User login
Fever, tachycardia, and tachypnea during a psychotic exacerbation
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.

Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.

EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
Psychotropic-induced hyponatremia

Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1

Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.

Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.

Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24

Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.

Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.

Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Psychiatry’s social impact: Pervasive and multifaceted
Psychiatry has an enormous swath of effects on the social structure of society, perhaps more than any other medical specialty. Its ramifications can be observed and experienced across medical, scientific, legal, financial, political, sexual, religious, cultural, sociological, and artistic aspects of the aggregate of humans living together that we call society.
And yet, despite its pervasive and significant consequences at multiple levels of human communities, psychiatry remains inadequately appreciated or understood. In fact, it is sometimes maligned in a manner that no other medical discipline ever has to face.
I will expound on what may sound like a sweeping statement, and let you decide if society is indeed influenced in myriad ways by the wide array of psychiatric brain disorders that impact various core components of society.
Consider the following major societal repercussions of psychiatric disorders:
")
- Twenty-five percent of the population suffers from a psychiatric disorder per the landmark Epidemiological Catchment Area (ECA) study,1,2 funded by the National Institutes of Health. This translates to 85 million children, adolescents, adults, and older adults. No other medical specialty comes close to affecting this massive number of individuals in society.
- According to the World Health Organization (WHO), 4 of the top 10 causes of disability across all medical conditions are psychiatric disorders (Table3). Depression, alcoholism, schizophrenia, and bipolar disorder account for the greatest proportion of individuals with disabilities. Obviously, the impact of psychiatry in society is more significant than any other medical specialty as far as functional disability is concerned.
- The jails and prisons of the country are brimming with psychiatric patients who are arrested, incarcerated, and criminalized because their brain disorder disrupts their behavior. This is one of the most serious (and frankly outrageous) legal problems in our society. It occurred after our society decided to shutter state-supported hospitals (asylums) where psychiatric patients used to be treated as medically ill persons by health care professionals such as physicians, nurses, psychologists, and social workers, not prison guards. Remember that in the 1960s, 50% of all hospital beds in the United States were occupied by psychiatric patients, which is another historical indication of the societal impact of psychiatry.
- Alcohol and drug abuse are undoubtedly one of society’s most intractable problems. They are not only psychiatric disorders, but are often associated with multiple other psychiatric comorbidities and can lead to a host of general medical and surgical consequences. They are not only costly in financial terms, but they also lead to an increase in crime and forensic problems. Premature death is a heavy toll for society due to alcohol and substance use, as the opioid epidemic clearly has demonstrated over the past few years.
- Homelessness is an endemic sociological cancer in the body of society and is very often driven by psychiatric disorders and addictions. Countless numbers of severely mentally ill patients became homeless when asylums were closed and they were “freed” from restrictive institutional settings. Homelessness and imprisonment became the heavy and shameful price of “freedom” for persons with disabling psychiatric disorders in our “advanced” society.
- Suicide, both completed and attempted, is intimately associated with psychiatric disorders. Approximately 47,000 deaths from suicide were reported in the United States in 2017.4 Given that more than 30 million Americans suffer from mood disorders, millions of suicide attempts take place, crowding the emergency rooms of the country with individuals who need to receive emergent health care. The tragic toll of suicide and the heavy medical care costs of suicide attempts are incalculable, and unfortunately have been growing steadily over the past 20 years.
- Homicide is sometimes committed by persons with a psychiatric disorder, most commonly antisocial personality disorder. The rate of homicide often is used as a measure of a city’s quality of life, and urban areas where access to psychiatric care is limited tend to have high homicide rates.
- School problems, whether due to attention-deficit/hyperactivity disorder, below-average intellectual abilities, conduct disorder, bullying, impulsive behavior, substance use, broken homes, or dysfunctional families (often due to addictive or psychiatric disorders), are a major societal problem. Whether the problem is truancy, school fights, or dropping out before getting a high school diploma, psychiatric illness is frequently the underlying reason.
- Sexual controversies, such as expanding and evolving gender identity issues and discrimination against non-cisgender individuals, have instigated both positive and negative initiatives in society. Sexual abuse of children and its grave psychiatric implications in adulthood continues to happen despite public outrage and law enforcement efforts, and is often driven by individuals with serious psychopathology. In addition, sexual addiction (and its many biopsychosocial complications) is often associated with neuropsychiatric disorders.
- Poverty and the perpetual underclass are often a result of psychiatric disorders, and represent an ongoing societal challenge that has proven impossible to fix just by throwing money at it. Whether the affected individuals are seriously mentally ill, addicted, cognitively impaired or challenged, or unmotivated because of a neuropsychiatric disorder, poverty is practically impossible to eliminate.
- One positive impact of psychiatry in society is that artistic abilities, writing talent, musical creativity, entrepreneurship, and high productivity are often associated with certain psychiatric conditions, such as bipolar disorder, autism, obsessive-compulsive disorder, and psychosis spectrum disorders. Society is enriched by the creative energy and out-of-the-box thinking of persons with mild to moderate neuropsychiatric disorders.
- The financial impact of psychiatry is massive. The direct and indirect costs of psychiatric and addictive disorders are estimated to be more than $400 billion/year. Even a single serious psychiatric disorder, such as schizophrenia, costs society approximately $70 billion/year. The same holds true for bipolar disorder and depression. Thus, psychiatry accounts for a substantial portion of the financial expenditures in society.
- And last but certainly not least are the impediments to psychiatric treatment for tens of millions of individuals in our society who need treatment the most: the lack of health insurance parity; the stigma of seeking psychiatric help; the serious shortage of psychiatrists, especially in inner-city areas and rural regions; the poor public understanding about psychiatric illness; and the fact that the success rate of psychiatric treatment is very similar to (and sometimes better than) that of serious cardiac, pulmonary, hepatic, or renal diseases. There are also many flawed religious, cultural, or philosophical belief systems that fail to accept that the mind is a product of brain biology and function and that psychiatric disorders are brain disorders that affect thought, mood impulses, cognition, and behavior, just as other brain disorders cause muscle weakness, epileptic seizures, or stroke. The public must understand that depression can be caused by stroke or multiple sclerosis, that Parkinson’s disease can cause hallucinations and delusions, and that brain tumors can cause personality changes.
Continue to: So, what should society do to address...
So, what should society do to address the multiple impacts of psychiatry on its structure and function? I have a brief answer: intensive research. If society would embark on a massive research effort to discover preventions and cures for psychiatric disorders, the return on investment would be tremendous in human and financial terms. Currently, only a miniscule amount of money (<0.5% of the annual cost of psychiatric disorders) is invested in psychiatric brain research. Society should embark on a BHAG (pronounced Bee Hag), an acronym for “Big Hairy Audacious Goal,” a term coined by Jim Collins and Jerry Poras, who authored the seminal book Built to Last: Successful Habits of Visionary Companies. The BHAG is an ambitious and visionary goal that steers a company (or in this case, society) to a much brighter future. It would be on the scale of the Manhattan Project in the 1940s, which developed the nuclear bomb that put an end to World War II. When it comes to psychiatry, society should do no less.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Regier DA, Myers JK, Kramer M, et al. The NIMH Epidemiologic Catchment Area program. Historical context, major objectives, and study population characteristics. Arch Gen Psychiatry. 1984;41(10):934-941.
2. Robins LN, Regier DA (eds). Psychiatric disorders in America: The Epidemiological Catchment Area Study. New York, NY: The Free Press; 1992.
3. World Health Organization. Global Burden of Disease (GBD) 2000 estimates. https://www.who.int/healthinfo/global_burden_disease/estimates_regional_2000/en/. Accessed January 17, 2019.
4. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Accessed January 18, 2019.
Psychiatry has an enormous swath of effects on the social structure of society, perhaps more than any other medical specialty. Its ramifications can be observed and experienced across medical, scientific, legal, financial, political, sexual, religious, cultural, sociological, and artistic aspects of the aggregate of humans living together that we call society.
And yet, despite its pervasive and significant consequences at multiple levels of human communities, psychiatry remains inadequately appreciated or understood. In fact, it is sometimes maligned in a manner that no other medical discipline ever has to face.
I will expound on what may sound like a sweeping statement, and let you decide if society is indeed influenced in myriad ways by the wide array of psychiatric brain disorders that impact various core components of society.
Consider the following major societal repercussions of psychiatric disorders:
- Twenty-five percent of the population suffers from a psychiatric disorder per the landmark Epidemiological Catchment Area (ECA) study,1,2 funded by the National Institutes of Health. This translates to 85 million children, adolescents, adults, and older adults. No other medical specialty comes close to affecting this massive number of individuals in society.
- According to the World Health Organization (WHO), 4 of the top 10 causes of disability across all medical conditions are psychiatric disorders (Table3). Depression, alcoholism, schizophrenia, and bipolar disorder account for the greatest proportion of individuals with disabilities. Obviously, the impact of psychiatry in society is more significant than any other medical specialty as far as functional disability is concerned.
- The jails and prisons of the country are brimming with psychiatric patients who are arrested, incarcerated, and criminalized because their brain disorder disrupts their behavior. This is one of the most serious (and frankly outrageous) legal problems in our society. It occurred after our society decided to shutter state-supported hospitals (asylums) where psychiatric patients used to be treated as medically ill persons by health care professionals such as physicians, nurses, psychologists, and social workers, not prison guards. Remember that in the 1960s, 50% of all hospital beds in the United States were occupied by psychiatric patients, which is another historical indication of the societal impact of psychiatry.
- Alcohol and drug abuse are undoubtedly one of society’s most intractable problems. They are not only psychiatric disorders, but are often associated with multiple other psychiatric comorbidities and can lead to a host of general medical and surgical consequences. They are not only costly in financial terms, but they also lead to an increase in crime and forensic problems. Premature death is a heavy toll for society due to alcohol and substance use, as the opioid epidemic clearly has demonstrated over the past few years.
- Homelessness is an endemic sociological cancer in the body of society and is very often driven by psychiatric disorders and addictions. Countless numbers of severely mentally ill patients became homeless when asylums were closed and they were “freed” from restrictive institutional settings. Homelessness and imprisonment became the heavy and shameful price of “freedom” for persons with disabling psychiatric disorders in our “advanced” society.
- Suicide, both completed and attempted, is intimately associated with psychiatric disorders. Approximately 47,000 deaths from suicide were reported in the United States in 2017.4 Given that more than 30 million Americans suffer from mood disorders, millions of suicide attempts take place, crowding the emergency rooms of the country with individuals who need to receive emergent health care. The tragic toll of suicide and the heavy medical care costs of suicide attempts are incalculable, and unfortunately have been growing steadily over the past 20 years.
- Homicide is sometimes committed by persons with a psychiatric disorder, most commonly antisocial personality disorder. The rate of homicide often is used as a measure of a city’s quality of life, and urban areas where access to psychiatric care is limited tend to have high homicide rates.
- School problems, whether due to attention-deficit/hyperactivity disorder, below-average intellectual abilities, conduct disorder, bullying, impulsive behavior, substance use, broken homes, or dysfunctional families (often due to addictive or psychiatric disorders), are a major societal problem. Whether the problem is truancy, school fights, or dropping out before getting a high school diploma, psychiatric illness is frequently the underlying reason.
- Sexual controversies, such as expanding and evolving gender identity issues and discrimination against non-cisgender individuals, have instigated both positive and negative initiatives in society. Sexual abuse of children and its grave psychiatric implications in adulthood continues to happen despite public outrage and law enforcement efforts, and is often driven by individuals with serious psychopathology. In addition, sexual addiction (and its many biopsychosocial complications) is often associated with neuropsychiatric disorders.
- Poverty and the perpetual underclass are often a result of psychiatric disorders, and represent an ongoing societal challenge that has proven impossible to fix just by throwing money at it. Whether the affected individuals are seriously mentally ill, addicted, cognitively impaired or challenged, or unmotivated because of a neuropsychiatric disorder, poverty is practically impossible to eliminate.
- One positive impact of psychiatry in society is that artistic abilities, writing talent, musical creativity, entrepreneurship, and high productivity are often associated with certain psychiatric conditions, such as bipolar disorder, autism, obsessive-compulsive disorder, and psychosis spectrum disorders. Society is enriched by the creative energy and out-of-the-box thinking of persons with mild to moderate neuropsychiatric disorders.
- The financial impact of psychiatry is massive. The direct and indirect costs of psychiatric and addictive disorders are estimated to be more than $400 billion/year. Even a single serious psychiatric disorder, such as schizophrenia, costs society approximately $70 billion/year. The same holds true for bipolar disorder and depression. Thus, psychiatry accounts for a substantial portion of the financial expenditures in society.
- And last but certainly not least are the impediments to psychiatric treatment for tens of millions of individuals in our society who need treatment the most: the lack of health insurance parity; the stigma of seeking psychiatric help; the serious shortage of psychiatrists, especially in inner-city areas and rural regions; the poor public understanding about psychiatric illness; and the fact that the success rate of psychiatric treatment is very similar to (and sometimes better than) that of serious cardiac, pulmonary, hepatic, or renal diseases. There are also many flawed religious, cultural, or philosophical belief systems that fail to accept that the mind is a product of brain biology and function and that psychiatric disorders are brain disorders that affect thought, mood impulses, cognition, and behavior, just as other brain disorders cause muscle weakness, epileptic seizures, or stroke. The public must understand that depression can be caused by stroke or multiple sclerosis, that Parkinson’s disease can cause hallucinations and delusions, and that brain tumors can cause personality changes.
Continue to: So, what should society do to address...
So, what should society do to address the multiple impacts of psychiatry on its structure and function? I have a brief answer: intensive research. If society would embark on a massive research effort to discover preventions and cures for psychiatric disorders, the return on investment would be tremendous in human and financial terms. Currently, only a miniscule amount of money (<0.5% of the annual cost of psychiatric disorders) is invested in psychiatric brain research. Society should embark on a BHAG (pronounced Bee Hag), an acronym for “Big Hairy Audacious Goal,” a term coined by Jim Collins and Jerry Poras, who authored the seminal book Built to Last: Successful Habits of Visionary Companies. The BHAG is an ambitious and visionary goal that steers a company (or in this case, society) to a much brighter future. It would be on the scale of the Manhattan Project in the 1940s, which developed the nuclear bomb that put an end to World War II. When it comes to psychiatry, society should do no less.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
Psychiatry has an enormous swath of effects on the social structure of society, perhaps more than any other medical specialty. Its ramifications can be observed and experienced across medical, scientific, legal, financial, political, sexual, religious, cultural, sociological, and artistic aspects of the aggregate of humans living together that we call society.
And yet, despite its pervasive and significant consequences at multiple levels of human communities, psychiatry remains inadequately appreciated or understood. In fact, it is sometimes maligned in a manner that no other medical discipline ever has to face.
I will expound on what may sound like a sweeping statement, and let you decide if society is indeed influenced in myriad ways by the wide array of psychiatric brain disorders that impact various core components of society.
Consider the following major societal repercussions of psychiatric disorders:
- Twenty-five percent of the population suffers from a psychiatric disorder per the landmark Epidemiological Catchment Area (ECA) study,1,2 funded by the National Institutes of Health. This translates to 85 million children, adolescents, adults, and older adults. No other medical specialty comes close to affecting this massive number of individuals in society.
- According to the World Health Organization (WHO), 4 of the top 10 causes of disability across all medical conditions are psychiatric disorders (Table3). Depression, alcoholism, schizophrenia, and bipolar disorder account for the greatest proportion of individuals with disabilities. Obviously, the impact of psychiatry in society is more significant than any other medical specialty as far as functional disability is concerned.
- The jails and prisons of the country are brimming with psychiatric patients who are arrested, incarcerated, and criminalized because their brain disorder disrupts their behavior. This is one of the most serious (and frankly outrageous) legal problems in our society. It occurred after our society decided to shutter state-supported hospitals (asylums) where psychiatric patients used to be treated as medically ill persons by health care professionals such as physicians, nurses, psychologists, and social workers, not prison guards. Remember that in the 1960s, 50% of all hospital beds in the United States were occupied by psychiatric patients, which is another historical indication of the societal impact of psychiatry.
- Alcohol and drug abuse are undoubtedly one of society’s most intractable problems. They are not only psychiatric disorders, but are often associated with multiple other psychiatric comorbidities and can lead to a host of general medical and surgical consequences. They are not only costly in financial terms, but they also lead to an increase in crime and forensic problems. Premature death is a heavy toll for society due to alcohol and substance use, as the opioid epidemic clearly has demonstrated over the past few years.
- Homelessness is an endemic sociological cancer in the body of society and is very often driven by psychiatric disorders and addictions. Countless numbers of severely mentally ill patients became homeless when asylums were closed and they were “freed” from restrictive institutional settings. Homelessness and imprisonment became the heavy and shameful price of “freedom” for persons with disabling psychiatric disorders in our “advanced” society.
- Suicide, both completed and attempted, is intimately associated with psychiatric disorders. Approximately 47,000 deaths from suicide were reported in the United States in 2017.4 Given that more than 30 million Americans suffer from mood disorders, millions of suicide attempts take place, crowding the emergency rooms of the country with individuals who need to receive emergent health care. The tragic toll of suicide and the heavy medical care costs of suicide attempts are incalculable, and unfortunately have been growing steadily over the past 20 years.
- Homicide is sometimes committed by persons with a psychiatric disorder, most commonly antisocial personality disorder. The rate of homicide often is used as a measure of a city’s quality of life, and urban areas where access to psychiatric care is limited tend to have high homicide rates.
- School problems, whether due to attention-deficit/hyperactivity disorder, below-average intellectual abilities, conduct disorder, bullying, impulsive behavior, substance use, broken homes, or dysfunctional families (often due to addictive or psychiatric disorders), are a major societal problem. Whether the problem is truancy, school fights, or dropping out before getting a high school diploma, psychiatric illness is frequently the underlying reason.
- Sexual controversies, such as expanding and evolving gender identity issues and discrimination against non-cisgender individuals, have instigated both positive and negative initiatives in society. Sexual abuse of children and its grave psychiatric implications in adulthood continues to happen despite public outrage and law enforcement efforts, and is often driven by individuals with serious psychopathology. In addition, sexual addiction (and its many biopsychosocial complications) is often associated with neuropsychiatric disorders.
- Poverty and the perpetual underclass are often a result of psychiatric disorders, and represent an ongoing societal challenge that has proven impossible to fix just by throwing money at it. Whether the affected individuals are seriously mentally ill, addicted, cognitively impaired or challenged, or unmotivated because of a neuropsychiatric disorder, poverty is practically impossible to eliminate.
- One positive impact of psychiatry in society is that artistic abilities, writing talent, musical creativity, entrepreneurship, and high productivity are often associated with certain psychiatric conditions, such as bipolar disorder, autism, obsessive-compulsive disorder, and psychosis spectrum disorders. Society is enriched by the creative energy and out-of-the-box thinking of persons with mild to moderate neuropsychiatric disorders.
- The financial impact of psychiatry is massive. The direct and indirect costs of psychiatric and addictive disorders are estimated to be more than $400 billion/year. Even a single serious psychiatric disorder, such as schizophrenia, costs society approximately $70 billion/year. The same holds true for bipolar disorder and depression. Thus, psychiatry accounts for a substantial portion of the financial expenditures in society.
- And last but certainly not least are the impediments to psychiatric treatment for tens of millions of individuals in our society who need treatment the most: the lack of health insurance parity; the stigma of seeking psychiatric help; the serious shortage of psychiatrists, especially in inner-city areas and rural regions; the poor public understanding about psychiatric illness; and the fact that the success rate of psychiatric treatment is very similar to (and sometimes better than) that of serious cardiac, pulmonary, hepatic, or renal diseases. There are also many flawed religious, cultural, or philosophical belief systems that fail to accept that the mind is a product of brain biology and function and that psychiatric disorders are brain disorders that affect thought, mood impulses, cognition, and behavior, just as other brain disorders cause muscle weakness, epileptic seizures, or stroke. The public must understand that depression can be caused by stroke or multiple sclerosis, that Parkinson’s disease can cause hallucinations and delusions, and that brain tumors can cause personality changes.
Continue to: So, what should society do to address...
So, what should society do to address the multiple impacts of psychiatry on its structure and function? I have a brief answer: intensive research. If society would embark on a massive research effort to discover preventions and cures for psychiatric disorders, the return on investment would be tremendous in human and financial terms. Currently, only a miniscule amount of money (<0.5% of the annual cost of psychiatric disorders) is invested in psychiatric brain research. Society should embark on a BHAG (pronounced Bee Hag), an acronym for “Big Hairy Audacious Goal,” a term coined by Jim Collins and Jerry Poras, who authored the seminal book Built to Last: Successful Habits of Visionary Companies. The BHAG is an ambitious and visionary goal that steers a company (or in this case, society) to a much brighter future. It would be on the scale of the Manhattan Project in the 1940s, which developed the nuclear bomb that put an end to World War II. When it comes to psychiatry, society should do no less.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Regier DA, Myers JK, Kramer M, et al. The NIMH Epidemiologic Catchment Area program. Historical context, major objectives, and study population characteristics. Arch Gen Psychiatry. 1984;41(10):934-941.
2. Robins LN, Regier DA (eds). Psychiatric disorders in America: The Epidemiological Catchment Area Study. New York, NY: The Free Press; 1992.
3. World Health Organization. Global Burden of Disease (GBD) 2000 estimates. https://www.who.int/healthinfo/global_burden_disease/estimates_regional_2000/en/. Accessed January 17, 2019.
4. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Accessed January 18, 2019.
1. Regier DA, Myers JK, Kramer M, et al. The NIMH Epidemiologic Catchment Area program. Historical context, major objectives, and study population characteristics. Arch Gen Psychiatry. 1984;41(10):934-941.
2. Robins LN, Regier DA (eds). Psychiatric disorders in America: The Epidemiological Catchment Area Study. New York, NY: The Free Press; 1992.
3. World Health Organization. Global Burden of Disease (GBD) 2000 estimates. https://www.who.int/healthinfo/global_burden_disease/estimates_regional_2000/en/. Accessed January 17, 2019.
4. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Accessed January 18, 2019.

The effect of collateral information on involuntary psychiatric commitment
Collateral information is a key component obtained during the psych
Here I describe a case in which collateral information obtained about a patient was a primary factor in that patient’s involuntary commitment. However, the patient’s subsequent behavior observed on an inpatient psychiatric unit was entirely inconsistent with those behaviors described by the collateral informant to be “continuous and dangerous.”
CASE
Mr. M, age 18, presented to an emergency psychiatric center for evaluation of dangerous and aggressive behavior. He had a history of autism spectrum disorder (ASD), which was well managed with oral risperidone. He was petitioned for an involuntary psychiatric admission by his foster mother, who reported that Mr. M was aggressive and dangerous, often punching holes in the walls of their home, and that he threatened to assault his foster siblings on several occasions. She detailed a progressively declining history for Mr. M and said that he was “constantly talking to voices in his head that absolutely consume him,” to the extent that Mr. M could not pay attention to his daily tasks. The admitting psychiatrist upheld the petition for involuntary admission, citing that based on the foster’s mother collateral information, Mr. M was deemed to be a danger to others and therefore fulfilled criteria for involuntary psychiatric admission.
Once admitted to the inpatient psychiatric unit, Mr. M was observed to be pleasant, cooperative, and fully engaged in the milieu. At no point during his 7-day admission was he observed to be internally preoccupied or remotely disorganized. Mr. M was switched from oral risperidone to oral haloperidol because he developed acute gynecomastia, and was discharged home.
Does collateral information lead to unfair bias?
The importance of collateral information on the psychiatric admission process must not be understated. It is an opportunity to hear a first-hand account of behaviors consistent with an acute psychiatric disturbance, and guides us in formulating a clinically appropriate assessment and plan. But what happens when our patients’ close contacts or informants provide misleading or unintentionally suboptimal collateral information? How must we reconcile the ethical and legal obligation we have to balance patient autonomy with beneficence?
Studies examining patients’ attitudes toward involuntary admissions have routinely found that patients are less likely than clinical staff to view the involuntary admission as clinically justified.2 Consistent with these findings, Mr. M did not view his admission as necessary. At first, he seemed to lack insight regarding the events precipitating his involuntary admission, describing himself not as responding to internal stimuli, but rather, “imaginative because I have autism.” As time went on, though, it was clear that his account of his behavior was in fact correct.
Mr. M’s diagnosis of ASD further complicated the over-reliance on misleading collateral information provided by his foster mother, because the admitting psychiatrist invariably perceived Mr. M as a poor historian. A study examining how subjective histories described by patients with neurologic or psychiatric disorders are perceived by clinicians found physicians had a tendency for negative stereotyping and placed less credence on those patients’ subjective histories.3 Other literature has similarly concluded that there is an urgent need to carefully weigh information supplied to us by collateral informants because the first-hand accounts of perceivably dangerous behavior often are incomplete or misleading.4-5
Continue to: Ideas for improvement...
Ideas for improvement: respecting patient autonomy
These issues underscore the need for a more thorough review of collateral information to ensure that patient autonomy is not unjustly violated. How do we implement these necessary ideas without creating further undue burden during the admission process? Certainly, I am not suggesting that we evaluate the collateral informant to the degree that we evaluate the patient. However, I have outlined some suggestions for ensuring we act in our patients’ best interest when processing collateral information during an admission:
- Until proven otherwise, the patient’s story is true. If our patient maintains descriptions of his behavior that exist in stark opposition to the collateral information we obtain, we should only not believe the patient if his presentation suggests he may be acutely impaired or a poor historian (such as profound disorganization, overt psychosis, or failing to have capacity).
- Treat symptoms, not diagnoses. In this case, Mr. M was described by his foster mother to be psychotic in addition to having ASD, and an inexperienced psychiatrist may have initiated a titration to a higher antipsychotic dose. However, in the absence of any observable signs of aggression or psychosis, there was simply no indication for further titration of his antipsychotic.
- Document, document, document. When collateral information is supplied to us, it is crucial that we maintain a detailed account of this information. If we have a reason to believe that a patient poses an immediate danger to himself or others, we should carefully document our reasoning so that changes in behavior (if any) can be observed on a day-to-day basis.
1. Testa M, West SG. Civil commitment in the United States. Psychiatry (Edgmont). 2010;7(10):30-40.
2. Roe D, Weishut DJ, Jaglom M, et al. Patients’ and staff members’ attitudes about the rights of hospitalized psychiatric patients. Psychiatr Serv. 2002;53(1):87-91.
3. Crichton P, Carel H, Kidd IJ. Epistemic injustice in psychiatry. BJPsych Bull. 2017;41(2):65-70.
4. Marett C, Mossman D. What is your liability for involuntary commitment based on fault information? Current Psychiatry. 2017;16(3):21-25,33.
5. Lincoln AL, Allen M. The influence of collateral information on access to inpatient psychiatric services. International Journal of Psychosocial Rehabilitation. 2002;6:99-108.

Dr. Witkin is a PGY-1 Psychiatry Resident, Department of Psychiatry, Albert Einstein Medical Center, Philadelphia, Pennsylvania.
Disclosure
The author reports no financial relationships with any companies whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Witkin is a PGY-1 Psychiatry Resident, Department of Psychiatry, Albert Einstein Medical Center, Philadelphia, Pennsylvania.
Disclosure
The author reports no financial relationships with any companies whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Witkin is a PGY-1 Psychiatry Resident, Department of Psychiatry, Albert Einstein Medical Center, Philadelphia, Pennsylvania.
Disclosure
The author reports no financial relationships with any companies whose products are mentioned in this article, or with manufacturers of competing products.
Collateral information is a key component obtained during the psych
Here I describe a case in which collateral information obtained about a patient was a primary factor in that patient’s involuntary commitment. However, the patient’s subsequent behavior observed on an inpatient psychiatric unit was entirely inconsistent with those behaviors described by the collateral informant to be “continuous and dangerous.”
CASE
Mr. M, age 18, presented to an emergency psychiatric center for evaluation of dangerous and aggressive behavior. He had a history of autism spectrum disorder (ASD), which was well managed with oral risperidone. He was petitioned for an involuntary psychiatric admission by his foster mother, who reported that Mr. M was aggressive and dangerous, often punching holes in the walls of their home, and that he threatened to assault his foster siblings on several occasions. She detailed a progressively declining history for Mr. M and said that he was “constantly talking to voices in his head that absolutely consume him,” to the extent that Mr. M could not pay attention to his daily tasks. The admitting psychiatrist upheld the petition for involuntary admission, citing that based on the foster’s mother collateral information, Mr. M was deemed to be a danger to others and therefore fulfilled criteria for involuntary psychiatric admission.
Once admitted to the inpatient psychiatric unit, Mr. M was observed to be pleasant, cooperative, and fully engaged in the milieu. At no point during his 7-day admission was he observed to be internally preoccupied or remotely disorganized. Mr. M was switched from oral risperidone to oral haloperidol because he developed acute gynecomastia, and was discharged home.
Does collateral information lead to unfair bias?
The importance of collateral information on the psychiatric admission process must not be understated. It is an opportunity to hear a first-hand account of behaviors consistent with an acute psychiatric disturbance, and guides us in formulating a clinically appropriate assessment and plan. But what happens when our patients’ close contacts or informants provide misleading or unintentionally suboptimal collateral information? How must we reconcile the ethical and legal obligation we have to balance patient autonomy with beneficence?
Studies examining patients’ attitudes toward involuntary admissions have routinely found that patients are less likely than clinical staff to view the involuntary admission as clinically justified.2 Consistent with these findings, Mr. M did not view his admission as necessary. At first, he seemed to lack insight regarding the events precipitating his involuntary admission, describing himself not as responding to internal stimuli, but rather, “imaginative because I have autism.” As time went on, though, it was clear that his account of his behavior was in fact correct.
Mr. M’s diagnosis of ASD further complicated the over-reliance on misleading collateral information provided by his foster mother, because the admitting psychiatrist invariably perceived Mr. M as a poor historian. A study examining how subjective histories described by patients with neurologic or psychiatric disorders are perceived by clinicians found physicians had a tendency for negative stereotyping and placed less credence on those patients’ subjective histories.3 Other literature has similarly concluded that there is an urgent need to carefully weigh information supplied to us by collateral informants because the first-hand accounts of perceivably dangerous behavior often are incomplete or misleading.4-5
Continue to: Ideas for improvement...
Ideas for improvement: respecting patient autonomy
These issues underscore the need for a more thorough review of collateral information to ensure that patient autonomy is not unjustly violated. How do we implement these necessary ideas without creating further undue burden during the admission process? Certainly, I am not suggesting that we evaluate the collateral informant to the degree that we evaluate the patient. However, I have outlined some suggestions for ensuring we act in our patients’ best interest when processing collateral information during an admission:
- Until proven otherwise, the patient’s story is true. If our patient maintains descriptions of his behavior that exist in stark opposition to the collateral information we obtain, we should only not believe the patient if his presentation suggests he may be acutely impaired or a poor historian (such as profound disorganization, overt psychosis, or failing to have capacity).
- Treat symptoms, not diagnoses. In this case, Mr. M was described by his foster mother to be psychotic in addition to having ASD, and an inexperienced psychiatrist may have initiated a titration to a higher antipsychotic dose. However, in the absence of any observable signs of aggression or psychosis, there was simply no indication for further titration of his antipsychotic.
- Document, document, document. When collateral information is supplied to us, it is crucial that we maintain a detailed account of this information. If we have a reason to believe that a patient poses an immediate danger to himself or others, we should carefully document our reasoning so that changes in behavior (if any) can be observed on a day-to-day basis.
Collateral information is a key component obtained during the psych
Here I describe a case in which collateral information obtained about a patient was a primary factor in that patient’s involuntary commitment. However, the patient’s subsequent behavior observed on an inpatient psychiatric unit was entirely inconsistent with those behaviors described by the collateral informant to be “continuous and dangerous.”
CASE
Mr. M, age 18, presented to an emergency psychiatric center for evaluation of dangerous and aggressive behavior. He had a history of autism spectrum disorder (ASD), which was well managed with oral risperidone. He was petitioned for an involuntary psychiatric admission by his foster mother, who reported that Mr. M was aggressive and dangerous, often punching holes in the walls of their home, and that he threatened to assault his foster siblings on several occasions. She detailed a progressively declining history for Mr. M and said that he was “constantly talking to voices in his head that absolutely consume him,” to the extent that Mr. M could not pay attention to his daily tasks. The admitting psychiatrist upheld the petition for involuntary admission, citing that based on the foster’s mother collateral information, Mr. M was deemed to be a danger to others and therefore fulfilled criteria for involuntary psychiatric admission.
Once admitted to the inpatient psychiatric unit, Mr. M was observed to be pleasant, cooperative, and fully engaged in the milieu. At no point during his 7-day admission was he observed to be internally preoccupied or remotely disorganized. Mr. M was switched from oral risperidone to oral haloperidol because he developed acute gynecomastia, and was discharged home.
Does collateral information lead to unfair bias?
The importance of collateral information on the psychiatric admission process must not be understated. It is an opportunity to hear a first-hand account of behaviors consistent with an acute psychiatric disturbance, and guides us in formulating a clinically appropriate assessment and plan. But what happens when our patients’ close contacts or informants provide misleading or unintentionally suboptimal collateral information? How must we reconcile the ethical and legal obligation we have to balance patient autonomy with beneficence?
Studies examining patients’ attitudes toward involuntary admissions have routinely found that patients are less likely than clinical staff to view the involuntary admission as clinically justified.2 Consistent with these findings, Mr. M did not view his admission as necessary. At first, he seemed to lack insight regarding the events precipitating his involuntary admission, describing himself not as responding to internal stimuli, but rather, “imaginative because I have autism.” As time went on, though, it was clear that his account of his behavior was in fact correct.
Mr. M’s diagnosis of ASD further complicated the over-reliance on misleading collateral information provided by his foster mother, because the admitting psychiatrist invariably perceived Mr. M as a poor historian. A study examining how subjective histories described by patients with neurologic or psychiatric disorders are perceived by clinicians found physicians had a tendency for negative stereotyping and placed less credence on those patients’ subjective histories.3 Other literature has similarly concluded that there is an urgent need to carefully weigh information supplied to us by collateral informants because the first-hand accounts of perceivably dangerous behavior often are incomplete or misleading.4-5
Continue to: Ideas for improvement...
Ideas for improvement: respecting patient autonomy
These issues underscore the need for a more thorough review of collateral information to ensure that patient autonomy is not unjustly violated. How do we implement these necessary ideas without creating further undue burden during the admission process? Certainly, I am not suggesting that we evaluate the collateral informant to the degree that we evaluate the patient. However, I have outlined some suggestions for ensuring we act in our patients’ best interest when processing collateral information during an admission:
- Until proven otherwise, the patient’s story is true. If our patient maintains descriptions of his behavior that exist in stark opposition to the collateral information we obtain, we should only not believe the patient if his presentation suggests he may be acutely impaired or a poor historian (such as profound disorganization, overt psychosis, or failing to have capacity).
- Treat symptoms, not diagnoses. In this case, Mr. M was described by his foster mother to be psychotic in addition to having ASD, and an inexperienced psychiatrist may have initiated a titration to a higher antipsychotic dose. However, in the absence of any observable signs of aggression or psychosis, there was simply no indication for further titration of his antipsychotic.
- Document, document, document. When collateral information is supplied to us, it is crucial that we maintain a detailed account of this information. If we have a reason to believe that a patient poses an immediate danger to himself or others, we should carefully document our reasoning so that changes in behavior (if any) can be observed on a day-to-day basis.
1. Testa M, West SG. Civil commitment in the United States. Psychiatry (Edgmont). 2010;7(10):30-40.
2. Roe D, Weishut DJ, Jaglom M, et al. Patients’ and staff members’ attitudes about the rights of hospitalized psychiatric patients. Psychiatr Serv. 2002;53(1):87-91.
3. Crichton P, Carel H, Kidd IJ. Epistemic injustice in psychiatry. BJPsych Bull. 2017;41(2):65-70.
4. Marett C, Mossman D. What is your liability for involuntary commitment based on fault information? Current Psychiatry. 2017;16(3):21-25,33.
5. Lincoln AL, Allen M. The influence of collateral information on access to inpatient psychiatric services. International Journal of Psychosocial Rehabilitation. 2002;6:99-108.
1. Testa M, West SG. Civil commitment in the United States. Psychiatry (Edgmont). 2010;7(10):30-40.
2. Roe D, Weishut DJ, Jaglom M, et al. Patients’ and staff members’ attitudes about the rights of hospitalized psychiatric patients. Psychiatr Serv. 2002;53(1):87-91.
3. Crichton P, Carel H, Kidd IJ. Epistemic injustice in psychiatry. BJPsych Bull. 2017;41(2):65-70.
4. Marett C, Mossman D. What is your liability for involuntary commitment based on fault information? Current Psychiatry. 2017;16(3):21-25,33.
5. Lincoln AL, Allen M. The influence of collateral information on access to inpatient psychiatric services. International Journal of Psychosocial Rehabilitation. 2002;6:99-108.
Differentiating serotonin syndrome and neuroleptic malignant syndrome
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).

It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.

Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.

Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.

Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.

Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.

Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
Serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) are each rare psychiatric emergencies that can lead to fatal outcomes. Their clinical presentations can overlap, which can make it difficult to differentiate between the 2 syndromes; however, their treatments are distinct, and it is imperative to know how to identify symptoms and accurately diagnose each of them to provide appropriate intervention. This article summarizes the 2 syndromes and their treatments, with a focus on how clinicians can distinguish them, provide prompt intervention, and prevent occurrence.
Serotonin syndrome
Mechanism. The decarboxylation and hydroxylation of tryptophan forms serotonin, also known as 5-hydroxytryptamine (5-HT), which can then be metabolized by monoamine oxidase-A (MAO-A) into 5-hydroxyindoleacetic acid (5-HIAA).1Medications can disrupt this pathway of serotonin production or its metabolism, and result in excessive levels of serotonin, which subsequently leads to an overactivation of central and peripheral serotonin receptors.1 Increased receptor activation leads to further upregulation, and ultimately more serotonin transmission. This can be caused by monotherapy or use of multiple serotonergic agents, polypharmacy with a combination of medication classes, drug interactions, or overdose. The wide variety of medications often prescribed by different clinicians can make identification of excessive serotonergic activity difficult, especially because mood stabilizers such as lithium,2 and non-psychiatric medications such as
- inhibition of serotonin uptake (seen with selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and tricyclic antidepressants [TCAs])
- inhibition of serotonin metabolism (seen with monoamine oxidase inhibitors [MAOIs])
- increased serotonin synthesis (seen with stimulants)
- increased serotonin release (seen with stimulants and opiates)
- activation of serotonin receptors (seen with lithium)
- inhibition of certain cytochrome P450 (CYP450) enzymes (seen with ciprofloxacin, fluconazole, etc.).
It is important to recognize that various serotonergic agents are involved in the CYP450 system. Inhibition of the CYP450 pathway by common antibiotics such as ciprofloxacin, or antifungals such as fluconazole, may result in an accumulation of serotonergic agents and place patients at increased risk for developing SS.
Clinical presentation. The clinical presentation of SS can range from mild to fatal. There is no specific laboratory test for diagnosis, although an elevation of the total creatine kinase (CK) and leukocyte count, as well as increased transaminase levels or lower bicarbonate levels, have been reported in the literature.4
Symptoms of SS generally present within 24 hours of starting/changing therapy and include a triad of mental status changes (altered mental status [AMS]), autonomic instability, and abnormalities of neuromuscular tone. Examples of AMS include agitation, anxiety, disorientation, and restlessness. Symptoms of autonomic instability include hypertension, tachycardia, tachypnea, hyperthermia, diaphoresis, flushed skin, vomiting, diarrhea, and arrhythmias. Symptoms stemming from changes in neuromuscular tone include tremors, clonus, hyperreflexia, and muscle rigidity.1 The multiple possible clinical presentations, as well as symptoms that overlap with those of other syndromes, can make SS difficult to recognize quickly in a clinical setting.
Diagnostic criteria. Sternbach’s diagnostic criteria for SS are defined as the presence of 3 or more of the 10 most common clinical features (Table 25). Due to concerns that Sternbach’s diagnostic criteria overemphasized an abnormal mental state (leading to possible confusion of SS with other AMS syndromes), the Hunter serotonin toxicity criteria6 (Figure6) were developed in 2003, and were found to be more sensitive and specific than Sternbach’s criteria. Both tools are often used in clinical practice.
Treatment. Treatment of SS begins with prompt discontinuation of all serotonergic agents. The intensity of treatment depends on the severity of the symptoms. Mild symptoms can be managed with supportive care,3 and in such cases, the syndrome generally resolves within 24 hours.7 Clinicians may use supportive care to normalize vital signs (oxygenation to maintain SpO2 >94%, IV fluids for volume depletion, cooling agents, antihypertensives, benzodiazepines for sedation or control of agitation, etc.). Patients who are more ill may require more aggressive treatment, such as the use of a serotonergic antagonist (ie, cyproheptadine) and those who are severely hyperthermic (temperature >41.1ºC) may require neuromuscular sedation, paralysis, and possibly endotracheal intubation.3
Continue to: Management pitfalls include...
Management pitfalls include misdiagnosis of SS, failure to recognize its rapid rate of progression, and adverse effects of pharmacologic therapy.3 The most effective treatment for SS is prevention. SS can be prevented by astute pharmacologic understanding, avoidance of polypharmacy, and physician education.3
Neuroleptic malignant syndrome
Possible mechanisms. Neuromuscular malignant syndrome is thought to result from dopamine receptor antagonism leading to a hypodopaminergic state in the striatum and hypothalamus.8 The pathophysiology behind NMS has not fully been elucidated; however, several hypotheses attempt to explain this life-threatening reaction. The first focuses on dopamine D2 receptor antagonism, because many of the neuroleptic (antipsychotic) medications that can precipitate NMS are involved in dopamine blockade. In this theory, blocking dopamine D2 receptors in the anterior hypothalamus explains the hyperthermia seen in NMS, while blockade in the corpus striatum is believed to lead to muscle rigidity.9
The second hypothesis suggests that neuroleptics may have a direct toxic effect to muscle cells. Neuroleptics influence calcium transport across the sarcoplasmic reticulum and can lead to increased calcium release, which may contribute to the muscle rigidity and hyperthermia seen in NMS.9
The third hypothesis involves hyperactivity of the sympathetic nervous system; it is thought that psychologic stressors alter frontal lobe function, with neuroleptics disrupting the inhibitory pathways of the sympathetic nervous system. The autonomic nervous system innervates multiple organ systems, so this excessively dysregulated sympathetic nervous system may be responsible for multiple NMS symptoms (hyperthermia, muscle rigidity, hypertension, diaphoresis, tachycardia, elevated CK.10
NMS can be caused by neuroleptic agents (both first- and second-generation antipsychotics) as well as antiemetics (Table 31). The time between use of these medications and onset of symptoms is highly variable. NMS can occur after a single dose, after a dose adjustment, or possibly after years of treatment with the same medication. It is not dose-dependent.11 In certain individuals, NMS may occur at therapeutic doses.
Continue to: Clinical presentation
Clinical presentation. Patients with NMS typically present with a tetrad of symptoms: mental status changes, muscular rigidity, hyperthermia, and autonomic instability.12 Mental status changes can include confusion and agitation, as well as catatonic signs and mutism. The muscular rigidity of NMS is characterized by “lead pipe rigidity” and may be accompanied by tremor, dystonia, or dyskinesias. Laboratory findings include elevated serum CK (from severe rigidity), often >1,000 U/L, although normal levels can be observed if rigidity has not yet developed.13
Treatment. The first step for treatment is to discontinue the causative medication.14 Initiate supportive therapy immediately to restrict the progression of symptoms. Interventions include cooling blankets, fluid resuscitation, and antihypertensives to maintain autonomic stability15 or benzodiazepines to control agitation. In severe cases, muscular rigidity may extend to the airways and intubation may be required. The severity of these symptoms may warrant admission to the ICU for close monitoring. Pharmacologic treatment with
Differentiating between SS and NMS
Differentiating between these 2 syndromes (Table 417) is critical to direct appropriate intervention. Table 517 outlines the treatment overview for SS and NMS.
Detailed history. A detailed history is imperative in making accurate diagnoses. Useful components of the history include a patient’s duration of symptoms and medication history (prescription medications as well as over-the-counter medications, supplements, and illicit drugs). Also assess for medical comorbidities, because certain medical diagnoses may alert the clinician that it is likely the patient had been prescribed serotonergic agents or neuroleptics, and renal or liver impairment may alert the clinician of decreased metabolism rates. Medication history is arguably the most useful piece of the interview, because serotonergic agents can cause SS, whereas dopamine blockers cause NMS. It should be noted that excess serotonin acts as a true toxidrome and is concentration-dependent in causing SS, whereas NMS is an idiosyncratic reaction to a drug.
Physical exam. Although there are many overlapping clinical manifestations, SS produces neuromuscular hyperactivity (ie, clonus, hyperreflexia), whereas NMS is characterized by more sluggish responses (ie, rigidity, bradyreflexia).18
Continue to: Laboratory findings
Laboratory findings. Overlap between NMS and SS also occurs with lab findings; both syndromes can result in leukocytosis, elevated CK from muscle damage, and low serum iron levels. However, these findings are more commonly associated with NMS and are seen in 75% of cases.17,19
Course of illness. Duration of symptoms can also help differentiate the 2 syndromes. SS typically develops within 24 hours of starting/changing therapy, whereas NMS symptoms can be present for days to weeks. Resolution of symptoms may also be helpful in differentiation because SS typically resolves within a few days of initiating treatment, whereas NMS resolves within 9 to 14 days of starting treatment.19
Bottom Line
The clinical presentations of serotonin syndrome (SS) and neuroleptic malignant syndrome (NMS) overlap, which can make them difficult to differentiate; however, they each have distinct approaches to treatment. Features in SS that are distinct from NMS include a history of serotonergic agents, rapid onset of symptoms, hyperreflexia, and clonus. NMS is slower in onset and can be found in patients who are prescribed dopamine antagonists, with distinct symptoms of rigidity and hyporeflexia.
Related Resources
- Kimmel R. Serotonin syndrome or NMS? Clues to diagnosis. Current Psychiatry. 2010;9(2):92.
- Strawn JR, Keck Jr PE, Caroff SN. Neuroleptic malignant syndrome: Answers to 6 tough questions. Current Psychiatry. 2008;7(1):95-101.
Drug Brand Names
Amantadine • Symmetrel
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Bromocriptine • Cycloset, Parlodel
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Ciprofloxacin • Cipro
Citalopram • Celexa
Clomipramine • Anafranil
Clozapine • Clozaril
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Dantrolene • Dantrium
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Dextromethorphan • Benylin, Dexalone
Dolasetron • Anzemet
Doxepin • Silenor
Droperidol • Inapsine
Duloxetine • Cymbalt
Escitalopram • Lexapro
Fentanyl • Actiq, Duragesic
Fluconazole • Diflucan
Fluoxetine • Prozac
Fluphenazine • Prolixin
Fluvoxamine • Luvox
Granisetron • Kytril
Haloperidol • Haldol
Isocarboxazid • Marplan
Levomilnacipran • Fetzima
Linezolid • Zyvox
Lithium • Eskalith, Lithobid
Meperidone • Demerol
Metoclopramide • Reglan
Milnacipran • Savella
Nefazodone • Serzone
Olanzapine • Zyprexa
Ondansetron • Zofran
Paliperidone • Invega
Palonosetron • Aloxi
Paroxetine • Paxil
Pentazocine • Talwin, Talacen
Perphenazine • Trilafon
Phenelzine • Nardil
Procarbazine • Matulane
Prochlorperazine • Compazine
Promethazine • Phenergan
Quetiapine • Seroquel
Rasagiline • Azilect
Risperidone • Risperdal
Safinamide • Xadago
Selegiline • Eldepryl, Zelapar
Sertraline • Zoloft
Sibutramine • Meridia
Tedizolid • Sivextro
Thioridazine • Mellaril
Tranylcypromine • Parnate
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Valproate • Depacon
Ziprasidone • Geodon
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.
1. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
2. Werneke U, Jamshidi F, Taylor D, et al. Conundrums in neurology: diagnosing serotonin syndrome – a meta-analysis of cases. BMC Neurol. 2016;16:97.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
4. Birmes P, Coppin D, Schmitt L, et al. Serotonin syndrome: a brief review. CMAJ. 2003;168(11):1439-1442.
5. Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705-713.
6. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003; 96(9):635-642.
7. Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021-1022.
8. Nisijima K. Serotonin syndrome overlapping with neuroleptic malignant syndrome: A case report and approaches for differentially diagnosing the two syndromes. Asian J Psychiatr. 2015;18:100-101.
9. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85(1):129-135.
10. Gurrera R. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
11. Pope HG Jr, Aizley HG, Keck PE Jr, et al. Neuroleptic malignant syndrome: long-term follow-up of 20 cases. J Clin Psychiatry. 1991;52(5):208-212.
12. Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168-173.
13. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77(1):185-202.
14. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome. Ann Pharmacother. 2016;50(11):973-981.
15. San Gabriel MC, Eddula-Changala B, Tan Y, et al. Electroconvulsive in a schizophrenic patient with neuroleptic malignant syndrome and rhabdomyolysis. J ECT. 2015;31(3):197-200.
16. Buggenhout S, Vandenberghe J, Sienaert P. Electroconvulsion therapy for neuroleptic malignant syndrome. Tijdschr Psychiatr. 2014;56(9):612-615.
17. Perry PJ, Wilborn CA. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry. 2012;24(2):155-162.
18. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin. 1997;13(4):763-783.
19. Dosi R, Ambaliya A, Joshi H, et al. Serotonin syndrome versus neuroleptic malignant syndrome: a challenge clinical quandary. BMJ Case Rep. 2014;2014:bcr201404154. doi:10.1136/bcr-2014-204154.

Older-age bipolar disorder: A case series
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
")
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.

A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
")
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.

Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.

The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.

Antidepressants for chronic pain
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.

Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88

Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.

Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
Approximately 55 years ago, tricyclic antidepressants (TCAs) began to be used to treat neuropathic pain.1 Eventually, clinical trials emerged suggesting the utility of TCAs for other chronic pain conditions, such as fibromyalgia (FM) and migraine prophylaxis. However, despite TCAs’ effectiveness in mitigating painful conditions, their adverse effects limited their use.
Pharmacologic advancements have led to the development of other antidepressant classes, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), and the use of these agents has come to eclipse that of TCAs. In the realm of pain management, such developments have raised the hope of possible alternative co-analgesic agents that could avoid the adverse effects associated with TCAs. Some of these agents have demonstrated efficacy for managing chronic pain states, while others have demonstrated only limited utility.
This article provides a synopsis of systematic reviews and meta-analyses examining the role of antidepressant therapy for managing several chronic pain conditions, including pain associated with neuropathy, FM, headache, and irritable bowel syndrome (IBS). Because the literature base is rapidly evolving, it is necessary to revisit the information gleaned from clinical data with respect to treatment effectiveness, and to clarify how antidepressants might be positioned in the management of chronic pain.
The effectiveness of antidepressants for pain
The pathophysiologic processes that precipitate and maintain chronic pain conditions are complex (Box 12-10). The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects and indirect effects (Box 22,3,8,10,11-33).
Box 1
The pathophysiologic processes precipitating and maintaining chronic pain conditions are complex. Persistent and chronic pain results from changes in sensitivity within both ascending pathways (relaying pain information from the periphery to the spinal cord and brain) and descending pain pathways (functioning to modulate ascending pain information).2,3 Tissue damage or peripheral nerve injury can lead to a cascade of neuroplastic changes within the CNS, resulting in hyperexcitability within the ascending pain pathways.
The descending pain pathways consist of the midbrain periaqueductal gray area (PGA), the rostroventral medulla (RVM), and the dorsolateral pontomesencephalic tegmentum (DLPT). The axons of the RVM (the outflow of which is serotonergic) and DLPT (the outflow of which is noradrenergic) terminate in the dorsal horn of the spinal cord,4 and thereby dampen pain signals arising from the periphery. Diminished output from descending pain pathways can heighten the pain experience. Input from the cortex, hypothalamus, and amygdala (among other structures) converges upon the PGA, RVM and DLPT, and can influence the degree of pain modulation emerging from descending pathways. In this way, thoughts, appraisals, and mood are believed to comprise cognitive and affective modifiers of pain experiences.
Devising effective chronic pain treatment becomes challenging; multimodal treatment approaches often are advocated, including pharmacologic treatment with analgesics in combination with co-analgesic medications such as antidepressants. Although a description of multimodal treatment is beyond the scope of this article, such treatment also would encompass physical therapy, occupational therapy, and psychotherapeutic interventions to augment rehabilitative efforts and the functional capabilities of patients who struggle with persisting pain.
Although the direct pain-mitigating effects of antidepressants are not fully understood, it is believed that augmentation of monoamine neurotransmission from supraspinal nuclei (ie, the RVM and DLPT) modulate pain transmission from the periphery.5,6 In addition, there is evidence that some effects of tricyclic antidepressants can modulate several other functions that impact peripheral and central sensitization.7-10
During the last several decades, antidepressants have been used to address—and have demonstrated clinical utility for—a variety of chronic pain states. However, antidepressants are not a panacea; some chronic pain conditions are more responsive to antidepressants than are others. The chronic painful states most amenable to antidepressants are those that result primarily from a process of neural sensitization, as opposed to acute somatic or visceral nociception. Hence, several meta-analyses and evidence-based reviews have long suggested the usefulness of antidepressants for mitigating pain associated with neuropathy,34,35 FM,36,37 headache,38 and IBS.39,40
Box 2
The pain-mitigating effects of antidepressants can be thought of in terms of direct analgesic effects (impacting neurotransmission of descending pathways independent of influences on mood) and indirect effects (presumably impacting cortical and limbic output to the periaqueductal gray area, the rostroventral medulla, and the dorsolateral pontomesencephalic tegmentum brought about by improvement in mood and/or cognitive appraisals) (Figure2,3,8,10,11,15,20,22,28,29). Support for the direct analgesic effects has been garnered from initial empirical work that demonstrated pain relief among patients with pain who are not depressed. Additionally, among patients who have depression and experience pain, analgesia reportedly often occurs within 2 weeks, which is before antidepressant effects are appreciated,11-15 and, at least for some antidepressants, occurs at doses far lower than those required to produce mood-elevating effects.11,12,16
On the other hand, it is well established that significant comorbidities exist between chronic pain states and psychiatric disorders (eg, depression and somatic symptom and related disorders).17-21 There may be common physiological substrates underlying chronic pain and depression.20,22 There are bidirectional influences of limbic (affective) systems and those CNS structures involved in pain processing and integration. The effects of pain and depression are reciprocal; the presence of one makes the management of the other more challenging.23-27 Mood disturbances can, therefore, impact pain processing by acting as affective and cognitive amplifiers of pain by leading to catastrophizing, pain severity augmentation, poor coping with pain-related stress, etc.28,29 It is plausible that the mood-elevating effects of antidepressants can improve pain by indirect effects, by modulating limbic activity, which in turn, impacts coping, cognitive appraisals of pain, etc.
Patients with somatoform disorders (using pre-DSM-5 terminology) frequently present with chronic pain, often in multiple sites.19 Such patients are characterized by hypervigilance for, and a predisposition to focus on, physical sensations and to appraise these sensations as reflecting a pathological state.30 Neuroimaging studies have begun to identify those neural circuits involved in somatoform disorders, many of which act as cognitive and affective amplifiers of visceral-somatic sensory processing. Many of these neural circuits overlap, and interact with, those involved in pain processing.31 Antidepressants can mitigate the severity of unexplained physical complaints, including pain, among patients who somatize32,33; however, due to the heterogeneity of studies upon which this claim is based, the quality of the evidence is reportedly low.33 There is uncertainty whether, or to what extent, antidepressant benefits among patients who somatize are due to a direct impact on pain modulation, or indirect effects on mood or cognitive appraisals/perceptions.
Despite the uncertainties about the exact mechanisms through which antidepressants exert analgesic effects, antidepressants can be appropriately used to treat patients with selected chronic pain syndromes, regardless of whether or not the patient has a psychiatric comorbidity. For those patients with pain and psychiatric comorbidities, the benefits may be brought about via direct mechanisms, indirect mechanisms, or a combination of both.
Continue to: Neuropathic pain
Neuropathic pain
Several treatment guidelines advocate for the use of antidepressants for neuropathic pain.41-44 For decades, TCAs have been employed off-label to successfully treat many patients with neuropathic pain states. Early investigations suggested that TCAs were robustly efficacious in managing patients with neuropathy.45-48 Calculated number-needed-to-treat (NNT) values for TCAs were quite low (ie, reflecting that few patients would need to be treated to yield a positive response in one patient compared with placebo), and were comparable to, if not slightly better than, the NNTs generated for anticonvulsants and α2-δ ligands, such as gabapentin or pregabalin.45-48
Unfortunately, early studies involving TCAs conducted many years ago do not meet contemporary standards of methodological rigor; they featured relatively small samples of patients assessed for brief post-treatment intervals with variable outcome measures. Thus, the NNT values obtained in meta-analyses based on these studies may overestimate treatment benefits.49 Further, NNT values derived from meta-analyses tended to combine all drugs within a particular antidepressant class (eg, amitriptyline, nortriptyline, desipramine, and imipramine among the TCAs) employed at diverse doses. Taken together, these limitations raise questions about the results of those meta-analyses.
Subsequent meta-analyses, which employed strict criteria to eliminate data from studies with potential sources of bias and used a primary outcome of frequencies of patients reporting at least 30% pain reduction compared with a placebo-controlled sample, suggest that the effectiveness of TCAs as a class for treating neuropathic pain is not as compelling as once was thought. Meta-analyses of studies employing specific TCAs revealed that there was little evidence to support the use of desipramine,50 imipramine,51 or nortriptyline52 in managing diabetic neuropathy or postherpetic neuralgia. Studies evaluating amitriptyline (dose range 12.5 to 150 mg/d), found low-level evidence of effectiveness; the benefit was expected to be present for a small subset (approximately 25%) of patients with neuropathic pain.53
There is moderate-quality evidence that duloxetine (60 to 120 mg/d) can produce a ≥50% improvement in pain severity ratings among patients with diabetic peripheral neuropathy.54 Although head-to-head studies with other antidepressants are limited, it appears that duloxetine and amitriptyline have comparable efficacy, even though the NNTs for amitriptyline were derived from lower-quality studies than those for duloxetine. Duloxetine is the only antidepressant to receive FDA approval for managing diabetic neuropathy. By contrast, studies assessing the utility of venlafaxine in neuropathic pain comprised small samples for brief durations, which limits the ability to draw clear (unbiased) support for its usefulness.55
Given the diversity of pathophysiologic processes underlying the disturbances that cause neuropathic pain disorders, it is unsurprising that the effectiveness of amitriptyline and duloxetine were not generalizable to all neuropathic pain states. Although amitriptyline produced pain-mitigating effects in patients with diabetic neuropathy and post-herpetic neuralgia, and duloxetine mitigated pain among patients with diabetic neuropathy, there was no evidence to suggest their effectiveness in phantom limb pain or human immunodeficiency virus-related and spinal cord-related neuropathies.35,53,54,56-58
Continue to: Fibromyalgia
Fibromyalgia
As with the issues encountered in interpreting the effectiveness of antidepressants in neuropathic pain, interpreting results gleaned from clinical trials of antidepressants for treating FM are fraught with similar difficulties. Although amitriptyline has been a first-line treatment for FM for many years, the evidence upon which such recommendations were based consisted of low-level studies that had a significant potential for bias.59 Large randomized trials would offer more compelling data regarding the efficacy of amitriptyline, but the prohibitive costs of such studies makes it unlikely they will be conducted. Amitriptyline (25 to 50 mg/d) was effective in mitigating FM-related pain in a small percentage of patients studied, with an estimated NNT of 4.59 Adverse effects, often contributing to treatment discontinuation, were encountered more frequently among patients who received amitriptyline compared with placebo.
Selective serotonin reuptake inhibitors failed to demonstrate significant pain relief (estimated NNT of 10), or improvement in fatigue or sleep problems, even though the studies upon which such conclusions were based were low-level studies with a high potential for bias.60 Although SSRIs have limited utility for mitigating pain, they are still quite useful for reducing depression among patients with FM.60
By contrast, the SNRIs duloxetine and milnacipran provided clinically relevant benefit over placebo in the frequency of patients reporting pain relief of ≥30%, as well as patients’ global impression of change.61 These agents, however, failed to provide clinically relevant benefit over placebo in improving health-related quality of life, reducing sleep problems, or improving fatigue. Nonetheless, duloxetine and milnacipran are FDA-approved for managing pain in FM. Studies assessing the efficacy of venlafaxine in the treatment of FM to date have been limited by small sample sizes, inconsistent dosing, lack of a placebo control, and lack of blinding, which limits the ability to clearly delineate the role of venlafaxine in managing FM.62
Mirtazapine (15 to 45 mg/d) showed a clinically relevant benefit compared with placebo for participant-reported pain relief of ≥30% and sleep disturbances. There was no benefit in terms of participant-reported improvement of quality of life, fatigue, or negative mood.63 The evidence was considered to be of low quality overall.
Headache
Amitriptyline has been employed off-label to address headache prophylaxis since 1964.64 Compared with placebo, it is efficacious in ameliorating migraine frequency and intensity as well as the frequency of tension headache.65,66 However, SSRIs and SNRIs (venlafaxine) failed to produce significant reductions in migraine frequency or severity or the frequencies of tension headache when compared with placebo.67,68
Continue to: Irritable bowel syndrome
Irritable bowel syndrome
Early studies addressing antidepressant efficacy in IBS reveal inconsistencies. For example, whereas some suggest that TCAs are effective in mitigating chronic, severe abdominal pain,39,40 others concluded that TCAs failed to demonstrate a significant analgesic benefit.69 A recent meta-analysis that restricted analysis of efficacy to randomized controlled trials (RCTs) with more rigorous methodological adherence found that pain relief in IBS is possible with both TCAs as well as SSRIs. However, adverse effects were more commonly encountered with TCAs than with SSRIs. Some of the inconsistencies in treatment efficacy reported in early studies may be due to variations in responsiveness of subsets of IBS patients. Specifically, the utility of TCAs appears to be best among patients with diarrheal-type (as opposed to constipation-type) IBS, presumably due to TCAs’ anticholinergic effects, whereas SSRIs may provide more of a benefit for patients with predominantly constipation-type IBS.40,70
Other chronic pain conditions
Antidepressants have been used to assist in the management of several other pain conditions, including oral-facial pain, interstitial cystitis, non-cardiac chest pain, and others. The role of antidepressants for such conditions remains unclear due to limitations in the prevailing empirical work, such as few trials, small sample sizes, variations in outcome measures, and insufficient randomization and blinding.71-76 The interpretation of results from systematic reviews and meta-analyses is limited because of these shortcomings.77 Hence, it has not always been possible to determine whether, and to what extent, patients with such conditions may benefit from antidepressants.
Neuromodulatory effects and efficacy for pain
The interplay of norepinephrine (NE) and serotonin (5-HT) neurotransmitter systems and cellular mechanisms involved in the descending modulation of pain pathways is complex. Experimental animal models of pain modulation suggest that 5-HT can both inhibit as well as promote pain perception by different physiological mechanisms, in contrast to NE, which is predominately inhibitory. While 5-HT in the descending modulating system can inhibit pain transmission ascending to the brain from the periphery, it appears that an intact noradrenergic system is necessary for the inhibitory influences of the serotonergic system to be appreciated.16,78,79 Deficiencies in one or both of these neurotransmitter systems may contribute to hyperactive pain processing, and thereby precipitate or maintain chronic pain.
Pain mitigation may be achieved best by enhancing both neurotransmitters simultaneously, less so by enhancing NE alone, and least by enhancing 5-HT alone.6 The ability to impact pain modulation would, therefore, depend on the degree to which an antidepressant capitalizes on both noradrenergic and serotonergic neurotransmission. Antidepressants commonly employed to manage pain are presented in Table 147,60,68,80-88 according to their primary neurotransmitter effects. Thus, the literature summarized above suggests that antidepressants that influence both NE and 5-HT transmission have greater analgesic effects than antidepressants with more specific effects, such as influencing 5-HT reuptake alone.80-85 It is unsurprising, therefore, that the SSRIs have not been demonstrated to be as consistently analgesic.47,60,68,80,86-88
Similarly, pharmacodynamic and pharmacokinetic differences within antidepressant classes may influence analgesic effectiveness. Simultaneous effects on NE and 5-HT are achieved at low doses with duloxetine and milnacipran. By contrast, 5-HT effects predominate at low doses for venlafaxine. To achieve pain-mitigating effects, higher doses of venlafaxine generally are required.89 Therefore, inconsistencies across studies regarding the analgesic benefits of venlafaxine may be attributable to variability in dosing; patients treated with lower doses may not have experienced sufficient NE effects to garner positive results.
Continue to: The differences in analgesic efficacy...
The differences in analgesic efficacy among specific TCAs may be understood in a similar fashion. Specifically, tertiary TCAs (imipramine and amitriptyline) inhibit both 5-HT and NE reuptake.6,90 Secondary amines (desipramine and nortriptyline) predominantly impact NE reuptake, possibly accounting for the lesser pain-mitigating benefit achieved with these agents, such as for treating neuropathic pain. Further, in vivo imipramine and amitriptyline are rapidly metabolized to secondary amines that are potent and selective NE reuptake inhibitors. In this way, the secondary amines may substantially lose the ability to modulate pain transmission because of the loss of concurrent 5-HT influences.90
Clinical pearls
The following practical points can help guide clinicians regarding the usefulness of antidepressants for pain management:
- Antidepressants can alleviate symptoms of depression and pain. The pain-mitigating effects of antidepressants are possible even among chronic pain patients who are not depressed. Antidepressants may confer benefits for chronic pain patients with depression and other comorbid conditions, such as somatic symptom and related disorders.
- Antidepressants are useful for select chronic pain states. Although the noradrenergic and serotonergic antidepressants (SNRIs and, to some extent, amitriptyline) appear to have efficacy for neuropathic pain and FM, the benefits of SSRIs appear to be less robust. On the other hand, SSRIs and TCAs may have potential benefit for patients with IBS. However, the results of meta-analyses are limited in the ability to provide information about which patients will best respond to which specific antidepressant or how well. Future research directed at identifying characteristics that can predict which patients are likely to benefit from one antidepressant vs another would help inform how best to tailor treatment to individual needs.
- The pain-mitigating effects of antidepressants often emerge early in the course of treatment (often before mood-elevating effects are observed). For example, in the case of amitriptyline, pain relief may be possible for some patients at doses generally lower than those required for mood-elevating effects. To date, there is limited information in the literature to determine what constitutes a sufficient duration of treatment, or when treatment should be modified.
- Failure to reduce pain should raise questions about whether the dose should be increased, an alternative agent should be tried, or combinations with other analgesic agents should be considered. Failure to achieve pain-mitigating effects with one antidepressant does not mean failure with others. Hence, failure to achieve desired effects with one agent might warrant an empirical trial with another agent. Presently, too few double-blind RCTs have been conducted to assess the pain-mitigating effects of other antidepressants (eg, bupropion and newer SNRIs such as desvenlafaxine and levomilnacipran). Meta-analysis of the analgesic effectiveness of these agents or comparisons to the efficacy of other antidepressant classes is, therefore, impossible at this time.
Because many chronic pain states are complex, patients will seldom experience clinically relevant benefit from any one intervention.53 The bigger implication for clinical research is to determine whether there is a sequence or combination of medication use that will provide overall better clinical effectiveness.53 Only limited data are available exploring the utility of combining pharmacologic approaches to address pain.91 For example, preliminary evidence suggests that combinations of complementary strategies, such as duloxetine combined with pregabalin, may result in significantly greater numbers of FM patients achieving ≥30% pain reduction compared with monotherapy with either agent alone or placebo.92
- Antidepressant selection may need to be based on medication-related adverse effect profiles and the potential for drug interactions. These factors are useful to consider in delineating multimodal treatment regimens for chronic pain in light of patients’ comorbidities and co-medication regimen. For example, the adverse effects of TCAs (anticholinergic and alpha-adrenergic influences) limit their utility for treating pain. Some of these effects can be more problematic in select populations, such as older adults or those with orthostatic difficulties, among others. TCAs are contraindicated in patients with closed-angle glaucoma, recent myocardial infarction, cardiac arrhythmias, poorly controlled seizures, or severe benign prostatic hypertrophy. Although the pain-mitigating effects of SNRIs have not been demonstrated to significantly exceed those of TCAs,68,93,94 SNRIs would offer an advantage of greater tolerability of adverse effects and relative safety in patients with comorbid medical conditions that would otherwise preclude TCA use. The adverse effects and common drug interactions associated with antidepressants are summarized in Table 295.
Conclusion
Chronic, nonmalignant pain conditions afflict many patients and significantly impair their ability to function. Because of heightened concerns related to the appropriateness of, and restricting inordinate access to, long-term opioid analgesics, clinicians need to explore the usefulness of co-analgesic agents, such as antidepressants. Significant comorbidities exist between psychiatric disorders and chronic pain, and psychiatrists are uniquely positioned to diagnose and treat psychiatric comorbidities, as well as pain, among their patients, especially since they understand the kinetics and dynamics of antidepressants.
Bottom Line
Antidepressants can alleviate symptoms of depression and pain. Noradrenergic and serotonergic antidepressants appear to have efficacy for pain associated with neuropathy and fibromyalgia, while selective serotonin reuptake inhibitors and tricyclic antidepressants may have benefit for patients with irritable bowel syndrome. However, evidence regarding which patients will best respond to which specific antidepressant is limited.
Continue to: Related Resources
Related Resources
- Williams AM, Knox ED. When to prescribe antidepressants to treat comorbid depression and pain disorders. Current Psychiatry. 2017;16(1):55-58.
- Maletic V, Demuri B. Chronic pain and depression: treatment of 2 culprits in common. Current Psychiatry. 2016;15(3):41,47-50,52.
Drug Brand Names
Amitriptyline • Elavil, Endep
Bupropion • Wellbutrin, Zyban
Carisoprodol • Rela, Soma
Cyclobenzaprine • Amrix, Flexeril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Duloxetine • Cymbalta
Fluoxetine • Prozac
Gabapentin • Horizant, Neurontin
Imipramine • Tofranil
Levomilnacipran • Fetzima
Methadone • Dolophine, Methadose
Milnacipran • Savella
Mirtazapine • Remeron
Nortriptyline • Pamelor
Paroxetine • Paxil
Pregabalin • Lyrica, Lyrica CR
Tapentadol • Nucynta
Tramadol • Ultram
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Warfarin • Coumadin, Jantoven
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.
1. Paoli F, Darcourt G, Cossa P. Preliminary note on the action of imipramine in painful states [in French]. Rev Neurol (Paris). 1960;102:503-504.
2. Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219-245.
3. Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2(2):83-91.
4. Lamont LA, Tranquilli WJ, Grimm KA. Physiology of pain. Vet Clin North Am Small Anim Pract. 2000;30(4):703-728, v.
5. Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M, eds. Wall and Melzack’s Textbook of Pain. 5th ed. Burlington, MA: Elsevier Health Sciences; 2005:125-142.
6. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009;7(4):331-336.
7. Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92(2):473-484.
8. Carter GT, Sullivan MD. Antidepressants in pain management. Curr Opin Investig Drugs. 2002;3(3):454-458.
9. Kawasaki Y, Kumamoto E, Furue H, et al. Alpha 2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98(3):682-689.
10. McCleane G. Antidepressants as analgesics. CNS Drugs. 2008;22(2):139-156.
11. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry. 2000;7(5):257-277.
12. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. 1990;10(4):262-270.
13. Fishbain DA. Evidence-based data on pain relief with antidepressants. Ann Med. 2000;32(5):305-316.
14. Fishbain DA, Detke MJ, Wernicke J, et al. The relationship between antidepressant and analgesic responses: findings from six placebo-controlled trials assessing the efficacy of duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2008;24(11):3105-3115.
15. Harada E, Tokuoka H, Fujikoshi S, et al. Is duloxetine’s effect on painful physical symptoms in depression an indirect result of improvement of depressive symptoms? Pooled analyses of three randomized controlled trials. Pain. 2016;157(3):577-584.
16. Kehoe WA. Antidepressants for chronic pain: selection and dosing considerations. Am J Pain Med. 1993;3(4):161-165.
17. Damush TM, Kroenke K, Bair MJ, et al. Pain self-management training increases self-efficacy, self-management behaviours and pain and depression outcomes. Eur J Pain. 2016;20(2):1070-1078.
18. DeVeaugh-Geiss AM, West SL, Miller WC, et al. The adverse effects of comorbid pain on depression outcomes in primary care patients: results from the ARTIST trial. Pain Medicine. 2010;11(5):732-741.
19. Egloff N, Cámara RJ, von Känel R, et al. Hypersensitivity and hyperalgesia in somatoform pain disorders. Gen Hosp Psychiatry. 2014;36(3):284-290.
20. Goesling J, Clauw DW, Hassett AL. Pain and depression: an integrative review of neurobiological and psychological factors. Curr Psych Reports. 2013;15(12):421.
21. Kroenke K, Wu J, Bair MJ, et al. Reciprocal relationship between pain and depression: a 12-Month longitudinal analysis in primary care. J Pain. 2011;12(9):964-973.
22. Leo RJ. Chronic pain and comorbid depression. Curr Treat Options Neurol. 2005;7(5):403-412.
23. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
24. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
25. Kroenke K, Shen J, Oxman TE, et al. Impact of pain on the outcomes of depression treatment: results from the RESPECT trial. Pain. 2008;134(1-2):209-215.
26. Mavandadi S, Ten Have TR, Katz IR, et al. Effect of depression treatment on depressive symptoms in older adulthood: the moderating role of pain. J Am Geriatr Soc. 2007;55(2):202-211.
27. Thielke SM, Fan MY, Sullivan M, et al. Pain limits the effectiveness of collaborative care for depression. Am J Geriatr Psychiatry. 2007;15(8):699-707.
28. Arnow BA, Hunkeler EM, Blasey CM, et al. Comorbid depression, chronic pain, and disability in primary care. Psychosom Med. 2006;68(2):262-268.
29. Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: Prevalence, work loss, and help seeking. J Affect Disord. 2006;92(2-3):185-193.
30. Nakao M, Barsky AJ. Clinical application of somatosensory amplification in psychosomatic medicine. Biopsychosoc Med. 2007;1:17.
31. Perez DL, Barsky AJ, Vago DR, et al. A neural circuit framework for somatosensory amplification in somatoform disorders. J Neuropsychiatry Clin Neurosci. 2015;27(1):e40-e50.
32. Fishbain DA, Cutler RB, Rosomoff HL, et al. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta-analysis. Psychosom Med. 1998;60(4):503-509.
33. Kleinstäuber M, Witthöft M, Steffanowski A, et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Database Syst Rev. 2014;(11):CD010628.
34. Collins SL, Moore RA, McQuay HJ, et al. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. J Pain Symptom Manage. 2000;20(6):449-458.
35. Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry. 2010;81(12):1372-1373.
36. Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics. 2000;41(2):104-113.
37. O’Malley PG, Balden E, Tomkins G, et al. Treatment of fibromyalgia with antidepressants: a meta-analysis. J Gen Intern Med. 2000;15(9):659-666.
38. Tomkins GE, Jackson JL, O’Malley PG, et al. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med. 2001;111(1):54-63.
39. Jackson JL, O’Malley PG, Tomkins G, et al. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med. 2000;108(1):65-72.
40. Lesbros-Pantoflickova D, Michetti P, Fried M et al. Meta-analysis: the treatment of irritable bowel syndrome. Aliment Pharmacol Ther. 2004;20(11-12):1253-1269.
41. Centre for Clinical Practice at NICE (UK). Neuropathic pain: the pharmacological management of neuropathic pain in adults in non-specialist settings. London, UK: National Institute for Health and Care Excellence, (UK); 2013.
42. O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009;122(suppl 10):S22-S32.
43. Moulin D, Boulanger A, Clark AJ, et al; Canadian Pain Society. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328-35.
44. Mu A, Weinberg E, Moulin DE, et al. Pharmacologic management of chronic neuropathic pain: Review of the Canadian Pain Society consensus statement. Can Fam Physician. 2017;63(11):844-852.
45. Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain. 2005;118(3):289-305.
46. Hempenstall K, Nurmikko TJ, Johnson RW, et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med. 2005;2(7):e164.
47. Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83(3):389-400.
48. Wu CL, Raja SN. An update on the treatment of postherpetic neuralgia. J Pain. 2008;9(suppl 1):S19-S30.
49. Kroenke K, Krebs EE, Bair MJ. Pharmacotherapy of chronic pain: a synthesis of recommendations from systematic reviews. Gen Hosp Psychiatry. 2009;31(3):206-219.
50. Hearn L, Moore RA, Derry S, et al. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(9):CD011003.
51. Hearn L, Derry S, Phillips T, et al. Imipramine for neuropathic pain in adults. Cochrane Database Syst Rev. 2014;(5):CD010769.
52. Derry S, Wiffen PJ, Aldington D, et al. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;1:CD011209.
53. Moore R, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242.
54. Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;(1):CD007115.
55. Gallagher HC, Gallagher RM, Butler M, et al. Venlafaxine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(8):CD011091.
56. Alviar MJ, Hale T, Dungca M. Pharmacologic interventions for treating phantom limb pain. Cochrane Database Syst Rev. 2016;10:CD006380.
57. Dinat N, Marinda E, Moch S, et al. Randomized, Double-Blind, Crossover Trial of Amitriptyline for Analgesia in Painful HIV-Associated Sensory Neuropathy. PLoS One. 2015;10(5):e0126297. doi: 10.1371/journal.pone.0126297.eCollection 2015.
58. Mehta S, McIntyre A, Janzen S, et al; Spinal Cord Injury Rehabilitation Evidence Team. Systematic review of pharmacologic treatments of pain after spinal cord injury: an update. Arch Phys Med Rehabil. 2016;97(8):1381-1391.e1.
59. Moore RA, Derry S, Aldington D, et al. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2012;(12):CD008242..
60. Walitt B, Urrútia G, Nishishinya MB, et al. Selective serotonin reuptake inhibitors for fibromyalgia syndrome. Cochrane Database Syst Rev. 2015;(6):CD011735.
61. Welsch P, Üçeyler N, Klose P, et al. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia. Cochrane Database Syst Rev. 2018;(2):CD010292.
62. VanderWeide LA, Smith SM, Trinkley KE. A systematic review of the efficacy of venlafaxine for the treatment of fibromyalgia. J Clin Pharm Ther. 2015;40(1):1-6.
63. Welsch P, Bernardy K, Derry S, et al. Mirtazapine for fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(8):CD012708.
64. Lance JW, Curran DA. Treatment of chronic tension headache. Lancet. 1964;283(7345):1236-1239.
65. Jackson JL, William S, Laura S, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222. doi: https://doi.org/10.1136/bmj.c5222
66. Xu XM, Liu Y, Dong MX, et al. Tricyclic antidepressants for preventing migraine in adults. Medicine. 2017;96(22):e6989. doi: 10.1097/MD.0000000000006989.
67. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of migraine in adults. Cochrane Database Syst Rev. 2015;(4):CD002919.
68. Banzi R, Cusi C, Randazzo C, et al. Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) for the prevention of tension-type headache in adults. Cochrane Database Syst Rev. 2015;(5):CD011681.
69. Quartero AO, Meineche-Schmidt V, Muris J, et al. Bulking agents, antispasmodic and antidepressant medication for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2005;(2):CD003460.
70. Ford AC, Talley NJ, Schoenfeld PS, et al. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2009;58(3):367-378.
71. Coss-Adame E, Erdogan A, Rao SS. Treatment of esophageal (noncardiac) chest pain: an expert review. Clin Gastroenterol Hepatol. 2014;12(8):1224-1245.
72. Kelada E, Jones A. Interstitial cystitis. Arch Gynecol Obstet. 2007;275(4):223-229.
73. Leo RJ, Dewani S. A systematic review of the utility of antidepressant pharmacotherapy in the treatment of vulvodynia pain. J Sex Med. 2013;10(10):2497-2505.
74. McMillan R, Forssell H, Buchanan JA, et al. Interventions for treating burning mouth syndrome. Cochrane Database Syst Rev. 2016;11:CD002779.
75. Patel DN. Inconclusive results of a systematic review of efficacy of antidepressants on orofacial pain disorders. Evid Based Dent. 2013;14(2):55-56.
76. Wang W, Sun YH, Wang YY, et al. Treatment of functional chest pain with antidepressants: a meta-analysis. Pain Physician. 2012;15(2):E131-E142.
77. Lavis JN. How can we support the use of systematic reviews in policymaking? PLoS Med. 2009;6(11):e1000141. doi: 10.1371/journal.pmed.1000141.
78. Sorkin L. Nociceptive transmission within the spinal cord. Mt Sinai J Med. 1991;58(3):208-216.
79. Yokogawa F, Kiuchi Y, Ishikawa Y, et al. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth Analg. 2002;95(1):163-168, table of contents.
80. Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. J Psychiatry Neurosci. 2001;26(1):30-36.
81. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35(suppl):S50-S53.
82. Max MB, Lynch SA, Muir J, et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med. 1992;326(19):1250-1256.
83. McQuay HJ, Tramèr M, Nye BA, et al. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68(2-3):217-227.
84. Mochizucki D. Serotonin and noradrenaline reuptake inhibitors in animal models of pain. Hum Psychopharmacol Clin Exp. 2004;19(suppl 1):15-19.
85. Sussman N. SNRIs versus SSRIs: mechanisms of action in treating depression and painful physical symptoms. Primary Care Companion J Clin Psychiatry. 2003;5(suppl 7):19-26.
86. Bundeff AW, Woodis CB. Selective serotonin reuptake inhibitors for the treatment of irritable bowel syndrome. Ann Pharmacother. 2014;48(6):777-784.
87. Jung AC, Staiger T, Sullivan M. The efficacy of selective serotonin reuptake inhibitors for the management of chronic pain. J Gen Intern Med. 1997;12(6):384-389.
88. Xie C, Tang Y, Wang Y, et al. Efficacy and safety of antidepressants for the treatment of irritable bowel syndrome: a meta-analysis. PLoS One. 2015;10(8):e0127815. doi: 10.1371/journal.pone.0127815. eCollection 2015.
89. Zijlstra TR , Barendregt PJ , van de Laar MA. Venlafaxine in fibromyalgia: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2002;46(suppl 9):S105.
90. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871-880.
91. Thorpe J, Shum B, Moore RA, et al. Combination pharmacotherapy for the treatment of fibromyalgia in adults. Cochrane Database Syst Rev. 2018;(2):CD010585.
92. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532-1540.
93. Häuser W, Petzke F, Üçeyler N, et al. Comparative efficacy and acceptability of amitriptyline, duloxetine and milnacipran in fibromyalgia syndrome: a systematic review with meta-analysis. Rheumatology (Oxford). 2011;50(3):532-543.
94. Hossain SM, Hussain SM, Ekram AR. Duloxetine in painful diabetic neuropathy: a systematic review. Clin J Pain. 2016;32(11):1005-1010.
95. Riediger C, Schuster T, Barlinn K, et al. Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front Neurol. 2017;8:307.

New concepts in the management of acute pancreatitis
Introduction
Acute pancreatitis (AP) is a major clinical and financial burden in the United States. Several major clinical guidelines provide evidence-based recommendations for the clinical management decisions in AP, including those from the American College of Gastroenterology (ACG; 2013),1 and the International Association of Pancreatology (IAP; 2013).2 More recently, the American Gastroenterological Association (AGA) released their own set of guidelines.3,4 In this update on AP, we review these guidelines and reference recent literature focused on epidemiology, risk factors, etiology, diagnosis, risk stratification, and recent advances in the early medical management of AP. Regarding the latter, we review six treatment interventions (pain management, intravenous fluid resuscitation, feeding, prophylactic antibiotics, probiotics, and timing of endoscopic retrograde cholangiopancreatography (ERCP) in acute biliary pancreatitis) and four preventive interventions (alcohol and smoking cessation, same-admission cholecystectomy for acute biliary pancreatitis, and chemoprevention and fluid administration for post-ERCP pancreatitis [PEP]). Updates on multidisciplinary management of (infected) pancreatic necrosis is beyond the scope of this review. Table 1 summarizes the concepts discussed in this article.

Recent advances in epidemiology and evaluation of AP
Epidemiology
AP is the third most common cause of gastrointestinal-related hospitalizations and fourth most common cause of readmission in 2014.5 Recent epidemiologic studies show conflicting trends for the incidence of AP, both increasing6 and decreasing,7 likely attributable to significant differences in study designs. Importantly, multiple studies have demonstrated that hospital length of stay, costs, and mortality have declined since 2009.6,8-10

Persistent organ failure (POF), defined as organ failure lasting more than 48 hours, is the major cause of death in AP. POF, if only a single organ during AP, is associated with 27%-36% mortality; if it is multiorgan, it is associated with 47% mortality.1,11 Other factors associated with increased hospital mortality include infected pancreatic necrosis,12-14 diabetes mellitus,15 hospital-acquired infection,16 advanced age (70 years and older),17 and obesity.18 Predictive factors of 1-year mortality include readmission within 30 days, higher Charlson Comorbidity Index, and longer hospitalization.19
Risk factors
We briefly highlight recent insights into risk factors for AP (Table 1) and refer to a recent review for further discussion.20 Current and former tobacco use are independent risk factors for AP.21 The dose-response relationship of alcohol to the risk of pancreatitis is complex,22 but five standard drinks per day for 5 years is a commonly used cut-off.1,23 New evidence suggests that the relationship between the dose of alcohol and risk of AP differs by sex, linearly in men but nonlinearly (J-shaped) in women.24 Risk of AP in women was decreased with alcohol consumption of up to 40 g/day (one standard drink contains 14 g of alcohol) and increased above this amount. Cannabis is a possible risk factor for toxin-induced AP and abstinence appears to abolish risk of recurrent attacks.25
Patients with inflammatory bowel disease (IBD) have a 2.9-fold higher risk for AP versus non-IBD cohorts26 with the most common etiologies are from gallstones and medications.27 In patients with end-stage renal disease (ESRD), the risk of AP is higher in those who receive peritoneal dialysis, compared with hemodialysis28-33 and who are women, older, or have cholelithiasis or liver disease.34As recently reviewed,35 pancreatic cancer appears to be associated with first-attack pancreatitis with few exceptions.36 In this setting, the overall incidence of pancreatic cancer is low (1.5%). The risk is greatest within the first year of the attack of AP, negligible below age 40 years but steadily rising through the fifth to eighth decades.37 Pancreatic cancer screening is a conditional recommendation of the ACG guidelines in patients with unexplained AP, particularly those aged 40 years or older.1
Etiology and diagnosis
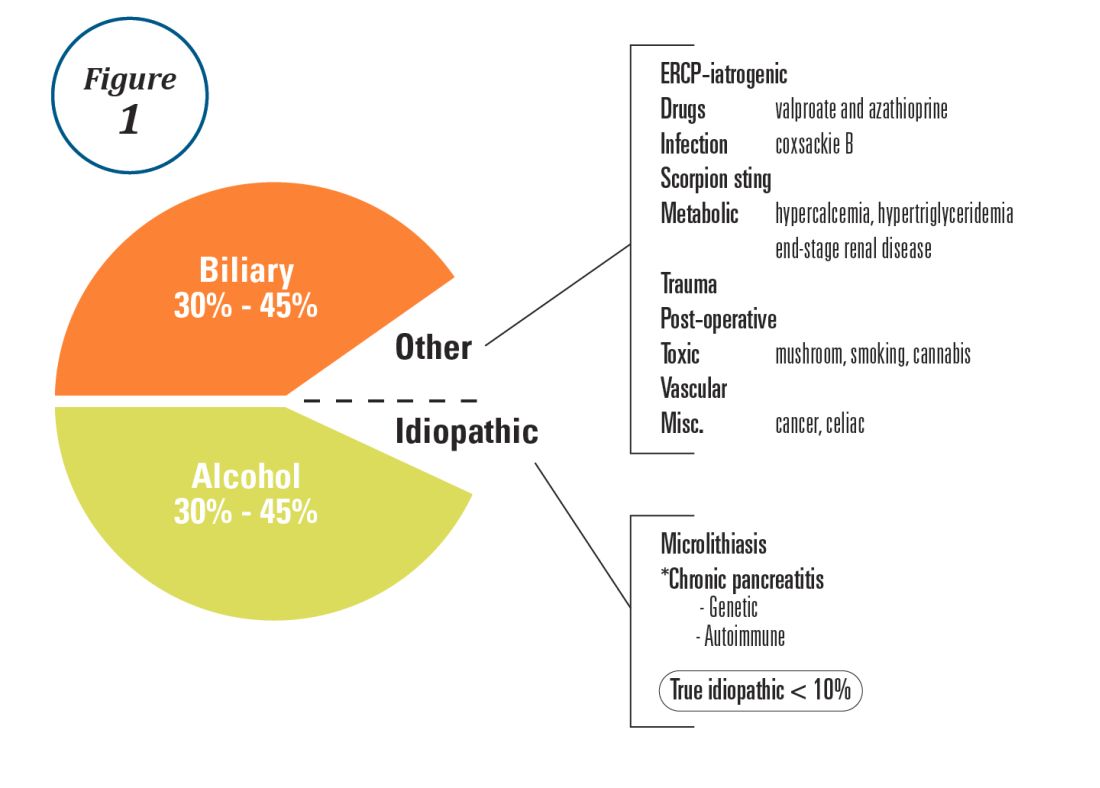
Alcohol and gallstones remain the most prevalent etiologies for AP.1 While hypertriglyceridemia accounted for 9% of AP in a systematic review of acute pancreatitis in 15 different countries,38 it is the second most common cause of acute pancreatitis in Asia (especially China).39 Figure 1 provides a breakdown of the etiologies and risk factors of pancreatitis. Importantly, it remains challenging to assign several toxic-metabolic etiologies as either a cause or risk factor for AP, particularly with regards to alcohol, smoking, and cannabis to name a few.
Guidelines and recent studies of AP raise questions about the threshold above which hypertriglyceridemia causes or poses as an important cofactor for AP. American and European societies define the threshold for triglycerides at 885-1,000 mg/dL.1,42,43 Pedersen et al. provide evidence of a graded risk of AP with hypertriglyceridemia: In multivariable analysis, adjusted hazard ratios for AP were much higher with nonfasting mild to moderately elevated plasma triglycerides (177-885 mg/dL), compared with normal values (below 89 mg/dL).44 Moreover, the risk of severe AP (developing POF) increases in proportion to triglyceride value, independent of the underlying cause of AP.45

Diagnosis of AP is derived from the revised Atlanta classification.46 The recommended timing and indications for offering cross-sectional imaging are after 48-72 hours in patients with no improvement to initial care.1 Endoscopic ultrasonography (EUS) has better diagnostic accuracy and sensitivity, compared with magnetic resonance cholangiopancreatography (MRCP) for choledocholithiasis, and has comparable specificity.47,48 Among noninvasive imaging modalities, MRCP is more sensitive than computed tomography (CT) for diagnosing choledocholithiasis.49 Despite guideline recommendations for more selective use of pancreatic imaging in the early assessment of AP, utilization of early CT or MRCP imaging (within the first 24 hours of care) remained high during 2014-2015, compared with 2006-2007.50

ERCP is not recommended as a pure diagnostic tool, owing to the availability of other diagnostic tests and a complication rate of 5%-10% with risks involving PEP, cholangitis, perforation, and hemorrhage.51 A recent systematic review of EUS and ERCP in acute biliary pancreatitis concluded that EUS had lower failure rates and had no complications, and the use of EUS avoided ERCP in 71.2% of cases.52
Risk stratification
The goals of using risk stratification tools in AP are to identify patients at risk for developing major outcomes, including POF, infected pancreatic necrosis, and death, and to ensure timely triaging of patients to an appropriate level of care. Existing prediction models have only moderate predictive value.53,54 Examples include simple risk stratification tools such as blood urea nitrogen (BUN) and hemoconcentration,55,56 disease-modifying patient variables (age, obesity, etc.), biomarkers (i.e., angiopoietin 2),57 and more complex clinical scoring systems such as Acute Physiology and Chronic Health Evaluation II (APACHE II), BISAP (BUN, impaired mental status, SIRS criteria, age, pleural effusion) score, early warning system (EWS), Glasgow-Imrie score, Japanese severity score, and recently the Pancreatitis Activity Scoring System (PASS).58 Two recent guidelines affirmed the importance of predicting the severity of AP, using one or more predictive tools.1,2 The recent 2018 AGA technical review does not debate this commonsense approach, but does highlight that there is no published observational study or randomized, controlled trial (RCT) investigating whether prediction tools affect clinical outcomes.4

Recent advances in early treatment of AP
Literature review and definitions
The AP literature contains heterogeneous definitions of severe AP and of what constitutes a major outcome in AP. Based on definitions of the 2013 revised Atlanta Criteria, the 2018 AGA technical review and clinical guidelines emphasized precise definitions of primary outcomes of clinical importance in AP, including death, persistent single organ failure, or persistent multiple organ failure, each requiring a duration of more than 48 hours, and infected pancreatic or peripancreatic necrosis or both (Table 2).3,4
Pain management
Management of pain in AP is complex and requires a detailed discussion beyond the scope of this review, but recent clinical and translational studies raise questions about the current practice of using opioids for pain management in AP. A provocative, multicenter, retrospective cohort study reported lower 30-day mortality among critically ill patients who received epidural analgesia versus standard care without epidural analgesia.59 The possible mechanism of protection and the drugs administered are unclear. An interesting hypothesis is that the epidural cohort may have received lower exposure to morphine, which may increase gut permeability, the risk of infectious complications, and severity of AP, based on a translational study in mice.60
Intravenous fluid administration
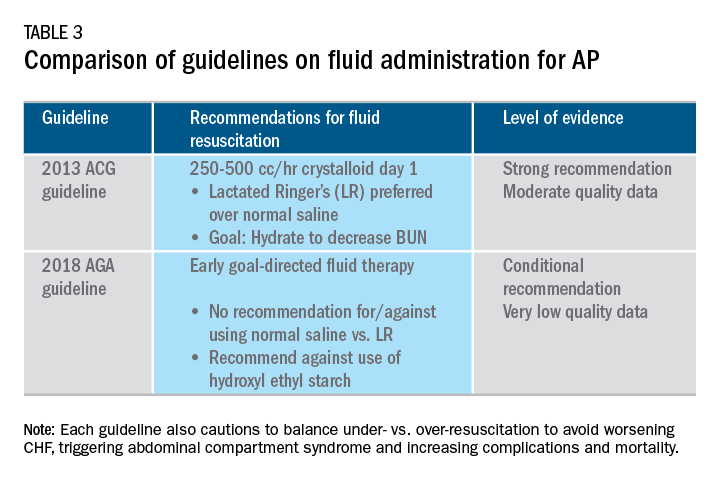
Supportive care with the use of IV fluid hydration is a mainstay of treatment for AP in the first 12-24 hours. Table 3 summarizes the guidelines in regards to IV fluid administration as delineated by the ACG and AGA guidelines on the management of pancreatitis.1,3 Guidelines advocate for early fluid resuscitation to correct intravascular depletion in order to reduce morbidity and mortality associated with AP.1,2,4 The 2018 AGA guidelines endorse a conditional recommendation for using goal-directed therapy for initial fluid management,3 do not recommend for or against normal saline versus lactated Ringer’s (LR), but do advise against the use of hydroxyethyl starch fluids.3 Consistent with these recommendations, two recent RCTs published subsequent to the prespecified time periods of the AGA technical review and guideline, observed no significant differences between LR and normal saline on clinically meaningful outcomes.61,62 The AGA guidelines acknowledge that evidence was of very-low quality in support of goal-directed therapy,3,4 which has not been shown to have a significant reduction in persistent multiple organ failure, mortality, or pancreatic necrosis, compared with usual care. As the authors noted, interpretation of the data was limited by the absence of other critical outcomes in these trials (infected pancreatic necrosis), lack of uniformity of specific outcomes and definitions of transient and POF, few trials, and risk of bias. There is a clear need for a large RCT to provide evidence to guide decision making with fluid resuscitation in AP, particularly in regard to fluid type, volume, rate, duration, endpoints, and clinical outcomes.
Feeding
More recently, the focus of nutrition in the management of AP has shifted away from patients remaining nil per os (NPO). Current guidelines advocate for early oral feeding (within 24 hours) in mild AP,3,4 in order to protect the gut-mucosal barrier. Remaining NPO when compared with early oral feeding has a 2.5-fold higher risk for interventions for necrosis.4 The recently published AGA technical review identified no significant impact on outcomes of early versus delayed oral feeding, which is consistent with observations of a landmark Dutch PYTHON trial entitled “Early versus on-demand nasoenteric tube feeding in acute pancreatitis.”4,63 There is no clear cutoff point for initiating feeding for those with severe AP. A suggested practical approach is to initiate feeding within 24-72 hours and offer enteral nutrition for those intolerant to oral feeds. In severe AP and moderately severe AP, enteral nutrition is recommended over parenteral nutrition.3,4 Enteral nutrition significantly reduces the risk of infected peripancreatic necrosis, single organ failure, and multiorgan failure.4 Finally, the AGA guidelines provide a conditional recommendation for providing enteral nutrition support through either the nasogastric or nasoenteric route.3 Further studies are required to determine the optimal timing, rate, and formulation of enteral nutrition in severe AP.
Antibiotics and probiotics
Current guidelines do not support the use of prophylactic antibiotics to prevent infection in necrotizing AP and severe AP.1-3 The AGA technical review reported that prophylactic antibiotics did not reduce infected pancreatic or peripancreatic necrosis, persistent single organ failure, or mortality.4 Guidelines advocate against the use of probiotics for severe AP, because of increased mortality risk.1
Timing of ERCP in acute biliary pancreatitis
There is universal agreement for offering urgent ERCP (within 24 hours) in biliary AP complicated by cholangitis.1-3,64 Figure 2 demonstrates an example of a cholangiogram completed within 24 hours of presentation of biliary AP complicated by cholangitis.
In the absence of cholangitis, the timing of ERCP for AP with persistent biliary obstruction is less clear.1-3 In line with recent guidelines, the 2018 AGA guidelines advocate against routine use of urgent ERCP for biliary AP without cholangitis,3 a conditional recommendation with overall low quality of data.4 The AGA technical review found that urgent ERCP, compared with conservative management in acute biliary pancreatitis without cholangitis had no significant effect on mortality, organ failure, infected pancreatic necrosis, and total necrotizing pancreatitis, but did significantly shorten hospital length of stay.4 There are limited data to guide decision making of when nonurgent ERCP should be performed in hospitalized patients with biliary AP with persistent obstruction and no cholangitis.3,64
Alcohol and smoking cessation
The AGA technical review advocates for brief alcohol intervention during hospitalization for alcohol-induced AP on the basis of one RCT that addresses the impact of alcohol counseling on recurrent bouts of AP4 plus evidence from a Cochrane review of alcohol-reduction strategies in primary care populations.65 Cessation of smoking – an established independent risk factor of AP – recurrent AP and chronic pancreatitis, should also be recommended as part of the management of AP.
Cholecystectomy
Evidence supports same-admission cholecystectomy for mild gallstone AP, a strong recommendation of published AGA guidelines.3 When compared with delayed cholecystectomy, same-admission cholecystectomy significantly reduced gallstone-related complications, readmissions for recurrent pancreatitis, and pancreaticobiliary complications, without having a significant impact on mortality during a 6-month follow-up period.66 Delaying cholecystectomy 6 weeks in patients with moderate-severe gallstone AP appears to reduce morbidity, including the development of infected collections, and mortality.4 An ongoing RCT, the APEC trial, aims to determine whether early ERCP with biliary sphincterotomy reduces major complications or death when compared with no intervention for biliary AP in patients at high risk of complications.67
Chemoprevention and IV fluid management of post-ERCP pancreatitis
Accumulating data support the effectiveness of chemoprevention, pancreatic stent placement, and fluid administration to prevent post-ERCP pancreatitis. Multiple RCTs, meta-analyses, and systematic reviews indicate that rectal NSAIDs) reduce post-ERCP pancreatitis onset68-71 and moderate-severe post-ERCP pancreatitis. Additionally, placement of a pancreatic duct stent may decrease the risk of severe post-ERCP pancreatitis in high-risk patients.3 Guidelines do not comment on fluid administrations for prevention of post-ERCP pancreatitis, but studies have shown that greater periprocedural IV fluid was an independent protective factor against moderate to severe PEP72 and was associated with shorter hospital length of stay.73 Recent meta-analyses and RCTs support using LR prior to ERCP to prevent PEP.74-77 Interestingly, a recent RCT shows that the combination of rectal indomethacin and LR, compared with combination placebo and normal saline reduced the risk of PEP in high-risk patients.78
Two ongoing multicenter RCTs will clarify the role of combination therapy. The Dutch FLUYT RCT aims to determine the optimal combination of rectal NSAIDs and periprocedural infusion of IV fluids to reduce the incidence of PEP and moderate-severe PEP79 and the Stent vs. Indomethacin (SVI) trial aims to determine the whether combination pancreatic stent placement plus rectal indomethacin is superior to monotherapy indomethacin for preventing post-ERCP pancreatitis in high-risk cases.80
Implications for clinical practice
The diagnosis and optimal management of AP require a systematic approach with multidisciplinary decision making. Morbidity and mortality in AP are driven by early or late POF, and the latter often is triggered by infected necrosis. Risk stratification of these patients at the point of contact is a commonsense approach to enable triaging of patients to the appropriate level of care. Regardless of pancreatitis severity, recommended treatment interventions include goal-directed IV fluid resuscitation, early feeding by mouth or enteral tube when necessary, avoidance of prophylactic antibiotics, avoidance of probiotics, and urgent ERCP for patients with acute biliary pancreatitis complicated by cholangitis. Key measures for preventing hospital readmission and pancreatitis include same-admission cholecystectomy for acute biliary pancreatitis and alcohol and smoking cessation. Preventive measures for post-ERCP pancreatitis in patients undergoing ERCP include rectal indomethacin, prophylactic pancreatic duct stent placement, and periprocedural fluid resuscitation.
Dr. Mandalia is a fellow, gastroenterology, department of internal medicine, division of gastroenterology, Michigan Medicine, Ann Arbor; Dr. DiMagno is associate professor of medicine, director, comprehensive pancreas program, department of internal medicine, division of gastroenterology, University of Michigan, Ann Arbor. Dr. Mandalia reports no conflicts of interest.
References
1. Tenner S et al. Am J Gastroenterol. 2013;108:1400.
2. Besseline M et al. Pancreatology. 2013;13(4, Supplement 2):e1-15.
3. Crockett SD et al. Gastroenterology. 2018;154(4):1096-101.
4. Vege SS et al. Gastroenterology. 2018;154(4):1103-39.
5. Peery AF et al. Gastroenterology. 2019 Jan;156(1):254-72.e11.
6. Krishna SG et al. Pancreas. 2017;46(4):482-8.
7. Sellers ZM et al. Gastroenterology. 2018;155(2):469-78.e1.
8. Brown A et al. JOP. 2008;9(4):408-14.
9. Fagenholz PJ et al. Ann Epidemiol. 2007;17(7):491.e1-.e8.
10. McNabb-Baltar J et al. Pancreas. 2014;43(5):687-91.
11. Johnson CD et al. Gut. 2004;53(9):1340-4.
12. Dellinger EP et al. Ann Surg. 2012;256(6):875-80.
13. Petrov MS et al. Gastroenterology. 2010;139(3):813-20.
14. Sternby H et al. Ann Surg. Apr 18. doi: 10.1097/SLA.0000000000002766.
15. Huh JH et al. J Clin Gastroenterol. 2018;52(2):178-83.
16. Wu BU et al. Gastroenterology. 2008;135(3):816-20.
17. Gardner TB et al. Clin Gastroenterol Hepatol. 2008;6(10):1070-6.
18. Krishna SG et al. Am J Gastroenterol. 2015;110(11):1608-19.
19. Lee PJ et al. Pancreas. 2016;45(4):561-4.
20. Mandalia A et al. F1000Research. 2018 Jun 28;7.
21. Majumder S et al. Pancreas. 2015;44(4):540-6.
22. DiMagno MJ. Clin Gastroenterol Hepatol. 2011;9(11):920-2.
23. Yadav D, Whitcomb DC. Nature Rev Gastroenterol Hepatol. 2010;7(3):131-45.
24. Samokhvalov AV et al. EBioMedicine. 2015;2(12):1996-2002.
25. Barkin JA et al. Pancreas. 2017;46(8):1035-8.
26. Chen Y-T et al. J Gastroenterol Hepatol. 2016;31(4):782-7.
27. Ramos LR et al. J Crohns Colitis. 2016;10(1):95-104.
28. Avram MM. Nephron. 1977;18(1):68-71.
29. Lankisch PG et al. Nephrol Dial Transplant. 2008;23(4):1401-5.
30. Owyang C et al. Mayo Clin Proc. 1979;54(12):769-73.
31. Owyang Cet al. Gut. 1982;23(5):357-61.
32. Quraishi ER et al. Am J Gastroenterol. 2005;100:2288.
33. Vaziri ND et al. Nephron. 1987;46(4):347-9.
34. Chen HJ et al. Nephrol Dial Transplant. 2017;32(10):1731-6.
35. Kirkegard J et al. Gastroenterology. 2018;May;154(6):1729-36.
36. Karlson BM, et al. Gastroenterology. 1997;113(2):587-92.
37. Munigala S et al. Clin Gastroenterol Hepatol. 2014;12(7):1143-50.e1.
38. Carr RA et al. Pancreatology. 2016;16(4):469-76.
39. Li X et al. BMC Gastroenterol. 2018;18(1):89.
40. Ahmed AU et al. Clin Gastroenterol Hepatol. 2016;14(5):738-46.
41. Sankaran SJ et al. Gastroenterology. 2015;149(6):1490-500.e1.
42. Berglund L et al. J Clin Endocrinol Metab. 2012;97(9):2969-89.
43. Catapano AL et al. Atherosclerosis. 2011;217(1):3-46.
44. Pedersen SB et al. JAMA Intern Med. 2016;176(12):1834-42.
45. Nawaz H et al. Am J Gastroenterol. 2015;110(10):1497-503.
46. Banks PA et al. Gut. 2013;62(1):102-11.
47. Kondo S et al. Eur J Radiol. 2005;54(2):271-5.
48. Meeralam Y et al. Gastrointest Endosc. 2017;86(6):986-93.
49. Stimac D et al. Am J Gastroenterol. 2007;102(5):997-1004.
50. Jin DX et al. Dig Dis Sci. 2017;62(10):2894-9.
51. Freeman ML. Gastrointest Endosc Clin N Am. 2012;22(3):567-86.
52. De Lisi S et al. Eur J Gastroenterol Hepatol. 2011;23(5):367-74.
53. Di MY et al. Ann Int Med. 2016;165(7):482-90.
54. Mounzer R et al. Gastroenterology. 2012;142(7):1476-82; quiz e15-6.
55. Koutroumpakis E et al. Am J Gastroenterol. 2015;110(12):1707-16.
56. Wu BU et al. Gastroenterology. 2009;137(1):129-35.
57. Buddingh KT et al. J Am Coll Surg. 2014;218(1):26-32.
58. Buxbaum J et al. Am J Gastroenterol. 2018;113(5):755-64.
59. Jabaudon M et al. Crit Car Med. 2018;46(3):e198-e205.
60. Barlass U et al. Gut. 2018;67(4):600-2.
61. Buxbaum JL et al. Am J Gastroenterol. 2017;112(5):797-803.
62. de-Madaria E et al. United Eur Gastroenterol J. 2018;6(1):63-72.
63. Bakker OJ et al. N Engl J Med. 2014;371(21):1983-93.
64. Tse F et al. Cochrane Database Syst Rev. 2012(5):Cd009779.
65. Kaner EFS et al. Cochrane Database Syst Rev. 2007(2):Cd004148.
66. da Costa DW et al. Lancet. 2015;386(10000):1261-8.
67. Schepers NJ et al. Trials. 2016;17:5.
68. Vadala di Prampero SF et al. Eur J Gastroenterol Hepatol. 2016;28(12):1415-24.
69. Kubiliun NM et al. Clin Gastroenterol Hepatol. 2015;13(7):1231-9; quiz e70-1.
70. Wan J et al. BMC Gastroenterol. 2017;17(1):43.
71. Yang C et al. Pancreatology. 2017;17(5):681-8.
72. DiMagno MJ et al. Pancreas. 2014;43(4):642-7.
73. Sagi SV et al. J Gastroenterol Hepatol. 2014;29(6):1316-20.
74. Choi JH et al. Clin Gastroenterol Hepatol. 2017;15(1):86-92.e1.
75. Wu D et al. J Clin Gastroenterol. 2017;51(8):e68-e76.
76. Zhang ZF et al. J Clin Gastroenterol. 2017;51(3):e17-e26.
77. Park CH et al. Endoscopy 2018 Apr;50(4):378-85.
78. Mok SRS et al. Gastrointest Endosc. 2017;85(5):1005-13.
79. Smeets XJN et al. Trials. 2018;19(1):207.
80. Elmunzer BJ et al. Trials. 2016;17(1):120.
Introduction
Acute pancreatitis (AP) is a major clinical and financial burden in the United States. Several major clinical guidelines provide evidence-based recommendations for the clinical management decisions in AP, including those from the American College of Gastroenterology (ACG; 2013),1 and the International Association of Pancreatology (IAP; 2013).2 More recently, the American Gastroenterological Association (AGA) released their own set of guidelines.3,4 In this update on AP, we review these guidelines and reference recent literature focused on epidemiology, risk factors, etiology, diagnosis, risk stratification, and recent advances in the early medical management of AP. Regarding the latter, we review six treatment interventions (pain management, intravenous fluid resuscitation, feeding, prophylactic antibiotics, probiotics, and timing of endoscopic retrograde cholangiopancreatography (ERCP) in acute biliary pancreatitis) and four preventive interventions (alcohol and smoking cessation, same-admission cholecystectomy for acute biliary pancreatitis, and chemoprevention and fluid administration for post-ERCP pancreatitis [PEP]). Updates on multidisciplinary management of (infected) pancreatic necrosis is beyond the scope of this review. Table 1 summarizes the concepts discussed in this article.
Recent advances in epidemiology and evaluation of AP
Epidemiology
AP is the third most common cause of gastrointestinal-related hospitalizations and fourth most common cause of readmission in 2014.5 Recent epidemiologic studies show conflicting trends for the incidence of AP, both increasing6 and decreasing,7 likely attributable to significant differences in study designs. Importantly, multiple studies have demonstrated that hospital length of stay, costs, and mortality have declined since 2009.6,8-10
Persistent organ failure (POF), defined as organ failure lasting more than 48 hours, is the major cause of death in AP. POF, if only a single organ during AP, is associated with 27%-36% mortality; if it is multiorgan, it is associated with 47% mortality.1,11 Other factors associated with increased hospital mortality include infected pancreatic necrosis,12-14 diabetes mellitus,15 hospital-acquired infection,16 advanced age (70 years and older),17 and obesity.18 Predictive factors of 1-year mortality include readmission within 30 days, higher Charlson Comorbidity Index, and longer hospitalization.19
Risk factors
We briefly highlight recent insights into risk factors for AP (Table 1) and refer to a recent review for further discussion.20 Current and former tobacco use are independent risk factors for AP.21 The dose-response relationship of alcohol to the risk of pancreatitis is complex,22 but five standard drinks per day for 5 years is a commonly used cut-off.1,23 New evidence suggests that the relationship between the dose of alcohol and risk of AP differs by sex, linearly in men but nonlinearly (J-shaped) in women.24 Risk of AP in women was decreased with alcohol consumption of up to 40 g/day (one standard drink contains 14 g of alcohol) and increased above this amount. Cannabis is a possible risk factor for toxin-induced AP and abstinence appears to abolish risk of recurrent attacks.25
Patients with inflammatory bowel disease (IBD) have a 2.9-fold higher risk for AP versus non-IBD cohorts26 with the most common etiologies are from gallstones and medications.27 In patients with end-stage renal disease (ESRD), the risk of AP is higher in those who receive peritoneal dialysis, compared with hemodialysis28-33 and who are women, older, or have cholelithiasis or liver disease.34As recently reviewed,35 pancreatic cancer appears to be associated with first-attack pancreatitis with few exceptions.36 In this setting, the overall incidence of pancreatic cancer is low (1.5%). The risk is greatest within the first year of the attack of AP, negligible below age 40 years but steadily rising through the fifth to eighth decades.37 Pancreatic cancer screening is a conditional recommendation of the ACG guidelines in patients with unexplained AP, particularly those aged 40 years or older.1
Etiology and diagnosis
Alcohol and gallstones remain the most prevalent etiologies for AP.1 While hypertriglyceridemia accounted for 9% of AP in a systematic review of acute pancreatitis in 15 different countries,38 it is the second most common cause of acute pancreatitis in Asia (especially China).39 Figure 1 provides a breakdown of the etiologies and risk factors of pancreatitis. Importantly, it remains challenging to assign several toxic-metabolic etiologies as either a cause or risk factor for AP, particularly with regards to alcohol, smoking, and cannabis to name a few.
Guidelines and recent studies of AP raise questions about the threshold above which hypertriglyceridemia causes or poses as an important cofactor for AP. American and European societies define the threshold for triglycerides at 885-1,000 mg/dL.1,42,43 Pedersen et al. provide evidence of a graded risk of AP with hypertriglyceridemia: In multivariable analysis, adjusted hazard ratios for AP were much higher with nonfasting mild to moderately elevated plasma triglycerides (177-885 mg/dL), compared with normal values (below 89 mg/dL).44 Moreover, the risk of severe AP (developing POF) increases in proportion to triglyceride value, independent of the underlying cause of AP.45
Diagnosis of AP is derived from the revised Atlanta classification.46 The recommended timing and indications for offering cross-sectional imaging are after 48-72 hours in patients with no improvement to initial care.1 Endoscopic ultrasonography (EUS) has better diagnostic accuracy and sensitivity, compared with magnetic resonance cholangiopancreatography (MRCP) for choledocholithiasis, and has comparable specificity.47,48 Among noninvasive imaging modalities, MRCP is more sensitive than computed tomography (CT) for diagnosing choledocholithiasis.49 Despite guideline recommendations for more selective use of pancreatic imaging in the early assessment of AP, utilization of early CT or MRCP imaging (within the first 24 hours of care) remained high during 2014-2015, compared with 2006-2007.50
ERCP is not recommended as a pure diagnostic tool, owing to the availability of other diagnostic tests and a complication rate of 5%-10% with risks involving PEP, cholangitis, perforation, and hemorrhage.51 A recent systematic review of EUS and ERCP in acute biliary pancreatitis concluded that EUS had lower failure rates and had no complications, and the use of EUS avoided ERCP in 71.2% of cases.52
Risk stratification
The goals of using risk stratification tools in AP are to identify patients at risk for developing major outcomes, including POF, infected pancreatic necrosis, and death, and to ensure timely triaging of patients to an appropriate level of care. Existing prediction models have only moderate predictive value.53,54 Examples include simple risk stratification tools such as blood urea nitrogen (BUN) and hemoconcentration,55,56 disease-modifying patient variables (age, obesity, etc.), biomarkers (i.e., angiopoietin 2),57 and more complex clinical scoring systems such as Acute Physiology and Chronic Health Evaluation II (APACHE II), BISAP (BUN, impaired mental status, SIRS criteria, age, pleural effusion) score, early warning system (EWS), Glasgow-Imrie score, Japanese severity score, and recently the Pancreatitis Activity Scoring System (PASS).58 Two recent guidelines affirmed the importance of predicting the severity of AP, using one or more predictive tools.1,2 The recent 2018 AGA technical review does not debate this commonsense approach, but does highlight that there is no published observational study or randomized, controlled trial (RCT) investigating whether prediction tools affect clinical outcomes.4
Recent advances in early treatment of AP
Literature review and definitions
The AP literature contains heterogeneous definitions of severe AP and of what constitutes a major outcome in AP. Based on definitions of the 2013 revised Atlanta Criteria, the 2018 AGA technical review and clinical guidelines emphasized precise definitions of primary outcomes of clinical importance in AP, including death, persistent single organ failure, or persistent multiple organ failure, each requiring a duration of more than 48 hours, and infected pancreatic or peripancreatic necrosis or both (Table 2).3,4
Pain management
Management of pain in AP is complex and requires a detailed discussion beyond the scope of this review, but recent clinical and translational studies raise questions about the current practice of using opioids for pain management in AP. A provocative, multicenter, retrospective cohort study reported lower 30-day mortality among critically ill patients who received epidural analgesia versus standard care without epidural analgesia.59 The possible mechanism of protection and the drugs administered are unclear. An interesting hypothesis is that the epidural cohort may have received lower exposure to morphine, which may increase gut permeability, the risk of infectious complications, and severity of AP, based on a translational study in mice.60
Intravenous fluid administration
Supportive care with the use of IV fluid hydration is a mainstay of treatment for AP in the first 12-24 hours. Table 3 summarizes the guidelines in regards to IV fluid administration as delineated by the ACG and AGA guidelines on the management of pancreatitis.1,3 Guidelines advocate for early fluid resuscitation to correct intravascular depletion in order to reduce morbidity and mortality associated with AP.1,2,4 The 2018 AGA guidelines endorse a conditional recommendation for using goal-directed therapy for initial fluid management,3 do not recommend for or against normal saline versus lactated Ringer’s (LR), but do advise against the use of hydroxyethyl starch fluids.3 Consistent with these recommendations, two recent RCTs published subsequent to the prespecified time periods of the AGA technical review and guideline, observed no significant differences between LR and normal saline on clinically meaningful outcomes.61,62 The AGA guidelines acknowledge that evidence was of very-low quality in support of goal-directed therapy,3,4 which has not been shown to have a significant reduction in persistent multiple organ failure, mortality, or pancreatic necrosis, compared with usual care. As the authors noted, interpretation of the data was limited by the absence of other critical outcomes in these trials (infected pancreatic necrosis), lack of uniformity of specific outcomes and definitions of transient and POF, few trials, and risk of bias. There is a clear need for a large RCT to provide evidence to guide decision making with fluid resuscitation in AP, particularly in regard to fluid type, volume, rate, duration, endpoints, and clinical outcomes.
Feeding
More recently, the focus of nutrition in the management of AP has shifted away from patients remaining nil per os (NPO). Current guidelines advocate for early oral feeding (within 24 hours) in mild AP,3,4 in order to protect the gut-mucosal barrier. Remaining NPO when compared with early oral feeding has a 2.5-fold higher risk for interventions for necrosis.4 The recently published AGA technical review identified no significant impact on outcomes of early versus delayed oral feeding, which is consistent with observations of a landmark Dutch PYTHON trial entitled “Early versus on-demand nasoenteric tube feeding in acute pancreatitis.”4,63 There is no clear cutoff point for initiating feeding for those with severe AP. A suggested practical approach is to initiate feeding within 24-72 hours and offer enteral nutrition for those intolerant to oral feeds. In severe AP and moderately severe AP, enteral nutrition is recommended over parenteral nutrition.3,4 Enteral nutrition significantly reduces the risk of infected peripancreatic necrosis, single organ failure, and multiorgan failure.4 Finally, the AGA guidelines provide a conditional recommendation for providing enteral nutrition support through either the nasogastric or nasoenteric route.3 Further studies are required to determine the optimal timing, rate, and formulation of enteral nutrition in severe AP.
Antibiotics and probiotics
Current guidelines do not support the use of prophylactic antibiotics to prevent infection in necrotizing AP and severe AP.1-3 The AGA technical review reported that prophylactic antibiotics did not reduce infected pancreatic or peripancreatic necrosis, persistent single organ failure, or mortality.4 Guidelines advocate against the use of probiotics for severe AP, because of increased mortality risk.1
Timing of ERCP in acute biliary pancreatitis
There is universal agreement for offering urgent ERCP (within 24 hours) in biliary AP complicated by cholangitis.1-3,64 Figure 2 demonstrates an example of a cholangiogram completed within 24 hours of presentation of biliary AP complicated by cholangitis.
In the absence of cholangitis, the timing of ERCP for AP with persistent biliary obstruction is less clear.1-3 In line with recent guidelines, the 2018 AGA guidelines advocate against routine use of urgent ERCP for biliary AP without cholangitis,3 a conditional recommendation with overall low quality of data.4 The AGA technical review found that urgent ERCP, compared with conservative management in acute biliary pancreatitis without cholangitis had no significant effect on mortality, organ failure, infected pancreatic necrosis, and total necrotizing pancreatitis, but did significantly shorten hospital length of stay.4 There are limited data to guide decision making of when nonurgent ERCP should be performed in hospitalized patients with biliary AP with persistent obstruction and no cholangitis.3,64
Alcohol and smoking cessation
The AGA technical review advocates for brief alcohol intervention during hospitalization for alcohol-induced AP on the basis of one RCT that addresses the impact of alcohol counseling on recurrent bouts of AP4 plus evidence from a Cochrane review of alcohol-reduction strategies in primary care populations.65 Cessation of smoking – an established independent risk factor of AP – recurrent AP and chronic pancreatitis, should also be recommended as part of the management of AP.
Cholecystectomy
Evidence supports same-admission cholecystectomy for mild gallstone AP, a strong recommendation of published AGA guidelines.3 When compared with delayed cholecystectomy, same-admission cholecystectomy significantly reduced gallstone-related complications, readmissions for recurrent pancreatitis, and pancreaticobiliary complications, without having a significant impact on mortality during a 6-month follow-up period.66 Delaying cholecystectomy 6 weeks in patients with moderate-severe gallstone AP appears to reduce morbidity, including the development of infected collections, and mortality.4 An ongoing RCT, the APEC trial, aims to determine whether early ERCP with biliary sphincterotomy reduces major complications or death when compared with no intervention for biliary AP in patients at high risk of complications.67
Chemoprevention and IV fluid management of post-ERCP pancreatitis
Accumulating data support the effectiveness of chemoprevention, pancreatic stent placement, and fluid administration to prevent post-ERCP pancreatitis. Multiple RCTs, meta-analyses, and systematic reviews indicate that rectal NSAIDs) reduce post-ERCP pancreatitis onset68-71 and moderate-severe post-ERCP pancreatitis. Additionally, placement of a pancreatic duct stent may decrease the risk of severe post-ERCP pancreatitis in high-risk patients.3 Guidelines do not comment on fluid administrations for prevention of post-ERCP pancreatitis, but studies have shown that greater periprocedural IV fluid was an independent protective factor against moderate to severe PEP72 and was associated with shorter hospital length of stay.73 Recent meta-analyses and RCTs support using LR prior to ERCP to prevent PEP.74-77 Interestingly, a recent RCT shows that the combination of rectal indomethacin and LR, compared with combination placebo and normal saline reduced the risk of PEP in high-risk patients.78
Two ongoing multicenter RCTs will clarify the role of combination therapy. The Dutch FLUYT RCT aims to determine the optimal combination of rectal NSAIDs and periprocedural infusion of IV fluids to reduce the incidence of PEP and moderate-severe PEP79 and the Stent vs. Indomethacin (SVI) trial aims to determine the whether combination pancreatic stent placement plus rectal indomethacin is superior to monotherapy indomethacin for preventing post-ERCP pancreatitis in high-risk cases.80
Implications for clinical practice
The diagnosis and optimal management of AP require a systematic approach with multidisciplinary decision making. Morbidity and mortality in AP are driven by early or late POF, and the latter often is triggered by infected necrosis. Risk stratification of these patients at the point of contact is a commonsense approach to enable triaging of patients to the appropriate level of care. Regardless of pancreatitis severity, recommended treatment interventions include goal-directed IV fluid resuscitation, early feeding by mouth or enteral tube when necessary, avoidance of prophylactic antibiotics, avoidance of probiotics, and urgent ERCP for patients with acute biliary pancreatitis complicated by cholangitis. Key measures for preventing hospital readmission and pancreatitis include same-admission cholecystectomy for acute biliary pancreatitis and alcohol and smoking cessation. Preventive measures for post-ERCP pancreatitis in patients undergoing ERCP include rectal indomethacin, prophylactic pancreatic duct stent placement, and periprocedural fluid resuscitation.
Dr. Mandalia is a fellow, gastroenterology, department of internal medicine, division of gastroenterology, Michigan Medicine, Ann Arbor; Dr. DiMagno is associate professor of medicine, director, comprehensive pancreas program, department of internal medicine, division of gastroenterology, University of Michigan, Ann Arbor. Dr. Mandalia reports no conflicts of interest.
References
1. Tenner S et al. Am J Gastroenterol. 2013;108:1400.
2. Besseline M et al. Pancreatology. 2013;13(4, Supplement 2):e1-15.
3. Crockett SD et al. Gastroenterology. 2018;154(4):1096-101.
4. Vege SS et al. Gastroenterology. 2018;154(4):1103-39.
5. Peery AF et al. Gastroenterology. 2019 Jan;156(1):254-72.e11.
6. Krishna SG et al. Pancreas. 2017;46(4):482-8.
7. Sellers ZM et al. Gastroenterology. 2018;155(2):469-78.e1.
8. Brown A et al. JOP. 2008;9(4):408-14.
9. Fagenholz PJ et al. Ann Epidemiol. 2007;17(7):491.e1-.e8.
10. McNabb-Baltar J et al. Pancreas. 2014;43(5):687-91.
11. Johnson CD et al. Gut. 2004;53(9):1340-4.
12. Dellinger EP et al. Ann Surg. 2012;256(6):875-80.
13. Petrov MS et al. Gastroenterology. 2010;139(3):813-20.
14. Sternby H et al. Ann Surg. Apr 18. doi: 10.1097/SLA.0000000000002766.
15. Huh JH et al. J Clin Gastroenterol. 2018;52(2):178-83.
16. Wu BU et al. Gastroenterology. 2008;135(3):816-20.
17. Gardner TB et al. Clin Gastroenterol Hepatol. 2008;6(10):1070-6.
18. Krishna SG et al. Am J Gastroenterol. 2015;110(11):1608-19.
19. Lee PJ et al. Pancreas. 2016;45(4):561-4.
20. Mandalia A et al. F1000Research. 2018 Jun 28;7.
21. Majumder S et al. Pancreas. 2015;44(4):540-6.
22. DiMagno MJ. Clin Gastroenterol Hepatol. 2011;9(11):920-2.
23. Yadav D, Whitcomb DC. Nature Rev Gastroenterol Hepatol. 2010;7(3):131-45.
24. Samokhvalov AV et al. EBioMedicine. 2015;2(12):1996-2002.
25. Barkin JA et al. Pancreas. 2017;46(8):1035-8.
26. Chen Y-T et al. J Gastroenterol Hepatol. 2016;31(4):782-7.
27. Ramos LR et al. J Crohns Colitis. 2016;10(1):95-104.
28. Avram MM. Nephron. 1977;18(1):68-71.
29. Lankisch PG et al. Nephrol Dial Transplant. 2008;23(4):1401-5.
30. Owyang C et al. Mayo Clin Proc. 1979;54(12):769-73.
31. Owyang Cet al. Gut. 1982;23(5):357-61.
32. Quraishi ER et al. Am J Gastroenterol. 2005;100:2288.
33. Vaziri ND et al. Nephron. 1987;46(4):347-9.
34. Chen HJ et al. Nephrol Dial Transplant. 2017;32(10):1731-6.
35. Kirkegard J et al. Gastroenterology. 2018;May;154(6):1729-36.
36. Karlson BM, et al. Gastroenterology. 1997;113(2):587-92.
37. Munigala S et al. Clin Gastroenterol Hepatol. 2014;12(7):1143-50.e1.
38. Carr RA et al. Pancreatology. 2016;16(4):469-76.
39. Li X et al. BMC Gastroenterol. 2018;18(1):89.
40. Ahmed AU et al. Clin Gastroenterol Hepatol. 2016;14(5):738-46.
41. Sankaran SJ et al. Gastroenterology. 2015;149(6):1490-500.e1.
42. Berglund L et al. J Clin Endocrinol Metab. 2012;97(9):2969-89.
43. Catapano AL et al. Atherosclerosis. 2011;217(1):3-46.
44. Pedersen SB et al. JAMA Intern Med. 2016;176(12):1834-42.
45. Nawaz H et al. Am J Gastroenterol. 2015;110(10):1497-503.
46. Banks PA et al. Gut. 2013;62(1):102-11.
47. Kondo S et al. Eur J Radiol. 2005;54(2):271-5.
48. Meeralam Y et al. Gastrointest Endosc. 2017;86(6):986-93.
49. Stimac D et al. Am J Gastroenterol. 2007;102(5):997-1004.
50. Jin DX et al. Dig Dis Sci. 2017;62(10):2894-9.
51. Freeman ML. Gastrointest Endosc Clin N Am. 2012;22(3):567-86.
52. De Lisi S et al. Eur J Gastroenterol Hepatol. 2011;23(5):367-74.
53. Di MY et al. Ann Int Med. 2016;165(7):482-90.
54. Mounzer R et al. Gastroenterology. 2012;142(7):1476-82; quiz e15-6.
55. Koutroumpakis E et al. Am J Gastroenterol. 2015;110(12):1707-16.
56. Wu BU et al. Gastroenterology. 2009;137(1):129-35.
57. Buddingh KT et al. J Am Coll Surg. 2014;218(1):26-32.
58. Buxbaum J et al. Am J Gastroenterol. 2018;113(5):755-64.
59. Jabaudon M et al. Crit Car Med. 2018;46(3):e198-e205.
60. Barlass U et al. Gut. 2018;67(4):600-2.
61. Buxbaum JL et al. Am J Gastroenterol. 2017;112(5):797-803.
62. de-Madaria E et al. United Eur Gastroenterol J. 2018;6(1):63-72.
63. Bakker OJ et al. N Engl J Med. 2014;371(21):1983-93.
64. Tse F et al. Cochrane Database Syst Rev. 2012(5):Cd009779.
65. Kaner EFS et al. Cochrane Database Syst Rev. 2007(2):Cd004148.
66. da Costa DW et al. Lancet. 2015;386(10000):1261-8.
67. Schepers NJ et al. Trials. 2016;17:5.
68. Vadala di Prampero SF et al. Eur J Gastroenterol Hepatol. 2016;28(12):1415-24.
69. Kubiliun NM et al. Clin Gastroenterol Hepatol. 2015;13(7):1231-9; quiz e70-1.
70. Wan J et al. BMC Gastroenterol. 2017;17(1):43.
71. Yang C et al. Pancreatology. 2017;17(5):681-8.
72. DiMagno MJ et al. Pancreas. 2014;43(4):642-7.
73. Sagi SV et al. J Gastroenterol Hepatol. 2014;29(6):1316-20.
74. Choi JH et al. Clin Gastroenterol Hepatol. 2017;15(1):86-92.e1.
75. Wu D et al. J Clin Gastroenterol. 2017;51(8):e68-e76.
76. Zhang ZF et al. J Clin Gastroenterol. 2017;51(3):e17-e26.
77. Park CH et al. Endoscopy 2018 Apr;50(4):378-85.
78. Mok SRS et al. Gastrointest Endosc. 2017;85(5):1005-13.
79. Smeets XJN et al. Trials. 2018;19(1):207.
80. Elmunzer BJ et al. Trials. 2016;17(1):120.
Introduction
Acute pancreatitis (AP) is a major clinical and financial burden in the United States. Several major clinical guidelines provide evidence-based recommendations for the clinical management decisions in AP, including those from the American College of Gastroenterology (ACG; 2013),1 and the International Association of Pancreatology (IAP; 2013).2 More recently, the American Gastroenterological Association (AGA) released their own set of guidelines.3,4 In this update on AP, we review these guidelines and reference recent literature focused on epidemiology, risk factors, etiology, diagnosis, risk stratification, and recent advances in the early medical management of AP. Regarding the latter, we review six treatment interventions (pain management, intravenous fluid resuscitation, feeding, prophylactic antibiotics, probiotics, and timing of endoscopic retrograde cholangiopancreatography (ERCP) in acute biliary pancreatitis) and four preventive interventions (alcohol and smoking cessation, same-admission cholecystectomy for acute biliary pancreatitis, and chemoprevention and fluid administration for post-ERCP pancreatitis [PEP]). Updates on multidisciplinary management of (infected) pancreatic necrosis is beyond the scope of this review. Table 1 summarizes the concepts discussed in this article.
Recent advances in epidemiology and evaluation of AP
Epidemiology
AP is the third most common cause of gastrointestinal-related hospitalizations and fourth most common cause of readmission in 2014.5 Recent epidemiologic studies show conflicting trends for the incidence of AP, both increasing6 and decreasing,7 likely attributable to significant differences in study designs. Importantly, multiple studies have demonstrated that hospital length of stay, costs, and mortality have declined since 2009.6,8-10
Persistent organ failure (POF), defined as organ failure lasting more than 48 hours, is the major cause of death in AP. POF, if only a single organ during AP, is associated with 27%-36% mortality; if it is multiorgan, it is associated with 47% mortality.1,11 Other factors associated with increased hospital mortality include infected pancreatic necrosis,12-14 diabetes mellitus,15 hospital-acquired infection,16 advanced age (70 years and older),17 and obesity.18 Predictive factors of 1-year mortality include readmission within 30 days, higher Charlson Comorbidity Index, and longer hospitalization.19
Risk factors
We briefly highlight recent insights into risk factors for AP (Table 1) and refer to a recent review for further discussion.20 Current and former tobacco use are independent risk factors for AP.21 The dose-response relationship of alcohol to the risk of pancreatitis is complex,22 but five standard drinks per day for 5 years is a commonly used cut-off.1,23 New evidence suggests that the relationship between the dose of alcohol and risk of AP differs by sex, linearly in men but nonlinearly (J-shaped) in women.24 Risk of AP in women was decreased with alcohol consumption of up to 40 g/day (one standard drink contains 14 g of alcohol) and increased above this amount. Cannabis is a possible risk factor for toxin-induced AP and abstinence appears to abolish risk of recurrent attacks.25
Patients with inflammatory bowel disease (IBD) have a 2.9-fold higher risk for AP versus non-IBD cohorts26 with the most common etiologies are from gallstones and medications.27 In patients with end-stage renal disease (ESRD), the risk of AP is higher in those who receive peritoneal dialysis, compared with hemodialysis28-33 and who are women, older, or have cholelithiasis or liver disease.34As recently reviewed,35 pancreatic cancer appears to be associated with first-attack pancreatitis with few exceptions.36 In this setting, the overall incidence of pancreatic cancer is low (1.5%). The risk is greatest within the first year of the attack of AP, negligible below age 40 years but steadily rising through the fifth to eighth decades.37 Pancreatic cancer screening is a conditional recommendation of the ACG guidelines in patients with unexplained AP, particularly those aged 40 years or older.1
Etiology and diagnosis
Alcohol and gallstones remain the most prevalent etiologies for AP.1 While hypertriglyceridemia accounted for 9% of AP in a systematic review of acute pancreatitis in 15 different countries,38 it is the second most common cause of acute pancreatitis in Asia (especially China).39 Figure 1 provides a breakdown of the etiologies and risk factors of pancreatitis. Importantly, it remains challenging to assign several toxic-metabolic etiologies as either a cause or risk factor for AP, particularly with regards to alcohol, smoking, and cannabis to name a few.
Guidelines and recent studies of AP raise questions about the threshold above which hypertriglyceridemia causes or poses as an important cofactor for AP. American and European societies define the threshold for triglycerides at 885-1,000 mg/dL.1,42,43 Pedersen et al. provide evidence of a graded risk of AP with hypertriglyceridemia: In multivariable analysis, adjusted hazard ratios for AP were much higher with nonfasting mild to moderately elevated plasma triglycerides (177-885 mg/dL), compared with normal values (below 89 mg/dL).44 Moreover, the risk of severe AP (developing POF) increases in proportion to triglyceride value, independent of the underlying cause of AP.45
Diagnosis of AP is derived from the revised Atlanta classification.46 The recommended timing and indications for offering cross-sectional imaging are after 48-72 hours in patients with no improvement to initial care.1 Endoscopic ultrasonography (EUS) has better diagnostic accuracy and sensitivity, compared with magnetic resonance cholangiopancreatography (MRCP) for choledocholithiasis, and has comparable specificity.47,48 Among noninvasive imaging modalities, MRCP is more sensitive than computed tomography (CT) for diagnosing choledocholithiasis.49 Despite guideline recommendations for more selective use of pancreatic imaging in the early assessment of AP, utilization of early CT or MRCP imaging (within the first 24 hours of care) remained high during 2014-2015, compared with 2006-2007.50
ERCP is not recommended as a pure diagnostic tool, owing to the availability of other diagnostic tests and a complication rate of 5%-10% with risks involving PEP, cholangitis, perforation, and hemorrhage.51 A recent systematic review of EUS and ERCP in acute biliary pancreatitis concluded that EUS had lower failure rates and had no complications, and the use of EUS avoided ERCP in 71.2% of cases.52
Risk stratification
The goals of using risk stratification tools in AP are to identify patients at risk for developing major outcomes, including POF, infected pancreatic necrosis, and death, and to ensure timely triaging of patients to an appropriate level of care. Existing prediction models have only moderate predictive value.53,54 Examples include simple risk stratification tools such as blood urea nitrogen (BUN) and hemoconcentration,55,56 disease-modifying patient variables (age, obesity, etc.), biomarkers (i.e., angiopoietin 2),57 and more complex clinical scoring systems such as Acute Physiology and Chronic Health Evaluation II (APACHE II), BISAP (BUN, impaired mental status, SIRS criteria, age, pleural effusion) score, early warning system (EWS), Glasgow-Imrie score, Japanese severity score, and recently the Pancreatitis Activity Scoring System (PASS).58 Two recent guidelines affirmed the importance of predicting the severity of AP, using one or more predictive tools.1,2 The recent 2018 AGA technical review does not debate this commonsense approach, but does highlight that there is no published observational study or randomized, controlled trial (RCT) investigating whether prediction tools affect clinical outcomes.4
Recent advances in early treatment of AP
Literature review and definitions
The AP literature contains heterogeneous definitions of severe AP and of what constitutes a major outcome in AP. Based on definitions of the 2013 revised Atlanta Criteria, the 2018 AGA technical review and clinical guidelines emphasized precise definitions of primary outcomes of clinical importance in AP, including death, persistent single organ failure, or persistent multiple organ failure, each requiring a duration of more than 48 hours, and infected pancreatic or peripancreatic necrosis or both (Table 2).3,4
Pain management
Management of pain in AP is complex and requires a detailed discussion beyond the scope of this review, but recent clinical and translational studies raise questions about the current practice of using opioids for pain management in AP. A provocative, multicenter, retrospective cohort study reported lower 30-day mortality among critically ill patients who received epidural analgesia versus standard care without epidural analgesia.59 The possible mechanism of protection and the drugs administered are unclear. An interesting hypothesis is that the epidural cohort may have received lower exposure to morphine, which may increase gut permeability, the risk of infectious complications, and severity of AP, based on a translational study in mice.60
Intravenous fluid administration
Supportive care with the use of IV fluid hydration is a mainstay of treatment for AP in the first 12-24 hours. Table 3 summarizes the guidelines in regards to IV fluid administration as delineated by the ACG and AGA guidelines on the management of pancreatitis.1,3 Guidelines advocate for early fluid resuscitation to correct intravascular depletion in order to reduce morbidity and mortality associated with AP.1,2,4 The 2018 AGA guidelines endorse a conditional recommendation for using goal-directed therapy for initial fluid management,3 do not recommend for or against normal saline versus lactated Ringer’s (LR), but do advise against the use of hydroxyethyl starch fluids.3 Consistent with these recommendations, two recent RCTs published subsequent to the prespecified time periods of the AGA technical review and guideline, observed no significant differences between LR and normal saline on clinically meaningful outcomes.61,62 The AGA guidelines acknowledge that evidence was of very-low quality in support of goal-directed therapy,3,4 which has not been shown to have a significant reduction in persistent multiple organ failure, mortality, or pancreatic necrosis, compared with usual care. As the authors noted, interpretation of the data was limited by the absence of other critical outcomes in these trials (infected pancreatic necrosis), lack of uniformity of specific outcomes and definitions of transient and POF, few trials, and risk of bias. There is a clear need for a large RCT to provide evidence to guide decision making with fluid resuscitation in AP, particularly in regard to fluid type, volume, rate, duration, endpoints, and clinical outcomes.
Feeding
More recently, the focus of nutrition in the management of AP has shifted away from patients remaining nil per os (NPO). Current guidelines advocate for early oral feeding (within 24 hours) in mild AP,3,4 in order to protect the gut-mucosal barrier. Remaining NPO when compared with early oral feeding has a 2.5-fold higher risk for interventions for necrosis.4 The recently published AGA technical review identified no significant impact on outcomes of early versus delayed oral feeding, which is consistent with observations of a landmark Dutch PYTHON trial entitled “Early versus on-demand nasoenteric tube feeding in acute pancreatitis.”4,63 There is no clear cutoff point for initiating feeding for those with severe AP. A suggested practical approach is to initiate feeding within 24-72 hours and offer enteral nutrition for those intolerant to oral feeds. In severe AP and moderately severe AP, enteral nutrition is recommended over parenteral nutrition.3,4 Enteral nutrition significantly reduces the risk of infected peripancreatic necrosis, single organ failure, and multiorgan failure.4 Finally, the AGA guidelines provide a conditional recommendation for providing enteral nutrition support through either the nasogastric or nasoenteric route.3 Further studies are required to determine the optimal timing, rate, and formulation of enteral nutrition in severe AP.
Antibiotics and probiotics
Current guidelines do not support the use of prophylactic antibiotics to prevent infection in necrotizing AP and severe AP.1-3 The AGA technical review reported that prophylactic antibiotics did not reduce infected pancreatic or peripancreatic necrosis, persistent single organ failure, or mortality.4 Guidelines advocate against the use of probiotics for severe AP, because of increased mortality risk.1
Timing of ERCP in acute biliary pancreatitis
There is universal agreement for offering urgent ERCP (within 24 hours) in biliary AP complicated by cholangitis.1-3,64 Figure 2 demonstrates an example of a cholangiogram completed within 24 hours of presentation of biliary AP complicated by cholangitis.
In the absence of cholangitis, the timing of ERCP for AP with persistent biliary obstruction is less clear.1-3 In line with recent guidelines, the 2018 AGA guidelines advocate against routine use of urgent ERCP for biliary AP without cholangitis,3 a conditional recommendation with overall low quality of data.4 The AGA technical review found that urgent ERCP, compared with conservative management in acute biliary pancreatitis without cholangitis had no significant effect on mortality, organ failure, infected pancreatic necrosis, and total necrotizing pancreatitis, but did significantly shorten hospital length of stay.4 There are limited data to guide decision making of when nonurgent ERCP should be performed in hospitalized patients with biliary AP with persistent obstruction and no cholangitis.3,64
Alcohol and smoking cessation
The AGA technical review advocates for brief alcohol intervention during hospitalization for alcohol-induced AP on the basis of one RCT that addresses the impact of alcohol counseling on recurrent bouts of AP4 plus evidence from a Cochrane review of alcohol-reduction strategies in primary care populations.65 Cessation of smoking – an established independent risk factor of AP – recurrent AP and chronic pancreatitis, should also be recommended as part of the management of AP.
Cholecystectomy
Evidence supports same-admission cholecystectomy for mild gallstone AP, a strong recommendation of published AGA guidelines.3 When compared with delayed cholecystectomy, same-admission cholecystectomy significantly reduced gallstone-related complications, readmissions for recurrent pancreatitis, and pancreaticobiliary complications, without having a significant impact on mortality during a 6-month follow-up period.66 Delaying cholecystectomy 6 weeks in patients with moderate-severe gallstone AP appears to reduce morbidity, including the development of infected collections, and mortality.4 An ongoing RCT, the APEC trial, aims to determine whether early ERCP with biliary sphincterotomy reduces major complications or death when compared with no intervention for biliary AP in patients at high risk of complications.67
Chemoprevention and IV fluid management of post-ERCP pancreatitis
Accumulating data support the effectiveness of chemoprevention, pancreatic stent placement, and fluid administration to prevent post-ERCP pancreatitis. Multiple RCTs, meta-analyses, and systematic reviews indicate that rectal NSAIDs) reduce post-ERCP pancreatitis onset68-71 and moderate-severe post-ERCP pancreatitis. Additionally, placement of a pancreatic duct stent may decrease the risk of severe post-ERCP pancreatitis in high-risk patients.3 Guidelines do not comment on fluid administrations for prevention of post-ERCP pancreatitis, but studies have shown that greater periprocedural IV fluid was an independent protective factor against moderate to severe PEP72 and was associated with shorter hospital length of stay.73 Recent meta-analyses and RCTs support using LR prior to ERCP to prevent PEP.74-77 Interestingly, a recent RCT shows that the combination of rectal indomethacin and LR, compared with combination placebo and normal saline reduced the risk of PEP in high-risk patients.78
Two ongoing multicenter RCTs will clarify the role of combination therapy. The Dutch FLUYT RCT aims to determine the optimal combination of rectal NSAIDs and periprocedural infusion of IV fluids to reduce the incidence of PEP and moderate-severe PEP79 and the Stent vs. Indomethacin (SVI) trial aims to determine the whether combination pancreatic stent placement plus rectal indomethacin is superior to monotherapy indomethacin for preventing post-ERCP pancreatitis in high-risk cases.80
Implications for clinical practice
The diagnosis and optimal management of AP require a systematic approach with multidisciplinary decision making. Morbidity and mortality in AP are driven by early or late POF, and the latter often is triggered by infected necrosis. Risk stratification of these patients at the point of contact is a commonsense approach to enable triaging of patients to the appropriate level of care. Regardless of pancreatitis severity, recommended treatment interventions include goal-directed IV fluid resuscitation, early feeding by mouth or enteral tube when necessary, avoidance of prophylactic antibiotics, avoidance of probiotics, and urgent ERCP for patients with acute biliary pancreatitis complicated by cholangitis. Key measures for preventing hospital readmission and pancreatitis include same-admission cholecystectomy for acute biliary pancreatitis and alcohol and smoking cessation. Preventive measures for post-ERCP pancreatitis in patients undergoing ERCP include rectal indomethacin, prophylactic pancreatic duct stent placement, and periprocedural fluid resuscitation.
Dr. Mandalia is a fellow, gastroenterology, department of internal medicine, division of gastroenterology, Michigan Medicine, Ann Arbor; Dr. DiMagno is associate professor of medicine, director, comprehensive pancreas program, department of internal medicine, division of gastroenterology, University of Michigan, Ann Arbor. Dr. Mandalia reports no conflicts of interest.
References
1. Tenner S et al. Am J Gastroenterol. 2013;108:1400.
2. Besseline M et al. Pancreatology. 2013;13(4, Supplement 2):e1-15.
3. Crockett SD et al. Gastroenterology. 2018;154(4):1096-101.
4. Vege SS et al. Gastroenterology. 2018;154(4):1103-39.
5. Peery AF et al. Gastroenterology. 2019 Jan;156(1):254-72.e11.
6. Krishna SG et al. Pancreas. 2017;46(4):482-8.
7. Sellers ZM et al. Gastroenterology. 2018;155(2):469-78.e1.
8. Brown A et al. JOP. 2008;9(4):408-14.
9. Fagenholz PJ et al. Ann Epidemiol. 2007;17(7):491.e1-.e8.
10. McNabb-Baltar J et al. Pancreas. 2014;43(5):687-91.
11. Johnson CD et al. Gut. 2004;53(9):1340-4.
12. Dellinger EP et al. Ann Surg. 2012;256(6):875-80.
13. Petrov MS et al. Gastroenterology. 2010;139(3):813-20.
14. Sternby H et al. Ann Surg. Apr 18. doi: 10.1097/SLA.0000000000002766.
15. Huh JH et al. J Clin Gastroenterol. 2018;52(2):178-83.
16. Wu BU et al. Gastroenterology. 2008;135(3):816-20.
17. Gardner TB et al. Clin Gastroenterol Hepatol. 2008;6(10):1070-6.
18. Krishna SG et al. Am J Gastroenterol. 2015;110(11):1608-19.
19. Lee PJ et al. Pancreas. 2016;45(4):561-4.
20. Mandalia A et al. F1000Research. 2018 Jun 28;7.
21. Majumder S et al. Pancreas. 2015;44(4):540-6.
22. DiMagno MJ. Clin Gastroenterol Hepatol. 2011;9(11):920-2.
23. Yadav D, Whitcomb DC. Nature Rev Gastroenterol Hepatol. 2010;7(3):131-45.
24. Samokhvalov AV et al. EBioMedicine. 2015;2(12):1996-2002.
25. Barkin JA et al. Pancreas. 2017;46(8):1035-8.
26. Chen Y-T et al. J Gastroenterol Hepatol. 2016;31(4):782-7.
27. Ramos LR et al. J Crohns Colitis. 2016;10(1):95-104.
28. Avram MM. Nephron. 1977;18(1):68-71.
29. Lankisch PG et al. Nephrol Dial Transplant. 2008;23(4):1401-5.
30. Owyang C et al. Mayo Clin Proc. 1979;54(12):769-73.
31. Owyang Cet al. Gut. 1982;23(5):357-61.
32. Quraishi ER et al. Am J Gastroenterol. 2005;100:2288.
33. Vaziri ND et al. Nephron. 1987;46(4):347-9.
34. Chen HJ et al. Nephrol Dial Transplant. 2017;32(10):1731-6.
35. Kirkegard J et al. Gastroenterology. 2018;May;154(6):1729-36.
36. Karlson BM, et al. Gastroenterology. 1997;113(2):587-92.
37. Munigala S et al. Clin Gastroenterol Hepatol. 2014;12(7):1143-50.e1.
38. Carr RA et al. Pancreatology. 2016;16(4):469-76.
39. Li X et al. BMC Gastroenterol. 2018;18(1):89.
40. Ahmed AU et al. Clin Gastroenterol Hepatol. 2016;14(5):738-46.
41. Sankaran SJ et al. Gastroenterology. 2015;149(6):1490-500.e1.
42. Berglund L et al. J Clin Endocrinol Metab. 2012;97(9):2969-89.
43. Catapano AL et al. Atherosclerosis. 2011;217(1):3-46.
44. Pedersen SB et al. JAMA Intern Med. 2016;176(12):1834-42.
45. Nawaz H et al. Am J Gastroenterol. 2015;110(10):1497-503.
46. Banks PA et al. Gut. 2013;62(1):102-11.
47. Kondo S et al. Eur J Radiol. 2005;54(2):271-5.
48. Meeralam Y et al. Gastrointest Endosc. 2017;86(6):986-93.
49. Stimac D et al. Am J Gastroenterol. 2007;102(5):997-1004.
50. Jin DX et al. Dig Dis Sci. 2017;62(10):2894-9.
51. Freeman ML. Gastrointest Endosc Clin N Am. 2012;22(3):567-86.
52. De Lisi S et al. Eur J Gastroenterol Hepatol. 2011;23(5):367-74.
53. Di MY et al. Ann Int Med. 2016;165(7):482-90.
54. Mounzer R et al. Gastroenterology. 2012;142(7):1476-82; quiz e15-6.
55. Koutroumpakis E et al. Am J Gastroenterol. 2015;110(12):1707-16.
56. Wu BU et al. Gastroenterology. 2009;137(1):129-35.
57. Buddingh KT et al. J Am Coll Surg. 2014;218(1):26-32.
58. Buxbaum J et al. Am J Gastroenterol. 2018;113(5):755-64.
59. Jabaudon M et al. Crit Car Med. 2018;46(3):e198-e205.
60. Barlass U et al. Gut. 2018;67(4):600-2.
61. Buxbaum JL et al. Am J Gastroenterol. 2017;112(5):797-803.
62. de-Madaria E et al. United Eur Gastroenterol J. 2018;6(1):63-72.
63. Bakker OJ et al. N Engl J Med. 2014;371(21):1983-93.
64. Tse F et al. Cochrane Database Syst Rev. 2012(5):Cd009779.
65. Kaner EFS et al. Cochrane Database Syst Rev. 2007(2):Cd004148.
66. da Costa DW et al. Lancet. 2015;386(10000):1261-8.
67. Schepers NJ et al. Trials. 2016;17:5.
68. Vadala di Prampero SF et al. Eur J Gastroenterol Hepatol. 2016;28(12):1415-24.
69. Kubiliun NM et al. Clin Gastroenterol Hepatol. 2015;13(7):1231-9; quiz e70-1.
70. Wan J et al. BMC Gastroenterol. 2017;17(1):43.
71. Yang C et al. Pancreatology. 2017;17(5):681-8.
72. DiMagno MJ et al. Pancreas. 2014;43(4):642-7.
73. Sagi SV et al. J Gastroenterol Hepatol. 2014;29(6):1316-20.
74. Choi JH et al. Clin Gastroenterol Hepatol. 2017;15(1):86-92.e1.
75. Wu D et al. J Clin Gastroenterol. 2017;51(8):e68-e76.
76. Zhang ZF et al. J Clin Gastroenterol. 2017;51(3):e17-e26.
77. Park CH et al. Endoscopy 2018 Apr;50(4):378-85.
78. Mok SRS et al. Gastrointest Endosc. 2017;85(5):1005-13.
79. Smeets XJN et al. Trials. 2018;19(1):207.
80. Elmunzer BJ et al. Trials. 2016;17(1):120.
A shutdown, a lawsuit, and drug prices
As I write this editorial, regarding maintenance of certification. Being a doctor, these days, is neither easy nor relaxing.

The government shutdown is cheered by some, but it has real consequences for 800,000 government workers who are not getting paid, and our scientific community, where grant applications, hiring, data collection, and other critical roles of government are needed, is at a standstill. The class action suit against ABIM is the latest action of physicians telling the ABIM that enough is enough. Read more and form your opinion from our page one article. The ACA continues to be attacked in a variety of ways. To those who want to abolish it, please have a reasonable alternative in place so that real people with real diseases are not left in a desperate situation.
Drug prices continue to make news. The most recent example is the enormous increase in the cost of insulin. I read a lot about the inner workings of the pharmaceutical industry and cannot fathom how such prices are justified. Perhaps we physicians and our medical societies should consider raising our voices for our patients.
In this month’s issue there are several articles about polyp detection and the long-term protective effect of colonoscopy. We are doing really important and excellent work to reduce the burden of colon cancer.
As a heads up, Digestive Disease Week® (DDW) returns to San Diego this year. Housing choices are opening up and in San Diego fill rapidly; visit www.DDW.org/registration for more information. This year’s science is ground-breaking and will continue to advance our knowledge about IBD, the microbiome, and other important topics.
John I. Allen, MD, MBA, AGAF
Editor in Chief
As I write this editorial, regarding maintenance of certification. Being a doctor, these days, is neither easy nor relaxing.
The government shutdown is cheered by some, but it has real consequences for 800,000 government workers who are not getting paid, and our scientific community, where grant applications, hiring, data collection, and other critical roles of government are needed, is at a standstill. The class action suit against ABIM is the latest action of physicians telling the ABIM that enough is enough. Read more and form your opinion from our page one article. The ACA continues to be attacked in a variety of ways. To those who want to abolish it, please have a reasonable alternative in place so that real people with real diseases are not left in a desperate situation.
Drug prices continue to make news. The most recent example is the enormous increase in the cost of insulin. I read a lot about the inner workings of the pharmaceutical industry and cannot fathom how such prices are justified. Perhaps we physicians and our medical societies should consider raising our voices for our patients.
In this month’s issue there are several articles about polyp detection and the long-term protective effect of colonoscopy. We are doing really important and excellent work to reduce the burden of colon cancer.
As a heads up, Digestive Disease Week® (DDW) returns to San Diego this year. Housing choices are opening up and in San Diego fill rapidly; visit www.DDW.org/registration for more information. This year’s science is ground-breaking and will continue to advance our knowledge about IBD, the microbiome, and other important topics.
John I. Allen, MD, MBA, AGAF
Editor in Chief
As I write this editorial, regarding maintenance of certification. Being a doctor, these days, is neither easy nor relaxing.
The government shutdown is cheered by some, but it has real consequences for 800,000 government workers who are not getting paid, and our scientific community, where grant applications, hiring, data collection, and other critical roles of government are needed, is at a standstill. The class action suit against ABIM is the latest action of physicians telling the ABIM that enough is enough. Read more and form your opinion from our page one article. The ACA continues to be attacked in a variety of ways. To those who want to abolish it, please have a reasonable alternative in place so that real people with real diseases are not left in a desperate situation.
Drug prices continue to make news. The most recent example is the enormous increase in the cost of insulin. I read a lot about the inner workings of the pharmaceutical industry and cannot fathom how such prices are justified. Perhaps we physicians and our medical societies should consider raising our voices for our patients.
In this month’s issue there are several articles about polyp detection and the long-term protective effect of colonoscopy. We are doing really important and excellent work to reduce the burden of colon cancer.
As a heads up, Digestive Disease Week® (DDW) returns to San Diego this year. Housing choices are opening up and in San Diego fill rapidly; visit www.DDW.org/registration for more information. This year’s science is ground-breaking and will continue to advance our knowledge about IBD, the microbiome, and other important topics.
John I. Allen, MD, MBA, AGAF
Editor in Chief
AGA Guideline: Treatment of mild to moderate ulcerative colitis
For patients with extensive mild to moderate ulcerative colitis, numerous randomized controlled trials support the use of either standard-dose mesalamine (2-3 grams per day) or diazo-bonded 5-aminosalicylic acid (ASA) instead of low-dose mesalamine, sulfasalazine, or no therapy, state new guidelines from the American Gastroenterological Association, published in Gastroenterology.
Sulfasalazine (2-4 grams per day) is less likely to be tolerated but remains a “reasonable option” for remitted patients who are already on it and for patients with prominent arthritis symptoms, especially if alternative treatments are cost prohibitive, wrote Cynthia W. Ko, MD, MS, of the University of Washington, Seattle, and her associates.
According to the guideline, patients with mild to moderate ulcerative colitis have less than four to six bowel movements per day, only mild or moderate rectal bleeding, no constitutional symptoms, and no high overall inflammatory burden or signs of high inflammatory activity on the Mayo Clinic score and Truelove and Witt’s criteria. These patients usually do not require colectomy, but this outcome is more likely when patients are diagnosed before age 40 years or have extensive disease or deep ulcers, extraintestinal manifestations, or elevated inflammatory markers. These higher-risk patients need more aggressive initial treatment and faster treatment intensification in cases of inadequate response, the guideline emphasizes. Even for cases of mild to moderate ulcerative colitis, treatment intensification is preferable to repeated courses of corticosteroids.
The guideline recommends adding rectal mesalamine to oral 5-ASA if patients have extensive or left-sided mild to moderate ulcerative colitis. In randomized controlled trials, this combination was significantly more likely to induce and maintain remission than was standard-dose oral mesalamine monotherapy, the authors noted. “In the maintenance trials, enemas were used twice per week or for 1 week per month. Both oral and topical mesalamine were well tolerated.”
For patients with moderate disease activity or a suboptimal response to standard-dose mesalamine or diazo-bonded 5-ASA, the guideline recommends adding rectal mesalamine to high-dose oral mesalamine (more than 3 grams daily). Combination therapy maximizes the delivery of mesalamine to the affected area of the colon, which optimizes the trial of 5-ASA before opting for treatment escalation, the authors noted. They recommend once-daily oral mesalamine dosing, since this is easier to adhere to and studies have found no benefit of more frequent dosing.
For inducing remission of mild to moderate ulcerative colitis, the guideline recommends standard-dose oral mesalamine or diazo-bonded 5-ASA over budesonide. “Overall, the budesonide preparations are not superior to mesalamine for induction of remission,” the authors wrote. Oral 5-ASAs are preferred, especially given the absence of data on the efficacy or safety of maintenance budesonide therapy.
For patients with mild to moderate ulcerative proctosigmoiditis or proctitis, the guideline conditionally recommends rectal mesalamine over oral mesalamine. Compared with placebo, rectal mesalamine suppositories were significantly more likely to induce remission in randomized trials of patients with mild to moderate ulcerative proctitis. If these patients cannot tolerate or are refractory to mesalamine suppositories, low-quality evidence supports rectal steroid therapy over no treatment, the guideline states. For patients with mild to moderate ulcerative proctosigmoiditis, moderate-quality evidence supports mesalamine enemas over rectal corticosteroids. If these patients want to avoid the difficulties of enemas, the guideline considers rectal corticosteroid foam a reasonable alternative.
Likewise, they cite low-quality evidence for adding oral prednisone or budesonide MMX to 5-ASA if patients are refractory to optimized 5-ASA therapy. No trials have directly compared rates of remission with budesonide MMX versus systemic corticosteroids. In just one placebo-controlled trial, adding budesonide MMX to 5-ASA slightly improved the chances of remission (risk ratio, 0.95; 95% confidence interval, 0.89-1.00). Furthermore, studies of other second-generation corticosteroids found they were better tolerated but no more likely to induce remission than oral prednisone or prednisolone.
Some patients with mild to moderate colitis respond inadequately to these recommended therapies and need systemic corticosteroids, immunomodulators, or biologic therapies to induce and maintain remission, the guideline authors noted. They make no recommendation on immunomodulators or biologics. Studies of probiotics, curcumin, and fecal microbiota transplantation are “urgently needed,” but for now, their use “risks delaying proven effective therapy, with the potential for worsening symptoms or complications,” they wrote. For patients without Clostridium difficile infections, they recommend against fecal microbiota transplantation except in the setting of a clinical trial.
The experts also noted the need for a tool to stratify patients with mild to moderate ulcerative colitis based on their risk of future progression and colectomy.
Finally, they call for studies on who will benefit most from high-dose mesalamine or topical mesalamine and on the relative safety and efficacy of budesonide and systemic corticosteroids in the event of an inadequate response to 5-ASAs.
All members were required to complete the disclosure statement. These statements are maintained at the American Gastroenterological Association headquarters in Bethesda, Maryland, and pertinent disclosures of conflict of interest are published with this report.
SOURCE: Crocket SD et al. Gastro 2019;156(2). doi: org/10.1053/j.gastro.2018.12.009.
For patients with extensive mild to moderate ulcerative colitis, numerous randomized controlled trials support the use of either standard-dose mesalamine (2-3 grams per day) or diazo-bonded 5-aminosalicylic acid (ASA) instead of low-dose mesalamine, sulfasalazine, or no therapy, state new guidelines from the American Gastroenterological Association, published in Gastroenterology.
Sulfasalazine (2-4 grams per day) is less likely to be tolerated but remains a “reasonable option” for remitted patients who are already on it and for patients with prominent arthritis symptoms, especially if alternative treatments are cost prohibitive, wrote Cynthia W. Ko, MD, MS, of the University of Washington, Seattle, and her associates.
According to the guideline, patients with mild to moderate ulcerative colitis have less than four to six bowel movements per day, only mild or moderate rectal bleeding, no constitutional symptoms, and no high overall inflammatory burden or signs of high inflammatory activity on the Mayo Clinic score and Truelove and Witt’s criteria. These patients usually do not require colectomy, but this outcome is more likely when patients are diagnosed before age 40 years or have extensive disease or deep ulcers, extraintestinal manifestations, or elevated inflammatory markers. These higher-risk patients need more aggressive initial treatment and faster treatment intensification in cases of inadequate response, the guideline emphasizes. Even for cases of mild to moderate ulcerative colitis, treatment intensification is preferable to repeated courses of corticosteroids.
The guideline recommends adding rectal mesalamine to oral 5-ASA if patients have extensive or left-sided mild to moderate ulcerative colitis. In randomized controlled trials, this combination was significantly more likely to induce and maintain remission than was standard-dose oral mesalamine monotherapy, the authors noted. “In the maintenance trials, enemas were used twice per week or for 1 week per month. Both oral and topical mesalamine were well tolerated.”
For patients with moderate disease activity or a suboptimal response to standard-dose mesalamine or diazo-bonded 5-ASA, the guideline recommends adding rectal mesalamine to high-dose oral mesalamine (more than 3 grams daily). Combination therapy maximizes the delivery of mesalamine to the affected area of the colon, which optimizes the trial of 5-ASA before opting for treatment escalation, the authors noted. They recommend once-daily oral mesalamine dosing, since this is easier to adhere to and studies have found no benefit of more frequent dosing.
For inducing remission of mild to moderate ulcerative colitis, the guideline recommends standard-dose oral mesalamine or diazo-bonded 5-ASA over budesonide. “Overall, the budesonide preparations are not superior to mesalamine for induction of remission,” the authors wrote. Oral 5-ASAs are preferred, especially given the absence of data on the efficacy or safety of maintenance budesonide therapy.
For patients with mild to moderate ulcerative proctosigmoiditis or proctitis, the guideline conditionally recommends rectal mesalamine over oral mesalamine. Compared with placebo, rectal mesalamine suppositories were significantly more likely to induce remission in randomized trials of patients with mild to moderate ulcerative proctitis. If these patients cannot tolerate or are refractory to mesalamine suppositories, low-quality evidence supports rectal steroid therapy over no treatment, the guideline states. For patients with mild to moderate ulcerative proctosigmoiditis, moderate-quality evidence supports mesalamine enemas over rectal corticosteroids. If these patients want to avoid the difficulties of enemas, the guideline considers rectal corticosteroid foam a reasonable alternative.
Likewise, they cite low-quality evidence for adding oral prednisone or budesonide MMX to 5-ASA if patients are refractory to optimized 5-ASA therapy. No trials have directly compared rates of remission with budesonide MMX versus systemic corticosteroids. In just one placebo-controlled trial, adding budesonide MMX to 5-ASA slightly improved the chances of remission (risk ratio, 0.95; 95% confidence interval, 0.89-1.00). Furthermore, studies of other second-generation corticosteroids found they were better tolerated but no more likely to induce remission than oral prednisone or prednisolone.
Some patients with mild to moderate colitis respond inadequately to these recommended therapies and need systemic corticosteroids, immunomodulators, or biologic therapies to induce and maintain remission, the guideline authors noted. They make no recommendation on immunomodulators or biologics. Studies of probiotics, curcumin, and fecal microbiota transplantation are “urgently needed,” but for now, their use “risks delaying proven effective therapy, with the potential for worsening symptoms or complications,” they wrote. For patients without Clostridium difficile infections, they recommend against fecal microbiota transplantation except in the setting of a clinical trial.
The experts also noted the need for a tool to stratify patients with mild to moderate ulcerative colitis based on their risk of future progression and colectomy.
Finally, they call for studies on who will benefit most from high-dose mesalamine or topical mesalamine and on the relative safety and efficacy of budesonide and systemic corticosteroids in the event of an inadequate response to 5-ASAs.
All members were required to complete the disclosure statement. These statements are maintained at the American Gastroenterological Association headquarters in Bethesda, Maryland, and pertinent disclosures of conflict of interest are published with this report.
SOURCE: Crocket SD et al. Gastro 2019;156(2). doi: org/10.1053/j.gastro.2018.12.009.
For patients with extensive mild to moderate ulcerative colitis, numerous randomized controlled trials support the use of either standard-dose mesalamine (2-3 grams per day) or diazo-bonded 5-aminosalicylic acid (ASA) instead of low-dose mesalamine, sulfasalazine, or no therapy, state new guidelines from the American Gastroenterological Association, published in Gastroenterology.
Sulfasalazine (2-4 grams per day) is less likely to be tolerated but remains a “reasonable option” for remitted patients who are already on it and for patients with prominent arthritis symptoms, especially if alternative treatments are cost prohibitive, wrote Cynthia W. Ko, MD, MS, of the University of Washington, Seattle, and her associates.
According to the guideline, patients with mild to moderate ulcerative colitis have less than four to six bowel movements per day, only mild or moderate rectal bleeding, no constitutional symptoms, and no high overall inflammatory burden or signs of high inflammatory activity on the Mayo Clinic score and Truelove and Witt’s criteria. These patients usually do not require colectomy, but this outcome is more likely when patients are diagnosed before age 40 years or have extensive disease or deep ulcers, extraintestinal manifestations, or elevated inflammatory markers. These higher-risk patients need more aggressive initial treatment and faster treatment intensification in cases of inadequate response, the guideline emphasizes. Even for cases of mild to moderate ulcerative colitis, treatment intensification is preferable to repeated courses of corticosteroids.
The guideline recommends adding rectal mesalamine to oral 5-ASA if patients have extensive or left-sided mild to moderate ulcerative colitis. In randomized controlled trials, this combination was significantly more likely to induce and maintain remission than was standard-dose oral mesalamine monotherapy, the authors noted. “In the maintenance trials, enemas were used twice per week or for 1 week per month. Both oral and topical mesalamine were well tolerated.”
For patients with moderate disease activity or a suboptimal response to standard-dose mesalamine or diazo-bonded 5-ASA, the guideline recommends adding rectal mesalamine to high-dose oral mesalamine (more than 3 grams daily). Combination therapy maximizes the delivery of mesalamine to the affected area of the colon, which optimizes the trial of 5-ASA before opting for treatment escalation, the authors noted. They recommend once-daily oral mesalamine dosing, since this is easier to adhere to and studies have found no benefit of more frequent dosing.
For inducing remission of mild to moderate ulcerative colitis, the guideline recommends standard-dose oral mesalamine or diazo-bonded 5-ASA over budesonide. “Overall, the budesonide preparations are not superior to mesalamine for induction of remission,” the authors wrote. Oral 5-ASAs are preferred, especially given the absence of data on the efficacy or safety of maintenance budesonide therapy.
For patients with mild to moderate ulcerative proctosigmoiditis or proctitis, the guideline conditionally recommends rectal mesalamine over oral mesalamine. Compared with placebo, rectal mesalamine suppositories were significantly more likely to induce remission in randomized trials of patients with mild to moderate ulcerative proctitis. If these patients cannot tolerate or are refractory to mesalamine suppositories, low-quality evidence supports rectal steroid therapy over no treatment, the guideline states. For patients with mild to moderate ulcerative proctosigmoiditis, moderate-quality evidence supports mesalamine enemas over rectal corticosteroids. If these patients want to avoid the difficulties of enemas, the guideline considers rectal corticosteroid foam a reasonable alternative.
Likewise, they cite low-quality evidence for adding oral prednisone or budesonide MMX to 5-ASA if patients are refractory to optimized 5-ASA therapy. No trials have directly compared rates of remission with budesonide MMX versus systemic corticosteroids. In just one placebo-controlled trial, adding budesonide MMX to 5-ASA slightly improved the chances of remission (risk ratio, 0.95; 95% confidence interval, 0.89-1.00). Furthermore, studies of other second-generation corticosteroids found they were better tolerated but no more likely to induce remission than oral prednisone or prednisolone.
Some patients with mild to moderate colitis respond inadequately to these recommended therapies and need systemic corticosteroids, immunomodulators, or biologic therapies to induce and maintain remission, the guideline authors noted. They make no recommendation on immunomodulators or biologics. Studies of probiotics, curcumin, and fecal microbiota transplantation are “urgently needed,” but for now, their use “risks delaying proven effective therapy, with the potential for worsening symptoms or complications,” they wrote. For patients without Clostridium difficile infections, they recommend against fecal microbiota transplantation except in the setting of a clinical trial.
The experts also noted the need for a tool to stratify patients with mild to moderate ulcerative colitis based on their risk of future progression and colectomy.
Finally, they call for studies on who will benefit most from high-dose mesalamine or topical mesalamine and on the relative safety and efficacy of budesonide and systemic corticosteroids in the event of an inadequate response to 5-ASAs.
All members were required to complete the disclosure statement. These statements are maintained at the American Gastroenterological Association headquarters in Bethesda, Maryland, and pertinent disclosures of conflict of interest are published with this report.
SOURCE: Crocket SD et al. Gastro 2019;156(2). doi: org/10.1053/j.gastro.2018.12.009.
FROM GASTROENTEROLOGY