User login
What's the diagnosis?
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
A 4-month male was referred to the pediatric dermatology clinic for a rash on the scalp, torso, and the diaper area since he was 2 months of age. He has been treated with nystatin, clotrimazole, and zinc oxide paste with partial improvement. After 2 months of partial improvement the rash worsened again, and he was referred to pediatric dermatology. The mother also reported asymptomatic left upper lateral eyebrow swelling noted a few weeks prior.
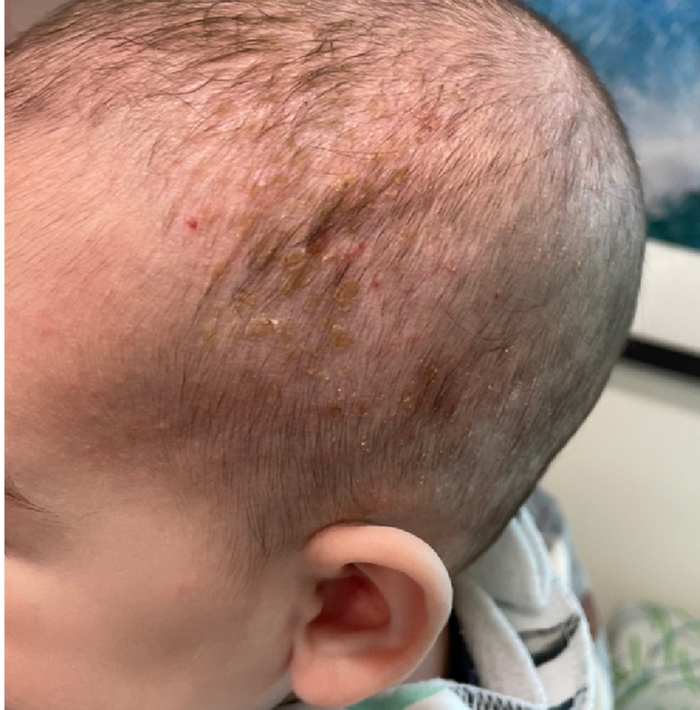
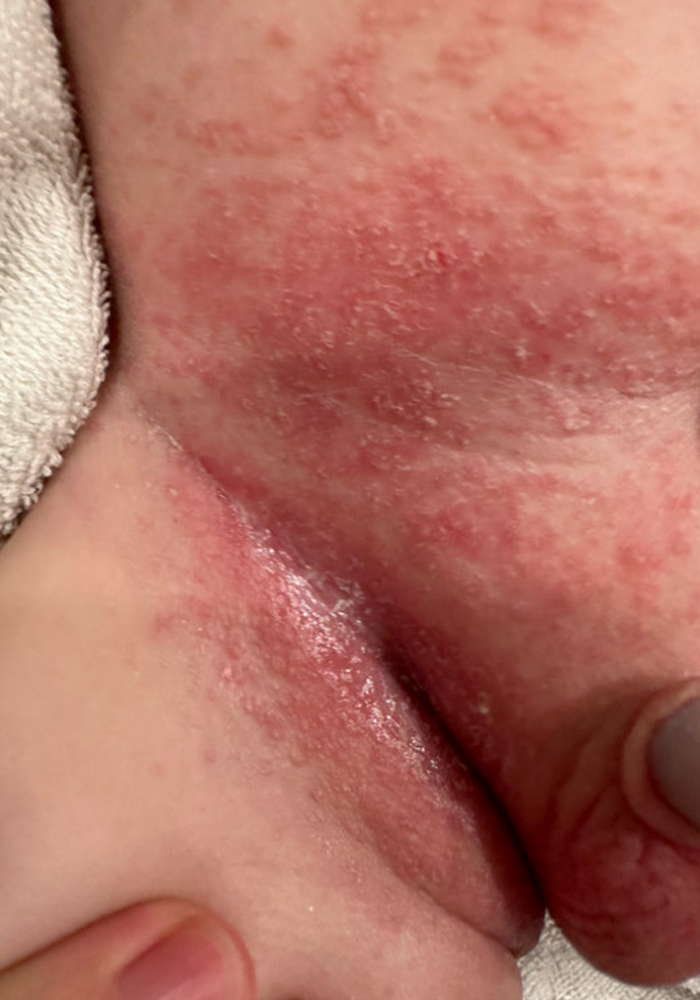
On the torso and diaper area, he had multiple scaly pink papules. On the groin he had eroded pink scaly plaques (Picture 2).
On his back he had a 3-mm yellow papule (Picture 3). 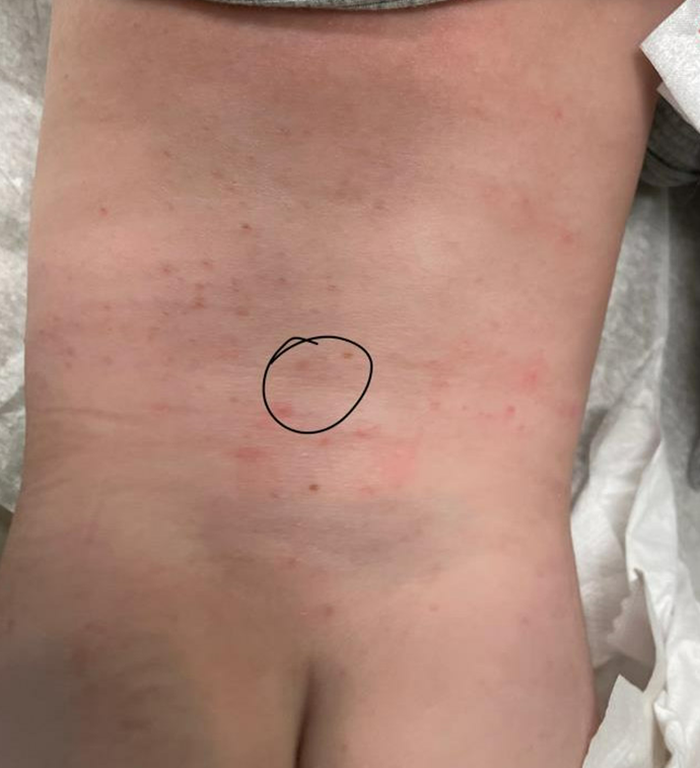
What's the diagnosis?
Given the characteristic clinical presentation, the most likely diagnosis is pilomatrixoma.
Pilomatrixomas are benign adnexal tumors that arise from immature matrix cells of the hair follicles located on dermal or subcutaneous tissue.
The cause of pilomatrixoma remains unclear. Recent studies have suggested that the development of pilomatrixoma are related to mutations in the Wnt signaling pathway, where beta-catenin gene (CTNNB1) mutation is the most frequently reported.1-4
Pilomatrixomas are more common in children and often present before 10 years of age.1,2,5 They commonly appear in head and neck, as well as upper extremities, trunk, and lower extremities.2,6
The clinical manifestations of pilomatrixomas are diverse and according to their appearance five classic clinical types are described: mass, pigmented, mixed, ulcerated, and keloid-like.2,3 The mass type is the predominant form, where it generally presents as a hard and freely mobile nodule covered by skin that may present a firm calcified protruding nodule. Other less common types include: lymphangiectasic, anetodermic, perforating, and bullous.2,6,7
Pilomatrixomas are mostly solitary, whereas multiple forms are reported to be associated with familial inheritance or syndromic conditions, such as myotonic dystrophy, Gardner syndrome, Turner’s syndrome, and Rubinstein-Taybi syndrome.2-4 However, children and adolescents occasionally present with multiple pilomatricomas with no associated syndrome.
On physical exam a helpful features for the diagnosis is the “teeter-totter sign,” which can be illustrated by pressing on one edge of the lesion that will cause the opposite edge to protrude from the skin. Another helpful tool is to use a light to transilluminate and the calcification produces a bluish opaque hue,8 as light cannot transmit through the calcification, often differentiating it from epidermal inclusion cysts or other noncalcified lesions.
What is the differential diagnosis?
Because of the diverse clinical presentations, pilomatrixomas are frequently misdiagnosed. The percentage of correct preoperative diagnosis reported is low, varying from 16% to 43% in different series.1,9-11 They most frequently are misdiagnosed as other types of cysts such as epidermal, dermoid, or sebaceous.2,3,5,12,13 Rapidly growing pilomatrixomas can be also be misdiagnosed as malignant soft-tissue tumors, cutaneous lymphoma, or sarcomas.5,13

When presenting with a classic history and physical features, diagnosis is clinical, and no further studies are recommended.14 To improve diagnostic accuracy when encountering unusual subtypes, imaging is recommended, including ultrasound. Ultrasound adds a high positive predictive value (95.56%).2 Generally, on ultrasound a pilomatrixoma is described as an oval, well-defined, heterogeneous, hyperechoic subcutaneous mass with or without posterior shadowing.2 The definitive diagnosis is, however, made by histopathologic examination.
Pilomatrixomas do not spontaneously regress, therefore complete surgical resection is the standard treatment. During the follow-up period, very low recurrence rates have been reported, varying from 1.5% to 2% which generally occurs because of incomplete resection.2,3
Plexiform neurofibromas are usually congenital tumors of peripheral nerve sheath associated with neurofibromatosis type 1, often with a “bag of worms” feel on palpation. Epidermoid cysts generally present as dermal nodules often with a visible puncture, mobile on soft and mobile on palpation. Dermatofibromas present as firm, usually hyperpigmented papule or nodules that are fixed to subcutaneous tissue, thus often “dimpling” when pitched. Dermatofibrosarcoma protuberans is a rare soft-tissue sarcoma which presents as a firm, slow growing indurated plaques growing over months to years.
Conclusion
Pilomatrixomas are a benign adnexal tumor that sometimes can present as atypical forms such as this case. Diagnosis is usually based on clinical diagnosis, and transillumination can be a bedside clue. When the clinical diagnosis remains obscure an ultrasound can be helpful. The main aim of this case is to improve awareness of the variable presentations of pilomatrixomas and the importance of high level of suspicion supported by careful clinical evaluation.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology, University of California, San Diego. Dr. Guelfand is a visiting dermatology resident in the division of pediaric and adolescent dermatology, University of Califonia, San Diego.
References
1. Jones CD et al. Am J Dermatopathol. 2018;40:631-41.
2. Hu JL et al. Arch Craniofac Surg. 2020;21(5):288-93.
3. Adhikari G and Jadhav GS. Cureus. 2022;14(2):22228.
4. Cóbar JP et al. J Surg Case Rep. 2023;2023(4):rjad182.
5. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
6. Kose D et al. J Cancer Res Ther. 2014;10(3):549-51.
7. Sabater-Abad J et al. Dermatol Online J. 2020;26(8):13030/qt4h16s45w.
8. Alkatan HM et al. Int J Surg Case Rep. 2021;84:106068.
9. Pant I et al. Indian J Dermatol. 2010;55:390-2.
10. Kaddu S et al. Am J Dermatopathol. 1996;18(4):333-8
11. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
12. Wang YN et al. Chin Med J (Engl). 2021;134(16):2011-2.
13. Yannoutsos A et al. Am J Dermatopathol. 2018;40(9):690-3.
14. Zhao A et al. Ear Nose Throat J. 2021;1455613211044778.
Given the characteristic clinical presentation, the most likely diagnosis is pilomatrixoma.
Pilomatrixomas are benign adnexal tumors that arise from immature matrix cells of the hair follicles located on dermal or subcutaneous tissue.
The cause of pilomatrixoma remains unclear. Recent studies have suggested that the development of pilomatrixoma are related to mutations in the Wnt signaling pathway, where beta-catenin gene (CTNNB1) mutation is the most frequently reported.1-4
Pilomatrixomas are more common in children and often present before 10 years of age.1,2,5 They commonly appear in head and neck, as well as upper extremities, trunk, and lower extremities.2,6
The clinical manifestations of pilomatrixomas are diverse and according to their appearance five classic clinical types are described: mass, pigmented, mixed, ulcerated, and keloid-like.2,3 The mass type is the predominant form, where it generally presents as a hard and freely mobile nodule covered by skin that may present a firm calcified protruding nodule. Other less common types include: lymphangiectasic, anetodermic, perforating, and bullous.2,6,7
Pilomatrixomas are mostly solitary, whereas multiple forms are reported to be associated with familial inheritance or syndromic conditions, such as myotonic dystrophy, Gardner syndrome, Turner’s syndrome, and Rubinstein-Taybi syndrome.2-4 However, children and adolescents occasionally present with multiple pilomatricomas with no associated syndrome.
On physical exam a helpful features for the diagnosis is the “teeter-totter sign,” which can be illustrated by pressing on one edge of the lesion that will cause the opposite edge to protrude from the skin. Another helpful tool is to use a light to transilluminate and the calcification produces a bluish opaque hue,8 as light cannot transmit through the calcification, often differentiating it from epidermal inclusion cysts or other noncalcified lesions.
What is the differential diagnosis?
Because of the diverse clinical presentations, pilomatrixomas are frequently misdiagnosed. The percentage of correct preoperative diagnosis reported is low, varying from 16% to 43% in different series.1,9-11 They most frequently are misdiagnosed as other types of cysts such as epidermal, dermoid, or sebaceous.2,3,5,12,13 Rapidly growing pilomatrixomas can be also be misdiagnosed as malignant soft-tissue tumors, cutaneous lymphoma, or sarcomas.5,13
When presenting with a classic history and physical features, diagnosis is clinical, and no further studies are recommended.14 To improve diagnostic accuracy when encountering unusual subtypes, imaging is recommended, including ultrasound. Ultrasound adds a high positive predictive value (95.56%).2 Generally, on ultrasound a pilomatrixoma is described as an oval, well-defined, heterogeneous, hyperechoic subcutaneous mass with or without posterior shadowing.2 The definitive diagnosis is, however, made by histopathologic examination.
Pilomatrixomas do not spontaneously regress, therefore complete surgical resection is the standard treatment. During the follow-up period, very low recurrence rates have been reported, varying from 1.5% to 2% which generally occurs because of incomplete resection.2,3
Plexiform neurofibromas are usually congenital tumors of peripheral nerve sheath associated with neurofibromatosis type 1, often with a “bag of worms” feel on palpation. Epidermoid cysts generally present as dermal nodules often with a visible puncture, mobile on soft and mobile on palpation. Dermatofibromas present as firm, usually hyperpigmented papule or nodules that are fixed to subcutaneous tissue, thus often “dimpling” when pitched. Dermatofibrosarcoma protuberans is a rare soft-tissue sarcoma which presents as a firm, slow growing indurated plaques growing over months to years.
Conclusion
Pilomatrixomas are a benign adnexal tumor that sometimes can present as atypical forms such as this case. Diagnosis is usually based on clinical diagnosis, and transillumination can be a bedside clue. When the clinical diagnosis remains obscure an ultrasound can be helpful. The main aim of this case is to improve awareness of the variable presentations of pilomatrixomas and the importance of high level of suspicion supported by careful clinical evaluation.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology, University of California, San Diego. Dr. Guelfand is a visiting dermatology resident in the division of pediaric and adolescent dermatology, University of Califonia, San Diego.
References
1. Jones CD et al. Am J Dermatopathol. 2018;40:631-41.
2. Hu JL et al. Arch Craniofac Surg. 2020;21(5):288-93.
3. Adhikari G and Jadhav GS. Cureus. 2022;14(2):22228.
4. Cóbar JP et al. J Surg Case Rep. 2023;2023(4):rjad182.
5. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
6. Kose D et al. J Cancer Res Ther. 2014;10(3):549-51.
7. Sabater-Abad J et al. Dermatol Online J. 2020;26(8):13030/qt4h16s45w.
8. Alkatan HM et al. Int J Surg Case Rep. 2021;84:106068.
9. Pant I et al. Indian J Dermatol. 2010;55:390-2.
10. Kaddu S et al. Am J Dermatopathol. 1996;18(4):333-8
11. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
12. Wang YN et al. Chin Med J (Engl). 2021;134(16):2011-2.
13. Yannoutsos A et al. Am J Dermatopathol. 2018;40(9):690-3.
14. Zhao A et al. Ear Nose Throat J. 2021;1455613211044778.
Given the characteristic clinical presentation, the most likely diagnosis is pilomatrixoma.
Pilomatrixomas are benign adnexal tumors that arise from immature matrix cells of the hair follicles located on dermal or subcutaneous tissue.
The cause of pilomatrixoma remains unclear. Recent studies have suggested that the development of pilomatrixoma are related to mutations in the Wnt signaling pathway, where beta-catenin gene (CTNNB1) mutation is the most frequently reported.1-4
Pilomatrixomas are more common in children and often present before 10 years of age.1,2,5 They commonly appear in head and neck, as well as upper extremities, trunk, and lower extremities.2,6
The clinical manifestations of pilomatrixomas are diverse and according to their appearance five classic clinical types are described: mass, pigmented, mixed, ulcerated, and keloid-like.2,3 The mass type is the predominant form, where it generally presents as a hard and freely mobile nodule covered by skin that may present a firm calcified protruding nodule. Other less common types include: lymphangiectasic, anetodermic, perforating, and bullous.2,6,7
Pilomatrixomas are mostly solitary, whereas multiple forms are reported to be associated with familial inheritance or syndromic conditions, such as myotonic dystrophy, Gardner syndrome, Turner’s syndrome, and Rubinstein-Taybi syndrome.2-4 However, children and adolescents occasionally present with multiple pilomatricomas with no associated syndrome.
On physical exam a helpful features for the diagnosis is the “teeter-totter sign,” which can be illustrated by pressing on one edge of the lesion that will cause the opposite edge to protrude from the skin. Another helpful tool is to use a light to transilluminate and the calcification produces a bluish opaque hue,8 as light cannot transmit through the calcification, often differentiating it from epidermal inclusion cysts or other noncalcified lesions.
What is the differential diagnosis?
Because of the diverse clinical presentations, pilomatrixomas are frequently misdiagnosed. The percentage of correct preoperative diagnosis reported is low, varying from 16% to 43% in different series.1,9-11 They most frequently are misdiagnosed as other types of cysts such as epidermal, dermoid, or sebaceous.2,3,5,12,13 Rapidly growing pilomatrixomas can be also be misdiagnosed as malignant soft-tissue tumors, cutaneous lymphoma, or sarcomas.5,13
When presenting with a classic history and physical features, diagnosis is clinical, and no further studies are recommended.14 To improve diagnostic accuracy when encountering unusual subtypes, imaging is recommended, including ultrasound. Ultrasound adds a high positive predictive value (95.56%).2 Generally, on ultrasound a pilomatrixoma is described as an oval, well-defined, heterogeneous, hyperechoic subcutaneous mass with or without posterior shadowing.2 The definitive diagnosis is, however, made by histopathologic examination.
Pilomatrixomas do not spontaneously regress, therefore complete surgical resection is the standard treatment. During the follow-up period, very low recurrence rates have been reported, varying from 1.5% to 2% which generally occurs because of incomplete resection.2,3
Plexiform neurofibromas are usually congenital tumors of peripheral nerve sheath associated with neurofibromatosis type 1, often with a “bag of worms” feel on palpation. Epidermoid cysts generally present as dermal nodules often with a visible puncture, mobile on soft and mobile on palpation. Dermatofibromas present as firm, usually hyperpigmented papule or nodules that are fixed to subcutaneous tissue, thus often “dimpling” when pitched. Dermatofibrosarcoma protuberans is a rare soft-tissue sarcoma which presents as a firm, slow growing indurated plaques growing over months to years.
Conclusion
Pilomatrixomas are a benign adnexal tumor that sometimes can present as atypical forms such as this case. Diagnosis is usually based on clinical diagnosis, and transillumination can be a bedside clue. When the clinical diagnosis remains obscure an ultrasound can be helpful. The main aim of this case is to improve awareness of the variable presentations of pilomatrixomas and the importance of high level of suspicion supported by careful clinical evaluation.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology, University of California, San Diego. Dr. Guelfand is a visiting dermatology resident in the division of pediaric and adolescent dermatology, University of Califonia, San Diego.
References
1. Jones CD et al. Am J Dermatopathol. 2018;40:631-41.
2. Hu JL et al. Arch Craniofac Surg. 2020;21(5):288-93.
3. Adhikari G and Jadhav GS. Cureus. 2022;14(2):22228.
4. Cóbar JP et al. J Surg Case Rep. 2023;2023(4):rjad182.
5. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
6. Kose D et al. J Cancer Res Ther. 2014;10(3):549-51.
7. Sabater-Abad J et al. Dermatol Online J. 2020;26(8):13030/qt4h16s45w.
8. Alkatan HM et al. Int J Surg Case Rep. 2021;84:106068.
9. Pant I et al. Indian J Dermatol. 2010;55:390-2.
10. Kaddu S et al. Am J Dermatopathol. 1996;18(4):333-8
11. Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016;85:148-53.
12. Wang YN et al. Chin Med J (Engl). 2021;134(16):2011-2.
13. Yannoutsos A et al. Am J Dermatopathol. 2018;40(9):690-3.
14. Zhao A et al. Ear Nose Throat J. 2021;1455613211044778.

On physical exam there was a well-circumscribed skin-colored nodule measuring 3.1 x 3 cm that was tender on palpation. The nodule was mobile, with a firm, stony feel, and no punctum was visualized. Transillumination revealed a subtle bluish hue within the nodule.
A 7-year-old male has a bumpy rash on the chin for several months
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
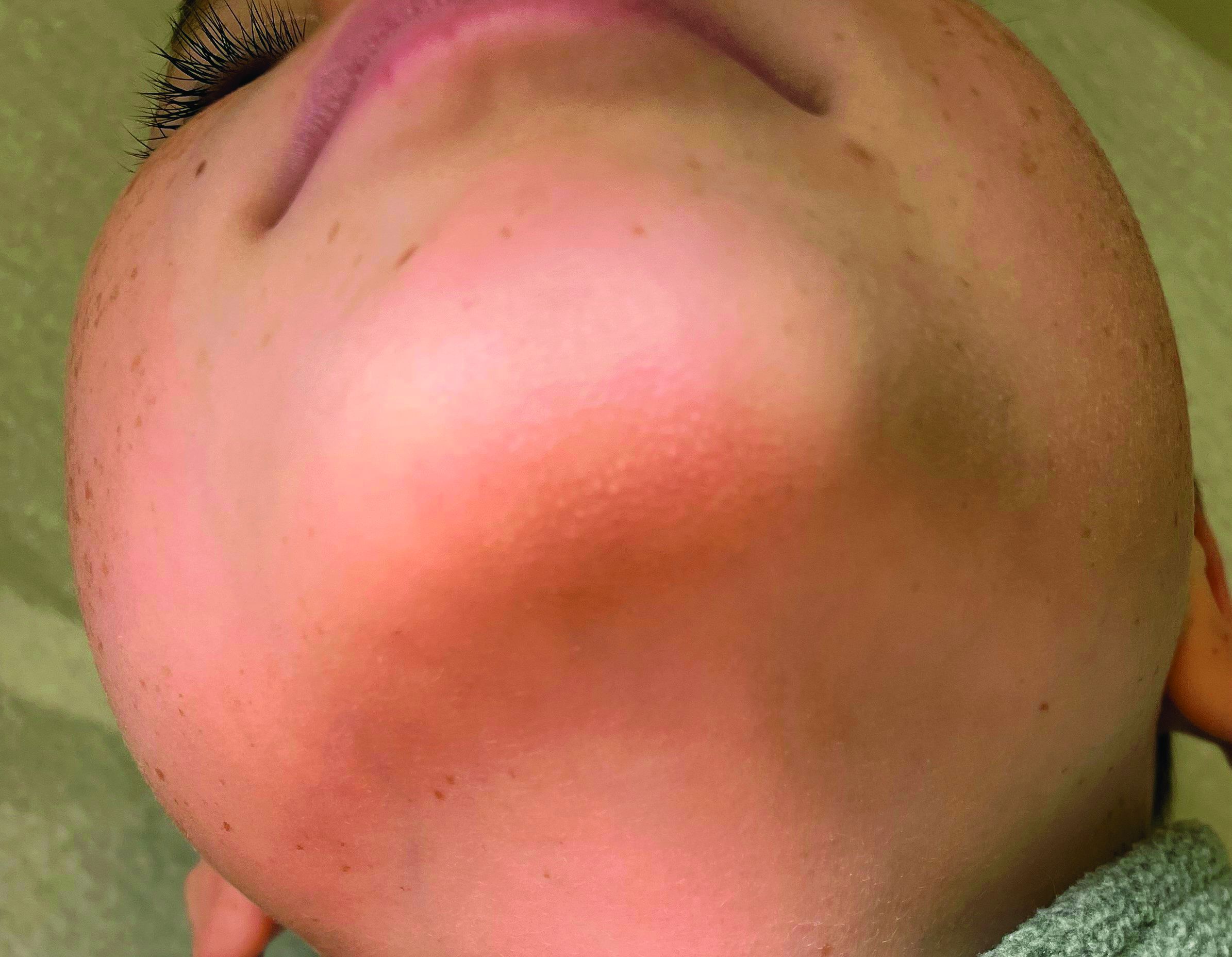
He is a healthy child with no past medical history. He is not taking any medications.
On physical exam he has follicular hyperkeratotic papules on the chin. No lesions on the axilla or thighs.
Scalp Nodule Associated With Hair Loss
The Diagnosis: Alopecic and Aseptic Nodule of the Scalp
Alopecic and aseptic nodule of the scalp (AANS) is an underdiagnosed condition presenting with one or few inflammatory nodules on the scalp with overlying nonscarring alopecia. The nodules can be soft, fluctuant, or firm and are characterized by negative fungal and bacterial stains as well as cultures.1 Trichoscopic features such as black or yellow dots, fine vellus hairs, and broken hairs have been reported.1-3 Dilated follicular openings may be seen and are termed the Eastern pancake sign, as they resemble the bubble cavities formed during the cooking of atayef.2 The histologic features of AANS often are nonspecific but show a nodular or pseudocystic, lymphohistiocytic to acute inflammatory component centered in the dermis.1 Granulomatous inflammation or isolated giant cells have been reported within the deep dermis.1,4 In our patient, histopathology revealed admixed acute and granulomatous inflammation within the deep dermis (Figure). Treatment of AANS includes oral antibiotics such as doxycycline, intralesional corticosteroids, or excision.1
. B, Admixed acute and granulomatous inflammation was present")
Although the etiology of AANS currently is unclear, a process of follicular plugging or a deep folliculitis sparing the bulge stem cells has been theorized. Young males are disproportionately affected.1 It is uncertain how much overlap there is, if any, between AANS and pseudocyst of the scalp, the latter of which primarily is reported in the Japanese literature and demonstrates alopecic nodules between the forehead and vertex of the scalp with pseudocystic architecture and granulomatous infiltration on histopathology.4-7
There are several clinical and histologic differences between AANS and other diagnoses in the differential. Dermoid cysts tend to present at birth, with 70% of cases presenting before the age of 6 years, and without overlying skin changes.8 They represent a benign entrapment of ectoderm along embryonic closure lines during development.9 Histologic examination typically will show a squamous-lined cyst within the dermis with associated adnexal structures.10 Cylindromas are benign neoplasms of eccrine sweat glands named after the histologic presentation of cylinder-shaped basaloid cell populations when cross-sectioned.11,12 When cylindromas coalesce on the scalp, they form a distinctive morphology sometimes loosely resembling a turban, giving them the previously more common name turban tumors.11,13 Cylindromas appear as slow-growing protuberant tumors that are erythematous or flesh colored. Cylindromas are 9 times more common in females.13 Pilar cysts have a stratified squamous epithelium lining with a palisaded outer layer and are derived from the outer root sheath of hair follicles.14 Clinically, pilar cysts are smooth mobile cysts that favor skin with a dense concentration of hair follicles.14,15 On palpation, pilar cysts are firm due to their keratinous contents and typically are nontender unless inflamed.15 Lipomas are benign mesenchymal tumors with mature adipocytes that often appear as subcutaneous nodules without overlying skin changes, though they can involve deep fascia. On palpation, lipomas generally are soft, mobile, and nontender.16
- Bellinato F, Maurelli M, Colato C, et al. Alopecic and aseptic nodules of the scalp: a new case with a systematic review of the literature [published online May 1, 2021]. Clin Case Rep. 2021;9:E04153. doi:10.1002/ccr3.4153
- Lázaro-Simó AI, Sancho MI, Quintana-Codina M, et al. Alopecic and aseptic nodules of the scalp with trichoscopic and ultrasonographic findings. Indian J Dermatol. 2017;62:515-518.
- Garrido-Colmenero C, Arias-Santiago S, Aneiros Fernández J, et al. Trichoscopy and ultrasonography features of aseptic and alopecic nodules of the scalp. J Eur Acad Dermatol Venereol. 2016;30:507-509. doi:10.1111/jdv.12903
- Seol JE, Park IH, Kim DH, et al. Alopecic and aseptic nodules of the scalp/pseudocyst of the scalp: clinicopathological and therapeutic analyses in 11 Korean patients. Dermatology. 2016;232:165-170.
- Lee SS, Kim SY, Im M, et al. Pseudocyst of the scalp. Ann Dermatol. 2011;23(suppl 2):S267-S269.
- Eisenberg EL. Alopecia-associated pseudocyst of the scalp. J Am Acad Dermatol. 2012;67:E114-E116.
- Tsuruta D, Hayashi A, Kobayashi H, et al. Pseudocyst of the scalp. Dermatology. 2005;210:333-335.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Julapalli MR, Cohen BA, Hollier LH, et al. Congenital, ill-defined, yellowish plaque: the nasal dermoid. Pediatr Dermatol. 2006;23:556-559.
- Reissis D, Pfaff MJ, Patel A, et al. Craniofacial dermoid cysts: histological analysis and inter-site comparison. Yale J Biol Med. 2014;87:349-357.
- Chauhan DS, Guruprasad Y. Dermal cylindroma of the scalp. Natl J Maxillofac Surg. 2012;3:59-61.
- Albores-Saavedra J, Heard SC, McLaren B, et al. Cylindroma (dermal analog tumor) of the breast: a comparison with cylindroma of the skin and adenoid cystic carcinoma of the breast. Am J Clin Pathol. 2005;123:866-873.
- Myers DJ, Fillman EP. Cylindroma. StatPearls. StatPearls Publishing; 2022.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128. doi:10.4103/1947-2714.107532
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2022. 16. Kolb L, Yarrarapu SNS, Ameer MA, et al. Lipoma. StatPearls. StatPearls Publishing; 2022.
The Diagnosis: Alopecic and Aseptic Nodule of the Scalp
Alopecic and aseptic nodule of the scalp (AANS) is an underdiagnosed condition presenting with one or few inflammatory nodules on the scalp with overlying nonscarring alopecia. The nodules can be soft, fluctuant, or firm and are characterized by negative fungal and bacterial stains as well as cultures.1 Trichoscopic features such as black or yellow dots, fine vellus hairs, and broken hairs have been reported.1-3 Dilated follicular openings may be seen and are termed the Eastern pancake sign, as they resemble the bubble cavities formed during the cooking of atayef.2 The histologic features of AANS often are nonspecific but show a nodular or pseudocystic, lymphohistiocytic to acute inflammatory component centered in the dermis.1 Granulomatous inflammation or isolated giant cells have been reported within the deep dermis.1,4 In our patient, histopathology revealed admixed acute and granulomatous inflammation within the deep dermis (Figure). Treatment of AANS includes oral antibiotics such as doxycycline, intralesional corticosteroids, or excision.1
Although the etiology of AANS currently is unclear, a process of follicular plugging or a deep folliculitis sparing the bulge stem cells has been theorized. Young males are disproportionately affected.1 It is uncertain how much overlap there is, if any, between AANS and pseudocyst of the scalp, the latter of which primarily is reported in the Japanese literature and demonstrates alopecic nodules between the forehead and vertex of the scalp with pseudocystic architecture and granulomatous infiltration on histopathology.4-7
There are several clinical and histologic differences between AANS and other diagnoses in the differential. Dermoid cysts tend to present at birth, with 70% of cases presenting before the age of 6 years, and without overlying skin changes.8 They represent a benign entrapment of ectoderm along embryonic closure lines during development.9 Histologic examination typically will show a squamous-lined cyst within the dermis with associated adnexal structures.10 Cylindromas are benign neoplasms of eccrine sweat glands named after the histologic presentation of cylinder-shaped basaloid cell populations when cross-sectioned.11,12 When cylindromas coalesce on the scalp, they form a distinctive morphology sometimes loosely resembling a turban, giving them the previously more common name turban tumors.11,13 Cylindromas appear as slow-growing protuberant tumors that are erythematous or flesh colored. Cylindromas are 9 times more common in females.13 Pilar cysts have a stratified squamous epithelium lining with a palisaded outer layer and are derived from the outer root sheath of hair follicles.14 Clinically, pilar cysts are smooth mobile cysts that favor skin with a dense concentration of hair follicles.14,15 On palpation, pilar cysts are firm due to their keratinous contents and typically are nontender unless inflamed.15 Lipomas are benign mesenchymal tumors with mature adipocytes that often appear as subcutaneous nodules without overlying skin changes, though they can involve deep fascia. On palpation, lipomas generally are soft, mobile, and nontender.16
The Diagnosis: Alopecic and Aseptic Nodule of the Scalp
Alopecic and aseptic nodule of the scalp (AANS) is an underdiagnosed condition presenting with one or few inflammatory nodules on the scalp with overlying nonscarring alopecia. The nodules can be soft, fluctuant, or firm and are characterized by negative fungal and bacterial stains as well as cultures.1 Trichoscopic features such as black or yellow dots, fine vellus hairs, and broken hairs have been reported.1-3 Dilated follicular openings may be seen and are termed the Eastern pancake sign, as they resemble the bubble cavities formed during the cooking of atayef.2 The histologic features of AANS often are nonspecific but show a nodular or pseudocystic, lymphohistiocytic to acute inflammatory component centered in the dermis.1 Granulomatous inflammation or isolated giant cells have been reported within the deep dermis.1,4 In our patient, histopathology revealed admixed acute and granulomatous inflammation within the deep dermis (Figure). Treatment of AANS includes oral antibiotics such as doxycycline, intralesional corticosteroids, or excision.1
Although the etiology of AANS currently is unclear, a process of follicular plugging or a deep folliculitis sparing the bulge stem cells has been theorized. Young males are disproportionately affected.1 It is uncertain how much overlap there is, if any, between AANS and pseudocyst of the scalp, the latter of which primarily is reported in the Japanese literature and demonstrates alopecic nodules between the forehead and vertex of the scalp with pseudocystic architecture and granulomatous infiltration on histopathology.4-7
There are several clinical and histologic differences between AANS and other diagnoses in the differential. Dermoid cysts tend to present at birth, with 70% of cases presenting before the age of 6 years, and without overlying skin changes.8 They represent a benign entrapment of ectoderm along embryonic closure lines during development.9 Histologic examination typically will show a squamous-lined cyst within the dermis with associated adnexal structures.10 Cylindromas are benign neoplasms of eccrine sweat glands named after the histologic presentation of cylinder-shaped basaloid cell populations when cross-sectioned.11,12 When cylindromas coalesce on the scalp, they form a distinctive morphology sometimes loosely resembling a turban, giving them the previously more common name turban tumors.11,13 Cylindromas appear as slow-growing protuberant tumors that are erythematous or flesh colored. Cylindromas are 9 times more common in females.13 Pilar cysts have a stratified squamous epithelium lining with a palisaded outer layer and are derived from the outer root sheath of hair follicles.14 Clinically, pilar cysts are smooth mobile cysts that favor skin with a dense concentration of hair follicles.14,15 On palpation, pilar cysts are firm due to their keratinous contents and typically are nontender unless inflamed.15 Lipomas are benign mesenchymal tumors with mature adipocytes that often appear as subcutaneous nodules without overlying skin changes, though they can involve deep fascia. On palpation, lipomas generally are soft, mobile, and nontender.16
- Bellinato F, Maurelli M, Colato C, et al. Alopecic and aseptic nodules of the scalp: a new case with a systematic review of the literature [published online May 1, 2021]. Clin Case Rep. 2021;9:E04153. doi:10.1002/ccr3.4153
- Lázaro-Simó AI, Sancho MI, Quintana-Codina M, et al. Alopecic and aseptic nodules of the scalp with trichoscopic and ultrasonographic findings. Indian J Dermatol. 2017;62:515-518.
- Garrido-Colmenero C, Arias-Santiago S, Aneiros Fernández J, et al. Trichoscopy and ultrasonography features of aseptic and alopecic nodules of the scalp. J Eur Acad Dermatol Venereol. 2016;30:507-509. doi:10.1111/jdv.12903
- Seol JE, Park IH, Kim DH, et al. Alopecic and aseptic nodules of the scalp/pseudocyst of the scalp: clinicopathological and therapeutic analyses in 11 Korean patients. Dermatology. 2016;232:165-170.
- Lee SS, Kim SY, Im M, et al. Pseudocyst of the scalp. Ann Dermatol. 2011;23(suppl 2):S267-S269.
- Eisenberg EL. Alopecia-associated pseudocyst of the scalp. J Am Acad Dermatol. 2012;67:E114-E116.
- Tsuruta D, Hayashi A, Kobayashi H, et al. Pseudocyst of the scalp. Dermatology. 2005;210:333-335.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Julapalli MR, Cohen BA, Hollier LH, et al. Congenital, ill-defined, yellowish plaque: the nasal dermoid. Pediatr Dermatol. 2006;23:556-559.
- Reissis D, Pfaff MJ, Patel A, et al. Craniofacial dermoid cysts: histological analysis and inter-site comparison. Yale J Biol Med. 2014;87:349-357.
- Chauhan DS, Guruprasad Y. Dermal cylindroma of the scalp. Natl J Maxillofac Surg. 2012;3:59-61.
- Albores-Saavedra J, Heard SC, McLaren B, et al. Cylindroma (dermal analog tumor) of the breast: a comparison with cylindroma of the skin and adenoid cystic carcinoma of the breast. Am J Clin Pathol. 2005;123:866-873.
- Myers DJ, Fillman EP. Cylindroma. StatPearls. StatPearls Publishing; 2022.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128. doi:10.4103/1947-2714.107532
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2022. 16. Kolb L, Yarrarapu SNS, Ameer MA, et al. Lipoma. StatPearls. StatPearls Publishing; 2022.
- Bellinato F, Maurelli M, Colato C, et al. Alopecic and aseptic nodules of the scalp: a new case with a systematic review of the literature [published online May 1, 2021]. Clin Case Rep. 2021;9:E04153. doi:10.1002/ccr3.4153
- Lázaro-Simó AI, Sancho MI, Quintana-Codina M, et al. Alopecic and aseptic nodules of the scalp with trichoscopic and ultrasonographic findings. Indian J Dermatol. 2017;62:515-518.
- Garrido-Colmenero C, Arias-Santiago S, Aneiros Fernández J, et al. Trichoscopy and ultrasonography features of aseptic and alopecic nodules of the scalp. J Eur Acad Dermatol Venereol. 2016;30:507-509. doi:10.1111/jdv.12903
- Seol JE, Park IH, Kim DH, et al. Alopecic and aseptic nodules of the scalp/pseudocyst of the scalp: clinicopathological and therapeutic analyses in 11 Korean patients. Dermatology. 2016;232:165-170.
- Lee SS, Kim SY, Im M, et al. Pseudocyst of the scalp. Ann Dermatol. 2011;23(suppl 2):S267-S269.
- Eisenberg EL. Alopecia-associated pseudocyst of the scalp. J Am Acad Dermatol. 2012;67:E114-E116.
- Tsuruta D, Hayashi A, Kobayashi H, et al. Pseudocyst of the scalp. Dermatology. 2005;210:333-335.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Julapalli MR, Cohen BA, Hollier LH, et al. Congenital, ill-defined, yellowish plaque: the nasal dermoid. Pediatr Dermatol. 2006;23:556-559.
- Reissis D, Pfaff MJ, Patel A, et al. Craniofacial dermoid cysts: histological analysis and inter-site comparison. Yale J Biol Med. 2014;87:349-357.
- Chauhan DS, Guruprasad Y. Dermal cylindroma of the scalp. Natl J Maxillofac Surg. 2012;3:59-61.
- Albores-Saavedra J, Heard SC, McLaren B, et al. Cylindroma (dermal analog tumor) of the breast: a comparison with cylindroma of the skin and adenoid cystic carcinoma of the breast. Am J Clin Pathol. 2005;123:866-873.
- Myers DJ, Fillman EP. Cylindroma. StatPearls. StatPearls Publishing; 2022.
- Ramaswamy AS, Manjunatha HK, Sunilkumar B, et al. Morphological spectrum of pilar cysts. N Am J Med Sci. 2013;5:124-128. doi:10.4103/1947-2714.107532
- Al Aboud DM, Yarrarapu SNS, Patel BC. Pilar cyst. StatPearls. StatPearls Publishing; 2022. 16. Kolb L, Yarrarapu SNS, Ameer MA, et al. Lipoma. StatPearls. StatPearls Publishing; 2022.
A 9-year-old boy presented with a soft subcutaneous nodule with overlying alopecia on the right parietal scalp of 5 months’ duration that had grown in size, became increasingly alopecic, and was complicated by intermittent pain. An excisional biopsy of the nodule revealed deep dermal mixed inflammation with scattered granulomas. No foreign material, definitive cystic spaces, or cyst wall lining was identified. Special stains including periodic acid– Schiff, Fite acid-fast, and Twort Gram were negative for infectious organisms. His postoperative course was uneventful, and no recurrence of the nodule was reported.


Scattered Red-Brown, Centrally Violaceous, Blanching Papules on an Infant
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
, neutrophils, and macrophages in the deep dermis (H&E, original magnification ×200).")
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
The Diagnosis: Neonatal-Onset Multisystem Inflammatory Disorder (NOMID)
The punch biopsy demonstrated a predominantly deep but somewhat superficial, periadnexal, neutrophilic and eosinophilic infiltrate (Figure). The eruption resolved 3 days later with supportive treatment, including appropriate wound care. Genetic analysis revealed an autosomal-dominant NLR family pyrin domain containing 3 gene, NLRP3, de novo variant associated with neonatal-onset multisystem inflammatory disorder (NOMID). Additional workup to characterize our patient’s inflammatory profile revealed elevated IL-18, CD3, CD4, S100A12, and S100A8/A9 levels. On day 48 of life, she was started on anakinra, an IL-1 inhibitor, at a dose of 1 mg/kg subcutaneously, which eventually was titrated to 10 mg/kg at hospital discharge. Hearing screenings were within normal limits.
Cryopyrin-associated periodic syndromes (CAPS) consist of 3 rare, IL-1–associated, autoinflammatory disorders, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and NOMID (also known as chronic infantile neurologic cutaneous and articular syndrome). These conditions result from a sporadic or autosomal-dominant gain-of-function mutations in a single gene, NLRP3, on chromosome 1q44. NLRP3 encodes for cryopyrin, an important component of an IL-1 and IL-18 activating inflammasome.1 The most severe manifestation of CAPS is NOMID, which typically presents at birth as a migratory urticarial eruption, growth failure, myalgia, fever, and abnormal facial features, including frontal bossing, saddle-shaped nose, and protruding eyes.2 The illness also can manifest with hepatosplenomegaly, lymphadenopathy, uveitis, sensorineural hearing loss, cerebral atrophy, and other neurologic manifestations.3 A diagnosis of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome was less likely given that our patient remained afebrile and did not show signs of lipodystrophy and persistent violaceous eyelid swelling. Both FCAS and MWS are less severe forms of CAPS when compared to NOMID. Familial cold autoinflammatory syndrome was less likely given the absence of the typical periodic fever pattern associated with the condition and severity of our patient’s symptoms. Muckle-Wells syndrome typically presents in adolescence with symptoms of FCAS, painful urticarial plaques, and progressive sensorinueral hearing loss. Tumor necrosis factor receptor–associated periodic fever (TRAPS) usually is associated with episodic fevers, abdominal pain, periorbital edema, migratory erythema, and arthralgia.1,3,4
Diagnostic criteria for CAPS include elevated inflammatory markers and serum amyloid, plus at least 2 of the typical CAPS symptoms: urticarial rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis, and skeletal abnormalities.4 The sensitivity and specificity of these diagnostic criteria are 84% and 91%, respectively. Additional findings that can be seen but are not part of the diagnostic criteria include intermittent fever, transient joint swelling, bony overgrowths, uveitis, optic disc edema, impaired growth, and hepatosplenomegaly.5 Laboratory findings may reveal leukocytosis, eosinophilia, anemia, and/or thrombocytopenia.3,5
Genetic testing, skin biopsies, ophthalmic examinations, neuroimaging, joint radiography, cerebrospinal fluid tests, and hearing examinations can be performed for confirmation of diagnosis and evaluation of systemic complications.4 A skin biopsy may reveal a neutrophilic infiltrate. Ophthalmic examination can demonstrate uveitis and optic disk edema. Neuroimaging may reveal cerebral atrophy or ventricular dilation. Lastly, joint radiography can be used to evaluate for the presence of premature long bone ossification or osseous overgrowth.4
In summary, NOMID is a multisystemic disorder with cutaneous manifestations. Early recognition of this entity is important given the severe sequelae and available efficacious therapy. Dermatologists should be aware of these manifestations, as dermatologic consultation and a skin biopsy may aid in diagnosis.
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31:596-609. doi:10.1016/j.berh.2017.12.001
- Hull KM, Shoham N, Jin Chae J, et al. The expanding spectrum of systemic autoinflammatory disorders and their rheumatic manifestations. Curr Opin Rheumatol. 2003;15:61-69. doi:10.1097/00002281-200301000-00011
- Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295-302. doi:10.1177/0194599811402296
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76:942-947. doi:10.1136/annrheumdis-2016-209686
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrinassociated autoinflammatory diseases. Arthritis Rheum. 2002; 46:3340-3348. doi:10.1002/art.10688
A 2-week-old infant girl was transferred to a specialty pediatric hospital where dermatology was consulted for evaluation of a diffuse eruption triggered by cold that was similar to an eruption present at birth. She was born at 31 weeks and 2 days’ gestation at an outside hospital via caesarean delivery. Early delivery was prompted by superimposed pre-eclampsia with severe hypertension after administration of antenatal steroids. At birth, the infant was cyanotic and apneic and had a documented skin eruption, according to the medical record. She had thrombocytopenia, elevated C-reactive protein, and an elevated temperature without fever. Extensive septic workup, including blood, urine, and cerebrospinal fluid cultures; herpes simplex virus and cytomegalovirus screening; and Toxoplasma polymerase chain reaction were negative. Magnetic resonance imaging of the brain revealed no evidence of intracranial congenital infection. Ampicillinsulbactam was initiated for presumed culture-negative sepsis. On day 2 of hospitalization, she developed conjunctival icterus, hepatomegaly, and jaundice. Direct hyperbilirubinemia; anemia; and elevated triglycerides, ferritin, and ammonia all were present. Coagulation studies were normal. Subsequent workup, including abdominal ultrasonography and hepatobiliary iminodiacetic acid scan, was concerning for biliary atresia. Despite appropriate treatment, her condition did not improve and she was transferred. Repeat abdominal ultrasonography on day 24 of life confirmed hepatomegaly but did not demonstrate other findings of biliary atresia. At the current presentation, physical examination revealed many scattered, redbrown and centrally violaceous, blanching papules measuring a few millimeters involving the trunk, arms, buttocks, and legs. A punch biopsy was obtained.


A 7-month-old male presents with pustules and inflamed papules on the scalp and extremities
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).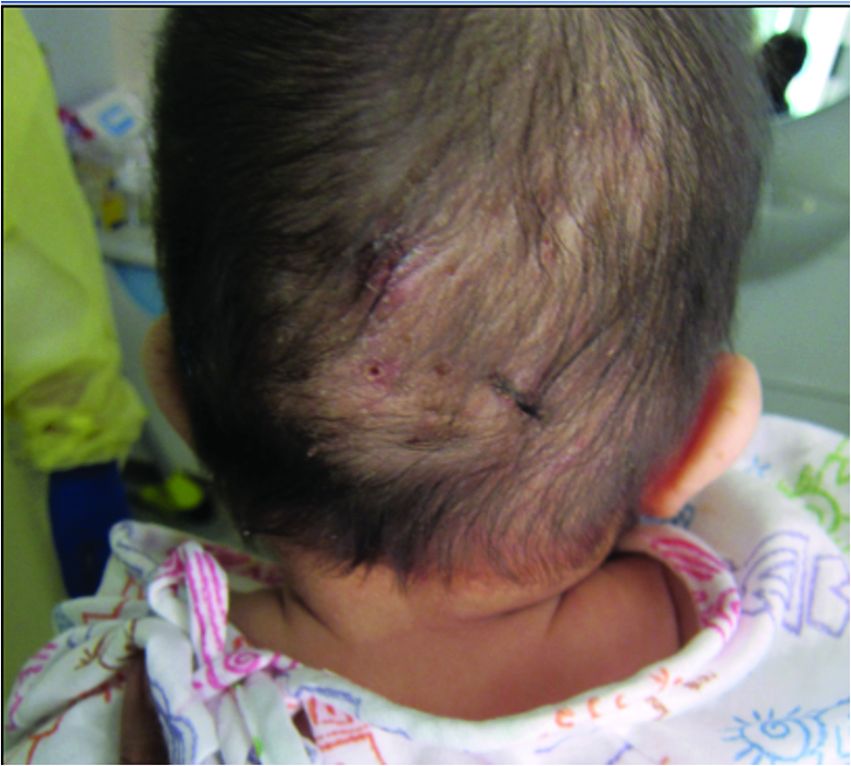
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
The bacterial, fungal, and atypical mycobacterial cultures from the lesions performed at the emergency department were all negative.
Pediatric dermatology was consulted and a punch biopsy of one of the lesions was done. Histopathologic examination showed a mixed perifollicular infiltrate of predominantly eosinophils with some neutrophils and associated microabscesses. Periodic acid Schiff and Fite stains failed to reveal any organisms. CD1 immunostain was negative. Fresh tissue cultures for bacteria, fungi, and atypical mycobacteria were negative.
Given the clinical presentation of chronic recurrent sterile pustules on an infant with associated eosinophilia and the reported histopathologic findings, the patient was diagnosed with eosinophilic pustular folliculitis of infancy (EPFI).
EPFI is a rare and idiopathic cutaneous disorder present in children. About 70% of the cases reported occur in the first 6 month of life and rarely present past 3 years of age. EPF encompasses a group of conditions including the classic adult form, or Ofuji disease. EPF is seen in immunosuppressed patients, mainly HIV positive, and EPF is also seen in infants and children.
In EPFI, males are most commonly affected. The condition presents, as it did in our patient, with recurrent crops of sterile papules and pustules mainly on the scalp, but they can occur in other parts of the body. The lesions go away within a few weeks to months without leaving any scars but it can take months to years to resolve. Histopathologic analysis of the lesions show an eosinophilic infiltrate which can be follicular, perifollicular, or periadnexal with associated flame figures in about 26% of cases.
Aggressive treatment is usually not needed as lesions are self-limited. Lesions can be treated with topical corticosteroids and oral antihistamine medications like cetirizine if symptomatic.
If the lesions start to present during the neonatal period, one may consider in the differential diagnosis, neonatal rashes like transient neonatal pustular melanosis and erythema toxicum neonatorum. Both of these neonatal conditions tend to resolve in the first month of life, compared with EPFI where lesions can come and go for months to years. EPFI lesions can be described as pustules and inflammatory papules, as well as furuncles and vesicles. All of the lesions may be seen in one patient at one time, which will not be typical for transient neonatal pustular melanosis or erythema toxicum. Eosinophils can be seen in erythema toxicum but folliculitis is not present. The inflammatory infiltrate seen in transient neonatal pustular melanosis is polymorphonuclear, not eosinophilic.
Early in the presentation, infectious conditions like staphylococcal or streptococcal folliculitis, cellulitis and furunculosis, tinea capitis, atypical mycobacterial infections, herpes simplex, and parasitic infections like scabies should be considered. In young infants, empiric antibiotic treatment may be started until cultures are finalized. If there is a family history of pruritic papules and pustules, scabies should be considered. A scabies prep can be done to rule out this entity.
Langerhans cell histiocytosis can also present with pustules and papules in early infancy and also has a predilection for the scalp. When this condition is in question, a skin biopsy should be performed which shows a CD1 positive histiocytic infiltrate.
In conclusion, EPFI is a benign rare condition that can present in infants as recurrent pustules and papules, mainly on the scalp, which are self-limited and if symptomatic can be treated with topical corticosteroids and antihistamines.
References
Alonso-Castro L et al. Dermatol Online J. 2012 Oct 15;18(10):6.
Frølunde AS et al. Clin Case Rep. 2021 May 11;9(5):e04167.
Hernández-Martín Á et al. J Am Acad Dermatol. 2013 Jan;68(1):150-5.
A 7-month-old male is brought to the emergency department for evaluation of pustules and inflamed papules on the scalp and extremities for several weeks of duration. The parents report the lesions started about a month prior and he has already been treated with cephalexin, clindamycin, and sulfamethoxazole without any improvement. Cultures sent prior by the child's pediatrician did not reveal any fungus or bacteria. The parents report a low-grade fever for about 3 days.
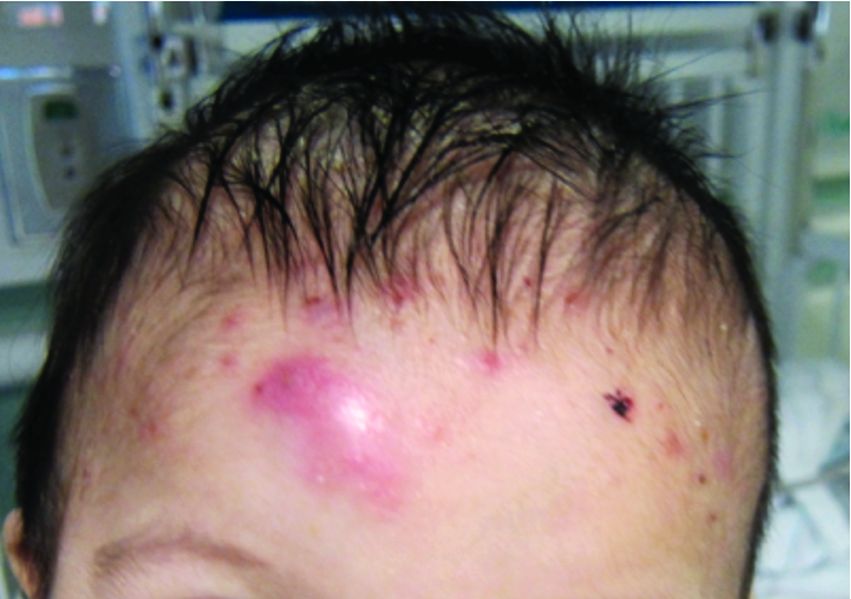
He was born via natural vaginal delivery with no instrumentation or external monitoring. Mom had prenatal care. Besides the skin lesions, the baby has been healthy and growing well. He has no history of eczema or severe infections. He has not been hospitalized before.
On physical examination the baby was not febrile. On the scalp and forehead, he had diffusely distributed pustules, erythematous papules, and nodules. He also presented with scattered, fine, small, crusted 1-2-mm pink papules on the trunk and extremities. He had no adenopathy or hepatosplenomegaly.
At the emergency department, samples from one of the pustules were sent for bacterial, fungal, and atypical mycobacteria cultures. Laboratory test showed a normal blood count with associated eosinophilia (2.8 x 109 L), and normal liver and kidney function. A head ultrasound showed three ill-defined hypoechoic foci within the scalp.
The patient was admitted for treatment with broad-spectrum antibiotics and dermatology was consulted.
A 9-year-old male presents with multiple thick scaly plaques on scalp, ears, and trunk
Given the characteristic clinical presentation, the most likely diagnosis is psoriasis.
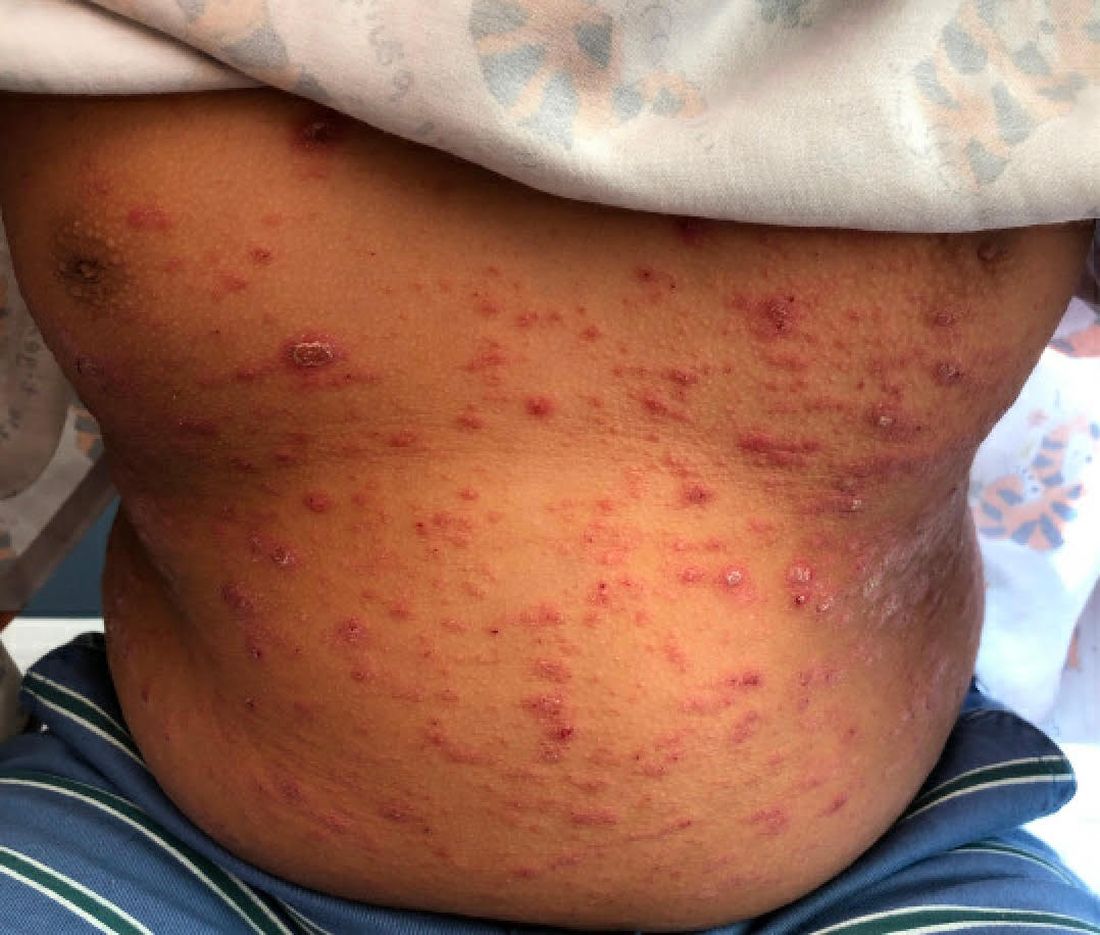
Psoriasis is a chronic immune-mediated disease that is characterized by well-demarcated thick scaly plaques on face, scalp, and intertriginous skin. Psoriasis is more common in adults than children, but the incidence of psoriasis in children has increased over time.1 Clinical presentation of psoriasis includes erythematous hyperkeratotic plaques, usually sharply demarcated. Pediatric patients may have multiple small papules and plaques less than 1 cm in size – “drop-size” – known as guttate lesions. Scalp and facial involvement are common in children. Chronic, inflamed plaques with coarse scale can involve ears, elbows, knees, and umbilicus, and nail changes can include pits, ridges, hyperkeratosis, and onycholysis or “oil spots.” While the diagnosis is clinical, biopsy can sometimes be useful to distinguish psoriasis from other papulosquamous conditions. Psoriasis in children is associated with obesity, higher rates of cardiovascular disease over a lifetime, as well as arthritis and mental health disorders.2
What’s the differential diagnosis?
The differential diagnosis for psoriasis can include papulosquamous diseases such as nummular eczema, pityriasis rosea, and pityriasis rubra pilaris. Tinea corporis may also be considered.

Nummular eczema, also known as “discoid eczema” is characterized by multiple pruritic, coin-shaped, eczematous lesions that may be actively oozing. The term “nummular” is derived from the Latin for “coin,” as lesions are distinct and annular. It is commonly associated with atopic dermatitis, and may be seen with contact dermatitis as well. Oozing, lichenification, hyperpigmentation and limited extent of skin coverage can help distinguish nummular dermatitis from psoriasis.
Pityriasis rosea is a common self-limited disease that is characterized by the appearance of acute, oval, papulosquamous patches on the trunk and proximal areas of the extremities. It usually begins with a characteristic “herald” patch, a single round or oval, sharply demarcated, pink lesion on the chest, neck, or back. Pityriasis rosea and guttate psoriasis may show similar clinical findings but the latter lacks a herald patch and is often preceded by streptococcal throat infection.
Pityriasis rubra pilaris is a rarer inflammatory disease characterized by follicular, hyperkeratotic papules, thick orange waxy palms (palmoplantar keratoderma), and erythroderma. It can also cause hair loss, nail changes, and itching. The rash shows areas with no involvement, “islands of sparing,” which is a signature characteristic of pityriasis rubra pilaris. Skin biopsies are an important diagnostic tool for pityriasis rubra pilaris. In the case of circumscribed pityriasis rubra pilaris, it may look similar to psoriasis, but it can be differentiated in that it is often accompanied by characteristic follicular papules and involvement of the palms, which are more waxy and orange in color.
When evaluating annular scaly patches, it is always important to consider tinea corporis. Tinea corporis will commonly have an annular border of scale with relative clearing in the center of lesions. In addition, when topical corticosteroids are used for prolonged periods, skin fungal infections can develop into “tinea incognito,” with paradoxical worsening since the immune response is suppressed and the fungal infection worsens.
Our patient had been previously treated with topical corticosteroids (medium to high strength) and topical calcineurin inhibitors without significant improvement. Other topical therapies for psoriasis include vitamin analogues, tazarotene, and newer therapies such as topical roflumilast (a phosphodiesterase-4 inhibitor approved for psoriasis in children over 12 years of age).3,4 In addition, as the indications for biological agents have been expanded, there are various options for treating psoriasis in children and adolescents when more active treatment is needed. Systemic therapies for more severe disease include traditional systemic immunosuppressives (for example, methotrexate, cyclosporine) and biologic agents. The four biologic agents currently approved for children are etanercept, ustekinumab, ixekizumab, and secukinumab. Our patient was treated with ustekinumab, which is an injectable biologic agent that blocks interleukin-12/23, with good response to date.
Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology; Dr. Choi is a visiting research physician in the division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice-chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California, San Diego, and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Tollefson MM et al. J Am Acad Dermatol. 2010;62(6):979-87.
2. Menter A et al. J Am Acad Dermatol. 2020;82(1):161-201.
3. Mark G et al. JAMA. 2022;328(11):1073-84.
4. Eichenfield LF et al. Pediatr Dermatol. 2018;35(2):170-81.
Given the characteristic clinical presentation, the most likely diagnosis is psoriasis.
Psoriasis is a chronic immune-mediated disease that is characterized by well-demarcated thick scaly plaques on face, scalp, and intertriginous skin. Psoriasis is more common in adults than children, but the incidence of psoriasis in children has increased over time.1 Clinical presentation of psoriasis includes erythematous hyperkeratotic plaques, usually sharply demarcated. Pediatric patients may have multiple small papules and plaques less than 1 cm in size – “drop-size” – known as guttate lesions. Scalp and facial involvement are common in children. Chronic, inflamed plaques with coarse scale can involve ears, elbows, knees, and umbilicus, and nail changes can include pits, ridges, hyperkeratosis, and onycholysis or “oil spots.” While the diagnosis is clinical, biopsy can sometimes be useful to distinguish psoriasis from other papulosquamous conditions. Psoriasis in children is associated with obesity, higher rates of cardiovascular disease over a lifetime, as well as arthritis and mental health disorders.2
What’s the differential diagnosis?
The differential diagnosis for psoriasis can include papulosquamous diseases such as nummular eczema, pityriasis rosea, and pityriasis rubra pilaris. Tinea corporis may also be considered.
Nummular eczema, also known as “discoid eczema” is characterized by multiple pruritic, coin-shaped, eczematous lesions that may be actively oozing. The term “nummular” is derived from the Latin for “coin,” as lesions are distinct and annular. It is commonly associated with atopic dermatitis, and may be seen with contact dermatitis as well. Oozing, lichenification, hyperpigmentation and limited extent of skin coverage can help distinguish nummular dermatitis from psoriasis.
Pityriasis rosea is a common self-limited disease that is characterized by the appearance of acute, oval, papulosquamous patches on the trunk and proximal areas of the extremities. It usually begins with a characteristic “herald” patch, a single round or oval, sharply demarcated, pink lesion on the chest, neck, or back. Pityriasis rosea and guttate psoriasis may show similar clinical findings but the latter lacks a herald patch and is often preceded by streptococcal throat infection.
Pityriasis rubra pilaris is a rarer inflammatory disease characterized by follicular, hyperkeratotic papules, thick orange waxy palms (palmoplantar keratoderma), and erythroderma. It can also cause hair loss, nail changes, and itching. The rash shows areas with no involvement, “islands of sparing,” which is a signature characteristic of pityriasis rubra pilaris. Skin biopsies are an important diagnostic tool for pityriasis rubra pilaris. In the case of circumscribed pityriasis rubra pilaris, it may look similar to psoriasis, but it can be differentiated in that it is often accompanied by characteristic follicular papules and involvement of the palms, which are more waxy and orange in color.
When evaluating annular scaly patches, it is always important to consider tinea corporis. Tinea corporis will commonly have an annular border of scale with relative clearing in the center of lesions. In addition, when topical corticosteroids are used for prolonged periods, skin fungal infections can develop into “tinea incognito,” with paradoxical worsening since the immune response is suppressed and the fungal infection worsens.
Our patient had been previously treated with topical corticosteroids (medium to high strength) and topical calcineurin inhibitors without significant improvement. Other topical therapies for psoriasis include vitamin analogues, tazarotene, and newer therapies such as topical roflumilast (a phosphodiesterase-4 inhibitor approved for psoriasis in children over 12 years of age).3,4 In addition, as the indications for biological agents have been expanded, there are various options for treating psoriasis in children and adolescents when more active treatment is needed. Systemic therapies for more severe disease include traditional systemic immunosuppressives (for example, methotrexate, cyclosporine) and biologic agents. The four biologic agents currently approved for children are etanercept, ustekinumab, ixekizumab, and secukinumab. Our patient was treated with ustekinumab, which is an injectable biologic agent that blocks interleukin-12/23, with good response to date.
Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology; Dr. Choi is a visiting research physician in the division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice-chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California, San Diego, and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Tollefson MM et al. J Am Acad Dermatol. 2010;62(6):979-87.
2. Menter A et al. J Am Acad Dermatol. 2020;82(1):161-201.
3. Mark G et al. JAMA. 2022;328(11):1073-84.
4. Eichenfield LF et al. Pediatr Dermatol. 2018;35(2):170-81.
Given the characteristic clinical presentation, the most likely diagnosis is psoriasis.
Psoriasis is a chronic immune-mediated disease that is characterized by well-demarcated thick scaly plaques on face, scalp, and intertriginous skin. Psoriasis is more common in adults than children, but the incidence of psoriasis in children has increased over time.1 Clinical presentation of psoriasis includes erythematous hyperkeratotic plaques, usually sharply demarcated. Pediatric patients may have multiple small papules and plaques less than 1 cm in size – “drop-size” – known as guttate lesions. Scalp and facial involvement are common in children. Chronic, inflamed plaques with coarse scale can involve ears, elbows, knees, and umbilicus, and nail changes can include pits, ridges, hyperkeratosis, and onycholysis or “oil spots.” While the diagnosis is clinical, biopsy can sometimes be useful to distinguish psoriasis from other papulosquamous conditions. Psoriasis in children is associated with obesity, higher rates of cardiovascular disease over a lifetime, as well as arthritis and mental health disorders.2
What’s the differential diagnosis?
The differential diagnosis for psoriasis can include papulosquamous diseases such as nummular eczema, pityriasis rosea, and pityriasis rubra pilaris. Tinea corporis may also be considered.
Nummular eczema, also known as “discoid eczema” is characterized by multiple pruritic, coin-shaped, eczematous lesions that may be actively oozing. The term “nummular” is derived from the Latin for “coin,” as lesions are distinct and annular. It is commonly associated with atopic dermatitis, and may be seen with contact dermatitis as well. Oozing, lichenification, hyperpigmentation and limited extent of skin coverage can help distinguish nummular dermatitis from psoriasis.
Pityriasis rosea is a common self-limited disease that is characterized by the appearance of acute, oval, papulosquamous patches on the trunk and proximal areas of the extremities. It usually begins with a characteristic “herald” patch, a single round or oval, sharply demarcated, pink lesion on the chest, neck, or back. Pityriasis rosea and guttate psoriasis may show similar clinical findings but the latter lacks a herald patch and is often preceded by streptococcal throat infection.
Pityriasis rubra pilaris is a rarer inflammatory disease characterized by follicular, hyperkeratotic papules, thick orange waxy palms (palmoplantar keratoderma), and erythroderma. It can also cause hair loss, nail changes, and itching. The rash shows areas with no involvement, “islands of sparing,” which is a signature characteristic of pityriasis rubra pilaris. Skin biopsies are an important diagnostic tool for pityriasis rubra pilaris. In the case of circumscribed pityriasis rubra pilaris, it may look similar to psoriasis, but it can be differentiated in that it is often accompanied by characteristic follicular papules and involvement of the palms, which are more waxy and orange in color.
When evaluating annular scaly patches, it is always important to consider tinea corporis. Tinea corporis will commonly have an annular border of scale with relative clearing in the center of lesions. In addition, when topical corticosteroids are used for prolonged periods, skin fungal infections can develop into “tinea incognito,” with paradoxical worsening since the immune response is suppressed and the fungal infection worsens.
Our patient had been previously treated with topical corticosteroids (medium to high strength) and topical calcineurin inhibitors without significant improvement. Other topical therapies for psoriasis include vitamin analogues, tazarotene, and newer therapies such as topical roflumilast (a phosphodiesterase-4 inhibitor approved for psoriasis in children over 12 years of age).3,4 In addition, as the indications for biological agents have been expanded, there are various options for treating psoriasis in children and adolescents when more active treatment is needed. Systemic therapies for more severe disease include traditional systemic immunosuppressives (for example, methotrexate, cyclosporine) and biologic agents. The four biologic agents currently approved for children are etanercept, ustekinumab, ixekizumab, and secukinumab. Our patient was treated with ustekinumab, which is an injectable biologic agent that blocks interleukin-12/23, with good response to date.
Dr. Al-Nabti is a clinical fellow in the division of pediatric and adolescent dermatology; Dr. Choi is a visiting research physician in the division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice-chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California, San Diego, and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Tollefson MM et al. J Am Acad Dermatol. 2010;62(6):979-87.
2. Menter A et al. J Am Acad Dermatol. 2020;82(1):161-201.
3. Mark G et al. JAMA. 2022;328(11):1073-84.
4. Eichenfield LF et al. Pediatr Dermatol. 2018;35(2):170-81.
A 9-year-old male is seen in the clinic with a 1-year history of multiple thick scaly plaques on scalp, ears, and trunk. He has been treated with hydrocortisone 1% ointment with no change in the lesions. He had upper respiratory tract symptoms 3 weeks prior to the visit.

Examination reveals erythematous, well-demarcated plaques of the anterior scalp with thick overlying micaceous scale with some extension onto the forehead and temples. Additionally, erythematous scaly patches on the ear, axilla, and umbilicus were noted. There was no palmar or plantar involvement. He denied joint swelling, stiffness, or pain in the morning.
Spreading Painful Lesions on the Legs
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
The Diagnosis: Cutaneous Leishmaniasis
A punch biopsy of the skin showed pseudoepitheliomatous hyperplasia of the epidermis with dermal granulomatous and suppurative inflammation; tissue cultures remained sterile. Polymerase chain reaction testing of the skin revealed the presence of Leishmania guyanensis complex. Leishmaniasis is a widespread parasitic disease transmitted via sandflies that often is seen in children and young adults.1 Although leishmaniasis is endemic to several countries within Southeast Asia, East Africa, and Latin America, an increase in international travel has brought the disease to nonendemic regions. Therefore, it is crucial to obtain a detailed history of travel and exposure to sandflies in patients who have recently returned from endemic regions.
Leishmaniasis may present in 3 forms: cutaneous, mucocutaneous, or visceral. Cutaneous clinical findings vary depending on disease stage, causative species, and host immune activation. Presentation following a sandfly bite typically includes a papule that progresses to an erythematous nodule. Cutaneous leishmaniasis commonly occurs in areas of the body that are easily accessible to sandflies, such as the face, neck, and limbs. Mucocutaneous leishmaniasis presents with nasal or oral involvement several years after the onset of cutaneous leishmaniasis; however, it can coexist with cutaneous involvement. Without treatment, mucocutaneous leishmaniasis may lead to perforation of the nasal septum, destruction of the mouth, and life-threatening airway obstruction.1 Determining the specific species is important due to the variation in treatment options and prognosis. Because Leishmania organisms are fastidious, obtaining a positive culture often is challenging. Polymerase chain reaction can be utilized for identification, with detection rates of 97%.1 Systemic treatment is indicated for patients with multiple or large lesions; lesions on the hands, feet, face, or joints; or immunocompromised patients. Antimonial drugs are the first-line treatment for most forms of leishmaniasis, though increasing resistance has led to a decrease in efficacy.1 Our patient ultimately was treated with 4 weeks of miltefosine 50 mg 3 times daily. She obtained full resolution of the lesions with no further treatment indicated.
Pemphigus vegetans may present with various clinical manifestations that often can lead to a delay in diagnosis. The Hallopeau subtype typically presents as pustular lesions, while the Neumann subtype may present as large vesiculobullous erosive lesions that rupture and form verrucous, crusted, vegetative plaques. The groin, inguinal folds, axillae, thighs, and flexural areas commonly are affected, but reports of nasal, vaginal, and conjunctival involvement also exist.2
Granuloma inguinale is a sexually transmitted ulcerative disease that is caused by infection with Klebsiella granulomatis. It typically is found in tropical and subtropical climates, including Australia, Brazil, India, and South Africa. The initial presentation includes a single papule or multiple papules or nodules in the genital area that progress to a painless ulcer. It can be diagnosed via biopsies or tissue smears, which will demonstrate the presence of inclusion bodies known as Donovan bodies.3
Cutaneous tuberculosis (TB) can have variable clinical presentations and may be acquired exogenously or endogenously. Cutaneous TB can be divided into 2 categories: exogenous TB caused by inoculation and endogenous TB due to direct spread or autoinoculation. Exogenous TB subtypes include tuberculous chancre and TB verrucosa cutis, while endogenous TB includes scrofuloderma, orificial TB, and lupus vulgaris.4 Patches and plaques are found in patients with lupus vulgaris and TB verrucosa cutis. Scrofuloderma, tuberculous chancre, and orificial TB can present as ulcerative or erosive lesions. Cutaneous TB infection can be diagnosed through a smear, culture, or polymerase chain reaction.4
Deep cutaneous fungal infections most commonly present in immunocompromised individuals, particularly those who are severely neutropenic and are receiving broad-spectrum systemic antimicrobial agents. Deep cutaneous fungal infections initially present as a papule and evolve into a pustule followed by a necrotic ulcer. The lesions typically are accompanied by a fever and/or vital sign abnormalities.5
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
- Pace D. Leishmaniasis [published online September 17, 2014]. J Infect. 2014;69(suppl 1):S10-S18. doi:10.1016/j.jinf.2014.07.016
- Messersmith L, Krauland K. Pemphigus vegetans. StatPearls. StatPearls Publishing; 2022.
- Ornelas J, Kiuru M, Konia T, et al. Granuloma inguinale in a 51-year-old man. Dermatol Online J. 2016;22:13030/qt52k0c4hj.
- Chen Q, Chen W, Hao F. Cutaneous tuberculosis: a great imitator. Clin Dermatol. 2019;37:192-199.
- Marcoux D, Jafarian F, Joncas V, et al. Deep cutaneous fungal infections in immunocompromised children. J Am Acad Dermatol. 2009;61:857-864.
A 14-year-old adolescent girl presented with spreading painful lesions on the legs and left forearm of 2 years’ duration. Her travel history included several countries in South and Central America, traversing the Colombian jungle on foot. Near the end of the jungle trip, she noted a skin lesion on the left forearm around the site of an insect bite. Within 1 month, the lesions spread to the legs. She was treated with topical corticosteroids without improvement. Physical examination revealed verrucous, reddish-brown plaques on the legs and left forearm. Intranasal examination revealed a red rounded lesion inside the left nostril.


An 11-year-old boy presents with small itchy bumps on the wrists, face, arms, and legs
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.
Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.
Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
The patient was diagnosed with lichen nitidus, given the characteristic clinical presentation.
Lichen nitidus is a rare chronic inflammatory condition of the skin that most commonly presents in children and young adults and does not seem to be restricted to any sex or race. The classic lesions are described as asymptomatic to slightly pruritic, small (1 mm), skin-colored to hypopigmented flat-topped papules.
Koebner phenomenon is usually seen in which the skin lesions appear in areas of traumatized healthy skin. The extremities, abdomen, chest, and penis are common locations for the lesions to occur. Rarely, the oral mucosa or nails can be involved. It has been described in patients with a diagnosis of Crohn’s disease, Niemann-Pick disease, Down syndrome, and HIV. The rare, generalized purpuric variant has been reported in a few cases associated with interferon and ribavirin treatment for hepatitis C infection and nivolumab treatment for cancer. The pathophysiology of lichen nitidus is unknown.
Lichen nitidus can occur in the presence of other skin conditions like lichen planus, atopic dermatitis, vitiligo, erythema nodosum, and lichen spinulosus. Histopathologic characteristics of lichen nitidus are described as a “ball and claw” of epidermal rete around a lymphohistiocytic infiltrate. Parakeratosis overlying epidermal atrophy and focal basal liquefaction degeneration is also seen.
The differential diagnosis of lichen nitidus includes flat warts, which can present as clusters of small flat-topped papules that can show a pseudo-Koebner phenomenon (where the virus is seeded in traumatized skin). The morphological difference between the condition is that lichen nitidus lesions are usually monomorphic, compared with flat warts, which usually present with different sizes and shapes.
Patients with a history of allergic contact dermatitis may present with a generalized monomorphic eruption of skin-colored papules (known as ID reaction) that can sometimes be very similar to lichen nitidus. Allergic contact dermatitis tends to respond fairly quickly to topical or systemic corticosteroids, unlike lichen nitidus. There are a few reports that consider lichen nitidus to be a variant of lichen planus, although they have different histopathologic findings. Lichen planus lesions are described as polygonal, pruritic, purple to pink papules most commonly seen on the wrists, lower back, and ankles. Lichen planus can be seen in patients with hepatitis C and may also occur secondary to medication.
Milia are small keratin cysts on the skin that are commonly seen in babies as primary milia and can be seen in older children secondary to trauma (commonly on the eyelids) or medications. Given their size and monomorphic appearance, they can sometimes be confused with lichen nitidus.
Lichen nitidus is often asymptomatic and the lesions resolve within a few months to years. Topical corticosteroids can be helpful to alleviate the symptoms in patients who present with pruritus. In more persistent and generalized cases, phototherapy, systemic corticosteroids, acitretin, isotretinoin, or cyclosporine can be considered.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Chu J and Lam JM. CMAJ. 2014 Dec 9;186(18):E688.
Lestringant G et al. Dermatology 1996;192:171-3.
Peterson JA et al. Proc (Bayl Univ Med Cent). 2021 Aug 25;35(1):70-2.
Schwartz C and Goodman MB. “Lichen nitidus,” in StatPearls. Treasure Island, Fla.: StatPearls Publishing, 2022.
An 11-year-old male with a prior history of atopic dermatitis as a young child, presents with 6 months of slightly itchy, small bumps on the wrists, face, arms, and legs. Has been treated with fluocinolone oil and hydrocortisone 2.5% for a month with no change in the lesions. Besides the use of topical corticosteroids, he has not been taking any other medications.
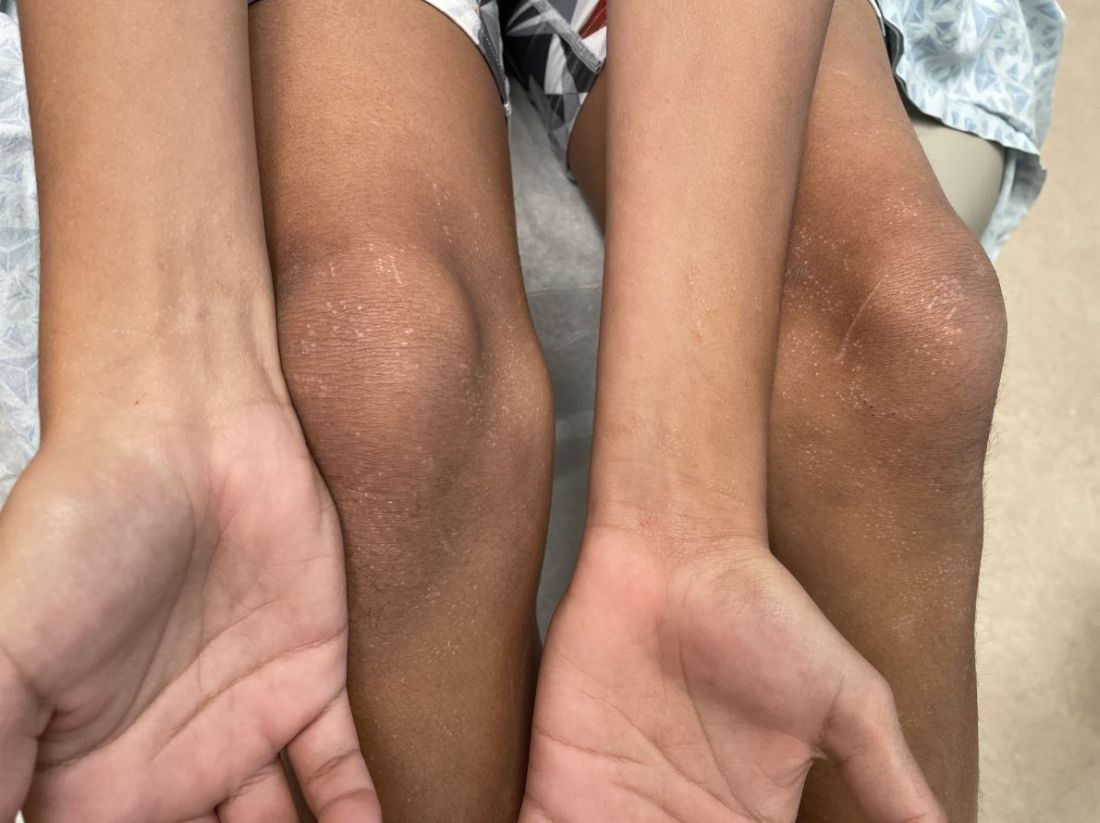
On physical examination he has multiple skin-colored, flat-topped papules that coalesce into plaques on the arms, legs, chest, and back (Photo 1). Koebner phenomenon was also seen on the knees and arms. There were no lesions in the mouth or on the nails.
A toddler presents with a dark line on a fingernail
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.

Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.

Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.
Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.
Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.
Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.
Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
Given the over 1-year history of an unchanging longitudinal band of pigment without extension to the proximal or lateral nailfolds or any other nail findings, the most likely diagnosis is benign longitudinal melanonychia.
Longitudinal melanonychia, also known as melanonychia striata, describes a brown to black streak of pigment extending from the nail matrix to the free edge of the nail.1,2
This disorder can occur secondary to a wide variety of benign and pathologic causes including lentigines, nevi, melanoma, chronic trauma, inflammatory skin diseases, systemic diseases, iatrogenic causes, and genetic syndromes.3 In melanocytic causes of longitudinal melanonychia, either melanocytic activation or hyperplasia drive pigmentary development leading to the brown to black band seen in the nail.4 Benign causes of longitudinal melanonychia include benign melanocyte activation, lentigo, and benign nevus.1
What’s the differential diagnosis?
The differential diagnosis for longitudinal melanonychia can include a wide variety of local and systemic causes. For our discussion, we will limit our differential to other locally involved disorders of the nail including subungual melanoma, subungual hematoma, onychomycosis, and glomus tumor.
Subungual melanoma is a rare subtype of acral lentiginous melanoma that most often presents as longitudinal melanonychia. Subungual melanoma is more common in those aged 50-70 years, individuals with personal or family history of melanoma or dysplastic nevus syndrome, and persons with African American, Native American, and Asian descent. Longitudinal melanonychia features that can be concerning for subungual melanoma include the presence of multiple colors, width greater than or equal to 3 mm, blurry borders, rapid increase in size, and extension to the proximal or lateral nailfolds (Hutchinson’s sign). Biopsy is required to make the diagnosis of subungual melanoma but is not necessary for melanonychia without atypical features.
Treatment of subungual melanoma depends on disease stage and can range from wide local excision of the nail apparatus to amputation of the affected digit and management with a medical oncologist. Given the absence of concerning neoplastic findings or personal or family history of melanoma, subungual melanoma is unlikely in this patient.
Subungual hematoma is an accumulation of blood underneath the nail plate that is typically the result of acute or chronic trauma to the distal phalanx. It can present as purple, red, pink, brown, or black discoloration under the nail plate and is most commonly found on the first toe. With acute trauma, pain is usually present upon initial injury. Subungual hematomas typically resolve on their own with normal nail growth. The absence of a history of trauma or pain, and the linear appearance of the lesion in our patient are inconsistent with a subungual hematoma.
Onychomycosis is a fungal infection of the nail caused by dermatophytes, nondermatophytes, or yeasts. It may present with longitudinal melanonychia; however, it more often presents with other nail abnormalities such as nail thickening, yellow discoloration, onycholysis, splitting, subungual hyperkeratosis, and nail plate destruction, which are not present in this patient. Furthermore, onychomycosis is more common in adults than children. Diagnosis is usually made with potassium hydroxide (KOH) preparations, histopathologic examination of nail clippings with a periodic acid-Schiff stain, fungal culture, or PCR.
Glomus tumor is a rare, benign neoplasm originating from cells of the glomus body. It is often found in the subungual region, in addition to other areas rich in glomus bodies such as the fingertips, palms, wrists, and forearms. Subungual glomus tumors present as a red, purple, or blueish lesions under the nail plate. Distal notching or an overlying longitudinal fissure may be present. Subungual glomus tumors are typically associated with pinpoint tenderness, paroxysmal pain, and cold sensitivity, features that are not present in our patient. The history and examination of our patient are much more consistent with benign longitudinal melanonychia.
It appears that melanoma associated with longitudinal melanonychia is very rare in children. According to one review published in 2020, only 12 cases of pediatric subungual melanoma have been reported.5 Recent series have observed longitudinal melanonychia in large sets of children, with findings that demonstrate that the vast majority of longitudinal melanonychia either stops progressing or regresses. These investigations therefore recommend serial observation of longitudinal melanonychia except in rare circumstances.6,7
Given the lack of troubling findings or concerning history, our patient was managed with observation. On follow-up 6 months later, he was found to have no change in his nail pigmentation.
Dr. Haft is an inflammatory skin disease fellow in the division of pediatric and adolescent dermatology; Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology; and Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics, all at the University of California and Rady Children’s Hospital, San Diego. They have no relevant disclosures.
References
1. Mannava KA et al. Hand Surg. 2013;18(1):133-9.
2. Leung AKC et al. Int J Dermatol. 2019;58(11):1239-45.
3. Andre J and Lateur N. Dermatol Clin. 2006;24(3):329-39.
4. Lee DK and Lipner SR. Ann Med. 2022;54(1):694-712.
5. Smith RJ and Rubin AI. Curr Opin Pediatr. 2020;32(4):506-15. .
6. Matsui Y et al. J Am Acad Dermatol. 2022;86(4):946-8.
7. Lee JS et al. J Am Acad Dermatol. 2022;87(2):366-72.
Examination findings reveal a 2-mm brown longitudinal band on the radial aspect of the right thumbnail that does not extend into the proximal or lateral nailfolds. The rest of the skin and nail exam is unremarkable.