User login
Postural Orthostatic Tachycardia Syndrome: A Consideration in Orthostatic Intolerance
CE/CME No: CR-1404
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define postural orthostatic tachycardia syndrome (POTS).
• List the presenting signs and symptoms of POTS.
• Differentiate POTS from other causes of orthostatic intolerance.
• Explain the common classifications of POTS.
• Enumerate the various referral and treatment options for the clinician to consider when a patient meets the criteria for POTS.
FACULTY
Molly Paulson is an Assistant Professor of Physician Assistant Studies in the College of Health Professions at Grand Valley State University, Grand Rapids, Michigan.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Postural orthostatic tachycardia syndrome may not be the first disorder that clinicians consider when they encounter a patient with orthostatic intolerance, but ignoring this possibility during a differential diagnosis can mean patients continue to experience unexplained dizziness, fatigue, syncope, and a variety of other related signs and symptoms. Arriving at the correct diagnosis will allow you to help patients manage the condition and return to the lives and activities they previously enjoyed.
Clinicians should consider postural orthostatic tachycardia syndrome (POTS; ICD-9 code 427.89) as part of the differential diagnosis of orthostatic intolerance (OI). Ignoring this possibility can delay proper diagnosis and treatment among patients who meet the criteria for POTS.
When patients present with symptoms of OI of unknown origin, it is important to consider a broad variety of conditions, including POTS, in the differential diagnosis. Clinicians should follow up with appropriate testing to determine if the criteria for any of these conditions are met. Once the cause of the OI is confirmed, appropriate initial care and referral will facilitate improved treatment for the patient.
A recent consensus statement defines POTS as an increase in heart rate of at least 30 beats/min or a heart rate of at least 120 beats/min upon standing from a supine position—or in response to a tilt-table test—that is sustained for 10 minutes without evidence of orthostatic hypotension.1,2 Although POTS can be seen in men, most cases occur in women of childbearing age, with a female-to-male ratio of 4:1 to 5:1.2 Patients most commonly present with symptoms similar to orthostatic hypotension.3 Typical symptoms include dizziness, near-syncope or syncope, fatigue, headache, visual changes, lightheadedness, weakness, and abdominal discomfort.4,5 Presenting symptoms also overlap with other, more prevalent disorders, leading to frequent misdiagnosis.
Examples of common initial misdiagnoses for patients with POTS include anxiety, chronic fatigue syndrome, fibromyalgia, menopause, orthostatic hypotension, unstable angina, and hyperadrenergic states.1,6 Inappropriate or delayed treatment of POTS can lead to significant daily functional impairment similar to that associated with chronic obstructive pulmonary disease or congestive heart failure.4,6,7
UNDERSTANDING ORTHOSTATIC INTOLERANCE
To better understand POTS, clinicians should have a clear understanding of OI. According to Stewart,8 OI is defined by symptoms of impending near-syncope or syncope, including dizziness, headache, fatigue, exercise intolerance, abdominal distress, or a sensation of feeling “hot” accompanied by sweating. These symptoms occur when a patient assumes an upright position and are relieved when the person returns to a supine position.8
Normally, when a person stands, gravitational forces cause a redistribution of 300 to 800 mL of blood in both the splanchnic and lower extremity circulation. This decreases systolic blood pressure and slows cardiac filling. The resultant decreased cardiac output stimulates increased sympathetic tone via the baroreceptors and other mechanisms to restore arterial blood pressure.1,9 In OI, this compensatory mechanism is compromised, and the patient experiences the symptoms of impending near-syncope or syncope, as discussed previously.
A broad array of medical conditions with multiple etiologies meet the criteria for OI.10 Although a comprehensive list of these conditions is beyond the scope of this article, it is essential for the clinician to look for primary and secondary causes of orthostasis by gathering a comprehensive history, followed by an extensive physical exam directed by the history. The patient should be asked about a history of allergies or autoimmune disease, cancers, eating disorders, infections, adverse effects of medication, and signs or symptoms of chronic sympathetic stimulation. It is also important to review the current medication list carefully and evaluate the patient for dysfunction of the cardiac, endocrine, renal, and nervous systems and for psychiatric disorders. A short list of conditions that may present with OI include anemia, anorexia, autoimmune disease, cancers, cardiac disease, diabetes, infections, paraneoplastic syndromes, vascular disease, and volume depletion.2,8,11,12
Hypotension is absent in POTS, but it otherwise also fits the profile of OI. POTS can also present with a host of other nonspecific, nonorthostatic symptoms, including nausea, vomiting, diarrhea, constipation, fatigue, migraine headaches, and chest pain.10 This unusual presentation can confound the clinician and complicate the medical picture, making a clear diagnosis difficult.
On the next page: History, epidemiology, and etiology >>
HISTORY, EPIDEMIOLOGY, AND ETIOLOGY
POTS was first recognized and described during the Civil War as irritable heart syndrome or soldier’s heart due to the frequency of the condition in combat soldiers. As more was published about the condition, the list of names grew to include Da Costa syndrome, anxiety neurosis, chronic orthostatic intolerance, effort syndrome, idiopathic hypovolemia, mitral valve prolapse syndrome, orthostatic tachycardia, positional tachycardia syndrome, and finally postural orthostatic tachycardia syndrome.5,12 Although initially described in men in the military, the condition is more frequently seen in young women from puberty to age 50. Some researchers have proposed that POTS is much more common than recent studies report; they suggest that the condition is underreported both because of the nonspecific nature of the symptoms and because it is not often included in the differential of OI.5
Although some researchers disagree,7,12 most of the literature supports multiple etiologies of POTS.1,2,13 Researchers have divided the syndrome into primary and secondary causes. They have further separated primary POTS into two broad categories based on neuropathic or hyperadrenergic pathology. In both subgroups, POTS is exacerbated by dependent vasodilatation and central hypovolemia.11
In the more common neuropathic POTS, a precipitating factor such as an infection, trauma, pregnancy, or surgery precedes the sudden onset of symptoms. In the majority of patients with neuropathic POTS, high normal to elevated serum norepinephrine (NE) levels are detected, as compared to “normal” controls,5 but known antibodies are not detected. However, in approximately 10% to 15% of patients with a reported antecedent infection, acetylcholine receptor antibodies have been detected.2,14 This form is described in the literature as immune-mediated POTS.2,11,14,15
The less common primary hyperadrenergic POTS presents more insidiously and may have a genetic component. As documented in some families, a point mutation in the NE transporter protein has been shown to slow clearance of NE from the synaptic cleft in the central nervous system. This leads to excessive central sympathetic stimulation of the periphery.12,13,16 Developmental POTS, which presents at puberty with lower extremity neuropathy, is another form of primary POTS and resolves with maturity (see Table 1).6
Secondary POTS can be triggered by several known conditions including alcoholism.12,16 Any disease or insult that damages the peripheral autonomic system may precipitate POTS. Examples include autoimmune diseases, diabetes, chemotherapy, and heavy metal poisoning.13 Genetic disorders have also been associated with or can contribute to secondary POTS. Joint hypermobility syndrome (JHS), also known as Ehlers-Danlos type III, is a genetic disorder characterized by defects in the structure of connective tissue.16 In JHS, the connective tissue in blood vessels is more elastic, leading to greater lower extremity pooling and decreased vasoconstrictive response to sympathetic stimulation on standing. The literature also notes that prolonged bed rest and any medications that contribute to central hypotension or interfere with compensatory peripheral vasoconstriction may precipitate POTS (see Table 2).
On the next page: Clinical presentation and differential diagnosis >>
CLINICAL PRESENTATION
Patients with POTS frequently present with a sudden onset of OI related to activity or changes from recumbency to standing. They often identify an illness, accident, or surgery preceding the onset of symptoms. Other clinical manifestations of POTS include acrocyanosis, fatigue, headaches, constitutional hypotension, sleep disturbances, and gastrointestinal symptoms.12,13 Frequently, patients come to the emergency department with chest pain, indigestion, fatigue, and heart palpitations,14,15 concerned that they are having a heart attack. Since symptoms resolve with recumbency and no clinical markers for acute coronary syndromes are found, no further workup is obtained, and patients are sent home with the recommendation to see their primary care provider.
The symptoms of POTS are complex, nonspecific, and debilitating. Fatigue, difficulty concentrating, and tachycardia when standing make activities of daily living such as work, home maintenance, and child care challenging. The unpredictable symptoms make life frustrating for patients and their families. The presenting symptoms, the normal results for routine blood tests, and the lack of cardiac abnormalities often lead providers to a diagnosis of anxiety or panic disorder.13 Since the symptoms of POTS are somewhat mitigated by treatment with selective serotonin reuptake inhibitors,16 patients and providers often will accept the diagnosis and not investigate the symptoms further.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of POTS begins with any condition that causes tachycardia or OI. Clinicians should also consider and rule out cardiac abnormalities such as inappropriate tachycardia, supraventricular tachycardia, Wolff-Parkinson-White syndrome, and other cardiac dysrhythmias. Other diagnoses that should be considered are endocrine abnormalities, including hyperthyroidism, hypoadrenalism, and pheochromocytoma; drug adverse effects and interactions, necessitating scrupulous evaluation of the patient’s medication list; changes in volume status; and kidney disease.1 Causes of autonomic dysfunction in both the peripheral and central nervous system, including Chiari I malformation, need to be scrutinized.17
Many conditions within the differential diagnosis can occur concomitantly with POTS and need to be treated, even as further evaluation continues. An extensive history and review of systems is essential to obtain a complete list of symptoms and lead the clinician toward an appropriate physical exam and necessary adjunct testing.
ASSOCIATED DISORDERS
Of the many conditions associated with POTS, JHS is the most common genetic disorder. In this disorder, a genetic mutation produces changes in the composition of the collagen, resulting in enhanced flexibility and compliance of any tissue with collagen in it.16 Mitral valve prolapse is also associated with JHS and POTS.12
POTS is present in 40% of patients diagnosed with chronic fatigue syndrome, as well as in a high percentage of those with fibromyalgia.6,7 The many related and comorbid ailments preclude a straightforward diagnosis. Testing for more common illnesses, as well as consideration of associated disorders, is essential in the care of the patient with POTS.
On the next page: Testing and referral >>
TESTING AND REFERRAL
An appropriate workup for POTS includes a complete and thorough history plus a comprehensive physical examination. This includes reviewing the patient’s medication list for any medications or combinations of medications that cause cerebral hypoperfusion, volume depletion, tachycardia, or peripheral vasodilation.2 It is essential to explore the family history for genetic disorders12 and to take a detailed social history, including lifestyle habits that may reproduce the symptoms of POTS.
The definitive diagnosis of POTS is made with a tilt-table test. Obtaining in-office orthostatic vitals can substantially increase the index of suspicion for the syndrome. The procedure used to obtain orthostatic vitals varies by institution. However, every protocol includes a period of rest in a supine position, after which the patient is asked to stand and the blood pressure and pulse are measured after a specified number of minutes. If the patient becomes symptomatic on standing, the blood pressure remains unchanged, and the pulse is elevated to at least 30 beats/min from the supine pulse or greater than 20 beats/min after 10 minutes, a diagnosis of POTS must be considered. Unless a more common cause of the tachycardia is found and treated to resolution, tilt-table testing should be ordered to evaluate these patients for POTS.
If POTS is suspected once the history and physical are completed, the patient should be referred to cardiology or neurology for tilt-table testing to confirm the diagnosis. Laboratory testing should include, but not be limited to, a complete blood count with differential, a comprehensive metabolic panel, Westergren sedimentation rate, and thyroid testing. Adrenal testing,11 a search for vitamin deficiencies, autoimmune diseases, and infective causes (eg, Lyme disease and cytomegalovirus), and urinalysis may also be appropriate based on the patient history and presentation.14
Broad ancillary testing and referral may be needed in patients with POTS. According to Giesken,14 an ECG, echocardiogram, cardiac event monitoring, and an evaluation by a cardiologist are essential. Pasupuleti and Vedre17 recommend referral to a neurologist for evaluation for peripheral and central lesions with electromyography and MRI. Depending on the practice, tilt-table testing may be available through either specialty and may guide the work-up. Referrals to an endocrinologist, a rheumatologist, and a nephrologist to evaluate hyperadrenergic states, autoimmune dysfunction, and kidney disease should be considered as well.14,16 Referrals and additional tests may be necessary, even if secondary causes of POTS are not identified. These tests, although extensive and expensive, may help determine the best and most appropriate treatment options to control the patient’s symptoms and restore quality of life.
On the next page: Treatment >>
TREATMENT
Once the diagnosis of POTS has been confirmed, symptomatic treatment tailored to the patient’s symptoms can begin. Secondary POTS often resolves with effective treatment of the causative disease. One of the most important aspects of effective treatment of primary POTS is to educate patients on how to control their symptoms. Patients must understand that primary POTS cannot be “cured” by current medical therapies. They need to be aware of the erratic symptoms and be proactive in preventing them from occurring. Both nonpharmacologic and pharmacologic treatments are helpful, but a multidisciplinary approach appears to be most efficacious for optimizing return to baseline function in patients.14,16
To prevent exacerbations, patients should avoid situations that precipitate symptoms, such as prolonged standing, inadequate water intake, and cold and hot environments. In addition, providers should advise patients to optimize their fluid status. The literature recommends adding up to 20 g of sodium to the diet daily, and patients should be instructed to drink a minimum of two liters of water during the course of the day.13,16 This includes drinking at least eight ounces of water before rising from a recumbent position. Patients should also be instructed to sleep on an incline7 and to gradually transition to standing when getting up in the morning or after a nap.12 Clinicians should also recommend waist-high graded elastic hose, an abdominal girdle, or both, to assist venous return from the lower extremities and to avoid splanchnic pooling.13
Exercise training has been shown to be effective in treating symptoms of POTS. A recent study published by Fu et al7 demonstrated improved oxygen uptake, increased blood volume, increased cardiac output, and increased left ventricular mass with graduated exercise over a period of several months. The study group demonstrated a lower average resting heart rate and improved exercise tolerance on completion of the trial. In fact, the study reported that after training, more than half of the study patients (10 of 19) no longer fulfilled the criteria for POTS, and all patients who underwent training experienced significant improvements in quality of life.7
PHARMACOLOGIC THERAPIES
Pharmacologic therapies for POTS abound. Treatments must be tailored to each patient’s needs based on the suspected or proven etiology of POTS, as well as any associated or comorbid conditions. Fludrocortisone will decrease salt loss and increase plasma volume in patients with hypovolemia. Midodrine improves vasoconstriction in the extremities. Beta-blockers slow the heart rate and prevent vasodilation. Clonidine can lower the blood pressure and decrease the heart rate by preventing central sympathetic stimulation.12 Alternatively, or in conjunction with the other treatment modalities, selective serotonin reuptake inhibitors may improve sleep, slow the heart rate,16 improve mood, and alleviate gastrointestinal symptoms. As with all medications, clinicians must discuss the risks, benefits, adverse effects, alternatives, and potential interactions with the patient prior to starting medications.
On the next page: Conclusion >>
CONCLUSION
POTS most often presents as OI, which is why investigation of OI should include consideration of POTS as part of the differential diagnosis. Untreated, the symptoms of POTS can prevent patients from participating in normal life activities, including recreation, school, and work, leading to dysfunction, disability, and depression.7 A prompt diagnosis, confirmed by tilt-table testing, will expedite appropriate referral, ancillary testing, and treatment. Optimal therapy has not been established, yet individualized patient education, along with the development of a personalized program to alleviate symptoms, will provide patients with a sense of hope and control over the syndrome. This approach will enable patients and their families to manage the condition and optimize their quality of life.
The author would like to thank Michael Whitehead, DHSc, MPAS, PA-C, DFAAPA, an adjunct professor at A. T. Still University in Mesa, Arizona, for his input on this article.
1. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome.Clin Auton Res. 2011;21:69-72.
2. Johnson JN, Mack KJ, Kuntz NL, et al. Postural orthostatic tachcardia syndrome: a clinical review. Pediatr Neurol. 2009;42:77-85.
3. US Department of Health and Human Services, National Institutes of Health, National Institiute of Neurological Disorders and Stroke. Postural Tachycardia Syndrome Information Page. 2011. www.ninds.nih.gov/disorders/postural_tachycardia_syndrome/postural_tachycardia_syndrome.htm. Accessed March 24, 2014.
4. Agarwal AK, Garg R, Ritch A, et al. Postural orthostatic tachycardia syndrome. Postgrad Med J. 2007;83:478-480.
5. Jacob G, Costa F, Shannon JR, et al. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343:1008-1014.
6. Staud R. Autonomic dysfunction in fibromyalgia syndrome: postural orthostatic tachycardia. Curr Rheumatol Rep. 2008;10:463-466.
7. Fu Q, VanGundy TB, Galbreath MM, et al. Cardiac origins of the postural orthostatic tachycardia syndrome. J Am Coll Cardiol. 2010;55:
2858-2868.
8. Stewart JM. Postural tachycardia syndrome and reflex syncope: similarities and differences. J Pediatr. 2009;154:481-485.
9. Lanier JB, Mote MB, Clay EC. Evaluation and management of orthostatic hypotension. Am Fam Physician. 2011;84:527-536.
10. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249.
11. Graham U, Ritchie KM. Reminder of important clinical lesson: postural orthostatic tachycardia syndrome. BMJ Case Rep. 2009;2009: bcr10.2008.1132. www.ncbi.nlm.nih.gov/pmc/articles/PMC3029273. Accessed March 24, 2014.
12. Mathias CJ, Low DA, Iodice V, et al. Postural tachycardia syndrome—current experience and concepts. Nat Rev Neurol. 2012;8:22-34.
13. Thanavaro JL, Thanavaro KL. Postural orthostatic tachycardia syndrome: diagnosis and treatment. Heart Lung. 2011;40:554-560.
14. Giesken B, Collins M. A 46-year-old woman with postural orthostatic tachycardia syndrome. JAAPA. 2013;26:30-34.
15. Low PA, Sandroni P, Joyner M, et al. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol. 2009;20:352-358.
16. Busmer L. Postural orthostatic tachycardia syndrome. Primary Health Care. 2011;21:16-20.
17. Pasupuleti DV, Vedre A. Postural orthostatic tachycardia warrants investigation of Chiari I malformation as a possible cause. Cardiology. 2005;103:55-56.
CE/CME No: CR-1404
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define postural orthostatic tachycardia syndrome (POTS).
• List the presenting signs and symptoms of POTS.
• Differentiate POTS from other causes of orthostatic intolerance.
• Explain the common classifications of POTS.
• Enumerate the various referral and treatment options for the clinician to consider when a patient meets the criteria for POTS.
FACULTY
Molly Paulson is an Assistant Professor of Physician Assistant Studies in the College of Health Professions at Grand Valley State University, Grand Rapids, Michigan.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Postural orthostatic tachycardia syndrome may not be the first disorder that clinicians consider when they encounter a patient with orthostatic intolerance, but ignoring this possibility during a differential diagnosis can mean patients continue to experience unexplained dizziness, fatigue, syncope, and a variety of other related signs and symptoms. Arriving at the correct diagnosis will allow you to help patients manage the condition and return to the lives and activities they previously enjoyed.
Clinicians should consider postural orthostatic tachycardia syndrome (POTS; ICD-9 code 427.89) as part of the differential diagnosis of orthostatic intolerance (OI). Ignoring this possibility can delay proper diagnosis and treatment among patients who meet the criteria for POTS.
When patients present with symptoms of OI of unknown origin, it is important to consider a broad variety of conditions, including POTS, in the differential diagnosis. Clinicians should follow up with appropriate testing to determine if the criteria for any of these conditions are met. Once the cause of the OI is confirmed, appropriate initial care and referral will facilitate improved treatment for the patient.
A recent consensus statement defines POTS as an increase in heart rate of at least 30 beats/min or a heart rate of at least 120 beats/min upon standing from a supine position—or in response to a tilt-table test—that is sustained for 10 minutes without evidence of orthostatic hypotension.1,2 Although POTS can be seen in men, most cases occur in women of childbearing age, with a female-to-male ratio of 4:1 to 5:1.2 Patients most commonly present with symptoms similar to orthostatic hypotension.3 Typical symptoms include dizziness, near-syncope or syncope, fatigue, headache, visual changes, lightheadedness, weakness, and abdominal discomfort.4,5 Presenting symptoms also overlap with other, more prevalent disorders, leading to frequent misdiagnosis.
Examples of common initial misdiagnoses for patients with POTS include anxiety, chronic fatigue syndrome, fibromyalgia, menopause, orthostatic hypotension, unstable angina, and hyperadrenergic states.1,6 Inappropriate or delayed treatment of POTS can lead to significant daily functional impairment similar to that associated with chronic obstructive pulmonary disease or congestive heart failure.4,6,7
UNDERSTANDING ORTHOSTATIC INTOLERANCE
To better understand POTS, clinicians should have a clear understanding of OI. According to Stewart,8 OI is defined by symptoms of impending near-syncope or syncope, including dizziness, headache, fatigue, exercise intolerance, abdominal distress, or a sensation of feeling “hot” accompanied by sweating. These symptoms occur when a patient assumes an upright position and are relieved when the person returns to a supine position.8
Normally, when a person stands, gravitational forces cause a redistribution of 300 to 800 mL of blood in both the splanchnic and lower extremity circulation. This decreases systolic blood pressure and slows cardiac filling. The resultant decreased cardiac output stimulates increased sympathetic tone via the baroreceptors and other mechanisms to restore arterial blood pressure.1,9 In OI, this compensatory mechanism is compromised, and the patient experiences the symptoms of impending near-syncope or syncope, as discussed previously.
A broad array of medical conditions with multiple etiologies meet the criteria for OI.10 Although a comprehensive list of these conditions is beyond the scope of this article, it is essential for the clinician to look for primary and secondary causes of orthostasis by gathering a comprehensive history, followed by an extensive physical exam directed by the history. The patient should be asked about a history of allergies or autoimmune disease, cancers, eating disorders, infections, adverse effects of medication, and signs or symptoms of chronic sympathetic stimulation. It is also important to review the current medication list carefully and evaluate the patient for dysfunction of the cardiac, endocrine, renal, and nervous systems and for psychiatric disorders. A short list of conditions that may present with OI include anemia, anorexia, autoimmune disease, cancers, cardiac disease, diabetes, infections, paraneoplastic syndromes, vascular disease, and volume depletion.2,8,11,12
Hypotension is absent in POTS, but it otherwise also fits the profile of OI. POTS can also present with a host of other nonspecific, nonorthostatic symptoms, including nausea, vomiting, diarrhea, constipation, fatigue, migraine headaches, and chest pain.10 This unusual presentation can confound the clinician and complicate the medical picture, making a clear diagnosis difficult.
On the next page: History, epidemiology, and etiology >>
HISTORY, EPIDEMIOLOGY, AND ETIOLOGY
POTS was first recognized and described during the Civil War as irritable heart syndrome or soldier’s heart due to the frequency of the condition in combat soldiers. As more was published about the condition, the list of names grew to include Da Costa syndrome, anxiety neurosis, chronic orthostatic intolerance, effort syndrome, idiopathic hypovolemia, mitral valve prolapse syndrome, orthostatic tachycardia, positional tachycardia syndrome, and finally postural orthostatic tachycardia syndrome.5,12 Although initially described in men in the military, the condition is more frequently seen in young women from puberty to age 50. Some researchers have proposed that POTS is much more common than recent studies report; they suggest that the condition is underreported both because of the nonspecific nature of the symptoms and because it is not often included in the differential of OI.5
Although some researchers disagree,7,12 most of the literature supports multiple etiologies of POTS.1,2,13 Researchers have divided the syndrome into primary and secondary causes. They have further separated primary POTS into two broad categories based on neuropathic or hyperadrenergic pathology. In both subgroups, POTS is exacerbated by dependent vasodilatation and central hypovolemia.11
In the more common neuropathic POTS, a precipitating factor such as an infection, trauma, pregnancy, or surgery precedes the sudden onset of symptoms. In the majority of patients with neuropathic POTS, high normal to elevated serum norepinephrine (NE) levels are detected, as compared to “normal” controls,5 but known antibodies are not detected. However, in approximately 10% to 15% of patients with a reported antecedent infection, acetylcholine receptor antibodies have been detected.2,14 This form is described in the literature as immune-mediated POTS.2,11,14,15
The less common primary hyperadrenergic POTS presents more insidiously and may have a genetic component. As documented in some families, a point mutation in the NE transporter protein has been shown to slow clearance of NE from the synaptic cleft in the central nervous system. This leads to excessive central sympathetic stimulation of the periphery.12,13,16 Developmental POTS, which presents at puberty with lower extremity neuropathy, is another form of primary POTS and resolves with maturity (see Table 1).6
Secondary POTS can be triggered by several known conditions including alcoholism.12,16 Any disease or insult that damages the peripheral autonomic system may precipitate POTS. Examples include autoimmune diseases, diabetes, chemotherapy, and heavy metal poisoning.13 Genetic disorders have also been associated with or can contribute to secondary POTS. Joint hypermobility syndrome (JHS), also known as Ehlers-Danlos type III, is a genetic disorder characterized by defects in the structure of connective tissue.16 In JHS, the connective tissue in blood vessels is more elastic, leading to greater lower extremity pooling and decreased vasoconstrictive response to sympathetic stimulation on standing. The literature also notes that prolonged bed rest and any medications that contribute to central hypotension or interfere with compensatory peripheral vasoconstriction may precipitate POTS (see Table 2).
On the next page: Clinical presentation and differential diagnosis >>
CLINICAL PRESENTATION
Patients with POTS frequently present with a sudden onset of OI related to activity or changes from recumbency to standing. They often identify an illness, accident, or surgery preceding the onset of symptoms. Other clinical manifestations of POTS include acrocyanosis, fatigue, headaches, constitutional hypotension, sleep disturbances, and gastrointestinal symptoms.12,13 Frequently, patients come to the emergency department with chest pain, indigestion, fatigue, and heart palpitations,14,15 concerned that they are having a heart attack. Since symptoms resolve with recumbency and no clinical markers for acute coronary syndromes are found, no further workup is obtained, and patients are sent home with the recommendation to see their primary care provider.
The symptoms of POTS are complex, nonspecific, and debilitating. Fatigue, difficulty concentrating, and tachycardia when standing make activities of daily living such as work, home maintenance, and child care challenging. The unpredictable symptoms make life frustrating for patients and their families. The presenting symptoms, the normal results for routine blood tests, and the lack of cardiac abnormalities often lead providers to a diagnosis of anxiety or panic disorder.13 Since the symptoms of POTS are somewhat mitigated by treatment with selective serotonin reuptake inhibitors,16 patients and providers often will accept the diagnosis and not investigate the symptoms further.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of POTS begins with any condition that causes tachycardia or OI. Clinicians should also consider and rule out cardiac abnormalities such as inappropriate tachycardia, supraventricular tachycardia, Wolff-Parkinson-White syndrome, and other cardiac dysrhythmias. Other diagnoses that should be considered are endocrine abnormalities, including hyperthyroidism, hypoadrenalism, and pheochromocytoma; drug adverse effects and interactions, necessitating scrupulous evaluation of the patient’s medication list; changes in volume status; and kidney disease.1 Causes of autonomic dysfunction in both the peripheral and central nervous system, including Chiari I malformation, need to be scrutinized.17
Many conditions within the differential diagnosis can occur concomitantly with POTS and need to be treated, even as further evaluation continues. An extensive history and review of systems is essential to obtain a complete list of symptoms and lead the clinician toward an appropriate physical exam and necessary adjunct testing.
ASSOCIATED DISORDERS
Of the many conditions associated with POTS, JHS is the most common genetic disorder. In this disorder, a genetic mutation produces changes in the composition of the collagen, resulting in enhanced flexibility and compliance of any tissue with collagen in it.16 Mitral valve prolapse is also associated with JHS and POTS.12
POTS is present in 40% of patients diagnosed with chronic fatigue syndrome, as well as in a high percentage of those with fibromyalgia.6,7 The many related and comorbid ailments preclude a straightforward diagnosis. Testing for more common illnesses, as well as consideration of associated disorders, is essential in the care of the patient with POTS.
On the next page: Testing and referral >>
TESTING AND REFERRAL
An appropriate workup for POTS includes a complete and thorough history plus a comprehensive physical examination. This includes reviewing the patient’s medication list for any medications or combinations of medications that cause cerebral hypoperfusion, volume depletion, tachycardia, or peripheral vasodilation.2 It is essential to explore the family history for genetic disorders12 and to take a detailed social history, including lifestyle habits that may reproduce the symptoms of POTS.
The definitive diagnosis of POTS is made with a tilt-table test. Obtaining in-office orthostatic vitals can substantially increase the index of suspicion for the syndrome. The procedure used to obtain orthostatic vitals varies by institution. However, every protocol includes a period of rest in a supine position, after which the patient is asked to stand and the blood pressure and pulse are measured after a specified number of minutes. If the patient becomes symptomatic on standing, the blood pressure remains unchanged, and the pulse is elevated to at least 30 beats/min from the supine pulse or greater than 20 beats/min after 10 minutes, a diagnosis of POTS must be considered. Unless a more common cause of the tachycardia is found and treated to resolution, tilt-table testing should be ordered to evaluate these patients for POTS.
If POTS is suspected once the history and physical are completed, the patient should be referred to cardiology or neurology for tilt-table testing to confirm the diagnosis. Laboratory testing should include, but not be limited to, a complete blood count with differential, a comprehensive metabolic panel, Westergren sedimentation rate, and thyroid testing. Adrenal testing,11 a search for vitamin deficiencies, autoimmune diseases, and infective causes (eg, Lyme disease and cytomegalovirus), and urinalysis may also be appropriate based on the patient history and presentation.14
Broad ancillary testing and referral may be needed in patients with POTS. According to Giesken,14 an ECG, echocardiogram, cardiac event monitoring, and an evaluation by a cardiologist are essential. Pasupuleti and Vedre17 recommend referral to a neurologist for evaluation for peripheral and central lesions with electromyography and MRI. Depending on the practice, tilt-table testing may be available through either specialty and may guide the work-up. Referrals to an endocrinologist, a rheumatologist, and a nephrologist to evaluate hyperadrenergic states, autoimmune dysfunction, and kidney disease should be considered as well.14,16 Referrals and additional tests may be necessary, even if secondary causes of POTS are not identified. These tests, although extensive and expensive, may help determine the best and most appropriate treatment options to control the patient’s symptoms and restore quality of life.
On the next page: Treatment >>
TREATMENT
Once the diagnosis of POTS has been confirmed, symptomatic treatment tailored to the patient’s symptoms can begin. Secondary POTS often resolves with effective treatment of the causative disease. One of the most important aspects of effective treatment of primary POTS is to educate patients on how to control their symptoms. Patients must understand that primary POTS cannot be “cured” by current medical therapies. They need to be aware of the erratic symptoms and be proactive in preventing them from occurring. Both nonpharmacologic and pharmacologic treatments are helpful, but a multidisciplinary approach appears to be most efficacious for optimizing return to baseline function in patients.14,16
To prevent exacerbations, patients should avoid situations that precipitate symptoms, such as prolonged standing, inadequate water intake, and cold and hot environments. In addition, providers should advise patients to optimize their fluid status. The literature recommends adding up to 20 g of sodium to the diet daily, and patients should be instructed to drink a minimum of two liters of water during the course of the day.13,16 This includes drinking at least eight ounces of water before rising from a recumbent position. Patients should also be instructed to sleep on an incline7 and to gradually transition to standing when getting up in the morning or after a nap.12 Clinicians should also recommend waist-high graded elastic hose, an abdominal girdle, or both, to assist venous return from the lower extremities and to avoid splanchnic pooling.13
Exercise training has been shown to be effective in treating symptoms of POTS. A recent study published by Fu et al7 demonstrated improved oxygen uptake, increased blood volume, increased cardiac output, and increased left ventricular mass with graduated exercise over a period of several months. The study group demonstrated a lower average resting heart rate and improved exercise tolerance on completion of the trial. In fact, the study reported that after training, more than half of the study patients (10 of 19) no longer fulfilled the criteria for POTS, and all patients who underwent training experienced significant improvements in quality of life.7
PHARMACOLOGIC THERAPIES
Pharmacologic therapies for POTS abound. Treatments must be tailored to each patient’s needs based on the suspected or proven etiology of POTS, as well as any associated or comorbid conditions. Fludrocortisone will decrease salt loss and increase plasma volume in patients with hypovolemia. Midodrine improves vasoconstriction in the extremities. Beta-blockers slow the heart rate and prevent vasodilation. Clonidine can lower the blood pressure and decrease the heart rate by preventing central sympathetic stimulation.12 Alternatively, or in conjunction with the other treatment modalities, selective serotonin reuptake inhibitors may improve sleep, slow the heart rate,16 improve mood, and alleviate gastrointestinal symptoms. As with all medications, clinicians must discuss the risks, benefits, adverse effects, alternatives, and potential interactions with the patient prior to starting medications.
On the next page: Conclusion >>
CONCLUSION
POTS most often presents as OI, which is why investigation of OI should include consideration of POTS as part of the differential diagnosis. Untreated, the symptoms of POTS can prevent patients from participating in normal life activities, including recreation, school, and work, leading to dysfunction, disability, and depression.7 A prompt diagnosis, confirmed by tilt-table testing, will expedite appropriate referral, ancillary testing, and treatment. Optimal therapy has not been established, yet individualized patient education, along with the development of a personalized program to alleviate symptoms, will provide patients with a sense of hope and control over the syndrome. This approach will enable patients and their families to manage the condition and optimize their quality of life.
The author would like to thank Michael Whitehead, DHSc, MPAS, PA-C, DFAAPA, an adjunct professor at A. T. Still University in Mesa, Arizona, for his input on this article.
CE/CME No: CR-1404
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define postural orthostatic tachycardia syndrome (POTS).
• List the presenting signs and symptoms of POTS.
• Differentiate POTS from other causes of orthostatic intolerance.
• Explain the common classifications of POTS.
• Enumerate the various referral and treatment options for the clinician to consider when a patient meets the criteria for POTS.
FACULTY
Molly Paulson is an Assistant Professor of Physician Assistant Studies in the College of Health Professions at Grand Valley State University, Grand Rapids, Michigan.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Postural orthostatic tachycardia syndrome may not be the first disorder that clinicians consider when they encounter a patient with orthostatic intolerance, but ignoring this possibility during a differential diagnosis can mean patients continue to experience unexplained dizziness, fatigue, syncope, and a variety of other related signs and symptoms. Arriving at the correct diagnosis will allow you to help patients manage the condition and return to the lives and activities they previously enjoyed.
Clinicians should consider postural orthostatic tachycardia syndrome (POTS; ICD-9 code 427.89) as part of the differential diagnosis of orthostatic intolerance (OI). Ignoring this possibility can delay proper diagnosis and treatment among patients who meet the criteria for POTS.
When patients present with symptoms of OI of unknown origin, it is important to consider a broad variety of conditions, including POTS, in the differential diagnosis. Clinicians should follow up with appropriate testing to determine if the criteria for any of these conditions are met. Once the cause of the OI is confirmed, appropriate initial care and referral will facilitate improved treatment for the patient.
A recent consensus statement defines POTS as an increase in heart rate of at least 30 beats/min or a heart rate of at least 120 beats/min upon standing from a supine position—or in response to a tilt-table test—that is sustained for 10 minutes without evidence of orthostatic hypotension.1,2 Although POTS can be seen in men, most cases occur in women of childbearing age, with a female-to-male ratio of 4:1 to 5:1.2 Patients most commonly present with symptoms similar to orthostatic hypotension.3 Typical symptoms include dizziness, near-syncope or syncope, fatigue, headache, visual changes, lightheadedness, weakness, and abdominal discomfort.4,5 Presenting symptoms also overlap with other, more prevalent disorders, leading to frequent misdiagnosis.
Examples of common initial misdiagnoses for patients with POTS include anxiety, chronic fatigue syndrome, fibromyalgia, menopause, orthostatic hypotension, unstable angina, and hyperadrenergic states.1,6 Inappropriate or delayed treatment of POTS can lead to significant daily functional impairment similar to that associated with chronic obstructive pulmonary disease or congestive heart failure.4,6,7
UNDERSTANDING ORTHOSTATIC INTOLERANCE
To better understand POTS, clinicians should have a clear understanding of OI. According to Stewart,8 OI is defined by symptoms of impending near-syncope or syncope, including dizziness, headache, fatigue, exercise intolerance, abdominal distress, or a sensation of feeling “hot” accompanied by sweating. These symptoms occur when a patient assumes an upright position and are relieved when the person returns to a supine position.8
Normally, when a person stands, gravitational forces cause a redistribution of 300 to 800 mL of blood in both the splanchnic and lower extremity circulation. This decreases systolic blood pressure and slows cardiac filling. The resultant decreased cardiac output stimulates increased sympathetic tone via the baroreceptors and other mechanisms to restore arterial blood pressure.1,9 In OI, this compensatory mechanism is compromised, and the patient experiences the symptoms of impending near-syncope or syncope, as discussed previously.
A broad array of medical conditions with multiple etiologies meet the criteria for OI.10 Although a comprehensive list of these conditions is beyond the scope of this article, it is essential for the clinician to look for primary and secondary causes of orthostasis by gathering a comprehensive history, followed by an extensive physical exam directed by the history. The patient should be asked about a history of allergies or autoimmune disease, cancers, eating disorders, infections, adverse effects of medication, and signs or symptoms of chronic sympathetic stimulation. It is also important to review the current medication list carefully and evaluate the patient for dysfunction of the cardiac, endocrine, renal, and nervous systems and for psychiatric disorders. A short list of conditions that may present with OI include anemia, anorexia, autoimmune disease, cancers, cardiac disease, diabetes, infections, paraneoplastic syndromes, vascular disease, and volume depletion.2,8,11,12
Hypotension is absent in POTS, but it otherwise also fits the profile of OI. POTS can also present with a host of other nonspecific, nonorthostatic symptoms, including nausea, vomiting, diarrhea, constipation, fatigue, migraine headaches, and chest pain.10 This unusual presentation can confound the clinician and complicate the medical picture, making a clear diagnosis difficult.
On the next page: History, epidemiology, and etiology >>
HISTORY, EPIDEMIOLOGY, AND ETIOLOGY
POTS was first recognized and described during the Civil War as irritable heart syndrome or soldier’s heart due to the frequency of the condition in combat soldiers. As more was published about the condition, the list of names grew to include Da Costa syndrome, anxiety neurosis, chronic orthostatic intolerance, effort syndrome, idiopathic hypovolemia, mitral valve prolapse syndrome, orthostatic tachycardia, positional tachycardia syndrome, and finally postural orthostatic tachycardia syndrome.5,12 Although initially described in men in the military, the condition is more frequently seen in young women from puberty to age 50. Some researchers have proposed that POTS is much more common than recent studies report; they suggest that the condition is underreported both because of the nonspecific nature of the symptoms and because it is not often included in the differential of OI.5
Although some researchers disagree,7,12 most of the literature supports multiple etiologies of POTS.1,2,13 Researchers have divided the syndrome into primary and secondary causes. They have further separated primary POTS into two broad categories based on neuropathic or hyperadrenergic pathology. In both subgroups, POTS is exacerbated by dependent vasodilatation and central hypovolemia.11
In the more common neuropathic POTS, a precipitating factor such as an infection, trauma, pregnancy, or surgery precedes the sudden onset of symptoms. In the majority of patients with neuropathic POTS, high normal to elevated serum norepinephrine (NE) levels are detected, as compared to “normal” controls,5 but known antibodies are not detected. However, in approximately 10% to 15% of patients with a reported antecedent infection, acetylcholine receptor antibodies have been detected.2,14 This form is described in the literature as immune-mediated POTS.2,11,14,15
The less common primary hyperadrenergic POTS presents more insidiously and may have a genetic component. As documented in some families, a point mutation in the NE transporter protein has been shown to slow clearance of NE from the synaptic cleft in the central nervous system. This leads to excessive central sympathetic stimulation of the periphery.12,13,16 Developmental POTS, which presents at puberty with lower extremity neuropathy, is another form of primary POTS and resolves with maturity (see Table 1).6
Secondary POTS can be triggered by several known conditions including alcoholism.12,16 Any disease or insult that damages the peripheral autonomic system may precipitate POTS. Examples include autoimmune diseases, diabetes, chemotherapy, and heavy metal poisoning.13 Genetic disorders have also been associated with or can contribute to secondary POTS. Joint hypermobility syndrome (JHS), also known as Ehlers-Danlos type III, is a genetic disorder characterized by defects in the structure of connective tissue.16 In JHS, the connective tissue in blood vessels is more elastic, leading to greater lower extremity pooling and decreased vasoconstrictive response to sympathetic stimulation on standing. The literature also notes that prolonged bed rest and any medications that contribute to central hypotension or interfere with compensatory peripheral vasoconstriction may precipitate POTS (see Table 2).
On the next page: Clinical presentation and differential diagnosis >>
CLINICAL PRESENTATION
Patients with POTS frequently present with a sudden onset of OI related to activity or changes from recumbency to standing. They often identify an illness, accident, or surgery preceding the onset of symptoms. Other clinical manifestations of POTS include acrocyanosis, fatigue, headaches, constitutional hypotension, sleep disturbances, and gastrointestinal symptoms.12,13 Frequently, patients come to the emergency department with chest pain, indigestion, fatigue, and heart palpitations,14,15 concerned that they are having a heart attack. Since symptoms resolve with recumbency and no clinical markers for acute coronary syndromes are found, no further workup is obtained, and patients are sent home with the recommendation to see their primary care provider.
The symptoms of POTS are complex, nonspecific, and debilitating. Fatigue, difficulty concentrating, and tachycardia when standing make activities of daily living such as work, home maintenance, and child care challenging. The unpredictable symptoms make life frustrating for patients and their families. The presenting symptoms, the normal results for routine blood tests, and the lack of cardiac abnormalities often lead providers to a diagnosis of anxiety or panic disorder.13 Since the symptoms of POTS are somewhat mitigated by treatment with selective serotonin reuptake inhibitors,16 patients and providers often will accept the diagnosis and not investigate the symptoms further.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of POTS begins with any condition that causes tachycardia or OI. Clinicians should also consider and rule out cardiac abnormalities such as inappropriate tachycardia, supraventricular tachycardia, Wolff-Parkinson-White syndrome, and other cardiac dysrhythmias. Other diagnoses that should be considered are endocrine abnormalities, including hyperthyroidism, hypoadrenalism, and pheochromocytoma; drug adverse effects and interactions, necessitating scrupulous evaluation of the patient’s medication list; changes in volume status; and kidney disease.1 Causes of autonomic dysfunction in both the peripheral and central nervous system, including Chiari I malformation, need to be scrutinized.17
Many conditions within the differential diagnosis can occur concomitantly with POTS and need to be treated, even as further evaluation continues. An extensive history and review of systems is essential to obtain a complete list of symptoms and lead the clinician toward an appropriate physical exam and necessary adjunct testing.
ASSOCIATED DISORDERS
Of the many conditions associated with POTS, JHS is the most common genetic disorder. In this disorder, a genetic mutation produces changes in the composition of the collagen, resulting in enhanced flexibility and compliance of any tissue with collagen in it.16 Mitral valve prolapse is also associated with JHS and POTS.12
POTS is present in 40% of patients diagnosed with chronic fatigue syndrome, as well as in a high percentage of those with fibromyalgia.6,7 The many related and comorbid ailments preclude a straightforward diagnosis. Testing for more common illnesses, as well as consideration of associated disorders, is essential in the care of the patient with POTS.
On the next page: Testing and referral >>
TESTING AND REFERRAL
An appropriate workup for POTS includes a complete and thorough history plus a comprehensive physical examination. This includes reviewing the patient’s medication list for any medications or combinations of medications that cause cerebral hypoperfusion, volume depletion, tachycardia, or peripheral vasodilation.2 It is essential to explore the family history for genetic disorders12 and to take a detailed social history, including lifestyle habits that may reproduce the symptoms of POTS.
The definitive diagnosis of POTS is made with a tilt-table test. Obtaining in-office orthostatic vitals can substantially increase the index of suspicion for the syndrome. The procedure used to obtain orthostatic vitals varies by institution. However, every protocol includes a period of rest in a supine position, after which the patient is asked to stand and the blood pressure and pulse are measured after a specified number of minutes. If the patient becomes symptomatic on standing, the blood pressure remains unchanged, and the pulse is elevated to at least 30 beats/min from the supine pulse or greater than 20 beats/min after 10 minutes, a diagnosis of POTS must be considered. Unless a more common cause of the tachycardia is found and treated to resolution, tilt-table testing should be ordered to evaluate these patients for POTS.
If POTS is suspected once the history and physical are completed, the patient should be referred to cardiology or neurology for tilt-table testing to confirm the diagnosis. Laboratory testing should include, but not be limited to, a complete blood count with differential, a comprehensive metabolic panel, Westergren sedimentation rate, and thyroid testing. Adrenal testing,11 a search for vitamin deficiencies, autoimmune diseases, and infective causes (eg, Lyme disease and cytomegalovirus), and urinalysis may also be appropriate based on the patient history and presentation.14
Broad ancillary testing and referral may be needed in patients with POTS. According to Giesken,14 an ECG, echocardiogram, cardiac event monitoring, and an evaluation by a cardiologist are essential. Pasupuleti and Vedre17 recommend referral to a neurologist for evaluation for peripheral and central lesions with electromyography and MRI. Depending on the practice, tilt-table testing may be available through either specialty and may guide the work-up. Referrals to an endocrinologist, a rheumatologist, and a nephrologist to evaluate hyperadrenergic states, autoimmune dysfunction, and kidney disease should be considered as well.14,16 Referrals and additional tests may be necessary, even if secondary causes of POTS are not identified. These tests, although extensive and expensive, may help determine the best and most appropriate treatment options to control the patient’s symptoms and restore quality of life.
On the next page: Treatment >>
TREATMENT
Once the diagnosis of POTS has been confirmed, symptomatic treatment tailored to the patient’s symptoms can begin. Secondary POTS often resolves with effective treatment of the causative disease. One of the most important aspects of effective treatment of primary POTS is to educate patients on how to control their symptoms. Patients must understand that primary POTS cannot be “cured” by current medical therapies. They need to be aware of the erratic symptoms and be proactive in preventing them from occurring. Both nonpharmacologic and pharmacologic treatments are helpful, but a multidisciplinary approach appears to be most efficacious for optimizing return to baseline function in patients.14,16
To prevent exacerbations, patients should avoid situations that precipitate symptoms, such as prolonged standing, inadequate water intake, and cold and hot environments. In addition, providers should advise patients to optimize their fluid status. The literature recommends adding up to 20 g of sodium to the diet daily, and patients should be instructed to drink a minimum of two liters of water during the course of the day.13,16 This includes drinking at least eight ounces of water before rising from a recumbent position. Patients should also be instructed to sleep on an incline7 and to gradually transition to standing when getting up in the morning or after a nap.12 Clinicians should also recommend waist-high graded elastic hose, an abdominal girdle, or both, to assist venous return from the lower extremities and to avoid splanchnic pooling.13
Exercise training has been shown to be effective in treating symptoms of POTS. A recent study published by Fu et al7 demonstrated improved oxygen uptake, increased blood volume, increased cardiac output, and increased left ventricular mass with graduated exercise over a period of several months. The study group demonstrated a lower average resting heart rate and improved exercise tolerance on completion of the trial. In fact, the study reported that after training, more than half of the study patients (10 of 19) no longer fulfilled the criteria for POTS, and all patients who underwent training experienced significant improvements in quality of life.7
PHARMACOLOGIC THERAPIES
Pharmacologic therapies for POTS abound. Treatments must be tailored to each patient’s needs based on the suspected or proven etiology of POTS, as well as any associated or comorbid conditions. Fludrocortisone will decrease salt loss and increase plasma volume in patients with hypovolemia. Midodrine improves vasoconstriction in the extremities. Beta-blockers slow the heart rate and prevent vasodilation. Clonidine can lower the blood pressure and decrease the heart rate by preventing central sympathetic stimulation.12 Alternatively, or in conjunction with the other treatment modalities, selective serotonin reuptake inhibitors may improve sleep, slow the heart rate,16 improve mood, and alleviate gastrointestinal symptoms. As with all medications, clinicians must discuss the risks, benefits, adverse effects, alternatives, and potential interactions with the patient prior to starting medications.
On the next page: Conclusion >>
CONCLUSION
POTS most often presents as OI, which is why investigation of OI should include consideration of POTS as part of the differential diagnosis. Untreated, the symptoms of POTS can prevent patients from participating in normal life activities, including recreation, school, and work, leading to dysfunction, disability, and depression.7 A prompt diagnosis, confirmed by tilt-table testing, will expedite appropriate referral, ancillary testing, and treatment. Optimal therapy has not been established, yet individualized patient education, along with the development of a personalized program to alleviate symptoms, will provide patients with a sense of hope and control over the syndrome. This approach will enable patients and their families to manage the condition and optimize their quality of life.
The author would like to thank Michael Whitehead, DHSc, MPAS, PA-C, DFAAPA, an adjunct professor at A. T. Still University in Mesa, Arizona, for his input on this article.
1. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome.Clin Auton Res. 2011;21:69-72.
2. Johnson JN, Mack KJ, Kuntz NL, et al. Postural orthostatic tachcardia syndrome: a clinical review. Pediatr Neurol. 2009;42:77-85.
3. US Department of Health and Human Services, National Institutes of Health, National Institiute of Neurological Disorders and Stroke. Postural Tachycardia Syndrome Information Page. 2011. www.ninds.nih.gov/disorders/postural_tachycardia_syndrome/postural_tachycardia_syndrome.htm. Accessed March 24, 2014.
4. Agarwal AK, Garg R, Ritch A, et al. Postural orthostatic tachycardia syndrome. Postgrad Med J. 2007;83:478-480.
5. Jacob G, Costa F, Shannon JR, et al. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343:1008-1014.
6. Staud R. Autonomic dysfunction in fibromyalgia syndrome: postural orthostatic tachycardia. Curr Rheumatol Rep. 2008;10:463-466.
7. Fu Q, VanGundy TB, Galbreath MM, et al. Cardiac origins of the postural orthostatic tachycardia syndrome. J Am Coll Cardiol. 2010;55:
2858-2868.
8. Stewart JM. Postural tachycardia syndrome and reflex syncope: similarities and differences. J Pediatr. 2009;154:481-485.
9. Lanier JB, Mote MB, Clay EC. Evaluation and management of orthostatic hypotension. Am Fam Physician. 2011;84:527-536.
10. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249.
11. Graham U, Ritchie KM. Reminder of important clinical lesson: postural orthostatic tachycardia syndrome. BMJ Case Rep. 2009;2009: bcr10.2008.1132. www.ncbi.nlm.nih.gov/pmc/articles/PMC3029273. Accessed March 24, 2014.
12. Mathias CJ, Low DA, Iodice V, et al. Postural tachycardia syndrome—current experience and concepts. Nat Rev Neurol. 2012;8:22-34.
13. Thanavaro JL, Thanavaro KL. Postural orthostatic tachycardia syndrome: diagnosis and treatment. Heart Lung. 2011;40:554-560.
14. Giesken B, Collins M. A 46-year-old woman with postural orthostatic tachycardia syndrome. JAAPA. 2013;26:30-34.
15. Low PA, Sandroni P, Joyner M, et al. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol. 2009;20:352-358.
16. Busmer L. Postural orthostatic tachycardia syndrome. Primary Health Care. 2011;21:16-20.
17. Pasupuleti DV, Vedre A. Postural orthostatic tachycardia warrants investigation of Chiari I malformation as a possible cause. Cardiology. 2005;103:55-56.
1. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome.Clin Auton Res. 2011;21:69-72.
2. Johnson JN, Mack KJ, Kuntz NL, et al. Postural orthostatic tachcardia syndrome: a clinical review. Pediatr Neurol. 2009;42:77-85.
3. US Department of Health and Human Services, National Institutes of Health, National Institiute of Neurological Disorders and Stroke. Postural Tachycardia Syndrome Information Page. 2011. www.ninds.nih.gov/disorders/postural_tachycardia_syndrome/postural_tachycardia_syndrome.htm. Accessed March 24, 2014.
4. Agarwal AK, Garg R, Ritch A, et al. Postural orthostatic tachycardia syndrome. Postgrad Med J. 2007;83:478-480.
5. Jacob G, Costa F, Shannon JR, et al. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343:1008-1014.
6. Staud R. Autonomic dysfunction in fibromyalgia syndrome: postural orthostatic tachycardia. Curr Rheumatol Rep. 2008;10:463-466.
7. Fu Q, VanGundy TB, Galbreath MM, et al. Cardiac origins of the postural orthostatic tachycardia syndrome. J Am Coll Cardiol. 2010;55:
2858-2868.
8. Stewart JM. Postural tachycardia syndrome and reflex syncope: similarities and differences. J Pediatr. 2009;154:481-485.
9. Lanier JB, Mote MB, Clay EC. Evaluation and management of orthostatic hypotension. Am Fam Physician. 2011;84:527-536.
10. Ojha A, McNeeley K, Heller E, et al. Orthostatic syndromes differ in syncope frequency. Am J Med. 2010;123:245-249.
11. Graham U, Ritchie KM. Reminder of important clinical lesson: postural orthostatic tachycardia syndrome. BMJ Case Rep. 2009;2009: bcr10.2008.1132. www.ncbi.nlm.nih.gov/pmc/articles/PMC3029273. Accessed March 24, 2014.
12. Mathias CJ, Low DA, Iodice V, et al. Postural tachycardia syndrome—current experience and concepts. Nat Rev Neurol. 2012;8:22-34.
13. Thanavaro JL, Thanavaro KL. Postural orthostatic tachycardia syndrome: diagnosis and treatment. Heart Lung. 2011;40:554-560.
14. Giesken B, Collins M. A 46-year-old woman with postural orthostatic tachycardia syndrome. JAAPA. 2013;26:30-34.
15. Low PA, Sandroni P, Joyner M, et al. Postural tachycardia syndrome (POTS). J Cardiovasc Electrophysiol. 2009;20:352-358.
16. Busmer L. Postural orthostatic tachycardia syndrome. Primary Health Care. 2011;21:16-20.
17. Pasupuleti DV, Vedre A. Postural orthostatic tachycardia warrants investigation of Chiari I malformation as a possible cause. Cardiology. 2005;103:55-56.
Allergic Rhinitis & Immunotherapy: Hope or Hype
CE/CME No: CR-1403
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the pathophysiology and etiology of allergic rhinitis (AR).
• Describe the prevalence and types of AR.
• List the differential diagnoses for AR.
• Describe the historical and physical examination findings that are typical
of AR.
• Explain the indications for and types of allergy testing.
• Discuss the types of allergy desensitization therapies/immunotherapies.
FACULTY
Randy D. Danielsen is a Professor and Dean of the Arizona School of Health Sciences, A.T. Still University in Mesa, Arizona, and a long-time PA with the Arizona Asthma & Allergy Institute. Linda S. MacConnell is an Assistant Professor in the Department of Physician Assistant Studies at the Arizona School of Health Sciences, A.T. Still University, and a formally trained otolaryngology PA.
The authors have no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Allergic rhinitis (AR), one of the most familiar complaints seen in primary care, is a common immunologic condition that occurs in genetically predisposed patients. AR is routinely treated through allergen avoidance and pharmacologic therapy. When these measures fail, however, immunologic treatment may be indicated. This review of AR and its treatment focuses on injection and oral immunotherapy.
Congestion; sneezing (particularly paroxysms); itchy nose, palate, and eyes; and runny nose are symptoms characteristic of allergic rhinitis (AR) seen every day in virtually all primary care offices. Patients are plagued not only by their symptoms, but also by AR-related sleep disturbances, resulting in fatigue and daytime sleepiness, irritability, and memory deficits. In children, these sleep disruptions may even cause behavioral disturbances. Depending on the cause, some patients may experience allergic symptoms only with outdoor environmental exposure and subsequent immunoglobulin E (IgE)–mediated responses to otherwise innocuous allergens, while others find that their symptoms are constant, occurring both indoors and out.
Although the prevalence of AR is increasing,1 allergies are certainly nothing new to humankind. In fact, hieroglyphics and Egyptian wall paintings have been discovered depicting a pharaoh dying from anaphylactic shock after receiving a wasp sting.2 In 1565, Leonardo Botallo described AR, calling it “rose catarrh” (mucous or phlegm) or “rose fever,” based on the mistaken idea that the symptoms were caused by rose pollen.2 John Babcock, an English physician, first diagnosed an upper respiratory disease that he called “hay fever” in 1819.
Seventy years later, Charles Blackley identified pollen as a cause of hay fever, documenting his findings in his 1873 book Experimental Researches on the Causes and Nature of Catarrhus Aestivus.2 Dr. Blackley performed the initial documented attempts at allergy desensitization treatments on himself—a willing patient, as he suffered from AR. He placed rye grass pollen onto his nasal mucosa, finding that after 30 minutes the nostril was completely occluded. He continued his experimentation by repeatedly exposing himself to pollen grains via abraded skin. Alas, he never noted any decrease in his symptoms.3
Presently, AR affects more than 55 million people in the United States4—approximately 10% to 30% of the adult population5 and more than 40% of children.6 The rising prevalence of AR is of concern in older adults, who tend to have related comorbidities (eg, chronic sinusitis, asthma, and otitis media). In fact, AR is the fifth most common chronic disease in the US.7
AR and its treatment impose a great economic burden on the health care system, critical in these days of affordable health care. In fact, in 2005 in the US, the overall (direct medical and indirect) cost of AR was $11.2 billion.8 Direct costs derive from office visits, diagnostic testing, and therapeutics. Costs are considerably higher when indirect expenses, including decreased productivity, missed school and missed workdays to care for children, and costs of travel to medical appointments, are included. In the US, approximately 3.5 million workdays and 2 million school days are lost each year due to AR.9 Decreases in productivity cost an estimated $600 per affected employee per year, all of which results in AR being the fifth costliest chronic disease.10,11
On the next page: Pathophysiology and examination >>
PATHOPHYSIOLOGY
AR is an immunologic disorder that occurs in genetically susceptible individuals who produce allergen-specific IgE antibody responses after environmental exposures. The IgE-mediated response causes inflammation of the nasal mucosa. Compared to controls, individuals with AR demonstrate increased amounts of IgE antibodies in the nasal mucosa.11 IgE binds to basophils in the bloodstream and mast cells in tissue. Allergens then attach to IgE on basophils and mast cells, which release histamines, prostaglandins, cytokines, and leukotrienes, with histamine being the most significant mediator in the inflammatory response.
In response to allergy-provoking substances, patients experience immediate- and late-phase symptoms. Symptoms of each stage are similar, but congestion is the hallmark of the late phase. While both phases are clinically important because of their contribution to the patient’s symptoms, most patients experience continued exposure to allergens, resulting in constant, overlapping symptoms.
HISTORY AND PHYSICAL EXAMINATION
Patients with AR relate a history of congestion, excessive mucous production, itchy, watery eyes, bouts of sneezing, and more systemic symptoms, such as headache, malaise, and excessive fatigue. It is important to evaluate the degree and duration of the symptoms, noting patterns and triggers, in an effort to confirm the diagnosis and to help the patient evaluate treatment options.
When taking the patient history, always review the family history, which is often notable for allergies and other atopic diseases. Be sure to ask about medications and recreational drug use; a number of substances have been implicated in the development of rhinitis, including anticholinergic medications, oxymetazoline (when overused), and cocaine. Also, question the patient about self-medication and treatment to determine what may or may not have provided relief. Further questioning may also reveal a history of comorbidities, including contact dermatitis, asthma, eczema, and chronic sinusitis.
The physical examination of the patient with rhinitis begins with observation of the patient’s outward appearance, which may reveal allergic shiners (dark to purplish areas under the eyes), conjunctivitis, an allergic salute (a transverse crease of the nose caused by upward rubbing of an itchy nose), mouth breathing, and a generally tired appearance. The nasal turbinates are swollen and often pale. Mucous secretions are usually thin and clear. Enlarged tonsils and posterior nasal drainage may be visualized. The types of AR are listed in Table 1.

On the next page: Diagnosis and treatment >>
DIAGNOSIS
Most of the clues needed to arrive at a diagnosis are discovered by taking a careful history and completing a physical examination. AR frequently underlies and/or coexists with acute upper respiratory infection (URI) and acute and chronic sinusitis. Differentiating acute URI and acute sinusitis from AR is usually relatively straightforward, based on the symptoms of the illness. The diagnosis of chronic sinusitis is made by radiologic imaging with CT scan.
Distinguishing nonallergic rhinitis (NAR) from AR can be far more difficult, because the symptoms of these conditions are similar and chronic in nature (see Table 2). Empiric treatment for AR may be attempted; however, further testing is often needed to differentiate the two. At this point, clinicians may choose to proceed with specific IgE blood tests. Alternatively, many medical practices are prepared to perform or refer for allergy skin testing.

TREATMENT
Avoidance of known triggers is the cornerstone of allergy treatment. Currently, the most effective pharmaceutical treatment for the majority of AR symptoms is inhaled nasal corticosteroids. Although less effective than corticosteroids, antihistamines—both nasal and oral—are a recommended addition to the regimen if the adverse effects and costs to the patient are tolerable. Other treatments include the leukotriene receptor antagonists, intranasal formulations of cromolyn, and the anticholinergic ipratropium bromide nasal spray, which is effective primarily on watery rhinorrhea. If symptoms are not controlled with medication, allergy immunotherapy (AI), the only known disease-modifying therapy for AR, may be indicated.
On the next page: Allergy testing >>
ALLERGY TESTING
In order to distinguish between AR and NAR and to direct treatment toward specific allergen avoidance and immunotherapy, providers have the choice of ordering in vitro blood IgE testing (to measure the antibodies that mediate an allergic response) or in vivo allergy skin testing (to measure the immune response to allergens that induces an allergic atopic reaction). Allergy testing is not a contemporary concept; the first allergy testing was documented in 1656 when Pierre Borel applied egg to a patient’s skin, which exhibited an allergic reaction.2
Allergy skin testing consists of applying multiple allergens to the skin of the patient’s forearms via tiny pinpricks while watching for immediate hypersensitivity reactions. The test begins with the placement of a drop of histamine to serve as a control. If after 10 minutes of watchful waiting the patient develops a reaction to the histamine (a positive test result), it is appropriate to test for antigens by placing drops of suspected allergen extracts on the skin.
After a period of time (usually 20 to 30 minutes), the area is inspected for allergic reaction. An immediate (early phase) wheal and flare (surrounding erythema) reaction may develop. This positive reaction indicates the presence of a mast cell–bound IgE antibody specific to the tested allergen. The size of the reactions is measured in millimeters, allowing for comparison to the histamine control.

A list of the commonly tested antigens in Arizona, as an example, is shown in Table 3. Antigens vary geographically and even from practice to practice. For up-to-date information on pollen counts by region, visit the American Academy of Allergy Asthma & Immunology Web site (see “Pollen Counts”).

On the next page: Patient selection and allergy immunotherapy >>
Patient Selection
After the clinician has determined that there is a high likelihood that the diagnosis is AR, allergy testing, needed to guide AI, is appropriate. Although not true of specific IgE testing, the accuracy of allergy skin testing results can be adversely affected by several medications. For example, some practitioners may choose to stop first-generation antihistamines two to three days before testing. It is generally accepted that the newer, second-generation antihistamines, which can affect skin-testing results longer, be stopped a week prior to testing.
Patients should be reminded that OTC sleep aids frequently contain antihistamines (particularly diphenhydramine) and that they must be discontinued prior to testing as well. Histamine H2-receptor antagonists such as cimetidine and ranitidine may be stopped a day or two before testing. Although β-blockers are only relatively contraindicated in both allergy testing and AI, many health care providers avoid testing and AI in patients taking oral and/or topical (eye drops) β-blocker therapy. Ultimately, the decision is made by individual health care practices.
In vivo allergy skin testing should not be performed on patients taking tricyclic antidepressants and monoamine oxidase inhibitors. Patients with significant cardiovascular disease should not undergo testing or treatment. Pregnancy is a relative contraindication, and allergy skin testing and AI are done only with obstetrician approval. Most allergists avoid allergy skin testing in pregnant women, however, because use of epinephrine, if required, introduces the risk for preterm labor.12,13 Special consideration should also be given to patients with immune deficiencies.
Setting for Allergy Evaluation and Treatment
Recently, many primary care practices have added allergy evaluation and management to the procedures and treatments they offer; however, evaluations and testing traditionally have been performed by allergy and immunology specialists, many of whom include PAs and NPs on their staff. PAs and NPs frequently manage the practices’ allergy programs. Allergists who are listed as American Board of Allergy and Immunology (ABAI)–certified have successfully passed the ABAI’s certifying examination. Other medical specialists, including otolaryngologists and the primary care specialties, are also well placed to evaluate patients with common allergy symptoms and provide appropriate treatment.
Allergy Immunotherapy
AI has now become more efficacious, safer, and more tolerable for the patient than when it was first introduced in 1911. At that time, Leonard Noon authored a brief article claiming that allergen-specific injections could modify AR. Unfortunately, Noon died at age 36 from tuberculosis, but his work was carried on by his associate, John Freeman. Together, they established the guidelines upon which contemporary AI is based, including the protocol to gradually increase the dose of allergen serum, starting with initial weekly to biweekly injections. They also voiced warnings about the potential for anaphylaxis.14 To this day, immunotherapy is still accomplished by the gradual administration of increasing amounts of the allergen to which the patient is sensitive. This tempers the abnormal immune response to that allergen, easing allergic symptoms.
In patients with IgE-mediated hypersensitivity reactions, confirmed by history, physical examination, and allergy skin testing, immunotherapy can be very effective. The results of immunotherapy may last for years and may even prevent the allergic march, the progression of allergic disease experienced by many patients that frequently begins early in life.15 This includes other allergy-related conditions, such as asthma and eczema (atopic dermatitis), and the acquisition of additional new allergies, including those to foods. AI also has been shown to decrease the frequency of comorbidities such as asthma.5 In addition, AI is used in carefully screened patients who desire to reduce the dosages of medication required to control their symptoms.
In a study published in 1999, Durham and colleagues clarified the questions surrounding the amount of time required for ongoing immunotherapy. They found that the desensitization and tolerance to allergens achieved by AI can last up to three years after a three- to four-year course of therapy. They also found that treatment should not start until an allergic component is identified by allergy skin testing or serum tests for allergen-specific IgE.16
As effective as immunotherapy has been documented to be, it is underused as a therapeutic modality. There are only 2 to 3 million patients receiving subcutaneous immunotherapy out of more than 55 million patients with AR.17
On the next page: Types of allergy immunotherapy >>
Types of AI
There are two categories of allergen desensitization therapy. The most common method is subcutaneous immunotherapy (SCIT), the so-called allergy shots. Less common and still somewhat controversial is sublingual immunotherapy (SLIT). Immunotherapy should be considered for patients who have secondary complications (eg, sinusitis, otitis) or asthma with allergies, or those in whom avoidance measures and medications fail. SCIT or SLIT may be desirable for patients with AR who have difficulty taking regular medications.
Subcutaneous immunotherapy. Currently, SCIT is the most recognized immunotherapy and the only one currently reimbursed by insurance. The procedure involves the subcutaneous injection of increasing doses of therapeutic solutions to which a patient has demonstrated sensitivity. The indications for this treatment are usually inhaled allergens, such as pollens and animal dander. SCIT may also be valuable in patients with asthma and atopic dermatitis.
The most common adverse reaction to allergy injections is a large localized reaction—primarily erythema, pruritus, discomfort, and possibly edema. Severe systemic reactions are extremely rare, with near-fatal to fatal reactions occurring at the rate of only 5.4 per million injections.11 The majority of these rare, albeit serious, complications are caused by higher-than-normal levels of pollen in the environment and dosing errors.11 Because of the uncommon but significant complications, patients undergoing immunotherapy should always receive injections in a medical office equipped with appropriate equipment and staff trained in handling anaphylaxis. It is standard protocol for patients to remain in the office for 30 minutes after administration for observation. Some clinicians prescribe epinephrine injectors (Epi-Pens) for patients to bring to every appointment as a condition for receiving their shot. Because of continuing controversy on this point, others only employ this requirement if the patient has a history of an adverse reaction.
Various protocols exist for the up-dosing of immunotherapy, most of which recommend weekly to twice-weekly injections prior to initiating maintenance therapy. Costs, risks, and benefits must be carefully considered and discussed with the patient prior to initiating immunotherapy.
Many insurance companies reimburse for immunotherapy, with varying copayments. Additionally, the time commitment may be taxing on the patient’s busy schedule. Weekly or biweekly appointments are required initially, and the patient must remain on site for half an hour. Although direct costs of SCIT are relatively easily measured and perhaps compensated, the indirect costs of time spent commuting and at the clinic are less tangible.
Success rates with SCIT may be more than 70% for certain allergens,18 but it is a long-term process with initial improvement often not seen until after six to 12 months of therapy. The benefits of therapy lead not only to reduction and suppression in symptoms (and medication), but also to reduction in comorbidity and lost school or workdays and improvement in quality of life.
Sublingual immunotherapy. Because of the possible safety concerns surrounding SCIT, along with problems relating to patient adherence to weekly office visits, alternative means of achieving allergen desensitization have been implemented. One of these methods, SLIT, has been increasingly supported by clinical evidence, especially in Europe. This therapy consists of applying aqueous allergen extract to the sublingual or oral mucosa, allowing it to be absorbed into the body. Subsequent swallowing of the extract allows the gut to respond with an increase in tolerance. The changes that result from this type of administration appear to be similar to those observed with SCIT.
SLIT has been in use for almost 30 years; the first published controlled study of this therapy was done in 1986 by Scadding and Brostoff.19 The World Allergy Organization recognized the safety and clinical efficacy of SLIT in 2009 after reviewing more than 60 controlled studies.20
A 2010 meta-analysis, reviewing documents from the prior 20 years of research, showed that SLIT decreased medication use and improved symptoms.21 Generally, SLIT was found to be more effective in adults than children. It was not as effective in patients with asthma as in those who were asthma-free. The timing of initiation of SLIT was also an important finding; when SLIT is used for grass pollen allergy, it should be started at least three months before the beginning of grass season.21 For other allergens, SLIT can be started at any time.
Because it is a home-based treatment, SLIT is far more convenient for the patient and therefore has become more popular in the US in the past decade. Its increased safety also contributes to its popularity. Although SLIT is commonly used in many parts of the world, at this writing, medications used in SLIT have not yet been approved by the US FDA. The FDA is currently reviewing two oral tablets for SLIT, and it is expected that its use in the US will increase once an approved product becomes available.22
It should be noted that no studies have directly compared SCIT and SLIT. Careful consultation between clinician and patient can help the patient arrive at the most appropriate modality for his or her condition based on symptomatology and lifestyle needs.
On the next page: Conclusion >>
CONCLUSION
For most patients, AR has little morbidity; however, for some whose rhinitis is moderate to severe, the complications can be a concern. If symptoms are not controlled with avoidance and/or medication, AI may be indicated. It comprises the building up of tolerance to the specific allergens as identified by allergy skin testing or in vitro specific IgE testing. AI, whether SCIT or SLIT, is the only means of altering the abnormal immune system response that underlies AR. Treatment may last as long as three to four years, which provides long-term efficacy of at least three years after cessation of therapy. The long-term prognosis for AR is excellent.
Appreciation is extended to Roxy Irestone, RN, Arizona Asthma & Allergy Institute, for her assistance is gathering factual information for this article.
1. Sibbald B, Rink E, D’Souza M. Is the prevalence of atopy increasing? Br J Gen Pract. 1990;40(337):338-340.
2. History of allergy. Allergy and Asthma Specialists Web site. www.allergyasthmaspecialist.com/allergy-and-asthma-clinic-of-kenosha-sc-education.htm. Accessed February 14, 2014.
3. Blackley CH. Hay Fever: Its Causes, Treatment, and Effective Prevention. London, England: Balliere, Tindall, & Cox; 1880.
4. Settipane RA. Rhinitis: a dose of epidemiological reality. Allergy Asthma Proc. 2003;24(3):147-154.
5. Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol. 2008;122(2 suppl):S1-S84.
6. Wright AL, Holberg CJ, Halonen M, et al. Epidemiology of physician-diagnosed allergic rhinitis in childhood. Pediatrics. 1994;94(6):895 -901.
7. Bernstein JA. Allergic and mixed rhinitis: epidemiology and natural history. Allergy Asthma Proc. 2010;31:365-369.
8. Meltzer EO, Bukstein DA. The economic impact of allergic rhinitis and current guidelines for treatment. Ann Allergy Asthma Immunol. 2011; 106(suppl 2):S12-S16.
9. American Academy of Allergy, Asthma & Immunology. Task force on allergic disorders: promoting best practice: raising the standard of care for patients with allergic disorders. Executive summary report. 1998.
10. Nathan RA. The burden of allergic rhinitis. Allergy Asthma Proc. 2007; 28:3-9.
11. Tran NP, Vickery J, Blaiss MS. Management of rhinitis: allergic and non-allergic. Allergy Asthma Immunol Res. 2011;3:148-156.
12. Disease Summaries: Allergic Diseases and Asthma in Pregnancy. World Allergy Organization. www.worldallergy.org/professional/allergic_
diseases_center/allergy_in_pregnancy/. Accessed February 14, 2014.
13. Simons FE, Ardusso LR, Dimov V, et al. World Allergy Organization Anaphylaxis Guidelines: 2013 update of the evidence base. Int Arch Allergy Immunol. 2013;162:193-204.
14. Noon L. Prophylactic inoculation against hay fever. Lancet. 1911;177:1572-1573.
15. Purello-D’Ambrosio F, Gangemi S, Merendino RA, et al. Prevention of new sensitizations in monosensitized subjects submitted to specific immunotherapy or not: a retrospective study. Clin & Exper Allergy. 2001;31:1295-1302.
16. Durham SR, Walker SM, Varga EM, et al. Long-term clinical efficacy of grass-pollen immunotherapy. N Engl J Med. 1999;341:468-475.
17. Mohapatra SS, Qazi M, Hellermann G. Immunotherapy for allergies and asthma: present and future. Curr Opin Pharmacol. 2010;10:276-288.
18. Calderon MA, Alves B, Jacobson M, et al. Allergen injection immunotherapy for seasonal allergic rhinitis. Cochrane Database Syst Rev. 2007; Iss 1, Art No: CD001936.
19. Scadding GK, Brostoff J. Low dose sublingual therapy in patients with allergic rhinitis due to house dust mite.Clin Allergy. 1986;16:483-491.
20. Canonica GW, Bousquet J, Casale T, et al. Sub-lingual immunotherapy: World Allergy Organization Position Paper 2009. Allergy. 2009;64 suppl 91:1-59.
21. Di Bona D, Plaia A, Scafidi V, et al. Efficacy of sublingual immunotherapy with grass allergens for seasonal allergic rhinitis: a systematic review and meta-analysis. J Allergy Clin Immunol. 2010;126:558-566.
22. Sikora JM, Tankersley MS; ACAAI Immunotherapy and Diagnostics Committee. Perception and practice of sublingual immunotherapy among practicing allergists in the United States: a follow-up survey. Ann Allergy Asthma Immunol. 2013;110:194-197.
CE/CME No: CR-1403
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the pathophysiology and etiology of allergic rhinitis (AR).
• Describe the prevalence and types of AR.
• List the differential diagnoses for AR.
• Describe the historical and physical examination findings that are typical
of AR.
• Explain the indications for and types of allergy testing.
• Discuss the types of allergy desensitization therapies/immunotherapies.
FACULTY
Randy D. Danielsen is a Professor and Dean of the Arizona School of Health Sciences, A.T. Still University in Mesa, Arizona, and a long-time PA with the Arizona Asthma & Allergy Institute. Linda S. MacConnell is an Assistant Professor in the Department of Physician Assistant Studies at the Arizona School of Health Sciences, A.T. Still University, and a formally trained otolaryngology PA.
The authors have no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Allergic rhinitis (AR), one of the most familiar complaints seen in primary care, is a common immunologic condition that occurs in genetically predisposed patients. AR is routinely treated through allergen avoidance and pharmacologic therapy. When these measures fail, however, immunologic treatment may be indicated. This review of AR and its treatment focuses on injection and oral immunotherapy.
Congestion; sneezing (particularly paroxysms); itchy nose, palate, and eyes; and runny nose are symptoms characteristic of allergic rhinitis (AR) seen every day in virtually all primary care offices. Patients are plagued not only by their symptoms, but also by AR-related sleep disturbances, resulting in fatigue and daytime sleepiness, irritability, and memory deficits. In children, these sleep disruptions may even cause behavioral disturbances. Depending on the cause, some patients may experience allergic symptoms only with outdoor environmental exposure and subsequent immunoglobulin E (IgE)–mediated responses to otherwise innocuous allergens, while others find that their symptoms are constant, occurring both indoors and out.
Although the prevalence of AR is increasing,1 allergies are certainly nothing new to humankind. In fact, hieroglyphics and Egyptian wall paintings have been discovered depicting a pharaoh dying from anaphylactic shock after receiving a wasp sting.2 In 1565, Leonardo Botallo described AR, calling it “rose catarrh” (mucous or phlegm) or “rose fever,” based on the mistaken idea that the symptoms were caused by rose pollen.2 John Babcock, an English physician, first diagnosed an upper respiratory disease that he called “hay fever” in 1819.
Seventy years later, Charles Blackley identified pollen as a cause of hay fever, documenting his findings in his 1873 book Experimental Researches on the Causes and Nature of Catarrhus Aestivus.2 Dr. Blackley performed the initial documented attempts at allergy desensitization treatments on himself—a willing patient, as he suffered from AR. He placed rye grass pollen onto his nasal mucosa, finding that after 30 minutes the nostril was completely occluded. He continued his experimentation by repeatedly exposing himself to pollen grains via abraded skin. Alas, he never noted any decrease in his symptoms.3
Presently, AR affects more than 55 million people in the United States4—approximately 10% to 30% of the adult population5 and more than 40% of children.6 The rising prevalence of AR is of concern in older adults, who tend to have related comorbidities (eg, chronic sinusitis, asthma, and otitis media). In fact, AR is the fifth most common chronic disease in the US.7
AR and its treatment impose a great economic burden on the health care system, critical in these days of affordable health care. In fact, in 2005 in the US, the overall (direct medical and indirect) cost of AR was $11.2 billion.8 Direct costs derive from office visits, diagnostic testing, and therapeutics. Costs are considerably higher when indirect expenses, including decreased productivity, missed school and missed workdays to care for children, and costs of travel to medical appointments, are included. In the US, approximately 3.5 million workdays and 2 million school days are lost each year due to AR.9 Decreases in productivity cost an estimated $600 per affected employee per year, all of which results in AR being the fifth costliest chronic disease.10,11
On the next page: Pathophysiology and examination >>
PATHOPHYSIOLOGY
AR is an immunologic disorder that occurs in genetically susceptible individuals who produce allergen-specific IgE antibody responses after environmental exposures. The IgE-mediated response causes inflammation of the nasal mucosa. Compared to controls, individuals with AR demonstrate increased amounts of IgE antibodies in the nasal mucosa.11 IgE binds to basophils in the bloodstream and mast cells in tissue. Allergens then attach to IgE on basophils and mast cells, which release histamines, prostaglandins, cytokines, and leukotrienes, with histamine being the most significant mediator in the inflammatory response.
In response to allergy-provoking substances, patients experience immediate- and late-phase symptoms. Symptoms of each stage are similar, but congestion is the hallmark of the late phase. While both phases are clinically important because of their contribution to the patient’s symptoms, most patients experience continued exposure to allergens, resulting in constant, overlapping symptoms.
HISTORY AND PHYSICAL EXAMINATION
Patients with AR relate a history of congestion, excessive mucous production, itchy, watery eyes, bouts of sneezing, and more systemic symptoms, such as headache, malaise, and excessive fatigue. It is important to evaluate the degree and duration of the symptoms, noting patterns and triggers, in an effort to confirm the diagnosis and to help the patient evaluate treatment options.
When taking the patient history, always review the family history, which is often notable for allergies and other atopic diseases. Be sure to ask about medications and recreational drug use; a number of substances have been implicated in the development of rhinitis, including anticholinergic medications, oxymetazoline (when overused), and cocaine. Also, question the patient about self-medication and treatment to determine what may or may not have provided relief. Further questioning may also reveal a history of comorbidities, including contact dermatitis, asthma, eczema, and chronic sinusitis.
The physical examination of the patient with rhinitis begins with observation of the patient’s outward appearance, which may reveal allergic shiners (dark to purplish areas under the eyes), conjunctivitis, an allergic salute (a transverse crease of the nose caused by upward rubbing of an itchy nose), mouth breathing, and a generally tired appearance. The nasal turbinates are swollen and often pale. Mucous secretions are usually thin and clear. Enlarged tonsils and posterior nasal drainage may be visualized. The types of AR are listed in Table 1.
On the next page: Diagnosis and treatment >>
DIAGNOSIS
Most of the clues needed to arrive at a diagnosis are discovered by taking a careful history and completing a physical examination. AR frequently underlies and/or coexists with acute upper respiratory infection (URI) and acute and chronic sinusitis. Differentiating acute URI and acute sinusitis from AR is usually relatively straightforward, based on the symptoms of the illness. The diagnosis of chronic sinusitis is made by radiologic imaging with CT scan.
Distinguishing nonallergic rhinitis (NAR) from AR can be far more difficult, because the symptoms of these conditions are similar and chronic in nature (see Table 2). Empiric treatment for AR may be attempted; however, further testing is often needed to differentiate the two. At this point, clinicians may choose to proceed with specific IgE blood tests. Alternatively, many medical practices are prepared to perform or refer for allergy skin testing.
TREATMENT
Avoidance of known triggers is the cornerstone of allergy treatment. Currently, the most effective pharmaceutical treatment for the majority of AR symptoms is inhaled nasal corticosteroids. Although less effective than corticosteroids, antihistamines—both nasal and oral—are a recommended addition to the regimen if the adverse effects and costs to the patient are tolerable. Other treatments include the leukotriene receptor antagonists, intranasal formulations of cromolyn, and the anticholinergic ipratropium bromide nasal spray, which is effective primarily on watery rhinorrhea. If symptoms are not controlled with medication, allergy immunotherapy (AI), the only known disease-modifying therapy for AR, may be indicated.
On the next page: Allergy testing >>
ALLERGY TESTING
In order to distinguish between AR and NAR and to direct treatment toward specific allergen avoidance and immunotherapy, providers have the choice of ordering in vitro blood IgE testing (to measure the antibodies that mediate an allergic response) or in vivo allergy skin testing (to measure the immune response to allergens that induces an allergic atopic reaction). Allergy testing is not a contemporary concept; the first allergy testing was documented in 1656 when Pierre Borel applied egg to a patient’s skin, which exhibited an allergic reaction.2
Allergy skin testing consists of applying multiple allergens to the skin of the patient’s forearms via tiny pinpricks while watching for immediate hypersensitivity reactions. The test begins with the placement of a drop of histamine to serve as a control. If after 10 minutes of watchful waiting the patient develops a reaction to the histamine (a positive test result), it is appropriate to test for antigens by placing drops of suspected allergen extracts on the skin.
After a period of time (usually 20 to 30 minutes), the area is inspected for allergic reaction. An immediate (early phase) wheal and flare (surrounding erythema) reaction may develop. This positive reaction indicates the presence of a mast cell–bound IgE antibody specific to the tested allergen. The size of the reactions is measured in millimeters, allowing for comparison to the histamine control.
A list of the commonly tested antigens in Arizona, as an example, is shown in Table 3. Antigens vary geographically and even from practice to practice. For up-to-date information on pollen counts by region, visit the American Academy of Allergy Asthma & Immunology Web site (see “Pollen Counts”).
On the next page: Patient selection and allergy immunotherapy >>
Patient Selection
After the clinician has determined that there is a high likelihood that the diagnosis is AR, allergy testing, needed to guide AI, is appropriate. Although not true of specific IgE testing, the accuracy of allergy skin testing results can be adversely affected by several medications. For example, some practitioners may choose to stop first-generation antihistamines two to three days before testing. It is generally accepted that the newer, second-generation antihistamines, which can affect skin-testing results longer, be stopped a week prior to testing.
Patients should be reminded that OTC sleep aids frequently contain antihistamines (particularly diphenhydramine) and that they must be discontinued prior to testing as well. Histamine H2-receptor antagonists such as cimetidine and ranitidine may be stopped a day or two before testing. Although β-blockers are only relatively contraindicated in both allergy testing and AI, many health care providers avoid testing and AI in patients taking oral and/or topical (eye drops) β-blocker therapy. Ultimately, the decision is made by individual health care practices.
In vivo allergy skin testing should not be performed on patients taking tricyclic antidepressants and monoamine oxidase inhibitors. Patients with significant cardiovascular disease should not undergo testing or treatment. Pregnancy is a relative contraindication, and allergy skin testing and AI are done only with obstetrician approval. Most allergists avoid allergy skin testing in pregnant women, however, because use of epinephrine, if required, introduces the risk for preterm labor.12,13 Special consideration should also be given to patients with immune deficiencies.
Setting for Allergy Evaluation and Treatment
Recently, many primary care practices have added allergy evaluation and management to the procedures and treatments they offer; however, evaluations and testing traditionally have been performed by allergy and immunology specialists, many of whom include PAs and NPs on their staff. PAs and NPs frequently manage the practices’ allergy programs. Allergists who are listed as American Board of Allergy and Immunology (ABAI)–certified have successfully passed the ABAI’s certifying examination. Other medical specialists, including otolaryngologists and the primary care specialties, are also well placed to evaluate patients with common allergy symptoms and provide appropriate treatment.
Allergy Immunotherapy
AI has now become more efficacious, safer, and more tolerable for the patient than when it was first introduced in 1911. At that time, Leonard Noon authored a brief article claiming that allergen-specific injections could modify AR. Unfortunately, Noon died at age 36 from tuberculosis, but his work was carried on by his associate, John Freeman. Together, they established the guidelines upon which contemporary AI is based, including the protocol to gradually increase the dose of allergen serum, starting with initial weekly to biweekly injections. They also voiced warnings about the potential for anaphylaxis.14 To this day, immunotherapy is still accomplished by the gradual administration of increasing amounts of the allergen to which the patient is sensitive. This tempers the abnormal immune response to that allergen, easing allergic symptoms.
In patients with IgE-mediated hypersensitivity reactions, confirmed by history, physical examination, and allergy skin testing, immunotherapy can be very effective. The results of immunotherapy may last for years and may even prevent the allergic march, the progression of allergic disease experienced by many patients that frequently begins early in life.15 This includes other allergy-related conditions, such as asthma and eczema (atopic dermatitis), and the acquisition of additional new allergies, including those to foods. AI also has been shown to decrease the frequency of comorbidities such as asthma.5 In addition, AI is used in carefully screened patients who desire to reduce the dosages of medication required to control their symptoms.
In a study published in 1999, Durham and colleagues clarified the questions surrounding the amount of time required for ongoing immunotherapy. They found that the desensitization and tolerance to allergens achieved by AI can last up to three years after a three- to four-year course of therapy. They also found that treatment should not start until an allergic component is identified by allergy skin testing or serum tests for allergen-specific IgE.16
As effective as immunotherapy has been documented to be, it is underused as a therapeutic modality. There are only 2 to 3 million patients receiving subcutaneous immunotherapy out of more than 55 million patients with AR.17
On the next page: Types of allergy immunotherapy >>
Types of AI
There are two categories of allergen desensitization therapy. The most common method is subcutaneous immunotherapy (SCIT), the so-called allergy shots. Less common and still somewhat controversial is sublingual immunotherapy (SLIT). Immunotherapy should be considered for patients who have secondary complications (eg, sinusitis, otitis) or asthma with allergies, or those in whom avoidance measures and medications fail. SCIT or SLIT may be desirable for patients with AR who have difficulty taking regular medications.
Subcutaneous immunotherapy. Currently, SCIT is the most recognized immunotherapy and the only one currently reimbursed by insurance. The procedure involves the subcutaneous injection of increasing doses of therapeutic solutions to which a patient has demonstrated sensitivity. The indications for this treatment are usually inhaled allergens, such as pollens and animal dander. SCIT may also be valuable in patients with asthma and atopic dermatitis.
The most common adverse reaction to allergy injections is a large localized reaction—primarily erythema, pruritus, discomfort, and possibly edema. Severe systemic reactions are extremely rare, with near-fatal to fatal reactions occurring at the rate of only 5.4 per million injections.11 The majority of these rare, albeit serious, complications are caused by higher-than-normal levels of pollen in the environment and dosing errors.11 Because of the uncommon but significant complications, patients undergoing immunotherapy should always receive injections in a medical office equipped with appropriate equipment and staff trained in handling anaphylaxis. It is standard protocol for patients to remain in the office for 30 minutes after administration for observation. Some clinicians prescribe epinephrine injectors (Epi-Pens) for patients to bring to every appointment as a condition for receiving their shot. Because of continuing controversy on this point, others only employ this requirement if the patient has a history of an adverse reaction.
Various protocols exist for the up-dosing of immunotherapy, most of which recommend weekly to twice-weekly injections prior to initiating maintenance therapy. Costs, risks, and benefits must be carefully considered and discussed with the patient prior to initiating immunotherapy.
Many insurance companies reimburse for immunotherapy, with varying copayments. Additionally, the time commitment may be taxing on the patient’s busy schedule. Weekly or biweekly appointments are required initially, and the patient must remain on site for half an hour. Although direct costs of SCIT are relatively easily measured and perhaps compensated, the indirect costs of time spent commuting and at the clinic are less tangible.
Success rates with SCIT may be more than 70% for certain allergens,18 but it is a long-term process with initial improvement often not seen until after six to 12 months of therapy. The benefits of therapy lead not only to reduction and suppression in symptoms (and medication), but also to reduction in comorbidity and lost school or workdays and improvement in quality of life.
Sublingual immunotherapy. Because of the possible safety concerns surrounding SCIT, along with problems relating to patient adherence to weekly office visits, alternative means of achieving allergen desensitization have been implemented. One of these methods, SLIT, has been increasingly supported by clinical evidence, especially in Europe. This therapy consists of applying aqueous allergen extract to the sublingual or oral mucosa, allowing it to be absorbed into the body. Subsequent swallowing of the extract allows the gut to respond with an increase in tolerance. The changes that result from this type of administration appear to be similar to those observed with SCIT.
SLIT has been in use for almost 30 years; the first published controlled study of this therapy was done in 1986 by Scadding and Brostoff.19 The World Allergy Organization recognized the safety and clinical efficacy of SLIT in 2009 after reviewing more than 60 controlled studies.20
A 2010 meta-analysis, reviewing documents from the prior 20 years of research, showed that SLIT decreased medication use and improved symptoms.21 Generally, SLIT was found to be more effective in adults than children. It was not as effective in patients with asthma as in those who were asthma-free. The timing of initiation of SLIT was also an important finding; when SLIT is used for grass pollen allergy, it should be started at least three months before the beginning of grass season.21 For other allergens, SLIT can be started at any time.
Because it is a home-based treatment, SLIT is far more convenient for the patient and therefore has become more popular in the US in the past decade. Its increased safety also contributes to its popularity. Although SLIT is commonly used in many parts of the world, at this writing, medications used in SLIT have not yet been approved by the US FDA. The FDA is currently reviewing two oral tablets for SLIT, and it is expected that its use in the US will increase once an approved product becomes available.22
It should be noted that no studies have directly compared SCIT and SLIT. Careful consultation between clinician and patient can help the patient arrive at the most appropriate modality for his or her condition based on symptomatology and lifestyle needs.
On the next page: Conclusion >>
CONCLUSION
For most patients, AR has little morbidity; however, for some whose rhinitis is moderate to severe, the complications can be a concern. If symptoms are not controlled with avoidance and/or medication, AI may be indicated. It comprises the building up of tolerance to the specific allergens as identified by allergy skin testing or in vitro specific IgE testing. AI, whether SCIT or SLIT, is the only means of altering the abnormal immune system response that underlies AR. Treatment may last as long as three to four years, which provides long-term efficacy of at least three years after cessation of therapy. The long-term prognosis for AR is excellent.
Appreciation is extended to Roxy Irestone, RN, Arizona Asthma & Allergy Institute, for her assistance is gathering factual information for this article.
CE/CME No: CR-1403
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the pathophysiology and etiology of allergic rhinitis (AR).
• Describe the prevalence and types of AR.
• List the differential diagnoses for AR.
• Describe the historical and physical examination findings that are typical
of AR.
• Explain the indications for and types of allergy testing.
• Discuss the types of allergy desensitization therapies/immunotherapies.
FACULTY
Randy D. Danielsen is a Professor and Dean of the Arizona School of Health Sciences, A.T. Still University in Mesa, Arizona, and a long-time PA with the Arizona Asthma & Allergy Institute. Linda S. MacConnell is an Assistant Professor in the Department of Physician Assistant Studies at the Arizona School of Health Sciences, A.T. Still University, and a formally trained otolaryngology PA.
The authors have no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Allergic rhinitis (AR), one of the most familiar complaints seen in primary care, is a common immunologic condition that occurs in genetically predisposed patients. AR is routinely treated through allergen avoidance and pharmacologic therapy. When these measures fail, however, immunologic treatment may be indicated. This review of AR and its treatment focuses on injection and oral immunotherapy.
Congestion; sneezing (particularly paroxysms); itchy nose, palate, and eyes; and runny nose are symptoms characteristic of allergic rhinitis (AR) seen every day in virtually all primary care offices. Patients are plagued not only by their symptoms, but also by AR-related sleep disturbances, resulting in fatigue and daytime sleepiness, irritability, and memory deficits. In children, these sleep disruptions may even cause behavioral disturbances. Depending on the cause, some patients may experience allergic symptoms only with outdoor environmental exposure and subsequent immunoglobulin E (IgE)–mediated responses to otherwise innocuous allergens, while others find that their symptoms are constant, occurring both indoors and out.
Although the prevalence of AR is increasing,1 allergies are certainly nothing new to humankind. In fact, hieroglyphics and Egyptian wall paintings have been discovered depicting a pharaoh dying from anaphylactic shock after receiving a wasp sting.2 In 1565, Leonardo Botallo described AR, calling it “rose catarrh” (mucous or phlegm) or “rose fever,” based on the mistaken idea that the symptoms were caused by rose pollen.2 John Babcock, an English physician, first diagnosed an upper respiratory disease that he called “hay fever” in 1819.
Seventy years later, Charles Blackley identified pollen as a cause of hay fever, documenting his findings in his 1873 book Experimental Researches on the Causes and Nature of Catarrhus Aestivus.2 Dr. Blackley performed the initial documented attempts at allergy desensitization treatments on himself—a willing patient, as he suffered from AR. He placed rye grass pollen onto his nasal mucosa, finding that after 30 minutes the nostril was completely occluded. He continued his experimentation by repeatedly exposing himself to pollen grains via abraded skin. Alas, he never noted any decrease in his symptoms.3
Presently, AR affects more than 55 million people in the United States4—approximately 10% to 30% of the adult population5 and more than 40% of children.6 The rising prevalence of AR is of concern in older adults, who tend to have related comorbidities (eg, chronic sinusitis, asthma, and otitis media). In fact, AR is the fifth most common chronic disease in the US.7
AR and its treatment impose a great economic burden on the health care system, critical in these days of affordable health care. In fact, in 2005 in the US, the overall (direct medical and indirect) cost of AR was $11.2 billion.8 Direct costs derive from office visits, diagnostic testing, and therapeutics. Costs are considerably higher when indirect expenses, including decreased productivity, missed school and missed workdays to care for children, and costs of travel to medical appointments, are included. In the US, approximately 3.5 million workdays and 2 million school days are lost each year due to AR.9 Decreases in productivity cost an estimated $600 per affected employee per year, all of which results in AR being the fifth costliest chronic disease.10,11
On the next page: Pathophysiology and examination >>
PATHOPHYSIOLOGY
AR is an immunologic disorder that occurs in genetically susceptible individuals who produce allergen-specific IgE antibody responses after environmental exposures. The IgE-mediated response causes inflammation of the nasal mucosa. Compared to controls, individuals with AR demonstrate increased amounts of IgE antibodies in the nasal mucosa.11 IgE binds to basophils in the bloodstream and mast cells in tissue. Allergens then attach to IgE on basophils and mast cells, which release histamines, prostaglandins, cytokines, and leukotrienes, with histamine being the most significant mediator in the inflammatory response.
In response to allergy-provoking substances, patients experience immediate- and late-phase symptoms. Symptoms of each stage are similar, but congestion is the hallmark of the late phase. While both phases are clinically important because of their contribution to the patient’s symptoms, most patients experience continued exposure to allergens, resulting in constant, overlapping symptoms.
HISTORY AND PHYSICAL EXAMINATION
Patients with AR relate a history of congestion, excessive mucous production, itchy, watery eyes, bouts of sneezing, and more systemic symptoms, such as headache, malaise, and excessive fatigue. It is important to evaluate the degree and duration of the symptoms, noting patterns and triggers, in an effort to confirm the diagnosis and to help the patient evaluate treatment options.
When taking the patient history, always review the family history, which is often notable for allergies and other atopic diseases. Be sure to ask about medications and recreational drug use; a number of substances have been implicated in the development of rhinitis, including anticholinergic medications, oxymetazoline (when overused), and cocaine. Also, question the patient about self-medication and treatment to determine what may or may not have provided relief. Further questioning may also reveal a history of comorbidities, including contact dermatitis, asthma, eczema, and chronic sinusitis.
The physical examination of the patient with rhinitis begins with observation of the patient’s outward appearance, which may reveal allergic shiners (dark to purplish areas under the eyes), conjunctivitis, an allergic salute (a transverse crease of the nose caused by upward rubbing of an itchy nose), mouth breathing, and a generally tired appearance. The nasal turbinates are swollen and often pale. Mucous secretions are usually thin and clear. Enlarged tonsils and posterior nasal drainage may be visualized. The types of AR are listed in Table 1.
On the next page: Diagnosis and treatment >>
DIAGNOSIS
Most of the clues needed to arrive at a diagnosis are discovered by taking a careful history and completing a physical examination. AR frequently underlies and/or coexists with acute upper respiratory infection (URI) and acute and chronic sinusitis. Differentiating acute URI and acute sinusitis from AR is usually relatively straightforward, based on the symptoms of the illness. The diagnosis of chronic sinusitis is made by radiologic imaging with CT scan.
Distinguishing nonallergic rhinitis (NAR) from AR can be far more difficult, because the symptoms of these conditions are similar and chronic in nature (see Table 2). Empiric treatment for AR may be attempted; however, further testing is often needed to differentiate the two. At this point, clinicians may choose to proceed with specific IgE blood tests. Alternatively, many medical practices are prepared to perform or refer for allergy skin testing.
TREATMENT
Avoidance of known triggers is the cornerstone of allergy treatment. Currently, the most effective pharmaceutical treatment for the majority of AR symptoms is inhaled nasal corticosteroids. Although less effective than corticosteroids, antihistamines—both nasal and oral—are a recommended addition to the regimen if the adverse effects and costs to the patient are tolerable. Other treatments include the leukotriene receptor antagonists, intranasal formulations of cromolyn, and the anticholinergic ipratropium bromide nasal spray, which is effective primarily on watery rhinorrhea. If symptoms are not controlled with medication, allergy immunotherapy (AI), the only known disease-modifying therapy for AR, may be indicated.
On the next page: Allergy testing >>
ALLERGY TESTING
In order to distinguish between AR and NAR and to direct treatment toward specific allergen avoidance and immunotherapy, providers have the choice of ordering in vitro blood IgE testing (to measure the antibodies that mediate an allergic response) or in vivo allergy skin testing (to measure the immune response to allergens that induces an allergic atopic reaction). Allergy testing is not a contemporary concept; the first allergy testing was documented in 1656 when Pierre Borel applied egg to a patient’s skin, which exhibited an allergic reaction.2
Allergy skin testing consists of applying multiple allergens to the skin of the patient’s forearms via tiny pinpricks while watching for immediate hypersensitivity reactions. The test begins with the placement of a drop of histamine to serve as a control. If after 10 minutes of watchful waiting the patient develops a reaction to the histamine (a positive test result), it is appropriate to test for antigens by placing drops of suspected allergen extracts on the skin.
After a period of time (usually 20 to 30 minutes), the area is inspected for allergic reaction. An immediate (early phase) wheal and flare (surrounding erythema) reaction may develop. This positive reaction indicates the presence of a mast cell–bound IgE antibody specific to the tested allergen. The size of the reactions is measured in millimeters, allowing for comparison to the histamine control.
A list of the commonly tested antigens in Arizona, as an example, is shown in Table 3. Antigens vary geographically and even from practice to practice. For up-to-date information on pollen counts by region, visit the American Academy of Allergy Asthma & Immunology Web site (see “Pollen Counts”).
On the next page: Patient selection and allergy immunotherapy >>
Patient Selection
After the clinician has determined that there is a high likelihood that the diagnosis is AR, allergy testing, needed to guide AI, is appropriate. Although not true of specific IgE testing, the accuracy of allergy skin testing results can be adversely affected by several medications. For example, some practitioners may choose to stop first-generation antihistamines two to three days before testing. It is generally accepted that the newer, second-generation antihistamines, which can affect skin-testing results longer, be stopped a week prior to testing.
Patients should be reminded that OTC sleep aids frequently contain antihistamines (particularly diphenhydramine) and that they must be discontinued prior to testing as well. Histamine H2-receptor antagonists such as cimetidine and ranitidine may be stopped a day or two before testing. Although β-blockers are only relatively contraindicated in both allergy testing and AI, many health care providers avoid testing and AI in patients taking oral and/or topical (eye drops) β-blocker therapy. Ultimately, the decision is made by individual health care practices.
In vivo allergy skin testing should not be performed on patients taking tricyclic antidepressants and monoamine oxidase inhibitors. Patients with significant cardiovascular disease should not undergo testing or treatment. Pregnancy is a relative contraindication, and allergy skin testing and AI are done only with obstetrician approval. Most allergists avoid allergy skin testing in pregnant women, however, because use of epinephrine, if required, introduces the risk for preterm labor.12,13 Special consideration should also be given to patients with immune deficiencies.
Setting for Allergy Evaluation and Treatment
Recently, many primary care practices have added allergy evaluation and management to the procedures and treatments they offer; however, evaluations and testing traditionally have been performed by allergy and immunology specialists, many of whom include PAs and NPs on their staff. PAs and NPs frequently manage the practices’ allergy programs. Allergists who are listed as American Board of Allergy and Immunology (ABAI)–certified have successfully passed the ABAI’s certifying examination. Other medical specialists, including otolaryngologists and the primary care specialties, are also well placed to evaluate patients with common allergy symptoms and provide appropriate treatment.
Allergy Immunotherapy
AI has now become more efficacious, safer, and more tolerable for the patient than when it was first introduced in 1911. At that time, Leonard Noon authored a brief article claiming that allergen-specific injections could modify AR. Unfortunately, Noon died at age 36 from tuberculosis, but his work was carried on by his associate, John Freeman. Together, they established the guidelines upon which contemporary AI is based, including the protocol to gradually increase the dose of allergen serum, starting with initial weekly to biweekly injections. They also voiced warnings about the potential for anaphylaxis.14 To this day, immunotherapy is still accomplished by the gradual administration of increasing amounts of the allergen to which the patient is sensitive. This tempers the abnormal immune response to that allergen, easing allergic symptoms.
In patients with IgE-mediated hypersensitivity reactions, confirmed by history, physical examination, and allergy skin testing, immunotherapy can be very effective. The results of immunotherapy may last for years and may even prevent the allergic march, the progression of allergic disease experienced by many patients that frequently begins early in life.15 This includes other allergy-related conditions, such as asthma and eczema (atopic dermatitis), and the acquisition of additional new allergies, including those to foods. AI also has been shown to decrease the frequency of comorbidities such as asthma.5 In addition, AI is used in carefully screened patients who desire to reduce the dosages of medication required to control their symptoms.
In a study published in 1999, Durham and colleagues clarified the questions surrounding the amount of time required for ongoing immunotherapy. They found that the desensitization and tolerance to allergens achieved by AI can last up to three years after a three- to four-year course of therapy. They also found that treatment should not start until an allergic component is identified by allergy skin testing or serum tests for allergen-specific IgE.16
As effective as immunotherapy has been documented to be, it is underused as a therapeutic modality. There are only 2 to 3 million patients receiving subcutaneous immunotherapy out of more than 55 million patients with AR.17
On the next page: Types of allergy immunotherapy >>
Types of AI
There are two categories of allergen desensitization therapy. The most common method is subcutaneous immunotherapy (SCIT), the so-called allergy shots. Less common and still somewhat controversial is sublingual immunotherapy (SLIT). Immunotherapy should be considered for patients who have secondary complications (eg, sinusitis, otitis) or asthma with allergies, or those in whom avoidance measures and medications fail. SCIT or SLIT may be desirable for patients with AR who have difficulty taking regular medications.
Subcutaneous immunotherapy. Currently, SCIT is the most recognized immunotherapy and the only one currently reimbursed by insurance. The procedure involves the subcutaneous injection of increasing doses of therapeutic solutions to which a patient has demonstrated sensitivity. The indications for this treatment are usually inhaled allergens, such as pollens and animal dander. SCIT may also be valuable in patients with asthma and atopic dermatitis.
The most common adverse reaction to allergy injections is a large localized reaction—primarily erythema, pruritus, discomfort, and possibly edema. Severe systemic reactions are extremely rare, with near-fatal to fatal reactions occurring at the rate of only 5.4 per million injections.11 The majority of these rare, albeit serious, complications are caused by higher-than-normal levels of pollen in the environment and dosing errors.11 Because of the uncommon but significant complications, patients undergoing immunotherapy should always receive injections in a medical office equipped with appropriate equipment and staff trained in handling anaphylaxis. It is standard protocol for patients to remain in the office for 30 minutes after administration for observation. Some clinicians prescribe epinephrine injectors (Epi-Pens) for patients to bring to every appointment as a condition for receiving their shot. Because of continuing controversy on this point, others only employ this requirement if the patient has a history of an adverse reaction.
Various protocols exist for the up-dosing of immunotherapy, most of which recommend weekly to twice-weekly injections prior to initiating maintenance therapy. Costs, risks, and benefits must be carefully considered and discussed with the patient prior to initiating immunotherapy.
Many insurance companies reimburse for immunotherapy, with varying copayments. Additionally, the time commitment may be taxing on the patient’s busy schedule. Weekly or biweekly appointments are required initially, and the patient must remain on site for half an hour. Although direct costs of SCIT are relatively easily measured and perhaps compensated, the indirect costs of time spent commuting and at the clinic are less tangible.
Success rates with SCIT may be more than 70% for certain allergens,18 but it is a long-term process with initial improvement often not seen until after six to 12 months of therapy. The benefits of therapy lead not only to reduction and suppression in symptoms (and medication), but also to reduction in comorbidity and lost school or workdays and improvement in quality of life.
Sublingual immunotherapy. Because of the possible safety concerns surrounding SCIT, along with problems relating to patient adherence to weekly office visits, alternative means of achieving allergen desensitization have been implemented. One of these methods, SLIT, has been increasingly supported by clinical evidence, especially in Europe. This therapy consists of applying aqueous allergen extract to the sublingual or oral mucosa, allowing it to be absorbed into the body. Subsequent swallowing of the extract allows the gut to respond with an increase in tolerance. The changes that result from this type of administration appear to be similar to those observed with SCIT.
SLIT has been in use for almost 30 years; the first published controlled study of this therapy was done in 1986 by Scadding and Brostoff.19 The World Allergy Organization recognized the safety and clinical efficacy of SLIT in 2009 after reviewing more than 60 controlled studies.20
A 2010 meta-analysis, reviewing documents from the prior 20 years of research, showed that SLIT decreased medication use and improved symptoms.21 Generally, SLIT was found to be more effective in adults than children. It was not as effective in patients with asthma as in those who were asthma-free. The timing of initiation of SLIT was also an important finding; when SLIT is used for grass pollen allergy, it should be started at least three months before the beginning of grass season.21 For other allergens, SLIT can be started at any time.
Because it is a home-based treatment, SLIT is far more convenient for the patient and therefore has become more popular in the US in the past decade. Its increased safety also contributes to its popularity. Although SLIT is commonly used in many parts of the world, at this writing, medications used in SLIT have not yet been approved by the US FDA. The FDA is currently reviewing two oral tablets for SLIT, and it is expected that its use in the US will increase once an approved product becomes available.22
It should be noted that no studies have directly compared SCIT and SLIT. Careful consultation between clinician and patient can help the patient arrive at the most appropriate modality for his or her condition based on symptomatology and lifestyle needs.
On the next page: Conclusion >>
CONCLUSION
For most patients, AR has little morbidity; however, for some whose rhinitis is moderate to severe, the complications can be a concern. If symptoms are not controlled with avoidance and/or medication, AI may be indicated. It comprises the building up of tolerance to the specific allergens as identified by allergy skin testing or in vitro specific IgE testing. AI, whether SCIT or SLIT, is the only means of altering the abnormal immune system response that underlies AR. Treatment may last as long as three to four years, which provides long-term efficacy of at least three years after cessation of therapy. The long-term prognosis for AR is excellent.
Appreciation is extended to Roxy Irestone, RN, Arizona Asthma & Allergy Institute, for her assistance is gathering factual information for this article.
1. Sibbald B, Rink E, D’Souza M. Is the prevalence of atopy increasing? Br J Gen Pract. 1990;40(337):338-340.
2. History of allergy. Allergy and Asthma Specialists Web site. www.allergyasthmaspecialist.com/allergy-and-asthma-clinic-of-kenosha-sc-education.htm. Accessed February 14, 2014.
3. Blackley CH. Hay Fever: Its Causes, Treatment, and Effective Prevention. London, England: Balliere, Tindall, & Cox; 1880.
4. Settipane RA. Rhinitis: a dose of epidemiological reality. Allergy Asthma Proc. 2003;24(3):147-154.
5. Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol. 2008;122(2 suppl):S1-S84.
6. Wright AL, Holberg CJ, Halonen M, et al. Epidemiology of physician-diagnosed allergic rhinitis in childhood. Pediatrics. 1994;94(6):895 -901.
7. Bernstein JA. Allergic and mixed rhinitis: epidemiology and natural history. Allergy Asthma Proc. 2010;31:365-369.
8. Meltzer EO, Bukstein DA. The economic impact of allergic rhinitis and current guidelines for treatment. Ann Allergy Asthma Immunol. 2011; 106(suppl 2):S12-S16.
9. American Academy of Allergy, Asthma & Immunology. Task force on allergic disorders: promoting best practice: raising the standard of care for patients with allergic disorders. Executive summary report. 1998.
10. Nathan RA. The burden of allergic rhinitis. Allergy Asthma Proc. 2007; 28:3-9.
11. Tran NP, Vickery J, Blaiss MS. Management of rhinitis: allergic and non-allergic. Allergy Asthma Immunol Res. 2011;3:148-156.
12. Disease Summaries: Allergic Diseases and Asthma in Pregnancy. World Allergy Organization. www.worldallergy.org/professional/allergic_
diseases_center/allergy_in_pregnancy/. Accessed February 14, 2014.
13. Simons FE, Ardusso LR, Dimov V, et al. World Allergy Organization Anaphylaxis Guidelines: 2013 update of the evidence base. Int Arch Allergy Immunol. 2013;162:193-204.
14. Noon L. Prophylactic inoculation against hay fever. Lancet. 1911;177:1572-1573.
15. Purello-D’Ambrosio F, Gangemi S, Merendino RA, et al. Prevention of new sensitizations in monosensitized subjects submitted to specific immunotherapy or not: a retrospective study. Clin & Exper Allergy. 2001;31:1295-1302.
16. Durham SR, Walker SM, Varga EM, et al. Long-term clinical efficacy of grass-pollen immunotherapy. N Engl J Med. 1999;341:468-475.
17. Mohapatra SS, Qazi M, Hellermann G. Immunotherapy for allergies and asthma: present and future. Curr Opin Pharmacol. 2010;10:276-288.
18. Calderon MA, Alves B, Jacobson M, et al. Allergen injection immunotherapy for seasonal allergic rhinitis. Cochrane Database Syst Rev. 2007; Iss 1, Art No: CD001936.
19. Scadding GK, Brostoff J. Low dose sublingual therapy in patients with allergic rhinitis due to house dust mite.Clin Allergy. 1986;16:483-491.
20. Canonica GW, Bousquet J, Casale T, et al. Sub-lingual immunotherapy: World Allergy Organization Position Paper 2009. Allergy. 2009;64 suppl 91:1-59.
21. Di Bona D, Plaia A, Scafidi V, et al. Efficacy of sublingual immunotherapy with grass allergens for seasonal allergic rhinitis: a systematic review and meta-analysis. J Allergy Clin Immunol. 2010;126:558-566.
22. Sikora JM, Tankersley MS; ACAAI Immunotherapy and Diagnostics Committee. Perception and practice of sublingual immunotherapy among practicing allergists in the United States: a follow-up survey. Ann Allergy Asthma Immunol. 2013;110:194-197.
1. Sibbald B, Rink E, D’Souza M. Is the prevalence of atopy increasing? Br J Gen Pract. 1990;40(337):338-340.
2. History of allergy. Allergy and Asthma Specialists Web site. www.allergyasthmaspecialist.com/allergy-and-asthma-clinic-of-kenosha-sc-education.htm. Accessed February 14, 2014.
3. Blackley CH. Hay Fever: Its Causes, Treatment, and Effective Prevention. London, England: Balliere, Tindall, & Cox; 1880.
4. Settipane RA. Rhinitis: a dose of epidemiological reality. Allergy Asthma Proc. 2003;24(3):147-154.
5. Wallace DV, Dykewicz MS, Bernstein DI, et al. The diagnosis and management of rhinitis: an updated practice parameter. J Allergy Clin Immunol. 2008;122(2 suppl):S1-S84.
6. Wright AL, Holberg CJ, Halonen M, et al. Epidemiology of physician-diagnosed allergic rhinitis in childhood. Pediatrics. 1994;94(6):895 -901.
7. Bernstein JA. Allergic and mixed rhinitis: epidemiology and natural history. Allergy Asthma Proc. 2010;31:365-369.
8. Meltzer EO, Bukstein DA. The economic impact of allergic rhinitis and current guidelines for treatment. Ann Allergy Asthma Immunol. 2011; 106(suppl 2):S12-S16.
9. American Academy of Allergy, Asthma & Immunology. Task force on allergic disorders: promoting best practice: raising the standard of care for patients with allergic disorders. Executive summary report. 1998.
10. Nathan RA. The burden of allergic rhinitis. Allergy Asthma Proc. 2007; 28:3-9.
11. Tran NP, Vickery J, Blaiss MS. Management of rhinitis: allergic and non-allergic. Allergy Asthma Immunol Res. 2011;3:148-156.
12. Disease Summaries: Allergic Diseases and Asthma in Pregnancy. World Allergy Organization. www.worldallergy.org/professional/allergic_
diseases_center/allergy_in_pregnancy/. Accessed February 14, 2014.
13. Simons FE, Ardusso LR, Dimov V, et al. World Allergy Organization Anaphylaxis Guidelines: 2013 update of the evidence base. Int Arch Allergy Immunol. 2013;162:193-204.
14. Noon L. Prophylactic inoculation against hay fever. Lancet. 1911;177:1572-1573.
15. Purello-D’Ambrosio F, Gangemi S, Merendino RA, et al. Prevention of new sensitizations in monosensitized subjects submitted to specific immunotherapy or not: a retrospective study. Clin & Exper Allergy. 2001;31:1295-1302.
16. Durham SR, Walker SM, Varga EM, et al. Long-term clinical efficacy of grass-pollen immunotherapy. N Engl J Med. 1999;341:468-475.
17. Mohapatra SS, Qazi M, Hellermann G. Immunotherapy for allergies and asthma: present and future. Curr Opin Pharmacol. 2010;10:276-288.
18. Calderon MA, Alves B, Jacobson M, et al. Allergen injection immunotherapy for seasonal allergic rhinitis. Cochrane Database Syst Rev. 2007; Iss 1, Art No: CD001936.
19. Scadding GK, Brostoff J. Low dose sublingual therapy in patients with allergic rhinitis due to house dust mite.Clin Allergy. 1986;16:483-491.
20. Canonica GW, Bousquet J, Casale T, et al. Sub-lingual immunotherapy: World Allergy Organization Position Paper 2009. Allergy. 2009;64 suppl 91:1-59.
21. Di Bona D, Plaia A, Scafidi V, et al. Efficacy of sublingual immunotherapy with grass allergens for seasonal allergic rhinitis: a systematic review and meta-analysis. J Allergy Clin Immunol. 2010;126:558-566.
22. Sikora JM, Tankersley MS; ACAAI Immunotherapy and Diagnostics Committee. Perception and practice of sublingual immunotherapy among practicing allergists in the United States: a follow-up survey. Ann Allergy Asthma Immunol. 2013;110:194-197.

Kidney Patients With Diabetes: Managing Their Medication
CE/CME No: CR-1402
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the incidence and staging of chronic kidney disease (CKD).
• Enumerate the treatment goals for the CKD patient with diabetes.
• Describe the hypoglycemic medications that can be used at each stage of CKD.
FACULTY
Cheryl Gilmartin is a Clinical Pharmacist in Nephrology and a Clinical Assistant Professor in the College of Pharmacy at the University of Illinois at Chicago. Jane S. Davis, a member of the Clinician Reviews editorial board, is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP). Kim Zuber, past chair of the NKF-CAP, is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because the vast majority of patients with chronic kidney disease (CKD) are diabetic and thus take hypoglycemic medications, knowledge of renal dosing for these medications, their mechanisms of action, and their safety profiles, as well as consideration of A1C goals, is vital for the practicing clinician. Management of the diabetic CKD patient, identified by stage of kidney disease, is outlined, with dosing regimens as determined by the glomerular filtration rate. Special attention is given to insulin management.
Diabetes is the most common cause of chronic kidney disease (CKD) throughout the world and leaves in its wake huge financial and social burdens.1 Diabetes has been the main cause of kidney failure in the United States for a number of years; as of 2012, diabetes became the most common cause of kidney failure in the world.2
The cost of caring for the rapidly increasing diabetes population in the US has been enormous. In 2012, the price tag for treating diabetes was $245 billion for medical care alone.3 The societal cost—loss in life years, loss of productivity caused by an increase in early retirement and disability, and burden of caregiving on families—is immeasurable.
In addition to CKD, diabetes is a major risk factor for heart disease and can lead to blindness and amputations. The rising incidence of diabetes has resulted in a new sense of urgency and has prompted the health care community to find new ways to reduce the burden on the patient, as well as encourage more aggressive research on halting progression of the disease, preventing end-stage kidney failure, and avoiding the need for dialysis.
Nearly two out of three (59%) adults in the US will be diagnosed with CKD stages 3 to 5 during their lifetimes.4 Some of this can be attributed to normal loss of kidney function associated with aging. However, much is due to the double burden of diabetes and hypertension.5 Patients with CKD require ongoing medical care, and much of it will be provided in primary care practices.6 This article discusses the Kidney Disease: Improving Global Outcomes (KDIGO) 2012 staging guidelines and explains which oral and injectable diabetes medications are acceptable at each stage of CKD (as defined by the glomerular filtration rate [GFR]) and how to simplify dosing.
On the next page: Staging CKD >>
STAGING CHRONIC KIDNEY DISEASE
The increasing incidence of CKD was first recognized in the 1990s. The need to care for the patient with CKD was understood, but there was a dearth of standardized management practices. Research was hindered by the many names used to identify kidney disease in the literature: from mildly elevated serum creatinine to low clearance to renal insufficiency.7 To rectify this, the National Kidney Foundation (NKF) convened an expert panel to develop a standard language and units of measurement to define kidney disease. The panel was tasked with developing classifications and an evaluation system for renal disease; identifying ways to attain more timely referrals to nephrology; clarifying research objectives; and delineating how to improve medication dosing. Ultimately, the main objective was to improve management of the patient with CKD.
In 2002, the NKF’s Dialysis Outcomes Quality Initiative (K/DOQI) defined the stages of kidney disease based on assessment of GFR and other kidney disease markers.8 GFR is a mathematical calculation for determining kidney function that corrects for loss of kidney function and incorporates the patient’s gender, race, age, and body size—the nonmodifiable risk factors known in 2001. GFR is expressed in mL/min/1.73 m2. Classifying patients with CKD according to the stage of kidney disease by GFR provides a mechanism for studying the efficacy and adverse reactions of medications while correlating them to each stage. This is of particular importance when treating patients with diabetes and kidney disease, as it can help to avoid adverse effects associated with inappropriate renal dosing. Many medications are renally eliminated; as kidney function diminishes, the pharmacokinetics of drugs are altered, potentially causing toxicity and an increased risk for drug interactions. As the GFR falls, even drugs not eliminated by the kidney may have altered pharmacokinetics and pharmacodynamics that may cause renal or systemic toxicity.1
Determining the precise method for renally dosing medication has been one of the most vigorously debated topics in CKD. Many medications were prescribed using the patient’s serum creatinine (SCr) concentration, a marker but not an exact measure of kidney disease.8 The Cockcroft-Gault equation, which provides an estimate of creatinine clearance (eCrCl; an alternate GFR measurement), was primarily used to determine renal drug dosing. 9
In 1999, a multisite study group worked to develop an equation to measure loss of kidney function in the patient with CKD.10,11 The new formula, known as the MDRD (Modification of Diet in Renal Disease) equation, standardized the measurement of loss of kidney function and provided an estimated GFR (eGFR) that was more accurate then eCrCl. At that time, the National Kidney Disease Education Program (NKDEP) decided to use the MDRD equation to assess renal function and use the eCrCl for dosing medications. The latter was chosen because most drugs were analyzed using eCrCl.9 More recently, the NKDEP stated that either the eCrCl or the eGFR is acceptable for renal drug dosing. Hudson and Nyman9 advise that when discrepancies occur between eGFR and eCrCl, clinical judgment should be applied in the elderly and in those with extremes in body weight. In those cases, therapeutic outcomes and risk versus benefit need to be considered when determining renal dosing.
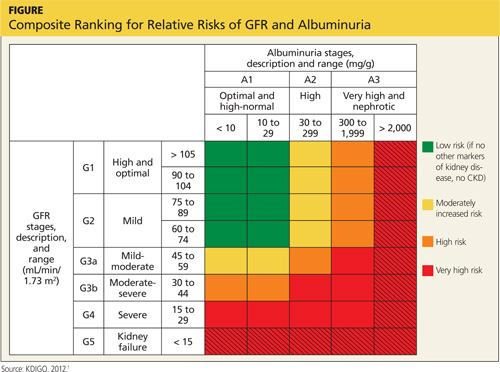
The publication of CKD stages by K/DOQI in 2002 sparked research that has uncovered additional criteria that might be useful in slowing the progression and improving the management of CKD. Thus, in 2013, new guidelines by KDIGO incorporating the latest international findings were published.1 KDIGO expands the definition of CKD to include albuminuria (ALB) as an added risk factor (see Figure) and has modified K/DOQI stages 3 and 5. ALB was included because it has predictive value for progression of CKD to kidney failure. In the new classification, there are three ranges defined for ALB measured in mg/g (see Table 1). The KDIGO guidelines, like K/DOQI, classify kidney disease using a 1-to-5 staging system; however, the stage numbers in the KDIGO classification are prefaced by a G (ie, G1, G2). Also, as mentioned above, stages G3 and G5 in the KDIGO classification have been further categorized.
CKD stage 3 was divided for practical reasons. It was found that the original CKD stage 3 was too large and diverse a classification to use for research studies, tracking of hospitalizations, and precise dosing of medications.12 K/DOQI stage 3 encompassed a GFR from 30 to 59 mL/min/1.73 m2. The KDIGO guideline divides K/DOQI stage 3 into G3a (GFR of 45 to 59 mL/min/1.73 m2) and G3b (GFR of 30 to 44 mL/min/1.73 m2). The staging is then further classified by the presence or absence of ALB. The patient with a GFR of 56 mL/min/1.73 m2 with ALB is in CKD stage 3aA3 (very high and nephrotic ALB) and has a greater risk for disease progression than the patient with a GFR of 56 mL/min/1.73 m2 without ALB. The latter patient is in CKD stage 3aA1(optimal and mildly high ALB). Finally, many medication dosages that were acceptable at a GFR of 56 mL/min/1.73 m2 had to be further adjusted for patients with a GFR of 30 mL/min/1.73 m2 to avoid toxicity. This further justified the KDIGO reclassification of stage 3.
Stage 5 from K/DOQI only reflected a GFR < 15 mL/min/1.73 m2. KDIGO utilizes the same GFR but splits the stage into patients not receiving dialysis and those receiving dialysis, G5 and G5D, respectively. The dosing and timing of medications due to drug dialyzability—including renal-specific medications—may be quite different for patients on dialysis as compared to those with a GFR < 15 mL/min/1.73 m2 not on dialysis, making the distinction beneficial.
On the next page: Diabetes management >>
DIABETES MANAGEMENT
The treatment of patients early in diabetic CKD is often the responsibility of primary care providers, who are faced with the daunting task of addressing the renal dosing of medications. The eGFR or eCrCl needs to be utilized to adjust diabetic medications, largely to avoid hypoglycemia. To alleviate some of the difficulty, the KDIGO CKD stage and the eCrCl and eGFR for diabetic medications are delineated in Table 2.1
Note that the guidelines are not meant to be strictly adhered to but rather are intended as a tool for managing the patient with CKD. Clinically significant patient factors need to be considered to adequately adjust drugs in CKD. Also, as new data become available from postmarketing reports, dosing adjustments, added precautions, or updated risk factors in these fragile patients may need to be incorporated into the guidelines. As new diabetic medications are approved by the FDA, kidney-specific dosing will continue to be a moving target.
While making lifestyle changes is very important early in diabetes, by the time diabetic nephropathy manifests, more aggressive action is warranted.12 A part of the diabetic microvascular triad (along with retinopathy and neuropathy), nephropathy signals existing and irreversible organ damage. Before instituting treatment to delay diabetic nephropathy progression, an A1C goal needs to be established. The long-standing goal has been an A1C < 7%, according to guidelines established by the American Diabetes Association, or 6.5%, as recognized by the American Association of Clinical Endocrinologists.13 The Diabetes Control and Complications Trial (DCCT) demonstrated microvascular benefit of tight glycemic control in type 1 diabetic patients when the A1C was maintained at an average of 7.2% (versus 9.1% in conventional therapy).14 The United Kingdom Prospective Diabetes Study similarly demonstrated microvascular benefit with intensive blood glucose control in type 2 diabetic patients who were maintained at a median A1C of 7% compared to the conventional treatment group maintained at an A1C of 7.9%.15
In January 2013, Perkovic and colleagues, in a post hoc analysis of the ADVANCE Trial, showed that maintaining an A1C between 6.5% and 7% in a patient with diabetes and macroalbuminuria (UACR > 300 μg/mg) slows progression to kidney failure.16 The time needed to treat with intensive glucose control was five years, and the number needed to treat was 41. Thus, for every 41 patients with diabetes, CKD, and ALB whose A1C is below 7%, one will not progress to kidney failure in five years of treatment.
The five-year lead time is troublesome, however. It discourages many patients from taking steps to achieve strict glycemic control; a significant number fail to follow their diabetic diets or take their medications until the damage is done. For the elderly kidney patient with diabetes and multiple other comorbidities, aggressively managing diabetes may result in hypoglycemia.12 On balance, the slight loss of kidney function five years hence is less problematic than the present risk for a fall and resulting hip fracture due to hypoglycemia. For this reason, the KDOQI glycemic guidelines suggest an A1C between 7% to 7.5% as the goal for those with significant comorbidities.12
On the next page: Diabetic medication dosing for CKD >>
DIABETIC MEDICATION DOSING FOR CKD
Glycemic control is important in delaying the progression of kidney failure in the patient with CKD. Hypoglycemic medications by definition may cause low blood glucose levels. This is of particular concern in patients with diminishing renal function, particularly the elderly and CKD patients. Antidiabetes drugs for patients in CKD stages 1 and 2 have few renal precautions, although care must be taken with metformin (see Table 3). Metformin is the drug of choice for the patient newly diagnosed with diabetes. It is inexpensive and effective, causes hypoglycemia only with intensive exercise, is taken orally, and is generally well tolerated. However, the package insert (PI) indicates stopping it in patients whose kidney disease has progressed to a GFR < 60 mL/min/1.73 m2 because it causes an increased risk for lactic acidosis.17,18
Metformin was developed prior to 1998, when the approval of the dosage regimen was tied to SCr levels rather than the presently accepted eGFR.19 As a result, the PI states that metformin should not be used in women with an SCr > 1.4 mg/dL or in men with an SCr > 1.5 mg/dL.17 However, as noted, SCr concentration alone is a very poor indicator of kidney disease (see Table 4).
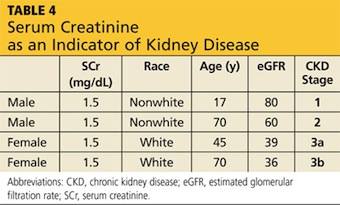
Currently, the KDIGO guidelines recommend assessing the GFR for metformin dosing. When the GFR is < 60 mL/min/1.73 m2, KDIGO recommends metformin be discontinued in patients with low muscle mass or serious concurrent illness, or those who are concurrently receiving renally eliminated or nephrotoxic drugs (this includes, but is not limited to, renin-angiotensin-aldosterone system [RAAS] inhibitors, NSAIDs, and diuretics).1 In those patients who do not have the exclusions delineated above, KDIGO recommends that metformin may be continued when the GFR is > 45 mL/min/1.73 m2 (stages G1 to G3a), closely monitored and reconsidered when the GFR is 30 to 44 mL/min/1.73 m2 (stage G3b), and discontinued when the GFR is < 30 mL/min/1.73 m2 (stages G4 to G5). The KDIGO differs from other sources that recommend metformin be stopped when the GFR is < 50 to 70 ml/min/1.73 m2.20-22 The metformin PI also recommends stopping metformin the day of or day before and two days after procedures in which radiographic iodinated contrast dye is administered, to avoid acute kidney failure. Finally, it is important to note that any combination medication formulated with metformin should be stopped when the GFR is 50 mL/min/1.73 m2 because lactic acidosis has also been seen in patients taking these medications.21
At stage 3a (GFR, 45 to 60 mL/min/1.73 m2), the sulfonylureas require dosing changes (see Table 2). Glipizide is often the oral medication of choice during CKD, and it may be used in all stages, including dialysis, but does require renal dosing and monitoring.23 Although the insulins are more effective than glipizide in glycemic control, many practitioners prescribe it for patients who will not accept an injectable medication. Glyburide is not used at this stage, as it can lead to complications in patients with a GFR < 60 mL/min/1.73 m2, particularly hypoglycemia.
Like the sulfonylureas, the meglitinides enhance insulin secretion.13,18 Repaglinide and nateglinide are fast-acting and need to be taken with meals. Because it is recommended that the dose of the meglitinides be omitted when a meal is skipped, they are good for patients who eat meals irregularly. The meglitinides require initiation at a low dose in severe renal insufficiency.
The thiazolidinediones (TDZs) are used with caution in patients with CKD, not only because of controversial adverse cardiac effects, but also for peripheral edema that interferes with CKD management.18 The TDZs (rosiglitazone and pioglitazone) require no dose adjustment and appear to have a beneficial effect in obese patients and in patients who experienced weight gain with other hypoglycemic agents.
The newest of the diabetic drugs, canagliflozin, requires a working kidney to be effective.24,25 It is used with precaution when GFR is 45 to 59 mL/min/1.73 m2 at stage G3a and is contraindicated when GFR is < 45 mL/min/1.73 m2 in stage G3b. Canagliflozin's effects are felt in early segment of proximal convoluted tubule where it blocks sodium-glucose co-transporter 2, thereby lowering reabsorption of glucose and increasing excretion of glucose in the urine. In practical terms, it can affect kidney function and, due to the inhibition of the glucose-sodium pathway, induce hyperkalemia.25 Monitoring of both the GFR and potassium is vital when administering this drug. As data from postmarketing reports come in and practitioners gain experience, use of canaglifozin in the patient with CKD will be further delineated.
At a GFR between 30 and 45 mL/min/1.73 m2, CKD stage 3b, alpha-glucosidase inhibitors acarbose and miglitol are contraindicated. The GLP-1 mimetic liraglutide needs to be avoided when the GFR is < 60 mL/min/1.73 m2 (stage G3a). While exenatide requires close monitoring and caution when the GFR is < 50 mL/min/1.73 m2, it needs to be discontinued at a GFR
< 30 mL/min/1.73 m2 (stage 4). DPP-4 inhibitors become problematic at a GFR < 60 mL/min/
1.73 m2 beginning at stage G3a. All except the DPP-4 inhibitor linagliptin require dosing changes as the loss of GFR continues. Exact dosing adjustments for the drug categories are found in Table 2.
In stage 4 CKD (GFR, 15 to 30 mL/min/1.73 m2), more medications are eliminated from consideration. The amylinomimetic pramlintide cannot be used for a patient with a GFR < 15 mL/min/1.73 m2 (see Table 2). Both GLP-1 mimetics, exenatide and liraglutide, must be stopped when the GFR drops to 30 mL/min/1.73 m2. DDP-4 inhibitors need to be dosed according to the GFR but are still acceptable at CKD stage 4. Ideally, patients with diabetes should be referred to nephrology at stage 3, but referral is vital by stage 4.
Although oral medications are not as commonly prescribed as insulins at CKD stages 4 and 5, in stage 5 CKD (GFR, < 15 mL/min/1.73 m2), a limited number of oral diabetic agents are available (see Table 2). Glipizide is inexpensive and generic; therefore, it is the most commonly used oral medication in this stage. A recent study found linagliptin was safe to use throughout all stages of kidney disease without adjustment for GFR.26 While nateglinide is still acceptable in stage 5, it is used less frequently than glipizide.
As the kidney fails, the need for diabetic medications also decreases and dosing needs to take into account any residual renal function. Because glucose is produced in the proximal tubules of the kidney (known as gluconeogenesis), the level of glucose falls as the kidney fails. Excretion of insulin and other diabetes medications decreases, and thus the amount of insulin remaining in circulation is increased. This, in combination with the reduced glucose, increases the patient’s risk for hypoglycemia.13 Although protocols for medication adjustment are tied to GFR levels, close follow-up by the practitioner is required because GFR is not a reliable measure of kidney function when creatinine levels change rapidly.27
On the next page: Insulins >>
INSULINS
Many patients start insulin prior to reaching CKD stage 4 or 5 because it is the best choice for managing diabetes through all stages of CKD.28 Insulin can slow the progression to kidney failure by providing better A1C control, and all patients should be encouraged to start insulin as soon as it is appropriate.29 Many patients are very reluctant to use injectables, but by stage 5 they are willing to use insulin if it will slow progression of disease and help prevent the need for dialysis.2
The long-acting basal insulins (detemir and glargine) or a combination of basal and oral medication can work quite well at stage 5. Many patients will require a low dose of the long-acting basal insulins and short-acting insulin with meals. As the kidney fails, the short-acting doses can often be discontinued.29 A protocol that mixes long- and short-acting insulin with the largest meal is very effective at this stage. The long-acting insulin dose will need to be decreased as the kidney continues to fail. The American College of Physicians protocol for insulin suggests a 25% decrease at stage 4 and a 50% decrease at stage 5. Each patient is different, however, and dosing must be determined individually.23
On the next page: Patient education >>
PATIENT EDUCATION
There is a correlation between patients who participate in kidney disease education programs and a decrease in the progression of kidney disease (see “Case Study: The Recalcitrant Patient With Diabetes"). These classes can be taught by a physician, physician assistant, nurse practitioner, or clinical nurse specialist. To alleviate the burden of end-stage kidney disease in the US, Medicare Part B pays for six outpatient kidney disease patient education classes per lifetime of the beneficiary.30

Since 2010, when kidney disease education programs were rolled out, over 10,000 classes have been taught.31 Many practitioners report that patients are more compliant and understand their disease better when they attend classes.32 Data are being gathered to determine the effectiveness of the classes on patient outcomes.
On the next page: Conclusion >>
CONCLUSION
Knowing how to manage the patient with both diabetes and kidney disease is increasingly vital as this patient population grows. Much of the management of these patients will fall to the primary care practitioner.6 The most effective way to start treatment is to identify which CKD stage the patient fits into.
Once the patient is properly categorized, safe and effective diabetic medications can be selected and dosed according to stage. Although the new classification system may be difficult to incorporate into some electronic medical record systems and practitioner behavior, ultimately, it will allow safer management of the patient with CKD and a better predictive power of outcome.
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Suppl. 2013;3:1-150.
2. Brazie M. Finding the sweet spot: trouble-shooting diabetic dilemmas. Oral presentation at NKF Spring Clinical Meetings; May 2012, Washington, DC.
3. Estimated cost of diabetes care $245 billion in U.S. in 2012. Renal & Urology News. March 9, 2013. www.renalandurologynews.com/estimated-cost-of-diabetes-245-billion-in-us-in-2012/article/283616/. Accessed January 17, 2013.
4. Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3-5 in the United States. Am J Kidney Dis. 2013;62(2):245-252.
5. Greenberg A, ed. National Kidney Foundation Primer of Kidney Diseases. 5th ed. Philadelphia, PA: Saunders Elsevier; 2009.
6. Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research study. Ann Fam Med. 2006;4(1):23-31.
7. Hsu CY, Chertow GM. Chronic renal confusion: insufficiency, failure, dysfunction, or disease. Am J Kidney Dis. 2000;49(3):482-496.
8. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification and stratification. Am J Kidney Dis. 2002;39(suppl 1):S1-S266.
9. Hudson JQ, Nyman HA. Use of the estimated glomerular filtration rate for drug dosing in the chronic kidney disease patient. Curr Opin Nephrol Hypertens. 2011;20:482-491.
10. Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461-470.
11. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612.
12. NKF-KDOQI clinical practice guideline for diabetes and CKD, guideline 2: management of hyperglycemia and general diabetes care in CKD. Am J Kidney Dis. 2012;60(5):850-886. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed January 2, 2014.
13. Abe M, Okada K, Soma M. Antidiabetic agents in patients with chronic kidney disease and end-stage renal disease on dialysis: metabolism and clinical practice. Curr Drug Metab. 2011;12(1):57-69.
14. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381-389.
15. Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405-411.
16. Perkovic V, Heerspink HL, Chalmers J, et al. Intensive glucose control improves kidney outcomes in patients with type 2 diabetes. Kidney Int. 2013;83(3):517-23.
17. Metformin [package insert]. US Department of Health and Human Services, US Food and Drug Administration. www.fda.gov/ohrms/dockets/dailys/02/May02/053102/800471e6.pdf. Accessed January 17, 2013.
18. Triplitt CL, Reasner CA. Chapter 83. Diabetes Mellitus. In: Wells BG, ed. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill; 2011.
19. FDA. Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulato ryInformation/Guidances/ucm072127.pdf. Accessed January 17, 2013.
20. Lacy CF, Armstrong LL, Goldman MP, Lance LL. Drug Information Handbook. 20th ed. Hudson, OH: Lexi-Comp, Inc.; 2011.
21. Rocha A, Almeida M, Santos J, Carvalho A. Metformin in patients with chronic kidney disease: strengths and weaknesses. J Nephrol. 2013; 26(10):55-60.
22. Gilbert SJ, Weiner DE, eds. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2014.
23. Aronoff GR, Bennett WM, Berns JS, et al. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults and Children. 5th ed. Philadelphia, PA: American College of Physicians; 2007.
24. Valentine V. The role of the kidney and the sodium-glucose co-transporter inhibition in diabetes management. Clin Diabetes. 2012;30(4):151-155.
25. Invokana [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2013.
26. McGill JB, Sloan L, Newman J, et al. Long-term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment. Diabetes Care. 2013;36(2):237-244.
27. National Kidney Disease and Education Program (NKDEP). Estimated glomerular filtration rate (eGFR) info sheet. NIH publication No.10-6286, March 2010.
28. Thummel K, Shen D, Isoherranen N, et al. Design and optimization of dosage regimens: pharmacokinetic data. In: Hardman J, Limbird L, Goodman G (eds). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006.
29. Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial. 2004;17(5):365-70.
30. Centers for Medicare and Medicaid Services. Medicare program: revisions to payment policies under the Physician Fee Schedule, Clinical Laboratory Fee Schedule & other revisions to Part B for CY 2014; Final Rule. Federal Register. 2009;74(226):61738-62188. www.gpo.gov/fdsys/pkg/FR-2009-11-25/html/E9-26502.htm. Accessed January 17, 2013.
31. Zuber K, Davis J. Kidney disease education: a niche for PAs and NPs. JAAPA. 2013;26(7):42-47.
32. Zuber K, Davis J. Stories from the trenches: the first year experience with kidney disease education. Nephrol News Issues. 2012;26(2):20-21.
CE/CME No: CR-1402
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the incidence and staging of chronic kidney disease (CKD).
• Enumerate the treatment goals for the CKD patient with diabetes.
• Describe the hypoglycemic medications that can be used at each stage of CKD.
FACULTY
Cheryl Gilmartin is a Clinical Pharmacist in Nephrology and a Clinical Assistant Professor in the College of Pharmacy at the University of Illinois at Chicago. Jane S. Davis, a member of the Clinician Reviews editorial board, is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP). Kim Zuber, past chair of the NKF-CAP, is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because the vast majority of patients with chronic kidney disease (CKD) are diabetic and thus take hypoglycemic medications, knowledge of renal dosing for these medications, their mechanisms of action, and their safety profiles, as well as consideration of A1C goals, is vital for the practicing clinician. Management of the diabetic CKD patient, identified by stage of kidney disease, is outlined, with dosing regimens as determined by the glomerular filtration rate. Special attention is given to insulin management.
Diabetes is the most common cause of chronic kidney disease (CKD) throughout the world and leaves in its wake huge financial and social burdens.1 Diabetes has been the main cause of kidney failure in the United States for a number of years; as of 2012, diabetes became the most common cause of kidney failure in the world.2
The cost of caring for the rapidly increasing diabetes population in the US has been enormous. In 2012, the price tag for treating diabetes was $245 billion for medical care alone.3 The societal cost—loss in life years, loss of productivity caused by an increase in early retirement and disability, and burden of caregiving on families—is immeasurable.
In addition to CKD, diabetes is a major risk factor for heart disease and can lead to blindness and amputations. The rising incidence of diabetes has resulted in a new sense of urgency and has prompted the health care community to find new ways to reduce the burden on the patient, as well as encourage more aggressive research on halting progression of the disease, preventing end-stage kidney failure, and avoiding the need for dialysis.
Nearly two out of three (59%) adults in the US will be diagnosed with CKD stages 3 to 5 during their lifetimes.4 Some of this can be attributed to normal loss of kidney function associated with aging. However, much is due to the double burden of diabetes and hypertension.5 Patients with CKD require ongoing medical care, and much of it will be provided in primary care practices.6 This article discusses the Kidney Disease: Improving Global Outcomes (KDIGO) 2012 staging guidelines and explains which oral and injectable diabetes medications are acceptable at each stage of CKD (as defined by the glomerular filtration rate [GFR]) and how to simplify dosing.
On the next page: Staging CKD >>
STAGING CHRONIC KIDNEY DISEASE
The increasing incidence of CKD was first recognized in the 1990s. The need to care for the patient with CKD was understood, but there was a dearth of standardized management practices. Research was hindered by the many names used to identify kidney disease in the literature: from mildly elevated serum creatinine to low clearance to renal insufficiency.7 To rectify this, the National Kidney Foundation (NKF) convened an expert panel to develop a standard language and units of measurement to define kidney disease. The panel was tasked with developing classifications and an evaluation system for renal disease; identifying ways to attain more timely referrals to nephrology; clarifying research objectives; and delineating how to improve medication dosing. Ultimately, the main objective was to improve management of the patient with CKD.
In 2002, the NKF’s Dialysis Outcomes Quality Initiative (K/DOQI) defined the stages of kidney disease based on assessment of GFR and other kidney disease markers.8 GFR is a mathematical calculation for determining kidney function that corrects for loss of kidney function and incorporates the patient’s gender, race, age, and body size—the nonmodifiable risk factors known in 2001. GFR is expressed in mL/min/1.73 m2. Classifying patients with CKD according to the stage of kidney disease by GFR provides a mechanism for studying the efficacy and adverse reactions of medications while correlating them to each stage. This is of particular importance when treating patients with diabetes and kidney disease, as it can help to avoid adverse effects associated with inappropriate renal dosing. Many medications are renally eliminated; as kidney function diminishes, the pharmacokinetics of drugs are altered, potentially causing toxicity and an increased risk for drug interactions. As the GFR falls, even drugs not eliminated by the kidney may have altered pharmacokinetics and pharmacodynamics that may cause renal or systemic toxicity.1
Determining the precise method for renally dosing medication has been one of the most vigorously debated topics in CKD. Many medications were prescribed using the patient’s serum creatinine (SCr) concentration, a marker but not an exact measure of kidney disease.8 The Cockcroft-Gault equation, which provides an estimate of creatinine clearance (eCrCl; an alternate GFR measurement), was primarily used to determine renal drug dosing. 9
In 1999, a multisite study group worked to develop an equation to measure loss of kidney function in the patient with CKD.10,11 The new formula, known as the MDRD (Modification of Diet in Renal Disease) equation, standardized the measurement of loss of kidney function and provided an estimated GFR (eGFR) that was more accurate then eCrCl. At that time, the National Kidney Disease Education Program (NKDEP) decided to use the MDRD equation to assess renal function and use the eCrCl for dosing medications. The latter was chosen because most drugs were analyzed using eCrCl.9 More recently, the NKDEP stated that either the eCrCl or the eGFR is acceptable for renal drug dosing. Hudson and Nyman9 advise that when discrepancies occur between eGFR and eCrCl, clinical judgment should be applied in the elderly and in those with extremes in body weight. In those cases, therapeutic outcomes and risk versus benefit need to be considered when determining renal dosing.
The publication of CKD stages by K/DOQI in 2002 sparked research that has uncovered additional criteria that might be useful in slowing the progression and improving the management of CKD. Thus, in 2013, new guidelines by KDIGO incorporating the latest international findings were published.1 KDIGO expands the definition of CKD to include albuminuria (ALB) as an added risk factor (see Figure) and has modified K/DOQI stages 3 and 5. ALB was included because it has predictive value for progression of CKD to kidney failure. In the new classification, there are three ranges defined for ALB measured in mg/g (see Table 1). The KDIGO guidelines, like K/DOQI, classify kidney disease using a 1-to-5 staging system; however, the stage numbers in the KDIGO classification are prefaced by a G (ie, G1, G2). Also, as mentioned above, stages G3 and G5 in the KDIGO classification have been further categorized.
CKD stage 3 was divided for practical reasons. It was found that the original CKD stage 3 was too large and diverse a classification to use for research studies, tracking of hospitalizations, and precise dosing of medications.12 K/DOQI stage 3 encompassed a GFR from 30 to 59 mL/min/1.73 m2. The KDIGO guideline divides K/DOQI stage 3 into G3a (GFR of 45 to 59 mL/min/1.73 m2) and G3b (GFR of 30 to 44 mL/min/1.73 m2). The staging is then further classified by the presence or absence of ALB. The patient with a GFR of 56 mL/min/1.73 m2 with ALB is in CKD stage 3aA3 (very high and nephrotic ALB) and has a greater risk for disease progression than the patient with a GFR of 56 mL/min/1.73 m2 without ALB. The latter patient is in CKD stage 3aA1(optimal and mildly high ALB). Finally, many medication dosages that were acceptable at a GFR of 56 mL/min/1.73 m2 had to be further adjusted for patients with a GFR of 30 mL/min/1.73 m2 to avoid toxicity. This further justified the KDIGO reclassification of stage 3.
Stage 5 from K/DOQI only reflected a GFR < 15 mL/min/1.73 m2. KDIGO utilizes the same GFR but splits the stage into patients not receiving dialysis and those receiving dialysis, G5 and G5D, respectively. The dosing and timing of medications due to drug dialyzability—including renal-specific medications—may be quite different for patients on dialysis as compared to those with a GFR < 15 mL/min/1.73 m2 not on dialysis, making the distinction beneficial.
On the next page: Diabetes management >>
DIABETES MANAGEMENT
The treatment of patients early in diabetic CKD is often the responsibility of primary care providers, who are faced with the daunting task of addressing the renal dosing of medications. The eGFR or eCrCl needs to be utilized to adjust diabetic medications, largely to avoid hypoglycemia. To alleviate some of the difficulty, the KDIGO CKD stage and the eCrCl and eGFR for diabetic medications are delineated in Table 2.1
Note that the guidelines are not meant to be strictly adhered to but rather are intended as a tool for managing the patient with CKD. Clinically significant patient factors need to be considered to adequately adjust drugs in CKD. Also, as new data become available from postmarketing reports, dosing adjustments, added precautions, or updated risk factors in these fragile patients may need to be incorporated into the guidelines. As new diabetic medications are approved by the FDA, kidney-specific dosing will continue to be a moving target.
While making lifestyle changes is very important early in diabetes, by the time diabetic nephropathy manifests, more aggressive action is warranted.12 A part of the diabetic microvascular triad (along with retinopathy and neuropathy), nephropathy signals existing and irreversible organ damage. Before instituting treatment to delay diabetic nephropathy progression, an A1C goal needs to be established. The long-standing goal has been an A1C < 7%, according to guidelines established by the American Diabetes Association, or 6.5%, as recognized by the American Association of Clinical Endocrinologists.13 The Diabetes Control and Complications Trial (DCCT) demonstrated microvascular benefit of tight glycemic control in type 1 diabetic patients when the A1C was maintained at an average of 7.2% (versus 9.1% in conventional therapy).14 The United Kingdom Prospective Diabetes Study similarly demonstrated microvascular benefit with intensive blood glucose control in type 2 diabetic patients who were maintained at a median A1C of 7% compared to the conventional treatment group maintained at an A1C of 7.9%.15
In January 2013, Perkovic and colleagues, in a post hoc analysis of the ADVANCE Trial, showed that maintaining an A1C between 6.5% and 7% in a patient with diabetes and macroalbuminuria (UACR > 300 μg/mg) slows progression to kidney failure.16 The time needed to treat with intensive glucose control was five years, and the number needed to treat was 41. Thus, for every 41 patients with diabetes, CKD, and ALB whose A1C is below 7%, one will not progress to kidney failure in five years of treatment.
The five-year lead time is troublesome, however. It discourages many patients from taking steps to achieve strict glycemic control; a significant number fail to follow their diabetic diets or take their medications until the damage is done. For the elderly kidney patient with diabetes and multiple other comorbidities, aggressively managing diabetes may result in hypoglycemia.12 On balance, the slight loss of kidney function five years hence is less problematic than the present risk for a fall and resulting hip fracture due to hypoglycemia. For this reason, the KDOQI glycemic guidelines suggest an A1C between 7% to 7.5% as the goal for those with significant comorbidities.12
On the next page: Diabetic medication dosing for CKD >>
DIABETIC MEDICATION DOSING FOR CKD
Glycemic control is important in delaying the progression of kidney failure in the patient with CKD. Hypoglycemic medications by definition may cause low blood glucose levels. This is of particular concern in patients with diminishing renal function, particularly the elderly and CKD patients. Antidiabetes drugs for patients in CKD stages 1 and 2 have few renal precautions, although care must be taken with metformin (see Table 3). Metformin is the drug of choice for the patient newly diagnosed with diabetes. It is inexpensive and effective, causes hypoglycemia only with intensive exercise, is taken orally, and is generally well tolerated. However, the package insert (PI) indicates stopping it in patients whose kidney disease has progressed to a GFR < 60 mL/min/1.73 m2 because it causes an increased risk for lactic acidosis.17,18
Metformin was developed prior to 1998, when the approval of the dosage regimen was tied to SCr levels rather than the presently accepted eGFR.19 As a result, the PI states that metformin should not be used in women with an SCr > 1.4 mg/dL or in men with an SCr > 1.5 mg/dL.17 However, as noted, SCr concentration alone is a very poor indicator of kidney disease (see Table 4).
Currently, the KDIGO guidelines recommend assessing the GFR for metformin dosing. When the GFR is < 60 mL/min/1.73 m2, KDIGO recommends metformin be discontinued in patients with low muscle mass or serious concurrent illness, or those who are concurrently receiving renally eliminated or nephrotoxic drugs (this includes, but is not limited to, renin-angiotensin-aldosterone system [RAAS] inhibitors, NSAIDs, and diuretics).1 In those patients who do not have the exclusions delineated above, KDIGO recommends that metformin may be continued when the GFR is > 45 mL/min/1.73 m2 (stages G1 to G3a), closely monitored and reconsidered when the GFR is 30 to 44 mL/min/1.73 m2 (stage G3b), and discontinued when the GFR is < 30 mL/min/1.73 m2 (stages G4 to G5). The KDIGO differs from other sources that recommend metformin be stopped when the GFR is < 50 to 70 ml/min/1.73 m2.20-22 The metformin PI also recommends stopping metformin the day of or day before and two days after procedures in which radiographic iodinated contrast dye is administered, to avoid acute kidney failure. Finally, it is important to note that any combination medication formulated with metformin should be stopped when the GFR is 50 mL/min/1.73 m2 because lactic acidosis has also been seen in patients taking these medications.21
At stage 3a (GFR, 45 to 60 mL/min/1.73 m2), the sulfonylureas require dosing changes (see Table 2). Glipizide is often the oral medication of choice during CKD, and it may be used in all stages, including dialysis, but does require renal dosing and monitoring.23 Although the insulins are more effective than glipizide in glycemic control, many practitioners prescribe it for patients who will not accept an injectable medication. Glyburide is not used at this stage, as it can lead to complications in patients with a GFR < 60 mL/min/1.73 m2, particularly hypoglycemia.
Like the sulfonylureas, the meglitinides enhance insulin secretion.13,18 Repaglinide and nateglinide are fast-acting and need to be taken with meals. Because it is recommended that the dose of the meglitinides be omitted when a meal is skipped, they are good for patients who eat meals irregularly. The meglitinides require initiation at a low dose in severe renal insufficiency.
The thiazolidinediones (TDZs) are used with caution in patients with CKD, not only because of controversial adverse cardiac effects, but also for peripheral edema that interferes with CKD management.18 The TDZs (rosiglitazone and pioglitazone) require no dose adjustment and appear to have a beneficial effect in obese patients and in patients who experienced weight gain with other hypoglycemic agents.
The newest of the diabetic drugs, canagliflozin, requires a working kidney to be effective.24,25 It is used with precaution when GFR is 45 to 59 mL/min/1.73 m2 at stage G3a and is contraindicated when GFR is < 45 mL/min/1.73 m2 in stage G3b. Canagliflozin's effects are felt in early segment of proximal convoluted tubule where it blocks sodium-glucose co-transporter 2, thereby lowering reabsorption of glucose and increasing excretion of glucose in the urine. In practical terms, it can affect kidney function and, due to the inhibition of the glucose-sodium pathway, induce hyperkalemia.25 Monitoring of both the GFR and potassium is vital when administering this drug. As data from postmarketing reports come in and practitioners gain experience, use of canaglifozin in the patient with CKD will be further delineated.
At a GFR between 30 and 45 mL/min/1.73 m2, CKD stage 3b, alpha-glucosidase inhibitors acarbose and miglitol are contraindicated. The GLP-1 mimetic liraglutide needs to be avoided when the GFR is < 60 mL/min/1.73 m2 (stage G3a). While exenatide requires close monitoring and caution when the GFR is < 50 mL/min/1.73 m2, it needs to be discontinued at a GFR
< 30 mL/min/1.73 m2 (stage 4). DPP-4 inhibitors become problematic at a GFR < 60 mL/min/
1.73 m2 beginning at stage G3a. All except the DPP-4 inhibitor linagliptin require dosing changes as the loss of GFR continues. Exact dosing adjustments for the drug categories are found in Table 2.
In stage 4 CKD (GFR, 15 to 30 mL/min/1.73 m2), more medications are eliminated from consideration. The amylinomimetic pramlintide cannot be used for a patient with a GFR < 15 mL/min/1.73 m2 (see Table 2). Both GLP-1 mimetics, exenatide and liraglutide, must be stopped when the GFR drops to 30 mL/min/1.73 m2. DDP-4 inhibitors need to be dosed according to the GFR but are still acceptable at CKD stage 4. Ideally, patients with diabetes should be referred to nephrology at stage 3, but referral is vital by stage 4.
Although oral medications are not as commonly prescribed as insulins at CKD stages 4 and 5, in stage 5 CKD (GFR, < 15 mL/min/1.73 m2), a limited number of oral diabetic agents are available (see Table 2). Glipizide is inexpensive and generic; therefore, it is the most commonly used oral medication in this stage. A recent study found linagliptin was safe to use throughout all stages of kidney disease without adjustment for GFR.26 While nateglinide is still acceptable in stage 5, it is used less frequently than glipizide.
As the kidney fails, the need for diabetic medications also decreases and dosing needs to take into account any residual renal function. Because glucose is produced in the proximal tubules of the kidney (known as gluconeogenesis), the level of glucose falls as the kidney fails. Excretion of insulin and other diabetes medications decreases, and thus the amount of insulin remaining in circulation is increased. This, in combination with the reduced glucose, increases the patient’s risk for hypoglycemia.13 Although protocols for medication adjustment are tied to GFR levels, close follow-up by the practitioner is required because GFR is not a reliable measure of kidney function when creatinine levels change rapidly.27
On the next page: Insulins >>
INSULINS
Many patients start insulin prior to reaching CKD stage 4 or 5 because it is the best choice for managing diabetes through all stages of CKD.28 Insulin can slow the progression to kidney failure by providing better A1C control, and all patients should be encouraged to start insulin as soon as it is appropriate.29 Many patients are very reluctant to use injectables, but by stage 5 they are willing to use insulin if it will slow progression of disease and help prevent the need for dialysis.2
The long-acting basal insulins (detemir and glargine) or a combination of basal and oral medication can work quite well at stage 5. Many patients will require a low dose of the long-acting basal insulins and short-acting insulin with meals. As the kidney fails, the short-acting doses can often be discontinued.29 A protocol that mixes long- and short-acting insulin with the largest meal is very effective at this stage. The long-acting insulin dose will need to be decreased as the kidney continues to fail. The American College of Physicians protocol for insulin suggests a 25% decrease at stage 4 and a 50% decrease at stage 5. Each patient is different, however, and dosing must be determined individually.23
On the next page: Patient education >>
PATIENT EDUCATION
There is a correlation between patients who participate in kidney disease education programs and a decrease in the progression of kidney disease (see “Case Study: The Recalcitrant Patient With Diabetes"). These classes can be taught by a physician, physician assistant, nurse practitioner, or clinical nurse specialist. To alleviate the burden of end-stage kidney disease in the US, Medicare Part B pays for six outpatient kidney disease patient education classes per lifetime of the beneficiary.30
Since 2010, when kidney disease education programs were rolled out, over 10,000 classes have been taught.31 Many practitioners report that patients are more compliant and understand their disease better when they attend classes.32 Data are being gathered to determine the effectiveness of the classes on patient outcomes.
On the next page: Conclusion >>
CONCLUSION
Knowing how to manage the patient with both diabetes and kidney disease is increasingly vital as this patient population grows. Much of the management of these patients will fall to the primary care practitioner.6 The most effective way to start treatment is to identify which CKD stage the patient fits into.
Once the patient is properly categorized, safe and effective diabetic medications can be selected and dosed according to stage. Although the new classification system may be difficult to incorporate into some electronic medical record systems and practitioner behavior, ultimately, it will allow safer management of the patient with CKD and a better predictive power of outcome.
CE/CME No: CR-1402
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the incidence and staging of chronic kidney disease (CKD).
• Enumerate the treatment goals for the CKD patient with diabetes.
• Describe the hypoglycemic medications that can be used at each stage of CKD.
FACULTY
Cheryl Gilmartin is a Clinical Pharmacist in Nephrology and a Clinical Assistant Professor in the College of Pharmacy at the University of Illinois at Chicago. Jane S. Davis, a member of the Clinician Reviews editorial board, is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP). Kim Zuber, past chair of the NKF-CAP, is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because the vast majority of patients with chronic kidney disease (CKD) are diabetic and thus take hypoglycemic medications, knowledge of renal dosing for these medications, their mechanisms of action, and their safety profiles, as well as consideration of A1C goals, is vital for the practicing clinician. Management of the diabetic CKD patient, identified by stage of kidney disease, is outlined, with dosing regimens as determined by the glomerular filtration rate. Special attention is given to insulin management.
Diabetes is the most common cause of chronic kidney disease (CKD) throughout the world and leaves in its wake huge financial and social burdens.1 Diabetes has been the main cause of kidney failure in the United States for a number of years; as of 2012, diabetes became the most common cause of kidney failure in the world.2
The cost of caring for the rapidly increasing diabetes population in the US has been enormous. In 2012, the price tag for treating diabetes was $245 billion for medical care alone.3 The societal cost—loss in life years, loss of productivity caused by an increase in early retirement and disability, and burden of caregiving on families—is immeasurable.
In addition to CKD, diabetes is a major risk factor for heart disease and can lead to blindness and amputations. The rising incidence of diabetes has resulted in a new sense of urgency and has prompted the health care community to find new ways to reduce the burden on the patient, as well as encourage more aggressive research on halting progression of the disease, preventing end-stage kidney failure, and avoiding the need for dialysis.
Nearly two out of three (59%) adults in the US will be diagnosed with CKD stages 3 to 5 during their lifetimes.4 Some of this can be attributed to normal loss of kidney function associated with aging. However, much is due to the double burden of diabetes and hypertension.5 Patients with CKD require ongoing medical care, and much of it will be provided in primary care practices.6 This article discusses the Kidney Disease: Improving Global Outcomes (KDIGO) 2012 staging guidelines and explains which oral and injectable diabetes medications are acceptable at each stage of CKD (as defined by the glomerular filtration rate [GFR]) and how to simplify dosing.
On the next page: Staging CKD >>
STAGING CHRONIC KIDNEY DISEASE
The increasing incidence of CKD was first recognized in the 1990s. The need to care for the patient with CKD was understood, but there was a dearth of standardized management practices. Research was hindered by the many names used to identify kidney disease in the literature: from mildly elevated serum creatinine to low clearance to renal insufficiency.7 To rectify this, the National Kidney Foundation (NKF) convened an expert panel to develop a standard language and units of measurement to define kidney disease. The panel was tasked with developing classifications and an evaluation system for renal disease; identifying ways to attain more timely referrals to nephrology; clarifying research objectives; and delineating how to improve medication dosing. Ultimately, the main objective was to improve management of the patient with CKD.
In 2002, the NKF’s Dialysis Outcomes Quality Initiative (K/DOQI) defined the stages of kidney disease based on assessment of GFR and other kidney disease markers.8 GFR is a mathematical calculation for determining kidney function that corrects for loss of kidney function and incorporates the patient’s gender, race, age, and body size—the nonmodifiable risk factors known in 2001. GFR is expressed in mL/min/1.73 m2. Classifying patients with CKD according to the stage of kidney disease by GFR provides a mechanism for studying the efficacy and adverse reactions of medications while correlating them to each stage. This is of particular importance when treating patients with diabetes and kidney disease, as it can help to avoid adverse effects associated with inappropriate renal dosing. Many medications are renally eliminated; as kidney function diminishes, the pharmacokinetics of drugs are altered, potentially causing toxicity and an increased risk for drug interactions. As the GFR falls, even drugs not eliminated by the kidney may have altered pharmacokinetics and pharmacodynamics that may cause renal or systemic toxicity.1
Determining the precise method for renally dosing medication has been one of the most vigorously debated topics in CKD. Many medications were prescribed using the patient’s serum creatinine (SCr) concentration, a marker but not an exact measure of kidney disease.8 The Cockcroft-Gault equation, which provides an estimate of creatinine clearance (eCrCl; an alternate GFR measurement), was primarily used to determine renal drug dosing. 9
In 1999, a multisite study group worked to develop an equation to measure loss of kidney function in the patient with CKD.10,11 The new formula, known as the MDRD (Modification of Diet in Renal Disease) equation, standardized the measurement of loss of kidney function and provided an estimated GFR (eGFR) that was more accurate then eCrCl. At that time, the National Kidney Disease Education Program (NKDEP) decided to use the MDRD equation to assess renal function and use the eCrCl for dosing medications. The latter was chosen because most drugs were analyzed using eCrCl.9 More recently, the NKDEP stated that either the eCrCl or the eGFR is acceptable for renal drug dosing. Hudson and Nyman9 advise that when discrepancies occur between eGFR and eCrCl, clinical judgment should be applied in the elderly and in those with extremes in body weight. In those cases, therapeutic outcomes and risk versus benefit need to be considered when determining renal dosing.
The publication of CKD stages by K/DOQI in 2002 sparked research that has uncovered additional criteria that might be useful in slowing the progression and improving the management of CKD. Thus, in 2013, new guidelines by KDIGO incorporating the latest international findings were published.1 KDIGO expands the definition of CKD to include albuminuria (ALB) as an added risk factor (see Figure) and has modified K/DOQI stages 3 and 5. ALB was included because it has predictive value for progression of CKD to kidney failure. In the new classification, there are three ranges defined for ALB measured in mg/g (see Table 1). The KDIGO guidelines, like K/DOQI, classify kidney disease using a 1-to-5 staging system; however, the stage numbers in the KDIGO classification are prefaced by a G (ie, G1, G2). Also, as mentioned above, stages G3 and G5 in the KDIGO classification have been further categorized.
CKD stage 3 was divided for practical reasons. It was found that the original CKD stage 3 was too large and diverse a classification to use for research studies, tracking of hospitalizations, and precise dosing of medications.12 K/DOQI stage 3 encompassed a GFR from 30 to 59 mL/min/1.73 m2. The KDIGO guideline divides K/DOQI stage 3 into G3a (GFR of 45 to 59 mL/min/1.73 m2) and G3b (GFR of 30 to 44 mL/min/1.73 m2). The staging is then further classified by the presence or absence of ALB. The patient with a GFR of 56 mL/min/1.73 m2 with ALB is in CKD stage 3aA3 (very high and nephrotic ALB) and has a greater risk for disease progression than the patient with a GFR of 56 mL/min/1.73 m2 without ALB. The latter patient is in CKD stage 3aA1(optimal and mildly high ALB). Finally, many medication dosages that were acceptable at a GFR of 56 mL/min/1.73 m2 had to be further adjusted for patients with a GFR of 30 mL/min/1.73 m2 to avoid toxicity. This further justified the KDIGO reclassification of stage 3.
Stage 5 from K/DOQI only reflected a GFR < 15 mL/min/1.73 m2. KDIGO utilizes the same GFR but splits the stage into patients not receiving dialysis and those receiving dialysis, G5 and G5D, respectively. The dosing and timing of medications due to drug dialyzability—including renal-specific medications—may be quite different for patients on dialysis as compared to those with a GFR < 15 mL/min/1.73 m2 not on dialysis, making the distinction beneficial.
On the next page: Diabetes management >>
DIABETES MANAGEMENT
The treatment of patients early in diabetic CKD is often the responsibility of primary care providers, who are faced with the daunting task of addressing the renal dosing of medications. The eGFR or eCrCl needs to be utilized to adjust diabetic medications, largely to avoid hypoglycemia. To alleviate some of the difficulty, the KDIGO CKD stage and the eCrCl and eGFR for diabetic medications are delineated in Table 2.1
Note that the guidelines are not meant to be strictly adhered to but rather are intended as a tool for managing the patient with CKD. Clinically significant patient factors need to be considered to adequately adjust drugs in CKD. Also, as new data become available from postmarketing reports, dosing adjustments, added precautions, or updated risk factors in these fragile patients may need to be incorporated into the guidelines. As new diabetic medications are approved by the FDA, kidney-specific dosing will continue to be a moving target.
While making lifestyle changes is very important early in diabetes, by the time diabetic nephropathy manifests, more aggressive action is warranted.12 A part of the diabetic microvascular triad (along with retinopathy and neuropathy), nephropathy signals existing and irreversible organ damage. Before instituting treatment to delay diabetic nephropathy progression, an A1C goal needs to be established. The long-standing goal has been an A1C < 7%, according to guidelines established by the American Diabetes Association, or 6.5%, as recognized by the American Association of Clinical Endocrinologists.13 The Diabetes Control and Complications Trial (DCCT) demonstrated microvascular benefit of tight glycemic control in type 1 diabetic patients when the A1C was maintained at an average of 7.2% (versus 9.1% in conventional therapy).14 The United Kingdom Prospective Diabetes Study similarly demonstrated microvascular benefit with intensive blood glucose control in type 2 diabetic patients who were maintained at a median A1C of 7% compared to the conventional treatment group maintained at an A1C of 7.9%.15
In January 2013, Perkovic and colleagues, in a post hoc analysis of the ADVANCE Trial, showed that maintaining an A1C between 6.5% and 7% in a patient with diabetes and macroalbuminuria (UACR > 300 μg/mg) slows progression to kidney failure.16 The time needed to treat with intensive glucose control was five years, and the number needed to treat was 41. Thus, for every 41 patients with diabetes, CKD, and ALB whose A1C is below 7%, one will not progress to kidney failure in five years of treatment.
The five-year lead time is troublesome, however. It discourages many patients from taking steps to achieve strict glycemic control; a significant number fail to follow their diabetic diets or take their medications until the damage is done. For the elderly kidney patient with diabetes and multiple other comorbidities, aggressively managing diabetes may result in hypoglycemia.12 On balance, the slight loss of kidney function five years hence is less problematic than the present risk for a fall and resulting hip fracture due to hypoglycemia. For this reason, the KDOQI glycemic guidelines suggest an A1C between 7% to 7.5% as the goal for those with significant comorbidities.12
On the next page: Diabetic medication dosing for CKD >>
DIABETIC MEDICATION DOSING FOR CKD
Glycemic control is important in delaying the progression of kidney failure in the patient with CKD. Hypoglycemic medications by definition may cause low blood glucose levels. This is of particular concern in patients with diminishing renal function, particularly the elderly and CKD patients. Antidiabetes drugs for patients in CKD stages 1 and 2 have few renal precautions, although care must be taken with metformin (see Table 3). Metformin is the drug of choice for the patient newly diagnosed with diabetes. It is inexpensive and effective, causes hypoglycemia only with intensive exercise, is taken orally, and is generally well tolerated. However, the package insert (PI) indicates stopping it in patients whose kidney disease has progressed to a GFR < 60 mL/min/1.73 m2 because it causes an increased risk for lactic acidosis.17,18
Metformin was developed prior to 1998, when the approval of the dosage regimen was tied to SCr levels rather than the presently accepted eGFR.19 As a result, the PI states that metformin should not be used in women with an SCr > 1.4 mg/dL or in men with an SCr > 1.5 mg/dL.17 However, as noted, SCr concentration alone is a very poor indicator of kidney disease (see Table 4).
Currently, the KDIGO guidelines recommend assessing the GFR for metformin dosing. When the GFR is < 60 mL/min/1.73 m2, KDIGO recommends metformin be discontinued in patients with low muscle mass or serious concurrent illness, or those who are concurrently receiving renally eliminated or nephrotoxic drugs (this includes, but is not limited to, renin-angiotensin-aldosterone system [RAAS] inhibitors, NSAIDs, and diuretics).1 In those patients who do not have the exclusions delineated above, KDIGO recommends that metformin may be continued when the GFR is > 45 mL/min/1.73 m2 (stages G1 to G3a), closely monitored and reconsidered when the GFR is 30 to 44 mL/min/1.73 m2 (stage G3b), and discontinued when the GFR is < 30 mL/min/1.73 m2 (stages G4 to G5). The KDIGO differs from other sources that recommend metformin be stopped when the GFR is < 50 to 70 ml/min/1.73 m2.20-22 The metformin PI also recommends stopping metformin the day of or day before and two days after procedures in which radiographic iodinated contrast dye is administered, to avoid acute kidney failure. Finally, it is important to note that any combination medication formulated with metformin should be stopped when the GFR is 50 mL/min/1.73 m2 because lactic acidosis has also been seen in patients taking these medications.21
At stage 3a (GFR, 45 to 60 mL/min/1.73 m2), the sulfonylureas require dosing changes (see Table 2). Glipizide is often the oral medication of choice during CKD, and it may be used in all stages, including dialysis, but does require renal dosing and monitoring.23 Although the insulins are more effective than glipizide in glycemic control, many practitioners prescribe it for patients who will not accept an injectable medication. Glyburide is not used at this stage, as it can lead to complications in patients with a GFR < 60 mL/min/1.73 m2, particularly hypoglycemia.
Like the sulfonylureas, the meglitinides enhance insulin secretion.13,18 Repaglinide and nateglinide are fast-acting and need to be taken with meals. Because it is recommended that the dose of the meglitinides be omitted when a meal is skipped, they are good for patients who eat meals irregularly. The meglitinides require initiation at a low dose in severe renal insufficiency.
The thiazolidinediones (TDZs) are used with caution in patients with CKD, not only because of controversial adverse cardiac effects, but also for peripheral edema that interferes with CKD management.18 The TDZs (rosiglitazone and pioglitazone) require no dose adjustment and appear to have a beneficial effect in obese patients and in patients who experienced weight gain with other hypoglycemic agents.
The newest of the diabetic drugs, canagliflozin, requires a working kidney to be effective.24,25 It is used with precaution when GFR is 45 to 59 mL/min/1.73 m2 at stage G3a and is contraindicated when GFR is < 45 mL/min/1.73 m2 in stage G3b. Canagliflozin's effects are felt in early segment of proximal convoluted tubule where it blocks sodium-glucose co-transporter 2, thereby lowering reabsorption of glucose and increasing excretion of glucose in the urine. In practical terms, it can affect kidney function and, due to the inhibition of the glucose-sodium pathway, induce hyperkalemia.25 Monitoring of both the GFR and potassium is vital when administering this drug. As data from postmarketing reports come in and practitioners gain experience, use of canaglifozin in the patient with CKD will be further delineated.
At a GFR between 30 and 45 mL/min/1.73 m2, CKD stage 3b, alpha-glucosidase inhibitors acarbose and miglitol are contraindicated. The GLP-1 mimetic liraglutide needs to be avoided when the GFR is < 60 mL/min/1.73 m2 (stage G3a). While exenatide requires close monitoring and caution when the GFR is < 50 mL/min/1.73 m2, it needs to be discontinued at a GFR
< 30 mL/min/1.73 m2 (stage 4). DPP-4 inhibitors become problematic at a GFR < 60 mL/min/
1.73 m2 beginning at stage G3a. All except the DPP-4 inhibitor linagliptin require dosing changes as the loss of GFR continues. Exact dosing adjustments for the drug categories are found in Table 2.
In stage 4 CKD (GFR, 15 to 30 mL/min/1.73 m2), more medications are eliminated from consideration. The amylinomimetic pramlintide cannot be used for a patient with a GFR < 15 mL/min/1.73 m2 (see Table 2). Both GLP-1 mimetics, exenatide and liraglutide, must be stopped when the GFR drops to 30 mL/min/1.73 m2. DDP-4 inhibitors need to be dosed according to the GFR but are still acceptable at CKD stage 4. Ideally, patients with diabetes should be referred to nephrology at stage 3, but referral is vital by stage 4.
Although oral medications are not as commonly prescribed as insulins at CKD stages 4 and 5, in stage 5 CKD (GFR, < 15 mL/min/1.73 m2), a limited number of oral diabetic agents are available (see Table 2). Glipizide is inexpensive and generic; therefore, it is the most commonly used oral medication in this stage. A recent study found linagliptin was safe to use throughout all stages of kidney disease without adjustment for GFR.26 While nateglinide is still acceptable in stage 5, it is used less frequently than glipizide.
As the kidney fails, the need for diabetic medications also decreases and dosing needs to take into account any residual renal function. Because glucose is produced in the proximal tubules of the kidney (known as gluconeogenesis), the level of glucose falls as the kidney fails. Excretion of insulin and other diabetes medications decreases, and thus the amount of insulin remaining in circulation is increased. This, in combination with the reduced glucose, increases the patient’s risk for hypoglycemia.13 Although protocols for medication adjustment are tied to GFR levels, close follow-up by the practitioner is required because GFR is not a reliable measure of kidney function when creatinine levels change rapidly.27
On the next page: Insulins >>
INSULINS
Many patients start insulin prior to reaching CKD stage 4 or 5 because it is the best choice for managing diabetes through all stages of CKD.28 Insulin can slow the progression to kidney failure by providing better A1C control, and all patients should be encouraged to start insulin as soon as it is appropriate.29 Many patients are very reluctant to use injectables, but by stage 5 they are willing to use insulin if it will slow progression of disease and help prevent the need for dialysis.2
The long-acting basal insulins (detemir and glargine) or a combination of basal and oral medication can work quite well at stage 5. Many patients will require a low dose of the long-acting basal insulins and short-acting insulin with meals. As the kidney fails, the short-acting doses can often be discontinued.29 A protocol that mixes long- and short-acting insulin with the largest meal is very effective at this stage. The long-acting insulin dose will need to be decreased as the kidney continues to fail. The American College of Physicians protocol for insulin suggests a 25% decrease at stage 4 and a 50% decrease at stage 5. Each patient is different, however, and dosing must be determined individually.23
On the next page: Patient education >>
PATIENT EDUCATION
There is a correlation between patients who participate in kidney disease education programs and a decrease in the progression of kidney disease (see “Case Study: The Recalcitrant Patient With Diabetes"). These classes can be taught by a physician, physician assistant, nurse practitioner, or clinical nurse specialist. To alleviate the burden of end-stage kidney disease in the US, Medicare Part B pays for six outpatient kidney disease patient education classes per lifetime of the beneficiary.30
Since 2010, when kidney disease education programs were rolled out, over 10,000 classes have been taught.31 Many practitioners report that patients are more compliant and understand their disease better when they attend classes.32 Data are being gathered to determine the effectiveness of the classes on patient outcomes.
On the next page: Conclusion >>
CONCLUSION
Knowing how to manage the patient with both diabetes and kidney disease is increasingly vital as this patient population grows. Much of the management of these patients will fall to the primary care practitioner.6 The most effective way to start treatment is to identify which CKD stage the patient fits into.
Once the patient is properly categorized, safe and effective diabetic medications can be selected and dosed according to stage. Although the new classification system may be difficult to incorporate into some electronic medical record systems and practitioner behavior, ultimately, it will allow safer management of the patient with CKD and a better predictive power of outcome.
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Suppl. 2013;3:1-150.
2. Brazie M. Finding the sweet spot: trouble-shooting diabetic dilemmas. Oral presentation at NKF Spring Clinical Meetings; May 2012, Washington, DC.
3. Estimated cost of diabetes care $245 billion in U.S. in 2012. Renal & Urology News. March 9, 2013. www.renalandurologynews.com/estimated-cost-of-diabetes-245-billion-in-us-in-2012/article/283616/. Accessed January 17, 2013.
4. Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3-5 in the United States. Am J Kidney Dis. 2013;62(2):245-252.
5. Greenberg A, ed. National Kidney Foundation Primer of Kidney Diseases. 5th ed. Philadelphia, PA: Saunders Elsevier; 2009.
6. Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research study. Ann Fam Med. 2006;4(1):23-31.
7. Hsu CY, Chertow GM. Chronic renal confusion: insufficiency, failure, dysfunction, or disease. Am J Kidney Dis. 2000;49(3):482-496.
8. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification and stratification. Am J Kidney Dis. 2002;39(suppl 1):S1-S266.
9. Hudson JQ, Nyman HA. Use of the estimated glomerular filtration rate for drug dosing in the chronic kidney disease patient. Curr Opin Nephrol Hypertens. 2011;20:482-491.
10. Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461-470.
11. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612.
12. NKF-KDOQI clinical practice guideline for diabetes and CKD, guideline 2: management of hyperglycemia and general diabetes care in CKD. Am J Kidney Dis. 2012;60(5):850-886. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed January 2, 2014.
13. Abe M, Okada K, Soma M. Antidiabetic agents in patients with chronic kidney disease and end-stage renal disease on dialysis: metabolism and clinical practice. Curr Drug Metab. 2011;12(1):57-69.
14. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381-389.
15. Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405-411.
16. Perkovic V, Heerspink HL, Chalmers J, et al. Intensive glucose control improves kidney outcomes in patients with type 2 diabetes. Kidney Int. 2013;83(3):517-23.
17. Metformin [package insert]. US Department of Health and Human Services, US Food and Drug Administration. www.fda.gov/ohrms/dockets/dailys/02/May02/053102/800471e6.pdf. Accessed January 17, 2013.
18. Triplitt CL, Reasner CA. Chapter 83. Diabetes Mellitus. In: Wells BG, ed. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill; 2011.
19. FDA. Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulato ryInformation/Guidances/ucm072127.pdf. Accessed January 17, 2013.
20. Lacy CF, Armstrong LL, Goldman MP, Lance LL. Drug Information Handbook. 20th ed. Hudson, OH: Lexi-Comp, Inc.; 2011.
21. Rocha A, Almeida M, Santos J, Carvalho A. Metformin in patients with chronic kidney disease: strengths and weaknesses. J Nephrol. 2013; 26(10):55-60.
22. Gilbert SJ, Weiner DE, eds. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2014.
23. Aronoff GR, Bennett WM, Berns JS, et al. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults and Children. 5th ed. Philadelphia, PA: American College of Physicians; 2007.
24. Valentine V. The role of the kidney and the sodium-glucose co-transporter inhibition in diabetes management. Clin Diabetes. 2012;30(4):151-155.
25. Invokana [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2013.
26. McGill JB, Sloan L, Newman J, et al. Long-term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment. Diabetes Care. 2013;36(2):237-244.
27. National Kidney Disease and Education Program (NKDEP). Estimated glomerular filtration rate (eGFR) info sheet. NIH publication No.10-6286, March 2010.
28. Thummel K, Shen D, Isoherranen N, et al. Design and optimization of dosage regimens: pharmacokinetic data. In: Hardman J, Limbird L, Goodman G (eds). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006.
29. Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial. 2004;17(5):365-70.
30. Centers for Medicare and Medicaid Services. Medicare program: revisions to payment policies under the Physician Fee Schedule, Clinical Laboratory Fee Schedule & other revisions to Part B for CY 2014; Final Rule. Federal Register. 2009;74(226):61738-62188. www.gpo.gov/fdsys/pkg/FR-2009-11-25/html/E9-26502.htm. Accessed January 17, 2013.
31. Zuber K, Davis J. Kidney disease education: a niche for PAs and NPs. JAAPA. 2013;26(7):42-47.
32. Zuber K, Davis J. Stories from the trenches: the first year experience with kidney disease education. Nephrol News Issues. 2012;26(2):20-21.
1. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Suppl. 2013;3:1-150.
2. Brazie M. Finding the sweet spot: trouble-shooting diabetic dilemmas. Oral presentation at NKF Spring Clinical Meetings; May 2012, Washington, DC.
3. Estimated cost of diabetes care $245 billion in U.S. in 2012. Renal & Urology News. March 9, 2013. www.renalandurologynews.com/estimated-cost-of-diabetes-245-billion-in-us-in-2012/article/283616/. Accessed January 17, 2013.
4. Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3-5 in the United States. Am J Kidney Dis. 2013;62(2):245-252.
5. Greenberg A, ed. National Kidney Foundation Primer of Kidney Diseases. 5th ed. Philadelphia, PA: Saunders Elsevier; 2009.
6. Spann SJ, Nutting PA, Galliher JM, et al. Management of type 2 diabetes in the primary care setting: a practice-based research study. Ann Fam Med. 2006;4(1):23-31.
7. Hsu CY, Chertow GM. Chronic renal confusion: insufficiency, failure, dysfunction, or disease. Am J Kidney Dis. 2000;49(3):482-496.
8. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification and stratification. Am J Kidney Dis. 2002;39(suppl 1):S1-S266.
9. Hudson JQ, Nyman HA. Use of the estimated glomerular filtration rate for drug dosing in the chronic kidney disease patient. Curr Opin Nephrol Hypertens. 2011;20:482-491.
10. Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461-470.
11. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612.
12. NKF-KDOQI clinical practice guideline for diabetes and CKD, guideline 2: management of hyperglycemia and general diabetes care in CKD. Am J Kidney Dis. 2012;60(5):850-886. www.kidney.org/professionals/kdoqi/guideline_diabetes/guide2.htm. Accessed January 2, 2014.
13. Abe M, Okada K, Soma M. Antidiabetic agents in patients with chronic kidney disease and end-stage renal disease on dialysis: metabolism and clinical practice. Curr Drug Metab. 2011;12(1):57-69.
14. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381-389.
15. Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405-411.
16. Perkovic V, Heerspink HL, Chalmers J, et al. Intensive glucose control improves kidney outcomes in patients with type 2 diabetes. Kidney Int. 2013;83(3):517-23.
17. Metformin [package insert]. US Department of Health and Human Services, US Food and Drug Administration. www.fda.gov/ohrms/dockets/dailys/02/May02/053102/800471e6.pdf. Accessed January 17, 2013.
18. Triplitt CL, Reasner CA. Chapter 83. Diabetes Mellitus. In: Wells BG, ed. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill; 2011.
19. FDA. Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulato ryInformation/Guidances/ucm072127.pdf. Accessed January 17, 2013.
20. Lacy CF, Armstrong LL, Goldman MP, Lance LL. Drug Information Handbook. 20th ed. Hudson, OH: Lexi-Comp, Inc.; 2011.
21. Rocha A, Almeida M, Santos J, Carvalho A. Metformin in patients with chronic kidney disease: strengths and weaknesses. J Nephrol. 2013; 26(10):55-60.
22. Gilbert SJ, Weiner DE, eds. National Kidney Foundation Primer on Kidney Diseases. 6th ed. Philadelphia, PA: Elsevier Saunders; 2014.
23. Aronoff GR, Bennett WM, Berns JS, et al. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults and Children. 5th ed. Philadelphia, PA: American College of Physicians; 2007.
24. Valentine V. The role of the kidney and the sodium-glucose co-transporter inhibition in diabetes management. Clin Diabetes. 2012;30(4):151-155.
25. Invokana [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2013.
26. McGill JB, Sloan L, Newman J, et al. Long-term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment. Diabetes Care. 2013;36(2):237-244.
27. National Kidney Disease and Education Program (NKDEP). Estimated glomerular filtration rate (eGFR) info sheet. NIH publication No.10-6286, March 2010.
28. Thummel K, Shen D, Isoherranen N, et al. Design and optimization of dosage regimens: pharmacokinetic data. In: Hardman J, Limbird L, Goodman G (eds). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006.
29. Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial. 2004;17(5):365-70.
30. Centers for Medicare and Medicaid Services. Medicare program: revisions to payment policies under the Physician Fee Schedule, Clinical Laboratory Fee Schedule & other revisions to Part B for CY 2014; Final Rule. Federal Register. 2009;74(226):61738-62188. www.gpo.gov/fdsys/pkg/FR-2009-11-25/html/E9-26502.htm. Accessed January 17, 2013.
31. Zuber K, Davis J. Kidney disease education: a niche for PAs and NPs. JAAPA. 2013;26(7):42-47.
32. Zuber K, Davis J. Stories from the trenches: the first year experience with kidney disease education. Nephrol News Issues. 2012;26(2):20-21.
Irritable Bowel Syndrome: Evidence-based Treatment
CE/CME No: CR-1401
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the criteria for diagnosis of irritable bowel syndrome (IBS).
• List three dietary interventions that may reduce symptoms of IBS.
• Discuss the role of probiotics in the treatment of IBS.
• List three classes of prescription medication that may reduce symptoms of IBS.
FACULTY
Suzanne Martin is an Assistant Professor at the University of Utah College of Nursing and works as a family nurse practitioner at the University of Utah Student Health Center in Salt Lake City.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Irritable bowel syndrome (IBS), a functional gastrointestinal disorder usually manifesting with abdominal pain and altered bowel movements, is often seen in primary care. With the recent advances in evidence-based knowledge, you can now more readily make a diagnosis and offer your patients with IBS a variety of treatment options tailored to their needs.
Irritable bowel syndrome (IBS) is the most common gastrointestinal complaint seen in both primary and gastroenterology clinics.1 Direct and indirect costs associated with IBS total more than $20 billion. Studies have shown that patients with IBS consume 50% more health care resources than matched controls. The prevalence of IBS ranges from less than 1% to more than 20%. Using strict criteria, the pooled prevalence rate of IBS in North America is 7%.1
IBS occurs more commonly in women and lower socioeconomic populations and is more likely to be diagnosed before age 50.1 Individuals with IBS report diminished health-related quality of life (HRQOL) scores compared to those without the disease. In some cases, decreased HRQOL can be severe, resulting in increased risk for suicidal behavior.1
The pathogenesis of IBS is not entirely understood. Contributing factors may include impaired gastrointestinal motility, visceral hypersensitivity, increased mucosal permeability, carbohydrate malabsorption, bacterial overgrowth, altered brain-gut axis, altered intestinal microbiota, psychosocial disturbances, and genetic predisposition.1,2
CLINICAL PRESENTATION
Hallmark symptoms of IBS are abdominal pain, dyspepsia, and altered bowel movements. The abdominal pain of IBS can be located anywhere and can range in severity from “annoying” to “debilitating” (see “Living with IBS,”). Eating and stress may aggravate pain, and a bowel movement may attenuate it. The pain is usually intermittent throughout the day; nocturnal pain is unusual and suggests an alternate cause.3

Dyspepsia (epigastric discomfort, postprandial fullness, early satiety) can occur in up to 87% of patients with IBS.4 Other features associated with IBS include gastroesophageal reflux disease nausea; noncardiac chest pain; bloating, belching and/or flatulence; sexual dysfunction; dyspareunia; urinary frequency and urgency; and fibromyalgia.
The patient with IBS will also experience a change in the type of bowel movement. Therefore, IBS is often categorized according to the predominant change: diarrhea (known as IBS-D), constipation (IBS-C), and alternating diarrhea and constipation (IBS-M). IBS-D is defined by diarrhea that is typically small to moderate in volume, occurs after meals or in the morning, and is associated with mucus 50% of the time.4 IBS-C is characterized by constipation with stools that are typically hard and pelletlike, accompanied by straining, with fewer than three bowel movements per week.
Patients with IBS may describe a sense of incomplete evacuation.4 Alarm signs and symptoms, which include progressive symptoms; nocturnal symptoms; weight loss, malnutrition, and anorexia; stools that are large in volume, bloody, or greasy; anemia, electrolyte disturbance, and elevated inflammatory markers; or a family history of inflammatory bowel disease, colorectal cancer, or celiac disease, should prompt evaluation for alternate etiologies.1,4
Continued on next page >>
DIAGNOSIS
Work-up
Although the patient with IBS may present with mild nonspecific abdominal pain, the physical exam is typically normal.4,5 Patients who meet the Rome III diagnostic criteria—in the absence of alarm signs and symptoms or worrisome family history—are candidates for a clinical diagnosis of IBS.6 These criteria, originally developed for research purposes but useful in a clinical setting, include
• Symptom onset at least six months prior to diagnosis
• Recurrent abdominal pain or discomfort for more than three days per month during the past three months, and at least two of the following features:
• Improvement of symptoms with defecation
• A change in stool frequency
• A change in stool form.1,4,6
Due to inherent difficulty in establishing the accuracy of such criteria, a simple and practical alternative clinical definition is suggested by the American College of Gastroenterology Task Force on IBS: IBS is characterized by abdominal pain or discomfort that occurs in association with altered bowel habits over a period of at least three months.1 Patients meeting clinical criteria without alarm symptoms or worrisome family history require no additional testing.1
Moderate evidence supports routine testing for celiac disease in individuals presenting with symptoms consistent with IBS-D or IBS-M.1 Lactose hydrogen breath testing is recommended for individuals reporting symptoms that suggest a correlation between the ingestion of lactose and onset of IBS symptoms.1 When a patient presents with alarm features, significant family history, refractory symptoms, or new concerning symptoms, further testing is indicated, with test selection based on specific symptoms and risk factors.1,4,5
Differential Diagnosis
In addition to IBS, there are numerous other conditions spanning several clinical categories that should be considered in patients who present with symptoms of IBS (see Table 1). In the cardiogenic group, abdominal angina, mesenteric artery thrombosis, and mesenteric artery venous thrombosis share common symptoms (eg, sudden stomach pain) with IBS, which may confuse the diagnostic picture. Gastrointestinal disorders such as bacterial overgrowth, celiac disease, gastroenteritis, inflammatory bowel disease, and anorectal dysfunction (incontinence) should be considered in patients who present with IBS symptoms. Pancreatic, ovarian, and colorectal cancer should also be investigated as a possible diagnosis.4,5,7
Continued on next page >>
TREATMENT
Evidence supports a number of therapeutic options, both lifestyle and pharmacologic (see Table 2). Lifestyle options include comprehensive patient education, dietary manipulation, cognitive behavioral therapy (CBT), and exercise. Pharmacologic options include antispasmodic agents, antidepressants, antidiarrheal agents, osmotic laxatives, alosetron, lubiprostone, rifaximin, and linaclotide.

Lifestyle changes
A therapeutic relationship that includes a nonjudgmental attitude, realistic expectations, and patient-directed care is the cornerstone of successful nonpharmacologic IBS management.4 In addition, patient education should emphasize management instead of cure, and the patient should be informed that IBS may wax and wane over a lifetime but will not shorten his/her lifespan.4
A six-week, single-blind, randomized trial (N = 262) attempted to assess the impact of patient–provider interaction on IBS symptoms and HRQOL. Participants were assigned to one of three groups: waiting list, limited intervention, and augmented intervention, with the last group receiving the greatest amount of education and patient–provider interaction. After three and six weeks, participants in the augmented group reported significant improvements in global IBS symptoms and HRQOL scores.8 Although these findings are limited by a short follow-up period, they underscore the importance of a strong therapeutic relationship in a successful outcome.
Diet. Since food sensitivities—to lactose; gluten; fiber; fermentable oligosaccharides, disaccharides, and monosaccharides, and polyols (FODMAP); and carbohydrates—are thought to be integral to the etiology of IBS, dietary manipulation plays a major role in managing symptoms.4
Due to overlapping symptoms, a lactose-restricted diet may be appropriate in a subset of IBS-D and IBS-M patients. A prospective clinical trial enrolled 70 adult participants with IBS, 24% of whom were lactose intolerant. All participants were given a lactose-restricted diet for six weeks, and a significant improvement of IBS symptoms in the lactose-intolerant subgroup was observed. This subgroup was advised to continue a lactose-restricted diet and was then followed for five years. Of the 14 participants who remained in the study for the five years, none reported IBS complaints.9 While limited by its small sample size, this study supports the ACG recommendation to consider lactose hydrogen breath testing to screen for lactose-intolerance in a select group of patients with IBS symptoms.
Gluten. It has been observed that gluten withdrawal may improve IBS symptoms. A recent four-week randomized controlled trial (RCT; N = 45) evaluated the effect of gluten restriction on subjective markers including stool frequency, form, and ease of passage, and on objective markers related to intestinal function in adults with known IBS-D who did not have gluten sensitivity or celiac disease.10 Participants on the gluten-restricted diet reported a significant reduction in stool frequency, especially among those with HLA DQ2 and DQ8 genotypes. There was no effect on stool form or ease of passage.10
An RCT conducted by Biesiekierski and colleagues enrolled 30 patients with IBS who reported symptom control on a gluten-free diet.11 Study participants were randomly assigned to either a continued gluten-free diet or to a gluten-containing diet. Over the six-week
follow-up period, significantly more participants on a gluten-containing diet reported inadequate control of IBS symptoms.11 The small sample size and short follow-up period led the researchers to conclude that additional studies are needed to confirm whether gluten restriction attenuates IBS symptoms.
Fiber. Data are inconclusive regarding efficacy of fiber in reducing IBS symptoms. A subgroup of patients with IBS report increased abdominal pain and bloating when increasing their fiber intake.1 This was supported by a systematic review that found no clinical improvement in IBS symptoms with bulking agents.12 That said, patients with IBS-C may benefit from a trial of dietary fiber, which should be introduced at a low dose (eg, 1/2 to 1 Tb of wheat bran or psyllium daily) and titrated up as needed and tolerated.1
FODMAP. A number of clinical trials have suggested that there is a benefit to consuming a diet low in FODMAP foods (see chart). Shepherd and colleagues conducted an RCT (N = 25) that compared the effects of high- and low-FODMAP diets on IBS symptoms over a two-week period. Of participants in the high-FODMAP group, 79% reported inadequate IBS symptom control, while only 14% in the low-FODMAP group reported inadequate IBS symptom control.13
In a second trial (N = 30) that had similar objectives, symptoms worsened significantly on a
high-FODMAP diet compared to a low one among the IBS participants. Healthy participants experienced more flatulence on the high-FODMAP diet compared to those on a low one.14
Carbohydrates. In a small clinical trial (N = 17), IBS-D participants consumed a standard diet for two weeks followed by a very low carbohydrate diet (< 20 g/d) for four weeks. They were assessed weekly for adequate relief of IBS symptoms. For purposes of the study, a patient was considered a responder if he/she experienced adequate relief of IBS symptoms during at least two of the four treatment weeks. In addition to symptom relief, secondary measures included abdominal pain, stool frequency, stool consistency, and quality of life.15
Of the 13 participants who completed the study, all were considered responders; 10 of 13 (77%) reported adequate relief of IBS symptoms for four out of four treatment weeks. There were significant improvements in abdominal pain, stool frequency, stool consistency, and quality of life during treatment weeks.15 Additional trials with larger sample sizes and longer follow-up periods are needed to confirm these preliminary findings.
Exercise. Regular exercise not only offers general health benefits but may also simultaneously reduce IBS symptoms. An RCT (N =102) assigned patients with IBS either to their normal daily activity or to 20 to 60 minutes of moderate-to-vigorous exercise three to five days a week. After 12 weeks, the exercise group showed decreased IBS symptom severity. They were also significantly less likely to report worsening IBS symptoms.16
Cognitive behavioral therapy. CBT may be helpful in reducing IBS symptoms. In an RCT (N = 75) that assigned patients with IBS to conventional CBT, self-administered CBT, or a control group, IBS symptoms improved significantly in those assigned to both CBT groups.17 A systematic review and meta-analysis reported a relative risk (RR) of persistent IBS symptoms with CBT of 0.67 and a number needed to treat (NNT) of 4.18
Continued on next page >>
Pharmacologic Treatment
Prescription and OTC medications serve an adjunctive role in the management of IBS.1 Efficacy trials in the IBS population are limited by disease heterogeneity, lack of disease markers, and high placebo response rates.19
Antispasmodics. Prescription antispasmodics, such as dicyclomine, have been shown to offer short-term relief of IBS-related abdominal pain; however, long-term outcomes are unknown. Possible adverse effects such as dizziness, dry mouth, blurred vision, and sluggishness may limit their use. Peppermint oil is considered an alternative to prescription antispasmodics and has been found to improve global IBS symptoms (RR, 2.25).12
Probiotics. A number of trials, systematic reviews, and meta-analyses have addressed the effect of probiotics on IBS symptoms. Meta-analyses found an RR of 0.72 to 0.77 for persistent IBS symptoms in patients using probiotics.20 Because probiotics can be beneficial, with relatively low cost and minimal associated risks, it is reasonable to consider a trial of probiotics, especially the specific strains offering the most promising results, such as Bifidobacterium infantis 35624 and Escherichia coli DSM 17252. Like most IBS treatments, it is unlikely that probiotics will benefit all IBS patients.20
Antidepressants. Tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) were found to be effective in reducing the risk for persistent IBS symptoms in adults (RR, 0.66; NNT, 4).12,18 TCAs can slow gastrointestinal transit time, which may be beneficial to patients with IBS-D but a drawback to those with IBS-C. SSRIs, such as fluoxetine, are especially appropriate for patients with concurrent anxiety or depression. Note that three to four weeks of treatment with antidepressants are required to see benefits.1
Others. Loperamide has been reported to significantly improve stool frequency and consistency in patients with IBS-D and IBS-M but not global IBS symptoms.1,2 Polyethylene glycol has been shown to be effective in treating adults with isolated constipation, but it was not superior to placebo in adults with IBS-C.21
A systematic review found that the 5-HT3 receptor antagonist alosetron offered clinical benefit to patients with IBS-D and IBS-M.22 However, due to reports of ischemic colitis and severe constipation, the FDA removed the drug from the US market in 2000. Ultimately, postmarketing data and patient demand brought the drug back onto the market in 2002, but it can be prescribed only with careful regulation and only for women with severe, refractory IBS-D.23,24
A locally acting chloride channel activator, lubiprostone, offers clinical benefit to patients with IBS-C and chronic constipation.25 Because of its unknown long-term effects, high expense, and lack of comparison data to other IBS-C treatments, this drug should only be given to women with severe refractory IBS-C.26
Two large multicenter RCTs found that a two-week course of rifaximin, a nonabsorbable antibiotic, reduced global IBS symptoms, especially bloating, in patients with IBS without constipation. The benefits continued through 10 weeks of follow-up.27 Prescribing rifaximin for the treatment of IBS is an off-label use and should be limited to patients with IBS-D who have not responded to currently available symptom-directed therapies. In addition, the lack of evidence for long-term benefits as well as the potential for development of antibiotic resistance should be borne in mind when using this drug.28
Linaclotide is approved for IBS-C, based on two large RCTs with 12- and 26-week follow-up periods. Treatment group participants reported substantial improvement in IBS-C symptoms. Approximately 5% of participants discontinued treatment due to diarrhea.29,30
Continued on next page >>
CONCLUSION
IBS is a commonly occurring disorder of heterogeneous nature and pathogenesis. Therefore, basing a diagnosis on the presenting symptoms is not ideal; using clinical criteria to make a determination is more accurate. Once the diagnosis is made, IBS can be categorized into subtypes according to bowel function: diarrhea, constipation, or mixed. Knowing the subtype may guide the clinician in recommending a treatment, eg, patients with IBS-D may benefit from loperamide and those with IBS-C find relief with lubiprostone. The range of treatment strategies includes making lifestyle changes (eg, diet and exercise); incorporating CBT; and prescribing neuromotility agents and probiotics.
1. Brandt LJ, Chey WD, Foxx-Orenstein AE, et al; American College of Gastroenterology Task Force on Irritable Bowel Syndrome. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(suppl 1):S1-S35.
2. Wald A. Irritable bowel syndrome—diarrhoea. Best Pract Res Clin Gastroenterol. 2012;26:573-580.
3. Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology. 2002;123:2108-2131.
4. World Gastroenterology Organization. Irritable bowel syndrome: a global perspective (2009). www.worldgastroenterology.org/assets/downloads/en/pdf/guidelines/20_irritable_bowel_syndrome.pdf. Accessed December 16, 2013.
5. Olden KW. Diagnosis of irritable bowel syndrome. Gastroenterology. 2002;122:1701-1714.
6. Engsbro AL, Begtrup LM, Kjeldsen J, et al. Patients suspected of irritable bowel syndrome—cross-sectional study exploring the sensitivity of Rome III criteria in primary care. Am J Gastroenterol. 2013;108:972-980.
7. Lehrer JK. Irritable bowel syndrome differential diagnoses. Medscape. http://emedicine.medscape.com/article/180389-overview. Accessed December 16, 2013.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336:999-1003.
9. Böhmer CJ, Tuynman HA. The effect of a lactose-restricted diet in patients with a positive lactose tolerance test, earlier diagnosed as irritable bowel syndrome: a 5-year follow-up study. Eur J Gastroenterol Hepatol. 2001; 13:941-944.
10. Vazquez-Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology. 2013;144:903-911.
11. Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol. 2011;106:508-514.
12. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;10(8):CD003460.
13. Shepherd SJ, Parker FC, Muir G, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence.Clin Gastroenterol Hepatol. 2008;6:765-771.
14. Ong DK, Mitchell SB, Barrett JS, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010;25:1366-1373.
15. Austin GL, Dalton CB, Hu Y, et al. A very low-carbohydrate diet improves symptoms and quality of life in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2009;7:706-708.
16. Johannesson E, Simrén M, Strid H, et al. Physical activity improves symptoms in irritable bowel syndrome: a randomized controlled trial. Am J Gastroenterol. 2011;106:915-922.
17. Lackner JM, Jaccard J, Krasner SS, et al. Self-administered cognitive behavior therapy for moderate to severe irritable bowel syndrome: clinical efficacy, tolerability, feasibility. Clin Gastroenterol Hepatol. 2008;6:899-906.
18. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58:367-378.
19. Jailwala J, Imperiale TF, Kroenke K. Pharmacologic treatment of the irritable bowel syndrome: a systematic review of randomized, controlled trials. Ann Intern Med. 2000;133:136-147.
20. Parkes GC, Sanderson JD, Whelan K. Treating irritable bowel syndrome with probiotics: the evidence. Proc Nutr Soc. 2010;69:187-194.
21. Awad RA, Camacho S. A randomized, double-blind, placebo-controlled trial of polyethylene glycol effects on fasting and postprandial rectal sensitivity and symptoms in hypersensitive constipation-predominant irritable bowel syndrome. Colorectal Dis. 2010;12:1131-1138.
22. Andresen V, Montori VM, Keller J, et al. Effects of 5-hydroxytryptamine (serotonin) type 3 antagonists on symptom relief and constipation in nonconstipated irritable bowel syndrome: a systematic review and meta-analysis of randomized controlled trials. Clin Gastroenterol Hepatol. 2008;6: 545-555.
23. FDA. Lotronex (alosetron hydrochloride) tablets. www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm172946.htm. Accessed December 16, 2013.
24. FDA. Lotronex (alosetron hydrochloride) information. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110450.htm. Accessed December 16, 2013.
25. Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329-341.
26. Lunsford TN, Harris LA. Lubiprostone: evaluation of the newest medication for the treatment of adult women with constipation-predominant irritable bowel syndrome. Int J Womens Health. 2010;2:361-374.
27. Pimentel M, Lembo A, Chey WD, et al; TARGET Study Group. Rifaximin therapy for patients with irritable bowel syndrome without constipation.
N Engl J Med. 2011;364:22-32.
28. Tack J. Antibiotic therapy for the irritable bowel syndrome.N Engl J Med. 2011;364:81-82.
29. Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol. 2012;107:1714-1724.
30. Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol. 2012;107:1702-1712.
31. Gibson PR, Shepherd SJ. Evidence-based dietary management of functional gastrointestinal symptoms: the FODMAP approach. J Gastroenterol Hepatol. 2010;25:252-258.
32. Magge S, Lembo A. Low-FODMAP diet for treatment of irritable bowel syndrome.Gastroenterol Hepatol. 2012;8:739-745.
CE/CME No: CR-1401
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the criteria for diagnosis of irritable bowel syndrome (IBS).
• List three dietary interventions that may reduce symptoms of IBS.
• Discuss the role of probiotics in the treatment of IBS.
• List three classes of prescription medication that may reduce symptoms of IBS.
FACULTY
Suzanne Martin is an Assistant Professor at the University of Utah College of Nursing and works as a family nurse practitioner at the University of Utah Student Health Center in Salt Lake City.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Irritable bowel syndrome (IBS), a functional gastrointestinal disorder usually manifesting with abdominal pain and altered bowel movements, is often seen in primary care. With the recent advances in evidence-based knowledge, you can now more readily make a diagnosis and offer your patients with IBS a variety of treatment options tailored to their needs.
Irritable bowel syndrome (IBS) is the most common gastrointestinal complaint seen in both primary and gastroenterology clinics.1 Direct and indirect costs associated with IBS total more than $20 billion. Studies have shown that patients with IBS consume 50% more health care resources than matched controls. The prevalence of IBS ranges from less than 1% to more than 20%. Using strict criteria, the pooled prevalence rate of IBS in North America is 7%.1
IBS occurs more commonly in women and lower socioeconomic populations and is more likely to be diagnosed before age 50.1 Individuals with IBS report diminished health-related quality of life (HRQOL) scores compared to those without the disease. In some cases, decreased HRQOL can be severe, resulting in increased risk for suicidal behavior.1
The pathogenesis of IBS is not entirely understood. Contributing factors may include impaired gastrointestinal motility, visceral hypersensitivity, increased mucosal permeability, carbohydrate malabsorption, bacterial overgrowth, altered brain-gut axis, altered intestinal microbiota, psychosocial disturbances, and genetic predisposition.1,2
CLINICAL PRESENTATION
Hallmark symptoms of IBS are abdominal pain, dyspepsia, and altered bowel movements. The abdominal pain of IBS can be located anywhere and can range in severity from “annoying” to “debilitating” (see “Living with IBS,”). Eating and stress may aggravate pain, and a bowel movement may attenuate it. The pain is usually intermittent throughout the day; nocturnal pain is unusual and suggests an alternate cause.3
Dyspepsia (epigastric discomfort, postprandial fullness, early satiety) can occur in up to 87% of patients with IBS.4 Other features associated with IBS include gastroesophageal reflux disease nausea; noncardiac chest pain; bloating, belching and/or flatulence; sexual dysfunction; dyspareunia; urinary frequency and urgency; and fibromyalgia.
The patient with IBS will also experience a change in the type of bowel movement. Therefore, IBS is often categorized according to the predominant change: diarrhea (known as IBS-D), constipation (IBS-C), and alternating diarrhea and constipation (IBS-M). IBS-D is defined by diarrhea that is typically small to moderate in volume, occurs after meals or in the morning, and is associated with mucus 50% of the time.4 IBS-C is characterized by constipation with stools that are typically hard and pelletlike, accompanied by straining, with fewer than three bowel movements per week.
Patients with IBS may describe a sense of incomplete evacuation.4 Alarm signs and symptoms, which include progressive symptoms; nocturnal symptoms; weight loss, malnutrition, and anorexia; stools that are large in volume, bloody, or greasy; anemia, electrolyte disturbance, and elevated inflammatory markers; or a family history of inflammatory bowel disease, colorectal cancer, or celiac disease, should prompt evaluation for alternate etiologies.1,4
Continued on next page >>
DIAGNOSIS
Work-up
Although the patient with IBS may present with mild nonspecific abdominal pain, the physical exam is typically normal.4,5 Patients who meet the Rome III diagnostic criteria—in the absence of alarm signs and symptoms or worrisome family history—are candidates for a clinical diagnosis of IBS.6 These criteria, originally developed for research purposes but useful in a clinical setting, include
• Symptom onset at least six months prior to diagnosis
• Recurrent abdominal pain or discomfort for more than three days per month during the past three months, and at least two of the following features:
• Improvement of symptoms with defecation
• A change in stool frequency
• A change in stool form.1,4,6
Due to inherent difficulty in establishing the accuracy of such criteria, a simple and practical alternative clinical definition is suggested by the American College of Gastroenterology Task Force on IBS: IBS is characterized by abdominal pain or discomfort that occurs in association with altered bowel habits over a period of at least three months.1 Patients meeting clinical criteria without alarm symptoms or worrisome family history require no additional testing.1
Moderate evidence supports routine testing for celiac disease in individuals presenting with symptoms consistent with IBS-D or IBS-M.1 Lactose hydrogen breath testing is recommended for individuals reporting symptoms that suggest a correlation between the ingestion of lactose and onset of IBS symptoms.1 When a patient presents with alarm features, significant family history, refractory symptoms, or new concerning symptoms, further testing is indicated, with test selection based on specific symptoms and risk factors.1,4,5
Differential Diagnosis
In addition to IBS, there are numerous other conditions spanning several clinical categories that should be considered in patients who present with symptoms of IBS (see Table 1). In the cardiogenic group, abdominal angina, mesenteric artery thrombosis, and mesenteric artery venous thrombosis share common symptoms (eg, sudden stomach pain) with IBS, which may confuse the diagnostic picture. Gastrointestinal disorders such as bacterial overgrowth, celiac disease, gastroenteritis, inflammatory bowel disease, and anorectal dysfunction (incontinence) should be considered in patients who present with IBS symptoms. Pancreatic, ovarian, and colorectal cancer should also be investigated as a possible diagnosis.4,5,7
Continued on next page >>
TREATMENT
Evidence supports a number of therapeutic options, both lifestyle and pharmacologic (see Table 2). Lifestyle options include comprehensive patient education, dietary manipulation, cognitive behavioral therapy (CBT), and exercise. Pharmacologic options include antispasmodic agents, antidepressants, antidiarrheal agents, osmotic laxatives, alosetron, lubiprostone, rifaximin, and linaclotide.
Lifestyle changes
A therapeutic relationship that includes a nonjudgmental attitude, realistic expectations, and patient-directed care is the cornerstone of successful nonpharmacologic IBS management.4 In addition, patient education should emphasize management instead of cure, and the patient should be informed that IBS may wax and wane over a lifetime but will not shorten his/her lifespan.4
A six-week, single-blind, randomized trial (N = 262) attempted to assess the impact of patient–provider interaction on IBS symptoms and HRQOL. Participants were assigned to one of three groups: waiting list, limited intervention, and augmented intervention, with the last group receiving the greatest amount of education and patient–provider interaction. After three and six weeks, participants in the augmented group reported significant improvements in global IBS symptoms and HRQOL scores.8 Although these findings are limited by a short follow-up period, they underscore the importance of a strong therapeutic relationship in a successful outcome.
Diet. Since food sensitivities—to lactose; gluten; fiber; fermentable oligosaccharides, disaccharides, and monosaccharides, and polyols (FODMAP); and carbohydrates—are thought to be integral to the etiology of IBS, dietary manipulation plays a major role in managing symptoms.4
Due to overlapping symptoms, a lactose-restricted diet may be appropriate in a subset of IBS-D and IBS-M patients. A prospective clinical trial enrolled 70 adult participants with IBS, 24% of whom were lactose intolerant. All participants were given a lactose-restricted diet for six weeks, and a significant improvement of IBS symptoms in the lactose-intolerant subgroup was observed. This subgroup was advised to continue a lactose-restricted diet and was then followed for five years. Of the 14 participants who remained in the study for the five years, none reported IBS complaints.9 While limited by its small sample size, this study supports the ACG recommendation to consider lactose hydrogen breath testing to screen for lactose-intolerance in a select group of patients with IBS symptoms.
Gluten. It has been observed that gluten withdrawal may improve IBS symptoms. A recent four-week randomized controlled trial (RCT; N = 45) evaluated the effect of gluten restriction on subjective markers including stool frequency, form, and ease of passage, and on objective markers related to intestinal function in adults with known IBS-D who did not have gluten sensitivity or celiac disease.10 Participants on the gluten-restricted diet reported a significant reduction in stool frequency, especially among those with HLA DQ2 and DQ8 genotypes. There was no effect on stool form or ease of passage.10
An RCT conducted by Biesiekierski and colleagues enrolled 30 patients with IBS who reported symptom control on a gluten-free diet.11 Study participants were randomly assigned to either a continued gluten-free diet or to a gluten-containing diet. Over the six-week
follow-up period, significantly more participants on a gluten-containing diet reported inadequate control of IBS symptoms.11 The small sample size and short follow-up period led the researchers to conclude that additional studies are needed to confirm whether gluten restriction attenuates IBS symptoms.
Fiber. Data are inconclusive regarding efficacy of fiber in reducing IBS symptoms. A subgroup of patients with IBS report increased abdominal pain and bloating when increasing their fiber intake.1 This was supported by a systematic review that found no clinical improvement in IBS symptoms with bulking agents.12 That said, patients with IBS-C may benefit from a trial of dietary fiber, which should be introduced at a low dose (eg, 1/2 to 1 Tb of wheat bran or psyllium daily) and titrated up as needed and tolerated.1
FODMAP. A number of clinical trials have suggested that there is a benefit to consuming a diet low in FODMAP foods (see chart). Shepherd and colleagues conducted an RCT (N = 25) that compared the effects of high- and low-FODMAP diets on IBS symptoms over a two-week period. Of participants in the high-FODMAP group, 79% reported inadequate IBS symptom control, while only 14% in the low-FODMAP group reported inadequate IBS symptom control.13
In a second trial (N = 30) that had similar objectives, symptoms worsened significantly on a
high-FODMAP diet compared to a low one among the IBS participants. Healthy participants experienced more flatulence on the high-FODMAP diet compared to those on a low one.14
Carbohydrates. In a small clinical trial (N = 17), IBS-D participants consumed a standard diet for two weeks followed by a very low carbohydrate diet (< 20 g/d) for four weeks. They were assessed weekly for adequate relief of IBS symptoms. For purposes of the study, a patient was considered a responder if he/she experienced adequate relief of IBS symptoms during at least two of the four treatment weeks. In addition to symptom relief, secondary measures included abdominal pain, stool frequency, stool consistency, and quality of life.15
Of the 13 participants who completed the study, all were considered responders; 10 of 13 (77%) reported adequate relief of IBS symptoms for four out of four treatment weeks. There were significant improvements in abdominal pain, stool frequency, stool consistency, and quality of life during treatment weeks.15 Additional trials with larger sample sizes and longer follow-up periods are needed to confirm these preliminary findings.
Exercise. Regular exercise not only offers general health benefits but may also simultaneously reduce IBS symptoms. An RCT (N =102) assigned patients with IBS either to their normal daily activity or to 20 to 60 minutes of moderate-to-vigorous exercise three to five days a week. After 12 weeks, the exercise group showed decreased IBS symptom severity. They were also significantly less likely to report worsening IBS symptoms.16
Cognitive behavioral therapy. CBT may be helpful in reducing IBS symptoms. In an RCT (N = 75) that assigned patients with IBS to conventional CBT, self-administered CBT, or a control group, IBS symptoms improved significantly in those assigned to both CBT groups.17 A systematic review and meta-analysis reported a relative risk (RR) of persistent IBS symptoms with CBT of 0.67 and a number needed to treat (NNT) of 4.18
Continued on next page >>
Pharmacologic Treatment
Prescription and OTC medications serve an adjunctive role in the management of IBS.1 Efficacy trials in the IBS population are limited by disease heterogeneity, lack of disease markers, and high placebo response rates.19
Antispasmodics. Prescription antispasmodics, such as dicyclomine, have been shown to offer short-term relief of IBS-related abdominal pain; however, long-term outcomes are unknown. Possible adverse effects such as dizziness, dry mouth, blurred vision, and sluggishness may limit their use. Peppermint oil is considered an alternative to prescription antispasmodics and has been found to improve global IBS symptoms (RR, 2.25).12
Probiotics. A number of trials, systematic reviews, and meta-analyses have addressed the effect of probiotics on IBS symptoms. Meta-analyses found an RR of 0.72 to 0.77 for persistent IBS symptoms in patients using probiotics.20 Because probiotics can be beneficial, with relatively low cost and minimal associated risks, it is reasonable to consider a trial of probiotics, especially the specific strains offering the most promising results, such as Bifidobacterium infantis 35624 and Escherichia coli DSM 17252. Like most IBS treatments, it is unlikely that probiotics will benefit all IBS patients.20
Antidepressants. Tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) were found to be effective in reducing the risk for persistent IBS symptoms in adults (RR, 0.66; NNT, 4).12,18 TCAs can slow gastrointestinal transit time, which may be beneficial to patients with IBS-D but a drawback to those with IBS-C. SSRIs, such as fluoxetine, are especially appropriate for patients with concurrent anxiety or depression. Note that three to four weeks of treatment with antidepressants are required to see benefits.1
Others. Loperamide has been reported to significantly improve stool frequency and consistency in patients with IBS-D and IBS-M but not global IBS symptoms.1,2 Polyethylene glycol has been shown to be effective in treating adults with isolated constipation, but it was not superior to placebo in adults with IBS-C.21
A systematic review found that the 5-HT3 receptor antagonist alosetron offered clinical benefit to patients with IBS-D and IBS-M.22 However, due to reports of ischemic colitis and severe constipation, the FDA removed the drug from the US market in 2000. Ultimately, postmarketing data and patient demand brought the drug back onto the market in 2002, but it can be prescribed only with careful regulation and only for women with severe, refractory IBS-D.23,24
A locally acting chloride channel activator, lubiprostone, offers clinical benefit to patients with IBS-C and chronic constipation.25 Because of its unknown long-term effects, high expense, and lack of comparison data to other IBS-C treatments, this drug should only be given to women with severe refractory IBS-C.26
Two large multicenter RCTs found that a two-week course of rifaximin, a nonabsorbable antibiotic, reduced global IBS symptoms, especially bloating, in patients with IBS without constipation. The benefits continued through 10 weeks of follow-up.27 Prescribing rifaximin for the treatment of IBS is an off-label use and should be limited to patients with IBS-D who have not responded to currently available symptom-directed therapies. In addition, the lack of evidence for long-term benefits as well as the potential for development of antibiotic resistance should be borne in mind when using this drug.28
Linaclotide is approved for IBS-C, based on two large RCTs with 12- and 26-week follow-up periods. Treatment group participants reported substantial improvement in IBS-C symptoms. Approximately 5% of participants discontinued treatment due to diarrhea.29,30
Continued on next page >>
CONCLUSION
IBS is a commonly occurring disorder of heterogeneous nature and pathogenesis. Therefore, basing a diagnosis on the presenting symptoms is not ideal; using clinical criteria to make a determination is more accurate. Once the diagnosis is made, IBS can be categorized into subtypes according to bowel function: diarrhea, constipation, or mixed. Knowing the subtype may guide the clinician in recommending a treatment, eg, patients with IBS-D may benefit from loperamide and those with IBS-C find relief with lubiprostone. The range of treatment strategies includes making lifestyle changes (eg, diet and exercise); incorporating CBT; and prescribing neuromotility agents and probiotics.
CE/CME No: CR-1401
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the criteria for diagnosis of irritable bowel syndrome (IBS).
• List three dietary interventions that may reduce symptoms of IBS.
• Discuss the role of probiotics in the treatment of IBS.
• List three classes of prescription medication that may reduce symptoms of IBS.
FACULTY
Suzanne Martin is an Assistant Professor at the University of Utah College of Nursing and works as a family nurse practitioner at the University of Utah Student Health Center in Salt Lake City.
The author has no financial disclosures to report.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Irritable bowel syndrome (IBS), a functional gastrointestinal disorder usually manifesting with abdominal pain and altered bowel movements, is often seen in primary care. With the recent advances in evidence-based knowledge, you can now more readily make a diagnosis and offer your patients with IBS a variety of treatment options tailored to their needs.
Irritable bowel syndrome (IBS) is the most common gastrointestinal complaint seen in both primary and gastroenterology clinics.1 Direct and indirect costs associated with IBS total more than $20 billion. Studies have shown that patients with IBS consume 50% more health care resources than matched controls. The prevalence of IBS ranges from less than 1% to more than 20%. Using strict criteria, the pooled prevalence rate of IBS in North America is 7%.1
IBS occurs more commonly in women and lower socioeconomic populations and is more likely to be diagnosed before age 50.1 Individuals with IBS report diminished health-related quality of life (HRQOL) scores compared to those without the disease. In some cases, decreased HRQOL can be severe, resulting in increased risk for suicidal behavior.1
The pathogenesis of IBS is not entirely understood. Contributing factors may include impaired gastrointestinal motility, visceral hypersensitivity, increased mucosal permeability, carbohydrate malabsorption, bacterial overgrowth, altered brain-gut axis, altered intestinal microbiota, psychosocial disturbances, and genetic predisposition.1,2
CLINICAL PRESENTATION
Hallmark symptoms of IBS are abdominal pain, dyspepsia, and altered bowel movements. The abdominal pain of IBS can be located anywhere and can range in severity from “annoying” to “debilitating” (see “Living with IBS,”). Eating and stress may aggravate pain, and a bowel movement may attenuate it. The pain is usually intermittent throughout the day; nocturnal pain is unusual and suggests an alternate cause.3
Dyspepsia (epigastric discomfort, postprandial fullness, early satiety) can occur in up to 87% of patients with IBS.4 Other features associated with IBS include gastroesophageal reflux disease nausea; noncardiac chest pain; bloating, belching and/or flatulence; sexual dysfunction; dyspareunia; urinary frequency and urgency; and fibromyalgia.
The patient with IBS will also experience a change in the type of bowel movement. Therefore, IBS is often categorized according to the predominant change: diarrhea (known as IBS-D), constipation (IBS-C), and alternating diarrhea and constipation (IBS-M). IBS-D is defined by diarrhea that is typically small to moderate in volume, occurs after meals or in the morning, and is associated with mucus 50% of the time.4 IBS-C is characterized by constipation with stools that are typically hard and pelletlike, accompanied by straining, with fewer than three bowel movements per week.
Patients with IBS may describe a sense of incomplete evacuation.4 Alarm signs and symptoms, which include progressive symptoms; nocturnal symptoms; weight loss, malnutrition, and anorexia; stools that are large in volume, bloody, or greasy; anemia, electrolyte disturbance, and elevated inflammatory markers; or a family history of inflammatory bowel disease, colorectal cancer, or celiac disease, should prompt evaluation for alternate etiologies.1,4
Continued on next page >>
DIAGNOSIS
Work-up
Although the patient with IBS may present with mild nonspecific abdominal pain, the physical exam is typically normal.4,5 Patients who meet the Rome III diagnostic criteria—in the absence of alarm signs and symptoms or worrisome family history—are candidates for a clinical diagnosis of IBS.6 These criteria, originally developed for research purposes but useful in a clinical setting, include
• Symptom onset at least six months prior to diagnosis
• Recurrent abdominal pain or discomfort for more than three days per month during the past three months, and at least two of the following features:
• Improvement of symptoms with defecation
• A change in stool frequency
• A change in stool form.1,4,6
Due to inherent difficulty in establishing the accuracy of such criteria, a simple and practical alternative clinical definition is suggested by the American College of Gastroenterology Task Force on IBS: IBS is characterized by abdominal pain or discomfort that occurs in association with altered bowel habits over a period of at least three months.1 Patients meeting clinical criteria without alarm symptoms or worrisome family history require no additional testing.1
Moderate evidence supports routine testing for celiac disease in individuals presenting with symptoms consistent with IBS-D or IBS-M.1 Lactose hydrogen breath testing is recommended for individuals reporting symptoms that suggest a correlation between the ingestion of lactose and onset of IBS symptoms.1 When a patient presents with alarm features, significant family history, refractory symptoms, or new concerning symptoms, further testing is indicated, with test selection based on specific symptoms and risk factors.1,4,5
Differential Diagnosis
In addition to IBS, there are numerous other conditions spanning several clinical categories that should be considered in patients who present with symptoms of IBS (see Table 1). In the cardiogenic group, abdominal angina, mesenteric artery thrombosis, and mesenteric artery venous thrombosis share common symptoms (eg, sudden stomach pain) with IBS, which may confuse the diagnostic picture. Gastrointestinal disorders such as bacterial overgrowth, celiac disease, gastroenteritis, inflammatory bowel disease, and anorectal dysfunction (incontinence) should be considered in patients who present with IBS symptoms. Pancreatic, ovarian, and colorectal cancer should also be investigated as a possible diagnosis.4,5,7
Continued on next page >>
TREATMENT
Evidence supports a number of therapeutic options, both lifestyle and pharmacologic (see Table 2). Lifestyle options include comprehensive patient education, dietary manipulation, cognitive behavioral therapy (CBT), and exercise. Pharmacologic options include antispasmodic agents, antidepressants, antidiarrheal agents, osmotic laxatives, alosetron, lubiprostone, rifaximin, and linaclotide.
Lifestyle changes
A therapeutic relationship that includes a nonjudgmental attitude, realistic expectations, and patient-directed care is the cornerstone of successful nonpharmacologic IBS management.4 In addition, patient education should emphasize management instead of cure, and the patient should be informed that IBS may wax and wane over a lifetime but will not shorten his/her lifespan.4
A six-week, single-blind, randomized trial (N = 262) attempted to assess the impact of patient–provider interaction on IBS symptoms and HRQOL. Participants were assigned to one of three groups: waiting list, limited intervention, and augmented intervention, with the last group receiving the greatest amount of education and patient–provider interaction. After three and six weeks, participants in the augmented group reported significant improvements in global IBS symptoms and HRQOL scores.8 Although these findings are limited by a short follow-up period, they underscore the importance of a strong therapeutic relationship in a successful outcome.
Diet. Since food sensitivities—to lactose; gluten; fiber; fermentable oligosaccharides, disaccharides, and monosaccharides, and polyols (FODMAP); and carbohydrates—are thought to be integral to the etiology of IBS, dietary manipulation plays a major role in managing symptoms.4
Due to overlapping symptoms, a lactose-restricted diet may be appropriate in a subset of IBS-D and IBS-M patients. A prospective clinical trial enrolled 70 adult participants with IBS, 24% of whom were lactose intolerant. All participants were given a lactose-restricted diet for six weeks, and a significant improvement of IBS symptoms in the lactose-intolerant subgroup was observed. This subgroup was advised to continue a lactose-restricted diet and was then followed for five years. Of the 14 participants who remained in the study for the five years, none reported IBS complaints.9 While limited by its small sample size, this study supports the ACG recommendation to consider lactose hydrogen breath testing to screen for lactose-intolerance in a select group of patients with IBS symptoms.
Gluten. It has been observed that gluten withdrawal may improve IBS symptoms. A recent four-week randomized controlled trial (RCT; N = 45) evaluated the effect of gluten restriction on subjective markers including stool frequency, form, and ease of passage, and on objective markers related to intestinal function in adults with known IBS-D who did not have gluten sensitivity or celiac disease.10 Participants on the gluten-restricted diet reported a significant reduction in stool frequency, especially among those with HLA DQ2 and DQ8 genotypes. There was no effect on stool form or ease of passage.10
An RCT conducted by Biesiekierski and colleagues enrolled 30 patients with IBS who reported symptom control on a gluten-free diet.11 Study participants were randomly assigned to either a continued gluten-free diet or to a gluten-containing diet. Over the six-week
follow-up period, significantly more participants on a gluten-containing diet reported inadequate control of IBS symptoms.11 The small sample size and short follow-up period led the researchers to conclude that additional studies are needed to confirm whether gluten restriction attenuates IBS symptoms.
Fiber. Data are inconclusive regarding efficacy of fiber in reducing IBS symptoms. A subgroup of patients with IBS report increased abdominal pain and bloating when increasing their fiber intake.1 This was supported by a systematic review that found no clinical improvement in IBS symptoms with bulking agents.12 That said, patients with IBS-C may benefit from a trial of dietary fiber, which should be introduced at a low dose (eg, 1/2 to 1 Tb of wheat bran or psyllium daily) and titrated up as needed and tolerated.1
FODMAP. A number of clinical trials have suggested that there is a benefit to consuming a diet low in FODMAP foods (see chart). Shepherd and colleagues conducted an RCT (N = 25) that compared the effects of high- and low-FODMAP diets on IBS symptoms over a two-week period. Of participants in the high-FODMAP group, 79% reported inadequate IBS symptom control, while only 14% in the low-FODMAP group reported inadequate IBS symptom control.13
In a second trial (N = 30) that had similar objectives, symptoms worsened significantly on a
high-FODMAP diet compared to a low one among the IBS participants. Healthy participants experienced more flatulence on the high-FODMAP diet compared to those on a low one.14
Carbohydrates. In a small clinical trial (N = 17), IBS-D participants consumed a standard diet for two weeks followed by a very low carbohydrate diet (< 20 g/d) for four weeks. They were assessed weekly for adequate relief of IBS symptoms. For purposes of the study, a patient was considered a responder if he/she experienced adequate relief of IBS symptoms during at least two of the four treatment weeks. In addition to symptom relief, secondary measures included abdominal pain, stool frequency, stool consistency, and quality of life.15
Of the 13 participants who completed the study, all were considered responders; 10 of 13 (77%) reported adequate relief of IBS symptoms for four out of four treatment weeks. There were significant improvements in abdominal pain, stool frequency, stool consistency, and quality of life during treatment weeks.15 Additional trials with larger sample sizes and longer follow-up periods are needed to confirm these preliminary findings.
Exercise. Regular exercise not only offers general health benefits but may also simultaneously reduce IBS symptoms. An RCT (N =102) assigned patients with IBS either to their normal daily activity or to 20 to 60 minutes of moderate-to-vigorous exercise three to five days a week. After 12 weeks, the exercise group showed decreased IBS symptom severity. They were also significantly less likely to report worsening IBS symptoms.16
Cognitive behavioral therapy. CBT may be helpful in reducing IBS symptoms. In an RCT (N = 75) that assigned patients with IBS to conventional CBT, self-administered CBT, or a control group, IBS symptoms improved significantly in those assigned to both CBT groups.17 A systematic review and meta-analysis reported a relative risk (RR) of persistent IBS symptoms with CBT of 0.67 and a number needed to treat (NNT) of 4.18
Continued on next page >>
Pharmacologic Treatment
Prescription and OTC medications serve an adjunctive role in the management of IBS.1 Efficacy trials in the IBS population are limited by disease heterogeneity, lack of disease markers, and high placebo response rates.19
Antispasmodics. Prescription antispasmodics, such as dicyclomine, have been shown to offer short-term relief of IBS-related abdominal pain; however, long-term outcomes are unknown. Possible adverse effects such as dizziness, dry mouth, blurred vision, and sluggishness may limit their use. Peppermint oil is considered an alternative to prescription antispasmodics and has been found to improve global IBS symptoms (RR, 2.25).12
Probiotics. A number of trials, systematic reviews, and meta-analyses have addressed the effect of probiotics on IBS symptoms. Meta-analyses found an RR of 0.72 to 0.77 for persistent IBS symptoms in patients using probiotics.20 Because probiotics can be beneficial, with relatively low cost and minimal associated risks, it is reasonable to consider a trial of probiotics, especially the specific strains offering the most promising results, such as Bifidobacterium infantis 35624 and Escherichia coli DSM 17252. Like most IBS treatments, it is unlikely that probiotics will benefit all IBS patients.20
Antidepressants. Tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs) were found to be effective in reducing the risk for persistent IBS symptoms in adults (RR, 0.66; NNT, 4).12,18 TCAs can slow gastrointestinal transit time, which may be beneficial to patients with IBS-D but a drawback to those with IBS-C. SSRIs, such as fluoxetine, are especially appropriate for patients with concurrent anxiety or depression. Note that three to four weeks of treatment with antidepressants are required to see benefits.1
Others. Loperamide has been reported to significantly improve stool frequency and consistency in patients with IBS-D and IBS-M but not global IBS symptoms.1,2 Polyethylene glycol has been shown to be effective in treating adults with isolated constipation, but it was not superior to placebo in adults with IBS-C.21
A systematic review found that the 5-HT3 receptor antagonist alosetron offered clinical benefit to patients with IBS-D and IBS-M.22 However, due to reports of ischemic colitis and severe constipation, the FDA removed the drug from the US market in 2000. Ultimately, postmarketing data and patient demand brought the drug back onto the market in 2002, but it can be prescribed only with careful regulation and only for women with severe, refractory IBS-D.23,24
A locally acting chloride channel activator, lubiprostone, offers clinical benefit to patients with IBS-C and chronic constipation.25 Because of its unknown long-term effects, high expense, and lack of comparison data to other IBS-C treatments, this drug should only be given to women with severe refractory IBS-C.26
Two large multicenter RCTs found that a two-week course of rifaximin, a nonabsorbable antibiotic, reduced global IBS symptoms, especially bloating, in patients with IBS without constipation. The benefits continued through 10 weeks of follow-up.27 Prescribing rifaximin for the treatment of IBS is an off-label use and should be limited to patients with IBS-D who have not responded to currently available symptom-directed therapies. In addition, the lack of evidence for long-term benefits as well as the potential for development of antibiotic resistance should be borne in mind when using this drug.28
Linaclotide is approved for IBS-C, based on two large RCTs with 12- and 26-week follow-up periods. Treatment group participants reported substantial improvement in IBS-C symptoms. Approximately 5% of participants discontinued treatment due to diarrhea.29,30
Continued on next page >>
CONCLUSION
IBS is a commonly occurring disorder of heterogeneous nature and pathogenesis. Therefore, basing a diagnosis on the presenting symptoms is not ideal; using clinical criteria to make a determination is more accurate. Once the diagnosis is made, IBS can be categorized into subtypes according to bowel function: diarrhea, constipation, or mixed. Knowing the subtype may guide the clinician in recommending a treatment, eg, patients with IBS-D may benefit from loperamide and those with IBS-C find relief with lubiprostone. The range of treatment strategies includes making lifestyle changes (eg, diet and exercise); incorporating CBT; and prescribing neuromotility agents and probiotics.
1. Brandt LJ, Chey WD, Foxx-Orenstein AE, et al; American College of Gastroenterology Task Force on Irritable Bowel Syndrome. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(suppl 1):S1-S35.
2. Wald A. Irritable bowel syndrome—diarrhoea. Best Pract Res Clin Gastroenterol. 2012;26:573-580.
3. Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology. 2002;123:2108-2131.
4. World Gastroenterology Organization. Irritable bowel syndrome: a global perspective (2009). www.worldgastroenterology.org/assets/downloads/en/pdf/guidelines/20_irritable_bowel_syndrome.pdf. Accessed December 16, 2013.
5. Olden KW. Diagnosis of irritable bowel syndrome. Gastroenterology. 2002;122:1701-1714.
6. Engsbro AL, Begtrup LM, Kjeldsen J, et al. Patients suspected of irritable bowel syndrome—cross-sectional study exploring the sensitivity of Rome III criteria in primary care. Am J Gastroenterol. 2013;108:972-980.
7. Lehrer JK. Irritable bowel syndrome differential diagnoses. Medscape. http://emedicine.medscape.com/article/180389-overview. Accessed December 16, 2013.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336:999-1003.
9. Böhmer CJ, Tuynman HA. The effect of a lactose-restricted diet in patients with a positive lactose tolerance test, earlier diagnosed as irritable bowel syndrome: a 5-year follow-up study. Eur J Gastroenterol Hepatol. 2001; 13:941-944.
10. Vazquez-Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology. 2013;144:903-911.
11. Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol. 2011;106:508-514.
12. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;10(8):CD003460.
13. Shepherd SJ, Parker FC, Muir G, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence.Clin Gastroenterol Hepatol. 2008;6:765-771.
14. Ong DK, Mitchell SB, Barrett JS, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010;25:1366-1373.
15. Austin GL, Dalton CB, Hu Y, et al. A very low-carbohydrate diet improves symptoms and quality of life in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2009;7:706-708.
16. Johannesson E, Simrén M, Strid H, et al. Physical activity improves symptoms in irritable bowel syndrome: a randomized controlled trial. Am J Gastroenterol. 2011;106:915-922.
17. Lackner JM, Jaccard J, Krasner SS, et al. Self-administered cognitive behavior therapy for moderate to severe irritable bowel syndrome: clinical efficacy, tolerability, feasibility. Clin Gastroenterol Hepatol. 2008;6:899-906.
18. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58:367-378.
19. Jailwala J, Imperiale TF, Kroenke K. Pharmacologic treatment of the irritable bowel syndrome: a systematic review of randomized, controlled trials. Ann Intern Med. 2000;133:136-147.
20. Parkes GC, Sanderson JD, Whelan K. Treating irritable bowel syndrome with probiotics: the evidence. Proc Nutr Soc. 2010;69:187-194.
21. Awad RA, Camacho S. A randomized, double-blind, placebo-controlled trial of polyethylene glycol effects on fasting and postprandial rectal sensitivity and symptoms in hypersensitive constipation-predominant irritable bowel syndrome. Colorectal Dis. 2010;12:1131-1138.
22. Andresen V, Montori VM, Keller J, et al. Effects of 5-hydroxytryptamine (serotonin) type 3 antagonists on symptom relief and constipation in nonconstipated irritable bowel syndrome: a systematic review and meta-analysis of randomized controlled trials. Clin Gastroenterol Hepatol. 2008;6: 545-555.
23. FDA. Lotronex (alosetron hydrochloride) tablets. www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm172946.htm. Accessed December 16, 2013.
24. FDA. Lotronex (alosetron hydrochloride) information. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110450.htm. Accessed December 16, 2013.
25. Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329-341.
26. Lunsford TN, Harris LA. Lubiprostone: evaluation of the newest medication for the treatment of adult women with constipation-predominant irritable bowel syndrome. Int J Womens Health. 2010;2:361-374.
27. Pimentel M, Lembo A, Chey WD, et al; TARGET Study Group. Rifaximin therapy for patients with irritable bowel syndrome without constipation.
N Engl J Med. 2011;364:22-32.
28. Tack J. Antibiotic therapy for the irritable bowel syndrome.N Engl J Med. 2011;364:81-82.
29. Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol. 2012;107:1714-1724.
30. Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol. 2012;107:1702-1712.
31. Gibson PR, Shepherd SJ. Evidence-based dietary management of functional gastrointestinal symptoms: the FODMAP approach. J Gastroenterol Hepatol. 2010;25:252-258.
32. Magge S, Lembo A. Low-FODMAP diet for treatment of irritable bowel syndrome.Gastroenterol Hepatol. 2012;8:739-745.
1. Brandt LJ, Chey WD, Foxx-Orenstein AE, et al; American College of Gastroenterology Task Force on Irritable Bowel Syndrome. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(suppl 1):S1-S35.
2. Wald A. Irritable bowel syndrome—diarrhoea. Best Pract Res Clin Gastroenterol. 2012;26:573-580.
3. Drossman DA, Camilleri M, Mayer EA, Whitehead WE. AGA technical review on irritable bowel syndrome. Gastroenterology. 2002;123:2108-2131.
4. World Gastroenterology Organization. Irritable bowel syndrome: a global perspective (2009). www.worldgastroenterology.org/assets/downloads/en/pdf/guidelines/20_irritable_bowel_syndrome.pdf. Accessed December 16, 2013.
5. Olden KW. Diagnosis of irritable bowel syndrome. Gastroenterology. 2002;122:1701-1714.
6. Engsbro AL, Begtrup LM, Kjeldsen J, et al. Patients suspected of irritable bowel syndrome—cross-sectional study exploring the sensitivity of Rome III criteria in primary care. Am J Gastroenterol. 2013;108:972-980.
7. Lehrer JK. Irritable bowel syndrome differential diagnoses. Medscape. http://emedicine.medscape.com/article/180389-overview. Accessed December 16, 2013.
8. Kaptchuk TJ, Kelley JM, Conboy LA, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ. 2008;336:999-1003.
9. Böhmer CJ, Tuynman HA. The effect of a lactose-restricted diet in patients with a positive lactose tolerance test, earlier diagnosed as irritable bowel syndrome: a 5-year follow-up study. Eur J Gastroenterol Hepatol. 2001; 13:941-944.
10. Vazquez-Roque MI, Camilleri M, Smyrk T, et al. A controlled trial of gluten-free diet in patients with irritable bowel syndrome-diarrhea: effects on bowel frequency and intestinal function. Gastroenterology. 2013;144:903-911.
11. Biesiekierski JR, Newnham ED, Irving PM, et al. Gluten causes gastrointestinal symptoms in subjects without celiac disease: a double-blind randomized placebo-controlled trial. Am J Gastroenterol. 2011;106:508-514.
12. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;10(8):CD003460.
13. Shepherd SJ, Parker FC, Muir G, Gibson PR. Dietary triggers of abdominal symptoms in patients with irritable bowel syndrome: randomized placebo-controlled evidence.Clin Gastroenterol Hepatol. 2008;6:765-771.
14. Ong DK, Mitchell SB, Barrett JS, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010;25:1366-1373.
15. Austin GL, Dalton CB, Hu Y, et al. A very low-carbohydrate diet improves symptoms and quality of life in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2009;7:706-708.
16. Johannesson E, Simrén M, Strid H, et al. Physical activity improves symptoms in irritable bowel syndrome: a randomized controlled trial. Am J Gastroenterol. 2011;106:915-922.
17. Lackner JM, Jaccard J, Krasner SS, et al. Self-administered cognitive behavior therapy for moderate to severe irritable bowel syndrome: clinical efficacy, tolerability, feasibility. Clin Gastroenterol Hepatol. 2008;6:899-906.
18. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58:367-378.
19. Jailwala J, Imperiale TF, Kroenke K. Pharmacologic treatment of the irritable bowel syndrome: a systematic review of randomized, controlled trials. Ann Intern Med. 2000;133:136-147.
20. Parkes GC, Sanderson JD, Whelan K. Treating irritable bowel syndrome with probiotics: the evidence. Proc Nutr Soc. 2010;69:187-194.
21. Awad RA, Camacho S. A randomized, double-blind, placebo-controlled trial of polyethylene glycol effects on fasting and postprandial rectal sensitivity and symptoms in hypersensitive constipation-predominant irritable bowel syndrome. Colorectal Dis. 2010;12:1131-1138.
22. Andresen V, Montori VM, Keller J, et al. Effects of 5-hydroxytryptamine (serotonin) type 3 antagonists on symptom relief and constipation in nonconstipated irritable bowel syndrome: a systematic review and meta-analysis of randomized controlled trials. Clin Gastroenterol Hepatol. 2008;6: 545-555.
23. FDA. Lotronex (alosetron hydrochloride) tablets. www.fda.gov/safety/medwatch/safetyinformation/safetyalertsforhumanmedicalproducts/ucm172946.htm. Accessed December 16, 2013.
24. FDA. Lotronex (alosetron hydrochloride) information. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm110450.htm. Accessed December 16, 2013.
25. Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329-341.
26. Lunsford TN, Harris LA. Lubiprostone: evaluation of the newest medication for the treatment of adult women with constipation-predominant irritable bowel syndrome. Int J Womens Health. 2010;2:361-374.
27. Pimentel M, Lembo A, Chey WD, et al; TARGET Study Group. Rifaximin therapy for patients with irritable bowel syndrome without constipation.
N Engl J Med. 2011;364:22-32.
28. Tack J. Antibiotic therapy for the irritable bowel syndrome.N Engl J Med. 2011;364:81-82.
29. Rao S, Lembo AJ, Shiff SJ, et al. A 12-week, randomized, controlled trial with a 4-week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol. 2012;107:1714-1724.
30. Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am J Gastroenterol. 2012;107:1702-1712.
31. Gibson PR, Shepherd SJ. Evidence-based dietary management of functional gastrointestinal symptoms: the FODMAP approach. J Gastroenterol Hepatol. 2010;25:252-258.
32. Magge S, Lembo A. Low-FODMAP diet for treatment of irritable bowel syndrome.Gastroenterol Hepatol. 2012;8:739-745.

Vertigo: Diagnosis and Management
CE/CME No: CR-1312
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the incidence, predisposing factors, and pathophysiology of benign paroxysmal positional vertigo (BPPV).
• Describe the typical presentation and history of symptoms in the patient with BPPV. Describe exam findings that may point to other causes of vertigo/dizziness.
• Describe how to perform the Dix-Hallpike test, the Epley maneuver, and the liberatory maneuver.
• Discuss diagnosis and management of BPPV based on current clinical practice guidelines. Describe positive Dix-Hallpike test results.
• Discuss evidence-based changes in the approach to patients with vertigo that are needed in primary care and the emergency department.
FACULTY
Mary Jo Howell Collie is a family nurse practitioner at Bland County Medical Clinic in Bastian, Virginia, and serves as a preceptor for nurse practitioner students.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Benign paroxysmal positional vertigo accounts for approximately 42% of cases of vertigo seen in primary care settings and is the single most common cause of vertigo in the United States. Our expert outlines an evidence-based approach to diagnosis, which results in an increase in desirable patient outcomes and a decrease in unnecessary tests and medications.
Dizziness is a common complaint of patients seen in both the primary care setting and the emergency department (ED). Dizziness can be classified as vertigo, disequilibrium, presyncope, and lightheadedness. Psychiatric disorders can be the cause in as many as 16% of patients who present with dizziness.1
Vestibular vertigo is the most common type of dizziness and can result from peripheral vestibular causes and central vestibular causes.1 Peripheral vestibular causes include benign paroxysmal positional vertigo (BPPV), Ménière’s disease, vestibular neuronitis, labyrinthitis, vestibular schwannoma, perilymphatic fistula, superior semicircular canal dehiscence syndrome, and trauma.2 Central vestibular causes of vertigo include vestibular migraine, vertebrobasilar ischemic stroke, and vertebrobasilar insufficiency (transient ischemic attack).2 (For more information, see Pearson T. Ménière’s disease: a lifelong merry-go-round. Clinician Reviews. 2013;23[10]:38-43.)
BPPV accounts for approximately 42% of cases of vertigo in nonspecialty settings such as primary care and is the single most common cause of vertigo in the United States.3,4 Other common causes include vestibular neuritis (41% of cases), Ménière’s disease (10%), and vascular disease (3%).4
BPPV is a disorder of the inner ear that is characterized by repeated episodes of positional vertigo, a spinning sensation produced by changes in head position relative to gravity. The term benign implies a form of positional vertigo not due to any serious central nervous system (CNS) disorder and carries an overall favorable prognosis. The term paroxysmal describes the sudden and rapid onset of vertigo.4
BPPV typically involves either the posterior semicircular canal (by far the most common) or the lateral (horizontal) semicircular canal.5 BPPV involving the posterior semicircular canal comprises 85% to 95% of all cases of BPPV and is the focus of this article.6
BPPV can be diagnosed and treated by multiple clinical disciplines, and there is considerable variation in the management of BPPV across disciplines.4 Delays in diagnosis and treatment can directly affect patients’ quality of life as well as increase health care costs. Patients with BPPV often receive inappropriately prescribed medications, such as vestibular suppressants, and potentially unnecessary diagnostic tests.4,7-9 In many cases, diagnostic and treatment decisions in regard to BPPV are not guided by current evidence.4
On the next page: Epidemiology >>
EPIDEMIOLOGY
The incidence of dizziness in the general population ranges from 20% to 30%, and with every five-year increase in age, there is a 10% increase in incidence of dizziness.10 Approximately 7.5 million patients are seen in ambulatory care annually with the chief complaint of dizziness.10 Further, with the increasing age of the US population, the incidence and prevalence of dizziness, and hence, of BPPV, will likely increase over the next 20 years.
BPPV is the most common vestibular disorder across the lifespan and most commonly presents between the fifth and seventh decade of life.4 The average age of onset of BPPV is 51 years,3 and it is rarely seen in those 35 and younger without a history of head trauma. The prevalence has been reported to range from 10.7 to 64 per 100,000 population, with a lifetime prevalence of 2.4%.4,11 It is estimated that 9% of elderly patients have unrecognized BPPV and experience greater risk for falls, depression, and interference with activities of daily living as a result.12 This, in turn, can lead to increased caregiver burden, decreased family productivity with resultant costs to society, and increased risk for nursing home placement. When not properly diagnosed and treated, BPPV can lead to significant morbidity, psychosocial problems, and increased medical costs.13
One of the main causes of BPPV is head trauma. Other predisposing factors include inactivity, major surgery, acute alcoholism, and CNS disease. BPPV is idiopathic in approximately 50% to 70% of cases.6 Spontaneous remission of BPPV can occur within days to months, or it can resolve after treatment and then recur.7 The recurrence rate of BPPV has been shown to be between 50% to 56% in some studies.11
Neuhauser et al13 evaluated the burden of dizziness within the general population in Germany, screening a cross-sectional sample of 4,869 participants for moderate or severe dizziness. The researchers estimated that 1.8% of adults seek medical care annually for new symptoms of moderate or severe dizziness or vertigo, and that vestibular vertigo accounts for approximately one third of cases of dizziness and vertigo seen in the medical setting. They commented that the latter finding is in line with other studies that have estimated that more than half of cases of dizziness in the medical setting (primary care, specialty care, and ED) are diagnosed as vestibular vertigo. The researchers also found that medical consultations and hospital visits were more frequent for vestibular vertigo than for nonvestibular dizziness. They concluded that more consideration should be given in primary care to common vestibular disorders, particularly BPPV, for which inexpensive and effective treatment with positioning maneuvers can be performed in the primary care setting.13
SUBOPTIMAL MANAGEMENT OF VERTIGO AND BPPV
Although vertigo can be debilitating and significantly reduce patients’ quality of life,13 40% to 80% of cases remain unexplained and therefore go untreated.14 BPPV not only affects patients physically but can also have serious effects on their emotional well-being.15 Anxiety has been found to be associated with BPPV in some cases. It is estimated that 86% of patients with BPPV symptoms experience problems with activities of daily living and experience work absences.11
According to clinical practice guidelines developed by the American Academy of Otolaryngology–Head and Neck Surgery (AAOHNS),health care costs associated with the diagnosis of BPPV alone approach $2 billion per year, and it costs approximately $2,000 per patient to arrive at a diagnosis of BPPV.4 A recent retrospective study examined 1,681 patients who presented to the ED with complaints of vertigo and dizziness over a three-year period.9 Nearly half the patients received a CT scan of the brain and head, resulting in a total cost of $988,200. However, fewer than 1% of the CTs revealed an underlying condition that required intervention. The researchers concluded that, for patients presenting with isolated dizziness, lightheadedness, or vertigo without other symptoms, the likelihood of finding an acute life-threatening abnormality on CT is low, and therefore CT is not helpful.
Newman-Toker et al8 studied 9,472 dizzy patients who visited the ED over a 13-year period; of the 7.4% who were diagnosed with a vestibular disorder, 84% had BPPV or acute peripheral vestibulopathy.8 Patients diagnosed with BPPV were more likely to undergo diagnostic imaging with CT and more likely to receive a prescription for the vestibular suppressant meclizine than nondizzy patients. The researchers concluded that these patients were not managed optimally, citing overuse of diagnostic imaging and prescription meclizine, which is not indicated for treating BPPV.8
The use of unnecessary diagnostic testing for the work-up of vertigo has been well documented in the literature. However, the trend of diagnostic imaging for vertigo and dizziness has continued, imposing an economic burden on the health care system. The reason for this may be twofold. First, primary care and ED providers may not feel confident in their ability to recognize and diagnose BPPV. Second, an underlying component may be the clinician’s perceived need to practice defensive medicine.
A report published by the Department of Health and Human Services included a physician survey regarding litigation.16 Of those surveyed, 79% responded that they had ordered more tests than they felt were needed, due to the fear of being sued. By extrapolation, it is reasonable to assume that similar practices and concerns may apply to nurse practitioners, of whom 70% to 80% work in primary care,17 as well as physician assistants in primary care.
Although medications are used frequently for treating dizziness, this practice is not supported by evidence-based criteria.7 Overuse of vestibular suppressants for treatment of BPPV has been identified in the literature in both primary care settings and the ED; in particular, use of meclizine for treatment of dizziness and vertigo needs to be reconsidered.4,8
On the next page: Pathophysiology and patient presentation >>
PATHOPHYSIOLOGY
BPPV most commonly is believed to result from calcium carbonate and protein crystals called otoconia becoming dislodged from the inner ear (utricle) and settling in one of the semicircular canals (most commonly the posterior); this theory is known as canalithiasis.18 When the patient moves certain ways, the otoconia shift and cause an abnormal stimulation of the motion sensor in the affected ear. This stimulation causes conflicting signals from the two labyrinths of the inner ear, resulting in brief, intense sensations of vertigo.18 A video describing the pathophysiology is available at www.youtube.com/watch?v=gDOrltSBvKI.
PATIENT PRESENTATION AND HISTORY
The patient’s description of symptoms is critical in the work-up for dizziness. It is important to ask patients to describe their symptoms using words other than “dizzy,” as the meaning of the word may vary from person to person.10
Dizziness includes a variety of symptoms such as vertigo, unsteadiness, weakness, presyncope, syncope, lightheadedness, or falling. Vertigo is the illusion of a rotational movement of one’s self or surroundings, a spinning sensation.19 True vertigo is most likely due to peripheral vestibular disorders. Complaints of disequilibrium and ataxia point to central pathology.2 Nonvertigo symptoms—generalized weakness, lightheadedness, imbalance, unsteadiness, and tilting sensations—can point to CNS, cardiovascular, and systemic diseases and require further investigation.4,10,18
A complete health history, including medications and assessing for head trauma, ear disease, or surgery, can be helpful in rendering a diagnosis. Patients should also be questioned about caffeine, nicotine, and alcohol use. A patient-centered questionnaire can be developed to guide the clinician in obtaining a thorough history of the patient’s perceptions of his/her symptoms. An important question to ask is, “Do you get dizzy when rolling over in bed?” A “yes” answer to this question raises the clinical suspicion for BPPV.
A typical description of BPPV symptoms includes a brief episode (less than a minute) of intense vertigo that can be brought about by positional changes associated with everyday activities such as rolling over in bed, tilting the head to look upward (eg, to place an object on a shelf higher than the head), or bending forward at the waist (eg, to tie shoes).4,18 This vertigo may occur frequently for weeks, disappear for months, and then begin again. Some patients may report that they were dizzy for hours or all day. On further questioning, however, the clinician may determine that the dizziness actually occurred in short, intense episodes,which, due to their severity, may be perceived as lasting longer than a minute.18 Commonly, patients may report periods of feeling imbalanced between BPPV episodes.4 Patients will sometimes report avoiding or modifying movements that commonly provoke symptoms in order to prevent an episode of vertigo.
Although the patient’s history can persuade the examiner to diagnose BPPV in a majority of cases, the AAOHNS guidelines state that history alone is insufficient to render an accurate diagnosis of BPPV.4
PHYSICAL EXAMINATION
The extent and focus of the physical examination is based on the patient’s history and symptoms. The goal of the exam is to reproduce the symptoms and to determine whether the patient has a benign cause of vertigo. Vital signs should always be obtained. A full head and neck exam should be performed to evaluate the ears, nose, and throat, since BPPV can occur secondary to other inner ear disorders.6 A complete neurologic exam should be done to assess for abnormalities in gait, coordination, and sensation. A thorough cardiovascular exam should be done to assess for carotid bruits and abnormal heart rate or rhythm. A carotid doppler, electrocardiogram, or Holter monitoring should be ordered only if abnormalities are found on the exam and/or there is a strong clinical suspicion of a cardiac cause based on the patient’s symptom history.20
The Dix-Hallpike maneuver should be performed in patients with vertigo to assess for posterior semicircular canal BPPV (see “Performing the Dix-Hallpike maneuver”).5 Although positive results from the Dix-Hallpike are the gold standard for diagnosing BPPV, negative results do not rule out BPPV since the patient may be asymptomatic on the day of the test.4,18 In patients with significant vascular disease, cervical stenosis and radiculopathies, severe kyphoscoliosis, Down syndrome, spinal cord injuries, low back dysfunction, ankylosing spondylitis, or morbid obesity, the Dix-Hallpike maneuver should be performed with caution.4 Obese patients may require an additional examiner for support.

If a positive response is observed on the initial side, no further testing is required; the examiner should immediately begin treatment with the canalith repositioning maneuver (described in the Treatment section). When the test response is negative, however, the maneuver should be repeated on the opposite side to confirm which ear is involved. Rarely is a response elicited in both the right and left ear-down positions with corresponding nystagmus; such a response is typically associated with head trauma.4
To rule out orthostatic hypotension, a possible source of “faintness” or “dizziness,” measure for changes in blood pressure (eg, decrease of 20 mm Hg systolic, decrease of 10 mm Hg diastolic) and pulse (eg, increase of 30 beats/min) from the supine to standing positions.20 These measurements should be performed after the Dix-Hallpike maneuver because the changes in patient position required to test BP may affect the results of the Dix-Hallpike maneuver. With the exception of positive results on the Dix-Hallpike test, the patient with BPPV will generally have unremarkable findings on the physical exam.3
On the next page: Lab work-up and imaging >>
LABORATORY WORK-UP AND IMAGING
There is no lab work that assists in making the diagnosis of BPPV. Radiographic imaging21 and laboratory testing are not beneficial and are in fact unnecessary and inappropriate in the patient with probable BPPV.6,8,20 There are no radiologic findings in the patient with BPPV alone.4,22 Clinical practice guidelines recommend against radiologic imaging in patients with BPPV, unless the diagnosis is uncertain or there are additional or unrelated exam findings or symptoms that justify testing.4
DIAGNOSIS
The Dix-Hallpike maneuver is considered the gold standard for diagnosing BPPV,4 although it is possible for the Dix-Hallpike test results to be negative in a patient with BPPV. If the test is negative and there is a strong clinical suspicion for BPPV, the patient may need to come back for a second visit to repeat the maneuver.4 The clinician may also consider referral to a specialist who performs vestibular function testing in order to decrease the time from the onset of symptoms to diagnosis and proper treatment.
According to the AAOHNS guidelines for BPPV, the specific diagnostic criteria for posterior canal BPPV include the patient history of repeated episodes of vertigo related to changes in head position; vertigo and nystagmus elicited on physical exam by the Dix-Hallpike test with a latency period between the onset and completion of the test; and an increase in intensity and then resolution within a minute of onset of the provoked vertigo and nystagmus.4
Patients should be questioned about associated hearing loss. Vertigo accompanied by hearing loss is typically not BPPV, but can be caused by Ménière’s disease or labyrinthitis. Patients presenting with Ménière’s disease commonly have sustained vertigo (lasting for hours), tinnitus, and fluctuating hearing loss.4 Vestibular labyrinthitis typically presents with severe vertigo that lasts from days to weeks (constant and not related to movement), severe nausea and vomiting, hearing loss, and tinnitus.4
Orthostatic hypotension, another cause of dizziness, should always be in the differential. Visual changes, ataxia, confusion, slurred speech, and numbness point to central causes of dizziness such as vertebrobasilar ischemic stroke or vertebrobasilar insufficiency.20
Once the BPPV diagnosis is made, it should be documented in the patient’s medical record. Using a diagnosis code for vertigo or dizziness is insufficient because it only describes the patient’s symptoms and is inadequate for follow-up and continuity of care. Nor does such a code provide the patient with the concise diagnosis needed to engage in self-care. Further, the incidence of the disease remains undocumented for purposes of research on BPPV.
On the next page: Treatment and management >>
TREATMENT AND MANAGEMENT
Particle repositioning maneuvers are the recommended treatment for BPPV. These maneuvers have a success rate of greater than 90%5,6 and usually provide immediate resolution of symptoms.23 The canalith repositioning maneuver (CRP; also called the Epley maneuver) and the liberatory maneuver (LM; also called the Semont maneuver) are effective treatments for posterior canal BPPV (see “The Epley Maneuver”).4 The CRP can be performed immediately following positive results on the Dix-Hallpike test.
![]()
The CRP depends on gravity to treat BPPV.24 The otoconia settle in the lowest part of the semicircular canals as the patient is moved through a series of positions. The maneuver requires the patient to be rotated 180° (through four positions) beginning with the affected side and then to the uninvolved side before returning to a sitting position.24 Each position is maintained for at least 30 s. Once the otoconia migrate out of the affected semicircular canal and back into the vestibule, the particles should dissolve.
The LM begins with the patient in a seated position with the head turned away from the affected side (see Figure 2). The clinician quickly moves the patient into a side-lying position toward the affected side, with the head turned upward and supported there for approximately 30 s (Step 1).The clinician then quickly moves the patient through the initial seated position (without pausing) to the opposite side-lying position without changing the head position (Step 2). With the head now facing downward, the patient remains motionless for another 30 s before the clinician brings the patient upright to the original seated position. Although patients are sometimes advised to remain upright for 24 to 48 h following in-office treatment (which is not believed to cause harm), there is insufficient evidence to support this recommendation.4
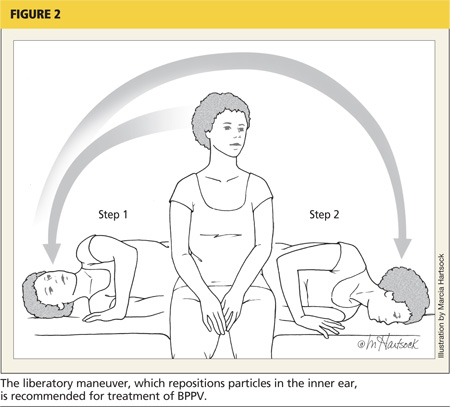
For ongoing care, current clinical guidelines recommend that practitioners offer either vestibular rehabilitation (performed by a clinician or self-administered by the patient) or provide for watchful waiting and follow-up based on the natural course of spontaneous resolution of symptoms.4
On the next page: Patient education and referral >>
PATIENT EDUCATION
It is important to remember that vertigo is a symptom, while BPPV is a diagnosis. Therefore, merely informing the patient that he or she has vertigo is not sufficient. The patient (and his/her family, if present) should be educated about the cause of the patient’s vertigo. The patient should be provided with information about BPPV that is delivered both verbally and through printed health education materials.
The chances and unpredictability of recurrence should be discussed. Ideally, the patient should be instructed to make a same-day follow-up appointment for treatment should symptoms recur. Patients, particularly the elderly, should be counseled about the risk for falls; fall risk assessment questionnaires with recommendations for prevention of injury are helpful.4
The patient with BPPV should be provided with instructions, including diagrams, of how to perform modified CRP exercises at home. Helpful videos are available on the Internet for patient use. For example, the University of Michigan has created videos for a patient diagnosed with BPPV of the right ear (www.youtube.com/watch?v=BY4UeRmTYmA) and the left ear (www.youtube.com/watch?v=lh72suV2p20).
In self-administered CRP, the patient moves through the same positions used for in-office CRP, except that the patient’s head is extended over the edge of a pillow.24 Patients should be instructed to stop the home exercises once they are symptom free for 24 h or more.
FOLLOW-UP AND REFERRAL
The need for follow-up varies depending on the patient’s response to treatment and the incidence of recurrence. Clinical practice guidelines recommend follow-up within a month of initial observation or treatment to reassess and confirm resolution of symptoms.4 Referral to specialists for treatment should be considered without delay if the primary care clinician does not feel confident treating BPPV and/or is unsure of the diagnosis based on the results of the Dix-Hallpike test, particularly if the patient’s quality of life is affected and safety is a concern.
On the next page: Conclusion >>
CONCLUSION
BPPV is a common disorder that presents most often in the primary care setting. However, most dizzy patients never see a specialist or clinician who is skilled in providing vestibular evaluation and treatment. To avoid missing the diagnosis or ordering unnecessary diagnostic and laboratory tests, clinicians can readily perform the Dix-Hallpike test and CRP in the office on patients with suspected BPPV.
CRP and vestibular rehabilitation have been proven effective in treating chronic disequilibrium and vertigo. Studies have reported that the mean wait time from initial presentation of symptoms to successful treatment was 92 weeks; 85% of these patients had immediate symptom resolution after the first treatment session with a specialist trained in CRP.25 Improvement in recognition of BPPV at the primary care level will markedly reduce the lag time to treatment.
The author would like to thank Alan L. Desmond, AuD, and Dr. Brian Collie, ENT, for their mentorship and expertise in vestibular disorders.
1. Kroenke K, Hoffman RM, Einstadter D. How common are various causes of dizziness? A critical review. Southern Med J. 2000;93:160-167.
2. Thompson TL, Amedee R. Vertigo: a review of common peripheral and central vestibular disorders.Ochsner J. 2009;9:20-26.
3. Li JC, Egan RE. Neurologic manifestations of benign positional vertigo (2012). Medscape. http://emedicine.medscape.com/article/1158940-overview. Accessed November 14, 2013.
4. Bhattacharyya N, Baugh RF, Orvidas L, et al. Clinical practice guideline: benign paroxysmal positional vertigo. Otolaryng Head Neck Surg. 2008; 139:S47-S81.
5. Steenerson RL, Cronin GW, Marbach PM. Effectiveness of treatment techniques in 923 cases of benign paroxysmal positional vertigo. Laryngoscope. 2005;115:226-231.
6. Parnes LS, Agrawal SK, Atlas J. Diagnosis and management of benign paroxysmal positional vertigo (BPPV). CMAJ. 2003;169:681-693.
7. Desmond AL. Vestibular Function: Clinical and Practice Management. 2nd ed. New York, NY: Thieme Medical Publishers; 2011:1-276.
8. Newman-Toker DE, Camargo CA, Hsieh Y, et al. Disconnect between charted vestibular diagnoses and emergency department management decisions: a cross-sectional analysis from a nationally representative sample. Acad Emerg Med. 2009;16:970-977.
9. Ahsan SF, Syamal MN, Yaremchuk K, et al. The costs and utility of imaging in evaluating dizzy patients in the emergency room. Laryngoscope. 2013;123:2250-2253.
10. Chan Y. Differential diagnosis of dizziness. Curr Opin Otolaryng Head Neck Surg. 2009;17:200-203.
11. von Brevern M, Radtke A, Lezius F, et al. Epidemiology of benign paroxysmal positional vertigo: a population based study. J Neurol Neurosurg Psychiatry. 2007;78:710-715.
12. Oghalai JS, Manolidis S, Barth JL, et al. Unrecognized benign paroxysmal positional vertigo in elderly patients. Otolaryng Head Neck Surg. 2000;122:630-634.
13. Neuhauser HK, Radtke A, von Brevern, et al. Burden of dizziness and vertigo in the community. Arch Intern Med. 2008;168:2118-2124.
14. Holmes S, Padgham ND. A review of the burden of vertigo. J Clin Nurs. 2011;20(19-20):2690-2701.
15. Pollak L, Segal P, Stryjer R, Stern HG. Beliefs and emotional reactions in patients with benign paroxysmal positional vertigo: a longitudinal study. Am J Otolaryng. 2012;33:221-225.
16. US Department of Health and Human Services. Confronting the new health care crisis: improving health care quality and lowering costs by fixing our medical liability system (2002). http://aspe.hhs.gov/daltcp/reports/litrefm.pdf. Accessed November 14, 2013.
17. Naylor MD, Kurtzman ET. The role of nurse practitioners in reinventing primary care. Health Affairs. 2010;29:893-899.
18. Desmond AL. Dizziness Reference Guide. Chatham, IL: Micromedical Technologies; 2009.
19. Goebel JA. The ten-minute examination of the dizzy patient. Semin Neurol. 2001;21:391-398.
20. Post RE, Dickerson LM. Dizziness: a diagnostic approach.Am Fam Physician. 2010;82:361-368.
21. Korres S, Riga M, Papacharalampous G, et al. Relative diagnostic importance of electronystagmography and magnetic resonance imaging in vestibular disorders. J Laryngol Otol. 2009;123:851-856.
22. American College of Radiology Expert Panel on Neurologic Imaging. American College of Radiology Appropriateness Criteria [report], 2008. www.acr.org/~/media/ACR/Documents/AppCriteria/Diagnostic/Vertigo HearingLoss.pdf. Accessed November 14, 2013.
23. Lee S-H, Kim JS. Benign paroxysmal positional vertigo. J Clin Neurol. 2010;6:51-63.
24. Helminski JO, Zee DS, Janssen I, Hain TC. Effectiveness of particle repositioning maneuvers in the treatment of benign paroxysmal positional vertigo: a systematic review. Phys Ther. 2010;90:663-678.
25. Fife D, Fitzgerald JE. Do patients with benign paroxysmal positional vertigo receive prompt treatment? Analysis of waiting times and human and financial costs associated with current practice. Int Journal Audiol. 2005;44:50-57.
CE/CME No: CR-1312
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the incidence, predisposing factors, and pathophysiology of benign paroxysmal positional vertigo (BPPV).
• Describe the typical presentation and history of symptoms in the patient with BPPV. Describe exam findings that may point to other causes of vertigo/dizziness.
• Describe how to perform the Dix-Hallpike test, the Epley maneuver, and the liberatory maneuver.
• Discuss diagnosis and management of BPPV based on current clinical practice guidelines. Describe positive Dix-Hallpike test results.
• Discuss evidence-based changes in the approach to patients with vertigo that are needed in primary care and the emergency department.
FACULTY
Mary Jo Howell Collie is a family nurse practitioner at Bland County Medical Clinic in Bastian, Virginia, and serves as a preceptor for nurse practitioner students.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Benign paroxysmal positional vertigo accounts for approximately 42% of cases of vertigo seen in primary care settings and is the single most common cause of vertigo in the United States. Our expert outlines an evidence-based approach to diagnosis, which results in an increase in desirable patient outcomes and a decrease in unnecessary tests and medications.
Dizziness is a common complaint of patients seen in both the primary care setting and the emergency department (ED). Dizziness can be classified as vertigo, disequilibrium, presyncope, and lightheadedness. Psychiatric disorders can be the cause in as many as 16% of patients who present with dizziness.1
Vestibular vertigo is the most common type of dizziness and can result from peripheral vestibular causes and central vestibular causes.1 Peripheral vestibular causes include benign paroxysmal positional vertigo (BPPV), Ménière’s disease, vestibular neuronitis, labyrinthitis, vestibular schwannoma, perilymphatic fistula, superior semicircular canal dehiscence syndrome, and trauma.2 Central vestibular causes of vertigo include vestibular migraine, vertebrobasilar ischemic stroke, and vertebrobasilar insufficiency (transient ischemic attack).2 (For more information, see Pearson T. Ménière’s disease: a lifelong merry-go-round. Clinician Reviews. 2013;23[10]:38-43.)
BPPV accounts for approximately 42% of cases of vertigo in nonspecialty settings such as primary care and is the single most common cause of vertigo in the United States.3,4 Other common causes include vestibular neuritis (41% of cases), Ménière’s disease (10%), and vascular disease (3%).4
BPPV is a disorder of the inner ear that is characterized by repeated episodes of positional vertigo, a spinning sensation produced by changes in head position relative to gravity. The term benign implies a form of positional vertigo not due to any serious central nervous system (CNS) disorder and carries an overall favorable prognosis. The term paroxysmal describes the sudden and rapid onset of vertigo.4
BPPV typically involves either the posterior semicircular canal (by far the most common) or the lateral (horizontal) semicircular canal.5 BPPV involving the posterior semicircular canal comprises 85% to 95% of all cases of BPPV and is the focus of this article.6
BPPV can be diagnosed and treated by multiple clinical disciplines, and there is considerable variation in the management of BPPV across disciplines.4 Delays in diagnosis and treatment can directly affect patients’ quality of life as well as increase health care costs. Patients with BPPV often receive inappropriately prescribed medications, such as vestibular suppressants, and potentially unnecessary diagnostic tests.4,7-9 In many cases, diagnostic and treatment decisions in regard to BPPV are not guided by current evidence.4
On the next page: Epidemiology >>
EPIDEMIOLOGY
The incidence of dizziness in the general population ranges from 20% to 30%, and with every five-year increase in age, there is a 10% increase in incidence of dizziness.10 Approximately 7.5 million patients are seen in ambulatory care annually with the chief complaint of dizziness.10 Further, with the increasing age of the US population, the incidence and prevalence of dizziness, and hence, of BPPV, will likely increase over the next 20 years.
BPPV is the most common vestibular disorder across the lifespan and most commonly presents between the fifth and seventh decade of life.4 The average age of onset of BPPV is 51 years,3 and it is rarely seen in those 35 and younger without a history of head trauma. The prevalence has been reported to range from 10.7 to 64 per 100,000 population, with a lifetime prevalence of 2.4%.4,11 It is estimated that 9% of elderly patients have unrecognized BPPV and experience greater risk for falls, depression, and interference with activities of daily living as a result.12 This, in turn, can lead to increased caregiver burden, decreased family productivity with resultant costs to society, and increased risk for nursing home placement. When not properly diagnosed and treated, BPPV can lead to significant morbidity, psychosocial problems, and increased medical costs.13
One of the main causes of BPPV is head trauma. Other predisposing factors include inactivity, major surgery, acute alcoholism, and CNS disease. BPPV is idiopathic in approximately 50% to 70% of cases.6 Spontaneous remission of BPPV can occur within days to months, or it can resolve after treatment and then recur.7 The recurrence rate of BPPV has been shown to be between 50% to 56% in some studies.11
Neuhauser et al13 evaluated the burden of dizziness within the general population in Germany, screening a cross-sectional sample of 4,869 participants for moderate or severe dizziness. The researchers estimated that 1.8% of adults seek medical care annually for new symptoms of moderate or severe dizziness or vertigo, and that vestibular vertigo accounts for approximately one third of cases of dizziness and vertigo seen in the medical setting. They commented that the latter finding is in line with other studies that have estimated that more than half of cases of dizziness in the medical setting (primary care, specialty care, and ED) are diagnosed as vestibular vertigo. The researchers also found that medical consultations and hospital visits were more frequent for vestibular vertigo than for nonvestibular dizziness. They concluded that more consideration should be given in primary care to common vestibular disorders, particularly BPPV, for which inexpensive and effective treatment with positioning maneuvers can be performed in the primary care setting.13
SUBOPTIMAL MANAGEMENT OF VERTIGO AND BPPV
Although vertigo can be debilitating and significantly reduce patients’ quality of life,13 40% to 80% of cases remain unexplained and therefore go untreated.14 BPPV not only affects patients physically but can also have serious effects on their emotional well-being.15 Anxiety has been found to be associated with BPPV in some cases. It is estimated that 86% of patients with BPPV symptoms experience problems with activities of daily living and experience work absences.11
According to clinical practice guidelines developed by the American Academy of Otolaryngology–Head and Neck Surgery (AAOHNS),health care costs associated with the diagnosis of BPPV alone approach $2 billion per year, and it costs approximately $2,000 per patient to arrive at a diagnosis of BPPV.4 A recent retrospective study examined 1,681 patients who presented to the ED with complaints of vertigo and dizziness over a three-year period.9 Nearly half the patients received a CT scan of the brain and head, resulting in a total cost of $988,200. However, fewer than 1% of the CTs revealed an underlying condition that required intervention. The researchers concluded that, for patients presenting with isolated dizziness, lightheadedness, or vertigo without other symptoms, the likelihood of finding an acute life-threatening abnormality on CT is low, and therefore CT is not helpful.
Newman-Toker et al8 studied 9,472 dizzy patients who visited the ED over a 13-year period; of the 7.4% who were diagnosed with a vestibular disorder, 84% had BPPV or acute peripheral vestibulopathy.8 Patients diagnosed with BPPV were more likely to undergo diagnostic imaging with CT and more likely to receive a prescription for the vestibular suppressant meclizine than nondizzy patients. The researchers concluded that these patients were not managed optimally, citing overuse of diagnostic imaging and prescription meclizine, which is not indicated for treating BPPV.8
The use of unnecessary diagnostic testing for the work-up of vertigo has been well documented in the literature. However, the trend of diagnostic imaging for vertigo and dizziness has continued, imposing an economic burden on the health care system. The reason for this may be twofold. First, primary care and ED providers may not feel confident in their ability to recognize and diagnose BPPV. Second, an underlying component may be the clinician’s perceived need to practice defensive medicine.
A report published by the Department of Health and Human Services included a physician survey regarding litigation.16 Of those surveyed, 79% responded that they had ordered more tests than they felt were needed, due to the fear of being sued. By extrapolation, it is reasonable to assume that similar practices and concerns may apply to nurse practitioners, of whom 70% to 80% work in primary care,17 as well as physician assistants in primary care.
Although medications are used frequently for treating dizziness, this practice is not supported by evidence-based criteria.7 Overuse of vestibular suppressants for treatment of BPPV has been identified in the literature in both primary care settings and the ED; in particular, use of meclizine for treatment of dizziness and vertigo needs to be reconsidered.4,8
On the next page: Pathophysiology and patient presentation >>
PATHOPHYSIOLOGY
BPPV most commonly is believed to result from calcium carbonate and protein crystals called otoconia becoming dislodged from the inner ear (utricle) and settling in one of the semicircular canals (most commonly the posterior); this theory is known as canalithiasis.18 When the patient moves certain ways, the otoconia shift and cause an abnormal stimulation of the motion sensor in the affected ear. This stimulation causes conflicting signals from the two labyrinths of the inner ear, resulting in brief, intense sensations of vertigo.18 A video describing the pathophysiology is available at www.youtube.com/watch?v=gDOrltSBvKI.
PATIENT PRESENTATION AND HISTORY
The patient’s description of symptoms is critical in the work-up for dizziness. It is important to ask patients to describe their symptoms using words other than “dizzy,” as the meaning of the word may vary from person to person.10
Dizziness includes a variety of symptoms such as vertigo, unsteadiness, weakness, presyncope, syncope, lightheadedness, or falling. Vertigo is the illusion of a rotational movement of one’s self or surroundings, a spinning sensation.19 True vertigo is most likely due to peripheral vestibular disorders. Complaints of disequilibrium and ataxia point to central pathology.2 Nonvertigo symptoms—generalized weakness, lightheadedness, imbalance, unsteadiness, and tilting sensations—can point to CNS, cardiovascular, and systemic diseases and require further investigation.4,10,18
A complete health history, including medications and assessing for head trauma, ear disease, or surgery, can be helpful in rendering a diagnosis. Patients should also be questioned about caffeine, nicotine, and alcohol use. A patient-centered questionnaire can be developed to guide the clinician in obtaining a thorough history of the patient’s perceptions of his/her symptoms. An important question to ask is, “Do you get dizzy when rolling over in bed?” A “yes” answer to this question raises the clinical suspicion for BPPV.
A typical description of BPPV symptoms includes a brief episode (less than a minute) of intense vertigo that can be brought about by positional changes associated with everyday activities such as rolling over in bed, tilting the head to look upward (eg, to place an object on a shelf higher than the head), or bending forward at the waist (eg, to tie shoes).4,18 This vertigo may occur frequently for weeks, disappear for months, and then begin again. Some patients may report that they were dizzy for hours or all day. On further questioning, however, the clinician may determine that the dizziness actually occurred in short, intense episodes,which, due to their severity, may be perceived as lasting longer than a minute.18 Commonly, patients may report periods of feeling imbalanced between BPPV episodes.4 Patients will sometimes report avoiding or modifying movements that commonly provoke symptoms in order to prevent an episode of vertigo.
Although the patient’s history can persuade the examiner to diagnose BPPV in a majority of cases, the AAOHNS guidelines state that history alone is insufficient to render an accurate diagnosis of BPPV.4
PHYSICAL EXAMINATION
The extent and focus of the physical examination is based on the patient’s history and symptoms. The goal of the exam is to reproduce the symptoms and to determine whether the patient has a benign cause of vertigo. Vital signs should always be obtained. A full head and neck exam should be performed to evaluate the ears, nose, and throat, since BPPV can occur secondary to other inner ear disorders.6 A complete neurologic exam should be done to assess for abnormalities in gait, coordination, and sensation. A thorough cardiovascular exam should be done to assess for carotid bruits and abnormal heart rate or rhythm. A carotid doppler, electrocardiogram, or Holter monitoring should be ordered only if abnormalities are found on the exam and/or there is a strong clinical suspicion of a cardiac cause based on the patient’s symptom history.20
The Dix-Hallpike maneuver should be performed in patients with vertigo to assess for posterior semicircular canal BPPV (see “Performing the Dix-Hallpike maneuver”).5 Although positive results from the Dix-Hallpike are the gold standard for diagnosing BPPV, negative results do not rule out BPPV since the patient may be asymptomatic on the day of the test.4,18 In patients with significant vascular disease, cervical stenosis and radiculopathies, severe kyphoscoliosis, Down syndrome, spinal cord injuries, low back dysfunction, ankylosing spondylitis, or morbid obesity, the Dix-Hallpike maneuver should be performed with caution.4 Obese patients may require an additional examiner for support.
If a positive response is observed on the initial side, no further testing is required; the examiner should immediately begin treatment with the canalith repositioning maneuver (described in the Treatment section). When the test response is negative, however, the maneuver should be repeated on the opposite side to confirm which ear is involved. Rarely is a response elicited in both the right and left ear-down positions with corresponding nystagmus; such a response is typically associated with head trauma.4
To rule out orthostatic hypotension, a possible source of “faintness” or “dizziness,” measure for changes in blood pressure (eg, decrease of 20 mm Hg systolic, decrease of 10 mm Hg diastolic) and pulse (eg, increase of 30 beats/min) from the supine to standing positions.20 These measurements should be performed after the Dix-Hallpike maneuver because the changes in patient position required to test BP may affect the results of the Dix-Hallpike maneuver. With the exception of positive results on the Dix-Hallpike test, the patient with BPPV will generally have unremarkable findings on the physical exam.3
On the next page: Lab work-up and imaging >>
LABORATORY WORK-UP AND IMAGING
There is no lab work that assists in making the diagnosis of BPPV. Radiographic imaging21 and laboratory testing are not beneficial and are in fact unnecessary and inappropriate in the patient with probable BPPV.6,8,20 There are no radiologic findings in the patient with BPPV alone.4,22 Clinical practice guidelines recommend against radiologic imaging in patients with BPPV, unless the diagnosis is uncertain or there are additional or unrelated exam findings or symptoms that justify testing.4
DIAGNOSIS
The Dix-Hallpike maneuver is considered the gold standard for diagnosing BPPV,4 although it is possible for the Dix-Hallpike test results to be negative in a patient with BPPV. If the test is negative and there is a strong clinical suspicion for BPPV, the patient may need to come back for a second visit to repeat the maneuver.4 The clinician may also consider referral to a specialist who performs vestibular function testing in order to decrease the time from the onset of symptoms to diagnosis and proper treatment.
According to the AAOHNS guidelines for BPPV, the specific diagnostic criteria for posterior canal BPPV include the patient history of repeated episodes of vertigo related to changes in head position; vertigo and nystagmus elicited on physical exam by the Dix-Hallpike test with a latency period between the onset and completion of the test; and an increase in intensity and then resolution within a minute of onset of the provoked vertigo and nystagmus.4
Patients should be questioned about associated hearing loss. Vertigo accompanied by hearing loss is typically not BPPV, but can be caused by Ménière’s disease or labyrinthitis. Patients presenting with Ménière’s disease commonly have sustained vertigo (lasting for hours), tinnitus, and fluctuating hearing loss.4 Vestibular labyrinthitis typically presents with severe vertigo that lasts from days to weeks (constant and not related to movement), severe nausea and vomiting, hearing loss, and tinnitus.4
Orthostatic hypotension, another cause of dizziness, should always be in the differential. Visual changes, ataxia, confusion, slurred speech, and numbness point to central causes of dizziness such as vertebrobasilar ischemic stroke or vertebrobasilar insufficiency.20
Once the BPPV diagnosis is made, it should be documented in the patient’s medical record. Using a diagnosis code for vertigo or dizziness is insufficient because it only describes the patient’s symptoms and is inadequate for follow-up and continuity of care. Nor does such a code provide the patient with the concise diagnosis needed to engage in self-care. Further, the incidence of the disease remains undocumented for purposes of research on BPPV.
On the next page: Treatment and management >>
TREATMENT AND MANAGEMENT
Particle repositioning maneuvers are the recommended treatment for BPPV. These maneuvers have a success rate of greater than 90%5,6 and usually provide immediate resolution of symptoms.23 The canalith repositioning maneuver (CRP; also called the Epley maneuver) and the liberatory maneuver (LM; also called the Semont maneuver) are effective treatments for posterior canal BPPV (see “The Epley Maneuver”).4 The CRP can be performed immediately following positive results on the Dix-Hallpike test.
![]()
The CRP depends on gravity to treat BPPV.24 The otoconia settle in the lowest part of the semicircular canals as the patient is moved through a series of positions. The maneuver requires the patient to be rotated 180° (through four positions) beginning with the affected side and then to the uninvolved side before returning to a sitting position.24 Each position is maintained for at least 30 s. Once the otoconia migrate out of the affected semicircular canal and back into the vestibule, the particles should dissolve.
The LM begins with the patient in a seated position with the head turned away from the affected side (see Figure 2). The clinician quickly moves the patient into a side-lying position toward the affected side, with the head turned upward and supported there for approximately 30 s (Step 1).The clinician then quickly moves the patient through the initial seated position (without pausing) to the opposite side-lying position without changing the head position (Step 2). With the head now facing downward, the patient remains motionless for another 30 s before the clinician brings the patient upright to the original seated position. Although patients are sometimes advised to remain upright for 24 to 48 h following in-office treatment (which is not believed to cause harm), there is insufficient evidence to support this recommendation.4
For ongoing care, current clinical guidelines recommend that practitioners offer either vestibular rehabilitation (performed by a clinician or self-administered by the patient) or provide for watchful waiting and follow-up based on the natural course of spontaneous resolution of symptoms.4
On the next page: Patient education and referral >>
PATIENT EDUCATION
It is important to remember that vertigo is a symptom, while BPPV is a diagnosis. Therefore, merely informing the patient that he or she has vertigo is not sufficient. The patient (and his/her family, if present) should be educated about the cause of the patient’s vertigo. The patient should be provided with information about BPPV that is delivered both verbally and through printed health education materials.
The chances and unpredictability of recurrence should be discussed. Ideally, the patient should be instructed to make a same-day follow-up appointment for treatment should symptoms recur. Patients, particularly the elderly, should be counseled about the risk for falls; fall risk assessment questionnaires with recommendations for prevention of injury are helpful.4
The patient with BPPV should be provided with instructions, including diagrams, of how to perform modified CRP exercises at home. Helpful videos are available on the Internet for patient use. For example, the University of Michigan has created videos for a patient diagnosed with BPPV of the right ear (www.youtube.com/watch?v=BY4UeRmTYmA) and the left ear (www.youtube.com/watch?v=lh72suV2p20).
In self-administered CRP, the patient moves through the same positions used for in-office CRP, except that the patient’s head is extended over the edge of a pillow.24 Patients should be instructed to stop the home exercises once they are symptom free for 24 h or more.
FOLLOW-UP AND REFERRAL
The need for follow-up varies depending on the patient’s response to treatment and the incidence of recurrence. Clinical practice guidelines recommend follow-up within a month of initial observation or treatment to reassess and confirm resolution of symptoms.4 Referral to specialists for treatment should be considered without delay if the primary care clinician does not feel confident treating BPPV and/or is unsure of the diagnosis based on the results of the Dix-Hallpike test, particularly if the patient’s quality of life is affected and safety is a concern.
On the next page: Conclusion >>
CONCLUSION
BPPV is a common disorder that presents most often in the primary care setting. However, most dizzy patients never see a specialist or clinician who is skilled in providing vestibular evaluation and treatment. To avoid missing the diagnosis or ordering unnecessary diagnostic and laboratory tests, clinicians can readily perform the Dix-Hallpike test and CRP in the office on patients with suspected BPPV.
CRP and vestibular rehabilitation have been proven effective in treating chronic disequilibrium and vertigo. Studies have reported that the mean wait time from initial presentation of symptoms to successful treatment was 92 weeks; 85% of these patients had immediate symptom resolution after the first treatment session with a specialist trained in CRP.25 Improvement in recognition of BPPV at the primary care level will markedly reduce the lag time to treatment.
The author would like to thank Alan L. Desmond, AuD, and Dr. Brian Collie, ENT, for their mentorship and expertise in vestibular disorders.
CE/CME No: CR-1312
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the incidence, predisposing factors, and pathophysiology of benign paroxysmal positional vertigo (BPPV).
• Describe the typical presentation and history of symptoms in the patient with BPPV. Describe exam findings that may point to other causes of vertigo/dizziness.
• Describe how to perform the Dix-Hallpike test, the Epley maneuver, and the liberatory maneuver.
• Discuss diagnosis and management of BPPV based on current clinical practice guidelines. Describe positive Dix-Hallpike test results.
• Discuss evidence-based changes in the approach to patients with vertigo that are needed in primary care and the emergency department.
FACULTY
Mary Jo Howell Collie is a family nurse practitioner at Bland County Medical Clinic in Bastian, Virginia, and serves as a preceptor for nurse practitioner students.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Benign paroxysmal positional vertigo accounts for approximately 42% of cases of vertigo seen in primary care settings and is the single most common cause of vertigo in the United States. Our expert outlines an evidence-based approach to diagnosis, which results in an increase in desirable patient outcomes and a decrease in unnecessary tests and medications.
Dizziness is a common complaint of patients seen in both the primary care setting and the emergency department (ED). Dizziness can be classified as vertigo, disequilibrium, presyncope, and lightheadedness. Psychiatric disorders can be the cause in as many as 16% of patients who present with dizziness.1
Vestibular vertigo is the most common type of dizziness and can result from peripheral vestibular causes and central vestibular causes.1 Peripheral vestibular causes include benign paroxysmal positional vertigo (BPPV), Ménière’s disease, vestibular neuronitis, labyrinthitis, vestibular schwannoma, perilymphatic fistula, superior semicircular canal dehiscence syndrome, and trauma.2 Central vestibular causes of vertigo include vestibular migraine, vertebrobasilar ischemic stroke, and vertebrobasilar insufficiency (transient ischemic attack).2 (For more information, see Pearson T. Ménière’s disease: a lifelong merry-go-round. Clinician Reviews. 2013;23[10]:38-43.)
BPPV accounts for approximately 42% of cases of vertigo in nonspecialty settings such as primary care and is the single most common cause of vertigo in the United States.3,4 Other common causes include vestibular neuritis (41% of cases), Ménière’s disease (10%), and vascular disease (3%).4
BPPV is a disorder of the inner ear that is characterized by repeated episodes of positional vertigo, a spinning sensation produced by changes in head position relative to gravity. The term benign implies a form of positional vertigo not due to any serious central nervous system (CNS) disorder and carries an overall favorable prognosis. The term paroxysmal describes the sudden and rapid onset of vertigo.4
BPPV typically involves either the posterior semicircular canal (by far the most common) or the lateral (horizontal) semicircular canal.5 BPPV involving the posterior semicircular canal comprises 85% to 95% of all cases of BPPV and is the focus of this article.6
BPPV can be diagnosed and treated by multiple clinical disciplines, and there is considerable variation in the management of BPPV across disciplines.4 Delays in diagnosis and treatment can directly affect patients’ quality of life as well as increase health care costs. Patients with BPPV often receive inappropriately prescribed medications, such as vestibular suppressants, and potentially unnecessary diagnostic tests.4,7-9 In many cases, diagnostic and treatment decisions in regard to BPPV are not guided by current evidence.4
On the next page: Epidemiology >>
EPIDEMIOLOGY
The incidence of dizziness in the general population ranges from 20% to 30%, and with every five-year increase in age, there is a 10% increase in incidence of dizziness.10 Approximately 7.5 million patients are seen in ambulatory care annually with the chief complaint of dizziness.10 Further, with the increasing age of the US population, the incidence and prevalence of dizziness, and hence, of BPPV, will likely increase over the next 20 years.
BPPV is the most common vestibular disorder across the lifespan and most commonly presents between the fifth and seventh decade of life.4 The average age of onset of BPPV is 51 years,3 and it is rarely seen in those 35 and younger without a history of head trauma. The prevalence has been reported to range from 10.7 to 64 per 100,000 population, with a lifetime prevalence of 2.4%.4,11 It is estimated that 9% of elderly patients have unrecognized BPPV and experience greater risk for falls, depression, and interference with activities of daily living as a result.12 This, in turn, can lead to increased caregiver burden, decreased family productivity with resultant costs to society, and increased risk for nursing home placement. When not properly diagnosed and treated, BPPV can lead to significant morbidity, psychosocial problems, and increased medical costs.13
One of the main causes of BPPV is head trauma. Other predisposing factors include inactivity, major surgery, acute alcoholism, and CNS disease. BPPV is idiopathic in approximately 50% to 70% of cases.6 Spontaneous remission of BPPV can occur within days to months, or it can resolve after treatment and then recur.7 The recurrence rate of BPPV has been shown to be between 50% to 56% in some studies.11
Neuhauser et al13 evaluated the burden of dizziness within the general population in Germany, screening a cross-sectional sample of 4,869 participants for moderate or severe dizziness. The researchers estimated that 1.8% of adults seek medical care annually for new symptoms of moderate or severe dizziness or vertigo, and that vestibular vertigo accounts for approximately one third of cases of dizziness and vertigo seen in the medical setting. They commented that the latter finding is in line with other studies that have estimated that more than half of cases of dizziness in the medical setting (primary care, specialty care, and ED) are diagnosed as vestibular vertigo. The researchers also found that medical consultations and hospital visits were more frequent for vestibular vertigo than for nonvestibular dizziness. They concluded that more consideration should be given in primary care to common vestibular disorders, particularly BPPV, for which inexpensive and effective treatment with positioning maneuvers can be performed in the primary care setting.13
SUBOPTIMAL MANAGEMENT OF VERTIGO AND BPPV
Although vertigo can be debilitating and significantly reduce patients’ quality of life,13 40% to 80% of cases remain unexplained and therefore go untreated.14 BPPV not only affects patients physically but can also have serious effects on their emotional well-being.15 Anxiety has been found to be associated with BPPV in some cases. It is estimated that 86% of patients with BPPV symptoms experience problems with activities of daily living and experience work absences.11
According to clinical practice guidelines developed by the American Academy of Otolaryngology–Head and Neck Surgery (AAOHNS),health care costs associated with the diagnosis of BPPV alone approach $2 billion per year, and it costs approximately $2,000 per patient to arrive at a diagnosis of BPPV.4 A recent retrospective study examined 1,681 patients who presented to the ED with complaints of vertigo and dizziness over a three-year period.9 Nearly half the patients received a CT scan of the brain and head, resulting in a total cost of $988,200. However, fewer than 1% of the CTs revealed an underlying condition that required intervention. The researchers concluded that, for patients presenting with isolated dizziness, lightheadedness, or vertigo without other symptoms, the likelihood of finding an acute life-threatening abnormality on CT is low, and therefore CT is not helpful.
Newman-Toker et al8 studied 9,472 dizzy patients who visited the ED over a 13-year period; of the 7.4% who were diagnosed with a vestibular disorder, 84% had BPPV or acute peripheral vestibulopathy.8 Patients diagnosed with BPPV were more likely to undergo diagnostic imaging with CT and more likely to receive a prescription for the vestibular suppressant meclizine than nondizzy patients. The researchers concluded that these patients were not managed optimally, citing overuse of diagnostic imaging and prescription meclizine, which is not indicated for treating BPPV.8
The use of unnecessary diagnostic testing for the work-up of vertigo has been well documented in the literature. However, the trend of diagnostic imaging for vertigo and dizziness has continued, imposing an economic burden on the health care system. The reason for this may be twofold. First, primary care and ED providers may not feel confident in their ability to recognize and diagnose BPPV. Second, an underlying component may be the clinician’s perceived need to practice defensive medicine.
A report published by the Department of Health and Human Services included a physician survey regarding litigation.16 Of those surveyed, 79% responded that they had ordered more tests than they felt were needed, due to the fear of being sued. By extrapolation, it is reasonable to assume that similar practices and concerns may apply to nurse practitioners, of whom 70% to 80% work in primary care,17 as well as physician assistants in primary care.
Although medications are used frequently for treating dizziness, this practice is not supported by evidence-based criteria.7 Overuse of vestibular suppressants for treatment of BPPV has been identified in the literature in both primary care settings and the ED; in particular, use of meclizine for treatment of dizziness and vertigo needs to be reconsidered.4,8
On the next page: Pathophysiology and patient presentation >>
PATHOPHYSIOLOGY
BPPV most commonly is believed to result from calcium carbonate and protein crystals called otoconia becoming dislodged from the inner ear (utricle) and settling in one of the semicircular canals (most commonly the posterior); this theory is known as canalithiasis.18 When the patient moves certain ways, the otoconia shift and cause an abnormal stimulation of the motion sensor in the affected ear. This stimulation causes conflicting signals from the two labyrinths of the inner ear, resulting in brief, intense sensations of vertigo.18 A video describing the pathophysiology is available at www.youtube.com/watch?v=gDOrltSBvKI.
PATIENT PRESENTATION AND HISTORY
The patient’s description of symptoms is critical in the work-up for dizziness. It is important to ask patients to describe their symptoms using words other than “dizzy,” as the meaning of the word may vary from person to person.10
Dizziness includes a variety of symptoms such as vertigo, unsteadiness, weakness, presyncope, syncope, lightheadedness, or falling. Vertigo is the illusion of a rotational movement of one’s self or surroundings, a spinning sensation.19 True vertigo is most likely due to peripheral vestibular disorders. Complaints of disequilibrium and ataxia point to central pathology.2 Nonvertigo symptoms—generalized weakness, lightheadedness, imbalance, unsteadiness, and tilting sensations—can point to CNS, cardiovascular, and systemic diseases and require further investigation.4,10,18
A complete health history, including medications and assessing for head trauma, ear disease, or surgery, can be helpful in rendering a diagnosis. Patients should also be questioned about caffeine, nicotine, and alcohol use. A patient-centered questionnaire can be developed to guide the clinician in obtaining a thorough history of the patient’s perceptions of his/her symptoms. An important question to ask is, “Do you get dizzy when rolling over in bed?” A “yes” answer to this question raises the clinical suspicion for BPPV.
A typical description of BPPV symptoms includes a brief episode (less than a minute) of intense vertigo that can be brought about by positional changes associated with everyday activities such as rolling over in bed, tilting the head to look upward (eg, to place an object on a shelf higher than the head), or bending forward at the waist (eg, to tie shoes).4,18 This vertigo may occur frequently for weeks, disappear for months, and then begin again. Some patients may report that they were dizzy for hours or all day. On further questioning, however, the clinician may determine that the dizziness actually occurred in short, intense episodes,which, due to their severity, may be perceived as lasting longer than a minute.18 Commonly, patients may report periods of feeling imbalanced between BPPV episodes.4 Patients will sometimes report avoiding or modifying movements that commonly provoke symptoms in order to prevent an episode of vertigo.
Although the patient’s history can persuade the examiner to diagnose BPPV in a majority of cases, the AAOHNS guidelines state that history alone is insufficient to render an accurate diagnosis of BPPV.4
PHYSICAL EXAMINATION
The extent and focus of the physical examination is based on the patient’s history and symptoms. The goal of the exam is to reproduce the symptoms and to determine whether the patient has a benign cause of vertigo. Vital signs should always be obtained. A full head and neck exam should be performed to evaluate the ears, nose, and throat, since BPPV can occur secondary to other inner ear disorders.6 A complete neurologic exam should be done to assess for abnormalities in gait, coordination, and sensation. A thorough cardiovascular exam should be done to assess for carotid bruits and abnormal heart rate or rhythm. A carotid doppler, electrocardiogram, or Holter monitoring should be ordered only if abnormalities are found on the exam and/or there is a strong clinical suspicion of a cardiac cause based on the patient’s symptom history.20
The Dix-Hallpike maneuver should be performed in patients with vertigo to assess for posterior semicircular canal BPPV (see “Performing the Dix-Hallpike maneuver”).5 Although positive results from the Dix-Hallpike are the gold standard for diagnosing BPPV, negative results do not rule out BPPV since the patient may be asymptomatic on the day of the test.4,18 In patients with significant vascular disease, cervical stenosis and radiculopathies, severe kyphoscoliosis, Down syndrome, spinal cord injuries, low back dysfunction, ankylosing spondylitis, or morbid obesity, the Dix-Hallpike maneuver should be performed with caution.4 Obese patients may require an additional examiner for support.
If a positive response is observed on the initial side, no further testing is required; the examiner should immediately begin treatment with the canalith repositioning maneuver (described in the Treatment section). When the test response is negative, however, the maneuver should be repeated on the opposite side to confirm which ear is involved. Rarely is a response elicited in both the right and left ear-down positions with corresponding nystagmus; such a response is typically associated with head trauma.4
To rule out orthostatic hypotension, a possible source of “faintness” or “dizziness,” measure for changes in blood pressure (eg, decrease of 20 mm Hg systolic, decrease of 10 mm Hg diastolic) and pulse (eg, increase of 30 beats/min) from the supine to standing positions.20 These measurements should be performed after the Dix-Hallpike maneuver because the changes in patient position required to test BP may affect the results of the Dix-Hallpike maneuver. With the exception of positive results on the Dix-Hallpike test, the patient with BPPV will generally have unremarkable findings on the physical exam.3
On the next page: Lab work-up and imaging >>
LABORATORY WORK-UP AND IMAGING
There is no lab work that assists in making the diagnosis of BPPV. Radiographic imaging21 and laboratory testing are not beneficial and are in fact unnecessary and inappropriate in the patient with probable BPPV.6,8,20 There are no radiologic findings in the patient with BPPV alone.4,22 Clinical practice guidelines recommend against radiologic imaging in patients with BPPV, unless the diagnosis is uncertain or there are additional or unrelated exam findings or symptoms that justify testing.4
DIAGNOSIS
The Dix-Hallpike maneuver is considered the gold standard for diagnosing BPPV,4 although it is possible for the Dix-Hallpike test results to be negative in a patient with BPPV. If the test is negative and there is a strong clinical suspicion for BPPV, the patient may need to come back for a second visit to repeat the maneuver.4 The clinician may also consider referral to a specialist who performs vestibular function testing in order to decrease the time from the onset of symptoms to diagnosis and proper treatment.
According to the AAOHNS guidelines for BPPV, the specific diagnostic criteria for posterior canal BPPV include the patient history of repeated episodes of vertigo related to changes in head position; vertigo and nystagmus elicited on physical exam by the Dix-Hallpike test with a latency period between the onset and completion of the test; and an increase in intensity and then resolution within a minute of onset of the provoked vertigo and nystagmus.4
Patients should be questioned about associated hearing loss. Vertigo accompanied by hearing loss is typically not BPPV, but can be caused by Ménière’s disease or labyrinthitis. Patients presenting with Ménière’s disease commonly have sustained vertigo (lasting for hours), tinnitus, and fluctuating hearing loss.4 Vestibular labyrinthitis typically presents with severe vertigo that lasts from days to weeks (constant and not related to movement), severe nausea and vomiting, hearing loss, and tinnitus.4
Orthostatic hypotension, another cause of dizziness, should always be in the differential. Visual changes, ataxia, confusion, slurred speech, and numbness point to central causes of dizziness such as vertebrobasilar ischemic stroke or vertebrobasilar insufficiency.20
Once the BPPV diagnosis is made, it should be documented in the patient’s medical record. Using a diagnosis code for vertigo or dizziness is insufficient because it only describes the patient’s symptoms and is inadequate for follow-up and continuity of care. Nor does such a code provide the patient with the concise diagnosis needed to engage in self-care. Further, the incidence of the disease remains undocumented for purposes of research on BPPV.
On the next page: Treatment and management >>
TREATMENT AND MANAGEMENT
Particle repositioning maneuvers are the recommended treatment for BPPV. These maneuvers have a success rate of greater than 90%5,6 and usually provide immediate resolution of symptoms.23 The canalith repositioning maneuver (CRP; also called the Epley maneuver) and the liberatory maneuver (LM; also called the Semont maneuver) are effective treatments for posterior canal BPPV (see “The Epley Maneuver”).4 The CRP can be performed immediately following positive results on the Dix-Hallpike test.
![]()
The CRP depends on gravity to treat BPPV.24 The otoconia settle in the lowest part of the semicircular canals as the patient is moved through a series of positions. The maneuver requires the patient to be rotated 180° (through four positions) beginning with the affected side and then to the uninvolved side before returning to a sitting position.24 Each position is maintained for at least 30 s. Once the otoconia migrate out of the affected semicircular canal and back into the vestibule, the particles should dissolve.
The LM begins with the patient in a seated position with the head turned away from the affected side (see Figure 2). The clinician quickly moves the patient into a side-lying position toward the affected side, with the head turned upward and supported there for approximately 30 s (Step 1).The clinician then quickly moves the patient through the initial seated position (without pausing) to the opposite side-lying position without changing the head position (Step 2). With the head now facing downward, the patient remains motionless for another 30 s before the clinician brings the patient upright to the original seated position. Although patients are sometimes advised to remain upright for 24 to 48 h following in-office treatment (which is not believed to cause harm), there is insufficient evidence to support this recommendation.4
For ongoing care, current clinical guidelines recommend that practitioners offer either vestibular rehabilitation (performed by a clinician or self-administered by the patient) or provide for watchful waiting and follow-up based on the natural course of spontaneous resolution of symptoms.4
On the next page: Patient education and referral >>
PATIENT EDUCATION
It is important to remember that vertigo is a symptom, while BPPV is a diagnosis. Therefore, merely informing the patient that he or she has vertigo is not sufficient. The patient (and his/her family, if present) should be educated about the cause of the patient’s vertigo. The patient should be provided with information about BPPV that is delivered both verbally and through printed health education materials.
The chances and unpredictability of recurrence should be discussed. Ideally, the patient should be instructed to make a same-day follow-up appointment for treatment should symptoms recur. Patients, particularly the elderly, should be counseled about the risk for falls; fall risk assessment questionnaires with recommendations for prevention of injury are helpful.4
The patient with BPPV should be provided with instructions, including diagrams, of how to perform modified CRP exercises at home. Helpful videos are available on the Internet for patient use. For example, the University of Michigan has created videos for a patient diagnosed with BPPV of the right ear (www.youtube.com/watch?v=BY4UeRmTYmA) and the left ear (www.youtube.com/watch?v=lh72suV2p20).
In self-administered CRP, the patient moves through the same positions used for in-office CRP, except that the patient’s head is extended over the edge of a pillow.24 Patients should be instructed to stop the home exercises once they are symptom free for 24 h or more.
FOLLOW-UP AND REFERRAL
The need for follow-up varies depending on the patient’s response to treatment and the incidence of recurrence. Clinical practice guidelines recommend follow-up within a month of initial observation or treatment to reassess and confirm resolution of symptoms.4 Referral to specialists for treatment should be considered without delay if the primary care clinician does not feel confident treating BPPV and/or is unsure of the diagnosis based on the results of the Dix-Hallpike test, particularly if the patient’s quality of life is affected and safety is a concern.
On the next page: Conclusion >>
CONCLUSION
BPPV is a common disorder that presents most often in the primary care setting. However, most dizzy patients never see a specialist or clinician who is skilled in providing vestibular evaluation and treatment. To avoid missing the diagnosis or ordering unnecessary diagnostic and laboratory tests, clinicians can readily perform the Dix-Hallpike test and CRP in the office on patients with suspected BPPV.
CRP and vestibular rehabilitation have been proven effective in treating chronic disequilibrium and vertigo. Studies have reported that the mean wait time from initial presentation of symptoms to successful treatment was 92 weeks; 85% of these patients had immediate symptom resolution after the first treatment session with a specialist trained in CRP.25 Improvement in recognition of BPPV at the primary care level will markedly reduce the lag time to treatment.
The author would like to thank Alan L. Desmond, AuD, and Dr. Brian Collie, ENT, for their mentorship and expertise in vestibular disorders.
1. Kroenke K, Hoffman RM, Einstadter D. How common are various causes of dizziness? A critical review. Southern Med J. 2000;93:160-167.
2. Thompson TL, Amedee R. Vertigo: a review of common peripheral and central vestibular disorders.Ochsner J. 2009;9:20-26.
3. Li JC, Egan RE. Neurologic manifestations of benign positional vertigo (2012). Medscape. http://emedicine.medscape.com/article/1158940-overview. Accessed November 14, 2013.
4. Bhattacharyya N, Baugh RF, Orvidas L, et al. Clinical practice guideline: benign paroxysmal positional vertigo. Otolaryng Head Neck Surg. 2008; 139:S47-S81.
5. Steenerson RL, Cronin GW, Marbach PM. Effectiveness of treatment techniques in 923 cases of benign paroxysmal positional vertigo. Laryngoscope. 2005;115:226-231.
6. Parnes LS, Agrawal SK, Atlas J. Diagnosis and management of benign paroxysmal positional vertigo (BPPV). CMAJ. 2003;169:681-693.
7. Desmond AL. Vestibular Function: Clinical and Practice Management. 2nd ed. New York, NY: Thieme Medical Publishers; 2011:1-276.
8. Newman-Toker DE, Camargo CA, Hsieh Y, et al. Disconnect between charted vestibular diagnoses and emergency department management decisions: a cross-sectional analysis from a nationally representative sample. Acad Emerg Med. 2009;16:970-977.
9. Ahsan SF, Syamal MN, Yaremchuk K, et al. The costs and utility of imaging in evaluating dizzy patients in the emergency room. Laryngoscope. 2013;123:2250-2253.
10. Chan Y. Differential diagnosis of dizziness. Curr Opin Otolaryng Head Neck Surg. 2009;17:200-203.
11. von Brevern M, Radtke A, Lezius F, et al. Epidemiology of benign paroxysmal positional vertigo: a population based study. J Neurol Neurosurg Psychiatry. 2007;78:710-715.
12. Oghalai JS, Manolidis S, Barth JL, et al. Unrecognized benign paroxysmal positional vertigo in elderly patients. Otolaryng Head Neck Surg. 2000;122:630-634.
13. Neuhauser HK, Radtke A, von Brevern, et al. Burden of dizziness and vertigo in the community. Arch Intern Med. 2008;168:2118-2124.
14. Holmes S, Padgham ND. A review of the burden of vertigo. J Clin Nurs. 2011;20(19-20):2690-2701.
15. Pollak L, Segal P, Stryjer R, Stern HG. Beliefs and emotional reactions in patients with benign paroxysmal positional vertigo: a longitudinal study. Am J Otolaryng. 2012;33:221-225.
16. US Department of Health and Human Services. Confronting the new health care crisis: improving health care quality and lowering costs by fixing our medical liability system (2002). http://aspe.hhs.gov/daltcp/reports/litrefm.pdf. Accessed November 14, 2013.
17. Naylor MD, Kurtzman ET. The role of nurse practitioners in reinventing primary care. Health Affairs. 2010;29:893-899.
18. Desmond AL. Dizziness Reference Guide. Chatham, IL: Micromedical Technologies; 2009.
19. Goebel JA. The ten-minute examination of the dizzy patient. Semin Neurol. 2001;21:391-398.
20. Post RE, Dickerson LM. Dizziness: a diagnostic approach.Am Fam Physician. 2010;82:361-368.
21. Korres S, Riga M, Papacharalampous G, et al. Relative diagnostic importance of electronystagmography and magnetic resonance imaging in vestibular disorders. J Laryngol Otol. 2009;123:851-856.
22. American College of Radiology Expert Panel on Neurologic Imaging. American College of Radiology Appropriateness Criteria [report], 2008. www.acr.org/~/media/ACR/Documents/AppCriteria/Diagnostic/Vertigo HearingLoss.pdf. Accessed November 14, 2013.
23. Lee S-H, Kim JS. Benign paroxysmal positional vertigo. J Clin Neurol. 2010;6:51-63.
24. Helminski JO, Zee DS, Janssen I, Hain TC. Effectiveness of particle repositioning maneuvers in the treatment of benign paroxysmal positional vertigo: a systematic review. Phys Ther. 2010;90:663-678.
25. Fife D, Fitzgerald JE. Do patients with benign paroxysmal positional vertigo receive prompt treatment? Analysis of waiting times and human and financial costs associated with current practice. Int Journal Audiol. 2005;44:50-57.
1. Kroenke K, Hoffman RM, Einstadter D. How common are various causes of dizziness? A critical review. Southern Med J. 2000;93:160-167.
2. Thompson TL, Amedee R. Vertigo: a review of common peripheral and central vestibular disorders.Ochsner J. 2009;9:20-26.
3. Li JC, Egan RE. Neurologic manifestations of benign positional vertigo (2012). Medscape. http://emedicine.medscape.com/article/1158940-overview. Accessed November 14, 2013.
4. Bhattacharyya N, Baugh RF, Orvidas L, et al. Clinical practice guideline: benign paroxysmal positional vertigo. Otolaryng Head Neck Surg. 2008; 139:S47-S81.
5. Steenerson RL, Cronin GW, Marbach PM. Effectiveness of treatment techniques in 923 cases of benign paroxysmal positional vertigo. Laryngoscope. 2005;115:226-231.
6. Parnes LS, Agrawal SK, Atlas J. Diagnosis and management of benign paroxysmal positional vertigo (BPPV). CMAJ. 2003;169:681-693.
7. Desmond AL. Vestibular Function: Clinical and Practice Management. 2nd ed. New York, NY: Thieme Medical Publishers; 2011:1-276.
8. Newman-Toker DE, Camargo CA, Hsieh Y, et al. Disconnect between charted vestibular diagnoses and emergency department management decisions: a cross-sectional analysis from a nationally representative sample. Acad Emerg Med. 2009;16:970-977.
9. Ahsan SF, Syamal MN, Yaremchuk K, et al. The costs and utility of imaging in evaluating dizzy patients in the emergency room. Laryngoscope. 2013;123:2250-2253.
10. Chan Y. Differential diagnosis of dizziness. Curr Opin Otolaryng Head Neck Surg. 2009;17:200-203.
11. von Brevern M, Radtke A, Lezius F, et al. Epidemiology of benign paroxysmal positional vertigo: a population based study. J Neurol Neurosurg Psychiatry. 2007;78:710-715.
12. Oghalai JS, Manolidis S, Barth JL, et al. Unrecognized benign paroxysmal positional vertigo in elderly patients. Otolaryng Head Neck Surg. 2000;122:630-634.
13. Neuhauser HK, Radtke A, von Brevern, et al. Burden of dizziness and vertigo in the community. Arch Intern Med. 2008;168:2118-2124.
14. Holmes S, Padgham ND. A review of the burden of vertigo. J Clin Nurs. 2011;20(19-20):2690-2701.
15. Pollak L, Segal P, Stryjer R, Stern HG. Beliefs and emotional reactions in patients with benign paroxysmal positional vertigo: a longitudinal study. Am J Otolaryng. 2012;33:221-225.
16. US Department of Health and Human Services. Confronting the new health care crisis: improving health care quality and lowering costs by fixing our medical liability system (2002). http://aspe.hhs.gov/daltcp/reports/litrefm.pdf. Accessed November 14, 2013.
17. Naylor MD, Kurtzman ET. The role of nurse practitioners in reinventing primary care. Health Affairs. 2010;29:893-899.
18. Desmond AL. Dizziness Reference Guide. Chatham, IL: Micromedical Technologies; 2009.
19. Goebel JA. The ten-minute examination of the dizzy patient. Semin Neurol. 2001;21:391-398.
20. Post RE, Dickerson LM. Dizziness: a diagnostic approach.Am Fam Physician. 2010;82:361-368.
21. Korres S, Riga M, Papacharalampous G, et al. Relative diagnostic importance of electronystagmography and magnetic resonance imaging in vestibular disorders. J Laryngol Otol. 2009;123:851-856.
22. American College of Radiology Expert Panel on Neurologic Imaging. American College of Radiology Appropriateness Criteria [report], 2008. www.acr.org/~/media/ACR/Documents/AppCriteria/Diagnostic/Vertigo HearingLoss.pdf. Accessed November 14, 2013.
23. Lee S-H, Kim JS. Benign paroxysmal positional vertigo. J Clin Neurol. 2010;6:51-63.
24. Helminski JO, Zee DS, Janssen I, Hain TC. Effectiveness of particle repositioning maneuvers in the treatment of benign paroxysmal positional vertigo: a systematic review. Phys Ther. 2010;90:663-678.
25. Fife D, Fitzgerald JE. Do patients with benign paroxysmal positional vertigo receive prompt treatment? Analysis of waiting times and human and financial costs associated with current practice. Int Journal Audiol. 2005;44:50-57.
Colorectal Cancer Screening: What’s Accurate and Cost-Effective?
CE/CME No: CR-13111
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the difference between indirect and direct screening methods for CRC and when to utilize each.
• Understand the age continuum of screening patients with and without increased risk factors for CRC.
• Decide which type of direct screening method is the best choice when looking at sensitivity and specificity and patient preference.
• Understand the risks, benefits, and limitations of each procedure available for CRC screening.
FACULTY
Carolyn Mueller, Molly Perry, and Lisa DeCicco are recent graduates of the Pace University–Lenox Hill Hospital Physician Assistant Program in New York. Ellen D. Mandel is a Clinical Professor in the Pace PA Program and an Associate Professor in the PA Program at Seton Hall University, South Orange, New Jersey.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because colorectal cancer is often asymptomatic, routine screening is essential to detect lesions at an early stage. The evolution of health care has brought new and improved screening methods for colorectal cancer, including CT colonography. This article weighs the pros and cons of the available screening methods used to detect colorectal cancer in the general population today.
Colorectal cancer (CRC) is the second-leading cause of death from cancer in Europe and the United States and the third most commonly diagnosed cancer in both men and women in the US.1-5 According to the American Cancer Society, 102,480 new cases of colon cancer and 40,340 new cases of rectal cancer will be diagnosed in 2013.5 Although screening rates remain low and the incidence of new CRC diagnoses rises annually, mortality rates are decreasing, most likely due to screening and improved treatment.5
CLINICAL PRESENTATION OF COLORECTAL CANCER
CRC is often asymptomatic until it reaches an advanced stage. At this point, symptoms include weight loss, night sweats, fever, loss of appetite, blood in the stool, pencil-shaped stools, diarrhea, constipation, anemia, and/or dizzy spells.6,7 On abdominal exam, a clinician may note dullness to percussion over the right or left lower quadrant, palpate a mass in the right or left lower quadrant, and/or elicit tenderness or guarding upon deep palpation. The clinician may be inclined to do a fecal occult blood test (FOBT) to confirm active GI bleeding.1,3
CURRENT SCREENING RECOMMENDATIONS
The US Preventive Services Task Force (USPSTF) currently recommends that screening for CRC—with FOBT, sigmoidoscopy, or colonoscopy—start at age 50 and continue through age 75.8 For patients whose first-degree relative has a history of CRC, initial screening should start at age 40.
It is recommended that people ages 76 to 85 make personalized, informed screening decisions in conjunction with their medical provider. Patients ages 85 or older should not be screened for CRC because of the estimated five-year time frame between the detection of cancer and the onset of symptoms or death. In the unlikely circumstance that the patient is screened and a lesion is found, the patient would not benefit but most likely experience harm from treatment efforts; however, this should be decided on a case-by-case basis.8
CONSIDERATIONS IN SCREENING THE ELDERLY
Much of the data on older populations are outdated, and thus more research needs to be conducted in this population. However, data from two studies dating from 2006 provide tools to help clinicians decide if screening in the older population is beneficial.
One study of the benefit of screening for CRC in the older population stated that clinicians must assess both the burden of chronic illness and the patient’s age as part of their evaluation. This study concluded that, because of the risks and costs that are associated with CRC screening, it is important to identify only those individuals who are likely to benefit from screening, rather than screening the elderly population in general.9
Another study examined the benefit of screening colonoscopy in two elderly groups (ages 75 to 79 and age ≥ 80) versus younger patients (ages 50 to 54; the control group). It was found that
• Screening in the older population may increase the risk for perforation and respiratory depression secondary to sedation,
• Screening may take longer to complete, and
• Screening may be challenging with less successful bowel preparation.10
Further, the prevalence of neoplasia was lowest in the control group (13.8%) compared to the 75-to-79-year-old (26.5%) and oldest (≥ 80; 28.6%) groups. The mean extension of life expectancy was much lower in the oldest group, compared to the control group (0.13 years versus 0.85 years), which represents a 6.5-fold difference. These results suggest that the benefit of colonoscopy screening in elderly persons ages 76 and older results in smaller gains in life expectancy and does not outweigh the risks. This test, therefore, should only be used when the patient expresses a preference for colonoscopy and the clinician feels it will significantly benefit the patient.10
On the next page: Screening methods >>
SCREENING METHODS
Current screening methods for CRC can be divided into two distinct categories: indirect and direct.1 Indirect screening tests include FOBT, fecal immunochemical testing, and stool DNA testing. Cancers are identified by detection of byproducts in the patient’s stool, such as blood or epithelial cells containing DNA of the adenomatous polyposis coli gene. These tests are simple to perform, have high specificity, and are relatively inexpensive, but they need to be repeated annually and have poor sensitivity.1 Positive test results of indirect screening often warrant further diagnostic testing, ultimately utilizing one of the direct screening methods.
Direct screening methods used to detect CRC—from least to most frequently employed—include barium enema (BE), CT colonography (CTC), and colonoscopy. Another direct method, flexible sigmoidoscopy, is not frequently used today, and when used, serves only as an intermediate step to colonoscopy. Direct screening provides visualization of the contour of the colon wall, the internal mucosa, and abnormal architecture. It is important to keep in mind that these tests require that the patient adhere to pretest preparation, may require patient sedation, and are more invasive and costly than indirect tests.1
Although the USPSTF has established recommendations for both test types, questions still remain about what constitutes the most cost-effective and accurate combination of screening tests for detecting CRC.8
On the next page: Barium enema >>
BARIUM ENEMA
Procedure
The BE, also known as a lower GI series, was the first screening test to allow the clinician to identify polyps or masses as outlined by barium sulfate.3 This test requires the patient to lie in an oblique position on his/her left side while barium sulfate (known as single-contrast BE, or SCBE), sometimes followed by air (known as double-contrast BE, or DCBE), is flowed through a tube inserted into the rectum. As the colon fills, the radiologist takes multiple overhead x-ray images. The patient is then required to roll on the table several times, causing the barium to coat the entire mucosa of the colon and rectum, which allows for visualization from various angles (see Figure 1).11
Patient Experience
Screening with the BE has some disadvantages to the patient. Patients may experience discomfort at multiple points: during the instillation of gas and barium into the colon, during the maintenance of the gas and barium levels, and during the maneuvering and holding positions of the procedure itself.2 Some patients, interviewed after a BE was performed, indicated feeling embarrassed during the procedure.6 Although the ability to evaluate images during the exam allows the radiologist to share preliminary results with the patient, the immediate disclosure of bad news is deemed somewhat inappropriate. If the procedure reveals positive findings, the radiologist must be sure to speak with the patient in a private area after the procedure. The patient is in a vulnerable state while in the exam room, and the exam room staff may not be adequately equipped to handle the emotional impact, properly address patient questions, or provide counseling.2,6
Advantages and Disadvantages
Few studies are now being done on the advantages and disadvantages of performing a BE compared to a colonoscopy or CTC—most likely due to the belief that colonoscopy is the better choice. BEs are considered to be one of the safer of the direct screening tests for CRC because sedation is not required and, compared to colonoscopy, the rate of colon perforation is lower (0.02% to 0.04% for BE versus 0.016% to 0.2% for colonoscopy).12,13
In a small study of 15 asymptomatic men age 71, it was found that BEs have a lower sensitivity for detecting CRC as compared with colonoscopy or CTC.2 Sensitivity for lesions ≥ 10 mm is only 48% and for lesions ≥ 6 mm is only 35%, proving the BE to be a highly ineffective screening test for CRC (see Table 1).3
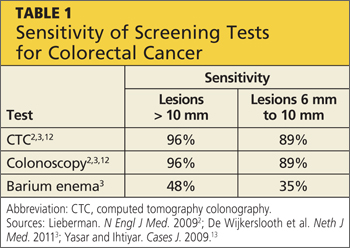
In a randomized study of 5,025 symptomatic patients with abnormal bowel movements and/or abdominal pain, BE has a detection rate of only 5% compared to the much higher sensitivity of CTC or colonoscopy.14
A third study calls attention to the risks associated with the DCBE exam, noting that it is less invasive and less dangerous than colonoscopy, as it does not require sedation and poses less risk for perforation of the lining of the colon.15 The study authors concluded that DCBE has a high sensitivity for clinically significant neoplasms (> 6 mm) but not for small polyps, which may be captured with other tests. DCBE may also supplement incomplete colonoscopy to rule out obstruction.
However, because of the loss of biopsy capabilities, further testing is required when abnormalities are found during the DCBE, diminishing the potential cost effectiveness of the exam. The study authors suggested that DCBEs may be used to screen those who are asymptomatic and seem to have minimal risk factors.15
Limitations
Limitations to successful BE screening include patient compliance and test result interpretation skills. With interpretation skills declining due to limited training of professionals to read BEs, results are becoming less accurate, and the test itself can be seen as less reliable. BEs are less popular, and therefore skills in reading the films are becoming outdated.2 If BE screening is to be used as the primary direct screening tool for CRC, it is imperative that radiologists and gastroenterology physicians and clinicians be well trained in this GI procedure.
Also, patients undergoing BE absorb about 15 mGy of radiation per procedure, versus 0.01 mGy to 0.15 mGy absorbed with a typical chest x-ray.16 For patients with a history of increased radiation exposure or if radiation exposure is a major concern of the patient, BE may not be an appropriate first choice.
On the next page: Colonoscopy >>
COLONOSCOPY
Colonoscopy is an endoscopic technique that allows internal inspection of the entire colorectal tract (see Figure 2).3 Although the most invasive of the exams being reviewed (and requiring extensive bowel prep), it is the gold standard for CRC screening.3
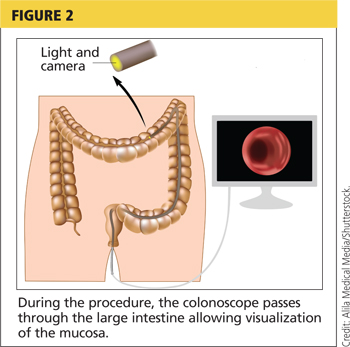
Current literature indicates that colonoscopy should be the screening method of choice for patients who have symptoms of colorectal cancer, positive results with an indirect screening exam such as FOBT, or who fall within a high-risk category. Persons at high risk for CRC include those with a significant family history, persons ages 50 and older, African Americans, and persons with an intestinal inflammatory condition, diabetes mellitus, obesity, sedentary lifestyle, positive smoking status, and low-fiber/high-fat diet.
Procedure
This procedure is performed on an outpatient basis; bowel prep is required one day prior to the procedure so that the bowel movements are clear of fecal matter. The patient receives short-lasting sedation, such as midazolam, via an IV line prior to being brought into the exam room. The procedure itself can take up to 30 minutes, with the patient comfortably sedated in an oblique position. The patient is allowed to recover for several hours, or until awake and able to pass flatus.
Patient Experience
The patient may experience discomfort during the bowel prep phase, as with BE preparation. The patient may be uncomfortable during insertion and manipulation of the colonoscope and also with gaseous insufflation that is used to improve visualization.
Advantages and Disadvantages
The advantages of colonoscopy over the other direct methods are the ability to immediately remove early cancer and colonic polyps and the ability to obtain histologic samples. A suspicious mass can be biopsied during the procedure and, depending on the size, may be completely excised. The histology of the biopsied tissue samples can aid in determining need for further treatment or establish an appropriate surveillance interval for the patient.3
The disadvantages of colonoscopy are related to the possible complications of the procedure, including colon perforation and postpolypectomy bleeding. The risk for these events is estimated to be between 0.1% and 0.3%.3 In addition, the short-lasting sedation that is used during the procedure poses the risk for possible respiratory collapse.6 Therefore, each patient requires medical clearance prior to administering sedation. The risk for a serious adverse event is 3 to 5 per 1,000 colonoscopies, and procedure-related mortality, while rare, has been reported.2,4,14
Limitations
Studies have found colonoscopy to be the most expensive of the direct screening tests, which may pose a problem for uninsured or underinsured patients.4,14
A collection of colonoscopy studies done on patients ages 50 to 66 showed an adenoma miss rate of 20% to 26% for any adenoma < 10 mm, and a 2.1% miss rate for adenomas ≥ 10 mm.3 Adenoma detection rates are dependent on optimal bowel preparation, complete examination of the colon, and the time the clinician spends examining all surfaces of the colon mucosa when withdrawing the colonoscope.3
Colonoscopy has the lowest adherence rate of all the CRC screening tests, which is not surprising since it is invasive, involves sedation, and requires thorough preparation. However, colonoscopies may be performed at longer intervals (up to 10 years) compared to other screening tests; the risk for developing CRC after a negative colonoscopy exam remains low.3
On the next page: CT colonography >>
CT COLONOGRAPHY
CTC is an emerging CRC screening test that is also known as virtual colonoscopy. According to available studies, CTC and colonoscopy might be equivalent for diagnosing cancer.14
Procedure
The preparation for CTC requires the patient to consume a low-residue diet one day prior to the procedure, which is considered to be an advantage over colonoscopy due to the decreased bowel preparation.3 With this procedure, a small rectal catheter is inserted into the anus and advanced to the rectum to allow carbon dioxide to be instilled for bowel insufflation. The patient lies supine on the table for a CT scan of the abdomen with the resulting 2D images visualizing polyps and CRC, if present.3 If necessary, 3D images can be compiled by a specialized software program to obtain a 360-degree view of the colon. In fact, recent studies show that 3D CTC is preferred to 2D because 3D polyp measurements are more representative of the true polyp size found on optic colonoscopy or surgery than are 2D measurements.17
Patient Experience
In one study, CTC screening was described as uncomfortable but not painful and was reported to be the most impersonal of all three tests because of less direct interaction, reducing patient embarrassment. This study reviewed qualitative interviews with patients regarding the fairly new CTC procedure and found that patients received little visual or verbal feedback and were confused regarding their test outcome immediately after the procedure.6
CTC was preferred by 72% of patients compared to colonoscopy and by 97% of patients compared to DCBE.18 In a study that evaluated the performance characteristics of CTC among 1,233 asymptomatic patients, 68% deemed CTC to be more convenient than colonoscopy, and more patients indicated that they preferred CTC over colonoscopy for screening (49.8% vs 41.1%; 9.2% had no preference).19
Advantages and Disadvantages
Compared with colonoscopy, CTC is comparably sensitive but safer and more acceptable to patients.2,6,14 CTC has a sensitivity of 96% for detecting lesions > 10 mm in diameter, but sensitivity decreases to 89% for lesions 6 mm to 10 mm in diameter (see Table 1).2,3,14
The follow-up screening intervals for CTC parallel those of colonoscopy. In a recent audit of 1,011 screening participants with a negative baseline CTC, a single carcinoma occurred during an average follow-up period of 4.73 ± 1.15 years.1
The current Colonography Reporting and Data System (C-RADS) guidelines for CTC interpretation recommend 6 mm as the minimum size for polyp reporting.17 This reporting threshold will result in a 77% reduction rate in invasive endoscopic procedures since it minimizes the number of cases that are sent for colonoscopy after CTC screening.17
A study performed by the American College of Radiology Imaging Network found there would be an approximate 12% referral rate for colonoscopy when using the current 6-mm polyp size threshold, but the referral rate would increase to 17% if a 5-mm threshold were used.17 The American Gastroenterological Association has stated that diminutive lesions—those measuring ≤ 5 mm—are of little to no clinical significance because only a fraction of them are neoplastic. Of these, fewer than 1% are histologically advanced, and essentially none are malignant.6 By not reporting diminutive lesions, there would be an incremental gain in the cost-effectiveness of the CTC scan and only a 1.3% loss in clinical CRC prevention efficacy.
It is widely accepted that any polyp ≥ 10 mm detected with CTC screening indicates a need for polypectomy via colonoscopy or surgery.20 For lesions ranging from 6.1 mm to 9.9 mm in diameter, CTC is suggested as a surveillance tool; patients may receive repeat CTC every 1 to 3 years until resection via colonoscopy is warranted.17
Limitations
CTC screening has many limitations, and evidence is lacking for a reduction of CRC incidence or mortality after CTC. Research has revealed a discrepancy between the polyp size measured by CTC versus the true polyp size seen on optic colonoscopy; CTC measurement can underestimate the size of a polyp by nearly 1.2 mm. Considering that C-RADS level 2 includes polyps 6.1 to 9.9 mm and level 3 includes polyps ≥ 10 mm, which automatically requires further investigation with colonoscopy, such a 1.2-mm discrepancy in measurement can make all the difference.17
CTC requires some bowel preparation, special resources, and expertise. The cost-effectiveness and risk profiles will vary, depending on whether referral for colonoscopy is required. Also, treatment recommendations for patients with polyps < 6 mm in diameter are uncertain.
CTC screening exposes the patient to increased amounts of ionizing radiation, which raises concern regarding risk for radiation-induced cancers. The effective dose to the whole body during a CTC is 6 to 20 mSv, compared to 0.02 mSv for a chest x-ray.16 One study estimated that performing CTC screening every five years from ages 50 to 80 would prevent the development of 24 CRC cases for every one radiation-induced cancer.21 Thus, the risks of radiation must be weighed against the benefits of screening, and the decision is often made by the individual patient.
Finally, the greatest limitation of CTC is that further testing may be required based on the preliminary results. Therefore, the patient may undergo two procedures rather than just one all-inclusive procedure, such as colonoscopy.
On the next page: Conclusion >>
CONCLUSION
CRC is the second leading cause of death from cancer among men and women in the United States despite the fact that it is largely preventable through diagnostic screening. Patients need education about the different types of CRC screening and about which method may be best for them, given their preferences, family history, personal history, age, and symptoms. All screening tests, direct or indirect, are cost-effective compared with no screening at all.20 Regardless of current recommendations, any screening that the patient is comfortable with should be encouraged.
BE was the first diagnostic tool to provide clinicians with the ability to visualize the patient’s lower gastrointestinal tract. It is becoming technologically outdated, however, and is no longer accepted as a primary diagnostic tool for CRC screening.
Colonoscopy, the most expensive direct screening test, provides complete visualization of the colon and allows for immediate biopsy and possible resection of a suspicious mass. This procedure is the most cost-effective direct screening method because it is comprehensive compared to BE and CTC, which may result in further investigation via colonoscopy if a mass is identified. Although colonoscopy is the most specific and sensitive for CRC screening, the outcome of the test strongly correlates with patient compliance with bowel preparation as well as clinician experience and expertise in performing a thorough exam.
While most US guidelines recommend colonoscopy as the gold standard diagnostic test, CTC is a reliable alternative for those patients who refuse colonoscopy. Future research on this newer method should consider altering the C-RADS threshold that necessitates follow-up with colonoscopy to account for the variation in polyp measurement. CTC is not a stand-alone replacement for the other direct CRC screening tests but is useful as an adjunct to increase overall patient compliance. Perhaps with time, this test may evolve to be a more prevailing recommendation for the preventive screening of CRC.
1. de Haan MC, Halligan S, Stoker J. Does CT colonography have a role for population-based colorectal cancer CRC screening? Eur Radiol. 2012;22: 1495-1503.
2. Lieberman D. Screening for colorectal cancer. N Engl J Med. 2009;361: 1179-1187.
3. de Wijkerslooth TR, Bossuyt PM, Dekker E. Strategies in screening for colon carcinoma. Neth J Med. 2011;69:112-119.
4. Whitlock E, Lin J, Liles E, et al. Screening for colorectal cancer: an updated systematic review. Evidence Synthesis No. 65, Part 1. AHRQ Publication No. 08-05124-EF-1. Rockville, Maryland, Agency for Healthcare Research and Quality, October 2008.
5. Colorectal cancer: What are the key statistics about colorectal cancer? American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics. Accessed October 14, 2013.
6. Von Wagner C, Knight K, Halligan S, et al. Patient experiences of colonoscopy, barium enema and CT colonography: a qualitative study. Br J Radiol. 2009;82:13-19.
7. Cappell MS. Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin N Am. 2008;37:1-24.
8. Screening for colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. US Preventive Services Task Force web site. www.uspreventiveservicestaskforce.org/uspstf/uspscolo.htm. Published 2008. Accessed October 14, 2013.
9. Gross CP, McAvay GJ, Krumholz HM, et al. The effect of age and chronic illness on life expectancy after a diagnosis of colorectal cancer: implications for screening. Ann Intern Med. 2006;145:646-653.
10. Lin OS, Kozarek RA, Schembre DB, et al. Screening colonoscopy in very elderly patients: prevalence of neoplasia and estimated impact on life expectancy. JAMA. 2006;295:2357-2365.
11. Colorectal cancer early detection: colorectal cancer screening tests. American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-screening-tests-used. Accessed October 14, 2013.
12. Lohsiriwat V. Colonoscopic perforation: incidence, risk factors, management and outcome. World J Gastroenterol 2010;16:425-430.
13. Yasar NF, Ihtiyar E. Colonic perforation during barium enema in a patient without known colonic disease: a case report. Cases J. 2009;2:6716.
14. Halligan S, Lilford RJ, Wardle J, et al. Design of a multicentre randomized trial to evaluate CT colonography versus colonoscopy or barium enema for diagnosis of colonic cancer in older symptomatic patients: The SIGGAR study. Trials. 2007;8:1-9.
15. Lohsiriwat V, Prapasrivorakul S, Suthikeeree W. Colorectal cancer screening by double contrast barium enema in Thai people.Asian Pacific J Cancer Prev. 2012;13:1273-1276.
16. Mayo JR, Aldrich J, Muller NL; Fleischner Society. Radiation exposure at chest CT: a statement of the Fleischner Society. Radiology. 2003;228:15-21.
17. Summers RM. Polyp size measurement at CT colonography: what do we know and what do we need to know? Radiology. 2010;255(3):707-720.
18. Gluecker TM, Johnson CD, Harmsen WS, et al. Colorectal cancer screening with CT colonography, colonoscopy, and double-contrast barium enema examination: prospective assessment of patient perceptions and p. Radiology. 2003;227:378-384.
19. Pickhardt PJ, Choi JR, Hwang I, et al. Computed tomographic virtual colonoscopy to screen for colorectal neoplasia in asymptomatic adults. N Engl J Med. 2003;349:2191-2200.
20. Pickhardt P, Hassan C, Laghi A, et al. Cost-effectiveness of colorectal cancer screening with computed tomography colonography. Cancer. 2007;109:2213-2221.
21. Berrington de Gonzalez A, Kim KP, Knudsen AB, et al. Radiation-related cancer risks from CT colongraphy screening: a risk-benefit analysis. AJR Am J Roentgenol. 2011;196:816-823.
CE/CME No: CR-13111
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the difference between indirect and direct screening methods for CRC and when to utilize each.
• Understand the age continuum of screening patients with and without increased risk factors for CRC.
• Decide which type of direct screening method is the best choice when looking at sensitivity and specificity and patient preference.
• Understand the risks, benefits, and limitations of each procedure available for CRC screening.
FACULTY
Carolyn Mueller, Molly Perry, and Lisa DeCicco are recent graduates of the Pace University–Lenox Hill Hospital Physician Assistant Program in New York. Ellen D. Mandel is a Clinical Professor in the Pace PA Program and an Associate Professor in the PA Program at Seton Hall University, South Orange, New Jersey.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because colorectal cancer is often asymptomatic, routine screening is essential to detect lesions at an early stage. The evolution of health care has brought new and improved screening methods for colorectal cancer, including CT colonography. This article weighs the pros and cons of the available screening methods used to detect colorectal cancer in the general population today.
Colorectal cancer (CRC) is the second-leading cause of death from cancer in Europe and the United States and the third most commonly diagnosed cancer in both men and women in the US.1-5 According to the American Cancer Society, 102,480 new cases of colon cancer and 40,340 new cases of rectal cancer will be diagnosed in 2013.5 Although screening rates remain low and the incidence of new CRC diagnoses rises annually, mortality rates are decreasing, most likely due to screening and improved treatment.5
CLINICAL PRESENTATION OF COLORECTAL CANCER
CRC is often asymptomatic until it reaches an advanced stage. At this point, symptoms include weight loss, night sweats, fever, loss of appetite, blood in the stool, pencil-shaped stools, diarrhea, constipation, anemia, and/or dizzy spells.6,7 On abdominal exam, a clinician may note dullness to percussion over the right or left lower quadrant, palpate a mass in the right or left lower quadrant, and/or elicit tenderness or guarding upon deep palpation. The clinician may be inclined to do a fecal occult blood test (FOBT) to confirm active GI bleeding.1,3
CURRENT SCREENING RECOMMENDATIONS
The US Preventive Services Task Force (USPSTF) currently recommends that screening for CRC—with FOBT, sigmoidoscopy, or colonoscopy—start at age 50 and continue through age 75.8 For patients whose first-degree relative has a history of CRC, initial screening should start at age 40.
It is recommended that people ages 76 to 85 make personalized, informed screening decisions in conjunction with their medical provider. Patients ages 85 or older should not be screened for CRC because of the estimated five-year time frame between the detection of cancer and the onset of symptoms or death. In the unlikely circumstance that the patient is screened and a lesion is found, the patient would not benefit but most likely experience harm from treatment efforts; however, this should be decided on a case-by-case basis.8
CONSIDERATIONS IN SCREENING THE ELDERLY
Much of the data on older populations are outdated, and thus more research needs to be conducted in this population. However, data from two studies dating from 2006 provide tools to help clinicians decide if screening in the older population is beneficial.
One study of the benefit of screening for CRC in the older population stated that clinicians must assess both the burden of chronic illness and the patient’s age as part of their evaluation. This study concluded that, because of the risks and costs that are associated with CRC screening, it is important to identify only those individuals who are likely to benefit from screening, rather than screening the elderly population in general.9
Another study examined the benefit of screening colonoscopy in two elderly groups (ages 75 to 79 and age ≥ 80) versus younger patients (ages 50 to 54; the control group). It was found that
• Screening in the older population may increase the risk for perforation and respiratory depression secondary to sedation,
• Screening may take longer to complete, and
• Screening may be challenging with less successful bowel preparation.10
Further, the prevalence of neoplasia was lowest in the control group (13.8%) compared to the 75-to-79-year-old (26.5%) and oldest (≥ 80; 28.6%) groups. The mean extension of life expectancy was much lower in the oldest group, compared to the control group (0.13 years versus 0.85 years), which represents a 6.5-fold difference. These results suggest that the benefit of colonoscopy screening in elderly persons ages 76 and older results in smaller gains in life expectancy and does not outweigh the risks. This test, therefore, should only be used when the patient expresses a preference for colonoscopy and the clinician feels it will significantly benefit the patient.10
On the next page: Screening methods >>
SCREENING METHODS
Current screening methods for CRC can be divided into two distinct categories: indirect and direct.1 Indirect screening tests include FOBT, fecal immunochemical testing, and stool DNA testing. Cancers are identified by detection of byproducts in the patient’s stool, such as blood or epithelial cells containing DNA of the adenomatous polyposis coli gene. These tests are simple to perform, have high specificity, and are relatively inexpensive, but they need to be repeated annually and have poor sensitivity.1 Positive test results of indirect screening often warrant further diagnostic testing, ultimately utilizing one of the direct screening methods.
Direct screening methods used to detect CRC—from least to most frequently employed—include barium enema (BE), CT colonography (CTC), and colonoscopy. Another direct method, flexible sigmoidoscopy, is not frequently used today, and when used, serves only as an intermediate step to colonoscopy. Direct screening provides visualization of the contour of the colon wall, the internal mucosa, and abnormal architecture. It is important to keep in mind that these tests require that the patient adhere to pretest preparation, may require patient sedation, and are more invasive and costly than indirect tests.1
Although the USPSTF has established recommendations for both test types, questions still remain about what constitutes the most cost-effective and accurate combination of screening tests for detecting CRC.8
On the next page: Barium enema >>
BARIUM ENEMA
Procedure
The BE, also known as a lower GI series, was the first screening test to allow the clinician to identify polyps or masses as outlined by barium sulfate.3 This test requires the patient to lie in an oblique position on his/her left side while barium sulfate (known as single-contrast BE, or SCBE), sometimes followed by air (known as double-contrast BE, or DCBE), is flowed through a tube inserted into the rectum. As the colon fills, the radiologist takes multiple overhead x-ray images. The patient is then required to roll on the table several times, causing the barium to coat the entire mucosa of the colon and rectum, which allows for visualization from various angles (see Figure 1).11
Patient Experience
Screening with the BE has some disadvantages to the patient. Patients may experience discomfort at multiple points: during the instillation of gas and barium into the colon, during the maintenance of the gas and barium levels, and during the maneuvering and holding positions of the procedure itself.2 Some patients, interviewed after a BE was performed, indicated feeling embarrassed during the procedure.6 Although the ability to evaluate images during the exam allows the radiologist to share preliminary results with the patient, the immediate disclosure of bad news is deemed somewhat inappropriate. If the procedure reveals positive findings, the radiologist must be sure to speak with the patient in a private area after the procedure. The patient is in a vulnerable state while in the exam room, and the exam room staff may not be adequately equipped to handle the emotional impact, properly address patient questions, or provide counseling.2,6
Advantages and Disadvantages
Few studies are now being done on the advantages and disadvantages of performing a BE compared to a colonoscopy or CTC—most likely due to the belief that colonoscopy is the better choice. BEs are considered to be one of the safer of the direct screening tests for CRC because sedation is not required and, compared to colonoscopy, the rate of colon perforation is lower (0.02% to 0.04% for BE versus 0.016% to 0.2% for colonoscopy).12,13
In a small study of 15 asymptomatic men age 71, it was found that BEs have a lower sensitivity for detecting CRC as compared with colonoscopy or CTC.2 Sensitivity for lesions ≥ 10 mm is only 48% and for lesions ≥ 6 mm is only 35%, proving the BE to be a highly ineffective screening test for CRC (see Table 1).3
In a randomized study of 5,025 symptomatic patients with abnormal bowel movements and/or abdominal pain, BE has a detection rate of only 5% compared to the much higher sensitivity of CTC or colonoscopy.14
A third study calls attention to the risks associated with the DCBE exam, noting that it is less invasive and less dangerous than colonoscopy, as it does not require sedation and poses less risk for perforation of the lining of the colon.15 The study authors concluded that DCBE has a high sensitivity for clinically significant neoplasms (> 6 mm) but not for small polyps, which may be captured with other tests. DCBE may also supplement incomplete colonoscopy to rule out obstruction.
However, because of the loss of biopsy capabilities, further testing is required when abnormalities are found during the DCBE, diminishing the potential cost effectiveness of the exam. The study authors suggested that DCBEs may be used to screen those who are asymptomatic and seem to have minimal risk factors.15
Limitations
Limitations to successful BE screening include patient compliance and test result interpretation skills. With interpretation skills declining due to limited training of professionals to read BEs, results are becoming less accurate, and the test itself can be seen as less reliable. BEs are less popular, and therefore skills in reading the films are becoming outdated.2 If BE screening is to be used as the primary direct screening tool for CRC, it is imperative that radiologists and gastroenterology physicians and clinicians be well trained in this GI procedure.
Also, patients undergoing BE absorb about 15 mGy of radiation per procedure, versus 0.01 mGy to 0.15 mGy absorbed with a typical chest x-ray.16 For patients with a history of increased radiation exposure or if radiation exposure is a major concern of the patient, BE may not be an appropriate first choice.
On the next page: Colonoscopy >>
COLONOSCOPY
Colonoscopy is an endoscopic technique that allows internal inspection of the entire colorectal tract (see Figure 2).3 Although the most invasive of the exams being reviewed (and requiring extensive bowel prep), it is the gold standard for CRC screening.3
Current literature indicates that colonoscopy should be the screening method of choice for patients who have symptoms of colorectal cancer, positive results with an indirect screening exam such as FOBT, or who fall within a high-risk category. Persons at high risk for CRC include those with a significant family history, persons ages 50 and older, African Americans, and persons with an intestinal inflammatory condition, diabetes mellitus, obesity, sedentary lifestyle, positive smoking status, and low-fiber/high-fat diet.
Procedure
This procedure is performed on an outpatient basis; bowel prep is required one day prior to the procedure so that the bowel movements are clear of fecal matter. The patient receives short-lasting sedation, such as midazolam, via an IV line prior to being brought into the exam room. The procedure itself can take up to 30 minutes, with the patient comfortably sedated in an oblique position. The patient is allowed to recover for several hours, or until awake and able to pass flatus.
Patient Experience
The patient may experience discomfort during the bowel prep phase, as with BE preparation. The patient may be uncomfortable during insertion and manipulation of the colonoscope and also with gaseous insufflation that is used to improve visualization.
Advantages and Disadvantages
The advantages of colonoscopy over the other direct methods are the ability to immediately remove early cancer and colonic polyps and the ability to obtain histologic samples. A suspicious mass can be biopsied during the procedure and, depending on the size, may be completely excised. The histology of the biopsied tissue samples can aid in determining need for further treatment or establish an appropriate surveillance interval for the patient.3
The disadvantages of colonoscopy are related to the possible complications of the procedure, including colon perforation and postpolypectomy bleeding. The risk for these events is estimated to be between 0.1% and 0.3%.3 In addition, the short-lasting sedation that is used during the procedure poses the risk for possible respiratory collapse.6 Therefore, each patient requires medical clearance prior to administering sedation. The risk for a serious adverse event is 3 to 5 per 1,000 colonoscopies, and procedure-related mortality, while rare, has been reported.2,4,14
Limitations
Studies have found colonoscopy to be the most expensive of the direct screening tests, which may pose a problem for uninsured or underinsured patients.4,14
A collection of colonoscopy studies done on patients ages 50 to 66 showed an adenoma miss rate of 20% to 26% for any adenoma < 10 mm, and a 2.1% miss rate for adenomas ≥ 10 mm.3 Adenoma detection rates are dependent on optimal bowel preparation, complete examination of the colon, and the time the clinician spends examining all surfaces of the colon mucosa when withdrawing the colonoscope.3
Colonoscopy has the lowest adherence rate of all the CRC screening tests, which is not surprising since it is invasive, involves sedation, and requires thorough preparation. However, colonoscopies may be performed at longer intervals (up to 10 years) compared to other screening tests; the risk for developing CRC after a negative colonoscopy exam remains low.3
On the next page: CT colonography >>
CT COLONOGRAPHY
CTC is an emerging CRC screening test that is also known as virtual colonoscopy. According to available studies, CTC and colonoscopy might be equivalent for diagnosing cancer.14
Procedure
The preparation for CTC requires the patient to consume a low-residue diet one day prior to the procedure, which is considered to be an advantage over colonoscopy due to the decreased bowel preparation.3 With this procedure, a small rectal catheter is inserted into the anus and advanced to the rectum to allow carbon dioxide to be instilled for bowel insufflation. The patient lies supine on the table for a CT scan of the abdomen with the resulting 2D images visualizing polyps and CRC, if present.3 If necessary, 3D images can be compiled by a specialized software program to obtain a 360-degree view of the colon. In fact, recent studies show that 3D CTC is preferred to 2D because 3D polyp measurements are more representative of the true polyp size found on optic colonoscopy or surgery than are 2D measurements.17
Patient Experience
In one study, CTC screening was described as uncomfortable but not painful and was reported to be the most impersonal of all three tests because of less direct interaction, reducing patient embarrassment. This study reviewed qualitative interviews with patients regarding the fairly new CTC procedure and found that patients received little visual or verbal feedback and were confused regarding their test outcome immediately after the procedure.6
CTC was preferred by 72% of patients compared to colonoscopy and by 97% of patients compared to DCBE.18 In a study that evaluated the performance characteristics of CTC among 1,233 asymptomatic patients, 68% deemed CTC to be more convenient than colonoscopy, and more patients indicated that they preferred CTC over colonoscopy for screening (49.8% vs 41.1%; 9.2% had no preference).19
Advantages and Disadvantages
Compared with colonoscopy, CTC is comparably sensitive but safer and more acceptable to patients.2,6,14 CTC has a sensitivity of 96% for detecting lesions > 10 mm in diameter, but sensitivity decreases to 89% for lesions 6 mm to 10 mm in diameter (see Table 1).2,3,14
The follow-up screening intervals for CTC parallel those of colonoscopy. In a recent audit of 1,011 screening participants with a negative baseline CTC, a single carcinoma occurred during an average follow-up period of 4.73 ± 1.15 years.1
The current Colonography Reporting and Data System (C-RADS) guidelines for CTC interpretation recommend 6 mm as the minimum size for polyp reporting.17 This reporting threshold will result in a 77% reduction rate in invasive endoscopic procedures since it minimizes the number of cases that are sent for colonoscopy after CTC screening.17
A study performed by the American College of Radiology Imaging Network found there would be an approximate 12% referral rate for colonoscopy when using the current 6-mm polyp size threshold, but the referral rate would increase to 17% if a 5-mm threshold were used.17 The American Gastroenterological Association has stated that diminutive lesions—those measuring ≤ 5 mm—are of little to no clinical significance because only a fraction of them are neoplastic. Of these, fewer than 1% are histologically advanced, and essentially none are malignant.6 By not reporting diminutive lesions, there would be an incremental gain in the cost-effectiveness of the CTC scan and only a 1.3% loss in clinical CRC prevention efficacy.
It is widely accepted that any polyp ≥ 10 mm detected with CTC screening indicates a need for polypectomy via colonoscopy or surgery.20 For lesions ranging from 6.1 mm to 9.9 mm in diameter, CTC is suggested as a surveillance tool; patients may receive repeat CTC every 1 to 3 years until resection via colonoscopy is warranted.17
Limitations
CTC screening has many limitations, and evidence is lacking for a reduction of CRC incidence or mortality after CTC. Research has revealed a discrepancy between the polyp size measured by CTC versus the true polyp size seen on optic colonoscopy; CTC measurement can underestimate the size of a polyp by nearly 1.2 mm. Considering that C-RADS level 2 includes polyps 6.1 to 9.9 mm and level 3 includes polyps ≥ 10 mm, which automatically requires further investigation with colonoscopy, such a 1.2-mm discrepancy in measurement can make all the difference.17
CTC requires some bowel preparation, special resources, and expertise. The cost-effectiveness and risk profiles will vary, depending on whether referral for colonoscopy is required. Also, treatment recommendations for patients with polyps < 6 mm in diameter are uncertain.
CTC screening exposes the patient to increased amounts of ionizing radiation, which raises concern regarding risk for radiation-induced cancers. The effective dose to the whole body during a CTC is 6 to 20 mSv, compared to 0.02 mSv for a chest x-ray.16 One study estimated that performing CTC screening every five years from ages 50 to 80 would prevent the development of 24 CRC cases for every one radiation-induced cancer.21 Thus, the risks of radiation must be weighed against the benefits of screening, and the decision is often made by the individual patient.
Finally, the greatest limitation of CTC is that further testing may be required based on the preliminary results. Therefore, the patient may undergo two procedures rather than just one all-inclusive procedure, such as colonoscopy.
On the next page: Conclusion >>
CONCLUSION
CRC is the second leading cause of death from cancer among men and women in the United States despite the fact that it is largely preventable through diagnostic screening. Patients need education about the different types of CRC screening and about which method may be best for them, given their preferences, family history, personal history, age, and symptoms. All screening tests, direct or indirect, are cost-effective compared with no screening at all.20 Regardless of current recommendations, any screening that the patient is comfortable with should be encouraged.
BE was the first diagnostic tool to provide clinicians with the ability to visualize the patient’s lower gastrointestinal tract. It is becoming technologically outdated, however, and is no longer accepted as a primary diagnostic tool for CRC screening.
Colonoscopy, the most expensive direct screening test, provides complete visualization of the colon and allows for immediate biopsy and possible resection of a suspicious mass. This procedure is the most cost-effective direct screening method because it is comprehensive compared to BE and CTC, which may result in further investigation via colonoscopy if a mass is identified. Although colonoscopy is the most specific and sensitive for CRC screening, the outcome of the test strongly correlates with patient compliance with bowel preparation as well as clinician experience and expertise in performing a thorough exam.
While most US guidelines recommend colonoscopy as the gold standard diagnostic test, CTC is a reliable alternative for those patients who refuse colonoscopy. Future research on this newer method should consider altering the C-RADS threshold that necessitates follow-up with colonoscopy to account for the variation in polyp measurement. CTC is not a stand-alone replacement for the other direct CRC screening tests but is useful as an adjunct to increase overall patient compliance. Perhaps with time, this test may evolve to be a more prevailing recommendation for the preventive screening of CRC.
CE/CME No: CR-13111
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the difference between indirect and direct screening methods for CRC and when to utilize each.
• Understand the age continuum of screening patients with and without increased risk factors for CRC.
• Decide which type of direct screening method is the best choice when looking at sensitivity and specificity and patient preference.
• Understand the risks, benefits, and limitations of each procedure available for CRC screening.
FACULTY
Carolyn Mueller, Molly Perry, and Lisa DeCicco are recent graduates of the Pace University–Lenox Hill Hospital Physician Assistant Program in New York. Ellen D. Mandel is a Clinical Professor in the Pace PA Program and an Associate Professor in the PA Program at Seton Hall University, South Orange, New Jersey.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Because colorectal cancer is often asymptomatic, routine screening is essential to detect lesions at an early stage. The evolution of health care has brought new and improved screening methods for colorectal cancer, including CT colonography. This article weighs the pros and cons of the available screening methods used to detect colorectal cancer in the general population today.
Colorectal cancer (CRC) is the second-leading cause of death from cancer in Europe and the United States and the third most commonly diagnosed cancer in both men and women in the US.1-5 According to the American Cancer Society, 102,480 new cases of colon cancer and 40,340 new cases of rectal cancer will be diagnosed in 2013.5 Although screening rates remain low and the incidence of new CRC diagnoses rises annually, mortality rates are decreasing, most likely due to screening and improved treatment.5
CLINICAL PRESENTATION OF COLORECTAL CANCER
CRC is often asymptomatic until it reaches an advanced stage. At this point, symptoms include weight loss, night sweats, fever, loss of appetite, blood in the stool, pencil-shaped stools, diarrhea, constipation, anemia, and/or dizzy spells.6,7 On abdominal exam, a clinician may note dullness to percussion over the right or left lower quadrant, palpate a mass in the right or left lower quadrant, and/or elicit tenderness or guarding upon deep palpation. The clinician may be inclined to do a fecal occult blood test (FOBT) to confirm active GI bleeding.1,3
CURRENT SCREENING RECOMMENDATIONS
The US Preventive Services Task Force (USPSTF) currently recommends that screening for CRC—with FOBT, sigmoidoscopy, or colonoscopy—start at age 50 and continue through age 75.8 For patients whose first-degree relative has a history of CRC, initial screening should start at age 40.
It is recommended that people ages 76 to 85 make personalized, informed screening decisions in conjunction with their medical provider. Patients ages 85 or older should not be screened for CRC because of the estimated five-year time frame between the detection of cancer and the onset of symptoms or death. In the unlikely circumstance that the patient is screened and a lesion is found, the patient would not benefit but most likely experience harm from treatment efforts; however, this should be decided on a case-by-case basis.8
CONSIDERATIONS IN SCREENING THE ELDERLY
Much of the data on older populations are outdated, and thus more research needs to be conducted in this population. However, data from two studies dating from 2006 provide tools to help clinicians decide if screening in the older population is beneficial.
One study of the benefit of screening for CRC in the older population stated that clinicians must assess both the burden of chronic illness and the patient’s age as part of their evaluation. This study concluded that, because of the risks and costs that are associated with CRC screening, it is important to identify only those individuals who are likely to benefit from screening, rather than screening the elderly population in general.9
Another study examined the benefit of screening colonoscopy in two elderly groups (ages 75 to 79 and age ≥ 80) versus younger patients (ages 50 to 54; the control group). It was found that
• Screening in the older population may increase the risk for perforation and respiratory depression secondary to sedation,
• Screening may take longer to complete, and
• Screening may be challenging with less successful bowel preparation.10
Further, the prevalence of neoplasia was lowest in the control group (13.8%) compared to the 75-to-79-year-old (26.5%) and oldest (≥ 80; 28.6%) groups. The mean extension of life expectancy was much lower in the oldest group, compared to the control group (0.13 years versus 0.85 years), which represents a 6.5-fold difference. These results suggest that the benefit of colonoscopy screening in elderly persons ages 76 and older results in smaller gains in life expectancy and does not outweigh the risks. This test, therefore, should only be used when the patient expresses a preference for colonoscopy and the clinician feels it will significantly benefit the patient.10
On the next page: Screening methods >>
SCREENING METHODS
Current screening methods for CRC can be divided into two distinct categories: indirect and direct.1 Indirect screening tests include FOBT, fecal immunochemical testing, and stool DNA testing. Cancers are identified by detection of byproducts in the patient’s stool, such as blood or epithelial cells containing DNA of the adenomatous polyposis coli gene. These tests are simple to perform, have high specificity, and are relatively inexpensive, but they need to be repeated annually and have poor sensitivity.1 Positive test results of indirect screening often warrant further diagnostic testing, ultimately utilizing one of the direct screening methods.
Direct screening methods used to detect CRC—from least to most frequently employed—include barium enema (BE), CT colonography (CTC), and colonoscopy. Another direct method, flexible sigmoidoscopy, is not frequently used today, and when used, serves only as an intermediate step to colonoscopy. Direct screening provides visualization of the contour of the colon wall, the internal mucosa, and abnormal architecture. It is important to keep in mind that these tests require that the patient adhere to pretest preparation, may require patient sedation, and are more invasive and costly than indirect tests.1
Although the USPSTF has established recommendations for both test types, questions still remain about what constitutes the most cost-effective and accurate combination of screening tests for detecting CRC.8
On the next page: Barium enema >>
BARIUM ENEMA
Procedure
The BE, also known as a lower GI series, was the first screening test to allow the clinician to identify polyps or masses as outlined by barium sulfate.3 This test requires the patient to lie in an oblique position on his/her left side while barium sulfate (known as single-contrast BE, or SCBE), sometimes followed by air (known as double-contrast BE, or DCBE), is flowed through a tube inserted into the rectum. As the colon fills, the radiologist takes multiple overhead x-ray images. The patient is then required to roll on the table several times, causing the barium to coat the entire mucosa of the colon and rectum, which allows for visualization from various angles (see Figure 1).11
Patient Experience
Screening with the BE has some disadvantages to the patient. Patients may experience discomfort at multiple points: during the instillation of gas and barium into the colon, during the maintenance of the gas and barium levels, and during the maneuvering and holding positions of the procedure itself.2 Some patients, interviewed after a BE was performed, indicated feeling embarrassed during the procedure.6 Although the ability to evaluate images during the exam allows the radiologist to share preliminary results with the patient, the immediate disclosure of bad news is deemed somewhat inappropriate. If the procedure reveals positive findings, the radiologist must be sure to speak with the patient in a private area after the procedure. The patient is in a vulnerable state while in the exam room, and the exam room staff may not be adequately equipped to handle the emotional impact, properly address patient questions, or provide counseling.2,6
Advantages and Disadvantages
Few studies are now being done on the advantages and disadvantages of performing a BE compared to a colonoscopy or CTC—most likely due to the belief that colonoscopy is the better choice. BEs are considered to be one of the safer of the direct screening tests for CRC because sedation is not required and, compared to colonoscopy, the rate of colon perforation is lower (0.02% to 0.04% for BE versus 0.016% to 0.2% for colonoscopy).12,13
In a small study of 15 asymptomatic men age 71, it was found that BEs have a lower sensitivity for detecting CRC as compared with colonoscopy or CTC.2 Sensitivity for lesions ≥ 10 mm is only 48% and for lesions ≥ 6 mm is only 35%, proving the BE to be a highly ineffective screening test for CRC (see Table 1).3
In a randomized study of 5,025 symptomatic patients with abnormal bowel movements and/or abdominal pain, BE has a detection rate of only 5% compared to the much higher sensitivity of CTC or colonoscopy.14
A third study calls attention to the risks associated with the DCBE exam, noting that it is less invasive and less dangerous than colonoscopy, as it does not require sedation and poses less risk for perforation of the lining of the colon.15 The study authors concluded that DCBE has a high sensitivity for clinically significant neoplasms (> 6 mm) but not for small polyps, which may be captured with other tests. DCBE may also supplement incomplete colonoscopy to rule out obstruction.
However, because of the loss of biopsy capabilities, further testing is required when abnormalities are found during the DCBE, diminishing the potential cost effectiveness of the exam. The study authors suggested that DCBEs may be used to screen those who are asymptomatic and seem to have minimal risk factors.15
Limitations
Limitations to successful BE screening include patient compliance and test result interpretation skills. With interpretation skills declining due to limited training of professionals to read BEs, results are becoming less accurate, and the test itself can be seen as less reliable. BEs are less popular, and therefore skills in reading the films are becoming outdated.2 If BE screening is to be used as the primary direct screening tool for CRC, it is imperative that radiologists and gastroenterology physicians and clinicians be well trained in this GI procedure.
Also, patients undergoing BE absorb about 15 mGy of radiation per procedure, versus 0.01 mGy to 0.15 mGy absorbed with a typical chest x-ray.16 For patients with a history of increased radiation exposure or if radiation exposure is a major concern of the patient, BE may not be an appropriate first choice.
On the next page: Colonoscopy >>
COLONOSCOPY
Colonoscopy is an endoscopic technique that allows internal inspection of the entire colorectal tract (see Figure 2).3 Although the most invasive of the exams being reviewed (and requiring extensive bowel prep), it is the gold standard for CRC screening.3
Current literature indicates that colonoscopy should be the screening method of choice for patients who have symptoms of colorectal cancer, positive results with an indirect screening exam such as FOBT, or who fall within a high-risk category. Persons at high risk for CRC include those with a significant family history, persons ages 50 and older, African Americans, and persons with an intestinal inflammatory condition, diabetes mellitus, obesity, sedentary lifestyle, positive smoking status, and low-fiber/high-fat diet.
Procedure
This procedure is performed on an outpatient basis; bowel prep is required one day prior to the procedure so that the bowel movements are clear of fecal matter. The patient receives short-lasting sedation, such as midazolam, via an IV line prior to being brought into the exam room. The procedure itself can take up to 30 minutes, with the patient comfortably sedated in an oblique position. The patient is allowed to recover for several hours, or until awake and able to pass flatus.
Patient Experience
The patient may experience discomfort during the bowel prep phase, as with BE preparation. The patient may be uncomfortable during insertion and manipulation of the colonoscope and also with gaseous insufflation that is used to improve visualization.
Advantages and Disadvantages
The advantages of colonoscopy over the other direct methods are the ability to immediately remove early cancer and colonic polyps and the ability to obtain histologic samples. A suspicious mass can be biopsied during the procedure and, depending on the size, may be completely excised. The histology of the biopsied tissue samples can aid in determining need for further treatment or establish an appropriate surveillance interval for the patient.3
The disadvantages of colonoscopy are related to the possible complications of the procedure, including colon perforation and postpolypectomy bleeding. The risk for these events is estimated to be between 0.1% and 0.3%.3 In addition, the short-lasting sedation that is used during the procedure poses the risk for possible respiratory collapse.6 Therefore, each patient requires medical clearance prior to administering sedation. The risk for a serious adverse event is 3 to 5 per 1,000 colonoscopies, and procedure-related mortality, while rare, has been reported.2,4,14
Limitations
Studies have found colonoscopy to be the most expensive of the direct screening tests, which may pose a problem for uninsured or underinsured patients.4,14
A collection of colonoscopy studies done on patients ages 50 to 66 showed an adenoma miss rate of 20% to 26% for any adenoma < 10 mm, and a 2.1% miss rate for adenomas ≥ 10 mm.3 Adenoma detection rates are dependent on optimal bowel preparation, complete examination of the colon, and the time the clinician spends examining all surfaces of the colon mucosa when withdrawing the colonoscope.3
Colonoscopy has the lowest adherence rate of all the CRC screening tests, which is not surprising since it is invasive, involves sedation, and requires thorough preparation. However, colonoscopies may be performed at longer intervals (up to 10 years) compared to other screening tests; the risk for developing CRC after a negative colonoscopy exam remains low.3
On the next page: CT colonography >>
CT COLONOGRAPHY
CTC is an emerging CRC screening test that is also known as virtual colonoscopy. According to available studies, CTC and colonoscopy might be equivalent for diagnosing cancer.14
Procedure
The preparation for CTC requires the patient to consume a low-residue diet one day prior to the procedure, which is considered to be an advantage over colonoscopy due to the decreased bowel preparation.3 With this procedure, a small rectal catheter is inserted into the anus and advanced to the rectum to allow carbon dioxide to be instilled for bowel insufflation. The patient lies supine on the table for a CT scan of the abdomen with the resulting 2D images visualizing polyps and CRC, if present.3 If necessary, 3D images can be compiled by a specialized software program to obtain a 360-degree view of the colon. In fact, recent studies show that 3D CTC is preferred to 2D because 3D polyp measurements are more representative of the true polyp size found on optic colonoscopy or surgery than are 2D measurements.17
Patient Experience
In one study, CTC screening was described as uncomfortable but not painful and was reported to be the most impersonal of all three tests because of less direct interaction, reducing patient embarrassment. This study reviewed qualitative interviews with patients regarding the fairly new CTC procedure and found that patients received little visual or verbal feedback and were confused regarding their test outcome immediately after the procedure.6
CTC was preferred by 72% of patients compared to colonoscopy and by 97% of patients compared to DCBE.18 In a study that evaluated the performance characteristics of CTC among 1,233 asymptomatic patients, 68% deemed CTC to be more convenient than colonoscopy, and more patients indicated that they preferred CTC over colonoscopy for screening (49.8% vs 41.1%; 9.2% had no preference).19
Advantages and Disadvantages
Compared with colonoscopy, CTC is comparably sensitive but safer and more acceptable to patients.2,6,14 CTC has a sensitivity of 96% for detecting lesions > 10 mm in diameter, but sensitivity decreases to 89% for lesions 6 mm to 10 mm in diameter (see Table 1).2,3,14
The follow-up screening intervals for CTC parallel those of colonoscopy. In a recent audit of 1,011 screening participants with a negative baseline CTC, a single carcinoma occurred during an average follow-up period of 4.73 ± 1.15 years.1
The current Colonography Reporting and Data System (C-RADS) guidelines for CTC interpretation recommend 6 mm as the minimum size for polyp reporting.17 This reporting threshold will result in a 77% reduction rate in invasive endoscopic procedures since it minimizes the number of cases that are sent for colonoscopy after CTC screening.17
A study performed by the American College of Radiology Imaging Network found there would be an approximate 12% referral rate for colonoscopy when using the current 6-mm polyp size threshold, but the referral rate would increase to 17% if a 5-mm threshold were used.17 The American Gastroenterological Association has stated that diminutive lesions—those measuring ≤ 5 mm—are of little to no clinical significance because only a fraction of them are neoplastic. Of these, fewer than 1% are histologically advanced, and essentially none are malignant.6 By not reporting diminutive lesions, there would be an incremental gain in the cost-effectiveness of the CTC scan and only a 1.3% loss in clinical CRC prevention efficacy.
It is widely accepted that any polyp ≥ 10 mm detected with CTC screening indicates a need for polypectomy via colonoscopy or surgery.20 For lesions ranging from 6.1 mm to 9.9 mm in diameter, CTC is suggested as a surveillance tool; patients may receive repeat CTC every 1 to 3 years until resection via colonoscopy is warranted.17
Limitations
CTC screening has many limitations, and evidence is lacking for a reduction of CRC incidence or mortality after CTC. Research has revealed a discrepancy between the polyp size measured by CTC versus the true polyp size seen on optic colonoscopy; CTC measurement can underestimate the size of a polyp by nearly 1.2 mm. Considering that C-RADS level 2 includes polyps 6.1 to 9.9 mm and level 3 includes polyps ≥ 10 mm, which automatically requires further investigation with colonoscopy, such a 1.2-mm discrepancy in measurement can make all the difference.17
CTC requires some bowel preparation, special resources, and expertise. The cost-effectiveness and risk profiles will vary, depending on whether referral for colonoscopy is required. Also, treatment recommendations for patients with polyps < 6 mm in diameter are uncertain.
CTC screening exposes the patient to increased amounts of ionizing radiation, which raises concern regarding risk for radiation-induced cancers. The effective dose to the whole body during a CTC is 6 to 20 mSv, compared to 0.02 mSv for a chest x-ray.16 One study estimated that performing CTC screening every five years from ages 50 to 80 would prevent the development of 24 CRC cases for every one radiation-induced cancer.21 Thus, the risks of radiation must be weighed against the benefits of screening, and the decision is often made by the individual patient.
Finally, the greatest limitation of CTC is that further testing may be required based on the preliminary results. Therefore, the patient may undergo two procedures rather than just one all-inclusive procedure, such as colonoscopy.
On the next page: Conclusion >>
CONCLUSION
CRC is the second leading cause of death from cancer among men and women in the United States despite the fact that it is largely preventable through diagnostic screening. Patients need education about the different types of CRC screening and about which method may be best for them, given their preferences, family history, personal history, age, and symptoms. All screening tests, direct or indirect, are cost-effective compared with no screening at all.20 Regardless of current recommendations, any screening that the patient is comfortable with should be encouraged.
BE was the first diagnostic tool to provide clinicians with the ability to visualize the patient’s lower gastrointestinal tract. It is becoming technologically outdated, however, and is no longer accepted as a primary diagnostic tool for CRC screening.
Colonoscopy, the most expensive direct screening test, provides complete visualization of the colon and allows for immediate biopsy and possible resection of a suspicious mass. This procedure is the most cost-effective direct screening method because it is comprehensive compared to BE and CTC, which may result in further investigation via colonoscopy if a mass is identified. Although colonoscopy is the most specific and sensitive for CRC screening, the outcome of the test strongly correlates with patient compliance with bowel preparation as well as clinician experience and expertise in performing a thorough exam.
While most US guidelines recommend colonoscopy as the gold standard diagnostic test, CTC is a reliable alternative for those patients who refuse colonoscopy. Future research on this newer method should consider altering the C-RADS threshold that necessitates follow-up with colonoscopy to account for the variation in polyp measurement. CTC is not a stand-alone replacement for the other direct CRC screening tests but is useful as an adjunct to increase overall patient compliance. Perhaps with time, this test may evolve to be a more prevailing recommendation for the preventive screening of CRC.
1. de Haan MC, Halligan S, Stoker J. Does CT colonography have a role for population-based colorectal cancer CRC screening? Eur Radiol. 2012;22: 1495-1503.
2. Lieberman D. Screening for colorectal cancer. N Engl J Med. 2009;361: 1179-1187.
3. de Wijkerslooth TR, Bossuyt PM, Dekker E. Strategies in screening for colon carcinoma. Neth J Med. 2011;69:112-119.
4. Whitlock E, Lin J, Liles E, et al. Screening for colorectal cancer: an updated systematic review. Evidence Synthesis No. 65, Part 1. AHRQ Publication No. 08-05124-EF-1. Rockville, Maryland, Agency for Healthcare Research and Quality, October 2008.
5. Colorectal cancer: What are the key statistics about colorectal cancer? American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics. Accessed October 14, 2013.
6. Von Wagner C, Knight K, Halligan S, et al. Patient experiences of colonoscopy, barium enema and CT colonography: a qualitative study. Br J Radiol. 2009;82:13-19.
7. Cappell MS. Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin N Am. 2008;37:1-24.
8. Screening for colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. US Preventive Services Task Force web site. www.uspreventiveservicestaskforce.org/uspstf/uspscolo.htm. Published 2008. Accessed October 14, 2013.
9. Gross CP, McAvay GJ, Krumholz HM, et al. The effect of age and chronic illness on life expectancy after a diagnosis of colorectal cancer: implications for screening. Ann Intern Med. 2006;145:646-653.
10. Lin OS, Kozarek RA, Schembre DB, et al. Screening colonoscopy in very elderly patients: prevalence of neoplasia and estimated impact on life expectancy. JAMA. 2006;295:2357-2365.
11. Colorectal cancer early detection: colorectal cancer screening tests. American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-screening-tests-used. Accessed October 14, 2013.
12. Lohsiriwat V. Colonoscopic perforation: incidence, risk factors, management and outcome. World J Gastroenterol 2010;16:425-430.
13. Yasar NF, Ihtiyar E. Colonic perforation during barium enema in a patient without known colonic disease: a case report. Cases J. 2009;2:6716.
14. Halligan S, Lilford RJ, Wardle J, et al. Design of a multicentre randomized trial to evaluate CT colonography versus colonoscopy or barium enema for diagnosis of colonic cancer in older symptomatic patients: The SIGGAR study. Trials. 2007;8:1-9.
15. Lohsiriwat V, Prapasrivorakul S, Suthikeeree W. Colorectal cancer screening by double contrast barium enema in Thai people.Asian Pacific J Cancer Prev. 2012;13:1273-1276.
16. Mayo JR, Aldrich J, Muller NL; Fleischner Society. Radiation exposure at chest CT: a statement of the Fleischner Society. Radiology. 2003;228:15-21.
17. Summers RM. Polyp size measurement at CT colonography: what do we know and what do we need to know? Radiology. 2010;255(3):707-720.
18. Gluecker TM, Johnson CD, Harmsen WS, et al. Colorectal cancer screening with CT colonography, colonoscopy, and double-contrast barium enema examination: prospective assessment of patient perceptions and p. Radiology. 2003;227:378-384.
19. Pickhardt PJ, Choi JR, Hwang I, et al. Computed tomographic virtual colonoscopy to screen for colorectal neoplasia in asymptomatic adults. N Engl J Med. 2003;349:2191-2200.
20. Pickhardt P, Hassan C, Laghi A, et al. Cost-effectiveness of colorectal cancer screening with computed tomography colonography. Cancer. 2007;109:2213-2221.
21. Berrington de Gonzalez A, Kim KP, Knudsen AB, et al. Radiation-related cancer risks from CT colongraphy screening: a risk-benefit analysis. AJR Am J Roentgenol. 2011;196:816-823.
1. de Haan MC, Halligan S, Stoker J. Does CT colonography have a role for population-based colorectal cancer CRC screening? Eur Radiol. 2012;22: 1495-1503.
2. Lieberman D. Screening for colorectal cancer. N Engl J Med. 2009;361: 1179-1187.
3. de Wijkerslooth TR, Bossuyt PM, Dekker E. Strategies in screening for colon carcinoma. Neth J Med. 2011;69:112-119.
4. Whitlock E, Lin J, Liles E, et al. Screening for colorectal cancer: an updated systematic review. Evidence Synthesis No. 65, Part 1. AHRQ Publication No. 08-05124-EF-1. Rockville, Maryland, Agency for Healthcare Research and Quality, October 2008.
5. Colorectal cancer: What are the key statistics about colorectal cancer? American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics. Accessed October 14, 2013.
6. Von Wagner C, Knight K, Halligan S, et al. Patient experiences of colonoscopy, barium enema and CT colonography: a qualitative study. Br J Radiol. 2009;82:13-19.
7. Cappell MS. Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin N Am. 2008;37:1-24.
8. Screening for colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. US Preventive Services Task Force web site. www.uspreventiveservicestaskforce.org/uspstf/uspscolo.htm. Published 2008. Accessed October 14, 2013.
9. Gross CP, McAvay GJ, Krumholz HM, et al. The effect of age and chronic illness on life expectancy after a diagnosis of colorectal cancer: implications for screening. Ann Intern Med. 2006;145:646-653.
10. Lin OS, Kozarek RA, Schembre DB, et al. Screening colonoscopy in very elderly patients: prevalence of neoplasia and estimated impact on life expectancy. JAMA. 2006;295:2357-2365.
11. Colorectal cancer early detection: colorectal cancer screening tests. American Cancer Society. www.cancer.org/cancer/colonandrectumcancer/moreinformation/colonandrectumcancerearlydetection/colorectal-cancer-early-detection-screening-tests-used. Accessed October 14, 2013.
12. Lohsiriwat V. Colonoscopic perforation: incidence, risk factors, management and outcome. World J Gastroenterol 2010;16:425-430.
13. Yasar NF, Ihtiyar E. Colonic perforation during barium enema in a patient without known colonic disease: a case report. Cases J. 2009;2:6716.
14. Halligan S, Lilford RJ, Wardle J, et al. Design of a multicentre randomized trial to evaluate CT colonography versus colonoscopy or barium enema for diagnosis of colonic cancer in older symptomatic patients: The SIGGAR study. Trials. 2007;8:1-9.
15. Lohsiriwat V, Prapasrivorakul S, Suthikeeree W. Colorectal cancer screening by double contrast barium enema in Thai people.Asian Pacific J Cancer Prev. 2012;13:1273-1276.
16. Mayo JR, Aldrich J, Muller NL; Fleischner Society. Radiation exposure at chest CT: a statement of the Fleischner Society. Radiology. 2003;228:15-21.
17. Summers RM. Polyp size measurement at CT colonography: what do we know and what do we need to know? Radiology. 2010;255(3):707-720.
18. Gluecker TM, Johnson CD, Harmsen WS, et al. Colorectal cancer screening with CT colonography, colonoscopy, and double-contrast barium enema examination: prospective assessment of patient perceptions and p. Radiology. 2003;227:378-384.
19. Pickhardt PJ, Choi JR, Hwang I, et al. Computed tomographic virtual colonoscopy to screen for colorectal neoplasia in asymptomatic adults. N Engl J Med. 2003;349:2191-2200.
20. Pickhardt P, Hassan C, Laghi A, et al. Cost-effectiveness of colorectal cancer screening with computed tomography colonography. Cancer. 2007;109:2213-2221.
21. Berrington de Gonzalez A, Kim KP, Knudsen AB, et al. Radiation-related cancer risks from CT colongraphy screening: a risk-benefit analysis. AJR Am J Roentgenol. 2011;196:816-823.
Ménière’s Disease: A Lifelong Merry-Go-Round
CE/CME No: CR-1310
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology of Ménière’s disease, as it is currently understood.
• Discuss the triad of symptoms that should prompt suspicion for Ménière’s disease in a primary care patient.
• List the diagnostic criteria for “definite” Ménière’s disease, as defined by the American Academy of Otolaryngology–Head and Neck Surgery.
• Review pharmacologic management, intratympanic injection and other nonoperative therapies, and surgical treatment for Ménière’s disease.
FACULTY
Tamera Pearson is an Associate Professor in the School of Nursing at Western Carolina University.
The author has no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Ménière’s disease is a complex disorder of intermittent vertigo, tinnitus, and hearing loss whose symptoms usually manifest between ages 20 and 60. Although this disorder is uncommon, its impact on a person’s quality of life can be significant. Here are the symptoms, criteria for diagnosis, and appropriate treatment or referrals for Ménière’s disease.
Ménière’s disease can significantly affect a person’s quality of life and is a challenge to diagnose and treat effectively. The French physician Prosper Ménière first described this disorder approximately 150 years ago. Yet, researchers are still uncertain of its exact etiology and underlying pathophysiology.1,2
Ménière’s disease is defined as a chronic condition with recurrent episodes of vertigo that are associated with sensorineural hearing loss, tinnitus, and/or a sensation of aural fullness.3,4 Thanks to researchers’ evolving knowledge of Ménière’s disease, a new definition has been proposed: a degenerating inner ear leading to impaired homoeostasis, hearing loss, and vertigo.5
In the United States, the prevalence of Ménière’s disease is estimated at 15 to 150 cases per 100,000 persons. (This wide variation in prevalence reflects a lack of standard diagnostic criteria, as well as differences based on geographic area.6,7) Many affected individuals experience symptoms significant enough to lead to disability.6,7 Patients with Ménière’s disease usually present between ages 20 and 60, with a peak incidence occurring between ages 40 and 50.1,4,6,8 This disease affects both genders, but is slightly more common in women.1,4,6,8
Diagnosis of Ménière’s disease is based on recognition of the clinical symptoms that characterize the disorder, and management is centered on heuristic treatment options. Thus, a person may experience mild to severe symptoms of Ménière’s disease for months to years before receiving either the diagnosis or first-line treatment. This article reviews the current understanding of the underlying physiologic mechanisms that cause Ménière’s disease and discusses the criteria for diagnosis and various treatment options.
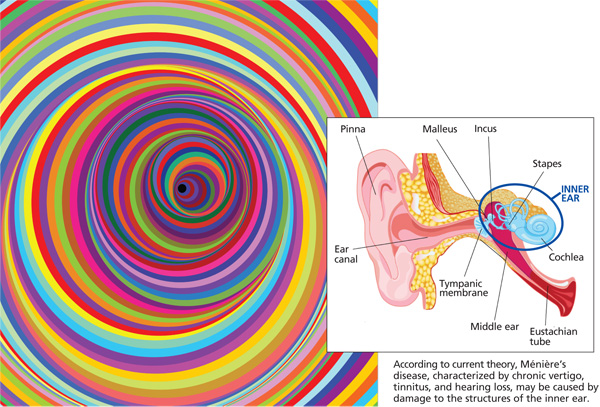
ETIOLOGY AND PATHOPHYSIOLOGY
The cause of Ménière’s disease and the subsequent mechanical, physiologic, and biochemical changes that occur are poorly understood, but several theories have been proposed. According to the current central theory, a buildup of fluid (endolymph) within the cochlear and saccular ducts in the inner ear causes distention of these structures into the endolymphatic space, resulting in the development of endolymphatic hydrops.4 Pressure from endolymphatic hydrops may cause damage to hair follicles and to the vestibular epithelium, resulting in symptoms of vertigo, tinnitus, and hearing loss.1,4 Researchers previously attributed the symptoms of Ménière’s disease completely to hydrops and focused on identifying anatomic abnormalities.7,9 However, studies now suggest that a range of pathophysiologic processes resulting from intrinsic and/or extrinsic factors may be responsible.7,9 While hydrops may develop, it is not always the definitive or only cause of Meniere’s disease symptoms.9
Recently recognized factors that contribute to the development of Meniere’s disease include autoimmune reactions, genetic irregularities, vascular abnormalities, and viral influences. Approximately one-third of Ménière’s disease cases can be attributed to an autoimmune origin.1,6 Researchers hypothesize that several immunologic processes may contribute to Ménière’s disease:
• Antibodies may cause inner ear damage,
• Injury to the inner ear may result in the release of cytokines which provoke immune reactions, and
• Certain genes may affect a person’s immune system and increase the probability of Ménière’s disease.1
The probability of a genetic influence is supported by the fact that one in 20 people with Ménière’s disease reports a positive family history of the disorder.4
Many patients with Ménière’s disease experience migraine headaches, and thus vascular abnormalities are another area of consideration among the etiologies of this disease.10 Researchers are also studying a potential viral cause in the development of Ménière’s disease.1,6
Regardless of the specific cause or physiologic changes that occur, the one common finding in patients with Ménière’s disease is a dysfunction of fluid homeostasis within the inner ear.
On the next page: Diagnosis >>
DIAGNOSIS
Establishing the diagnosis of Ménière’s disease can be difficult and time-consuming because the symptoms of the disorder are nonspecific and variable. Ménière’s disease is a clinical diagnosis, and thus the clinician must conduct a thorough physical exam and elicit a very specific history, including a detailed description of vertigo incidents and associated symptoms. Often, the greatest challenge is encouraging patients to articulate the details of their episodes. Patients may not keep a record of the variations of episodes, nor do they always know what information is needed. Thus, the provider needs to elicit specific information by asking questions regarding frequency and duration of episodes, as well as fluctuation of hearing loss, nausea, and tinnitus. Symptoms associated with vertigo during a Ménière’s episode may include nausea, vomiting, gait imbalance, and tinnitus. Most vertigo attacks from Ménière’s disease occur in clusters, but they may also occur sporadically.6
An additional challenge for clinicians is that other potential diagnoses related to vertigo must be excluded before the diagnosis of Ménière’s disease can be made. Also, it is important to note that specialists may differentiate Ménière’s disease, an idiopathic condition, from Ménière’s syndrome, which results from known causes of damage to the inner ear. In the literature, however, this distinction in terminology is not always clear.7
Specific diagnostic criteria for Ménière’s disease, defined in 1995 by the American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS), remain the gold standard for diagnosis.3 A “definite” diagnosis of Ménière’s disease is based on:
• A history of two or more episodes of spontaneous vertigo lasting 20 minutes or longer,
• Hearing loss documented by audiometry at least once,
• Presence of tinnitus, and/or
• A sensation of aural fullness.2,6,7
The AAO-HNS diagnostic criteria also define categories of “probable” and “possible” Ménière’s disease based on the frequency of vertigo episodes or the presence of a combination of associated symptoms (see Table 1).3,7
Patients with Ménière’s disease may experience different patterns of symptoms. “Auditory dominant” Ménière’s disease produces more hearing loss changes than vertigo, while “vestibular dominant” causes frequent episodes of severe vertigo and less severe hearing changes. A “mixed” pattern of Ménière’s disease manifests with both hearing fluctuations and vertigo.5
Unilateral symptoms are most common; however, bilateral disorder occurs in approximately 25% of patients, either at onset or with changing symptomatology over time.5,8,9
On the next page: History and examination >>
HISTORY AND EXAMINATION
Obtaining a detailed history from the patient and completing thorough neurologic and otologic examinations are essential components of the diagnostic process. Audiometry should be completed to evaluate neurosensory hearing loss, as audiometrically documented hearing loss is part of the AAO-HSN diagnostic criteria for Ménière’s disease.6
Based on findings from the patient’s history, physical exam, and audiometric testing, a tentative diagnosis can be made. The role and inclusion of adjunctive tests in the diagnostic process varies considerably by region in the US. While not required for the diagnosis of Ménière’s disease, electrical vestibular stimulation and videonystagmography are useful tests to assess abnormalities in vestibular function and monitor disease progression, which may help determine intervention options.6 Additional diagnostic tests may be suggested due to the essential need to exclude other potential causes of vertigo prior to determining the final diagnosis of Ménière’s disease.
Triggers of Vertigo
Selected triggers of vertigo that must be considered are benign paroxysmal positional vertigo (BPPV), labyrinthitis, acoustic neuroma, migraine with vertigo, and cerebral vascular events.6 Diagnostic tests are indicated to rule out certain problems, such as MRI to exclude a tumor or an acoustic neuroma. Distinct differences noted during a complete assessment may help eliminate certain disorders. BPPV is triggered by a change in physical position and usually lasts less than one minute; the diagnosis can be confirmed by the Dix-Hallpike maneuver.4 Labyrinthitis is characterized by acute vertigo associated with continuing imbalance, while instability with walking resolves completely between vertigo episodes in Ménière’s disease.4 If abnormal neurologic manifestations are noted during the exam or reported in a patient’s account of a vertigo episode, then a transient ischemic attack or stroke must be ruled out by more detailed diagnostic testing.
TREATMENT OPTIONS
Presently, no evidence-based guidelines exist for the treatment of Ménière’s disease, and the evidence supporting the efficacy of currently used therapies is inconsistent. However, several medicines and treatments are useful in relieving symptoms and improving a patient’s quality of life.
Primary care clinicians can initiate treatment for Ménière’s disease through lifestyle recommendations and prescription of specific medications. Everyday adjustments that incorporate dietary changes, stress reduction, adequate sleep, and regular exercise have been shown to improve vertigo symptoms in 60% of patients with Ménière’s disease.5,9
On the next page: Lifestyle changes >>
Lifestyle Changes
Dietary changes. Patients diagnosed with Ménière’s disease may benefit from following a low-sodium diet, limiting their daily sodium intake to between 1,000 and 2,000 mg.2,7,11 A low-sodium diet is believed to have a positive impact on inner ear fluid homeostasis by decreasing fluid retention and reducing the endolymphatic hydrops.2,7,11 Decreasing alcohol and caffeine consumption is also routinely recommended as part of the treatment of Ménière’s disease.2,5
Researchers have recently suggested a different approach to dietary changes for Ménière’s disease that reflects the underlying loss of ability to regulate fluid in the inner ear. This alternate method of dietary regulation aims to maintain fluid homeostasis by avoiding variations in the daily intake of sodium, caffeine, or alcohol, rather than limiting daily consumption.5
The goal of any proposed dietary changes is to limit fluid and electrolyte shifts that could disrupt the delicate fluid balance in the inner ear.9 When caring for patients with Ménière’s disease, clinicians need to keep in mind that dietary changes may be difficult and will probably require ongoing encouragement.
Stress reduction. Stress is associated with the occurrence of Ménière’s disease and often is the trigger for an acute episode of symptom exacerbation.5 Thus, clinicians should encourage stress management as a way to reduce the impact of Ménière’s disease on a patient’s life. Stress reduction techniques that can be recommended include progressive relaxation, meditation with deep breathing, yoga, and exercise.
Although studies of the effect of stress reduction methods on Ménière’s disease are not available in the current literature, the association of stress with Ménière’s disease is well documented.5 By avoiding stress, it is hoped, patients may experience a reduction in the frequency and severity of Ménière’s disease–associated episodes of vertigo. Researchers also suggest that stress reduction and patient education may help alleviate patients’ feelings of frustration resulting from misinformation about their condition.2,11
Oral Medications for Acute Relief
Acute attacks of vertigo associated with Ménière’s disease can be treated with benzodiazepines, antiemetics, or anticholinergic medications.4,6 Alleviation of symptoms is achieved through different physiologic pathways, based on the drug category prescribed. If a patient reports typical symptoms of Ménière’s disease but has not undergone audiometry, the plausible diagnosis may lead to tentative treatment for acute episodes if other causes of vertigo have been ruled out.
Antihistamines, such as meclizine or dimenhydrinate, may help reduce vertigo symptoms and associated nausea by blocking the effects of histamine.4,6 One of the most common side effects of antihistamines is drowsiness, so patients must be cautioned to avoid certain activities while taking this medication. Antihistamines should not be given to patients with glaucoma or prostate disease due to the potentially strong anticholinergic effects of these drugs.4,6
Scopolamineis a belladonna alkaloid that can be applied topically on the tissue just behind the ear to help reduce nausea and vomiting related to vertigo.11
Another option for treatment of acute vertigo is a benzodiazepine, such as alprazolam, to suppress active cerebellar responses; this agent may also reduce anxiety associated with an acute episode of vertigo.6,11 Benzodiazepines should be started at the lowest dose and increased as needed to the maximum recommended for individual medications based on symptom relief and side effects.6 Although caution needs to be used when prescribing benzodiazepines, studies show that they can be effective for persons with Meniere’s disease.11
Other antiemetic medications, such as promethazine or ondansetron, may be needed to treat severe nausea, but these agents should be used cautiously with other medications due a potential side effect of sedation.
Long-Term Oral Medication
Medication for long-term management of Ménière’s disease can promote improvement in symptoms and reduce the frequency of vertigo episodes. A mild diuretic, such as hydrochlorothiazide with or without triamterene, taken on a regular basis reduces extracellular fluids and may decrease pressure from endolymphatic hydrops.2,7 While strong evidence regarding the efficacy of diuretics is lacking, the majority of patients with Ménière’s disease who are treated with diuretics do experience improvement in vertigo.2,5
Betahistine hydrochloride, a vasodilator and histamine receptor antagonist, is another medication to consider for management of Ménière’s disease.1 This agent is not approved by the FDA; however, the FDA classifies betahistine as an inert chemical, so it is available in compounding pharmacies in the United States. The efficacy of betahistine has not been clearly or consistently established in research studies, but it has been and continues to be widely used to treat Ménière’s disease in Europe, with good results. Betahistine affects the microcirculation in the inner ear and inhibits the vestibular nuclei, which may reduce the frequency of vertigo episodes and improve tinnitus associated with Ménière’s disease.2,8,11
On the next page: Intratympanic medication >>
Intratympanic Medication
Patients who do not respond well to the previously described management should be referred to a specialist for additional treatment options. An otolaryngology specialist may administer intratympanic medications to patients with Ménière’s disease who have not responded to primary medical therapy.
Patients in the US who have not responded positively to lifestyle or diuretic medication are commonly offered treatment with intratympanic dexamethasone. The primary goal of this therapy is to improve vertigo without affecting a patient’s hearing; an added effect may be a potential positive impact on the immune system.11 Studies show that intratympanic steroid injection results in control of vertigo in patients with Ménière’s disease, but up to four injection treatments may be required for optimal effectiveness.2,7 Improvement of vertigo is achieved in more than 80% of patients who undergo intratympanic steroid injections.9
An option reserved for patients with severe, frequent vertigo related to Ménière’s disease is a type of chemical ablation of the labyrinth induced by injecting gentamicin into the middle ear.2 Gentamicin has a toxic effect on the vestibular hair cells in the inner ear, resulting in elimination of vestibular function.2 Intratympanic gentamicin is reported to reduce symptoms from Ménière’s disease, but this treatment is only recommended for patients with unilateral disease because it may induce permanent hearing loss.11
The primary care clinician needs to be aware of these intratympanic procedures and encourage patients to follow up with the specialist if additional treatments are indicated.
Portable Pressure Device
Use of the Meniett device is a minimally invasive treatment for Ménière’s disease based on the principle of using alternating pressure to stimulate the flow of endolymph.11 This handheld device delivers low-pressure pulses within the inner ear through a standard ventilation tube in order to increase exchange of fluids and improve homeostasis.8 The Meniett device should be used for five-minute intervals three times per day.12
Several studies have shown excellent results in patients who use the Meniett device routinely for several weeks.2,12 As noted, the use of this device, which is obtained by prescription from an otolaryngologist, requires placement of ventilation tubes.
Acupuncture
A traditional Chinese medical approach, acupuncture is one complementary and alternative medicine therapy that has been studied as a treatment option for Ménière’s disease. Studies on the use of acupuncture to treat vertigo demonstrate a beneficial effect for persons with this disease. While the optimal number and frequency of treatments has not been determined, all types of acupuncture studied showed benefit. Acupuncture has a positive effect in both acute episodes of vertigo in those without Ménière’s disease and in patients who have had Ménière’s disease for many years.13
Vestibular Rehabilitation
An additional adjunctive treatment option to consider for patients with residual disequilibrium is vestibular rehabilitation. Vestibular rehabilitation is designed to desensitize or retrain the balance system response through a series of exercises and activities supervised by a physical or occupational therapist. This rehabilitation may improve balance in patients with Ménière’s disease who have undergone medical or surgical intervention used to treat vertigo. Patients who have significant balance problems occurring between acute vertigo episodes may also benefit from vestibular rehabilitation.6
Surgical Treatment
Surgical intervention should be the last resort to treat Ménière’s disease due to the higher risk involved with any surgical procedure and the potential adverse effect on hearing. Endolymphatic sac decompression surgery involves removing a portion of the mastoid bone, resulting in decompression of the sac adjacent to the sigmoid sinus. This procedure has been used for more than 40 years to control vertigo and has the advantage of preserving hearing.7,9 However, the benefit of this procedure is now somewhat controversial and possibly related to a placebo effect.6 Researchers also report positive results with the use of tenotomy surgery, which involves severing tendons to the stapedius and tensor tympani muscles in the middle ear.14
No surgical procedure should be considered without the recommendation of an otolaryngology specialist. The decision should be made based on the severity of the disease and its effect on the patient, weighed against the risks involved in such an invasive treatment option.
On the next page: Conclusion >>
CONCLUSION
Ménière’s disease is a complex disorder that can significantly alter a person’s quality of life. While neither the exact cause nor pathophysiology underlying Ménière’s disease is well understood, several solid theories are being investigated and contribute to the current understanding of treatment options. Primary care clinicians can help determine this clinical diagnosis based on a detailed history and comprehensive assessment of recurrent vertigo with tinnitus, hearing loss, and possibly a sensation of aural fullness. Establishing the diagnosis of Ménière’s disease requires ruling out other possible causes of vertigo.
Lifestyle changes that improve the consistency of dietary intake of sodium, caffeine, and alcohol as well as reduction of stress are ongoing recommendations for patients with Ménière’s disease. Oral medications from a range of drug categories may be used to improve acute and chronic symptoms, including antiemetics, anticholinergics, antihistamines, benzodiazepines, and mild diuretics. Additionally, a compounded substance with vasodilator and histamine- receptor–antagonist properties (betahistine) can be used for treatment of Meniere’s.
Patients who do not respond well to conservative therapy should be referred to an otolaryngologist for possible intratympanic medications, ventilation tube placement with a prescription for pulse pressure therapy (ie, Meniett device), or surgical intervention. Primary care clinicians can initiate treatment for Ménière’s disease by recommending lifestyle changes, prescribing oral medications, providing patient education, and recognizing indications for referral.
1. Greco A, Gallo A, Fusconi M, et al. Ménière’s disease might be an autoimmune condition? Autoimmun Rev. 2012;11:731-738.
2. Greenberg SL, Nedzelski JM. Medical and noninvasive therapy for Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1081-1090.
3. American Academy of Otolaryngology–Head and Neck Foundation, Inc. Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Ménière’s disease. Otolaryngol Head Neck Surg. 1995;113:181-185.
4. Syed I, Aldren C. Ménière’s disease: an evidence based approach to assessment and management. Int J Clin Pract. 2012;66:166-170.
5. Rauch SD. Clinical hints and precipitating factors in patients suffering from Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1011-1017.
6. Dinces EA, Rauch SS. Ménière’s disease. In: UpToDate. Deschler DG, Lin FH, eds. 2012:November 27,2012.
7. Semaan MT, Megerian CA. Ménière’s disease: a challenging and relentless disorder. Otolaryngol Clin North Am. 2011;44:383-403.
8. Martin González C, González FM, Trinidad A, et al. Medical management of Ménière’s disease: a 10-year case series and review of literature. Eur Arch Otorhinolaryngol. 2010;267:1371-1376.
9. Berlinger NT. Ménière’s disease: new concepts, new treatments. Minn Med. 2011;94:33-36.
10. von Brevern M, Neuhauser H. Epidemiological evidence for a link between vertigo and migraine. J Vestib Res. 2011;21:299-304.
11. Coelho DH, Lalwani AK. Medical management of Ménière’s disease. Laryngoscope. 2008;118:1099-1108.
12. Gates GA, Green JD Jr, Tucci DL, Telian SA. The effects of transtympanic micropressure treatment in people with unilateral Ménière’s disease. Arch Otolaryngol Head Neck Surg. 2004;130:718-725.
13. Long A, Xing M, Morgan K, Brettle A. Exploring the evidence base for acupuncture in the treatment of Ménière’s syndrome-a systematic review. Evid Based Complement Alternat Med. 2011;2011:1-13.
14. Loader B, Beicht D, Hamzavi JS, Franz P. Tenotomy of the middle ear muscles causes a dramatic reduction in vertigo attacks and improves audiological function in definite Ménière’s disease. Acta Otolaryngol. 2012;132:491-497.
CE/CME No: CR-1310
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology of Ménière’s disease, as it is currently understood.
• Discuss the triad of symptoms that should prompt suspicion for Ménière’s disease in a primary care patient.
• List the diagnostic criteria for “definite” Ménière’s disease, as defined by the American Academy of Otolaryngology–Head and Neck Surgery.
• Review pharmacologic management, intratympanic injection and other nonoperative therapies, and surgical treatment for Ménière’s disease.
FACULTY
Tamera Pearson is an Associate Professor in the School of Nursing at Western Carolina University.
The author has no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Ménière’s disease is a complex disorder of intermittent vertigo, tinnitus, and hearing loss whose symptoms usually manifest between ages 20 and 60. Although this disorder is uncommon, its impact on a person’s quality of life can be significant. Here are the symptoms, criteria for diagnosis, and appropriate treatment or referrals for Ménière’s disease.
Ménière’s disease can significantly affect a person’s quality of life and is a challenge to diagnose and treat effectively. The French physician Prosper Ménière first described this disorder approximately 150 years ago. Yet, researchers are still uncertain of its exact etiology and underlying pathophysiology.1,2
Ménière’s disease is defined as a chronic condition with recurrent episodes of vertigo that are associated with sensorineural hearing loss, tinnitus, and/or a sensation of aural fullness.3,4 Thanks to researchers’ evolving knowledge of Ménière’s disease, a new definition has been proposed: a degenerating inner ear leading to impaired homoeostasis, hearing loss, and vertigo.5
In the United States, the prevalence of Ménière’s disease is estimated at 15 to 150 cases per 100,000 persons. (This wide variation in prevalence reflects a lack of standard diagnostic criteria, as well as differences based on geographic area.6,7) Many affected individuals experience symptoms significant enough to lead to disability.6,7 Patients with Ménière’s disease usually present between ages 20 and 60, with a peak incidence occurring between ages 40 and 50.1,4,6,8 This disease affects both genders, but is slightly more common in women.1,4,6,8
Diagnosis of Ménière’s disease is based on recognition of the clinical symptoms that characterize the disorder, and management is centered on heuristic treatment options. Thus, a person may experience mild to severe symptoms of Ménière’s disease for months to years before receiving either the diagnosis or first-line treatment. This article reviews the current understanding of the underlying physiologic mechanisms that cause Ménière’s disease and discusses the criteria for diagnosis and various treatment options.
ETIOLOGY AND PATHOPHYSIOLOGY
The cause of Ménière’s disease and the subsequent mechanical, physiologic, and biochemical changes that occur are poorly understood, but several theories have been proposed. According to the current central theory, a buildup of fluid (endolymph) within the cochlear and saccular ducts in the inner ear causes distention of these structures into the endolymphatic space, resulting in the development of endolymphatic hydrops.4 Pressure from endolymphatic hydrops may cause damage to hair follicles and to the vestibular epithelium, resulting in symptoms of vertigo, tinnitus, and hearing loss.1,4 Researchers previously attributed the symptoms of Ménière’s disease completely to hydrops and focused on identifying anatomic abnormalities.7,9 However, studies now suggest that a range of pathophysiologic processes resulting from intrinsic and/or extrinsic factors may be responsible.7,9 While hydrops may develop, it is not always the definitive or only cause of Meniere’s disease symptoms.9
Recently recognized factors that contribute to the development of Meniere’s disease include autoimmune reactions, genetic irregularities, vascular abnormalities, and viral influences. Approximately one-third of Ménière’s disease cases can be attributed to an autoimmune origin.1,6 Researchers hypothesize that several immunologic processes may contribute to Ménière’s disease:
• Antibodies may cause inner ear damage,
• Injury to the inner ear may result in the release of cytokines which provoke immune reactions, and
• Certain genes may affect a person’s immune system and increase the probability of Ménière’s disease.1
The probability of a genetic influence is supported by the fact that one in 20 people with Ménière’s disease reports a positive family history of the disorder.4
Many patients with Ménière’s disease experience migraine headaches, and thus vascular abnormalities are another area of consideration among the etiologies of this disease.10 Researchers are also studying a potential viral cause in the development of Ménière’s disease.1,6
Regardless of the specific cause or physiologic changes that occur, the one common finding in patients with Ménière’s disease is a dysfunction of fluid homeostasis within the inner ear.
On the next page: Diagnosis >>
DIAGNOSIS
Establishing the diagnosis of Ménière’s disease can be difficult and time-consuming because the symptoms of the disorder are nonspecific and variable. Ménière’s disease is a clinical diagnosis, and thus the clinician must conduct a thorough physical exam and elicit a very specific history, including a detailed description of vertigo incidents and associated symptoms. Often, the greatest challenge is encouraging patients to articulate the details of their episodes. Patients may not keep a record of the variations of episodes, nor do they always know what information is needed. Thus, the provider needs to elicit specific information by asking questions regarding frequency and duration of episodes, as well as fluctuation of hearing loss, nausea, and tinnitus. Symptoms associated with vertigo during a Ménière’s episode may include nausea, vomiting, gait imbalance, and tinnitus. Most vertigo attacks from Ménière’s disease occur in clusters, but they may also occur sporadically.6
An additional challenge for clinicians is that other potential diagnoses related to vertigo must be excluded before the diagnosis of Ménière’s disease can be made. Also, it is important to note that specialists may differentiate Ménière’s disease, an idiopathic condition, from Ménière’s syndrome, which results from known causes of damage to the inner ear. In the literature, however, this distinction in terminology is not always clear.7
Specific diagnostic criteria for Ménière’s disease, defined in 1995 by the American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS), remain the gold standard for diagnosis.3 A “definite” diagnosis of Ménière’s disease is based on:
• A history of two or more episodes of spontaneous vertigo lasting 20 minutes or longer,
• Hearing loss documented by audiometry at least once,
• Presence of tinnitus, and/or
• A sensation of aural fullness.2,6,7
The AAO-HNS diagnostic criteria also define categories of “probable” and “possible” Ménière’s disease based on the frequency of vertigo episodes or the presence of a combination of associated symptoms (see Table 1).3,7
Patients with Ménière’s disease may experience different patterns of symptoms. “Auditory dominant” Ménière’s disease produces more hearing loss changes than vertigo, while “vestibular dominant” causes frequent episodes of severe vertigo and less severe hearing changes. A “mixed” pattern of Ménière’s disease manifests with both hearing fluctuations and vertigo.5
Unilateral symptoms are most common; however, bilateral disorder occurs in approximately 25% of patients, either at onset or with changing symptomatology over time.5,8,9
On the next page: History and examination >>
HISTORY AND EXAMINATION
Obtaining a detailed history from the patient and completing thorough neurologic and otologic examinations are essential components of the diagnostic process. Audiometry should be completed to evaluate neurosensory hearing loss, as audiometrically documented hearing loss is part of the AAO-HSN diagnostic criteria for Ménière’s disease.6
Based on findings from the patient’s history, physical exam, and audiometric testing, a tentative diagnosis can be made. The role and inclusion of adjunctive tests in the diagnostic process varies considerably by region in the US. While not required for the diagnosis of Ménière’s disease, electrical vestibular stimulation and videonystagmography are useful tests to assess abnormalities in vestibular function and monitor disease progression, which may help determine intervention options.6 Additional diagnostic tests may be suggested due to the essential need to exclude other potential causes of vertigo prior to determining the final diagnosis of Ménière’s disease.
Triggers of Vertigo
Selected triggers of vertigo that must be considered are benign paroxysmal positional vertigo (BPPV), labyrinthitis, acoustic neuroma, migraine with vertigo, and cerebral vascular events.6 Diagnostic tests are indicated to rule out certain problems, such as MRI to exclude a tumor or an acoustic neuroma. Distinct differences noted during a complete assessment may help eliminate certain disorders. BPPV is triggered by a change in physical position and usually lasts less than one minute; the diagnosis can be confirmed by the Dix-Hallpike maneuver.4 Labyrinthitis is characterized by acute vertigo associated with continuing imbalance, while instability with walking resolves completely between vertigo episodes in Ménière’s disease.4 If abnormal neurologic manifestations are noted during the exam or reported in a patient’s account of a vertigo episode, then a transient ischemic attack or stroke must be ruled out by more detailed diagnostic testing.
TREATMENT OPTIONS
Presently, no evidence-based guidelines exist for the treatment of Ménière’s disease, and the evidence supporting the efficacy of currently used therapies is inconsistent. However, several medicines and treatments are useful in relieving symptoms and improving a patient’s quality of life.
Primary care clinicians can initiate treatment for Ménière’s disease through lifestyle recommendations and prescription of specific medications. Everyday adjustments that incorporate dietary changes, stress reduction, adequate sleep, and regular exercise have been shown to improve vertigo symptoms in 60% of patients with Ménière’s disease.5,9
On the next page: Lifestyle changes >>
Lifestyle Changes
Dietary changes. Patients diagnosed with Ménière’s disease may benefit from following a low-sodium diet, limiting their daily sodium intake to between 1,000 and 2,000 mg.2,7,11 A low-sodium diet is believed to have a positive impact on inner ear fluid homeostasis by decreasing fluid retention and reducing the endolymphatic hydrops.2,7,11 Decreasing alcohol and caffeine consumption is also routinely recommended as part of the treatment of Ménière’s disease.2,5
Researchers have recently suggested a different approach to dietary changes for Ménière’s disease that reflects the underlying loss of ability to regulate fluid in the inner ear. This alternate method of dietary regulation aims to maintain fluid homeostasis by avoiding variations in the daily intake of sodium, caffeine, or alcohol, rather than limiting daily consumption.5
The goal of any proposed dietary changes is to limit fluid and electrolyte shifts that could disrupt the delicate fluid balance in the inner ear.9 When caring for patients with Ménière’s disease, clinicians need to keep in mind that dietary changes may be difficult and will probably require ongoing encouragement.
Stress reduction. Stress is associated with the occurrence of Ménière’s disease and often is the trigger for an acute episode of symptom exacerbation.5 Thus, clinicians should encourage stress management as a way to reduce the impact of Ménière’s disease on a patient’s life. Stress reduction techniques that can be recommended include progressive relaxation, meditation with deep breathing, yoga, and exercise.
Although studies of the effect of stress reduction methods on Ménière’s disease are not available in the current literature, the association of stress with Ménière’s disease is well documented.5 By avoiding stress, it is hoped, patients may experience a reduction in the frequency and severity of Ménière’s disease–associated episodes of vertigo. Researchers also suggest that stress reduction and patient education may help alleviate patients’ feelings of frustration resulting from misinformation about their condition.2,11
Oral Medications for Acute Relief
Acute attacks of vertigo associated with Ménière’s disease can be treated with benzodiazepines, antiemetics, or anticholinergic medications.4,6 Alleviation of symptoms is achieved through different physiologic pathways, based on the drug category prescribed. If a patient reports typical symptoms of Ménière’s disease but has not undergone audiometry, the plausible diagnosis may lead to tentative treatment for acute episodes if other causes of vertigo have been ruled out.
Antihistamines, such as meclizine or dimenhydrinate, may help reduce vertigo symptoms and associated nausea by blocking the effects of histamine.4,6 One of the most common side effects of antihistamines is drowsiness, so patients must be cautioned to avoid certain activities while taking this medication. Antihistamines should not be given to patients with glaucoma or prostate disease due to the potentially strong anticholinergic effects of these drugs.4,6
Scopolamineis a belladonna alkaloid that can be applied topically on the tissue just behind the ear to help reduce nausea and vomiting related to vertigo.11
Another option for treatment of acute vertigo is a benzodiazepine, such as alprazolam, to suppress active cerebellar responses; this agent may also reduce anxiety associated with an acute episode of vertigo.6,11 Benzodiazepines should be started at the lowest dose and increased as needed to the maximum recommended for individual medications based on symptom relief and side effects.6 Although caution needs to be used when prescribing benzodiazepines, studies show that they can be effective for persons with Meniere’s disease.11
Other antiemetic medications, such as promethazine or ondansetron, may be needed to treat severe nausea, but these agents should be used cautiously with other medications due a potential side effect of sedation.
Long-Term Oral Medication
Medication for long-term management of Ménière’s disease can promote improvement in symptoms and reduce the frequency of vertigo episodes. A mild diuretic, such as hydrochlorothiazide with or without triamterene, taken on a regular basis reduces extracellular fluids and may decrease pressure from endolymphatic hydrops.2,7 While strong evidence regarding the efficacy of diuretics is lacking, the majority of patients with Ménière’s disease who are treated with diuretics do experience improvement in vertigo.2,5
Betahistine hydrochloride, a vasodilator and histamine receptor antagonist, is another medication to consider for management of Ménière’s disease.1 This agent is not approved by the FDA; however, the FDA classifies betahistine as an inert chemical, so it is available in compounding pharmacies in the United States. The efficacy of betahistine has not been clearly or consistently established in research studies, but it has been and continues to be widely used to treat Ménière’s disease in Europe, with good results. Betahistine affects the microcirculation in the inner ear and inhibits the vestibular nuclei, which may reduce the frequency of vertigo episodes and improve tinnitus associated with Ménière’s disease.2,8,11
On the next page: Intratympanic medication >>
Intratympanic Medication
Patients who do not respond well to the previously described management should be referred to a specialist for additional treatment options. An otolaryngology specialist may administer intratympanic medications to patients with Ménière’s disease who have not responded to primary medical therapy.
Patients in the US who have not responded positively to lifestyle or diuretic medication are commonly offered treatment with intratympanic dexamethasone. The primary goal of this therapy is to improve vertigo without affecting a patient’s hearing; an added effect may be a potential positive impact on the immune system.11 Studies show that intratympanic steroid injection results in control of vertigo in patients with Ménière’s disease, but up to four injection treatments may be required for optimal effectiveness.2,7 Improvement of vertigo is achieved in more than 80% of patients who undergo intratympanic steroid injections.9
An option reserved for patients with severe, frequent vertigo related to Ménière’s disease is a type of chemical ablation of the labyrinth induced by injecting gentamicin into the middle ear.2 Gentamicin has a toxic effect on the vestibular hair cells in the inner ear, resulting in elimination of vestibular function.2 Intratympanic gentamicin is reported to reduce symptoms from Ménière’s disease, but this treatment is only recommended for patients with unilateral disease because it may induce permanent hearing loss.11
The primary care clinician needs to be aware of these intratympanic procedures and encourage patients to follow up with the specialist if additional treatments are indicated.
Portable Pressure Device
Use of the Meniett device is a minimally invasive treatment for Ménière’s disease based on the principle of using alternating pressure to stimulate the flow of endolymph.11 This handheld device delivers low-pressure pulses within the inner ear through a standard ventilation tube in order to increase exchange of fluids and improve homeostasis.8 The Meniett device should be used for five-minute intervals three times per day.12
Several studies have shown excellent results in patients who use the Meniett device routinely for several weeks.2,12 As noted, the use of this device, which is obtained by prescription from an otolaryngologist, requires placement of ventilation tubes.
Acupuncture
A traditional Chinese medical approach, acupuncture is one complementary and alternative medicine therapy that has been studied as a treatment option for Ménière’s disease. Studies on the use of acupuncture to treat vertigo demonstrate a beneficial effect for persons with this disease. While the optimal number and frequency of treatments has not been determined, all types of acupuncture studied showed benefit. Acupuncture has a positive effect in both acute episodes of vertigo in those without Ménière’s disease and in patients who have had Ménière’s disease for many years.13
Vestibular Rehabilitation
An additional adjunctive treatment option to consider for patients with residual disequilibrium is vestibular rehabilitation. Vestibular rehabilitation is designed to desensitize or retrain the balance system response through a series of exercises and activities supervised by a physical or occupational therapist. This rehabilitation may improve balance in patients with Ménière’s disease who have undergone medical or surgical intervention used to treat vertigo. Patients who have significant balance problems occurring between acute vertigo episodes may also benefit from vestibular rehabilitation.6
Surgical Treatment
Surgical intervention should be the last resort to treat Ménière’s disease due to the higher risk involved with any surgical procedure and the potential adverse effect on hearing. Endolymphatic sac decompression surgery involves removing a portion of the mastoid bone, resulting in decompression of the sac adjacent to the sigmoid sinus. This procedure has been used for more than 40 years to control vertigo and has the advantage of preserving hearing.7,9 However, the benefit of this procedure is now somewhat controversial and possibly related to a placebo effect.6 Researchers also report positive results with the use of tenotomy surgery, which involves severing tendons to the stapedius and tensor tympani muscles in the middle ear.14
No surgical procedure should be considered without the recommendation of an otolaryngology specialist. The decision should be made based on the severity of the disease and its effect on the patient, weighed against the risks involved in such an invasive treatment option.
On the next page: Conclusion >>
CONCLUSION
Ménière’s disease is a complex disorder that can significantly alter a person’s quality of life. While neither the exact cause nor pathophysiology underlying Ménière’s disease is well understood, several solid theories are being investigated and contribute to the current understanding of treatment options. Primary care clinicians can help determine this clinical diagnosis based on a detailed history and comprehensive assessment of recurrent vertigo with tinnitus, hearing loss, and possibly a sensation of aural fullness. Establishing the diagnosis of Ménière’s disease requires ruling out other possible causes of vertigo.
Lifestyle changes that improve the consistency of dietary intake of sodium, caffeine, and alcohol as well as reduction of stress are ongoing recommendations for patients with Ménière’s disease. Oral medications from a range of drug categories may be used to improve acute and chronic symptoms, including antiemetics, anticholinergics, antihistamines, benzodiazepines, and mild diuretics. Additionally, a compounded substance with vasodilator and histamine- receptor–antagonist properties (betahistine) can be used for treatment of Meniere’s.
Patients who do not respond well to conservative therapy should be referred to an otolaryngologist for possible intratympanic medications, ventilation tube placement with a prescription for pulse pressure therapy (ie, Meniett device), or surgical intervention. Primary care clinicians can initiate treatment for Ménière’s disease by recommending lifestyle changes, prescribing oral medications, providing patient education, and recognizing indications for referral.
CE/CME No: CR-1310
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology of Ménière’s disease, as it is currently understood.
• Discuss the triad of symptoms that should prompt suspicion for Ménière’s disease in a primary care patient.
• List the diagnostic criteria for “definite” Ménière’s disease, as defined by the American Academy of Otolaryngology–Head and Neck Surgery.
• Review pharmacologic management, intratympanic injection and other nonoperative therapies, and surgical treatment for Ménière’s disease.
FACULTY
Tamera Pearson is an Associate Professor in the School of Nursing at Western Carolina University.
The author has no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Ménière’s disease is a complex disorder of intermittent vertigo, tinnitus, and hearing loss whose symptoms usually manifest between ages 20 and 60. Although this disorder is uncommon, its impact on a person’s quality of life can be significant. Here are the symptoms, criteria for diagnosis, and appropriate treatment or referrals for Ménière’s disease.
Ménière’s disease can significantly affect a person’s quality of life and is a challenge to diagnose and treat effectively. The French physician Prosper Ménière first described this disorder approximately 150 years ago. Yet, researchers are still uncertain of its exact etiology and underlying pathophysiology.1,2
Ménière’s disease is defined as a chronic condition with recurrent episodes of vertigo that are associated with sensorineural hearing loss, tinnitus, and/or a sensation of aural fullness.3,4 Thanks to researchers’ evolving knowledge of Ménière’s disease, a new definition has been proposed: a degenerating inner ear leading to impaired homoeostasis, hearing loss, and vertigo.5
In the United States, the prevalence of Ménière’s disease is estimated at 15 to 150 cases per 100,000 persons. (This wide variation in prevalence reflects a lack of standard diagnostic criteria, as well as differences based on geographic area.6,7) Many affected individuals experience symptoms significant enough to lead to disability.6,7 Patients with Ménière’s disease usually present between ages 20 and 60, with a peak incidence occurring between ages 40 and 50.1,4,6,8 This disease affects both genders, but is slightly more common in women.1,4,6,8
Diagnosis of Ménière’s disease is based on recognition of the clinical symptoms that characterize the disorder, and management is centered on heuristic treatment options. Thus, a person may experience mild to severe symptoms of Ménière’s disease for months to years before receiving either the diagnosis or first-line treatment. This article reviews the current understanding of the underlying physiologic mechanisms that cause Ménière’s disease and discusses the criteria for diagnosis and various treatment options.
ETIOLOGY AND PATHOPHYSIOLOGY
The cause of Ménière’s disease and the subsequent mechanical, physiologic, and biochemical changes that occur are poorly understood, but several theories have been proposed. According to the current central theory, a buildup of fluid (endolymph) within the cochlear and saccular ducts in the inner ear causes distention of these structures into the endolymphatic space, resulting in the development of endolymphatic hydrops.4 Pressure from endolymphatic hydrops may cause damage to hair follicles and to the vestibular epithelium, resulting in symptoms of vertigo, tinnitus, and hearing loss.1,4 Researchers previously attributed the symptoms of Ménière’s disease completely to hydrops and focused on identifying anatomic abnormalities.7,9 However, studies now suggest that a range of pathophysiologic processes resulting from intrinsic and/or extrinsic factors may be responsible.7,9 While hydrops may develop, it is not always the definitive or only cause of Meniere’s disease symptoms.9
Recently recognized factors that contribute to the development of Meniere’s disease include autoimmune reactions, genetic irregularities, vascular abnormalities, and viral influences. Approximately one-third of Ménière’s disease cases can be attributed to an autoimmune origin.1,6 Researchers hypothesize that several immunologic processes may contribute to Ménière’s disease:
• Antibodies may cause inner ear damage,
• Injury to the inner ear may result in the release of cytokines which provoke immune reactions, and
• Certain genes may affect a person’s immune system and increase the probability of Ménière’s disease.1
The probability of a genetic influence is supported by the fact that one in 20 people with Ménière’s disease reports a positive family history of the disorder.4
Many patients with Ménière’s disease experience migraine headaches, and thus vascular abnormalities are another area of consideration among the etiologies of this disease.10 Researchers are also studying a potential viral cause in the development of Ménière’s disease.1,6
Regardless of the specific cause or physiologic changes that occur, the one common finding in patients with Ménière’s disease is a dysfunction of fluid homeostasis within the inner ear.
On the next page: Diagnosis >>
DIAGNOSIS
Establishing the diagnosis of Ménière’s disease can be difficult and time-consuming because the symptoms of the disorder are nonspecific and variable. Ménière’s disease is a clinical diagnosis, and thus the clinician must conduct a thorough physical exam and elicit a very specific history, including a detailed description of vertigo incidents and associated symptoms. Often, the greatest challenge is encouraging patients to articulate the details of their episodes. Patients may not keep a record of the variations of episodes, nor do they always know what information is needed. Thus, the provider needs to elicit specific information by asking questions regarding frequency and duration of episodes, as well as fluctuation of hearing loss, nausea, and tinnitus. Symptoms associated with vertigo during a Ménière’s episode may include nausea, vomiting, gait imbalance, and tinnitus. Most vertigo attacks from Ménière’s disease occur in clusters, but they may also occur sporadically.6
An additional challenge for clinicians is that other potential diagnoses related to vertigo must be excluded before the diagnosis of Ménière’s disease can be made. Also, it is important to note that specialists may differentiate Ménière’s disease, an idiopathic condition, from Ménière’s syndrome, which results from known causes of damage to the inner ear. In the literature, however, this distinction in terminology is not always clear.7
Specific diagnostic criteria for Ménière’s disease, defined in 1995 by the American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS), remain the gold standard for diagnosis.3 A “definite” diagnosis of Ménière’s disease is based on:
• A history of two or more episodes of spontaneous vertigo lasting 20 minutes or longer,
• Hearing loss documented by audiometry at least once,
• Presence of tinnitus, and/or
• A sensation of aural fullness.2,6,7
The AAO-HNS diagnostic criteria also define categories of “probable” and “possible” Ménière’s disease based on the frequency of vertigo episodes or the presence of a combination of associated symptoms (see Table 1).3,7
Patients with Ménière’s disease may experience different patterns of symptoms. “Auditory dominant” Ménière’s disease produces more hearing loss changes than vertigo, while “vestibular dominant” causes frequent episodes of severe vertigo and less severe hearing changes. A “mixed” pattern of Ménière’s disease manifests with both hearing fluctuations and vertigo.5
Unilateral symptoms are most common; however, bilateral disorder occurs in approximately 25% of patients, either at onset or with changing symptomatology over time.5,8,9
On the next page: History and examination >>
HISTORY AND EXAMINATION
Obtaining a detailed history from the patient and completing thorough neurologic and otologic examinations are essential components of the diagnostic process. Audiometry should be completed to evaluate neurosensory hearing loss, as audiometrically documented hearing loss is part of the AAO-HSN diagnostic criteria for Ménière’s disease.6
Based on findings from the patient’s history, physical exam, and audiometric testing, a tentative diagnosis can be made. The role and inclusion of adjunctive tests in the diagnostic process varies considerably by region in the US. While not required for the diagnosis of Ménière’s disease, electrical vestibular stimulation and videonystagmography are useful tests to assess abnormalities in vestibular function and monitor disease progression, which may help determine intervention options.6 Additional diagnostic tests may be suggested due to the essential need to exclude other potential causes of vertigo prior to determining the final diagnosis of Ménière’s disease.
Triggers of Vertigo
Selected triggers of vertigo that must be considered are benign paroxysmal positional vertigo (BPPV), labyrinthitis, acoustic neuroma, migraine with vertigo, and cerebral vascular events.6 Diagnostic tests are indicated to rule out certain problems, such as MRI to exclude a tumor or an acoustic neuroma. Distinct differences noted during a complete assessment may help eliminate certain disorders. BPPV is triggered by a change in physical position and usually lasts less than one minute; the diagnosis can be confirmed by the Dix-Hallpike maneuver.4 Labyrinthitis is characterized by acute vertigo associated with continuing imbalance, while instability with walking resolves completely between vertigo episodes in Ménière’s disease.4 If abnormal neurologic manifestations are noted during the exam or reported in a patient’s account of a vertigo episode, then a transient ischemic attack or stroke must be ruled out by more detailed diagnostic testing.
TREATMENT OPTIONS
Presently, no evidence-based guidelines exist for the treatment of Ménière’s disease, and the evidence supporting the efficacy of currently used therapies is inconsistent. However, several medicines and treatments are useful in relieving symptoms and improving a patient’s quality of life.
Primary care clinicians can initiate treatment for Ménière’s disease through lifestyle recommendations and prescription of specific medications. Everyday adjustments that incorporate dietary changes, stress reduction, adequate sleep, and regular exercise have been shown to improve vertigo symptoms in 60% of patients with Ménière’s disease.5,9
On the next page: Lifestyle changes >>
Lifestyle Changes
Dietary changes. Patients diagnosed with Ménière’s disease may benefit from following a low-sodium diet, limiting their daily sodium intake to between 1,000 and 2,000 mg.2,7,11 A low-sodium diet is believed to have a positive impact on inner ear fluid homeostasis by decreasing fluid retention and reducing the endolymphatic hydrops.2,7,11 Decreasing alcohol and caffeine consumption is also routinely recommended as part of the treatment of Ménière’s disease.2,5
Researchers have recently suggested a different approach to dietary changes for Ménière’s disease that reflects the underlying loss of ability to regulate fluid in the inner ear. This alternate method of dietary regulation aims to maintain fluid homeostasis by avoiding variations in the daily intake of sodium, caffeine, or alcohol, rather than limiting daily consumption.5
The goal of any proposed dietary changes is to limit fluid and electrolyte shifts that could disrupt the delicate fluid balance in the inner ear.9 When caring for patients with Ménière’s disease, clinicians need to keep in mind that dietary changes may be difficult and will probably require ongoing encouragement.
Stress reduction. Stress is associated with the occurrence of Ménière’s disease and often is the trigger for an acute episode of symptom exacerbation.5 Thus, clinicians should encourage stress management as a way to reduce the impact of Ménière’s disease on a patient’s life. Stress reduction techniques that can be recommended include progressive relaxation, meditation with deep breathing, yoga, and exercise.
Although studies of the effect of stress reduction methods on Ménière’s disease are not available in the current literature, the association of stress with Ménière’s disease is well documented.5 By avoiding stress, it is hoped, patients may experience a reduction in the frequency and severity of Ménière’s disease–associated episodes of vertigo. Researchers also suggest that stress reduction and patient education may help alleviate patients’ feelings of frustration resulting from misinformation about their condition.2,11
Oral Medications for Acute Relief
Acute attacks of vertigo associated with Ménière’s disease can be treated with benzodiazepines, antiemetics, or anticholinergic medications.4,6 Alleviation of symptoms is achieved through different physiologic pathways, based on the drug category prescribed. If a patient reports typical symptoms of Ménière’s disease but has not undergone audiometry, the plausible diagnosis may lead to tentative treatment for acute episodes if other causes of vertigo have been ruled out.
Antihistamines, such as meclizine or dimenhydrinate, may help reduce vertigo symptoms and associated nausea by blocking the effects of histamine.4,6 One of the most common side effects of antihistamines is drowsiness, so patients must be cautioned to avoid certain activities while taking this medication. Antihistamines should not be given to patients with glaucoma or prostate disease due to the potentially strong anticholinergic effects of these drugs.4,6
Scopolamineis a belladonna alkaloid that can be applied topically on the tissue just behind the ear to help reduce nausea and vomiting related to vertigo.11
Another option for treatment of acute vertigo is a benzodiazepine, such as alprazolam, to suppress active cerebellar responses; this agent may also reduce anxiety associated with an acute episode of vertigo.6,11 Benzodiazepines should be started at the lowest dose and increased as needed to the maximum recommended for individual medications based on symptom relief and side effects.6 Although caution needs to be used when prescribing benzodiazepines, studies show that they can be effective for persons with Meniere’s disease.11
Other antiemetic medications, such as promethazine or ondansetron, may be needed to treat severe nausea, but these agents should be used cautiously with other medications due a potential side effect of sedation.
Long-Term Oral Medication
Medication for long-term management of Ménière’s disease can promote improvement in symptoms and reduce the frequency of vertigo episodes. A mild diuretic, such as hydrochlorothiazide with or without triamterene, taken on a regular basis reduces extracellular fluids and may decrease pressure from endolymphatic hydrops.2,7 While strong evidence regarding the efficacy of diuretics is lacking, the majority of patients with Ménière’s disease who are treated with diuretics do experience improvement in vertigo.2,5
Betahistine hydrochloride, a vasodilator and histamine receptor antagonist, is another medication to consider for management of Ménière’s disease.1 This agent is not approved by the FDA; however, the FDA classifies betahistine as an inert chemical, so it is available in compounding pharmacies in the United States. The efficacy of betahistine has not been clearly or consistently established in research studies, but it has been and continues to be widely used to treat Ménière’s disease in Europe, with good results. Betahistine affects the microcirculation in the inner ear and inhibits the vestibular nuclei, which may reduce the frequency of vertigo episodes and improve tinnitus associated with Ménière’s disease.2,8,11
On the next page: Intratympanic medication >>
Intratympanic Medication
Patients who do not respond well to the previously described management should be referred to a specialist for additional treatment options. An otolaryngology specialist may administer intratympanic medications to patients with Ménière’s disease who have not responded to primary medical therapy.
Patients in the US who have not responded positively to lifestyle or diuretic medication are commonly offered treatment with intratympanic dexamethasone. The primary goal of this therapy is to improve vertigo without affecting a patient’s hearing; an added effect may be a potential positive impact on the immune system.11 Studies show that intratympanic steroid injection results in control of vertigo in patients with Ménière’s disease, but up to four injection treatments may be required for optimal effectiveness.2,7 Improvement of vertigo is achieved in more than 80% of patients who undergo intratympanic steroid injections.9
An option reserved for patients with severe, frequent vertigo related to Ménière’s disease is a type of chemical ablation of the labyrinth induced by injecting gentamicin into the middle ear.2 Gentamicin has a toxic effect on the vestibular hair cells in the inner ear, resulting in elimination of vestibular function.2 Intratympanic gentamicin is reported to reduce symptoms from Ménière’s disease, but this treatment is only recommended for patients with unilateral disease because it may induce permanent hearing loss.11
The primary care clinician needs to be aware of these intratympanic procedures and encourage patients to follow up with the specialist if additional treatments are indicated.
Portable Pressure Device
Use of the Meniett device is a minimally invasive treatment for Ménière’s disease based on the principle of using alternating pressure to stimulate the flow of endolymph.11 This handheld device delivers low-pressure pulses within the inner ear through a standard ventilation tube in order to increase exchange of fluids and improve homeostasis.8 The Meniett device should be used for five-minute intervals three times per day.12
Several studies have shown excellent results in patients who use the Meniett device routinely for several weeks.2,12 As noted, the use of this device, which is obtained by prescription from an otolaryngologist, requires placement of ventilation tubes.
Acupuncture
A traditional Chinese medical approach, acupuncture is one complementary and alternative medicine therapy that has been studied as a treatment option for Ménière’s disease. Studies on the use of acupuncture to treat vertigo demonstrate a beneficial effect for persons with this disease. While the optimal number and frequency of treatments has not been determined, all types of acupuncture studied showed benefit. Acupuncture has a positive effect in both acute episodes of vertigo in those without Ménière’s disease and in patients who have had Ménière’s disease for many years.13
Vestibular Rehabilitation
An additional adjunctive treatment option to consider for patients with residual disequilibrium is vestibular rehabilitation. Vestibular rehabilitation is designed to desensitize or retrain the balance system response through a series of exercises and activities supervised by a physical or occupational therapist. This rehabilitation may improve balance in patients with Ménière’s disease who have undergone medical or surgical intervention used to treat vertigo. Patients who have significant balance problems occurring between acute vertigo episodes may also benefit from vestibular rehabilitation.6
Surgical Treatment
Surgical intervention should be the last resort to treat Ménière’s disease due to the higher risk involved with any surgical procedure and the potential adverse effect on hearing. Endolymphatic sac decompression surgery involves removing a portion of the mastoid bone, resulting in decompression of the sac adjacent to the sigmoid sinus. This procedure has been used for more than 40 years to control vertigo and has the advantage of preserving hearing.7,9 However, the benefit of this procedure is now somewhat controversial and possibly related to a placebo effect.6 Researchers also report positive results with the use of tenotomy surgery, which involves severing tendons to the stapedius and tensor tympani muscles in the middle ear.14
No surgical procedure should be considered without the recommendation of an otolaryngology specialist. The decision should be made based on the severity of the disease and its effect on the patient, weighed against the risks involved in such an invasive treatment option.
On the next page: Conclusion >>
CONCLUSION
Ménière’s disease is a complex disorder that can significantly alter a person’s quality of life. While neither the exact cause nor pathophysiology underlying Ménière’s disease is well understood, several solid theories are being investigated and contribute to the current understanding of treatment options. Primary care clinicians can help determine this clinical diagnosis based on a detailed history and comprehensive assessment of recurrent vertigo with tinnitus, hearing loss, and possibly a sensation of aural fullness. Establishing the diagnosis of Ménière’s disease requires ruling out other possible causes of vertigo.
Lifestyle changes that improve the consistency of dietary intake of sodium, caffeine, and alcohol as well as reduction of stress are ongoing recommendations for patients with Ménière’s disease. Oral medications from a range of drug categories may be used to improve acute and chronic symptoms, including antiemetics, anticholinergics, antihistamines, benzodiazepines, and mild diuretics. Additionally, a compounded substance with vasodilator and histamine- receptor–antagonist properties (betahistine) can be used for treatment of Meniere’s.
Patients who do not respond well to conservative therapy should be referred to an otolaryngologist for possible intratympanic medications, ventilation tube placement with a prescription for pulse pressure therapy (ie, Meniett device), or surgical intervention. Primary care clinicians can initiate treatment for Ménière’s disease by recommending lifestyle changes, prescribing oral medications, providing patient education, and recognizing indications for referral.
1. Greco A, Gallo A, Fusconi M, et al. Ménière’s disease might be an autoimmune condition? Autoimmun Rev. 2012;11:731-738.
2. Greenberg SL, Nedzelski JM. Medical and noninvasive therapy for Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1081-1090.
3. American Academy of Otolaryngology–Head and Neck Foundation, Inc. Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Ménière’s disease. Otolaryngol Head Neck Surg. 1995;113:181-185.
4. Syed I, Aldren C. Ménière’s disease: an evidence based approach to assessment and management. Int J Clin Pract. 2012;66:166-170.
5. Rauch SD. Clinical hints and precipitating factors in patients suffering from Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1011-1017.
6. Dinces EA, Rauch SS. Ménière’s disease. In: UpToDate. Deschler DG, Lin FH, eds. 2012:November 27,2012.
7. Semaan MT, Megerian CA. Ménière’s disease: a challenging and relentless disorder. Otolaryngol Clin North Am. 2011;44:383-403.
8. Martin González C, González FM, Trinidad A, et al. Medical management of Ménière’s disease: a 10-year case series and review of literature. Eur Arch Otorhinolaryngol. 2010;267:1371-1376.
9. Berlinger NT. Ménière’s disease: new concepts, new treatments. Minn Med. 2011;94:33-36.
10. von Brevern M, Neuhauser H. Epidemiological evidence for a link between vertigo and migraine. J Vestib Res. 2011;21:299-304.
11. Coelho DH, Lalwani AK. Medical management of Ménière’s disease. Laryngoscope. 2008;118:1099-1108.
12. Gates GA, Green JD Jr, Tucci DL, Telian SA. The effects of transtympanic micropressure treatment in people with unilateral Ménière’s disease. Arch Otolaryngol Head Neck Surg. 2004;130:718-725.
13. Long A, Xing M, Morgan K, Brettle A. Exploring the evidence base for acupuncture in the treatment of Ménière’s syndrome-a systematic review. Evid Based Complement Alternat Med. 2011;2011:1-13.
14. Loader B, Beicht D, Hamzavi JS, Franz P. Tenotomy of the middle ear muscles causes a dramatic reduction in vertigo attacks and improves audiological function in definite Ménière’s disease. Acta Otolaryngol. 2012;132:491-497.
1. Greco A, Gallo A, Fusconi M, et al. Ménière’s disease might be an autoimmune condition? Autoimmun Rev. 2012;11:731-738.
2. Greenberg SL, Nedzelski JM. Medical and noninvasive therapy for Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1081-1090.
3. American Academy of Otolaryngology–Head and Neck Foundation, Inc. Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Ménière’s disease. Otolaryngol Head Neck Surg. 1995;113:181-185.
4. Syed I, Aldren C. Ménière’s disease: an evidence based approach to assessment and management. Int J Clin Pract. 2012;66:166-170.
5. Rauch SD. Clinical hints and precipitating factors in patients suffering from Ménière’s disease. Otolaryngol Clin North Am. 2010;43:1011-1017.
6. Dinces EA, Rauch SS. Ménière’s disease. In: UpToDate. Deschler DG, Lin FH, eds. 2012:November 27,2012.
7. Semaan MT, Megerian CA. Ménière’s disease: a challenging and relentless disorder. Otolaryngol Clin North Am. 2011;44:383-403.
8. Martin González C, González FM, Trinidad A, et al. Medical management of Ménière’s disease: a 10-year case series and review of literature. Eur Arch Otorhinolaryngol. 2010;267:1371-1376.
9. Berlinger NT. Ménière’s disease: new concepts, new treatments. Minn Med. 2011;94:33-36.
10. von Brevern M, Neuhauser H. Epidemiological evidence for a link between vertigo and migraine. J Vestib Res. 2011;21:299-304.
11. Coelho DH, Lalwani AK. Medical management of Ménière’s disease. Laryngoscope. 2008;118:1099-1108.
12. Gates GA, Green JD Jr, Tucci DL, Telian SA. The effects of transtympanic micropressure treatment in people with unilateral Ménière’s disease. Arch Otolaryngol Head Neck Surg. 2004;130:718-725.
13. Long A, Xing M, Morgan K, Brettle A. Exploring the evidence base for acupuncture in the treatment of Ménière’s syndrome-a systematic review. Evid Based Complement Alternat Med. 2011;2011:1-13.
14. Loader B, Beicht D, Hamzavi JS, Franz P. Tenotomy of the middle ear muscles causes a dramatic reduction in vertigo attacks and improves audiological function in definite Ménière’s disease. Acta Otolaryngol. 2012;132:491-497.

HPV Infection and Cervical Cancer Prevention
CE/CME No: CR-1309
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the central role that human papillomavirus (HPV) infection plays in the development of cervical intraepithelial neoplasia (CIN) and cervical cancer.
• Instruct patients on contributing cofactors of HPV infection and cervical cancer, including tobacco use, parity, use of oral contraceptives, co-infection with HIV, and immunosuppression.
• Describe the current recommendations for cervical cancer screening from professional societies, national health organizations, and federal agencies, including age-appropriate screening for cytology and high-risk HPV.
• Explain the timing and administration schedules of the currently available HPV vaccines and the patient populations for which the vaccines have been approved.
• Discuss the ablative and excisional procedures used to treat CIN and the treatment options for cervical cancer.
FACULTY
Heather P. Adams is an Assistant Professor/Clinical Coordinator in the Physician Assistant Program at Gannon University in Erie, Pennsylvania; in the program, Erica L. Carnright is a Physician Assistant student on clinical rotations.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Improved understanding of the central role of human papillomavirus (HPV) in cervical carcinogenesis has led to the development of vaccines and DNA testing for high-risk HPV subtypes. But age-appropriate cytologic screening remains the cornerstone in the prevention and early detection of cervical intraepithelial neoplasia (CIN) and cervical cancer. With prompt diagnosis and treatment of both CIN and early-stage cervical cancer, the prognosis for this disease is excellent.
Cervical cancer is the third most common cancer in women worldwide.1 More than 12,000 women are diagnosed with cervical cancer annually in the United States, with nearly 4,000 cervical cancer deaths reported each year.2-4 Globally, cervical cancer accounts for more than 529,000 new cancer cases and more than 275,000 cancer deaths in women annually, with more than 80% of these cases occurring in developing countries.1,5-7 Over the past 50 years, overall cervical cancer incidence and mortality have declined by more than 70% in the US, largely due to the development of the Papanicolaou screening test (Pap smear).7
Cervical cancer develops from precancerous, or neoplastic, cells of the cervix. Known as cervical intraepithelial neoplasia (CIN), these precancerous lesions are caused by infection with the human papillomavirus (HPV), the main etiologic factor in the development of cervical cancer. The progression from CIN to invasive cervical cancer is typically slow. Because of the long natural history of cervical cancer, screening and preventive measures, when appropriately implemented, can identify precancerous cells before they progress to cervical cancer.
EPIDEMIOLOGY AND RISK FACTORS
In 2013, approximately 12,340 new cases of cervical cancer will be diagnosed in the US, and 4,030 US women will die as a result of cervical cancer, according to National Cancer Institute estimates.8 Although the incidence rate of cervical cancer in the US has declined significantly, medically underserved populations remain disproportionately affected, with more than 60% of new cases occurring in underserved areas or underscreened populations.6 Epidemiologic studies have also shown higher incidence and mortality among minority populations.9 The incidence of cervical cancer is 30% higher among African American than white women, and the mortality rate is twice as high.9 The incidence of cervical cancer is highest among Hispanics.8
Although many risk factors contribute to the development of cervical cancer, HPV infection is the most important (see Table 12,7,9). HPV has been detected in up to 99% of all invasive cervical cancers, and its presence is necessary for oncogenesis to occur.10 HPV is the most common type of sexually transmitted infection (STI) in the US, with 75% to 80% of sexually active adults acquiring an HPV infection before age 50.11 It is estimated that a woman has a 10% lifetime risk for being infected with HPV, and this risk increases by 15% to 25% with each new sexual partner.7
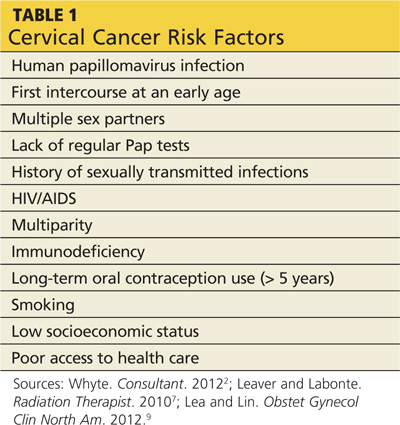
Studies have shown that cigarette smoking contributes to the development of cervical cancer, with both previous and current smokers having a two- to threefold increased risk for high-grade CIN and cervical cancer, compared with never-smokers.5,9,12 Although the exact cause of this association has not been determined, carcinogens from cigarettes have been found within the cervical mucosa, which may cause alterations in the tissue.2,7
Long-term (≥ 5 years) use of oral contraceptive pills (OCP) by patients with an HPV cervical infection has been established as a risk factor for cervical cancer. It has been shown that the use of OCP causes an increase in HPV gene expression.5 In a study that compared two groups of women who tested positive for HPV DNA, the group that used OCP had a fourfold greater risk for cervical cancer than the group that did not use OCP.2,9,13
Parity, particularly multiparity, and HIV co-infection have also been implicated as risk factors for cervical cancer. The relationship between parity and cervical cancer likely results from the presence of the transformation zone on the ectocervix and increased squamous metaplasia during pregnancy, both of which make cervical tissue more vulnerable to HPV infection.14 HIV co-infection increases the risk for cervical cancer secondary to its immunosuppressive effects; women with both HIV and high-risk HPV infections are four times more likely to develop cervical cancer.15
On the next page: Cervical anatomy >>
CERVICAL ANATOMY
The cervix is the narrow, fibromuscular neck that makes up the lower portion of the uterus. The endocervix is the cervical canal, and the ectocervix extends inferiorly into the vagina.7 The internal os is the opening of the cervix into the uterus, and the external os is the opening of the cervix into the vagina. The endocervix is lined with glandular columnar epithelium. This tissue can extend beyond the external os onto the ectocervix, and when this occurs it is known as cervical ectopy.16 The surface of the cervix lying between the glandular tissue and the vaginal wall is comprised of stratified nonkeratinizing squamous epithelium.7 The squamocolumnar junction (SCJ) is defined as the location where the squamous and glandular cells meet.17 Adjacent to the SCJ is the transformation zone, an area vulnerable to HPV infection where glandular cells are actively undergoing squamous metaplasia.17
The location of the SCJ varies depending on age and hormonal changes. In prepubertal females, the SCJ is close to the external os, but in women of reproductive age, the SCJ moves away from the external os onto the surface of the ectocervix. This change in the location of the SCJ occurs due to increased estrogen levels following menarche, which cause the endocervical canal to elongate.7,17 A satisfactory Pap smear for cervical cytology includes cells from the transformation zone, as well as the ectocervix and endocervix.
HUMAN PAPILLOMAVIRUS
The human papillomavirus is a nonenveloped, double-stranded DNA virus that is known to cause abnormalities in skin cells. This virus is predominantly spread through sexual contact via skin-to-skin transmission. There are more than 100 known subtypes in the HPV family, of which 40 subtypes have been recognized to cause genital tract disease. Of these 40 subtypes, 15 have been identified to be oncogenic.9,18,19
HPV subtypes are separated into low-risk and high-risk categories (see Table 25,7,18). The low-risk subtypes cause either no cellular change, low-grade intraepithelial neoplasia, or condyloma. The high-risk subtypes are associated with the development of CIN and cancer. Of the 15 high-risk subtypes, HPV-16 and HPV-18 are the most oncogenic, followed by HPV-31 and HPV-45. HPV-16 is implicated in up to 70% of cervical cancers, HPV-18 in up to 20%, and HPV-31 and HPV-45 in up to 10%.5,9,20 Of the low-risk HPV subtypes, HPV-6 and HPV-11 cause 90% of condyloma infections.16,21
After HPV infection is contracted, the virus migrates into the squamous epithelial mucosa of the cervix, where it is capable of altering cells and causing the development of precancerous properties. This typically happens within the transformation zone, which is vulnerable to HPV infection.17 Depending on the state of the host’s immune system, many of these HPV infections clear without intervention. It is estimated that approximately 70% of HPV infections resolve spontaneously within one year, and 90% resolve within two years.5 HPV infections that persist beyond two years are more likely to lead to the development of CIN and, ultimately, cervical cancer, if left untreated.22 There is an association between genetic amplification (extra copies) of gene 3q26 and progression of CIN to cervical cancer, but ultimately a high-risk HPV subtype must be involved for cervical cancer to develop.23
On the next page: Clinical manifestations, screening, and prevention >>
CLINICAL MANIFESTATIONS
Due to the widespread use of cervical cytology, many cases of CIN and cervical cancer are diagnosed well before symptoms develop. In the early stages of cervical cancer, most women are asymptomatic, although some women may present with a watery, blood-tinged, or malodorous vaginal discharge.7 Any type of abnormal bleeding, whether it is between menstrual cycles, postcoital, or postmenopause, warrants an evaluation for pathologic processes, including but not limited to cervical cancer. Patients with late-stage cervical cancer may present with complaints of pelvic pain and painful intercourse due to the enlargement of a cervical mass. Vaginal discharge may change in consistency from watery to purulent and become foul-smelling due to cervical tissue necrosis.7,9,22 In some cases of advanced invasive cervical cancer, patients will experience hematuria or rectal bleeding secondary to tumor invasion through the bladder or rectal wall.9 Other nonspecific signs and symptoms of cervical cancer include unexplained weight loss accompanied by nausea or vomiting, as well as loss of appetite.7,9
SCREENING AND PREVENTION
Widespread use of the Pap test since the 1950s has led to marked reductions in the incidence of cervical cancer. The Pap test enables clinicians to screen for and detect CIN. More recently, liquid-based cytology has been utilized for the same purpose. The Pap smear involves placing the sample cells directly onto a microscopic slide. With liquid-based cytology, the sample is placed into a vial,2,7 where a portion is processed in preservative liquid under light microscopy for review by a cytologist.7 The remainder of the cell sample can be tested for HPV and STIs.
DNA testing for high-risk HPV subtypes is available and has been incorporated into the cervical cancer screening guidelines. The guidelines recommend HPV DNA testing as a co-test to be performed with cytology in women older than 30.2,24 The recommendation for HPV DNA testing in this age-group reflects greater concern that older women’s immune systems will not clear the virus, leaving these women at higher risk for developing CIN and cancer.25,26 HPV-16 and HPV-18 genotyping is available and is recommended in women 30 or older whose co-testing reveals normal cytology in the presence of a high-risk HPV subtype.27
Additionally, HPV DNA testing is used to triage cytology that reveals atypical squamous cells of undetermined significance. HPV DNA testing is not performed routinely in younger women because of the high likelihood that their immune systems will clear the virus. Genetic testing for the 3q26 gene amplification is available but has not yet been incorporated into the screening guidelines.27
Screening Guidelines
In September 2012, the American Cancer Society, American Society for Colposcopy and Cervical Pathology (ASCCP), and the American Society for Clinical Pathology developed a new set of cervical cancer screening guidelines.28 The US Preventive Services Task Force (USPSTF) and American College of Obstetricians and Gynecologists (ACOG) have also created standard recommendations for cervical cancer screening (see Table 32,28-34). Although ACOG’s standards have varied from those of other organizations in the past, their 2012 guidelines now align closely.
All these organizations recommend that cervical screening begin at age 21 for all women, regardless of the age of sexual initiation.31 Many young women who acquire an HPV infection will clear the infection within the first one to two years. Eliminating screening of women younger than 21 will help to avoid unnecessary biopsies and invasive treatment.29-32
All the guidelines include general recommendations for screening; however, each organization has made exceptions for patients at higher risk for developing cervical cancer.2,28-34 ASCCP has also developed algorithms for management of the multitude of abnormal cervical cancer screening results. These algorithms are available through the ASCCP Web site (www.asccp.org).
Vaccinations
HPV vaccines developed in recent years are now helping to prevent HPV infections from occurring. Currently, the FDA has approved the use of two HPV vaccines: a quadrivalent recombinant HPV vaccine (Gardasil) in 2006 and a bivalent HPV vaccine in 2009 (Cervarix).2 The quadrivalent vaccine was approved for girls and women ages 9 to 26 and protects against HPV subtypes 6 and 11—two low-risk HPV subtypes that can cause genital warts—and subtypes 16 and 18—two high-risk HPV subtypes that can cause cervical cancer. This vaccine is given in a three-dose series at months 0, 2, and 6, and should be initiated before women become sexually active for maximum effectiveness.22,35 The quadrivalent vaccine received additional FDA approval for administration in boys and men ages 9 to 26 in 2010.36
The bivalent vaccine is given in a three-dose series as well and is approved for girls and women ages 9 to 25.37 This vaccine offers protection against high-risk HPV subtypes 16 and 18.38
Recent trials have shown that both the quadrivalent and bivalent vaccines are more than 90% effective in preventing the development of precancerous cells from HPV subtypes 16 and 18.2 Although these vaccines provide protection against the two most oncogenic HPV subtypes, routine cytologic testing is still recommended in patients who receive the vaccines because they do not protect against all high-risk subtypes.2,35,38
On the next page: Diagnosis and staging >>
DIAGNOSIS AND STAGING
The Bethesda staging system provides uniform terminology to classify all abnormal Pap smears and cytology reports (see Table 434). Patients with abnormal cytology typically undergo a colposcopic examination to further evaluate the abnormality. During a colposcopy, the clinician examines the cervix with the use of a lighted microscope, known as a colposcope, which magnifies the cells of the cervix and allows for localized sampling of abnormal-appearing areas through punch biopsy.7,9 The area of focus is the transformation zone because of its known vulnerability to HPV infections. A colposcopy is considered inadequate if the SCJ, and therefore the transformation zone, is not fully visualized.
Acetic acid is applied to the cervix during colposcopy, and any dysplastic cells present take up the acid and turn white, a process known as acetowhitening. Indications for a punch biopsy include acetowhitening, leukoplakia, and abnormal vasculature marked by punctation, bizarre-appearing vessels, or the appearance of a mosaic pattern.16 Indications for sampling of the endocervical canal through endocervical curettage include inadequate colposcopy, presence of a lesion that extends beyond the view of the colposcope, or atypical glandular cells on cytology.16 The biopsy results help to determine if a diagnostic conization is needed for further evaluation of the lesion.16
After a cervical biopsy is performed, the abnormal cells may be classified as CIN 1, 2, or 3 or carcinoma in situ (see Table 516). These stages of intraepithelial neoplasia are determined by the depth to which abnormal, immature cells have invaded the cervical epithelium.16 CIN 2 and 3 are more likely to develop into cervical cancer if they are not properly treated.5,16,17 The majority of CIN 1 lesions do not progress into cancer and typically resolve on their own or are cleared by the patient’s immune system. While most CIN 1 lesions are caused by high-risk HPV, these may be less oncogenic subtypes.7,17
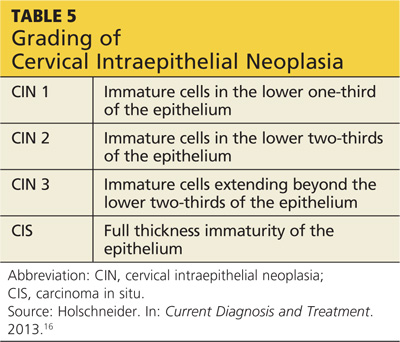
The two most common histologic subtypes of cervical cancer are squamous cell carcinoma and adenocarcinoma. Squamous cell carcinoma comprises more than 70% of cervical cancers, while adenocarcinoma makes up approximately 25%.39 Neuroendocrine, small cell, and mixed-cell carcinomas make up the remainder of cervical cancers.9 Squamous cell carcinoma arises from the squamocolumnar junction or ectocervix, and adenocarcinoma tends to arise from the glandular cells of the endocervix.7,9,17 Studies show that adenocarcinoma is slowly becoming more prevalent in the US than squamous cell carcinoma.9
Once a diagnosis of cervical cancer has been established, additional testing and procedures are performed to rule out lymph node and organ involvement.9,16 The International Federation of Gynecology and Oncology system is then used to clinically stage the cervical cancer40 (see Table 67,40,41). This staging system, updated in 2009, is based solely on clinical examination findings. Once cervical cancer is staged, measures are taken to assess which treatment option is most appropriate.
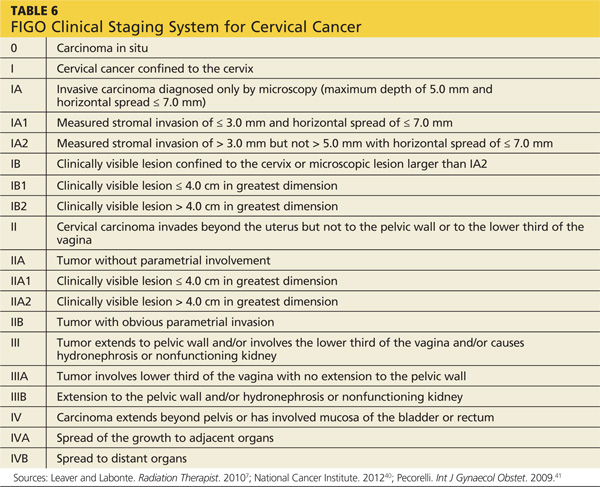
On the next page: Treatment modalities >>
TREATMENT MODALITIES
Treatment strategies for CIN correspond to the degree of neoplasia and adequacy of colposcopy, and tend to vary among clinicians. For invasive cervical cancer, findings such as lymph node dissemination and adjacent organ involvement are key factors in determining which therapy will be selected.16
Cervical Intraepithelial Neoplasia
The treatment options for CIN can be divided into two categories, ablative and excisional. These techniques may be used only when invasive cervical cancer has been excluded.16,42 Cryotherapy and laser ablation are two common ablative techniques. One disadvantage of ablative techniques is that abnormal tissue is destroyed during the procedure, making it impossible for a tissue specimen to be collected and sent for additional evaluation.42,43 Cold knife conization, laser cone excision, and loop electrosurgical excision procedure (LEEP) are all excisional treatments that allow tissue to be collected and further evaluated.44 Although all these procedures can be used for any stage of CIN, excisional procedures are preferred for CIN 2 and CIN 3 lesions because moderate and severe intraepithelial neoplasias have a higher incidence of undetected microinvasive disease within the endocervical canal. Ablative therapy is generally reserved for treating low-grade CIN or small, focal high-grade lesions.42
Ablative techniques. Cryotherapy was the first outpatient treatment developed for CIN and is still widely used due to its ease, low cost, and low complication rate.44 It is an in-office procedure that utilizes nitrous oxide and carbon dioxide to freeze and ablate abnormal cells on the ectocervix. The procedure involves a freeze-thaw-freeze technique that has improved the efficacy of this treatment.16 The main side effects of cryotherapy are mild cramping and copious watery discharge that can last for up to 4 weeks postprocedure.42,45
Laser ablation is not used as commonly as cryosurgery, but is a viable option for women with large CIN lesions or women unwilling to undergo a LEEP. This procedure is performed under either local or general anesthesia and involves the use of a carbon dioxide laser under colposcopic guidance to ablate the transformation zone of the cervix.45 Laser ablation completely destroys the lesion while causing minimal damage to the surrounding, unaffected tissue. After treatment, patients may experience vaginal discharge for approximately 1 to 2 weeks.44,45 Overall, this technique is very expensive and requires a substantial amount of training to achieve maximum effectiveness.44
Excisional techniques. The LEEP is the procedure of choice for treating moderate and high-grade lesions. LEEP uses a wire loop electrode to excise the transformation zone of the cervix. Acetic acid or an iodine solution known as Lugol’s solution is applied to the cervix to delineate the margins of the lesion.42 The majority of these procedures can be done under local anesthesia, but on occasion sedation may be required.44 Complications, although rare, include post-treatment bleeding, infection, cervical stenosis, and cervical incompetence.45
Excisional conization is a procedure that can be performed with a scalpel (cold knife conization) or with a laser (laser cone excision).45 This procedure involves the removal of a cone-shaped portion of the cervix. Following the conization, endocervical curettage may be performed to acquire tissue samples and assess the endocervical canal. Also known as a cone biopsy, the procedure is typically performed under sedation. The types of complications associated with excisional conization are the same as those for LEEP, but the complication rate is higher when cone biopsy is performed.16,45
CERVICAL CANCER
Treatment methods for cervical cancer can be classified according to one of three disease stages: early-stage (IA2-IIA2), locally advanced (IIB-IVA), and advanced disease (IVB). Common treatment options for cervical cancer include radical hysterectomy with pelvic lymphadenectomy, radiation, and chemotherapy.9,17 Patients diagnosed with early-stage disease typically undergo a radical hysterectomy with pelvic lymphadenectomy. Cervical conization is an alternative option for young women who want to preserve fertility and wish to avoid a hysterectomy.45 Women who undergo surgery and are found to have more extensive disease or parametrial invasion are offered adjuvant radiation therapy.9
Patients diagnosed with locally advanced cancer are typically treated with radiation, chemotherapy, or both.17,45 The combination of radiation and chemotherapy in cervical cancer has been shown to increase survival rates in late-stage disease.7 Those diagnosed with advanced or disseminated disease are treated with palliative radiation. Overall, the five-year survival rates for patients diagnosed with early-stage cervical cancer who receive appropriate treatment have proven to be excellent7 (see Table 77,40,46).
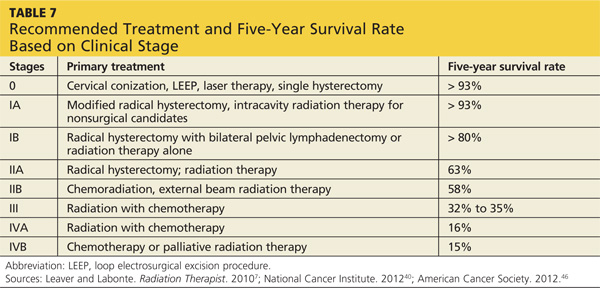
On the next page: Conclusion >>
CONCLUSION
The incidence and prevalence of cervical cancer continue to decline in the US; the majority of cases that arise are due to the lack of knowledge and availability of screening services in underserved areas. Increasing awareness and developing clinics for these under-screened populations would most likely contribute to a further decline in the incidence of CIN and cervical cancer.
Once diagnosed with an HPV infection, women should be informed that the body generally clears these infections within 1 to 2 years. Persistence of a high-risk HPV infection greatly increases the risk for neoplastic tissue transformation. These women need to be followed diligently so that treatment can be implemented at the first signs of a CIN 2 or higher grade lesion. Ultimately, implementing adequate screening techniques allows for early detection of CIN and prevention of cervical cancer.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. Cancer J Clin. 2011;61:69-90.
2. Whyte J. HPV and cervical cancer: latest developments. Consultant. 2012;52:555-560.
3. CDC. Cervical cancer statistics. www.cdc.gov/cancer/cervical/statistics/. Accessed July 23, 2013.
4. National Cancer Institute. Human papillomavirus (HPV) vaccines. www.cancer.gov/cancertopics/factsheet/prevention/HPV-vaccine. Accessed July 23, 2013.
5. de Freitas AC, Gurgel AP, Chagas BS, et al. Susceptibility to cervical cancer: an overview. Gynecol Oncol. 2012;126:304-311.
6. Scarinci IC, Garcia FA, Kobetz E, et al. Cervical cancer prevention: new tools and old barriers. Cancer. 2010;116:2531-2542.
7. Leaver D, Labonte G. HPV and cervical cancer. Radiation Therapist. 2010;19:27-45.
8. National Cancer Institute. SEER stat facts sheets: Cervix uteri cancer. http://seer.cancer.gov/statfacts/html/cervix.html#incidence-mortality. Accessed July 5, 2013.
9. Lea JS, Lin KY. Cervical cancer. Obstet Gynecol Clin North Am. 2012;39:233-253.
10. McKeever AE. Cervical cancer risk among college-age women: a review of the literature. SGNO J. 2010;20:6-12.
11. Frumovitz M. Invasive cervical cancer: epidemiology, risk factors, clinical manifestations, and diagnosis. UpToDate. Goff B, Falk SJ, eds. UpToDate, Waltham. MA, 2013. www.uptodate.com/contents/invasive-cervical-cancer-epidemiology-risk-factors-clinical-manifestations-and-diagnosis?detectedLanguage=en&source=search_result&search=Lifetime+prevalence+of+HPV+infection&selectedTitle=10%7E150&provider=noProvider. Accessed July 23, 2013.
12. Collins S, Rollason TP, Young LS, Woodman CB. Cigarette smoking is an independent risk factor for cervical intraepithelial neoplasia in young women: a longitudinal study. Eur J Cancer. 2010;46:405-411.
13. Moreno V, Bosch FX, Muñoz N, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085-1092.
14. Muñoz N, Franceschi S, Bosetti C, et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359(9312):1093-1101.
15. Kalra S. Studies examine relationship between HIV and cervical cancer in women. The AIDS Beacon. 2010. www.aidsbeacon.com/news/2010/07/23/studies-examine-relationship-between-hiv-and-cervical-cancer-in-women-aids-2010/. Accessed July 6, 2013.
16. Holschneider CH. Premalignant and malignant disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics and Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:807-808, 811-816, 820.
17. Rajaram S, Chitrathara K, Maheshwari A. Cervical Cancer: Contemporary Management. 1st ed. New Dehli, India: Jaypee Brothers Medical Publishers; 2012.
18. Carter JR, Ding Z, Rose BR. HPV infection and cervical disease: a review. Aust N Z J Obstet Gynaecol. 2011;51:103-108.
19. Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. 2010;117(2 Suppl):S5-10.
20. Khachikyan I, Stratton P. Benign disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics & Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:650.
21. CDC. 2010 sexually transmitted disease surveillance. www.cdc.gov/std/stats10/other.htm. Accessed July 23, 2013.
22. Juckett G, Hartman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82:1209-1214.
23. Verri A, Jalali GR, Cecchini G, et al. Significant progression of uterine cervical epithelial lesion accompanied by marked increase in 3q26 gene amplification. Lab Med. 2010;42:134-136.
24. Schwaiger C, Aruda M, LaCoursiere S, Rubin R. Current guidelines for cervical cancer screening. J Am Acad Nurse Prac. 2012;24:417-424.
25. CDC. Cervical cancer screening with the HPV test and the Pap test in women ages 30 and older. www.cdc.gov/cancer/hpv/basic_info/screening/cervical_screening.htm. Accessed July 5, 2013.
26. Schiffman M, Wentzensen N, Wacholder S, et al. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103: 368-383.
27. Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updates consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis. 2013;17:S1-S27.
28. American Society for Colposcopy and Cervical Pathology. Cervical cancer screening recommendations, 2012. www.asccp.org/Portals/9/docs/pdfs/Practice%20Management/ASCCP_Cervical_Cancer_Screening_Recommendations.pdf#zoom=80. Accessed July 6, 2013.
29. American Cancer Society. New screening guidelines for cervical cancer. March 14, 2012. www.cancer.org/cancer/news/news/new-screening-guidelines-for-cervical-cancer. Accessed July 23, 2013.
30. American Congress of Obstetricians and Gynecologists. USPSTF updated cervical cancer screening. www.acog.org/~/media/Districts/District%20II/PDFs/USPSTF_Cervical_Ca_Screening_Guidelines.pdf?dmc=1&ts=20130723T1510069242. Accessed July 23, 2013.
31. CDC. Cervical cancer screening guidelines for average-risk women. www.cdc.gov/cancer/cervical/pdf/guidelines.pdf. Accessed July 5, 2013.
32. Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;156:880-891.
33. US Preventive Services Task Force. Screening for cervical cancer. Current recommendation. www.uspreventiveservicestaskforce.org/uspstf11/cervcancer/cervcancerrs.htm#clinical. Accessed July 6, 2013.
34. American College of Obstetricians and Gynecologists; Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin Number 131: Screening for cervical cancer. Obstet Gynecol. 2012;120:1222-1238.
35. Warman J. Cervical cancer screening in young women: saving lives with prevention and detection. Oncol Nurs Forum. 2010;37:33-38.
36. Gardasil [package insert]. Whitehouse Station, NJ: Merck & Co; 2011.
37. Cervarix [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
38. Lowy DR, Schiller JT. Reducing HPV-associated cancer globally. Cancer Prev Res (Phila). 2012;5:18-23.
39. Morrison RS, Moody R, Shelton M. Pap smear rates: predictor of cervical cancer mortality disparity? Online J Rural Nurs Health Care. 2010;10:21-27.
40. National Cancer Institute. Cervical cancer treatment. www.cancer.gov/cancertopics/pdq/treatment/cervical/HealthProfessional/page1/AllPages#2.
41. Pecorelli S. Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet. 2009;105:103-104.
42. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 99. management of abnormal cervical cytology and histology. J Obstet Gynecol. 2008;112:1419-1444.
43. Bowring J, Tulloch I, Phadnis SV, et al. Secondary excision for cervical intraepithelial neoplasia: an evaluation of two treatment methods. J Obstet Gynaecol. 2010;30:511-514.
44. Jhingran A. Neoplasms of the cervix. In: Hong KW, Bast RC Jr, Hait WN, et al, eds. Holland-Frei Cancer Medicine. 8th ed. Ontario, Canada: BC Decker; 2010:1304-1312.
45. Kim SH. Preinvasive disease of the lower genital tract. In: Chu CS, Rubin SC, eds. Manual of Gynecologic Oncology. 1st ed. Hackensack, NJ: World Scientific Publishing Company; 2011:80-87.
46. American Cancer Society. Cervical cancer key statistics. http://www.cancer.org/cancer/cervicalcancer/detailedguide/cervical-cancer-key-statistics. Accessed July 5, 2013.
CE/CME No: CR-1309
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the central role that human papillomavirus (HPV) infection plays in the development of cervical intraepithelial neoplasia (CIN) and cervical cancer.
• Instruct patients on contributing cofactors of HPV infection and cervical cancer, including tobacco use, parity, use of oral contraceptives, co-infection with HIV, and immunosuppression.
• Describe the current recommendations for cervical cancer screening from professional societies, national health organizations, and federal agencies, including age-appropriate screening for cytology and high-risk HPV.
• Explain the timing and administration schedules of the currently available HPV vaccines and the patient populations for which the vaccines have been approved.
• Discuss the ablative and excisional procedures used to treat CIN and the treatment options for cervical cancer.
FACULTY
Heather P. Adams is an Assistant Professor/Clinical Coordinator in the Physician Assistant Program at Gannon University in Erie, Pennsylvania; in the program, Erica L. Carnright is a Physician Assistant student on clinical rotations.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Improved understanding of the central role of human papillomavirus (HPV) in cervical carcinogenesis has led to the development of vaccines and DNA testing for high-risk HPV subtypes. But age-appropriate cytologic screening remains the cornerstone in the prevention and early detection of cervical intraepithelial neoplasia (CIN) and cervical cancer. With prompt diagnosis and treatment of both CIN and early-stage cervical cancer, the prognosis for this disease is excellent.
Cervical cancer is the third most common cancer in women worldwide.1 More than 12,000 women are diagnosed with cervical cancer annually in the United States, with nearly 4,000 cervical cancer deaths reported each year.2-4 Globally, cervical cancer accounts for more than 529,000 new cancer cases and more than 275,000 cancer deaths in women annually, with more than 80% of these cases occurring in developing countries.1,5-7 Over the past 50 years, overall cervical cancer incidence and mortality have declined by more than 70% in the US, largely due to the development of the Papanicolaou screening test (Pap smear).7
Cervical cancer develops from precancerous, or neoplastic, cells of the cervix. Known as cervical intraepithelial neoplasia (CIN), these precancerous lesions are caused by infection with the human papillomavirus (HPV), the main etiologic factor in the development of cervical cancer. The progression from CIN to invasive cervical cancer is typically slow. Because of the long natural history of cervical cancer, screening and preventive measures, when appropriately implemented, can identify precancerous cells before they progress to cervical cancer.
EPIDEMIOLOGY AND RISK FACTORS
In 2013, approximately 12,340 new cases of cervical cancer will be diagnosed in the US, and 4,030 US women will die as a result of cervical cancer, according to National Cancer Institute estimates.8 Although the incidence rate of cervical cancer in the US has declined significantly, medically underserved populations remain disproportionately affected, with more than 60% of new cases occurring in underserved areas or underscreened populations.6 Epidemiologic studies have also shown higher incidence and mortality among minority populations.9 The incidence of cervical cancer is 30% higher among African American than white women, and the mortality rate is twice as high.9 The incidence of cervical cancer is highest among Hispanics.8
Although many risk factors contribute to the development of cervical cancer, HPV infection is the most important (see Table 12,7,9). HPV has been detected in up to 99% of all invasive cervical cancers, and its presence is necessary for oncogenesis to occur.10 HPV is the most common type of sexually transmitted infection (STI) in the US, with 75% to 80% of sexually active adults acquiring an HPV infection before age 50.11 It is estimated that a woman has a 10% lifetime risk for being infected with HPV, and this risk increases by 15% to 25% with each new sexual partner.7
Studies have shown that cigarette smoking contributes to the development of cervical cancer, with both previous and current smokers having a two- to threefold increased risk for high-grade CIN and cervical cancer, compared with never-smokers.5,9,12 Although the exact cause of this association has not been determined, carcinogens from cigarettes have been found within the cervical mucosa, which may cause alterations in the tissue.2,7
Long-term (≥ 5 years) use of oral contraceptive pills (OCP) by patients with an HPV cervical infection has been established as a risk factor for cervical cancer. It has been shown that the use of OCP causes an increase in HPV gene expression.5 In a study that compared two groups of women who tested positive for HPV DNA, the group that used OCP had a fourfold greater risk for cervical cancer than the group that did not use OCP.2,9,13
Parity, particularly multiparity, and HIV co-infection have also been implicated as risk factors for cervical cancer. The relationship between parity and cervical cancer likely results from the presence of the transformation zone on the ectocervix and increased squamous metaplasia during pregnancy, both of which make cervical tissue more vulnerable to HPV infection.14 HIV co-infection increases the risk for cervical cancer secondary to its immunosuppressive effects; women with both HIV and high-risk HPV infections are four times more likely to develop cervical cancer.15
On the next page: Cervical anatomy >>
CERVICAL ANATOMY
The cervix is the narrow, fibromuscular neck that makes up the lower portion of the uterus. The endocervix is the cervical canal, and the ectocervix extends inferiorly into the vagina.7 The internal os is the opening of the cervix into the uterus, and the external os is the opening of the cervix into the vagina. The endocervix is lined with glandular columnar epithelium. This tissue can extend beyond the external os onto the ectocervix, and when this occurs it is known as cervical ectopy.16 The surface of the cervix lying between the glandular tissue and the vaginal wall is comprised of stratified nonkeratinizing squamous epithelium.7 The squamocolumnar junction (SCJ) is defined as the location where the squamous and glandular cells meet.17 Adjacent to the SCJ is the transformation zone, an area vulnerable to HPV infection where glandular cells are actively undergoing squamous metaplasia.17
The location of the SCJ varies depending on age and hormonal changes. In prepubertal females, the SCJ is close to the external os, but in women of reproductive age, the SCJ moves away from the external os onto the surface of the ectocervix. This change in the location of the SCJ occurs due to increased estrogen levels following menarche, which cause the endocervical canal to elongate.7,17 A satisfactory Pap smear for cervical cytology includes cells from the transformation zone, as well as the ectocervix and endocervix.
HUMAN PAPILLOMAVIRUS
The human papillomavirus is a nonenveloped, double-stranded DNA virus that is known to cause abnormalities in skin cells. This virus is predominantly spread through sexual contact via skin-to-skin transmission. There are more than 100 known subtypes in the HPV family, of which 40 subtypes have been recognized to cause genital tract disease. Of these 40 subtypes, 15 have been identified to be oncogenic.9,18,19
HPV subtypes are separated into low-risk and high-risk categories (see Table 25,7,18). The low-risk subtypes cause either no cellular change, low-grade intraepithelial neoplasia, or condyloma. The high-risk subtypes are associated with the development of CIN and cancer. Of the 15 high-risk subtypes, HPV-16 and HPV-18 are the most oncogenic, followed by HPV-31 and HPV-45. HPV-16 is implicated in up to 70% of cervical cancers, HPV-18 in up to 20%, and HPV-31 and HPV-45 in up to 10%.5,9,20 Of the low-risk HPV subtypes, HPV-6 and HPV-11 cause 90% of condyloma infections.16,21
After HPV infection is contracted, the virus migrates into the squamous epithelial mucosa of the cervix, where it is capable of altering cells and causing the development of precancerous properties. This typically happens within the transformation zone, which is vulnerable to HPV infection.17 Depending on the state of the host’s immune system, many of these HPV infections clear without intervention. It is estimated that approximately 70% of HPV infections resolve spontaneously within one year, and 90% resolve within two years.5 HPV infections that persist beyond two years are more likely to lead to the development of CIN and, ultimately, cervical cancer, if left untreated.22 There is an association between genetic amplification (extra copies) of gene 3q26 and progression of CIN to cervical cancer, but ultimately a high-risk HPV subtype must be involved for cervical cancer to develop.23
On the next page: Clinical manifestations, screening, and prevention >>
CLINICAL MANIFESTATIONS
Due to the widespread use of cervical cytology, many cases of CIN and cervical cancer are diagnosed well before symptoms develop. In the early stages of cervical cancer, most women are asymptomatic, although some women may present with a watery, blood-tinged, or malodorous vaginal discharge.7 Any type of abnormal bleeding, whether it is between menstrual cycles, postcoital, or postmenopause, warrants an evaluation for pathologic processes, including but not limited to cervical cancer. Patients with late-stage cervical cancer may present with complaints of pelvic pain and painful intercourse due to the enlargement of a cervical mass. Vaginal discharge may change in consistency from watery to purulent and become foul-smelling due to cervical tissue necrosis.7,9,22 In some cases of advanced invasive cervical cancer, patients will experience hematuria or rectal bleeding secondary to tumor invasion through the bladder or rectal wall.9 Other nonspecific signs and symptoms of cervical cancer include unexplained weight loss accompanied by nausea or vomiting, as well as loss of appetite.7,9
SCREENING AND PREVENTION
Widespread use of the Pap test since the 1950s has led to marked reductions in the incidence of cervical cancer. The Pap test enables clinicians to screen for and detect CIN. More recently, liquid-based cytology has been utilized for the same purpose. The Pap smear involves placing the sample cells directly onto a microscopic slide. With liquid-based cytology, the sample is placed into a vial,2,7 where a portion is processed in preservative liquid under light microscopy for review by a cytologist.7 The remainder of the cell sample can be tested for HPV and STIs.
DNA testing for high-risk HPV subtypes is available and has been incorporated into the cervical cancer screening guidelines. The guidelines recommend HPV DNA testing as a co-test to be performed with cytology in women older than 30.2,24 The recommendation for HPV DNA testing in this age-group reflects greater concern that older women’s immune systems will not clear the virus, leaving these women at higher risk for developing CIN and cancer.25,26 HPV-16 and HPV-18 genotyping is available and is recommended in women 30 or older whose co-testing reveals normal cytology in the presence of a high-risk HPV subtype.27
Additionally, HPV DNA testing is used to triage cytology that reveals atypical squamous cells of undetermined significance. HPV DNA testing is not performed routinely in younger women because of the high likelihood that their immune systems will clear the virus. Genetic testing for the 3q26 gene amplification is available but has not yet been incorporated into the screening guidelines.27
Screening Guidelines
In September 2012, the American Cancer Society, American Society for Colposcopy and Cervical Pathology (ASCCP), and the American Society for Clinical Pathology developed a new set of cervical cancer screening guidelines.28 The US Preventive Services Task Force (USPSTF) and American College of Obstetricians and Gynecologists (ACOG) have also created standard recommendations for cervical cancer screening (see Table 32,28-34). Although ACOG’s standards have varied from those of other organizations in the past, their 2012 guidelines now align closely.
All these organizations recommend that cervical screening begin at age 21 for all women, regardless of the age of sexual initiation.31 Many young women who acquire an HPV infection will clear the infection within the first one to two years. Eliminating screening of women younger than 21 will help to avoid unnecessary biopsies and invasive treatment.29-32
All the guidelines include general recommendations for screening; however, each organization has made exceptions for patients at higher risk for developing cervical cancer.2,28-34 ASCCP has also developed algorithms for management of the multitude of abnormal cervical cancer screening results. These algorithms are available through the ASCCP Web site (www.asccp.org).
Vaccinations
HPV vaccines developed in recent years are now helping to prevent HPV infections from occurring. Currently, the FDA has approved the use of two HPV vaccines: a quadrivalent recombinant HPV vaccine (Gardasil) in 2006 and a bivalent HPV vaccine in 2009 (Cervarix).2 The quadrivalent vaccine was approved for girls and women ages 9 to 26 and protects against HPV subtypes 6 and 11—two low-risk HPV subtypes that can cause genital warts—and subtypes 16 and 18—two high-risk HPV subtypes that can cause cervical cancer. This vaccine is given in a three-dose series at months 0, 2, and 6, and should be initiated before women become sexually active for maximum effectiveness.22,35 The quadrivalent vaccine received additional FDA approval for administration in boys and men ages 9 to 26 in 2010.36
The bivalent vaccine is given in a three-dose series as well and is approved for girls and women ages 9 to 25.37 This vaccine offers protection against high-risk HPV subtypes 16 and 18.38
Recent trials have shown that both the quadrivalent and bivalent vaccines are more than 90% effective in preventing the development of precancerous cells from HPV subtypes 16 and 18.2 Although these vaccines provide protection against the two most oncogenic HPV subtypes, routine cytologic testing is still recommended in patients who receive the vaccines because they do not protect against all high-risk subtypes.2,35,38
On the next page: Diagnosis and staging >>
DIAGNOSIS AND STAGING
The Bethesda staging system provides uniform terminology to classify all abnormal Pap smears and cytology reports (see Table 434). Patients with abnormal cytology typically undergo a colposcopic examination to further evaluate the abnormality. During a colposcopy, the clinician examines the cervix with the use of a lighted microscope, known as a colposcope, which magnifies the cells of the cervix and allows for localized sampling of abnormal-appearing areas through punch biopsy.7,9 The area of focus is the transformation zone because of its known vulnerability to HPV infections. A colposcopy is considered inadequate if the SCJ, and therefore the transformation zone, is not fully visualized.
Acetic acid is applied to the cervix during colposcopy, and any dysplastic cells present take up the acid and turn white, a process known as acetowhitening. Indications for a punch biopsy include acetowhitening, leukoplakia, and abnormal vasculature marked by punctation, bizarre-appearing vessels, or the appearance of a mosaic pattern.16 Indications for sampling of the endocervical canal through endocervical curettage include inadequate colposcopy, presence of a lesion that extends beyond the view of the colposcope, or atypical glandular cells on cytology.16 The biopsy results help to determine if a diagnostic conization is needed for further evaluation of the lesion.16
After a cervical biopsy is performed, the abnormal cells may be classified as CIN 1, 2, or 3 or carcinoma in situ (see Table 516). These stages of intraepithelial neoplasia are determined by the depth to which abnormal, immature cells have invaded the cervical epithelium.16 CIN 2 and 3 are more likely to develop into cervical cancer if they are not properly treated.5,16,17 The majority of CIN 1 lesions do not progress into cancer and typically resolve on their own or are cleared by the patient’s immune system. While most CIN 1 lesions are caused by high-risk HPV, these may be less oncogenic subtypes.7,17
The two most common histologic subtypes of cervical cancer are squamous cell carcinoma and adenocarcinoma. Squamous cell carcinoma comprises more than 70% of cervical cancers, while adenocarcinoma makes up approximately 25%.39 Neuroendocrine, small cell, and mixed-cell carcinomas make up the remainder of cervical cancers.9 Squamous cell carcinoma arises from the squamocolumnar junction or ectocervix, and adenocarcinoma tends to arise from the glandular cells of the endocervix.7,9,17 Studies show that adenocarcinoma is slowly becoming more prevalent in the US than squamous cell carcinoma.9
Once a diagnosis of cervical cancer has been established, additional testing and procedures are performed to rule out lymph node and organ involvement.9,16 The International Federation of Gynecology and Oncology system is then used to clinically stage the cervical cancer40 (see Table 67,40,41). This staging system, updated in 2009, is based solely on clinical examination findings. Once cervical cancer is staged, measures are taken to assess which treatment option is most appropriate.
On the next page: Treatment modalities >>
TREATMENT MODALITIES
Treatment strategies for CIN correspond to the degree of neoplasia and adequacy of colposcopy, and tend to vary among clinicians. For invasive cervical cancer, findings such as lymph node dissemination and adjacent organ involvement are key factors in determining which therapy will be selected.16
Cervical Intraepithelial Neoplasia
The treatment options for CIN can be divided into two categories, ablative and excisional. These techniques may be used only when invasive cervical cancer has been excluded.16,42 Cryotherapy and laser ablation are two common ablative techniques. One disadvantage of ablative techniques is that abnormal tissue is destroyed during the procedure, making it impossible for a tissue specimen to be collected and sent for additional evaluation.42,43 Cold knife conization, laser cone excision, and loop electrosurgical excision procedure (LEEP) are all excisional treatments that allow tissue to be collected and further evaluated.44 Although all these procedures can be used for any stage of CIN, excisional procedures are preferred for CIN 2 and CIN 3 lesions because moderate and severe intraepithelial neoplasias have a higher incidence of undetected microinvasive disease within the endocervical canal. Ablative therapy is generally reserved for treating low-grade CIN or small, focal high-grade lesions.42
Ablative techniques. Cryotherapy was the first outpatient treatment developed for CIN and is still widely used due to its ease, low cost, and low complication rate.44 It is an in-office procedure that utilizes nitrous oxide and carbon dioxide to freeze and ablate abnormal cells on the ectocervix. The procedure involves a freeze-thaw-freeze technique that has improved the efficacy of this treatment.16 The main side effects of cryotherapy are mild cramping and copious watery discharge that can last for up to 4 weeks postprocedure.42,45
Laser ablation is not used as commonly as cryosurgery, but is a viable option for women with large CIN lesions or women unwilling to undergo a LEEP. This procedure is performed under either local or general anesthesia and involves the use of a carbon dioxide laser under colposcopic guidance to ablate the transformation zone of the cervix.45 Laser ablation completely destroys the lesion while causing minimal damage to the surrounding, unaffected tissue. After treatment, patients may experience vaginal discharge for approximately 1 to 2 weeks.44,45 Overall, this technique is very expensive and requires a substantial amount of training to achieve maximum effectiveness.44
Excisional techniques. The LEEP is the procedure of choice for treating moderate and high-grade lesions. LEEP uses a wire loop electrode to excise the transformation zone of the cervix. Acetic acid or an iodine solution known as Lugol’s solution is applied to the cervix to delineate the margins of the lesion.42 The majority of these procedures can be done under local anesthesia, but on occasion sedation may be required.44 Complications, although rare, include post-treatment bleeding, infection, cervical stenosis, and cervical incompetence.45
Excisional conization is a procedure that can be performed with a scalpel (cold knife conization) or with a laser (laser cone excision).45 This procedure involves the removal of a cone-shaped portion of the cervix. Following the conization, endocervical curettage may be performed to acquire tissue samples and assess the endocervical canal. Also known as a cone biopsy, the procedure is typically performed under sedation. The types of complications associated with excisional conization are the same as those for LEEP, but the complication rate is higher when cone biopsy is performed.16,45
CERVICAL CANCER
Treatment methods for cervical cancer can be classified according to one of three disease stages: early-stage (IA2-IIA2), locally advanced (IIB-IVA), and advanced disease (IVB). Common treatment options for cervical cancer include radical hysterectomy with pelvic lymphadenectomy, radiation, and chemotherapy.9,17 Patients diagnosed with early-stage disease typically undergo a radical hysterectomy with pelvic lymphadenectomy. Cervical conization is an alternative option for young women who want to preserve fertility and wish to avoid a hysterectomy.45 Women who undergo surgery and are found to have more extensive disease or parametrial invasion are offered adjuvant radiation therapy.9
Patients diagnosed with locally advanced cancer are typically treated with radiation, chemotherapy, or both.17,45 The combination of radiation and chemotherapy in cervical cancer has been shown to increase survival rates in late-stage disease.7 Those diagnosed with advanced or disseminated disease are treated with palliative radiation. Overall, the five-year survival rates for patients diagnosed with early-stage cervical cancer who receive appropriate treatment have proven to be excellent7 (see Table 77,40,46).
On the next page: Conclusion >>
CONCLUSION
The incidence and prevalence of cervical cancer continue to decline in the US; the majority of cases that arise are due to the lack of knowledge and availability of screening services in underserved areas. Increasing awareness and developing clinics for these under-screened populations would most likely contribute to a further decline in the incidence of CIN and cervical cancer.
Once diagnosed with an HPV infection, women should be informed that the body generally clears these infections within 1 to 2 years. Persistence of a high-risk HPV infection greatly increases the risk for neoplastic tissue transformation. These women need to be followed diligently so that treatment can be implemented at the first signs of a CIN 2 or higher grade lesion. Ultimately, implementing adequate screening techniques allows for early detection of CIN and prevention of cervical cancer.
CE/CME No: CR-1309
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the central role that human papillomavirus (HPV) infection plays in the development of cervical intraepithelial neoplasia (CIN) and cervical cancer.
• Instruct patients on contributing cofactors of HPV infection and cervical cancer, including tobacco use, parity, use of oral contraceptives, co-infection with HIV, and immunosuppression.
• Describe the current recommendations for cervical cancer screening from professional societies, national health organizations, and federal agencies, including age-appropriate screening for cytology and high-risk HPV.
• Explain the timing and administration schedules of the currently available HPV vaccines and the patient populations for which the vaccines have been approved.
• Discuss the ablative and excisional procedures used to treat CIN and the treatment options for cervical cancer.
FACULTY
Heather P. Adams is an Assistant Professor/Clinical Coordinator in the Physician Assistant Program at Gannon University in Erie, Pennsylvania; in the program, Erica L. Carnright is a Physician Assistant student on clinical rotations.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Improved understanding of the central role of human papillomavirus (HPV) in cervical carcinogenesis has led to the development of vaccines and DNA testing for high-risk HPV subtypes. But age-appropriate cytologic screening remains the cornerstone in the prevention and early detection of cervical intraepithelial neoplasia (CIN) and cervical cancer. With prompt diagnosis and treatment of both CIN and early-stage cervical cancer, the prognosis for this disease is excellent.
Cervical cancer is the third most common cancer in women worldwide.1 More than 12,000 women are diagnosed with cervical cancer annually in the United States, with nearly 4,000 cervical cancer deaths reported each year.2-4 Globally, cervical cancer accounts for more than 529,000 new cancer cases and more than 275,000 cancer deaths in women annually, with more than 80% of these cases occurring in developing countries.1,5-7 Over the past 50 years, overall cervical cancer incidence and mortality have declined by more than 70% in the US, largely due to the development of the Papanicolaou screening test (Pap smear).7
Cervical cancer develops from precancerous, or neoplastic, cells of the cervix. Known as cervical intraepithelial neoplasia (CIN), these precancerous lesions are caused by infection with the human papillomavirus (HPV), the main etiologic factor in the development of cervical cancer. The progression from CIN to invasive cervical cancer is typically slow. Because of the long natural history of cervical cancer, screening and preventive measures, when appropriately implemented, can identify precancerous cells before they progress to cervical cancer.
EPIDEMIOLOGY AND RISK FACTORS
In 2013, approximately 12,340 new cases of cervical cancer will be diagnosed in the US, and 4,030 US women will die as a result of cervical cancer, according to National Cancer Institute estimates.8 Although the incidence rate of cervical cancer in the US has declined significantly, medically underserved populations remain disproportionately affected, with more than 60% of new cases occurring in underserved areas or underscreened populations.6 Epidemiologic studies have also shown higher incidence and mortality among minority populations.9 The incidence of cervical cancer is 30% higher among African American than white women, and the mortality rate is twice as high.9 The incidence of cervical cancer is highest among Hispanics.8
Although many risk factors contribute to the development of cervical cancer, HPV infection is the most important (see Table 12,7,9). HPV has been detected in up to 99% of all invasive cervical cancers, and its presence is necessary for oncogenesis to occur.10 HPV is the most common type of sexually transmitted infection (STI) in the US, with 75% to 80% of sexually active adults acquiring an HPV infection before age 50.11 It is estimated that a woman has a 10% lifetime risk for being infected with HPV, and this risk increases by 15% to 25% with each new sexual partner.7
Studies have shown that cigarette smoking contributes to the development of cervical cancer, with both previous and current smokers having a two- to threefold increased risk for high-grade CIN and cervical cancer, compared with never-smokers.5,9,12 Although the exact cause of this association has not been determined, carcinogens from cigarettes have been found within the cervical mucosa, which may cause alterations in the tissue.2,7
Long-term (≥ 5 years) use of oral contraceptive pills (OCP) by patients with an HPV cervical infection has been established as a risk factor for cervical cancer. It has been shown that the use of OCP causes an increase in HPV gene expression.5 In a study that compared two groups of women who tested positive for HPV DNA, the group that used OCP had a fourfold greater risk for cervical cancer than the group that did not use OCP.2,9,13
Parity, particularly multiparity, and HIV co-infection have also been implicated as risk factors for cervical cancer. The relationship between parity and cervical cancer likely results from the presence of the transformation zone on the ectocervix and increased squamous metaplasia during pregnancy, both of which make cervical tissue more vulnerable to HPV infection.14 HIV co-infection increases the risk for cervical cancer secondary to its immunosuppressive effects; women with both HIV and high-risk HPV infections are four times more likely to develop cervical cancer.15
On the next page: Cervical anatomy >>
CERVICAL ANATOMY
The cervix is the narrow, fibromuscular neck that makes up the lower portion of the uterus. The endocervix is the cervical canal, and the ectocervix extends inferiorly into the vagina.7 The internal os is the opening of the cervix into the uterus, and the external os is the opening of the cervix into the vagina. The endocervix is lined with glandular columnar epithelium. This tissue can extend beyond the external os onto the ectocervix, and when this occurs it is known as cervical ectopy.16 The surface of the cervix lying between the glandular tissue and the vaginal wall is comprised of stratified nonkeratinizing squamous epithelium.7 The squamocolumnar junction (SCJ) is defined as the location where the squamous and glandular cells meet.17 Adjacent to the SCJ is the transformation zone, an area vulnerable to HPV infection where glandular cells are actively undergoing squamous metaplasia.17
The location of the SCJ varies depending on age and hormonal changes. In prepubertal females, the SCJ is close to the external os, but in women of reproductive age, the SCJ moves away from the external os onto the surface of the ectocervix. This change in the location of the SCJ occurs due to increased estrogen levels following menarche, which cause the endocervical canal to elongate.7,17 A satisfactory Pap smear for cervical cytology includes cells from the transformation zone, as well as the ectocervix and endocervix.
HUMAN PAPILLOMAVIRUS
The human papillomavirus is a nonenveloped, double-stranded DNA virus that is known to cause abnormalities in skin cells. This virus is predominantly spread through sexual contact via skin-to-skin transmission. There are more than 100 known subtypes in the HPV family, of which 40 subtypes have been recognized to cause genital tract disease. Of these 40 subtypes, 15 have been identified to be oncogenic.9,18,19
HPV subtypes are separated into low-risk and high-risk categories (see Table 25,7,18). The low-risk subtypes cause either no cellular change, low-grade intraepithelial neoplasia, or condyloma. The high-risk subtypes are associated with the development of CIN and cancer. Of the 15 high-risk subtypes, HPV-16 and HPV-18 are the most oncogenic, followed by HPV-31 and HPV-45. HPV-16 is implicated in up to 70% of cervical cancers, HPV-18 in up to 20%, and HPV-31 and HPV-45 in up to 10%.5,9,20 Of the low-risk HPV subtypes, HPV-6 and HPV-11 cause 90% of condyloma infections.16,21
After HPV infection is contracted, the virus migrates into the squamous epithelial mucosa of the cervix, where it is capable of altering cells and causing the development of precancerous properties. This typically happens within the transformation zone, which is vulnerable to HPV infection.17 Depending on the state of the host’s immune system, many of these HPV infections clear without intervention. It is estimated that approximately 70% of HPV infections resolve spontaneously within one year, and 90% resolve within two years.5 HPV infections that persist beyond two years are more likely to lead to the development of CIN and, ultimately, cervical cancer, if left untreated.22 There is an association between genetic amplification (extra copies) of gene 3q26 and progression of CIN to cervical cancer, but ultimately a high-risk HPV subtype must be involved for cervical cancer to develop.23
On the next page: Clinical manifestations, screening, and prevention >>
CLINICAL MANIFESTATIONS
Due to the widespread use of cervical cytology, many cases of CIN and cervical cancer are diagnosed well before symptoms develop. In the early stages of cervical cancer, most women are asymptomatic, although some women may present with a watery, blood-tinged, or malodorous vaginal discharge.7 Any type of abnormal bleeding, whether it is between menstrual cycles, postcoital, or postmenopause, warrants an evaluation for pathologic processes, including but not limited to cervical cancer. Patients with late-stage cervical cancer may present with complaints of pelvic pain and painful intercourse due to the enlargement of a cervical mass. Vaginal discharge may change in consistency from watery to purulent and become foul-smelling due to cervical tissue necrosis.7,9,22 In some cases of advanced invasive cervical cancer, patients will experience hematuria or rectal bleeding secondary to tumor invasion through the bladder or rectal wall.9 Other nonspecific signs and symptoms of cervical cancer include unexplained weight loss accompanied by nausea or vomiting, as well as loss of appetite.7,9
SCREENING AND PREVENTION
Widespread use of the Pap test since the 1950s has led to marked reductions in the incidence of cervical cancer. The Pap test enables clinicians to screen for and detect CIN. More recently, liquid-based cytology has been utilized for the same purpose. The Pap smear involves placing the sample cells directly onto a microscopic slide. With liquid-based cytology, the sample is placed into a vial,2,7 where a portion is processed in preservative liquid under light microscopy for review by a cytologist.7 The remainder of the cell sample can be tested for HPV and STIs.
DNA testing for high-risk HPV subtypes is available and has been incorporated into the cervical cancer screening guidelines. The guidelines recommend HPV DNA testing as a co-test to be performed with cytology in women older than 30.2,24 The recommendation for HPV DNA testing in this age-group reflects greater concern that older women’s immune systems will not clear the virus, leaving these women at higher risk for developing CIN and cancer.25,26 HPV-16 and HPV-18 genotyping is available and is recommended in women 30 or older whose co-testing reveals normal cytology in the presence of a high-risk HPV subtype.27
Additionally, HPV DNA testing is used to triage cytology that reveals atypical squamous cells of undetermined significance. HPV DNA testing is not performed routinely in younger women because of the high likelihood that their immune systems will clear the virus. Genetic testing for the 3q26 gene amplification is available but has not yet been incorporated into the screening guidelines.27
Screening Guidelines
In September 2012, the American Cancer Society, American Society for Colposcopy and Cervical Pathology (ASCCP), and the American Society for Clinical Pathology developed a new set of cervical cancer screening guidelines.28 The US Preventive Services Task Force (USPSTF) and American College of Obstetricians and Gynecologists (ACOG) have also created standard recommendations for cervical cancer screening (see Table 32,28-34). Although ACOG’s standards have varied from those of other organizations in the past, their 2012 guidelines now align closely.
All these organizations recommend that cervical screening begin at age 21 for all women, regardless of the age of sexual initiation.31 Many young women who acquire an HPV infection will clear the infection within the first one to two years. Eliminating screening of women younger than 21 will help to avoid unnecessary biopsies and invasive treatment.29-32
All the guidelines include general recommendations for screening; however, each organization has made exceptions for patients at higher risk for developing cervical cancer.2,28-34 ASCCP has also developed algorithms for management of the multitude of abnormal cervical cancer screening results. These algorithms are available through the ASCCP Web site (www.asccp.org).
Vaccinations
HPV vaccines developed in recent years are now helping to prevent HPV infections from occurring. Currently, the FDA has approved the use of two HPV vaccines: a quadrivalent recombinant HPV vaccine (Gardasil) in 2006 and a bivalent HPV vaccine in 2009 (Cervarix).2 The quadrivalent vaccine was approved for girls and women ages 9 to 26 and protects against HPV subtypes 6 and 11—two low-risk HPV subtypes that can cause genital warts—and subtypes 16 and 18—two high-risk HPV subtypes that can cause cervical cancer. This vaccine is given in a three-dose series at months 0, 2, and 6, and should be initiated before women become sexually active for maximum effectiveness.22,35 The quadrivalent vaccine received additional FDA approval for administration in boys and men ages 9 to 26 in 2010.36
The bivalent vaccine is given in a three-dose series as well and is approved for girls and women ages 9 to 25.37 This vaccine offers protection against high-risk HPV subtypes 16 and 18.38
Recent trials have shown that both the quadrivalent and bivalent vaccines are more than 90% effective in preventing the development of precancerous cells from HPV subtypes 16 and 18.2 Although these vaccines provide protection against the two most oncogenic HPV subtypes, routine cytologic testing is still recommended in patients who receive the vaccines because they do not protect against all high-risk subtypes.2,35,38
On the next page: Diagnosis and staging >>
DIAGNOSIS AND STAGING
The Bethesda staging system provides uniform terminology to classify all abnormal Pap smears and cytology reports (see Table 434). Patients with abnormal cytology typically undergo a colposcopic examination to further evaluate the abnormality. During a colposcopy, the clinician examines the cervix with the use of a lighted microscope, known as a colposcope, which magnifies the cells of the cervix and allows for localized sampling of abnormal-appearing areas through punch biopsy.7,9 The area of focus is the transformation zone because of its known vulnerability to HPV infections. A colposcopy is considered inadequate if the SCJ, and therefore the transformation zone, is not fully visualized.
Acetic acid is applied to the cervix during colposcopy, and any dysplastic cells present take up the acid and turn white, a process known as acetowhitening. Indications for a punch biopsy include acetowhitening, leukoplakia, and abnormal vasculature marked by punctation, bizarre-appearing vessels, or the appearance of a mosaic pattern.16 Indications for sampling of the endocervical canal through endocervical curettage include inadequate colposcopy, presence of a lesion that extends beyond the view of the colposcope, or atypical glandular cells on cytology.16 The biopsy results help to determine if a diagnostic conization is needed for further evaluation of the lesion.16
After a cervical biopsy is performed, the abnormal cells may be classified as CIN 1, 2, or 3 or carcinoma in situ (see Table 516). These stages of intraepithelial neoplasia are determined by the depth to which abnormal, immature cells have invaded the cervical epithelium.16 CIN 2 and 3 are more likely to develop into cervical cancer if they are not properly treated.5,16,17 The majority of CIN 1 lesions do not progress into cancer and typically resolve on their own or are cleared by the patient’s immune system. While most CIN 1 lesions are caused by high-risk HPV, these may be less oncogenic subtypes.7,17
The two most common histologic subtypes of cervical cancer are squamous cell carcinoma and adenocarcinoma. Squamous cell carcinoma comprises more than 70% of cervical cancers, while adenocarcinoma makes up approximately 25%.39 Neuroendocrine, small cell, and mixed-cell carcinomas make up the remainder of cervical cancers.9 Squamous cell carcinoma arises from the squamocolumnar junction or ectocervix, and adenocarcinoma tends to arise from the glandular cells of the endocervix.7,9,17 Studies show that adenocarcinoma is slowly becoming more prevalent in the US than squamous cell carcinoma.9
Once a diagnosis of cervical cancer has been established, additional testing and procedures are performed to rule out lymph node and organ involvement.9,16 The International Federation of Gynecology and Oncology system is then used to clinically stage the cervical cancer40 (see Table 67,40,41). This staging system, updated in 2009, is based solely on clinical examination findings. Once cervical cancer is staged, measures are taken to assess which treatment option is most appropriate.
On the next page: Treatment modalities >>
TREATMENT MODALITIES
Treatment strategies for CIN correspond to the degree of neoplasia and adequacy of colposcopy, and tend to vary among clinicians. For invasive cervical cancer, findings such as lymph node dissemination and adjacent organ involvement are key factors in determining which therapy will be selected.16
Cervical Intraepithelial Neoplasia
The treatment options for CIN can be divided into two categories, ablative and excisional. These techniques may be used only when invasive cervical cancer has been excluded.16,42 Cryotherapy and laser ablation are two common ablative techniques. One disadvantage of ablative techniques is that abnormal tissue is destroyed during the procedure, making it impossible for a tissue specimen to be collected and sent for additional evaluation.42,43 Cold knife conization, laser cone excision, and loop electrosurgical excision procedure (LEEP) are all excisional treatments that allow tissue to be collected and further evaluated.44 Although all these procedures can be used for any stage of CIN, excisional procedures are preferred for CIN 2 and CIN 3 lesions because moderate and severe intraepithelial neoplasias have a higher incidence of undetected microinvasive disease within the endocervical canal. Ablative therapy is generally reserved for treating low-grade CIN or small, focal high-grade lesions.42
Ablative techniques. Cryotherapy was the first outpatient treatment developed for CIN and is still widely used due to its ease, low cost, and low complication rate.44 It is an in-office procedure that utilizes nitrous oxide and carbon dioxide to freeze and ablate abnormal cells on the ectocervix. The procedure involves a freeze-thaw-freeze technique that has improved the efficacy of this treatment.16 The main side effects of cryotherapy are mild cramping and copious watery discharge that can last for up to 4 weeks postprocedure.42,45
Laser ablation is not used as commonly as cryosurgery, but is a viable option for women with large CIN lesions or women unwilling to undergo a LEEP. This procedure is performed under either local or general anesthesia and involves the use of a carbon dioxide laser under colposcopic guidance to ablate the transformation zone of the cervix.45 Laser ablation completely destroys the lesion while causing minimal damage to the surrounding, unaffected tissue. After treatment, patients may experience vaginal discharge for approximately 1 to 2 weeks.44,45 Overall, this technique is very expensive and requires a substantial amount of training to achieve maximum effectiveness.44
Excisional techniques. The LEEP is the procedure of choice for treating moderate and high-grade lesions. LEEP uses a wire loop electrode to excise the transformation zone of the cervix. Acetic acid or an iodine solution known as Lugol’s solution is applied to the cervix to delineate the margins of the lesion.42 The majority of these procedures can be done under local anesthesia, but on occasion sedation may be required.44 Complications, although rare, include post-treatment bleeding, infection, cervical stenosis, and cervical incompetence.45
Excisional conization is a procedure that can be performed with a scalpel (cold knife conization) or with a laser (laser cone excision).45 This procedure involves the removal of a cone-shaped portion of the cervix. Following the conization, endocervical curettage may be performed to acquire tissue samples and assess the endocervical canal. Also known as a cone biopsy, the procedure is typically performed under sedation. The types of complications associated with excisional conization are the same as those for LEEP, but the complication rate is higher when cone biopsy is performed.16,45
CERVICAL CANCER
Treatment methods for cervical cancer can be classified according to one of three disease stages: early-stage (IA2-IIA2), locally advanced (IIB-IVA), and advanced disease (IVB). Common treatment options for cervical cancer include radical hysterectomy with pelvic lymphadenectomy, radiation, and chemotherapy.9,17 Patients diagnosed with early-stage disease typically undergo a radical hysterectomy with pelvic lymphadenectomy. Cervical conization is an alternative option for young women who want to preserve fertility and wish to avoid a hysterectomy.45 Women who undergo surgery and are found to have more extensive disease or parametrial invasion are offered adjuvant radiation therapy.9
Patients diagnosed with locally advanced cancer are typically treated with radiation, chemotherapy, or both.17,45 The combination of radiation and chemotherapy in cervical cancer has been shown to increase survival rates in late-stage disease.7 Those diagnosed with advanced or disseminated disease are treated with palliative radiation. Overall, the five-year survival rates for patients diagnosed with early-stage cervical cancer who receive appropriate treatment have proven to be excellent7 (see Table 77,40,46).
On the next page: Conclusion >>
CONCLUSION
The incidence and prevalence of cervical cancer continue to decline in the US; the majority of cases that arise are due to the lack of knowledge and availability of screening services in underserved areas. Increasing awareness and developing clinics for these under-screened populations would most likely contribute to a further decline in the incidence of CIN and cervical cancer.
Once diagnosed with an HPV infection, women should be informed that the body generally clears these infections within 1 to 2 years. Persistence of a high-risk HPV infection greatly increases the risk for neoplastic tissue transformation. These women need to be followed diligently so that treatment can be implemented at the first signs of a CIN 2 or higher grade lesion. Ultimately, implementing adequate screening techniques allows for early detection of CIN and prevention of cervical cancer.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. Cancer J Clin. 2011;61:69-90.
2. Whyte J. HPV and cervical cancer: latest developments. Consultant. 2012;52:555-560.
3. CDC. Cervical cancer statistics. www.cdc.gov/cancer/cervical/statistics/. Accessed July 23, 2013.
4. National Cancer Institute. Human papillomavirus (HPV) vaccines. www.cancer.gov/cancertopics/factsheet/prevention/HPV-vaccine. Accessed July 23, 2013.
5. de Freitas AC, Gurgel AP, Chagas BS, et al. Susceptibility to cervical cancer: an overview. Gynecol Oncol. 2012;126:304-311.
6. Scarinci IC, Garcia FA, Kobetz E, et al. Cervical cancer prevention: new tools and old barriers. Cancer. 2010;116:2531-2542.
7. Leaver D, Labonte G. HPV and cervical cancer. Radiation Therapist. 2010;19:27-45.
8. National Cancer Institute. SEER stat facts sheets: Cervix uteri cancer. http://seer.cancer.gov/statfacts/html/cervix.html#incidence-mortality. Accessed July 5, 2013.
9. Lea JS, Lin KY. Cervical cancer. Obstet Gynecol Clin North Am. 2012;39:233-253.
10. McKeever AE. Cervical cancer risk among college-age women: a review of the literature. SGNO J. 2010;20:6-12.
11. Frumovitz M. Invasive cervical cancer: epidemiology, risk factors, clinical manifestations, and diagnosis. UpToDate. Goff B, Falk SJ, eds. UpToDate, Waltham. MA, 2013. www.uptodate.com/contents/invasive-cervical-cancer-epidemiology-risk-factors-clinical-manifestations-and-diagnosis?detectedLanguage=en&source=search_result&search=Lifetime+prevalence+of+HPV+infection&selectedTitle=10%7E150&provider=noProvider. Accessed July 23, 2013.
12. Collins S, Rollason TP, Young LS, Woodman CB. Cigarette smoking is an independent risk factor for cervical intraepithelial neoplasia in young women: a longitudinal study. Eur J Cancer. 2010;46:405-411.
13. Moreno V, Bosch FX, Muñoz N, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085-1092.
14. Muñoz N, Franceschi S, Bosetti C, et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359(9312):1093-1101.
15. Kalra S. Studies examine relationship between HIV and cervical cancer in women. The AIDS Beacon. 2010. www.aidsbeacon.com/news/2010/07/23/studies-examine-relationship-between-hiv-and-cervical-cancer-in-women-aids-2010/. Accessed July 6, 2013.
16. Holschneider CH. Premalignant and malignant disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics and Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:807-808, 811-816, 820.
17. Rajaram S, Chitrathara K, Maheshwari A. Cervical Cancer: Contemporary Management. 1st ed. New Dehli, India: Jaypee Brothers Medical Publishers; 2012.
18. Carter JR, Ding Z, Rose BR. HPV infection and cervical disease: a review. Aust N Z J Obstet Gynaecol. 2011;51:103-108.
19. Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. 2010;117(2 Suppl):S5-10.
20. Khachikyan I, Stratton P. Benign disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics & Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:650.
21. CDC. 2010 sexually transmitted disease surveillance. www.cdc.gov/std/stats10/other.htm. Accessed July 23, 2013.
22. Juckett G, Hartman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82:1209-1214.
23. Verri A, Jalali GR, Cecchini G, et al. Significant progression of uterine cervical epithelial lesion accompanied by marked increase in 3q26 gene amplification. Lab Med. 2010;42:134-136.
24. Schwaiger C, Aruda M, LaCoursiere S, Rubin R. Current guidelines for cervical cancer screening. J Am Acad Nurse Prac. 2012;24:417-424.
25. CDC. Cervical cancer screening with the HPV test and the Pap test in women ages 30 and older. www.cdc.gov/cancer/hpv/basic_info/screening/cervical_screening.htm. Accessed July 5, 2013.
26. Schiffman M, Wentzensen N, Wacholder S, et al. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103: 368-383.
27. Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updates consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis. 2013;17:S1-S27.
28. American Society for Colposcopy and Cervical Pathology. Cervical cancer screening recommendations, 2012. www.asccp.org/Portals/9/docs/pdfs/Practice%20Management/ASCCP_Cervical_Cancer_Screening_Recommendations.pdf#zoom=80. Accessed July 6, 2013.
29. American Cancer Society. New screening guidelines for cervical cancer. March 14, 2012. www.cancer.org/cancer/news/news/new-screening-guidelines-for-cervical-cancer. Accessed July 23, 2013.
30. American Congress of Obstetricians and Gynecologists. USPSTF updated cervical cancer screening. www.acog.org/~/media/Districts/District%20II/PDFs/USPSTF_Cervical_Ca_Screening_Guidelines.pdf?dmc=1&ts=20130723T1510069242. Accessed July 23, 2013.
31. CDC. Cervical cancer screening guidelines for average-risk women. www.cdc.gov/cancer/cervical/pdf/guidelines.pdf. Accessed July 5, 2013.
32. Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;156:880-891.
33. US Preventive Services Task Force. Screening for cervical cancer. Current recommendation. www.uspreventiveservicestaskforce.org/uspstf11/cervcancer/cervcancerrs.htm#clinical. Accessed July 6, 2013.
34. American College of Obstetricians and Gynecologists; Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin Number 131: Screening for cervical cancer. Obstet Gynecol. 2012;120:1222-1238.
35. Warman J. Cervical cancer screening in young women: saving lives with prevention and detection. Oncol Nurs Forum. 2010;37:33-38.
36. Gardasil [package insert]. Whitehouse Station, NJ: Merck & Co; 2011.
37. Cervarix [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
38. Lowy DR, Schiller JT. Reducing HPV-associated cancer globally. Cancer Prev Res (Phila). 2012;5:18-23.
39. Morrison RS, Moody R, Shelton M. Pap smear rates: predictor of cervical cancer mortality disparity? Online J Rural Nurs Health Care. 2010;10:21-27.
40. National Cancer Institute. Cervical cancer treatment. www.cancer.gov/cancertopics/pdq/treatment/cervical/HealthProfessional/page1/AllPages#2.
41. Pecorelli S. Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet. 2009;105:103-104.
42. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 99. management of abnormal cervical cytology and histology. J Obstet Gynecol. 2008;112:1419-1444.
43. Bowring J, Tulloch I, Phadnis SV, et al. Secondary excision for cervical intraepithelial neoplasia: an evaluation of two treatment methods. J Obstet Gynaecol. 2010;30:511-514.
44. Jhingran A. Neoplasms of the cervix. In: Hong KW, Bast RC Jr, Hait WN, et al, eds. Holland-Frei Cancer Medicine. 8th ed. Ontario, Canada: BC Decker; 2010:1304-1312.
45. Kim SH. Preinvasive disease of the lower genital tract. In: Chu CS, Rubin SC, eds. Manual of Gynecologic Oncology. 1st ed. Hackensack, NJ: World Scientific Publishing Company; 2011:80-87.
46. American Cancer Society. Cervical cancer key statistics. http://www.cancer.org/cancer/cervicalcancer/detailedguide/cervical-cancer-key-statistics. Accessed July 5, 2013.
1. Jemal A, Bray F, Center MM, et al. Global cancer statistics. Cancer J Clin. 2011;61:69-90.
2. Whyte J. HPV and cervical cancer: latest developments. Consultant. 2012;52:555-560.
3. CDC. Cervical cancer statistics. www.cdc.gov/cancer/cervical/statistics/. Accessed July 23, 2013.
4. National Cancer Institute. Human papillomavirus (HPV) vaccines. www.cancer.gov/cancertopics/factsheet/prevention/HPV-vaccine. Accessed July 23, 2013.
5. de Freitas AC, Gurgel AP, Chagas BS, et al. Susceptibility to cervical cancer: an overview. Gynecol Oncol. 2012;126:304-311.
6. Scarinci IC, Garcia FA, Kobetz E, et al. Cervical cancer prevention: new tools and old barriers. Cancer. 2010;116:2531-2542.
7. Leaver D, Labonte G. HPV and cervical cancer. Radiation Therapist. 2010;19:27-45.
8. National Cancer Institute. SEER stat facts sheets: Cervix uteri cancer. http://seer.cancer.gov/statfacts/html/cervix.html#incidence-mortality. Accessed July 5, 2013.
9. Lea JS, Lin KY. Cervical cancer. Obstet Gynecol Clin North Am. 2012;39:233-253.
10. McKeever AE. Cervical cancer risk among college-age women: a review of the literature. SGNO J. 2010;20:6-12.
11. Frumovitz M. Invasive cervical cancer: epidemiology, risk factors, clinical manifestations, and diagnosis. UpToDate. Goff B, Falk SJ, eds. UpToDate, Waltham. MA, 2013. www.uptodate.com/contents/invasive-cervical-cancer-epidemiology-risk-factors-clinical-manifestations-and-diagnosis?detectedLanguage=en&source=search_result&search=Lifetime+prevalence+of+HPV+infection&selectedTitle=10%7E150&provider=noProvider. Accessed July 23, 2013.
12. Collins S, Rollason TP, Young LS, Woodman CB. Cigarette smoking is an independent risk factor for cervical intraepithelial neoplasia in young women: a longitudinal study. Eur J Cancer. 2010;46:405-411.
13. Moreno V, Bosch FX, Muñoz N, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085-1092.
14. Muñoz N, Franceschi S, Bosetti C, et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359(9312):1093-1101.
15. Kalra S. Studies examine relationship between HIV and cervical cancer in women. The AIDS Beacon. 2010. www.aidsbeacon.com/news/2010/07/23/studies-examine-relationship-between-hiv-and-cervical-cancer-in-women-aids-2010/. Accessed July 6, 2013.
16. Holschneider CH. Premalignant and malignant disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics and Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:807-808, 811-816, 820.
17. Rajaram S, Chitrathara K, Maheshwari A. Cervical Cancer: Contemporary Management. 1st ed. New Dehli, India: Jaypee Brothers Medical Publishers; 2012.
18. Carter JR, Ding Z, Rose BR. HPV infection and cervical disease: a review. Aust N Z J Obstet Gynaecol. 2011;51:103-108.
19. Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. 2010;117(2 Suppl):S5-10.
20. Khachikyan I, Stratton P. Benign disorders of the uterine cervix. In: Decherney AH, Nathan L, Laufer N, Roman AS, eds. Current Diagnosis and Treatment: Obstetrics & Gynecology. 11th ed. New York, NY: McGraw-Hill; 2013:650.
21. CDC. 2010 sexually transmitted disease surveillance. www.cdc.gov/std/stats10/other.htm. Accessed July 23, 2013.
22. Juckett G, Hartman-Adams H. Human papillomavirus: clinical manifestations and prevention. Am Fam Physician. 2010;82:1209-1214.
23. Verri A, Jalali GR, Cecchini G, et al. Significant progression of uterine cervical epithelial lesion accompanied by marked increase in 3q26 gene amplification. Lab Med. 2010;42:134-136.
24. Schwaiger C, Aruda M, LaCoursiere S, Rubin R. Current guidelines for cervical cancer screening. J Am Acad Nurse Prac. 2012;24:417-424.
25. CDC. Cervical cancer screening with the HPV test and the Pap test in women ages 30 and older. www.cdc.gov/cancer/hpv/basic_info/screening/cervical_screening.htm. Accessed July 5, 2013.
26. Schiffman M, Wentzensen N, Wacholder S, et al. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103: 368-383.
27. Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updates consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis. 2013;17:S1-S27.
28. American Society for Colposcopy and Cervical Pathology. Cervical cancer screening recommendations, 2012. www.asccp.org/Portals/9/docs/pdfs/Practice%20Management/ASCCP_Cervical_Cancer_Screening_Recommendations.pdf#zoom=80. Accessed July 6, 2013.
29. American Cancer Society. New screening guidelines for cervical cancer. March 14, 2012. www.cancer.org/cancer/news/news/new-screening-guidelines-for-cervical-cancer. Accessed July 23, 2013.
30. American Congress of Obstetricians and Gynecologists. USPSTF updated cervical cancer screening. www.acog.org/~/media/Districts/District%20II/PDFs/USPSTF_Cervical_Ca_Screening_Guidelines.pdf?dmc=1&ts=20130723T1510069242. Accessed July 23, 2013.
31. CDC. Cervical cancer screening guidelines for average-risk women. www.cdc.gov/cancer/cervical/pdf/guidelines.pdf. Accessed July 5, 2013.
32. Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2012;156:880-891.
33. US Preventive Services Task Force. Screening for cervical cancer. Current recommendation. www.uspreventiveservicestaskforce.org/uspstf11/cervcancer/cervcancerrs.htm#clinical. Accessed July 6, 2013.
34. American College of Obstetricians and Gynecologists; Committee on Practice Bulletins—Gynecology. ACOG Practice Bulletin Number 131: Screening for cervical cancer. Obstet Gynecol. 2012;120:1222-1238.
35. Warman J. Cervical cancer screening in young women: saving lives with prevention and detection. Oncol Nurs Forum. 2010;37:33-38.
36. Gardasil [package insert]. Whitehouse Station, NJ: Merck & Co; 2011.
37. Cervarix [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2012.
38. Lowy DR, Schiller JT. Reducing HPV-associated cancer globally. Cancer Prev Res (Phila). 2012;5:18-23.
39. Morrison RS, Moody R, Shelton M. Pap smear rates: predictor of cervical cancer mortality disparity? Online J Rural Nurs Health Care. 2010;10:21-27.
40. National Cancer Institute. Cervical cancer treatment. www.cancer.gov/cancertopics/pdq/treatment/cervical/HealthProfessional/page1/AllPages#2.
41. Pecorelli S. Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet. 2009;105:103-104.
42. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 99. management of abnormal cervical cytology and histology. J Obstet Gynecol. 2008;112:1419-1444.
43. Bowring J, Tulloch I, Phadnis SV, et al. Secondary excision for cervical intraepithelial neoplasia: an evaluation of two treatment methods. J Obstet Gynaecol. 2010;30:511-514.
44. Jhingran A. Neoplasms of the cervix. In: Hong KW, Bast RC Jr, Hait WN, et al, eds. Holland-Frei Cancer Medicine. 8th ed. Ontario, Canada: BC Decker; 2010:1304-1312.
45. Kim SH. Preinvasive disease of the lower genital tract. In: Chu CS, Rubin SC, eds. Manual of Gynecologic Oncology. 1st ed. Hackensack, NJ: World Scientific Publishing Company; 2011:80-87.
46. American Cancer Society. Cervical cancer key statistics. http://www.cancer.org/cancer/cervicalcancer/detailedguide/cervical-cancer-key-statistics. Accessed July 5, 2013.
Herpes Zoster Infection
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
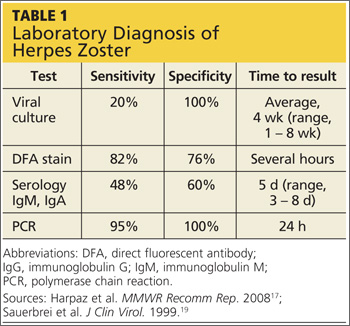
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
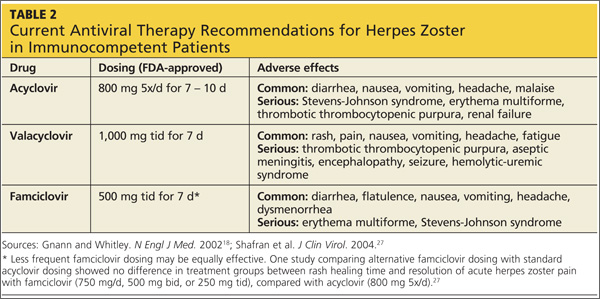
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
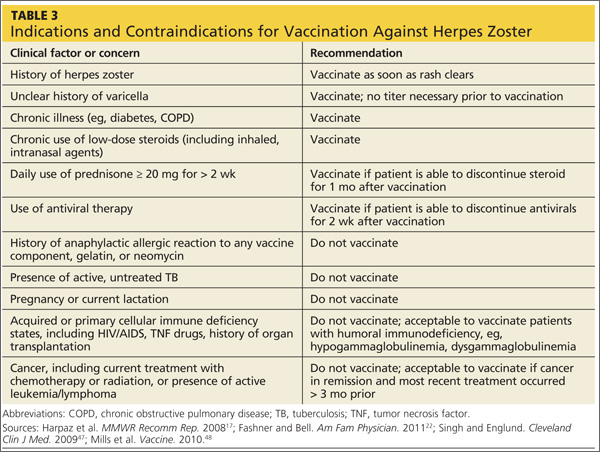
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.

1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.
1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
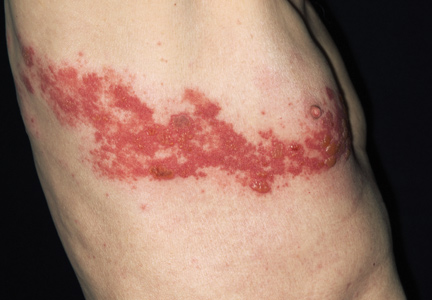
Interstitial Cystitis: A Painful Syndrome
CE/CME No: CR-1307
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
•
Describe the pathophysiology
of interstitial cystitis/bladder
pain syndrome (IC/BPS), as
it is currently understood.
•
Discuss urogenital signs and
symptoms that should prompt suspicion for IC/BPS in a primary care patient.
•
Explain the clinical diagnosis of
IC/BPS and key considerations for referral.
•
Review medical management, nonoperative therapy, and surgical treatment of IC/BPS.
FACULTY
LaToya M. Haynes practices at the Carolinas Pain Institute and the Center for Clinical Research in Winston-Salem, North Carolina, and is a preceptor for PA students. Kelly Bilello is a PA at Genitourinary Surgical Consultants in Denver. Jade Breeback practices at Cone Health Primary Care in Kernersville, North Carolina. Jessica Cain is a PA in emergency medicine at the University of Cincinnati Medical Center. Jennifer Wenninger is a cardiothoracic and vascular surgery PA at Bellin Health Care Systems in Green Bay, Wisconsin. M. Jane McDaniel is an Instructor in the Department of Physician Assistant Studies at Wake Forest School of Medicine in Winston-Salem.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Interstitial cystitis/bladder pain syndrome (IC/BPS) is a common, painful disease of the urinary bladder. Difficult to diagnose and frequently misdiagnosed as another common urologic disorder, IC/BPS challenges health care providers to identify it early and implement current treatment algorithms that may simplify management and improve quality of life for affected patients.
Interstitial cystitis (IC), or bladder pain syndrome (BPS), is a clinical condition characterized by bladder pain, urinary frequency and urgency, and increased nighttime urination (nocturia).1 More specifically, IC/BPS is defined as an unpleasant sensation in the bladder, abdomen, or pelvis (ie, pain, pressure, burning, and/or other discomfort) perceived to be originating in the urinary bladder. The condition is associated with lower urinary tract symptoms of more than six weeks’ duration, with no infection or other identifiable cause present.2
IC/BPS lacks a single known etiology; rather, it most likely results from multiple contributing factors that cascade into a painful and potentially debilitating syndrome. The condition was first described more than a century ago,3,4 but its complex nature and conflicting theories about its pathogenesis present both diagnostic and therapeutic challenges for health care professionals. Frequent misdiagnosis of IC/BPS as another common urologic disorder can make timely, appropriate treatment elusive.
Without a clearly described pathophysiology, IC/BPS has always been difficult to define using standardized diagnostic criteria and precise terminology. The definition of the condition was revised in 2002 and again in 2008, when the nomenclature bladder pain syndrome was introduced.1,5,6
Less than 10 years ago, US researchers described IC as a subgroup of BPS,7 while in Europe, BPS is used as the broader term, with IC still considered a well-defined subgroup that usually involves ulceration.6 The future may find IC, BPS, and painful bladder syndrome (PBS) used as interchangeable terms—or as unique diagnoses. A better understanding of the pathophysiology of IC/BPS/PBS would contribute not only to resolving issues of nomenclature, but also to establishing an accurate diagnosis earlier in the disease process and providing more efficient, effective treatment.
THE PROBLEM OF EPIDEMIOLOGY
Inconsistencies in the terminology, definitions, and diagnostic criteria of IC/BPS have made epidemiology difficult to establish.1 It has been suggested that IC/BPS is underdiagnosed in the United States and that its prevalence is much greater than generally reported.8
According to one study of IC in a managed care population, its prevalence in 2005 was 197 per 100,000 women and 41 per 100,000 men, with the female-to-male ratio estimated at 5:1.9 In 2011, researchers for the RAND Corporation published what they called the first population-based “symptom prevalence estimate” among US women older than 18, based on more than 100,000 screening interviews conducted by phone. According to their findings, between 3.3 and 7.9 million US women meet the stated criteria for IC/BPS (ie, between 3,113 and 7,453 women per 100,000).10 These conflicting data exemplify the range of epidemiologic conclusions that exist regarding this condition.
On the next page: Proposed pathophysiology >>
THE PROPOSED PATHOPHYSIOLOGY
IC/BPS is thought to begin with an initial insult to the bladder that leads to dysfunction of the epithelial layer. This insult may be the result of a neurogenic inflammation, autoimmunity, subclinical or chronic infection, or bladder urothelial defects.1 Dysfunction in the epithelial layer includes altered bladder epithelial expression of human leukocyte antigen I and II; decreased expression of uroplakin (an antitoxic protein in the bladder), and a defective glycosaminoglycan mucus layer.4 This damage to the epithelial layer alters the permeability of the bladder, allowing potassium ions to enter the urothelium and depolarize motor and sensory nerves. This potassium leak then activates the mast cells, causing mastocytosis and the release of histamine.11 These processes disrupt the homeostasis of the urinary tract and allow the development of inflammation—a main cause of the pelvic pain associated with IC/BPS4,12,13 (see Figure 114).
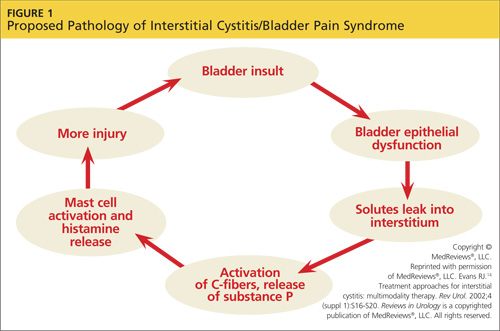
Other factors that exacerbate the primary inflammation in the bladder are C-fibers and nerve growth factor (NGF). C-fibers are afferent fibers found in the peripheral nerves of the somatic sensory system that convey input signals from the periphery to the central nervous system.3 In patients with IC/BPS, initial inflammation activates C-fibers, which produce substance P, nociceptor, and other inflammatory mediators. These mediators exacerbate existing inflammation and further facilitate mast cell activation.3
NGF is a protein that is critical for the maintenance of sympathetic and sensory neurons; it is important not only in the urinary tract but in all organ systems. Increased levels of NGF, a prevalent finding in patients with IC/BPS, is an indicator of inflammation in the body. The precise mechanism that causes elevated NGF in patients with IC/BPS is not well understood, but its presence supports the theory that inflammation is a cause of pelvic pain in IC/BPS.12
The urinary urgency and frequency experienced by patients with IC/BPS is in part due to the role nitric oxide (NO) plays in bladder activity. Patients with IC have decreased levels of urinary NO (a reduction thought to be the result of a decrease in L-arginine) and urinary NO synthase.12,15,16 Ordinarily, NO synthase converts L-arginine to NO, which helps to control relaxation of the bladder smooth muscle, allowing more urine to be stored. In patients with IC/BPS, NO insufficiency leads to bladder overactivity.15
On the next page: Patient history and presentation >>
PATIENT HISTORY AND PRESENTATION
A detailed patient history is imperative in establishing the diagnosis of IC/BPS. Symptoms that should prompt the clinician to consider IC/BPS include:
• Pelvic or bladder pain relieved with voiding
• Dyspareunia
• Increased frequency of urination with no infection present
• Urinary urgency with pain, and
• Increased nocturia.17,18
Early IC presents variably, and pain, though a common symptom, is not always present.19 Chronic pain is defined by duration of at least six months, with the discomfort perceived as originating in the bladder.8 In addition to patients who experience pain, those who void several times during the night should also be considered for further evaluation.19
Many patients describe their symptoms in terms of flares and periods of remission. Some patients associate flares with stress, seasonal allergies, sexual activity, consumption of certain foods, and the premenstrual week.17,20 Patients with IC/BPS are commonly misdiagnosed with recurrent urinary tract infections; hence the need to standardize the criteria for diagnosis of IC/BPS.17
DIAGNOSIS
There are currently three available sets of diagnostic criteria for patients with IC/BPS. These are the National Institute for Diabetes and Diseases of the Kidney (NIDDK) definition (1990),21 the International Continence Society (ICS) definition of painful bladder disorders (2002),5 and the European Society for the Study of IC/BPS (ESSIC) definition (2008).6 In particular, the ESSIC criteria were formulated to help identify IC/BPS earlier in the disease course.
The 1990 NIDDK protocol, developed for research purposes,12 featured inclusion and exclusion criteria. Exclusion criteria included age younger than 18 years and presence of benign bladder tumors, radiation cystitis, tuberculosis cystitis, bacterial cystitis, vaginitis, symptomatic urethral diverticulum, uterine/cervical/vaginal cancers, and/or active herpes; urinary frequency of less than five episodes in 12 hours; and less than two episodes of nocturia per night.19
NIDDK inclusion criteria required two or more of the following: Hunner’s ulcer, pain on bladder filling, general pelvic pain, glomerulations on endoscopy, and decreased bladder compliance on cystometrogram.19
This protocol proved to be excessively restrictive for clinical use and was widely replaced by the ICS criteria in 2002. The ICS criteria5 allowed more varied patient presentations; the exclusions featured in the NIDDK guideline, it has been estimated, could have eliminated at least one-third of patients who would reasonably be considered to have IC/BPS.21
In contrast to the NIDDK criteria, the ICS criteria5 defined BPS as “the complaint of suprapubic pain related to bladder filling, accompanied by other symptoms such as increased daytime and nighttime urinary frequency in the absence of proven urinary tract infection or other obvious pathology.”5 Additionally, the ICS document restricts the diagnosis of IC to patients with painful bladder syndrome in addition to “typical cystoscopic and histologic features.”6
According to the ESSIC proposal on diagnostic criteria, classification, and nomenclature,6 diagnosing IC/BPS requires symptoms of chronic pain related to the urinary bladder accompanied by at least one other urinary symptom, such as daytime and nighttime frequency and exclusion of confusable diseases and cystoscopy with hydrodistention and biopsy, if indicated. The new statement does not include the required absence of UTI or other pathology identified in the previous (ICS) criteria; these, too, overlooked a portion of the population who would be considered to have IC/BPS. Therefore, the ESSIC classification provides the most comprehensive criteria for diagnosing IC/BPS and has been determined as best for diagnostic purposes in the early disease stages.12
On the next page: Applying the criteria and referral >>
Applying the Criteria
IC/BPS remains a diagnosis of exclusion.12 The most common disorders seen in the differential diagnosis for IC/BPS (ie, “confusable diseases”6) include bacterial cystitis, vaginitis, pelvic pain, vulvodynia, urinary tract infections, yeast infections, sexually transmitted infections, endometriosis, overactive bladder, and genitourinary malignancies.5,12
Biopsy or cystoscopy with short-duration, low-pressure hydrodistention can be performed on patients who present with persistent pelvic pain and urinary symptoms.2,12 Common cystoscopic findings in patients with IC/BPS include Hunner’s lesions, glomerulations, and inflammatory infiltrates on biopsy.12,21 Hunner’s lesions are described as “patches of red mucosa exhibiting small vessels radiating to a central pale scar.”21 These lesions may also be referred to as Hunner’s ulcers.12 Not always visible on cystoscopy, Hunner’s lesions may be seen only after hydrodistention of the bladder under anesthesia.
Cystoscopic findings can be misleading for providers, as not all stages of IC/BPS manifest in the same manner. No single laboratory finding will identify IC/BPS. The only way to diagnose this disease is to rule out all other diseases with similar presentations.18
When to Refer
Specific findings that may indicate the need for referral include severe pain, hematuria, chronic UTI, and pyuria. Generally, however, the decision to refer the patient with IC/BPS to a urologist or urogynecologist depends on the primary care provider’s comfort level. Some providers choose to refer as soon as identifying symptoms of IC/BPS have been confirmed, whereas others may wish to proceed with further evaluation and/or treatment before referring.2,22
Even if the provider decides to refer immediately after identifying symptoms, it is important to initiate some patient education: for example, explaining that the patient will likely require further tests, including cystoscopy and possibly urodynamic evaluation.2,23 Smokers and other patients at high risk for bladder cancer should be referred for cystoscopy.2
If the primary care provider chooses to proceed with evaluation and treatment before referring the patient, follow-up is typically recommended at one-month intervals for the first three months, then every three months thereafter.18 This allows the clinician to monitor a patient’s progress and address concerns that may develop. Symptoms may be slow to respond to treatment, so it is essential to encourage the patient to adhere to the prescribed regimen. If three to six months of first-line treatment yield no response, further consultation and evaluation are warranted. Overall, a multidisciplinary approach that includes the participation of a urologist, a gynecologist, or other appropriate specialist will help ensure optimal treatment and care.18
A good tool that is often used to gauge the patient’s progress is the O’Leary/Sant Voiding and Pain Indices23-25 (see Figure 224). Reviewing patient responses to this questionnaire, with its precise numerical system, at each follow-up appointment can be especially helpful.
On the next page: Treatment >>
TREATMENT
Management of IC/BPS can be challenging, because it is such a multifaceted disorder. Patient education beginning shortly after diagnosis is crucial, as treatment regimens may involve complex multimodal therapy over long periods of time, oftentimes with a very gradual response (see “For Your Patient”).

Lifestyle changes for patients with IC/BPS are considered an important component of treatment. Dietary changes—specifically, reducing intake of foods with high acidic content (citrus fruits, tomatoes), alcoholic beverages, spices, and potassium—have been found helpful.5 Reducing stress and anxiety, whenever possible, has also been noted to alleviate symptoms.24
Another nonpharmacologic option is physical therapy, including biofeedback and bladder retraining.12,13 Biofeedback is particularly useful in patients who experience pelvic pain attributed to spasms of the pelvic floor.12 Bladder retraining can be used to reduce urinary frequency through techniques that include scheduled voiding. Physical therapy strategies should be revisited regularly to maintain their therapeutic benefits.5,12
Oral Medications
The mainstay of pharmacologic treatment, and the one most thoroughly studied, is oral pentosan polysulfate (PPS), which belongs to the class of heparins or heparinoids.2,26 PPS is thought to attach to the mucosa of the bladder, reestablishing its glycosaminoglycan layer and restoring normal function of this permeable barrier.14 Overall, this drug is well tolerated and relieves the symptoms of pain, urgency, and frequency. Patients may start to experience improvement in symptoms after four weeks of treatment; however, it can take six months or longer to achieve the full benefit of this therapy.13,25
Other pharmacologic agents used in the treatment of IC/BPS include antihistamines, tricyclic antidepressants, and some antiepileptic medications. Some patients with IC/BPS experience symptoms attributable to bladder mastocytosis and mast cell activation, explaining the efficacy of antihistamines for these particular patients.27 Among the antihistamines, hydroxyzine, an H1-receptor antagonist, is a common pharmacologic option. Similarly, cetirizine can be used in patients for whom the sedating effects of hydroxyzine may prove hazardous.28
Antidepressants, especially tricyclic antidepressants (TCAs, eg, amitriptyline), can also provide some relief for patients, including alleviation of pain, possible antihistamine effects, and mild anticholinergic action, leading to decreased urinary urgency and frequency.2,26,29 Of note, the TCA imipramine should be avoided in patients with IC/BPS, as it has a sympathomimetic effect that can worsen symptoms of dysfunctional voiding in this patient population.14
Gabapentin, an antiepileptic, is used for improvement of severe, persistent pain. Alternatives to gabapentin include, but are not limited to, phenytoin, carbamazepine, and valproic acid.14 The effectiveness of these medications in the treatment of IC/BPS lend credence to the theory that, in addition to bladder mucosa dysfunction, symptoms are also mediated through an inflammatory neurogenic pathway.
Patients should be encouraged to continue use of PPS or other prescribed pharmacologic treatments even if there is no immediate relief of symptoms.25 According to a treatment algorithm from the American Urological Association,2 however, ineffective treatments should be stopped and diagnosis should be reconsidered if there is no improvement within a “clinically meaningful time frame.”
Additional Pharmacologic Options
Intravesical therapy is another mode of pharmacologic treatment.2,29 This treatment is usually reserved for IC/BPS flares and management of cases lacking the desired response to oral medications. Dimethyl sulfoxide (DMSO) is a commonly used intravesical agent. DMSO acts to provide pain relief and reduce inflammation, in addition to effecting histamine release from mast cells.27 Intravesical heparinoids essentially employ the same mechanism of action as oral PPS to maintain and enhance the bladder’s mucosal lining. This treatment is also commonly used in patients who need to discontinue use of oral PPS due to side effects.27,30
Treatment options for refractory IC/BPS include immunosuppression (and surgical therapy, below). Prednisone and cyclosporine have been shown to be effective immunosuppressive agents.23 Side effects make the use of these medications less desirable; also, symptoms have been shown to return in many patients after treatment is stopped.23
The FDA has recently approved the use of onabotulinum toxin A (Botox) injections into the bladder for treatment of urinary urgency and frequency that are not responsive to standard medical therapy. Since patients with IC often experience such symptoms with no relief from standard therapy, intratrigonal and periurethral injections of Botox are being administered for treatment of IC in some patients with moderate success.31-33 Although intradetrusor Botox use is recommended as a fifth-line treatment in the AUA guidelines,2 it is important to note that this agent is not FDA-approved specifically for IC, but rather for any refractory condition presenting with urinary frequency and urgency.
Surgical Therapy
Surgical intervention (a sixth-line treatment option, according to the AUA guidelines2) is rarely indicated except in cases of severe IC/BPS that have been refractory to all other treatment options and in which spontaneous remission of symptoms seems unlikely. Supravesical urinary diversion, usually through the creation of an ileal conduit, is the procedure of choice and is often performed in conjunction with a cystectomy. Unfortunately in some cases, pelvic pain has been noted to continue postcystectomy, a finding that also supports a neurogenic etiology for IC/BPS.14
On the Horizon
Although IC/PBS is difficult to treat, new data suggest that use of extended diagnostics, including molecular markers to detect the disease early and guide effective treatment, may greatly improve current therapeutic options.34
Prescribing selective anticholinergic and antihistamine pharmacotherapy based on the patient’s specific muscarinic and histamine receptor profile, respectively, may provide greater symptom relief.34 Maintaining the appropriate, individualized therapy could represent a significant advance in treatment for IC/BPS. However, further research on the topic is needed.
On the next page: Conclusion >>
CONCLUSION
IC/BPS is a complex multifactorial syndrome that may manifest with disabling pain. Further research on this disease is warranted to help facilitate an earlier, more consistent diagnosis and produce more effective treatment options. Early diagnosis of IC/BPS is essential for successful therapy. Once the diagnosis is made, a cautious regimen of different treatments, following the American Urological Association’s clinical practice guidelines for interstitial cystitis, should be implemented. Patients should also be encouraged to consider specific dietary changes and other lifestyle adjustments under a clinician’s supervision.
The authors wish to thank Carol Hildebrandt for her help in preparing this manuscript and Robert J. Evans, MD, Associate Professor of Urology, Wake Forest Baptist Medical Center, for his editorial expertise.
1. Dasgupta J, Tincello DG. Interstitial cystitis/bladder pain syndrome: an update. Maturitas. 2009;64:212-217.
2. Hanno PM, Burks DA, Clemens JQ, et al. Diagnosis and treatment of interstitial cystitis/bladder pain syndrome: American Urological Association (AUA) Guideline (2011). www.auanet.org/education/guidelines/ic-bladder-pain-syndrome.cfm. Accessed June 5, 2013.
3. Evans RJ; University of Tennessee Advanced Studies in Pharmacy. Pathophysiology and clinical presentation of interstitial cystitis (2005). www.utasip.com/files/articlefiles/pdf/XASIP_Issue_Mar_p8_14.pdf. Accessed June 5, 2013.
4. Sant GR. Etiology, pathogenesis, and diagnosis of interstitial cystitis. Rev Urol. 2002;(4 suppl 1):S9-S15.
5. Abrams P, Cardozo L, Fall M, et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167-178.
6. van de Merwe JP, Nordling J, Bouchelouche P, et al. Diagnostic criteria, classification, and nomenclature for painful bladder syndrome/interstitial cystitis: an ESSIC proposal. Eur Urol. 2008;53:60-67.
7. Bogart LM, Berry SH, Clemens JQ. Symptoms of interstitial cystitis, painful bladder syndrome and similar diseases in women: a systematic review. J Urol. 2007;177:450-456.
8. Kusek JW, Nyberg LM. The epidemiology of interstitial cystitis: is it time to expand our definition? Urology. 2001;57(6 suppl 1):95-99.
9. Clemens JQ, Meenan RT, Rosetti MC, et al. Prevalence and incidence of interstitial cystitis in a managed care population. J Urol. 2005;173:98-102.
10. Berry SH, Elliott MN, Suttorp M, et al. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol. 2011;186:540-544.
11. Rosamilia A, Dwyer PL. Pathophysiology of interstitial cystitis. Curr Opin Obstet Gynecol. 2000;12:405-410.
12. Grover S, Srivastava A, Lee R, et al. Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol. 2011;3:19-33.
13. Rosenberg MT, Page S, Hazzard MA. Prevalence of interstitial cystitis in a primary care setting. Urology. 2007;69(4 suppl):S48-S52.
14. Evans RJ. Treatment approaches for interstitial cystitis: multimodality therapy. Rev Urol. 2002;(4 suppl 1):S16-S20.
15. Wesselmann U. Interstitial cystitis: a chronic visceral pain syndrome. Urology. 2001;57(6 suppl 1):102.
16. Hosseini A, Ehrén I, Wiklund NP. Nitric oxide as an objective marker for evaluation of treatment response in patients with classic interstitial cystitis. J Urol. 2004;172(6 pt 1):2261-2265.
17. Ho MH, Bhatia NN, Khorram O. Physiologic role of nitric oxide and nitric oxide synthase in female lower urinary tract. Curr Opin Obstet Gynecol. 2004;16:423-429.
18. Rosenberg MT, Newman DK, Page SA. Interstitial cystitis/painful bladder syndrome: symptom recognition is key to early identification, treatment. Cleve Clin J Med. 2007;74:854-862.
19. Driscoll A, Teichman JM. How do patients with interstitial cystitis present? J Urol. 2001;166:2118-2120.
20. Parsons CL. Interstitial cystitis: epidemiology and clinical presentation. Clin Obstet Gynecol. 2002;45:242-249.
21. Hanno PM, Landis JR, Matthews-Cook Y, et al. The diagnosis of interstitial cystitis revisited: lessons learned from the National Institutes of Health Interstitial Cystitis Database study. J Urol. 1999;161:553-557.
22. Whitmore KE, Theoharides TC. When to suspect interstitial cystitis. J Fam Pract. 2011;60:340-348.
23. Butrick CW, Howard FM, Sand PK. Diagnosis and treatment of interstitial cystitis/painful bladder syndrome: a review. J Womens Health (Larchmt). 2010;19:1185-1193.
24. O’Leary MP, Sant GR, Fowler FJ Jr, et al. The interstitial cystitis symptom index and problem index. Urology. 1997;49(5A suppl):58-63.
25. Nickel JC. Forensic dissection of a clinical trial: lessons learned in understanding and managing interstitial cystitis. Rev Urol. 2010;12:e78-e85.
26. Anger JT, Zabihi N, Clemens JQ, et al. Treatment choice, duration, and cost in patients with interstitial cystitis and painful bladder syndrome. Int Urogynecol J. 2011;22:395-400.
27. Nickel JC. Interstitial cystitis: characterization and management of an enigmatic urologic syndrome. Rev Urol. 2002;4:112-121.
28. Dell JR. Interstitial cystitis/painful bladder syndrome: appropriate diagnosis and management. J Womens Health (Larchmt). 2007;16:1181-1187.
29. National Kidney and Urologic Diseases Information Clearinghouse. Interstitial cystitis/painful bladder syndrome (2011). NIH Publication No. 11–3220. http://kidney.niddk.nih.gov/kudiseases/pubs/interstitialcystitis/IC_PBS_T_508.pdf. Accessed June 5, 2013.
30. Davis EL, El Khoudary SR, Talbott EO, et al. Safety and efficacy of the use of intravesical and oral pentosan polysulfate sodium for interstitial cystitis: a randomized double-blind clinical trial. J Urol. 2008;179:177-185.
31. Pinto R, Lopes T, Silva J, et al. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol. 2013;189:548-553.
32. Pinto R, Lopoes T, Frias B, et al. Trigonal injection of botulinum toxin A in patients with refractory bladder pain syndrome/interstitial cystitis. Eur Urol. 2010;58:360-365.
33. Gottsch HP, Miller JL, Yang CC, Berger RE. A pilot study of botulinum toxin for interstitial cystitis/painful bladder syndrome. Neurourol Urodyn. 2011; 30:93-96.
34. Neuhaus J, Schwalenberg T, Horn LC, et al. New aspects in the differential diagnosis and therapy of bladder pain syndrome/interstitial cystitis. Adv Urol. 2011;2011:639479.
CE/CME No: CR-1307
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
•
Describe the pathophysiology
of interstitial cystitis/bladder
pain syndrome (IC/BPS), as
it is currently understood.
•
Discuss urogenital signs and
symptoms that should prompt suspicion for IC/BPS in a primary care patient.
•
Explain the clinical diagnosis of
IC/BPS and key considerations for referral.
•
Review medical management, nonoperative therapy, and surgical treatment of IC/BPS.
FACULTY
LaToya M. Haynes practices at the Carolinas Pain Institute and the Center for Clinical Research in Winston-Salem, North Carolina, and is a preceptor for PA students. Kelly Bilello is a PA at Genitourinary Surgical Consultants in Denver. Jade Breeback practices at Cone Health Primary Care in Kernersville, North Carolina. Jessica Cain is a PA in emergency medicine at the University of Cincinnati Medical Center. Jennifer Wenninger is a cardiothoracic and vascular surgery PA at Bellin Health Care Systems in Green Bay, Wisconsin. M. Jane McDaniel is an Instructor in the Department of Physician Assistant Studies at Wake Forest School of Medicine in Winston-Salem.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Interstitial cystitis/bladder pain syndrome (IC/BPS) is a common, painful disease of the urinary bladder. Difficult to diagnose and frequently misdiagnosed as another common urologic disorder, IC/BPS challenges health care providers to identify it early and implement current treatment algorithms that may simplify management and improve quality of life for affected patients.
Interstitial cystitis (IC), or bladder pain syndrome (BPS), is a clinical condition characterized by bladder pain, urinary frequency and urgency, and increased nighttime urination (nocturia).1 More specifically, IC/BPS is defined as an unpleasant sensation in the bladder, abdomen, or pelvis (ie, pain, pressure, burning, and/or other discomfort) perceived to be originating in the urinary bladder. The condition is associated with lower urinary tract symptoms of more than six weeks’ duration, with no infection or other identifiable cause present.2
IC/BPS lacks a single known etiology; rather, it most likely results from multiple contributing factors that cascade into a painful and potentially debilitating syndrome. The condition was first described more than a century ago,3,4 but its complex nature and conflicting theories about its pathogenesis present both diagnostic and therapeutic challenges for health care professionals. Frequent misdiagnosis of IC/BPS as another common urologic disorder can make timely, appropriate treatment elusive.
Without a clearly described pathophysiology, IC/BPS has always been difficult to define using standardized diagnostic criteria and precise terminology. The definition of the condition was revised in 2002 and again in 2008, when the nomenclature bladder pain syndrome was introduced.1,5,6
Less than 10 years ago, US researchers described IC as a subgroup of BPS,7 while in Europe, BPS is used as the broader term, with IC still considered a well-defined subgroup that usually involves ulceration.6 The future may find IC, BPS, and painful bladder syndrome (PBS) used as interchangeable terms—or as unique diagnoses. A better understanding of the pathophysiology of IC/BPS/PBS would contribute not only to resolving issues of nomenclature, but also to establishing an accurate diagnosis earlier in the disease process and providing more efficient, effective treatment.
THE PROBLEM OF EPIDEMIOLOGY
Inconsistencies in the terminology, definitions, and diagnostic criteria of IC/BPS have made epidemiology difficult to establish.1 It has been suggested that IC/BPS is underdiagnosed in the United States and that its prevalence is much greater than generally reported.8
According to one study of IC in a managed care population, its prevalence in 2005 was 197 per 100,000 women and 41 per 100,000 men, with the female-to-male ratio estimated at 5:1.9 In 2011, researchers for the RAND Corporation published what they called the first population-based “symptom prevalence estimate” among US women older than 18, based on more than 100,000 screening interviews conducted by phone. According to their findings, between 3.3 and 7.9 million US women meet the stated criteria for IC/BPS (ie, between 3,113 and 7,453 women per 100,000).10 These conflicting data exemplify the range of epidemiologic conclusions that exist regarding this condition.
On the next page: Proposed pathophysiology >>
THE PROPOSED PATHOPHYSIOLOGY
IC/BPS is thought to begin with an initial insult to the bladder that leads to dysfunction of the epithelial layer. This insult may be the result of a neurogenic inflammation, autoimmunity, subclinical or chronic infection, or bladder urothelial defects.1 Dysfunction in the epithelial layer includes altered bladder epithelial expression of human leukocyte antigen I and II; decreased expression of uroplakin (an antitoxic protein in the bladder), and a defective glycosaminoglycan mucus layer.4 This damage to the epithelial layer alters the permeability of the bladder, allowing potassium ions to enter the urothelium and depolarize motor and sensory nerves. This potassium leak then activates the mast cells, causing mastocytosis and the release of histamine.11 These processes disrupt the homeostasis of the urinary tract and allow the development of inflammation—a main cause of the pelvic pain associated with IC/BPS4,12,13 (see Figure 114).
Other factors that exacerbate the primary inflammation in the bladder are C-fibers and nerve growth factor (NGF). C-fibers are afferent fibers found in the peripheral nerves of the somatic sensory system that convey input signals from the periphery to the central nervous system.3 In patients with IC/BPS, initial inflammation activates C-fibers, which produce substance P, nociceptor, and other inflammatory mediators. These mediators exacerbate existing inflammation and further facilitate mast cell activation.3
NGF is a protein that is critical for the maintenance of sympathetic and sensory neurons; it is important not only in the urinary tract but in all organ systems. Increased levels of NGF, a prevalent finding in patients with IC/BPS, is an indicator of inflammation in the body. The precise mechanism that causes elevated NGF in patients with IC/BPS is not well understood, but its presence supports the theory that inflammation is a cause of pelvic pain in IC/BPS.12
The urinary urgency and frequency experienced by patients with IC/BPS is in part due to the role nitric oxide (NO) plays in bladder activity. Patients with IC have decreased levels of urinary NO (a reduction thought to be the result of a decrease in L-arginine) and urinary NO synthase.12,15,16 Ordinarily, NO synthase converts L-arginine to NO, which helps to control relaxation of the bladder smooth muscle, allowing more urine to be stored. In patients with IC/BPS, NO insufficiency leads to bladder overactivity.15
On the next page: Patient history and presentation >>
PATIENT HISTORY AND PRESENTATION
A detailed patient history is imperative in establishing the diagnosis of IC/BPS. Symptoms that should prompt the clinician to consider IC/BPS include:
• Pelvic or bladder pain relieved with voiding
• Dyspareunia
• Increased frequency of urination with no infection present
• Urinary urgency with pain, and
• Increased nocturia.17,18
Early IC presents variably, and pain, though a common symptom, is not always present.19 Chronic pain is defined by duration of at least six months, with the discomfort perceived as originating in the bladder.8 In addition to patients who experience pain, those who void several times during the night should also be considered for further evaluation.19
Many patients describe their symptoms in terms of flares and periods of remission. Some patients associate flares with stress, seasonal allergies, sexual activity, consumption of certain foods, and the premenstrual week.17,20 Patients with IC/BPS are commonly misdiagnosed with recurrent urinary tract infections; hence the need to standardize the criteria for diagnosis of IC/BPS.17
DIAGNOSIS
There are currently three available sets of diagnostic criteria for patients with IC/BPS. These are the National Institute for Diabetes and Diseases of the Kidney (NIDDK) definition (1990),21 the International Continence Society (ICS) definition of painful bladder disorders (2002),5 and the European Society for the Study of IC/BPS (ESSIC) definition (2008).6 In particular, the ESSIC criteria were formulated to help identify IC/BPS earlier in the disease course.
The 1990 NIDDK protocol, developed for research purposes,12 featured inclusion and exclusion criteria. Exclusion criteria included age younger than 18 years and presence of benign bladder tumors, radiation cystitis, tuberculosis cystitis, bacterial cystitis, vaginitis, symptomatic urethral diverticulum, uterine/cervical/vaginal cancers, and/or active herpes; urinary frequency of less than five episodes in 12 hours; and less than two episodes of nocturia per night.19
NIDDK inclusion criteria required two or more of the following: Hunner’s ulcer, pain on bladder filling, general pelvic pain, glomerulations on endoscopy, and decreased bladder compliance on cystometrogram.19
This protocol proved to be excessively restrictive for clinical use and was widely replaced by the ICS criteria in 2002. The ICS criteria5 allowed more varied patient presentations; the exclusions featured in the NIDDK guideline, it has been estimated, could have eliminated at least one-third of patients who would reasonably be considered to have IC/BPS.21
In contrast to the NIDDK criteria, the ICS criteria5 defined BPS as “the complaint of suprapubic pain related to bladder filling, accompanied by other symptoms such as increased daytime and nighttime urinary frequency in the absence of proven urinary tract infection or other obvious pathology.”5 Additionally, the ICS document restricts the diagnosis of IC to patients with painful bladder syndrome in addition to “typical cystoscopic and histologic features.”6
According to the ESSIC proposal on diagnostic criteria, classification, and nomenclature,6 diagnosing IC/BPS requires symptoms of chronic pain related to the urinary bladder accompanied by at least one other urinary symptom, such as daytime and nighttime frequency and exclusion of confusable diseases and cystoscopy with hydrodistention and biopsy, if indicated. The new statement does not include the required absence of UTI or other pathology identified in the previous (ICS) criteria; these, too, overlooked a portion of the population who would be considered to have IC/BPS. Therefore, the ESSIC classification provides the most comprehensive criteria for diagnosing IC/BPS and has been determined as best for diagnostic purposes in the early disease stages.12
On the next page: Applying the criteria and referral >>
Applying the Criteria
IC/BPS remains a diagnosis of exclusion.12 The most common disorders seen in the differential diagnosis for IC/BPS (ie, “confusable diseases”6) include bacterial cystitis, vaginitis, pelvic pain, vulvodynia, urinary tract infections, yeast infections, sexually transmitted infections, endometriosis, overactive bladder, and genitourinary malignancies.5,12
Biopsy or cystoscopy with short-duration, low-pressure hydrodistention can be performed on patients who present with persistent pelvic pain and urinary symptoms.2,12 Common cystoscopic findings in patients with IC/BPS include Hunner’s lesions, glomerulations, and inflammatory infiltrates on biopsy.12,21 Hunner’s lesions are described as “patches of red mucosa exhibiting small vessels radiating to a central pale scar.”21 These lesions may also be referred to as Hunner’s ulcers.12 Not always visible on cystoscopy, Hunner’s lesions may be seen only after hydrodistention of the bladder under anesthesia.
Cystoscopic findings can be misleading for providers, as not all stages of IC/BPS manifest in the same manner. No single laboratory finding will identify IC/BPS. The only way to diagnose this disease is to rule out all other diseases with similar presentations.18
When to Refer
Specific findings that may indicate the need for referral include severe pain, hematuria, chronic UTI, and pyuria. Generally, however, the decision to refer the patient with IC/BPS to a urologist or urogynecologist depends on the primary care provider’s comfort level. Some providers choose to refer as soon as identifying symptoms of IC/BPS have been confirmed, whereas others may wish to proceed with further evaluation and/or treatment before referring.2,22
Even if the provider decides to refer immediately after identifying symptoms, it is important to initiate some patient education: for example, explaining that the patient will likely require further tests, including cystoscopy and possibly urodynamic evaluation.2,23 Smokers and other patients at high risk for bladder cancer should be referred for cystoscopy.2
If the primary care provider chooses to proceed with evaluation and treatment before referring the patient, follow-up is typically recommended at one-month intervals for the first three months, then every three months thereafter.18 This allows the clinician to monitor a patient’s progress and address concerns that may develop. Symptoms may be slow to respond to treatment, so it is essential to encourage the patient to adhere to the prescribed regimen. If three to six months of first-line treatment yield no response, further consultation and evaluation are warranted. Overall, a multidisciplinary approach that includes the participation of a urologist, a gynecologist, or other appropriate specialist will help ensure optimal treatment and care.18
A good tool that is often used to gauge the patient’s progress is the O’Leary/Sant Voiding and Pain Indices23-25 (see Figure 224). Reviewing patient responses to this questionnaire, with its precise numerical system, at each follow-up appointment can be especially helpful.
On the next page: Treatment >>
TREATMENT
Management of IC/BPS can be challenging, because it is such a multifaceted disorder. Patient education beginning shortly after diagnosis is crucial, as treatment regimens may involve complex multimodal therapy over long periods of time, oftentimes with a very gradual response (see “For Your Patient”).
Lifestyle changes for patients with IC/BPS are considered an important component of treatment. Dietary changes—specifically, reducing intake of foods with high acidic content (citrus fruits, tomatoes), alcoholic beverages, spices, and potassium—have been found helpful.5 Reducing stress and anxiety, whenever possible, has also been noted to alleviate symptoms.24
Another nonpharmacologic option is physical therapy, including biofeedback and bladder retraining.12,13 Biofeedback is particularly useful in patients who experience pelvic pain attributed to spasms of the pelvic floor.12 Bladder retraining can be used to reduce urinary frequency through techniques that include scheduled voiding. Physical therapy strategies should be revisited regularly to maintain their therapeutic benefits.5,12
Oral Medications
The mainstay of pharmacologic treatment, and the one most thoroughly studied, is oral pentosan polysulfate (PPS), which belongs to the class of heparins or heparinoids.2,26 PPS is thought to attach to the mucosa of the bladder, reestablishing its glycosaminoglycan layer and restoring normal function of this permeable barrier.14 Overall, this drug is well tolerated and relieves the symptoms of pain, urgency, and frequency. Patients may start to experience improvement in symptoms after four weeks of treatment; however, it can take six months or longer to achieve the full benefit of this therapy.13,25
Other pharmacologic agents used in the treatment of IC/BPS include antihistamines, tricyclic antidepressants, and some antiepileptic medications. Some patients with IC/BPS experience symptoms attributable to bladder mastocytosis and mast cell activation, explaining the efficacy of antihistamines for these particular patients.27 Among the antihistamines, hydroxyzine, an H1-receptor antagonist, is a common pharmacologic option. Similarly, cetirizine can be used in patients for whom the sedating effects of hydroxyzine may prove hazardous.28
Antidepressants, especially tricyclic antidepressants (TCAs, eg, amitriptyline), can also provide some relief for patients, including alleviation of pain, possible antihistamine effects, and mild anticholinergic action, leading to decreased urinary urgency and frequency.2,26,29 Of note, the TCA imipramine should be avoided in patients with IC/BPS, as it has a sympathomimetic effect that can worsen symptoms of dysfunctional voiding in this patient population.14
Gabapentin, an antiepileptic, is used for improvement of severe, persistent pain. Alternatives to gabapentin include, but are not limited to, phenytoin, carbamazepine, and valproic acid.14 The effectiveness of these medications in the treatment of IC/BPS lend credence to the theory that, in addition to bladder mucosa dysfunction, symptoms are also mediated through an inflammatory neurogenic pathway.
Patients should be encouraged to continue use of PPS or other prescribed pharmacologic treatments even if there is no immediate relief of symptoms.25 According to a treatment algorithm from the American Urological Association,2 however, ineffective treatments should be stopped and diagnosis should be reconsidered if there is no improvement within a “clinically meaningful time frame.”
Additional Pharmacologic Options
Intravesical therapy is another mode of pharmacologic treatment.2,29 This treatment is usually reserved for IC/BPS flares and management of cases lacking the desired response to oral medications. Dimethyl sulfoxide (DMSO) is a commonly used intravesical agent. DMSO acts to provide pain relief and reduce inflammation, in addition to effecting histamine release from mast cells.27 Intravesical heparinoids essentially employ the same mechanism of action as oral PPS to maintain and enhance the bladder’s mucosal lining. This treatment is also commonly used in patients who need to discontinue use of oral PPS due to side effects.27,30
Treatment options for refractory IC/BPS include immunosuppression (and surgical therapy, below). Prednisone and cyclosporine have been shown to be effective immunosuppressive agents.23 Side effects make the use of these medications less desirable; also, symptoms have been shown to return in many patients after treatment is stopped.23
The FDA has recently approved the use of onabotulinum toxin A (Botox) injections into the bladder for treatment of urinary urgency and frequency that are not responsive to standard medical therapy. Since patients with IC often experience such symptoms with no relief from standard therapy, intratrigonal and periurethral injections of Botox are being administered for treatment of IC in some patients with moderate success.31-33 Although intradetrusor Botox use is recommended as a fifth-line treatment in the AUA guidelines,2 it is important to note that this agent is not FDA-approved specifically for IC, but rather for any refractory condition presenting with urinary frequency and urgency.
Surgical Therapy
Surgical intervention (a sixth-line treatment option, according to the AUA guidelines2) is rarely indicated except in cases of severe IC/BPS that have been refractory to all other treatment options and in which spontaneous remission of symptoms seems unlikely. Supravesical urinary diversion, usually through the creation of an ileal conduit, is the procedure of choice and is often performed in conjunction with a cystectomy. Unfortunately in some cases, pelvic pain has been noted to continue postcystectomy, a finding that also supports a neurogenic etiology for IC/BPS.14
On the Horizon
Although IC/PBS is difficult to treat, new data suggest that use of extended diagnostics, including molecular markers to detect the disease early and guide effective treatment, may greatly improve current therapeutic options.34
Prescribing selective anticholinergic and antihistamine pharmacotherapy based on the patient’s specific muscarinic and histamine receptor profile, respectively, may provide greater symptom relief.34 Maintaining the appropriate, individualized therapy could represent a significant advance in treatment for IC/BPS. However, further research on the topic is needed.
On the next page: Conclusion >>
CONCLUSION
IC/BPS is a complex multifactorial syndrome that may manifest with disabling pain. Further research on this disease is warranted to help facilitate an earlier, more consistent diagnosis and produce more effective treatment options. Early diagnosis of IC/BPS is essential for successful therapy. Once the diagnosis is made, a cautious regimen of different treatments, following the American Urological Association’s clinical practice guidelines for interstitial cystitis, should be implemented. Patients should also be encouraged to consider specific dietary changes and other lifestyle adjustments under a clinician’s supervision.
The authors wish to thank Carol Hildebrandt for her help in preparing this manuscript and Robert J. Evans, MD, Associate Professor of Urology, Wake Forest Baptist Medical Center, for his editorial expertise.
CE/CME No: CR-1307
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
•
Describe the pathophysiology
of interstitial cystitis/bladder
pain syndrome (IC/BPS), as
it is currently understood.
•
Discuss urogenital signs and
symptoms that should prompt suspicion for IC/BPS in a primary care patient.
•
Explain the clinical diagnosis of
IC/BPS and key considerations for referral.
•
Review medical management, nonoperative therapy, and surgical treatment of IC/BPS.
FACULTY
LaToya M. Haynes practices at the Carolinas Pain Institute and the Center for Clinical Research in Winston-Salem, North Carolina, and is a preceptor for PA students. Kelly Bilello is a PA at Genitourinary Surgical Consultants in Denver. Jade Breeback practices at Cone Health Primary Care in Kernersville, North Carolina. Jessica Cain is a PA in emergency medicine at the University of Cincinnati Medical Center. Jennifer Wenninger is a cardiothoracic and vascular surgery PA at Bellin Health Care Systems in Green Bay, Wisconsin. M. Jane McDaniel is an Instructor in the Department of Physician Assistant Studies at Wake Forest School of Medicine in Winston-Salem.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Interstitial cystitis/bladder pain syndrome (IC/BPS) is a common, painful disease of the urinary bladder. Difficult to diagnose and frequently misdiagnosed as another common urologic disorder, IC/BPS challenges health care providers to identify it early and implement current treatment algorithms that may simplify management and improve quality of life for affected patients.
Interstitial cystitis (IC), or bladder pain syndrome (BPS), is a clinical condition characterized by bladder pain, urinary frequency and urgency, and increased nighttime urination (nocturia).1 More specifically, IC/BPS is defined as an unpleasant sensation in the bladder, abdomen, or pelvis (ie, pain, pressure, burning, and/or other discomfort) perceived to be originating in the urinary bladder. The condition is associated with lower urinary tract symptoms of more than six weeks’ duration, with no infection or other identifiable cause present.2
IC/BPS lacks a single known etiology; rather, it most likely results from multiple contributing factors that cascade into a painful and potentially debilitating syndrome. The condition was first described more than a century ago,3,4 but its complex nature and conflicting theories about its pathogenesis present both diagnostic and therapeutic challenges for health care professionals. Frequent misdiagnosis of IC/BPS as another common urologic disorder can make timely, appropriate treatment elusive.
Without a clearly described pathophysiology, IC/BPS has always been difficult to define using standardized diagnostic criteria and precise terminology. The definition of the condition was revised in 2002 and again in 2008, when the nomenclature bladder pain syndrome was introduced.1,5,6
Less than 10 years ago, US researchers described IC as a subgroup of BPS,7 while in Europe, BPS is used as the broader term, with IC still considered a well-defined subgroup that usually involves ulceration.6 The future may find IC, BPS, and painful bladder syndrome (PBS) used as interchangeable terms—or as unique diagnoses. A better understanding of the pathophysiology of IC/BPS/PBS would contribute not only to resolving issues of nomenclature, but also to establishing an accurate diagnosis earlier in the disease process and providing more efficient, effective treatment.
THE PROBLEM OF EPIDEMIOLOGY
Inconsistencies in the terminology, definitions, and diagnostic criteria of IC/BPS have made epidemiology difficult to establish.1 It has been suggested that IC/BPS is underdiagnosed in the United States and that its prevalence is much greater than generally reported.8
According to one study of IC in a managed care population, its prevalence in 2005 was 197 per 100,000 women and 41 per 100,000 men, with the female-to-male ratio estimated at 5:1.9 In 2011, researchers for the RAND Corporation published what they called the first population-based “symptom prevalence estimate” among US women older than 18, based on more than 100,000 screening interviews conducted by phone. According to their findings, between 3.3 and 7.9 million US women meet the stated criteria for IC/BPS (ie, between 3,113 and 7,453 women per 100,000).10 These conflicting data exemplify the range of epidemiologic conclusions that exist regarding this condition.
On the next page: Proposed pathophysiology >>
THE PROPOSED PATHOPHYSIOLOGY
IC/BPS is thought to begin with an initial insult to the bladder that leads to dysfunction of the epithelial layer. This insult may be the result of a neurogenic inflammation, autoimmunity, subclinical or chronic infection, or bladder urothelial defects.1 Dysfunction in the epithelial layer includes altered bladder epithelial expression of human leukocyte antigen I and II; decreased expression of uroplakin (an antitoxic protein in the bladder), and a defective glycosaminoglycan mucus layer.4 This damage to the epithelial layer alters the permeability of the bladder, allowing potassium ions to enter the urothelium and depolarize motor and sensory nerves. This potassium leak then activates the mast cells, causing mastocytosis and the release of histamine.11 These processes disrupt the homeostasis of the urinary tract and allow the development of inflammation—a main cause of the pelvic pain associated with IC/BPS4,12,13 (see Figure 114).
Other factors that exacerbate the primary inflammation in the bladder are C-fibers and nerve growth factor (NGF). C-fibers are afferent fibers found in the peripheral nerves of the somatic sensory system that convey input signals from the periphery to the central nervous system.3 In patients with IC/BPS, initial inflammation activates C-fibers, which produce substance P, nociceptor, and other inflammatory mediators. These mediators exacerbate existing inflammation and further facilitate mast cell activation.3
NGF is a protein that is critical for the maintenance of sympathetic and sensory neurons; it is important not only in the urinary tract but in all organ systems. Increased levels of NGF, a prevalent finding in patients with IC/BPS, is an indicator of inflammation in the body. The precise mechanism that causes elevated NGF in patients with IC/BPS is not well understood, but its presence supports the theory that inflammation is a cause of pelvic pain in IC/BPS.12
The urinary urgency and frequency experienced by patients with IC/BPS is in part due to the role nitric oxide (NO) plays in bladder activity. Patients with IC have decreased levels of urinary NO (a reduction thought to be the result of a decrease in L-arginine) and urinary NO synthase.12,15,16 Ordinarily, NO synthase converts L-arginine to NO, which helps to control relaxation of the bladder smooth muscle, allowing more urine to be stored. In patients with IC/BPS, NO insufficiency leads to bladder overactivity.15
On the next page: Patient history and presentation >>
PATIENT HISTORY AND PRESENTATION
A detailed patient history is imperative in establishing the diagnosis of IC/BPS. Symptoms that should prompt the clinician to consider IC/BPS include:
• Pelvic or bladder pain relieved with voiding
• Dyspareunia
• Increased frequency of urination with no infection present
• Urinary urgency with pain, and
• Increased nocturia.17,18
Early IC presents variably, and pain, though a common symptom, is not always present.19 Chronic pain is defined by duration of at least six months, with the discomfort perceived as originating in the bladder.8 In addition to patients who experience pain, those who void several times during the night should also be considered for further evaluation.19
Many patients describe their symptoms in terms of flares and periods of remission. Some patients associate flares with stress, seasonal allergies, sexual activity, consumption of certain foods, and the premenstrual week.17,20 Patients with IC/BPS are commonly misdiagnosed with recurrent urinary tract infections; hence the need to standardize the criteria for diagnosis of IC/BPS.17
DIAGNOSIS
There are currently three available sets of diagnostic criteria for patients with IC/BPS. These are the National Institute for Diabetes and Diseases of the Kidney (NIDDK) definition (1990),21 the International Continence Society (ICS) definition of painful bladder disorders (2002),5 and the European Society for the Study of IC/BPS (ESSIC) definition (2008).6 In particular, the ESSIC criteria were formulated to help identify IC/BPS earlier in the disease course.
The 1990 NIDDK protocol, developed for research purposes,12 featured inclusion and exclusion criteria. Exclusion criteria included age younger than 18 years and presence of benign bladder tumors, radiation cystitis, tuberculosis cystitis, bacterial cystitis, vaginitis, symptomatic urethral diverticulum, uterine/cervical/vaginal cancers, and/or active herpes; urinary frequency of less than five episodes in 12 hours; and less than two episodes of nocturia per night.19
NIDDK inclusion criteria required two or more of the following: Hunner’s ulcer, pain on bladder filling, general pelvic pain, glomerulations on endoscopy, and decreased bladder compliance on cystometrogram.19
This protocol proved to be excessively restrictive for clinical use and was widely replaced by the ICS criteria in 2002. The ICS criteria5 allowed more varied patient presentations; the exclusions featured in the NIDDK guideline, it has been estimated, could have eliminated at least one-third of patients who would reasonably be considered to have IC/BPS.21
In contrast to the NIDDK criteria, the ICS criteria5 defined BPS as “the complaint of suprapubic pain related to bladder filling, accompanied by other symptoms such as increased daytime and nighttime urinary frequency in the absence of proven urinary tract infection or other obvious pathology.”5 Additionally, the ICS document restricts the diagnosis of IC to patients with painful bladder syndrome in addition to “typical cystoscopic and histologic features.”6
According to the ESSIC proposal on diagnostic criteria, classification, and nomenclature,6 diagnosing IC/BPS requires symptoms of chronic pain related to the urinary bladder accompanied by at least one other urinary symptom, such as daytime and nighttime frequency and exclusion of confusable diseases and cystoscopy with hydrodistention and biopsy, if indicated. The new statement does not include the required absence of UTI or other pathology identified in the previous (ICS) criteria; these, too, overlooked a portion of the population who would be considered to have IC/BPS. Therefore, the ESSIC classification provides the most comprehensive criteria for diagnosing IC/BPS and has been determined as best for diagnostic purposes in the early disease stages.12
On the next page: Applying the criteria and referral >>
Applying the Criteria
IC/BPS remains a diagnosis of exclusion.12 The most common disorders seen in the differential diagnosis for IC/BPS (ie, “confusable diseases”6) include bacterial cystitis, vaginitis, pelvic pain, vulvodynia, urinary tract infections, yeast infections, sexually transmitted infections, endometriosis, overactive bladder, and genitourinary malignancies.5,12
Biopsy or cystoscopy with short-duration, low-pressure hydrodistention can be performed on patients who present with persistent pelvic pain and urinary symptoms.2,12 Common cystoscopic findings in patients with IC/BPS include Hunner’s lesions, glomerulations, and inflammatory infiltrates on biopsy.12,21 Hunner’s lesions are described as “patches of red mucosa exhibiting small vessels radiating to a central pale scar.”21 These lesions may also be referred to as Hunner’s ulcers.12 Not always visible on cystoscopy, Hunner’s lesions may be seen only after hydrodistention of the bladder under anesthesia.
Cystoscopic findings can be misleading for providers, as not all stages of IC/BPS manifest in the same manner. No single laboratory finding will identify IC/BPS. The only way to diagnose this disease is to rule out all other diseases with similar presentations.18
When to Refer
Specific findings that may indicate the need for referral include severe pain, hematuria, chronic UTI, and pyuria. Generally, however, the decision to refer the patient with IC/BPS to a urologist or urogynecologist depends on the primary care provider’s comfort level. Some providers choose to refer as soon as identifying symptoms of IC/BPS have been confirmed, whereas others may wish to proceed with further evaluation and/or treatment before referring.2,22
Even if the provider decides to refer immediately after identifying symptoms, it is important to initiate some patient education: for example, explaining that the patient will likely require further tests, including cystoscopy and possibly urodynamic evaluation.2,23 Smokers and other patients at high risk for bladder cancer should be referred for cystoscopy.2
If the primary care provider chooses to proceed with evaluation and treatment before referring the patient, follow-up is typically recommended at one-month intervals for the first three months, then every three months thereafter.18 This allows the clinician to monitor a patient’s progress and address concerns that may develop. Symptoms may be slow to respond to treatment, so it is essential to encourage the patient to adhere to the prescribed regimen. If three to six months of first-line treatment yield no response, further consultation and evaluation are warranted. Overall, a multidisciplinary approach that includes the participation of a urologist, a gynecologist, or other appropriate specialist will help ensure optimal treatment and care.18
A good tool that is often used to gauge the patient’s progress is the O’Leary/Sant Voiding and Pain Indices23-25 (see Figure 224). Reviewing patient responses to this questionnaire, with its precise numerical system, at each follow-up appointment can be especially helpful.
On the next page: Treatment >>
TREATMENT
Management of IC/BPS can be challenging, because it is such a multifaceted disorder. Patient education beginning shortly after diagnosis is crucial, as treatment regimens may involve complex multimodal therapy over long periods of time, oftentimes with a very gradual response (see “For Your Patient”).
Lifestyle changes for patients with IC/BPS are considered an important component of treatment. Dietary changes—specifically, reducing intake of foods with high acidic content (citrus fruits, tomatoes), alcoholic beverages, spices, and potassium—have been found helpful.5 Reducing stress and anxiety, whenever possible, has also been noted to alleviate symptoms.24
Another nonpharmacologic option is physical therapy, including biofeedback and bladder retraining.12,13 Biofeedback is particularly useful in patients who experience pelvic pain attributed to spasms of the pelvic floor.12 Bladder retraining can be used to reduce urinary frequency through techniques that include scheduled voiding. Physical therapy strategies should be revisited regularly to maintain their therapeutic benefits.5,12
Oral Medications
The mainstay of pharmacologic treatment, and the one most thoroughly studied, is oral pentosan polysulfate (PPS), which belongs to the class of heparins or heparinoids.2,26 PPS is thought to attach to the mucosa of the bladder, reestablishing its glycosaminoglycan layer and restoring normal function of this permeable barrier.14 Overall, this drug is well tolerated and relieves the symptoms of pain, urgency, and frequency. Patients may start to experience improvement in symptoms after four weeks of treatment; however, it can take six months or longer to achieve the full benefit of this therapy.13,25
Other pharmacologic agents used in the treatment of IC/BPS include antihistamines, tricyclic antidepressants, and some antiepileptic medications. Some patients with IC/BPS experience symptoms attributable to bladder mastocytosis and mast cell activation, explaining the efficacy of antihistamines for these particular patients.27 Among the antihistamines, hydroxyzine, an H1-receptor antagonist, is a common pharmacologic option. Similarly, cetirizine can be used in patients for whom the sedating effects of hydroxyzine may prove hazardous.28
Antidepressants, especially tricyclic antidepressants (TCAs, eg, amitriptyline), can also provide some relief for patients, including alleviation of pain, possible antihistamine effects, and mild anticholinergic action, leading to decreased urinary urgency and frequency.2,26,29 Of note, the TCA imipramine should be avoided in patients with IC/BPS, as it has a sympathomimetic effect that can worsen symptoms of dysfunctional voiding in this patient population.14
Gabapentin, an antiepileptic, is used for improvement of severe, persistent pain. Alternatives to gabapentin include, but are not limited to, phenytoin, carbamazepine, and valproic acid.14 The effectiveness of these medications in the treatment of IC/BPS lend credence to the theory that, in addition to bladder mucosa dysfunction, symptoms are also mediated through an inflammatory neurogenic pathway.
Patients should be encouraged to continue use of PPS or other prescribed pharmacologic treatments even if there is no immediate relief of symptoms.25 According to a treatment algorithm from the American Urological Association,2 however, ineffective treatments should be stopped and diagnosis should be reconsidered if there is no improvement within a “clinically meaningful time frame.”
Additional Pharmacologic Options
Intravesical therapy is another mode of pharmacologic treatment.2,29 This treatment is usually reserved for IC/BPS flares and management of cases lacking the desired response to oral medications. Dimethyl sulfoxide (DMSO) is a commonly used intravesical agent. DMSO acts to provide pain relief and reduce inflammation, in addition to effecting histamine release from mast cells.27 Intravesical heparinoids essentially employ the same mechanism of action as oral PPS to maintain and enhance the bladder’s mucosal lining. This treatment is also commonly used in patients who need to discontinue use of oral PPS due to side effects.27,30
Treatment options for refractory IC/BPS include immunosuppression (and surgical therapy, below). Prednisone and cyclosporine have been shown to be effective immunosuppressive agents.23 Side effects make the use of these medications less desirable; also, symptoms have been shown to return in many patients after treatment is stopped.23
The FDA has recently approved the use of onabotulinum toxin A (Botox) injections into the bladder for treatment of urinary urgency and frequency that are not responsive to standard medical therapy. Since patients with IC often experience such symptoms with no relief from standard therapy, intratrigonal and periurethral injections of Botox are being administered for treatment of IC in some patients with moderate success.31-33 Although intradetrusor Botox use is recommended as a fifth-line treatment in the AUA guidelines,2 it is important to note that this agent is not FDA-approved specifically for IC, but rather for any refractory condition presenting with urinary frequency and urgency.
Surgical Therapy
Surgical intervention (a sixth-line treatment option, according to the AUA guidelines2) is rarely indicated except in cases of severe IC/BPS that have been refractory to all other treatment options and in which spontaneous remission of symptoms seems unlikely. Supravesical urinary diversion, usually through the creation of an ileal conduit, is the procedure of choice and is often performed in conjunction with a cystectomy. Unfortunately in some cases, pelvic pain has been noted to continue postcystectomy, a finding that also supports a neurogenic etiology for IC/BPS.14
On the Horizon
Although IC/PBS is difficult to treat, new data suggest that use of extended diagnostics, including molecular markers to detect the disease early and guide effective treatment, may greatly improve current therapeutic options.34
Prescribing selective anticholinergic and antihistamine pharmacotherapy based on the patient’s specific muscarinic and histamine receptor profile, respectively, may provide greater symptom relief.34 Maintaining the appropriate, individualized therapy could represent a significant advance in treatment for IC/BPS. However, further research on the topic is needed.
On the next page: Conclusion >>
CONCLUSION
IC/BPS is a complex multifactorial syndrome that may manifest with disabling pain. Further research on this disease is warranted to help facilitate an earlier, more consistent diagnosis and produce more effective treatment options. Early diagnosis of IC/BPS is essential for successful therapy. Once the diagnosis is made, a cautious regimen of different treatments, following the American Urological Association’s clinical practice guidelines for interstitial cystitis, should be implemented. Patients should also be encouraged to consider specific dietary changes and other lifestyle adjustments under a clinician’s supervision.
The authors wish to thank Carol Hildebrandt for her help in preparing this manuscript and Robert J. Evans, MD, Associate Professor of Urology, Wake Forest Baptist Medical Center, for his editorial expertise.
1. Dasgupta J, Tincello DG. Interstitial cystitis/bladder pain syndrome: an update. Maturitas. 2009;64:212-217.
2. Hanno PM, Burks DA, Clemens JQ, et al. Diagnosis and treatment of interstitial cystitis/bladder pain syndrome: American Urological Association (AUA) Guideline (2011). www.auanet.org/education/guidelines/ic-bladder-pain-syndrome.cfm. Accessed June 5, 2013.
3. Evans RJ; University of Tennessee Advanced Studies in Pharmacy. Pathophysiology and clinical presentation of interstitial cystitis (2005). www.utasip.com/files/articlefiles/pdf/XASIP_Issue_Mar_p8_14.pdf. Accessed June 5, 2013.
4. Sant GR. Etiology, pathogenesis, and diagnosis of interstitial cystitis. Rev Urol. 2002;(4 suppl 1):S9-S15.
5. Abrams P, Cardozo L, Fall M, et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167-178.
6. van de Merwe JP, Nordling J, Bouchelouche P, et al. Diagnostic criteria, classification, and nomenclature for painful bladder syndrome/interstitial cystitis: an ESSIC proposal. Eur Urol. 2008;53:60-67.
7. Bogart LM, Berry SH, Clemens JQ. Symptoms of interstitial cystitis, painful bladder syndrome and similar diseases in women: a systematic review. J Urol. 2007;177:450-456.
8. Kusek JW, Nyberg LM. The epidemiology of interstitial cystitis: is it time to expand our definition? Urology. 2001;57(6 suppl 1):95-99.
9. Clemens JQ, Meenan RT, Rosetti MC, et al. Prevalence and incidence of interstitial cystitis in a managed care population. J Urol. 2005;173:98-102.
10. Berry SH, Elliott MN, Suttorp M, et al. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol. 2011;186:540-544.
11. Rosamilia A, Dwyer PL. Pathophysiology of interstitial cystitis. Curr Opin Obstet Gynecol. 2000;12:405-410.
12. Grover S, Srivastava A, Lee R, et al. Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol. 2011;3:19-33.
13. Rosenberg MT, Page S, Hazzard MA. Prevalence of interstitial cystitis in a primary care setting. Urology. 2007;69(4 suppl):S48-S52.
14. Evans RJ. Treatment approaches for interstitial cystitis: multimodality therapy. Rev Urol. 2002;(4 suppl 1):S16-S20.
15. Wesselmann U. Interstitial cystitis: a chronic visceral pain syndrome. Urology. 2001;57(6 suppl 1):102.
16. Hosseini A, Ehrén I, Wiklund NP. Nitric oxide as an objective marker for evaluation of treatment response in patients with classic interstitial cystitis. J Urol. 2004;172(6 pt 1):2261-2265.
17. Ho MH, Bhatia NN, Khorram O. Physiologic role of nitric oxide and nitric oxide synthase in female lower urinary tract. Curr Opin Obstet Gynecol. 2004;16:423-429.
18. Rosenberg MT, Newman DK, Page SA. Interstitial cystitis/painful bladder syndrome: symptom recognition is key to early identification, treatment. Cleve Clin J Med. 2007;74:854-862.
19. Driscoll A, Teichman JM. How do patients with interstitial cystitis present? J Urol. 2001;166:2118-2120.
20. Parsons CL. Interstitial cystitis: epidemiology and clinical presentation. Clin Obstet Gynecol. 2002;45:242-249.
21. Hanno PM, Landis JR, Matthews-Cook Y, et al. The diagnosis of interstitial cystitis revisited: lessons learned from the National Institutes of Health Interstitial Cystitis Database study. J Urol. 1999;161:553-557.
22. Whitmore KE, Theoharides TC. When to suspect interstitial cystitis. J Fam Pract. 2011;60:340-348.
23. Butrick CW, Howard FM, Sand PK. Diagnosis and treatment of interstitial cystitis/painful bladder syndrome: a review. J Womens Health (Larchmt). 2010;19:1185-1193.
24. O’Leary MP, Sant GR, Fowler FJ Jr, et al. The interstitial cystitis symptom index and problem index. Urology. 1997;49(5A suppl):58-63.
25. Nickel JC. Forensic dissection of a clinical trial: lessons learned in understanding and managing interstitial cystitis. Rev Urol. 2010;12:e78-e85.
26. Anger JT, Zabihi N, Clemens JQ, et al. Treatment choice, duration, and cost in patients with interstitial cystitis and painful bladder syndrome. Int Urogynecol J. 2011;22:395-400.
27. Nickel JC. Interstitial cystitis: characterization and management of an enigmatic urologic syndrome. Rev Urol. 2002;4:112-121.
28. Dell JR. Interstitial cystitis/painful bladder syndrome: appropriate diagnosis and management. J Womens Health (Larchmt). 2007;16:1181-1187.
29. National Kidney and Urologic Diseases Information Clearinghouse. Interstitial cystitis/painful bladder syndrome (2011). NIH Publication No. 11–3220. http://kidney.niddk.nih.gov/kudiseases/pubs/interstitialcystitis/IC_PBS_T_508.pdf. Accessed June 5, 2013.
30. Davis EL, El Khoudary SR, Talbott EO, et al. Safety and efficacy of the use of intravesical and oral pentosan polysulfate sodium for interstitial cystitis: a randomized double-blind clinical trial. J Urol. 2008;179:177-185.
31. Pinto R, Lopes T, Silva J, et al. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol. 2013;189:548-553.
32. Pinto R, Lopoes T, Frias B, et al. Trigonal injection of botulinum toxin A in patients with refractory bladder pain syndrome/interstitial cystitis. Eur Urol. 2010;58:360-365.
33. Gottsch HP, Miller JL, Yang CC, Berger RE. A pilot study of botulinum toxin for interstitial cystitis/painful bladder syndrome. Neurourol Urodyn. 2011; 30:93-96.
34. Neuhaus J, Schwalenberg T, Horn LC, et al. New aspects in the differential diagnosis and therapy of bladder pain syndrome/interstitial cystitis. Adv Urol. 2011;2011:639479.
1. Dasgupta J, Tincello DG. Interstitial cystitis/bladder pain syndrome: an update. Maturitas. 2009;64:212-217.
2. Hanno PM, Burks DA, Clemens JQ, et al. Diagnosis and treatment of interstitial cystitis/bladder pain syndrome: American Urological Association (AUA) Guideline (2011). www.auanet.org/education/guidelines/ic-bladder-pain-syndrome.cfm. Accessed June 5, 2013.
3. Evans RJ; University of Tennessee Advanced Studies in Pharmacy. Pathophysiology and clinical presentation of interstitial cystitis (2005). www.utasip.com/files/articlefiles/pdf/XASIP_Issue_Mar_p8_14.pdf. Accessed June 5, 2013.
4. Sant GR. Etiology, pathogenesis, and diagnosis of interstitial cystitis. Rev Urol. 2002;(4 suppl 1):S9-S15.
5. Abrams P, Cardozo L, Fall M, et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167-178.
6. van de Merwe JP, Nordling J, Bouchelouche P, et al. Diagnostic criteria, classification, and nomenclature for painful bladder syndrome/interstitial cystitis: an ESSIC proposal. Eur Urol. 2008;53:60-67.
7. Bogart LM, Berry SH, Clemens JQ. Symptoms of interstitial cystitis, painful bladder syndrome and similar diseases in women: a systematic review. J Urol. 2007;177:450-456.
8. Kusek JW, Nyberg LM. The epidemiology of interstitial cystitis: is it time to expand our definition? Urology. 2001;57(6 suppl 1):95-99.
9. Clemens JQ, Meenan RT, Rosetti MC, et al. Prevalence and incidence of interstitial cystitis in a managed care population. J Urol. 2005;173:98-102.
10. Berry SH, Elliott MN, Suttorp M, et al. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol. 2011;186:540-544.
11. Rosamilia A, Dwyer PL. Pathophysiology of interstitial cystitis. Curr Opin Obstet Gynecol. 2000;12:405-410.
12. Grover S, Srivastava A, Lee R, et al. Role of inflammation in bladder function and interstitial cystitis. Ther Adv Urol. 2011;3:19-33.
13. Rosenberg MT, Page S, Hazzard MA. Prevalence of interstitial cystitis in a primary care setting. Urology. 2007;69(4 suppl):S48-S52.
14. Evans RJ. Treatment approaches for interstitial cystitis: multimodality therapy. Rev Urol. 2002;(4 suppl 1):S16-S20.
15. Wesselmann U. Interstitial cystitis: a chronic visceral pain syndrome. Urology. 2001;57(6 suppl 1):102.
16. Hosseini A, Ehrén I, Wiklund NP. Nitric oxide as an objective marker for evaluation of treatment response in patients with classic interstitial cystitis. J Urol. 2004;172(6 pt 1):2261-2265.
17. Ho MH, Bhatia NN, Khorram O. Physiologic role of nitric oxide and nitric oxide synthase in female lower urinary tract. Curr Opin Obstet Gynecol. 2004;16:423-429.
18. Rosenberg MT, Newman DK, Page SA. Interstitial cystitis/painful bladder syndrome: symptom recognition is key to early identification, treatment. Cleve Clin J Med. 2007;74:854-862.
19. Driscoll A, Teichman JM. How do patients with interstitial cystitis present? J Urol. 2001;166:2118-2120.
20. Parsons CL. Interstitial cystitis: epidemiology and clinical presentation. Clin Obstet Gynecol. 2002;45:242-249.
21. Hanno PM, Landis JR, Matthews-Cook Y, et al. The diagnosis of interstitial cystitis revisited: lessons learned from the National Institutes of Health Interstitial Cystitis Database study. J Urol. 1999;161:553-557.
22. Whitmore KE, Theoharides TC. When to suspect interstitial cystitis. J Fam Pract. 2011;60:340-348.
23. Butrick CW, Howard FM, Sand PK. Diagnosis and treatment of interstitial cystitis/painful bladder syndrome: a review. J Womens Health (Larchmt). 2010;19:1185-1193.
24. O’Leary MP, Sant GR, Fowler FJ Jr, et al. The interstitial cystitis symptom index and problem index. Urology. 1997;49(5A suppl):58-63.
25. Nickel JC. Forensic dissection of a clinical trial: lessons learned in understanding and managing interstitial cystitis. Rev Urol. 2010;12:e78-e85.
26. Anger JT, Zabihi N, Clemens JQ, et al. Treatment choice, duration, and cost in patients with interstitial cystitis and painful bladder syndrome. Int Urogynecol J. 2011;22:395-400.
27. Nickel JC. Interstitial cystitis: characterization and management of an enigmatic urologic syndrome. Rev Urol. 2002;4:112-121.
28. Dell JR. Interstitial cystitis/painful bladder syndrome: appropriate diagnosis and management. J Womens Health (Larchmt). 2007;16:1181-1187.
29. National Kidney and Urologic Diseases Information Clearinghouse. Interstitial cystitis/painful bladder syndrome (2011). NIH Publication No. 11–3220. http://kidney.niddk.nih.gov/kudiseases/pubs/interstitialcystitis/IC_PBS_T_508.pdf. Accessed June 5, 2013.
30. Davis EL, El Khoudary SR, Talbott EO, et al. Safety and efficacy of the use of intravesical and oral pentosan polysulfate sodium for interstitial cystitis: a randomized double-blind clinical trial. J Urol. 2008;179:177-185.
31. Pinto R, Lopes T, Silva J, et al. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol. 2013;189:548-553.
32. Pinto R, Lopoes T, Frias B, et al. Trigonal injection of botulinum toxin A in patients with refractory bladder pain syndrome/interstitial cystitis. Eur Urol. 2010;58:360-365.
33. Gottsch HP, Miller JL, Yang CC, Berger RE. A pilot study of botulinum toxin for interstitial cystitis/painful bladder syndrome. Neurourol Urodyn. 2011; 30:93-96.
34. Neuhaus J, Schwalenberg T, Horn LC, et al. New aspects in the differential diagnosis and therapy of bladder pain syndrome/interstitial cystitis. Adv Urol. 2011;2011:639479.