User login
MAT access helps address opioid use disorder
PHILADELPHIA –

That was the key takeaway from a presentation given by Charles Reznikoff, MD, at the annual meeting of the American College of Physicians.
“There is fairly robust evidence on medication assisted therapy for OUD [opioid use disorder],” said Dr. Reznikoff of the University of Minnesota, Minneapolis. “MAT lowers mortality 70% in people with OUD all-cause mortality.”
He mentioned some examples in Europe, where access to medication assisted therapy has had a dramatic effect on opioid overdoses and deaths.
For example, any doctor in France since 1995 can prescribe drugs to help with OUD treatment and most buprenorphine prescriptions are written by primary care physicians. As a result, there has been a tenfold increase in the number of patients suffering from OUD receiving medication assisted treatment. In addition, there has been an 80% drop in the overdose death rate.
But Dr. Reznikoff’s recommendation on the need for more MAT was more about methadone and Suboxone, which contains a combination of buprenorphine and naloxone, than the current drive to expand the distribution of naloxone alone. He noted that naloxone is good but it really serves as a short-term fix that slows the rate of overdose deaths and provides time to implement other long-term fixes. Many states are providing naloxone to first responders to help treat patients who are overdosing on opioids, but little is being done to expand access to other medication assisted treatments, he said.
“I am bringing it up to try to illustrate what is happening in American policy making that we are reaching for naloxone first and we are not reaching for MAT first,” Dr. Reznikoff said, noting that you can give naloxone to patients without changing the underlying system of care.
He also stressed the need to get more medication assisted treatment into the penal system.
“If you have opioid use disorder [and] you are incarcerated, almost nowhere in America can you get medication assisted therapy while incarcerated,” Dr. Reznikoff said. “You lose your tolerance, but you still have addiction. You are released from incarceration and your rate of death is 20-fold the average OUD rate of death in the first 2 weeks after release.”
As a counter example, he noted that Portugal in 2001 switched its approach to OUD from criminal to a public health issue and stopped incarcerating people with opioid addiction. This led to a decrease by 75% in active heroin users, and the country now boasts the lowest rate of drug-related death in Western Europe, 1/50th of the rate in America.
“We pretty strongly believe that if they were given medication while incarcerated or just not incarcerated, they would not have that 20-fold risk of death after release,” he said. “Incarceration is actually an accelerant in the opioid epidemic and we are not going to get out of this without addressing that tough issue.”
Another effective policy option that should be expanded further is the use of prescription drug monitoring programs, which can be effective in controlling the amount of prescriptions written, said Rebecca Haffajee, PhD, assistant professor, health management and policy, University of Michigan School of Public Health, Ann Arbor.
“We do have good evidence on the prescribing outcomes and particularly attributable to a few key features, [such as] use mandates, registration mandates, delegate access, all of those features have been shown across different types of patient populations – Medicaid, Medicare, commercially insured – to reduce overall opioid prescribing and some high risk measures, polypharmacy, doctor shopping, pharmacy shopping, those sorts of things,” noted Dr. Haffajee.
Twenty percent of state laws enacted to address the opioid crisis have involved prescription drug monitoring programs, she added.
There is not enough evidence to determine whether other laws, such as those that limit quantity and dosage levels, are effective in the fight against opioid use disorder, added Dr. Reznikoff.
The speakers did not report any conflicts.
PHILADELPHIA –
That was the key takeaway from a presentation given by Charles Reznikoff, MD, at the annual meeting of the American College of Physicians.
“There is fairly robust evidence on medication assisted therapy for OUD [opioid use disorder],” said Dr. Reznikoff of the University of Minnesota, Minneapolis. “MAT lowers mortality 70% in people with OUD all-cause mortality.”
He mentioned some examples in Europe, where access to medication assisted therapy has had a dramatic effect on opioid overdoses and deaths.
For example, any doctor in France since 1995 can prescribe drugs to help with OUD treatment and most buprenorphine prescriptions are written by primary care physicians. As a result, there has been a tenfold increase in the number of patients suffering from OUD receiving medication assisted treatment. In addition, there has been an 80% drop in the overdose death rate.
But Dr. Reznikoff’s recommendation on the need for more MAT was more about methadone and Suboxone, which contains a combination of buprenorphine and naloxone, than the current drive to expand the distribution of naloxone alone. He noted that naloxone is good but it really serves as a short-term fix that slows the rate of overdose deaths and provides time to implement other long-term fixes. Many states are providing naloxone to first responders to help treat patients who are overdosing on opioids, but little is being done to expand access to other medication assisted treatments, he said.
“I am bringing it up to try to illustrate what is happening in American policy making that we are reaching for naloxone first and we are not reaching for MAT first,” Dr. Reznikoff said, noting that you can give naloxone to patients without changing the underlying system of care.
He also stressed the need to get more medication assisted treatment into the penal system.
“If you have opioid use disorder [and] you are incarcerated, almost nowhere in America can you get medication assisted therapy while incarcerated,” Dr. Reznikoff said. “You lose your tolerance, but you still have addiction. You are released from incarceration and your rate of death is 20-fold the average OUD rate of death in the first 2 weeks after release.”
As a counter example, he noted that Portugal in 2001 switched its approach to OUD from criminal to a public health issue and stopped incarcerating people with opioid addiction. This led to a decrease by 75% in active heroin users, and the country now boasts the lowest rate of drug-related death in Western Europe, 1/50th of the rate in America.
“We pretty strongly believe that if they were given medication while incarcerated or just not incarcerated, they would not have that 20-fold risk of death after release,” he said. “Incarceration is actually an accelerant in the opioid epidemic and we are not going to get out of this without addressing that tough issue.”
Another effective policy option that should be expanded further is the use of prescription drug monitoring programs, which can be effective in controlling the amount of prescriptions written, said Rebecca Haffajee, PhD, assistant professor, health management and policy, University of Michigan School of Public Health, Ann Arbor.
“We do have good evidence on the prescribing outcomes and particularly attributable to a few key features, [such as] use mandates, registration mandates, delegate access, all of those features have been shown across different types of patient populations – Medicaid, Medicare, commercially insured – to reduce overall opioid prescribing and some high risk measures, polypharmacy, doctor shopping, pharmacy shopping, those sorts of things,” noted Dr. Haffajee.
Twenty percent of state laws enacted to address the opioid crisis have involved prescription drug monitoring programs, she added.
There is not enough evidence to determine whether other laws, such as those that limit quantity and dosage levels, are effective in the fight against opioid use disorder, added Dr. Reznikoff.
The speakers did not report any conflicts.
PHILADELPHIA –
That was the key takeaway from a presentation given by Charles Reznikoff, MD, at the annual meeting of the American College of Physicians.
“There is fairly robust evidence on medication assisted therapy for OUD [opioid use disorder],” said Dr. Reznikoff of the University of Minnesota, Minneapolis. “MAT lowers mortality 70% in people with OUD all-cause mortality.”
He mentioned some examples in Europe, where access to medication assisted therapy has had a dramatic effect on opioid overdoses and deaths.
For example, any doctor in France since 1995 can prescribe drugs to help with OUD treatment and most buprenorphine prescriptions are written by primary care physicians. As a result, there has been a tenfold increase in the number of patients suffering from OUD receiving medication assisted treatment. In addition, there has been an 80% drop in the overdose death rate.
But Dr. Reznikoff’s recommendation on the need for more MAT was more about methadone and Suboxone, which contains a combination of buprenorphine and naloxone, than the current drive to expand the distribution of naloxone alone. He noted that naloxone is good but it really serves as a short-term fix that slows the rate of overdose deaths and provides time to implement other long-term fixes. Many states are providing naloxone to first responders to help treat patients who are overdosing on opioids, but little is being done to expand access to other medication assisted treatments, he said.
“I am bringing it up to try to illustrate what is happening in American policy making that we are reaching for naloxone first and we are not reaching for MAT first,” Dr. Reznikoff said, noting that you can give naloxone to patients without changing the underlying system of care.
He also stressed the need to get more medication assisted treatment into the penal system.
“If you have opioid use disorder [and] you are incarcerated, almost nowhere in America can you get medication assisted therapy while incarcerated,” Dr. Reznikoff said. “You lose your tolerance, but you still have addiction. You are released from incarceration and your rate of death is 20-fold the average OUD rate of death in the first 2 weeks after release.”
As a counter example, he noted that Portugal in 2001 switched its approach to OUD from criminal to a public health issue and stopped incarcerating people with opioid addiction. This led to a decrease by 75% in active heroin users, and the country now boasts the lowest rate of drug-related death in Western Europe, 1/50th of the rate in America.
“We pretty strongly believe that if they were given medication while incarcerated or just not incarcerated, they would not have that 20-fold risk of death after release,” he said. “Incarceration is actually an accelerant in the opioid epidemic and we are not going to get out of this without addressing that tough issue.”
Another effective policy option that should be expanded further is the use of prescription drug monitoring programs, which can be effective in controlling the amount of prescriptions written, said Rebecca Haffajee, PhD, assistant professor, health management and policy, University of Michigan School of Public Health, Ann Arbor.
“We do have good evidence on the prescribing outcomes and particularly attributable to a few key features, [such as] use mandates, registration mandates, delegate access, all of those features have been shown across different types of patient populations – Medicaid, Medicare, commercially insured – to reduce overall opioid prescribing and some high risk measures, polypharmacy, doctor shopping, pharmacy shopping, those sorts of things,” noted Dr. Haffajee.
Twenty percent of state laws enacted to address the opioid crisis have involved prescription drug monitoring programs, she added.
There is not enough evidence to determine whether other laws, such as those that limit quantity and dosage levels, are effective in the fight against opioid use disorder, added Dr. Reznikoff.
The speakers did not report any conflicts.
REPORTING FROM INTERNAL MEDICINE 2019
A Critical Review of Periodic Health Screening Using Specific Screening Criteria: Part 2: Selected Endocrine, Metabolic, and Gastrointestinal Diseases
C3 inhibitor shows potential in PNH and AIHA
GLASGOW – APL-2, a complement factor 3 (C3) inhibitor, may be a future treatment option for paroxysmal nocturnal hemoglobinuria (PNH) and autoimmune hemolytic anemia (AIHA), according to investigators from two separate studies.

Early results from the phase 1b PADDOCK trial for PNH and the phase 2 PLAUDIT trial for AIHA showed that APL-2 significantly increased hemoglobin levels, with additional improvements reported in lactate dehydrogenase (LDH), absolute reticulocyte count, and bilirubin. The findings were presented at the annual meeting of the British Society for Haematology.
By blocking C3, APL-2 acts further upstream than approved C5 inhibitors eculizumab and ravulizumab, thereby controlling extravascular hemolysis in addition to intravascular hemolysis. This broader level of control is needed for some patients, the investigators said, such as those with PNH who have inadequate responses to C5 inhibition.
PNH
“Even in PNH patients treated with eculizumab, up to 70% may have suboptimal hemoglobin responses and about 30% may still require blood transfusions,” said lead author of the PADDOCK trial, Raymond Wong, MD, of the Prince of Wales Hospital in Hong Kong.
PNH patients included in the open-label, dose-escalation PADDOCK study had greater than 10% white blood cell clones, LDH that was at least twice the upper limit of normal, at least one transfusion within the past year, a platelet count below 30,000/mm3, and an absolute neutrophil count greater than 500 x 109/L.
Dr. Wong described experiences with a cohort of 20 patients who received 270 mg APL-2 subcutaneously daily for at least 28 days, with the option to continue treatment for up to 2 years thereafter, if desired.
From these 20 patients, 2 patients completed the initial 28-day period but did not elect to continue and 2 patients withdrew because of adverse events (ovarian cancer and severe aplastic anemia), leaving 16 patients in the present analysis. Before treatment, these individuals were transfusion dependent, with an average transfusion rate of 8.7 transfusions per year.
Results showed that mean hemoglobin increased from 8.0 g/dL at baseline to 10.8 g/dL at day 29 and 12.2 g/dL at day 85. LDH dropped 900%, from 2,416 U/L (9 times the upper limit of normal) to 271 U/L (0.9 times the upper limit of normal). Absolute reticulocyte count and bilirubin also normalized.
Overall, these improvements led to a meaningful clinical impact, Dr. Wong said, with fatigue scores improving and most patients becoming transfusion independent on maintenance therapy, with the exception of one patient who developed severe aplastic anemia after 1 year. No significant infections or thromboses occurred.
When asked where APL-2 might fit in with current treatment paradigm, Dr. Wong said that multiple applications for PNH are being investigated, including first-line therapy and after failure of eculizumab.
AIHA
Results from the phase 2 PLAUDIT trial, presented by Bruno Fattizzo, MD, of the University of Milan, offered a glimpse at APL-2 in a different setting: AIHA.

Eligibility required hemoglobin levels of less than 11 g/dL, signs of hemolysis, and positive direct antiglobulin test for IgG and/or complement C3.
Dr. Fattizzo discussed results from five patients with cold agglutinin disease and five patients with C3-positive warm AIHA who had received 56 days of therapy.
Among the five patients with cold agglutinin disease, mean hemoglobin increased from 8.7 g/dL to 12.1 g/dL, while patients with warm C3-positive AIHA had a mean increase from 9.3 g/dL to 11.3 g/dL. As with the PNH study, absolute reticulocyte count, LDH, and indirect bilirubin normalized across all 10 patients.
“Some of the patients included in the trial have already reached more than 48 weeks, something like 64 weeks in the study, and they are still doing well,” Dr. Fattizzo said. “So it really seems that those who are do respond really keep the response with ongoing treatment.”
Nine out of 12 patients with cold agglutinin disease (75%) and 8 out of 9 patients (89%) with warm AIHA experienced adverse events, although these were mostly grade 1 or 2 and deemed unrelated to APL-2 by the investigators.
Five grade 3 adverse events in six patients included oral squamous carcinoma, hemolytic flare, pneumonia, purpura, and acute kidney injury. Five grade 4 adverse events in two patients included high calcium, high creatinine, hypoxia, and hemolytic flare, causing these two patients to withdraw from the study. No grade 3 or 4 adverse events were considered related to APL-2.
“APL-2 appears to be well tolerated and safe,” Dr. Fattizzo said, adding that a phase 3 trial for cold agglutinin disease and C3-positive warm AIHA C3+ is planned.
Both studies are sponsored by Apellis Pharmaceuticals. Dr. Wong and his colleagues reported financial relationships with Alexion Pharmaceuticals, Apellis, Celgene, Janssen, and other companies. Dr. Fattizzo reported having no conflicts of interest.
GLASGOW – APL-2, a complement factor 3 (C3) inhibitor, may be a future treatment option for paroxysmal nocturnal hemoglobinuria (PNH) and autoimmune hemolytic anemia (AIHA), according to investigators from two separate studies.
Early results from the phase 1b PADDOCK trial for PNH and the phase 2 PLAUDIT trial for AIHA showed that APL-2 significantly increased hemoglobin levels, with additional improvements reported in lactate dehydrogenase (LDH), absolute reticulocyte count, and bilirubin. The findings were presented at the annual meeting of the British Society for Haematology.
By blocking C3, APL-2 acts further upstream than approved C5 inhibitors eculizumab and ravulizumab, thereby controlling extravascular hemolysis in addition to intravascular hemolysis. This broader level of control is needed for some patients, the investigators said, such as those with PNH who have inadequate responses to C5 inhibition.
PNH
“Even in PNH patients treated with eculizumab, up to 70% may have suboptimal hemoglobin responses and about 30% may still require blood transfusions,” said lead author of the PADDOCK trial, Raymond Wong, MD, of the Prince of Wales Hospital in Hong Kong.
PNH patients included in the open-label, dose-escalation PADDOCK study had greater than 10% white blood cell clones, LDH that was at least twice the upper limit of normal, at least one transfusion within the past year, a platelet count below 30,000/mm3, and an absolute neutrophil count greater than 500 x 109/L.
Dr. Wong described experiences with a cohort of 20 patients who received 270 mg APL-2 subcutaneously daily for at least 28 days, with the option to continue treatment for up to 2 years thereafter, if desired.
From these 20 patients, 2 patients completed the initial 28-day period but did not elect to continue and 2 patients withdrew because of adverse events (ovarian cancer and severe aplastic anemia), leaving 16 patients in the present analysis. Before treatment, these individuals were transfusion dependent, with an average transfusion rate of 8.7 transfusions per year.
Results showed that mean hemoglobin increased from 8.0 g/dL at baseline to 10.8 g/dL at day 29 and 12.2 g/dL at day 85. LDH dropped 900%, from 2,416 U/L (9 times the upper limit of normal) to 271 U/L (0.9 times the upper limit of normal). Absolute reticulocyte count and bilirubin also normalized.
Overall, these improvements led to a meaningful clinical impact, Dr. Wong said, with fatigue scores improving and most patients becoming transfusion independent on maintenance therapy, with the exception of one patient who developed severe aplastic anemia after 1 year. No significant infections or thromboses occurred.
When asked where APL-2 might fit in with current treatment paradigm, Dr. Wong said that multiple applications for PNH are being investigated, including first-line therapy and after failure of eculizumab.
AIHA
Results from the phase 2 PLAUDIT trial, presented by Bruno Fattizzo, MD, of the University of Milan, offered a glimpse at APL-2 in a different setting: AIHA.
Eligibility required hemoglobin levels of less than 11 g/dL, signs of hemolysis, and positive direct antiglobulin test for IgG and/or complement C3.
Dr. Fattizzo discussed results from five patients with cold agglutinin disease and five patients with C3-positive warm AIHA who had received 56 days of therapy.
Among the five patients with cold agglutinin disease, mean hemoglobin increased from 8.7 g/dL to 12.1 g/dL, while patients with warm C3-positive AIHA had a mean increase from 9.3 g/dL to 11.3 g/dL. As with the PNH study, absolute reticulocyte count, LDH, and indirect bilirubin normalized across all 10 patients.
“Some of the patients included in the trial have already reached more than 48 weeks, something like 64 weeks in the study, and they are still doing well,” Dr. Fattizzo said. “So it really seems that those who are do respond really keep the response with ongoing treatment.”
Nine out of 12 patients with cold agglutinin disease (75%) and 8 out of 9 patients (89%) with warm AIHA experienced adverse events, although these were mostly grade 1 or 2 and deemed unrelated to APL-2 by the investigators.
Five grade 3 adverse events in six patients included oral squamous carcinoma, hemolytic flare, pneumonia, purpura, and acute kidney injury. Five grade 4 adverse events in two patients included high calcium, high creatinine, hypoxia, and hemolytic flare, causing these two patients to withdraw from the study. No grade 3 or 4 adverse events were considered related to APL-2.
“APL-2 appears to be well tolerated and safe,” Dr. Fattizzo said, adding that a phase 3 trial for cold agglutinin disease and C3-positive warm AIHA C3+ is planned.
Both studies are sponsored by Apellis Pharmaceuticals. Dr. Wong and his colleagues reported financial relationships with Alexion Pharmaceuticals, Apellis, Celgene, Janssen, and other companies. Dr. Fattizzo reported having no conflicts of interest.
GLASGOW – APL-2, a complement factor 3 (C3) inhibitor, may be a future treatment option for paroxysmal nocturnal hemoglobinuria (PNH) and autoimmune hemolytic anemia (AIHA), according to investigators from two separate studies.
Early results from the phase 1b PADDOCK trial for PNH and the phase 2 PLAUDIT trial for AIHA showed that APL-2 significantly increased hemoglobin levels, with additional improvements reported in lactate dehydrogenase (LDH), absolute reticulocyte count, and bilirubin. The findings were presented at the annual meeting of the British Society for Haematology.
By blocking C3, APL-2 acts further upstream than approved C5 inhibitors eculizumab and ravulizumab, thereby controlling extravascular hemolysis in addition to intravascular hemolysis. This broader level of control is needed for some patients, the investigators said, such as those with PNH who have inadequate responses to C5 inhibition.
PNH
“Even in PNH patients treated with eculizumab, up to 70% may have suboptimal hemoglobin responses and about 30% may still require blood transfusions,” said lead author of the PADDOCK trial, Raymond Wong, MD, of the Prince of Wales Hospital in Hong Kong.
PNH patients included in the open-label, dose-escalation PADDOCK study had greater than 10% white blood cell clones, LDH that was at least twice the upper limit of normal, at least one transfusion within the past year, a platelet count below 30,000/mm3, and an absolute neutrophil count greater than 500 x 109/L.
Dr. Wong described experiences with a cohort of 20 patients who received 270 mg APL-2 subcutaneously daily for at least 28 days, with the option to continue treatment for up to 2 years thereafter, if desired.
From these 20 patients, 2 patients completed the initial 28-day period but did not elect to continue and 2 patients withdrew because of adverse events (ovarian cancer and severe aplastic anemia), leaving 16 patients in the present analysis. Before treatment, these individuals were transfusion dependent, with an average transfusion rate of 8.7 transfusions per year.
Results showed that mean hemoglobin increased from 8.0 g/dL at baseline to 10.8 g/dL at day 29 and 12.2 g/dL at day 85. LDH dropped 900%, from 2,416 U/L (9 times the upper limit of normal) to 271 U/L (0.9 times the upper limit of normal). Absolute reticulocyte count and bilirubin also normalized.
Overall, these improvements led to a meaningful clinical impact, Dr. Wong said, with fatigue scores improving and most patients becoming transfusion independent on maintenance therapy, with the exception of one patient who developed severe aplastic anemia after 1 year. No significant infections or thromboses occurred.
When asked where APL-2 might fit in with current treatment paradigm, Dr. Wong said that multiple applications for PNH are being investigated, including first-line therapy and after failure of eculizumab.
AIHA
Results from the phase 2 PLAUDIT trial, presented by Bruno Fattizzo, MD, of the University of Milan, offered a glimpse at APL-2 in a different setting: AIHA.
Eligibility required hemoglobin levels of less than 11 g/dL, signs of hemolysis, and positive direct antiglobulin test for IgG and/or complement C3.
Dr. Fattizzo discussed results from five patients with cold agglutinin disease and five patients with C3-positive warm AIHA who had received 56 days of therapy.
Among the five patients with cold agglutinin disease, mean hemoglobin increased from 8.7 g/dL to 12.1 g/dL, while patients with warm C3-positive AIHA had a mean increase from 9.3 g/dL to 11.3 g/dL. As with the PNH study, absolute reticulocyte count, LDH, and indirect bilirubin normalized across all 10 patients.
“Some of the patients included in the trial have already reached more than 48 weeks, something like 64 weeks in the study, and they are still doing well,” Dr. Fattizzo said. “So it really seems that those who are do respond really keep the response with ongoing treatment.”
Nine out of 12 patients with cold agglutinin disease (75%) and 8 out of 9 patients (89%) with warm AIHA experienced adverse events, although these were mostly grade 1 or 2 and deemed unrelated to APL-2 by the investigators.
Five grade 3 adverse events in six patients included oral squamous carcinoma, hemolytic flare, pneumonia, purpura, and acute kidney injury. Five grade 4 adverse events in two patients included high calcium, high creatinine, hypoxia, and hemolytic flare, causing these two patients to withdraw from the study. No grade 3 or 4 adverse events were considered related to APL-2.
“APL-2 appears to be well tolerated and safe,” Dr. Fattizzo said, adding that a phase 3 trial for cold agglutinin disease and C3-positive warm AIHA C3+ is planned.
Both studies are sponsored by Apellis Pharmaceuticals. Dr. Wong and his colleagues reported financial relationships with Alexion Pharmaceuticals, Apellis, Celgene, Janssen, and other companies. Dr. Fattizzo reported having no conflicts of interest.
REPORTING FROM BSH 2019
Reviews of Self-Instructional Materials
Metabolite levels might help differentiate schizophrenia, bipolar
ORLANDO – Profiling metabolites found in the plasma could be a way to get an earlier handle on how a person’s mental illness will progress, researchers said the annual congress of the Schizophrenia International Research Society.

Investigators at the University of São Paulo found that certain substances – such as phospholipids and sphingolipids – were found in differing levels at the time of a first episode of psychosis, allowing them to accurately identify bipolar disorder or schizophrenia or healthy controls 87% of the time.
The findings suggest a way to help get patients more targeted treatment earlier in their disease course, said Helena Joaquim, MD, PhD, a postdoctoral fellow in the university’s psychiatry department.
“The levels of these metabolites can be a biomarker for psychosis, as well as a diagnostic marker for schizophrenia and bipolar disorder, aiding clinical practice,” the researchers wrote in a poster presentation.
At the time of a first episode of psychosis, the actual diagnosis can be difficult to pinpoint, making the illness more difficult to treat. Since lipid metabolism is altered in neuropsychiatric diseases, the researchers turned to the metabolite levels of patients to pin down a specific diagnosis sooner.
They collected plasma from 28 schizophrenia patients, 27 bipolar patients, and 30 healthy controls. They looked specifically at acylcarnitines, phospholipids, lysophospholipids and sphingolipids. They found that phospholipids were elevated in schizophrenia patients, compared with controls, and even more elevated in patients with bipolar disorder. A similar pattern was seen with lysophospholipids. Sphingolipids were found at lower levels in schizophrenia and elevated in bipolar disorder, compared with controls.
Levels of those three metabolites allowed researchers 87.1% of the time to correctly identify patients as a healthy control or as having schizophrenia or bipolar disorder. Acylcarnitines were not found to be differentiated between schizophrenia and bipolar disorder, the researchers found.
The most interesting part of their efforts so far, Dr. Joaquim said, was a shotgun analysis, an untargeted approach in which about 186 metabolites were measured and plotted on a graph. The compounds have not yet all been identified, but researchers found that levels of these substances were clustered in conspicuous ways.
“It seems like the negative schizophrenia patients are more like the depressive bipolar, and the positive symptoms are more like manic bipolar,” Dr. Joaquim said.
. For example, looking at metabolite levels might help determine whether schizophrenia patients are more likely to be troubled by positive symptoms, such as hallucinations.
“We have to look back,” Dr. Joaquim said, “and try to identify those metabolites that are the most different between the groups.”
Dr. Joaquim reported no relevant disclosures.
ORLANDO – Profiling metabolites found in the plasma could be a way to get an earlier handle on how a person’s mental illness will progress, researchers said the annual congress of the Schizophrenia International Research Society.
Investigators at the University of São Paulo found that certain substances – such as phospholipids and sphingolipids – were found in differing levels at the time of a first episode of psychosis, allowing them to accurately identify bipolar disorder or schizophrenia or healthy controls 87% of the time.
The findings suggest a way to help get patients more targeted treatment earlier in their disease course, said Helena Joaquim, MD, PhD, a postdoctoral fellow in the university’s psychiatry department.
“The levels of these metabolites can be a biomarker for psychosis, as well as a diagnostic marker for schizophrenia and bipolar disorder, aiding clinical practice,” the researchers wrote in a poster presentation.
At the time of a first episode of psychosis, the actual diagnosis can be difficult to pinpoint, making the illness more difficult to treat. Since lipid metabolism is altered in neuropsychiatric diseases, the researchers turned to the metabolite levels of patients to pin down a specific diagnosis sooner.
They collected plasma from 28 schizophrenia patients, 27 bipolar patients, and 30 healthy controls. They looked specifically at acylcarnitines, phospholipids, lysophospholipids and sphingolipids. They found that phospholipids were elevated in schizophrenia patients, compared with controls, and even more elevated in patients with bipolar disorder. A similar pattern was seen with lysophospholipids. Sphingolipids were found at lower levels in schizophrenia and elevated in bipolar disorder, compared with controls.
Levels of those three metabolites allowed researchers 87.1% of the time to correctly identify patients as a healthy control or as having schizophrenia or bipolar disorder. Acylcarnitines were not found to be differentiated between schizophrenia and bipolar disorder, the researchers found.
The most interesting part of their efforts so far, Dr. Joaquim said, was a shotgun analysis, an untargeted approach in which about 186 metabolites were measured and plotted on a graph. The compounds have not yet all been identified, but researchers found that levels of these substances were clustered in conspicuous ways.
“It seems like the negative schizophrenia patients are more like the depressive bipolar, and the positive symptoms are more like manic bipolar,” Dr. Joaquim said.
. For example, looking at metabolite levels might help determine whether schizophrenia patients are more likely to be troubled by positive symptoms, such as hallucinations.
“We have to look back,” Dr. Joaquim said, “and try to identify those metabolites that are the most different between the groups.”
Dr. Joaquim reported no relevant disclosures.
ORLANDO – Profiling metabolites found in the plasma could be a way to get an earlier handle on how a person’s mental illness will progress, researchers said the annual congress of the Schizophrenia International Research Society.
Investigators at the University of São Paulo found that certain substances – such as phospholipids and sphingolipids – were found in differing levels at the time of a first episode of psychosis, allowing them to accurately identify bipolar disorder or schizophrenia or healthy controls 87% of the time.
The findings suggest a way to help get patients more targeted treatment earlier in their disease course, said Helena Joaquim, MD, PhD, a postdoctoral fellow in the university’s psychiatry department.
“The levels of these metabolites can be a biomarker for psychosis, as well as a diagnostic marker for schizophrenia and bipolar disorder, aiding clinical practice,” the researchers wrote in a poster presentation.
At the time of a first episode of psychosis, the actual diagnosis can be difficult to pinpoint, making the illness more difficult to treat. Since lipid metabolism is altered in neuropsychiatric diseases, the researchers turned to the metabolite levels of patients to pin down a specific diagnosis sooner.
They collected plasma from 28 schizophrenia patients, 27 bipolar patients, and 30 healthy controls. They looked specifically at acylcarnitines, phospholipids, lysophospholipids and sphingolipids. They found that phospholipids were elevated in schizophrenia patients, compared with controls, and even more elevated in patients with bipolar disorder. A similar pattern was seen with lysophospholipids. Sphingolipids were found at lower levels in schizophrenia and elevated in bipolar disorder, compared with controls.
Levels of those three metabolites allowed researchers 87.1% of the time to correctly identify patients as a healthy control or as having schizophrenia or bipolar disorder. Acylcarnitines were not found to be differentiated between schizophrenia and bipolar disorder, the researchers found.
The most interesting part of their efforts so far, Dr. Joaquim said, was a shotgun analysis, an untargeted approach in which about 186 metabolites were measured and plotted on a graph. The compounds have not yet all been identified, but researchers found that levels of these substances were clustered in conspicuous ways.
“It seems like the negative schizophrenia patients are more like the depressive bipolar, and the positive symptoms are more like manic bipolar,” Dr. Joaquim said.
. For example, looking at metabolite levels might help determine whether schizophrenia patients are more likely to be troubled by positive symptoms, such as hallucinations.
“We have to look back,” Dr. Joaquim said, “and try to identify those metabolites that are the most different between the groups.”
Dr. Joaquim reported no relevant disclosures.
REPORTING FROM SIRS 2019
Sunscreen Regulations and Advice for Your Patients
If by now you have not had a patient ask, “Doctor, what sunscreen should I use NOW?” you will soon.
The US Food and Drug Administration (FDA) recently published a press release detailing a proposed rule on how manufacturers will be required to test and label sunscreens in the United States.1,2 Although the press release was complicated and contained much information, the media specifically latched onto the FDA’s consideration of only 2 active sunscreen ingredients—zinc oxide and titanium dioxide—as generally recognized as safe and effective (GRASE). In response, some patients may assume that most sunscreens on the market are dangerous.
How did this new proposed rule come about? To understand the process, it takes some explanation of the history of the FDA’s regulation of sunscreens.
How are sunscreens regulated by the FDA?
The regulatory process for sunscreens in the United States is complicated. The FDA regulates sunscreens as over-the-counter (OTC) drugs rather than as cosmetics, which is how they are regulated in most of the rest of the world.
The US sunscreen regulation process began in 1978 with an advance notice of proposed rulemaking from the FDA that included recommendations from an advisory review panel on the safe and effective use of OTC sunscreen products.3 At that time, 21 active sunscreen ingredients and their maximum use concentrations were listed and determined to be safe, or GRASE. It also gave manufacturers guidance on how to test for efficacy with the methodology for determining the sun protection factor (SPF) as well as various labeling requirements. Over the years, the FDA has issued a number of other sunscreen guidelines, such as removing padimate A and adding avobenzone and zinc oxide to the list of GRASE ingredients in the 1990s.4,5
In 1999, the FDA issued a final rule that listed 16 active sunscreen ingredients and concentrations as GRASE.6 There were some restrictions as to certain combinations of ingredients that could not be used in a finished product. Labeling requirements, including a maximum SPF of 30, also were put in place. This final rule established a final sunscreen monograph that was supposed to have been effective by 2002; however, in 2001 the agency delayed the effective date indefinitely because they had not yet established broad-spectrum (UVA) protection testing and labeling.7
The FDA published a proposed rule in 2007 as well as a final rule in 2011 that again listed the same 16 ingredients as GRASE and specified labeling and testing methods for establishing SPF, broad-spectrum protection, and water-resistance claims.8,9 The final rule limited product labels to a maximum SPF of 50+; provided directions for use with regard to other labeling elements (eg, warnings); and identified specific claims that would not be allowed on product labels, such as “waterproof” and “all-day protection.”9
Nevertheless, an effective final OTC monograph for sunscreen products has not yet been published.
What is the Sunscreen Innovation Act?
In 2014, the US Congress enacted the Sunscreen Innovation Act10 primarily to mandate that the FDA develop a more efficient way to determine the safety and efficacy of new active sunscreen ingredients that were commonly used in Europe and other parts of the world at the time. Many of these agents were thought to be more protective in the UVA and/or UVB spectrum, and if added to the list of GRASE ingredients available to US manufacturers, they would lead to the development of products that would improve the protection offered by sunscreens marketed to US consumers. The time and extent application (TEA) was established, a method that allowed manufacturers to apply for FDA approval of specific agents. The TEA also suggested allowing data generated in other countries where these agents were already in use for years to be considered in the FDA’s evaluation of the agents as GRASE. In addition, Congress mandated that a final monograph on OTC sunscreens be published by the end of 2019. A number of manufacturers have submitted TEAs for new active sunscreen ingredients, and so far, all have been rejected.
Why is the FDA interested in more safety data?
Since then, the FDA has become concerned not only with the safety and efficacy of newly proposed agents through the TEA but also with the original 16 active sunscreen ingredients listed as GRASE in the 2011 final rule. In the 1970s and 1980s, sunscreen use was limited to beach vacations or outdoor sporting events, but sun-protective behaviors have changed dramatically since that time, with health care providers now becoming cognizant of the growing threats of skin cancer and melanoma as well as the cosmetic concerns of photoaging, thereby recommending daily sunscreen use to their patients. In addition, the science behind sunscreens with higher concentrations of active ingredients intended to achieve higher and higher SPFs and their respective penetration of the skin has evolved, leading to new concerns about systemic toxicity. Early limited research frequently touted by the lay media has suggested that some of these agents might lead to hormonal changes, reproductive toxicity, and carcinogenicity.
In November 2016, the FDA issued a guidance for manufacturers that outlined the safety data that would be required to establish an OTC sunscreen active ingredient as GRASE.11 It also provided detailed information about both clinical and nonclinical safety testing, including human irritation and sensitization studies as well as human photosafety studies. In vitro dermal and systemic carcinogenicity studies and animal developmental and reproductive toxicity studies also were required as well studies regarding safety in children.
Many of these recommendations were already being utilized by manufacturers; however, one important change was the requirement for human absorption studies by a maximal usage trial, which more accurately addresses the absorption of sunscreen agents according to actual use. Such studies will be required at the highest allowable concentration of an agent in multiple vehicles and over large body surface areas for considerable exposure times.
This guidance to sunscreen manufacturers was announced to the public in a press release in May 2018.12
What are the new regulations?
All of this has culminated in the recent proposed rule, which includes several important proposals2:
- Of the 16 currently marketed active sunscreen ingredients, only 2—zinc oxide and titanium dioxide—are considered GRASE. Two ingredients—trolamine salicylate and para-aminobenzoic acid—are considered non-GRASE, but there is not enough information at this time to determine if the remaining 12 ingredients are GRASE. The FDA is working with manufacturers to obtain sufficient information to make this determination.
- Approved dosage formulations include sprays, oils, lotions, creams, gels, butters, pastes, ointments, and sticks. Further information is needed regarding powders before they can be considered.
- The maximum SPF will be increased from 50+ to 60+.
- Sunscreens with an SPF of 15 or higher are required to provide broad-spectrum protection commensurate with the SPF, expanding on critical wavelength testing.
- There are new labeling changes, including a requirement that active ingredients be listed on the front of the packaging.
- Sunscreen products that contain insect repellents are considered non-GRASE.
What’s next?
The process for the proposed final rule has now entered a 90-day public comment period that will end on May 27, 2019; however, it is unlikely that a final monograph as mandated by Congress will be produced by the end of this year.
Sunscreen manufacturers currently are coordinating a response to the proposed rule through the Personal Care Products Council and the Consumer Healthcare Products Association Sunscreen Task Force. It is likely that the new required testing will be costly, with estimates exceeding tens or even hundreds of millions of dollars. In all likelihood, the number of active ingredients that the industry will agree to support with costly testing will be fewer than the 12 that are now on the list. It also is likely that this process will lead to fewer sunscreen products for consumers to choose from and almost certainly at a higher cost.
What do we tell patients in the meantime?
According to the FDA’s rules, it was necessary that this process was made public, but it will almost certainly concern our patients as to the safety of the sunscreen products they have been using. We should be concerned that some of our patients may limit their use of sunscreens because of safety concerns.
There is no question that, as physicians, we want to “first, do no harm,” so we should all be interested in assuring our patients that our sunscreen recommendations are safe and we support the FDA proposal for additional data. The good news is that when this process is completed, a large number of agents will likely be found to be GRASE. When the FDA finally gives its imprimatur to sunscreens, it will hopefully help to silence those naysayers who report that sunscreens are dangerous for consumers; however, it has been suggested by some in industry that the new testing required may take at least 5 years.
What should dermatologists do when we are asked, “What sunscreen should I use NOW?” For most patients, I would explain the regulatory process and assure them that the risk-benefit ratio at this point suggests they should continue using the same sunscreens that they are currently using. For special situations such as pregnant women and children, it may be best to suggest products that contain only the 2 GRASE inorganic agents.
- FDA advances new proposed regulation to make sure that sunscreens are safe and effective [news release]. Silver Spring, MD: US Food and Drug Administration; February 21, 2019. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm631736.htm. Accessed April 4, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 1978;43(166):38206-38269. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph. Fed Registr. 1996;60(180):48645-48655. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph; enforcement policy. Fed Registr. 1998;63(204):56584-56589. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; final monograph. Fed Registr. 1999;64(98):27666-27693. To be codified at 21 CFR §310, 352, 700, and 740.
- Sunscreen drug products for over-the-counter human use; final monograph; partial stay; final rule. Fed Registr. 2001;66:67485-67487. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Registr. 2007;72(165):49069-49122. To be codified at 21 CFR §347 and 352.
- Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Registr. 2011;76(117):35619-35665. To be codified at 21 CFR §201 and 310.
- Sunscreen Innovation Act, S 2141, 113th Cong, 2nd Sess (2014).
- Nonprescription sunscreen drug products-safety and effectiveness data; guidance for industry; availability. Fed Registr. 2016;81(226):84594-84595.
- Statement from Commissioner Scott Gottlieb, MD, on new FDA actions to keep consumers safe from the harmful effects of sun exposure, and ensure the long-term safety and benefits of sunscreens [news release]. Silver Spring, MD: US Food and Drug Administration; May 22, 2018. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm608499.htm. Accessed April 5, 2019.
If by now you have not had a patient ask, “Doctor, what sunscreen should I use NOW?” you will soon.
The US Food and Drug Administration (FDA) recently published a press release detailing a proposed rule on how manufacturers will be required to test and label sunscreens in the United States.1,2 Although the press release was complicated and contained much information, the media specifically latched onto the FDA’s consideration of only 2 active sunscreen ingredients—zinc oxide and titanium dioxide—as generally recognized as safe and effective (GRASE). In response, some patients may assume that most sunscreens on the market are dangerous.
How did this new proposed rule come about? To understand the process, it takes some explanation of the history of the FDA’s regulation of sunscreens.
How are sunscreens regulated by the FDA?
The regulatory process for sunscreens in the United States is complicated. The FDA regulates sunscreens as over-the-counter (OTC) drugs rather than as cosmetics, which is how they are regulated in most of the rest of the world.
The US sunscreen regulation process began in 1978 with an advance notice of proposed rulemaking from the FDA that included recommendations from an advisory review panel on the safe and effective use of OTC sunscreen products.3 At that time, 21 active sunscreen ingredients and their maximum use concentrations were listed and determined to be safe, or GRASE. It also gave manufacturers guidance on how to test for efficacy with the methodology for determining the sun protection factor (SPF) as well as various labeling requirements. Over the years, the FDA has issued a number of other sunscreen guidelines, such as removing padimate A and adding avobenzone and zinc oxide to the list of GRASE ingredients in the 1990s.4,5
In 1999, the FDA issued a final rule that listed 16 active sunscreen ingredients and concentrations as GRASE.6 There were some restrictions as to certain combinations of ingredients that could not be used in a finished product. Labeling requirements, including a maximum SPF of 30, also were put in place. This final rule established a final sunscreen monograph that was supposed to have been effective by 2002; however, in 2001 the agency delayed the effective date indefinitely because they had not yet established broad-spectrum (UVA) protection testing and labeling.7
The FDA published a proposed rule in 2007 as well as a final rule in 2011 that again listed the same 16 ingredients as GRASE and specified labeling and testing methods for establishing SPF, broad-spectrum protection, and water-resistance claims.8,9 The final rule limited product labels to a maximum SPF of 50+; provided directions for use with regard to other labeling elements (eg, warnings); and identified specific claims that would not be allowed on product labels, such as “waterproof” and “all-day protection.”9
Nevertheless, an effective final OTC monograph for sunscreen products has not yet been published.
What is the Sunscreen Innovation Act?
In 2014, the US Congress enacted the Sunscreen Innovation Act10 primarily to mandate that the FDA develop a more efficient way to determine the safety and efficacy of new active sunscreen ingredients that were commonly used in Europe and other parts of the world at the time. Many of these agents were thought to be more protective in the UVA and/or UVB spectrum, and if added to the list of GRASE ingredients available to US manufacturers, they would lead to the development of products that would improve the protection offered by sunscreens marketed to US consumers. The time and extent application (TEA) was established, a method that allowed manufacturers to apply for FDA approval of specific agents. The TEA also suggested allowing data generated in other countries where these agents were already in use for years to be considered in the FDA’s evaluation of the agents as GRASE. In addition, Congress mandated that a final monograph on OTC sunscreens be published by the end of 2019. A number of manufacturers have submitted TEAs for new active sunscreen ingredients, and so far, all have been rejected.
Why is the FDA interested in more safety data?
Since then, the FDA has become concerned not only with the safety and efficacy of newly proposed agents through the TEA but also with the original 16 active sunscreen ingredients listed as GRASE in the 2011 final rule. In the 1970s and 1980s, sunscreen use was limited to beach vacations or outdoor sporting events, but sun-protective behaviors have changed dramatically since that time, with health care providers now becoming cognizant of the growing threats of skin cancer and melanoma as well as the cosmetic concerns of photoaging, thereby recommending daily sunscreen use to their patients. In addition, the science behind sunscreens with higher concentrations of active ingredients intended to achieve higher and higher SPFs and their respective penetration of the skin has evolved, leading to new concerns about systemic toxicity. Early limited research frequently touted by the lay media has suggested that some of these agents might lead to hormonal changes, reproductive toxicity, and carcinogenicity.
In November 2016, the FDA issued a guidance for manufacturers that outlined the safety data that would be required to establish an OTC sunscreen active ingredient as GRASE.11 It also provided detailed information about both clinical and nonclinical safety testing, including human irritation and sensitization studies as well as human photosafety studies. In vitro dermal and systemic carcinogenicity studies and animal developmental and reproductive toxicity studies also were required as well studies regarding safety in children.
Many of these recommendations were already being utilized by manufacturers; however, one important change was the requirement for human absorption studies by a maximal usage trial, which more accurately addresses the absorption of sunscreen agents according to actual use. Such studies will be required at the highest allowable concentration of an agent in multiple vehicles and over large body surface areas for considerable exposure times.
This guidance to sunscreen manufacturers was announced to the public in a press release in May 2018.12
What are the new regulations?
All of this has culminated in the recent proposed rule, which includes several important proposals2:
- Of the 16 currently marketed active sunscreen ingredients, only 2—zinc oxide and titanium dioxide—are considered GRASE. Two ingredients—trolamine salicylate and para-aminobenzoic acid—are considered non-GRASE, but there is not enough information at this time to determine if the remaining 12 ingredients are GRASE. The FDA is working with manufacturers to obtain sufficient information to make this determination.
- Approved dosage formulations include sprays, oils, lotions, creams, gels, butters, pastes, ointments, and sticks. Further information is needed regarding powders before they can be considered.
- The maximum SPF will be increased from 50+ to 60+.
- Sunscreens with an SPF of 15 or higher are required to provide broad-spectrum protection commensurate with the SPF, expanding on critical wavelength testing.
- There are new labeling changes, including a requirement that active ingredients be listed on the front of the packaging.
- Sunscreen products that contain insect repellents are considered non-GRASE.
What’s next?
The process for the proposed final rule has now entered a 90-day public comment period that will end on May 27, 2019; however, it is unlikely that a final monograph as mandated by Congress will be produced by the end of this year.
Sunscreen manufacturers currently are coordinating a response to the proposed rule through the Personal Care Products Council and the Consumer Healthcare Products Association Sunscreen Task Force. It is likely that the new required testing will be costly, with estimates exceeding tens or even hundreds of millions of dollars. In all likelihood, the number of active ingredients that the industry will agree to support with costly testing will be fewer than the 12 that are now on the list. It also is likely that this process will lead to fewer sunscreen products for consumers to choose from and almost certainly at a higher cost.
What do we tell patients in the meantime?
According to the FDA’s rules, it was necessary that this process was made public, but it will almost certainly concern our patients as to the safety of the sunscreen products they have been using. We should be concerned that some of our patients may limit their use of sunscreens because of safety concerns.
There is no question that, as physicians, we want to “first, do no harm,” so we should all be interested in assuring our patients that our sunscreen recommendations are safe and we support the FDA proposal for additional data. The good news is that when this process is completed, a large number of agents will likely be found to be GRASE. When the FDA finally gives its imprimatur to sunscreens, it will hopefully help to silence those naysayers who report that sunscreens are dangerous for consumers; however, it has been suggested by some in industry that the new testing required may take at least 5 years.
What should dermatologists do when we are asked, “What sunscreen should I use NOW?” For most patients, I would explain the regulatory process and assure them that the risk-benefit ratio at this point suggests they should continue using the same sunscreens that they are currently using. For special situations such as pregnant women and children, it may be best to suggest products that contain only the 2 GRASE inorganic agents.
If by now you have not had a patient ask, “Doctor, what sunscreen should I use NOW?” you will soon.
The US Food and Drug Administration (FDA) recently published a press release detailing a proposed rule on how manufacturers will be required to test and label sunscreens in the United States.1,2 Although the press release was complicated and contained much information, the media specifically latched onto the FDA’s consideration of only 2 active sunscreen ingredients—zinc oxide and titanium dioxide—as generally recognized as safe and effective (GRASE). In response, some patients may assume that most sunscreens on the market are dangerous.
How did this new proposed rule come about? To understand the process, it takes some explanation of the history of the FDA’s regulation of sunscreens.
How are sunscreens regulated by the FDA?
The regulatory process for sunscreens in the United States is complicated. The FDA regulates sunscreens as over-the-counter (OTC) drugs rather than as cosmetics, which is how they are regulated in most of the rest of the world.
The US sunscreen regulation process began in 1978 with an advance notice of proposed rulemaking from the FDA that included recommendations from an advisory review panel on the safe and effective use of OTC sunscreen products.3 At that time, 21 active sunscreen ingredients and their maximum use concentrations were listed and determined to be safe, or GRASE. It also gave manufacturers guidance on how to test for efficacy with the methodology for determining the sun protection factor (SPF) as well as various labeling requirements. Over the years, the FDA has issued a number of other sunscreen guidelines, such as removing padimate A and adding avobenzone and zinc oxide to the list of GRASE ingredients in the 1990s.4,5
In 1999, the FDA issued a final rule that listed 16 active sunscreen ingredients and concentrations as GRASE.6 There were some restrictions as to certain combinations of ingredients that could not be used in a finished product. Labeling requirements, including a maximum SPF of 30, also were put in place. This final rule established a final sunscreen monograph that was supposed to have been effective by 2002; however, in 2001 the agency delayed the effective date indefinitely because they had not yet established broad-spectrum (UVA) protection testing and labeling.7
The FDA published a proposed rule in 2007 as well as a final rule in 2011 that again listed the same 16 ingredients as GRASE and specified labeling and testing methods for establishing SPF, broad-spectrum protection, and water-resistance claims.8,9 The final rule limited product labels to a maximum SPF of 50+; provided directions for use with regard to other labeling elements (eg, warnings); and identified specific claims that would not be allowed on product labels, such as “waterproof” and “all-day protection.”9
Nevertheless, an effective final OTC monograph for sunscreen products has not yet been published.
What is the Sunscreen Innovation Act?
In 2014, the US Congress enacted the Sunscreen Innovation Act10 primarily to mandate that the FDA develop a more efficient way to determine the safety and efficacy of new active sunscreen ingredients that were commonly used in Europe and other parts of the world at the time. Many of these agents were thought to be more protective in the UVA and/or UVB spectrum, and if added to the list of GRASE ingredients available to US manufacturers, they would lead to the development of products that would improve the protection offered by sunscreens marketed to US consumers. The time and extent application (TEA) was established, a method that allowed manufacturers to apply for FDA approval of specific agents. The TEA also suggested allowing data generated in other countries where these agents were already in use for years to be considered in the FDA’s evaluation of the agents as GRASE. In addition, Congress mandated that a final monograph on OTC sunscreens be published by the end of 2019. A number of manufacturers have submitted TEAs for new active sunscreen ingredients, and so far, all have been rejected.
Why is the FDA interested in more safety data?
Since then, the FDA has become concerned not only with the safety and efficacy of newly proposed agents through the TEA but also with the original 16 active sunscreen ingredients listed as GRASE in the 2011 final rule. In the 1970s and 1980s, sunscreen use was limited to beach vacations or outdoor sporting events, but sun-protective behaviors have changed dramatically since that time, with health care providers now becoming cognizant of the growing threats of skin cancer and melanoma as well as the cosmetic concerns of photoaging, thereby recommending daily sunscreen use to their patients. In addition, the science behind sunscreens with higher concentrations of active ingredients intended to achieve higher and higher SPFs and their respective penetration of the skin has evolved, leading to new concerns about systemic toxicity. Early limited research frequently touted by the lay media has suggested that some of these agents might lead to hormonal changes, reproductive toxicity, and carcinogenicity.
In November 2016, the FDA issued a guidance for manufacturers that outlined the safety data that would be required to establish an OTC sunscreen active ingredient as GRASE.11 It also provided detailed information about both clinical and nonclinical safety testing, including human irritation and sensitization studies as well as human photosafety studies. In vitro dermal and systemic carcinogenicity studies and animal developmental and reproductive toxicity studies also were required as well studies regarding safety in children.
Many of these recommendations were already being utilized by manufacturers; however, one important change was the requirement for human absorption studies by a maximal usage trial, which more accurately addresses the absorption of sunscreen agents according to actual use. Such studies will be required at the highest allowable concentration of an agent in multiple vehicles and over large body surface areas for considerable exposure times.
This guidance to sunscreen manufacturers was announced to the public in a press release in May 2018.12
What are the new regulations?
All of this has culminated in the recent proposed rule, which includes several important proposals2:
- Of the 16 currently marketed active sunscreen ingredients, only 2—zinc oxide and titanium dioxide—are considered GRASE. Two ingredients—trolamine salicylate and para-aminobenzoic acid—are considered non-GRASE, but there is not enough information at this time to determine if the remaining 12 ingredients are GRASE. The FDA is working with manufacturers to obtain sufficient information to make this determination.
- Approved dosage formulations include sprays, oils, lotions, creams, gels, butters, pastes, ointments, and sticks. Further information is needed regarding powders before they can be considered.
- The maximum SPF will be increased from 50+ to 60+.
- Sunscreens with an SPF of 15 or higher are required to provide broad-spectrum protection commensurate with the SPF, expanding on critical wavelength testing.
- There are new labeling changes, including a requirement that active ingredients be listed on the front of the packaging.
- Sunscreen products that contain insect repellents are considered non-GRASE.
What’s next?
The process for the proposed final rule has now entered a 90-day public comment period that will end on May 27, 2019; however, it is unlikely that a final monograph as mandated by Congress will be produced by the end of this year.
Sunscreen manufacturers currently are coordinating a response to the proposed rule through the Personal Care Products Council and the Consumer Healthcare Products Association Sunscreen Task Force. It is likely that the new required testing will be costly, with estimates exceeding tens or even hundreds of millions of dollars. In all likelihood, the number of active ingredients that the industry will agree to support with costly testing will be fewer than the 12 that are now on the list. It also is likely that this process will lead to fewer sunscreen products for consumers to choose from and almost certainly at a higher cost.
What do we tell patients in the meantime?
According to the FDA’s rules, it was necessary that this process was made public, but it will almost certainly concern our patients as to the safety of the sunscreen products they have been using. We should be concerned that some of our patients may limit their use of sunscreens because of safety concerns.
There is no question that, as physicians, we want to “first, do no harm,” so we should all be interested in assuring our patients that our sunscreen recommendations are safe and we support the FDA proposal for additional data. The good news is that when this process is completed, a large number of agents will likely be found to be GRASE. When the FDA finally gives its imprimatur to sunscreens, it will hopefully help to silence those naysayers who report that sunscreens are dangerous for consumers; however, it has been suggested by some in industry that the new testing required may take at least 5 years.
What should dermatologists do when we are asked, “What sunscreen should I use NOW?” For most patients, I would explain the regulatory process and assure them that the risk-benefit ratio at this point suggests they should continue using the same sunscreens that they are currently using. For special situations such as pregnant women and children, it may be best to suggest products that contain only the 2 GRASE inorganic agents.
- FDA advances new proposed regulation to make sure that sunscreens are safe and effective [news release]. Silver Spring, MD: US Food and Drug Administration; February 21, 2019. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm631736.htm. Accessed April 4, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 1978;43(166):38206-38269. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph. Fed Registr. 1996;60(180):48645-48655. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph; enforcement policy. Fed Registr. 1998;63(204):56584-56589. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; final monograph. Fed Registr. 1999;64(98):27666-27693. To be codified at 21 CFR §310, 352, 700, and 740.
- Sunscreen drug products for over-the-counter human use; final monograph; partial stay; final rule. Fed Registr. 2001;66:67485-67487. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Registr. 2007;72(165):49069-49122. To be codified at 21 CFR §347 and 352.
- Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Registr. 2011;76(117):35619-35665. To be codified at 21 CFR §201 and 310.
- Sunscreen Innovation Act, S 2141, 113th Cong, 2nd Sess (2014).
- Nonprescription sunscreen drug products-safety and effectiveness data; guidance for industry; availability. Fed Registr. 2016;81(226):84594-84595.
- Statement from Commissioner Scott Gottlieb, MD, on new FDA actions to keep consumers safe from the harmful effects of sun exposure, and ensure the long-term safety and benefits of sunscreens [news release]. Silver Spring, MD: US Food and Drug Administration; May 22, 2018. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm608499.htm. Accessed April 5, 2019.
- FDA advances new proposed regulation to make sure that sunscreens are safe and effective [news release]. Silver Spring, MD: US Food and Drug Administration; February 21, 2019. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm631736.htm. Accessed April 4, 2019.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 2019;84(38):6204-6275. To be codified at 21 CFR §201, 310, 347, and 352.
- Sunscreen drug products for over-the-counter human use. Fed Registr. 1978;43(166):38206-38269. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph. Fed Registr. 1996;60(180):48645-48655. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; amendment to the tentative final monograph; enforcement policy. Fed Registr. 1998;63(204):56584-56589. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; final monograph. Fed Registr. 1999;64(98):27666-27693. To be codified at 21 CFR §310, 352, 700, and 740.
- Sunscreen drug products for over-the-counter human use; final monograph; partial stay; final rule. Fed Registr. 2001;66:67485-67487. To be codified at 21 CFR §352.
- Sunscreen drug products for over-the-counter human use; proposed amendment of final monograph. Fed Registr. 2007;72(165):49069-49122. To be codified at 21 CFR §347 and 352.
- Labeling and effectiveness testing; sunscreen drug products for over-the-counter human use. Fed Registr. 2011;76(117):35619-35665. To be codified at 21 CFR §201 and 310.
- Sunscreen Innovation Act, S 2141, 113th Cong, 2nd Sess (2014).
- Nonprescription sunscreen drug products-safety and effectiveness data; guidance for industry; availability. Fed Registr. 2016;81(226):84594-84595.
- Statement from Commissioner Scott Gottlieb, MD, on new FDA actions to keep consumers safe from the harmful effects of sun exposure, and ensure the long-term safety and benefits of sunscreens [news release]. Silver Spring, MD: US Food and Drug Administration; May 22, 2018. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm608499.htm. Accessed April 5, 2019.

An Integrated System for the Recording and Retrieval of Medical Data in a Primary Care Setting: Part 7: The Encounter Form and the Minimum Basic Data Set
A Critical Review of Periodic Health Screening Using Specific Screening Criteria: Part 1: Selected Diseases of Respiratory, Cardiovascular, and Central Nervous System
Toward a Research Base in Family Practice
Filamentous bacteriophage linked to lung infections in patients with cystic fibrosis
according to a study of the prevalence and clinical relevance of Pf phage in two patient cohorts.
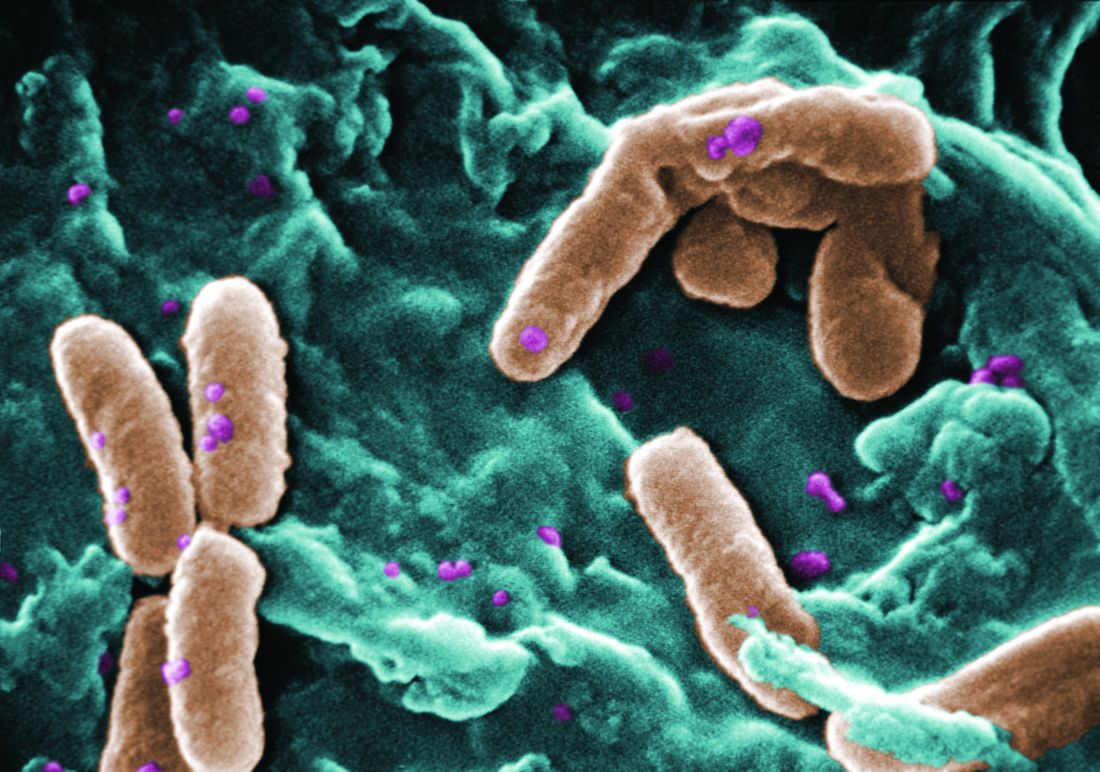
“Our data from both the Stanford and Danish CF cohorts together suggest that either patients with CF acquire Pf phage–producing strains of P. aeruginosa or the Pf phage–negative P. aeruginosa become infected with Pf phage as patients age and their disease progresses,” wrote Elizabeth B. Burgener, MD, of Stanford (Calif.) University, and her coauthors. The study was published in Science Translational Medicine.The study analyzed a previous Danish longitudinal cohort of 34 patients and a prospective cross-sectional cohort of 76 patients at Stanford, 58 of which had P. aeruginosa. The researchers also reviewed a collection of genetic sequences called the Pseudomonas Genome Database to determine the prevalence of Pf phage, finding evidence in 1,159 of 2,226 P. aeruginosa sequences (52.1%).
In the Danish cohort, 21 of the 34 CF patients (61.8%; 95% confidence interval, 43.6%-77.8%) had at least one P. aeruginosa isolate containing Pf phage; 9 (26.5%) of the patients were found to be consistently positive for Pf phage. Those who were consistently positive were also older than those who never had Pf phage detected (19.1 years vs. 13.9 years; P = .046), suggesting that “there may be a tendency for P. aeruginosa strains that produce Pf phage to dominate in the sputum of individual patients with CF over time.”
In the Stanford cohort, the prevalence of Pf phage was 36.2% (21 of 58; 95% CI, 24.0%-49.9%) in patients with P. aeruginosa infection and 27.6% (21 of 76; 95% CI, 18.0%-39.1%) in all patients. No Pf phage was detected in any P. aeruginosa–negative samples. Patients positive for Pf phage in this cohort were also older than patients who were negative.
The authors acknowledged their study’s limitations, describing the methods used to collect and sequence the analyzed samples as “highly heterogeneous.” In addition, the two cohorts were CF specific while the Genome Database is not. Finally, the CF cohorts only had a single dominant strain sampled, though multiple P. aeruginosa lineages are often present in CF patients.
One author reported receiving grants from the Cystic Fibrosis Foundation, clinical trial support, and consulting fees from industry. Two other authors are inventors on a patent application that covers the development of a vaccine that targets Pf phage. The others reported no conflicts of interest.
SOURCE: Burgener EB et al. Sci Transl Med. 2019 Apr 17. doi: 10.1126/scitranslmed.aau9748.
according to a study of the prevalence and clinical relevance of Pf phage in two patient cohorts.
“Our data from both the Stanford and Danish CF cohorts together suggest that either patients with CF acquire Pf phage–producing strains of P. aeruginosa or the Pf phage–negative P. aeruginosa become infected with Pf phage as patients age and their disease progresses,” wrote Elizabeth B. Burgener, MD, of Stanford (Calif.) University, and her coauthors. The study was published in Science Translational Medicine.The study analyzed a previous Danish longitudinal cohort of 34 patients and a prospective cross-sectional cohort of 76 patients at Stanford, 58 of which had P. aeruginosa. The researchers also reviewed a collection of genetic sequences called the Pseudomonas Genome Database to determine the prevalence of Pf phage, finding evidence in 1,159 of 2,226 P. aeruginosa sequences (52.1%).
In the Danish cohort, 21 of the 34 CF patients (61.8%; 95% confidence interval, 43.6%-77.8%) had at least one P. aeruginosa isolate containing Pf phage; 9 (26.5%) of the patients were found to be consistently positive for Pf phage. Those who were consistently positive were also older than those who never had Pf phage detected (19.1 years vs. 13.9 years; P = .046), suggesting that “there may be a tendency for P. aeruginosa strains that produce Pf phage to dominate in the sputum of individual patients with CF over time.”
In the Stanford cohort, the prevalence of Pf phage was 36.2% (21 of 58; 95% CI, 24.0%-49.9%) in patients with P. aeruginosa infection and 27.6% (21 of 76; 95% CI, 18.0%-39.1%) in all patients. No Pf phage was detected in any P. aeruginosa–negative samples. Patients positive for Pf phage in this cohort were also older than patients who were negative.
The authors acknowledged their study’s limitations, describing the methods used to collect and sequence the analyzed samples as “highly heterogeneous.” In addition, the two cohorts were CF specific while the Genome Database is not. Finally, the CF cohorts only had a single dominant strain sampled, though multiple P. aeruginosa lineages are often present in CF patients.
One author reported receiving grants from the Cystic Fibrosis Foundation, clinical trial support, and consulting fees from industry. Two other authors are inventors on a patent application that covers the development of a vaccine that targets Pf phage. The others reported no conflicts of interest.
SOURCE: Burgener EB et al. Sci Transl Med. 2019 Apr 17. doi: 10.1126/scitranslmed.aau9748.
according to a study of the prevalence and clinical relevance of Pf phage in two patient cohorts.
“Our data from both the Stanford and Danish CF cohorts together suggest that either patients with CF acquire Pf phage–producing strains of P. aeruginosa or the Pf phage–negative P. aeruginosa become infected with Pf phage as patients age and their disease progresses,” wrote Elizabeth B. Burgener, MD, of Stanford (Calif.) University, and her coauthors. The study was published in Science Translational Medicine.The study analyzed a previous Danish longitudinal cohort of 34 patients and a prospective cross-sectional cohort of 76 patients at Stanford, 58 of which had P. aeruginosa. The researchers also reviewed a collection of genetic sequences called the Pseudomonas Genome Database to determine the prevalence of Pf phage, finding evidence in 1,159 of 2,226 P. aeruginosa sequences (52.1%).
In the Danish cohort, 21 of the 34 CF patients (61.8%; 95% confidence interval, 43.6%-77.8%) had at least one P. aeruginosa isolate containing Pf phage; 9 (26.5%) of the patients were found to be consistently positive for Pf phage. Those who were consistently positive were also older than those who never had Pf phage detected (19.1 years vs. 13.9 years; P = .046), suggesting that “there may be a tendency for P. aeruginosa strains that produce Pf phage to dominate in the sputum of individual patients with CF over time.”
In the Stanford cohort, the prevalence of Pf phage was 36.2% (21 of 58; 95% CI, 24.0%-49.9%) in patients with P. aeruginosa infection and 27.6% (21 of 76; 95% CI, 18.0%-39.1%) in all patients. No Pf phage was detected in any P. aeruginosa–negative samples. Patients positive for Pf phage in this cohort were also older than patients who were negative.
The authors acknowledged their study’s limitations, describing the methods used to collect and sequence the analyzed samples as “highly heterogeneous.” In addition, the two cohorts were CF specific while the Genome Database is not. Finally, the CF cohorts only had a single dominant strain sampled, though multiple P. aeruginosa lineages are often present in CF patients.
One author reported receiving grants from the Cystic Fibrosis Foundation, clinical trial support, and consulting fees from industry. Two other authors are inventors on a patent application that covers the development of a vaccine that targets Pf phage. The others reported no conflicts of interest.
SOURCE: Burgener EB et al. Sci Transl Med. 2019 Apr 17. doi: 10.1126/scitranslmed.aau9748.
FROM SCIENCE TRANSLATIONAL MEDICINE