User login
Inpatient management of diabetes: An increasing challenge to the hospitalist physician
In this supplement, Avoiding Complications in the Hospitalized Patient: The Case for Tight Glycemic Control, Dr. Susan S. Braithwaite defines specific populations, disorders, and hospital settings for which there now is strong evidence supporting the belief that short‐term glycemic control will affect outcomes during the course of hospital treatment.1 She provides a comprehensive summary of key studies showing the benefits of tight glycemic control in hospitalized patients. Dr. James S. Krinsley reviews the evidence that supports more intensive glucose control, along with a real‐world success story that demonstrates how to apply the new glycemic targets in a multidisciplinary performance improvement project.2 He discusses important issues surrounding the successful implementation of a tight glycemic control protocol, including barriers to implementation, setting the glycemic target, and tips for choosing the right protocol. Dr. Franklin Michota describes a practical guideline for how to implement a more physiologic and sensible insulin regimen for management of inpatient hyperglycemia.3 He reports on the disadvantages of the sliding scale and recommends the implementation of a standardized subcutaneous insulin order set with the use of scheduled basal and nutritional insulin in the inpatient management of diabetes. Drs. Asudani and Calles‐Escandon focus on the management of noncritically ill patients with hyperglycemia in medical and surgical units.4 They propose a successful insulin regimen to be used in non‐ICU settings that is based on the combined use of basal, alimentary (prandial), and corrective insulin. This supplement provides the hospitalist physician with the necessary tools to implement glycemic control programs in critical care and noncritical care units and can be summarized as follows.
Hyperglycemia in hospitalized patients is a common, serious, and costly health care problem with profound medical consequences. Thirty‐eight percent of patients admitted to the hospital have hyperglycemia, about one third of whom have no history of diabetes before admission.5 Increasing evidence indicates that the development of hyperglycemia during acute medical or surgical illness is not a physiologic or benign condition but is a marker of poor clinical outcome and mortality.510 Evidence from observational studies indicates that the development of hyperglycemia in critical illness is associated with an increased risk of complications and mortality, a longer hospital stay, a higher rate of admission to the ICU, and a higher likelihood that transitional or nursing home care after hospital discharge will be required.5, 7, 914 Prospective randomized trials with critical care patients have shown that aggressive glycemic control reduces short‐ and long‐term mortality, multiorgan failure, systemic infections, and length of hospital and ICU stays7, 911 and lower the total cost of hospitalization.15 Controlling hyperglycemia is also important for adult patients admitted to general surgical and medical wards. In such patients, the presence of hyperglycemia is associated with prolonged hospital stay, infection, disability after hospital discharge, and death.5, 11, 16
Insulin, given either intravenously as a continuous infusion or subcutaneously, is currently the only available agent for effectively controlling glycemia in the hospital. In the critical care setting, a variety of intravenous infusion protocols have been shown to be effective in achieving glycemic control with a low‐rate of hypoglycemic events and in improving hospital outcomes.1723 However, no prospective and randomized interventional studies have focused on the optimal management of hyperglycemia and its effect on clinical outcome among noncritically ill patients admitted to general medicine services. Fear of hypoglycemia leads physicians to inappropriately hold to their patients' previous outpatient diabetic regimens and to initiate sliding‐scale insulin coverage, a practice associated with limited therapeutic success.20, 24, 25 The most physiologic and effective insulin therapy provides both basal and nutritional insulin.11 The basal insulin requirement is the amount of exogenous insulin necessary to regulate hepatic glucose production and peripheral glucose uptake and to prevent ketogenesis. The nutritional, or prandial, insulin requirement is the amount of insulin necessary to cover meals and the administration of intravenous dextrose, TPN, and enteral feedings. Prandial or mealtime insulin replacement has its main effect on peripheral glucose disposal. In addition to the basal and nutritional insulin requirements, patients often require supplemental or correction doses of insulin to treat unexpected hyperglycemia. The supplemental algorithm should not be confused with the sliding scale, which traditionally has been used alone, with no scheduled dose. Insulins used for basal requirements are NPH (which is intermediate acting) and long‐acting insulin analogues (glargine and detemir). To cover nutritional need, regular insulin or rapid‐acting analogues (lispro, aspart, glulisine) can be used. Although no inpatient controlled trials using the basal‐nutritional insulin regimen have been reported, the use of basal and nutritional insulin regimen may be a better alternative to the use of intermediate insulin (NPH) and regular insulin in hospitalized patients.
Hypoglycemia in hospitalized patients with diabetes is a concern, and it has been a major barrier to aggressive treatment of hyperglycemia in the hospital. Severe hypoglycemia, defined as a glucose level less than 40 mg/dL, occurred at least once in 5.1% of patients in the intensively treated group in Van den Berghe's surgical ICU study, versus 0.8% of patients in the conventionally treated group.19 The incidence of severe hypoglycemia (<40 mg/dL) reported by Krinsley et al. prior to institution of the intensified protocol was 0.35% of all values obtained, compared to 0.34% of those obtained during the treatment period, again without any overt adverse consequences.26 Factors that increase the risk of hypoglycemia in the hospital include inadequate glucose monitoring, lack of clear communication or coordination between the dietary team, transportation, and nursing staff, and indecipherable orders. Clear algorithms for insulin orders and clear hypoglycemia protocols are critical to preventing hypoglycemia.
What should the target blood glucose level be in noncritically ill patients with diabetes? A recent position statement of the American Association of Clinical Endocrinology with cosponsorship by the American Diabetes Association, the American Heart Association, the American Society of Anesthesiologists, the Endocrine Society, the Society of Critical Care Medicine, the Society of Hospital Medicine, the Society of Thoracic Surgeons, and the American Association of Diabetes Educators27 recommended a glycemic target between 80 and 110 mg/dL for hospitalized patients in the intensive care unit and a preprandial glucose goal of less than 110 mg/dL and a random glucose less than 180 mg/dL for patients in noncritical care settings. The Joint Commission on Accreditation of Healthcare Organization recently proposed tight glucose control for the critically ill as a core quality of care measure for all U.S. hospitals that participate in the Medicare program (
- .Defining the benefits of euglycemia in the hospitalized patient.J Hosp Med.2007;2(suppl 1):5–12.
- .Translating evidence into practice in managing inpatient hyperglycemia.J Hosp Med.2007;2(suppl 1):13–19.
- .What are the disadvantages of sliding‐scale insulin?J Hosp Med.2007;2(suppl 1):20–22.
- ,.Inpatient hyperglycemia: Slide through the scale but cover the bases first.J Hosp Med.2007;2(suppl 1):23–32.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet2000;355:773–778.
- ,,,:Glucose control and mortality in critically ill patients.JAMA.2003;290:2041–2047.
- ,:Hospital management of diabetes.Endocrinol Metab Clin North Am.2000;29:745–770.
- ,,,,,.Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era?J Am Coll Cardiol.2002;40:1748–1754.
- ,,.Admission plasma glucose. Independent risk factor for long‐term prognosis after myocardial infarction even in nondiabetic patients.Diabetes Care.1999;22:1827–1831.
- ,,,,,,.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.27:553–597,2004
- ,,.Hyperglycemia in acutely ill patients.JAMA.2002;288:2167–2169.
- ,,,,,,.Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus.Arch Intern Med.2004;164:982–988.
- ,.ICU care for patients with diabetes.Current Opinions Endocrinol.2004;11:75–81.
- ,.Cost analysis of intensive glycemic control in critically ill adult patients.Chest.2006;129:644–650.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.JPEN J Parenter Enteral Nutr.1998;22:77–81.
- ,,:Feasibility of insulin‐glucose infusion in diabetic patients with acute myocardial infarction. A report from the multicenter trial: DIGAMI.Diabetes Care.1994;17:1007–1014.
- ,,,,.Hyperglycemic crises in urban blacks.Arch Intern Med.1997;157:669–675.
- ,,, et al.Intensive insulin therapy in the critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,.Intravenous insulin nomogram improves blood glucose control in the critically ill.Crit Care Med.2001;29:1714–1719.
- ,,, et al.Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,,.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–360; discussion360–352.
- ,,, et al.Implementation of a safe and effective insulin infusion protocol in a medical intensive care unit.Diabetes Care.2004;27:461–467.
- ,,.Glycemic control and sliding scale insulin use in medical inpatients with diabetes mellitus.Arch Intern Med.1997;157:545–552.
- ,,,,.Efficacy of sliding‐scale insulin therapy: a comparison with prospective regimens.Fam Pract Res J.1994;14:313–322.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(suppl 2):4–9.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979.
In this supplement, Avoiding Complications in the Hospitalized Patient: The Case for Tight Glycemic Control, Dr. Susan S. Braithwaite defines specific populations, disorders, and hospital settings for which there now is strong evidence supporting the belief that short‐term glycemic control will affect outcomes during the course of hospital treatment.1 She provides a comprehensive summary of key studies showing the benefits of tight glycemic control in hospitalized patients. Dr. James S. Krinsley reviews the evidence that supports more intensive glucose control, along with a real‐world success story that demonstrates how to apply the new glycemic targets in a multidisciplinary performance improvement project.2 He discusses important issues surrounding the successful implementation of a tight glycemic control protocol, including barriers to implementation, setting the glycemic target, and tips for choosing the right protocol. Dr. Franklin Michota describes a practical guideline for how to implement a more physiologic and sensible insulin regimen for management of inpatient hyperglycemia.3 He reports on the disadvantages of the sliding scale and recommends the implementation of a standardized subcutaneous insulin order set with the use of scheduled basal and nutritional insulin in the inpatient management of diabetes. Drs. Asudani and Calles‐Escandon focus on the management of noncritically ill patients with hyperglycemia in medical and surgical units.4 They propose a successful insulin regimen to be used in non‐ICU settings that is based on the combined use of basal, alimentary (prandial), and corrective insulin. This supplement provides the hospitalist physician with the necessary tools to implement glycemic control programs in critical care and noncritical care units and can be summarized as follows.
Hyperglycemia in hospitalized patients is a common, serious, and costly health care problem with profound medical consequences. Thirty‐eight percent of patients admitted to the hospital have hyperglycemia, about one third of whom have no history of diabetes before admission.5 Increasing evidence indicates that the development of hyperglycemia during acute medical or surgical illness is not a physiologic or benign condition but is a marker of poor clinical outcome and mortality.510 Evidence from observational studies indicates that the development of hyperglycemia in critical illness is associated with an increased risk of complications and mortality, a longer hospital stay, a higher rate of admission to the ICU, and a higher likelihood that transitional or nursing home care after hospital discharge will be required.5, 7, 914 Prospective randomized trials with critical care patients have shown that aggressive glycemic control reduces short‐ and long‐term mortality, multiorgan failure, systemic infections, and length of hospital and ICU stays7, 911 and lower the total cost of hospitalization.15 Controlling hyperglycemia is also important for adult patients admitted to general surgical and medical wards. In such patients, the presence of hyperglycemia is associated with prolonged hospital stay, infection, disability after hospital discharge, and death.5, 11, 16
Insulin, given either intravenously as a continuous infusion or subcutaneously, is currently the only available agent for effectively controlling glycemia in the hospital. In the critical care setting, a variety of intravenous infusion protocols have been shown to be effective in achieving glycemic control with a low‐rate of hypoglycemic events and in improving hospital outcomes.1723 However, no prospective and randomized interventional studies have focused on the optimal management of hyperglycemia and its effect on clinical outcome among noncritically ill patients admitted to general medicine services. Fear of hypoglycemia leads physicians to inappropriately hold to their patients' previous outpatient diabetic regimens and to initiate sliding‐scale insulin coverage, a practice associated with limited therapeutic success.20, 24, 25 The most physiologic and effective insulin therapy provides both basal and nutritional insulin.11 The basal insulin requirement is the amount of exogenous insulin necessary to regulate hepatic glucose production and peripheral glucose uptake and to prevent ketogenesis. The nutritional, or prandial, insulin requirement is the amount of insulin necessary to cover meals and the administration of intravenous dextrose, TPN, and enteral feedings. Prandial or mealtime insulin replacement has its main effect on peripheral glucose disposal. In addition to the basal and nutritional insulin requirements, patients often require supplemental or correction doses of insulin to treat unexpected hyperglycemia. The supplemental algorithm should not be confused with the sliding scale, which traditionally has been used alone, with no scheduled dose. Insulins used for basal requirements are NPH (which is intermediate acting) and long‐acting insulin analogues (glargine and detemir). To cover nutritional need, regular insulin or rapid‐acting analogues (lispro, aspart, glulisine) can be used. Although no inpatient controlled trials using the basal‐nutritional insulin regimen have been reported, the use of basal and nutritional insulin regimen may be a better alternative to the use of intermediate insulin (NPH) and regular insulin in hospitalized patients.
Hypoglycemia in hospitalized patients with diabetes is a concern, and it has been a major barrier to aggressive treatment of hyperglycemia in the hospital. Severe hypoglycemia, defined as a glucose level less than 40 mg/dL, occurred at least once in 5.1% of patients in the intensively treated group in Van den Berghe's surgical ICU study, versus 0.8% of patients in the conventionally treated group.19 The incidence of severe hypoglycemia (<40 mg/dL) reported by Krinsley et al. prior to institution of the intensified protocol was 0.35% of all values obtained, compared to 0.34% of those obtained during the treatment period, again without any overt adverse consequences.26 Factors that increase the risk of hypoglycemia in the hospital include inadequate glucose monitoring, lack of clear communication or coordination between the dietary team, transportation, and nursing staff, and indecipherable orders. Clear algorithms for insulin orders and clear hypoglycemia protocols are critical to preventing hypoglycemia.
What should the target blood glucose level be in noncritically ill patients with diabetes? A recent position statement of the American Association of Clinical Endocrinology with cosponsorship by the American Diabetes Association, the American Heart Association, the American Society of Anesthesiologists, the Endocrine Society, the Society of Critical Care Medicine, the Society of Hospital Medicine, the Society of Thoracic Surgeons, and the American Association of Diabetes Educators27 recommended a glycemic target between 80 and 110 mg/dL for hospitalized patients in the intensive care unit and a preprandial glucose goal of less than 110 mg/dL and a random glucose less than 180 mg/dL for patients in noncritical care settings. The Joint Commission on Accreditation of Healthcare Organization recently proposed tight glucose control for the critically ill as a core quality of care measure for all U.S. hospitals that participate in the Medicare program (
In this supplement, Avoiding Complications in the Hospitalized Patient: The Case for Tight Glycemic Control, Dr. Susan S. Braithwaite defines specific populations, disorders, and hospital settings for which there now is strong evidence supporting the belief that short‐term glycemic control will affect outcomes during the course of hospital treatment.1 She provides a comprehensive summary of key studies showing the benefits of tight glycemic control in hospitalized patients. Dr. James S. Krinsley reviews the evidence that supports more intensive glucose control, along with a real‐world success story that demonstrates how to apply the new glycemic targets in a multidisciplinary performance improvement project.2 He discusses important issues surrounding the successful implementation of a tight glycemic control protocol, including barriers to implementation, setting the glycemic target, and tips for choosing the right protocol. Dr. Franklin Michota describes a practical guideline for how to implement a more physiologic and sensible insulin regimen for management of inpatient hyperglycemia.3 He reports on the disadvantages of the sliding scale and recommends the implementation of a standardized subcutaneous insulin order set with the use of scheduled basal and nutritional insulin in the inpatient management of diabetes. Drs. Asudani and Calles‐Escandon focus on the management of noncritically ill patients with hyperglycemia in medical and surgical units.4 They propose a successful insulin regimen to be used in non‐ICU settings that is based on the combined use of basal, alimentary (prandial), and corrective insulin. This supplement provides the hospitalist physician with the necessary tools to implement glycemic control programs in critical care and noncritical care units and can be summarized as follows.
Hyperglycemia in hospitalized patients is a common, serious, and costly health care problem with profound medical consequences. Thirty‐eight percent of patients admitted to the hospital have hyperglycemia, about one third of whom have no history of diabetes before admission.5 Increasing evidence indicates that the development of hyperglycemia during acute medical or surgical illness is not a physiologic or benign condition but is a marker of poor clinical outcome and mortality.510 Evidence from observational studies indicates that the development of hyperglycemia in critical illness is associated with an increased risk of complications and mortality, a longer hospital stay, a higher rate of admission to the ICU, and a higher likelihood that transitional or nursing home care after hospital discharge will be required.5, 7, 914 Prospective randomized trials with critical care patients have shown that aggressive glycemic control reduces short‐ and long‐term mortality, multiorgan failure, systemic infections, and length of hospital and ICU stays7, 911 and lower the total cost of hospitalization.15 Controlling hyperglycemia is also important for adult patients admitted to general surgical and medical wards. In such patients, the presence of hyperglycemia is associated with prolonged hospital stay, infection, disability after hospital discharge, and death.5, 11, 16
Insulin, given either intravenously as a continuous infusion or subcutaneously, is currently the only available agent for effectively controlling glycemia in the hospital. In the critical care setting, a variety of intravenous infusion protocols have been shown to be effective in achieving glycemic control with a low‐rate of hypoglycemic events and in improving hospital outcomes.1723 However, no prospective and randomized interventional studies have focused on the optimal management of hyperglycemia and its effect on clinical outcome among noncritically ill patients admitted to general medicine services. Fear of hypoglycemia leads physicians to inappropriately hold to their patients' previous outpatient diabetic regimens and to initiate sliding‐scale insulin coverage, a practice associated with limited therapeutic success.20, 24, 25 The most physiologic and effective insulin therapy provides both basal and nutritional insulin.11 The basal insulin requirement is the amount of exogenous insulin necessary to regulate hepatic glucose production and peripheral glucose uptake and to prevent ketogenesis. The nutritional, or prandial, insulin requirement is the amount of insulin necessary to cover meals and the administration of intravenous dextrose, TPN, and enteral feedings. Prandial or mealtime insulin replacement has its main effect on peripheral glucose disposal. In addition to the basal and nutritional insulin requirements, patients often require supplemental or correction doses of insulin to treat unexpected hyperglycemia. The supplemental algorithm should not be confused with the sliding scale, which traditionally has been used alone, with no scheduled dose. Insulins used for basal requirements are NPH (which is intermediate acting) and long‐acting insulin analogues (glargine and detemir). To cover nutritional need, regular insulin or rapid‐acting analogues (lispro, aspart, glulisine) can be used. Although no inpatient controlled trials using the basal‐nutritional insulin regimen have been reported, the use of basal and nutritional insulin regimen may be a better alternative to the use of intermediate insulin (NPH) and regular insulin in hospitalized patients.
Hypoglycemia in hospitalized patients with diabetes is a concern, and it has been a major barrier to aggressive treatment of hyperglycemia in the hospital. Severe hypoglycemia, defined as a glucose level less than 40 mg/dL, occurred at least once in 5.1% of patients in the intensively treated group in Van den Berghe's surgical ICU study, versus 0.8% of patients in the conventionally treated group.19 The incidence of severe hypoglycemia (<40 mg/dL) reported by Krinsley et al. prior to institution of the intensified protocol was 0.35% of all values obtained, compared to 0.34% of those obtained during the treatment period, again without any overt adverse consequences.26 Factors that increase the risk of hypoglycemia in the hospital include inadequate glucose monitoring, lack of clear communication or coordination between the dietary team, transportation, and nursing staff, and indecipherable orders. Clear algorithms for insulin orders and clear hypoglycemia protocols are critical to preventing hypoglycemia.
What should the target blood glucose level be in noncritically ill patients with diabetes? A recent position statement of the American Association of Clinical Endocrinology with cosponsorship by the American Diabetes Association, the American Heart Association, the American Society of Anesthesiologists, the Endocrine Society, the Society of Critical Care Medicine, the Society of Hospital Medicine, the Society of Thoracic Surgeons, and the American Association of Diabetes Educators27 recommended a glycemic target between 80 and 110 mg/dL for hospitalized patients in the intensive care unit and a preprandial glucose goal of less than 110 mg/dL and a random glucose less than 180 mg/dL for patients in noncritical care settings. The Joint Commission on Accreditation of Healthcare Organization recently proposed tight glucose control for the critically ill as a core quality of care measure for all U.S. hospitals that participate in the Medicare program (
- .Defining the benefits of euglycemia in the hospitalized patient.J Hosp Med.2007;2(suppl 1):5–12.
- .Translating evidence into practice in managing inpatient hyperglycemia.J Hosp Med.2007;2(suppl 1):13–19.
- .What are the disadvantages of sliding‐scale insulin?J Hosp Med.2007;2(suppl 1):20–22.
- ,.Inpatient hyperglycemia: Slide through the scale but cover the bases first.J Hosp Med.2007;2(suppl 1):23–32.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet2000;355:773–778.
- ,,,:Glucose control and mortality in critically ill patients.JAMA.2003;290:2041–2047.
- ,:Hospital management of diabetes.Endocrinol Metab Clin North Am.2000;29:745–770.
- ,,,,,.Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era?J Am Coll Cardiol.2002;40:1748–1754.
- ,,.Admission plasma glucose. Independent risk factor for long‐term prognosis after myocardial infarction even in nondiabetic patients.Diabetes Care.1999;22:1827–1831.
- ,,,,,,.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.27:553–597,2004
- ,,.Hyperglycemia in acutely ill patients.JAMA.2002;288:2167–2169.
- ,,,,,,.Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus.Arch Intern Med.2004;164:982–988.
- ,.ICU care for patients with diabetes.Current Opinions Endocrinol.2004;11:75–81.
- ,.Cost analysis of intensive glycemic control in critically ill adult patients.Chest.2006;129:644–650.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.JPEN J Parenter Enteral Nutr.1998;22:77–81.
- ,,:Feasibility of insulin‐glucose infusion in diabetic patients with acute myocardial infarction. A report from the multicenter trial: DIGAMI.Diabetes Care.1994;17:1007–1014.
- ,,,,.Hyperglycemic crises in urban blacks.Arch Intern Med.1997;157:669–675.
- ,,, et al.Intensive insulin therapy in the critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,.Intravenous insulin nomogram improves blood glucose control in the critically ill.Crit Care Med.2001;29:1714–1719.
- ,,, et al.Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,,.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–360; discussion360–352.
- ,,, et al.Implementation of a safe and effective insulin infusion protocol in a medical intensive care unit.Diabetes Care.2004;27:461–467.
- ,,.Glycemic control and sliding scale insulin use in medical inpatients with diabetes mellitus.Arch Intern Med.1997;157:545–552.
- ,,,,.Efficacy of sliding‐scale insulin therapy: a comparison with prospective regimens.Fam Pract Res J.1994;14:313–322.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(suppl 2):4–9.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979.
- .Defining the benefits of euglycemia in the hospitalized patient.J Hosp Med.2007;2(suppl 1):5–12.
- .Translating evidence into practice in managing inpatient hyperglycemia.J Hosp Med.2007;2(suppl 1):13–19.
- .What are the disadvantages of sliding‐scale insulin?J Hosp Med.2007;2(suppl 1):20–22.
- ,.Inpatient hyperglycemia: Slide through the scale but cover the bases first.J Hosp Med.2007;2(suppl 1):23–32.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet2000;355:773–778.
- ,,,:Glucose control and mortality in critically ill patients.JAMA.2003;290:2041–2047.
- ,:Hospital management of diabetes.Endocrinol Metab Clin North Am.2000;29:745–770.
- ,,,,,.Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era?J Am Coll Cardiol.2002;40:1748–1754.
- ,,.Admission plasma glucose. Independent risk factor for long‐term prognosis after myocardial infarction even in nondiabetic patients.Diabetes Care.1999;22:1827–1831.
- ,,,,,,.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.27:553–597,2004
- ,,.Hyperglycemia in acutely ill patients.JAMA.2002;288:2167–2169.
- ,,,,,,.Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus.Arch Intern Med.2004;164:982–988.
- ,.ICU care for patients with diabetes.Current Opinions Endocrinol.2004;11:75–81.
- ,.Cost analysis of intensive glycemic control in critically ill adult patients.Chest.2006;129:644–650.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.JPEN J Parenter Enteral Nutr.1998;22:77–81.
- ,,:Feasibility of insulin‐glucose infusion in diabetic patients with acute myocardial infarction. A report from the multicenter trial: DIGAMI.Diabetes Care.1994;17:1007–1014.
- ,,,,.Hyperglycemic crises in urban blacks.Arch Intern Med.1997;157:669–675.
- ,,, et al.Intensive insulin therapy in the critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,.Intravenous insulin nomogram improves blood glucose control in the critically ill.Crit Care Med.2001;29:1714–1719.
- ,,, et al.Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting.J Thorac Cardiovasc Surg.2003;125:1007–1021.
- ,,,.Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures.Ann Thorac Surg.1999;67:352–360; discussion360–352.
- ,,, et al.Implementation of a safe and effective insulin infusion protocol in a medical intensive care unit.Diabetes Care.2004;27:461–467.
- ,,.Glycemic control and sliding scale insulin use in medical inpatients with diabetes mellitus.Arch Intern Med.1997;157:545–552.
- ,,,,.Efficacy of sliding‐scale insulin therapy: a comparison with prospective regimens.Fam Pract Res J.1994;14:313–322.
- .Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients.Mayo Clin Proc.2003;78:1471–1478.
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(suppl 2):4–9.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979.
Inpatient Hyperglycemia
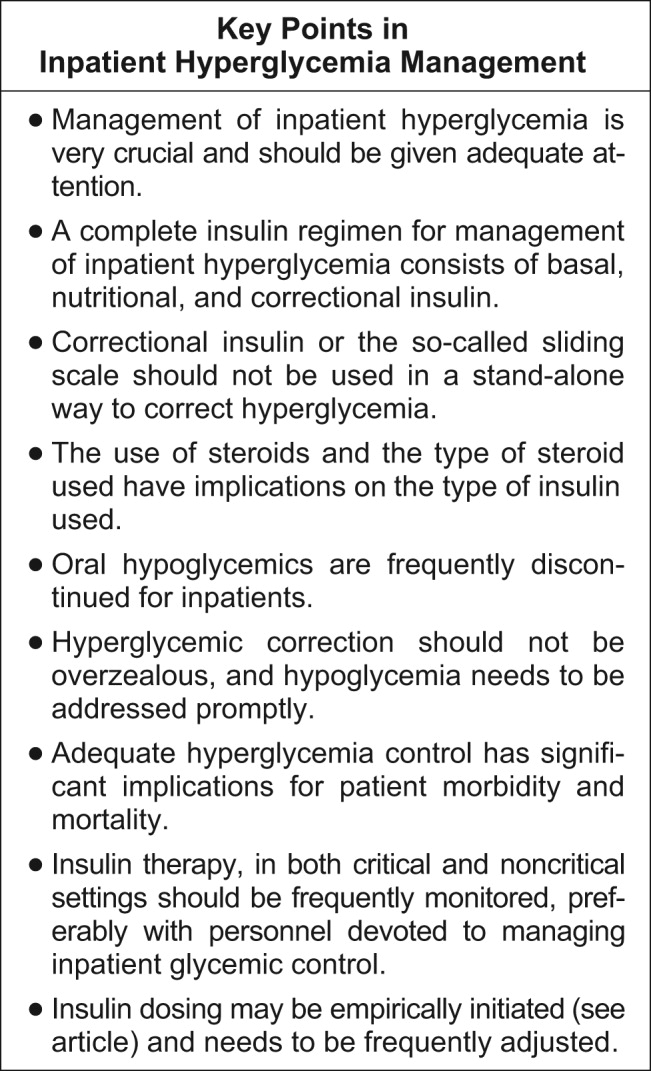
A very compelling and growing body of evidence highlights the benefits to hospitalized patients of intensive (insulin‐based) glycemic control. However, we have a tendency to attend to patients' acute problems during inpatient stays, and glycemic control frequently takes a backseat. As hospitalists, we frequently come across patients with diabetes admitted for various other reasons, as well as patients who develop hyperglycemia while hospitalized. During a hospital stay, it is usually not recommended that an oral hypoglycemic regimen be continued, and insulin use is necessary to more reliably control blood glucose. In this article, we emphasize the need to better manage inpatient hyperglycemia and to make a conscious effort to prescribe insulin in a more rational manner. We propose that insulin orders for an inpatient address: (1) basal insulinization, (2) meal or prandial insulin, and (3) corrective insulin. In this schema, the supplemental boluses of insulin administered to correct a blood glucose level that exceeds a set value are viewed as an adjunct to a basal/bolus insulin regimen. We also recognize the practical limitations of attaining stringent glucose targets and pinpoint those areas in need of further research.
BACKGROUND
It is not entirely clear how and when the use of the very popular insulin sliding scale as the sole approach to controlling inpatient hyperglycemia became such a widespread practice. However, the sliding scale has been passed along to subsequent generations as gospel. Despite receiving much criticism, the regular insulin sliding scale remains sacred to medical practitioners. Unfortunately, the sliding scale is very frequently the sole therapeutic tool used to control hyperglycemia, and not as a complement to a more physiologically complete (basal/bolus) insulin regimen. As attractive as the use of continuous intravenous insulin infusion is to endocrinologists, it is not frequently used outside intensive care units for many reasons. Where there is apparent agreement is in the need to improve inpatient management of hyperglycemia.
THE PROBLEM: HYPERGLYCEMIC INPATIENT
Hyperglycemia is defined as a fasting glucose level greater than 126 mg/dL or 2 or more random blood glucose levels greater than 200 mg/dL.1 Not infrequently, patients admitted to our ward have a history of diabetes; however, a good proportion of admitted patients have no such history. In a retrospective analysis of more than 2000 consecutive hospital admissions, hyperglycemia was found in as many as 38% of the patients in whom blood glucose was measured and documented in the chart, about a third of which did not previously carry the diagnosis of diabetes. Hyperglycemia in this specific setting, dubbed stress hyperglycemia,1 is quite frequently found in hospitalized patients and has been shown to increase the risk of death, congestive heart failure, and cardiogenic shock after myocardial infarction.2 Acute insulin resistance is also seen frequently in an acutely ill patient and is attributed to the release and metabolic actions of counterregulatory hormones and cytokine excess.3 Patients often require increased amounts of insulin to maintain glucose at an acceptable level. Iatrogenic hyperglycemia may occur as a consequence of glucocorticoids or excessive infusion of dextrose. In critically ill patients, vasopressors may also be associated with iatrogenic hyperglycemia. Inpatient hyperglycemia is associated with nosocomial infections, increased mortality, increased length of stay, and poor overall outcome.4 Of interest is that stress hyperglycemia was associated with more adverse outcome than was hyperglycemia in a patient with known diabetes.1, 2 We are not sure if this phenomenon of stress hyperglycemia is pathogenic or serves as a marker of disease severity.
Is Hyperglycemia Really a Problem?
Compelling evidence that control of hyperglycemia improves the outcomes of patients undergoing cardiothoracic surgery was provided by the Portland trial. Although this study was not randomized and its glycemia targets were not well defined, it demonstrated that better control of blood glucose levels drastically reduces the incidence of chest wall infections and the need for transfusions and significantly shortens hospital length of stay (LOS).5
The results of the Diabetes Mellitus Insulin‐Glucose in Acute Myocardial Infarction (DIGAMI) study showed that hyperglycemic patients with acute myocardial infarction had improved outcomes when intravenous administration of insulin was used to aggressively control glycemia.6 Van den Berghe et al. found significantly lower mortality and morbidity rates in surgical intensive care unit patients in whom aggressive glycemic control was attained with continuous intravenous insulin infusion. The study also identified reduced requirement of antibiotics, red cell transfusions, dialysis, and ventilatory support with aggressive glycemic control.7 It was also shown that there was significantly reduced morbidity in all patients in the medical ICU receiving intensive insulin therapy.8 Another meta‐analysis found that insulin therapy initiated in hospitalized critically ill patients in different clinical settings had a beneficial effect on short‐term mortality.9 Krinsley observed hyperglycemia to be associated with adverse outcomes in acutely ill adult patients and that its treatment has been shown to improve mortality and morbidity in a variety of settings.10 In their study of adults with diabetes, Golden et al. identified hyperglycemia as an independent risk factor for surgical infection of diabetic patients undergoing cardiac surgery.11 A meta‐analysis by Capes et al. showed a 3‐fold higher risk of poor functional recovery in nondiabetic hyperglycemic patients compared to that of nondiabetic euglycemic patients.2 A recent retrospective analysis found that patients with hyperglycemia treated for acute exacerbation of chronic obstructive pulmonary disease had poor outcomes.12
It is possible to give an account and references of only a limited number of such studies. The prevailing message conveyed in all these studies is that patients with poorly managed hyperglycemia have a poor overall outcome. Hence, the need to better manage inpatient hyperglycemia cannot be overemphasized.13
After an extensive search, we could not find well‐designed prospective randomized studies of patients who are not acutely ill or are outside the perisurgical period. However, the DIGAMI, Van den Berghe, and Portland trials generated a powerful and large momentum that has created interest in establishing protocols for keeping the blood glucose of patients in most medical and surgical critical care units in the suggested range.57, 13 Moreover, extrapolation of the data to noncritical and nonsurgical patients made possible a consensus conference organized by the American Association of Clinical Endocrinologists (AACE) that garnered support from many other medical associations. The position paper published by the AACE calls for tighter glycemic control in hospitalized patients. The AACE recommends that blood glucose concentrations for intensive care unit patients be maintained below 110 mg/dL. In noncritically ill patients, the preprandial glucose level should not exceed 110 mg/dL, and maximum glucose should not exceed 180 mg/dL.14 The American Diabetes Association (ADA) does not recommend any target glucose values for noncritical patients but does believe there is a need to have better inpatient hyperglycemic management. Some authorities believe that until the amount of scientific data increases, it is prudent to stay within the ADA‐recommended ambulatory guidelines for a preprandial plasma glucose level of 90‐130 mg/dL15 and a postprandial blood glucose level not to exceed 180 mg/dL.
Additionally, due attention must be paid to hypoglycemia secondary to aggressive glycemic control.
Because of the absence of evidence‐based information, it is not surprising that opinions conflict about the optimal level of blood glucose for an inpatient. We believe that in the absence of definitive evidence, it is prudent to adhere to the targets recommended by these associations.
A SOLUTION: WHAT TO DO AND HOW TO DO IT
Ideally, a system should be established to attain euglycemia without the attendant risk of hypoglycemia. The Joint Commission on Accreditation of Healthcare Organizations recently showed recognition of this need this by collaborating with the American Diabetes Association to establish a program to certify inpatient diabetes care center programs that meet national standards. The program must be carried out in all inpatient settings and should include the following elements16:
-
Specific staff education requirements;
-
Written blood glucosemonitoring protocols;
-
Plans for the treatment of hypoglycemia and hyperglycemia;
-
Collection of data on the incidence of hypoglycemia;
-
Education of patients on self‐managing their diabetes; and
-
An identified program champion or program champion team.
The Joint Commission's Advanced Inpatient Diabetes Certification Program is based on the ADA guidelines; the scope of this manuscript does not cover all the elements required to receive certification.16 In the rest of the article, we focus on the basic principles of the use of insulin to control hyperglycemia in the hospital setting.
The normal system that regulates glycemia encompasses a very complex system of hormonal and metabolic regulators. At the core of this system, insulin is the key regulator. Therapeutic insulin is therefore the best resource available for controlling hyperglycemia in the hospital setting.
Of the other currently available therapies, none offers the power and rapidity that insulin has to control blood glucose level. The biguanides are usually contraindicated in the hospital setting because most patients with hyperglycemia and/or diabetes are acutely ill and hence at risk of lactic acidosis. Furthermore, in a large number of these patients radio‐contrast agents are used; hence, transient renal failure is common, posing yet another risk factor for lactic acidosis. The thiazolidinediones (TZDs) are slow to act and not as powerful in controlling acute hyperglycemia and thus are not the optimal tool available when the metabolic situation changes drastically as occurs in hospitalized patients. Precaution needs to be taken when using TZD to treat patients who have congestive heart failure or hepatic insufficiency. The action of the sulfonylureas (SUs) imparts a high risk of hypoglycemia and/or poor insulinemic response during stress to patients being treated with them; therefore, it is usually recommended that patients in a hospital setting not be treated with SUs, except for selected very stable patients. The new emerging therapies (incretin mimetics, dipeptidyl peptidase‐IV inhibitors, amylin) have never been tested in the hospital setting, and hence no recommendation can be made at this stage. Thus, we believe that the main tool available for treating the hospitalized patient with hyperglycemia is insulin coupled with proper nutrition and a system of information to monitor therapeutic progress, which allows for proper and timely adjustments as well as for treatment of hypoglycemia.
Within this setting a conceptual frame for insulin administration has been proposed. Exogenous insulin needs to be provided to mimic as closely as possible the physiological pattern of endogenous insulin secretion. The latter is broadly thought to be composed of 2 secretory components: a basal component and a prandial, or alimentary, component. The basal component of insulin secretion represents the rate of insulin produced independent of meal ingestion, which is mainly governed by the prevailing concentrations of arterial blood glucose and other hormonal and metabolic regulators. Prandial insulin is the increase in insulin secretion that occurs after eating, which occurs as a complex pattern of pulses. Roughly, prandial insulin secretion is mainly determined by the quantity and composition of the meal ingested, especially the quantity of carbohydrate.
Thus, the insulin dose that an inpatient requires may be thought as consisting of basal and nutritional insulin requirements. To these 2 components we also add a third component: a correctional insulin component.
The basal insulin requirement of a given patient can be estimated by taking into consideration the type of diabetes and body weight. The nutritional insulin requirement refers to the insulin required to cover nutritional intake, which in a hospital setting may correspond to regular meals, intravenous dextrose, nutritional supplements, enteral feedings, or parenteral nutrition. Because our estimates are not very accurate, corrective insulin is required to correct elevated concentrations of plasma glucose (usually measured with finger sticks) A scale or table of corrective insulin can be constructed on the basis of type of diabetes, body weight, and/or total amount of daily doses of basal and nutritional insulin. Obviously, many will think of corrective insulin as a sliding scale. It is import to remember that this scale is complementary to prescribed basal and nutritional insulin doses and not a substitute for them. 0
How can insulin be prescribed in the hospital to cover the 3 facets of insulin (basal, alimentary, and corrective)? The following is a pragmatic approach that we found useful and uses the above considerations as an underpinning. We will first consider the general medicine or surgical ward and then the intensive care setting.
General Medical and Surgical Wards
Basal insulin
The activity of the ideal insulin preparation for this task should not show any peak, instead should remain in a steady state for 24 hours. Currently, three insulin preparations can be considered for this purpose. Glargine insulin is an analogue of insulin that has a stronger capacity to form and maintain hexamers of insulin and its rate of absorption from the subcutaneous depot, which allows for quasi‐steady‐state action for 24 hours. However, variability is sometime noted clinically in the length of duration and the absence or presence of a peak. Neutralized protamine insulin (NPH) is a mixture of protamine and human insulin in which the complexing of the 2 proteins retards absorption of insulin. The action profile of NPH insulin definitively displays a peak (between 6 and 10 hours after injection); however, the timing of this peak varies from patient to patient and (in the same patient) from day to day. There is also variability in the widely quoted duration of action (12‐18 hours). Despite these shortcomings, NPH has been used for several decades and has widespread acceptance among physicians, especially because it costs less than glargine insulin. Detemir insulin is a new analogue of insulin. The insulin molecule has been complexed with a fatty acid. This modification protracts absorption from the subcutaneous depot and also within the blood compartment because the acylated insulin binds to albumin, which then acts as a reservoir. There is very little experience with detemir in clinical scenarios and none in the hospital setting.
Our preferred basal insulin, given current knowledge and experience, is glargine, except in those special cases in which insulin that has a shorter action is needed (ie, patients with tube feeds, use of steroids), listed below in the Special Considerations section.
Alimentary or nutritional or prandial or bolus insulin
The ideal insulin to cover the prandial period should have rapid onset and rapid dissipation of activity. Two types of insulin preparation have these characteristics. The insulin traditionally used in this setting is regular human insulin. Unfortunately, the onset of action of this preparation is not as rapid (20‐30 minutes), forcing it to be prescribed as a preprandial insulin. In a hospital setting, where the dynamics of meal serving and NPO periods are highly unpredictable, this creates a serious risk of hypoglycemia when preprandial insulin is the choice. The activity of regular human insulin lasts 4‐6 hours, and thus there is also the risk of stacking multiple prandial doses and hence an increase in the risk of hypoglycemia. So, although this insulin has been used for many decades, it has been rapidly replaced by the new analogues of insulin (lispro, aspart, and glulisine), which have a much faster onset of activity (within 15 minutes of injection) because of its rapid absorption from the subcutaneous depot. Moreover, the dissipation of insulin action of these preparations is faster (3‐4 hours). The minor decrease in glycemic power at equivalent doses compared to preprandial administration can usually be easily overcome with a minimal increase in dosage. It is becoming widely accepted because it allows for flexibility in the timing of administration (which can be made contingent on meal ingestion) and also in dosing because it can be better tailored to the amount of food consumed. Overall, our preference is for the use of analogues in the hospital setting. The only drawback is cost.
Corrective insulin
The type of insulin used for the correction of glycemia that exceeds the target follows from the same considerations as those used for the alimentary or prandial insulin. For simplicity, the same type of insulin chosen for alimentary insulin should be the one selected for corrective insulin.
How to dose the insulin?
Once the type of insulin preparations has been chosen, dosing is the next task. The hospitalist should remember that the initial prescription or dosage of insulin will need to be reevaluated daily to allow for glycemia management and to avoid hypoglycemia. As in many other fields of medicine, a single approach does not fit all scenarios. The following is a list of scenarios commonly encountered in our inpatient population. There is no definitive way to suggest how successfully they are managed as outpatients. It may be reasonable to assume glycemic control with HbA1C of less than 7% as being highly successful, HbA1c between 7.1% and 8.5% as being moderately successful, and HbA1C greater than 8.5% as being unsuccessful. These HbA1C levels help to guide us through decision making and have been very helpful in our practice. Recognition of hyperglycemia either on admission or during an in‐hospital stay warrants consideration of insulin‐based management. For several reasons, HbA1c should be tested in patients who are found to have hyperglycemia in the hospital. Elevated glycated hemoglobin enables the recognition of previously undiagnosed diabetes and helps in the identification of patients with poorly controlled diabetes; hence, the hospital stay can be an opportunity to change treatment approaches or emphasize compliance. Likewise, in a hyperglycemic patient with normal HbA1C, it should be considered whether stress hyperglycemia has developed.
-
Patient is using a highly successful regimen with oral agents. Recommendation: continue using oral agents if no contraindications exist, the patient is unlikely to receive contrast dye tests, and admission is for a minor indication requiring a short inpatient stay. Oral agents may need to be stopped if poor glycemic control after hospitalization or any contraindication is identified.
-
Patient is using a regimen of insulin that has been very successful. Recommendation: follow the same regimen and add to it the table for correction insulin.
-
Patient is using a regimen of insulin that is moderately successful. Recommendation: keep the same regimen but increase doses and add the correction insulin table.
-
Patient is using an unsuccessful oral regimen. Recommendation: discontinue oral agents and start basal, alimentary, and corrective insulin
-
Patient is using a very unsuccessful regimen of insulin. Recommendation: reevaluate and prescribe a basal, alimentary, and correction insulin regimen.
-
Patient is recently or newly diagnosed with diabetes. Recommendation: while in the hospital use a basal, alimentary, and corrective insulin regimen.
-
Patient has type 1 diabetes. Recommendation: prescribe full insulin coverage with basal, nutritional, and correctional insulin.
-
Patient is receiving an IV drip of insulin and is no longer critical and tolerating po intake. Recommendation: overlap IV drip with subcutaneous insulin for at least 4 hours and then continue subcutaneous insulin.
Empirical calculation of basal insulin.
Once you have decided which insulin to use as basal insulin, the following may be used to calculate empirical doses. Suggested insulin types for basal include glargine and NPH insulin.
For scenarios 4, 5, 6, and 7 we use a simple formula to estimate the basal insulin requirements. Longer‐acting insulin requirements may be calculated as:
-
For type 2 diabetes: 0.4 units/kg/day of basal insulin.
-
For type 1 diabetes: 0.2 units/kg/day of basal insulin.
-
The adjustments should be made every 48 hours 2‐5 units at a time or 10% of the dose.
-
For a regimen based on glargine insulin: full dose is administered daily.
-
For NPH: two thirds is given AM and one third PM.
-
For scenario 8: basal insulin is estimated as total dose of insulin drip per hour for the last 6 hours 0.8 24. We recommend an overlap of IV drip and SQ insulin of at least 4 hours.
Empirical calculation of alimentary or nutritional insulin.
Once the type of insulin has been chosen, the insulin doses may be calculated empirically. Suggested insulin choices include lispro, aspart, and glusiline insulin.
As a convenient tool, the total daily alimentary or nutritional insulin requirement is nearly equal to total daily basal insulin. This dose estimation may then be divided into various premeal doses on the basis of the carbohydrate content of the meal. Provision of 1 unit of short‐acting insulin should be made for every 15 units of carbohydrate intake. The following rough estimation may be used to calculate the premeal alimentary insulin dose.
-
For type 2 diabetes: Empirically, 0.1, 0.15, and 0.15 units of short‐acting insulin/kg for breakfast, lunch, and dinner, respectively.
-
For type 1 diabetes: Empirically, we suggest 0.05‐0.1 units of rapid‐acting insulin/kg, to be administered before meals.
The premeal dose requirement of an individual patient may be significantly different. If patient is NPO, then alimentary insulin is not prescribed; specific doses need to be suspended if patient is made NPO and resumed when PO is restored.
Total daily dose of insulin may vary according to body weight, endogenous insulin secretory capacity, and degree of insulin resistance. Variation tends to be greater for those with type 2 diabetes.
We encourage administration of alimentary insulin at or immediately after meal ingestion. This implies a system of alert for patients to let nurses know when they have finished eating.
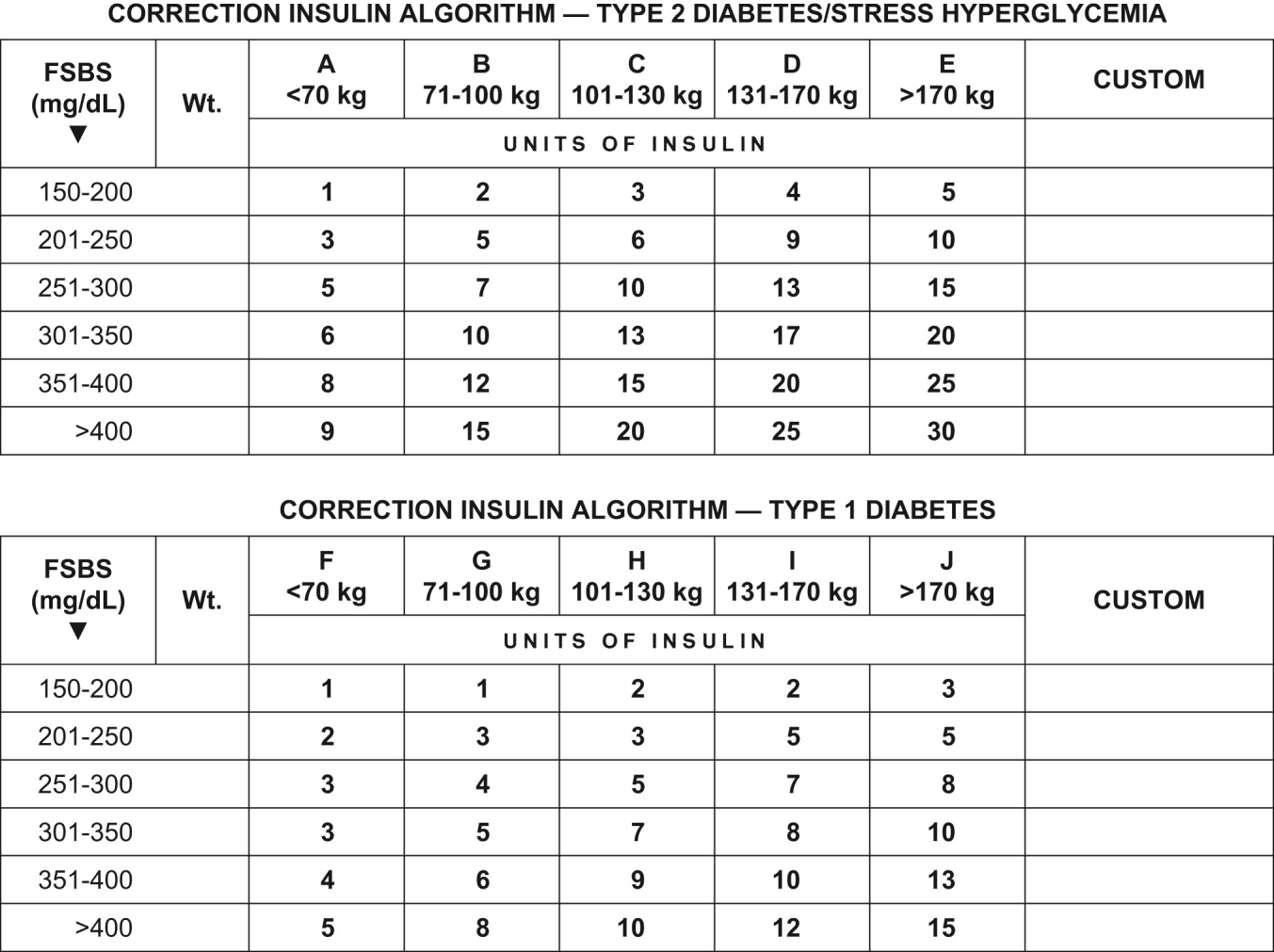
Empirical calculation of correctional insulin (sliding scale).
Although every institution relies on its own guidelines, we reproduce here the correction table we use at Wake Forest UniversityBaptist Medical Center (WFUBMC), which was generated based on type of diabetes and body weight:
In our inpatient practice this correction table has been quite helpful. Bear in mind that there are several correctional insulin dose algorithms, and the one most suitable to local needs should be adopted.
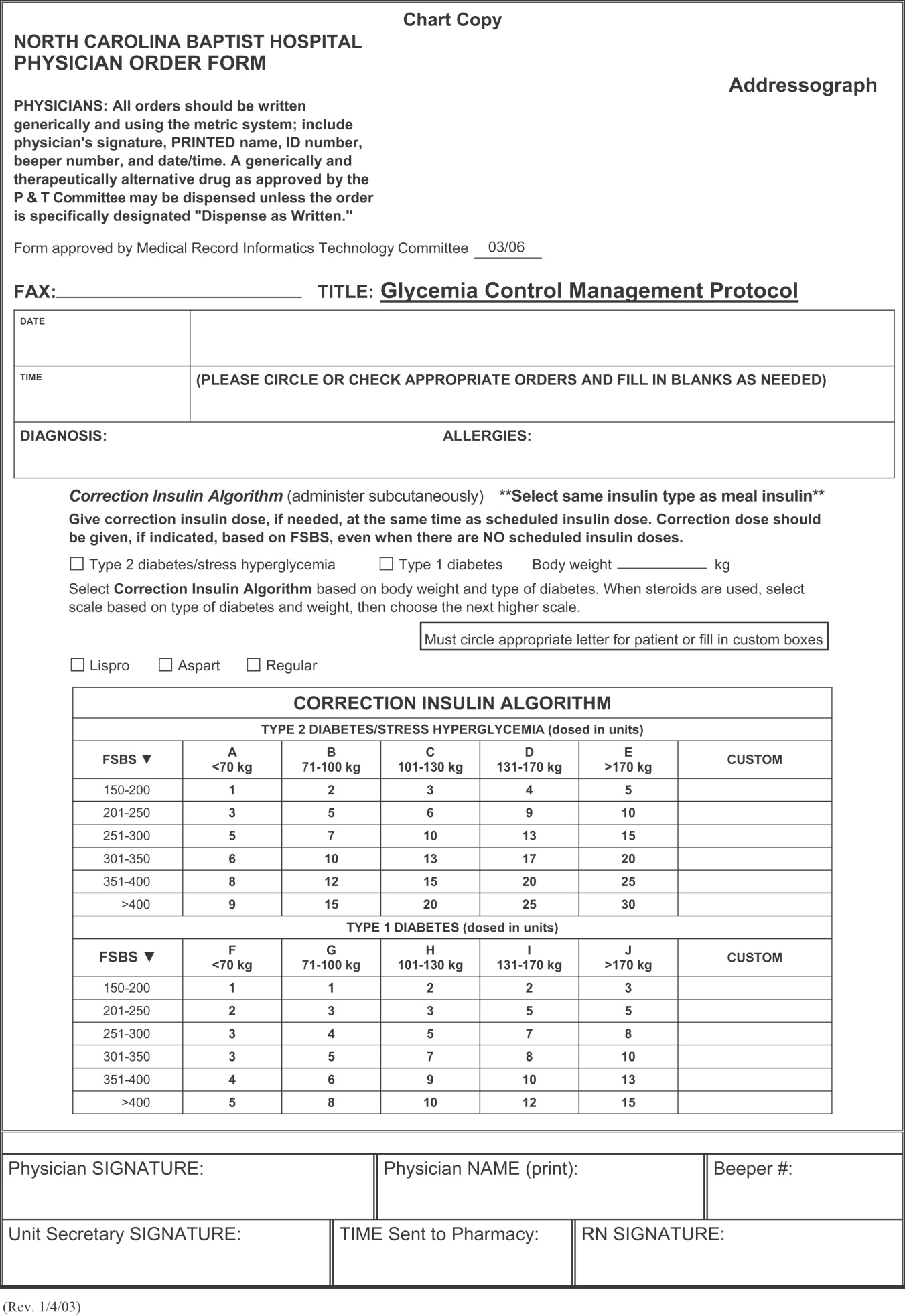
Once the insulin to be used for correctional insulin has been chosen, the algorithm shown in Figure 2 may be used.
To ensure clarity about the prescribed regimen, we encourage the use of preformatted templates or (preferably) computerized orders. Figure 3 shows an example, an order template that we use at WFUBMC.
Intensive Medical and Surgical Care Unit
Continuous intravenous insulin infusion (IV insulin drip) is the most suitable way to administer insulin to critically ill patients. For continuous intravenous insulin infusion, regular insulin is most commonly used and in fact is the only type of insulin studied in prospective randomized trials. This requires adequate staffing, frequent monitoring, and frequent dose adjustments. Such stringent glycemic control is appropriate for patients in critical care units. The American College of Endocrinology recommends using intravenous insulin therapy in the subset of inpatients who have diabetic ketoacidosis, are before major surgical procedures, are undergoing fasting for more than 12 hours and have type 1 diabetes, are critically ill, are undergoing labor and delivery, are being treated for myocardial infarction, have just had organ transplantation, are being maintained on total parenteral nutrition, or have other illnesses requiring prompt glucose control.14
The use of continuous intravenous insulin infusion on a regular basis on all medical floors is not routinely recommended because there is not adequate scientific data to support its use from both clinical and financial perspectives. There are various protocols for attaining recommended levels, and every institution must adopt or develop a protocol that both suits its needs and is feasible. Again, aggressive glycemic control using intravenous insulin requires a well‐monitored setup and is ideal for intensive care units.
Management of hypoglycemia
The therapeutic window between insulin effectiveness and insulin‐associated hypoglycemia is very narrow; hence, proper management of blood glucose needs to be embedded within a system in which every member of the team taking care of a patient with diabetes has appropriate knowledge of the task at hand. Of equal importance is the development of a protocol to treat hypoglycemia minimizing the overzealous treatment that leads to severe hyperglycemia. This protocol also assumes that all oral hypoglycemic agents have been discontinued.
Hypoglycemia protocol (FSBS < 70 mg/dL)
-
Patient conscious and able to eat (select one):
-
Provide patient with 15 g of carbohydrate (120 cc of fruit juice or 180 cc of regular soda or 240 cc of skim milk or 3 glucose tablets).
-
Recheck fingerstick blood sugar (FSBS) in 15 minutes.
-
Repeat above if FSBS still <70 mg/dL; continue cycle until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and IV access available
-
Administer 15 mL of 50% dextrose IV (mix in 25 mL of NS) and call HO
-
Check FSBS in 15 minutes.
-
Repeat IV 50% dextrose until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and no IV access available
-
Administer glucagon 1 mg IM.
-
Turn patient on side (to avoid broncho‐aspiration) and call HO.
-
Check FSBS in 15 minutes.
-
If still <70 mg/dL, start IV line and follow protocol for an unconscious patient with IV access available.
Special considerations
Steroid use.
No clinical trials have been conducted to define a quantitative approach to managing hyperglycemia induced by steroids (in those patients without previous diabetes) or to understand adjustments in insulin dose for diabetic patients who will undergo treatment with steroids.
Empirically, we recommend the use of NPH insulin and to adjust the dose calculation 20% higher for low‐dose prednisone (10‐20 mg/day), 30% higher for medium‐dose prednisone (21‐40 mg/day), and 50% higher for high‐dose prednisone (>41 mg/day). We do not recommend the use of glargine insulin in this setting since the half‐life of prednisone is less than 24 hours; hence, the risk of hypoglycemia is high when using very long‐acting insulin. Also, empirically, we make the recommendation to maximize the NPH AM dose and minimize the PMdose, possibly dividing the calculated dose into three quarters AM, one quarter PM. We also caution that this approach would not be appropriate when a patient is using other steroids (prednisolone, methylprednisolone, and dexamethasone) because the half‐life of these steroids exceeds 24 hours, and in such cases glargine insulin may be suitable.
Enteral and parenteral feeding.
Frequently patients need enteral or parenteral feeding. The former may be given as continuous or discontinuous infusion; hence, in this particular setting, a specific insulin regimen must be customized in close collaboration with the dietician. For example, for those patients who are chronically fed enterally and for whom a system of bolus has been established, the use of a basal insulin may be warranted. However, for the patient who is being fed nocturnally only, we would probably choose NPH as the insulin regimen. Good success has been found in some hospitals with the use of premixed insulin preparations when enteral feeding is continuous for 24 hours. Patients fed parenterally may receive their basal and alimentary insulin as an addition to the nutrition bag, complemented with correction insulin administered subcutaneously.
CONCLUSIONS
In conclusion, it is the responsibility of hospitalists to make a conscious effort to manage hyperglycemia in patients who are previously diabetic or become hyperglycemic during hospitalization in order to improve their clinical outcome. Hospitals need to realize that this task is far from being the lone duty of physicians; hence, systems for hyperglycemia management that engage multidisciplinary teams must be established.
Although the ideal way to do so in the critical care setting is continuous intensive insulin infusion therapy, this may not always be practical. In such cases, basal and alimentary insulin with appropriate insulin sliding scales should be used. Using the sliding scales alone should be strongly discouraged, as they tend to only troubleshoot a situation and allow the damage caused to the patients on a molecular level to be camouflaged in objective ways. Appropriate attention should be paid to the risk of developing hypoglycemia as a sequela of overzealous correction of hyperglycemia. This leaves us with the therapeutically desirable band of the glycemic spectrum. However, this band is wide enough to make it possible to achieve better performance. Although the target glycemic range definitively needs to be determined, it is reasonable to have 80‐110 mg/dL as the target range for critically ill patients, as generally agreed by both the ACE and ADA, and a glycemic range of preprandial glucose between 90 and 130 mg/dL for ambulatory patients. The maximal blood glucose should not exceed 180 mg/dL. Having realized the adverse impact on patients of uncontrolled hyperglycemia, at the next morning report, it is appropriate for the nurse to say, Mr. Smith's finger‐stick glucoses are better controlled now, and he required only 2 units of additional insulin coverage yesterday. If we still hear high glucose numbers and keep fixing this problem with sliding‐scale insulin alone, we are not doing a good job.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet.2000;355:773–778.
- .Insulin therapy for the critically ill patient.Clin Cornerstone.2003;5(2):56–63.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.J Parenter Enter Nutr.1998;22:77–81.
- ,,,.Metabolism.1994;43:279–284.
- ,,, et al.Randomized trial of insulin‐glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): effects on mortality at 1 year.J Am Coll Cardiol.1995;26:57–65.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461
- ,,.Insulin therapy for critically ill hospitalized patients: a meta‐analysis of randomized controlled trials.Arch Intern Med.2004;164:2005–2011.
- .Perioperative glucose control.Curr Opin Anaesthesiol.2006;19(2):111–116.
- ,,,:Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes.Diabetes Care.1999;22:1408–1414.
- ,,, et al.Hyperglycaemia is associated with poor outcomes in patients admitted to hospital with acute exacerbations of chronic obstructive pulmonary disease.Thorax.2006;61:284–289.
- ,,, et al.;American Diabetes Association Diabetes in Hospitals Writing Committee.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591. Errata in: Diabetes Care. 2004;27:856. Hirsh, Irl B [corrected to Hirsch, Irl B]; dosage error in text; and Diabetes Care. 2004;27:1255
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(1):77–82.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979
- Joint Commission on Accreditation of Health Organizations Web site. Avail at: http://www.jointcommission.org/NewsRoom/NewsReleases/jc_nr_072006.htm. Accessed September 29,2006.
A very compelling and growing body of evidence highlights the benefits to hospitalized patients of intensive (insulin‐based) glycemic control. However, we have a tendency to attend to patients' acute problems during inpatient stays, and glycemic control frequently takes a backseat. As hospitalists, we frequently come across patients with diabetes admitted for various other reasons, as well as patients who develop hyperglycemia while hospitalized. During a hospital stay, it is usually not recommended that an oral hypoglycemic regimen be continued, and insulin use is necessary to more reliably control blood glucose. In this article, we emphasize the need to better manage inpatient hyperglycemia and to make a conscious effort to prescribe insulin in a more rational manner. We propose that insulin orders for an inpatient address: (1) basal insulinization, (2) meal or prandial insulin, and (3) corrective insulin. In this schema, the supplemental boluses of insulin administered to correct a blood glucose level that exceeds a set value are viewed as an adjunct to a basal/bolus insulin regimen. We also recognize the practical limitations of attaining stringent glucose targets and pinpoint those areas in need of further research.
BACKGROUND
It is not entirely clear how and when the use of the very popular insulin sliding scale as the sole approach to controlling inpatient hyperglycemia became such a widespread practice. However, the sliding scale has been passed along to subsequent generations as gospel. Despite receiving much criticism, the regular insulin sliding scale remains sacred to medical practitioners. Unfortunately, the sliding scale is very frequently the sole therapeutic tool used to control hyperglycemia, and not as a complement to a more physiologically complete (basal/bolus) insulin regimen. As attractive as the use of continuous intravenous insulin infusion is to endocrinologists, it is not frequently used outside intensive care units for many reasons. Where there is apparent agreement is in the need to improve inpatient management of hyperglycemia.
THE PROBLEM: HYPERGLYCEMIC INPATIENT
Hyperglycemia is defined as a fasting glucose level greater than 126 mg/dL or 2 or more random blood glucose levels greater than 200 mg/dL.1 Not infrequently, patients admitted to our ward have a history of diabetes; however, a good proportion of admitted patients have no such history. In a retrospective analysis of more than 2000 consecutive hospital admissions, hyperglycemia was found in as many as 38% of the patients in whom blood glucose was measured and documented in the chart, about a third of which did not previously carry the diagnosis of diabetes. Hyperglycemia in this specific setting, dubbed stress hyperglycemia,1 is quite frequently found in hospitalized patients and has been shown to increase the risk of death, congestive heart failure, and cardiogenic shock after myocardial infarction.2 Acute insulin resistance is also seen frequently in an acutely ill patient and is attributed to the release and metabolic actions of counterregulatory hormones and cytokine excess.3 Patients often require increased amounts of insulin to maintain glucose at an acceptable level. Iatrogenic hyperglycemia may occur as a consequence of glucocorticoids or excessive infusion of dextrose. In critically ill patients, vasopressors may also be associated with iatrogenic hyperglycemia. Inpatient hyperglycemia is associated with nosocomial infections, increased mortality, increased length of stay, and poor overall outcome.4 Of interest is that stress hyperglycemia was associated with more adverse outcome than was hyperglycemia in a patient with known diabetes.1, 2 We are not sure if this phenomenon of stress hyperglycemia is pathogenic or serves as a marker of disease severity.
Is Hyperglycemia Really a Problem?
Compelling evidence that control of hyperglycemia improves the outcomes of patients undergoing cardiothoracic surgery was provided by the Portland trial. Although this study was not randomized and its glycemia targets were not well defined, it demonstrated that better control of blood glucose levels drastically reduces the incidence of chest wall infections and the need for transfusions and significantly shortens hospital length of stay (LOS).5
The results of the Diabetes Mellitus Insulin‐Glucose in Acute Myocardial Infarction (DIGAMI) study showed that hyperglycemic patients with acute myocardial infarction had improved outcomes when intravenous administration of insulin was used to aggressively control glycemia.6 Van den Berghe et al. found significantly lower mortality and morbidity rates in surgical intensive care unit patients in whom aggressive glycemic control was attained with continuous intravenous insulin infusion. The study also identified reduced requirement of antibiotics, red cell transfusions, dialysis, and ventilatory support with aggressive glycemic control.7 It was also shown that there was significantly reduced morbidity in all patients in the medical ICU receiving intensive insulin therapy.8 Another meta‐analysis found that insulin therapy initiated in hospitalized critically ill patients in different clinical settings had a beneficial effect on short‐term mortality.9 Krinsley observed hyperglycemia to be associated with adverse outcomes in acutely ill adult patients and that its treatment has been shown to improve mortality and morbidity in a variety of settings.10 In their study of adults with diabetes, Golden et al. identified hyperglycemia as an independent risk factor for surgical infection of diabetic patients undergoing cardiac surgery.11 A meta‐analysis by Capes et al. showed a 3‐fold higher risk of poor functional recovery in nondiabetic hyperglycemic patients compared to that of nondiabetic euglycemic patients.2 A recent retrospective analysis found that patients with hyperglycemia treated for acute exacerbation of chronic obstructive pulmonary disease had poor outcomes.12
It is possible to give an account and references of only a limited number of such studies. The prevailing message conveyed in all these studies is that patients with poorly managed hyperglycemia have a poor overall outcome. Hence, the need to better manage inpatient hyperglycemia cannot be overemphasized.13
After an extensive search, we could not find well‐designed prospective randomized studies of patients who are not acutely ill or are outside the perisurgical period. However, the DIGAMI, Van den Berghe, and Portland trials generated a powerful and large momentum that has created interest in establishing protocols for keeping the blood glucose of patients in most medical and surgical critical care units in the suggested range.57, 13 Moreover, extrapolation of the data to noncritical and nonsurgical patients made possible a consensus conference organized by the American Association of Clinical Endocrinologists (AACE) that garnered support from many other medical associations. The position paper published by the AACE calls for tighter glycemic control in hospitalized patients. The AACE recommends that blood glucose concentrations for intensive care unit patients be maintained below 110 mg/dL. In noncritically ill patients, the preprandial glucose level should not exceed 110 mg/dL, and maximum glucose should not exceed 180 mg/dL.14 The American Diabetes Association (ADA) does not recommend any target glucose values for noncritical patients but does believe there is a need to have better inpatient hyperglycemic management. Some authorities believe that until the amount of scientific data increases, it is prudent to stay within the ADA‐recommended ambulatory guidelines for a preprandial plasma glucose level of 90‐130 mg/dL15 and a postprandial blood glucose level not to exceed 180 mg/dL.
Additionally, due attention must be paid to hypoglycemia secondary to aggressive glycemic control.
Because of the absence of evidence‐based information, it is not surprising that opinions conflict about the optimal level of blood glucose for an inpatient. We believe that in the absence of definitive evidence, it is prudent to adhere to the targets recommended by these associations.
A SOLUTION: WHAT TO DO AND HOW TO DO IT
Ideally, a system should be established to attain euglycemia without the attendant risk of hypoglycemia. The Joint Commission on Accreditation of Healthcare Organizations recently showed recognition of this need this by collaborating with the American Diabetes Association to establish a program to certify inpatient diabetes care center programs that meet national standards. The program must be carried out in all inpatient settings and should include the following elements16:
-
Specific staff education requirements;
-
Written blood glucosemonitoring protocols;
-
Plans for the treatment of hypoglycemia and hyperglycemia;
-
Collection of data on the incidence of hypoglycemia;
-
Education of patients on self‐managing their diabetes; and
-
An identified program champion or program champion team.
The Joint Commission's Advanced Inpatient Diabetes Certification Program is based on the ADA guidelines; the scope of this manuscript does not cover all the elements required to receive certification.16 In the rest of the article, we focus on the basic principles of the use of insulin to control hyperglycemia in the hospital setting.
The normal system that regulates glycemia encompasses a very complex system of hormonal and metabolic regulators. At the core of this system, insulin is the key regulator. Therapeutic insulin is therefore the best resource available for controlling hyperglycemia in the hospital setting.
Of the other currently available therapies, none offers the power and rapidity that insulin has to control blood glucose level. The biguanides are usually contraindicated in the hospital setting because most patients with hyperglycemia and/or diabetes are acutely ill and hence at risk of lactic acidosis. Furthermore, in a large number of these patients radio‐contrast agents are used; hence, transient renal failure is common, posing yet another risk factor for lactic acidosis. The thiazolidinediones (TZDs) are slow to act and not as powerful in controlling acute hyperglycemia and thus are not the optimal tool available when the metabolic situation changes drastically as occurs in hospitalized patients. Precaution needs to be taken when using TZD to treat patients who have congestive heart failure or hepatic insufficiency. The action of the sulfonylureas (SUs) imparts a high risk of hypoglycemia and/or poor insulinemic response during stress to patients being treated with them; therefore, it is usually recommended that patients in a hospital setting not be treated with SUs, except for selected very stable patients. The new emerging therapies (incretin mimetics, dipeptidyl peptidase‐IV inhibitors, amylin) have never been tested in the hospital setting, and hence no recommendation can be made at this stage. Thus, we believe that the main tool available for treating the hospitalized patient with hyperglycemia is insulin coupled with proper nutrition and a system of information to monitor therapeutic progress, which allows for proper and timely adjustments as well as for treatment of hypoglycemia.
Within this setting a conceptual frame for insulin administration has been proposed. Exogenous insulin needs to be provided to mimic as closely as possible the physiological pattern of endogenous insulin secretion. The latter is broadly thought to be composed of 2 secretory components: a basal component and a prandial, or alimentary, component. The basal component of insulin secretion represents the rate of insulin produced independent of meal ingestion, which is mainly governed by the prevailing concentrations of arterial blood glucose and other hormonal and metabolic regulators. Prandial insulin is the increase in insulin secretion that occurs after eating, which occurs as a complex pattern of pulses. Roughly, prandial insulin secretion is mainly determined by the quantity and composition of the meal ingested, especially the quantity of carbohydrate.
Thus, the insulin dose that an inpatient requires may be thought as consisting of basal and nutritional insulin requirements. To these 2 components we also add a third component: a correctional insulin component.
The basal insulin requirement of a given patient can be estimated by taking into consideration the type of diabetes and body weight. The nutritional insulin requirement refers to the insulin required to cover nutritional intake, which in a hospital setting may correspond to regular meals, intravenous dextrose, nutritional supplements, enteral feedings, or parenteral nutrition. Because our estimates are not very accurate, corrective insulin is required to correct elevated concentrations of plasma glucose (usually measured with finger sticks) A scale or table of corrective insulin can be constructed on the basis of type of diabetes, body weight, and/or total amount of daily doses of basal and nutritional insulin. Obviously, many will think of corrective insulin as a sliding scale. It is import to remember that this scale is complementary to prescribed basal and nutritional insulin doses and not a substitute for them. 0
How can insulin be prescribed in the hospital to cover the 3 facets of insulin (basal, alimentary, and corrective)? The following is a pragmatic approach that we found useful and uses the above considerations as an underpinning. We will first consider the general medicine or surgical ward and then the intensive care setting.
General Medical and Surgical Wards
Basal insulin
The activity of the ideal insulin preparation for this task should not show any peak, instead should remain in a steady state for 24 hours. Currently, three insulin preparations can be considered for this purpose. Glargine insulin is an analogue of insulin that has a stronger capacity to form and maintain hexamers of insulin and its rate of absorption from the subcutaneous depot, which allows for quasi‐steady‐state action for 24 hours. However, variability is sometime noted clinically in the length of duration and the absence or presence of a peak. Neutralized protamine insulin (NPH) is a mixture of protamine and human insulin in which the complexing of the 2 proteins retards absorption of insulin. The action profile of NPH insulin definitively displays a peak (between 6 and 10 hours after injection); however, the timing of this peak varies from patient to patient and (in the same patient) from day to day. There is also variability in the widely quoted duration of action (12‐18 hours). Despite these shortcomings, NPH has been used for several decades and has widespread acceptance among physicians, especially because it costs less than glargine insulin. Detemir insulin is a new analogue of insulin. The insulin molecule has been complexed with a fatty acid. This modification protracts absorption from the subcutaneous depot and also within the blood compartment because the acylated insulin binds to albumin, which then acts as a reservoir. There is very little experience with detemir in clinical scenarios and none in the hospital setting.
Our preferred basal insulin, given current knowledge and experience, is glargine, except in those special cases in which insulin that has a shorter action is needed (ie, patients with tube feeds, use of steroids), listed below in the Special Considerations section.
Alimentary or nutritional or prandial or bolus insulin
The ideal insulin to cover the prandial period should have rapid onset and rapid dissipation of activity. Two types of insulin preparation have these characteristics. The insulin traditionally used in this setting is regular human insulin. Unfortunately, the onset of action of this preparation is not as rapid (20‐30 minutes), forcing it to be prescribed as a preprandial insulin. In a hospital setting, where the dynamics of meal serving and NPO periods are highly unpredictable, this creates a serious risk of hypoglycemia when preprandial insulin is the choice. The activity of regular human insulin lasts 4‐6 hours, and thus there is also the risk of stacking multiple prandial doses and hence an increase in the risk of hypoglycemia. So, although this insulin has been used for many decades, it has been rapidly replaced by the new analogues of insulin (lispro, aspart, and glulisine), which have a much faster onset of activity (within 15 minutes of injection) because of its rapid absorption from the subcutaneous depot. Moreover, the dissipation of insulin action of these preparations is faster (3‐4 hours). The minor decrease in glycemic power at equivalent doses compared to preprandial administration can usually be easily overcome with a minimal increase in dosage. It is becoming widely accepted because it allows for flexibility in the timing of administration (which can be made contingent on meal ingestion) and also in dosing because it can be better tailored to the amount of food consumed. Overall, our preference is for the use of analogues in the hospital setting. The only drawback is cost.
Corrective insulin
The type of insulin used for the correction of glycemia that exceeds the target follows from the same considerations as those used for the alimentary or prandial insulin. For simplicity, the same type of insulin chosen for alimentary insulin should be the one selected for corrective insulin.
How to dose the insulin?
Once the type of insulin preparations has been chosen, dosing is the next task. The hospitalist should remember that the initial prescription or dosage of insulin will need to be reevaluated daily to allow for glycemia management and to avoid hypoglycemia. As in many other fields of medicine, a single approach does not fit all scenarios. The following is a list of scenarios commonly encountered in our inpatient population. There is no definitive way to suggest how successfully they are managed as outpatients. It may be reasonable to assume glycemic control with HbA1C of less than 7% as being highly successful, HbA1c between 7.1% and 8.5% as being moderately successful, and HbA1C greater than 8.5% as being unsuccessful. These HbA1C levels help to guide us through decision making and have been very helpful in our practice. Recognition of hyperglycemia either on admission or during an in‐hospital stay warrants consideration of insulin‐based management. For several reasons, HbA1c should be tested in patients who are found to have hyperglycemia in the hospital. Elevated glycated hemoglobin enables the recognition of previously undiagnosed diabetes and helps in the identification of patients with poorly controlled diabetes; hence, the hospital stay can be an opportunity to change treatment approaches or emphasize compliance. Likewise, in a hyperglycemic patient with normal HbA1C, it should be considered whether stress hyperglycemia has developed.
-
Patient is using a highly successful regimen with oral agents. Recommendation: continue using oral agents if no contraindications exist, the patient is unlikely to receive contrast dye tests, and admission is for a minor indication requiring a short inpatient stay. Oral agents may need to be stopped if poor glycemic control after hospitalization or any contraindication is identified.
-
Patient is using a regimen of insulin that has been very successful. Recommendation: follow the same regimen and add to it the table for correction insulin.
-
Patient is using a regimen of insulin that is moderately successful. Recommendation: keep the same regimen but increase doses and add the correction insulin table.
-
Patient is using an unsuccessful oral regimen. Recommendation: discontinue oral agents and start basal, alimentary, and corrective insulin
-
Patient is using a very unsuccessful regimen of insulin. Recommendation: reevaluate and prescribe a basal, alimentary, and correction insulin regimen.
-
Patient is recently or newly diagnosed with diabetes. Recommendation: while in the hospital use a basal, alimentary, and corrective insulin regimen.
-
Patient has type 1 diabetes. Recommendation: prescribe full insulin coverage with basal, nutritional, and correctional insulin.
-
Patient is receiving an IV drip of insulin and is no longer critical and tolerating po intake. Recommendation: overlap IV drip with subcutaneous insulin for at least 4 hours and then continue subcutaneous insulin.
Empirical calculation of basal insulin.
Once you have decided which insulin to use as basal insulin, the following may be used to calculate empirical doses. Suggested insulin types for basal include glargine and NPH insulin.
For scenarios 4, 5, 6, and 7 we use a simple formula to estimate the basal insulin requirements. Longer‐acting insulin requirements may be calculated as:
-
For type 2 diabetes: 0.4 units/kg/day of basal insulin.
-
For type 1 diabetes: 0.2 units/kg/day of basal insulin.
-
The adjustments should be made every 48 hours 2‐5 units at a time or 10% of the dose.
-
For a regimen based on glargine insulin: full dose is administered daily.
-
For NPH: two thirds is given AM and one third PM.
-
For scenario 8: basal insulin is estimated as total dose of insulin drip per hour for the last 6 hours 0.8 24. We recommend an overlap of IV drip and SQ insulin of at least 4 hours.
Empirical calculation of alimentary or nutritional insulin.
Once the type of insulin has been chosen, the insulin doses may be calculated empirically. Suggested insulin choices include lispro, aspart, and glusiline insulin.
As a convenient tool, the total daily alimentary or nutritional insulin requirement is nearly equal to total daily basal insulin. This dose estimation may then be divided into various premeal doses on the basis of the carbohydrate content of the meal. Provision of 1 unit of short‐acting insulin should be made for every 15 units of carbohydrate intake. The following rough estimation may be used to calculate the premeal alimentary insulin dose.
-
For type 2 diabetes: Empirically, 0.1, 0.15, and 0.15 units of short‐acting insulin/kg for breakfast, lunch, and dinner, respectively.
-
For type 1 diabetes: Empirically, we suggest 0.05‐0.1 units of rapid‐acting insulin/kg, to be administered before meals.
The premeal dose requirement of an individual patient may be significantly different. If patient is NPO, then alimentary insulin is not prescribed; specific doses need to be suspended if patient is made NPO and resumed when PO is restored.
Total daily dose of insulin may vary according to body weight, endogenous insulin secretory capacity, and degree of insulin resistance. Variation tends to be greater for those with type 2 diabetes.
We encourage administration of alimentary insulin at or immediately after meal ingestion. This implies a system of alert for patients to let nurses know when they have finished eating.
Empirical calculation of correctional insulin (sliding scale).
Although every institution relies on its own guidelines, we reproduce here the correction table we use at Wake Forest UniversityBaptist Medical Center (WFUBMC), which was generated based on type of diabetes and body weight:
In our inpatient practice this correction table has been quite helpful. Bear in mind that there are several correctional insulin dose algorithms, and the one most suitable to local needs should be adopted.
Once the insulin to be used for correctional insulin has been chosen, the algorithm shown in Figure 2 may be used.
To ensure clarity about the prescribed regimen, we encourage the use of preformatted templates or (preferably) computerized orders. Figure 3 shows an example, an order template that we use at WFUBMC.
Intensive Medical and Surgical Care Unit
Continuous intravenous insulin infusion (IV insulin drip) is the most suitable way to administer insulin to critically ill patients. For continuous intravenous insulin infusion, regular insulin is most commonly used and in fact is the only type of insulin studied in prospective randomized trials. This requires adequate staffing, frequent monitoring, and frequent dose adjustments. Such stringent glycemic control is appropriate for patients in critical care units. The American College of Endocrinology recommends using intravenous insulin therapy in the subset of inpatients who have diabetic ketoacidosis, are before major surgical procedures, are undergoing fasting for more than 12 hours and have type 1 diabetes, are critically ill, are undergoing labor and delivery, are being treated for myocardial infarction, have just had organ transplantation, are being maintained on total parenteral nutrition, or have other illnesses requiring prompt glucose control.14
The use of continuous intravenous insulin infusion on a regular basis on all medical floors is not routinely recommended because there is not adequate scientific data to support its use from both clinical and financial perspectives. There are various protocols for attaining recommended levels, and every institution must adopt or develop a protocol that both suits its needs and is feasible. Again, aggressive glycemic control using intravenous insulin requires a well‐monitored setup and is ideal for intensive care units.
Management of hypoglycemia
The therapeutic window between insulin effectiveness and insulin‐associated hypoglycemia is very narrow; hence, proper management of blood glucose needs to be embedded within a system in which every member of the team taking care of a patient with diabetes has appropriate knowledge of the task at hand. Of equal importance is the development of a protocol to treat hypoglycemia minimizing the overzealous treatment that leads to severe hyperglycemia. This protocol also assumes that all oral hypoglycemic agents have been discontinued.
Hypoglycemia protocol (FSBS < 70 mg/dL)
-
Patient conscious and able to eat (select one):
-
Provide patient with 15 g of carbohydrate (120 cc of fruit juice or 180 cc of regular soda or 240 cc of skim milk or 3 glucose tablets).
-
Recheck fingerstick blood sugar (FSBS) in 15 minutes.
-
Repeat above if FSBS still <70 mg/dL; continue cycle until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and IV access available
-
Administer 15 mL of 50% dextrose IV (mix in 25 mL of NS) and call HO
-
Check FSBS in 15 minutes.
-
Repeat IV 50% dextrose until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and no IV access available
-
Administer glucagon 1 mg IM.
-
Turn patient on side (to avoid broncho‐aspiration) and call HO.
-
Check FSBS in 15 minutes.
-
If still <70 mg/dL, start IV line and follow protocol for an unconscious patient with IV access available.
Special considerations
Steroid use.
No clinical trials have been conducted to define a quantitative approach to managing hyperglycemia induced by steroids (in those patients without previous diabetes) or to understand adjustments in insulin dose for diabetic patients who will undergo treatment with steroids.
Empirically, we recommend the use of NPH insulin and to adjust the dose calculation 20% higher for low‐dose prednisone (10‐20 mg/day), 30% higher for medium‐dose prednisone (21‐40 mg/day), and 50% higher for high‐dose prednisone (>41 mg/day). We do not recommend the use of glargine insulin in this setting since the half‐life of prednisone is less than 24 hours; hence, the risk of hypoglycemia is high when using very long‐acting insulin. Also, empirically, we make the recommendation to maximize the NPH AM dose and minimize the PMdose, possibly dividing the calculated dose into three quarters AM, one quarter PM. We also caution that this approach would not be appropriate when a patient is using other steroids (prednisolone, methylprednisolone, and dexamethasone) because the half‐life of these steroids exceeds 24 hours, and in such cases glargine insulin may be suitable.
Enteral and parenteral feeding.
Frequently patients need enteral or parenteral feeding. The former may be given as continuous or discontinuous infusion; hence, in this particular setting, a specific insulin regimen must be customized in close collaboration with the dietician. For example, for those patients who are chronically fed enterally and for whom a system of bolus has been established, the use of a basal insulin may be warranted. However, for the patient who is being fed nocturnally only, we would probably choose NPH as the insulin regimen. Good success has been found in some hospitals with the use of premixed insulin preparations when enteral feeding is continuous for 24 hours. Patients fed parenterally may receive their basal and alimentary insulin as an addition to the nutrition bag, complemented with correction insulin administered subcutaneously.
CONCLUSIONS
In conclusion, it is the responsibility of hospitalists to make a conscious effort to manage hyperglycemia in patients who are previously diabetic or become hyperglycemic during hospitalization in order to improve their clinical outcome. Hospitals need to realize that this task is far from being the lone duty of physicians; hence, systems for hyperglycemia management that engage multidisciplinary teams must be established.
Although the ideal way to do so in the critical care setting is continuous intensive insulin infusion therapy, this may not always be practical. In such cases, basal and alimentary insulin with appropriate insulin sliding scales should be used. Using the sliding scales alone should be strongly discouraged, as they tend to only troubleshoot a situation and allow the damage caused to the patients on a molecular level to be camouflaged in objective ways. Appropriate attention should be paid to the risk of developing hypoglycemia as a sequela of overzealous correction of hyperglycemia. This leaves us with the therapeutically desirable band of the glycemic spectrum. However, this band is wide enough to make it possible to achieve better performance. Although the target glycemic range definitively needs to be determined, it is reasonable to have 80‐110 mg/dL as the target range for critically ill patients, as generally agreed by both the ACE and ADA, and a glycemic range of preprandial glucose between 90 and 130 mg/dL for ambulatory patients. The maximal blood glucose should not exceed 180 mg/dL. Having realized the adverse impact on patients of uncontrolled hyperglycemia, at the next morning report, it is appropriate for the nurse to say, Mr. Smith's finger‐stick glucoses are better controlled now, and he required only 2 units of additional insulin coverage yesterday. If we still hear high glucose numbers and keep fixing this problem with sliding‐scale insulin alone, we are not doing a good job.
A very compelling and growing body of evidence highlights the benefits to hospitalized patients of intensive (insulin‐based) glycemic control. However, we have a tendency to attend to patients' acute problems during inpatient stays, and glycemic control frequently takes a backseat. As hospitalists, we frequently come across patients with diabetes admitted for various other reasons, as well as patients who develop hyperglycemia while hospitalized. During a hospital stay, it is usually not recommended that an oral hypoglycemic regimen be continued, and insulin use is necessary to more reliably control blood glucose. In this article, we emphasize the need to better manage inpatient hyperglycemia and to make a conscious effort to prescribe insulin in a more rational manner. We propose that insulin orders for an inpatient address: (1) basal insulinization, (2) meal or prandial insulin, and (3) corrective insulin. In this schema, the supplemental boluses of insulin administered to correct a blood glucose level that exceeds a set value are viewed as an adjunct to a basal/bolus insulin regimen. We also recognize the practical limitations of attaining stringent glucose targets and pinpoint those areas in need of further research.
BACKGROUND
It is not entirely clear how and when the use of the very popular insulin sliding scale as the sole approach to controlling inpatient hyperglycemia became such a widespread practice. However, the sliding scale has been passed along to subsequent generations as gospel. Despite receiving much criticism, the regular insulin sliding scale remains sacred to medical practitioners. Unfortunately, the sliding scale is very frequently the sole therapeutic tool used to control hyperglycemia, and not as a complement to a more physiologically complete (basal/bolus) insulin regimen. As attractive as the use of continuous intravenous insulin infusion is to endocrinologists, it is not frequently used outside intensive care units for many reasons. Where there is apparent agreement is in the need to improve inpatient management of hyperglycemia.
THE PROBLEM: HYPERGLYCEMIC INPATIENT
Hyperglycemia is defined as a fasting glucose level greater than 126 mg/dL or 2 or more random blood glucose levels greater than 200 mg/dL.1 Not infrequently, patients admitted to our ward have a history of diabetes; however, a good proportion of admitted patients have no such history. In a retrospective analysis of more than 2000 consecutive hospital admissions, hyperglycemia was found in as many as 38% of the patients in whom blood glucose was measured and documented in the chart, about a third of which did not previously carry the diagnosis of diabetes. Hyperglycemia in this specific setting, dubbed stress hyperglycemia,1 is quite frequently found in hospitalized patients and has been shown to increase the risk of death, congestive heart failure, and cardiogenic shock after myocardial infarction.2 Acute insulin resistance is also seen frequently in an acutely ill patient and is attributed to the release and metabolic actions of counterregulatory hormones and cytokine excess.3 Patients often require increased amounts of insulin to maintain glucose at an acceptable level. Iatrogenic hyperglycemia may occur as a consequence of glucocorticoids or excessive infusion of dextrose. In critically ill patients, vasopressors may also be associated with iatrogenic hyperglycemia. Inpatient hyperglycemia is associated with nosocomial infections, increased mortality, increased length of stay, and poor overall outcome.4 Of interest is that stress hyperglycemia was associated with more adverse outcome than was hyperglycemia in a patient with known diabetes.1, 2 We are not sure if this phenomenon of stress hyperglycemia is pathogenic or serves as a marker of disease severity.
Is Hyperglycemia Really a Problem?
Compelling evidence that control of hyperglycemia improves the outcomes of patients undergoing cardiothoracic surgery was provided by the Portland trial. Although this study was not randomized and its glycemia targets were not well defined, it demonstrated that better control of blood glucose levels drastically reduces the incidence of chest wall infections and the need for transfusions and significantly shortens hospital length of stay (LOS).5
The results of the Diabetes Mellitus Insulin‐Glucose in Acute Myocardial Infarction (DIGAMI) study showed that hyperglycemic patients with acute myocardial infarction had improved outcomes when intravenous administration of insulin was used to aggressively control glycemia.6 Van den Berghe et al. found significantly lower mortality and morbidity rates in surgical intensive care unit patients in whom aggressive glycemic control was attained with continuous intravenous insulin infusion. The study also identified reduced requirement of antibiotics, red cell transfusions, dialysis, and ventilatory support with aggressive glycemic control.7 It was also shown that there was significantly reduced morbidity in all patients in the medical ICU receiving intensive insulin therapy.8 Another meta‐analysis found that insulin therapy initiated in hospitalized critically ill patients in different clinical settings had a beneficial effect on short‐term mortality.9 Krinsley observed hyperglycemia to be associated with adverse outcomes in acutely ill adult patients and that its treatment has been shown to improve mortality and morbidity in a variety of settings.10 In their study of adults with diabetes, Golden et al. identified hyperglycemia as an independent risk factor for surgical infection of diabetic patients undergoing cardiac surgery.11 A meta‐analysis by Capes et al. showed a 3‐fold higher risk of poor functional recovery in nondiabetic hyperglycemic patients compared to that of nondiabetic euglycemic patients.2 A recent retrospective analysis found that patients with hyperglycemia treated for acute exacerbation of chronic obstructive pulmonary disease had poor outcomes.12
It is possible to give an account and references of only a limited number of such studies. The prevailing message conveyed in all these studies is that patients with poorly managed hyperglycemia have a poor overall outcome. Hence, the need to better manage inpatient hyperglycemia cannot be overemphasized.13
After an extensive search, we could not find well‐designed prospective randomized studies of patients who are not acutely ill or are outside the perisurgical period. However, the DIGAMI, Van den Berghe, and Portland trials generated a powerful and large momentum that has created interest in establishing protocols for keeping the blood glucose of patients in most medical and surgical critical care units in the suggested range.57, 13 Moreover, extrapolation of the data to noncritical and nonsurgical patients made possible a consensus conference organized by the American Association of Clinical Endocrinologists (AACE) that garnered support from many other medical associations. The position paper published by the AACE calls for tighter glycemic control in hospitalized patients. The AACE recommends that blood glucose concentrations for intensive care unit patients be maintained below 110 mg/dL. In noncritically ill patients, the preprandial glucose level should not exceed 110 mg/dL, and maximum glucose should not exceed 180 mg/dL.14 The American Diabetes Association (ADA) does not recommend any target glucose values for noncritical patients but does believe there is a need to have better inpatient hyperglycemic management. Some authorities believe that until the amount of scientific data increases, it is prudent to stay within the ADA‐recommended ambulatory guidelines for a preprandial plasma glucose level of 90‐130 mg/dL15 and a postprandial blood glucose level not to exceed 180 mg/dL.
Additionally, due attention must be paid to hypoglycemia secondary to aggressive glycemic control.
Because of the absence of evidence‐based information, it is not surprising that opinions conflict about the optimal level of blood glucose for an inpatient. We believe that in the absence of definitive evidence, it is prudent to adhere to the targets recommended by these associations.
A SOLUTION: WHAT TO DO AND HOW TO DO IT
Ideally, a system should be established to attain euglycemia without the attendant risk of hypoglycemia. The Joint Commission on Accreditation of Healthcare Organizations recently showed recognition of this need this by collaborating with the American Diabetes Association to establish a program to certify inpatient diabetes care center programs that meet national standards. The program must be carried out in all inpatient settings and should include the following elements16:
-
Specific staff education requirements;
-
Written blood glucosemonitoring protocols;
-
Plans for the treatment of hypoglycemia and hyperglycemia;
-
Collection of data on the incidence of hypoglycemia;
-
Education of patients on self‐managing their diabetes; and
-
An identified program champion or program champion team.
The Joint Commission's Advanced Inpatient Diabetes Certification Program is based on the ADA guidelines; the scope of this manuscript does not cover all the elements required to receive certification.16 In the rest of the article, we focus on the basic principles of the use of insulin to control hyperglycemia in the hospital setting.
The normal system that regulates glycemia encompasses a very complex system of hormonal and metabolic regulators. At the core of this system, insulin is the key regulator. Therapeutic insulin is therefore the best resource available for controlling hyperglycemia in the hospital setting.
Of the other currently available therapies, none offers the power and rapidity that insulin has to control blood glucose level. The biguanides are usually contraindicated in the hospital setting because most patients with hyperglycemia and/or diabetes are acutely ill and hence at risk of lactic acidosis. Furthermore, in a large number of these patients radio‐contrast agents are used; hence, transient renal failure is common, posing yet another risk factor for lactic acidosis. The thiazolidinediones (TZDs) are slow to act and not as powerful in controlling acute hyperglycemia and thus are not the optimal tool available when the metabolic situation changes drastically as occurs in hospitalized patients. Precaution needs to be taken when using TZD to treat patients who have congestive heart failure or hepatic insufficiency. The action of the sulfonylureas (SUs) imparts a high risk of hypoglycemia and/or poor insulinemic response during stress to patients being treated with them; therefore, it is usually recommended that patients in a hospital setting not be treated with SUs, except for selected very stable patients. The new emerging therapies (incretin mimetics, dipeptidyl peptidase‐IV inhibitors, amylin) have never been tested in the hospital setting, and hence no recommendation can be made at this stage. Thus, we believe that the main tool available for treating the hospitalized patient with hyperglycemia is insulin coupled with proper nutrition and a system of information to monitor therapeutic progress, which allows for proper and timely adjustments as well as for treatment of hypoglycemia.
Within this setting a conceptual frame for insulin administration has been proposed. Exogenous insulin needs to be provided to mimic as closely as possible the physiological pattern of endogenous insulin secretion. The latter is broadly thought to be composed of 2 secretory components: a basal component and a prandial, or alimentary, component. The basal component of insulin secretion represents the rate of insulin produced independent of meal ingestion, which is mainly governed by the prevailing concentrations of arterial blood glucose and other hormonal and metabolic regulators. Prandial insulin is the increase in insulin secretion that occurs after eating, which occurs as a complex pattern of pulses. Roughly, prandial insulin secretion is mainly determined by the quantity and composition of the meal ingested, especially the quantity of carbohydrate.
Thus, the insulin dose that an inpatient requires may be thought as consisting of basal and nutritional insulin requirements. To these 2 components we also add a third component: a correctional insulin component.
The basal insulin requirement of a given patient can be estimated by taking into consideration the type of diabetes and body weight. The nutritional insulin requirement refers to the insulin required to cover nutritional intake, which in a hospital setting may correspond to regular meals, intravenous dextrose, nutritional supplements, enteral feedings, or parenteral nutrition. Because our estimates are not very accurate, corrective insulin is required to correct elevated concentrations of plasma glucose (usually measured with finger sticks) A scale or table of corrective insulin can be constructed on the basis of type of diabetes, body weight, and/or total amount of daily doses of basal and nutritional insulin. Obviously, many will think of corrective insulin as a sliding scale. It is import to remember that this scale is complementary to prescribed basal and nutritional insulin doses and not a substitute for them. 0
How can insulin be prescribed in the hospital to cover the 3 facets of insulin (basal, alimentary, and corrective)? The following is a pragmatic approach that we found useful and uses the above considerations as an underpinning. We will first consider the general medicine or surgical ward and then the intensive care setting.
General Medical and Surgical Wards
Basal insulin
The activity of the ideal insulin preparation for this task should not show any peak, instead should remain in a steady state for 24 hours. Currently, three insulin preparations can be considered for this purpose. Glargine insulin is an analogue of insulin that has a stronger capacity to form and maintain hexamers of insulin and its rate of absorption from the subcutaneous depot, which allows for quasi‐steady‐state action for 24 hours. However, variability is sometime noted clinically in the length of duration and the absence or presence of a peak. Neutralized protamine insulin (NPH) is a mixture of protamine and human insulin in which the complexing of the 2 proteins retards absorption of insulin. The action profile of NPH insulin definitively displays a peak (between 6 and 10 hours after injection); however, the timing of this peak varies from patient to patient and (in the same patient) from day to day. There is also variability in the widely quoted duration of action (12‐18 hours). Despite these shortcomings, NPH has been used for several decades and has widespread acceptance among physicians, especially because it costs less than glargine insulin. Detemir insulin is a new analogue of insulin. The insulin molecule has been complexed with a fatty acid. This modification protracts absorption from the subcutaneous depot and also within the blood compartment because the acylated insulin binds to albumin, which then acts as a reservoir. There is very little experience with detemir in clinical scenarios and none in the hospital setting.
Our preferred basal insulin, given current knowledge and experience, is glargine, except in those special cases in which insulin that has a shorter action is needed (ie, patients with tube feeds, use of steroids), listed below in the Special Considerations section.
Alimentary or nutritional or prandial or bolus insulin
The ideal insulin to cover the prandial period should have rapid onset and rapid dissipation of activity. Two types of insulin preparation have these characteristics. The insulin traditionally used in this setting is regular human insulin. Unfortunately, the onset of action of this preparation is not as rapid (20‐30 minutes), forcing it to be prescribed as a preprandial insulin. In a hospital setting, where the dynamics of meal serving and NPO periods are highly unpredictable, this creates a serious risk of hypoglycemia when preprandial insulin is the choice. The activity of regular human insulin lasts 4‐6 hours, and thus there is also the risk of stacking multiple prandial doses and hence an increase in the risk of hypoglycemia. So, although this insulin has been used for many decades, it has been rapidly replaced by the new analogues of insulin (lispro, aspart, and glulisine), which have a much faster onset of activity (within 15 minutes of injection) because of its rapid absorption from the subcutaneous depot. Moreover, the dissipation of insulin action of these preparations is faster (3‐4 hours). The minor decrease in glycemic power at equivalent doses compared to preprandial administration can usually be easily overcome with a minimal increase in dosage. It is becoming widely accepted because it allows for flexibility in the timing of administration (which can be made contingent on meal ingestion) and also in dosing because it can be better tailored to the amount of food consumed. Overall, our preference is for the use of analogues in the hospital setting. The only drawback is cost.
Corrective insulin
The type of insulin used for the correction of glycemia that exceeds the target follows from the same considerations as those used for the alimentary or prandial insulin. For simplicity, the same type of insulin chosen for alimentary insulin should be the one selected for corrective insulin.
How to dose the insulin?
Once the type of insulin preparations has been chosen, dosing is the next task. The hospitalist should remember that the initial prescription or dosage of insulin will need to be reevaluated daily to allow for glycemia management and to avoid hypoglycemia. As in many other fields of medicine, a single approach does not fit all scenarios. The following is a list of scenarios commonly encountered in our inpatient population. There is no definitive way to suggest how successfully they are managed as outpatients. It may be reasonable to assume glycemic control with HbA1C of less than 7% as being highly successful, HbA1c between 7.1% and 8.5% as being moderately successful, and HbA1C greater than 8.5% as being unsuccessful. These HbA1C levels help to guide us through decision making and have been very helpful in our practice. Recognition of hyperglycemia either on admission or during an in‐hospital stay warrants consideration of insulin‐based management. For several reasons, HbA1c should be tested in patients who are found to have hyperglycemia in the hospital. Elevated glycated hemoglobin enables the recognition of previously undiagnosed diabetes and helps in the identification of patients with poorly controlled diabetes; hence, the hospital stay can be an opportunity to change treatment approaches or emphasize compliance. Likewise, in a hyperglycemic patient with normal HbA1C, it should be considered whether stress hyperglycemia has developed.
-
Patient is using a highly successful regimen with oral agents. Recommendation: continue using oral agents if no contraindications exist, the patient is unlikely to receive contrast dye tests, and admission is for a minor indication requiring a short inpatient stay. Oral agents may need to be stopped if poor glycemic control after hospitalization or any contraindication is identified.
-
Patient is using a regimen of insulin that has been very successful. Recommendation: follow the same regimen and add to it the table for correction insulin.
-
Patient is using a regimen of insulin that is moderately successful. Recommendation: keep the same regimen but increase doses and add the correction insulin table.
-
Patient is using an unsuccessful oral regimen. Recommendation: discontinue oral agents and start basal, alimentary, and corrective insulin
-
Patient is using a very unsuccessful regimen of insulin. Recommendation: reevaluate and prescribe a basal, alimentary, and correction insulin regimen.
-
Patient is recently or newly diagnosed with diabetes. Recommendation: while in the hospital use a basal, alimentary, and corrective insulin regimen.
-
Patient has type 1 diabetes. Recommendation: prescribe full insulin coverage with basal, nutritional, and correctional insulin.
-
Patient is receiving an IV drip of insulin and is no longer critical and tolerating po intake. Recommendation: overlap IV drip with subcutaneous insulin for at least 4 hours and then continue subcutaneous insulin.
Empirical calculation of basal insulin.
Once you have decided which insulin to use as basal insulin, the following may be used to calculate empirical doses. Suggested insulin types for basal include glargine and NPH insulin.
For scenarios 4, 5, 6, and 7 we use a simple formula to estimate the basal insulin requirements. Longer‐acting insulin requirements may be calculated as:
-
For type 2 diabetes: 0.4 units/kg/day of basal insulin.
-
For type 1 diabetes: 0.2 units/kg/day of basal insulin.
-
The adjustments should be made every 48 hours 2‐5 units at a time or 10% of the dose.
-
For a regimen based on glargine insulin: full dose is administered daily.
-
For NPH: two thirds is given AM and one third PM.
-
For scenario 8: basal insulin is estimated as total dose of insulin drip per hour for the last 6 hours 0.8 24. We recommend an overlap of IV drip and SQ insulin of at least 4 hours.
Empirical calculation of alimentary or nutritional insulin.
Once the type of insulin has been chosen, the insulin doses may be calculated empirically. Suggested insulin choices include lispro, aspart, and glusiline insulin.
As a convenient tool, the total daily alimentary or nutritional insulin requirement is nearly equal to total daily basal insulin. This dose estimation may then be divided into various premeal doses on the basis of the carbohydrate content of the meal. Provision of 1 unit of short‐acting insulin should be made for every 15 units of carbohydrate intake. The following rough estimation may be used to calculate the premeal alimentary insulin dose.
-
For type 2 diabetes: Empirically, 0.1, 0.15, and 0.15 units of short‐acting insulin/kg for breakfast, lunch, and dinner, respectively.
-
For type 1 diabetes: Empirically, we suggest 0.05‐0.1 units of rapid‐acting insulin/kg, to be administered before meals.
The premeal dose requirement of an individual patient may be significantly different. If patient is NPO, then alimentary insulin is not prescribed; specific doses need to be suspended if patient is made NPO and resumed when PO is restored.
Total daily dose of insulin may vary according to body weight, endogenous insulin secretory capacity, and degree of insulin resistance. Variation tends to be greater for those with type 2 diabetes.
We encourage administration of alimentary insulin at or immediately after meal ingestion. This implies a system of alert for patients to let nurses know when they have finished eating.
Empirical calculation of correctional insulin (sliding scale).
Although every institution relies on its own guidelines, we reproduce here the correction table we use at Wake Forest UniversityBaptist Medical Center (WFUBMC), which was generated based on type of diabetes and body weight:
In our inpatient practice this correction table has been quite helpful. Bear in mind that there are several correctional insulin dose algorithms, and the one most suitable to local needs should be adopted.
Once the insulin to be used for correctional insulin has been chosen, the algorithm shown in Figure 2 may be used.
To ensure clarity about the prescribed regimen, we encourage the use of preformatted templates or (preferably) computerized orders. Figure 3 shows an example, an order template that we use at WFUBMC.
Intensive Medical and Surgical Care Unit
Continuous intravenous insulin infusion (IV insulin drip) is the most suitable way to administer insulin to critically ill patients. For continuous intravenous insulin infusion, regular insulin is most commonly used and in fact is the only type of insulin studied in prospective randomized trials. This requires adequate staffing, frequent monitoring, and frequent dose adjustments. Such stringent glycemic control is appropriate for patients in critical care units. The American College of Endocrinology recommends using intravenous insulin therapy in the subset of inpatients who have diabetic ketoacidosis, are before major surgical procedures, are undergoing fasting for more than 12 hours and have type 1 diabetes, are critically ill, are undergoing labor and delivery, are being treated for myocardial infarction, have just had organ transplantation, are being maintained on total parenteral nutrition, or have other illnesses requiring prompt glucose control.14
The use of continuous intravenous insulin infusion on a regular basis on all medical floors is not routinely recommended because there is not adequate scientific data to support its use from both clinical and financial perspectives. There are various protocols for attaining recommended levels, and every institution must adopt or develop a protocol that both suits its needs and is feasible. Again, aggressive glycemic control using intravenous insulin requires a well‐monitored setup and is ideal for intensive care units.
Management of hypoglycemia
The therapeutic window between insulin effectiveness and insulin‐associated hypoglycemia is very narrow; hence, proper management of blood glucose needs to be embedded within a system in which every member of the team taking care of a patient with diabetes has appropriate knowledge of the task at hand. Of equal importance is the development of a protocol to treat hypoglycemia minimizing the overzealous treatment that leads to severe hyperglycemia. This protocol also assumes that all oral hypoglycemic agents have been discontinued.
Hypoglycemia protocol (FSBS < 70 mg/dL)
-
Patient conscious and able to eat (select one):
-
Provide patient with 15 g of carbohydrate (120 cc of fruit juice or 180 cc of regular soda or 240 cc of skim milk or 3 glucose tablets).
-
Recheck fingerstick blood sugar (FSBS) in 15 minutes.
-
Repeat above if FSBS still <70 mg/dL; continue cycle until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and IV access available
-
Administer 15 mL of 50% dextrose IV (mix in 25 mL of NS) and call HO
-
Check FSBS in 15 minutes.
-
Repeat IV 50% dextrose until FSBS is >70 mg/dL.
-
Once FSBS is >70 mg/dL, recheck FSBS in 1 hour; if it is <70 mg/dL, repeat above cycle and call HO.
-
Patient NPO or unconscious and no IV access available
-
Administer glucagon 1 mg IM.
-
Turn patient on side (to avoid broncho‐aspiration) and call HO.
-
Check FSBS in 15 minutes.
-
If still <70 mg/dL, start IV line and follow protocol for an unconscious patient with IV access available.
Special considerations
Steroid use.
No clinical trials have been conducted to define a quantitative approach to managing hyperglycemia induced by steroids (in those patients without previous diabetes) or to understand adjustments in insulin dose for diabetic patients who will undergo treatment with steroids.
Empirically, we recommend the use of NPH insulin and to adjust the dose calculation 20% higher for low‐dose prednisone (10‐20 mg/day), 30% higher for medium‐dose prednisone (21‐40 mg/day), and 50% higher for high‐dose prednisone (>41 mg/day). We do not recommend the use of glargine insulin in this setting since the half‐life of prednisone is less than 24 hours; hence, the risk of hypoglycemia is high when using very long‐acting insulin. Also, empirically, we make the recommendation to maximize the NPH AM dose and minimize the PMdose, possibly dividing the calculated dose into three quarters AM, one quarter PM. We also caution that this approach would not be appropriate when a patient is using other steroids (prednisolone, methylprednisolone, and dexamethasone) because the half‐life of these steroids exceeds 24 hours, and in such cases glargine insulin may be suitable.
Enteral and parenteral feeding.
Frequently patients need enteral or parenteral feeding. The former may be given as continuous or discontinuous infusion; hence, in this particular setting, a specific insulin regimen must be customized in close collaboration with the dietician. For example, for those patients who are chronically fed enterally and for whom a system of bolus has been established, the use of a basal insulin may be warranted. However, for the patient who is being fed nocturnally only, we would probably choose NPH as the insulin regimen. Good success has been found in some hospitals with the use of premixed insulin preparations when enteral feeding is continuous for 24 hours. Patients fed parenterally may receive their basal and alimentary insulin as an addition to the nutrition bag, complemented with correction insulin administered subcutaneously.
CONCLUSIONS
In conclusion, it is the responsibility of hospitalists to make a conscious effort to manage hyperglycemia in patients who are previously diabetic or become hyperglycemic during hospitalization in order to improve their clinical outcome. Hospitals need to realize that this task is far from being the lone duty of physicians; hence, systems for hyperglycemia management that engage multidisciplinary teams must be established.
Although the ideal way to do so in the critical care setting is continuous intensive insulin infusion therapy, this may not always be practical. In such cases, basal and alimentary insulin with appropriate insulin sliding scales should be used. Using the sliding scales alone should be strongly discouraged, as they tend to only troubleshoot a situation and allow the damage caused to the patients on a molecular level to be camouflaged in objective ways. Appropriate attention should be paid to the risk of developing hypoglycemia as a sequela of overzealous correction of hyperglycemia. This leaves us with the therapeutically desirable band of the glycemic spectrum. However, this band is wide enough to make it possible to achieve better performance. Although the target glycemic range definitively needs to be determined, it is reasonable to have 80‐110 mg/dL as the target range for critically ill patients, as generally agreed by both the ACE and ADA, and a glycemic range of preprandial glucose between 90 and 130 mg/dL for ambulatory patients. The maximal blood glucose should not exceed 180 mg/dL. Having realized the adverse impact on patients of uncontrolled hyperglycemia, at the next morning report, it is appropriate for the nurse to say, Mr. Smith's finger‐stick glucoses are better controlled now, and he required only 2 units of additional insulin coverage yesterday. If we still hear high glucose numbers and keep fixing this problem with sliding‐scale insulin alone, we are not doing a good job.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet.2000;355:773–778.
- .Insulin therapy for the critically ill patient.Clin Cornerstone.2003;5(2):56–63.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.J Parenter Enter Nutr.1998;22:77–81.
- ,,,.Metabolism.1994;43:279–284.
- ,,, et al.Randomized trial of insulin‐glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): effects on mortality at 1 year.J Am Coll Cardiol.1995;26:57–65.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461
- ,,.Insulin therapy for critically ill hospitalized patients: a meta‐analysis of randomized controlled trials.Arch Intern Med.2004;164:2005–2011.
- .Perioperative glucose control.Curr Opin Anaesthesiol.2006;19(2):111–116.
- ,,,:Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes.Diabetes Care.1999;22:1408–1414.
- ,,, et al.Hyperglycaemia is associated with poor outcomes in patients admitted to hospital with acute exacerbations of chronic obstructive pulmonary disease.Thorax.2006;61:284–289.
- ,,, et al.;American Diabetes Association Diabetes in Hospitals Writing Committee.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591. Errata in: Diabetes Care. 2004;27:856. Hirsh, Irl B [corrected to Hirsch, Irl B]; dosage error in text; and Diabetes Care. 2004;27:1255
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(1):77–82.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979
- Joint Commission on Accreditation of Health Organizations Web site. Avail at: http://www.jointcommission.org/NewsRoom/NewsReleases/jc_nr_072006.htm. Accessed September 29,2006.
- ,,,,,.Hyperglycemia: an independent marker of in‐hospital mortality in patients with undiagnosed diabetes.J Clin Endocrinol Metab.2002;87:978–982.
- ,,,.Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview.Lancet.2000;355:773–778.
- .Insulin therapy for the critically ill patient.Clin Cornerstone.2003;5(2):56–63.
- ,,, et al.Early postoperative glucose control predicts nosocomial infection rate in diabetic patients.J Parenter Enter Nutr.1998;22:77–81.
- ,,,.Metabolism.1994;43:279–284.
- ,,, et al.Randomized trial of insulin‐glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): effects on mortality at 1 year.J Am Coll Cardiol.1995;26:57–65.
- ,,, et al.Intensive insulin therapy in critically ill patients.N Engl J Med.2001;345:1359–1367.
- ,,, et al.Intensive insulin therapy in the medical ICU.N Engl J Med.2006;354:449–461
- ,,.Insulin therapy for critically ill hospitalized patients: a meta‐analysis of randomized controlled trials.Arch Intern Med.2004;164:2005–2011.
- .Perioperative glucose control.Curr Opin Anaesthesiol.2006;19(2):111–116.
- ,,,:Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes.Diabetes Care.1999;22:1408–1414.
- ,,, et al.Hyperglycaemia is associated with poor outcomes in patients admitted to hospital with acute exacerbations of chronic obstructive pulmonary disease.Thorax.2006;61:284–289.
- ,,, et al.;American Diabetes Association Diabetes in Hospitals Writing Committee.Management of diabetes and hyperglycemia in hospitals.Diabetes Care.2004;27:553–591. Errata in: Diabetes Care. 2004;27:856. Hirsh, Irl B [corrected to Hirsch, Irl B]; dosage error in text; and Diabetes Care. 2004;27:1255
- ,,, et al.American College of Endocrinology position statement on inpatient diabetes and metabolic control.Endocr Pract.2004;10(1):77–82.
- ,.Counterpoint: inpatient glucose management: a premature call to arms?Diabetes Care.2005;28:976–979
- Joint Commission on Accreditation of Health Organizations Web site. Avail at: http://www.jointcommission.org/NewsRoom/NewsReleases/jc_nr_072006.htm. Accessed September 29,2006.
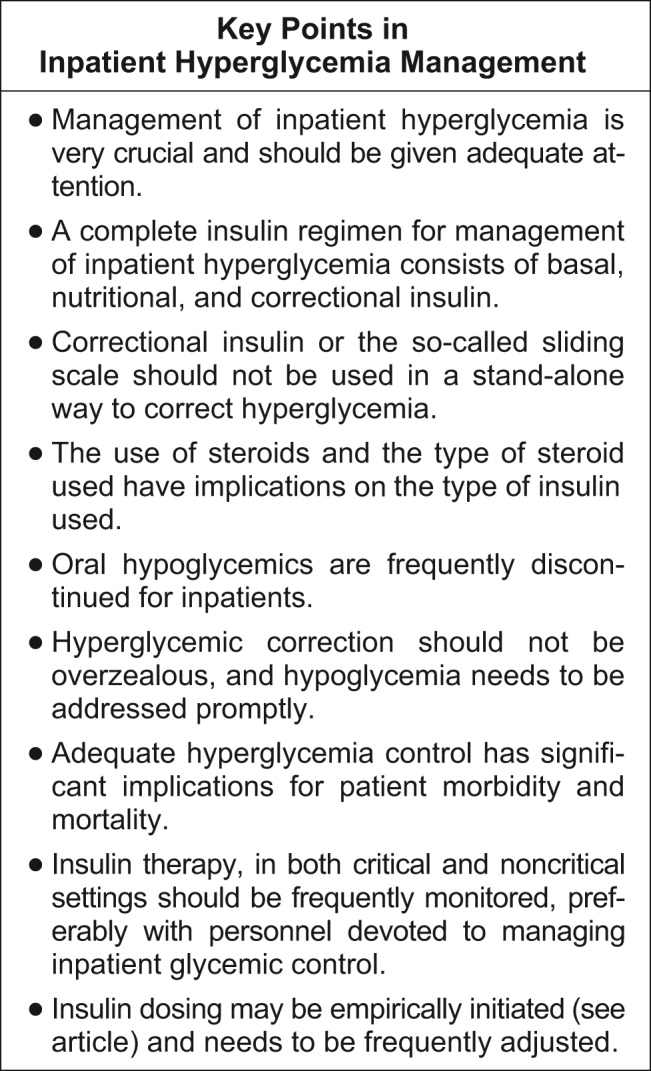
The Hospitalist and Stroke Prevention
Prevention has the greatest potential to reduce the societal burden from stroke.1 Several therapies that specifically target the underlying atherosclerotic disease process have been shown in clinical trials to markedly lower the risk of recurrent vascular events including stroke.2 However, there is great variability in how clinical trial data are implemented in clinical practice for ischemic stroke prevention.35 This has led to a knowledge‐implementation‐practice gap, possibly because of the limited awareness of the scientific evidence supporting various treatments, as well as the lack of a systematic approach to hospital stroke care.3 Our review discusses the evidence for reducing vascular risk after ischemic stroke and successful models of systematic interventions initiated during stroke hospitalization, with the goal of narrowing the stroke hospitalization evidencepractice gap.
Societal Burden
Stroke is the third‐leading cause of death in the United States and the leading cause of serious long‐term disability.6 Approximately 700,000 Americans have a new stroke or recurrent strokes every year, whereas nearly 5 million live with the consequences of stroke; nearly all stroke survivors (90%) have some residual functional deficit, and approximately 40% experience moderate to severe impairment.6 Stroke mortality is substantial, with a 30‐day case fatality rate after first stroke (of any cause) of about 25%.7, 8 Indeed, four‐fifths of patients do not survive for 10 years after stroke, and approximately one‐third of all case fatalities occur in the first year after a stroke.8 The estimated economic impact in 2006, US$57.9 billion, further underscores the substantial mortality and morbidity of stroke.6 Given the limited options for acute stroke therapies,9 stroke prevention remains an important therapeutic goal, especially because fewer than 5% of acute stroke patients in the United States currently receive the only Food and Drug Administrationapproved treatmentintravenous tissue plasminogen activator.10 It is obvious that additional strategies are urgently needed to reduce the devastating consequences of stroke.
Why Involve the Hospitalist?
The Hospitalist system in the United States is rapidly growing.11 Tthe Society of Hospital Medicine projects that by 2010 there will be approximately 30,000 hospitalists in the United States.11 A member census conducted by the American Academy of Neurology in 2000 found 13,500 practicing neurologists, most of whom are concentrated in urban and metropolitan areas.12 As such, with more than 700,000 strokes occurring each year,6 most stroke patients in the United States will not be seen or evaluated by a neurologist. Indeed, one study indicated that only 11.3% of stroke patients are attended exclusively by a neurologist.13 Furthermore, it is not uncommon for stroke patients to have numerous other medical issues that require attention and multidisciplinary care coordination during the hospital stay, an area where hospitalists excel. Conceivably, the ability to promptly identify and treat these non‐neurological comorbidities, which account for at least 30% of the deaths from acute ischemic stroke,14 could go a long way toward improving stroke outcomes.
Hospitalists are in the forefront of developing strategies for improving the quality of acute care and patient satisfaction, reducing medical errors, and focusing on efficient resource utilization. Translating evidence‐based strategies for acute stroke care into actual practice is a mechanism for improving the quality of care, ensuring that basic care does not deviate from provider to provider or from day to day (weekdays compared to weekend days/holidays) while at the same time allowing for the individualization of care appropriate to a patient's unique needs.15 After the acute treatment of stroke or TIA, additional measures must be initiated as soon as it is safe to do so in order to begin the process of limiting stroke progression and preventing recurrence. Secondary prevention measures require a coordinated transition in order to ensure continuation of care and follow‐up as needed. After a thorough risk assessment is complete, hospitalists will need to consider a 3‐pronged approach to secondary prevention that follows the national guidelines described above: pharmacotherapy, behavior modification, and, in some cases, surgical intervention.
Secondary Stroke
Secondary or recurrent strokes are strokes that occur after a first stroke or TIA,2 and the single biggest risk factor for having a stroke is already having had one.2 Because hospitalists generally see patients after ischemic cerebrovascular events have already happened, their opportunities to intervene are mostly geared toward reducing the risk of secondary stroke (beyond enhancing the prevention of complications from the index event). Recent community‐based data indicate that the short‐term risk of secondary stroke is high.16, 17 After a minor stroke or TIA, the risk of recurrent stroke or TIA increases over time8%‐12% within 7 days, 12%‐15% within 30 days, and 17%‐19% within 90 days.18 In the largest study of short‐term risk following TIA,19 there was an 11% risk of stroke (51% of which occurred in the 48 hours after TIA), an 13% risk of TIA, and a 25% risk of any adverse event within 90 days of the TIA.
Overall, the risk of a second cerebrovascular event is highest in the first year after a stroke/TIA (12%), declining to about 5% annually thereafter.7 The effects of secondary stroke are more devastating than those of the primary stroke: the 30‐day fatality rate after a first recurrent stroke is almost double that after the first‐ever stroke (41% versus 22%).20 The pathological factors that lead to TIA and stroke, such as platelet aggregation and subsequent thrombosis or the systolic stroke of blood against stenotic carotid plaques, are one and the same. As such, the short‐ and long‐term risks of recurrent events after both first stroke or first TIA necessitate investigation into a patient's vascular risk and early initiation of appropriate stroke prevention strategies.21
Cross Risk
Because the atherothrombotic disease process is systemic in nature with a variety of manifestations, stroke patients with atherosclerosis frequently have coexistent coronary artery disease and peripheral artery disease,22 and as such, are at risk for vascular events emanating from any of these beds in addition to that of the cervicocephalic arterial tree.23, 24 For instance, in a study of individuals in a long‐term care facility, among the patients with ischemic stroke, 56% had overlapping coronary artery disease, 28% had peripheral artery disease,25 and 38% of the patients had at least 2 manifestations of their atherosclerotic disease. The take‐home message here is that hospitalists also have the opportunity while treating patients hospitalized following stroke to prevent other vascular events by identifying and treating stroke patients who have systemic atherosclerosis.
Risk Factors
The first step in any approach to stroke prevention is the identification of predisposing risk factors. Several of the known biological and lifestyle risk factors associated with cerebrovascular disease were identified decades ago from large longitudinal studies.2 Certain stroke risk factors are nonmodifiable and therefore cannot be the target of intervention. 26 Treatment of the various stroke risk factors could have a substantial impact on reducing the burden of stroke. Table 1 shows the number needed to treat to prevent one stroke per year by modification of the individual stroke risk factor.
| Treatment | Relative risk reduction | Number needed to treat (1 stroke/year) |
|---|---|---|
| ||
| Antihypertensives | 28% | 51 |
| Statins | 25% | 57 |
| Aspirin | 28% | 77 |
| Smoking cessation | 33% | 43 |
| Carotid endarterectomy | 44% | 26 |
Guidelines for Secondary Stroke Prevention
Several organizations have published guidelines for the prevention of secondary stroke based on clinical evidence and expert consensus. Key guidelines include those published by the American Stroke Association (ASA),2 American College of Chest Physicians (ACCP),27 and the National Stroke Association. Although these guidelines are broadaddressing many components of stroke prevention and careeach contains recommendations specifically applicable to secondary prevention in most stroke patients who the hospitalist will encounter. Some provide hospital‐based guidelines that focus on care protocols and systems processes (ie, ASA Stroke Systems Guidelines), whereas others are therapy‐based guidelines (i.e, ACCP Guidelines on Antithrombotic Therapy for Ischemic Stroke). In the next few sections, we discuss common risk factors for and causes of secondary stroke and the prevailing guideline recommendations for modifying them. Discussion of the management of rare causes of ischemic stroke such as arterial dissection, vasculitis, patent foramen ovale, and so forth is beyond the scope of this article.
Hypertension, Dyslipidemia, and Diabetes
Table 2 shows the current national guideline recommendations for the management of premier vascular risk factorshypertension, dyslipidemia, and diabetesin ischemic stroke and TIA patients.2 Antihypertensive therapy is recommended for the prevention of secondary stroke and other vascular events in patients who have experienced an ischemic stroke or TIA and are beyond the hyperacute period.28, 29 Such treatment should be considered for all ischemic stroke and TIA patients regardless of history of hypertension.28 Although available data support the use of diuretics and the combination of diuretics plus an angiotensin‐converting enzyme inhibitor,28, 30 selection of specific medications should be individualized according to a patient's comorbid conditions.29 It is also important to note that despite the proven benefit of beta blockers in the secondary prevention of recurrent cardiac events, current evidence shows no clear benefit from the use of beta blockers in the prevention of stroke.29, 31
| Risk Factor | Recommendation |
|---|---|
| |
| Hypertension | Antihypertensive beyond hyperacute stroke period60 |
| Data support diuretic or diuretic + ACEI,2830 but individualize based on patient characteristics | |
| Antihypertensive in all patients regardless of history of hypertension28 | |
| Aim for average reduction of 10/5 mm HG or blood pressure < 120/80 mm Hg28 | |
| Encourage reduced intake of dietary salt | |
| Dyslipidemia | Statin for LDL‐C goal < 100 mg/dL in those with CAD or symptomatic atherosclerosis33, 34 |
| Target LDL‐C < 70 mg/dL for very high‐risk persons61 | |
| Statin for stroke or TIA because of atherosclerosis regardless of LDL‐C level33, 34 | |
| Niacin or gemfibrozil for patients with low HDL‐C62, 63 | |
| Diabetes | ACEIs and ARBs should be first‐choice blood pressure drugs37, 38a |
| Glucose control to near normoglycemic levels39 | |
| Target glycosylated hemoglobin 7%64 | |
For ischemic cerebrovascular disease patients with dyslipidemia or symptomatic atherosclerosis, cholesterol management should be according to the current Adult Treatment Panel (ATP) guidelines.32 Statins should be the first‐line treatment.33, 34 Ischemic stroke or TIA patients whose underlying stroke mechanism is presumed to be atherosclerosis should be considered for statin therapy even if they have normal cholesterol levels and no evidence of atherosclerosis.33, 34 The recent Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study was the first study to specifically investigate the effect of statins in patients with a prior stroke but with normal cholesterol levels and no evidence of coronary heart disease. It found that treatment with atorvastatin 80 mg/day (vs. placebo) was associated with a 16% reduction in relative risk of recurrent stroke.34
The care of an ischemic stroke or TIA patient who has diabetes warrants more rigorous control of blood pressure and lipids.35, 36 Such patients usually require more than one antihypertensive drug. ACEIs and angiotensin receptor blockers (ARBs) are more effective in reducing the progression of renal disease and are the recommended first‐choice medications for these patients.37, 38 The target for glucose control should be reaching near‐normoglycemic levels.39
Large‐Artery Atherosclerosis
In selected at‐risk stroke patients, surgical techniques (eg, carotid endarterectomy [CEA], carotid angioplasty and/or stenting [CAS]) may reduce the rate of recurrent stroke.4044 For patients who have had ischemic cerebrovascular events in the preceding 6 months and who have ipsilateral severe (70%‐99%) cervical carotid artery stenosis, CEA done by a surgeon is recommended; it has a perioperative morbidity and mortality of less than 6%.40 For those with ipsilateral moderate (50%‐69%) cervical carotid stenosis, CEA should be considered, and whether to operate should be decided on the basis of the patient's age, sex, comorbidities, and severity of initial symptoms.41 Analyses of endarterectomy trials indicated that the benefit from CEA is greatest if performed within 2 weeks of a patient's last ischemic event, the advantage it confers rapidly falling with increasing delay.45 From the hospitalist's standpoint, it is of prime importance to ensure that patients admitted to the hospital with a TIA or ischemic stroke are not discharged before it has been established whether have severe carotid stenosis that requires a revascularization procedure. If carotid stenosis is less than 50%, CEA is not recommended.41
A newer, less invasive form of carotid artery revascularization is CAS,46 which is performed by operators with established periprocedural morbidity and mortality rates of 4%‐6% and may be considered in those with:
-
Symptomatic severe stenosis (>70%) that is difficult to access surgically.2
-
Medical issues that greatly increase the risks of surgery, such as clinically significant cardiac disease, severe pulmonary disease, contralateral carotid occlusion, contralateral laryngeal nerve palsy, radiation‐induced stenosis or restenosis after carotid endarterectomy, and more than 80 years old.43
Angioplasty and/or stenting may also be considered when patients with symptomatic extracranial vertebral stenosis are having symptoms despite optimal medical risk factor treatments.2 Among those with hemodynamically significant stenosis of the major intracranial vasculature (basilar, middle cerebrals, distal carotids, and vertebrals) experiencing symptoms despite optimal medical risk factor treatments, angioplasty and/or stenting is considered experimental.2
The degree of arterial stenosis can be assessed by ultrasound, magnetic resonance angiogram (MRA), computed tomography angiogram (CTA), and conventional catheter angiogram, the last of which remains the gold standard. A carotid ultrasound performed at a certified vascular laboratory or by an experienced radiology technologist that shows less than 50% stenosis need not be followed up with another neuroimaging test. Generally, MRA tends to overestimate the degree of arterial stenosis but is a useful screening tool. In the event that an MRA reveals more than 50% stenosis, another diagnostic modality such as a carotid duplex, CTA, or conventional catheter angiogram should be performed to confirm this finding.
Antithrombotic Treatment
Cardioembolic Stroke Mechanism
Although it can sometimes be difficult to determine the precise mechanism underlying a patient's stroke or TIA, those who have a high‐risk source of cardiogenic embolism should generally be treated with anticoagulant medications to prevent recurrence.2 Among ischemic cerebrovascular event patients with persistent or paroxysmal atrial fibrillation, anticoagulation with adjusted‐dose warfarin (target international normalized ratio [INR] of 2.5; range, 2.0‐3.0) should be administered.47 The ASA recommends initiating oral anticoagulation within 2 weeks of an ischemic stroke or TIA but indicates that further delays may be appropriate for patients with large infarcts or uncontrolled hypertension.2 For patients unable to take oral anticoagulants, aspirin 325 mg/day should be given instead. Among patients who suffered an ischemic stroke or TIA because of an acute myocardial infarction in whom left ventricular mural thrombus is identified by echocardiography or another form of cardiac imaging, oral anticoagulation should be considered, aiming for an INR of 2.0‐3.0 for at least 3 months and up to 1 year.2 Patients receiving oral anticoagulation who also have ischemic coronary artery disease should be prescribed aspirin as well, in doses up to 162 mg/day.2
Noncardioembolic Stroke Mechanism
For ischemic stroke or TIA patients who have no high‐risk source of cardiogenic embolism, antiplatelet agents rather than oral anticoagulation are generally recommended to reduce the risk of recurrent stroke and other cardiovascular events.4850 Acceptable options for initial therapy include:
-
Aspirin (50 to 325 mg/day)48;
-
Combination of aspirin (50 mg) and extended‐release dipyridamole (400 mg) daily49, 51;
-
Clopidogrel (75 mg) daily.50
The combination of aspirin and extended‐release dipyridamole is suggested instead of aspirin alone, and clopidogrel may be considered instead of aspirin alone.49, 51 However, currently there is not enough data to make evidence‐based recommendations for choosing between antiplatelet drugs beyond aspirin.2 Furthermore, there is no evidence that increasing the dose of aspirin for patients who have had an ischemic stroke while taking aspirin provides additional benefit.2 The selection of an antiplatelet agent must be individualized, giving due consideration to a patient's presumed stroke mechanism, risk factor profile, and tolerance.
Other antiplatelet guidelines for noncardioembolic stroke/TIA patients include that:
Education for Behavior Modification
It is crucial to discharge patients with the tools they need to make important lifestyle changes. Patients can significantly reduce their stroke risk by making changes in their everyday patterns of behavior. As much education as possible about smoking cessation, exercise, diet, and the warning signs of stroke should be provided often as possible during hospitalization for a stroke and need not be left to nurses. Stroke education is extremely important so patients understand the need to call for emergency medical services immediately if they even suspect they are having stroke symptoms because of the very narrow window of opportunity for treatment of an acute stroke.54 All patients should be encouraged to make lifestyle adjustments such as ceasing smoking, reducing alcohol intake, and controlling weight. Smoking cessation appears to be effective in preventing secondary stroke (33% reduction in relative risk),44 and initiating smoking cessation counseling during hospitalization for stroke may result in a high rate of adherence to smoking cessation, at least in the short term.55 Table 3 displays current national guideline recommendations on lifestyle modification approaches.2
| Risk Factor | Recommendation |
|---|---|
| |
| Smoking | Smoking cessation |
| Avoid environmental smoke | |
| Counseling, nicotine products, and oral smoking cessation medications | |
| Alcohol | Eliminate or reduce alcohol consumption |
| Light to moderate levels2 drinks/day for men, 1 drink/day for nonpregnant women may be considered | |
| Obesity | Weight reduction goal: BMI 18.5‐24.9 kg/m2 and waist circumference < 35 inches for women, < 40 inches for men |
| Encourage weight management through balance of caloric intake, physical activity, behavioral counseling | |
| Physical Activity | At least 30 minutes of moderate‐intensity physical exercise most days of the week |
| Supervised therapeutic exercise regimen for those with residual disability | |
EvidencePractice Gap
There are now many secondary stroke prevention modalities, and there is a copious amount of data validating the efficacy of quite a few of them.2 Yet there is a large gap in implementing evidence‐based secondary prevention strategies.35 TIA and ischemic stroke patients are often discharged from the hospital without being prescribed any preventive medications, despite the data supporting the use of antiplatelet agents, anticoagulants, and antihypertensives for prevention of secondary stroke.4 In addition, several behavioral interventions could help patients to avoid stroke recurrence,2 but quite often stroke patients are not educated about them during the acute care period.4 Poor discharge treatment utilization limits the effectiveness of proven therapies, resulting in lost opportunities to reduce the burden of secondary stroke.
The reasons for these care gaps are multifactorial and can be traced to patient and provider issues as well as to health care delivery processes. Our understanding of the reasons for this gap is improving. Generally speaking, preventive services are used less frequently than those services or treatment modalities that provide immediate relief or economic benefit. The benefit of most preventive services is more readily seen at a population level than at a individual level and accrues slowly over time. It becomes more difficult to stress prevention in a health care system driven by technology‐based acute care.3
Current clinical management of acute stroke patients has stroke specialists and hospital physicians focusing on the acute management and diagnostic workup during hospitalization. Initiation of long‐term treatment is often deferred to after discharge, when the patient resumes long‐term primary care follow‐up.54 This deferred approach may result in therapy not being initiated or being initiated less efficiently and at a time (weeks or months after the initial presentation) when the stroke event and underlying atherosclerotic disease may no longer be the focus of either the patient or the primary care physician.54
Initiating medications during the acute stroke hospitalization phase sends the patient the message that these therapies are important for preventing recurrence and are an essential part of their treatment.54 More important, hospital initiation of secondary prevention therapies has been shown to be a strong predictor of these therapies continuing to be used after discharge56 and is associated with better clinical outcomes.5759 Table 4 shows some of the resources available to assist hospitalists in overcoming the evidencepractice gap in stroke treatment.
| Tool | Description |
|---|---|
| |
| AHA Get with the GuidelinesStroke ( |
Focuses on care team protocols to facilitate appropriate in‐hospital and discharge stroke treatment utilization |
| Identifies champions to lead, develop, and mobilize teams to optimally implement evidence‐based stroke treatment in acute care hospitals | |
| Utilizes standardized admission orders, patient educational materials, data monitoring | |
| Provides resources to help hospitals obtain JCAHO certification | |
| UCLA Stroke PROTECT (Preventing Recurrence of Thromboembolic Events through Coordinated Treatment) program ( |
Integrates 8 proven secondary stroke prevention measures into standard stroke care provided during hospitalization |
| Applies quality improvement measures through preprinted admission orders, care maps, discharge protocols, educational materials, patient self‐assessment logs, and data monitoring tools | |
| JCAHO Disease Specific Certification for acute stroke care ( |
Designates eligible hospitals as primary stroke centers |
| Promotes compliance with consensus‐based national standards | |
| Encourages effective use of established clinical practice guidelines to manage and optimize stroke care | |
| Fosters an organized approach to performance measurement and improvement activities | |
CONCLUSIONS
The acute stroke hospitalization setting provides the ideal opportunity for hospitalists to not only institute evidence‐based prevention therapies for recurrent stroke but also to have the undivided attention of patients and their families. Furthermore, it may be risky to assume that relevant therapy when deferred will be initiated in a timely fashion, if at all, after hospital discharge. As part of an effective continuum of care, hospitalists have an important role not just in the management of acute ischemic stroke, but also in long‐term reduction of vascular risk.
- .Stroke prevention: windows of opportunity and failed expectations? A discussion of modifiable cardiovascular risk factors and a prevention proposal.Neuroepidemiology.1997;16(4):163–173.
- ,,, et al.Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co‐sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline.Stroke.2006;37:577–617.
- ,,.Stroke prevention: narrowing the evidence‐practice gap.Neurology.2000;54:1899–1906.
- ,,, et al.Acute stroke care in the US: results from 4 pilot prototypes of the Paul Coverdell National Acute Stroke Registry.Stroke.2005;36:1232–1240.
- ,,.,.Lipid Assessment and treatment patterns in hospitalized TIA and ischemic stroke patients.J Hosp Med.2006;1:214–220.
- ,,, et al.Heart disease and stroke statistics—2006 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee.Circulation.2006;113(6):e85–e151.
- .Long‐term outcome after ischaemic stroke/transient ischaemic attack.Cerebrovasc Dis.2003;16(Suppl 1):14–19.
- ,,,,.Ten‐year survival after first‐ever stroke in the perth community stroke study.Stroke.2003;34:1842–1846.
- NINDS rt‐PA Stroke Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,,,,.Assessing patterns of t‐PA use in acute stroke.Stroke.2002;33:354.
- Growth of hospital medicine nationwide. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/Media/GrowthofHospitalMedicineNationwide/Growth_of_ Hospital_M.htm. Accessed April 12,2006.
- American Academy of Neurology. Available at: http://www.aan.com/students/medical/faq.cfm. Accessed June 25,2006.
- .The neurologist's role in stroke management.Stroke.1996;27:1935–1936.
- .Prevention and management of medical complications of the hospitalized elderly stroke patient.Clin Geriatr Med.1991;7:475–482.
- ,,, et al.The Stroke Prevention Policy Model: linking evidence and clinical decisions.Ann Intern Med.1997;127:704–711.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,, et al.Incidence and short‐term prognosis of transient ischemic attack in a population‐based study.Stroke.2005;36:720–723.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,,.Short‐term prognosis after emergency department diagnosis of TIA.JAMA.2000;284:2901–2906.
- ,,,,.Ten‐year risk of first recurrent stroke and disability after first‐ever stroke in the Perth Community Stroke Study.Stroke.2004;35:731–735.
- .New strategies for prevention of ischemic stroke: the LIFE study.Curr Neurol Neurosci Rep.2003;3(1):46–51.
- ,,, et al.[Manifestations of atherosclerosis in various vascular regions. Similarities and differences regarding epidemiology, etiology and prognosis].Med Klin.2002;97(4):221–228.
- ,,,,,.Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences.Ann Intern Med.2001;134(3):224–238.
- .Prevention of strokes and recurrent strokes.J Neurol Neurosurg Psychiatry.1998;64:716.
- ,.Prevalence of coexistence of coronary artery disease, ischemic stroke, and peripheral arterial disease in older persons, mean age 80 years, in an academic hospital‐based geriatrics practice.J Am Geriatr Soc.1999;47:1255–1256.
- ,,.Risk factors and their management for stroke prevention: outlook for 1999 and beyond.Neurology.1999;53(7 Suppl 4):S15–S24.
- ,,,,.Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest. Sep2004;126(3 Suppl):483S–512S.
- Randomised trial of a perindopril‐based blood‐pressure‐lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,.Blood pressure reduction and secondary prevention of stroke and other vascular events: a systematic review.Stroke.2003;34:2741–2748.
- Post‐stroke antihypertensive treatment study. A preliminary result.PATS Collaborating Group.Chin Med J (Engl).1995;108:710–717.
- ,,.Should beta blockers remain first choice in the treatment of primary hypertension? A meta‐analysis.Lancet.2005;366:1545–1553.
- ,,, et al.Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines.Circulation.2004;110(2):227–239.
- Heart Protection Study Collaborative Group.Effects of cholesterol‐lowering with Simvastatin on stroke and other major vascular events in 20, 536 people with cerebrovascular disease or other high‐risk conditions.Lancet.2004;363:757–767.
- , et al.The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study. Presented at the 15th European Stroke Conference, Brussels, Belgium, May 16‐19,2006.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38: UK Prospective Diabetes Study Group.BMJ.1998;317:703–713.
- ,,,,, for theHeart Protection Study Collaborative Group.MRC/BHF Heart Protection Study of cholesterol‐lowering with simvastatin in 5963 people with diabetes: a randomised placebo‐controlled trial.Lancet.2003;361:2005–2016.
- ,,,.The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy: the Collaborative Study Group.N Engl J Med.1993;329:1456–1462.
- ,,, for theCollaborative Study Group.Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy.N Engl J Med.2001;345:861–869.
- ,,., et al.Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non‐insulin‐dependent diabetes mellitus: a randomized prospective 6‐year study.Diabetes Res Clin Pract.1995;28:103–117.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high‐grade carotid stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1991;325:445–453.
- ,,, et al.Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1998;339:1415–1425.
- Endarterectomy for asymptomatic carotid artery stenosis.Executive Committee for the Asymptomatic Carotid Atherosclerosis Study.JAMA.1995;273:1421–1428.
- ,,, et al.Protected carotid‐artery stenting versus endarterectomy in high‐risk patients.N Engl J Med.2004;351:1493–1501.
- ,,.New evidence for stroke prevention: scientific review.JAMA.2002;288:1388–1395.
- ,,,,.Endarterectomy for symptomatic carotid stenosis in relation to clinical subgroups and timing of surgery.Lancet.2004;363:915–924.
- .Carotid artery surgery vs. stent: a cardiovascular perspective.Catheter Cardiovasc Interv.2004;63:377–384.
- ,,, et al.Warfarin anticoagulation and outcomes in patients with atrial fibrillation: a systematic review and metaanalysis.Chest.2004;126:1938–1945.
- Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients.BMJ.2002;324:71–86.
- ,,,,,.European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke.J Neurol Sci.1996;143(1‐2):1–13.
- A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE).CAPRIE Steering Committee.Lancet.1996;348:1329–1339.
- ,,,,.Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial.Lancet.2006;367:1665–1673.
- ,,, et al.Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high‐risk patients (MATCH): randomised, double‐blind, placebo‐controlled trial.Lancet.2004;364:331–337.
- ,,, et al.Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events.N Engl J Med. Apr 202006;354(16):1706–1717.
- ,,, et al.PROTECT: A Coordinated Stroke Treatment Program to Prevent Recurrent Thromboembolic Events.Neurology. Vol63;2004:1217–1222.
- ,,, et al.In‐hospital initiation of secondary stroke prevention therapies yields high rates of adherence at follow up.Stroke.2004;35:2879–2883.
- ,,, et al.In‐hospital initiation of lipid‐lowering therapy after coronary intervention as a predictor of long‐term utilization: a propensity analysis.Arch Intern Med.2003;163:2576–2582.
- ,,, et al.Early effects of statins in patients with coronary artery disease and high C‐reactive protein.Am J Cardiol.2004;94:1107–1112.
- ,,,.Improved treatment of coronary heart disease by implementation of a Cardiac Hospitalization Atherosclerosis Management Program (CHAMP).Am J Cardiol. Apr 12001;87(7):819–822.
- ,,.Importance of in‐hospital initiation of evidence‐based medical therapies for heart failure‐a review.Am J Cardiol.2004;94:1155–1160.
- ,,,.Blood pressure and stroke: an overview of published reviews.Stroke.2004;35:1024.
- ,,, et al.Intensive versus moderate lipid lowering with statins after acute coronary syndromes.N Engl J Med.2004;350:1495–1504.
- ,,.Niacin therapy in atherosclerosis.Curr Opin Lipidol.2004;15:659–665.
- ,,, et al.Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: The Veterans Affairs HDL Intervention Trial (VA‐HIT).Circulation.2001;103:2828–2833.
- ,,.The effect of long‐term intensified insulin treatment on the development of microvascular complications of diabetes mellitus.N Engl J Med.1993;329:304–309.
Prevention has the greatest potential to reduce the societal burden from stroke.1 Several therapies that specifically target the underlying atherosclerotic disease process have been shown in clinical trials to markedly lower the risk of recurrent vascular events including stroke.2 However, there is great variability in how clinical trial data are implemented in clinical practice for ischemic stroke prevention.35 This has led to a knowledge‐implementation‐practice gap, possibly because of the limited awareness of the scientific evidence supporting various treatments, as well as the lack of a systematic approach to hospital stroke care.3 Our review discusses the evidence for reducing vascular risk after ischemic stroke and successful models of systematic interventions initiated during stroke hospitalization, with the goal of narrowing the stroke hospitalization evidencepractice gap.
Societal Burden
Stroke is the third‐leading cause of death in the United States and the leading cause of serious long‐term disability.6 Approximately 700,000 Americans have a new stroke or recurrent strokes every year, whereas nearly 5 million live with the consequences of stroke; nearly all stroke survivors (90%) have some residual functional deficit, and approximately 40% experience moderate to severe impairment.6 Stroke mortality is substantial, with a 30‐day case fatality rate after first stroke (of any cause) of about 25%.7, 8 Indeed, four‐fifths of patients do not survive for 10 years after stroke, and approximately one‐third of all case fatalities occur in the first year after a stroke.8 The estimated economic impact in 2006, US$57.9 billion, further underscores the substantial mortality and morbidity of stroke.6 Given the limited options for acute stroke therapies,9 stroke prevention remains an important therapeutic goal, especially because fewer than 5% of acute stroke patients in the United States currently receive the only Food and Drug Administrationapproved treatmentintravenous tissue plasminogen activator.10 It is obvious that additional strategies are urgently needed to reduce the devastating consequences of stroke.
Why Involve the Hospitalist?
The Hospitalist system in the United States is rapidly growing.11 Tthe Society of Hospital Medicine projects that by 2010 there will be approximately 30,000 hospitalists in the United States.11 A member census conducted by the American Academy of Neurology in 2000 found 13,500 practicing neurologists, most of whom are concentrated in urban and metropolitan areas.12 As such, with more than 700,000 strokes occurring each year,6 most stroke patients in the United States will not be seen or evaluated by a neurologist. Indeed, one study indicated that only 11.3% of stroke patients are attended exclusively by a neurologist.13 Furthermore, it is not uncommon for stroke patients to have numerous other medical issues that require attention and multidisciplinary care coordination during the hospital stay, an area where hospitalists excel. Conceivably, the ability to promptly identify and treat these non‐neurological comorbidities, which account for at least 30% of the deaths from acute ischemic stroke,14 could go a long way toward improving stroke outcomes.
Hospitalists are in the forefront of developing strategies for improving the quality of acute care and patient satisfaction, reducing medical errors, and focusing on efficient resource utilization. Translating evidence‐based strategies for acute stroke care into actual practice is a mechanism for improving the quality of care, ensuring that basic care does not deviate from provider to provider or from day to day (weekdays compared to weekend days/holidays) while at the same time allowing for the individualization of care appropriate to a patient's unique needs.15 After the acute treatment of stroke or TIA, additional measures must be initiated as soon as it is safe to do so in order to begin the process of limiting stroke progression and preventing recurrence. Secondary prevention measures require a coordinated transition in order to ensure continuation of care and follow‐up as needed. After a thorough risk assessment is complete, hospitalists will need to consider a 3‐pronged approach to secondary prevention that follows the national guidelines described above: pharmacotherapy, behavior modification, and, in some cases, surgical intervention.
Secondary Stroke
Secondary or recurrent strokes are strokes that occur after a first stroke or TIA,2 and the single biggest risk factor for having a stroke is already having had one.2 Because hospitalists generally see patients after ischemic cerebrovascular events have already happened, their opportunities to intervene are mostly geared toward reducing the risk of secondary stroke (beyond enhancing the prevention of complications from the index event). Recent community‐based data indicate that the short‐term risk of secondary stroke is high.16, 17 After a minor stroke or TIA, the risk of recurrent stroke or TIA increases over time8%‐12% within 7 days, 12%‐15% within 30 days, and 17%‐19% within 90 days.18 In the largest study of short‐term risk following TIA,19 there was an 11% risk of stroke (51% of which occurred in the 48 hours after TIA), an 13% risk of TIA, and a 25% risk of any adverse event within 90 days of the TIA.
Overall, the risk of a second cerebrovascular event is highest in the first year after a stroke/TIA (12%), declining to about 5% annually thereafter.7 The effects of secondary stroke are more devastating than those of the primary stroke: the 30‐day fatality rate after a first recurrent stroke is almost double that after the first‐ever stroke (41% versus 22%).20 The pathological factors that lead to TIA and stroke, such as platelet aggregation and subsequent thrombosis or the systolic stroke of blood against stenotic carotid plaques, are one and the same. As such, the short‐ and long‐term risks of recurrent events after both first stroke or first TIA necessitate investigation into a patient's vascular risk and early initiation of appropriate stroke prevention strategies.21
Cross Risk
Because the atherothrombotic disease process is systemic in nature with a variety of manifestations, stroke patients with atherosclerosis frequently have coexistent coronary artery disease and peripheral artery disease,22 and as such, are at risk for vascular events emanating from any of these beds in addition to that of the cervicocephalic arterial tree.23, 24 For instance, in a study of individuals in a long‐term care facility, among the patients with ischemic stroke, 56% had overlapping coronary artery disease, 28% had peripheral artery disease,25 and 38% of the patients had at least 2 manifestations of their atherosclerotic disease. The take‐home message here is that hospitalists also have the opportunity while treating patients hospitalized following stroke to prevent other vascular events by identifying and treating stroke patients who have systemic atherosclerosis.
Risk Factors
The first step in any approach to stroke prevention is the identification of predisposing risk factors. Several of the known biological and lifestyle risk factors associated with cerebrovascular disease were identified decades ago from large longitudinal studies.2 Certain stroke risk factors are nonmodifiable and therefore cannot be the target of intervention. 26 Treatment of the various stroke risk factors could have a substantial impact on reducing the burden of stroke. Table 1 shows the number needed to treat to prevent one stroke per year by modification of the individual stroke risk factor.
| Treatment | Relative risk reduction | Number needed to treat (1 stroke/year) |
|---|---|---|
| ||
| Antihypertensives | 28% | 51 |
| Statins | 25% | 57 |
| Aspirin | 28% | 77 |
| Smoking cessation | 33% | 43 |
| Carotid endarterectomy | 44% | 26 |
Guidelines for Secondary Stroke Prevention
Several organizations have published guidelines for the prevention of secondary stroke based on clinical evidence and expert consensus. Key guidelines include those published by the American Stroke Association (ASA),2 American College of Chest Physicians (ACCP),27 and the National Stroke Association. Although these guidelines are broadaddressing many components of stroke prevention and careeach contains recommendations specifically applicable to secondary prevention in most stroke patients who the hospitalist will encounter. Some provide hospital‐based guidelines that focus on care protocols and systems processes (ie, ASA Stroke Systems Guidelines), whereas others are therapy‐based guidelines (i.e, ACCP Guidelines on Antithrombotic Therapy for Ischemic Stroke). In the next few sections, we discuss common risk factors for and causes of secondary stroke and the prevailing guideline recommendations for modifying them. Discussion of the management of rare causes of ischemic stroke such as arterial dissection, vasculitis, patent foramen ovale, and so forth is beyond the scope of this article.
Hypertension, Dyslipidemia, and Diabetes
Table 2 shows the current national guideline recommendations for the management of premier vascular risk factorshypertension, dyslipidemia, and diabetesin ischemic stroke and TIA patients.2 Antihypertensive therapy is recommended for the prevention of secondary stroke and other vascular events in patients who have experienced an ischemic stroke or TIA and are beyond the hyperacute period.28, 29 Such treatment should be considered for all ischemic stroke and TIA patients regardless of history of hypertension.28 Although available data support the use of diuretics and the combination of diuretics plus an angiotensin‐converting enzyme inhibitor,28, 30 selection of specific medications should be individualized according to a patient's comorbid conditions.29 It is also important to note that despite the proven benefit of beta blockers in the secondary prevention of recurrent cardiac events, current evidence shows no clear benefit from the use of beta blockers in the prevention of stroke.29, 31
| Risk Factor | Recommendation |
|---|---|
| |
| Hypertension | Antihypertensive beyond hyperacute stroke period60 |
| Data support diuretic or diuretic + ACEI,2830 but individualize based on patient characteristics | |
| Antihypertensive in all patients regardless of history of hypertension28 | |
| Aim for average reduction of 10/5 mm HG or blood pressure < 120/80 mm Hg28 | |
| Encourage reduced intake of dietary salt | |
| Dyslipidemia | Statin for LDL‐C goal < 100 mg/dL in those with CAD or symptomatic atherosclerosis33, 34 |
| Target LDL‐C < 70 mg/dL for very high‐risk persons61 | |
| Statin for stroke or TIA because of atherosclerosis regardless of LDL‐C level33, 34 | |
| Niacin or gemfibrozil for patients with low HDL‐C62, 63 | |
| Diabetes | ACEIs and ARBs should be first‐choice blood pressure drugs37, 38a |
| Glucose control to near normoglycemic levels39 | |
| Target glycosylated hemoglobin 7%64 | |
For ischemic cerebrovascular disease patients with dyslipidemia or symptomatic atherosclerosis, cholesterol management should be according to the current Adult Treatment Panel (ATP) guidelines.32 Statins should be the first‐line treatment.33, 34 Ischemic stroke or TIA patients whose underlying stroke mechanism is presumed to be atherosclerosis should be considered for statin therapy even if they have normal cholesterol levels and no evidence of atherosclerosis.33, 34 The recent Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study was the first study to specifically investigate the effect of statins in patients with a prior stroke but with normal cholesterol levels and no evidence of coronary heart disease. It found that treatment with atorvastatin 80 mg/day (vs. placebo) was associated with a 16% reduction in relative risk of recurrent stroke.34
The care of an ischemic stroke or TIA patient who has diabetes warrants more rigorous control of blood pressure and lipids.35, 36 Such patients usually require more than one antihypertensive drug. ACEIs and angiotensin receptor blockers (ARBs) are more effective in reducing the progression of renal disease and are the recommended first‐choice medications for these patients.37, 38 The target for glucose control should be reaching near‐normoglycemic levels.39
Large‐Artery Atherosclerosis
In selected at‐risk stroke patients, surgical techniques (eg, carotid endarterectomy [CEA], carotid angioplasty and/or stenting [CAS]) may reduce the rate of recurrent stroke.4044 For patients who have had ischemic cerebrovascular events in the preceding 6 months and who have ipsilateral severe (70%‐99%) cervical carotid artery stenosis, CEA done by a surgeon is recommended; it has a perioperative morbidity and mortality of less than 6%.40 For those with ipsilateral moderate (50%‐69%) cervical carotid stenosis, CEA should be considered, and whether to operate should be decided on the basis of the patient's age, sex, comorbidities, and severity of initial symptoms.41 Analyses of endarterectomy trials indicated that the benefit from CEA is greatest if performed within 2 weeks of a patient's last ischemic event, the advantage it confers rapidly falling with increasing delay.45 From the hospitalist's standpoint, it is of prime importance to ensure that patients admitted to the hospital with a TIA or ischemic stroke are not discharged before it has been established whether have severe carotid stenosis that requires a revascularization procedure. If carotid stenosis is less than 50%, CEA is not recommended.41
A newer, less invasive form of carotid artery revascularization is CAS,46 which is performed by operators with established periprocedural morbidity and mortality rates of 4%‐6% and may be considered in those with:
-
Symptomatic severe stenosis (>70%) that is difficult to access surgically.2
-
Medical issues that greatly increase the risks of surgery, such as clinically significant cardiac disease, severe pulmonary disease, contralateral carotid occlusion, contralateral laryngeal nerve palsy, radiation‐induced stenosis or restenosis after carotid endarterectomy, and more than 80 years old.43
Angioplasty and/or stenting may also be considered when patients with symptomatic extracranial vertebral stenosis are having symptoms despite optimal medical risk factor treatments.2 Among those with hemodynamically significant stenosis of the major intracranial vasculature (basilar, middle cerebrals, distal carotids, and vertebrals) experiencing symptoms despite optimal medical risk factor treatments, angioplasty and/or stenting is considered experimental.2
The degree of arterial stenosis can be assessed by ultrasound, magnetic resonance angiogram (MRA), computed tomography angiogram (CTA), and conventional catheter angiogram, the last of which remains the gold standard. A carotid ultrasound performed at a certified vascular laboratory or by an experienced radiology technologist that shows less than 50% stenosis need not be followed up with another neuroimaging test. Generally, MRA tends to overestimate the degree of arterial stenosis but is a useful screening tool. In the event that an MRA reveals more than 50% stenosis, another diagnostic modality such as a carotid duplex, CTA, or conventional catheter angiogram should be performed to confirm this finding.
Antithrombotic Treatment
Cardioembolic Stroke Mechanism
Although it can sometimes be difficult to determine the precise mechanism underlying a patient's stroke or TIA, those who have a high‐risk source of cardiogenic embolism should generally be treated with anticoagulant medications to prevent recurrence.2 Among ischemic cerebrovascular event patients with persistent or paroxysmal atrial fibrillation, anticoagulation with adjusted‐dose warfarin (target international normalized ratio [INR] of 2.5; range, 2.0‐3.0) should be administered.47 The ASA recommends initiating oral anticoagulation within 2 weeks of an ischemic stroke or TIA but indicates that further delays may be appropriate for patients with large infarcts or uncontrolled hypertension.2 For patients unable to take oral anticoagulants, aspirin 325 mg/day should be given instead. Among patients who suffered an ischemic stroke or TIA because of an acute myocardial infarction in whom left ventricular mural thrombus is identified by echocardiography or another form of cardiac imaging, oral anticoagulation should be considered, aiming for an INR of 2.0‐3.0 for at least 3 months and up to 1 year.2 Patients receiving oral anticoagulation who also have ischemic coronary artery disease should be prescribed aspirin as well, in doses up to 162 mg/day.2
Noncardioembolic Stroke Mechanism
For ischemic stroke or TIA patients who have no high‐risk source of cardiogenic embolism, antiplatelet agents rather than oral anticoagulation are generally recommended to reduce the risk of recurrent stroke and other cardiovascular events.4850 Acceptable options for initial therapy include:
-
Aspirin (50 to 325 mg/day)48;
-
Combination of aspirin (50 mg) and extended‐release dipyridamole (400 mg) daily49, 51;
-
Clopidogrel (75 mg) daily.50
The combination of aspirin and extended‐release dipyridamole is suggested instead of aspirin alone, and clopidogrel may be considered instead of aspirin alone.49, 51 However, currently there is not enough data to make evidence‐based recommendations for choosing between antiplatelet drugs beyond aspirin.2 Furthermore, there is no evidence that increasing the dose of aspirin for patients who have had an ischemic stroke while taking aspirin provides additional benefit.2 The selection of an antiplatelet agent must be individualized, giving due consideration to a patient's presumed stroke mechanism, risk factor profile, and tolerance.
Other antiplatelet guidelines for noncardioembolic stroke/TIA patients include that:
Education for Behavior Modification
It is crucial to discharge patients with the tools they need to make important lifestyle changes. Patients can significantly reduce their stroke risk by making changes in their everyday patterns of behavior. As much education as possible about smoking cessation, exercise, diet, and the warning signs of stroke should be provided often as possible during hospitalization for a stroke and need not be left to nurses. Stroke education is extremely important so patients understand the need to call for emergency medical services immediately if they even suspect they are having stroke symptoms because of the very narrow window of opportunity for treatment of an acute stroke.54 All patients should be encouraged to make lifestyle adjustments such as ceasing smoking, reducing alcohol intake, and controlling weight. Smoking cessation appears to be effective in preventing secondary stroke (33% reduction in relative risk),44 and initiating smoking cessation counseling during hospitalization for stroke may result in a high rate of adherence to smoking cessation, at least in the short term.55 Table 3 displays current national guideline recommendations on lifestyle modification approaches.2
| Risk Factor | Recommendation |
|---|---|
| |
| Smoking | Smoking cessation |
| Avoid environmental smoke | |
| Counseling, nicotine products, and oral smoking cessation medications | |
| Alcohol | Eliminate or reduce alcohol consumption |
| Light to moderate levels2 drinks/day for men, 1 drink/day for nonpregnant women may be considered | |
| Obesity | Weight reduction goal: BMI 18.5‐24.9 kg/m2 and waist circumference < 35 inches for women, < 40 inches for men |
| Encourage weight management through balance of caloric intake, physical activity, behavioral counseling | |
| Physical Activity | At least 30 minutes of moderate‐intensity physical exercise most days of the week |
| Supervised therapeutic exercise regimen for those with residual disability | |
EvidencePractice Gap
There are now many secondary stroke prevention modalities, and there is a copious amount of data validating the efficacy of quite a few of them.2 Yet there is a large gap in implementing evidence‐based secondary prevention strategies.35 TIA and ischemic stroke patients are often discharged from the hospital without being prescribed any preventive medications, despite the data supporting the use of antiplatelet agents, anticoagulants, and antihypertensives for prevention of secondary stroke.4 In addition, several behavioral interventions could help patients to avoid stroke recurrence,2 but quite often stroke patients are not educated about them during the acute care period.4 Poor discharge treatment utilization limits the effectiveness of proven therapies, resulting in lost opportunities to reduce the burden of secondary stroke.
The reasons for these care gaps are multifactorial and can be traced to patient and provider issues as well as to health care delivery processes. Our understanding of the reasons for this gap is improving. Generally speaking, preventive services are used less frequently than those services or treatment modalities that provide immediate relief or economic benefit. The benefit of most preventive services is more readily seen at a population level than at a individual level and accrues slowly over time. It becomes more difficult to stress prevention in a health care system driven by technology‐based acute care.3
Current clinical management of acute stroke patients has stroke specialists and hospital physicians focusing on the acute management and diagnostic workup during hospitalization. Initiation of long‐term treatment is often deferred to after discharge, when the patient resumes long‐term primary care follow‐up.54 This deferred approach may result in therapy not being initiated or being initiated less efficiently and at a time (weeks or months after the initial presentation) when the stroke event and underlying atherosclerotic disease may no longer be the focus of either the patient or the primary care physician.54
Initiating medications during the acute stroke hospitalization phase sends the patient the message that these therapies are important for preventing recurrence and are an essential part of their treatment.54 More important, hospital initiation of secondary prevention therapies has been shown to be a strong predictor of these therapies continuing to be used after discharge56 and is associated with better clinical outcomes.5759 Table 4 shows some of the resources available to assist hospitalists in overcoming the evidencepractice gap in stroke treatment.
| Tool | Description |
|---|---|
| |
| AHA Get with the GuidelinesStroke ( |
Focuses on care team protocols to facilitate appropriate in‐hospital and discharge stroke treatment utilization |
| Identifies champions to lead, develop, and mobilize teams to optimally implement evidence‐based stroke treatment in acute care hospitals | |
| Utilizes standardized admission orders, patient educational materials, data monitoring | |
| Provides resources to help hospitals obtain JCAHO certification | |
| UCLA Stroke PROTECT (Preventing Recurrence of Thromboembolic Events through Coordinated Treatment) program ( |
Integrates 8 proven secondary stroke prevention measures into standard stroke care provided during hospitalization |
| Applies quality improvement measures through preprinted admission orders, care maps, discharge protocols, educational materials, patient self‐assessment logs, and data monitoring tools | |
| JCAHO Disease Specific Certification for acute stroke care ( |
Designates eligible hospitals as primary stroke centers |
| Promotes compliance with consensus‐based national standards | |
| Encourages effective use of established clinical practice guidelines to manage and optimize stroke care | |
| Fosters an organized approach to performance measurement and improvement activities | |
CONCLUSIONS
The acute stroke hospitalization setting provides the ideal opportunity for hospitalists to not only institute evidence‐based prevention therapies for recurrent stroke but also to have the undivided attention of patients and their families. Furthermore, it may be risky to assume that relevant therapy when deferred will be initiated in a timely fashion, if at all, after hospital discharge. As part of an effective continuum of care, hospitalists have an important role not just in the management of acute ischemic stroke, but also in long‐term reduction of vascular risk.
Prevention has the greatest potential to reduce the societal burden from stroke.1 Several therapies that specifically target the underlying atherosclerotic disease process have been shown in clinical trials to markedly lower the risk of recurrent vascular events including stroke.2 However, there is great variability in how clinical trial data are implemented in clinical practice for ischemic stroke prevention.35 This has led to a knowledge‐implementation‐practice gap, possibly because of the limited awareness of the scientific evidence supporting various treatments, as well as the lack of a systematic approach to hospital stroke care.3 Our review discusses the evidence for reducing vascular risk after ischemic stroke and successful models of systematic interventions initiated during stroke hospitalization, with the goal of narrowing the stroke hospitalization evidencepractice gap.
Societal Burden
Stroke is the third‐leading cause of death in the United States and the leading cause of serious long‐term disability.6 Approximately 700,000 Americans have a new stroke or recurrent strokes every year, whereas nearly 5 million live with the consequences of stroke; nearly all stroke survivors (90%) have some residual functional deficit, and approximately 40% experience moderate to severe impairment.6 Stroke mortality is substantial, with a 30‐day case fatality rate after first stroke (of any cause) of about 25%.7, 8 Indeed, four‐fifths of patients do not survive for 10 years after stroke, and approximately one‐third of all case fatalities occur in the first year after a stroke.8 The estimated economic impact in 2006, US$57.9 billion, further underscores the substantial mortality and morbidity of stroke.6 Given the limited options for acute stroke therapies,9 stroke prevention remains an important therapeutic goal, especially because fewer than 5% of acute stroke patients in the United States currently receive the only Food and Drug Administrationapproved treatmentintravenous tissue plasminogen activator.10 It is obvious that additional strategies are urgently needed to reduce the devastating consequences of stroke.
Why Involve the Hospitalist?
The Hospitalist system in the United States is rapidly growing.11 Tthe Society of Hospital Medicine projects that by 2010 there will be approximately 30,000 hospitalists in the United States.11 A member census conducted by the American Academy of Neurology in 2000 found 13,500 practicing neurologists, most of whom are concentrated in urban and metropolitan areas.12 As such, with more than 700,000 strokes occurring each year,6 most stroke patients in the United States will not be seen or evaluated by a neurologist. Indeed, one study indicated that only 11.3% of stroke patients are attended exclusively by a neurologist.13 Furthermore, it is not uncommon for stroke patients to have numerous other medical issues that require attention and multidisciplinary care coordination during the hospital stay, an area where hospitalists excel. Conceivably, the ability to promptly identify and treat these non‐neurological comorbidities, which account for at least 30% of the deaths from acute ischemic stroke,14 could go a long way toward improving stroke outcomes.
Hospitalists are in the forefront of developing strategies for improving the quality of acute care and patient satisfaction, reducing medical errors, and focusing on efficient resource utilization. Translating evidence‐based strategies for acute stroke care into actual practice is a mechanism for improving the quality of care, ensuring that basic care does not deviate from provider to provider or from day to day (weekdays compared to weekend days/holidays) while at the same time allowing for the individualization of care appropriate to a patient's unique needs.15 After the acute treatment of stroke or TIA, additional measures must be initiated as soon as it is safe to do so in order to begin the process of limiting stroke progression and preventing recurrence. Secondary prevention measures require a coordinated transition in order to ensure continuation of care and follow‐up as needed. After a thorough risk assessment is complete, hospitalists will need to consider a 3‐pronged approach to secondary prevention that follows the national guidelines described above: pharmacotherapy, behavior modification, and, in some cases, surgical intervention.
Secondary Stroke
Secondary or recurrent strokes are strokes that occur after a first stroke or TIA,2 and the single biggest risk factor for having a stroke is already having had one.2 Because hospitalists generally see patients after ischemic cerebrovascular events have already happened, their opportunities to intervene are mostly geared toward reducing the risk of secondary stroke (beyond enhancing the prevention of complications from the index event). Recent community‐based data indicate that the short‐term risk of secondary stroke is high.16, 17 After a minor stroke or TIA, the risk of recurrent stroke or TIA increases over time8%‐12% within 7 days, 12%‐15% within 30 days, and 17%‐19% within 90 days.18 In the largest study of short‐term risk following TIA,19 there was an 11% risk of stroke (51% of which occurred in the 48 hours after TIA), an 13% risk of TIA, and a 25% risk of any adverse event within 90 days of the TIA.
Overall, the risk of a second cerebrovascular event is highest in the first year after a stroke/TIA (12%), declining to about 5% annually thereafter.7 The effects of secondary stroke are more devastating than those of the primary stroke: the 30‐day fatality rate after a first recurrent stroke is almost double that after the first‐ever stroke (41% versus 22%).20 The pathological factors that lead to TIA and stroke, such as platelet aggregation and subsequent thrombosis or the systolic stroke of blood against stenotic carotid plaques, are one and the same. As such, the short‐ and long‐term risks of recurrent events after both first stroke or first TIA necessitate investigation into a patient's vascular risk and early initiation of appropriate stroke prevention strategies.21
Cross Risk
Because the atherothrombotic disease process is systemic in nature with a variety of manifestations, stroke patients with atherosclerosis frequently have coexistent coronary artery disease and peripheral artery disease,22 and as such, are at risk for vascular events emanating from any of these beds in addition to that of the cervicocephalic arterial tree.23, 24 For instance, in a study of individuals in a long‐term care facility, among the patients with ischemic stroke, 56% had overlapping coronary artery disease, 28% had peripheral artery disease,25 and 38% of the patients had at least 2 manifestations of their atherosclerotic disease. The take‐home message here is that hospitalists also have the opportunity while treating patients hospitalized following stroke to prevent other vascular events by identifying and treating stroke patients who have systemic atherosclerosis.
Risk Factors
The first step in any approach to stroke prevention is the identification of predisposing risk factors. Several of the known biological and lifestyle risk factors associated with cerebrovascular disease were identified decades ago from large longitudinal studies.2 Certain stroke risk factors are nonmodifiable and therefore cannot be the target of intervention. 26 Treatment of the various stroke risk factors could have a substantial impact on reducing the burden of stroke. Table 1 shows the number needed to treat to prevent one stroke per year by modification of the individual stroke risk factor.
| Treatment | Relative risk reduction | Number needed to treat (1 stroke/year) |
|---|---|---|
| ||
| Antihypertensives | 28% | 51 |
| Statins | 25% | 57 |
| Aspirin | 28% | 77 |
| Smoking cessation | 33% | 43 |
| Carotid endarterectomy | 44% | 26 |
Guidelines for Secondary Stroke Prevention
Several organizations have published guidelines for the prevention of secondary stroke based on clinical evidence and expert consensus. Key guidelines include those published by the American Stroke Association (ASA),2 American College of Chest Physicians (ACCP),27 and the National Stroke Association. Although these guidelines are broadaddressing many components of stroke prevention and careeach contains recommendations specifically applicable to secondary prevention in most stroke patients who the hospitalist will encounter. Some provide hospital‐based guidelines that focus on care protocols and systems processes (ie, ASA Stroke Systems Guidelines), whereas others are therapy‐based guidelines (i.e, ACCP Guidelines on Antithrombotic Therapy for Ischemic Stroke). In the next few sections, we discuss common risk factors for and causes of secondary stroke and the prevailing guideline recommendations for modifying them. Discussion of the management of rare causes of ischemic stroke such as arterial dissection, vasculitis, patent foramen ovale, and so forth is beyond the scope of this article.
Hypertension, Dyslipidemia, and Diabetes
Table 2 shows the current national guideline recommendations for the management of premier vascular risk factorshypertension, dyslipidemia, and diabetesin ischemic stroke and TIA patients.2 Antihypertensive therapy is recommended for the prevention of secondary stroke and other vascular events in patients who have experienced an ischemic stroke or TIA and are beyond the hyperacute period.28, 29 Such treatment should be considered for all ischemic stroke and TIA patients regardless of history of hypertension.28 Although available data support the use of diuretics and the combination of diuretics plus an angiotensin‐converting enzyme inhibitor,28, 30 selection of specific medications should be individualized according to a patient's comorbid conditions.29 It is also important to note that despite the proven benefit of beta blockers in the secondary prevention of recurrent cardiac events, current evidence shows no clear benefit from the use of beta blockers in the prevention of stroke.29, 31
| Risk Factor | Recommendation |
|---|---|
| |
| Hypertension | Antihypertensive beyond hyperacute stroke period60 |
| Data support diuretic or diuretic + ACEI,2830 but individualize based on patient characteristics | |
| Antihypertensive in all patients regardless of history of hypertension28 | |
| Aim for average reduction of 10/5 mm HG or blood pressure < 120/80 mm Hg28 | |
| Encourage reduced intake of dietary salt | |
| Dyslipidemia | Statin for LDL‐C goal < 100 mg/dL in those with CAD or symptomatic atherosclerosis33, 34 |
| Target LDL‐C < 70 mg/dL for very high‐risk persons61 | |
| Statin for stroke or TIA because of atherosclerosis regardless of LDL‐C level33, 34 | |
| Niacin or gemfibrozil for patients with low HDL‐C62, 63 | |
| Diabetes | ACEIs and ARBs should be first‐choice blood pressure drugs37, 38a |
| Glucose control to near normoglycemic levels39 | |
| Target glycosylated hemoglobin 7%64 | |
For ischemic cerebrovascular disease patients with dyslipidemia or symptomatic atherosclerosis, cholesterol management should be according to the current Adult Treatment Panel (ATP) guidelines.32 Statins should be the first‐line treatment.33, 34 Ischemic stroke or TIA patients whose underlying stroke mechanism is presumed to be atherosclerosis should be considered for statin therapy even if they have normal cholesterol levels and no evidence of atherosclerosis.33, 34 The recent Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study was the first study to specifically investigate the effect of statins in patients with a prior stroke but with normal cholesterol levels and no evidence of coronary heart disease. It found that treatment with atorvastatin 80 mg/day (vs. placebo) was associated with a 16% reduction in relative risk of recurrent stroke.34
The care of an ischemic stroke or TIA patient who has diabetes warrants more rigorous control of blood pressure and lipids.35, 36 Such patients usually require more than one antihypertensive drug. ACEIs and angiotensin receptor blockers (ARBs) are more effective in reducing the progression of renal disease and are the recommended first‐choice medications for these patients.37, 38 The target for glucose control should be reaching near‐normoglycemic levels.39
Large‐Artery Atherosclerosis
In selected at‐risk stroke patients, surgical techniques (eg, carotid endarterectomy [CEA], carotid angioplasty and/or stenting [CAS]) may reduce the rate of recurrent stroke.4044 For patients who have had ischemic cerebrovascular events in the preceding 6 months and who have ipsilateral severe (70%‐99%) cervical carotid artery stenosis, CEA done by a surgeon is recommended; it has a perioperative morbidity and mortality of less than 6%.40 For those with ipsilateral moderate (50%‐69%) cervical carotid stenosis, CEA should be considered, and whether to operate should be decided on the basis of the patient's age, sex, comorbidities, and severity of initial symptoms.41 Analyses of endarterectomy trials indicated that the benefit from CEA is greatest if performed within 2 weeks of a patient's last ischemic event, the advantage it confers rapidly falling with increasing delay.45 From the hospitalist's standpoint, it is of prime importance to ensure that patients admitted to the hospital with a TIA or ischemic stroke are not discharged before it has been established whether have severe carotid stenosis that requires a revascularization procedure. If carotid stenosis is less than 50%, CEA is not recommended.41
A newer, less invasive form of carotid artery revascularization is CAS,46 which is performed by operators with established periprocedural morbidity and mortality rates of 4%‐6% and may be considered in those with:
-
Symptomatic severe stenosis (>70%) that is difficult to access surgically.2
-
Medical issues that greatly increase the risks of surgery, such as clinically significant cardiac disease, severe pulmonary disease, contralateral carotid occlusion, contralateral laryngeal nerve palsy, radiation‐induced stenosis or restenosis after carotid endarterectomy, and more than 80 years old.43
Angioplasty and/or stenting may also be considered when patients with symptomatic extracranial vertebral stenosis are having symptoms despite optimal medical risk factor treatments.2 Among those with hemodynamically significant stenosis of the major intracranial vasculature (basilar, middle cerebrals, distal carotids, and vertebrals) experiencing symptoms despite optimal medical risk factor treatments, angioplasty and/or stenting is considered experimental.2
The degree of arterial stenosis can be assessed by ultrasound, magnetic resonance angiogram (MRA), computed tomography angiogram (CTA), and conventional catheter angiogram, the last of which remains the gold standard. A carotid ultrasound performed at a certified vascular laboratory or by an experienced radiology technologist that shows less than 50% stenosis need not be followed up with another neuroimaging test. Generally, MRA tends to overestimate the degree of arterial stenosis but is a useful screening tool. In the event that an MRA reveals more than 50% stenosis, another diagnostic modality such as a carotid duplex, CTA, or conventional catheter angiogram should be performed to confirm this finding.
Antithrombotic Treatment
Cardioembolic Stroke Mechanism
Although it can sometimes be difficult to determine the precise mechanism underlying a patient's stroke or TIA, those who have a high‐risk source of cardiogenic embolism should generally be treated with anticoagulant medications to prevent recurrence.2 Among ischemic cerebrovascular event patients with persistent or paroxysmal atrial fibrillation, anticoagulation with adjusted‐dose warfarin (target international normalized ratio [INR] of 2.5; range, 2.0‐3.0) should be administered.47 The ASA recommends initiating oral anticoagulation within 2 weeks of an ischemic stroke or TIA but indicates that further delays may be appropriate for patients with large infarcts or uncontrolled hypertension.2 For patients unable to take oral anticoagulants, aspirin 325 mg/day should be given instead. Among patients who suffered an ischemic stroke or TIA because of an acute myocardial infarction in whom left ventricular mural thrombus is identified by echocardiography or another form of cardiac imaging, oral anticoagulation should be considered, aiming for an INR of 2.0‐3.0 for at least 3 months and up to 1 year.2 Patients receiving oral anticoagulation who also have ischemic coronary artery disease should be prescribed aspirin as well, in doses up to 162 mg/day.2
Noncardioembolic Stroke Mechanism
For ischemic stroke or TIA patients who have no high‐risk source of cardiogenic embolism, antiplatelet agents rather than oral anticoagulation are generally recommended to reduce the risk of recurrent stroke and other cardiovascular events.4850 Acceptable options for initial therapy include:
-
Aspirin (50 to 325 mg/day)48;
-
Combination of aspirin (50 mg) and extended‐release dipyridamole (400 mg) daily49, 51;
-
Clopidogrel (75 mg) daily.50
The combination of aspirin and extended‐release dipyridamole is suggested instead of aspirin alone, and clopidogrel may be considered instead of aspirin alone.49, 51 However, currently there is not enough data to make evidence‐based recommendations for choosing between antiplatelet drugs beyond aspirin.2 Furthermore, there is no evidence that increasing the dose of aspirin for patients who have had an ischemic stroke while taking aspirin provides additional benefit.2 The selection of an antiplatelet agent must be individualized, giving due consideration to a patient's presumed stroke mechanism, risk factor profile, and tolerance.
Other antiplatelet guidelines for noncardioembolic stroke/TIA patients include that:
Education for Behavior Modification
It is crucial to discharge patients with the tools they need to make important lifestyle changes. Patients can significantly reduce their stroke risk by making changes in their everyday patterns of behavior. As much education as possible about smoking cessation, exercise, diet, and the warning signs of stroke should be provided often as possible during hospitalization for a stroke and need not be left to nurses. Stroke education is extremely important so patients understand the need to call for emergency medical services immediately if they even suspect they are having stroke symptoms because of the very narrow window of opportunity for treatment of an acute stroke.54 All patients should be encouraged to make lifestyle adjustments such as ceasing smoking, reducing alcohol intake, and controlling weight. Smoking cessation appears to be effective in preventing secondary stroke (33% reduction in relative risk),44 and initiating smoking cessation counseling during hospitalization for stroke may result in a high rate of adherence to smoking cessation, at least in the short term.55 Table 3 displays current national guideline recommendations on lifestyle modification approaches.2
| Risk Factor | Recommendation |
|---|---|
| |
| Smoking | Smoking cessation |
| Avoid environmental smoke | |
| Counseling, nicotine products, and oral smoking cessation medications | |
| Alcohol | Eliminate or reduce alcohol consumption |
| Light to moderate levels2 drinks/day for men, 1 drink/day for nonpregnant women may be considered | |
| Obesity | Weight reduction goal: BMI 18.5‐24.9 kg/m2 and waist circumference < 35 inches for women, < 40 inches for men |
| Encourage weight management through balance of caloric intake, physical activity, behavioral counseling | |
| Physical Activity | At least 30 minutes of moderate‐intensity physical exercise most days of the week |
| Supervised therapeutic exercise regimen for those with residual disability | |
EvidencePractice Gap
There are now many secondary stroke prevention modalities, and there is a copious amount of data validating the efficacy of quite a few of them.2 Yet there is a large gap in implementing evidence‐based secondary prevention strategies.35 TIA and ischemic stroke patients are often discharged from the hospital without being prescribed any preventive medications, despite the data supporting the use of antiplatelet agents, anticoagulants, and antihypertensives for prevention of secondary stroke.4 In addition, several behavioral interventions could help patients to avoid stroke recurrence,2 but quite often stroke patients are not educated about them during the acute care period.4 Poor discharge treatment utilization limits the effectiveness of proven therapies, resulting in lost opportunities to reduce the burden of secondary stroke.
The reasons for these care gaps are multifactorial and can be traced to patient and provider issues as well as to health care delivery processes. Our understanding of the reasons for this gap is improving. Generally speaking, preventive services are used less frequently than those services or treatment modalities that provide immediate relief or economic benefit. The benefit of most preventive services is more readily seen at a population level than at a individual level and accrues slowly over time. It becomes more difficult to stress prevention in a health care system driven by technology‐based acute care.3
Current clinical management of acute stroke patients has stroke specialists and hospital physicians focusing on the acute management and diagnostic workup during hospitalization. Initiation of long‐term treatment is often deferred to after discharge, when the patient resumes long‐term primary care follow‐up.54 This deferred approach may result in therapy not being initiated or being initiated less efficiently and at a time (weeks or months after the initial presentation) when the stroke event and underlying atherosclerotic disease may no longer be the focus of either the patient or the primary care physician.54
Initiating medications during the acute stroke hospitalization phase sends the patient the message that these therapies are important for preventing recurrence and are an essential part of their treatment.54 More important, hospital initiation of secondary prevention therapies has been shown to be a strong predictor of these therapies continuing to be used after discharge56 and is associated with better clinical outcomes.5759 Table 4 shows some of the resources available to assist hospitalists in overcoming the evidencepractice gap in stroke treatment.
| Tool | Description |
|---|---|
| |
| AHA Get with the GuidelinesStroke ( |
Focuses on care team protocols to facilitate appropriate in‐hospital and discharge stroke treatment utilization |
| Identifies champions to lead, develop, and mobilize teams to optimally implement evidence‐based stroke treatment in acute care hospitals | |
| Utilizes standardized admission orders, patient educational materials, data monitoring | |
| Provides resources to help hospitals obtain JCAHO certification | |
| UCLA Stroke PROTECT (Preventing Recurrence of Thromboembolic Events through Coordinated Treatment) program ( |
Integrates 8 proven secondary stroke prevention measures into standard stroke care provided during hospitalization |
| Applies quality improvement measures through preprinted admission orders, care maps, discharge protocols, educational materials, patient self‐assessment logs, and data monitoring tools | |
| JCAHO Disease Specific Certification for acute stroke care ( |
Designates eligible hospitals as primary stroke centers |
| Promotes compliance with consensus‐based national standards | |
| Encourages effective use of established clinical practice guidelines to manage and optimize stroke care | |
| Fosters an organized approach to performance measurement and improvement activities | |
CONCLUSIONS
The acute stroke hospitalization setting provides the ideal opportunity for hospitalists to not only institute evidence‐based prevention therapies for recurrent stroke but also to have the undivided attention of patients and their families. Furthermore, it may be risky to assume that relevant therapy when deferred will be initiated in a timely fashion, if at all, after hospital discharge. As part of an effective continuum of care, hospitalists have an important role not just in the management of acute ischemic stroke, but also in long‐term reduction of vascular risk.
- .Stroke prevention: windows of opportunity and failed expectations? A discussion of modifiable cardiovascular risk factors and a prevention proposal.Neuroepidemiology.1997;16(4):163–173.
- ,,, et al.Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co‐sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline.Stroke.2006;37:577–617.
- ,,.Stroke prevention: narrowing the evidence‐practice gap.Neurology.2000;54:1899–1906.
- ,,, et al.Acute stroke care in the US: results from 4 pilot prototypes of the Paul Coverdell National Acute Stroke Registry.Stroke.2005;36:1232–1240.
- ,,.,.Lipid Assessment and treatment patterns in hospitalized TIA and ischemic stroke patients.J Hosp Med.2006;1:214–220.
- ,,, et al.Heart disease and stroke statistics—2006 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee.Circulation.2006;113(6):e85–e151.
- .Long‐term outcome after ischaemic stroke/transient ischaemic attack.Cerebrovasc Dis.2003;16(Suppl 1):14–19.
- ,,,,.Ten‐year survival after first‐ever stroke in the perth community stroke study.Stroke.2003;34:1842–1846.
- NINDS rt‐PA Stroke Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,,,,.Assessing patterns of t‐PA use in acute stroke.Stroke.2002;33:354.
- Growth of hospital medicine nationwide. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/Media/GrowthofHospitalMedicineNationwide/Growth_of_ Hospital_M.htm. Accessed April 12,2006.
- American Academy of Neurology. Available at: http://www.aan.com/students/medical/faq.cfm. Accessed June 25,2006.
- .The neurologist's role in stroke management.Stroke.1996;27:1935–1936.
- .Prevention and management of medical complications of the hospitalized elderly stroke patient.Clin Geriatr Med.1991;7:475–482.
- ,,, et al.The Stroke Prevention Policy Model: linking evidence and clinical decisions.Ann Intern Med.1997;127:704–711.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,, et al.Incidence and short‐term prognosis of transient ischemic attack in a population‐based study.Stroke.2005;36:720–723.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,,.Short‐term prognosis after emergency department diagnosis of TIA.JAMA.2000;284:2901–2906.
- ,,,,.Ten‐year risk of first recurrent stroke and disability after first‐ever stroke in the Perth Community Stroke Study.Stroke.2004;35:731–735.
- .New strategies for prevention of ischemic stroke: the LIFE study.Curr Neurol Neurosci Rep.2003;3(1):46–51.
- ,,, et al.[Manifestations of atherosclerosis in various vascular regions. Similarities and differences regarding epidemiology, etiology and prognosis].Med Klin.2002;97(4):221–228.
- ,,,,,.Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences.Ann Intern Med.2001;134(3):224–238.
- .Prevention of strokes and recurrent strokes.J Neurol Neurosurg Psychiatry.1998;64:716.
- ,.Prevalence of coexistence of coronary artery disease, ischemic stroke, and peripheral arterial disease in older persons, mean age 80 years, in an academic hospital‐based geriatrics practice.J Am Geriatr Soc.1999;47:1255–1256.
- ,,.Risk factors and their management for stroke prevention: outlook for 1999 and beyond.Neurology.1999;53(7 Suppl 4):S15–S24.
- ,,,,.Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest. Sep2004;126(3 Suppl):483S–512S.
- Randomised trial of a perindopril‐based blood‐pressure‐lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,.Blood pressure reduction and secondary prevention of stroke and other vascular events: a systematic review.Stroke.2003;34:2741–2748.
- Post‐stroke antihypertensive treatment study. A preliminary result.PATS Collaborating Group.Chin Med J (Engl).1995;108:710–717.
- ,,.Should beta blockers remain first choice in the treatment of primary hypertension? A meta‐analysis.Lancet.2005;366:1545–1553.
- ,,, et al.Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines.Circulation.2004;110(2):227–239.
- Heart Protection Study Collaborative Group.Effects of cholesterol‐lowering with Simvastatin on stroke and other major vascular events in 20, 536 people with cerebrovascular disease or other high‐risk conditions.Lancet.2004;363:757–767.
- , et al.The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study. Presented at the 15th European Stroke Conference, Brussels, Belgium, May 16‐19,2006.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38: UK Prospective Diabetes Study Group.BMJ.1998;317:703–713.
- ,,,,, for theHeart Protection Study Collaborative Group.MRC/BHF Heart Protection Study of cholesterol‐lowering with simvastatin in 5963 people with diabetes: a randomised placebo‐controlled trial.Lancet.2003;361:2005–2016.
- ,,,.The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy: the Collaborative Study Group.N Engl J Med.1993;329:1456–1462.
- ,,, for theCollaborative Study Group.Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy.N Engl J Med.2001;345:861–869.
- ,,., et al.Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non‐insulin‐dependent diabetes mellitus: a randomized prospective 6‐year study.Diabetes Res Clin Pract.1995;28:103–117.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high‐grade carotid stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1991;325:445–453.
- ,,, et al.Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1998;339:1415–1425.
- Endarterectomy for asymptomatic carotid artery stenosis.Executive Committee for the Asymptomatic Carotid Atherosclerosis Study.JAMA.1995;273:1421–1428.
- ,,, et al.Protected carotid‐artery stenting versus endarterectomy in high‐risk patients.N Engl J Med.2004;351:1493–1501.
- ,,.New evidence for stroke prevention: scientific review.JAMA.2002;288:1388–1395.
- ,,,,.Endarterectomy for symptomatic carotid stenosis in relation to clinical subgroups and timing of surgery.Lancet.2004;363:915–924.
- .Carotid artery surgery vs. stent: a cardiovascular perspective.Catheter Cardiovasc Interv.2004;63:377–384.
- ,,, et al.Warfarin anticoagulation and outcomes in patients with atrial fibrillation: a systematic review and metaanalysis.Chest.2004;126:1938–1945.
- Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients.BMJ.2002;324:71–86.
- ,,,,,.European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke.J Neurol Sci.1996;143(1‐2):1–13.
- A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE).CAPRIE Steering Committee.Lancet.1996;348:1329–1339.
- ,,,,.Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial.Lancet.2006;367:1665–1673.
- ,,, et al.Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high‐risk patients (MATCH): randomised, double‐blind, placebo‐controlled trial.Lancet.2004;364:331–337.
- ,,, et al.Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events.N Engl J Med. Apr 202006;354(16):1706–1717.
- ,,, et al.PROTECT: A Coordinated Stroke Treatment Program to Prevent Recurrent Thromboembolic Events.Neurology. Vol63;2004:1217–1222.
- ,,, et al.In‐hospital initiation of secondary stroke prevention therapies yields high rates of adherence at follow up.Stroke.2004;35:2879–2883.
- ,,, et al.In‐hospital initiation of lipid‐lowering therapy after coronary intervention as a predictor of long‐term utilization: a propensity analysis.Arch Intern Med.2003;163:2576–2582.
- ,,, et al.Early effects of statins in patients with coronary artery disease and high C‐reactive protein.Am J Cardiol.2004;94:1107–1112.
- ,,,.Improved treatment of coronary heart disease by implementation of a Cardiac Hospitalization Atherosclerosis Management Program (CHAMP).Am J Cardiol. Apr 12001;87(7):819–822.
- ,,.Importance of in‐hospital initiation of evidence‐based medical therapies for heart failure‐a review.Am J Cardiol.2004;94:1155–1160.
- ,,,.Blood pressure and stroke: an overview of published reviews.Stroke.2004;35:1024.
- ,,, et al.Intensive versus moderate lipid lowering with statins after acute coronary syndromes.N Engl J Med.2004;350:1495–1504.
- ,,.Niacin therapy in atherosclerosis.Curr Opin Lipidol.2004;15:659–665.
- ,,, et al.Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: The Veterans Affairs HDL Intervention Trial (VA‐HIT).Circulation.2001;103:2828–2833.
- ,,.The effect of long‐term intensified insulin treatment on the development of microvascular complications of diabetes mellitus.N Engl J Med.1993;329:304–309.
- .Stroke prevention: windows of opportunity and failed expectations? A discussion of modifiable cardiovascular risk factors and a prevention proposal.Neuroepidemiology.1997;16(4):163–173.
- ,,, et al.Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co‐sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline.Stroke.2006;37:577–617.
- ,,.Stroke prevention: narrowing the evidence‐practice gap.Neurology.2000;54:1899–1906.
- ,,, et al.Acute stroke care in the US: results from 4 pilot prototypes of the Paul Coverdell National Acute Stroke Registry.Stroke.2005;36:1232–1240.
- ,,.,.Lipid Assessment and treatment patterns in hospitalized TIA and ischemic stroke patients.J Hosp Med.2006;1:214–220.
- ,,, et al.Heart disease and stroke statistics—2006 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee.Circulation.2006;113(6):e85–e151.
- .Long‐term outcome after ischaemic stroke/transient ischaemic attack.Cerebrovasc Dis.2003;16(Suppl 1):14–19.
- ,,,,.Ten‐year survival after first‐ever stroke in the perth community stroke study.Stroke.2003;34:1842–1846.
- NINDS rt‐PA Stroke Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,,,,.Assessing patterns of t‐PA use in acute stroke.Stroke.2002;33:354.
- Growth of hospital medicine nationwide. Available at: http://www.hospitalmedicine.org/Content/NavigationMenu/Media/GrowthofHospitalMedicineNationwide/Growth_of_ Hospital_M.htm. Accessed April 12,2006.
- American Academy of Neurology. Available at: http://www.aan.com/students/medical/faq.cfm. Accessed June 25,2006.
- .The neurologist's role in stroke management.Stroke.1996;27:1935–1936.
- .Prevention and management of medical complications of the hospitalized elderly stroke patient.Clin Geriatr Med.1991;7:475–482.
- ,,, et al.The Stroke Prevention Policy Model: linking evidence and clinical decisions.Ann Intern Med.1997;127:704–711.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,, et al.Incidence and short‐term prognosis of transient ischemic attack in a population‐based study.Stroke.2005;36:720–723.
- ,,.Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services.BMJ.2004;328:326.
- ,,,.Short‐term prognosis after emergency department diagnosis of TIA.JAMA.2000;284:2901–2906.
- ,,,,.Ten‐year risk of first recurrent stroke and disability after first‐ever stroke in the Perth Community Stroke Study.Stroke.2004;35:731–735.
- .New strategies for prevention of ischemic stroke: the LIFE study.Curr Neurol Neurosci Rep.2003;3(1):46–51.
- ,,, et al.[Manifestations of atherosclerosis in various vascular regions. Similarities and differences regarding epidemiology, etiology and prognosis].Med Klin.2002;97(4):221–228.
- ,,,,,.Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences.Ann Intern Med.2001;134(3):224–238.
- .Prevention of strokes and recurrent strokes.J Neurol Neurosurg Psychiatry.1998;64:716.
- ,.Prevalence of coexistence of coronary artery disease, ischemic stroke, and peripheral arterial disease in older persons, mean age 80 years, in an academic hospital‐based geriatrics practice.J Am Geriatr Soc.1999;47:1255–1256.
- ,,.Risk factors and their management for stroke prevention: outlook for 1999 and beyond.Neurology.1999;53(7 Suppl 4):S15–S24.
- ,,,,.Antithrombotic and thrombolytic therapy for ischemic stroke: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.Chest. Sep2004;126(3 Suppl):483S–512S.
- Randomised trial of a perindopril‐based blood‐pressure‐lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,.Blood pressure reduction and secondary prevention of stroke and other vascular events: a systematic review.Stroke.2003;34:2741–2748.
- Post‐stroke antihypertensive treatment study. A preliminary result.PATS Collaborating Group.Chin Med J (Engl).1995;108:710–717.
- ,,.Should beta blockers remain first choice in the treatment of primary hypertension? A meta‐analysis.Lancet.2005;366:1545–1553.
- ,,, et al.Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines.Circulation.2004;110(2):227–239.
- Heart Protection Study Collaborative Group.Effects of cholesterol‐lowering with Simvastatin on stroke and other major vascular events in 20, 536 people with cerebrovascular disease or other high‐risk conditions.Lancet.2004;363:757–767.
- , et al.The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study. Presented at the 15th European Stroke Conference, Brussels, Belgium, May 16‐19,2006.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38: UK Prospective Diabetes Study Group.BMJ.1998;317:703–713.
- ,,,,, for theHeart Protection Study Collaborative Group.MRC/BHF Heart Protection Study of cholesterol‐lowering with simvastatin in 5963 people with diabetes: a randomised placebo‐controlled trial.Lancet.2003;361:2005–2016.
- ,,,.The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy: the Collaborative Study Group.N Engl J Med.1993;329:1456–1462.
- ,,, for theCollaborative Study Group.Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy.N Engl J Med.2001;345:861–869.
- ,,., et al.Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non‐insulin‐dependent diabetes mellitus: a randomized prospective 6‐year study.Diabetes Res Clin Pract.1995;28:103–117.
- Beneficial effect of carotid endarterectomy in symptomatic patients with high‐grade carotid stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1991;325:445–453.
- ,,, et al.Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis.North American Symptomatic Carotid Endarterectomy Trial Collaborators.N Engl J Med.1998;339:1415–1425.
- Endarterectomy for asymptomatic carotid artery stenosis.Executive Committee for the Asymptomatic Carotid Atherosclerosis Study.JAMA.1995;273:1421–1428.
- ,,, et al.Protected carotid‐artery stenting versus endarterectomy in high‐risk patients.N Engl J Med.2004;351:1493–1501.
- ,,.New evidence for stroke prevention: scientific review.JAMA.2002;288:1388–1395.
- ,,,,.Endarterectomy for symptomatic carotid stenosis in relation to clinical subgroups and timing of surgery.Lancet.2004;363:915–924.
- .Carotid artery surgery vs. stent: a cardiovascular perspective.Catheter Cardiovasc Interv.2004;63:377–384.
- ,,, et al.Warfarin anticoagulation and outcomes in patients with atrial fibrillation: a systematic review and metaanalysis.Chest.2004;126:1938–1945.
- Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients.BMJ.2002;324:71–86.
- ,,,,,.European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke.J Neurol Sci.1996;143(1‐2):1–13.
- A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE).CAPRIE Steering Committee.Lancet.1996;348:1329–1339.
- ,,,,.Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): randomised controlled trial.Lancet.2006;367:1665–1673.
- ,,, et al.Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high‐risk patients (MATCH): randomised, double‐blind, placebo‐controlled trial.Lancet.2004;364:331–337.
- ,,, et al.Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events.N Engl J Med. Apr 202006;354(16):1706–1717.
- ,,, et al.PROTECT: A Coordinated Stroke Treatment Program to Prevent Recurrent Thromboembolic Events.Neurology. Vol63;2004:1217–1222.
- ,,, et al.In‐hospital initiation of secondary stroke prevention therapies yields high rates of adherence at follow up.Stroke.2004;35:2879–2883.
- ,,, et al.In‐hospital initiation of lipid‐lowering therapy after coronary intervention as a predictor of long‐term utilization: a propensity analysis.Arch Intern Med.2003;163:2576–2582.
- ,,, et al.Early effects of statins in patients with coronary artery disease and high C‐reactive protein.Am J Cardiol.2004;94:1107–1112.
- ,,,.Improved treatment of coronary heart disease by implementation of a Cardiac Hospitalization Atherosclerosis Management Program (CHAMP).Am J Cardiol. Apr 12001;87(7):819–822.
- ,,.Importance of in‐hospital initiation of evidence‐based medical therapies for heart failure‐a review.Am J Cardiol.2004;94:1155–1160.
- ,,,.Blood pressure and stroke: an overview of published reviews.Stroke.2004;35:1024.
- ,,, et al.Intensive versus moderate lipid lowering with statins after acute coronary syndromes.N Engl J Med.2004;350:1495–1504.
- ,,.Niacin therapy in atherosclerosis.Curr Opin Lipidol.2004;15:659–665.
- ,,, et al.Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: The Veterans Affairs HDL Intervention Trial (VA‐HIT).Circulation.2001;103:2828–2833.
- ,,.The effect of long‐term intensified insulin treatment on the development of microvascular complications of diabetes mellitus.N Engl J Med.1993;329:304–309.
Embracing, with strengthened spirits
Embracing, with strengthened spirits
What do you think of aging? I cautiously asked her.
She smiled, highlighting her wrinkles.
And said, I have grown out of the embarrassment,
Of being incontinent,
And forgetting my neighbors' names.
Embarrassment of not being able,
To recall life's many precious moments.
It bothered me until recently,
How can I miss those traffic lights,
And pay for the same grocery twice.
I explained to myself: Honey, we all age.
We all age, she continued.
And there is a distinct joy.
A joy of aging.
The wrinkles on my face,
Tell all the moments I smiled.
Doc, You know how do I take it? she asked.
And she continued, I am not decaying.
But,
I am Aging, gracefully.
It is like embracing a weakening body,
With strengthened spirits.
She smiled, adding another wrinkle to her face, gracefully.
I guess. It is all about perspective.
The attitude that matters.
Shades of Her Life
Which color do you prefer?
He asked her.
As she stood between a wide choice.
He asked her again, Ma'am! Which shade do you like?
A flashback revisiteda state of reverie.
Life has offered her so many colors, she pondered.
It is always funto choose your favorite colors.
The unmeasured joy of having her favorite crayons,
And the unparalleled delight of choosing a blue dress and the silver car.
Flashing the pink friendship band as a young girl,
The sobriety of black interview attire,
The pleasure of counting rainbow colors, after a drizzle,
The eye catchy fluorescence of tender years,
The compelling need to match her nails with her dress,
Highlighting the hair with different shades,
Oh so many colors have shaded her life.
It is amazing!
She chose colors at every moment in her life.
Colors and more colorsand the joy they brought in her life
Excuse me! The man interrupted her train of thoughts.
May I help you choose one? he asked again.
Do you want to try one, ma'am? he continued.
She startled and fumbled.
Holding back her tears, she strengthened her femininity once again,
Like she did after every cycle of drenching chemotherapy
For her maligned breasts.
She regained her composureshe regained her strength,
Oh, am sorry, sir, she said and pointed towards the golden brown wig,
Perched on a mannequin.
Can I have that shade, please? she gently smiled.
Embracing, with strengthened spirits
What do you think of aging? I cautiously asked her.
She smiled, highlighting her wrinkles.
And said, I have grown out of the embarrassment,
Of being incontinent,
And forgetting my neighbors' names.
Embarrassment of not being able,
To recall life's many precious moments.
It bothered me until recently,
How can I miss those traffic lights,
And pay for the same grocery twice.
I explained to myself: Honey, we all age.
We all age, she continued.
And there is a distinct joy.
A joy of aging.
The wrinkles on my face,
Tell all the moments I smiled.
Doc, You know how do I take it? she asked.
And she continued, I am not decaying.
But,
I am Aging, gracefully.
It is like embracing a weakening body,
With strengthened spirits.
She smiled, adding another wrinkle to her face, gracefully.
I guess. It is all about perspective.
The attitude that matters.
Shades of Her Life
Which color do you prefer?
He asked her.
As she stood between a wide choice.
He asked her again, Ma'am! Which shade do you like?
A flashback revisiteda state of reverie.
Life has offered her so many colors, she pondered.
It is always funto choose your favorite colors.
The unmeasured joy of having her favorite crayons,
And the unparalleled delight of choosing a blue dress and the silver car.
Flashing the pink friendship band as a young girl,
The sobriety of black interview attire,
The pleasure of counting rainbow colors, after a drizzle,
The eye catchy fluorescence of tender years,
The compelling need to match her nails with her dress,
Highlighting the hair with different shades,
Oh so many colors have shaded her life.
It is amazing!
She chose colors at every moment in her life.
Colors and more colorsand the joy they brought in her life
Excuse me! The man interrupted her train of thoughts.
May I help you choose one? he asked again.
Do you want to try one, ma'am? he continued.
She startled and fumbled.
Holding back her tears, she strengthened her femininity once again,
Like she did after every cycle of drenching chemotherapy
For her maligned breasts.
She regained her composureshe regained her strength,
Oh, am sorry, sir, she said and pointed towards the golden brown wig,
Perched on a mannequin.
Can I have that shade, please? she gently smiled.
Embracing, with strengthened spirits
What do you think of aging? I cautiously asked her.
She smiled, highlighting her wrinkles.
And said, I have grown out of the embarrassment,
Of being incontinent,
And forgetting my neighbors' names.
Embarrassment of not being able,
To recall life's many precious moments.
It bothered me until recently,
How can I miss those traffic lights,
And pay for the same grocery twice.
I explained to myself: Honey, we all age.
We all age, she continued.
And there is a distinct joy.
A joy of aging.
The wrinkles on my face,
Tell all the moments I smiled.
Doc, You know how do I take it? she asked.
And she continued, I am not decaying.
But,
I am Aging, gracefully.
It is like embracing a weakening body,
With strengthened spirits.
She smiled, adding another wrinkle to her face, gracefully.
I guess. It is all about perspective.
The attitude that matters.
Shades of Her Life
Which color do you prefer?
He asked her.
As she stood between a wide choice.
He asked her again, Ma'am! Which shade do you like?
A flashback revisiteda state of reverie.
Life has offered her so many colors, she pondered.
It is always funto choose your favorite colors.
The unmeasured joy of having her favorite crayons,
And the unparalleled delight of choosing a blue dress and the silver car.
Flashing the pink friendship band as a young girl,
The sobriety of black interview attire,
The pleasure of counting rainbow colors, after a drizzle,
The eye catchy fluorescence of tender years,
The compelling need to match her nails with her dress,
Highlighting the hair with different shades,
Oh so many colors have shaded her life.
It is amazing!
She chose colors at every moment in her life.
Colors and more colorsand the joy they brought in her life
Excuse me! The man interrupted her train of thoughts.
May I help you choose one? he asked again.
Do you want to try one, ma'am? he continued.
She startled and fumbled.
Holding back her tears, she strengthened her femininity once again,
Like she did after every cycle of drenching chemotherapy
For her maligned breasts.
She regained her composureshe regained her strength,
Oh, am sorry, sir, she said and pointed towards the golden brown wig,
Perched on a mannequin.
Can I have that shade, please? she gently smiled.
Effective methods for preventing pressure ulcers
-
CLINICAL QUESTION: Which interventions are effective for the prevention of pressure ulcers?
-
BOTTOM LINE: Effective strategies for preventing pressure ulcers include the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, are also effective. (LOE = 1a‐)
-
REFERENCE: Reddy M, Gill SS. Rochon PA. Preventing pressure ulcers: a systematic review. JAMA 2006;296:974‐984.
-
STUDY DESIGN: Systematic review
-
FUNDING: Government
-
SETTING: Various (meta‐analysis)
-
SYNOPSIS: Multiple preventive approaches are used in the management of pressure ulcers. These authors systematically searched multiple evidence‐based databases including the Cochrane Registry, bibliographies of identified articles, and scientific meeting abstracts for randomized controlled trials (RCTs) evaluating preventive measures for pressure ulcers. No language restrictions were applied. They used standard methods to critically appraise individual RCTs. The search strategy identified 763 citations, from which 59 trials meeting eligibility criteria were selected. The methodologic quality of the RCTs was generally suboptimal. Interventions were grouped into 3 categories: those addressing impairments in (1) mobility, (2) nutrition, and (3) skin health. Effective strategies for those with impaired mobility included the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning (every 2 vs every 4 hours) is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, were helpful, but the incremental benefit of other specific topical agents is minimal.
-
CLINICAL QUESTION: Which interventions are effective for the prevention of pressure ulcers?
-
BOTTOM LINE: Effective strategies for preventing pressure ulcers include the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, are also effective. (LOE = 1a‐)
-
REFERENCE: Reddy M, Gill SS. Rochon PA. Preventing pressure ulcers: a systematic review. JAMA 2006;296:974‐984.
-
STUDY DESIGN: Systematic review
-
FUNDING: Government
-
SETTING: Various (meta‐analysis)
-
SYNOPSIS: Multiple preventive approaches are used in the management of pressure ulcers. These authors systematically searched multiple evidence‐based databases including the Cochrane Registry, bibliographies of identified articles, and scientific meeting abstracts for randomized controlled trials (RCTs) evaluating preventive measures for pressure ulcers. No language restrictions were applied. They used standard methods to critically appraise individual RCTs. The search strategy identified 763 citations, from which 59 trials meeting eligibility criteria were selected. The methodologic quality of the RCTs was generally suboptimal. Interventions were grouped into 3 categories: those addressing impairments in (1) mobility, (2) nutrition, and (3) skin health. Effective strategies for those with impaired mobility included the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning (every 2 vs every 4 hours) is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, were helpful, but the incremental benefit of other specific topical agents is minimal.
-
CLINICAL QUESTION: Which interventions are effective for the prevention of pressure ulcers?
-
BOTTOM LINE: Effective strategies for preventing pressure ulcers include the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, are also effective. (LOE = 1a‐)
-
REFERENCE: Reddy M, Gill SS. Rochon PA. Preventing pressure ulcers: a systematic review. JAMA 2006;296:974‐984.
-
STUDY DESIGN: Systematic review
-
FUNDING: Government
-
SETTING: Various (meta‐analysis)
-
SYNOPSIS: Multiple preventive approaches are used in the management of pressure ulcers. These authors systematically searched multiple evidence‐based databases including the Cochrane Registry, bibliographies of identified articles, and scientific meeting abstracts for randomized controlled trials (RCTs) evaluating preventive measures for pressure ulcers. No language restrictions were applied. They used standard methods to critically appraise individual RCTs. The search strategy identified 763 citations, from which 59 trials meeting eligibility criteria were selected. The methodologic quality of the RCTs was generally suboptimal. Interventions were grouped into 3 categories: those addressing impairments in (1) mobility, (2) nutrition, and (3) skin health. Effective strategies for those with impaired mobility included the use of support surfaces (mattresses, beds, and cushions), mattress overlays on operating tables, and specialized foam and sheepskin overlays. Frequent repositioning is effective, but the optimal schedule for turning (every 2 vs every 4 hours) is uncertain. Nutritional supplements are beneficial in patients with impaired nutrition. Simple skin moisturizers, specifically to the sacral area, were helpful, but the incremental benefit of other specific topical agents is minimal.
Clinical Conundrum
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
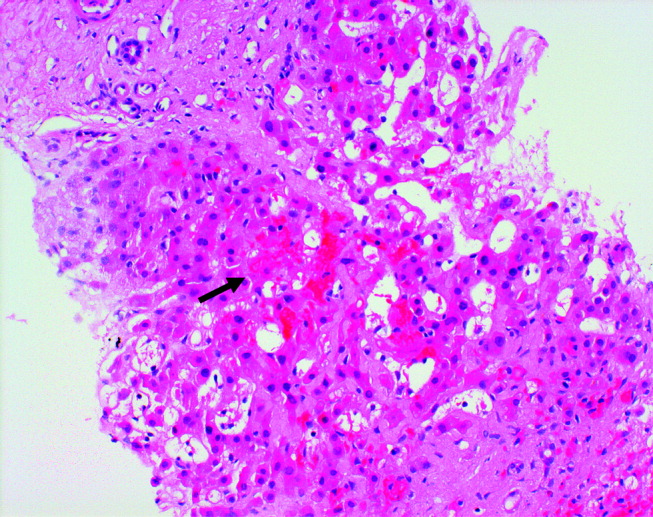
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
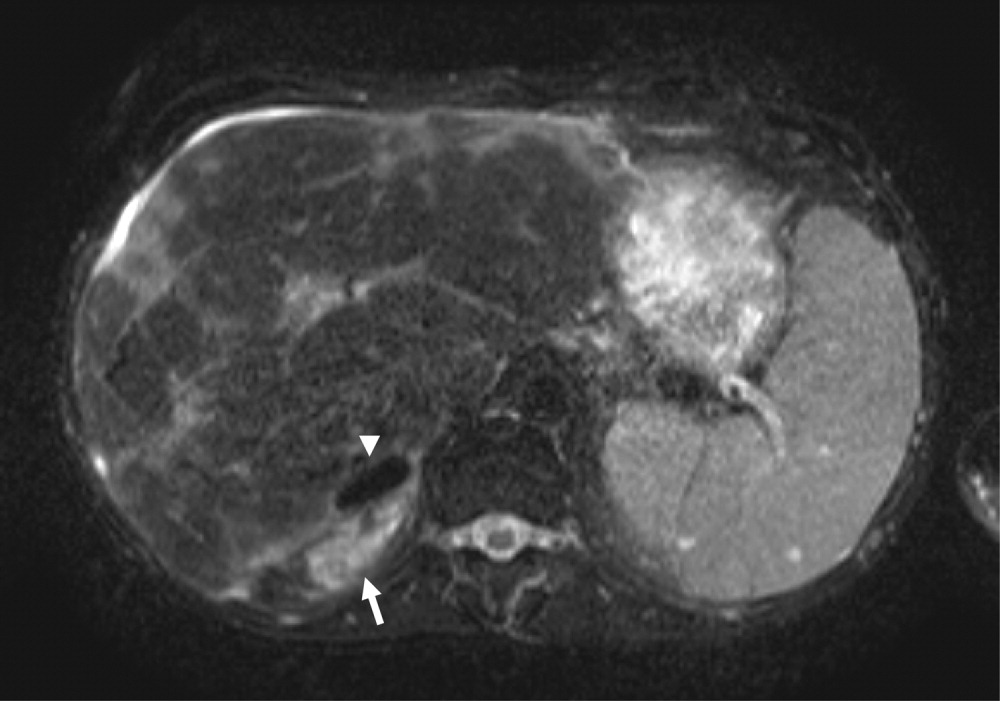
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range < 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
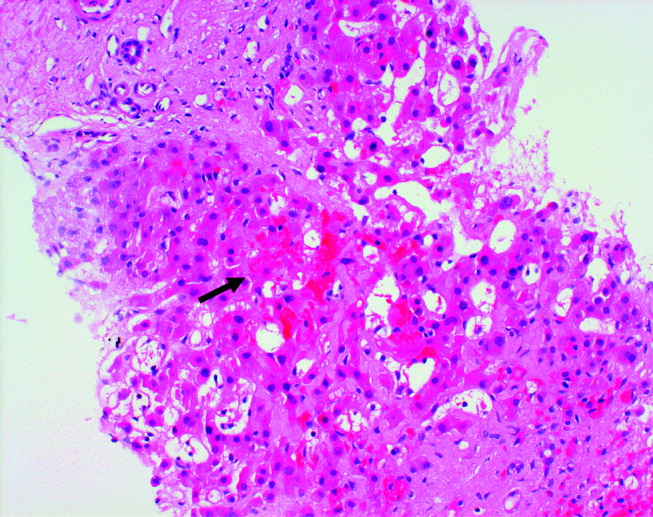
Annual reviewers list
We deeply appreciate the involvement of our reviewers who made the Journal of Hospital Medicine so successful in its first year. Listed below are the many reviewers and volume of their contributions. They have our sincere gratitude.
Reviewed 4 or More Articles
Eric Alper (6)
David Anthony (4)
Vineet Arora (4)
Thomas E. Baudendistel (11)
Daniel J. Brotman (7)
Vincent W. Chiang (5)
Eugene Shu‐Sen Chu (5)
Gurpreet Dhaliwal (7)
Lorenzo Di Francesco (4)
Andras Fenyves (4)
Stacy Fischer (5)
Kathlyn Fletcher (5)
Philip H. Goodman (4)
Carolyn Gould (4)
Jeffrey L. Greenwald (6)
Lakshmi Halasyamani (4)
Brian Harte (13)
Christopher P. Landrigan (5)
Peter K. Lindenauer (4)
Greg Maynard (7)
Sylvia Cheney McKean (4)
Thomas Aquinas Murphy (4)
James C. Pile (9)
Thomas Price (4)
Sumant Ranji (8)
Bradley Allen Sharpe (4)
Jason Stein (7)
Robin Tricoles (9)
Guillermo E. Umpierrez (5)
Arpana Vidyarthi (6)
Heidi Wald (7)
David Wesorick (4)
Reviewed 3 Articles
Ron G. Angus
Paul Aronowitz
Vanitha Bala
Jennifer Best
Cynthia Jean Brown
Gregory Bump
Hugo Quinny Cheng
Eva Chittenden
Eric Coleman
Curtiss B. Cook
Edward Etchells
Alan John Forster
Roma Y. Gianchandani
Leslie W. Hall
Jennifer Hanrahan
Amir K. Jaffer
Peter John Kaboli
Jennifer Kapo
Dennis Manning
Constantine Manthous
Janet Nagamine
Kevin J. O'Leary
Brian Michael Pate
Robert C. Pendleton
Jeffrey Lawrence Schnipper
Hasan Shabbir
James Edwin Stone
Chad Whelan
Audrey Young
Reviewed 2 Articles
Drew Abernathy
Stephen J. Bekanich
Paul Cantey
Kerry Cho
Patrick Conway
Jasminka Criley
Catherine Curley
Jennifer Daru
Catherine F. Decker
Andrew Paul DeFilippis
Daniel J. DiBona
Mark Earnest
Douglas Einstadter
Margaret Fang
Jonathan M. Flacker
Bradley Evan Flansbaum
Michael Frankel
Jeffrey Glasheen
Amir H. Hamrahian
Karen E. Hauer
Eric Edwin Howell
Carlos Manuel Isada
Christopher Seoung Kim
Sunil Kripalani
Jean S. Kutner
Cindy Lai
Janet Larson
David Likosky
David Ling
Michelle Magee
Navneet Majhail
Michael Matheny
George Mathew
Govardhanan Nagaiah
James Newman
Christopher Ohl
Shawn Ralston
Daniel A. Rauch
John James Ross
Joel Rubenstein
David Schulman
Kaveh G. Shojania
Gregory Randall Smith Jr.
Peter Youngers Watson
Chad T. Whelan
Neil Winawer
Scott Wright
Reviewed 1 Article
Adebola Adesanya
Nasim Afsarmanesh
Richard Keith Albert
Mel L. Anderson, III
Wendy Artrong
Thomas W. Barrett
David Bar‐Shain
Marc Baskin
Brent Beasley
Thomas Bookwalter
Susan S. Braithwaite
Beril Caker
Douglas Carlson
Alison Chantal Caviness
Steven L. Cohn
Yvette Marie Cua
Russ Cucina
Ethan Ulysses Cumbler
Mellar Davis
Allan S. Detsky
Jeffrey Randolph Dichter
Thomas Donner
Daniel David Dressler
Erin Egan
Matthew Eisen
Kenneth Richard Epstein
Leslie Fall
Shaun Uiglas Frost
Michael Sebastian Galin
Matthew Garber
Rajesh Garg
Raminder Singh Gill
Jackie Glover
Adrienne Green
Paul Hain
Braden Hale
Sajeev Handa
Julie Hauer
Michael Heisler
Jeanne M. Huddleston
Alan J. Hunter
Kevin Hwang
Brian Jack
Ian Harold Jenkins
Kurien John
Daniel Johnson
Todd Joyner
Deepa Kabirdas
Allen Kachalia
Abel Ngo Kho
Steven Jay Kravet
Marco Aurelio Ladino
Robert Lash
Joshua Lee
Sei Lee
Arthur Jefferson Lesesne
Marcia Levetown
Luci Leykum
Joshua David Liberman
Jonathan Mansbach
Brian Markoff
David Meltzer
Anna Leco Merca
Barbara Messinger‐Rapport
Gregory Misky
William Moran
Brahmajee Nallamothu
Theore Elliott Nash
Heather Nye
Timothy O'Brien
Bruce Ovbiagele
Thomas Andrew Owens
Mary Pak
Steven Zvi Pantilat
Vikas Parekh
Kimberly Rask
Michael Rothberg
Hilary F. Ryder
Wael Saber
Sanjay Saint
Rene Salazar
Kaycee Sink
N. Smith
Malathi Srinivasin
Raj Srivastava
Erin Stucky
Alexander Turchin
Bobbak Vahid
Robert Wachter
Robert L. Wears
Howard Weitz
Winthrop Whitcomb
Mark V. Williams
Sherrie Williams
David Woods
We deeply appreciate the involvement of our reviewers who made the Journal of Hospital Medicine so successful in its first year. Listed below are the many reviewers and volume of their contributions. They have our sincere gratitude.
Reviewed 4 or More Articles
Eric Alper (6)
David Anthony (4)
Vineet Arora (4)
Thomas E. Baudendistel (11)
Daniel J. Brotman (7)
Vincent W. Chiang (5)
Eugene Shu‐Sen Chu (5)
Gurpreet Dhaliwal (7)
Lorenzo Di Francesco (4)
Andras Fenyves (4)
Stacy Fischer (5)
Kathlyn Fletcher (5)
Philip H. Goodman (4)
Carolyn Gould (4)
Jeffrey L. Greenwald (6)
Lakshmi Halasyamani (4)
Brian Harte (13)
Christopher P. Landrigan (5)
Peter K. Lindenauer (4)
Greg Maynard (7)
Sylvia Cheney McKean (4)
Thomas Aquinas Murphy (4)
James C. Pile (9)
Thomas Price (4)
Sumant Ranji (8)
Bradley Allen Sharpe (4)
Jason Stein (7)
Robin Tricoles (9)
Guillermo E. Umpierrez (5)
Arpana Vidyarthi (6)
Heidi Wald (7)
David Wesorick (4)
Reviewed 3 Articles
Ron G. Angus
Paul Aronowitz
Vanitha Bala
Jennifer Best
Cynthia Jean Brown
Gregory Bump
Hugo Quinny Cheng
Eva Chittenden
Eric Coleman
Curtiss B. Cook
Edward Etchells
Alan John Forster
Roma Y. Gianchandani
Leslie W. Hall
Jennifer Hanrahan
Amir K. Jaffer
Peter John Kaboli
Jennifer Kapo
Dennis Manning
Constantine Manthous
Janet Nagamine
Kevin J. O'Leary
Brian Michael Pate
Robert C. Pendleton
Jeffrey Lawrence Schnipper
Hasan Shabbir
James Edwin Stone
Chad Whelan
Audrey Young
Reviewed 2 Articles
Drew Abernathy
Stephen J. Bekanich
Paul Cantey
Kerry Cho
Patrick Conway
Jasminka Criley
Catherine Curley
Jennifer Daru
Catherine F. Decker
Andrew Paul DeFilippis
Daniel J. DiBona
Mark Earnest
Douglas Einstadter
Margaret Fang
Jonathan M. Flacker
Bradley Evan Flansbaum
Michael Frankel
Jeffrey Glasheen
Amir H. Hamrahian
Karen E. Hauer
Eric Edwin Howell
Carlos Manuel Isada
Christopher Seoung Kim
Sunil Kripalani
Jean S. Kutner
Cindy Lai
Janet Larson
David Likosky
David Ling
Michelle Magee
Navneet Majhail
Michael Matheny
George Mathew
Govardhanan Nagaiah
James Newman
Christopher Ohl
Shawn Ralston
Daniel A. Rauch
John James Ross
Joel Rubenstein
David Schulman
Kaveh G. Shojania
Gregory Randall Smith Jr.
Peter Youngers Watson
Chad T. Whelan
Neil Winawer
Scott Wright
Reviewed 1 Article
Adebola Adesanya
Nasim Afsarmanesh
Richard Keith Albert
Mel L. Anderson, III
Wendy Artrong
Thomas W. Barrett
David Bar‐Shain
Marc Baskin
Brent Beasley
Thomas Bookwalter
Susan S. Braithwaite
Beril Caker
Douglas Carlson
Alison Chantal Caviness
Steven L. Cohn
Yvette Marie Cua
Russ Cucina
Ethan Ulysses Cumbler
Mellar Davis
Allan S. Detsky
Jeffrey Randolph Dichter
Thomas Donner
Daniel David Dressler
Erin Egan
Matthew Eisen
Kenneth Richard Epstein
Leslie Fall
Shaun Uiglas Frost
Michael Sebastian Galin
Matthew Garber
Rajesh Garg
Raminder Singh Gill
Jackie Glover
Adrienne Green
Paul Hain
Braden Hale
Sajeev Handa
Julie Hauer
Michael Heisler
Jeanne M. Huddleston
Alan J. Hunter
Kevin Hwang
Brian Jack
Ian Harold Jenkins
Kurien John
Daniel Johnson
Todd Joyner
Deepa Kabirdas
Allen Kachalia
Abel Ngo Kho
Steven Jay Kravet
Marco Aurelio Ladino
Robert Lash
Joshua Lee
Sei Lee
Arthur Jefferson Lesesne
Marcia Levetown
Luci Leykum
Joshua David Liberman
Jonathan Mansbach
Brian Markoff
David Meltzer
Anna Leco Merca
Barbara Messinger‐Rapport
Gregory Misky
William Moran
Brahmajee Nallamothu
Theore Elliott Nash
Heather Nye
Timothy O'Brien
Bruce Ovbiagele
Thomas Andrew Owens
Mary Pak
Steven Zvi Pantilat
Vikas Parekh
Kimberly Rask
Michael Rothberg
Hilary F. Ryder
Wael Saber
Sanjay Saint
Rene Salazar
Kaycee Sink
N. Smith
Malathi Srinivasin
Raj Srivastava
Erin Stucky
Alexander Turchin
Bobbak Vahid
Robert Wachter
Robert L. Wears
Howard Weitz
Winthrop Whitcomb
Mark V. Williams
Sherrie Williams
David Woods
We deeply appreciate the involvement of our reviewers who made the Journal of Hospital Medicine so successful in its first year. Listed below are the many reviewers and volume of their contributions. They have our sincere gratitude.
Reviewed 4 or More Articles
Eric Alper (6)
David Anthony (4)
Vineet Arora (4)
Thomas E. Baudendistel (11)
Daniel J. Brotman (7)
Vincent W. Chiang (5)
Eugene Shu‐Sen Chu (5)
Gurpreet Dhaliwal (7)
Lorenzo Di Francesco (4)
Andras Fenyves (4)
Stacy Fischer (5)
Kathlyn Fletcher (5)
Philip H. Goodman (4)
Carolyn Gould (4)
Jeffrey L. Greenwald (6)
Lakshmi Halasyamani (4)
Brian Harte (13)
Christopher P. Landrigan (5)
Peter K. Lindenauer (4)
Greg Maynard (7)
Sylvia Cheney McKean (4)
Thomas Aquinas Murphy (4)
James C. Pile (9)
Thomas Price (4)
Sumant Ranji (8)
Bradley Allen Sharpe (4)
Jason Stein (7)
Robin Tricoles (9)
Guillermo E. Umpierrez (5)
Arpana Vidyarthi (6)
Heidi Wald (7)
David Wesorick (4)
Reviewed 3 Articles
Ron G. Angus
Paul Aronowitz
Vanitha Bala
Jennifer Best
Cynthia Jean Brown
Gregory Bump
Hugo Quinny Cheng
Eva Chittenden
Eric Coleman
Curtiss B. Cook
Edward Etchells
Alan John Forster
Roma Y. Gianchandani
Leslie W. Hall
Jennifer Hanrahan
Amir K. Jaffer
Peter John Kaboli
Jennifer Kapo
Dennis Manning
Constantine Manthous
Janet Nagamine
Kevin J. O'Leary
Brian Michael Pate
Robert C. Pendleton
Jeffrey Lawrence Schnipper
Hasan Shabbir
James Edwin Stone
Chad Whelan
Audrey Young
Reviewed 2 Articles
Drew Abernathy
Stephen J. Bekanich
Paul Cantey
Kerry Cho
Patrick Conway
Jasminka Criley
Catherine Curley
Jennifer Daru
Catherine F. Decker
Andrew Paul DeFilippis
Daniel J. DiBona
Mark Earnest
Douglas Einstadter
Margaret Fang
Jonathan M. Flacker
Bradley Evan Flansbaum
Michael Frankel
Jeffrey Glasheen
Amir H. Hamrahian
Karen E. Hauer
Eric Edwin Howell
Carlos Manuel Isada
Christopher Seoung Kim
Sunil Kripalani
Jean S. Kutner
Cindy Lai
Janet Larson
David Likosky
David Ling
Michelle Magee
Navneet Majhail
Michael Matheny
George Mathew
Govardhanan Nagaiah
James Newman
Christopher Ohl
Shawn Ralston
Daniel A. Rauch
John James Ross
Joel Rubenstein
David Schulman
Kaveh G. Shojania
Gregory Randall Smith Jr.
Peter Youngers Watson
Chad T. Whelan
Neil Winawer
Scott Wright
Reviewed 1 Article
Adebola Adesanya
Nasim Afsarmanesh
Richard Keith Albert
Mel L. Anderson, III
Wendy Artrong
Thomas W. Barrett
David Bar‐Shain
Marc Baskin
Brent Beasley
Thomas Bookwalter
Susan S. Braithwaite
Beril Caker
Douglas Carlson
Alison Chantal Caviness
Steven L. Cohn
Yvette Marie Cua
Russ Cucina
Ethan Ulysses Cumbler
Mellar Davis
Allan S. Detsky
Jeffrey Randolph Dichter
Thomas Donner
Daniel David Dressler
Erin Egan
Matthew Eisen
Kenneth Richard Epstein
Leslie Fall
Shaun Uiglas Frost
Michael Sebastian Galin
Matthew Garber
Rajesh Garg
Raminder Singh Gill
Jackie Glover
Adrienne Green
Paul Hain
Braden Hale
Sajeev Handa
Julie Hauer
Michael Heisler
Jeanne M. Huddleston
Alan J. Hunter
Kevin Hwang
Brian Jack
Ian Harold Jenkins
Kurien John
Daniel Johnson
Todd Joyner
Deepa Kabirdas
Allen Kachalia
Abel Ngo Kho
Steven Jay Kravet
Marco Aurelio Ladino
Robert Lash
Joshua Lee
Sei Lee
Arthur Jefferson Lesesne
Marcia Levetown
Luci Leykum
Joshua David Liberman
Jonathan Mansbach
Brian Markoff
David Meltzer
Anna Leco Merca
Barbara Messinger‐Rapport
Gregory Misky
William Moran
Brahmajee Nallamothu
Theore Elliott Nash
Heather Nye
Timothy O'Brien
Bruce Ovbiagele
Thomas Andrew Owens
Mary Pak
Steven Zvi Pantilat
Vikas Parekh
Kimberly Rask
Michael Rothberg
Hilary F. Ryder
Wael Saber
Sanjay Saint
Rene Salazar
Kaycee Sink
N. Smith
Malathi Srinivasin
Raj Srivastava
Erin Stucky
Alexander Turchin
Bobbak Vahid
Robert Wachter
Robert L. Wears
Howard Weitz
Winthrop Whitcomb
Mark V. Williams
Sherrie Williams
David Woods
Fixed‐dose, subcutaneous, unfractionated heparin effective for VTE
-
CLINICAL QUESTION: How safe and effective is fixed‐dose subcutaneous unfractionated heparin in the treatment of venous thromboembolism?
-
BOTTOM LINE: In this study, fixed‐dose weight‐adjusted unfractionated heparin (UFH) administered subcutaneously was as safe and effective as low‐molecular‐weight heparin (LMWH) in the treatment of venous thromboembolism (VTE). Estimated drug costs for a 6‐day course are $712 for LMWH and $37 for UFH. Most clinicians will want to see similar results from at least 1 additional well‐done clinical trial, including more patients with symptomatic pulmonary embolism, before routinely treating VTE with subcutaneous UFH. (LOE = 1b)
-
REFERENCE: Kearon C, Ginsberg JS, Julian JA, et al, for the Fixed‐Dose Heparin (FIDO) Investigators. Comparison of fixed‐dose weight‐adjusted unfractionated heparin and low‐molecular‐weight heparin for acute treatment of venous thromboembolism. JAMA 2006;296:935‐942.
-
STUDY DESIGN: Randomized controlled trial (single‐blinded)
-
FUNDING: Foundation
-
SETTING: Outpatient (any)
-
ALLOCATION: Concealed
-
SYNOPSIS: These investigators randomly assigned (concealed allocation assignment) 708 patients, 18 years or older, with acute VTE to subcutaneous UFH (initial dose of 333 U/kg, followed by a fixed dose of 250 U/kg every 12 hours) or LMWH (dalteparin or enoxaparin, 100 IU/kg every 12 hours). The dose of subcutaneous UFH remained fixed for individual patients and was not changed during treatment as a result of anticoagulation profiles. The diagnosis of VTE included patients with acute deep vein thrombosis of the legs (81%) or symptomatic pulmonary embolism (19%). Oral warfarin was usually started on the same day as heparin in both groups and continued for a minimum of 3 months with doses adjusted to achieve an international normalized ratio (INR) of between 2.0 and 3.0. Heparin was continued for at least 5 days and until the INR was 2.0 or higher for 2 consecutive days. Individuals unaware of treatment group assignment assessed all outcomes, including study eligibility criteria. Follow‐up occurred for more than 98% of subjects for 3 months. All eligible and consenting patients underwent final data analysis. The risk of recurrent VTE in the first 3 months after treatment was not significantly different between patients in the UFH group (3.8%) and those in the LMWH group (3.4%). The risk of major bleeding during the first 10 days of treatment was also similar between the UFH group (1.1%) and LMWH group (1.4%). Approximately 70% of patients in both groups received treatment entirely out of hospital. Overall, there were 18 deaths in the UFH group and 22 deaths in the LMWH group (difference not significant). Adverse events were unrelated to whether subjects were subtherapeutic or supratherapeutic.
-
CLINICAL QUESTION: How safe and effective is fixed‐dose subcutaneous unfractionated heparin in the treatment of venous thromboembolism?
-
BOTTOM LINE: In this study, fixed‐dose weight‐adjusted unfractionated heparin (UFH) administered subcutaneously was as safe and effective as low‐molecular‐weight heparin (LMWH) in the treatment of venous thromboembolism (VTE). Estimated drug costs for a 6‐day course are $712 for LMWH and $37 for UFH. Most clinicians will want to see similar results from at least 1 additional well‐done clinical trial, including more patients with symptomatic pulmonary embolism, before routinely treating VTE with subcutaneous UFH. (LOE = 1b)
-
REFERENCE: Kearon C, Ginsberg JS, Julian JA, et al, for the Fixed‐Dose Heparin (FIDO) Investigators. Comparison of fixed‐dose weight‐adjusted unfractionated heparin and low‐molecular‐weight heparin for acute treatment of venous thromboembolism. JAMA 2006;296:935‐942.
-
STUDY DESIGN: Randomized controlled trial (single‐blinded)
-
FUNDING: Foundation
-
SETTING: Outpatient (any)
-
ALLOCATION: Concealed
-
SYNOPSIS: These investigators randomly assigned (concealed allocation assignment) 708 patients, 18 years or older, with acute VTE to subcutaneous UFH (initial dose of 333 U/kg, followed by a fixed dose of 250 U/kg every 12 hours) or LMWH (dalteparin or enoxaparin, 100 IU/kg every 12 hours). The dose of subcutaneous UFH remained fixed for individual patients and was not changed during treatment as a result of anticoagulation profiles. The diagnosis of VTE included patients with acute deep vein thrombosis of the legs (81%) or symptomatic pulmonary embolism (19%). Oral warfarin was usually started on the same day as heparin in both groups and continued for a minimum of 3 months with doses adjusted to achieve an international normalized ratio (INR) of between 2.0 and 3.0. Heparin was continued for at least 5 days and until the INR was 2.0 or higher for 2 consecutive days. Individuals unaware of treatment group assignment assessed all outcomes, including study eligibility criteria. Follow‐up occurred for more than 98% of subjects for 3 months. All eligible and consenting patients underwent final data analysis. The risk of recurrent VTE in the first 3 months after treatment was not significantly different between patients in the UFH group (3.8%) and those in the LMWH group (3.4%). The risk of major bleeding during the first 10 days of treatment was also similar between the UFH group (1.1%) and LMWH group (1.4%). Approximately 70% of patients in both groups received treatment entirely out of hospital. Overall, there were 18 deaths in the UFH group and 22 deaths in the LMWH group (difference not significant). Adverse events were unrelated to whether subjects were subtherapeutic or supratherapeutic.
-
CLINICAL QUESTION: How safe and effective is fixed‐dose subcutaneous unfractionated heparin in the treatment of venous thromboembolism?
-
BOTTOM LINE: In this study, fixed‐dose weight‐adjusted unfractionated heparin (UFH) administered subcutaneously was as safe and effective as low‐molecular‐weight heparin (LMWH) in the treatment of venous thromboembolism (VTE). Estimated drug costs for a 6‐day course are $712 for LMWH and $37 for UFH. Most clinicians will want to see similar results from at least 1 additional well‐done clinical trial, including more patients with symptomatic pulmonary embolism, before routinely treating VTE with subcutaneous UFH. (LOE = 1b)
-
REFERENCE: Kearon C, Ginsberg JS, Julian JA, et al, for the Fixed‐Dose Heparin (FIDO) Investigators. Comparison of fixed‐dose weight‐adjusted unfractionated heparin and low‐molecular‐weight heparin for acute treatment of venous thromboembolism. JAMA 2006;296:935‐942.
-
STUDY DESIGN: Randomized controlled trial (single‐blinded)
-
FUNDING: Foundation
-
SETTING: Outpatient (any)
-
ALLOCATION: Concealed
-
SYNOPSIS: These investigators randomly assigned (concealed allocation assignment) 708 patients, 18 years or older, with acute VTE to subcutaneous UFH (initial dose of 333 U/kg, followed by a fixed dose of 250 U/kg every 12 hours) or LMWH (dalteparin or enoxaparin, 100 IU/kg every 12 hours). The dose of subcutaneous UFH remained fixed for individual patients and was not changed during treatment as a result of anticoagulation profiles. The diagnosis of VTE included patients with acute deep vein thrombosis of the legs (81%) or symptomatic pulmonary embolism (19%). Oral warfarin was usually started on the same day as heparin in both groups and continued for a minimum of 3 months with doses adjusted to achieve an international normalized ratio (INR) of between 2.0 and 3.0. Heparin was continued for at least 5 days and until the INR was 2.0 or higher for 2 consecutive days. Individuals unaware of treatment group assignment assessed all outcomes, including study eligibility criteria. Follow‐up occurred for more than 98% of subjects for 3 months. All eligible and consenting patients underwent final data analysis. The risk of recurrent VTE in the first 3 months after treatment was not significantly different between patients in the UFH group (3.8%) and those in the LMWH group (3.4%). The risk of major bleeding during the first 10 days of treatment was also similar between the UFH group (1.1%) and LMWH group (1.4%). Approximately 70% of patients in both groups received treatment entirely out of hospital. Overall, there were 18 deaths in the UFH group and 22 deaths in the LMWH group (difference not significant). Adverse events were unrelated to whether subjects were subtherapeutic or supratherapeutic.
Disseminated histoplasmosis
A previously healthy 27‐year‐old El Salvadoran immigrant presented with a 2‐week history of cough, fever, rigors, prostration, anorexia, weight loss, and scant hemoptysis. Physical examination revealed a thin, febrile, toxic‐appearing man in respiratory distress with bibasilar rales and scattered wheezes. Laboratory data showed a sodium of 126 mEq/L, lactate dehydrogenase of 617 U/L, ferritin of 3570 ng/mL, and liver test abnormalities suggestive of cholestasis. Chest film (Fig. 1) and computed tomography (Fig. 2) demonstrated a diffuse miliary air space pattern. Sputum smears for mycobacterium tuberculosis were negative. A urine histoplasmosis antigen level was markedly positive (7.6 EIA units), and bone marrow cultures eventually grew Histoplasma capsulatum. The HIV test result was positive, and his CD4 count was 34 cells/mm3. He was successfully treated with liposomal amphotericin B followed by itraconazole.
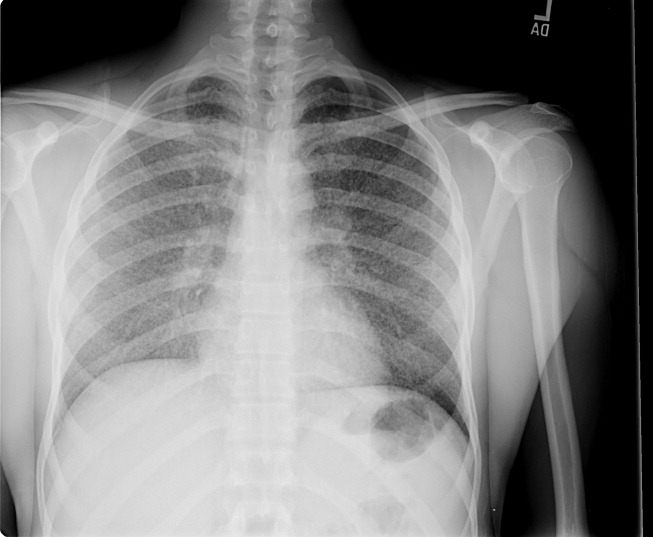
Histoplasmosis is the most prevalent endemic mycosis in Latin America. Most infections are asymptomatic or self‐limited, but immunodeficient individuals may develop acute pulmonary or severe, progressive disseminated infection, usually from reactivation of latent disease. Although nonspecific, the serum lactate dehydrogenase and ferritin levels are often markedly elevated. Chest imaging may be normal or show a diffuse reticulonodular pattern (with nodules less than 3 mm in diameter), indistinguishable from miliary tuberculosis. In HIV‐infected individuals, disseminated histoplasmosis usually develops when the CD4 count is less than 75 cells/mm3. Treatment is generally lifelong.
A previously healthy 27‐year‐old El Salvadoran immigrant presented with a 2‐week history of cough, fever, rigors, prostration, anorexia, weight loss, and scant hemoptysis. Physical examination revealed a thin, febrile, toxic‐appearing man in respiratory distress with bibasilar rales and scattered wheezes. Laboratory data showed a sodium of 126 mEq/L, lactate dehydrogenase of 617 U/L, ferritin of 3570 ng/mL, and liver test abnormalities suggestive of cholestasis. Chest film (Fig. 1) and computed tomography (Fig. 2) demonstrated a diffuse miliary air space pattern. Sputum smears for mycobacterium tuberculosis were negative. A urine histoplasmosis antigen level was markedly positive (7.6 EIA units), and bone marrow cultures eventually grew Histoplasma capsulatum. The HIV test result was positive, and his CD4 count was 34 cells/mm3. He was successfully treated with liposomal amphotericin B followed by itraconazole.
Histoplasmosis is the most prevalent endemic mycosis in Latin America. Most infections are asymptomatic or self‐limited, but immunodeficient individuals may develop acute pulmonary or severe, progressive disseminated infection, usually from reactivation of latent disease. Although nonspecific, the serum lactate dehydrogenase and ferritin levels are often markedly elevated. Chest imaging may be normal or show a diffuse reticulonodular pattern (with nodules less than 3 mm in diameter), indistinguishable from miliary tuberculosis. In HIV‐infected individuals, disseminated histoplasmosis usually develops when the CD4 count is less than 75 cells/mm3. Treatment is generally lifelong.
A previously healthy 27‐year‐old El Salvadoran immigrant presented with a 2‐week history of cough, fever, rigors, prostration, anorexia, weight loss, and scant hemoptysis. Physical examination revealed a thin, febrile, toxic‐appearing man in respiratory distress with bibasilar rales and scattered wheezes. Laboratory data showed a sodium of 126 mEq/L, lactate dehydrogenase of 617 U/L, ferritin of 3570 ng/mL, and liver test abnormalities suggestive of cholestasis. Chest film (Fig. 1) and computed tomography (Fig. 2) demonstrated a diffuse miliary air space pattern. Sputum smears for mycobacterium tuberculosis were negative. A urine histoplasmosis antigen level was markedly positive (7.6 EIA units), and bone marrow cultures eventually grew Histoplasma capsulatum. The HIV test result was positive, and his CD4 count was 34 cells/mm3. He was successfully treated with liposomal amphotericin B followed by itraconazole.
Histoplasmosis is the most prevalent endemic mycosis in Latin America. Most infections are asymptomatic or self‐limited, but immunodeficient individuals may develop acute pulmonary or severe, progressive disseminated infection, usually from reactivation of latent disease. Although nonspecific, the serum lactate dehydrogenase and ferritin levels are often markedly elevated. Chest imaging may be normal or show a diffuse reticulonodular pattern (with nodules less than 3 mm in diameter), indistinguishable from miliary tuberculosis. In HIV‐infected individuals, disseminated histoplasmosis usually develops when the CD4 count is less than 75 cells/mm3. Treatment is generally lifelong.
At Home

It was 8 p.m. on a cold Minnesota night, and my vision was obscured by flurries of snow and decomposing wiper blades. I was late for home. When I arrived, 11 sets of eyes turned toward me. (Twelve, really, counting my boxer Chocky Locky). They were sitting in the dining room, and the evening meal had just started. I washed quickly and took my seat at the table.
We have a large dining table that seats 12, but our current dish service—due to an unforeseen disaster—has only 11 settings. Thus, our service was at its maximum capacity. Tonight’s crowd included my wife and me, a grandmother, four offspring of various genders and a female teenager of unknown origin, a male preteen well-known to the household, and two young females who had sat at the table before.
As per standard operating procedure, each person at the table began to speak at the same time. Eventually order was established, though this was at best a transitory phenomenon. We received reports on each participant’s day, with highlights of lunch hour mayhem, recess riots, and general curricular boredom.
I began to question the unknown teen: name, age, place of origin, habits, and so on, but my history taking was interrupted. My younger son wanted to relay the results of an important test he had taken. He had passed and was now certified to use punctuation. I turned to resume my history taking, but made the important physical exam observation of intense eye rolling on the part of my daughter and her friend. This is a well-known physical finding in this age group and one that generally signals a pre-seizure threshold that I did not want to further induce.
After an intense nutritional session that included all major food groups and several minor ones, there were several short, unscheduled presentations. The grandmother gave a long and interesting family history with highlights of a great-great grandfather, who had been a freelance horse thief for both the Polish and Russian armies, and his son, who had been—alternatively—a gambler, a rabbi, a communist, and a union organizer.
After this history lesson, we received a fascinating report from one of my male offspring entitled, “proper placement of the hand and axillae, combined with repetitive flapping movements of the arm, to elicit an auditory stimulus similar to flatulence.” Much hilarity ensued.

It was unclear whose turn it was to clear the table. The schedule was not available, and several of the offspring cited work limits they would hate to see abused. Eventually the job was done with only minimal threats of withdrawal of privileges. As I prepared to resume my reading, a call went out for transportation services. It was time to discharge one of the visiting children to her abode. I was happy to decrease the numbers in house, though I would have been happier to see our numbers go even lower. Our length of stay seemed to be rising daily. As I attempted to initiate the transportation home, I realized we had to go through the checklist. Do you have your scarf, your gloves, and your shoes? Did you have a good time? I considered a policy of no readmission in 30 days, but it was voted down in a team meeting.
I returned from transportation duties, and I sat quietly for a moment and looked at the Times and the Post-Bulletin. These were papers I had been waiting to review—especially the comics. But something always seems to come up when one has papers to review. My youngest daughter and her friends needed my guidance on an art project. I had hoped they would see one, do one, teach one, but I had to repeatedly sketch the face outlines for them to color.
As I sat down again to the papers, the doorbell rang. Nobody else appeared to be on doorbell duty, so I went to the front of the house. There was a young man in a white shirt and black tie. He was proselytizing for a religious group. I informed him that our house had adopted a “no religious rep policy.” This helps us to avoid being unduly influenced by reps, though I must admit I still use my Taoist pen. It’s a Zen-Pen: one side writes, the other doesn’t.
Finally, I returned to my seat. At that point, the room plunged into darkness. A moment of panic. Quickly, I took my own pulse, a technique I had learned from the medical literature. Once calmed, I went through the differential. The power might be out for the whole city or just the house. Perhaps a fuse had blown from one too many electronic entertainment devices. Or maybe mice had chewed through a power cable. As I pondered my next course of diagnostic action and reviewed my alternatives in an evidence-based and allorhythmic approach, I auscultated a series of breath sounds that might be interpreted as gasps or giggles.
I intuitively realized that it was a severe case of “little girls playing a joke on Dad by switching off the light.” I had two choices. The first was to be crabby and bellow for them to quit playing with the switch. I felt this to be a harsh choice with bad potential side effects. Instead, I ducked under the table and silently hid. When proper lighting was restored, they were amazed to see I had vanished, and when they came to investigate, I revealed myself and uttered the key phrase: “Boo!” A riotous wrestling match ensued, which led to the injury of a family heirloom and my spine.
It was time for bed, despite my elder son’s complaint of a type of chronic insomnia only remediable by late-night cartoon observation. Evening reading began. Such important journals as Click Clack Moo, P.J. Funnybunny Camps Out, and the Stinky Cheese Man and Other Fairly Stupid Tales were on the agenda.
As I drifted off to sleep, the phone rang. It was a wrong number. Several hours later, an emergency call for supplemental H2O was answered. Two hours after that, I awoke to a hideous scream; it was a nightmare. My alarm rang at 6:15, and I jumped from bed refreshed. No, that’s a lie. I stumbled from bed after hitting the snooze button four times. Morning nutrition rounds were a stale toaster pastry and coffee. Team Newman noted that there were last-minute reports not finished for school and preparation for a day of testing.
I drove to work exhausted after a night of Home. TH
Dr. Newman is the physician editor of The Hospitalist. He’s also consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine, Rochester, Minn.
It was 8 p.m. on a cold Minnesota night, and my vision was obscured by flurries of snow and decomposing wiper blades. I was late for home. When I arrived, 11 sets of eyes turned toward me. (Twelve, really, counting my boxer Chocky Locky). They were sitting in the dining room, and the evening meal had just started. I washed quickly and took my seat at the table.
We have a large dining table that seats 12, but our current dish service—due to an unforeseen disaster—has only 11 settings. Thus, our service was at its maximum capacity. Tonight’s crowd included my wife and me, a grandmother, four offspring of various genders and a female teenager of unknown origin, a male preteen well-known to the household, and two young females who had sat at the table before.
As per standard operating procedure, each person at the table began to speak at the same time. Eventually order was established, though this was at best a transitory phenomenon. We received reports on each participant’s day, with highlights of lunch hour mayhem, recess riots, and general curricular boredom.
I began to question the unknown teen: name, age, place of origin, habits, and so on, but my history taking was interrupted. My younger son wanted to relay the results of an important test he had taken. He had passed and was now certified to use punctuation. I turned to resume my history taking, but made the important physical exam observation of intense eye rolling on the part of my daughter and her friend. This is a well-known physical finding in this age group and one that generally signals a pre-seizure threshold that I did not want to further induce.
After an intense nutritional session that included all major food groups and several minor ones, there were several short, unscheduled presentations. The grandmother gave a long and interesting family history with highlights of a great-great grandfather, who had been a freelance horse thief for both the Polish and Russian armies, and his son, who had been—alternatively—a gambler, a rabbi, a communist, and a union organizer.
After this history lesson, we received a fascinating report from one of my male offspring entitled, “proper placement of the hand and axillae, combined with repetitive flapping movements of the arm, to elicit an auditory stimulus similar to flatulence.” Much hilarity ensued.
It was unclear whose turn it was to clear the table. The schedule was not available, and several of the offspring cited work limits they would hate to see abused. Eventually the job was done with only minimal threats of withdrawal of privileges. As I prepared to resume my reading, a call went out for transportation services. It was time to discharge one of the visiting children to her abode. I was happy to decrease the numbers in house, though I would have been happier to see our numbers go even lower. Our length of stay seemed to be rising daily. As I attempted to initiate the transportation home, I realized we had to go through the checklist. Do you have your scarf, your gloves, and your shoes? Did you have a good time? I considered a policy of no readmission in 30 days, but it was voted down in a team meeting.
I returned from transportation duties, and I sat quietly for a moment and looked at the Times and the Post-Bulletin. These were papers I had been waiting to review—especially the comics. But something always seems to come up when one has papers to review. My youngest daughter and her friends needed my guidance on an art project. I had hoped they would see one, do one, teach one, but I had to repeatedly sketch the face outlines for them to color.
As I sat down again to the papers, the doorbell rang. Nobody else appeared to be on doorbell duty, so I went to the front of the house. There was a young man in a white shirt and black tie. He was proselytizing for a religious group. I informed him that our house had adopted a “no religious rep policy.” This helps us to avoid being unduly influenced by reps, though I must admit I still use my Taoist pen. It’s a Zen-Pen: one side writes, the other doesn’t.
Finally, I returned to my seat. At that point, the room plunged into darkness. A moment of panic. Quickly, I took my own pulse, a technique I had learned from the medical literature. Once calmed, I went through the differential. The power might be out for the whole city or just the house. Perhaps a fuse had blown from one too many electronic entertainment devices. Or maybe mice had chewed through a power cable. As I pondered my next course of diagnostic action and reviewed my alternatives in an evidence-based and allorhythmic approach, I auscultated a series of breath sounds that might be interpreted as gasps or giggles.
I intuitively realized that it was a severe case of “little girls playing a joke on Dad by switching off the light.” I had two choices. The first was to be crabby and bellow for them to quit playing with the switch. I felt this to be a harsh choice with bad potential side effects. Instead, I ducked under the table and silently hid. When proper lighting was restored, they were amazed to see I had vanished, and when they came to investigate, I revealed myself and uttered the key phrase: “Boo!” A riotous wrestling match ensued, which led to the injury of a family heirloom and my spine.
It was time for bed, despite my elder son’s complaint of a type of chronic insomnia only remediable by late-night cartoon observation. Evening reading began. Such important journals as Click Clack Moo, P.J. Funnybunny Camps Out, and the Stinky Cheese Man and Other Fairly Stupid Tales were on the agenda.
As I drifted off to sleep, the phone rang. It was a wrong number. Several hours later, an emergency call for supplemental H2O was answered. Two hours after that, I awoke to a hideous scream; it was a nightmare. My alarm rang at 6:15, and I jumped from bed refreshed. No, that’s a lie. I stumbled from bed after hitting the snooze button four times. Morning nutrition rounds were a stale toaster pastry and coffee. Team Newman noted that there were last-minute reports not finished for school and preparation for a day of testing.
I drove to work exhausted after a night of Home. TH
Dr. Newman is the physician editor of The Hospitalist. He’s also consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine, Rochester, Minn.
It was 8 p.m. on a cold Minnesota night, and my vision was obscured by flurries of snow and decomposing wiper blades. I was late for home. When I arrived, 11 sets of eyes turned toward me. (Twelve, really, counting my boxer Chocky Locky). They were sitting in the dining room, and the evening meal had just started. I washed quickly and took my seat at the table.
We have a large dining table that seats 12, but our current dish service—due to an unforeseen disaster—has only 11 settings. Thus, our service was at its maximum capacity. Tonight’s crowd included my wife and me, a grandmother, four offspring of various genders and a female teenager of unknown origin, a male preteen well-known to the household, and two young females who had sat at the table before.
As per standard operating procedure, each person at the table began to speak at the same time. Eventually order was established, though this was at best a transitory phenomenon. We received reports on each participant’s day, with highlights of lunch hour mayhem, recess riots, and general curricular boredom.
I began to question the unknown teen: name, age, place of origin, habits, and so on, but my history taking was interrupted. My younger son wanted to relay the results of an important test he had taken. He had passed and was now certified to use punctuation. I turned to resume my history taking, but made the important physical exam observation of intense eye rolling on the part of my daughter and her friend. This is a well-known physical finding in this age group and one that generally signals a pre-seizure threshold that I did not want to further induce.
After an intense nutritional session that included all major food groups and several minor ones, there were several short, unscheduled presentations. The grandmother gave a long and interesting family history with highlights of a great-great grandfather, who had been a freelance horse thief for both the Polish and Russian armies, and his son, who had been—alternatively—a gambler, a rabbi, a communist, and a union organizer.
After this history lesson, we received a fascinating report from one of my male offspring entitled, “proper placement of the hand and axillae, combined with repetitive flapping movements of the arm, to elicit an auditory stimulus similar to flatulence.” Much hilarity ensued.
It was unclear whose turn it was to clear the table. The schedule was not available, and several of the offspring cited work limits they would hate to see abused. Eventually the job was done with only minimal threats of withdrawal of privileges. As I prepared to resume my reading, a call went out for transportation services. It was time to discharge one of the visiting children to her abode. I was happy to decrease the numbers in house, though I would have been happier to see our numbers go even lower. Our length of stay seemed to be rising daily. As I attempted to initiate the transportation home, I realized we had to go through the checklist. Do you have your scarf, your gloves, and your shoes? Did you have a good time? I considered a policy of no readmission in 30 days, but it was voted down in a team meeting.
I returned from transportation duties, and I sat quietly for a moment and looked at the Times and the Post-Bulletin. These were papers I had been waiting to review—especially the comics. But something always seems to come up when one has papers to review. My youngest daughter and her friends needed my guidance on an art project. I had hoped they would see one, do one, teach one, but I had to repeatedly sketch the face outlines for them to color.
As I sat down again to the papers, the doorbell rang. Nobody else appeared to be on doorbell duty, so I went to the front of the house. There was a young man in a white shirt and black tie. He was proselytizing for a religious group. I informed him that our house had adopted a “no religious rep policy.” This helps us to avoid being unduly influenced by reps, though I must admit I still use my Taoist pen. It’s a Zen-Pen: one side writes, the other doesn’t.
Finally, I returned to my seat. At that point, the room plunged into darkness. A moment of panic. Quickly, I took my own pulse, a technique I had learned from the medical literature. Once calmed, I went through the differential. The power might be out for the whole city or just the house. Perhaps a fuse had blown from one too many electronic entertainment devices. Or maybe mice had chewed through a power cable. As I pondered my next course of diagnostic action and reviewed my alternatives in an evidence-based and allorhythmic approach, I auscultated a series of breath sounds that might be interpreted as gasps or giggles.
I intuitively realized that it was a severe case of “little girls playing a joke on Dad by switching off the light.” I had two choices. The first was to be crabby and bellow for them to quit playing with the switch. I felt this to be a harsh choice with bad potential side effects. Instead, I ducked under the table and silently hid. When proper lighting was restored, they were amazed to see I had vanished, and when they came to investigate, I revealed myself and uttered the key phrase: “Boo!” A riotous wrestling match ensued, which led to the injury of a family heirloom and my spine.
It was time for bed, despite my elder son’s complaint of a type of chronic insomnia only remediable by late-night cartoon observation. Evening reading began. Such important journals as Click Clack Moo, P.J. Funnybunny Camps Out, and the Stinky Cheese Man and Other Fairly Stupid Tales were on the agenda.
As I drifted off to sleep, the phone rang. It was a wrong number. Several hours later, an emergency call for supplemental H2O was answered. Two hours after that, I awoke to a hideous scream; it was a nightmare. My alarm rang at 6:15, and I jumped from bed refreshed. No, that’s a lie. I stumbled from bed after hitting the snooze button four times. Morning nutrition rounds were a stale toaster pastry and coffee. Team Newman noted that there were last-minute reports not finished for school and preparation for a day of testing.
I drove to work exhausted after a night of Home. TH
Dr. Newman is the physician editor of The Hospitalist. He’s also consultant, Hospital Internal Medicine, and assistant professor of internal medicine and medical history, Mayo Clinic College of Medicine, Rochester, Minn.