User login
Thrombotic thrombocytopenic purpura: 2008 Update
Thrombotic thrombocytopenic purpura (TTP) is one of the few hematologic emergencies. Untreated, most patients die, but prompt and appropriate treatment allows most patients not only to survive but to recover, frequently without long-term sequelae.
TTP is rare. The estimated annual incidence of all TTP syndromes is about 11 cases per million in the general population, and the incidence of severe ADAMTS13 deficiency (see discussion below) is about 2 per million. Therefore, even large medical centers typically see only one or two cases each year. The syndromes are much more common in women, and the incidence among blacks is nine times higher than the incidence among non-blacks. Nevertheless, despite the rarity of this disease, good evidence exists to help guide patient care, thanks to national registries and research organizations, such as the Canadian Apheresis Study Group and the Oklahoma TTP-Hemolytic Uremic Syndrome (HUS) Registry.
This article reviews the physiologic basis of TTP, how to recognize it, and how best to treat it. We also discuss other conditions that clinically resemble TTP but probably have different underlying causes.
A YOUNG WOMAN WITH ARM WEAKNESS
A 24-year-old black woman presents to a community hospital with weakness in her left arm, which began about 30 minutes previously. She has had progressive dyspnea over the last several weeks, but has otherwise been completely well and has had no medical problems in the past other than being obese.
Physical examination reveals weakness in her left arm as well as mild dysarthria, which was not previously noted by the patient or her family. Her laboratory findings:
- White blood cell count 16.7 × 109/L (reference range 4.5–11.0)
- Platelet count 32 × 109/L (150–350)
- Hemoglobin concentration 6.5 g/dL (1.4–17.5)
- Peripheral blood smear: normal white cells, rare platelets, red cells normo-chromic with many fragments
- Lactate dehydrogenase (LDH) concentration 2,300 U/L (100–200).
In view of her symptoms and laboratory values, the physician suspects she may have TTP and refers her to McMaster University Medical Center in Hamilton, Ontario, Canada. Plasma exchange is started immediately; one plasma volume is removed and replaced with fresh frozen plasma. Nevertheless, the patient’s condition deteriorates overnight, she becomes more confused and cannot protect her airway, her LDH concentration rises further, and her hemoglobin concentration falls. She is transferred to the intensive care unit. Her plasma exchange prescription is increased to 1.5 volumes twice daily (although little evidence exists that plasma exchange twice daily is more effective than once daily).
On the third day of her stay, she becomes completely paralyzed on the left side. In addition to her twice-daily plasma exchange procedures, a plasma infusion and corticosteroid therapy are initiated. Her platelet count stabilizes at about 20 × 109/L.
The patient next develops renal insufficiency and requires three acute hemodialysis treatments. (Plasma infusion frequently leads to volume overload in critically ill patients. Some intravascular volume can be removed with plasma exchange; however, significant volume overload with significant renal insufficiency can only be treated with renal replacement therapy.)
The patient undergoes 28 consecutive days of twice-daily plasma exchange and gradually improves, as measured by increasing platelet counts, a gradual fall in the LDH concentration, and stabilization of—and ultimately an increase in—the hemoglobin level. She is weaned off plasma infusions, and then plasma exchange is tapered to once a day and then to alternate days.
She is completely well at the time of discharge 4 weeks after her initial admission, with no residual deficits.
Comment. This case shows that even patients with apparently devastating compromise and neurologic deficits can completely recover with aggressive plasma exchange and other therapies. One child treated at the Hospital for Sick Children, affiliated with the University of Toronto, developed TTP and had 120 consecutive days of plasma exchange: she was unconscious and comatose for much of that time, but she ultimately recovered and is now completely well without residual neurologic deficits.
TTP MAY BE DUE TO ADAMTS13 DEFICIENCY
Twenty-five years ago, little was known about TTP except for its clinical manifestations. Now, it is known to be caused in some patients by an acquired deficiency of a circulating metalloproteinase. In very rare cases a hereditary deficiency of ADAMTS13 causes TTP. In addition, a number of conditions share clinical features with TTP but have other underlying causes.
In acquired TTP, an autoantibody forms against ADAMTS13, a zinc-containing metalloproteinase that is also known as von Willebrand factor-cleaving protease. Normally, von Willebrand factor circulates in plasma as multimers that allow platelets to adhere to vascular surfaces. When von Willebrand factor is initially released from endothelial cells, it exists as large multimers, which are more adhesive for platelets than normal. These large multimers are normally cleaved into smaller units by ADAMTS13. If ADAMTS13 is lacking, the very-high-molecular-weight von Willebrand factor multimers accumulate, causing platelet agglutination and the vascular occlusion that results in the manifestations of TTP.
In 1994, ADAMTS13, the gene of which is on the ninth chromosome, was shown to cleave von Willebrand factor under conditions of high shear stress. In 1996, a congenital homozygous deficiency of ADAMTS13 was found to be associated with platelet microthrombi. Afterwards, some patients with TTP were shown to have low or undetectable levels of ADAMTS13, owing to immunoglobulin G antibodies directed against the enzyme.
TTP AND RELATED SYNDROMES
Clinically, TTP encompasses a number of different but related syndromes, some of which have different physiologic bases.
TTP
TTP is characterized by moderate to severe thrombocytopenia, red cell fragmentation, and elevated LDH levels (due to red cell destruction and also muscle and organ ischemia). The pentad of features classically associated with TTP in the era before effective treatment (thrombocytopenia, fever, renal failure, neurologic deficit, and microangiopathic hemolytic anemia) is rarely seen in countries with advanced medical care: renal insufficiency and neurologic events are end-stage manifestations, and the disease should be recognizable well before these manifestations occur. Otherwise unexplained thrombocytopenia, microangiopathic hemolytic anemia, and an elevated LDH should strongly suggest TTP. TTP is the appropriate designation for adults with these clinical features, even in the presence of renal failure. TTP is uncommon in children.
Most patients present with nonspecific constitutional symptoms, such as weakness, abdominal pain, nausea, and vomiting. Typically, the family physician orders a complete blood cell count and finds that the platelet count and hemoglobin are low. Red cell fragments are noted in the peripheral blood smear. Further testing reveals an elevated LDH concentration.
HUS
HUS was initially described 30 years after TTP in children with acute renal failure in addition to thrombocytopenia and microangiopathic hemolytic anemia. The term “HUS” is currently used primarily to describe the condition in children.
In children, two forms of HUS exist:
Diarrhea-associated HUS is associated with diarrhea that is commonly bloody, due to an enterotoxin produced by Escherichia coli O157:H7.
Endemic diarrhea-associated HUS is much more common than HUS associated with epidemics. Endemic cases are caused by E coli O157:H7 present in the environment. Other patients present with clinically apparent HUS but the causal bacterium cannot be detected. The kidney transplant program at our center often sees young patients with this disease who do not have E coli O157:H7 infection, and the pathogenesis is not understood. Epidemic cases are less common but the outbreaks are dramatic. About 10 years ago, E coli O157:H7 entered the water supply in the small city of Walkerton, Ontario, and many people developed the epidemic form of HUS over a period of several weeks. Most such patients spontaneously recovered without plasma exchange, although many were left with impaired renal function.
Atypical HUS. Less often, HUS in children is not associated with a prodrome of diarrhea and is referred to as “atypical” HUS. These children often have a more prolonged and complicated course and resemble adults with TTP.
Familial TTP-HUS
Familial TTP-HUS is very rare. It may present with hemolysis and thrombocytopenia in childhood or early adulthood. Many patients present with renal insufficiency, and only careful evaluation reveals hemolysis and thrombocytopenia. The disease typically manifests acutely: a patient may have an upper respiratory tract infection and subsequently develop an episode of TTP-HUS. Episodes tend to recur, and multiple family members may also be affected.
Plasma infusion is an effective treatment, and plasma exchange is usually not required. Since more patients are now surviving well into adulthood, some are being seen to develop antibodies to the ADAMTS13 in the infused plasma, analogous to patients with severe hemophilia developing inhibitors to factor VIII. The disease may progress despite treatment: we have been treating a young woman who has had a series of catastrophic complications and now has chronic renal failure requiring hemodialysis (see discussion below).
Post-transplant microangiopathy
Post-transplant microangiopathy is most likely to develop after solid-organ or stem-cell allograft transplantation. Manifestations resemble those of TTP, but the mechanism is probably quite different. Multiple causes probably exist, depending on the setting.
Post-transplant microangiopathy does not respond to the usual therapies for TTP, although we treat it, like TTP, with corticosteroids, antiplatelet agents, and plasma exchange. Other centers do not use plasma exchange for these patients. Most patients have a poor prognosis, especially those with a transplant other than a kidney.
A spectrum of related syndromes
A number of diseases clinically resemble TTP. Enhanced diagnostic capacity and better molecular biologic techniques are revealing that they often have very different underlying causes and that in some cases they require different treatment.

TOWARD DIAGNOSTIC CRITERIA
Ruutu et al,1 in a consensus conference, used rigorous methods to establish diagnostic criteria for microangiopathy associated with stem cell transplantation:
- More than 4% red blood cell fragments in the peripheral blood. A laboratory report that states that “few fragments” are present is not nearly as useful as one that estimates the quantity; eg, 1% fragments would have very different implications than 6% fragments.
- Thrombocytopenia—a platelet count of less than 50 × 109/L or more than a 50% reduction from previous counts
- Increased LDH concentration
- Reduced hemoglobin concentration or increased transfusion requirement
- Decrease in serum haptoglobin, which, like red blood cell fragments, is a marker of hemolysis rather than of reduced synthesis.
The ADAMTS13 level need not be assessed. Metalloproteinase deficiency need not be proved to diagnose TTP. Although our hospital is a TTP referral center, we do not routinely offer the test. Too often the test results cause confusion: a patient can have a normal level of ADAMTS13 and still have TTP that responds to plasma exchange, and levels can be low in conditions other than TTP.
THE CHALLENGES OF TREATMENT
Plasma exchange is the primary treatment for TTP
Rock et al2 performed a randomized trial in which 102 patients with TTP received either a 1.5-volume plasma exchange daily for 3 days and then 1-volume plasma exchanges as needed to control the disease or plasma infusion. Patients who received plasma exchange had a better initial response, a higher survival rate, and a lower rate of relapse than patients receiving plasma infusion. These findings established plasma exchange as the treatment of choice for TTP.
However, the trial had some inherent problems: patients who had plasma infusions tended to develop renal insufficiency and as a result did not receive as much plasma because they could not tolerate as much volume as those who had plasma exchange. Plasma exchange probably worked better because it could deliver more plasma over a fixed period of time, enabling patients to obtain more of the ADAMTS13 enzyme, rather than because it was an intrinsically better treatment. This interpretation is the basis for our occasional use of twice-daily plasma exchange in critically ill patients.
TTP is different from other autoimmune diseases such as idiopathic thrombocytopenia purpura, in which the primary treatments are immunosuppressive agents. Some evidence exists for treating TTP with immunosuppressive agents, but the primary treatment should be plasma exchange.
Plasma infusion is useful in some cases
Although small case series and our own experience provide evidence for the benefit of treating TTP with high-dose plasma infusions (25 mL/kg/day, or about 1.5 to 2.0 L/day for an average-sized adult), problems will likely arise with volume overload if the patient has any significant renal insufficiency. Dialysis or ultrafiltration may be used to treat volume overload; however, it is difficult to remove the large volumes of fluid required for high-volume plasma infusion.
Plasma infusion should be reserved for two situations:
- If plasma exchange cannot be promptly started
- For patients with very severe or refractory disease, between plasma exchange sessions.
Benefit of cryoprecipitate-poor plasma is uncertain
Fresh frozen plasma is believed to contain nearly physiologic levels of all of the plasma proteins. When plasma is cooled to around 4°C, a precipitate forms that contains a variety of substances, including the higher molecular weight multimers of von Willebrand factor. Because TTP involves excess large multimers, giving plasma in which the high molecular weight multimers have been removed should in theory be better. In many centers, such cryoprecipitate-poor plasma is routinely used to treat TTP.
However, evidence that cryoprecipitate-poor plasma is better is lacking. A large study in Canada evaluating this question was terminated because of a lack of patient accrual, a common fate of clinical trials of rare diseases. A randomized study in 17 patients failed to show an advantage of cryoprecipitate-poor plasma over regular plasma, but the study was too small to draw firm conclusions given large confidence intervals about the point estimate of the treatment effect.
Cryoprecipitate-poor plasma is more expensive than regular plasma and is not as available. We do not routinely use it in our center to initially manage patients with TTP, but we do use it for patients who are refractory to standard treatment.
Scott et al3 measured the concentration of ADAMTS13 in a variety of plasma products and found that there are significant amounts in cryoprecipitate. Although giving cryoprecipitate-poor plasma provides less of the high molecular weight multimers, which is desirable for patients with TTP, it also provides less ADAMTS13, which is not desirable.
Do antiplatelet agents have a role in acute TTP?
Most algorithms for managing acute TTP include the use of aspirin or dipyridamole (Persantine) or both, and there is some evidence in favor of this approach,4 but whether antiplatelet therapy should be used for inpatients with severe thrombocytopenia remains controversial. In our practice, we usually provide antiplatelet therapy even for patients with severe thrombocytopenia because we believe TTP involves platelet-mediated hypercoagulability rather than increased bleeding risk.
Do corticosteroids have a role?
Corticosteroids were widely used to treat TTP even before the disease was discovered to be immune mediated. In our center we routinely use them.
Unfortunately, few data exist on the efficacy of steroid therapy for TTP. As a result, we can only make a weak recommendation for its use: using the American College of Chest Physicians rating system for the strength of clinical evidence, it would receive a 2C recommendation. This is the weakest possible recommendation, being based on widespread use but poor-quality data.
Stopping vs tapering plasma exchange
Whether plasma exchange should be tapered or simply stopped is also controversial and not well studied. Nevertheless, a widespread clinical practice—once the platelet count returns to 200 × 109/L or higher and the patient looks and feels well—is to reduce the plasma exchange sessions to once every 3 days, then to once every 7 days, and then to once every 2 weeks.
In our practice, we taper plasma exchange in this fashion for a minimum of two treatments beyond what we think the patient really needs. As a result, we tend to treat about once every 2 weeks for weeks or even months after the acute illness.
Rituximab may help
Rituximab (Rituxan), a monoclonal antibody against mature B cells, is increasingly being used in treating TTP. Past and present treatments for TTP, including splenectomy, corticosteroids, and plasma exchange are immunomodulatory, so the use of rituximab may be justified. Case reports provided the rationale for a large, multicenter, randomized controlled trial, which is currently under way.5
CONDITIONS THAT ARE NOT TTP
Some conditions may be confused with TTP but are clearly something different:
Patients with isolated thrombocytopenia and normal blood smear findings and no coagulopathy most likely have idiopathic thrombocytopenia purpura or, in the correct clinical circumstance, heparin-induced thrombocytopenia.
A patient with an extremely low platelet count but no fragments or very few fragments with microangiopathic hemolytic anemia may have either drug-associated thrombocytopenia or disseminated intravascular coagulopathy, particularly if there is concomitant coagulopathy.
Many pregnancy-associated microangiopathies resemble TTP, and it may be difficult to differentiate them from TTP; if confusion as to the diagnosis exists, the patient should be treated with plasma exchange, as this therapy may be life-saving.
Many rheumatologic conditions are characterized by an acute illness with nonspecific findings, such as low-grade hemolysis and thrombocytopenia. For example, Wegener granulomatosis can present with evidence of hemolysis, thrombocytopenia, and renal impairment.
Systemic lupus erythematosus can also initially present with an “early-TTP”-like picture. Evidence of glomerulonephritis is not consistent with TTP, and urinary red cell casts makes the diagnosis of lupus more likely. Helmet cell fragments in the peripheral blood smear are supposedly more characteristic of TTP, but their presence is not diagnostic.
Scleroderma renal crisis can present like TTP, but because it is unlikely that a patient with known scleroderma would have a second rare disease, it is best to treat it as scleroderma, which does not require plasma exchange or plasma infusion.
In general, if the diagnosis is uncertain, the safest course is to treat the patient with plasma exchange, then try to establish the diagnosis, because TTP is fatal if not promptly treated. Although plasma exchange is probably overused, it is more innocuous than untreated TTP.
- Ruutu T, Barosi G, Benjamin RJ, et al; European Group for Blood and Marrow Transplantation; European LeukemiaNet. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica 2007; 92:95–100.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Scott EA, Puca KE, Pietz BC, Duchateau BK, Friedman KD. Comparison and stability of ADAMTS13 activity in therapeutic plasma products. Transfusion 2007; 47:120–125.
- Bobbio-Pallavicini E, Gugliotta L, Centurioni R, et al. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica 1997; 82:429–435.
- George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher 2006; 21:49–56.
Thrombotic thrombocytopenic purpura (TTP) is one of the few hematologic emergencies. Untreated, most patients die, but prompt and appropriate treatment allows most patients not only to survive but to recover, frequently without long-term sequelae.
TTP is rare. The estimated annual incidence of all TTP syndromes is about 11 cases per million in the general population, and the incidence of severe ADAMTS13 deficiency (see discussion below) is about 2 per million. Therefore, even large medical centers typically see only one or two cases each year. The syndromes are much more common in women, and the incidence among blacks is nine times higher than the incidence among non-blacks. Nevertheless, despite the rarity of this disease, good evidence exists to help guide patient care, thanks to national registries and research organizations, such as the Canadian Apheresis Study Group and the Oklahoma TTP-Hemolytic Uremic Syndrome (HUS) Registry.
This article reviews the physiologic basis of TTP, how to recognize it, and how best to treat it. We also discuss other conditions that clinically resemble TTP but probably have different underlying causes.
A YOUNG WOMAN WITH ARM WEAKNESS
A 24-year-old black woman presents to a community hospital with weakness in her left arm, which began about 30 minutes previously. She has had progressive dyspnea over the last several weeks, but has otherwise been completely well and has had no medical problems in the past other than being obese.
Physical examination reveals weakness in her left arm as well as mild dysarthria, which was not previously noted by the patient or her family. Her laboratory findings:
- White blood cell count 16.7 × 109/L (reference range 4.5–11.0)
- Platelet count 32 × 109/L (150–350)
- Hemoglobin concentration 6.5 g/dL (1.4–17.5)
- Peripheral blood smear: normal white cells, rare platelets, red cells normo-chromic with many fragments
- Lactate dehydrogenase (LDH) concentration 2,300 U/L (100–200).
In view of her symptoms and laboratory values, the physician suspects she may have TTP and refers her to McMaster University Medical Center in Hamilton, Ontario, Canada. Plasma exchange is started immediately; one plasma volume is removed and replaced with fresh frozen plasma. Nevertheless, the patient’s condition deteriorates overnight, she becomes more confused and cannot protect her airway, her LDH concentration rises further, and her hemoglobin concentration falls. She is transferred to the intensive care unit. Her plasma exchange prescription is increased to 1.5 volumes twice daily (although little evidence exists that plasma exchange twice daily is more effective than once daily).
On the third day of her stay, she becomes completely paralyzed on the left side. In addition to her twice-daily plasma exchange procedures, a plasma infusion and corticosteroid therapy are initiated. Her platelet count stabilizes at about 20 × 109/L.
The patient next develops renal insufficiency and requires three acute hemodialysis treatments. (Plasma infusion frequently leads to volume overload in critically ill patients. Some intravascular volume can be removed with plasma exchange; however, significant volume overload with significant renal insufficiency can only be treated with renal replacement therapy.)
The patient undergoes 28 consecutive days of twice-daily plasma exchange and gradually improves, as measured by increasing platelet counts, a gradual fall in the LDH concentration, and stabilization of—and ultimately an increase in—the hemoglobin level. She is weaned off plasma infusions, and then plasma exchange is tapered to once a day and then to alternate days.
She is completely well at the time of discharge 4 weeks after her initial admission, with no residual deficits.
Comment. This case shows that even patients with apparently devastating compromise and neurologic deficits can completely recover with aggressive plasma exchange and other therapies. One child treated at the Hospital for Sick Children, affiliated with the University of Toronto, developed TTP and had 120 consecutive days of plasma exchange: she was unconscious and comatose for much of that time, but she ultimately recovered and is now completely well without residual neurologic deficits.
TTP MAY BE DUE TO ADAMTS13 DEFICIENCY
Twenty-five years ago, little was known about TTP except for its clinical manifestations. Now, it is known to be caused in some patients by an acquired deficiency of a circulating metalloproteinase. In very rare cases a hereditary deficiency of ADAMTS13 causes TTP. In addition, a number of conditions share clinical features with TTP but have other underlying causes.
In acquired TTP, an autoantibody forms against ADAMTS13, a zinc-containing metalloproteinase that is also known as von Willebrand factor-cleaving protease. Normally, von Willebrand factor circulates in plasma as multimers that allow platelets to adhere to vascular surfaces. When von Willebrand factor is initially released from endothelial cells, it exists as large multimers, which are more adhesive for platelets than normal. These large multimers are normally cleaved into smaller units by ADAMTS13. If ADAMTS13 is lacking, the very-high-molecular-weight von Willebrand factor multimers accumulate, causing platelet agglutination and the vascular occlusion that results in the manifestations of TTP.
In 1994, ADAMTS13, the gene of which is on the ninth chromosome, was shown to cleave von Willebrand factor under conditions of high shear stress. In 1996, a congenital homozygous deficiency of ADAMTS13 was found to be associated with platelet microthrombi. Afterwards, some patients with TTP were shown to have low or undetectable levels of ADAMTS13, owing to immunoglobulin G antibodies directed against the enzyme.
TTP AND RELATED SYNDROMES
Clinically, TTP encompasses a number of different but related syndromes, some of which have different physiologic bases.
TTP
TTP is characterized by moderate to severe thrombocytopenia, red cell fragmentation, and elevated LDH levels (due to red cell destruction and also muscle and organ ischemia). The pentad of features classically associated with TTP in the era before effective treatment (thrombocytopenia, fever, renal failure, neurologic deficit, and microangiopathic hemolytic anemia) is rarely seen in countries with advanced medical care: renal insufficiency and neurologic events are end-stage manifestations, and the disease should be recognizable well before these manifestations occur. Otherwise unexplained thrombocytopenia, microangiopathic hemolytic anemia, and an elevated LDH should strongly suggest TTP. TTP is the appropriate designation for adults with these clinical features, even in the presence of renal failure. TTP is uncommon in children.
Most patients present with nonspecific constitutional symptoms, such as weakness, abdominal pain, nausea, and vomiting. Typically, the family physician orders a complete blood cell count and finds that the platelet count and hemoglobin are low. Red cell fragments are noted in the peripheral blood smear. Further testing reveals an elevated LDH concentration.
HUS
HUS was initially described 30 years after TTP in children with acute renal failure in addition to thrombocytopenia and microangiopathic hemolytic anemia. The term “HUS” is currently used primarily to describe the condition in children.
In children, two forms of HUS exist:
Diarrhea-associated HUS is associated with diarrhea that is commonly bloody, due to an enterotoxin produced by Escherichia coli O157:H7.
Endemic diarrhea-associated HUS is much more common than HUS associated with epidemics. Endemic cases are caused by E coli O157:H7 present in the environment. Other patients present with clinically apparent HUS but the causal bacterium cannot be detected. The kidney transplant program at our center often sees young patients with this disease who do not have E coli O157:H7 infection, and the pathogenesis is not understood. Epidemic cases are less common but the outbreaks are dramatic. About 10 years ago, E coli O157:H7 entered the water supply in the small city of Walkerton, Ontario, and many people developed the epidemic form of HUS over a period of several weeks. Most such patients spontaneously recovered without plasma exchange, although many were left with impaired renal function.
Atypical HUS. Less often, HUS in children is not associated with a prodrome of diarrhea and is referred to as “atypical” HUS. These children often have a more prolonged and complicated course and resemble adults with TTP.
Familial TTP-HUS
Familial TTP-HUS is very rare. It may present with hemolysis and thrombocytopenia in childhood or early adulthood. Many patients present with renal insufficiency, and only careful evaluation reveals hemolysis and thrombocytopenia. The disease typically manifests acutely: a patient may have an upper respiratory tract infection and subsequently develop an episode of TTP-HUS. Episodes tend to recur, and multiple family members may also be affected.
Plasma infusion is an effective treatment, and plasma exchange is usually not required. Since more patients are now surviving well into adulthood, some are being seen to develop antibodies to the ADAMTS13 in the infused plasma, analogous to patients with severe hemophilia developing inhibitors to factor VIII. The disease may progress despite treatment: we have been treating a young woman who has had a series of catastrophic complications and now has chronic renal failure requiring hemodialysis (see discussion below).
Post-transplant microangiopathy
Post-transplant microangiopathy is most likely to develop after solid-organ or stem-cell allograft transplantation. Manifestations resemble those of TTP, but the mechanism is probably quite different. Multiple causes probably exist, depending on the setting.
Post-transplant microangiopathy does not respond to the usual therapies for TTP, although we treat it, like TTP, with corticosteroids, antiplatelet agents, and plasma exchange. Other centers do not use plasma exchange for these patients. Most patients have a poor prognosis, especially those with a transplant other than a kidney.
A spectrum of related syndromes
A number of diseases clinically resemble TTP. Enhanced diagnostic capacity and better molecular biologic techniques are revealing that they often have very different underlying causes and that in some cases they require different treatment.
TOWARD DIAGNOSTIC CRITERIA
Ruutu et al,1 in a consensus conference, used rigorous methods to establish diagnostic criteria for microangiopathy associated with stem cell transplantation:
- More than 4% red blood cell fragments in the peripheral blood. A laboratory report that states that “few fragments” are present is not nearly as useful as one that estimates the quantity; eg, 1% fragments would have very different implications than 6% fragments.
- Thrombocytopenia—a platelet count of less than 50 × 109/L or more than a 50% reduction from previous counts
- Increased LDH concentration
- Reduced hemoglobin concentration or increased transfusion requirement
- Decrease in serum haptoglobin, which, like red blood cell fragments, is a marker of hemolysis rather than of reduced synthesis.
The ADAMTS13 level need not be assessed. Metalloproteinase deficiency need not be proved to diagnose TTP. Although our hospital is a TTP referral center, we do not routinely offer the test. Too often the test results cause confusion: a patient can have a normal level of ADAMTS13 and still have TTP that responds to plasma exchange, and levels can be low in conditions other than TTP.
THE CHALLENGES OF TREATMENT
Plasma exchange is the primary treatment for TTP
Rock et al2 performed a randomized trial in which 102 patients with TTP received either a 1.5-volume plasma exchange daily for 3 days and then 1-volume plasma exchanges as needed to control the disease or plasma infusion. Patients who received plasma exchange had a better initial response, a higher survival rate, and a lower rate of relapse than patients receiving plasma infusion. These findings established plasma exchange as the treatment of choice for TTP.
However, the trial had some inherent problems: patients who had plasma infusions tended to develop renal insufficiency and as a result did not receive as much plasma because they could not tolerate as much volume as those who had plasma exchange. Plasma exchange probably worked better because it could deliver more plasma over a fixed period of time, enabling patients to obtain more of the ADAMTS13 enzyme, rather than because it was an intrinsically better treatment. This interpretation is the basis for our occasional use of twice-daily plasma exchange in critically ill patients.
TTP is different from other autoimmune diseases such as idiopathic thrombocytopenia purpura, in which the primary treatments are immunosuppressive agents. Some evidence exists for treating TTP with immunosuppressive agents, but the primary treatment should be plasma exchange.
Plasma infusion is useful in some cases
Although small case series and our own experience provide evidence for the benefit of treating TTP with high-dose plasma infusions (25 mL/kg/day, or about 1.5 to 2.0 L/day for an average-sized adult), problems will likely arise with volume overload if the patient has any significant renal insufficiency. Dialysis or ultrafiltration may be used to treat volume overload; however, it is difficult to remove the large volumes of fluid required for high-volume plasma infusion.
Plasma infusion should be reserved for two situations:
- If plasma exchange cannot be promptly started
- For patients with very severe or refractory disease, between plasma exchange sessions.
Benefit of cryoprecipitate-poor plasma is uncertain
Fresh frozen plasma is believed to contain nearly physiologic levels of all of the plasma proteins. When plasma is cooled to around 4°C, a precipitate forms that contains a variety of substances, including the higher molecular weight multimers of von Willebrand factor. Because TTP involves excess large multimers, giving plasma in which the high molecular weight multimers have been removed should in theory be better. In many centers, such cryoprecipitate-poor plasma is routinely used to treat TTP.
However, evidence that cryoprecipitate-poor plasma is better is lacking. A large study in Canada evaluating this question was terminated because of a lack of patient accrual, a common fate of clinical trials of rare diseases. A randomized study in 17 patients failed to show an advantage of cryoprecipitate-poor plasma over regular plasma, but the study was too small to draw firm conclusions given large confidence intervals about the point estimate of the treatment effect.
Cryoprecipitate-poor plasma is more expensive than regular plasma and is not as available. We do not routinely use it in our center to initially manage patients with TTP, but we do use it for patients who are refractory to standard treatment.
Scott et al3 measured the concentration of ADAMTS13 in a variety of plasma products and found that there are significant amounts in cryoprecipitate. Although giving cryoprecipitate-poor plasma provides less of the high molecular weight multimers, which is desirable for patients with TTP, it also provides less ADAMTS13, which is not desirable.
Do antiplatelet agents have a role in acute TTP?
Most algorithms for managing acute TTP include the use of aspirin or dipyridamole (Persantine) or both, and there is some evidence in favor of this approach,4 but whether antiplatelet therapy should be used for inpatients with severe thrombocytopenia remains controversial. In our practice, we usually provide antiplatelet therapy even for patients with severe thrombocytopenia because we believe TTP involves platelet-mediated hypercoagulability rather than increased bleeding risk.
Do corticosteroids have a role?
Corticosteroids were widely used to treat TTP even before the disease was discovered to be immune mediated. In our center we routinely use them.
Unfortunately, few data exist on the efficacy of steroid therapy for TTP. As a result, we can only make a weak recommendation for its use: using the American College of Chest Physicians rating system for the strength of clinical evidence, it would receive a 2C recommendation. This is the weakest possible recommendation, being based on widespread use but poor-quality data.
Stopping vs tapering plasma exchange
Whether plasma exchange should be tapered or simply stopped is also controversial and not well studied. Nevertheless, a widespread clinical practice—once the platelet count returns to 200 × 109/L or higher and the patient looks and feels well—is to reduce the plasma exchange sessions to once every 3 days, then to once every 7 days, and then to once every 2 weeks.
In our practice, we taper plasma exchange in this fashion for a minimum of two treatments beyond what we think the patient really needs. As a result, we tend to treat about once every 2 weeks for weeks or even months after the acute illness.
Rituximab may help
Rituximab (Rituxan), a monoclonal antibody against mature B cells, is increasingly being used in treating TTP. Past and present treatments for TTP, including splenectomy, corticosteroids, and plasma exchange are immunomodulatory, so the use of rituximab may be justified. Case reports provided the rationale for a large, multicenter, randomized controlled trial, which is currently under way.5
CONDITIONS THAT ARE NOT TTP
Some conditions may be confused with TTP but are clearly something different:
Patients with isolated thrombocytopenia and normal blood smear findings and no coagulopathy most likely have idiopathic thrombocytopenia purpura or, in the correct clinical circumstance, heparin-induced thrombocytopenia.
A patient with an extremely low platelet count but no fragments or very few fragments with microangiopathic hemolytic anemia may have either drug-associated thrombocytopenia or disseminated intravascular coagulopathy, particularly if there is concomitant coagulopathy.
Many pregnancy-associated microangiopathies resemble TTP, and it may be difficult to differentiate them from TTP; if confusion as to the diagnosis exists, the patient should be treated with plasma exchange, as this therapy may be life-saving.
Many rheumatologic conditions are characterized by an acute illness with nonspecific findings, such as low-grade hemolysis and thrombocytopenia. For example, Wegener granulomatosis can present with evidence of hemolysis, thrombocytopenia, and renal impairment.
Systemic lupus erythematosus can also initially present with an “early-TTP”-like picture. Evidence of glomerulonephritis is not consistent with TTP, and urinary red cell casts makes the diagnosis of lupus more likely. Helmet cell fragments in the peripheral blood smear are supposedly more characteristic of TTP, but their presence is not diagnostic.
Scleroderma renal crisis can present like TTP, but because it is unlikely that a patient with known scleroderma would have a second rare disease, it is best to treat it as scleroderma, which does not require plasma exchange or plasma infusion.
In general, if the diagnosis is uncertain, the safest course is to treat the patient with plasma exchange, then try to establish the diagnosis, because TTP is fatal if not promptly treated. Although plasma exchange is probably overused, it is more innocuous than untreated TTP.
Thrombotic thrombocytopenic purpura (TTP) is one of the few hematologic emergencies. Untreated, most patients die, but prompt and appropriate treatment allows most patients not only to survive but to recover, frequently without long-term sequelae.
TTP is rare. The estimated annual incidence of all TTP syndromes is about 11 cases per million in the general population, and the incidence of severe ADAMTS13 deficiency (see discussion below) is about 2 per million. Therefore, even large medical centers typically see only one or two cases each year. The syndromes are much more common in women, and the incidence among blacks is nine times higher than the incidence among non-blacks. Nevertheless, despite the rarity of this disease, good evidence exists to help guide patient care, thanks to national registries and research organizations, such as the Canadian Apheresis Study Group and the Oklahoma TTP-Hemolytic Uremic Syndrome (HUS) Registry.
This article reviews the physiologic basis of TTP, how to recognize it, and how best to treat it. We also discuss other conditions that clinically resemble TTP but probably have different underlying causes.
A YOUNG WOMAN WITH ARM WEAKNESS
A 24-year-old black woman presents to a community hospital with weakness in her left arm, which began about 30 minutes previously. She has had progressive dyspnea over the last several weeks, but has otherwise been completely well and has had no medical problems in the past other than being obese.
Physical examination reveals weakness in her left arm as well as mild dysarthria, which was not previously noted by the patient or her family. Her laboratory findings:
- White blood cell count 16.7 × 109/L (reference range 4.5–11.0)
- Platelet count 32 × 109/L (150–350)
- Hemoglobin concentration 6.5 g/dL (1.4–17.5)
- Peripheral blood smear: normal white cells, rare platelets, red cells normo-chromic with many fragments
- Lactate dehydrogenase (LDH) concentration 2,300 U/L (100–200).
In view of her symptoms and laboratory values, the physician suspects she may have TTP and refers her to McMaster University Medical Center in Hamilton, Ontario, Canada. Plasma exchange is started immediately; one plasma volume is removed and replaced with fresh frozen plasma. Nevertheless, the patient’s condition deteriorates overnight, she becomes more confused and cannot protect her airway, her LDH concentration rises further, and her hemoglobin concentration falls. She is transferred to the intensive care unit. Her plasma exchange prescription is increased to 1.5 volumes twice daily (although little evidence exists that plasma exchange twice daily is more effective than once daily).
On the third day of her stay, she becomes completely paralyzed on the left side. In addition to her twice-daily plasma exchange procedures, a plasma infusion and corticosteroid therapy are initiated. Her platelet count stabilizes at about 20 × 109/L.
The patient next develops renal insufficiency and requires three acute hemodialysis treatments. (Plasma infusion frequently leads to volume overload in critically ill patients. Some intravascular volume can be removed with plasma exchange; however, significant volume overload with significant renal insufficiency can only be treated with renal replacement therapy.)
The patient undergoes 28 consecutive days of twice-daily plasma exchange and gradually improves, as measured by increasing platelet counts, a gradual fall in the LDH concentration, and stabilization of—and ultimately an increase in—the hemoglobin level. She is weaned off plasma infusions, and then plasma exchange is tapered to once a day and then to alternate days.
She is completely well at the time of discharge 4 weeks after her initial admission, with no residual deficits.
Comment. This case shows that even patients with apparently devastating compromise and neurologic deficits can completely recover with aggressive plasma exchange and other therapies. One child treated at the Hospital for Sick Children, affiliated with the University of Toronto, developed TTP and had 120 consecutive days of plasma exchange: she was unconscious and comatose for much of that time, but she ultimately recovered and is now completely well without residual neurologic deficits.
TTP MAY BE DUE TO ADAMTS13 DEFICIENCY
Twenty-five years ago, little was known about TTP except for its clinical manifestations. Now, it is known to be caused in some patients by an acquired deficiency of a circulating metalloproteinase. In very rare cases a hereditary deficiency of ADAMTS13 causes TTP. In addition, a number of conditions share clinical features with TTP but have other underlying causes.
In acquired TTP, an autoantibody forms against ADAMTS13, a zinc-containing metalloproteinase that is also known as von Willebrand factor-cleaving protease. Normally, von Willebrand factor circulates in plasma as multimers that allow platelets to adhere to vascular surfaces. When von Willebrand factor is initially released from endothelial cells, it exists as large multimers, which are more adhesive for platelets than normal. These large multimers are normally cleaved into smaller units by ADAMTS13. If ADAMTS13 is lacking, the very-high-molecular-weight von Willebrand factor multimers accumulate, causing platelet agglutination and the vascular occlusion that results in the manifestations of TTP.
In 1994, ADAMTS13, the gene of which is on the ninth chromosome, was shown to cleave von Willebrand factor under conditions of high shear stress. In 1996, a congenital homozygous deficiency of ADAMTS13 was found to be associated with platelet microthrombi. Afterwards, some patients with TTP were shown to have low or undetectable levels of ADAMTS13, owing to immunoglobulin G antibodies directed against the enzyme.
TTP AND RELATED SYNDROMES
Clinically, TTP encompasses a number of different but related syndromes, some of which have different physiologic bases.
TTP
TTP is characterized by moderate to severe thrombocytopenia, red cell fragmentation, and elevated LDH levels (due to red cell destruction and also muscle and organ ischemia). The pentad of features classically associated with TTP in the era before effective treatment (thrombocytopenia, fever, renal failure, neurologic deficit, and microangiopathic hemolytic anemia) is rarely seen in countries with advanced medical care: renal insufficiency and neurologic events are end-stage manifestations, and the disease should be recognizable well before these manifestations occur. Otherwise unexplained thrombocytopenia, microangiopathic hemolytic anemia, and an elevated LDH should strongly suggest TTP. TTP is the appropriate designation for adults with these clinical features, even in the presence of renal failure. TTP is uncommon in children.
Most patients present with nonspecific constitutional symptoms, such as weakness, abdominal pain, nausea, and vomiting. Typically, the family physician orders a complete blood cell count and finds that the platelet count and hemoglobin are low. Red cell fragments are noted in the peripheral blood smear. Further testing reveals an elevated LDH concentration.
HUS
HUS was initially described 30 years after TTP in children with acute renal failure in addition to thrombocytopenia and microangiopathic hemolytic anemia. The term “HUS” is currently used primarily to describe the condition in children.
In children, two forms of HUS exist:
Diarrhea-associated HUS is associated with diarrhea that is commonly bloody, due to an enterotoxin produced by Escherichia coli O157:H7.
Endemic diarrhea-associated HUS is much more common than HUS associated with epidemics. Endemic cases are caused by E coli O157:H7 present in the environment. Other patients present with clinically apparent HUS but the causal bacterium cannot be detected. The kidney transplant program at our center often sees young patients with this disease who do not have E coli O157:H7 infection, and the pathogenesis is not understood. Epidemic cases are less common but the outbreaks are dramatic. About 10 years ago, E coli O157:H7 entered the water supply in the small city of Walkerton, Ontario, and many people developed the epidemic form of HUS over a period of several weeks. Most such patients spontaneously recovered without plasma exchange, although many were left with impaired renal function.
Atypical HUS. Less often, HUS in children is not associated with a prodrome of diarrhea and is referred to as “atypical” HUS. These children often have a more prolonged and complicated course and resemble adults with TTP.
Familial TTP-HUS
Familial TTP-HUS is very rare. It may present with hemolysis and thrombocytopenia in childhood or early adulthood. Many patients present with renal insufficiency, and only careful evaluation reveals hemolysis and thrombocytopenia. The disease typically manifests acutely: a patient may have an upper respiratory tract infection and subsequently develop an episode of TTP-HUS. Episodes tend to recur, and multiple family members may also be affected.
Plasma infusion is an effective treatment, and plasma exchange is usually not required. Since more patients are now surviving well into adulthood, some are being seen to develop antibodies to the ADAMTS13 in the infused plasma, analogous to patients with severe hemophilia developing inhibitors to factor VIII. The disease may progress despite treatment: we have been treating a young woman who has had a series of catastrophic complications and now has chronic renal failure requiring hemodialysis (see discussion below).
Post-transplant microangiopathy
Post-transplant microangiopathy is most likely to develop after solid-organ or stem-cell allograft transplantation. Manifestations resemble those of TTP, but the mechanism is probably quite different. Multiple causes probably exist, depending on the setting.
Post-transplant microangiopathy does not respond to the usual therapies for TTP, although we treat it, like TTP, with corticosteroids, antiplatelet agents, and plasma exchange. Other centers do not use plasma exchange for these patients. Most patients have a poor prognosis, especially those with a transplant other than a kidney.
A spectrum of related syndromes
A number of diseases clinically resemble TTP. Enhanced diagnostic capacity and better molecular biologic techniques are revealing that they often have very different underlying causes and that in some cases they require different treatment.
TOWARD DIAGNOSTIC CRITERIA
Ruutu et al,1 in a consensus conference, used rigorous methods to establish diagnostic criteria for microangiopathy associated with stem cell transplantation:
- More than 4% red blood cell fragments in the peripheral blood. A laboratory report that states that “few fragments” are present is not nearly as useful as one that estimates the quantity; eg, 1% fragments would have very different implications than 6% fragments.
- Thrombocytopenia—a platelet count of less than 50 × 109/L or more than a 50% reduction from previous counts
- Increased LDH concentration
- Reduced hemoglobin concentration or increased transfusion requirement
- Decrease in serum haptoglobin, which, like red blood cell fragments, is a marker of hemolysis rather than of reduced synthesis.
The ADAMTS13 level need not be assessed. Metalloproteinase deficiency need not be proved to diagnose TTP. Although our hospital is a TTP referral center, we do not routinely offer the test. Too often the test results cause confusion: a patient can have a normal level of ADAMTS13 and still have TTP that responds to plasma exchange, and levels can be low in conditions other than TTP.
THE CHALLENGES OF TREATMENT
Plasma exchange is the primary treatment for TTP
Rock et al2 performed a randomized trial in which 102 patients with TTP received either a 1.5-volume plasma exchange daily for 3 days and then 1-volume plasma exchanges as needed to control the disease or plasma infusion. Patients who received plasma exchange had a better initial response, a higher survival rate, and a lower rate of relapse than patients receiving plasma infusion. These findings established plasma exchange as the treatment of choice for TTP.
However, the trial had some inherent problems: patients who had plasma infusions tended to develop renal insufficiency and as a result did not receive as much plasma because they could not tolerate as much volume as those who had plasma exchange. Plasma exchange probably worked better because it could deliver more plasma over a fixed period of time, enabling patients to obtain more of the ADAMTS13 enzyme, rather than because it was an intrinsically better treatment. This interpretation is the basis for our occasional use of twice-daily plasma exchange in critically ill patients.
TTP is different from other autoimmune diseases such as idiopathic thrombocytopenia purpura, in which the primary treatments are immunosuppressive agents. Some evidence exists for treating TTP with immunosuppressive agents, but the primary treatment should be plasma exchange.
Plasma infusion is useful in some cases
Although small case series and our own experience provide evidence for the benefit of treating TTP with high-dose plasma infusions (25 mL/kg/day, or about 1.5 to 2.0 L/day for an average-sized adult), problems will likely arise with volume overload if the patient has any significant renal insufficiency. Dialysis or ultrafiltration may be used to treat volume overload; however, it is difficult to remove the large volumes of fluid required for high-volume plasma infusion.
Plasma infusion should be reserved for two situations:
- If plasma exchange cannot be promptly started
- For patients with very severe or refractory disease, between plasma exchange sessions.
Benefit of cryoprecipitate-poor plasma is uncertain
Fresh frozen plasma is believed to contain nearly physiologic levels of all of the plasma proteins. When plasma is cooled to around 4°C, a precipitate forms that contains a variety of substances, including the higher molecular weight multimers of von Willebrand factor. Because TTP involves excess large multimers, giving plasma in which the high molecular weight multimers have been removed should in theory be better. In many centers, such cryoprecipitate-poor plasma is routinely used to treat TTP.
However, evidence that cryoprecipitate-poor plasma is better is lacking. A large study in Canada evaluating this question was terminated because of a lack of patient accrual, a common fate of clinical trials of rare diseases. A randomized study in 17 patients failed to show an advantage of cryoprecipitate-poor plasma over regular plasma, but the study was too small to draw firm conclusions given large confidence intervals about the point estimate of the treatment effect.
Cryoprecipitate-poor plasma is more expensive than regular plasma and is not as available. We do not routinely use it in our center to initially manage patients with TTP, but we do use it for patients who are refractory to standard treatment.
Scott et al3 measured the concentration of ADAMTS13 in a variety of plasma products and found that there are significant amounts in cryoprecipitate. Although giving cryoprecipitate-poor plasma provides less of the high molecular weight multimers, which is desirable for patients with TTP, it also provides less ADAMTS13, which is not desirable.
Do antiplatelet agents have a role in acute TTP?
Most algorithms for managing acute TTP include the use of aspirin or dipyridamole (Persantine) or both, and there is some evidence in favor of this approach,4 but whether antiplatelet therapy should be used for inpatients with severe thrombocytopenia remains controversial. In our practice, we usually provide antiplatelet therapy even for patients with severe thrombocytopenia because we believe TTP involves platelet-mediated hypercoagulability rather than increased bleeding risk.
Do corticosteroids have a role?
Corticosteroids were widely used to treat TTP even before the disease was discovered to be immune mediated. In our center we routinely use them.
Unfortunately, few data exist on the efficacy of steroid therapy for TTP. As a result, we can only make a weak recommendation for its use: using the American College of Chest Physicians rating system for the strength of clinical evidence, it would receive a 2C recommendation. This is the weakest possible recommendation, being based on widespread use but poor-quality data.
Stopping vs tapering plasma exchange
Whether plasma exchange should be tapered or simply stopped is also controversial and not well studied. Nevertheless, a widespread clinical practice—once the platelet count returns to 200 × 109/L or higher and the patient looks and feels well—is to reduce the plasma exchange sessions to once every 3 days, then to once every 7 days, and then to once every 2 weeks.
In our practice, we taper plasma exchange in this fashion for a minimum of two treatments beyond what we think the patient really needs. As a result, we tend to treat about once every 2 weeks for weeks or even months after the acute illness.
Rituximab may help
Rituximab (Rituxan), a monoclonal antibody against mature B cells, is increasingly being used in treating TTP. Past and present treatments for TTP, including splenectomy, corticosteroids, and plasma exchange are immunomodulatory, so the use of rituximab may be justified. Case reports provided the rationale for a large, multicenter, randomized controlled trial, which is currently under way.5
CONDITIONS THAT ARE NOT TTP
Some conditions may be confused with TTP but are clearly something different:
Patients with isolated thrombocytopenia and normal blood smear findings and no coagulopathy most likely have idiopathic thrombocytopenia purpura or, in the correct clinical circumstance, heparin-induced thrombocytopenia.
A patient with an extremely low platelet count but no fragments or very few fragments with microangiopathic hemolytic anemia may have either drug-associated thrombocytopenia or disseminated intravascular coagulopathy, particularly if there is concomitant coagulopathy.
Many pregnancy-associated microangiopathies resemble TTP, and it may be difficult to differentiate them from TTP; if confusion as to the diagnosis exists, the patient should be treated with plasma exchange, as this therapy may be life-saving.
Many rheumatologic conditions are characterized by an acute illness with nonspecific findings, such as low-grade hemolysis and thrombocytopenia. For example, Wegener granulomatosis can present with evidence of hemolysis, thrombocytopenia, and renal impairment.
Systemic lupus erythematosus can also initially present with an “early-TTP”-like picture. Evidence of glomerulonephritis is not consistent with TTP, and urinary red cell casts makes the diagnosis of lupus more likely. Helmet cell fragments in the peripheral blood smear are supposedly more characteristic of TTP, but their presence is not diagnostic.
Scleroderma renal crisis can present like TTP, but because it is unlikely that a patient with known scleroderma would have a second rare disease, it is best to treat it as scleroderma, which does not require plasma exchange or plasma infusion.
In general, if the diagnosis is uncertain, the safest course is to treat the patient with plasma exchange, then try to establish the diagnosis, because TTP is fatal if not promptly treated. Although plasma exchange is probably overused, it is more innocuous than untreated TTP.
- Ruutu T, Barosi G, Benjamin RJ, et al; European Group for Blood and Marrow Transplantation; European LeukemiaNet. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica 2007; 92:95–100.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Scott EA, Puca KE, Pietz BC, Duchateau BK, Friedman KD. Comparison and stability of ADAMTS13 activity in therapeutic plasma products. Transfusion 2007; 47:120–125.
- Bobbio-Pallavicini E, Gugliotta L, Centurioni R, et al. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica 1997; 82:429–435.
- George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher 2006; 21:49–56.
- Ruutu T, Barosi G, Benjamin RJ, et al; European Group for Blood and Marrow Transplantation; European LeukemiaNet. Diagnostic criteria for hematopoietic stem cell transplant-associated microangiopathy: results of a consensus process by an International Working Group. Haematologica 2007; 92:95–100.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Scott EA, Puca KE, Pietz BC, Duchateau BK, Friedman KD. Comparison and stability of ADAMTS13 activity in therapeutic plasma products. Transfusion 2007; 47:120–125.
- Bobbio-Pallavicini E, Gugliotta L, Centurioni R, et al. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica 1997; 82:429–435.
- George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher 2006; 21:49–56.
KEY POINTS
- Strokes and renal insufficiency are end-stage manifestations of TTP; the condition is usually diagnosed before they occur.
- Classic TTP should be rapidly and aggressively treated with plasma exchange. Plasma infusion therapy plays a role for patients who cannot promptly receive plasma exchange or for patients with severe disease between episodes of plasma exchange.
- Antiplatelet therapy may be appropriate along with plasma exchange for patients without severe thrombocytopenia.
- If a renal transplant recipient develops systemic symptoms with TTP-like disease, one should consider modifying or withdrawing the immunosuppressive therapy, although this may result in loss of function and the need for transplant nephrectomy.
An elderly woman with shortness of breath
A frail 75-year-old woman with diabetes, hyperlipidemia, and a history of rheumatic fever in childhood presents to the emergency department of a community hospital with complaints of chest pressure, shortness of breath on exertion, and easy fatigability. Her shortness of breath started 6 months ago but has become much worse over the past few days.
On examination, her pulse is 110 and irregular, and she has markedly distended neck veins and evidence of pulmonary edema. She has a systolic murmur, but it is difficult to characterize due to tachycardia. Electrocardiography shows atrial fibrillation with rapid ventricular response and right axis deviation. Chest radiography shows bilateral pleural effusions.
The patient is given a diuretic, anticoagulation is started to prevent thromboembolism, and she undergoes cardioversion for the atrial fibrillation.
Transthoracic echocardiography is performed and reveals biatrial enlargement, anterior mitral valve leaflet thickening, mitral valve calcification, and moderate mitral regurgitation. Her ejection fraction is normal, but she has mild right ventricular systolic dysfunction with moderate tricuspid regurgitation and an estimated right ventricular systolic pressure of 90 mm Hg (normal range 15–30 mm Hg).
After an uneventful hospital course, she is discharged on a stable dose of a diuretic and oral anticoagulation. Despite adequate diuresis and maintenance of normal sinus rhythm, however, she continues to experience severe dyspnea, which limits her ability to perform simple tasks, such as dusting the furniture in her home. She is referred to Cleveland Clinic for further evaluation.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. What was the most likely cause of this patient’s initial acute presentation to the emergency department?
- An acute decrease in mitral valve area
- Rheumatic mitral valve stenosis
- Acute coronary syndrome
- Atrial fibrillation
Although an acute decrease in mitral valve area caused by an atrial myxoma, thrombus, or vegetation is a possibility, this patient’s symptoms gradually increased over a period of months. She also did not have signs of infection, nor did she have any signs of embolic phenomena to suggest myxoma, thrombus, or vegetation, all of which frequently present with emboli.
Given her history of rheumatic fever and her echocardiographic findings, rheumatic mitral valve stenosis is high on the list of differential diagnoses. Rheumatic mitral valve stenosis is a chronic process in which the valve area decreases at a rate of about 0.1 cm2/year.1,2
Acute coronary syndrome is a possibility in this elderly woman with multiple risk factors for coronary artery disease. An evaluation for coronary insufficiency should be considered, but the most striking finding on her initial electrocardiogram is atrial fibrillation with a rapid ventricular response.
The onset of atrial fibrillation in the setting of valvular heart disease is the most likely cause of her acute decompensation with signs and symptoms of congestive heart failure. In severe mitral stenosis, because the mitral valve orifice is narrowed, a higher left atrial pressure and longer ventricular filling time are required to maintain forward flow. Now add atrial fibrillation to this situation: the left atrium no longer contracts properly, so less blood is forced through the narrowed valve, and with the rapid heart rate the left ventricle has less time to fill. These two conditions result in elevated left atrial and pulmonary venous pressures, which in turn result in pulmonary edema and congestive heart failure. Thus, patients with mitral stenosis tolerate atrial fibrillation poorly.
WHAT IS THE NEXT STEP?
2. What would be the most appropriate next step for our patient?
- Transesophageal echocardiography
- Right heart catheterization
- An exercise treadmill stress test0
Transesophageal echocardiography is a reasonable option, as mitral stenosis is strongly suspected but transthoracic echocardiography did not reveal severe mitral valve disease. The use of transesophageal echocardiography for the diagnosis of mitral stenosis in this situation is a class IC recommendation (ie, the procedure is recommended, although very few trials have been done) in the American College of Cardiology and American Heart Association Valvular Disease guidelines.3
Right heart catheterization is the diagnostic test of choice in this situation, as the patient has evidence of biventricular heart failure and her right ventricular systolic pressure of 90 mm Hg is consistent with severe pulmonary hypertension. Right heart catheterization would help differentiate primary pulmonary hypertension causing dyspnea from pulmonary hypertension secondary to elevated left-sided pressures. It would also provide a direct hemodynamic estimate of her cardiac function.
Exercise treadmill testing. Evaluation for myocardial ischemia is a reasonable option, as our patient is elderly and has hypertension and diabetes, both of which are risk factors for coronary artery disease. Moreover, in a patient with diabetes, myocardial ischemia can present as dyspnea without typical anginal chest pain. Because of her age and severely limiting dyspnea, however, she would be unlikely to achieve an adequate heart rate during exercise treadmill testing, so this may not be the optimal type of stress test.
Although asymptomatic patients with moderate or severe mitral stenosis should undergo evaluation of exercise capacity and change in pulmonary artery pressures with exercise to determine the need for percutaneous balloon mitral valvuloplasty (see below),3 our patient is symptomatic and already has evidence of severe pulmonary hypertension on transthoracic echocardiography.
Other options for evaluating for myocardial ischemia include pharmacologic stress testing with imaging (eg, dobutamine echocardiography or adenosine nuclear imaging) or proceeding directly to coronary angiography.
CASE CONTINUED: RIGHT HEART CATHETERIZATION, CORONARY ANGIOGRAPHY
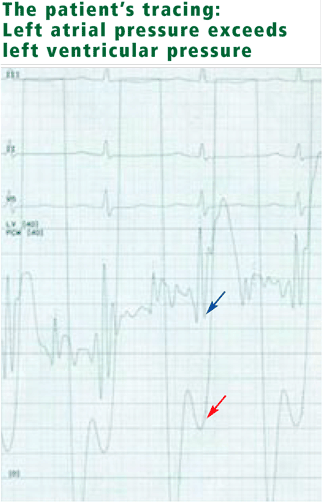
- Mean right atrial pressure 6 mm Hg (normal 2–7 mm Hg)
- Right ventricular pressure 102/6 mm Hg (consistent with severe pulmonary hypertension) (normal 15–30/1–7 mm Hg)
- Pulmonary artery pressure 102/40 mm Hg (normal 15–30/4–12 mm Hg)
- Mean pulmonary capillary wedge pressure 25 mm Hg (normal 4–12 mm Hg)
- Cardiac output 3.16 L/min (normal 4–8 L/min)
- Cardiac index 2.10 L/min/m2 (normal 2.5–4.2 L/min/m2)
- v waves are not prominent.
Because her symptoms raise concern for ischemia, coronary angiography is also performed and shows minimal, nonobstructive coronary artery disease. Her left ventricular end-diastolic pressure is 8 mm Hg (normal 5–12 mm Hg).
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis?
- Tricuspid stenosis
- Pulmonic stenosis
- Mitral stenosis
- Mitral regurgitation
Tricuspid stenosis would result in a higher pressure in the right atrium than in the right ventricle. In our patient, the right atrial pressure and the right ventricular diastolic pressure are both 6 mm Hg, eliminating this possibility.
Similarly, pulmonic stenosis would result in a higher pressure in the right ventricle than in the pulmonary artery. In our patient both the right ventricular systolic pressure and the pulmonary artery systolic pressures are 102 mm Hg.
Acute mitral regurgitation may result in increased wedge pressure and tall v waves (reflecting left atrial filling during ventricular systole). In chronic mitral regurgitation, however, the wedge pressure may be normal and the patient may have relatively normal-appearing v waves.4
Mitral stenosis results in a marked gradient between the pulmonary capillary wedge pressure and the left ventricular diastolic pressure in the absence of pulmonary veno-occlusive disease. This gradient can be measured by simultaneous catheterization of the right heart (to measure the wedge pressure, which is an indirect measure of left atrial pressure) and the left heart (to measure the left ventricular diastolic pressure). If the patient does not have significant mitral stenosis, the wedge pressure should be approximately equal to the left ventricular diastolic pressure. In our patient, the wedge pressure (and therefore the left atrial pressure) is 25 mm Hg, and the left ventricular end-diastolic pressure is 8 mm Hg—a difference of 17 mm Hg, consistent with significant mitral stenosis.
CASE CONTINUED: TRANSESOPHAGEAL ECHOCARDIOGRAPHY
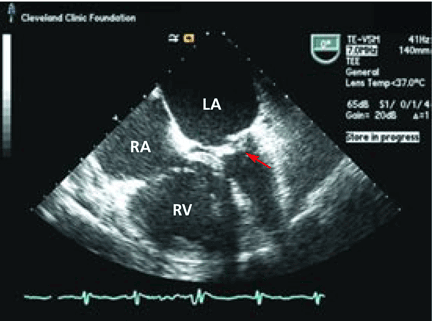
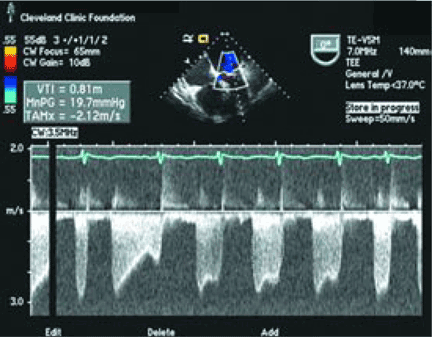
WHAT IS THE TREATMENT?
4. Which of the following is the preferred technique for correcting mitral stenosis in this patient?
- Percutaneous balloon mitral valvuloplasty
- Mitral valve surgery
- Percutaneous mitral valve replacement
Although there are several options for mechanical treatment of mitral stenosis, percutaneous balloon mitral valvuloplasty by experienced operators is the procedure of choice for patients who have symptomatic moderate-to-severe mitral stenosis with favorable valve morphology but do not have significant mitral regurgitation or left atrial thrombus.3 The hemodynamic and symptomatic improvement that can be expected after this procedure can be predicted using several echocardiographic criteria, including valve mobility, subvalvular thickening, valve leaflet thickening, and valve leaflet calcification,5 as well as the degree of commissural calcification or commissural fusion.6 Success rates are better if the valve is relatively more mobile and has lesser degrees of valvular and subvalvular thickening, calcification, and commissural fusion.
Mitral valve surgery (repair if possible) is indicated in patients with acceptable operative risk who have symptomatic (New York Heart Association class III or IV) moderate-to-severe mitral stenosis if percutaneous balloon mitral valvuloplasty is unavailable, in cases in which an atrial thrombus or moderate-to-severe mitral regurgitation precludes balloon valvuloplasty, or when the valve morphology is not favorable for balloon valvulo-plasty.3
Although she has moderate mitral regurgitation and poor valve morphology, our patient is a poor surgical candidate because of her advanced age, severe pulmonary hypertension, and poor functional status. Patients with moderate-to-severe mitral stenosis and class III or IV symptoms who have nonpliable, calcified valves but are not candidates for open heart surgery have a class IIb indication for percutaneous balloon mitral valvuloplasty—ie, the procedure may be considered.3
In addition, the procedure also carries a class IIb recommendation in patients with moderate-to-severe mitral stenosis and new-onset atrial fibrillation (provided that they do not have a thrombus in the left atrium or moderate-to-severe mitral regurgitation), even without symptoms.3
Percutaneous mitral valve replacement is not available in clinical practice, although this is an active area of clinical research and may be available in the future.
CASE CONTINUED: THE PATIENT UNDERGOES BALLOON VALVULOPLASTY
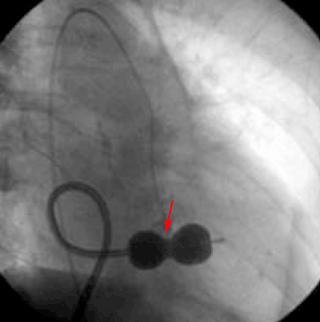
Transthoracic echocardiography performed 4 months later shows moderate mitral stenosis with a mean gradient of 9.0 mm Hg, a mitral valve area of 1.7 cm2, and moderate mitral regurgitation. Her right ventricular systolic pressure is estimated to be 74 mm Hg. The patient reports less dyspnea during her housework and now has New York Heart Association class II symptoms. Her treatment regimen includes warfarin (Coumadin) for atrial fibrillation, a beta-blocker to control her heart rate in atrial fibrillation and increase her left ventricular filling time, and a low-dose diuretic.
- Gordon SP, Douglas PS, Come PC, et al. Two-dimensional and Doppler echocardiographic determinants of the natural history of mitral valve narrowing in patients with rheumatic mitral stenosis: implications for follow-up. J Am Coll Cardiol 1992; 19:968–973.
- Sagie A, Freitas N, Padial LR, et al. Doppler echocardiographic assessment of long-term progression of mitral stenosis in 103 patients: valve area and right heart disease. J Am Coll Cardiol 1996; 28:472–479.
- Bonow ROC, Blase A, Chatterjee K, et al. ACC/AHA 2006 Practice Guidelines for the Management of Patients With Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease) Developed in Collaboration With the Society of Cardiovascular Anesthesiologists Endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2006; 48:598–675.
- Braunwald E. The syndrome of severe mitral regurgitation with normal left atrial pressure. Circulation 1963; 27:29–35.
- Wilkins GT, Weyman AE, Abascal VM, et al. Percutaneous balloon dilatation of the mitral valve: an analysis of echocardiographic variables related to outcome and the mechanism of dilatation. Br Heart J 1988; 60:299–308.
- Cannan CR, Nishimura RA, Reeder GS, et al. Echocardiographic assessment of commissural calcium: a simple predictor of outcome after percutaneous mitral balloon valvotomy. J Am Coll Cardiol 1997; 29:175–180.
A frail 75-year-old woman with diabetes, hyperlipidemia, and a history of rheumatic fever in childhood presents to the emergency department of a community hospital with complaints of chest pressure, shortness of breath on exertion, and easy fatigability. Her shortness of breath started 6 months ago but has become much worse over the past few days.
On examination, her pulse is 110 and irregular, and she has markedly distended neck veins and evidence of pulmonary edema. She has a systolic murmur, but it is difficult to characterize due to tachycardia. Electrocardiography shows atrial fibrillation with rapid ventricular response and right axis deviation. Chest radiography shows bilateral pleural effusions.
The patient is given a diuretic, anticoagulation is started to prevent thromboembolism, and she undergoes cardioversion for the atrial fibrillation.
Transthoracic echocardiography is performed and reveals biatrial enlargement, anterior mitral valve leaflet thickening, mitral valve calcification, and moderate mitral regurgitation. Her ejection fraction is normal, but she has mild right ventricular systolic dysfunction with moderate tricuspid regurgitation and an estimated right ventricular systolic pressure of 90 mm Hg (normal range 15–30 mm Hg).
After an uneventful hospital course, she is discharged on a stable dose of a diuretic and oral anticoagulation. Despite adequate diuresis and maintenance of normal sinus rhythm, however, she continues to experience severe dyspnea, which limits her ability to perform simple tasks, such as dusting the furniture in her home. She is referred to Cleveland Clinic for further evaluation.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. What was the most likely cause of this patient’s initial acute presentation to the emergency department?
- An acute decrease in mitral valve area
- Rheumatic mitral valve stenosis
- Acute coronary syndrome
- Atrial fibrillation
Although an acute decrease in mitral valve area caused by an atrial myxoma, thrombus, or vegetation is a possibility, this patient’s symptoms gradually increased over a period of months. She also did not have signs of infection, nor did she have any signs of embolic phenomena to suggest myxoma, thrombus, or vegetation, all of which frequently present with emboli.
Given her history of rheumatic fever and her echocardiographic findings, rheumatic mitral valve stenosis is high on the list of differential diagnoses. Rheumatic mitral valve stenosis is a chronic process in which the valve area decreases at a rate of about 0.1 cm2/year.1,2
Acute coronary syndrome is a possibility in this elderly woman with multiple risk factors for coronary artery disease. An evaluation for coronary insufficiency should be considered, but the most striking finding on her initial electrocardiogram is atrial fibrillation with a rapid ventricular response.
The onset of atrial fibrillation in the setting of valvular heart disease is the most likely cause of her acute decompensation with signs and symptoms of congestive heart failure. In severe mitral stenosis, because the mitral valve orifice is narrowed, a higher left atrial pressure and longer ventricular filling time are required to maintain forward flow. Now add atrial fibrillation to this situation: the left atrium no longer contracts properly, so less blood is forced through the narrowed valve, and with the rapid heart rate the left ventricle has less time to fill. These two conditions result in elevated left atrial and pulmonary venous pressures, which in turn result in pulmonary edema and congestive heart failure. Thus, patients with mitral stenosis tolerate atrial fibrillation poorly.
WHAT IS THE NEXT STEP?
2. What would be the most appropriate next step for our patient?
- Transesophageal echocardiography
- Right heart catheterization
- An exercise treadmill stress test0
Transesophageal echocardiography is a reasonable option, as mitral stenosis is strongly suspected but transthoracic echocardiography did not reveal severe mitral valve disease. The use of transesophageal echocardiography for the diagnosis of mitral stenosis in this situation is a class IC recommendation (ie, the procedure is recommended, although very few trials have been done) in the American College of Cardiology and American Heart Association Valvular Disease guidelines.3
Right heart catheterization is the diagnostic test of choice in this situation, as the patient has evidence of biventricular heart failure and her right ventricular systolic pressure of 90 mm Hg is consistent with severe pulmonary hypertension. Right heart catheterization would help differentiate primary pulmonary hypertension causing dyspnea from pulmonary hypertension secondary to elevated left-sided pressures. It would also provide a direct hemodynamic estimate of her cardiac function.
Exercise treadmill testing. Evaluation for myocardial ischemia is a reasonable option, as our patient is elderly and has hypertension and diabetes, both of which are risk factors for coronary artery disease. Moreover, in a patient with diabetes, myocardial ischemia can present as dyspnea without typical anginal chest pain. Because of her age and severely limiting dyspnea, however, she would be unlikely to achieve an adequate heart rate during exercise treadmill testing, so this may not be the optimal type of stress test.
Although asymptomatic patients with moderate or severe mitral stenosis should undergo evaluation of exercise capacity and change in pulmonary artery pressures with exercise to determine the need for percutaneous balloon mitral valvuloplasty (see below),3 our patient is symptomatic and already has evidence of severe pulmonary hypertension on transthoracic echocardiography.
Other options for evaluating for myocardial ischemia include pharmacologic stress testing with imaging (eg, dobutamine echocardiography or adenosine nuclear imaging) or proceeding directly to coronary angiography.
CASE CONTINUED: RIGHT HEART CATHETERIZATION, CORONARY ANGIOGRAPHY
- Mean right atrial pressure 6 mm Hg (normal 2–7 mm Hg)
- Right ventricular pressure 102/6 mm Hg (consistent with severe pulmonary hypertension) (normal 15–30/1–7 mm Hg)
- Pulmonary artery pressure 102/40 mm Hg (normal 15–30/4–12 mm Hg)
- Mean pulmonary capillary wedge pressure 25 mm Hg (normal 4–12 mm Hg)
- Cardiac output 3.16 L/min (normal 4–8 L/min)
- Cardiac index 2.10 L/min/m2 (normal 2.5–4.2 L/min/m2)
- v waves are not prominent.
Because her symptoms raise concern for ischemia, coronary angiography is also performed and shows minimal, nonobstructive coronary artery disease. Her left ventricular end-diastolic pressure is 8 mm Hg (normal 5–12 mm Hg).
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis?
- Tricuspid stenosis
- Pulmonic stenosis
- Mitral stenosis
- Mitral regurgitation
Tricuspid stenosis would result in a higher pressure in the right atrium than in the right ventricle. In our patient, the right atrial pressure and the right ventricular diastolic pressure are both 6 mm Hg, eliminating this possibility.
Similarly, pulmonic stenosis would result in a higher pressure in the right ventricle than in the pulmonary artery. In our patient both the right ventricular systolic pressure and the pulmonary artery systolic pressures are 102 mm Hg.
Acute mitral regurgitation may result in increased wedge pressure and tall v waves (reflecting left atrial filling during ventricular systole). In chronic mitral regurgitation, however, the wedge pressure may be normal and the patient may have relatively normal-appearing v waves.4
Mitral stenosis results in a marked gradient between the pulmonary capillary wedge pressure and the left ventricular diastolic pressure in the absence of pulmonary veno-occlusive disease. This gradient can be measured by simultaneous catheterization of the right heart (to measure the wedge pressure, which is an indirect measure of left atrial pressure) and the left heart (to measure the left ventricular diastolic pressure). If the patient does not have significant mitral stenosis, the wedge pressure should be approximately equal to the left ventricular diastolic pressure. In our patient, the wedge pressure (and therefore the left atrial pressure) is 25 mm Hg, and the left ventricular end-diastolic pressure is 8 mm Hg—a difference of 17 mm Hg, consistent with significant mitral stenosis.
CASE CONTINUED: TRANSESOPHAGEAL ECHOCARDIOGRAPHY
WHAT IS THE TREATMENT?
4. Which of the following is the preferred technique for correcting mitral stenosis in this patient?
- Percutaneous balloon mitral valvuloplasty
- Mitral valve surgery
- Percutaneous mitral valve replacement
Although there are several options for mechanical treatment of mitral stenosis, percutaneous balloon mitral valvuloplasty by experienced operators is the procedure of choice for patients who have symptomatic moderate-to-severe mitral stenosis with favorable valve morphology but do not have significant mitral regurgitation or left atrial thrombus.3 The hemodynamic and symptomatic improvement that can be expected after this procedure can be predicted using several echocardiographic criteria, including valve mobility, subvalvular thickening, valve leaflet thickening, and valve leaflet calcification,5 as well as the degree of commissural calcification or commissural fusion.6 Success rates are better if the valve is relatively more mobile and has lesser degrees of valvular and subvalvular thickening, calcification, and commissural fusion.
Mitral valve surgery (repair if possible) is indicated in patients with acceptable operative risk who have symptomatic (New York Heart Association class III or IV) moderate-to-severe mitral stenosis if percutaneous balloon mitral valvuloplasty is unavailable, in cases in which an atrial thrombus or moderate-to-severe mitral regurgitation precludes balloon valvuloplasty, or when the valve morphology is not favorable for balloon valvulo-plasty.3
Although she has moderate mitral regurgitation and poor valve morphology, our patient is a poor surgical candidate because of her advanced age, severe pulmonary hypertension, and poor functional status. Patients with moderate-to-severe mitral stenosis and class III or IV symptoms who have nonpliable, calcified valves but are not candidates for open heart surgery have a class IIb indication for percutaneous balloon mitral valvuloplasty—ie, the procedure may be considered.3
In addition, the procedure also carries a class IIb recommendation in patients with moderate-to-severe mitral stenosis and new-onset atrial fibrillation (provided that they do not have a thrombus in the left atrium or moderate-to-severe mitral regurgitation), even without symptoms.3
Percutaneous mitral valve replacement is not available in clinical practice, although this is an active area of clinical research and may be available in the future.
CASE CONTINUED: THE PATIENT UNDERGOES BALLOON VALVULOPLASTY
Transthoracic echocardiography performed 4 months later shows moderate mitral stenosis with a mean gradient of 9.0 mm Hg, a mitral valve area of 1.7 cm2, and moderate mitral regurgitation. Her right ventricular systolic pressure is estimated to be 74 mm Hg. The patient reports less dyspnea during her housework and now has New York Heart Association class II symptoms. Her treatment regimen includes warfarin (Coumadin) for atrial fibrillation, a beta-blocker to control her heart rate in atrial fibrillation and increase her left ventricular filling time, and a low-dose diuretic.
A frail 75-year-old woman with diabetes, hyperlipidemia, and a history of rheumatic fever in childhood presents to the emergency department of a community hospital with complaints of chest pressure, shortness of breath on exertion, and easy fatigability. Her shortness of breath started 6 months ago but has become much worse over the past few days.
On examination, her pulse is 110 and irregular, and she has markedly distended neck veins and evidence of pulmonary edema. She has a systolic murmur, but it is difficult to characterize due to tachycardia. Electrocardiography shows atrial fibrillation with rapid ventricular response and right axis deviation. Chest radiography shows bilateral pleural effusions.
The patient is given a diuretic, anticoagulation is started to prevent thromboembolism, and she undergoes cardioversion for the atrial fibrillation.
Transthoracic echocardiography is performed and reveals biatrial enlargement, anterior mitral valve leaflet thickening, mitral valve calcification, and moderate mitral regurgitation. Her ejection fraction is normal, but she has mild right ventricular systolic dysfunction with moderate tricuspid regurgitation and an estimated right ventricular systolic pressure of 90 mm Hg (normal range 15–30 mm Hg).
After an uneventful hospital course, she is discharged on a stable dose of a diuretic and oral anticoagulation. Despite adequate diuresis and maintenance of normal sinus rhythm, however, she continues to experience severe dyspnea, which limits her ability to perform simple tasks, such as dusting the furniture in her home. She is referred to Cleveland Clinic for further evaluation.
WHAT IS THE CAUSE OF HER SYMPTOMS?
1. What was the most likely cause of this patient’s initial acute presentation to the emergency department?
- An acute decrease in mitral valve area
- Rheumatic mitral valve stenosis
- Acute coronary syndrome
- Atrial fibrillation
Although an acute decrease in mitral valve area caused by an atrial myxoma, thrombus, or vegetation is a possibility, this patient’s symptoms gradually increased over a period of months. She also did not have signs of infection, nor did she have any signs of embolic phenomena to suggest myxoma, thrombus, or vegetation, all of which frequently present with emboli.
Given her history of rheumatic fever and her echocardiographic findings, rheumatic mitral valve stenosis is high on the list of differential diagnoses. Rheumatic mitral valve stenosis is a chronic process in which the valve area decreases at a rate of about 0.1 cm2/year.1,2
Acute coronary syndrome is a possibility in this elderly woman with multiple risk factors for coronary artery disease. An evaluation for coronary insufficiency should be considered, but the most striking finding on her initial electrocardiogram is atrial fibrillation with a rapid ventricular response.
The onset of atrial fibrillation in the setting of valvular heart disease is the most likely cause of her acute decompensation with signs and symptoms of congestive heart failure. In severe mitral stenosis, because the mitral valve orifice is narrowed, a higher left atrial pressure and longer ventricular filling time are required to maintain forward flow. Now add atrial fibrillation to this situation: the left atrium no longer contracts properly, so less blood is forced through the narrowed valve, and with the rapid heart rate the left ventricle has less time to fill. These two conditions result in elevated left atrial and pulmonary venous pressures, which in turn result in pulmonary edema and congestive heart failure. Thus, patients with mitral stenosis tolerate atrial fibrillation poorly.
WHAT IS THE NEXT STEP?
2. What would be the most appropriate next step for our patient?
- Transesophageal echocardiography
- Right heart catheterization
- An exercise treadmill stress test0
Transesophageal echocardiography is a reasonable option, as mitral stenosis is strongly suspected but transthoracic echocardiography did not reveal severe mitral valve disease. The use of transesophageal echocardiography for the diagnosis of mitral stenosis in this situation is a class IC recommendation (ie, the procedure is recommended, although very few trials have been done) in the American College of Cardiology and American Heart Association Valvular Disease guidelines.3
Right heart catheterization is the diagnostic test of choice in this situation, as the patient has evidence of biventricular heart failure and her right ventricular systolic pressure of 90 mm Hg is consistent with severe pulmonary hypertension. Right heart catheterization would help differentiate primary pulmonary hypertension causing dyspnea from pulmonary hypertension secondary to elevated left-sided pressures. It would also provide a direct hemodynamic estimate of her cardiac function.
Exercise treadmill testing. Evaluation for myocardial ischemia is a reasonable option, as our patient is elderly and has hypertension and diabetes, both of which are risk factors for coronary artery disease. Moreover, in a patient with diabetes, myocardial ischemia can present as dyspnea without typical anginal chest pain. Because of her age and severely limiting dyspnea, however, she would be unlikely to achieve an adequate heart rate during exercise treadmill testing, so this may not be the optimal type of stress test.
Although asymptomatic patients with moderate or severe mitral stenosis should undergo evaluation of exercise capacity and change in pulmonary artery pressures with exercise to determine the need for percutaneous balloon mitral valvuloplasty (see below),3 our patient is symptomatic and already has evidence of severe pulmonary hypertension on transthoracic echocardiography.
Other options for evaluating for myocardial ischemia include pharmacologic stress testing with imaging (eg, dobutamine echocardiography or adenosine nuclear imaging) or proceeding directly to coronary angiography.
CASE CONTINUED: RIGHT HEART CATHETERIZATION, CORONARY ANGIOGRAPHY
- Mean right atrial pressure 6 mm Hg (normal 2–7 mm Hg)
- Right ventricular pressure 102/6 mm Hg (consistent with severe pulmonary hypertension) (normal 15–30/1–7 mm Hg)
- Pulmonary artery pressure 102/40 mm Hg (normal 15–30/4–12 mm Hg)
- Mean pulmonary capillary wedge pressure 25 mm Hg (normal 4–12 mm Hg)
- Cardiac output 3.16 L/min (normal 4–8 L/min)
- Cardiac index 2.10 L/min/m2 (normal 2.5–4.2 L/min/m2)
- v waves are not prominent.
Because her symptoms raise concern for ischemia, coronary angiography is also performed and shows minimal, nonobstructive coronary artery disease. Her left ventricular end-diastolic pressure is 8 mm Hg (normal 5–12 mm Hg).
WHAT IS THE DIAGNOSIS?
3. What is the most likely diagnosis?
- Tricuspid stenosis
- Pulmonic stenosis
- Mitral stenosis
- Mitral regurgitation
Tricuspid stenosis would result in a higher pressure in the right atrium than in the right ventricle. In our patient, the right atrial pressure and the right ventricular diastolic pressure are both 6 mm Hg, eliminating this possibility.
Similarly, pulmonic stenosis would result in a higher pressure in the right ventricle than in the pulmonary artery. In our patient both the right ventricular systolic pressure and the pulmonary artery systolic pressures are 102 mm Hg.
Acute mitral regurgitation may result in increased wedge pressure and tall v waves (reflecting left atrial filling during ventricular systole). In chronic mitral regurgitation, however, the wedge pressure may be normal and the patient may have relatively normal-appearing v waves.4
Mitral stenosis results in a marked gradient between the pulmonary capillary wedge pressure and the left ventricular diastolic pressure in the absence of pulmonary veno-occlusive disease. This gradient can be measured by simultaneous catheterization of the right heart (to measure the wedge pressure, which is an indirect measure of left atrial pressure) and the left heart (to measure the left ventricular diastolic pressure). If the patient does not have significant mitral stenosis, the wedge pressure should be approximately equal to the left ventricular diastolic pressure. In our patient, the wedge pressure (and therefore the left atrial pressure) is 25 mm Hg, and the left ventricular end-diastolic pressure is 8 mm Hg—a difference of 17 mm Hg, consistent with significant mitral stenosis.
CASE CONTINUED: TRANSESOPHAGEAL ECHOCARDIOGRAPHY
WHAT IS THE TREATMENT?
4. Which of the following is the preferred technique for correcting mitral stenosis in this patient?
- Percutaneous balloon mitral valvuloplasty
- Mitral valve surgery
- Percutaneous mitral valve replacement
Although there are several options for mechanical treatment of mitral stenosis, percutaneous balloon mitral valvuloplasty by experienced operators is the procedure of choice for patients who have symptomatic moderate-to-severe mitral stenosis with favorable valve morphology but do not have significant mitral regurgitation or left atrial thrombus.3 The hemodynamic and symptomatic improvement that can be expected after this procedure can be predicted using several echocardiographic criteria, including valve mobility, subvalvular thickening, valve leaflet thickening, and valve leaflet calcification,5 as well as the degree of commissural calcification or commissural fusion.6 Success rates are better if the valve is relatively more mobile and has lesser degrees of valvular and subvalvular thickening, calcification, and commissural fusion.
Mitral valve surgery (repair if possible) is indicated in patients with acceptable operative risk who have symptomatic (New York Heart Association class III or IV) moderate-to-severe mitral stenosis if percutaneous balloon mitral valvuloplasty is unavailable, in cases in which an atrial thrombus or moderate-to-severe mitral regurgitation precludes balloon valvuloplasty, or when the valve morphology is not favorable for balloon valvulo-plasty.3
Although she has moderate mitral regurgitation and poor valve morphology, our patient is a poor surgical candidate because of her advanced age, severe pulmonary hypertension, and poor functional status. Patients with moderate-to-severe mitral stenosis and class III or IV symptoms who have nonpliable, calcified valves but are not candidates for open heart surgery have a class IIb indication for percutaneous balloon mitral valvuloplasty—ie, the procedure may be considered.3
In addition, the procedure also carries a class IIb recommendation in patients with moderate-to-severe mitral stenosis and new-onset atrial fibrillation (provided that they do not have a thrombus in the left atrium or moderate-to-severe mitral regurgitation), even without symptoms.3
Percutaneous mitral valve replacement is not available in clinical practice, although this is an active area of clinical research and may be available in the future.
CASE CONTINUED: THE PATIENT UNDERGOES BALLOON VALVULOPLASTY
Transthoracic echocardiography performed 4 months later shows moderate mitral stenosis with a mean gradient of 9.0 mm Hg, a mitral valve area of 1.7 cm2, and moderate mitral regurgitation. Her right ventricular systolic pressure is estimated to be 74 mm Hg. The patient reports less dyspnea during her housework and now has New York Heart Association class II symptoms. Her treatment regimen includes warfarin (Coumadin) for atrial fibrillation, a beta-blocker to control her heart rate in atrial fibrillation and increase her left ventricular filling time, and a low-dose diuretic.
- Gordon SP, Douglas PS, Come PC, et al. Two-dimensional and Doppler echocardiographic determinants of the natural history of mitral valve narrowing in patients with rheumatic mitral stenosis: implications for follow-up. J Am Coll Cardiol 1992; 19:968–973.
- Sagie A, Freitas N, Padial LR, et al. Doppler echocardiographic assessment of long-term progression of mitral stenosis in 103 patients: valve area and right heart disease. J Am Coll Cardiol 1996; 28:472–479.
- Bonow ROC, Blase A, Chatterjee K, et al. ACC/AHA 2006 Practice Guidelines for the Management of Patients With Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease) Developed in Collaboration With the Society of Cardiovascular Anesthesiologists Endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2006; 48:598–675.
- Braunwald E. The syndrome of severe mitral regurgitation with normal left atrial pressure. Circulation 1963; 27:29–35.
- Wilkins GT, Weyman AE, Abascal VM, et al. Percutaneous balloon dilatation of the mitral valve: an analysis of echocardiographic variables related to outcome and the mechanism of dilatation. Br Heart J 1988; 60:299–308.
- Cannan CR, Nishimura RA, Reeder GS, et al. Echocardiographic assessment of commissural calcium: a simple predictor of outcome after percutaneous mitral balloon valvotomy. J Am Coll Cardiol 1997; 29:175–180.
- Gordon SP, Douglas PS, Come PC, et al. Two-dimensional and Doppler echocardiographic determinants of the natural history of mitral valve narrowing in patients with rheumatic mitral stenosis: implications for follow-up. J Am Coll Cardiol 1992; 19:968–973.
- Sagie A, Freitas N, Padial LR, et al. Doppler echocardiographic assessment of long-term progression of mitral stenosis in 103 patients: valve area and right heart disease. J Am Coll Cardiol 1996; 28:472–479.
- Bonow ROC, Blase A, Chatterjee K, et al. ACC/AHA 2006 Practice Guidelines for the Management of Patients With Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease) Developed in Collaboration With the Society of Cardiovascular Anesthesiologists Endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2006; 48:598–675.
- Braunwald E. The syndrome of severe mitral regurgitation with normal left atrial pressure. Circulation 1963; 27:29–35.
- Wilkins GT, Weyman AE, Abascal VM, et al. Percutaneous balloon dilatation of the mitral valve: an analysis of echocardiographic variables related to outcome and the mechanism of dilatation. Br Heart J 1988; 60:299–308.
- Cannan CR, Nishimura RA, Reeder GS, et al. Echocardiographic assessment of commissural calcium: a simple predictor of outcome after percutaneous mitral balloon valvotomy. J Am Coll Cardiol 1997; 29:175–180.
Perioperative management of bariatric surgery patients: Focus on metabolic bone disease
A 56-year-old woman who underwent Roux-en-y bariatric surgery because of morbid obesity 6 years ago presents to her primary care physician with vague complaints of fatigue, myalgias, arthralgias, and weakness that have slowly been getting worse. Before surgery she weighed 340 pounds (154 kg), and in the first 2 years afterward she lost 160 pounds (72.5 kg). She is postmenopausal, has no history of fractures, nephrolithiasis, or thyroid disease, and does not smoke or consume alcohol. She gives herself monthly intramuscular vitamin B12 injections, and takes a multivitamin tablet, calcium carbonate 500 mg, and vitamin D 400 IU daily.
After her surgery she returned for her first two postoperative appointments, but because she was feeling well, was losing weight, and had returned to work full-time, she cancelled all subsequent appointments with the surgeon, bariatrician, and dietitian.
On physical examination, the patient’s weight is stable at 187 pounds (84.5 kg), her height is 165.1 cm, and her body mass index is 31. Her head, eyes, ears, nose, throat, heart, lungs, and abdomen are normal. Her upper legs are weak, requiring her to use her arms in rising from a chair, and she feels discomfort when the proximal muscles of her arms and legs are palpated. She has mild osteoarthritis of the hands and knees. Her neurologic examination is normal.
Pertinent laboratory data:
- Calcium 8.1 mg/dL (reference range 8.5–10.5)
- Albumin 3.7 g/dL (3.4–4.7)
- Magnesium 1.9 mg/dL (1.7–2.6)
- Phosphorus 2.7 mg/dL (2.4–4.5)
- Alkaline phosphatase 240 U/L (40–150)
- Intact parathyroid hormone 215 pg/mL (10–60)
- 25-hydroxyvitamin D < 7.0 ng/mL (31–80)
- 24-hour urine volume 2,310 mL
- Urine creatinine normal
- Urine calcium 25.4 mg/24 hours (100–300).
Dual-energy x-ray absorptiometry (DXA) data, lumbar spine:
- Bone mineral density 0.933 g/cm2
- T score –2.0
- Z score –0.8.
Left total hip:
- Bone mineral density 0.628 g/cm2
- T score –2.6
- Z score –2.4.
METABOLIC BONE DISEASE: A CASE IN POINT
This is a classic presentation of metabolic bone disease in a bariatric surgery patient lost to follow-up. Many patients have non-specific and vague symptoms for many months or years that are often incorrectly diagnosed as fibromyalgia, rheumatoid arthritis, polymyalgia rheumatica, Paget disease, or depression.1 They typically have low serum and urine calcium levels, very low or undetectable 25-hydroxyvitamin D levels, high alkaline phosphatase levels, secondary hyperparathyroidism, and a clinical picture consistent with both osteomalacia and osteoporosis.1
This case underscores the importance of monitoring nutrients and biochemical markers at baseline and on an ongoing basis to detect early indicators of malabsorption and ultimately prevent the development of metabolic bone disease and fragility fracture, with its risks of disability and even death. It also illustrates the essential role that primary care physicians play in the continuing care of these patients.
THE OBESITY-BONE CONNECTION
Although we used to think that morbid obesity protected against metabolic bone disease, in fact, vitamin D and calcium deficiencies and elevated parathyroid hormone (PTH) levels are common in extremely obese people, placing them at risk of low bone mass.2–6 More than 60% of candidates for weight-loss surgery are deficient in vitamin D,5,7 and 25% to 48% have elevated PTH levels.5,6
And that is before bariatric surgery: afterward, severely restricted oral intake and significant weight loss, coupled with a procedure that bypasses the major site of calcium absorption, place many patients at extremely high risk.5,8
After combination restrictive and malabsorptive procedures (eg, the popular Rouxen-y procedure, in which the stomach is reduced in size—”restricted”—and the proximal duodenum is bypassed so that less food is absorbed), as patients lose weight their PTH levels rise and 25-hydroxyvitamin D levels decrease, although corrected calcium levels usually remain within normal limits.3,9 Secondary hyperparathyroidism has been documented as soon as 8 weeks after bariatric surgery, and osteomalacia after gastric bypass surgery is not uncommon.1,7,10–13
Exclusively restrictive procedures such as gastric banding, formerly presumed not to alter bone metabolism, now also appear to place patients at risk of metabolic bone disease due to inadequate intake of calcium and vitamin D in the immediate postoperative period.10
Numerous reported cases further illustrate the ever-present risk of metabolic bone disease in this population if adequate supplementation of calcium and vitamin D is not given. In these cases, significant bone disease occurred from 8 weeks to 32 years after bariatric surgery, often with devastating consequences.1,4,8,11–14
Voluntary weight loss, involuntary bone loss
When overweight or obese people lose weight—whether by dieting or by bariatric surgery—they also lose bone: a voluntary loss of approximately 10% of body weight results in a loss of 1% to 2% of bone at all sites. This loss appears to vary among populations: premenopausal women younger than 45 years may be able to lose a moderate amount of weight without a significant increase in fracture risk, while a study of overweight men found that a 7% weight loss resulted in a 1% bone loss.15
The percentage of bone lost correlates strongly with how fast the weight is lost. A recent study found that losing 0.7 kg/week was more detrimental to bone than a slower loss of 0.3 kg/week, due to the activation of the calcium-PTH axis.16
After bariatric surgery, many patients rapidly lose 50 kg—some even lose 100 kg or more. This rapid weight loss, combined with severely restricted oral intake, decreased calcium absorption, and vitamin D deficiency places these patients at extremely high risk of rapidly developing metabolic bone disease.3,8,9 In one large study, metabolic bone disease developed in more than 70% of patients who underwent a malabsorptive procedure, while in a second study, markers of bone resorption were elevated as soon as 8 weeks after bariatric surgery, regardless of whether the patient underwent a malabsorptive or restrictive bariatric procedure.13 Yet another study found that 48% of patients had a statistically significant bone mineral reduction of more than 3% 12 months after undergoing gastric banding.10
ESSENTIAL NUTRIENTS FOR BONE HEALTH

Protein
Dietary protein is needed to maintain bone structure, and although there is a link between high protein intake, calciuria, and fracture risk, the potentially harmful effects appear to be ameliorated when high protein intake is coupled with adequate calcium.17–20 This fact is of particular importance after bariatric surgery because once the patient can consume enough fluids to maintain hemodynamic stability, he or she is given a relatively high-protein diet to prevent protein malnutrition.21
Inadequate protein intake also has a detrimental effect on bone; therefore, it is essential to assess postoperative protein intake.22 Rizzoli and Bonjour23 noted that markers of bone turnover were higher with a low-protein diet (0.7 g protein per kg body weight) than with a diet containing 2.1 g protein per kg. In two trials examining graded levels of protein ingestion (0.7, 0.8, 0.9, and 1.0 g protein per kg body weight), decreased calcium absorption and an acute rise in PTH were noted by day 4 of the 0.7- and 0.8-g/kg diets but not during the 0.9- or 1.0-g/kg diets.24,25 And a systematic review of protein and bone health concluded that diets containing 1.0 to 1.5 g protein/kg are best for bone health.26 This is particularly worrisome, since the current recommended dietary allowance for protein is only 0.8 g/kg, which may be insufficient to promote calcium homeostasis.26,27
Vitamin D
Vitamin D is essential for calcium absorption, stimulation of osteoblast activity, and normal bone mineralization throughout the life span.27 Dietary vitamin D is mainly absorbed by passive diffusion in the proximal and mid small intestine in a process that is highly dependent on bile salts.28–30 Dietary sources of vitamin D are clinically important because exposure to ultraviolet B radiation is often insufficient, especially in northern latitudes.27
Up to 84% of morbidly obese patients have vitamin D deficiency.2,3,5,2,29,31 The mechanism of vitamin D deficiency and secondary hyperparathyroidism in the morbidly obese remains unclear, although one study concluded they were likely due to sequestration of vitamin D in adipose tissue and subsequent limited bioavailability.32
Correction of vitamin D deficiency requires more than just an over-the-counter multivitamin, but standard multivitamins also contain vitamin A, so taking more than one tablet a day increases the risk of vitamin A excess.33 Repletion can often be safely achieved orally by giving 50,000 IU of vitamin D weekly for 8 weeks, followed by a maintenance dose of one 50,000 IU tablet every 2 weeks. If a repeat serum level shows suboptimal repletion (less than 32 ng/mL), an additional 8-week course is recommended.29 For patients who cannot tolerate or adequately absorb oral supplements, exposure to sunlight is still the best source of vitamin D and is an effective alternative.29,33
Calcium
Dietary calcium deficiency is a well-established risk factor for osteoporosis and fragility fractures. Therefore, supplemental calcium should be prescribed for patients who do not meet their defined need.34,35 Of note: a normal serum calcium level does not imply adequate calcium intake or absorption. Calcium homeostasis is tightly regulated and is maintained by a combination of gut absorption, bone resorption, and renal reabsorption. If dietary intake is inadequate, calcium is resorbed from the bone.
The duodenum is the major site of active calcium uptake, while the rest of the small intestine and the colon appear to absorb some calcium passively. When the physiologic need for calcium is increased, active transport appears to take place throughout the duodenum, the ileum, and, to a lesser degree, the jejunum and the colon.36
In the normal gastrointestinal tract, 20% to 60% of dietary calcium is absorbed.36–38 Patients who have lost absorptive surface area (eg, after Roux-en-y bariatric surgery) need to have their calcium intake optimized. However, optimal dosing based on the type of surgical procedure is currently undefined.
Judicious monitoring for compliance and adequate absorption is recommended. Some patients will stop taking their calcium supplement due to gastrointestinal side effects such as gas, bloating, or constipation. And for some patients, a standard calcium supplement may be insufficient to promote adequate calcium absorption. Measuring urinary calcium in a 24-hour sample can help in assessing the adequacy of calcium intake: abnormally low urine calcium in the presence of normal renal function suggests inadequate absorption. For patients reporting gastrointestinal side effects or those with a history of calcium oxalate renal stones, calcium citrate supplements are better tolerated, alter urine acidity, and often prevent further stone formation.
Vitamin B12 (cobalamin)
Vitamin B12 deficiency is associated with increased fracture risk, and it may be an important modifiable risk factor for osteoporosis.39–41 After surgery, malabsorption of vitamin B12 is commonly the result of altered gut function in the gastric pouch or sleeve, but malabsorption also occurs when more than 60 to 100 cm of terminal ileum has been bypassed.42
Vitamin B12 supplementation is recommended for all patients after bariatric surgery, because deficiency is common.42 Patients with relatively mild malabsorption can maintain their B12 level by taking 350 μg orally; however, many patients require lifelong subcutaneous injections.7,39,42–45
Magnesium
Magnesium appears to affect bone remodeling and strength, to have a positive association with hip bone mineral density, and to play an important role in calcium and bone metabolism.
Magnesium is absorbed in the distal small intestine by carrier-mediated and paracellular routes.46 When the distal small intestine is bypassed, magnesium deficiency occurs as a result of reduced absorption and chelation with unabsorbed fatty acids in the bowel lumen.42 Chronic hypomagnesemia impairs PTH secretion, resulting in altered calcium metabolism, hypocalcemia, and vitamin D abnormalities, further decreasing jejunal magnesium absorption.26,42,47
Few well-designed studies have investigated the effect of magnesium intake on bone health, and although there is evidence that postmenopausal women may benefit from magnesium supplementation, studies of magnesium supplementation after bariatric surgery are lacking.47,48
A prevailing misconception promoted by manufacturers of calcium-magnesium supplements and others is that magnesium is necessary for calcium absorption and efficacy. In fact, magnesium deficiency typically must be severe to impair calcium absorption. With usual dietary intake of magnesium and normal serum magnesium levels, no such relationship exists.49–53
THE ROLE OF DXA IN THE CARE OF THE BARIATRIC SURGERY PATIENT
DXA is the gold standard for measuring bone density. The results are reported as a T score and as a Z score.
The T score is the bone density in an area of interest expressed in standard deviations from the mean value of a reference database of young adults. The World Health Organization defines normal as a T score greater than or equal to –1, low bone mass (previously called osteopenia) as a score between –1 and –2.5, and osteoporosis as a score of less than or equal to –2.5. (If a fragility fracture has occurred, “established” or “severe” osteoporosis is present.) Of note: these criteria only apply to DXA of the posterior-anterior spine, femoral neck, and the proximal (33%) radius in post-menopausal women and men over the age of 50 years.54,55 The International Society of Clinical Densitometry has extended the criteria to include total hip measurements.56
The Z score should be used instead of the T score for premenopausal women and men younger than 50 years.56 The Z score is the patient’s bone mineral density expressed in standard deviations from the mean in a reference population matched for sex and age. A Z score greater than –2.0 is “within the expected range for age,” and –2.0 or lower is “below the expected range for age.” There are separate guidelines for DXA reporting in the diagnosis of metabolic bone disease in people younger than 20 years, and this topic is beyond the scope of this article.

Who should undergo DXA?
According to the International Society of Clinical Densitometry, bone density testing is indicated in the general population in women 65 years of age and older, postmenopausal women younger than 65 with risk factors, men 70 and older, adults with fragility fractures, adults taking a medication or having a disease or condition associated with low bone mass or bone loss, any patient being treated for low bone mass (to monitor the treatment effect), and any person in whom evidence of bone loss would affect treatment decisions.58
The National Osteoporosis Foundation recommends initiating therapy to reduce fracture risk in postmenopausal women with a central DXA T score below –2 in the absence of risk factors, and in women with T scores below –1.5 if one or more risk factors is present.34 Therefore, in view of the known risks, the likely need for interventions before surgery, and the ability to prevent the illness and death associated with metabolic bone disease, we recommend that all bariatric surgery patients undergo DXA at baseline as part of the preoperative evaluation.
Improvements in DXA technology
Newer DXA machines can accommodate patients weighing up to 450 pounds (the limit with older machines was 275 pounds for central measurements). In addition to measuring bone density, they also can map the distribution of fat in the body—patients with an android (apple-shaped) distribution are at higher risk of cardiovascular disease than those with a gynecoid (pear-shaped) distribution.59–63 For those patients who cannot be accommodated on a DXA table, DXA of the forearm can be used to assess bone density and fracture risk.
How often should DXA be repeated?
The estimated monitoring time interval is derived from the statistically defined least significant change divided by the anticipated change in bone density over time.64 When estimating the monitoring time interval for changes in body composition, the rate of weight loss and the psychological impact on the patient must be taken into consideration.
In general, DXA testing more frequently than every 2 years remains controversial unless one is initiating, monitoring, or changing therapy or monitoring conditions associated with rapid bone loss such as glucocorticoid therapy. In the bariatric surgery population, however, there is convincing evidence that significant changes may be detected after 12 months that would influence clinical decisions, particularly in the year immediately after surgery.8,9,13,21,27,65
Anabolic and antiresorptive bone drugs
Prescribed medications for the prevention and treatment of osteoporosis should also be an integral part of the treatment plan for at-risk morbidly obese patients. But the decision to prescribe an antiresorptive or bone-forming medication must take into consideration the patient’s risk-benefit profile, including the likelihood of gastrointestinal side effects and his or her ability and willingness to follow specific dosing instructions. Intravenous preparations are now available for patients who cannot absorb or tolerate oral antiresorptive medications. However, specific recommendations about the use of anabolic or antiresorptive bone medications in perioperative bariatric patients have yet to be elucidated.
RECOMMENDATIONS
Although a variety of recommendations have been published, there are no established guidelines for perioperative screening, risk stratification, or management of metabolic bone disease in bariatric surgery patients.7,44,65–67 And the literature remains inconclusive on key issues such as when to start supplements, which biochemical indices should be checked before surgery, whether baseline and annual DXA should be done, and whether antiresorptive agents such as bisphosphonates should be used prophylactically during rapid weight loss.
However, numerous studies and case reports cited here and elsewhere further underscore the ever-present risk of metabolic bone disease in this patient population, and the need for meticulous perioperative and long-term monitoring.44,65–67

With these caveats in mind, we offer our recommendations (Table 3).
Preoperative assessment
We recommend obtaining baseline biochemical indices, including albumin, 25-hydroxy-vitamin D, calcium, magnesium, phosphorus, alkaline phosphatase, folate, vitamin B12, thyroid-stimulating hormone, and PTH levels, and DXA in all bariatric surgery candidates. These indices should be used to assess for primary and secondary metabolic bone disease, to enable prompt presurgical interventions, and to guide the clinician in selecting appropriate postoperative interventions and surveillance.
We recommend starting a multivitamin with minerals at the first preoperative visit. A calcium supplement that provides calcium and vitamin D appropriate to the patient’s age and sex is also recommended until surgery. After surgery, and while rapid weight loss is occurring, a minimum of 1,800 mg of calcium and 800 to 1,000 IU of vitamin D is recommended, keeping in mind that the required level of supplemental vitamin D during periods of rapid weight loss remains unclear.68,69
However, before prescribing supplementation, one should thoroughly review the patient’s nutrition history, including the use of homeopathic medications, herbal preparations, and supplements. Many over-the-counter and over-the-Internet supplements are touted as being good for bone health, and some may indeed be beneficial, but others can be detrimental and need to be discontinued.27,47 Furthermore, in a patient with a severely restricted stomach capacity, it is important to ensure that less efficacious supplements do not compromise the intake of essential fluids, protein, and prescribed medications.
Immediate postoperative period
Hospitalization and surgery result in nutrient deficiencies. In bariatric surgery patients, particularly those who have preoperative nutritional deficiencies, repletion in the immediate postoperative period is believed to be of benefit. Therefore, in the immediate postoperative period we recommend infusing a standard-dose multivitamin with minerals daily along with adequate intravenous hydration until the patient can resume oral feeding. Once the patient can tolerate liquids, presurgical supplementation needs to be resumed, preferably in a liquid or chewable form to facilitate tolerance and absorption.
Short-term and long-term follow-up
Follow-up visits with a bariatric specialist should start 4 weeks after surgery and should be repeated every 3 to 4 months for the first year. If the patient continues to do well, annual visits may be sufficient thereafter. Compliance with supplements should be checked as indicated, as should nutritional indices. DXA should be repeated every 1 to 2 years, depending on the patient’s risk profile.
WHAT SHOULD BE DONE FOR OUR PATIENT?
Initial treatment for the patient described at the beginning of this article should include vitamin D repletion with cholecalciferol 50,000 IU, calcium supplements of at least 1,200 mg daily, and addressing of modifiable risk factors for fracture, including the risk of falling due to her proximal weakness. Her laboratory studies should be repeated in 6 to 12 weeks, with calcium and vitamin D supplement dosages adjusted on the basis of her response. Once the serum calcium level has normalized, we would consider the use of a bisphosphonate. DXA should be repeated in 1 to 2 years to monitor the effectiveness of the prescribed interventions.
- De Prisco C, Levine SN. Metabolic bone disease after gastric bypass surgery for obesity. Am J Med Sci 2005; 329:57–61.
- Ybarra J, Sanchez-Harnandez J, Gich I, et al. Unchanged hypovitaminosis D and secondary hyperparathyroidism in morbid obesity after bariatric surgery. Obes Surg 2005; 15:330–335.
- Hamoui N, Anthone G, Crookes F. Calcium metabolism in the morbidly obese. Obes Surg 2004; 14:9–12.
- Parikh SJ, Edelman M, Uwaifo GI, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab 2004; 89:1196–1199.
- Carlin AM, Rao DS, Meslemani AM, et al. Prevalence of vitamin D depletion among morbidly obese patients seeking gastric bypass surgery. Surg Obes Rel Dis 2006; 2:98–103.
- Puzziferri N, Blankenship J, Wolfe BM. Surgical treatment of obesity. Endocrine 2006; 29:11–19.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Haria DM, Sibonga JD, Taylor HC. Hypocalcemia, hypovitaminosis D osteopathy, osteopenia, and secondary hyperparathyroidism 32 years after jejunoileal bypass. Endocr Pract 2005; 11:335–340.
- Newbery L, Dolan K, Hatzifotis M, et al. Calcium and vitamin D depletion and elevated parathyroid hormone following biliopancreatic diversion. Obes Surg 2003; 13:893–895.
- Pugnale N, Guisti V, Suter M, et al. Bone metabolism and risk of secondary hyperparathyroidism 12 months after gastric banding in obese pre-menopausal women. Int J Obesity 2003; 27:110–116.
- Goldner WS, O’Dorisio TM, Dillon JS, Mason EE. Severe metabolic bone disease as a long-term complication of obesity surgery. Obes Surg 2002; 12:685–692.
- Atreja A, Abacan C, Licata A. A 51-year-old woman with debilitating cramps 12 years after bariatric surgery. Cleve Clin J Med 2003; 70:417–426.
- Collazo-Clavell ML, Jimenez A, Hodgson SF, et al. Osteomalacia after Rouxen-Y gastric bypass. Endocr Pract 2004; 10:287–288.
- Dennisson E. Osteoporosis. In:Pinchera A, Bertagna X, Fischer J, editors. Endocrinology and Metabolism. London: McGraw-Hill International (UK) Ltd; 2001:271–282.
- Shapses SA, Cifuentes M. Body weight/composition and weight change: effects on bone health. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health New Jersey: Humana Press; 2004:549–573.
- Shapses SA, Cifuentes M, Sherrell R, et al. Rate of weight loss influences calcium absorption. J Bone Miner Res 2002; 17:S471.
- Munger RG, Cerhan JR, Chiu BC. Prospective study of dietary protein intake and risk of hip fracture in postmenopausal women. Am J Clin Nutr 1999; 69:147–152.
- Weikert C, Walter D, Hoffmann K, et al. The relation between dietary protein, calcium and bone health in women: results from the EPIC-Potsdam cohort. Ann Nutr Metab 2005; 49:312–318.
- Whiting SJ, Boyle JL, Thompson A. Dietary protein, phosphorus and potassium are beneficial to bone mineral density in adult men consuming adequate dietary calcium. J Am Col Nutr 2002; 21:402–409.
- Teegarden D, Lyle RM, McCabe GP, et al. Dietary calcium, protein, and phosphorus are related to bone mineral density and content in young women. Am J Clin Nutr 1998; 68:749–754.
- Bloomberg RD, Fleishman A, Nalle JE, et al. Nutritional deficiencies following bariatric surgery: what have we learned? Obes Surg 2005; 15:145–154.
- Greenspan SL, Resnick NM. Geriatric endocrinology. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7 New York: McGraw-Hill; 2004:842–866.
- Rizzoli R, Bonjour JP. Dietary protein and bone health. J Bone Miner Res 2004; 19:527–531.
- Kerstetter J, Svastisalee C, Caseria D, et al. A threshold for low-protein-diet-induced elevations in parathyroid hormone. Am J Clin Nutr 2000; 72:168–173.
- Giannini S, Nobile M, Sartori L, et al. Acute effects of moderate dietary protein restriction in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Am J Clin Nutr 1999; 69:267–271.
- Ilich JZ, Kerstetter JE. Nutrition in bone health revisited: a story beyond calcium. J Am Coll Nutr 2000; 19:715–737.
- Williams S, Seidner D. Metabolic bone disease in gastrointestinal illness. Gastroenterol Clin North Am 2007; 36 1:161–190.
- Shoback D, Marcus R, Bilke D. Metabolic bone disease. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7. New York: McGraw-Hill; 2004:295–361.
- Holick MF. Vitamin D. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:329–346.
- Rosen CJ. Vitamin D and bone health in adults and the elderly. In:Holick MF. Vitamin D: Physiology, Molecular Biology, and Clinical Applications. New Jersey: Humana Press; 1999:287–306.
- Buffington C, Walker B, Cowan GS, et al. Vitamin D deficiency in the morbidly obese. Obes Surg 1993; 3:421–424.
- Wortsman J, Matsuoka L, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 2000; 72:690–693.
- Holick MF, Garabedian M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:106–114.
- National Osteoporosis Foundation. Physician’s Guide to Prevention and Treatment of Osteoporosis. http://www.nof.org/physguide. Washington D.C. 1998.
- National Academy of Sciences Executive Summary: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington DC. 1997.
- Favus MJ, Bushinsky DA, Lemann J. Regulation of calcium, magnesium and phosphate metabolism. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:76–83.
- Weaver CM, Heaney RP. Calcium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins, 1999:141–155.
- Gueguen L, Oiubtukkart A. The bioavailability of dietary calcium. J Am Coll Nutr 2000; 19:119S–136S.
- Tucker KL, Hannan MT, Qiao N, et al. Low plasma vitamin B12 is associated with lower BMD: the Framingham osteoporosis study. J Bone Miner Res 2005; 20:152–158.
- Goerss JB, Kim CH, Atkinson EJ, et al. Risk of fractures in patients with pernicious anemia. J Bone Miner Res 1992; 7:573–579.
- Eastell R, Vieira NE, Yergey AL, et al. Pernicious anemia is a risk factor for osteoporosis. Clin Sci 1992; 82:681–685.
- Nightingale JMD, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006; 55:1–12.
- Rhode BM, Tamin H, Gilfix BM, et al. Treatment of vitamin B12 deficiency after gastric surgery for severe obesity. Obes Surg 1995; 5:154–158.
- Brethauer SA, Chand C, Schauer PR. Risks and benefits of bariatric surgery: current evidence. Cleve Clin J Med 2006; 73:993–1007.
- Weir DG, Scott JW. Vitamin B12 “cobalamin”. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:447–458.
- Shils ME. Magnesium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:169–192.
- National Osteoporosis Foundation. Osteoporosis Clinical Updates: Over-the-counter products & osteoporosis: Case discussions. 2002; Vol IIIIssue 2. Washington DC.
- Stendig-Lindenberg G, Tepper R, Leicher I. Trabecular bone density in a two year controlled trial of personal magnesium in osteoporosis. Magnes Res 1993:155–163.
- Heaney RP. Sodium, potassium, phosphorus, and magnesium. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health. New Jersey: Humana Press; 2004:327–344.
- Spencer H, Fuller H, Norris C, et al. Effect of magnesium on the intestinal absorption of calcium in man. J Am Coll Nutr 1994; 13:483–492.
- Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327:1637–1642.
- Dawson-Hughes B, Harris SS, Krall EA, et al. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age and older. N Engl J Med 1997; 337:670–676.
- Rude RK, Olerich M. Magnesium deficiency: possible role in osteoporosis associated with gluten-sensitive enteropathy. Osteoporos Int 1996; 6:453–461.
- Report of a WHO Study Group. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. 1994; 843:1–129.
- Kanis JA, Melton LJ, Christiansen C, et al. The diagnosis of osteoporosis. J Bone Miner Res 1994; 9:1137–1141.
- Official Positions of the International Society for Clinical Densitometry. West Hartford, CT. September, 2005. http://www.iscd.org/Visitors/positions/OfficialPositionsText.cfm.
- Licata A. Diagnosing primary osteoporosis: It’s more than a T score. Cleve Clin J Med 2006; 73:473–476.
- Writing group for the ISCD Position Development Conference. Indications and reporting for dual-energy x-ray absorptiometry. J Clin Densitom 2004; 7:37–44.
- Park YW, Heymsfield SB, Gallagher D. Are dual-energy X-ray absorptiometry regional estimates associated with visceral adipose tissue mass? Int J Obes Relat Metab Disord 2002; 26:978–983.
- Glickman SG, Marn CS, Supiano MA, et al. Validity and reliability of dual-energy x-ray absorptiometry for the assessment of abdominal adiposity. J Appl Physiol 2004; 97:509–514.
- Hull HR, Hester CN, Fields DA. The effect of the holiday season on body weight and composition in college students. Nutr Metab (Lond) 2006; 3:44–51.
- Aubertin-Leheudre M, Goulet EDB, Khalil A, et al. Effect of sarcopenia on cardiovascular disease risk factors in obese post-menopausal women. Obesity 2006; 14:2277–2283.
- Lubrano C, Cornoldi A, Pili M, et al. Reduction of risk factors for cardiovascular diseases in morbid-obese patients following biliary-intestinal bypass: 3 years’ follow-up. Int J Obes Relat Metab Disord 2004; 28:1600–1606.
- Bonnick SL, Johnston CC, Kleerekoper M, et al. Importance of precision in bone density measurements. J Clin Densitom 2001; 4:105–110.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Hensrud DD, McMahon MM. Bariatric surgery in adults with extreme (not morbid) obesity. Mayo Clin Proc 2006; 81 suppl:S3–S4.
- McGlinch BP, Que FG, Nelson JL, et al. Perioperative care of patients undergoing bariatric surgery. Mayo Clin Proc 2006; 81 10, suppl:S25–S33.
- Ricci TA, Chowdhury HA, Heymsfield SB, et al. Calcium supplementation suppresses bone turnover during weight reduction in postmenopausal women. J Bone Miner Res 1998; 13:1045–1050.
- Jensen LB, Kollerup G, Quaade F, et al. Bone mineral changes in obese women during moderate weight loss with and without calcium supplementation. J Bone Miner Res 2001; 16:141–147.
A 56-year-old woman who underwent Roux-en-y bariatric surgery because of morbid obesity 6 years ago presents to her primary care physician with vague complaints of fatigue, myalgias, arthralgias, and weakness that have slowly been getting worse. Before surgery she weighed 340 pounds (154 kg), and in the first 2 years afterward she lost 160 pounds (72.5 kg). She is postmenopausal, has no history of fractures, nephrolithiasis, or thyroid disease, and does not smoke or consume alcohol. She gives herself monthly intramuscular vitamin B12 injections, and takes a multivitamin tablet, calcium carbonate 500 mg, and vitamin D 400 IU daily.
After her surgery she returned for her first two postoperative appointments, but because she was feeling well, was losing weight, and had returned to work full-time, she cancelled all subsequent appointments with the surgeon, bariatrician, and dietitian.
On physical examination, the patient’s weight is stable at 187 pounds (84.5 kg), her height is 165.1 cm, and her body mass index is 31. Her head, eyes, ears, nose, throat, heart, lungs, and abdomen are normal. Her upper legs are weak, requiring her to use her arms in rising from a chair, and she feels discomfort when the proximal muscles of her arms and legs are palpated. She has mild osteoarthritis of the hands and knees. Her neurologic examination is normal.
Pertinent laboratory data:
- Calcium 8.1 mg/dL (reference range 8.5–10.5)
- Albumin 3.7 g/dL (3.4–4.7)
- Magnesium 1.9 mg/dL (1.7–2.6)
- Phosphorus 2.7 mg/dL (2.4–4.5)
- Alkaline phosphatase 240 U/L (40–150)
- Intact parathyroid hormone 215 pg/mL (10–60)
- 25-hydroxyvitamin D < 7.0 ng/mL (31–80)
- 24-hour urine volume 2,310 mL
- Urine creatinine normal
- Urine calcium 25.4 mg/24 hours (100–300).
Dual-energy x-ray absorptiometry (DXA) data, lumbar spine:
- Bone mineral density 0.933 g/cm2
- T score –2.0
- Z score –0.8.
Left total hip:
- Bone mineral density 0.628 g/cm2
- T score –2.6
- Z score –2.4.
METABOLIC BONE DISEASE: A CASE IN POINT
This is a classic presentation of metabolic bone disease in a bariatric surgery patient lost to follow-up. Many patients have non-specific and vague symptoms for many months or years that are often incorrectly diagnosed as fibromyalgia, rheumatoid arthritis, polymyalgia rheumatica, Paget disease, or depression.1 They typically have low serum and urine calcium levels, very low or undetectable 25-hydroxyvitamin D levels, high alkaline phosphatase levels, secondary hyperparathyroidism, and a clinical picture consistent with both osteomalacia and osteoporosis.1
This case underscores the importance of monitoring nutrients and biochemical markers at baseline and on an ongoing basis to detect early indicators of malabsorption and ultimately prevent the development of metabolic bone disease and fragility fracture, with its risks of disability and even death. It also illustrates the essential role that primary care physicians play in the continuing care of these patients.
THE OBESITY-BONE CONNECTION
Although we used to think that morbid obesity protected against metabolic bone disease, in fact, vitamin D and calcium deficiencies and elevated parathyroid hormone (PTH) levels are common in extremely obese people, placing them at risk of low bone mass.2–6 More than 60% of candidates for weight-loss surgery are deficient in vitamin D,5,7 and 25% to 48% have elevated PTH levels.5,6
And that is before bariatric surgery: afterward, severely restricted oral intake and significant weight loss, coupled with a procedure that bypasses the major site of calcium absorption, place many patients at extremely high risk.5,8
After combination restrictive and malabsorptive procedures (eg, the popular Rouxen-y procedure, in which the stomach is reduced in size—”restricted”—and the proximal duodenum is bypassed so that less food is absorbed), as patients lose weight their PTH levels rise and 25-hydroxyvitamin D levels decrease, although corrected calcium levels usually remain within normal limits.3,9 Secondary hyperparathyroidism has been documented as soon as 8 weeks after bariatric surgery, and osteomalacia after gastric bypass surgery is not uncommon.1,7,10–13
Exclusively restrictive procedures such as gastric banding, formerly presumed not to alter bone metabolism, now also appear to place patients at risk of metabolic bone disease due to inadequate intake of calcium and vitamin D in the immediate postoperative period.10
Numerous reported cases further illustrate the ever-present risk of metabolic bone disease in this population if adequate supplementation of calcium and vitamin D is not given. In these cases, significant bone disease occurred from 8 weeks to 32 years after bariatric surgery, often with devastating consequences.1,4,8,11–14
Voluntary weight loss, involuntary bone loss
When overweight or obese people lose weight—whether by dieting or by bariatric surgery—they also lose bone: a voluntary loss of approximately 10% of body weight results in a loss of 1% to 2% of bone at all sites. This loss appears to vary among populations: premenopausal women younger than 45 years may be able to lose a moderate amount of weight without a significant increase in fracture risk, while a study of overweight men found that a 7% weight loss resulted in a 1% bone loss.15
The percentage of bone lost correlates strongly with how fast the weight is lost. A recent study found that losing 0.7 kg/week was more detrimental to bone than a slower loss of 0.3 kg/week, due to the activation of the calcium-PTH axis.16
After bariatric surgery, many patients rapidly lose 50 kg—some even lose 100 kg or more. This rapid weight loss, combined with severely restricted oral intake, decreased calcium absorption, and vitamin D deficiency places these patients at extremely high risk of rapidly developing metabolic bone disease.3,8,9 In one large study, metabolic bone disease developed in more than 70% of patients who underwent a malabsorptive procedure, while in a second study, markers of bone resorption were elevated as soon as 8 weeks after bariatric surgery, regardless of whether the patient underwent a malabsorptive or restrictive bariatric procedure.13 Yet another study found that 48% of patients had a statistically significant bone mineral reduction of more than 3% 12 months after undergoing gastric banding.10
ESSENTIAL NUTRIENTS FOR BONE HEALTH
Protein
Dietary protein is needed to maintain bone structure, and although there is a link between high protein intake, calciuria, and fracture risk, the potentially harmful effects appear to be ameliorated when high protein intake is coupled with adequate calcium.17–20 This fact is of particular importance after bariatric surgery because once the patient can consume enough fluids to maintain hemodynamic stability, he or she is given a relatively high-protein diet to prevent protein malnutrition.21
Inadequate protein intake also has a detrimental effect on bone; therefore, it is essential to assess postoperative protein intake.22 Rizzoli and Bonjour23 noted that markers of bone turnover were higher with a low-protein diet (0.7 g protein per kg body weight) than with a diet containing 2.1 g protein per kg. In two trials examining graded levels of protein ingestion (0.7, 0.8, 0.9, and 1.0 g protein per kg body weight), decreased calcium absorption and an acute rise in PTH were noted by day 4 of the 0.7- and 0.8-g/kg diets but not during the 0.9- or 1.0-g/kg diets.24,25 And a systematic review of protein and bone health concluded that diets containing 1.0 to 1.5 g protein/kg are best for bone health.26 This is particularly worrisome, since the current recommended dietary allowance for protein is only 0.8 g/kg, which may be insufficient to promote calcium homeostasis.26,27
Vitamin D
Vitamin D is essential for calcium absorption, stimulation of osteoblast activity, and normal bone mineralization throughout the life span.27 Dietary vitamin D is mainly absorbed by passive diffusion in the proximal and mid small intestine in a process that is highly dependent on bile salts.28–30 Dietary sources of vitamin D are clinically important because exposure to ultraviolet B radiation is often insufficient, especially in northern latitudes.27
Up to 84% of morbidly obese patients have vitamin D deficiency.2,3,5,2,29,31 The mechanism of vitamin D deficiency and secondary hyperparathyroidism in the morbidly obese remains unclear, although one study concluded they were likely due to sequestration of vitamin D in adipose tissue and subsequent limited bioavailability.32
Correction of vitamin D deficiency requires more than just an over-the-counter multivitamin, but standard multivitamins also contain vitamin A, so taking more than one tablet a day increases the risk of vitamin A excess.33 Repletion can often be safely achieved orally by giving 50,000 IU of vitamin D weekly for 8 weeks, followed by a maintenance dose of one 50,000 IU tablet every 2 weeks. If a repeat serum level shows suboptimal repletion (less than 32 ng/mL), an additional 8-week course is recommended.29 For patients who cannot tolerate or adequately absorb oral supplements, exposure to sunlight is still the best source of vitamin D and is an effective alternative.29,33
Calcium
Dietary calcium deficiency is a well-established risk factor for osteoporosis and fragility fractures. Therefore, supplemental calcium should be prescribed for patients who do not meet their defined need.34,35 Of note: a normal serum calcium level does not imply adequate calcium intake or absorption. Calcium homeostasis is tightly regulated and is maintained by a combination of gut absorption, bone resorption, and renal reabsorption. If dietary intake is inadequate, calcium is resorbed from the bone.
The duodenum is the major site of active calcium uptake, while the rest of the small intestine and the colon appear to absorb some calcium passively. When the physiologic need for calcium is increased, active transport appears to take place throughout the duodenum, the ileum, and, to a lesser degree, the jejunum and the colon.36
In the normal gastrointestinal tract, 20% to 60% of dietary calcium is absorbed.36–38 Patients who have lost absorptive surface area (eg, after Roux-en-y bariatric surgery) need to have their calcium intake optimized. However, optimal dosing based on the type of surgical procedure is currently undefined.
Judicious monitoring for compliance and adequate absorption is recommended. Some patients will stop taking their calcium supplement due to gastrointestinal side effects such as gas, bloating, or constipation. And for some patients, a standard calcium supplement may be insufficient to promote adequate calcium absorption. Measuring urinary calcium in a 24-hour sample can help in assessing the adequacy of calcium intake: abnormally low urine calcium in the presence of normal renal function suggests inadequate absorption. For patients reporting gastrointestinal side effects or those with a history of calcium oxalate renal stones, calcium citrate supplements are better tolerated, alter urine acidity, and often prevent further stone formation.
Vitamin B12 (cobalamin)
Vitamin B12 deficiency is associated with increased fracture risk, and it may be an important modifiable risk factor for osteoporosis.39–41 After surgery, malabsorption of vitamin B12 is commonly the result of altered gut function in the gastric pouch or sleeve, but malabsorption also occurs when more than 60 to 100 cm of terminal ileum has been bypassed.42
Vitamin B12 supplementation is recommended for all patients after bariatric surgery, because deficiency is common.42 Patients with relatively mild malabsorption can maintain their B12 level by taking 350 μg orally; however, many patients require lifelong subcutaneous injections.7,39,42–45
Magnesium
Magnesium appears to affect bone remodeling and strength, to have a positive association with hip bone mineral density, and to play an important role in calcium and bone metabolism.
Magnesium is absorbed in the distal small intestine by carrier-mediated and paracellular routes.46 When the distal small intestine is bypassed, magnesium deficiency occurs as a result of reduced absorption and chelation with unabsorbed fatty acids in the bowel lumen.42 Chronic hypomagnesemia impairs PTH secretion, resulting in altered calcium metabolism, hypocalcemia, and vitamin D abnormalities, further decreasing jejunal magnesium absorption.26,42,47
Few well-designed studies have investigated the effect of magnesium intake on bone health, and although there is evidence that postmenopausal women may benefit from magnesium supplementation, studies of magnesium supplementation after bariatric surgery are lacking.47,48
A prevailing misconception promoted by manufacturers of calcium-magnesium supplements and others is that magnesium is necessary for calcium absorption and efficacy. In fact, magnesium deficiency typically must be severe to impair calcium absorption. With usual dietary intake of magnesium and normal serum magnesium levels, no such relationship exists.49–53
THE ROLE OF DXA IN THE CARE OF THE BARIATRIC SURGERY PATIENT
DXA is the gold standard for measuring bone density. The results are reported as a T score and as a Z score.
The T score is the bone density in an area of interest expressed in standard deviations from the mean value of a reference database of young adults. The World Health Organization defines normal as a T score greater than or equal to –1, low bone mass (previously called osteopenia) as a score between –1 and –2.5, and osteoporosis as a score of less than or equal to –2.5. (If a fragility fracture has occurred, “established” or “severe” osteoporosis is present.) Of note: these criteria only apply to DXA of the posterior-anterior spine, femoral neck, and the proximal (33%) radius in post-menopausal women and men over the age of 50 years.54,55 The International Society of Clinical Densitometry has extended the criteria to include total hip measurements.56
The Z score should be used instead of the T score for premenopausal women and men younger than 50 years.56 The Z score is the patient’s bone mineral density expressed in standard deviations from the mean in a reference population matched for sex and age. A Z score greater than –2.0 is “within the expected range for age,” and –2.0 or lower is “below the expected range for age.” There are separate guidelines for DXA reporting in the diagnosis of metabolic bone disease in people younger than 20 years, and this topic is beyond the scope of this article.
Who should undergo DXA?
According to the International Society of Clinical Densitometry, bone density testing is indicated in the general population in women 65 years of age and older, postmenopausal women younger than 65 with risk factors, men 70 and older, adults with fragility fractures, adults taking a medication or having a disease or condition associated with low bone mass or bone loss, any patient being treated for low bone mass (to monitor the treatment effect), and any person in whom evidence of bone loss would affect treatment decisions.58
The National Osteoporosis Foundation recommends initiating therapy to reduce fracture risk in postmenopausal women with a central DXA T score below –2 in the absence of risk factors, and in women with T scores below –1.5 if one or more risk factors is present.34 Therefore, in view of the known risks, the likely need for interventions before surgery, and the ability to prevent the illness and death associated with metabolic bone disease, we recommend that all bariatric surgery patients undergo DXA at baseline as part of the preoperative evaluation.
Improvements in DXA technology
Newer DXA machines can accommodate patients weighing up to 450 pounds (the limit with older machines was 275 pounds for central measurements). In addition to measuring bone density, they also can map the distribution of fat in the body—patients with an android (apple-shaped) distribution are at higher risk of cardiovascular disease than those with a gynecoid (pear-shaped) distribution.59–63 For those patients who cannot be accommodated on a DXA table, DXA of the forearm can be used to assess bone density and fracture risk.
How often should DXA be repeated?
The estimated monitoring time interval is derived from the statistically defined least significant change divided by the anticipated change in bone density over time.64 When estimating the monitoring time interval for changes in body composition, the rate of weight loss and the psychological impact on the patient must be taken into consideration.
In general, DXA testing more frequently than every 2 years remains controversial unless one is initiating, monitoring, or changing therapy or monitoring conditions associated with rapid bone loss such as glucocorticoid therapy. In the bariatric surgery population, however, there is convincing evidence that significant changes may be detected after 12 months that would influence clinical decisions, particularly in the year immediately after surgery.8,9,13,21,27,65
Anabolic and antiresorptive bone drugs
Prescribed medications for the prevention and treatment of osteoporosis should also be an integral part of the treatment plan for at-risk morbidly obese patients. But the decision to prescribe an antiresorptive or bone-forming medication must take into consideration the patient’s risk-benefit profile, including the likelihood of gastrointestinal side effects and his or her ability and willingness to follow specific dosing instructions. Intravenous preparations are now available for patients who cannot absorb or tolerate oral antiresorptive medications. However, specific recommendations about the use of anabolic or antiresorptive bone medications in perioperative bariatric patients have yet to be elucidated.
RECOMMENDATIONS
Although a variety of recommendations have been published, there are no established guidelines for perioperative screening, risk stratification, or management of metabolic bone disease in bariatric surgery patients.7,44,65–67 And the literature remains inconclusive on key issues such as when to start supplements, which biochemical indices should be checked before surgery, whether baseline and annual DXA should be done, and whether antiresorptive agents such as bisphosphonates should be used prophylactically during rapid weight loss.
However, numerous studies and case reports cited here and elsewhere further underscore the ever-present risk of metabolic bone disease in this patient population, and the need for meticulous perioperative and long-term monitoring.44,65–67
With these caveats in mind, we offer our recommendations (Table 3).
Preoperative assessment
We recommend obtaining baseline biochemical indices, including albumin, 25-hydroxy-vitamin D, calcium, magnesium, phosphorus, alkaline phosphatase, folate, vitamin B12, thyroid-stimulating hormone, and PTH levels, and DXA in all bariatric surgery candidates. These indices should be used to assess for primary and secondary metabolic bone disease, to enable prompt presurgical interventions, and to guide the clinician in selecting appropriate postoperative interventions and surveillance.
We recommend starting a multivitamin with minerals at the first preoperative visit. A calcium supplement that provides calcium and vitamin D appropriate to the patient’s age and sex is also recommended until surgery. After surgery, and while rapid weight loss is occurring, a minimum of 1,800 mg of calcium and 800 to 1,000 IU of vitamin D is recommended, keeping in mind that the required level of supplemental vitamin D during periods of rapid weight loss remains unclear.68,69
However, before prescribing supplementation, one should thoroughly review the patient’s nutrition history, including the use of homeopathic medications, herbal preparations, and supplements. Many over-the-counter and over-the-Internet supplements are touted as being good for bone health, and some may indeed be beneficial, but others can be detrimental and need to be discontinued.27,47 Furthermore, in a patient with a severely restricted stomach capacity, it is important to ensure that less efficacious supplements do not compromise the intake of essential fluids, protein, and prescribed medications.
Immediate postoperative period
Hospitalization and surgery result in nutrient deficiencies. In bariatric surgery patients, particularly those who have preoperative nutritional deficiencies, repletion in the immediate postoperative period is believed to be of benefit. Therefore, in the immediate postoperative period we recommend infusing a standard-dose multivitamin with minerals daily along with adequate intravenous hydration until the patient can resume oral feeding. Once the patient can tolerate liquids, presurgical supplementation needs to be resumed, preferably in a liquid or chewable form to facilitate tolerance and absorption.
Short-term and long-term follow-up
Follow-up visits with a bariatric specialist should start 4 weeks after surgery and should be repeated every 3 to 4 months for the first year. If the patient continues to do well, annual visits may be sufficient thereafter. Compliance with supplements should be checked as indicated, as should nutritional indices. DXA should be repeated every 1 to 2 years, depending on the patient’s risk profile.
WHAT SHOULD BE DONE FOR OUR PATIENT?
Initial treatment for the patient described at the beginning of this article should include vitamin D repletion with cholecalciferol 50,000 IU, calcium supplements of at least 1,200 mg daily, and addressing of modifiable risk factors for fracture, including the risk of falling due to her proximal weakness. Her laboratory studies should be repeated in 6 to 12 weeks, with calcium and vitamin D supplement dosages adjusted on the basis of her response. Once the serum calcium level has normalized, we would consider the use of a bisphosphonate. DXA should be repeated in 1 to 2 years to monitor the effectiveness of the prescribed interventions.
A 56-year-old woman who underwent Roux-en-y bariatric surgery because of morbid obesity 6 years ago presents to her primary care physician with vague complaints of fatigue, myalgias, arthralgias, and weakness that have slowly been getting worse. Before surgery she weighed 340 pounds (154 kg), and in the first 2 years afterward she lost 160 pounds (72.5 kg). She is postmenopausal, has no history of fractures, nephrolithiasis, or thyroid disease, and does not smoke or consume alcohol. She gives herself monthly intramuscular vitamin B12 injections, and takes a multivitamin tablet, calcium carbonate 500 mg, and vitamin D 400 IU daily.
After her surgery she returned for her first two postoperative appointments, but because she was feeling well, was losing weight, and had returned to work full-time, she cancelled all subsequent appointments with the surgeon, bariatrician, and dietitian.
On physical examination, the patient’s weight is stable at 187 pounds (84.5 kg), her height is 165.1 cm, and her body mass index is 31. Her head, eyes, ears, nose, throat, heart, lungs, and abdomen are normal. Her upper legs are weak, requiring her to use her arms in rising from a chair, and she feels discomfort when the proximal muscles of her arms and legs are palpated. She has mild osteoarthritis of the hands and knees. Her neurologic examination is normal.
Pertinent laboratory data:
- Calcium 8.1 mg/dL (reference range 8.5–10.5)
- Albumin 3.7 g/dL (3.4–4.7)
- Magnesium 1.9 mg/dL (1.7–2.6)
- Phosphorus 2.7 mg/dL (2.4–4.5)
- Alkaline phosphatase 240 U/L (40–150)
- Intact parathyroid hormone 215 pg/mL (10–60)
- 25-hydroxyvitamin D < 7.0 ng/mL (31–80)
- 24-hour urine volume 2,310 mL
- Urine creatinine normal
- Urine calcium 25.4 mg/24 hours (100–300).
Dual-energy x-ray absorptiometry (DXA) data, lumbar spine:
- Bone mineral density 0.933 g/cm2
- T score –2.0
- Z score –0.8.
Left total hip:
- Bone mineral density 0.628 g/cm2
- T score –2.6
- Z score –2.4.
METABOLIC BONE DISEASE: A CASE IN POINT
This is a classic presentation of metabolic bone disease in a bariatric surgery patient lost to follow-up. Many patients have non-specific and vague symptoms for many months or years that are often incorrectly diagnosed as fibromyalgia, rheumatoid arthritis, polymyalgia rheumatica, Paget disease, or depression.1 They typically have low serum and urine calcium levels, very low or undetectable 25-hydroxyvitamin D levels, high alkaline phosphatase levels, secondary hyperparathyroidism, and a clinical picture consistent with both osteomalacia and osteoporosis.1
This case underscores the importance of monitoring nutrients and biochemical markers at baseline and on an ongoing basis to detect early indicators of malabsorption and ultimately prevent the development of metabolic bone disease and fragility fracture, with its risks of disability and even death. It also illustrates the essential role that primary care physicians play in the continuing care of these patients.
THE OBESITY-BONE CONNECTION
Although we used to think that morbid obesity protected against metabolic bone disease, in fact, vitamin D and calcium deficiencies and elevated parathyroid hormone (PTH) levels are common in extremely obese people, placing them at risk of low bone mass.2–6 More than 60% of candidates for weight-loss surgery are deficient in vitamin D,5,7 and 25% to 48% have elevated PTH levels.5,6
And that is before bariatric surgery: afterward, severely restricted oral intake and significant weight loss, coupled with a procedure that bypasses the major site of calcium absorption, place many patients at extremely high risk.5,8
After combination restrictive and malabsorptive procedures (eg, the popular Rouxen-y procedure, in which the stomach is reduced in size—”restricted”—and the proximal duodenum is bypassed so that less food is absorbed), as patients lose weight their PTH levels rise and 25-hydroxyvitamin D levels decrease, although corrected calcium levels usually remain within normal limits.3,9 Secondary hyperparathyroidism has been documented as soon as 8 weeks after bariatric surgery, and osteomalacia after gastric bypass surgery is not uncommon.1,7,10–13
Exclusively restrictive procedures such as gastric banding, formerly presumed not to alter bone metabolism, now also appear to place patients at risk of metabolic bone disease due to inadequate intake of calcium and vitamin D in the immediate postoperative period.10
Numerous reported cases further illustrate the ever-present risk of metabolic bone disease in this population if adequate supplementation of calcium and vitamin D is not given. In these cases, significant bone disease occurred from 8 weeks to 32 years after bariatric surgery, often with devastating consequences.1,4,8,11–14
Voluntary weight loss, involuntary bone loss
When overweight or obese people lose weight—whether by dieting or by bariatric surgery—they also lose bone: a voluntary loss of approximately 10% of body weight results in a loss of 1% to 2% of bone at all sites. This loss appears to vary among populations: premenopausal women younger than 45 years may be able to lose a moderate amount of weight without a significant increase in fracture risk, while a study of overweight men found that a 7% weight loss resulted in a 1% bone loss.15
The percentage of bone lost correlates strongly with how fast the weight is lost. A recent study found that losing 0.7 kg/week was more detrimental to bone than a slower loss of 0.3 kg/week, due to the activation of the calcium-PTH axis.16
After bariatric surgery, many patients rapidly lose 50 kg—some even lose 100 kg or more. This rapid weight loss, combined with severely restricted oral intake, decreased calcium absorption, and vitamin D deficiency places these patients at extremely high risk of rapidly developing metabolic bone disease.3,8,9 In one large study, metabolic bone disease developed in more than 70% of patients who underwent a malabsorptive procedure, while in a second study, markers of bone resorption were elevated as soon as 8 weeks after bariatric surgery, regardless of whether the patient underwent a malabsorptive or restrictive bariatric procedure.13 Yet another study found that 48% of patients had a statistically significant bone mineral reduction of more than 3% 12 months after undergoing gastric banding.10
ESSENTIAL NUTRIENTS FOR BONE HEALTH
Protein
Dietary protein is needed to maintain bone structure, and although there is a link between high protein intake, calciuria, and fracture risk, the potentially harmful effects appear to be ameliorated when high protein intake is coupled with adequate calcium.17–20 This fact is of particular importance after bariatric surgery because once the patient can consume enough fluids to maintain hemodynamic stability, he or she is given a relatively high-protein diet to prevent protein malnutrition.21
Inadequate protein intake also has a detrimental effect on bone; therefore, it is essential to assess postoperative protein intake.22 Rizzoli and Bonjour23 noted that markers of bone turnover were higher with a low-protein diet (0.7 g protein per kg body weight) than with a diet containing 2.1 g protein per kg. In two trials examining graded levels of protein ingestion (0.7, 0.8, 0.9, and 1.0 g protein per kg body weight), decreased calcium absorption and an acute rise in PTH were noted by day 4 of the 0.7- and 0.8-g/kg diets but not during the 0.9- or 1.0-g/kg diets.24,25 And a systematic review of protein and bone health concluded that diets containing 1.0 to 1.5 g protein/kg are best for bone health.26 This is particularly worrisome, since the current recommended dietary allowance for protein is only 0.8 g/kg, which may be insufficient to promote calcium homeostasis.26,27
Vitamin D
Vitamin D is essential for calcium absorption, stimulation of osteoblast activity, and normal bone mineralization throughout the life span.27 Dietary vitamin D is mainly absorbed by passive diffusion in the proximal and mid small intestine in a process that is highly dependent on bile salts.28–30 Dietary sources of vitamin D are clinically important because exposure to ultraviolet B radiation is often insufficient, especially in northern latitudes.27
Up to 84% of morbidly obese patients have vitamin D deficiency.2,3,5,2,29,31 The mechanism of vitamin D deficiency and secondary hyperparathyroidism in the morbidly obese remains unclear, although one study concluded they were likely due to sequestration of vitamin D in adipose tissue and subsequent limited bioavailability.32
Correction of vitamin D deficiency requires more than just an over-the-counter multivitamin, but standard multivitamins also contain vitamin A, so taking more than one tablet a day increases the risk of vitamin A excess.33 Repletion can often be safely achieved orally by giving 50,000 IU of vitamin D weekly for 8 weeks, followed by a maintenance dose of one 50,000 IU tablet every 2 weeks. If a repeat serum level shows suboptimal repletion (less than 32 ng/mL), an additional 8-week course is recommended.29 For patients who cannot tolerate or adequately absorb oral supplements, exposure to sunlight is still the best source of vitamin D and is an effective alternative.29,33
Calcium
Dietary calcium deficiency is a well-established risk factor for osteoporosis and fragility fractures. Therefore, supplemental calcium should be prescribed for patients who do not meet their defined need.34,35 Of note: a normal serum calcium level does not imply adequate calcium intake or absorption. Calcium homeostasis is tightly regulated and is maintained by a combination of gut absorption, bone resorption, and renal reabsorption. If dietary intake is inadequate, calcium is resorbed from the bone.
The duodenum is the major site of active calcium uptake, while the rest of the small intestine and the colon appear to absorb some calcium passively. When the physiologic need for calcium is increased, active transport appears to take place throughout the duodenum, the ileum, and, to a lesser degree, the jejunum and the colon.36
In the normal gastrointestinal tract, 20% to 60% of dietary calcium is absorbed.36–38 Patients who have lost absorptive surface area (eg, after Roux-en-y bariatric surgery) need to have their calcium intake optimized. However, optimal dosing based on the type of surgical procedure is currently undefined.
Judicious monitoring for compliance and adequate absorption is recommended. Some patients will stop taking their calcium supplement due to gastrointestinal side effects such as gas, bloating, or constipation. And for some patients, a standard calcium supplement may be insufficient to promote adequate calcium absorption. Measuring urinary calcium in a 24-hour sample can help in assessing the adequacy of calcium intake: abnormally low urine calcium in the presence of normal renal function suggests inadequate absorption. For patients reporting gastrointestinal side effects or those with a history of calcium oxalate renal stones, calcium citrate supplements are better tolerated, alter urine acidity, and often prevent further stone formation.
Vitamin B12 (cobalamin)
Vitamin B12 deficiency is associated with increased fracture risk, and it may be an important modifiable risk factor for osteoporosis.39–41 After surgery, malabsorption of vitamin B12 is commonly the result of altered gut function in the gastric pouch or sleeve, but malabsorption also occurs when more than 60 to 100 cm of terminal ileum has been bypassed.42
Vitamin B12 supplementation is recommended for all patients after bariatric surgery, because deficiency is common.42 Patients with relatively mild malabsorption can maintain their B12 level by taking 350 μg orally; however, many patients require lifelong subcutaneous injections.7,39,42–45
Magnesium
Magnesium appears to affect bone remodeling and strength, to have a positive association with hip bone mineral density, and to play an important role in calcium and bone metabolism.
Magnesium is absorbed in the distal small intestine by carrier-mediated and paracellular routes.46 When the distal small intestine is bypassed, magnesium deficiency occurs as a result of reduced absorption and chelation with unabsorbed fatty acids in the bowel lumen.42 Chronic hypomagnesemia impairs PTH secretion, resulting in altered calcium metabolism, hypocalcemia, and vitamin D abnormalities, further decreasing jejunal magnesium absorption.26,42,47
Few well-designed studies have investigated the effect of magnesium intake on bone health, and although there is evidence that postmenopausal women may benefit from magnesium supplementation, studies of magnesium supplementation after bariatric surgery are lacking.47,48
A prevailing misconception promoted by manufacturers of calcium-magnesium supplements and others is that magnesium is necessary for calcium absorption and efficacy. In fact, magnesium deficiency typically must be severe to impair calcium absorption. With usual dietary intake of magnesium and normal serum magnesium levels, no such relationship exists.49–53
THE ROLE OF DXA IN THE CARE OF THE BARIATRIC SURGERY PATIENT
DXA is the gold standard for measuring bone density. The results are reported as a T score and as a Z score.
The T score is the bone density in an area of interest expressed in standard deviations from the mean value of a reference database of young adults. The World Health Organization defines normal as a T score greater than or equal to –1, low bone mass (previously called osteopenia) as a score between –1 and –2.5, and osteoporosis as a score of less than or equal to –2.5. (If a fragility fracture has occurred, “established” or “severe” osteoporosis is present.) Of note: these criteria only apply to DXA of the posterior-anterior spine, femoral neck, and the proximal (33%) radius in post-menopausal women and men over the age of 50 years.54,55 The International Society of Clinical Densitometry has extended the criteria to include total hip measurements.56
The Z score should be used instead of the T score for premenopausal women and men younger than 50 years.56 The Z score is the patient’s bone mineral density expressed in standard deviations from the mean in a reference population matched for sex and age. A Z score greater than –2.0 is “within the expected range for age,” and –2.0 or lower is “below the expected range for age.” There are separate guidelines for DXA reporting in the diagnosis of metabolic bone disease in people younger than 20 years, and this topic is beyond the scope of this article.
Who should undergo DXA?
According to the International Society of Clinical Densitometry, bone density testing is indicated in the general population in women 65 years of age and older, postmenopausal women younger than 65 with risk factors, men 70 and older, adults with fragility fractures, adults taking a medication or having a disease or condition associated with low bone mass or bone loss, any patient being treated for low bone mass (to monitor the treatment effect), and any person in whom evidence of bone loss would affect treatment decisions.58
The National Osteoporosis Foundation recommends initiating therapy to reduce fracture risk in postmenopausal women with a central DXA T score below –2 in the absence of risk factors, and in women with T scores below –1.5 if one or more risk factors is present.34 Therefore, in view of the known risks, the likely need for interventions before surgery, and the ability to prevent the illness and death associated with metabolic bone disease, we recommend that all bariatric surgery patients undergo DXA at baseline as part of the preoperative evaluation.
Improvements in DXA technology
Newer DXA machines can accommodate patients weighing up to 450 pounds (the limit with older machines was 275 pounds for central measurements). In addition to measuring bone density, they also can map the distribution of fat in the body—patients with an android (apple-shaped) distribution are at higher risk of cardiovascular disease than those with a gynecoid (pear-shaped) distribution.59–63 For those patients who cannot be accommodated on a DXA table, DXA of the forearm can be used to assess bone density and fracture risk.
How often should DXA be repeated?
The estimated monitoring time interval is derived from the statistically defined least significant change divided by the anticipated change in bone density over time.64 When estimating the monitoring time interval for changes in body composition, the rate of weight loss and the psychological impact on the patient must be taken into consideration.
In general, DXA testing more frequently than every 2 years remains controversial unless one is initiating, monitoring, or changing therapy or monitoring conditions associated with rapid bone loss such as glucocorticoid therapy. In the bariatric surgery population, however, there is convincing evidence that significant changes may be detected after 12 months that would influence clinical decisions, particularly in the year immediately after surgery.8,9,13,21,27,65
Anabolic and antiresorptive bone drugs
Prescribed medications for the prevention and treatment of osteoporosis should also be an integral part of the treatment plan for at-risk morbidly obese patients. But the decision to prescribe an antiresorptive or bone-forming medication must take into consideration the patient’s risk-benefit profile, including the likelihood of gastrointestinal side effects and his or her ability and willingness to follow specific dosing instructions. Intravenous preparations are now available for patients who cannot absorb or tolerate oral antiresorptive medications. However, specific recommendations about the use of anabolic or antiresorptive bone medications in perioperative bariatric patients have yet to be elucidated.
RECOMMENDATIONS
Although a variety of recommendations have been published, there are no established guidelines for perioperative screening, risk stratification, or management of metabolic bone disease in bariatric surgery patients.7,44,65–67 And the literature remains inconclusive on key issues such as when to start supplements, which biochemical indices should be checked before surgery, whether baseline and annual DXA should be done, and whether antiresorptive agents such as bisphosphonates should be used prophylactically during rapid weight loss.
However, numerous studies and case reports cited here and elsewhere further underscore the ever-present risk of metabolic bone disease in this patient population, and the need for meticulous perioperative and long-term monitoring.44,65–67
With these caveats in mind, we offer our recommendations (Table 3).
Preoperative assessment
We recommend obtaining baseline biochemical indices, including albumin, 25-hydroxy-vitamin D, calcium, magnesium, phosphorus, alkaline phosphatase, folate, vitamin B12, thyroid-stimulating hormone, and PTH levels, and DXA in all bariatric surgery candidates. These indices should be used to assess for primary and secondary metabolic bone disease, to enable prompt presurgical interventions, and to guide the clinician in selecting appropriate postoperative interventions and surveillance.
We recommend starting a multivitamin with minerals at the first preoperative visit. A calcium supplement that provides calcium and vitamin D appropriate to the patient’s age and sex is also recommended until surgery. After surgery, and while rapid weight loss is occurring, a minimum of 1,800 mg of calcium and 800 to 1,000 IU of vitamin D is recommended, keeping in mind that the required level of supplemental vitamin D during periods of rapid weight loss remains unclear.68,69
However, before prescribing supplementation, one should thoroughly review the patient’s nutrition history, including the use of homeopathic medications, herbal preparations, and supplements. Many over-the-counter and over-the-Internet supplements are touted as being good for bone health, and some may indeed be beneficial, but others can be detrimental and need to be discontinued.27,47 Furthermore, in a patient with a severely restricted stomach capacity, it is important to ensure that less efficacious supplements do not compromise the intake of essential fluids, protein, and prescribed medications.
Immediate postoperative period
Hospitalization and surgery result in nutrient deficiencies. In bariatric surgery patients, particularly those who have preoperative nutritional deficiencies, repletion in the immediate postoperative period is believed to be of benefit. Therefore, in the immediate postoperative period we recommend infusing a standard-dose multivitamin with minerals daily along with adequate intravenous hydration until the patient can resume oral feeding. Once the patient can tolerate liquids, presurgical supplementation needs to be resumed, preferably in a liquid or chewable form to facilitate tolerance and absorption.
Short-term and long-term follow-up
Follow-up visits with a bariatric specialist should start 4 weeks after surgery and should be repeated every 3 to 4 months for the first year. If the patient continues to do well, annual visits may be sufficient thereafter. Compliance with supplements should be checked as indicated, as should nutritional indices. DXA should be repeated every 1 to 2 years, depending on the patient’s risk profile.
WHAT SHOULD BE DONE FOR OUR PATIENT?
Initial treatment for the patient described at the beginning of this article should include vitamin D repletion with cholecalciferol 50,000 IU, calcium supplements of at least 1,200 mg daily, and addressing of modifiable risk factors for fracture, including the risk of falling due to her proximal weakness. Her laboratory studies should be repeated in 6 to 12 weeks, with calcium and vitamin D supplement dosages adjusted on the basis of her response. Once the serum calcium level has normalized, we would consider the use of a bisphosphonate. DXA should be repeated in 1 to 2 years to monitor the effectiveness of the prescribed interventions.
- De Prisco C, Levine SN. Metabolic bone disease after gastric bypass surgery for obesity. Am J Med Sci 2005; 329:57–61.
- Ybarra J, Sanchez-Harnandez J, Gich I, et al. Unchanged hypovitaminosis D and secondary hyperparathyroidism in morbid obesity after bariatric surgery. Obes Surg 2005; 15:330–335.
- Hamoui N, Anthone G, Crookes F. Calcium metabolism in the morbidly obese. Obes Surg 2004; 14:9–12.
- Parikh SJ, Edelman M, Uwaifo GI, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab 2004; 89:1196–1199.
- Carlin AM, Rao DS, Meslemani AM, et al. Prevalence of vitamin D depletion among morbidly obese patients seeking gastric bypass surgery. Surg Obes Rel Dis 2006; 2:98–103.
- Puzziferri N, Blankenship J, Wolfe BM. Surgical treatment of obesity. Endocrine 2006; 29:11–19.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Haria DM, Sibonga JD, Taylor HC. Hypocalcemia, hypovitaminosis D osteopathy, osteopenia, and secondary hyperparathyroidism 32 years after jejunoileal bypass. Endocr Pract 2005; 11:335–340.
- Newbery L, Dolan K, Hatzifotis M, et al. Calcium and vitamin D depletion and elevated parathyroid hormone following biliopancreatic diversion. Obes Surg 2003; 13:893–895.
- Pugnale N, Guisti V, Suter M, et al. Bone metabolism and risk of secondary hyperparathyroidism 12 months after gastric banding in obese pre-menopausal women. Int J Obesity 2003; 27:110–116.
- Goldner WS, O’Dorisio TM, Dillon JS, Mason EE. Severe metabolic bone disease as a long-term complication of obesity surgery. Obes Surg 2002; 12:685–692.
- Atreja A, Abacan C, Licata A. A 51-year-old woman with debilitating cramps 12 years after bariatric surgery. Cleve Clin J Med 2003; 70:417–426.
- Collazo-Clavell ML, Jimenez A, Hodgson SF, et al. Osteomalacia after Rouxen-Y gastric bypass. Endocr Pract 2004; 10:287–288.
- Dennisson E. Osteoporosis. In:Pinchera A, Bertagna X, Fischer J, editors. Endocrinology and Metabolism. London: McGraw-Hill International (UK) Ltd; 2001:271–282.
- Shapses SA, Cifuentes M. Body weight/composition and weight change: effects on bone health. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health New Jersey: Humana Press; 2004:549–573.
- Shapses SA, Cifuentes M, Sherrell R, et al. Rate of weight loss influences calcium absorption. J Bone Miner Res 2002; 17:S471.
- Munger RG, Cerhan JR, Chiu BC. Prospective study of dietary protein intake and risk of hip fracture in postmenopausal women. Am J Clin Nutr 1999; 69:147–152.
- Weikert C, Walter D, Hoffmann K, et al. The relation between dietary protein, calcium and bone health in women: results from the EPIC-Potsdam cohort. Ann Nutr Metab 2005; 49:312–318.
- Whiting SJ, Boyle JL, Thompson A. Dietary protein, phosphorus and potassium are beneficial to bone mineral density in adult men consuming adequate dietary calcium. J Am Col Nutr 2002; 21:402–409.
- Teegarden D, Lyle RM, McCabe GP, et al. Dietary calcium, protein, and phosphorus are related to bone mineral density and content in young women. Am J Clin Nutr 1998; 68:749–754.
- Bloomberg RD, Fleishman A, Nalle JE, et al. Nutritional deficiencies following bariatric surgery: what have we learned? Obes Surg 2005; 15:145–154.
- Greenspan SL, Resnick NM. Geriatric endocrinology. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7 New York: McGraw-Hill; 2004:842–866.
- Rizzoli R, Bonjour JP. Dietary protein and bone health. J Bone Miner Res 2004; 19:527–531.
- Kerstetter J, Svastisalee C, Caseria D, et al. A threshold for low-protein-diet-induced elevations in parathyroid hormone. Am J Clin Nutr 2000; 72:168–173.
- Giannini S, Nobile M, Sartori L, et al. Acute effects of moderate dietary protein restriction in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Am J Clin Nutr 1999; 69:267–271.
- Ilich JZ, Kerstetter JE. Nutrition in bone health revisited: a story beyond calcium. J Am Coll Nutr 2000; 19:715–737.
- Williams S, Seidner D. Metabolic bone disease in gastrointestinal illness. Gastroenterol Clin North Am 2007; 36 1:161–190.
- Shoback D, Marcus R, Bilke D. Metabolic bone disease. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7. New York: McGraw-Hill; 2004:295–361.
- Holick MF. Vitamin D. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:329–346.
- Rosen CJ. Vitamin D and bone health in adults and the elderly. In:Holick MF. Vitamin D: Physiology, Molecular Biology, and Clinical Applications. New Jersey: Humana Press; 1999:287–306.
- Buffington C, Walker B, Cowan GS, et al. Vitamin D deficiency in the morbidly obese. Obes Surg 1993; 3:421–424.
- Wortsman J, Matsuoka L, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 2000; 72:690–693.
- Holick MF, Garabedian M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:106–114.
- National Osteoporosis Foundation. Physician’s Guide to Prevention and Treatment of Osteoporosis. http://www.nof.org/physguide. Washington D.C. 1998.
- National Academy of Sciences Executive Summary: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington DC. 1997.
- Favus MJ, Bushinsky DA, Lemann J. Regulation of calcium, magnesium and phosphate metabolism. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:76–83.
- Weaver CM, Heaney RP. Calcium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins, 1999:141–155.
- Gueguen L, Oiubtukkart A. The bioavailability of dietary calcium. J Am Coll Nutr 2000; 19:119S–136S.
- Tucker KL, Hannan MT, Qiao N, et al. Low plasma vitamin B12 is associated with lower BMD: the Framingham osteoporosis study. J Bone Miner Res 2005; 20:152–158.
- Goerss JB, Kim CH, Atkinson EJ, et al. Risk of fractures in patients with pernicious anemia. J Bone Miner Res 1992; 7:573–579.
- Eastell R, Vieira NE, Yergey AL, et al. Pernicious anemia is a risk factor for osteoporosis. Clin Sci 1992; 82:681–685.
- Nightingale JMD, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006; 55:1–12.
- Rhode BM, Tamin H, Gilfix BM, et al. Treatment of vitamin B12 deficiency after gastric surgery for severe obesity. Obes Surg 1995; 5:154–158.
- Brethauer SA, Chand C, Schauer PR. Risks and benefits of bariatric surgery: current evidence. Cleve Clin J Med 2006; 73:993–1007.
- Weir DG, Scott JW. Vitamin B12 “cobalamin”. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:447–458.
- Shils ME. Magnesium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:169–192.
- National Osteoporosis Foundation. Osteoporosis Clinical Updates: Over-the-counter products & osteoporosis: Case discussions. 2002; Vol IIIIssue 2. Washington DC.
- Stendig-Lindenberg G, Tepper R, Leicher I. Trabecular bone density in a two year controlled trial of personal magnesium in osteoporosis. Magnes Res 1993:155–163.
- Heaney RP. Sodium, potassium, phosphorus, and magnesium. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health. New Jersey: Humana Press; 2004:327–344.
- Spencer H, Fuller H, Norris C, et al. Effect of magnesium on the intestinal absorption of calcium in man. J Am Coll Nutr 1994; 13:483–492.
- Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327:1637–1642.
- Dawson-Hughes B, Harris SS, Krall EA, et al. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age and older. N Engl J Med 1997; 337:670–676.
- Rude RK, Olerich M. Magnesium deficiency: possible role in osteoporosis associated with gluten-sensitive enteropathy. Osteoporos Int 1996; 6:453–461.
- Report of a WHO Study Group. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. 1994; 843:1–129.
- Kanis JA, Melton LJ, Christiansen C, et al. The diagnosis of osteoporosis. J Bone Miner Res 1994; 9:1137–1141.
- Official Positions of the International Society for Clinical Densitometry. West Hartford, CT. September, 2005. http://www.iscd.org/Visitors/positions/OfficialPositionsText.cfm.
- Licata A. Diagnosing primary osteoporosis: It’s more than a T score. Cleve Clin J Med 2006; 73:473–476.
- Writing group for the ISCD Position Development Conference. Indications and reporting for dual-energy x-ray absorptiometry. J Clin Densitom 2004; 7:37–44.
- Park YW, Heymsfield SB, Gallagher D. Are dual-energy X-ray absorptiometry regional estimates associated with visceral adipose tissue mass? Int J Obes Relat Metab Disord 2002; 26:978–983.
- Glickman SG, Marn CS, Supiano MA, et al. Validity and reliability of dual-energy x-ray absorptiometry for the assessment of abdominal adiposity. J Appl Physiol 2004; 97:509–514.
- Hull HR, Hester CN, Fields DA. The effect of the holiday season on body weight and composition in college students. Nutr Metab (Lond) 2006; 3:44–51.
- Aubertin-Leheudre M, Goulet EDB, Khalil A, et al. Effect of sarcopenia on cardiovascular disease risk factors in obese post-menopausal women. Obesity 2006; 14:2277–2283.
- Lubrano C, Cornoldi A, Pili M, et al. Reduction of risk factors for cardiovascular diseases in morbid-obese patients following biliary-intestinal bypass: 3 years’ follow-up. Int J Obes Relat Metab Disord 2004; 28:1600–1606.
- Bonnick SL, Johnston CC, Kleerekoper M, et al. Importance of precision in bone density measurements. J Clin Densitom 2001; 4:105–110.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Hensrud DD, McMahon MM. Bariatric surgery in adults with extreme (not morbid) obesity. Mayo Clin Proc 2006; 81 suppl:S3–S4.
- McGlinch BP, Que FG, Nelson JL, et al. Perioperative care of patients undergoing bariatric surgery. Mayo Clin Proc 2006; 81 10, suppl:S25–S33.
- Ricci TA, Chowdhury HA, Heymsfield SB, et al. Calcium supplementation suppresses bone turnover during weight reduction in postmenopausal women. J Bone Miner Res 1998; 13:1045–1050.
- Jensen LB, Kollerup G, Quaade F, et al. Bone mineral changes in obese women during moderate weight loss with and without calcium supplementation. J Bone Miner Res 2001; 16:141–147.
- De Prisco C, Levine SN. Metabolic bone disease after gastric bypass surgery for obesity. Am J Med Sci 2005; 329:57–61.
- Ybarra J, Sanchez-Harnandez J, Gich I, et al. Unchanged hypovitaminosis D and secondary hyperparathyroidism in morbid obesity after bariatric surgery. Obes Surg 2005; 15:330–335.
- Hamoui N, Anthone G, Crookes F. Calcium metabolism in the morbidly obese. Obes Surg 2004; 14:9–12.
- Parikh SJ, Edelman M, Uwaifo GI, et al. Gastric bypass surgery for morbid obesity leads to an increase in bone turnover and a decrease in bone mass. J Clin Endocrinol Metab 2004; 89:1196–1199.
- Carlin AM, Rao DS, Meslemani AM, et al. Prevalence of vitamin D depletion among morbidly obese patients seeking gastric bypass surgery. Surg Obes Rel Dis 2006; 2:98–103.
- Puzziferri N, Blankenship J, Wolfe BM. Surgical treatment of obesity. Endocrine 2006; 29:11–19.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Haria DM, Sibonga JD, Taylor HC. Hypocalcemia, hypovitaminosis D osteopathy, osteopenia, and secondary hyperparathyroidism 32 years after jejunoileal bypass. Endocr Pract 2005; 11:335–340.
- Newbery L, Dolan K, Hatzifotis M, et al. Calcium and vitamin D depletion and elevated parathyroid hormone following biliopancreatic diversion. Obes Surg 2003; 13:893–895.
- Pugnale N, Guisti V, Suter M, et al. Bone metabolism and risk of secondary hyperparathyroidism 12 months after gastric banding in obese pre-menopausal women. Int J Obesity 2003; 27:110–116.
- Goldner WS, O’Dorisio TM, Dillon JS, Mason EE. Severe metabolic bone disease as a long-term complication of obesity surgery. Obes Surg 2002; 12:685–692.
- Atreja A, Abacan C, Licata A. A 51-year-old woman with debilitating cramps 12 years after bariatric surgery. Cleve Clin J Med 2003; 70:417–426.
- Collazo-Clavell ML, Jimenez A, Hodgson SF, et al. Osteomalacia after Rouxen-Y gastric bypass. Endocr Pract 2004; 10:287–288.
- Dennisson E. Osteoporosis. In:Pinchera A, Bertagna X, Fischer J, editors. Endocrinology and Metabolism. London: McGraw-Hill International (UK) Ltd; 2001:271–282.
- Shapses SA, Cifuentes M. Body weight/composition and weight change: effects on bone health. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health New Jersey: Humana Press; 2004:549–573.
- Shapses SA, Cifuentes M, Sherrell R, et al. Rate of weight loss influences calcium absorption. J Bone Miner Res 2002; 17:S471.
- Munger RG, Cerhan JR, Chiu BC. Prospective study of dietary protein intake and risk of hip fracture in postmenopausal women. Am J Clin Nutr 1999; 69:147–152.
- Weikert C, Walter D, Hoffmann K, et al. The relation between dietary protein, calcium and bone health in women: results from the EPIC-Potsdam cohort. Ann Nutr Metab 2005; 49:312–318.
- Whiting SJ, Boyle JL, Thompson A. Dietary protein, phosphorus and potassium are beneficial to bone mineral density in adult men consuming adequate dietary calcium. J Am Col Nutr 2002; 21:402–409.
- Teegarden D, Lyle RM, McCabe GP, et al. Dietary calcium, protein, and phosphorus are related to bone mineral density and content in young women. Am J Clin Nutr 1998; 68:749–754.
- Bloomberg RD, Fleishman A, Nalle JE, et al. Nutritional deficiencies following bariatric surgery: what have we learned? Obes Surg 2005; 15:145–154.
- Greenspan SL, Resnick NM. Geriatric endocrinology. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7 New York: McGraw-Hill; 2004:842–866.
- Rizzoli R, Bonjour JP. Dietary protein and bone health. J Bone Miner Res 2004; 19:527–531.
- Kerstetter J, Svastisalee C, Caseria D, et al. A threshold for low-protein-diet-induced elevations in parathyroid hormone. Am J Clin Nutr 2000; 72:168–173.
- Giannini S, Nobile M, Sartori L, et al. Acute effects of moderate dietary protein restriction in patients with idiopathic hypercalciuria and calcium nephrolithiasis. Am J Clin Nutr 1999; 69:267–271.
- Ilich JZ, Kerstetter JE. Nutrition in bone health revisited: a story beyond calcium. J Am Coll Nutr 2000; 19:715–737.
- Williams S, Seidner D. Metabolic bone disease in gastrointestinal illness. Gastroenterol Clin North Am 2007; 36 1:161–190.
- Shoback D, Marcus R, Bilke D. Metabolic bone disease. In:Greenspan FS, Gardner DG, editors. Basic and Clinical Endocrinology 7. New York: McGraw-Hill; 2004:295–361.
- Holick MF. Vitamin D. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:329–346.
- Rosen CJ. Vitamin D and bone health in adults and the elderly. In:Holick MF. Vitamin D: Physiology, Molecular Biology, and Clinical Applications. New Jersey: Humana Press; 1999:287–306.
- Buffington C, Walker B, Cowan GS, et al. Vitamin D deficiency in the morbidly obese. Obes Surg 1993; 3:421–424.
- Wortsman J, Matsuoka L, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 2000; 72:690–693.
- Holick MF, Garabedian M. Vitamin D: photobiology, metabolism, mechanism of action, and clinical applications. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:106–114.
- National Osteoporosis Foundation. Physician’s Guide to Prevention and Treatment of Osteoporosis. http://www.nof.org/physguide. Washington D.C. 1998.
- National Academy of Sciences Executive Summary: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington DC. 1997.
- Favus MJ, Bushinsky DA, Lemann J. Regulation of calcium, magnesium and phosphate metabolism. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism 6. Washington DC. American Society for Bone and Mineral Research, 2006:76–83.
- Weaver CM, Heaney RP. Calcium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins, 1999:141–155.
- Gueguen L, Oiubtukkart A. The bioavailability of dietary calcium. J Am Coll Nutr 2000; 19:119S–136S.
- Tucker KL, Hannan MT, Qiao N, et al. Low plasma vitamin B12 is associated with lower BMD: the Framingham osteoporosis study. J Bone Miner Res 2005; 20:152–158.
- Goerss JB, Kim CH, Atkinson EJ, et al. Risk of fractures in patients with pernicious anemia. J Bone Miner Res 1992; 7:573–579.
- Eastell R, Vieira NE, Yergey AL, et al. Pernicious anemia is a risk factor for osteoporosis. Clin Sci 1992; 82:681–685.
- Nightingale JMD, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006; 55:1–12.
- Rhode BM, Tamin H, Gilfix BM, et al. Treatment of vitamin B12 deficiency after gastric surgery for severe obesity. Obes Surg 1995; 5:154–158.
- Brethauer SA, Chand C, Schauer PR. Risks and benefits of bariatric surgery: current evidence. Cleve Clin J Med 2006; 73:993–1007.
- Weir DG, Scott JW. Vitamin B12 “cobalamin”. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:447–458.
- Shils ME. Magnesium. In:Shils ME, Olsen JA, Shine M, et al, editors. Modern Nutrition in Health and Disease 9. Philadelphia: Lippincott Williams & Wilkins; 1999:169–192.
- National Osteoporosis Foundation. Osteoporosis Clinical Updates: Over-the-counter products & osteoporosis: Case discussions. 2002; Vol IIIIssue 2. Washington DC.
- Stendig-Lindenberg G, Tepper R, Leicher I. Trabecular bone density in a two year controlled trial of personal magnesium in osteoporosis. Magnes Res 1993:155–163.
- Heaney RP. Sodium, potassium, phosphorus, and magnesium. In:Holick MF, Dawson-Hughes B, editors. Nutrition and Bone Health. New Jersey: Humana Press; 2004:327–344.
- Spencer H, Fuller H, Norris C, et al. Effect of magnesium on the intestinal absorption of calcium in man. J Am Coll Nutr 1994; 13:483–492.
- Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D and calcium to prevent hip fractures in elderly women. N Engl J Med 1992; 327:1637–1642.
- Dawson-Hughes B, Harris SS, Krall EA, et al. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age and older. N Engl J Med 1997; 337:670–676.
- Rude RK, Olerich M. Magnesium deficiency: possible role in osteoporosis associated with gluten-sensitive enteropathy. Osteoporos Int 1996; 6:453–461.
- Report of a WHO Study Group. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. 1994; 843:1–129.
- Kanis JA, Melton LJ, Christiansen C, et al. The diagnosis of osteoporosis. J Bone Miner Res 1994; 9:1137–1141.
- Official Positions of the International Society for Clinical Densitometry. West Hartford, CT. September, 2005. http://www.iscd.org/Visitors/positions/OfficialPositionsText.cfm.
- Licata A. Diagnosing primary osteoporosis: It’s more than a T score. Cleve Clin J Med 2006; 73:473–476.
- Writing group for the ISCD Position Development Conference. Indications and reporting for dual-energy x-ray absorptiometry. J Clin Densitom 2004; 7:37–44.
- Park YW, Heymsfield SB, Gallagher D. Are dual-energy X-ray absorptiometry regional estimates associated with visceral adipose tissue mass? Int J Obes Relat Metab Disord 2002; 26:978–983.
- Glickman SG, Marn CS, Supiano MA, et al. Validity and reliability of dual-energy x-ray absorptiometry for the assessment of abdominal adiposity. J Appl Physiol 2004; 97:509–514.
- Hull HR, Hester CN, Fields DA. The effect of the holiday season on body weight and composition in college students. Nutr Metab (Lond) 2006; 3:44–51.
- Aubertin-Leheudre M, Goulet EDB, Khalil A, et al. Effect of sarcopenia on cardiovascular disease risk factors in obese post-menopausal women. Obesity 2006; 14:2277–2283.
- Lubrano C, Cornoldi A, Pili M, et al. Reduction of risk factors for cardiovascular diseases in morbid-obese patients following biliary-intestinal bypass: 3 years’ follow-up. Int J Obes Relat Metab Disord 2004; 28:1600–1606.
- Bonnick SL, Johnston CC, Kleerekoper M, et al. Importance of precision in bone density measurements. J Clin Densitom 2001; 4:105–110.
- Mason EM, Jalagani H, Vinik AI. Metabolic complications of bariatric surgery: diagnosis and management issues. Gastroenterol Clin North Am 2006 34:25–33.
- Hensrud DD, McMahon MM. Bariatric surgery in adults with extreme (not morbid) obesity. Mayo Clin Proc 2006; 81 suppl:S3–S4.
- McGlinch BP, Que FG, Nelson JL, et al. Perioperative care of patients undergoing bariatric surgery. Mayo Clin Proc 2006; 81 10, suppl:S25–S33.
- Ricci TA, Chowdhury HA, Heymsfield SB, et al. Calcium supplementation suppresses bone turnover during weight reduction in postmenopausal women. J Bone Miner Res 1998; 13:1045–1050.
- Jensen LB, Kollerup G, Quaade F, et al. Bone mineral changes in obese women during moderate weight loss with and without calcium supplementation. J Bone Miner Res 2001; 16:141–147.
KEY POINTS
- Metabolic bone disease in obese patients is multifactorial: causes include sequestration of vitamin D in the adipocytes, inadequate nutrition due to chronic dieting, and lack of physical activity.
- Before bariatric surgery, one must look for and treat preexisting nutritional deficiencies.
- In the immediate postoperative period, aggressive strategies (ie, giving multivitamins and minerals intravenously and orally) can prevent nutritional deficiencies and secondary bone disease.
- Postoperatively, many bariatric patients require chewable or liquid supplements to facilitate adequate absorption.
- Clinical suspicion, timely interventions, and lifelong monitoring can prevent metabolic bone disease in bariatric surgery patients.
Anemia of chronic kidney disease: When normalcy becomes undesirable
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
KEY POINTS
- ESAs reduce the need for blood transfusions and possibly improve quality of life.
- It is unclear if higher hemoglobin levels per se actually caused the adverse events in these trials. Event rates were highest in patients who responded poorly to ESAs.
- We concur with the FDA’s recommendation that the hemoglobin level be raised to no higher than 12 g/dL with ESAs in patients with chronic kidney disease or renal failure.
- Transient excursions of the hemoglobin level above 12 g/dL should not be a cause for panic. Rather, the next ESA dose should be reduced.
Testosterone Replacement Therapy in the VA Setting
Larva Currens in a Patient Scheduled for Renal Transplant
Case Report
A 54-year-old woman with polycystic disease of kidneys was scheduled for renal transplant and presented with a 2-week history of an extremely pruritic rash that primarily affected her torso, buttocks, shoulders, and thighs. She described the lesions as red, raised, and linear, typically lasting less than 24 hours at a time. She had been previously treated with a 5-day course of prednisone by another physician, without improvement. Her only new medication was glucosamine and chondroitin sulfate, which she had started one week prior to the eruption. Raloxifene hydrochloride and cetirizine hydrochloride were long-term medications.
The patient reported one similar episode many years ago. She denied any recent changes in her health and reported no gastrointestinal tract or pulmonary symptoms. She was raised in Panama and had visited there in the past year. She specifically denied walking without shoes.
On physical examination, the patient had a pink, serpiginous, urticarial plaque on the right side of the trunk that was surrounded by a few red serpiginous patches (Figure). Her white blood cell count was 6.5X103/µL (reference range, 3.5–10.5X103/µL), with 19.2% eosinophils (reference, 2.7%). Her absolute eosinophil count was elevated at 1200/µL (reference range, 0–450/µL). A review of prior laboratory test results indicated that her absolute eosinophil count also had been elevated 6 months prior to presentation. Serologic evaluation by enzyme-linked immunosorbent assay was positive for Strongyloides. Results of stool studies did not reveal ova and parasites.
PLEASE REFER TO THE PDF TO VIEW THE FIGURE
The patient was treated with oral thiabendazole 1500 mg twice daily for 2 days and her transplant was postponed. Her rash resolved, but 2 weeks later, her white blood cell count was 5.6X103/µL, with 11.6% eosinophils. She was subsequently treated with a single dose of 200 µg/kg of ivermectin. Results of a complete blood count obtained 2 weeks later demonstrated that her eosinophil count was within reference range and she was able to proceed with the transplant. The patient's sister (the donor) also was born in Panama and had negative serologic evaluation results for Strongyloides. The patient did well following the transplant and the results of repeat serologic evaluations performed 4 months after the transplant were negative for Strongyloides.
Comment
Strongyloides stercoralis is an intestinal nematode primarily found in tropical or subtropical countries. Humans are infected by filariform larvae that dwell in the soil. Larvae penetrate intact skin, gain access to the venous system, pass through the heart to the lungs, enter the pulmonary alveoli, migrate up the tracheobronchial tree, and are swallowed, thereby entering the gastrointestinal tract.1 The larvae mature into adult females that penetrate the mucosa of the small intestine and deposit eggs. The eggs hatch into rhabditiform larvae that are passed in the stool to the soil where transformation into the infective form (filariform) occurs. Autoinfection may take place when this transformation to the infective-stage larvae occurs within the gastrointestinal tract, enabling the infective larvae to invade the lower large bowel or perianal skin and begin the migratory pathway. Autoinfection can allow the persistence of infection for long periods of time and also can allow chronic infections to persist in climates where free-living larvae cannot survive.2
Uncomplicated infection with S stercoralis can cause cutaneous, gastrointestinal tract, and pulmonary symptoms corresponding to the involvement of organs during the parasite's life cycle. Rash is uncommon in acute infection, though it is common in chronic disease. Maculopapular eruptions and chronic urticaria have been reported in up to two-thirds of patients.3 Larva currens is a migratory, rapidly extending, serpiginous, urticarial lesion that is pathognomonic for chronic strongyloidiasis. The rash typically lasts from several hours to several days. It most commonly affects the buttocks, perineum, and thighs, and is secondary to invasion of perianal skin by filariform larvae from the patient's intestine. Arthur and Shelley4 proposed the term larva currens (running larva) because the larvae and subsequent rash can move up to 10 cm per hour.
In a healthy host, the cellular immune system seems to limit parasite invasion of mucosal tissues.5 If immunosuppression occurs, individuals with strongyloidiasis can develop a hyperinfective syndrome and massive numbers of larvae can invade any organ of the body, with a mortality rate of 70% to 90%.6,7 The cutaneous manifestation of disseminated strongyloidiasis is the rapid onset of a petechial and purpuric eruption that typically involves the proximal extremities and trunk and results from massive invasion of the skin by filariform larvae. The "thumbprint sign" refers to a pattern of periumbilical ecchymoses resembling multiple thumbprints that can occur in hyperinfection.8
Gastrointestinal tract symptoms predominate in acute infection. Diarrhea and midepigastric pain that may mimic peptic ulcer disease are common. Diarrhea also can alternate with constipation. Other gastrointestinal tract symptoms include nausea, vomiting, anorexia, pruritus ani, and bloating.1 Some severe cases can have malabsorption and evidence of a protein-losing enteropathy.9,10
Pulmonary symptoms in acute infection can occur and include wheezing, coughing, and shortness of breath.1 Larval migration through the lungs also can lead to transient pulmonary infiltrates. Some patients have presented with asthma.11 Patients with chronic disease may have gastrointestinal tract and pulmonary symptoms, though chronic infection tends to be indolent and patients may be asymptomatic.
Diagnosis can be difficult, as results from stool samples often are negative and multiple samples may be required. Results of biopsies performed on larva currens specimens usually do not reveal larvae, though biopsy results of the petechial and purpuric eruptions of disseminated disease will reveal larvae. Serologic testing with enzyme-linked immunosorbent assay has a sensitivity of approximately 90%.12,13
Traditionally, thiabendazole has been used to treat this infection, though in approximately 30% of cases, the parasite is not eradicated from the feces. Ivermectin has been found to be more effective for treating uncomplicated chronic disease.14
In most cases of disseminated disease, patients were receiving corticosteroids or other immunosuppressive drugs or had an underlying illness, such as malignancy or AIDS.1,2,15,16 Our patient was scheduled to undergo a renal transplant and fatal disseminated strongyloidiasis has been reported in patients undergoing renal transplant.2,16 Morgan et al2 reviewed 29 cases of strongyloidiasis complicating renal transplants; 15 patients died.
Infection in the immunocompromised patient can be complicated by the fact that invasive larvae can transport gram-negative bacilli from the intestine to sites of migration, such as the pulmonary and central nervous systems.5 Gram-negative sepsis, meningitis, or pneumonia can result. Diagnosis can be difficult because eosinophilia often is absent in immunocompromised patients with disseminated disease.5
Although common in tropical and subtropical countries, other geographic regions of endemic Strongyloides are recognized. The climate and soil of the southeastern United States favor the survival of the organism,5 and the parasite was reported in 3% (N=561) of a group of rural Kentucky schoolchildren17; similar findings were reported in another study conducted in Kentucky.18Strongyloides also was the most commonly detected parasite in a review of stool samples examined at the University of Kentucky Medical Center.19 Ex–prisoners of war who served in Southeast Asia during World War II also constitute an at-risk group in the United States.20-22
It is imperative to rule out the presence of this parasite prior to transplant in patients with a geographic history predisposing them to infection, a history of eosinophilia, or symptoms of chronic strongyloidiasis.1 Many transplantation centers routinely screen for this parasite as part of the pretransplant evaluation. Although uncommon in acute infections, cutaneous involvement often is present in chronic strongyloidiasis.1 It also is important to follow patients already treated for larva currens closely posttransplant, as therapeutic failures occur.
- Longworth DL, Weller PF. Hyperinfection syndrome with strongyloidiasis. In: Remington JS, Swartz MN, eds. Current Clinical Topics in Infectious Diseases. New York, NY: McGraw-Hill; 1986:1-26.
- Morgan JS, Schaffner W, Sone WJ. Opportunistic strongyloidiasis in renal transplant recipients. Transplantation. 1986;42:518-524.
- Grove DI. Strongyloidiasis in allied ex–prisoners of war in south-east Asia. Br Med J. 1980;280:598-601.
- Arthur RP, Shelley WB. Larva currens; a distinctive variant of cutaneous larva migrans due to Strongyloides stercoralis. AMA Arch Derm. 1958;78:186-190.
- Zygmunt DJ. Strongyloides stercoralis. Infect Control Hosp Epidemiol. 1990;11:495-497.
- Singh S. Human strongyloidiasis in AIDS era: its zoonotic importance. J Assoc Physicians India. 2002;50:415-422.
- Rothenberg ME. Eosinophilia. N Engl J Med. 1998;338:1592-1600.
- Bank DE, Grossman ME, Kohn SR, et al. The thumbprint sign: rapid diagnosis of disseminated strongyloidiasis. J Am Acad Dermatol. 1990;23(2, pt 1):324-326.
- Milner PF, Irvine RA, Barton CJ, et al. Intestinal malabsorption in Strongyloides stercoralis infestation. Gut. 1965;6:574-581.
- O'Brien W. Intestinal malabsorption in acute infection with Strongyloides stercoralis. Trans R Soc Trop Med Hyg. 1975;69:69-77.
- Nwokolo C, Imohiosen EA. Strongyloidiasis of respiratory tract presenting as "asthma". Br Med J. 1973;2:153-154.
- Neva FA, Gam AA, Burke J. Comparison of larval antigens in an enzyme-linked immunosorbent assay for strongyloidiasis in humans. J Infect Dis. 1981;144:427-432.
- Genta RM. Strongyloidiasis. In: Walls KW, Schantz PM, eds. Immunodiagnosis of Parasitic Diseases. Vol 1. Orlando, FL: Academic Press Inc; 1986:183-199.
- Igual-Adell R, Oltra-Alcaraz C, Soler-Company E, et al. Efficacy and safety of ivermectin and thiabendazole in the treatment of strongyloidiasis. Expert Opin Pharmacother. 2004;5:2615-2619.
- Maayan S, Wormser GP, Widerhorn J, et al. Strongyloides stercoralis hyperinfection in a patient with the acquired immune deficiency syndrome. Am J Med. 1987;83:945-948.
- Weller IV, Copland P, Gabriel R. Strongyloides stercoralis infection in renal transplant recipients [letter]. Br Med J (Clin Res Ed). 1981;282:524.
- Walzer PD, Milder JE, Banwell JG, et al. Epidemiologic features of Strongyloides stercoralis infection in an endemic area of the United States. Am J Trop Med Hyg. 1982;31:313-319.
- Fulmer HS, Huempfner HR. Intestinal helminths in eastern Kentucky: a survey in three rural counties. Am J Trop Med Hyg. 1965;14:269-275.
- Milder JE, Walzer PD, Kilgore G, et al. Clinical features of Strongyloides stercoralis infection in an endemic area of the United States. Gastroenterology. 1981;80:1481-1488.
- Genta RM, Weesner R, Douce RW, et al. Strongyloidiasis in US veterans of the Vietnam and other wars. JAMA. 1987;258:49-52.
- Gill GV, Welch E, Bailey JW, et al. Chronic Strongyloides stercoralis infection in former British Far East prisoners of war. QJM. 2004;97:789-795.
- Pelletier LL Jr. Chronic strongyloidiasis in World War II Far East ex–prisoners of war. Am J Trop Med Hyg. 1984;33:55-61.
Case Report
A 54-year-old woman with polycystic disease of kidneys was scheduled for renal transplant and presented with a 2-week history of an extremely pruritic rash that primarily affected her torso, buttocks, shoulders, and thighs. She described the lesions as red, raised, and linear, typically lasting less than 24 hours at a time. She had been previously treated with a 5-day course of prednisone by another physician, without improvement. Her only new medication was glucosamine and chondroitin sulfate, which she had started one week prior to the eruption. Raloxifene hydrochloride and cetirizine hydrochloride were long-term medications.
The patient reported one similar episode many years ago. She denied any recent changes in her health and reported no gastrointestinal tract or pulmonary symptoms. She was raised in Panama and had visited there in the past year. She specifically denied walking without shoes.
On physical examination, the patient had a pink, serpiginous, urticarial plaque on the right side of the trunk that was surrounded by a few red serpiginous patches (Figure). Her white blood cell count was 6.5X103/µL (reference range, 3.5–10.5X103/µL), with 19.2% eosinophils (reference, 2.7%). Her absolute eosinophil count was elevated at 1200/µL (reference range, 0–450/µL). A review of prior laboratory test results indicated that her absolute eosinophil count also had been elevated 6 months prior to presentation. Serologic evaluation by enzyme-linked immunosorbent assay was positive for Strongyloides. Results of stool studies did not reveal ova and parasites.
PLEASE REFER TO THE PDF TO VIEW THE FIGURE
The patient was treated with oral thiabendazole 1500 mg twice daily for 2 days and her transplant was postponed. Her rash resolved, but 2 weeks later, her white blood cell count was 5.6X103/µL, with 11.6% eosinophils. She was subsequently treated with a single dose of 200 µg/kg of ivermectin. Results of a complete blood count obtained 2 weeks later demonstrated that her eosinophil count was within reference range and she was able to proceed with the transplant. The patient's sister (the donor) also was born in Panama and had negative serologic evaluation results for Strongyloides. The patient did well following the transplant and the results of repeat serologic evaluations performed 4 months after the transplant were negative for Strongyloides.
Comment
Strongyloides stercoralis is an intestinal nematode primarily found in tropical or subtropical countries. Humans are infected by filariform larvae that dwell in the soil. Larvae penetrate intact skin, gain access to the venous system, pass through the heart to the lungs, enter the pulmonary alveoli, migrate up the tracheobronchial tree, and are swallowed, thereby entering the gastrointestinal tract.1 The larvae mature into adult females that penetrate the mucosa of the small intestine and deposit eggs. The eggs hatch into rhabditiform larvae that are passed in the stool to the soil where transformation into the infective form (filariform) occurs. Autoinfection may take place when this transformation to the infective-stage larvae occurs within the gastrointestinal tract, enabling the infective larvae to invade the lower large bowel or perianal skin and begin the migratory pathway. Autoinfection can allow the persistence of infection for long periods of time and also can allow chronic infections to persist in climates where free-living larvae cannot survive.2
Uncomplicated infection with S stercoralis can cause cutaneous, gastrointestinal tract, and pulmonary symptoms corresponding to the involvement of organs during the parasite's life cycle. Rash is uncommon in acute infection, though it is common in chronic disease. Maculopapular eruptions and chronic urticaria have been reported in up to two-thirds of patients.3 Larva currens is a migratory, rapidly extending, serpiginous, urticarial lesion that is pathognomonic for chronic strongyloidiasis. The rash typically lasts from several hours to several days. It most commonly affects the buttocks, perineum, and thighs, and is secondary to invasion of perianal skin by filariform larvae from the patient's intestine. Arthur and Shelley4 proposed the term larva currens (running larva) because the larvae and subsequent rash can move up to 10 cm per hour.
In a healthy host, the cellular immune system seems to limit parasite invasion of mucosal tissues.5 If immunosuppression occurs, individuals with strongyloidiasis can develop a hyperinfective syndrome and massive numbers of larvae can invade any organ of the body, with a mortality rate of 70% to 90%.6,7 The cutaneous manifestation of disseminated strongyloidiasis is the rapid onset of a petechial and purpuric eruption that typically involves the proximal extremities and trunk and results from massive invasion of the skin by filariform larvae. The "thumbprint sign" refers to a pattern of periumbilical ecchymoses resembling multiple thumbprints that can occur in hyperinfection.8
Gastrointestinal tract symptoms predominate in acute infection. Diarrhea and midepigastric pain that may mimic peptic ulcer disease are common. Diarrhea also can alternate with constipation. Other gastrointestinal tract symptoms include nausea, vomiting, anorexia, pruritus ani, and bloating.1 Some severe cases can have malabsorption and evidence of a protein-losing enteropathy.9,10
Pulmonary symptoms in acute infection can occur and include wheezing, coughing, and shortness of breath.1 Larval migration through the lungs also can lead to transient pulmonary infiltrates. Some patients have presented with asthma.11 Patients with chronic disease may have gastrointestinal tract and pulmonary symptoms, though chronic infection tends to be indolent and patients may be asymptomatic.
Diagnosis can be difficult, as results from stool samples often are negative and multiple samples may be required. Results of biopsies performed on larva currens specimens usually do not reveal larvae, though biopsy results of the petechial and purpuric eruptions of disseminated disease will reveal larvae. Serologic testing with enzyme-linked immunosorbent assay has a sensitivity of approximately 90%.12,13
Traditionally, thiabendazole has been used to treat this infection, though in approximately 30% of cases, the parasite is not eradicated from the feces. Ivermectin has been found to be more effective for treating uncomplicated chronic disease.14
In most cases of disseminated disease, patients were receiving corticosteroids or other immunosuppressive drugs or had an underlying illness, such as malignancy or AIDS.1,2,15,16 Our patient was scheduled to undergo a renal transplant and fatal disseminated strongyloidiasis has been reported in patients undergoing renal transplant.2,16 Morgan et al2 reviewed 29 cases of strongyloidiasis complicating renal transplants; 15 patients died.
Infection in the immunocompromised patient can be complicated by the fact that invasive larvae can transport gram-negative bacilli from the intestine to sites of migration, such as the pulmonary and central nervous systems.5 Gram-negative sepsis, meningitis, or pneumonia can result. Diagnosis can be difficult because eosinophilia often is absent in immunocompromised patients with disseminated disease.5
Although common in tropical and subtropical countries, other geographic regions of endemic Strongyloides are recognized. The climate and soil of the southeastern United States favor the survival of the organism,5 and the parasite was reported in 3% (N=561) of a group of rural Kentucky schoolchildren17; similar findings were reported in another study conducted in Kentucky.18Strongyloides also was the most commonly detected parasite in a review of stool samples examined at the University of Kentucky Medical Center.19 Ex–prisoners of war who served in Southeast Asia during World War II also constitute an at-risk group in the United States.20-22
It is imperative to rule out the presence of this parasite prior to transplant in patients with a geographic history predisposing them to infection, a history of eosinophilia, or symptoms of chronic strongyloidiasis.1 Many transplantation centers routinely screen for this parasite as part of the pretransplant evaluation. Although uncommon in acute infections, cutaneous involvement often is present in chronic strongyloidiasis.1 It also is important to follow patients already treated for larva currens closely posttransplant, as therapeutic failures occur.
Case Report
A 54-year-old woman with polycystic disease of kidneys was scheduled for renal transplant and presented with a 2-week history of an extremely pruritic rash that primarily affected her torso, buttocks, shoulders, and thighs. She described the lesions as red, raised, and linear, typically lasting less than 24 hours at a time. She had been previously treated with a 5-day course of prednisone by another physician, without improvement. Her only new medication was glucosamine and chondroitin sulfate, which she had started one week prior to the eruption. Raloxifene hydrochloride and cetirizine hydrochloride were long-term medications.
The patient reported one similar episode many years ago. She denied any recent changes in her health and reported no gastrointestinal tract or pulmonary symptoms. She was raised in Panama and had visited there in the past year. She specifically denied walking without shoes.
On physical examination, the patient had a pink, serpiginous, urticarial plaque on the right side of the trunk that was surrounded by a few red serpiginous patches (Figure). Her white blood cell count was 6.5X103/µL (reference range, 3.5–10.5X103/µL), with 19.2% eosinophils (reference, 2.7%). Her absolute eosinophil count was elevated at 1200/µL (reference range, 0–450/µL). A review of prior laboratory test results indicated that her absolute eosinophil count also had been elevated 6 months prior to presentation. Serologic evaluation by enzyme-linked immunosorbent assay was positive for Strongyloides. Results of stool studies did not reveal ova and parasites.
PLEASE REFER TO THE PDF TO VIEW THE FIGURE
The patient was treated with oral thiabendazole 1500 mg twice daily for 2 days and her transplant was postponed. Her rash resolved, but 2 weeks later, her white blood cell count was 5.6X103/µL, with 11.6% eosinophils. She was subsequently treated with a single dose of 200 µg/kg of ivermectin. Results of a complete blood count obtained 2 weeks later demonstrated that her eosinophil count was within reference range and she was able to proceed with the transplant. The patient's sister (the donor) also was born in Panama and had negative serologic evaluation results for Strongyloides. The patient did well following the transplant and the results of repeat serologic evaluations performed 4 months after the transplant were negative for Strongyloides.
Comment
Strongyloides stercoralis is an intestinal nematode primarily found in tropical or subtropical countries. Humans are infected by filariform larvae that dwell in the soil. Larvae penetrate intact skin, gain access to the venous system, pass through the heart to the lungs, enter the pulmonary alveoli, migrate up the tracheobronchial tree, and are swallowed, thereby entering the gastrointestinal tract.1 The larvae mature into adult females that penetrate the mucosa of the small intestine and deposit eggs. The eggs hatch into rhabditiform larvae that are passed in the stool to the soil where transformation into the infective form (filariform) occurs. Autoinfection may take place when this transformation to the infective-stage larvae occurs within the gastrointestinal tract, enabling the infective larvae to invade the lower large bowel or perianal skin and begin the migratory pathway. Autoinfection can allow the persistence of infection for long periods of time and also can allow chronic infections to persist in climates where free-living larvae cannot survive.2
Uncomplicated infection with S stercoralis can cause cutaneous, gastrointestinal tract, and pulmonary symptoms corresponding to the involvement of organs during the parasite's life cycle. Rash is uncommon in acute infection, though it is common in chronic disease. Maculopapular eruptions and chronic urticaria have been reported in up to two-thirds of patients.3 Larva currens is a migratory, rapidly extending, serpiginous, urticarial lesion that is pathognomonic for chronic strongyloidiasis. The rash typically lasts from several hours to several days. It most commonly affects the buttocks, perineum, and thighs, and is secondary to invasion of perianal skin by filariform larvae from the patient's intestine. Arthur and Shelley4 proposed the term larva currens (running larva) because the larvae and subsequent rash can move up to 10 cm per hour.
In a healthy host, the cellular immune system seems to limit parasite invasion of mucosal tissues.5 If immunosuppression occurs, individuals with strongyloidiasis can develop a hyperinfective syndrome and massive numbers of larvae can invade any organ of the body, with a mortality rate of 70% to 90%.6,7 The cutaneous manifestation of disseminated strongyloidiasis is the rapid onset of a petechial and purpuric eruption that typically involves the proximal extremities and trunk and results from massive invasion of the skin by filariform larvae. The "thumbprint sign" refers to a pattern of periumbilical ecchymoses resembling multiple thumbprints that can occur in hyperinfection.8
Gastrointestinal tract symptoms predominate in acute infection. Diarrhea and midepigastric pain that may mimic peptic ulcer disease are common. Diarrhea also can alternate with constipation. Other gastrointestinal tract symptoms include nausea, vomiting, anorexia, pruritus ani, and bloating.1 Some severe cases can have malabsorption and evidence of a protein-losing enteropathy.9,10
Pulmonary symptoms in acute infection can occur and include wheezing, coughing, and shortness of breath.1 Larval migration through the lungs also can lead to transient pulmonary infiltrates. Some patients have presented with asthma.11 Patients with chronic disease may have gastrointestinal tract and pulmonary symptoms, though chronic infection tends to be indolent and patients may be asymptomatic.
Diagnosis can be difficult, as results from stool samples often are negative and multiple samples may be required. Results of biopsies performed on larva currens specimens usually do not reveal larvae, though biopsy results of the petechial and purpuric eruptions of disseminated disease will reveal larvae. Serologic testing with enzyme-linked immunosorbent assay has a sensitivity of approximately 90%.12,13
Traditionally, thiabendazole has been used to treat this infection, though in approximately 30% of cases, the parasite is not eradicated from the feces. Ivermectin has been found to be more effective for treating uncomplicated chronic disease.14
In most cases of disseminated disease, patients were receiving corticosteroids or other immunosuppressive drugs or had an underlying illness, such as malignancy or AIDS.1,2,15,16 Our patient was scheduled to undergo a renal transplant and fatal disseminated strongyloidiasis has been reported in patients undergoing renal transplant.2,16 Morgan et al2 reviewed 29 cases of strongyloidiasis complicating renal transplants; 15 patients died.
Infection in the immunocompromised patient can be complicated by the fact that invasive larvae can transport gram-negative bacilli from the intestine to sites of migration, such as the pulmonary and central nervous systems.5 Gram-negative sepsis, meningitis, or pneumonia can result. Diagnosis can be difficult because eosinophilia often is absent in immunocompromised patients with disseminated disease.5
Although common in tropical and subtropical countries, other geographic regions of endemic Strongyloides are recognized. The climate and soil of the southeastern United States favor the survival of the organism,5 and the parasite was reported in 3% (N=561) of a group of rural Kentucky schoolchildren17; similar findings were reported in another study conducted in Kentucky.18Strongyloides also was the most commonly detected parasite in a review of stool samples examined at the University of Kentucky Medical Center.19 Ex–prisoners of war who served in Southeast Asia during World War II also constitute an at-risk group in the United States.20-22
It is imperative to rule out the presence of this parasite prior to transplant in patients with a geographic history predisposing them to infection, a history of eosinophilia, or symptoms of chronic strongyloidiasis.1 Many transplantation centers routinely screen for this parasite as part of the pretransplant evaluation. Although uncommon in acute infections, cutaneous involvement often is present in chronic strongyloidiasis.1 It also is important to follow patients already treated for larva currens closely posttransplant, as therapeutic failures occur.
- Longworth DL, Weller PF. Hyperinfection syndrome with strongyloidiasis. In: Remington JS, Swartz MN, eds. Current Clinical Topics in Infectious Diseases. New York, NY: McGraw-Hill; 1986:1-26.
- Morgan JS, Schaffner W, Sone WJ. Opportunistic strongyloidiasis in renal transplant recipients. Transplantation. 1986;42:518-524.
- Grove DI. Strongyloidiasis in allied ex–prisoners of war in south-east Asia. Br Med J. 1980;280:598-601.
- Arthur RP, Shelley WB. Larva currens; a distinctive variant of cutaneous larva migrans due to Strongyloides stercoralis. AMA Arch Derm. 1958;78:186-190.
- Zygmunt DJ. Strongyloides stercoralis. Infect Control Hosp Epidemiol. 1990;11:495-497.
- Singh S. Human strongyloidiasis in AIDS era: its zoonotic importance. J Assoc Physicians India. 2002;50:415-422.
- Rothenberg ME. Eosinophilia. N Engl J Med. 1998;338:1592-1600.
- Bank DE, Grossman ME, Kohn SR, et al. The thumbprint sign: rapid diagnosis of disseminated strongyloidiasis. J Am Acad Dermatol. 1990;23(2, pt 1):324-326.
- Milner PF, Irvine RA, Barton CJ, et al. Intestinal malabsorption in Strongyloides stercoralis infestation. Gut. 1965;6:574-581.
- O'Brien W. Intestinal malabsorption in acute infection with Strongyloides stercoralis. Trans R Soc Trop Med Hyg. 1975;69:69-77.
- Nwokolo C, Imohiosen EA. Strongyloidiasis of respiratory tract presenting as "asthma". Br Med J. 1973;2:153-154.
- Neva FA, Gam AA, Burke J. Comparison of larval antigens in an enzyme-linked immunosorbent assay for strongyloidiasis in humans. J Infect Dis. 1981;144:427-432.
- Genta RM. Strongyloidiasis. In: Walls KW, Schantz PM, eds. Immunodiagnosis of Parasitic Diseases. Vol 1. Orlando, FL: Academic Press Inc; 1986:183-199.
- Igual-Adell R, Oltra-Alcaraz C, Soler-Company E, et al. Efficacy and safety of ivermectin and thiabendazole in the treatment of strongyloidiasis. Expert Opin Pharmacother. 2004;5:2615-2619.
- Maayan S, Wormser GP, Widerhorn J, et al. Strongyloides stercoralis hyperinfection in a patient with the acquired immune deficiency syndrome. Am J Med. 1987;83:945-948.
- Weller IV, Copland P, Gabriel R. Strongyloides stercoralis infection in renal transplant recipients [letter]. Br Med J (Clin Res Ed). 1981;282:524.
- Walzer PD, Milder JE, Banwell JG, et al. Epidemiologic features of Strongyloides stercoralis infection in an endemic area of the United States. Am J Trop Med Hyg. 1982;31:313-319.
- Fulmer HS, Huempfner HR. Intestinal helminths in eastern Kentucky: a survey in three rural counties. Am J Trop Med Hyg. 1965;14:269-275.
- Milder JE, Walzer PD, Kilgore G, et al. Clinical features of Strongyloides stercoralis infection in an endemic area of the United States. Gastroenterology. 1981;80:1481-1488.
- Genta RM, Weesner R, Douce RW, et al. Strongyloidiasis in US veterans of the Vietnam and other wars. JAMA. 1987;258:49-52.
- Gill GV, Welch E, Bailey JW, et al. Chronic Strongyloides stercoralis infection in former British Far East prisoners of war. QJM. 2004;97:789-795.
- Pelletier LL Jr. Chronic strongyloidiasis in World War II Far East ex–prisoners of war. Am J Trop Med Hyg. 1984;33:55-61.
- Longworth DL, Weller PF. Hyperinfection syndrome with strongyloidiasis. In: Remington JS, Swartz MN, eds. Current Clinical Topics in Infectious Diseases. New York, NY: McGraw-Hill; 1986:1-26.
- Morgan JS, Schaffner W, Sone WJ. Opportunistic strongyloidiasis in renal transplant recipients. Transplantation. 1986;42:518-524.
- Grove DI. Strongyloidiasis in allied ex–prisoners of war in south-east Asia. Br Med J. 1980;280:598-601.
- Arthur RP, Shelley WB. Larva currens; a distinctive variant of cutaneous larva migrans due to Strongyloides stercoralis. AMA Arch Derm. 1958;78:186-190.
- Zygmunt DJ. Strongyloides stercoralis. Infect Control Hosp Epidemiol. 1990;11:495-497.
- Singh S. Human strongyloidiasis in AIDS era: its zoonotic importance. J Assoc Physicians India. 2002;50:415-422.
- Rothenberg ME. Eosinophilia. N Engl J Med. 1998;338:1592-1600.
- Bank DE, Grossman ME, Kohn SR, et al. The thumbprint sign: rapid diagnosis of disseminated strongyloidiasis. J Am Acad Dermatol. 1990;23(2, pt 1):324-326.
- Milner PF, Irvine RA, Barton CJ, et al. Intestinal malabsorption in Strongyloides stercoralis infestation. Gut. 1965;6:574-581.
- O'Brien W. Intestinal malabsorption in acute infection with Strongyloides stercoralis. Trans R Soc Trop Med Hyg. 1975;69:69-77.
- Nwokolo C, Imohiosen EA. Strongyloidiasis of respiratory tract presenting as "asthma". Br Med J. 1973;2:153-154.
- Neva FA, Gam AA, Burke J. Comparison of larval antigens in an enzyme-linked immunosorbent assay for strongyloidiasis in humans. J Infect Dis. 1981;144:427-432.
- Genta RM. Strongyloidiasis. In: Walls KW, Schantz PM, eds. Immunodiagnosis of Parasitic Diseases. Vol 1. Orlando, FL: Academic Press Inc; 1986:183-199.
- Igual-Adell R, Oltra-Alcaraz C, Soler-Company E, et al. Efficacy and safety of ivermectin and thiabendazole in the treatment of strongyloidiasis. Expert Opin Pharmacother. 2004;5:2615-2619.
- Maayan S, Wormser GP, Widerhorn J, et al. Strongyloides stercoralis hyperinfection in a patient with the acquired immune deficiency syndrome. Am J Med. 1987;83:945-948.
- Weller IV, Copland P, Gabriel R. Strongyloides stercoralis infection in renal transplant recipients [letter]. Br Med J (Clin Res Ed). 1981;282:524.
- Walzer PD, Milder JE, Banwell JG, et al. Epidemiologic features of Strongyloides stercoralis infection in an endemic area of the United States. Am J Trop Med Hyg. 1982;31:313-319.
- Fulmer HS, Huempfner HR. Intestinal helminths in eastern Kentucky: a survey in three rural counties. Am J Trop Med Hyg. 1965;14:269-275.
- Milder JE, Walzer PD, Kilgore G, et al. Clinical features of Strongyloides stercoralis infection in an endemic area of the United States. Gastroenterology. 1981;80:1481-1488.
- Genta RM, Weesner R, Douce RW, et al. Strongyloidiasis in US veterans of the Vietnam and other wars. JAMA. 1987;258:49-52.
- Gill GV, Welch E, Bailey JW, et al. Chronic Strongyloides stercoralis infection in former British Far East prisoners of war. QJM. 2004;97:789-795.
- Pelletier LL Jr. Chronic strongyloidiasis in World War II Far East ex–prisoners of war. Am J Trop Med Hyg. 1984;33:55-61.
Clindamycin Phosphate 1.2%–Tretinoin 0.025% Gel: Vehicle Characteristics, Stability, and Tolerability
Prevalence of Mood and Sleep Problems in Chronic Skin Diseases: A Pilot Study
What Is Your Diagnosis? Cutaneous Mastocytosis (Urticaria Pigmentosa)
Adviser ONLY on the Web
BSO for breast Ca patient—OK to code as CIS surgery?
In any case, your primary diagnosis would be V50.42 (prophylactic organ removal, ovary), followed by V10.3, then followed by V86.1 because she is probably estrogen-receptor positive (meaning that taking anti-estrogens will not prevent the return of cancer).
If she is still being treated for cancer in situ, then 233.0 is correct but V50.42 needs to be the primary diagnosis because, otherwise, you get a mismatch between the diagnosis and the surgery (i.e., it appears that you are performing an oophorectomy because of breast cancer).
BSO for breast Ca patient—OK to code as CIS surgery?
In any case, your primary diagnosis would be V50.42 (prophylactic organ removal, ovary), followed by V10.3, then followed by V86.1 because she is probably estrogen-receptor positive (meaning that taking anti-estrogens will not prevent the return of cancer).
If she is still being treated for cancer in situ, then 233.0 is correct but V50.42 needs to be the primary diagnosis because, otherwise, you get a mismatch between the diagnosis and the surgery (i.e., it appears that you are performing an oophorectomy because of breast cancer).
BSO for breast Ca patient—OK to code as CIS surgery?
In any case, your primary diagnosis would be V50.42 (prophylactic organ removal, ovary), followed by V10.3, then followed by V86.1 because she is probably estrogen-receptor positive (meaning that taking anti-estrogens will not prevent the return of cancer).
If she is still being treated for cancer in situ, then 233.0 is correct but V50.42 needs to be the primary diagnosis because, otherwise, you get a mismatch between the diagnosis and the surgery (i.e., it appears that you are performing an oophorectomy because of breast cancer).