment to esketamine hydrochloride or placebo. The average age of the patients was 46 years, and 66% were female.
among the 176 patients who achieved stable remission. In addition, relapse occurred in 25.8% in the esketamine and antidepressant group and 57.6 in the antidepressant and placebo among the patients who achieved stable response.
The median duration esketamine use during the maintenance phase was 17.7 weeks among patients who achieved stable remission and 19.4 among those who achieved stable response.
The study was designed to include a 4-week screening and prospective observation phase, a 4-week open-label induction phase, a 12-week optimization phase, a maintenance phase, and a 2-week posttreatment follow-up phase.
The most common adverse events reported by esketamine patients were transient dysgeusia, vertigo, dissociation, somnolence, and dizziness. Most of these effects were moderate; no cases of respiratory depression, interstitial cystitis, or death were reported.
The results were limited by several factors, including the transient effects of esketamine that made blinding difficult, the researchers noted. However, the findings support the safety of the spray and “significant, clinically meaningful superiority” of the spray, compared with placebo, for relapse prevention in patients with treatment-resistant depression, they concluded.
Janssen Research and Development funded the study. Dr. Daly and several coauthors are employees of the company.
SOURCE: Daly E et al. JAMA Psychiatry. 2019 Jun 5. doi: 10.1001/jamapsychiatry.2019.1189.
Esketamine nasal spray used with an oral antidepressant was significantly more effective at delaying a relapse of depression compared with placebo, based on data from 297 adults in remission.
Patients with treatment-resistant depression are more likely to relapse, wrote Ella J. Daly, MD, of Janssen Research and Development, Titusville, N.J., and colleagues.
In the SUSTAIN-1 study, published in JAMA Psychiatry, the researchers randomized 297 adults in the maintenance phase of depression treatment to esketamine hydrochloride or placebo. The average age of the patients was 46 years, and 66% were female.
among the 176 patients who achieved stable remission. In addition, relapse occurred in 25.8% in the esketamine and antidepressant group and 57.6 in the antidepressant and placebo among the patients who achieved stable response.
The median duration esketamine use during the maintenance phase was 17.7 weeks among patients who achieved stable remission and 19.4 among those who achieved stable response.
The study was designed to include a 4-week screening and prospective observation phase, a 4-week open-label induction phase, a 12-week optimization phase, a maintenance phase, and a 2-week posttreatment follow-up phase.
The most common adverse events reported by esketamine patients were transient dysgeusia, vertigo, dissociation, somnolence, and dizziness. Most of these effects were moderate; no cases of respiratory depression, interstitial cystitis, or death were reported.
The results were limited by several factors, including the transient effects of esketamine that made blinding difficult, the researchers noted. However, the findings support the safety of the spray and “significant, clinically meaningful superiority” of the spray, compared with placebo, for relapse prevention in patients with treatment-resistant depression, they concluded.
Janssen Research and Development funded the study. Dr. Daly and several coauthors are employees of the company.
SOURCE: Daly E et al. JAMA Psychiatry. 2019 Jun 5. doi: 10.1001/jamapsychiatry.2019.1189.
FROM JAMA PSYCHIATRY
Novel genetic therapy reduces key protein in Huntington’s disease
according to a study published in the New England Journal of Medicine.
Huntington’s disease is an autosomal-dominant neurodegenerative disease caused by CAG trinucleotide repeat expansion in HTT, resulting in a mutant huntingtin protein. No disease-modifying treatment currently exists. The experimental therapy tested in this trial, developed by Ionis Pharmaceuticals and licensed to Roche as HTTRx, is an antisense oligonucleotide that inhibits HTT messenger RNA signaling specific to the production of the mutant huntingtin protein implicated in Huntington’s disease. Whether HTTRx, which is delivered intrathecally, can produce functional or cognitive improvement is yet unclear, as this randomized, double-blinded, multiple-ascending-dose, placebo-controlled trial, which enrolled 46 patients in Canada, Germany, and the United Kingdom, was primarily a safety study.
For the phase 1-2a trial, lead author Sarah J. Tabrizi, MB, ChB, PhD, of University College London and colleagues assigned patients with early Huntington’s disease to monthly intrathecal injections of one of five different doses of HTTRx (10, 30, 60, 90 or 120 mg), or placebo. Most patients (n = 34) received active drug. After the 85-day treatment period, in which four doses were delivered, patients were followed for 4 months.
The treatment groups saw a mean dose-dependent reduction from baseline in the concentration of CSF mutant huntingtin of between –20% and –42% at 28 days post dosing, while the placebo arm saw an increase of a mean 10%. The most common adverse events seen in the trial were procedure-related pain and headache following spinal puncture.
Other endpoints in the study included concentrations of mutant huntingtin in plasma, the effect of treatment on other neurodegenerative biomarkers, and cognitive scores.
The median peak plasma concentrations of HTTRx were reached within 4 hours after the bolus intrathecal administration and declined to less than 30% of the peak concentration by 24 hours after administration. There was no evidence of accumulation of concentration in plasma 24 hours after dose administration.
Functional, cognitive, psychiatric, and neurologic clinical outcomes were generally unchanged at the dose-group level during the trial, and no meaningful differences were observed between patients who received placebo and patients who received active treatment, regardless of the dose level.
An open-label, follow-up study in the same group of patients, all of whom have been assigned to the 120-mg dose monthly or every other month, is expected to end in October 2019. While the extension study is also mainly a safety study, it will also look at biomarkers and cognitive scores over a longer treatment period.
The study was funded by Ionis Pharmaceuticals and F. Hoffmann–La Roche, and most of the authors, including Dr. Tabrizi, reported financial relationships with one or both entities.
SOURCE: Tabrizi SJ et al. N Eng J Med. 2019:380;2307-16.
according to a study published in the New England Journal of Medicine.
Huntington’s disease is an autosomal-dominant neurodegenerative disease caused by CAG trinucleotide repeat expansion in HTT, resulting in a mutant huntingtin protein. No disease-modifying treatment currently exists. The experimental therapy tested in this trial, developed by Ionis Pharmaceuticals and licensed to Roche as HTTRx, is an antisense oligonucleotide that inhibits HTT messenger RNA signaling specific to the production of the mutant huntingtin protein implicated in Huntington’s disease. Whether HTTRx, which is delivered intrathecally, can produce functional or cognitive improvement is yet unclear, as this randomized, double-blinded, multiple-ascending-dose, placebo-controlled trial, which enrolled 46 patients in Canada, Germany, and the United Kingdom, was primarily a safety study.
For the phase 1-2a trial, lead author Sarah J. Tabrizi, MB, ChB, PhD, of University College London and colleagues assigned patients with early Huntington’s disease to monthly intrathecal injections of one of five different doses of HTTRx (10, 30, 60, 90 or 120 mg), or placebo. Most patients (n = 34) received active drug. After the 85-day treatment period, in which four doses were delivered, patients were followed for 4 months.
The treatment groups saw a mean dose-dependent reduction from baseline in the concentration of CSF mutant huntingtin of between –20% and –42% at 28 days post dosing, while the placebo arm saw an increase of a mean 10%. The most common adverse events seen in the trial were procedure-related pain and headache following spinal puncture.
Other endpoints in the study included concentrations of mutant huntingtin in plasma, the effect of treatment on other neurodegenerative biomarkers, and cognitive scores.
The median peak plasma concentrations of HTTRx were reached within 4 hours after the bolus intrathecal administration and declined to less than 30% of the peak concentration by 24 hours after administration. There was no evidence of accumulation of concentration in plasma 24 hours after dose administration.
Functional, cognitive, psychiatric, and neurologic clinical outcomes were generally unchanged at the dose-group level during the trial, and no meaningful differences were observed between patients who received placebo and patients who received active treatment, regardless of the dose level.
An open-label, follow-up study in the same group of patients, all of whom have been assigned to the 120-mg dose monthly or every other month, is expected to end in October 2019. While the extension study is also mainly a safety study, it will also look at biomarkers and cognitive scores over a longer treatment period.
The study was funded by Ionis Pharmaceuticals and F. Hoffmann–La Roche, and most of the authors, including Dr. Tabrizi, reported financial relationships with one or both entities.
SOURCE: Tabrizi SJ et al. N Eng J Med. 2019:380;2307-16.
according to a study published in the New England Journal of Medicine.
Huntington’s disease is an autosomal-dominant neurodegenerative disease caused by CAG trinucleotide repeat expansion in HTT, resulting in a mutant huntingtin protein. No disease-modifying treatment currently exists. The experimental therapy tested in this trial, developed by Ionis Pharmaceuticals and licensed to Roche as HTTRx, is an antisense oligonucleotide that inhibits HTT messenger RNA signaling specific to the production of the mutant huntingtin protein implicated in Huntington’s disease. Whether HTTRx, which is delivered intrathecally, can produce functional or cognitive improvement is yet unclear, as this randomized, double-blinded, multiple-ascending-dose, placebo-controlled trial, which enrolled 46 patients in Canada, Germany, and the United Kingdom, was primarily a safety study.
For the phase 1-2a trial, lead author Sarah J. Tabrizi, MB, ChB, PhD, of University College London and colleagues assigned patients with early Huntington’s disease to monthly intrathecal injections of one of five different doses of HTTRx (10, 30, 60, 90 or 120 mg), or placebo. Most patients (n = 34) received active drug. After the 85-day treatment period, in which four doses were delivered, patients were followed for 4 months.
The treatment groups saw a mean dose-dependent reduction from baseline in the concentration of CSF mutant huntingtin of between –20% and –42% at 28 days post dosing, while the placebo arm saw an increase of a mean 10%. The most common adverse events seen in the trial were procedure-related pain and headache following spinal puncture.
Other endpoints in the study included concentrations of mutant huntingtin in plasma, the effect of treatment on other neurodegenerative biomarkers, and cognitive scores.
The median peak plasma concentrations of HTTRx were reached within 4 hours after the bolus intrathecal administration and declined to less than 30% of the peak concentration by 24 hours after administration. There was no evidence of accumulation of concentration in plasma 24 hours after dose administration.
Functional, cognitive, psychiatric, and neurologic clinical outcomes were generally unchanged at the dose-group level during the trial, and no meaningful differences were observed between patients who received placebo and patients who received active treatment, regardless of the dose level.
An open-label, follow-up study in the same group of patients, all of whom have been assigned to the 120-mg dose monthly or every other month, is expected to end in October 2019. While the extension study is also mainly a safety study, it will also look at biomarkers and cognitive scores over a longer treatment period.
The study was funded by Ionis Pharmaceuticals and F. Hoffmann–La Roche, and most of the authors, including Dr. Tabrizi, reported financial relationships with one or both entities.
SOURCE: Tabrizi SJ et al. N Eng J Med. 2019:380;2307-16.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
KRISTINE: Three-year data help forge path to T-DM1-based deescalation in HER2+ BC
CHICAGO – Combining trastuzumab emtansine (T-DM1) and pertuzumab (P) reduced grade 3+ toxicity in patients with HER2-positive stage I-III breast cancer in the KRISTINE trial, but led to lower event-free survival (EFS) and pathological complete response (pCR) rates vs. standard chemotherapy plus dual HER2 blockade, according to the preplanned 3-year final data analysis.

The EFS rate among participants in the randomized, phase 3 study who completed follow-up was 94.2% in 189 patients who received neoadjuvant T-DM1+P treatment and 85.3% in 196 patients who received docetaxel, carboplatin, and trastuzumab (TCH) plus pertuzumab (hazard ratio, 2.61). The difference was due to more locoregional progression events before surgery (15 [6.7%] vs. 0 in the groups, respectively), Dr. Sara A. Hurvitz, MD, reported at the annual meeting of the American Association of Clinical Oncology.
The curves separated early, prior to surgery, without much change after surgery, noted Dr. Hurvitz, a medical oncologist at the University of California, Los Angeles, where she also serves as director of the Breast Cancer Clinical Trials Program.
Additional analysis showed that low HER2 expression by mRNA or immunohistochemistry (IHC), and HER2 heterogeneity “tended to correlate with locoregional progression.”
Invasive disease-free survival (IDFS) risk, however, was similar with the two treatments (93% and 92%, respectively; HR, 1.11), and, as has been shown “many times over,” experiencing a pCR was associated with reduced risk of an IDFS event (HR, 0.24), regardless of treatment arm, Dr. Hurvitz said.
The previously reported primary results of the study, which failed to reach its primary endpoint, showed a pCR of 44% vs. 56% in 223 women who received TDM-1+P and 221 who received TCH+P, respectively. (Lancet Oncol. 2018 Jan;19[1]:115-126. doi: 10.1016/S1470-2045[17]30716-7).
Of note, additional data reported in a poster at the 2016 San Antonio Breast Cancer Symposium showed that pCR rates “were higher with TCH+P in those tumors with IHC2+ HER2 staining (20% vs. 7% in the T-DM1 arm), or IHC3+ HER2 staining (61% vs. 50%),” she said (SABCS 2016 P6-07-09).
“During neoadjuvant treatment, however, it’s not surprising that the T-DM1+P arm had a more favorable safety profile with a lower incidence of grade 3-4 events, lower incidence of [serious adverse events], and lower incidence of AEs leading to treatment discontinuation,” she said.
The overall rate of grade 3 or greater AEs was 31.8% vs. 67.6% with T-DM1+P vs. TCH+P, but the T-DM1 regimen was associated with more grade 3+ AEs during adjuvant treatment (24.5% vs. 9.9%), and with more adverse events leading to treatment discontinuation – both overall (20.2% vs. 11.0%) and during adjuvant therapy (18.4% vs. 3.8%), said Dr. Hurvitz, noting, however, that 50 patients in the T-DM1+P arm received cytotoxic chemotherapy in the adjuvant phase as allowed by study protocol.
Patient-reported outcomes favored T-DM1+P during the neoadjuvant phase, but were similar in the two groups during the adjuvant phase.
Adverse events occurring substantially more often with TCH+P (2% or greater difference in incidence between the groups) mainly included neutropenia, diarrhea, febrile neutropenia, and anemia, but peripheral neuropathy was a bit higher in the T-DM1 arm, she said.
“Standard-of-care neoadjuvant therapy for HER2-positive breast cancer is chemotherapy plus dual HER2 blockade with trastuzumab and pertuzumab, followed by continued HER2 blockade in the adjuvant setting,” Dr. Hurvitz said, noting that rates of pCR, which is associated with prolonged survival, range from 46% to 62%. “Despite the good outcomes ... 15% of patients will relapse or die; moreover, our standard cytotoxic approaches are associated with systemic toxicity, so there still is a need for effective, less toxic therapies.”
The antibody drug conjugate (ADC) T-DM1 is associated with a lower incidence of AEs typically associated with cytotoxic chemotherapy due to its targeted nature, and in the German ADAPT study it has shown some evidence of efficacy as monotherapy or with endocrine therapy in the neoadjuvant setting in HER2-positive, hormone receptor-positive breast cancer.
“So when we designed this clinical trial we thought that combining T-DM1 with pertuzumab might be an efficacious therapy that would provide patients with a less toxic regimen,” she said.
Participants had centrally-confirmed HER2-positive breast cancer over 2 cm and were randomly assigned 1:1 to T-DM1+P or TCH+P every 3 weeks for six cycles prior to surgery. Those who received T-DM1+P continued adjuvant T-DM1+P for 12 cycles, and those who received TCH+P received adjuvant trastuzumab plus pertuzumab for 12 cycles.
Those in the T-DM1 arm were allowed to receive standard adjuvant chemotherapy at physician discretion – and were encouraged to do so if they had residual disease in the breast greater than 1 cm or lymph node-positive disease. They then went on to receive T-DM1+P for 12 cycles, she said.
“We know that patients who achieve a pathologic complete response have a very good 3-year [IDFS], and for our study, for either arm, it was around 97%. Patients with residual disease have a lower 3-year IDFS in the mid [80% range] representing an unmet need,” she said.
In addition, the similar overall risk of an IDFS event with T-DM1+P and TCH+P in this study suggests that systemic chemotherapy might be unnecessary for some patients.
“But, of course, identification of these patients is going to be critical in determining who can have a deescalation approach, and the clinical utility of chemotherapy-sparing regimens must be confirmed in prospective studies, hopefully using biomarkers,” she concluded.
In a companion article published June 3 in the Journal of Clinical Oncology, Dr. Hurvitz and her colleagues further noted that “the role of T-DM1 in early HER2-positive breast cancer is evolving, with two trials evaluating this agent in the adjuvant setting.”
These include the KATHERINE trial, which showed a lower risk of invasive breast cancer recurrence or death with adjuvant T-DM1 vs. adjuvant trastuzumab in patients with residual disease after neoadjuvant systemic chemotherapy plus single or dual HER-directed therapy (HR, 0.50), and the ongoing KAITLIN trial, which is comparing T-DM1+P with taxane plus trastuzumab after anthracyclines as adjuvant therapy in patients who have not received prior neoadjuvant therapy.
“Data from KAITLIN will further define the clinical utility of adjuvant T-DM1+P in patients with HER2-positive early breast cancer,” they wrote.
During a discussion of the KRISTINE study findings and other related data presented at ASCO 2019, Mark D. Pegram, MD, a medical oncologist and professor at Stanford (Calif.) University, said that T-DM1-based neoadjuvant regimens appear, based on peer-reviewed published data from KRISTINE and other studies (such as the Swedish PREDIX HER2 trial, which was also discussed during the session), to be clinically active and well tolerated in HER2-positive early breast cancer.
“Early adopters may consider neoadjuvant T-DM1 in patients who are perhaps not candidates for chemotherapy due to comorbidities, age, et cetera, or those patients who frankly refuse chemotherapy, of which we all have a few,” said Dr. Pegram, who also is the first director of the Breast Cancer Oncology Program at Stanford Women’s Cancer Center. “The burden is on us to identify molecular, genetic, or perhaps imaging markers to identify patients who are most suitable for consideration of deescalation strategies with T-DM1 or newer HER2 antibody drug conjugates [in development].”
Dr. Pegram also highlighted the KRISTINE EFS finding on locoregional progression prior to surgery.
“Sara showed you that the ... event-free survival outcomes that are deleterious happen prior to surgery, which is, I think, fascinating, and if we could identify those patients prospectively, it could be very powerful in maximally exploiting the potential of deescalation with T-DM1 or T-DM1-based regimens,” he said. “But we’re not there yet, obviously.”
The KRISTINE study was funded by F. Hoffmann-La Roche and Genentech. Dr. Hurvitz reported research funding to her institution from Ambryx, Amgen, Bayer, Biomarin, Boehringer Ingelheim, Cascadian Therapeutics, Daiichi Sankyo, Dignitana, Genentech/Roche, GlaxoSmithKline, Lilly, Macrogenics, Medivation, Merrimack, Novartis, OBI Pharma, Pfizer, Puma Biotechnology, Sanofi, and Seattle Genetics, and travel/accommodations/expenses from Lilly, Novartis, and OBI Pharma. Dr. Pegram reported relationships (honoraria; consulting/advisory roles) with Daiichi Sankyo, Genentech/Roche, Macrogenics, and Seattle Genetics.
SOURCE: Hurvitz S et al. ASCO 2019: Abstract 500.
CHICAGO – Combining trastuzumab emtansine (T-DM1) and pertuzumab (P) reduced grade 3+ toxicity in patients with HER2-positive stage I-III breast cancer in the KRISTINE trial, but led to lower event-free survival (EFS) and pathological complete response (pCR) rates vs. standard chemotherapy plus dual HER2 blockade, according to the preplanned 3-year final data analysis.
The EFS rate among participants in the randomized, phase 3 study who completed follow-up was 94.2% in 189 patients who received neoadjuvant T-DM1+P treatment and 85.3% in 196 patients who received docetaxel, carboplatin, and trastuzumab (TCH) plus pertuzumab (hazard ratio, 2.61). The difference was due to more locoregional progression events before surgery (15 [6.7%] vs. 0 in the groups, respectively), Dr. Sara A. Hurvitz, MD, reported at the annual meeting of the American Association of Clinical Oncology.
The curves separated early, prior to surgery, without much change after surgery, noted Dr. Hurvitz, a medical oncologist at the University of California, Los Angeles, where she also serves as director of the Breast Cancer Clinical Trials Program.
Additional analysis showed that low HER2 expression by mRNA or immunohistochemistry (IHC), and HER2 heterogeneity “tended to correlate with locoregional progression.”
Invasive disease-free survival (IDFS) risk, however, was similar with the two treatments (93% and 92%, respectively; HR, 1.11), and, as has been shown “many times over,” experiencing a pCR was associated with reduced risk of an IDFS event (HR, 0.24), regardless of treatment arm, Dr. Hurvitz said.
The previously reported primary results of the study, which failed to reach its primary endpoint, showed a pCR of 44% vs. 56% in 223 women who received TDM-1+P and 221 who received TCH+P, respectively. (Lancet Oncol. 2018 Jan;19[1]:115-126. doi: 10.1016/S1470-2045[17]30716-7).
Of note, additional data reported in a poster at the 2016 San Antonio Breast Cancer Symposium showed that pCR rates “were higher with TCH+P in those tumors with IHC2+ HER2 staining (20% vs. 7% in the T-DM1 arm), or IHC3+ HER2 staining (61% vs. 50%),” she said (SABCS 2016 P6-07-09).
“During neoadjuvant treatment, however, it’s not surprising that the T-DM1+P arm had a more favorable safety profile with a lower incidence of grade 3-4 events, lower incidence of [serious adverse events], and lower incidence of AEs leading to treatment discontinuation,” she said.
The overall rate of grade 3 or greater AEs was 31.8% vs. 67.6% with T-DM1+P vs. TCH+P, but the T-DM1 regimen was associated with more grade 3+ AEs during adjuvant treatment (24.5% vs. 9.9%), and with more adverse events leading to treatment discontinuation – both overall (20.2% vs. 11.0%) and during adjuvant therapy (18.4% vs. 3.8%), said Dr. Hurvitz, noting, however, that 50 patients in the T-DM1+P arm received cytotoxic chemotherapy in the adjuvant phase as allowed by study protocol.
Patient-reported outcomes favored T-DM1+P during the neoadjuvant phase, but were similar in the two groups during the adjuvant phase.
Adverse events occurring substantially more often with TCH+P (2% or greater difference in incidence between the groups) mainly included neutropenia, diarrhea, febrile neutropenia, and anemia, but peripheral neuropathy was a bit higher in the T-DM1 arm, she said.
“Standard-of-care neoadjuvant therapy for HER2-positive breast cancer is chemotherapy plus dual HER2 blockade with trastuzumab and pertuzumab, followed by continued HER2 blockade in the adjuvant setting,” Dr. Hurvitz said, noting that rates of pCR, which is associated with prolonged survival, range from 46% to 62%. “Despite the good outcomes ... 15% of patients will relapse or die; moreover, our standard cytotoxic approaches are associated with systemic toxicity, so there still is a need for effective, less toxic therapies.”
The antibody drug conjugate (ADC) T-DM1 is associated with a lower incidence of AEs typically associated with cytotoxic chemotherapy due to its targeted nature, and in the German ADAPT study it has shown some evidence of efficacy as monotherapy or with endocrine therapy in the neoadjuvant setting in HER2-positive, hormone receptor-positive breast cancer.
“So when we designed this clinical trial we thought that combining T-DM1 with pertuzumab might be an efficacious therapy that would provide patients with a less toxic regimen,” she said.
Participants had centrally-confirmed HER2-positive breast cancer over 2 cm and were randomly assigned 1:1 to T-DM1+P or TCH+P every 3 weeks for six cycles prior to surgery. Those who received T-DM1+P continued adjuvant T-DM1+P for 12 cycles, and those who received TCH+P received adjuvant trastuzumab plus pertuzumab for 12 cycles.
Those in the T-DM1 arm were allowed to receive standard adjuvant chemotherapy at physician discretion – and were encouraged to do so if they had residual disease in the breast greater than 1 cm or lymph node-positive disease. They then went on to receive T-DM1+P for 12 cycles, she said.
“We know that patients who achieve a pathologic complete response have a very good 3-year [IDFS], and for our study, for either arm, it was around 97%. Patients with residual disease have a lower 3-year IDFS in the mid [80% range] representing an unmet need,” she said.
In addition, the similar overall risk of an IDFS event with T-DM1+P and TCH+P in this study suggests that systemic chemotherapy might be unnecessary for some patients.
“But, of course, identification of these patients is going to be critical in determining who can have a deescalation approach, and the clinical utility of chemotherapy-sparing regimens must be confirmed in prospective studies, hopefully using biomarkers,” she concluded.
In a companion article published June 3 in the Journal of Clinical Oncology, Dr. Hurvitz and her colleagues further noted that “the role of T-DM1 in early HER2-positive breast cancer is evolving, with two trials evaluating this agent in the adjuvant setting.”
These include the KATHERINE trial, which showed a lower risk of invasive breast cancer recurrence or death with adjuvant T-DM1 vs. adjuvant trastuzumab in patients with residual disease after neoadjuvant systemic chemotherapy plus single or dual HER-directed therapy (HR, 0.50), and the ongoing KAITLIN trial, which is comparing T-DM1+P with taxane plus trastuzumab after anthracyclines as adjuvant therapy in patients who have not received prior neoadjuvant therapy.
“Data from KAITLIN will further define the clinical utility of adjuvant T-DM1+P in patients with HER2-positive early breast cancer,” they wrote.
During a discussion of the KRISTINE study findings and other related data presented at ASCO 2019, Mark D. Pegram, MD, a medical oncologist and professor at Stanford (Calif.) University, said that T-DM1-based neoadjuvant regimens appear, based on peer-reviewed published data from KRISTINE and other studies (such as the Swedish PREDIX HER2 trial, which was also discussed during the session), to be clinically active and well tolerated in HER2-positive early breast cancer.
“Early adopters may consider neoadjuvant T-DM1 in patients who are perhaps not candidates for chemotherapy due to comorbidities, age, et cetera, or those patients who frankly refuse chemotherapy, of which we all have a few,” said Dr. Pegram, who also is the first director of the Breast Cancer Oncology Program at Stanford Women’s Cancer Center. “The burden is on us to identify molecular, genetic, or perhaps imaging markers to identify patients who are most suitable for consideration of deescalation strategies with T-DM1 or newer HER2 antibody drug conjugates [in development].”
Dr. Pegram also highlighted the KRISTINE EFS finding on locoregional progression prior to surgery.
“Sara showed you that the ... event-free survival outcomes that are deleterious happen prior to surgery, which is, I think, fascinating, and if we could identify those patients prospectively, it could be very powerful in maximally exploiting the potential of deescalation with T-DM1 or T-DM1-based regimens,” he said. “But we’re not there yet, obviously.”
The KRISTINE study was funded by F. Hoffmann-La Roche and Genentech. Dr. Hurvitz reported research funding to her institution from Ambryx, Amgen, Bayer, Biomarin, Boehringer Ingelheim, Cascadian Therapeutics, Daiichi Sankyo, Dignitana, Genentech/Roche, GlaxoSmithKline, Lilly, Macrogenics, Medivation, Merrimack, Novartis, OBI Pharma, Pfizer, Puma Biotechnology, Sanofi, and Seattle Genetics, and travel/accommodations/expenses from Lilly, Novartis, and OBI Pharma. Dr. Pegram reported relationships (honoraria; consulting/advisory roles) with Daiichi Sankyo, Genentech/Roche, Macrogenics, and Seattle Genetics.
SOURCE: Hurvitz S et al. ASCO 2019: Abstract 500.
CHICAGO – Combining trastuzumab emtansine (T-DM1) and pertuzumab (P) reduced grade 3+ toxicity in patients with HER2-positive stage I-III breast cancer in the KRISTINE trial, but led to lower event-free survival (EFS) and pathological complete response (pCR) rates vs. standard chemotherapy plus dual HER2 blockade, according to the preplanned 3-year final data analysis.
The EFS rate among participants in the randomized, phase 3 study who completed follow-up was 94.2% in 189 patients who received neoadjuvant T-DM1+P treatment and 85.3% in 196 patients who received docetaxel, carboplatin, and trastuzumab (TCH) plus pertuzumab (hazard ratio, 2.61). The difference was due to more locoregional progression events before surgery (15 [6.7%] vs. 0 in the groups, respectively), Dr. Sara A. Hurvitz, MD, reported at the annual meeting of the American Association of Clinical Oncology.
The curves separated early, prior to surgery, without much change after surgery, noted Dr. Hurvitz, a medical oncologist at the University of California, Los Angeles, where she also serves as director of the Breast Cancer Clinical Trials Program.
Additional analysis showed that low HER2 expression by mRNA or immunohistochemistry (IHC), and HER2 heterogeneity “tended to correlate with locoregional progression.”
Invasive disease-free survival (IDFS) risk, however, was similar with the two treatments (93% and 92%, respectively; HR, 1.11), and, as has been shown “many times over,” experiencing a pCR was associated with reduced risk of an IDFS event (HR, 0.24), regardless of treatment arm, Dr. Hurvitz said.
The previously reported primary results of the study, which failed to reach its primary endpoint, showed a pCR of 44% vs. 56% in 223 women who received TDM-1+P and 221 who received TCH+P, respectively. (Lancet Oncol. 2018 Jan;19[1]:115-126. doi: 10.1016/S1470-2045[17]30716-7).
Of note, additional data reported in a poster at the 2016 San Antonio Breast Cancer Symposium showed that pCR rates “were higher with TCH+P in those tumors with IHC2+ HER2 staining (20% vs. 7% in the T-DM1 arm), or IHC3+ HER2 staining (61% vs. 50%),” she said (SABCS 2016 P6-07-09).
“During neoadjuvant treatment, however, it’s not surprising that the T-DM1+P arm had a more favorable safety profile with a lower incidence of grade 3-4 events, lower incidence of [serious adverse events], and lower incidence of AEs leading to treatment discontinuation,” she said.
The overall rate of grade 3 or greater AEs was 31.8% vs. 67.6% with T-DM1+P vs. TCH+P, but the T-DM1 regimen was associated with more grade 3+ AEs during adjuvant treatment (24.5% vs. 9.9%), and with more adverse events leading to treatment discontinuation – both overall (20.2% vs. 11.0%) and during adjuvant therapy (18.4% vs. 3.8%), said Dr. Hurvitz, noting, however, that 50 patients in the T-DM1+P arm received cytotoxic chemotherapy in the adjuvant phase as allowed by study protocol.
Patient-reported outcomes favored T-DM1+P during the neoadjuvant phase, but were similar in the two groups during the adjuvant phase.
Adverse events occurring substantially more often with TCH+P (2% or greater difference in incidence between the groups) mainly included neutropenia, diarrhea, febrile neutropenia, and anemia, but peripheral neuropathy was a bit higher in the T-DM1 arm, she said.
“Standard-of-care neoadjuvant therapy for HER2-positive breast cancer is chemotherapy plus dual HER2 blockade with trastuzumab and pertuzumab, followed by continued HER2 blockade in the adjuvant setting,” Dr. Hurvitz said, noting that rates of pCR, which is associated with prolonged survival, range from 46% to 62%. “Despite the good outcomes ... 15% of patients will relapse or die; moreover, our standard cytotoxic approaches are associated with systemic toxicity, so there still is a need for effective, less toxic therapies.”
The antibody drug conjugate (ADC) T-DM1 is associated with a lower incidence of AEs typically associated with cytotoxic chemotherapy due to its targeted nature, and in the German ADAPT study it has shown some evidence of efficacy as monotherapy or with endocrine therapy in the neoadjuvant setting in HER2-positive, hormone receptor-positive breast cancer.
“So when we designed this clinical trial we thought that combining T-DM1 with pertuzumab might be an efficacious therapy that would provide patients with a less toxic regimen,” she said.
Participants had centrally-confirmed HER2-positive breast cancer over 2 cm and were randomly assigned 1:1 to T-DM1+P or TCH+P every 3 weeks for six cycles prior to surgery. Those who received T-DM1+P continued adjuvant T-DM1+P for 12 cycles, and those who received TCH+P received adjuvant trastuzumab plus pertuzumab for 12 cycles.
Those in the T-DM1 arm were allowed to receive standard adjuvant chemotherapy at physician discretion – and were encouraged to do so if they had residual disease in the breast greater than 1 cm or lymph node-positive disease. They then went on to receive T-DM1+P for 12 cycles, she said.
“We know that patients who achieve a pathologic complete response have a very good 3-year [IDFS], and for our study, for either arm, it was around 97%. Patients with residual disease have a lower 3-year IDFS in the mid [80% range] representing an unmet need,” she said.
In addition, the similar overall risk of an IDFS event with T-DM1+P and TCH+P in this study suggests that systemic chemotherapy might be unnecessary for some patients.
“But, of course, identification of these patients is going to be critical in determining who can have a deescalation approach, and the clinical utility of chemotherapy-sparing regimens must be confirmed in prospective studies, hopefully using biomarkers,” she concluded.
In a companion article published June 3 in the Journal of Clinical Oncology, Dr. Hurvitz and her colleagues further noted that “the role of T-DM1 in early HER2-positive breast cancer is evolving, with two trials evaluating this agent in the adjuvant setting.”
These include the KATHERINE trial, which showed a lower risk of invasive breast cancer recurrence or death with adjuvant T-DM1 vs. adjuvant trastuzumab in patients with residual disease after neoadjuvant systemic chemotherapy plus single or dual HER-directed therapy (HR, 0.50), and the ongoing KAITLIN trial, which is comparing T-DM1+P with taxane plus trastuzumab after anthracyclines as adjuvant therapy in patients who have not received prior neoadjuvant therapy.
“Data from KAITLIN will further define the clinical utility of adjuvant T-DM1+P in patients with HER2-positive early breast cancer,” they wrote.
During a discussion of the KRISTINE study findings and other related data presented at ASCO 2019, Mark D. Pegram, MD, a medical oncologist and professor at Stanford (Calif.) University, said that T-DM1-based neoadjuvant regimens appear, based on peer-reviewed published data from KRISTINE and other studies (such as the Swedish PREDIX HER2 trial, which was also discussed during the session), to be clinically active and well tolerated in HER2-positive early breast cancer.
“Early adopters may consider neoadjuvant T-DM1 in patients who are perhaps not candidates for chemotherapy due to comorbidities, age, et cetera, or those patients who frankly refuse chemotherapy, of which we all have a few,” said Dr. Pegram, who also is the first director of the Breast Cancer Oncology Program at Stanford Women’s Cancer Center. “The burden is on us to identify molecular, genetic, or perhaps imaging markers to identify patients who are most suitable for consideration of deescalation strategies with T-DM1 or newer HER2 antibody drug conjugates [in development].”
Dr. Pegram also highlighted the KRISTINE EFS finding on locoregional progression prior to surgery.
“Sara showed you that the ... event-free survival outcomes that are deleterious happen prior to surgery, which is, I think, fascinating, and if we could identify those patients prospectively, it could be very powerful in maximally exploiting the potential of deescalation with T-DM1 or T-DM1-based regimens,” he said. “But we’re not there yet, obviously.”
The KRISTINE study was funded by F. Hoffmann-La Roche and Genentech. Dr. Hurvitz reported research funding to her institution from Ambryx, Amgen, Bayer, Biomarin, Boehringer Ingelheim, Cascadian Therapeutics, Daiichi Sankyo, Dignitana, Genentech/Roche, GlaxoSmithKline, Lilly, Macrogenics, Medivation, Merrimack, Novartis, OBI Pharma, Pfizer, Puma Biotechnology, Sanofi, and Seattle Genetics, and travel/accommodations/expenses from Lilly, Novartis, and OBI Pharma. Dr. Pegram reported relationships (honoraria; consulting/advisory roles) with Daiichi Sankyo, Genentech/Roche, Macrogenics, and Seattle Genetics.
SOURCE: Hurvitz S et al. ASCO 2019: Abstract 500.
REPORTING FROM ASCO 2019
Judge bars contraceptive mandate from being enforced

In a June 5, 2019, opinion, U.S. District Judge Reed O’Connor granted a permanent injunction on the contraceptive mandate, ruling that both the mandate and the accommodation process violate the Religious Freedom Restoration Act. The injunction applies to all individuals and employers – regardless of size or nonprofit status – that oppose contraceptive coverage based on religious beliefs.
In his ruling, Judge O’Connor said the contraceptive mandate substantially burdens the plaintiffs’ religious exercise.
“The point of the contraceptive mandate is to ensure all ACA-compliant insurance plans include cost-free coverage of all FDA [Food and Drug Administration]-approved contraceptive methods [and] the point of the individual mandate is to ensure individuals purchase ACA-compliant insurance plans,” Judge O’Conner wrote. “The result? The individual plaintiffs are forced out of either the health insurance market or their religious exercise. And by choosing to adhere to their religious beliefs, not only are the individual plaintiffs excluded from the insurance market, they are forced to violate federal law. That the contraceptive mandate systematically discriminates against the individual class by blocking members’ entrance into the marketplace – due to religious exercise – is a substantial burden of the highest order.”
The case, DeOtte v. Azar, started with an October 2018 legal challenge by several Texas residents and a business over having to comply with the Affordable Care Act mandate. The plaintiffs argued the requirement violates their religious freedom, and that the court should strike it down as unconstitutional. The current Justice Department has largely chosen not to defend the case, agreeing that forcing people and employers with religious objections to comply with the contraceptive mandate violates the Religious Freedom Restoration Act. In 2018, the department issued new rules expanding exemptions to the ACA’s contraceptive mandate on moral or religious grounds.
Legal challenges against the expanded exemptions continue through the courts. Judges in California and Pennsylvania have temporarily banned the rules from taking effect. Analysts say the final answer on the contraceptive mandate could come from the U.S. Supreme Court.
In a June 5, 2019, opinion, U.S. District Judge Reed O’Connor granted a permanent injunction on the contraceptive mandate, ruling that both the mandate and the accommodation process violate the Religious Freedom Restoration Act. The injunction applies to all individuals and employers – regardless of size or nonprofit status – that oppose contraceptive coverage based on religious beliefs.
In his ruling, Judge O’Connor said the contraceptive mandate substantially burdens the plaintiffs’ religious exercise.
“The point of the contraceptive mandate is to ensure all ACA-compliant insurance plans include cost-free coverage of all FDA [Food and Drug Administration]-approved contraceptive methods [and] the point of the individual mandate is to ensure individuals purchase ACA-compliant insurance plans,” Judge O’Conner wrote. “The result? The individual plaintiffs are forced out of either the health insurance market or their religious exercise. And by choosing to adhere to their religious beliefs, not only are the individual plaintiffs excluded from the insurance market, they are forced to violate federal law. That the contraceptive mandate systematically discriminates against the individual class by blocking members’ entrance into the marketplace – due to religious exercise – is a substantial burden of the highest order.”
The case, DeOtte v. Azar, started with an October 2018 legal challenge by several Texas residents and a business over having to comply with the Affordable Care Act mandate. The plaintiffs argued the requirement violates their religious freedom, and that the court should strike it down as unconstitutional. The current Justice Department has largely chosen not to defend the case, agreeing that forcing people and employers with religious objections to comply with the contraceptive mandate violates the Religious Freedom Restoration Act. In 2018, the department issued new rules expanding exemptions to the ACA’s contraceptive mandate on moral or religious grounds.
Legal challenges against the expanded exemptions continue through the courts. Judges in California and Pennsylvania have temporarily banned the rules from taking effect. Analysts say the final answer on the contraceptive mandate could come from the U.S. Supreme Court.
In a June 5, 2019, opinion, U.S. District Judge Reed O’Connor granted a permanent injunction on the contraceptive mandate, ruling that both the mandate and the accommodation process violate the Religious Freedom Restoration Act. The injunction applies to all individuals and employers – regardless of size or nonprofit status – that oppose contraceptive coverage based on religious beliefs.
In his ruling, Judge O’Connor said the contraceptive mandate substantially burdens the plaintiffs’ religious exercise.
“The point of the contraceptive mandate is to ensure all ACA-compliant insurance plans include cost-free coverage of all FDA [Food and Drug Administration]-approved contraceptive methods [and] the point of the individual mandate is to ensure individuals purchase ACA-compliant insurance plans,” Judge O’Conner wrote. “The result? The individual plaintiffs are forced out of either the health insurance market or their religious exercise. And by choosing to adhere to their religious beliefs, not only are the individual plaintiffs excluded from the insurance market, they are forced to violate federal law. That the contraceptive mandate systematically discriminates against the individual class by blocking members’ entrance into the marketplace – due to religious exercise – is a substantial burden of the highest order.”
The case, DeOtte v. Azar, started with an October 2018 legal challenge by several Texas residents and a business over having to comply with the Affordable Care Act mandate. The plaintiffs argued the requirement violates their religious freedom, and that the court should strike it down as unconstitutional. The current Justice Department has largely chosen not to defend the case, agreeing that forcing people and employers with religious objections to comply with the contraceptive mandate violates the Religious Freedom Restoration Act. In 2018, the department issued new rules expanding exemptions to the ACA’s contraceptive mandate on moral or religious grounds.
Legal challenges against the expanded exemptions continue through the courts. Judges in California and Pennsylvania have temporarily banned the rules from taking effect. Analysts say the final answer on the contraceptive mandate could come from the U.S. Supreme Court.
Reducing adverse drug reactions
Easing the inpatient/outpatient transition
Adverse drug reactions are a problem hospitalists encounter often. An estimated 9% of hospital admissions in older adults are the result of adverse drug reactions, and up to one in five adults experience an adverse drug reaction during hospitalization.
“Many interventions have been tried to solve this problem, and certain of them have worked, but to date we don’t have any great solutions that meaningfully impact the rate of these events in a way that’s feasible in most health care environments, so any efforts to reduce the burden of these problems in older adults could be hugely beneficial,” said Michael Steinman, MD, author of an editorial highlighting a new approach.
His editorial in BMJ Quality & Safety cites research on the Pharm2Pharm program, implemented in six Hawaiian hospitals, in which hospital-based pharmacists identified inpatients at high risk of medication misadventures with criteria such as use of multiple medications, presence of high-risk medications such as warfarin or glucose-lowering drugs, and a history of previous acute care use resulting from medication-related problems. The hospital pharmacist would then meet with the patient to reconcile medications and facilitate a coordinated hand-off to a community pharmacist, who would meet with the patient after discharge.
In addition to a 36% reduction in the rate of medication-related hospitalizations, the intervention generated an estimated savings of $6.6 million per year in avoided hospitalizations.
There are two major takeaways, said Dr. Steinman, who is based in the division of geriatrics at the University of California, San Francisco: It’s critical to focus on transitions and coordination between inpatient and outpatient care to address medication-related problems, and pharmacists can be extremely helpful in that.
“Decisions about drug therapy in the hospital may seem reasonable in the short term but often won’t stick in the long term unless there is a coordinated care that can help ensure appropriate follow-through once patients return home,” Dr. Steinman said. “The study that the editorial references is a systems intervention that hospitalists can advocate for in their own institutions, but in the immediate day-to-day, trying to ensure solid coordination of medication management from the inpatient to outpatient setting is likely to be very helpful for their patients.”
The long-term outcomes of hospitalized patients are largely influenced by getting them set up with appropriate community resources and supports once they leave the hospital, he added, and the hospital can play a critical role in putting these pieces into place.
Reference
1. Steinman MA. Reducing hospital admissions for adverse drug events through coordinated pharmacist care: learning from Hawai’i without a field trip. BMJ Qual Saf. Epub 2018 Nov 24. doi: 10.1136/bmjqs-2018-008815. Accessed Dec. 11, 2018.
Easing the inpatient/outpatient transition
Easing the inpatient/outpatient transition
Adverse drug reactions are a problem hospitalists encounter often. An estimated 9% of hospital admissions in older adults are the result of adverse drug reactions, and up to one in five adults experience an adverse drug reaction during hospitalization.
“Many interventions have been tried to solve this problem, and certain of them have worked, but to date we don’t have any great solutions that meaningfully impact the rate of these events in a way that’s feasible in most health care environments, so any efforts to reduce the burden of these problems in older adults could be hugely beneficial,” said Michael Steinman, MD, author of an editorial highlighting a new approach.
His editorial in BMJ Quality & Safety cites research on the Pharm2Pharm program, implemented in six Hawaiian hospitals, in which hospital-based pharmacists identified inpatients at high risk of medication misadventures with criteria such as use of multiple medications, presence of high-risk medications such as warfarin or glucose-lowering drugs, and a history of previous acute care use resulting from medication-related problems. The hospital pharmacist would then meet with the patient to reconcile medications and facilitate a coordinated hand-off to a community pharmacist, who would meet with the patient after discharge.
In addition to a 36% reduction in the rate of medication-related hospitalizations, the intervention generated an estimated savings of $6.6 million per year in avoided hospitalizations.
There are two major takeaways, said Dr. Steinman, who is based in the division of geriatrics at the University of California, San Francisco: It’s critical to focus on transitions and coordination between inpatient and outpatient care to address medication-related problems, and pharmacists can be extremely helpful in that.
“Decisions about drug therapy in the hospital may seem reasonable in the short term but often won’t stick in the long term unless there is a coordinated care that can help ensure appropriate follow-through once patients return home,” Dr. Steinman said. “The study that the editorial references is a systems intervention that hospitalists can advocate for in their own institutions, but in the immediate day-to-day, trying to ensure solid coordination of medication management from the inpatient to outpatient setting is likely to be very helpful for their patients.”
The long-term outcomes of hospitalized patients are largely influenced by getting them set up with appropriate community resources and supports once they leave the hospital, he added, and the hospital can play a critical role in putting these pieces into place.
Reference
1. Steinman MA. Reducing hospital admissions for adverse drug events through coordinated pharmacist care: learning from Hawai’i without a field trip. BMJ Qual Saf. Epub 2018 Nov 24. doi: 10.1136/bmjqs-2018-008815. Accessed Dec. 11, 2018.
Adverse drug reactions are a problem hospitalists encounter often. An estimated 9% of hospital admissions in older adults are the result of adverse drug reactions, and up to one in five adults experience an adverse drug reaction during hospitalization.
“Many interventions have been tried to solve this problem, and certain of them have worked, but to date we don’t have any great solutions that meaningfully impact the rate of these events in a way that’s feasible in most health care environments, so any efforts to reduce the burden of these problems in older adults could be hugely beneficial,” said Michael Steinman, MD, author of an editorial highlighting a new approach.
His editorial in BMJ Quality & Safety cites research on the Pharm2Pharm program, implemented in six Hawaiian hospitals, in which hospital-based pharmacists identified inpatients at high risk of medication misadventures with criteria such as use of multiple medications, presence of high-risk medications such as warfarin or glucose-lowering drugs, and a history of previous acute care use resulting from medication-related problems. The hospital pharmacist would then meet with the patient to reconcile medications and facilitate a coordinated hand-off to a community pharmacist, who would meet with the patient after discharge.
In addition to a 36% reduction in the rate of medication-related hospitalizations, the intervention generated an estimated savings of $6.6 million per year in avoided hospitalizations.
There are two major takeaways, said Dr. Steinman, who is based in the division of geriatrics at the University of California, San Francisco: It’s critical to focus on transitions and coordination between inpatient and outpatient care to address medication-related problems, and pharmacists can be extremely helpful in that.
“Decisions about drug therapy in the hospital may seem reasonable in the short term but often won’t stick in the long term unless there is a coordinated care that can help ensure appropriate follow-through once patients return home,” Dr. Steinman said. “The study that the editorial references is a systems intervention that hospitalists can advocate for in their own institutions, but in the immediate day-to-day, trying to ensure solid coordination of medication management from the inpatient to outpatient setting is likely to be very helpful for their patients.”
The long-term outcomes of hospitalized patients are largely influenced by getting them set up with appropriate community resources and supports once they leave the hospital, he added, and the hospital can play a critical role in putting these pieces into place.
Reference
1. Steinman MA. Reducing hospital admissions for adverse drug events through coordinated pharmacist care: learning from Hawai’i without a field trip. BMJ Qual Saf. Epub 2018 Nov 24. doi: 10.1136/bmjqs-2018-008815. Accessed Dec. 11, 2018.