User login
Administrative burden and burnout
In May 2019, SHM sent a letter to U.S. Senators Tina Smith and Bill Cassidy in support of the Reducing Administrative Costs and Burdens in Health Care Act of 2019. In excerpts from the letter below, the society details the link between administrative burdens and physician burnout.

Providers and hospital systems expend countless resources, both time and dollars, adhering to unnecessary and excessive administrative burdens instead of investing those resources in providing quality patient care. National data suggests that more than 50 percent of the physician workforce is burned out. Excessive administrative burden is a major contributor to physician burnout, which negatively affects quality and safety within the hospital and further increases health care costs. Notably, the Reducing Administrative Costs and Burdens in Health Care Act calls for a 50% reduction of unnecessary administrative costs from the Department of Health and Human Services within the next ten years.
Hospitalists are front-line clinicians in America's acute care hospitals whose professional focus is the general medical care of hospitalized patients. Their unique position in the healthcare system affords hospitalists a distinct perspective and systems-based approach to confronting and solving challenges at the individual provider and overall institutional level of the hospital. In this capacity, hospitalists experience multiple examples of administrative requirements directly detracting from patient care and redirecting finite resources away from care to meet compliance demands.
By way of example, navigating the administrative rules around inpatient admissions and outpatient observation care, for example, requires a significant shift of healthcare resources away from patient care. While patients admitted under observation receive nearly identical care to those admitted as an inpatient, hospitalists report that, in addition to themselves as the direct healthcare provider, status determinations between inpatient admissions and outpatient observation care require the input of a myriad of staff including nursing, coding/compliance teams, utilization review, case managers and external review organizations. A recent study in the Journal of Hospital Medicine indicated that an average of 5.1 full time employees, not including case managers, are required to navigate the audit and appeals process associated with hospital stay status determinations. These are resources that should be directly used for patient care, but are redirected towards regulation compliance, increasing cost of care without increasing quality.
To read the entire letter, visit https://www.hospitalmedicine.org/policy--advocacy/letters/shm-supports-the-reducing-administrative-costs-and-burdens-in-health-care-act-of-2019/.
In May 2019, SHM sent a letter to U.S. Senators Tina Smith and Bill Cassidy in support of the Reducing Administrative Costs and Burdens in Health Care Act of 2019. In excerpts from the letter below, the society details the link between administrative burdens and physician burnout.
Providers and hospital systems expend countless resources, both time and dollars, adhering to unnecessary and excessive administrative burdens instead of investing those resources in providing quality patient care. National data suggests that more than 50 percent of the physician workforce is burned out. Excessive administrative burden is a major contributor to physician burnout, which negatively affects quality and safety within the hospital and further increases health care costs. Notably, the Reducing Administrative Costs and Burdens in Health Care Act calls for a 50% reduction of unnecessary administrative costs from the Department of Health and Human Services within the next ten years.
Hospitalists are front-line clinicians in America's acute care hospitals whose professional focus is the general medical care of hospitalized patients. Their unique position in the healthcare system affords hospitalists a distinct perspective and systems-based approach to confronting and solving challenges at the individual provider and overall institutional level of the hospital. In this capacity, hospitalists experience multiple examples of administrative requirements directly detracting from patient care and redirecting finite resources away from care to meet compliance demands.
By way of example, navigating the administrative rules around inpatient admissions and outpatient observation care, for example, requires a significant shift of healthcare resources away from patient care. While patients admitted under observation receive nearly identical care to those admitted as an inpatient, hospitalists report that, in addition to themselves as the direct healthcare provider, status determinations between inpatient admissions and outpatient observation care require the input of a myriad of staff including nursing, coding/compliance teams, utilization review, case managers and external review organizations. A recent study in the Journal of Hospital Medicine indicated that an average of 5.1 full time employees, not including case managers, are required to navigate the audit and appeals process associated with hospital stay status determinations. These are resources that should be directly used for patient care, but are redirected towards regulation compliance, increasing cost of care without increasing quality.
To read the entire letter, visit https://www.hospitalmedicine.org/policy--advocacy/letters/shm-supports-the-reducing-administrative-costs-and-burdens-in-health-care-act-of-2019/.
In May 2019, SHM sent a letter to U.S. Senators Tina Smith and Bill Cassidy in support of the Reducing Administrative Costs and Burdens in Health Care Act of 2019. In excerpts from the letter below, the society details the link between administrative burdens and physician burnout.
Providers and hospital systems expend countless resources, both time and dollars, adhering to unnecessary and excessive administrative burdens instead of investing those resources in providing quality patient care. National data suggests that more than 50 percent of the physician workforce is burned out. Excessive administrative burden is a major contributor to physician burnout, which negatively affects quality and safety within the hospital and further increases health care costs. Notably, the Reducing Administrative Costs and Burdens in Health Care Act calls for a 50% reduction of unnecessary administrative costs from the Department of Health and Human Services within the next ten years.
Hospitalists are front-line clinicians in America's acute care hospitals whose professional focus is the general medical care of hospitalized patients. Their unique position in the healthcare system affords hospitalists a distinct perspective and systems-based approach to confronting and solving challenges at the individual provider and overall institutional level of the hospital. In this capacity, hospitalists experience multiple examples of administrative requirements directly detracting from patient care and redirecting finite resources away from care to meet compliance demands.
By way of example, navigating the administrative rules around inpatient admissions and outpatient observation care, for example, requires a significant shift of healthcare resources away from patient care. While patients admitted under observation receive nearly identical care to those admitted as an inpatient, hospitalists report that, in addition to themselves as the direct healthcare provider, status determinations between inpatient admissions and outpatient observation care require the input of a myriad of staff including nursing, coding/compliance teams, utilization review, case managers and external review organizations. A recent study in the Journal of Hospital Medicine indicated that an average of 5.1 full time employees, not including case managers, are required to navigate the audit and appeals process associated with hospital stay status determinations. These are resources that should be directly used for patient care, but are redirected towards regulation compliance, increasing cost of care without increasing quality.
To read the entire letter, visit https://www.hospitalmedicine.org/policy--advocacy/letters/shm-supports-the-reducing-administrative-costs-and-burdens-in-health-care-act-of-2019/.
Community pediatric care is diminishing
The mantra of community hospital administrators is that pediatric care does not pay. Neonatal intensive care pays. For pediatrics, it is similar to how football programs (Medicare patients) support minor sports (pediatrics and obstetrics) at colleges. However, fewer even mildly sick newborns are cared for at community hospitals, which has led to a centralization of neonatal and pediatric care and a loss of pediatric expertise at the affected hospitals.

Pediatric hospitalists are hired to cover the pediatric floor, the emergency department, and labor and delivery, then fired over empty pediatric beds. The rationale expressed is that pediatricians have done such a good job in preventive care that children rarely need hospitalization, so why have a pediatric inpatient unit? It is true that preventive care has been an integral part of primary care for children. Significantly less that 1% of child office visits result in hospitalization.
Advocate Health Care has closed inpatient pediatric units at Illinois Masonic, on Chicago’s North Side, Good Samaritan in Downers Grove, and Good Shepherd in Barrington. Units also have been closed at Mount Sinai in North Lawndale, Norwegian American on Chicago’s West Side, Little Company of Mary in Evergreen Park, and Alexian Brothers in Elk Grove.

As a Chicago-area pediatrician for more than 30 years, I have learned several things about community-based pediatric care:
1. Pediatrics is a geographic specialty. Parents will travel to shop, but would rather walk or have a short ride to their children’s medical providers. Secondary care should be community based, and hospitalization, if necessary, should be close by as well.
2. Hospitals that ceased delivering pediatric inpatient care lost their child-friendliness and pediatric competence, becoming uncomfortable delivering almost any care for children (e.g., sedated MRIs and EEGs, x-rays and ultrasounds, ECGs and echocardiograms, and emergency care).
3. In almost all hospitals, after pediatrics was gone eventually so passed obstetrics (another less remunerative specialty). Sick newborns need immediate, competent care. Most pediatric hospitalizations are short term, often overnight. Delaying newborn care is a medicolegal nightmare. and exposes the child and his or her family to a potentially dangerous drive or helicopter ride.
4. As pediatric subspecialty care becomes more centralized, parents are asked to travel for hours to see a pediatric specialist. There are times when that is necessary (e.g., cardiovascular surgery). Pediatric subspecialists, such as pediatric otolaryngologists, then leave community hospitals, forcing even minor surgeries (e.g., ear ventilation tubes) to be done at a center. In rural areas, this could mean hours of travel, lost work days, and family disruption.
5. Children’s hospitals get uninsured and publicly insured children sent hundreds of miles, because there were no subspecialists in the community who would care for these children.
What is the solution, in our profit-focused health care system?
1. Hospitals’ Certificates of Need could include a mandate for pediatric care.
2. Children’s hospitals could be made responsible for community-based care within their geographic catchment areas.
3. The state or the federal government could mandate and financially support community-based hospital care.
4. Deciding what level of care might be appropriate for each community could depend upon closeness to a pediatric hospital, health problems in the community, and the availability of pediatric specialists.
5. A condition for medical licensure might be that a community-based pediatric subspecialist is required to care for a proportion of the uninsured or publicly insured children in his or her area.
6. Reimbursements for pediatric care need to rise enough to make caring for children worth it.
The major decision point regarding care for children cannot be financial, but must instead embrace the needs of each affected community. If quality health care is a right, and not a privilege, then it is time to stop closing pediatric inpatient units, and, instead, look for creative ways to better care for our children.
This process has led to pediatric care being available only in designated centers. The centralization of pediatric care has progressed from 30 years ago, when most community hospitals had inpatient pediatric units, to the search for innovative ways to fill pediatric beds in the mid-90s (sick day care, flex- or shared pediatric units), to the wholesale closure of community pediatric inpatient beds, from 2000 to the present. I have, unfortunately, seen this firsthand, watching the rise of pediatric mega-hospitals and the demise of community pediatrics. It is a simple financial argument. Care for children simply does not pay nearly as well as does care for adults, especially Medicaid patients. Pediatricians are the poorest paid practicing doctors (public health doctors are paid less).
It is true that pediatricians always have been at the forefront of preventive medicine, and that pediatric patients almost always get better, in spite of our best-intentioned interventions. So community-based pediatricians admit very few patients.
With the loss of pediatric units, community hospitals lose their comfort caring for children. This includes phlebotomy, x-ray, trauma, surgery, and behavioral health. And eroding community hospital pediatric expertise has catastrophic implications for rural hospitals, where parents may have to drive for hours to find a child-friendly emergency department.
Is there an answer?
1. Hospitals are responsible for the patients they serve, including children. Why should a hospital be able to close pediatric services so easily?
2. Every hospital that sees children, through the emergency department, needs to have a pediatrician available to evaluate a child, 24/7.
3. There needs to be an observation unit for children, with pediatric staffing, for overnight stays.
4. Pediatric hospitalists should be staffing community hospitals.
5. Pediatric behavioral health resources need to be available, e.g., inpatient psychiatry, partial hospitalization programs, intensive outpatient programs.
6. Telehealth communication is not adequate to address acute care problems, because the hospital caring for the child has to have the proper equipment and adequate expertise to carry out the recommendations of the teleconsultant.
If we accept that our children will shape the future, we must allow them to survive and thrive. Is health care a right or a privilege, and is it just for adults or for children, too?
Dr. Ochs is in private practice at Ravenswood Pediatrics in Chicago. He said he had no relevant financial disclosures. Email him at pdnews@mdedge.com.
The mantra of community hospital administrators is that pediatric care does not pay. Neonatal intensive care pays. For pediatrics, it is similar to how football programs (Medicare patients) support minor sports (pediatrics and obstetrics) at colleges. However, fewer even mildly sick newborns are cared for at community hospitals, which has led to a centralization of neonatal and pediatric care and a loss of pediatric expertise at the affected hospitals.
Pediatric hospitalists are hired to cover the pediatric floor, the emergency department, and labor and delivery, then fired over empty pediatric beds. The rationale expressed is that pediatricians have done such a good job in preventive care that children rarely need hospitalization, so why have a pediatric inpatient unit? It is true that preventive care has been an integral part of primary care for children. Significantly less that 1% of child office visits result in hospitalization.
Advocate Health Care has closed inpatient pediatric units at Illinois Masonic, on Chicago’s North Side, Good Samaritan in Downers Grove, and Good Shepherd in Barrington. Units also have been closed at Mount Sinai in North Lawndale, Norwegian American on Chicago’s West Side, Little Company of Mary in Evergreen Park, and Alexian Brothers in Elk Grove.
As a Chicago-area pediatrician for more than 30 years, I have learned several things about community-based pediatric care:
1. Pediatrics is a geographic specialty. Parents will travel to shop, but would rather walk or have a short ride to their children’s medical providers. Secondary care should be community based, and hospitalization, if necessary, should be close by as well.
2. Hospitals that ceased delivering pediatric inpatient care lost their child-friendliness and pediatric competence, becoming uncomfortable delivering almost any care for children (e.g., sedated MRIs and EEGs, x-rays and ultrasounds, ECGs and echocardiograms, and emergency care).
3. In almost all hospitals, after pediatrics was gone eventually so passed obstetrics (another less remunerative specialty). Sick newborns need immediate, competent care. Most pediatric hospitalizations are short term, often overnight. Delaying newborn care is a medicolegal nightmare. and exposes the child and his or her family to a potentially dangerous drive or helicopter ride.
4. As pediatric subspecialty care becomes more centralized, parents are asked to travel for hours to see a pediatric specialist. There are times when that is necessary (e.g., cardiovascular surgery). Pediatric subspecialists, such as pediatric otolaryngologists, then leave community hospitals, forcing even minor surgeries (e.g., ear ventilation tubes) to be done at a center. In rural areas, this could mean hours of travel, lost work days, and family disruption.
5. Children’s hospitals get uninsured and publicly insured children sent hundreds of miles, because there were no subspecialists in the community who would care for these children.
What is the solution, in our profit-focused health care system?
1. Hospitals’ Certificates of Need could include a mandate for pediatric care.
2. Children’s hospitals could be made responsible for community-based care within their geographic catchment areas.
3. The state or the federal government could mandate and financially support community-based hospital care.
4. Deciding what level of care might be appropriate for each community could depend upon closeness to a pediatric hospital, health problems in the community, and the availability of pediatric specialists.
5. A condition for medical licensure might be that a community-based pediatric subspecialist is required to care for a proportion of the uninsured or publicly insured children in his or her area.
6. Reimbursements for pediatric care need to rise enough to make caring for children worth it.
The major decision point regarding care for children cannot be financial, but must instead embrace the needs of each affected community. If quality health care is a right, and not a privilege, then it is time to stop closing pediatric inpatient units, and, instead, look for creative ways to better care for our children.
This process has led to pediatric care being available only in designated centers. The centralization of pediatric care has progressed from 30 years ago, when most community hospitals had inpatient pediatric units, to the search for innovative ways to fill pediatric beds in the mid-90s (sick day care, flex- or shared pediatric units), to the wholesale closure of community pediatric inpatient beds, from 2000 to the present. I have, unfortunately, seen this firsthand, watching the rise of pediatric mega-hospitals and the demise of community pediatrics. It is a simple financial argument. Care for children simply does not pay nearly as well as does care for adults, especially Medicaid patients. Pediatricians are the poorest paid practicing doctors (public health doctors are paid less).
It is true that pediatricians always have been at the forefront of preventive medicine, and that pediatric patients almost always get better, in spite of our best-intentioned interventions. So community-based pediatricians admit very few patients.
With the loss of pediatric units, community hospitals lose their comfort caring for children. This includes phlebotomy, x-ray, trauma, surgery, and behavioral health. And eroding community hospital pediatric expertise has catastrophic implications for rural hospitals, where parents may have to drive for hours to find a child-friendly emergency department.
Is there an answer?
1. Hospitals are responsible for the patients they serve, including children. Why should a hospital be able to close pediatric services so easily?
2. Every hospital that sees children, through the emergency department, needs to have a pediatrician available to evaluate a child, 24/7.
3. There needs to be an observation unit for children, with pediatric staffing, for overnight stays.
4. Pediatric hospitalists should be staffing community hospitals.
5. Pediatric behavioral health resources need to be available, e.g., inpatient psychiatry, partial hospitalization programs, intensive outpatient programs.
6. Telehealth communication is not adequate to address acute care problems, because the hospital caring for the child has to have the proper equipment and adequate expertise to carry out the recommendations of the teleconsultant.
If we accept that our children will shape the future, we must allow them to survive and thrive. Is health care a right or a privilege, and is it just for adults or for children, too?
Dr. Ochs is in private practice at Ravenswood Pediatrics in Chicago. He said he had no relevant financial disclosures. Email him at pdnews@mdedge.com.
The mantra of community hospital administrators is that pediatric care does not pay. Neonatal intensive care pays. For pediatrics, it is similar to how football programs (Medicare patients) support minor sports (pediatrics and obstetrics) at colleges. However, fewer even mildly sick newborns are cared for at community hospitals, which has led to a centralization of neonatal and pediatric care and a loss of pediatric expertise at the affected hospitals.
Pediatric hospitalists are hired to cover the pediatric floor, the emergency department, and labor and delivery, then fired over empty pediatric beds. The rationale expressed is that pediatricians have done such a good job in preventive care that children rarely need hospitalization, so why have a pediatric inpatient unit? It is true that preventive care has been an integral part of primary care for children. Significantly less that 1% of child office visits result in hospitalization.
Advocate Health Care has closed inpatient pediatric units at Illinois Masonic, on Chicago’s North Side, Good Samaritan in Downers Grove, and Good Shepherd in Barrington. Units also have been closed at Mount Sinai in North Lawndale, Norwegian American on Chicago’s West Side, Little Company of Mary in Evergreen Park, and Alexian Brothers in Elk Grove.
As a Chicago-area pediatrician for more than 30 years, I have learned several things about community-based pediatric care:
1. Pediatrics is a geographic specialty. Parents will travel to shop, but would rather walk or have a short ride to their children’s medical providers. Secondary care should be community based, and hospitalization, if necessary, should be close by as well.
2. Hospitals that ceased delivering pediatric inpatient care lost their child-friendliness and pediatric competence, becoming uncomfortable delivering almost any care for children (e.g., sedated MRIs and EEGs, x-rays and ultrasounds, ECGs and echocardiograms, and emergency care).
3. In almost all hospitals, after pediatrics was gone eventually so passed obstetrics (another less remunerative specialty). Sick newborns need immediate, competent care. Most pediatric hospitalizations are short term, often overnight. Delaying newborn care is a medicolegal nightmare. and exposes the child and his or her family to a potentially dangerous drive or helicopter ride.
4. As pediatric subspecialty care becomes more centralized, parents are asked to travel for hours to see a pediatric specialist. There are times when that is necessary (e.g., cardiovascular surgery). Pediatric subspecialists, such as pediatric otolaryngologists, then leave community hospitals, forcing even minor surgeries (e.g., ear ventilation tubes) to be done at a center. In rural areas, this could mean hours of travel, lost work days, and family disruption.
5. Children’s hospitals get uninsured and publicly insured children sent hundreds of miles, because there were no subspecialists in the community who would care for these children.
What is the solution, in our profit-focused health care system?
1. Hospitals’ Certificates of Need could include a mandate for pediatric care.
2. Children’s hospitals could be made responsible for community-based care within their geographic catchment areas.
3. The state or the federal government could mandate and financially support community-based hospital care.
4. Deciding what level of care might be appropriate for each community could depend upon closeness to a pediatric hospital, health problems in the community, and the availability of pediatric specialists.
5. A condition for medical licensure might be that a community-based pediatric subspecialist is required to care for a proportion of the uninsured or publicly insured children in his or her area.
6. Reimbursements for pediatric care need to rise enough to make caring for children worth it.
The major decision point regarding care for children cannot be financial, but must instead embrace the needs of each affected community. If quality health care is a right, and not a privilege, then it is time to stop closing pediatric inpatient units, and, instead, look for creative ways to better care for our children.
This process has led to pediatric care being available only in designated centers. The centralization of pediatric care has progressed from 30 years ago, when most community hospitals had inpatient pediatric units, to the search for innovative ways to fill pediatric beds in the mid-90s (sick day care, flex- or shared pediatric units), to the wholesale closure of community pediatric inpatient beds, from 2000 to the present. I have, unfortunately, seen this firsthand, watching the rise of pediatric mega-hospitals and the demise of community pediatrics. It is a simple financial argument. Care for children simply does not pay nearly as well as does care for adults, especially Medicaid patients. Pediatricians are the poorest paid practicing doctors (public health doctors are paid less).
It is true that pediatricians always have been at the forefront of preventive medicine, and that pediatric patients almost always get better, in spite of our best-intentioned interventions. So community-based pediatricians admit very few patients.
With the loss of pediatric units, community hospitals lose their comfort caring for children. This includes phlebotomy, x-ray, trauma, surgery, and behavioral health. And eroding community hospital pediatric expertise has catastrophic implications for rural hospitals, where parents may have to drive for hours to find a child-friendly emergency department.
Is there an answer?
1. Hospitals are responsible for the patients they serve, including children. Why should a hospital be able to close pediatric services so easily?
2. Every hospital that sees children, through the emergency department, needs to have a pediatrician available to evaluate a child, 24/7.
3. There needs to be an observation unit for children, with pediatric staffing, for overnight stays.
4. Pediatric hospitalists should be staffing community hospitals.
5. Pediatric behavioral health resources need to be available, e.g., inpatient psychiatry, partial hospitalization programs, intensive outpatient programs.
6. Telehealth communication is not adequate to address acute care problems, because the hospital caring for the child has to have the proper equipment and adequate expertise to carry out the recommendations of the teleconsultant.
If we accept that our children will shape the future, we must allow them to survive and thrive. Is health care a right or a privilege, and is it just for adults or for children, too?
Dr. Ochs is in private practice at Ravenswood Pediatrics in Chicago. He said he had no relevant financial disclosures. Email him at pdnews@mdedge.com.

RECOVERY: Early SAVR benefits asymptomatic severe AS patients
PHILADELPHIA – Early aortic valve replacement surgery for patients with asymptomatic, severe aortic stenosis has been a controversial strategy, but the first randomized trial comparing early surgery with conservative management found that early-surgery patients had a 17-fold improved survival at 8 years, according to trial results reported at the American Heart Association scientific sessions.

Albeit small – 145 patients randomized to early surgery or conservative therapy – the RECOVERY trial results provide important evidence to support early preemptive aortic valve replacement for patients with asymptomatic but severe aortic stenosis, said Duk-Hyun Kang, MD, of the division of cardiology, Asan Medical Center in Seoul, South Korea.
The trial randomized 73 patients to early surgical aortic valve replacement (SAVR) and 72 to conventional treatment. Baseline characteristics were similar between the two groups, with a mean EuroSCORE II of 0.9, peak aortic-valve jet velocity (Vmax) of 5.1 m/sec, and mean aortic-valve area of 0.63 cm2. Fifty-three conventional treatment patients went on to have SAVR when symptoms developed during follow-up. There were no operation-related deaths in either group, although one early surgery patient had a stroke and one conventional treatment patients had an MI during the operative period.
At a median follow-up of 6.2 years, 1 early-surgery patient (1.4%) and 11 conventional-therapy patients (15.3%) died from operative or cardiovascular death, Dr. Kang said (P = 0.003). “The number needed to treat to prevent one cardiovascular death within 4 years was 20 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidences of operative CV or death were 1.4% for both in the early-surgery group and 5.7% and 25.5% in the conventional-treatment group, Dr. Kang said (P = 0.003).
Rates of all-cause mortality were 6.8% and 20.8% (P = 0.030) in the respective groups. “The number needed to treat to save one life in 4 years was 16 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidence of any-cause death was 4.1% and 10.2% in the early-surgery patients and 9.7% and 31.8% in the conventional group (P = 0.018).
Among the trial limitations Dr. Kang acknowledged were that the population had severe AS with aortic velocity of 4.5 m/sec or greater. “The benefit of early surgery may be relatively smaller in asymptomatic patients with less severe aortic stenosis,” he said.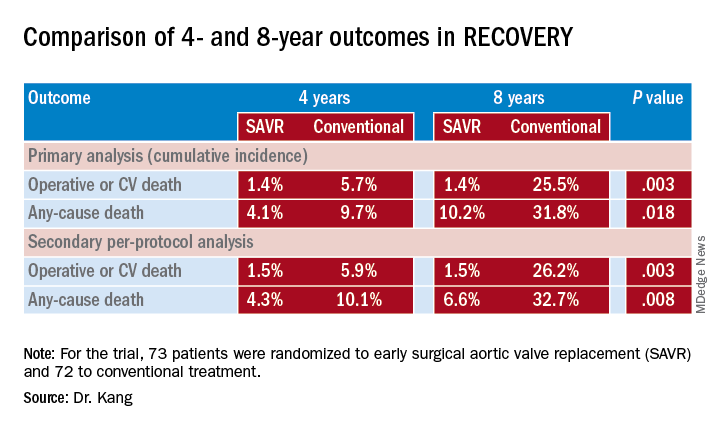
Not all patients had an exercise test to confirm their asymptomatic status, but as discussant Robert Bonow, MD, of Northwestern University in Chicago pointed out, “this is what we’re dealing with clinically.” Also the mean age of the patient population – 64.9 years – is relatively young, and they had a high incidence of bicuspid aortic valve, few comorbidities, and low operative risk, Dr. Kang said. “Thus, our study population is quite different from the populations enrolled in the TAVR [transcatheter AVR] trials, and the results of our study cannot be directly applied to early TAVR for asymptomatic severe aortic stenosis,” he said.
The mean age in both the PARTNER 3 (N Engl J Med. 2019;380:1695-705) and EVOLUT (N Engl J Med. 2019;380:1706-15) trials comparing TAVR and SAVR in low-risk patients was 73.6 years, and they had higher rates of comorbidities than did the RECOVERY patients.
Dr. Bonow said the RECOVERY findings add to the PARTNER 3 and EVOLUT findings and raise the question about rethinking guideline indications for AVR in asymptomatic severe AS patients. “Should we be talking about moving our Vmax threshold down to 4.5 m/sec, or maybe increasing the Class IIa indications to Class I?” he said. “I think we need to wait to see more data to support these excellent results.”
However, most of these asymptomatic patients do eventually have surgery “in a very short period of time,” Dr. Bonow said. “So from a clinical management point of view, I think we already have the data suggesting that we could move the ball forward, and now we have these excellent outcome data from Korea as well.”
Results were published simultaneously in the New England Journal of Medicine (2019 Nov 16. doi: 10.1056/NEJMoa1912846).
Dr. Kang and Dr. Bonow had no financial relationships to disclose.
PHILADELPHIA – Early aortic valve replacement surgery for patients with asymptomatic, severe aortic stenosis has been a controversial strategy, but the first randomized trial comparing early surgery with conservative management found that early-surgery patients had a 17-fold improved survival at 8 years, according to trial results reported at the American Heart Association scientific sessions.
Albeit small – 145 patients randomized to early surgery or conservative therapy – the RECOVERY trial results provide important evidence to support early preemptive aortic valve replacement for patients with asymptomatic but severe aortic stenosis, said Duk-Hyun Kang, MD, of the division of cardiology, Asan Medical Center in Seoul, South Korea.
The trial randomized 73 patients to early surgical aortic valve replacement (SAVR) and 72 to conventional treatment. Baseline characteristics were similar between the two groups, with a mean EuroSCORE II of 0.9, peak aortic-valve jet velocity (Vmax) of 5.1 m/sec, and mean aortic-valve area of 0.63 cm2. Fifty-three conventional treatment patients went on to have SAVR when symptoms developed during follow-up. There were no operation-related deaths in either group, although one early surgery patient had a stroke and one conventional treatment patients had an MI during the operative period.
At a median follow-up of 6.2 years, 1 early-surgery patient (1.4%) and 11 conventional-therapy patients (15.3%) died from operative or cardiovascular death, Dr. Kang said (P = 0.003). “The number needed to treat to prevent one cardiovascular death within 4 years was 20 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidences of operative CV or death were 1.4% for both in the early-surgery group and 5.7% and 25.5% in the conventional-treatment group, Dr. Kang said (P = 0.003).
Rates of all-cause mortality were 6.8% and 20.8% (P = 0.030) in the respective groups. “The number needed to treat to save one life in 4 years was 16 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidence of any-cause death was 4.1% and 10.2% in the early-surgery patients and 9.7% and 31.8% in the conventional group (P = 0.018).
Among the trial limitations Dr. Kang acknowledged were that the population had severe AS with aortic velocity of 4.5 m/sec or greater. “The benefit of early surgery may be relatively smaller in asymptomatic patients with less severe aortic stenosis,” he said.
Not all patients had an exercise test to confirm their asymptomatic status, but as discussant Robert Bonow, MD, of Northwestern University in Chicago pointed out, “this is what we’re dealing with clinically.” Also the mean age of the patient population – 64.9 years – is relatively young, and they had a high incidence of bicuspid aortic valve, few comorbidities, and low operative risk, Dr. Kang said. “Thus, our study population is quite different from the populations enrolled in the TAVR [transcatheter AVR] trials, and the results of our study cannot be directly applied to early TAVR for asymptomatic severe aortic stenosis,” he said.
The mean age in both the PARTNER 3 (N Engl J Med. 2019;380:1695-705) and EVOLUT (N Engl J Med. 2019;380:1706-15) trials comparing TAVR and SAVR in low-risk patients was 73.6 years, and they had higher rates of comorbidities than did the RECOVERY patients.
Dr. Bonow said the RECOVERY findings add to the PARTNER 3 and EVOLUT findings and raise the question about rethinking guideline indications for AVR in asymptomatic severe AS patients. “Should we be talking about moving our Vmax threshold down to 4.5 m/sec, or maybe increasing the Class IIa indications to Class I?” he said. “I think we need to wait to see more data to support these excellent results.”
However, most of these asymptomatic patients do eventually have surgery “in a very short period of time,” Dr. Bonow said. “So from a clinical management point of view, I think we already have the data suggesting that we could move the ball forward, and now we have these excellent outcome data from Korea as well.”
Results were published simultaneously in the New England Journal of Medicine (2019 Nov 16. doi: 10.1056/NEJMoa1912846).
Dr. Kang and Dr. Bonow had no financial relationships to disclose.
PHILADELPHIA – Early aortic valve replacement surgery for patients with asymptomatic, severe aortic stenosis has been a controversial strategy, but the first randomized trial comparing early surgery with conservative management found that early-surgery patients had a 17-fold improved survival at 8 years, according to trial results reported at the American Heart Association scientific sessions.
Albeit small – 145 patients randomized to early surgery or conservative therapy – the RECOVERY trial results provide important evidence to support early preemptive aortic valve replacement for patients with asymptomatic but severe aortic stenosis, said Duk-Hyun Kang, MD, of the division of cardiology, Asan Medical Center in Seoul, South Korea.
The trial randomized 73 patients to early surgical aortic valve replacement (SAVR) and 72 to conventional treatment. Baseline characteristics were similar between the two groups, with a mean EuroSCORE II of 0.9, peak aortic-valve jet velocity (Vmax) of 5.1 m/sec, and mean aortic-valve area of 0.63 cm2. Fifty-three conventional treatment patients went on to have SAVR when symptoms developed during follow-up. There were no operation-related deaths in either group, although one early surgery patient had a stroke and one conventional treatment patients had an MI during the operative period.
At a median follow-up of 6.2 years, 1 early-surgery patient (1.4%) and 11 conventional-therapy patients (15.3%) died from operative or cardiovascular death, Dr. Kang said (P = 0.003). “The number needed to treat to prevent one cardiovascular death within 4 years was 20 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidences of operative CV or death were 1.4% for both in the early-surgery group and 5.7% and 25.5% in the conventional-treatment group, Dr. Kang said (P = 0.003).
Rates of all-cause mortality were 6.8% and 20.8% (P = 0.030) in the respective groups. “The number needed to treat to save one life in 4 years was 16 patients,” Dr. Kang said. At 4 and 8 years, the cumulative incidence of any-cause death was 4.1% and 10.2% in the early-surgery patients and 9.7% and 31.8% in the conventional group (P = 0.018).
Among the trial limitations Dr. Kang acknowledged were that the population had severe AS with aortic velocity of 4.5 m/sec or greater. “The benefit of early surgery may be relatively smaller in asymptomatic patients with less severe aortic stenosis,” he said.
Not all patients had an exercise test to confirm their asymptomatic status, but as discussant Robert Bonow, MD, of Northwestern University in Chicago pointed out, “this is what we’re dealing with clinically.” Also the mean age of the patient population – 64.9 years – is relatively young, and they had a high incidence of bicuspid aortic valve, few comorbidities, and low operative risk, Dr. Kang said. “Thus, our study population is quite different from the populations enrolled in the TAVR [transcatheter AVR] trials, and the results of our study cannot be directly applied to early TAVR for asymptomatic severe aortic stenosis,” he said.
The mean age in both the PARTNER 3 (N Engl J Med. 2019;380:1695-705) and EVOLUT (N Engl J Med. 2019;380:1706-15) trials comparing TAVR and SAVR in low-risk patients was 73.6 years, and they had higher rates of comorbidities than did the RECOVERY patients.
Dr. Bonow said the RECOVERY findings add to the PARTNER 3 and EVOLUT findings and raise the question about rethinking guideline indications for AVR in asymptomatic severe AS patients. “Should we be talking about moving our Vmax threshold down to 4.5 m/sec, or maybe increasing the Class IIa indications to Class I?” he said. “I think we need to wait to see more data to support these excellent results.”
However, most of these asymptomatic patients do eventually have surgery “in a very short period of time,” Dr. Bonow said. “So from a clinical management point of view, I think we already have the data suggesting that we could move the ball forward, and now we have these excellent outcome data from Korea as well.”
Results were published simultaneously in the New England Journal of Medicine (2019 Nov 16. doi: 10.1056/NEJMoa1912846).
Dr. Kang and Dr. Bonow had no financial relationships to disclose.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
Promising early efficacy of venetoclax/navitoclax in r/r acute lymphoblastic leukemia
ORLANDO – A combination of venetoclax (Venclexta) and the experimental BCL-2 inhibitor navitoclax showed good activity and acceptable safety in both children and adults with relapsed or refractory acute lymphoblastic leukemia or lymphoblastic lymphoma, investigators in a phase 1 trial reported.

The overall rate of combined complete response (CR), CR with incomplete marrow recovery (CRi) or incomplete platelet recovery (CRp) was 49% among 45 patients: 24 with acute lymphoblastic leukemia of B-cell lineage (B-ALL), 18 with T-cell lineage ALL (T-ALL), and 3 with lymphoblastic lymphoma, reported Norman J. Lacayo, MD, from Stanford (Calif.) University.
“Venetoclax, navitoclax, and chemotherapy is well tolerated with few discontinuations for dose reductions due to adverse events in patients with relapsed/refractory ALL or lymphoblastic lymphoma. The preliminary efficacy was promising in this heavily pretreated population of patients, including pediatric patients and patients with prior stem cell transplantation of CAR [chimeric antigen receptor] T-cell therapy,” he said at the annual meeting of the American Society of Hematology.
Venetoclax is a highly selective inhibitor of the B-cell lymphoma 2 (BCL-2) pathway. Navitoclax inhibits BCL-2, the BCL–extra large (BCL-XL) transmembrane molecule, and the apoptotic protein BCL-W, but was associated with dose-limiting toxicities when used at standard doses in monotherapy, he noted.
To see whether adding venetoclax to low-dose navitoclax could be safe and have synergistic activity against BCL-2, Dr. Lacayo and colleagues are conducting a phase 1, open-label, dose-escalation study of patients aged 4 years and older with relapsed/refractory ALL or lymphoblastic lymphoma.
Patients receive the weight-adjusted equivalent of 200 mg venetoclax on day 1, and 400 mg equivalent daily thereafter. Beginning on day 3, patients receive oral navitoclax daily at doses of 25, 50, or 100 mg for patients weighing 45 kg or more, or 25 or 50 mg for patients weighing from 20 to less than 45 kg.
Patients can also receive two cycles of chemotherapy with asparaginase, vincristine, and dexamethasone, with additional cycles allowed at the investigators’ discretion.
The patients reported on at ASH 2019 had received a median of 4 prior lines of therapy (range, 1-10).
After a median time on study of 8 months, preliminary efficacy – a secondary endpoint of this phase 1 trial – was promising, Dr. Lacayo said. The CR rate was 25% among patients with B-ALL, 11% in patients with T-ALL, and 67% (two of three patients) with lymphoblastic lymphoma. The respective CRi rates were 13%, 17%, and 0%, and respective CRp rates were 17%, 11%, and 0%.
In addition, 3 of 24 patients (13%) with B-ALL had a partial response, as did 1 patient with lymphoblastic lymphoma.
The median time to first response was about 1.1 months. The median duration of response was 9.1 months for patients with B-ALL, 4.2 months for patients with T-ALL, and had not been reached among patients with lymphoblastic leukemia.
The median overall survival was 9.7 months for patients with B-ALL, 6.8 months for those with T-ALL, and not reached for those with lymphoblastic leukemia.
In all, 6 of 24 patients with B-ALL, 3 of 18 with T-ALL, and 2 of 3 with lymphoblastic leukemia survived long enough to proceed to stem cell transplantation or CAR T-cell therapy.
Analysis of the combination’s safety, the primary endpoint, showed that 58% of patients had grade 3-4 adverse events (AEs) related to venetoclax, and 42% had grade 3-4 AEs related to navitoclax. Four patients had to discontinue the combination because of treatment-related AEs.
Dose-limiting toxicities, which occurred in seven patients, included delayed count recovery, drug-induced liver injury, intestinal ischemia, and increase in serum bilirubin.
One patient died from an intestinal ischemic event deemed to be related to the combination, and seven other patients had fatal adverse events considered not related to the study drugs. The causes of death included sepsis, septic shock, cardiac arrest, and neurotoxicity.
The investigators are enrolling an expansion cohort to see whether a 21-day dosing schedule of venetoclax with 50 mg navitoclax, or 25 mg for patients under 45 kg, could improve count recovery time.
The study was funded by AbbVie. Dr. Lacayo reported having no conflict of interests to disclose. Several coauthors are AbbVie employees.
SOURCE: Lacayo NJ et al. ASH 2019, Abstract 285.
ORLANDO – A combination of venetoclax (Venclexta) and the experimental BCL-2 inhibitor navitoclax showed good activity and acceptable safety in both children and adults with relapsed or refractory acute lymphoblastic leukemia or lymphoblastic lymphoma, investigators in a phase 1 trial reported.
The overall rate of combined complete response (CR), CR with incomplete marrow recovery (CRi) or incomplete platelet recovery (CRp) was 49% among 45 patients: 24 with acute lymphoblastic leukemia of B-cell lineage (B-ALL), 18 with T-cell lineage ALL (T-ALL), and 3 with lymphoblastic lymphoma, reported Norman J. Lacayo, MD, from Stanford (Calif.) University.
“Venetoclax, navitoclax, and chemotherapy is well tolerated with few discontinuations for dose reductions due to adverse events in patients with relapsed/refractory ALL or lymphoblastic lymphoma. The preliminary efficacy was promising in this heavily pretreated population of patients, including pediatric patients and patients with prior stem cell transplantation of CAR [chimeric antigen receptor] T-cell therapy,” he said at the annual meeting of the American Society of Hematology.
Venetoclax is a highly selective inhibitor of the B-cell lymphoma 2 (BCL-2) pathway. Navitoclax inhibits BCL-2, the BCL–extra large (BCL-XL) transmembrane molecule, and the apoptotic protein BCL-W, but was associated with dose-limiting toxicities when used at standard doses in monotherapy, he noted.
To see whether adding venetoclax to low-dose navitoclax could be safe and have synergistic activity against BCL-2, Dr. Lacayo and colleagues are conducting a phase 1, open-label, dose-escalation study of patients aged 4 years and older with relapsed/refractory ALL or lymphoblastic lymphoma.
Patients receive the weight-adjusted equivalent of 200 mg venetoclax on day 1, and 400 mg equivalent daily thereafter. Beginning on day 3, patients receive oral navitoclax daily at doses of 25, 50, or 100 mg for patients weighing 45 kg or more, or 25 or 50 mg for patients weighing from 20 to less than 45 kg.
Patients can also receive two cycles of chemotherapy with asparaginase, vincristine, and dexamethasone, with additional cycles allowed at the investigators’ discretion.
The patients reported on at ASH 2019 had received a median of 4 prior lines of therapy (range, 1-10).
After a median time on study of 8 months, preliminary efficacy – a secondary endpoint of this phase 1 trial – was promising, Dr. Lacayo said. The CR rate was 25% among patients with B-ALL, 11% in patients with T-ALL, and 67% (two of three patients) with lymphoblastic lymphoma. The respective CRi rates were 13%, 17%, and 0%, and respective CRp rates were 17%, 11%, and 0%.
In addition, 3 of 24 patients (13%) with B-ALL had a partial response, as did 1 patient with lymphoblastic lymphoma.
The median time to first response was about 1.1 months. The median duration of response was 9.1 months for patients with B-ALL, 4.2 months for patients with T-ALL, and had not been reached among patients with lymphoblastic leukemia.
The median overall survival was 9.7 months for patients with B-ALL, 6.8 months for those with T-ALL, and not reached for those with lymphoblastic leukemia.
In all, 6 of 24 patients with B-ALL, 3 of 18 with T-ALL, and 2 of 3 with lymphoblastic leukemia survived long enough to proceed to stem cell transplantation or CAR T-cell therapy.
Analysis of the combination’s safety, the primary endpoint, showed that 58% of patients had grade 3-4 adverse events (AEs) related to venetoclax, and 42% had grade 3-4 AEs related to navitoclax. Four patients had to discontinue the combination because of treatment-related AEs.
Dose-limiting toxicities, which occurred in seven patients, included delayed count recovery, drug-induced liver injury, intestinal ischemia, and increase in serum bilirubin.
One patient died from an intestinal ischemic event deemed to be related to the combination, and seven other patients had fatal adverse events considered not related to the study drugs. The causes of death included sepsis, septic shock, cardiac arrest, and neurotoxicity.
The investigators are enrolling an expansion cohort to see whether a 21-day dosing schedule of venetoclax with 50 mg navitoclax, or 25 mg for patients under 45 kg, could improve count recovery time.
The study was funded by AbbVie. Dr. Lacayo reported having no conflict of interests to disclose. Several coauthors are AbbVie employees.
SOURCE: Lacayo NJ et al. ASH 2019, Abstract 285.
ORLANDO – A combination of venetoclax (Venclexta) and the experimental BCL-2 inhibitor navitoclax showed good activity and acceptable safety in both children and adults with relapsed or refractory acute lymphoblastic leukemia or lymphoblastic lymphoma, investigators in a phase 1 trial reported.
The overall rate of combined complete response (CR), CR with incomplete marrow recovery (CRi) or incomplete platelet recovery (CRp) was 49% among 45 patients: 24 with acute lymphoblastic leukemia of B-cell lineage (B-ALL), 18 with T-cell lineage ALL (T-ALL), and 3 with lymphoblastic lymphoma, reported Norman J. Lacayo, MD, from Stanford (Calif.) University.
“Venetoclax, navitoclax, and chemotherapy is well tolerated with few discontinuations for dose reductions due to adverse events in patients with relapsed/refractory ALL or lymphoblastic lymphoma. The preliminary efficacy was promising in this heavily pretreated population of patients, including pediatric patients and patients with prior stem cell transplantation of CAR [chimeric antigen receptor] T-cell therapy,” he said at the annual meeting of the American Society of Hematology.
Venetoclax is a highly selective inhibitor of the B-cell lymphoma 2 (BCL-2) pathway. Navitoclax inhibits BCL-2, the BCL–extra large (BCL-XL) transmembrane molecule, and the apoptotic protein BCL-W, but was associated with dose-limiting toxicities when used at standard doses in monotherapy, he noted.
To see whether adding venetoclax to low-dose navitoclax could be safe and have synergistic activity against BCL-2, Dr. Lacayo and colleagues are conducting a phase 1, open-label, dose-escalation study of patients aged 4 years and older with relapsed/refractory ALL or lymphoblastic lymphoma.
Patients receive the weight-adjusted equivalent of 200 mg venetoclax on day 1, and 400 mg equivalent daily thereafter. Beginning on day 3, patients receive oral navitoclax daily at doses of 25, 50, or 100 mg for patients weighing 45 kg or more, or 25 or 50 mg for patients weighing from 20 to less than 45 kg.
Patients can also receive two cycles of chemotherapy with asparaginase, vincristine, and dexamethasone, with additional cycles allowed at the investigators’ discretion.
The patients reported on at ASH 2019 had received a median of 4 prior lines of therapy (range, 1-10).
After a median time on study of 8 months, preliminary efficacy – a secondary endpoint of this phase 1 trial – was promising, Dr. Lacayo said. The CR rate was 25% among patients with B-ALL, 11% in patients with T-ALL, and 67% (two of three patients) with lymphoblastic lymphoma. The respective CRi rates were 13%, 17%, and 0%, and respective CRp rates were 17%, 11%, and 0%.
In addition, 3 of 24 patients (13%) with B-ALL had a partial response, as did 1 patient with lymphoblastic lymphoma.
The median time to first response was about 1.1 months. The median duration of response was 9.1 months for patients with B-ALL, 4.2 months for patients with T-ALL, and had not been reached among patients with lymphoblastic leukemia.
The median overall survival was 9.7 months for patients with B-ALL, 6.8 months for those with T-ALL, and not reached for those with lymphoblastic leukemia.
In all, 6 of 24 patients with B-ALL, 3 of 18 with T-ALL, and 2 of 3 with lymphoblastic leukemia survived long enough to proceed to stem cell transplantation or CAR T-cell therapy.
Analysis of the combination’s safety, the primary endpoint, showed that 58% of patients had grade 3-4 adverse events (AEs) related to venetoclax, and 42% had grade 3-4 AEs related to navitoclax. Four patients had to discontinue the combination because of treatment-related AEs.
Dose-limiting toxicities, which occurred in seven patients, included delayed count recovery, drug-induced liver injury, intestinal ischemia, and increase in serum bilirubin.
One patient died from an intestinal ischemic event deemed to be related to the combination, and seven other patients had fatal adverse events considered not related to the study drugs. The causes of death included sepsis, septic shock, cardiac arrest, and neurotoxicity.
The investigators are enrolling an expansion cohort to see whether a 21-day dosing schedule of venetoclax with 50 mg navitoclax, or 25 mg for patients under 45 kg, could improve count recovery time.
The study was funded by AbbVie. Dr. Lacayo reported having no conflict of interests to disclose. Several coauthors are AbbVie employees.
SOURCE: Lacayo NJ et al. ASH 2019, Abstract 285.
REPORTING FROM ASH 2019
Testosterone gel increases LV mass in older men
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.

“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.
“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.
“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
REPORTING FROM AHA 2019
Turoctocog alfa prevented, treated bleeds in previously untreated pediatric patients with severe hemophilia A
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.

Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.
Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.
Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
FROM HAEMOPHILIA
Coronary function testing during angiography boosts 1-year angina outcomes
PHILADELPHIA – Longer and more extensive follow-up of the positive CorMicA trial confirmed that clinicians who receive added information on the presence or absence of microvascular coronary disease or coronary vasospasm in patients with chronic, stable angina but without detectable coronary obstruction adjust patient treatment in ways that produce better outcomes.

For example, expanded 1-year follow-up of the 151-patient, multicenter, CorMicA randomized study showed that stable angina patients without coronary obstruction treated by clinicians aware of microvascular or vasospastic coronary disease had blood pressures that averaged about 12/5 mm Hg lower than those of patients in the control arm, in which this information was kept blinded.
Clinicians in the study who received information about diagnoses of microvascular angina, vasospastic angina, both, or neither, generally better tailored the treatments they gave these patients, were more likely to prescribe cardiac rehabilitation to these patients, and successfully capped increases in blood pressure that occurred in the control patients, Colin Berry, MD, said at the American Heart Association scientific sessions. The investigators made the diagnoses of microvascular and vasospastic angina using an “invasive diagnostic procedure” that involved placing a wire with pressure and temperature sensors in a selected coronary artery and measuring flow patterns that resulted from a systemic infusion of adenosine and from incremental, intracoronary infusions of acetylcholine.
The CorMicA results “tell us that, if we use existing drugs and nonpharmacologic treatments, we can improve patients’ quality of life,” said Dr. Berry, co-lead investigator of the study and professor of cardiology and imaging at the University of Glasgow. “There were probably two drivers of change in treatment” between the intervention arm where clinicians learned whether their patients had microvascular angina or vasospasm, and the control arm where this information was kept blinded. Some patients reclassified as having microvascular disease or vasospasm were restarted on standard agents for treatment of coronary artery disease such as statins and ACE inhibitors that had previously been stopped, noted Dr. Berry. The identification of a specific type of vascular disorder also helped guide treatment. For example, patients identified as having vasospasm were taken off of beta-blockers, which are contraindicated in these patients, and instead began treatment with a calcium channel antagonist, he said.
“CorMicA was a landmark trial. The interventional cardiologists at my center believe that the testing [for microvascular and vasospastic angina] should be used routinely, based on the reported data,” said Harmony R. Reynolds, MD, a cardiologist at NYU Langone Health in New York.
The CorMicA (Coronary Microvascular Angina) trial enrolled 391 patients at either of two hospitals in Scotland during November 2016–November 2017. All patients were scheduled for clinically indicated, elective diagnostic angiography for suspected stable angina. Clinicians identified a coronary obstruction in 206 patients that excluded them from the main CorMicA analysis. They focused on the 181 without coronary obstruction visible by angiography, and specifically the 151 of these patients who underwent the invasive diagnostic procedure, with 76 randomized to have their diagnostic-procedure results relayed to their treating physicians and 75 patients whose diagnostic results remained blinded. The study’s primary endpoint was the mean difference in angina severity at 6 months after treatment assessed by the Seattle Angina Questionnaire (SAQ) summary score. After 6 months, the between-group difference in average SAQ summary scores was 11.7 units, a statistically significant as well as clinically meaningful benefit that linked with knowledge of the coronary function of patients based on invasive testing (J Am Coll Cardiol. 2018 Dec 11;72[23 Part A]:2841-55).
The prespecified 1-year follow-up with another SAQ took place for 142 of the 151 randomized patients, and showed ongoing separation of SAQ scores between the intervention and control patients, reaching a statistically significant mean difference of 13.6 units, Dr. Berry reported. Other benefits that linked with dissemination of results from the invasive diagnostic procedure included maintenance of the quality-of-life benefit seen after 6 months, an incremental further reduction in illness perception from 6 to 12 months, and a substantial further improvement in global treatment satisfaction, which grew from a 30% higher level in the intervention group compared with controls at 6 months to a 44% between-group difference after 12 months. Concurrently with Dr. Berry’s report, the results appeared online (JACC: Cardiovasc Interv. 2020 Jan 13;13[1] 33-45).
Additional findings for various clinical measures that were not part of the 6-month findings provided further insight into the impact that clinician knowledge about microvascular disease or vasospasm had on patients. These metrics suggested that the benefits in the intervention patients may have largely resulted from their avoiding elevations in risk markers that occurred among controls who received routine care.
For example, after 12 months, average 12/5 mm Hg differences in systolic and diastolic blood pressures between the two arms was nearly all because of pressure increases from baseline in the control patients with only very small drops in pressure from baseline among the intervention patients. Weight measurements after 1 year showed that the control patients were on average 1.3 kg heavier than the intervention patients, again mostly because of weight increases among the controls. Enrollment in a cardiac rehabilitation program at 12 months had occurred for 40% of the intervention patients and 16% of the controls, a 73% relative increase in the intervention arm, Dr. Berry reported.
The invasive, coronary, physiological assessments used in CorMicA led to stratified medical therapy and created an opportunity for better long-term angina treatment in patients without obstructive coronary disease, Dr. Berry concluded. He stressed the need to further test this hypothesis in larger, prospective studies, and to assess its cost effectiveness. But the proof-of-concept demonstrated by the CorMicA results indicate a role for more thorough investigation of coronary dysfunction when obstructive disease is absent, with the opportunity to use this information to better tailor treatment. The findings also highlight the potential for new drug development that can address nonobstructive microvascular or vasospastic coronary disease, he said.
Dr. Berry has been a speaker on behalf of Abbott Vascular, and he has received research funding from AstraZeneca, GlaxoSmithKline, HeartFlow, Novartis, and Siemens Healthcare. Dr. Reynolds had no disclosures.
SOURCE: Ford TJ. AHA 2019, session FS.AOS.03 abstract.
PHILADELPHIA – Longer and more extensive follow-up of the positive CorMicA trial confirmed that clinicians who receive added information on the presence or absence of microvascular coronary disease or coronary vasospasm in patients with chronic, stable angina but without detectable coronary obstruction adjust patient treatment in ways that produce better outcomes.
For example, expanded 1-year follow-up of the 151-patient, multicenter, CorMicA randomized study showed that stable angina patients without coronary obstruction treated by clinicians aware of microvascular or vasospastic coronary disease had blood pressures that averaged about 12/5 mm Hg lower than those of patients in the control arm, in which this information was kept blinded.
Clinicians in the study who received information about diagnoses of microvascular angina, vasospastic angina, both, or neither, generally better tailored the treatments they gave these patients, were more likely to prescribe cardiac rehabilitation to these patients, and successfully capped increases in blood pressure that occurred in the control patients, Colin Berry, MD, said at the American Heart Association scientific sessions. The investigators made the diagnoses of microvascular and vasospastic angina using an “invasive diagnostic procedure” that involved placing a wire with pressure and temperature sensors in a selected coronary artery and measuring flow patterns that resulted from a systemic infusion of adenosine and from incremental, intracoronary infusions of acetylcholine.
The CorMicA results “tell us that, if we use existing drugs and nonpharmacologic treatments, we can improve patients’ quality of life,” said Dr. Berry, co-lead investigator of the study and professor of cardiology and imaging at the University of Glasgow. “There were probably two drivers of change in treatment” between the intervention arm where clinicians learned whether their patients had microvascular angina or vasospasm, and the control arm where this information was kept blinded. Some patients reclassified as having microvascular disease or vasospasm were restarted on standard agents for treatment of coronary artery disease such as statins and ACE inhibitors that had previously been stopped, noted Dr. Berry. The identification of a specific type of vascular disorder also helped guide treatment. For example, patients identified as having vasospasm were taken off of beta-blockers, which are contraindicated in these patients, and instead began treatment with a calcium channel antagonist, he said.
“CorMicA was a landmark trial. The interventional cardiologists at my center believe that the testing [for microvascular and vasospastic angina] should be used routinely, based on the reported data,” said Harmony R. Reynolds, MD, a cardiologist at NYU Langone Health in New York.
The CorMicA (Coronary Microvascular Angina) trial enrolled 391 patients at either of two hospitals in Scotland during November 2016–November 2017. All patients were scheduled for clinically indicated, elective diagnostic angiography for suspected stable angina. Clinicians identified a coronary obstruction in 206 patients that excluded them from the main CorMicA analysis. They focused on the 181 without coronary obstruction visible by angiography, and specifically the 151 of these patients who underwent the invasive diagnostic procedure, with 76 randomized to have their diagnostic-procedure results relayed to their treating physicians and 75 patients whose diagnostic results remained blinded. The study’s primary endpoint was the mean difference in angina severity at 6 months after treatment assessed by the Seattle Angina Questionnaire (SAQ) summary score. After 6 months, the between-group difference in average SAQ summary scores was 11.7 units, a statistically significant as well as clinically meaningful benefit that linked with knowledge of the coronary function of patients based on invasive testing (J Am Coll Cardiol. 2018 Dec 11;72[23 Part A]:2841-55).
The prespecified 1-year follow-up with another SAQ took place for 142 of the 151 randomized patients, and showed ongoing separation of SAQ scores between the intervention and control patients, reaching a statistically significant mean difference of 13.6 units, Dr. Berry reported. Other benefits that linked with dissemination of results from the invasive diagnostic procedure included maintenance of the quality-of-life benefit seen after 6 months, an incremental further reduction in illness perception from 6 to 12 months, and a substantial further improvement in global treatment satisfaction, which grew from a 30% higher level in the intervention group compared with controls at 6 months to a 44% between-group difference after 12 months. Concurrently with Dr. Berry’s report, the results appeared online (JACC: Cardiovasc Interv. 2020 Jan 13;13[1] 33-45).
Additional findings for various clinical measures that were not part of the 6-month findings provided further insight into the impact that clinician knowledge about microvascular disease or vasospasm had on patients. These metrics suggested that the benefits in the intervention patients may have largely resulted from their avoiding elevations in risk markers that occurred among controls who received routine care.
For example, after 12 months, average 12/5 mm Hg differences in systolic and diastolic blood pressures between the two arms was nearly all because of pressure increases from baseline in the control patients with only very small drops in pressure from baseline among the intervention patients. Weight measurements after 1 year showed that the control patients were on average 1.3 kg heavier than the intervention patients, again mostly because of weight increases among the controls. Enrollment in a cardiac rehabilitation program at 12 months had occurred for 40% of the intervention patients and 16% of the controls, a 73% relative increase in the intervention arm, Dr. Berry reported.
The invasive, coronary, physiological assessments used in CorMicA led to stratified medical therapy and created an opportunity for better long-term angina treatment in patients without obstructive coronary disease, Dr. Berry concluded. He stressed the need to further test this hypothesis in larger, prospective studies, and to assess its cost effectiveness. But the proof-of-concept demonstrated by the CorMicA results indicate a role for more thorough investigation of coronary dysfunction when obstructive disease is absent, with the opportunity to use this information to better tailor treatment. The findings also highlight the potential for new drug development that can address nonobstructive microvascular or vasospastic coronary disease, he said.
Dr. Berry has been a speaker on behalf of Abbott Vascular, and he has received research funding from AstraZeneca, GlaxoSmithKline, HeartFlow, Novartis, and Siemens Healthcare. Dr. Reynolds had no disclosures.
SOURCE: Ford TJ. AHA 2019, session FS.AOS.03 abstract.
PHILADELPHIA – Longer and more extensive follow-up of the positive CorMicA trial confirmed that clinicians who receive added information on the presence or absence of microvascular coronary disease or coronary vasospasm in patients with chronic, stable angina but without detectable coronary obstruction adjust patient treatment in ways that produce better outcomes.
For example, expanded 1-year follow-up of the 151-patient, multicenter, CorMicA randomized study showed that stable angina patients without coronary obstruction treated by clinicians aware of microvascular or vasospastic coronary disease had blood pressures that averaged about 12/5 mm Hg lower than those of patients in the control arm, in which this information was kept blinded.
Clinicians in the study who received information about diagnoses of microvascular angina, vasospastic angina, both, or neither, generally better tailored the treatments they gave these patients, were more likely to prescribe cardiac rehabilitation to these patients, and successfully capped increases in blood pressure that occurred in the control patients, Colin Berry, MD, said at the American Heart Association scientific sessions. The investigators made the diagnoses of microvascular and vasospastic angina using an “invasive diagnostic procedure” that involved placing a wire with pressure and temperature sensors in a selected coronary artery and measuring flow patterns that resulted from a systemic infusion of adenosine and from incremental, intracoronary infusions of acetylcholine.
The CorMicA results “tell us that, if we use existing drugs and nonpharmacologic treatments, we can improve patients’ quality of life,” said Dr. Berry, co-lead investigator of the study and professor of cardiology and imaging at the University of Glasgow. “There were probably two drivers of change in treatment” between the intervention arm where clinicians learned whether their patients had microvascular angina or vasospasm, and the control arm where this information was kept blinded. Some patients reclassified as having microvascular disease or vasospasm were restarted on standard agents for treatment of coronary artery disease such as statins and ACE inhibitors that had previously been stopped, noted Dr. Berry. The identification of a specific type of vascular disorder also helped guide treatment. For example, patients identified as having vasospasm were taken off of beta-blockers, which are contraindicated in these patients, and instead began treatment with a calcium channel antagonist, he said.
“CorMicA was a landmark trial. The interventional cardiologists at my center believe that the testing [for microvascular and vasospastic angina] should be used routinely, based on the reported data,” said Harmony R. Reynolds, MD, a cardiologist at NYU Langone Health in New York.
The CorMicA (Coronary Microvascular Angina) trial enrolled 391 patients at either of two hospitals in Scotland during November 2016–November 2017. All patients were scheduled for clinically indicated, elective diagnostic angiography for suspected stable angina. Clinicians identified a coronary obstruction in 206 patients that excluded them from the main CorMicA analysis. They focused on the 181 without coronary obstruction visible by angiography, and specifically the 151 of these patients who underwent the invasive diagnostic procedure, with 76 randomized to have their diagnostic-procedure results relayed to their treating physicians and 75 patients whose diagnostic results remained blinded. The study’s primary endpoint was the mean difference in angina severity at 6 months after treatment assessed by the Seattle Angina Questionnaire (SAQ) summary score. After 6 months, the between-group difference in average SAQ summary scores was 11.7 units, a statistically significant as well as clinically meaningful benefit that linked with knowledge of the coronary function of patients based on invasive testing (J Am Coll Cardiol. 2018 Dec 11;72[23 Part A]:2841-55).
The prespecified 1-year follow-up with another SAQ took place for 142 of the 151 randomized patients, and showed ongoing separation of SAQ scores between the intervention and control patients, reaching a statistically significant mean difference of 13.6 units, Dr. Berry reported. Other benefits that linked with dissemination of results from the invasive diagnostic procedure included maintenance of the quality-of-life benefit seen after 6 months, an incremental further reduction in illness perception from 6 to 12 months, and a substantial further improvement in global treatment satisfaction, which grew from a 30% higher level in the intervention group compared with controls at 6 months to a 44% between-group difference after 12 months. Concurrently with Dr. Berry’s report, the results appeared online (JACC: Cardiovasc Interv. 2020 Jan 13;13[1] 33-45).
Additional findings for various clinical measures that were not part of the 6-month findings provided further insight into the impact that clinician knowledge about microvascular disease or vasospasm had on patients. These metrics suggested that the benefits in the intervention patients may have largely resulted from their avoiding elevations in risk markers that occurred among controls who received routine care.
For example, after 12 months, average 12/5 mm Hg differences in systolic and diastolic blood pressures between the two arms was nearly all because of pressure increases from baseline in the control patients with only very small drops in pressure from baseline among the intervention patients. Weight measurements after 1 year showed that the control patients were on average 1.3 kg heavier than the intervention patients, again mostly because of weight increases among the controls. Enrollment in a cardiac rehabilitation program at 12 months had occurred for 40% of the intervention patients and 16% of the controls, a 73% relative increase in the intervention arm, Dr. Berry reported.
The invasive, coronary, physiological assessments used in CorMicA led to stratified medical therapy and created an opportunity for better long-term angina treatment in patients without obstructive coronary disease, Dr. Berry concluded. He stressed the need to further test this hypothesis in larger, prospective studies, and to assess its cost effectiveness. But the proof-of-concept demonstrated by the CorMicA results indicate a role for more thorough investigation of coronary dysfunction when obstructive disease is absent, with the opportunity to use this information to better tailor treatment. The findings also highlight the potential for new drug development that can address nonobstructive microvascular or vasospastic coronary disease, he said.
Dr. Berry has been a speaker on behalf of Abbott Vascular, and he has received research funding from AstraZeneca, GlaxoSmithKline, HeartFlow, Novartis, and Siemens Healthcare. Dr. Reynolds had no disclosures.
SOURCE: Ford TJ. AHA 2019, session FS.AOS.03 abstract.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
REGN1979 shows good activity in pretreated aggressive B-NHL
ORLANDO – A novel bispecific antibody directed against CD20 and CD3 was associated in a phase 1 trial with a high overall response rate among patients with relapsed or refractory B-cell non-Hodgkin lymphomas in early clinical trials, including patients with diffuse large B-cell lymphoma (DLBCL) that had progressed following chimeric antigen receptor (CAR) T-cell therapy.

Among 22 patients previously treated for relapsed/refractory follicular lymphoma of grade 1-3a, there were 21 responses (95%), including 17 complete responses (CR) and 4 partial responses (PR), with the remaining patient having stable disease at 12 weeks of follow-up, reported Rajat Bannerji, MD, PhD, from the Rutgers Cancer Institute of New Jersey in New Brunswick.
“We had activity that was fairly robust in this heavily pretreated population with follicular lymphoma, large-cell patients who had not received CAR T and large-cell patients who had received CAR T, mantle cell, and marginal zone [lymphoma],” he said at the annual meeting of the American Society of Hematology.
REGN1979 is an anti-CD20 and anti-CD3 bispecific IgG4 antibody. It is designed to cross-link and activate CD3-expressing T cells on contact with CD20-positive B cells to kill CD20-positive tumor cells independent of T-cell receptor recognition.
The antibody is administered via an escalating dose schedule consisting of initial, intermediate, and step-up doses.
In addition to the follicular lymphoma response rates noted before, patients with heavily pretreated DLBCL who received the antibody at a dose of 80 mg or higher had an overall response rate of 57.9% (11 patients) including 42.1% CR (8 patients), and 15.8% PR (3 patients). Two patients had stable disease at the 12-week assessment, three had disease progression, and three were not available for assessment.
Among seven patients with DLBCL treated at 80 mg or above who had not received CAR T therapy, five had a CR, one had stable disease, and one had disease progression. Of 12 patients with prior CAR T exposure, 3 had complete responses, 3 had partial responses, 1 had stable disease, 2 had progressive disease, and 3 were not available for assessment.
Among six patients with mantle cell lymphoma and six with marginal zone lymphoma treated across all disease levels, the ORR in each cohort was 67%, with two of six patients in each cohort having a complete response, and two having a partial response.
The safety analysis of all 110 patients enrolled showed that no patients experience a dose-limiting toxicity during the escalation phase, and no maximum tolerated doses were identified.
The most common treatment-related adverse events (AEs) were pyrexia in 88 patients, cytokine release syndrome in 65, chills in 56, fatigue in 40, and anemia in 39.
The most common grade 3-4 AEs were anemia in 24, and hypophosphatemia, lymphopenia, and neutropenia in 21 patients each.
Neurologic AEs were transient and did not require treatment discontinuation, and there were no grade 4 neurologic AEs or deaths from neurologic side effects.
Six patients discontinued the study drug because of treatment-related AEs that included cytomegalovirus infection, grade 3 hemolysis, fatigue, pneumonia, and toxoplasmosis.
A total of 15 patients died during the study, 10 of which were caused by progressive disease, with other deaths caused by gastric perforation, cardiac arrest, lung infection, pneumonia, and 1 from fungal pneumonia 7 months after treatment discontinuation. In addition, after the data cutoff, one patient with mantle cell lymphoma blastoid variant with bone-marrow involvement and bulky disease who was enrolled in an expansion cohort died from tumor lysis syndrome.
The dose-escalation portion of the trial has been completed and expansion cohorts are being enrolled. In addition, REGN1979 is being investigated in a phase 2 global multiarm trial.
The study is supported by Regeneron Pharmaceuticals. Dr. Bannerji reported research funding, travel support, and consulting fees from Regeneron and others.
SOURCE: Bannerji R et al. ASH 2019, Abstract 762.
ORLANDO – A novel bispecific antibody directed against CD20 and CD3 was associated in a phase 1 trial with a high overall response rate among patients with relapsed or refractory B-cell non-Hodgkin lymphomas in early clinical trials, including patients with diffuse large B-cell lymphoma (DLBCL) that had progressed following chimeric antigen receptor (CAR) T-cell therapy.
Among 22 patients previously treated for relapsed/refractory follicular lymphoma of grade 1-3a, there were 21 responses (95%), including 17 complete responses (CR) and 4 partial responses (PR), with the remaining patient having stable disease at 12 weeks of follow-up, reported Rajat Bannerji, MD, PhD, from the Rutgers Cancer Institute of New Jersey in New Brunswick.
“We had activity that was fairly robust in this heavily pretreated population with follicular lymphoma, large-cell patients who had not received CAR T and large-cell patients who had received CAR T, mantle cell, and marginal zone [lymphoma],” he said at the annual meeting of the American Society of Hematology.
REGN1979 is an anti-CD20 and anti-CD3 bispecific IgG4 antibody. It is designed to cross-link and activate CD3-expressing T cells on contact with CD20-positive B cells to kill CD20-positive tumor cells independent of T-cell receptor recognition.
The antibody is administered via an escalating dose schedule consisting of initial, intermediate, and step-up doses.
In addition to the follicular lymphoma response rates noted before, patients with heavily pretreated DLBCL who received the antibody at a dose of 80 mg or higher had an overall response rate of 57.9% (11 patients) including 42.1% CR (8 patients), and 15.8% PR (3 patients). Two patients had stable disease at the 12-week assessment, three had disease progression, and three were not available for assessment.
Among seven patients with DLBCL treated at 80 mg or above who had not received CAR T therapy, five had a CR, one had stable disease, and one had disease progression. Of 12 patients with prior CAR T exposure, 3 had complete responses, 3 had partial responses, 1 had stable disease, 2 had progressive disease, and 3 were not available for assessment.
Among six patients with mantle cell lymphoma and six with marginal zone lymphoma treated across all disease levels, the ORR in each cohort was 67%, with two of six patients in each cohort having a complete response, and two having a partial response.
The safety analysis of all 110 patients enrolled showed that no patients experience a dose-limiting toxicity during the escalation phase, and no maximum tolerated doses were identified.
The most common treatment-related adverse events (AEs) were pyrexia in 88 patients, cytokine release syndrome in 65, chills in 56, fatigue in 40, and anemia in 39.
The most common grade 3-4 AEs were anemia in 24, and hypophosphatemia, lymphopenia, and neutropenia in 21 patients each.
Neurologic AEs were transient and did not require treatment discontinuation, and there were no grade 4 neurologic AEs or deaths from neurologic side effects.
Six patients discontinued the study drug because of treatment-related AEs that included cytomegalovirus infection, grade 3 hemolysis, fatigue, pneumonia, and toxoplasmosis.
A total of 15 patients died during the study, 10 of which were caused by progressive disease, with other deaths caused by gastric perforation, cardiac arrest, lung infection, pneumonia, and 1 from fungal pneumonia 7 months after treatment discontinuation. In addition, after the data cutoff, one patient with mantle cell lymphoma blastoid variant with bone-marrow involvement and bulky disease who was enrolled in an expansion cohort died from tumor lysis syndrome.
The dose-escalation portion of the trial has been completed and expansion cohorts are being enrolled. In addition, REGN1979 is being investigated in a phase 2 global multiarm trial.
The study is supported by Regeneron Pharmaceuticals. Dr. Bannerji reported research funding, travel support, and consulting fees from Regeneron and others.
SOURCE: Bannerji R et al. ASH 2019, Abstract 762.
ORLANDO – A novel bispecific antibody directed against CD20 and CD3 was associated in a phase 1 trial with a high overall response rate among patients with relapsed or refractory B-cell non-Hodgkin lymphomas in early clinical trials, including patients with diffuse large B-cell lymphoma (DLBCL) that had progressed following chimeric antigen receptor (CAR) T-cell therapy.
Among 22 patients previously treated for relapsed/refractory follicular lymphoma of grade 1-3a, there were 21 responses (95%), including 17 complete responses (CR) and 4 partial responses (PR), with the remaining patient having stable disease at 12 weeks of follow-up, reported Rajat Bannerji, MD, PhD, from the Rutgers Cancer Institute of New Jersey in New Brunswick.
“We had activity that was fairly robust in this heavily pretreated population with follicular lymphoma, large-cell patients who had not received CAR T and large-cell patients who had received CAR T, mantle cell, and marginal zone [lymphoma],” he said at the annual meeting of the American Society of Hematology.
REGN1979 is an anti-CD20 and anti-CD3 bispecific IgG4 antibody. It is designed to cross-link and activate CD3-expressing T cells on contact with CD20-positive B cells to kill CD20-positive tumor cells independent of T-cell receptor recognition.
The antibody is administered via an escalating dose schedule consisting of initial, intermediate, and step-up doses.
In addition to the follicular lymphoma response rates noted before, patients with heavily pretreated DLBCL who received the antibody at a dose of 80 mg or higher had an overall response rate of 57.9% (11 patients) including 42.1% CR (8 patients), and 15.8% PR (3 patients). Two patients had stable disease at the 12-week assessment, three had disease progression, and three were not available for assessment.
Among seven patients with DLBCL treated at 80 mg or above who had not received CAR T therapy, five had a CR, one had stable disease, and one had disease progression. Of 12 patients with prior CAR T exposure, 3 had complete responses, 3 had partial responses, 1 had stable disease, 2 had progressive disease, and 3 were not available for assessment.
Among six patients with mantle cell lymphoma and six with marginal zone lymphoma treated across all disease levels, the ORR in each cohort was 67%, with two of six patients in each cohort having a complete response, and two having a partial response.
The safety analysis of all 110 patients enrolled showed that no patients experience a dose-limiting toxicity during the escalation phase, and no maximum tolerated doses were identified.
The most common treatment-related adverse events (AEs) were pyrexia in 88 patients, cytokine release syndrome in 65, chills in 56, fatigue in 40, and anemia in 39.
The most common grade 3-4 AEs were anemia in 24, and hypophosphatemia, lymphopenia, and neutropenia in 21 patients each.
Neurologic AEs were transient and did not require treatment discontinuation, and there were no grade 4 neurologic AEs or deaths from neurologic side effects.
Six patients discontinued the study drug because of treatment-related AEs that included cytomegalovirus infection, grade 3 hemolysis, fatigue, pneumonia, and toxoplasmosis.
A total of 15 patients died during the study, 10 of which were caused by progressive disease, with other deaths caused by gastric perforation, cardiac arrest, lung infection, pneumonia, and 1 from fungal pneumonia 7 months after treatment discontinuation. In addition, after the data cutoff, one patient with mantle cell lymphoma blastoid variant with bone-marrow involvement and bulky disease who was enrolled in an expansion cohort died from tumor lysis syndrome.
The dose-escalation portion of the trial has been completed and expansion cohorts are being enrolled. In addition, REGN1979 is being investigated in a phase 2 global multiarm trial.
The study is supported by Regeneron Pharmaceuticals. Dr. Bannerji reported research funding, travel support, and consulting fees from Regeneron and others.
SOURCE: Bannerji R et al. ASH 2019, Abstract 762.
REPORTING FROM ASH 2019
FDA committee rejects oxycodegol, new opioid designed for less abuse
A Food and Drug Administration advisory committee voted 27-0 on Jan. 14 against recommending agency approval of oxycodegol, a new type of opioid molecule designed to slow passage of the drug through the blood-brain barrier and hence cause less of a “high” and potentially blunt the drug’s abuse potential.

But the panel, which includes members from both the Anesthetic and Analgesic Drug Products Advisory Committee and the Drug Safety and Risk Management Advisory Committee, unanimously shared the opinion that the data submitted by the drug developer, Nektar, failed to clearly demonstrate the drug’s safety and efficacy.
“There was a lot of ambiguity [in the data] about safety and efficacy, and this is too important an issue to have so much ambiguity,” summed up Ronald S. Litman, DO, professor of anesthesiology and critical care at the University of Pennsylvania in Philadelphia and chair of the combined committee.
“The subliminal message” that would surround oxycodegol if it received FDA approval would be that it is a “safer” opioid, “and that would make it a blockbuster drug, and for that to happen you need data that [prove] the benefits outweigh risks, but we didn’t see those data. It was all speculation, and we can’t approve a potential blockbuster opioid in the middle of a public health opioid crisis based on speculation,” commented Steve B. Meisel, PharmD, system director of medication safety for Fairview Health Services in Minneapolis and a committee member.
According to Nektar, oxycodegol is a new molecular entity that combines a morphinan pharmacophore, oxycodol, with a six-unit chain of polyethylene glycol, a design that slows movement of the drug into the brain by about an hour after an oral dose when compared with oxycodone. It works as a full mu opioid receptor agonist that inhibits the nociceptive pathways while it reduces reinforcing or euphoric effects and other negative CNS-mediated side effects because of its slowed entry into the central nervous system.
But, as became apparent during the course of the day’s presentations and questions, while the clinical evidence in general supported this mechanism, the data were flawed by many limitations and caveats. FDA staffers critiqued the company’s data set for a variety of issues, including testing dosages that were “arbitrary and flawed,” evidence for “high oral abuse potential” in one study, including “a high rate of euphoria” of 17% even at the recommended dosage level (and a higher euphoria incidence at a higher dosage), limited information on additional pain medications that enrolled patients had used both before enrollment and during the study, and possible enrollment bias. Many panel members also raised concerns that included the efficacy data coming from a single randomized study, incomplete data on possible hepatic toxicity, no data to address potential abuse via injection or inhalation, a very modest demonstrated increment in pain relief, and abuse potential studies that relied on a single dose of the drug.
On the other hand, many committee members, and others in the pain field, applauded the general concept behind oxycodegol and urged the developer to run additional studies and collect more data for a future FDA application.
There has been “a lot of controversy” about oxycodegol, often centered on the question “why do we need another opioid,” commented Steven P. Cohen, MD, professor of anesthesiology, neurology, and pain medicine and chief of pain medicine at Johns Hopkins Health in Baltimore. “Having another opioid on the market with a different pain indication and possibly lower abuse potential” would be welcome and “almost certainly would not result in more prescriptions or abuse,” said Dr. Cohen, who was not a committee member but addressed in an interview the need for new and effective pain medications that offer an alternative to conventional opioids. “Patients who might otherwise be prescribed a different opioid might receive oxycodegol instead,” he suggested. But first, the developing company will need to collect a lot more clinical data than it has reported so far.
A Food and Drug Administration advisory committee voted 27-0 on Jan. 14 against recommending agency approval of oxycodegol, a new type of opioid molecule designed to slow passage of the drug through the blood-brain barrier and hence cause less of a “high” and potentially blunt the drug’s abuse potential.
But the panel, which includes members from both the Anesthetic and Analgesic Drug Products Advisory Committee and the Drug Safety and Risk Management Advisory Committee, unanimously shared the opinion that the data submitted by the drug developer, Nektar, failed to clearly demonstrate the drug’s safety and efficacy.
“There was a lot of ambiguity [in the data] about safety and efficacy, and this is too important an issue to have so much ambiguity,” summed up Ronald S. Litman, DO, professor of anesthesiology and critical care at the University of Pennsylvania in Philadelphia and chair of the combined committee.
“The subliminal message” that would surround oxycodegol if it received FDA approval would be that it is a “safer” opioid, “and that would make it a blockbuster drug, and for that to happen you need data that [prove] the benefits outweigh risks, but we didn’t see those data. It was all speculation, and we can’t approve a potential blockbuster opioid in the middle of a public health opioid crisis based on speculation,” commented Steve B. Meisel, PharmD, system director of medication safety for Fairview Health Services in Minneapolis and a committee member.
According to Nektar, oxycodegol is a new molecular entity that combines a morphinan pharmacophore, oxycodol, with a six-unit chain of polyethylene glycol, a design that slows movement of the drug into the brain by about an hour after an oral dose when compared with oxycodone. It works as a full mu opioid receptor agonist that inhibits the nociceptive pathways while it reduces reinforcing or euphoric effects and other negative CNS-mediated side effects because of its slowed entry into the central nervous system.
But, as became apparent during the course of the day’s presentations and questions, while the clinical evidence in general supported this mechanism, the data were flawed by many limitations and caveats. FDA staffers critiqued the company’s data set for a variety of issues, including testing dosages that were “arbitrary and flawed,” evidence for “high oral abuse potential” in one study, including “a high rate of euphoria” of 17% even at the recommended dosage level (and a higher euphoria incidence at a higher dosage), limited information on additional pain medications that enrolled patients had used both before enrollment and during the study, and possible enrollment bias. Many panel members also raised concerns that included the efficacy data coming from a single randomized study, incomplete data on possible hepatic toxicity, no data to address potential abuse via injection or inhalation, a very modest demonstrated increment in pain relief, and abuse potential studies that relied on a single dose of the drug.
On the other hand, many committee members, and others in the pain field, applauded the general concept behind oxycodegol and urged the developer to run additional studies and collect more data for a future FDA application.
There has been “a lot of controversy” about oxycodegol, often centered on the question “why do we need another opioid,” commented Steven P. Cohen, MD, professor of anesthesiology, neurology, and pain medicine and chief of pain medicine at Johns Hopkins Health in Baltimore. “Having another opioid on the market with a different pain indication and possibly lower abuse potential” would be welcome and “almost certainly would not result in more prescriptions or abuse,” said Dr. Cohen, who was not a committee member but addressed in an interview the need for new and effective pain medications that offer an alternative to conventional opioids. “Patients who might otherwise be prescribed a different opioid might receive oxycodegol instead,” he suggested. But first, the developing company will need to collect a lot more clinical data than it has reported so far.
A Food and Drug Administration advisory committee voted 27-0 on Jan. 14 against recommending agency approval of oxycodegol, a new type of opioid molecule designed to slow passage of the drug through the blood-brain barrier and hence cause less of a “high” and potentially blunt the drug’s abuse potential.
But the panel, which includes members from both the Anesthetic and Analgesic Drug Products Advisory Committee and the Drug Safety and Risk Management Advisory Committee, unanimously shared the opinion that the data submitted by the drug developer, Nektar, failed to clearly demonstrate the drug’s safety and efficacy.
“There was a lot of ambiguity [in the data] about safety and efficacy, and this is too important an issue to have so much ambiguity,” summed up Ronald S. Litman, DO, professor of anesthesiology and critical care at the University of Pennsylvania in Philadelphia and chair of the combined committee.
“The subliminal message” that would surround oxycodegol if it received FDA approval would be that it is a “safer” opioid, “and that would make it a blockbuster drug, and for that to happen you need data that [prove] the benefits outweigh risks, but we didn’t see those data. It was all speculation, and we can’t approve a potential blockbuster opioid in the middle of a public health opioid crisis based on speculation,” commented Steve B. Meisel, PharmD, system director of medication safety for Fairview Health Services in Minneapolis and a committee member.
According to Nektar, oxycodegol is a new molecular entity that combines a morphinan pharmacophore, oxycodol, with a six-unit chain of polyethylene glycol, a design that slows movement of the drug into the brain by about an hour after an oral dose when compared with oxycodone. It works as a full mu opioid receptor agonist that inhibits the nociceptive pathways while it reduces reinforcing or euphoric effects and other negative CNS-mediated side effects because of its slowed entry into the central nervous system.
But, as became apparent during the course of the day’s presentations and questions, while the clinical evidence in general supported this mechanism, the data were flawed by many limitations and caveats. FDA staffers critiqued the company’s data set for a variety of issues, including testing dosages that were “arbitrary and flawed,” evidence for “high oral abuse potential” in one study, including “a high rate of euphoria” of 17% even at the recommended dosage level (and a higher euphoria incidence at a higher dosage), limited information on additional pain medications that enrolled patients had used both before enrollment and during the study, and possible enrollment bias. Many panel members also raised concerns that included the efficacy data coming from a single randomized study, incomplete data on possible hepatic toxicity, no data to address potential abuse via injection or inhalation, a very modest demonstrated increment in pain relief, and abuse potential studies that relied on a single dose of the drug.
On the other hand, many committee members, and others in the pain field, applauded the general concept behind oxycodegol and urged the developer to run additional studies and collect more data for a future FDA application.
There has been “a lot of controversy” about oxycodegol, often centered on the question “why do we need another opioid,” commented Steven P. Cohen, MD, professor of anesthesiology, neurology, and pain medicine and chief of pain medicine at Johns Hopkins Health in Baltimore. “Having another opioid on the market with a different pain indication and possibly lower abuse potential” would be welcome and “almost certainly would not result in more prescriptions or abuse,” said Dr. Cohen, who was not a committee member but addressed in an interview the need for new and effective pain medications that offer an alternative to conventional opioids. “Patients who might otherwise be prescribed a different opioid might receive oxycodegol instead,” he suggested. But first, the developing company will need to collect a lot more clinical data than it has reported so far.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING
Your colleague appears depressed. Now what?
Encountering a colleague who appears depressed or suicidal can be challenging, but experts say the worst thing you can do is turn away.
, professor of clinical psychiatry at State University of New York, Brooklyn. “Don’t leave it to ‘the other guy.’ ”
Below, Dr. Myers and other experts offer advice on how to approach a fellow physician who seems depressed or might be in need of professional help.
1. Reach out to the colleague, and find a private, quiet space to talk.
2. Let them know you’re there for them, and do not pass judgment.
3. Share a short list of reasons for your concerns. For example, “It seems you have lost some weight,” or “You seem sad a lot of the time.”
4. Encourage them to accept help. Refrain from giving them a list of names for professional help. Instead, make the phone call for them or set up the appointment, if necessary.
5. Ask specifically about suicidal ideation. For instance, “Are you having thoughts of hurting yourself?” or “Are you having thoughts of ending your life?” People who are suicidal often want someone to ask and help.
6. Stay with the colleague until he or she has worked out a plan of safety – for example, returned to family/loved ones, visited a physician or mental health professional, gone to an emergency department, or called for an ambulance or other assistance.
7. Accompany the colleague to the emergency department or call 911.
8. Remember these two goals: Inquire about safety/danger. Ensure safety of the individual.
Sources: Dr. Myers, Dr. Zisook, and Dr. Yellowlees.
For more information, contact the American Foundation for Suicide Prevention (www.afsp.org), and the 24-hour crisis line: 1-800-273-TALK (8255). Late last year, the Federal Communications Commission approved plans to designate 988 as a suicide prevention hotline number. Implementation of the proposal is expected to take several months.
Encountering a colleague who appears depressed or suicidal can be challenging, but experts say the worst thing you can do is turn away.
, professor of clinical psychiatry at State University of New York, Brooklyn. “Don’t leave it to ‘the other guy.’ ”
Below, Dr. Myers and other experts offer advice on how to approach a fellow physician who seems depressed or might be in need of professional help.
1. Reach out to the colleague, and find a private, quiet space to talk.
2. Let them know you’re there for them, and do not pass judgment.
3. Share a short list of reasons for your concerns. For example, “It seems you have lost some weight,” or “You seem sad a lot of the time.”
4. Encourage them to accept help. Refrain from giving them a list of names for professional help. Instead, make the phone call for them or set up the appointment, if necessary.
5. Ask specifically about suicidal ideation. For instance, “Are you having thoughts of hurting yourself?” or “Are you having thoughts of ending your life?” People who are suicidal often want someone to ask and help.
6. Stay with the colleague until he or she has worked out a plan of safety – for example, returned to family/loved ones, visited a physician or mental health professional, gone to an emergency department, or called for an ambulance or other assistance.
7. Accompany the colleague to the emergency department or call 911.
8. Remember these two goals: Inquire about safety/danger. Ensure safety of the individual.
Sources: Dr. Myers, Dr. Zisook, and Dr. Yellowlees.
For more information, contact the American Foundation for Suicide Prevention (www.afsp.org), and the 24-hour crisis line: 1-800-273-TALK (8255). Late last year, the Federal Communications Commission approved plans to designate 988 as a suicide prevention hotline number. Implementation of the proposal is expected to take several months.
Encountering a colleague who appears depressed or suicidal can be challenging, but experts say the worst thing you can do is turn away.
, professor of clinical psychiatry at State University of New York, Brooklyn. “Don’t leave it to ‘the other guy.’ ”
Below, Dr. Myers and other experts offer advice on how to approach a fellow physician who seems depressed or might be in need of professional help.
1. Reach out to the colleague, and find a private, quiet space to talk.
2. Let them know you’re there for them, and do not pass judgment.
3. Share a short list of reasons for your concerns. For example, “It seems you have lost some weight,” or “You seem sad a lot of the time.”
4. Encourage them to accept help. Refrain from giving them a list of names for professional help. Instead, make the phone call for them or set up the appointment, if necessary.
5. Ask specifically about suicidal ideation. For instance, “Are you having thoughts of hurting yourself?” or “Are you having thoughts of ending your life?” People who are suicidal often want someone to ask and help.
6. Stay with the colleague until he or she has worked out a plan of safety – for example, returned to family/loved ones, visited a physician or mental health professional, gone to an emergency department, or called for an ambulance or other assistance.
7. Accompany the colleague to the emergency department or call 911.
8. Remember these two goals: Inquire about safety/danger. Ensure safety of the individual.
Sources: Dr. Myers, Dr. Zisook, and Dr. Yellowlees.
For more information, contact the American Foundation for Suicide Prevention (www.afsp.org), and the 24-hour crisis line: 1-800-273-TALK (8255). Late last year, the Federal Communications Commission approved plans to designate 988 as a suicide prevention hotline number. Implementation of the proposal is expected to take several months.