User login
How to explain physician compounding to legislators
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com
This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com
This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
In Ohio, new limits on drug compounding in physicians’ offices went into effect in April and have become a real hindrance to care for dermatology patients. The State of Ohio Board of Pharmacy has defined compounding as combining two or more prescription drugs and has required that physicians who perform this “compounding” must obtain a “Terminal Distributor of Dangerous Drugs” license. Ohio is the “test state,” and these rules, unless vigorously opposed, will be coming to your state.
[polldaddy:9779752]
The rules state that “compounded” drugs used within 6 hours of preparation must be prepared in a designated clean medication area with proper hand hygiene and the use of powder-free gloves. “Compounded” drugs that are used more than 6 hours after preparation, require a designated clean room with access limited to authorized personnel, environmental control devices such as a laminar flow hood, and additional equipment and training of personnel to maintain an aseptic environment. A separate license is required for each office location.
The state pharmacy boards are eager to restrict physicians – as well as dentists and veterinarians – and to collect annual licensing fees. Additionally, according to an article from the Ohio State Medical Association, noncompliant physicians can be fined by the pharmacy board.
We are talking big money, power, and dreams of clinical relevancy (and billable activities) here.
What can dermatologists do to prevent this regulatory overreach? I encourage you to plan a visit to your state representative, where you can demonstrate how these restrictions affect you and your patients – an exercise that should be both fun and compelling. All you need to illustrate your case is a simple kit that includes a syringe (but no needles in the statehouse!), a bottle of lidocaine with epinephrine, a bottle of 8.4% bicarbonate, alcohol pads, and gloves.
First, explain to your audience that there is a skin cancer epidemic with more than 5.4 million new cases a year and that, over the past 20 years, the incidence of skin cancer has doubled and is projected to double again over the next 20 years. Further, explain that dermatologists treat more than 70% of these cases in the office setting, under local anesthesia, at a huge cost savings to the public and government (it costs an average of 12 times as much to remove these cancers in the outpatient department at the hospital). Remember, states foot most of the bill for Medicaid and Medicare gap indigent coverage.
Take the bottle of lidocaine with epinephrine and open the syringe pack (Staffers love this demonstration; everyone is fascinated with shots.). Put on your gloves, wipe the top of the lidocaine bottle with an alcohol swab, and explain that this medicine is the anesthetic preferred for skin cancer surgery. Explain how it not only numbs the skin, but also causes vasoconstriction, so that the cancer can be easily and safely removed in the office.
Then explain that, in order for the epinephrine to be stable, the solution has to be very acidic (a pH of 4.2, in fact). Explain that this makes it burn like hell unless you add 0.1 cc per cc of 8.4% bicarbonate, in which case the perceived pain on a 10-point scale will drop from 8 to 2. Then pick up the bottle of bicarbonate and explain that you will no longer be able to mix these two components anymore without a “Terminal Distributor of Dangerous Drugs” license because your state pharmacy board considers this compounding. Your representative is likely to give you looks of astonishment, disbelief, and then a dawning realization of the absurdity of the situation.
Follow-up questions may include “Why can’t you buy buffered lidocaine with epinephrine from the compounding pharmacy?” Easy answer: because each patient needs an individual prescription, and you may not know in advance which patient will need it, and how much the patient will need, and it becomes unstable once it has been buffered. It also will cost the patient $45 per 5-cc syringe, and it will be degraded by the time the patient returns from the compounding pharmacy. Explain further that it costs you only 84 cents to make a 5-cc syringe of buffered lidocaine; that some patients may need as many as 10 syringes; and that these costs are all included in the surgery (free!) if the physician draws it up in the office.
A simple summary is – less pain, less cost – and no history of infections or complications.
It is an eye-opener when you demonstrate how ridiculous the compounding rules being imposed are for physicians and patients. I’ve used this demonstration at the state and federal legislative level, and more recently, at the Food and Drug Administration.
If you get the chance, when a state legislator is in your office, become an advocate for your patients and fellow physicians. Make sure physician offices are excluded from these definitions of com
This column was updated June 22, 2017.
Dr. Coldiron is in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. He is a past president of the American Academy of Dermatology. Write to him at dermnews@frontlinemedcom.com.
Mepolizumab improves asthma after 1 year despite comorbidities
Adults with asthma who were newly prescribed mepolizumab showed significant improvement in symptoms after 1 year regardless of comorbidities, based on data from 822 individuals.
Comorbidities including chronic rhinosinusitis with polyps (CRSwNP), gastroesophageal reflux disease GERD), anxiety and depression, and chronic obstructive pulmonary disorder (COPD) are common in patients with severe asthma and add to the disease burden, wrote Mark C. Liu, MD, of Johns Hopkins University, Baltimore, and colleagues.
“Some comorbidities, such as CRSwNP, share pathophysiological mechanisms with severe asthma, with interleukin-5 (IL-5),” and treatments targeting IL-5 could improve outcomes, they said.
In the real-world REALITI-A study, mepolizumab, a humanized monoclonal antibody that targets IL-5, significantly reduced asthma exacerbation and oral corticosteroid use in severe asthma patients, they said.
To assess the impact of mepolizumab on patients with comorbidities, the researchers conducted a post hoc analysis of 822 adults with severe asthma, including 321 with CRSwNP, 309 with GERD, 203 with depression/anxiety, and 81 with COPD. The findings were published in the Journal of Allergy and Clinical Immunology: In Practice.
The main outcomes were the rate of clinically significant asthma exacerbations (CSEs) between the 12 months before and after mepolizumab initiation, and the changes from baseline in the daily maintenance use of oral corticosteroids (OCS).
Across all comorbidities, the rate of CSEs decreased significantly from the pretreatment period to the follow-up period, from 4.28 events per year to 1.23 events per year.
“A numerically greater reduction in the rate of CSEs was reported for patients with versus without CRSwNP, whereas the reverse was reported for patients with versus without COPD and depression/anxiety, although the confidence intervals were large for the with COPD subgroup,” the researchers wrote.
The median maintenance dose of oral corticosteroids decreased by at least 50% across all comorbidities after mepolizumab treatment; patients with CRSwNP had the greatest reduction (83%).
In addition, scores on the Asthma Control Questionnaire–5 decreased by at least 0.63 points, and least squared (LS) mean changes in forced expiratory volume per second (FEV1) increased from baseline across all comorbidities after mepolizumab treatment by at least 74 mL.
Although patients with versus without CRSwNP had greater improvements, patients without GERD, depression/anxiety, and COPD had greater improvements than did those without the respective conditions with the exception of greater FEV1 improvement in patients with vs. without COPD.
“Patients with severe asthma and comorbid CRSwNP are recognized as having a high disease burden, as demonstrated by more frequent exacerbations,” the researchers wrote in their discussion. “Mepolizumab may serve to reduce the disease burden of this high-risk group by targeting the common pathophysiological pathway of IL-5 and eosinophilic-driven inflammation because it has proven clinical benefits in treating asthma and CRSwNP separately and together,” and the current study findings support the use of mepolizumab for this population in particular, they said.
The findings were limited by several factors including the incomplete data for voluntary assessments, the post hoc design and relatively small numbers of patients in various subgroups, notably COPD, and the potential inaccurate diagnosis of COPD, the researchers noted.
“Nevertheless, because the amount of improvement in each outcome following mepolizumab treatment differed depending on the comorbidity in question, our findings highlight the impact that comorbidities and their prevalence and severity have on outcomes,” and the overall success of mepolizumab across clinical characteristics and comorbidities supports the generalizability of the findings to the larger population of adults with severe asthma, they concluded.
The study was supported by GlaxoSmithKline. Dr. Liu disclosed research funding from GSK, Boehringer Ingelheim, and Gossamer Bio, and participation on advisory boards for AstraZeneca, GSK, and Gossamer Bio.
Adults with asthma who were newly prescribed mepolizumab showed significant improvement in symptoms after 1 year regardless of comorbidities, based on data from 822 individuals.
Comorbidities including chronic rhinosinusitis with polyps (CRSwNP), gastroesophageal reflux disease GERD), anxiety and depression, and chronic obstructive pulmonary disorder (COPD) are common in patients with severe asthma and add to the disease burden, wrote Mark C. Liu, MD, of Johns Hopkins University, Baltimore, and colleagues.
“Some comorbidities, such as CRSwNP, share pathophysiological mechanisms with severe asthma, with interleukin-5 (IL-5),” and treatments targeting IL-5 could improve outcomes, they said.
In the real-world REALITI-A study, mepolizumab, a humanized monoclonal antibody that targets IL-5, significantly reduced asthma exacerbation and oral corticosteroid use in severe asthma patients, they said.
To assess the impact of mepolizumab on patients with comorbidities, the researchers conducted a post hoc analysis of 822 adults with severe asthma, including 321 with CRSwNP, 309 with GERD, 203 with depression/anxiety, and 81 with COPD. The findings were published in the Journal of Allergy and Clinical Immunology: In Practice.
The main outcomes were the rate of clinically significant asthma exacerbations (CSEs) between the 12 months before and after mepolizumab initiation, and the changes from baseline in the daily maintenance use of oral corticosteroids (OCS).
Across all comorbidities, the rate of CSEs decreased significantly from the pretreatment period to the follow-up period, from 4.28 events per year to 1.23 events per year.
“A numerically greater reduction in the rate of CSEs was reported for patients with versus without CRSwNP, whereas the reverse was reported for patients with versus without COPD and depression/anxiety, although the confidence intervals were large for the with COPD subgroup,” the researchers wrote.
The median maintenance dose of oral corticosteroids decreased by at least 50% across all comorbidities after mepolizumab treatment; patients with CRSwNP had the greatest reduction (83%).
In addition, scores on the Asthma Control Questionnaire–5 decreased by at least 0.63 points, and least squared (LS) mean changes in forced expiratory volume per second (FEV1) increased from baseline across all comorbidities after mepolizumab treatment by at least 74 mL.
Although patients with versus without CRSwNP had greater improvements, patients without GERD, depression/anxiety, and COPD had greater improvements than did those without the respective conditions with the exception of greater FEV1 improvement in patients with vs. without COPD.
“Patients with severe asthma and comorbid CRSwNP are recognized as having a high disease burden, as demonstrated by more frequent exacerbations,” the researchers wrote in their discussion. “Mepolizumab may serve to reduce the disease burden of this high-risk group by targeting the common pathophysiological pathway of IL-5 and eosinophilic-driven inflammation because it has proven clinical benefits in treating asthma and CRSwNP separately and together,” and the current study findings support the use of mepolizumab for this population in particular, they said.
The findings were limited by several factors including the incomplete data for voluntary assessments, the post hoc design and relatively small numbers of patients in various subgroups, notably COPD, and the potential inaccurate diagnosis of COPD, the researchers noted.
“Nevertheless, because the amount of improvement in each outcome following mepolizumab treatment differed depending on the comorbidity in question, our findings highlight the impact that comorbidities and their prevalence and severity have on outcomes,” and the overall success of mepolizumab across clinical characteristics and comorbidities supports the generalizability of the findings to the larger population of adults with severe asthma, they concluded.
The study was supported by GlaxoSmithKline. Dr. Liu disclosed research funding from GSK, Boehringer Ingelheim, and Gossamer Bio, and participation on advisory boards for AstraZeneca, GSK, and Gossamer Bio.
Adults with asthma who were newly prescribed mepolizumab showed significant improvement in symptoms after 1 year regardless of comorbidities, based on data from 822 individuals.
Comorbidities including chronic rhinosinusitis with polyps (CRSwNP), gastroesophageal reflux disease GERD), anxiety and depression, and chronic obstructive pulmonary disorder (COPD) are common in patients with severe asthma and add to the disease burden, wrote Mark C. Liu, MD, of Johns Hopkins University, Baltimore, and colleagues.
“Some comorbidities, such as CRSwNP, share pathophysiological mechanisms with severe asthma, with interleukin-5 (IL-5),” and treatments targeting IL-5 could improve outcomes, they said.
In the real-world REALITI-A study, mepolizumab, a humanized monoclonal antibody that targets IL-5, significantly reduced asthma exacerbation and oral corticosteroid use in severe asthma patients, they said.
To assess the impact of mepolizumab on patients with comorbidities, the researchers conducted a post hoc analysis of 822 adults with severe asthma, including 321 with CRSwNP, 309 with GERD, 203 with depression/anxiety, and 81 with COPD. The findings were published in the Journal of Allergy and Clinical Immunology: In Practice.
The main outcomes were the rate of clinically significant asthma exacerbations (CSEs) between the 12 months before and after mepolizumab initiation, and the changes from baseline in the daily maintenance use of oral corticosteroids (OCS).
Across all comorbidities, the rate of CSEs decreased significantly from the pretreatment period to the follow-up period, from 4.28 events per year to 1.23 events per year.
“A numerically greater reduction in the rate of CSEs was reported for patients with versus without CRSwNP, whereas the reverse was reported for patients with versus without COPD and depression/anxiety, although the confidence intervals were large for the with COPD subgroup,” the researchers wrote.
The median maintenance dose of oral corticosteroids decreased by at least 50% across all comorbidities after mepolizumab treatment; patients with CRSwNP had the greatest reduction (83%).
In addition, scores on the Asthma Control Questionnaire–5 decreased by at least 0.63 points, and least squared (LS) mean changes in forced expiratory volume per second (FEV1) increased from baseline across all comorbidities after mepolizumab treatment by at least 74 mL.
Although patients with versus without CRSwNP had greater improvements, patients without GERD, depression/anxiety, and COPD had greater improvements than did those without the respective conditions with the exception of greater FEV1 improvement in patients with vs. without COPD.
“Patients with severe asthma and comorbid CRSwNP are recognized as having a high disease burden, as demonstrated by more frequent exacerbations,” the researchers wrote in their discussion. “Mepolizumab may serve to reduce the disease burden of this high-risk group by targeting the common pathophysiological pathway of IL-5 and eosinophilic-driven inflammation because it has proven clinical benefits in treating asthma and CRSwNP separately and together,” and the current study findings support the use of mepolizumab for this population in particular, they said.
The findings were limited by several factors including the incomplete data for voluntary assessments, the post hoc design and relatively small numbers of patients in various subgroups, notably COPD, and the potential inaccurate diagnosis of COPD, the researchers noted.
“Nevertheless, because the amount of improvement in each outcome following mepolizumab treatment differed depending on the comorbidity in question, our findings highlight the impact that comorbidities and their prevalence and severity have on outcomes,” and the overall success of mepolizumab across clinical characteristics and comorbidities supports the generalizability of the findings to the larger population of adults with severe asthma, they concluded.
The study was supported by GlaxoSmithKline. Dr. Liu disclosed research funding from GSK, Boehringer Ingelheim, and Gossamer Bio, and participation on advisory boards for AstraZeneca, GSK, and Gossamer Bio.
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY: IN PRACTICE
FDA, FTC uniting to promote biosimilars
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.

The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.
“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.
“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
The Food and Drug Administration is collaborating with the Federal Trade Commission (FTC) to expand the biosimilars market.
The two agencies signed a joint statement on Feb. 3, 2020, outlining four sets of goals aimed at creating meaningful competition from biosimilars against their reference biologic products.
“Competition is key for helping American patients have access to affordable medicines,” FDA Commissioner Stephen Hahn, MD, said in a statement. “Strengthening efforts to curtail and discourage anticompetitive behavior is key for facilitating robust competition for patients in the biologics marketplace, including through biosimilars, bringing down the costs of these crucial products for patients.”
“We appreciate and applaud the FDA and FTC in recognizing that biosimilar development and approval has not been as robust as many stakeholders had hoped,” said Colin Edgerton, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “We continue to see anticompetitive activities that prevent manufacturers from developing biosimilar products. We hope that a greater focus on these practices will pave the way for more biosimilars to be developed.”
The statement highlighted four goals. First is that the agencies will coordinate to promote greater competition in the biologic market, including the development of materials to educate the market about biosimilars. The FDA and FTC also will be sponsoring a public workshop on March 9 to discuss competition for biologics.
“This workshop is the first step,” Dr. Edgerton said. “ACR will continue to work with other organizations and patient groups to help educate providers and patients on the scientific rigor that is required in developing and approving biosimilars. Additionally, we look forward to working with the FDA and FTC to continue this conversation on ways to encourage more development of biosimilar products and greater education for the providers and patients.”
The second goal has the FDA and FTC working together “to deter behavior that impedes access to samples needed for the development of biologics, including biosimilars,” the joint statement notes.
Third, the agencies will crack down on “false or misleading communications about biologics, including biosimilars, within their respective authorities,” according to the joint statement.
“FDA and FTC, as authorized by their respective statutes, will work together to address false or misleading communications about biologics, including biosimilars,” the statement continues. “In particular, if a communication makes a false or misleading comparison between a reference product and a biosimilar in a manner that misrepresents the safety or efficacy of biosimilars, deceives consumers, or deters competition, FDA and FTC intend to take appropriate action within their respective authorities. FDA intends to take appropriate action to address such communications where those communications have the potential to impact public health.”
Finally, the FTC committed to review patent settlement agreements involving biologics, including biosimilars, for antitrust violations.
Dr. Edgerton highlighted why this agreement between the two agencies is so important.
“Biologics are life-changing treatments for many of our patients,” he said. “Due to the high cost of discovery and development, the cost of biologics has resulted in delayed access and financial hardships for so many. It has always been our hope that biosimilars would offer the same life-changing treatment for patients at a lower price point. A robust biosimilars market is imperative to allow greater access to these treatments that can help patients to have a better quality of life.”
Separately, the FDA issued a draft guidance document for comment on manufacturers seeking licensure of biosimilar products that do not cover all the approved uses of the reference product, as well as how to add uses over time that were not part of the initial license of the biosimilar product. The draft guidance covers licensure of products, labeling of biosimilars with fewer indications than the reference product, supplemental applications for indications not on the initial biosimilar application but covered by the reference product, and the timing of applications.
The FDA notes in the draft guidance that this is needed to cover situations such as when some indications on the reference product are covered by exclusivity, although it does encourage a biosimilar manufacturer to seek licensure for all indications that the reference product does have.
Pregnant women in clinical trials: FDA questions how to include them
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”
Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.

The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”
Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.
The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Pregnant women are rarely included in clinical drug trials, creating a significant and potentially dangerous gap in knowledge. Now, a new draft guidance from the Food and Drug Administration broadens the discussion about these trials, suggesting issues to consider – including ethics and risks – when testing medications in pregnant women.
“The guidance opens the possibility of ethical conduct of trials in pregnant women but carefully lays out the caveats to be considered,” Christina Chambers, PhD, a perinatal epidemiologist at the University of California, San Diego, said in an interview. “With proper planning and thoughtful consultation with the relevant experts, this change in regulatory limitations will benefit pregnant women and their children.”
Attitudes have evolved toward more acceptance of including pregnant women in drug trials, according to a 2015 committee opinion from the American College of Obstetricians and Gynecologists. Still, “concerns about the potential for pregnancy in research trial participants have led to practices involving overly burdensome contraception requirements,” the opinion states. “Although changes have been made to encourage and recruit more women into research studies, a gap still exists in the available data on health and disease in women, including those who are pregnant” (Obstet Gynecol 2015;126:e100-7).
[polldaddy:9979976]
The draft guidance, released April 6 by the FDA, is “intended to advance scientific research in pregnant women, and discusses issues that should be considered within the framework of human subject protection regulations,” according to posting comments in the Federal Register.
The draft notes that in some cases, the lack of data about drugs may harm pregnant women and their fetuses by leading physicians to be fearful about prescribing medication. Conversely, physicians and pregnant women are often in the dark about the risks and benefits of medications that are prescribed and used, according to the draft.
In terms of research going forward, the guidance says “development of accessible treatment options for the pregnant population is a significant public health issue.”
The guidance, which recommends that clinical trial sponsors consider enlisting ethicists to take part in drug development program, offers these guidelines, among others, to drugmakers:
- It is “ethically justifiable” to include pregnant women in clinical trials under specific circumstances. “Sponsors should consider meeting with the appropriate FDA review division early in the development phase to discuss when and how to include pregnant women in the drug development plan. These discussions should involve FDA experts in bioethics and maternal health.”
- “Pregnant women can be enrolled in clinical trials that involve greater than minimal risk to the fetuses if the trials offer the potential for direct clinical benefit to the enrolled pregnant women and/or their fetuses.”
- A new pregnancy during a randomized, blinded clinical trial should prompt unblinding “so that counseling may be offered based on whether the fetus has been exposed to the investigational drug, placebo, or control.”
- The pregnant woman may continue the trial if potential benefits outweigh the risks.
- In general, pregnant women should not be enrolled in phase 1 and phase 2 clinical trials. Instead, those trials should be completed first “in a nonpregnant population that include females of reproductive potential.”
- Several types of events may call for the cessation of a clinical trial that includes pregnant women, such as serious maternal or fetal adverse events.
The draft guidance should take note of the fact that birth defects often don’t appear for months or even longer, according to Gerald Briggs, BPharm, FCCP, clinical professor of pharmacy at the University of California, San Francisco. “Until first year of life or later, the babies need to be monitored,” he said in an interview.
Mr. Briggs, who led a 2015 report examining the role of pregnant women in phase 4 clinical drug trials, added that the document should take note of recommendations from clinical teratologists regarding the design of animal studies that should be performed prior to human trials (Am J Obstet Gynecol. 2015;213(6):810-5).
Comments on the draft guidance can be made at www.federalregister.gov and are due by June 8, 2018.
Dr. Chambers and Mr. Briggs reported no relevant disclosures.
Preprint publishing challenges the status quo in medicine
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.

Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”
The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.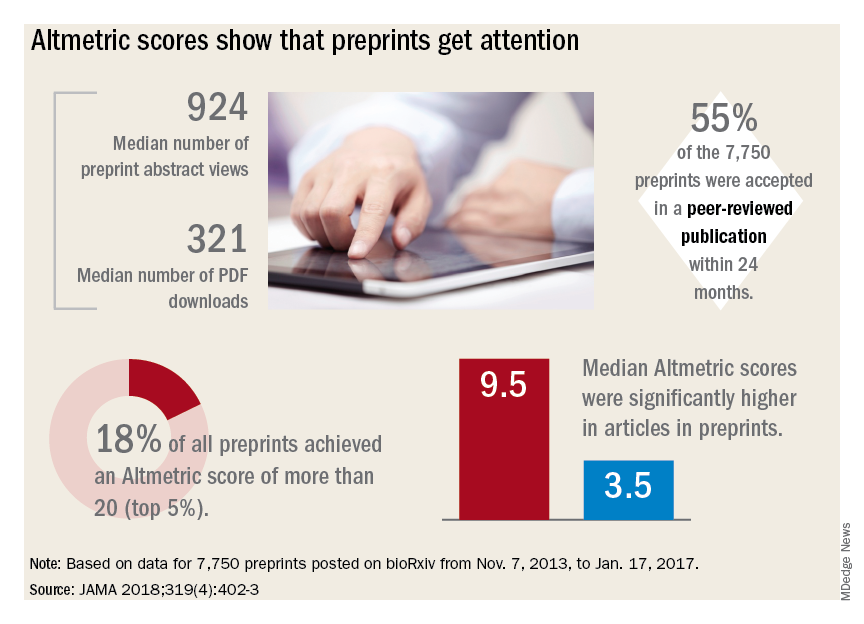
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.
But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.
But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
It’s another beautiful day on the upper east side of Manhattan. The sun shines through the window shades, my 2-year-old daughter sings to herself as she wakes up, my wife has just returned from an early-morning workout – all is right as rain.
My phone buzzes. My stomach clenches. It buzzes again. My Twitter alerts are here. I dread this part of my morning ritual – finding out if I’ve been scooped overnight by the massive inflow of scientific manuscripts reported to me by my army of scientific literature–searching Twitter bots.
But this massive data dump now has a #fakenews problem. It’s not Russian election meddling, it’s open source “preprint” publications. Nearly half of my morning list of Twitter alerts now are sourced from the latest uploads to bioRxiv. BioRxiv is an online site run by scientists at Cold Spring Harbor Laboratory and is composed of posting manuscripts without undergoing a peer-review process. Now, most commonly, these manuscripts are concurrently under review in the bona fide peer-review process elsewhere, but unrevised, they are uploaded directly for public consumption.
There was one recent tweet that highlighted some interesting logistical considerations for bioRxiv manuscripts in the peer-review process. The tweet from an unnamed laboratory complains that a peer reviewer is displeased with the authors citing their own bioRxiv paper, while the tweeter contends that all referenced information, online or otherwise, must be cited. Moreover, the reviewer brings up an accusation of self-plagiarism as the submitted manuscript is identical to the one on bioRxiv. While the latter just seems like a misunderstanding of the bioRxiv platform, the former is a really interesting question of whether bioRxiv represents data that can/should be referenced.
Proponents of the platform are excited that data is accessible sooner, that one’s latest and greatest scientific finding can be “scoop proof” by getting it online and marking one’s territory. Naysayers contend that, without peer review, the work cannot truly be part of the scientific literature and should be taken with great caution.
There is undoubtedly danger. Online media sources Gizmodo and the Motley Fool both reported that a January 2018 bioRxiv preprint resulted in a nearly 20% drop in stock prices of CRISPR biotechnology firms Editas Medicine and Intellia Therapeutics. The manuscript warned of the potential immunogenicity of CRISPR, suggesting that preexisting antibodies might limit its clinical application. Far more cynically, this highlights how a stock price could theoretically be artificially manipulated through preprint data.
The preprint is an open market response to the long, arduous process that peer review has become, but undoubtedly, peer review is an essential part of how we maintain transparency and accountability in science and medicine. It remains to be seen exactly how journal editors intend to use bioRxiv submissions in the appraisal of “novelty.”
How will the scientific community vet and referee the works, and will the title and conclusions of a scientifically flawed work permeate misleading information into the field and lay public? Would you let it influence your research or clinical practice? We will be finding out one tweet at a time.
Aaron D. Viny, MD, is with the Memorial Sloan Kettering Cancer Center, N.Y., where he is a clinical instructor, is on the staff of the leukemia service, and is a clinical researcher in the Ross Levine Lab. He reported having no relevant financial disclosures. Contact him on Twitter @TheDoctorIsVin.
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.
Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”
The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
Like an upstart quick-draw challenging a grizzled gunslinger, preprint servers are muscling in on the once-exclusive territory of scientific journals.
These online venues sidestep the time-honored but lengthy peer-review process in favor of instant data dissemination. By directly posting unreviewed papers, authors escape the months-long drudgery of peer review, stake an immediate claim on new ideas, and connect instantly with like-minded scientists whose feedback can mold this new idea into a sound scientific contribution.
“The caveat, of course, is that it may be crap.”
That’s the unvarnished truth of preprint publishing, said John Inglis, PhD – and he should know. As the cofounder of Cold Spring Harbor Laboratory’s bioRxiv, the largest-to-date preprint server for the biological sciences, he gives equal billing to both the lofty and the low, and lets them soar or sink by their own merit.
And many of them do soar, Dr. Inglis said. Of the more than 20,000 papers published since bioRxiv’s modest beginning in 2013, slightly more than 60% have gone on to peer-reviewed publication. The four most prolific sources of bioRxiv preprints are the research powerhouses of Stanford, Cambridge, Oxford, and Harvard. The twitterverse is virtually awash with #bioRxiv tags, which alert bioRxiv’s 18,000 followers to new papers in any of 27 subject areas. “We gave up counting 2 years ago, when we reached 100,000,” Dr. Inglis said.
BioRxiv, pronounced “bioarchive,” may be the largest preprint server for the biological sciences, but it’s not the only one. The Center for Open Science has created a preprint server search engine, which lists 25 such servers, a number of them in the life sciences.
PeerJ Preprints also offers a home for unreviewed papers, accepting “drafts of an article, abstract, or poster that has not yet been peer reviewed for formal publication.” Authors can submit a draft, incomplete, or final version, which can be online within 24 hours.
The bioRxiv model is coming to medicine, too. A new preprint server – to be called medRxiv – is expected to launch later in 2018 and will accept a wide range of papers on health and medicine, including clinical trial results.
Brand new or rebrand?
Preprint – or at least the concept of it – is nothing new, Dr. Inglis said. It’s simply the extension into the digital space of something that has been happening for many decades in the physical space.
Scientists have always written drafts of their papers and sent them out to friends and colleagues for feedback before unveiling them publicly. In the early 1990s, UC Berkeley astrophysicist Joanne Cohn began emailing unreviewed physics papers to colleagues. Within a couple of years, physicist Paul Ginsparg, PhD, of Cornell University, created a central repository for these papers at the Los Alamos National Laboratory. This repository became aRxiv, a central component of communication in the physical sciences, and the progenitor of the preprint servers now in existence.
The biological sciences were far behind this curve of open sharing, Dr. Inglis said. “I think some biologists were always aware of aRxiv and intrigued by it, but most were unconvinced that the habits and behaviors of research biologists would support a similar process.”
The competition inherent in research biology was likely a large driver of that lag. “Biological experiments are complicated, it takes a long time for ideas to evolve and results to arrive, and people are possessive of their data and ideas. They have always shared information through conferences, but there was a lot of hesitation about making this information available in an uncontrolled way, beyond the audiences at those meetings,” he said.
[polldaddy:9970002]
Nature Publishing Group first floated the preprint notion among biologists in 2006, with Nature Precedings. It published more than 2,000 papers before folding, rather suddenly, in 2012. A publisher’s statement simply said that the effort was “unsustainable as originally conceived.”
Commentators suspected the model was a financial bust, and indeed, preprint servers aren’t money machines. BioRxiv, proudly not for profit, was founded with financial support from Cold Spring Harbor Laboratory and survives largely on private grants. In April 2017, it received a grant for an undisclosed amount from the Chan Zuckerberg Initiative, established by Facebook founder Mark Zuckerberg and his wife, Priscilla Chan.
Who’s minding the data?
The screening process at bioRxiv is minimal, Dr. Inglis said. An in-house staff checks each paper for obvious flaws, like plagiarism, irrelevance, unacceptable article type, and offensive language. Then they’re sent out to a committee of affiliate scientists, which confirms that the manuscript is a research paper and that it contains science, without judging the quality of that science. Papers aren’t edited before being posted online.
Each bioRxiv paper gets a DOI link, and appears with the following disclaimer detailing the risks inherent in reading “unrefereed” science: “Because [peer review] can be lengthy, authors use the bioRxiv service to make their manuscripts available as ‘preprints’ before peer review, allowing other scientists to see, discuss, and comment on the findings immediately. Readers should therefore be aware that articles on bioRxiv have not been finalized by authors, might contain errors, and report information that has not yet been accepted or endorsed in any way by the scientific or medical community.”
From biology to medicine
The bioRxiv team is poised to jump into a different pool now – medical science. Although the launch date isn’t firm yet, medRxiv will go live sometime very soon, Dr. Inglis said. It’s a proposed partnership between Cold Spring Harbor Laboratory, the Yale-based YODA Project (Yale University Open Data Access Project), and BMJ. The medRxiv papers, like those posted to bioRxiv, will be screened but not peer reviewed or scrutinized for trial design, methodology, or interpretation of results.
The benefits of medRxiv will be more rapid communication of research results, increased opportunities for collaboration, the sharing of hard-to-publish outputs like quality innovations in health care, and greater transparency of clinical trials data, Dr. Inglis said. Despite this, he expects the same kind of push-back bioRxiv initially encountered, at least in the beginning.
“I expect we will be turning the clock back 5 years and find a lot of people who think this is potentially a bad thing, a risk that poor information or misinformation is going to be disseminated to a wider audience, which is exactly what we heard about bioRxiv,” he said. “But we hope that when medRxiv launches, it will demonstrate the same kind of gradual acceptance as people get more and more familiar with the preprint platform.”
The founders intend to build into the server policies to mitigate the risk from medically relevant information that hasn’t been peer reviewed, such as not accepting case studies or editorials and opinion pieces, he added.
While many find the preprint disclaimer acceptable on papers that have no immediate clinical impact, there is concern about applying it to papers that discuss patient treatment.
Howard Bauchner, MD, JAMA’s editor in chief, addressed it in an editorial published in September 2017. Although not explicitly directed at bioRxiv, Dr. Bauchner took a firm stance against shortcutting the evaluation of evidence that is often years in the making.
“New interest in preprint servers in clinical medicine increases the likelihood of premature dissemination and public consumption of clinical research findings prior to rigorous evaluation and peer review,” Dr. Bauchner wrote. “For most articles, public consumption of research findings prior to peer review will have little influence on health, but for some articles, the effect could be devastating for some patients if the results made public prior to peer review are wrong or incorrectly interpreted.”
Dr. Bauchner did not overstate the potential influence of unvetted science, as a January 2018 bioRxiv study on CRISPR gene editing clearly demonstrated. The paper by Carsten Charlesworth, a doctoral student at Stanford (Calif.) University, found that up to 79% of humans could already be immune to Crispr-Cas9, the gene-editing protein derived from Staphylococcus aureus and S. pyogenes. More than science geeks were reading: The report initially sent CRISPR stocks tumbling.
Aaron D. Viny, MD, is in general a hesitant fan of bioRxiv’s preprint platform. But he raised an eyebrow when he learned about medRxiv.
“The only pressure that I can see in regulating these reports is social media,” said Dr. Viny, a hematologic oncologist at Memorial Sloan Kettering, in New York. “The fear is that it will be misused in two different realms. The most dangerous and worrisome, of course, is for patients using the data to influence their care plan, when the data haven’t been vetted appropriately. But secondarily, how could it influence the economics of clinical trials? There is no shortage of hedge fund managers in biotech. These data could misinform a consultant who might know the area in a way that artificially exploits early research data. Could that permit someone to submit disingenuous data to manipulate the stock of a given pharmaceutical company? I don’t know how you police that kind of thing.”
Who’s loving it – and why?
There are plenty of reasons to support a thriving preprint community, said Jessica Polka, PhD, director of ASAPbio, (Accelerating Science and Publication in biology), a group that bills itself as a scientist-driven initiative to promote the productive use of preprints in the life sciences.
“Preprinting complements traditional journal publishing by allowing researchers to rapidly communicate their findings to the scientific community,” she said. “This, in turn, provides them with opportunities for earlier and broader feedback and a way to transparently demonstrate progress on a project. More importantly, the whole community benefits by having earlier access to research findings, which can accelerate the pace of discovery.”
The disclosures applied to every preprint paper are the publisher’s way of assuring this same awareness, she said. And preprints do need to be approached with some skepticism, as should peer-reviewed literature.
“The veracity of published papers is not always a given. An example is the 1998 vaccine paper [published in the Lancet] by Dr. Andrew Wakefield,” which launched the antivaccine movement. “But the answer to problems of reliability is to provide more information about the research and how it has been verified and evaluated, not less information. For example, confirmation bias can make it difficult to refute work that has been published. The current incentives for publishing negative results in a journal are not strong enough to reveal all of the information that could be useful to other researchers, but preprinting reduces the barrier to sharing negative results,” she said.
Swimming up the (main)stream
Universal peer-reviewed acceptance of preprints isn’t a done deal, Dr. Polka said. Journals are tussling with how to handle these papers. The Lancet clearly states that preprints don’t constitute prior publication and are welcome. The New England Journal of Medicine offers an uncontestable “no way.”
JAMA discourages submitting preprints, and will consider one only if the submitted version offers “meaningful new information” above what the preprint disseminated.
Cell Press has a slightly different take. They will consider papers previously posted on preprint services, but the policy applies only to the original submitted version of the paper. “We do not support posting of revisions that respond to editorial input and peer review or the final published version to preprint servers,” the policy notes.
In an interview, Deborah Sweet, PhD, the group’s vice president of editorial, elaborated on the policy. “In our view, one of the most important purposes of preprint posting is to gather feedback from the scientific community before a formal submission to a journal,” she said. “The ‘original submission’ term in our guidelines refers to the first version of the paper submitted to [Cell Press], which could include revisions made in response to community feedback on a preprint. After formal submission, we think it is most appropriate to incorporate and represent the value of the editorial and peer-review evaluation process in the final published journal article so that is clearly identifiable as the version of record.”
bioRxiv has made substantial inroads with dozens of other peer-reviewed journals. More than 100 – including a number of publications by EMBO Press and PLOS (Public Library of Science) – participate in bioRxiv’s B2J (BioRxiv-to-journal) direct-submission program.
With a few clicks, authors can transmit their bioRxiv manuscript files directly to these journals, without having to prepare separate submissions, Dr. Sweet said. Last year, Cell Press added two publications – Cell Reports and Structure – to the B2J program. “Once the paper is sent, it moves behind the scenes to the journal system and reappears as a formal submission,” she said. “In our process, before transferring the paper to the journal editors, authors have a chance to update the files (for example, to add a cover letter) and answer the standard questions that we ask, including ones about reviewer suggestions and exclusion requests. Once that step is done, the paper is handed over to the editorial team, and it’s ready to go for consideration in the same way as any other submission.”
Who’s reading?
Regardless of whether peer-review journals grant them legitimacy, preprints are getting a lot of views. A recent research letter, published in JAMA, looked at readership and online attention in 7,750 preprints posted from November 2013 to January 2017.
Primary author Stylianos Serghiou then selected 776 papers that had first appeared in bioRxiv, and matched them with 3,647 peer-reviewed articles lacking preprint exposure. He examined several publishing metrics for the papers, including views and downloads, citations in other sources, and Altmetric scores.
Altmetric tracks digital attention to scientific papers: Wikipedia citations, mentions in policy documents, blog discussions, and social media mentions including Facebook, Reddit, and Twitter. An Altmetric “attention score” of more than 20 corresponds to articles in the top 5% of readership, he said in an interview.
“Almost one in five of the bioRxiv preprints were getting these very high Almetric scores – much higher scores than articles that had no preprint posting,” Mr. Serghiou said in an interview.
Other findings include:
- The median number of preprint abstract views was 924, and the median number of PDF downloads was 321.
- In total, 18% of the preprints achieved an Altmetric score of more than 20.
- Of 7,750 preprints, 55% were accepted in a peer-reviewed publication within 24 months.
- Altmetric scores were significantly higher in articles in preprints (median 9.5 vs. 3.5).
The differences are probably related, at least in part, to the digital media savvy of preprint authors, Mr. Serghiou suggested. “We speculate that people who publish in bioRxiv may be more familiar with social media methods of making others aware of their work. They tend to be very good at using platforms like Twitter and Facebook to promote their results.”
Despite the high exposure scores, only 10% of bioRxiv articles get any posted comments or feedback – a key raison d’être for using a preprint service.
“Ten percent doesn’t sound like a very robust [feedback], but most journal articles get no comments whatsoever,” Dr. Inglis said. “And if they do, especially on the weekly magazines of science, comments may be from someone who has an ax to grind, or who doesn’t know much about the subject.”
What isn’t measured, in either volume or import, is the private communication a preprint engenders, Dr. Inglis said. “Feedback comes directly and privately to the author through email or at meetings or on the phone. We hear time and again that authors get hundreds of downloads after posting, and receive numerous contacts from colleagues who want to know more, to point out weaknesses, or request collaborations. These are the advantages we see from this potentially anxiety-provoking process of putting a manuscript out that has not been approved for publication. The entire purpose is to accelerate the speed of research by accelerating the speed of communication.”
Dr. Inglis, Dr. Sweet, and Dr. Polka are all employees of their respective companies. Dr. Viny and Mr. Serghiou both reported having no financial disclosures relevant to this article.
FDA proposes lower nicotine levels in cigarettes
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
Dermatology residency match: Is the glut of applications for limited positions corrupting the process?
SAN DIEGO – Whether was the topic of discussion at a session on dermatoethics at the annual meeting of the American Academy of Dermatology.
“I think it is unethical and we need to address it,” Jane M. Grant-Kels, MD, said during the session. “What we are doing through the process of physicians getting into dermatology residency programs is telling them to lie to us and to do well on a single examination,” the United States Medical Licensing Examination.
Lionel G. Bercovitch, MD, is among the dermatologists who acknowledge the unarguable fact that application rates are high but don’t see it as a crisis of credibility.
“I don’t believe that dermatology match is broken, unethical, or unfair. The match is not perfect, but it’s fair,” contended Dr. Bercovitch, professor of dermatology at Brown University, Providence, R.I. “The problem [of an application glut] is real, but it’s not an ethical issue.”
Dr. Grant-Kels sees it in ethical terms because, in her view, “everyone is gaming the system. It makes applicants liars” when they profess interest in moving to a remote location or planning to practice a certain type of dermatology.
The “extremely competitive” process leads a majority of applicants to “shot gun” their filings to many programs such that dermatology residency programs are “deluged” with applications, Dr. Grant-Kels said. Data from the Association of American Medical Colleges for 2017 showed an average of just over 500 applications received by each U.S. dermatology residency program.
As a result, residency programs feel forced to apply blind filters that generally cull out more than a third of the applications received. Dr. Grant-Kels decried the need for programs to impose arbitrary barriers to entering dermatology based on a score from a single examination or other criteria like membership in Alpha Omega Alpha or current location.
“Blanket screening methods run the risk of excluding genuinely interested and qualified candidates who do not fall above a threshold. This violates the principal of nonmaleficence,” she said. “Screens are unfair.”
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/dermatology-residency-match?iframe=1"}]
Dr. Grant-Kels proposed a pair of potential remedies: putting a cap on the number of applications someone can make and – a more realistic approach – mentors’ giving guidance to prospective applicants.
“It’s a problem that kids are applying to dermatology programs who have no business applying, who really don’t have a chance,” she said.
Dr. Bercovitch noted that most dermatology residency programs are too small and that, while the number of residency slots has been rising, it has not kept pace with increasing demand from physicians seeking residency slots. He saw no ethical reason for physicians to feel they should rein in the number of applications they file, and he said the only obligations for residency programs are to strictly adhere to the Match rules and both federal and state civil rights and labor laws and to be nondiscriminatory and avoid nepotism and conflicts of interest. Because programs cannot seriously consider nor interview several hundred applicants each year, some type of filtering is needed, and no filter is fair or perfect, he conceded.
“Filters are inherently unfair” to certain applicants, “but how else to effectively screen” hundreds of applications, Dr. Bercovitch asked.
“We need to talk about this. It’s not a good system. If we don’t talk about it, it will never change,” Dr. Grant-Kels said.
Dr. Grant-Kels and Dr. Bercovitch had no disclosures.
SAN DIEGO – Whether was the topic of discussion at a session on dermatoethics at the annual meeting of the American Academy of Dermatology.
“I think it is unethical and we need to address it,” Jane M. Grant-Kels, MD, said during the session. “What we are doing through the process of physicians getting into dermatology residency programs is telling them to lie to us and to do well on a single examination,” the United States Medical Licensing Examination.
Lionel G. Bercovitch, MD, is among the dermatologists who acknowledge the unarguable fact that application rates are high but don’t see it as a crisis of credibility.
“I don’t believe that dermatology match is broken, unethical, or unfair. The match is not perfect, but it’s fair,” contended Dr. Bercovitch, professor of dermatology at Brown University, Providence, R.I. “The problem [of an application glut] is real, but it’s not an ethical issue.”
Dr. Grant-Kels sees it in ethical terms because, in her view, “everyone is gaming the system. It makes applicants liars” when they profess interest in moving to a remote location or planning to practice a certain type of dermatology.
The “extremely competitive” process leads a majority of applicants to “shot gun” their filings to many programs such that dermatology residency programs are “deluged” with applications, Dr. Grant-Kels said. Data from the Association of American Medical Colleges for 2017 showed an average of just over 500 applications received by each U.S. dermatology residency program.
As a result, residency programs feel forced to apply blind filters that generally cull out more than a third of the applications received. Dr. Grant-Kels decried the need for programs to impose arbitrary barriers to entering dermatology based on a score from a single examination or other criteria like membership in Alpha Omega Alpha or current location.
“Blanket screening methods run the risk of excluding genuinely interested and qualified candidates who do not fall above a threshold. This violates the principal of nonmaleficence,” she said. “Screens are unfair.”
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/dermatology-residency-match?iframe=1"}]
Dr. Grant-Kels proposed a pair of potential remedies: putting a cap on the number of applications someone can make and – a more realistic approach – mentors’ giving guidance to prospective applicants.
“It’s a problem that kids are applying to dermatology programs who have no business applying, who really don’t have a chance,” she said.
Dr. Bercovitch noted that most dermatology residency programs are too small and that, while the number of residency slots has been rising, it has not kept pace with increasing demand from physicians seeking residency slots. He saw no ethical reason for physicians to feel they should rein in the number of applications they file, and he said the only obligations for residency programs are to strictly adhere to the Match rules and both federal and state civil rights and labor laws and to be nondiscriminatory and avoid nepotism and conflicts of interest. Because programs cannot seriously consider nor interview several hundred applicants each year, some type of filtering is needed, and no filter is fair or perfect, he conceded.
“Filters are inherently unfair” to certain applicants, “but how else to effectively screen” hundreds of applications, Dr. Bercovitch asked.
“We need to talk about this. It’s not a good system. If we don’t talk about it, it will never change,” Dr. Grant-Kels said.
Dr. Grant-Kels and Dr. Bercovitch had no disclosures.
SAN DIEGO – Whether was the topic of discussion at a session on dermatoethics at the annual meeting of the American Academy of Dermatology.
“I think it is unethical and we need to address it,” Jane M. Grant-Kels, MD, said during the session. “What we are doing through the process of physicians getting into dermatology residency programs is telling them to lie to us and to do well on a single examination,” the United States Medical Licensing Examination.
Lionel G. Bercovitch, MD, is among the dermatologists who acknowledge the unarguable fact that application rates are high but don’t see it as a crisis of credibility.
“I don’t believe that dermatology match is broken, unethical, or unfair. The match is not perfect, but it’s fair,” contended Dr. Bercovitch, professor of dermatology at Brown University, Providence, R.I. “The problem [of an application glut] is real, but it’s not an ethical issue.”
Dr. Grant-Kels sees it in ethical terms because, in her view, “everyone is gaming the system. It makes applicants liars” when they profess interest in moving to a remote location or planning to practice a certain type of dermatology.
The “extremely competitive” process leads a majority of applicants to “shot gun” their filings to many programs such that dermatology residency programs are “deluged” with applications, Dr. Grant-Kels said. Data from the Association of American Medical Colleges for 2017 showed an average of just over 500 applications received by each U.S. dermatology residency program.
As a result, residency programs feel forced to apply blind filters that generally cull out more than a third of the applications received. Dr. Grant-Kels decried the need for programs to impose arbitrary barriers to entering dermatology based on a score from a single examination or other criteria like membership in Alpha Omega Alpha or current location.
“Blanket screening methods run the risk of excluding genuinely interested and qualified candidates who do not fall above a threshold. This violates the principal of nonmaleficence,” she said. “Screens are unfair.”
[polldaddy:{"method":"iframe","type":"survey","src":"//newspolls2017.polldaddy.com/s/dermatology-residency-match?iframe=1"}]
Dr. Grant-Kels proposed a pair of potential remedies: putting a cap on the number of applications someone can make and – a more realistic approach – mentors’ giving guidance to prospective applicants.
“It’s a problem that kids are applying to dermatology programs who have no business applying, who really don’t have a chance,” she said.
Dr. Bercovitch noted that most dermatology residency programs are too small and that, while the number of residency slots has been rising, it has not kept pace with increasing demand from physicians seeking residency slots. He saw no ethical reason for physicians to feel they should rein in the number of applications they file, and he said the only obligations for residency programs are to strictly adhere to the Match rules and both federal and state civil rights and labor laws and to be nondiscriminatory and avoid nepotism and conflicts of interest. Because programs cannot seriously consider nor interview several hundred applicants each year, some type of filtering is needed, and no filter is fair or perfect, he conceded.
“Filters are inherently unfair” to certain applicants, “but how else to effectively screen” hundreds of applications, Dr. Bercovitch asked.
“We need to talk about this. It’s not a good system. If we don’t talk about it, it will never change,” Dr. Grant-Kels said.
Dr. Grant-Kels and Dr. Bercovitch had no disclosures.
EXPERT ANALYSIS FROM AAD 18
Health care panhandlers: A symptom of our system’s baked-in pressures?
A few nights a week after work I have to stop by the store for this or that.
In the last 1-2 months there’s always been a couple at the parking lot exit, both in wheelchairs, with a big sign asking for money to help one of them beat cancer. They even have the amount listed.
But, by the same token, they could be quite legitimate. The American health care system is full of cracks that seriously ill people can slip through. One recent survey found that about 30% of Americans had trouble paying their medical bills.
It’s easy to look at people like this and think, “I’ll never let that happen to me.” We assume they must be smokers, or irresponsible spenders, or some other reason that makes us feel we won’t stumble into the same pitfalls. That’s reassuring, and sometimes true, but not always. And probably more often than we want to realize.
The world is full of people and families devastated by bad luck. Through no fault of their own, they develop a terrible medical condition or suffer grievous injuries, and suddenly, decent, hard-working, previously healthy people are facing foreclosure and financial ruin. It could, quite literally, be any of us.
Case in point: My family has good insurance and has averaged $10,000 in out-of-pocket medical expenses per year for the last several years. That’s for routine stuff: meeting deductibles, copays on medications, tests, and doctor visits, a few ER trips, etc. The only real “surprise” in there was when my wife broke her leg and needed surgery.
If the panhandlers really did have legitimate medical issues, I might be willing to help out. I give to charity. My grandmother and parents stressed that value to me, and I try to teach it to my kids. But, sadly, we live in a world full of con artists who try to make money by taking advantage of caring peoples’ feelings. Look at all the scams that immediately cropped up following the recent hurricane and wildfire disasters. Without knowing the truth, I’d rather give to an organization like the Salvation Army or Red Cross, hoping they have more experience than I do in sorting out who’s really in need.
As a doctor, I also try to justify it by thinking about how much care I do for “free.” This includes uninsured hospital patients we all see on call, knowing we’ll end up writing their bill off as a loss, and bounced checks for copays and deductible portions that we know we’ll never see.
But, no matter how I try to rationalize it, it still bothers me when I see them sitting there as I leave the store. I don’t know if they’re legitimate. But if they are, they aren’t alone, and there’s something seriously wrong with our health care system.
[polldaddy:9876776]
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
A few nights a week after work I have to stop by the store for this or that.
In the last 1-2 months there’s always been a couple at the parking lot exit, both in wheelchairs, with a big sign asking for money to help one of them beat cancer. They even have the amount listed.
But, by the same token, they could be quite legitimate. The American health care system is full of cracks that seriously ill people can slip through. One recent survey found that about 30% of Americans had trouble paying their medical bills.
It’s easy to look at people like this and think, “I’ll never let that happen to me.” We assume they must be smokers, or irresponsible spenders, or some other reason that makes us feel we won’t stumble into the same pitfalls. That’s reassuring, and sometimes true, but not always. And probably more often than we want to realize.
The world is full of people and families devastated by bad luck. Through no fault of their own, they develop a terrible medical condition or suffer grievous injuries, and suddenly, decent, hard-working, previously healthy people are facing foreclosure and financial ruin. It could, quite literally, be any of us.
Case in point: My family has good insurance and has averaged $10,000 in out-of-pocket medical expenses per year for the last several years. That’s for routine stuff: meeting deductibles, copays on medications, tests, and doctor visits, a few ER trips, etc. The only real “surprise” in there was when my wife broke her leg and needed surgery.
If the panhandlers really did have legitimate medical issues, I might be willing to help out. I give to charity. My grandmother and parents stressed that value to me, and I try to teach it to my kids. But, sadly, we live in a world full of con artists who try to make money by taking advantage of caring peoples’ feelings. Look at all the scams that immediately cropped up following the recent hurricane and wildfire disasters. Without knowing the truth, I’d rather give to an organization like the Salvation Army or Red Cross, hoping they have more experience than I do in sorting out who’s really in need.
As a doctor, I also try to justify it by thinking about how much care I do for “free.” This includes uninsured hospital patients we all see on call, knowing we’ll end up writing their bill off as a loss, and bounced checks for copays and deductible portions that we know we’ll never see.
But, no matter how I try to rationalize it, it still bothers me when I see them sitting there as I leave the store. I don’t know if they’re legitimate. But if they are, they aren’t alone, and there’s something seriously wrong with our health care system.
[polldaddy:9876776]
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
A few nights a week after work I have to stop by the store for this or that.
In the last 1-2 months there’s always been a couple at the parking lot exit, both in wheelchairs, with a big sign asking for money to help one of them beat cancer. They even have the amount listed.
But, by the same token, they could be quite legitimate. The American health care system is full of cracks that seriously ill people can slip through. One recent survey found that about 30% of Americans had trouble paying their medical bills.
It’s easy to look at people like this and think, “I’ll never let that happen to me.” We assume they must be smokers, or irresponsible spenders, or some other reason that makes us feel we won’t stumble into the same pitfalls. That’s reassuring, and sometimes true, but not always. And probably more often than we want to realize.
The world is full of people and families devastated by bad luck. Through no fault of their own, they develop a terrible medical condition or suffer grievous injuries, and suddenly, decent, hard-working, previously healthy people are facing foreclosure and financial ruin. It could, quite literally, be any of us.
Case in point: My family has good insurance and has averaged $10,000 in out-of-pocket medical expenses per year for the last several years. That’s for routine stuff: meeting deductibles, copays on medications, tests, and doctor visits, a few ER trips, etc. The only real “surprise” in there was when my wife broke her leg and needed surgery.
If the panhandlers really did have legitimate medical issues, I might be willing to help out. I give to charity. My grandmother and parents stressed that value to me, and I try to teach it to my kids. But, sadly, we live in a world full of con artists who try to make money by taking advantage of caring peoples’ feelings. Look at all the scams that immediately cropped up following the recent hurricane and wildfire disasters. Without knowing the truth, I’d rather give to an organization like the Salvation Army or Red Cross, hoping they have more experience than I do in sorting out who’s really in need.
As a doctor, I also try to justify it by thinking about how much care I do for “free.” This includes uninsured hospital patients we all see on call, knowing we’ll end up writing their bill off as a loss, and bounced checks for copays and deductible portions that we know we’ll never see.
But, no matter how I try to rationalize it, it still bothers me when I see them sitting there as I leave the store. I don’t know if they’re legitimate. But if they are, they aren’t alone, and there’s something seriously wrong with our health care system.
[polldaddy:9876776]
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Burnout
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at dermnews@frontlinemedcom.com
Adherence boon, or Big Brother loom?
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]