User login
ASH to recognize researchers at annual meeting
The American Society of Hematology (ASH) plans to honor 10 researchers with awards and lectures at this year’s annual meeting, scheduled to take place Dec. 7-10 in Orlando.

Richard Aster, MD, will receive the 2019 Wallace H. Coulter Award for Lifetime Achievement in Hematology. Dr. Aster, of the Medical College of Wisconsin and Versiti Blood Center of Wisconsin in Milwaukee, “will be honored for his significant contributions to hematology through research, mentorship, and education throughout his 62-year career,” according to ASH.
Dr. Aster is known for his research on immune diseases that affect blood cells, particularly platelets. His work has led to improvements in platelet preparation, storage, and matching. In addition, he and his team developed the standard techniques for diagnosing immune thrombocytopenia and heparin-induced thrombocytopenia-thrombosis.
Other ASH awardees include William Eaton, MD, PhD, and Richard A. Larson, MD, who will receive the 2019 Henry M. Stratton Medal “for their seminal contributions to basic and clinical/translational hematology research, respectively.”
Dr. Eaton, of the National Institute of Diabetes and Digestive and Kidney Diseases conducts research on sickle cell disease, and his work contributed to the development of hydroxyurea. Dr. Larson, of the University of Chicago is the namesake of the Larson regimen (CALGB 8811) for acute lymphoblastic leukemia, and he played a key role in research that led to the U.S. approval of midostaurin.
Philip Greenberg, MD, will deliver the 2019 E. Donnall Thomas Lecture in recognition of “his outstanding contributions to the field of immunotherapy.” Dr. Greenberg, of theFred Hutchinson Cancer Research Center and the University of Washington in Seattle, is known for contributing to the development of T-cell adoptive immune therapy. His lecture will focus on the engineering of T cells to target acute myeloid leukemia and other malignancies.
Sriram Krishnaswamy, PhD, and Jeffrey I. Weitz, MD, will deliver the 2019 Ernest Beutler Lecture in recognition of “their significant research contributions to the understanding and treatment of blood clots.” The researchers will each deliver one part of the lecture at the meeting, and both will discuss research related to novel anticoagulants.
Dr. Krishnaswamy, of the University of Pennsylvania and Children’s Hospital of Philadelphia, is considered an authority on the function of surface-dependent coagulation complexes. Dr. Weitz, of McMaster University in Hamilton, Ont., has conducted research that led to the development of novel anticoagulants.
Emmanuelle Passegué, PhD, will receive the 2019 William Dameshek Prize “for her outstanding contributions to the understanding of hematopoietic stem cells.” Dr. Passegué, of Columbia University Irving Medical Center in New York, conducts research focused on changes to hematopoietic stem cells in the contexts of myeloid malignancies and physiological aging.
Griffin Rodgers, MD, will receive the ASH Award for Leadership in Promoting Diversity “for his extraordinary commitment to diversity and inclusion in hematology.” As director of the National Institute of Diabetes and Digestive and Kidney Diseases, Dr. Rodgers has worked to promote diversity in the scientific workforce and in clinical trials.
Leonard Zon, MD, and Michael R. DeBaun, MD, will receive the 2019 ASH Mentor Award “for their sustained, outstanding commitment to the training and career development of early career hematologists.”
Dr. DeBaun, of Vanderbilt University School of Medicine in Nashville, Tenn., has mentees ranging from high school students with sickle cell disease to tenured faculty members at medical schools. Dr. Zon, of Harvard University and Boston Children’s Hospital, organizes mentoring events for postdocs, graduate students, and technicians.
The American Society of Hematology (ASH) plans to honor 10 researchers with awards and lectures at this year’s annual meeting, scheduled to take place Dec. 7-10 in Orlando.
Richard Aster, MD, will receive the 2019 Wallace H. Coulter Award for Lifetime Achievement in Hematology. Dr. Aster, of the Medical College of Wisconsin and Versiti Blood Center of Wisconsin in Milwaukee, “will be honored for his significant contributions to hematology through research, mentorship, and education throughout his 62-year career,” according to ASH.
Dr. Aster is known for his research on immune diseases that affect blood cells, particularly platelets. His work has led to improvements in platelet preparation, storage, and matching. In addition, he and his team developed the standard techniques for diagnosing immune thrombocytopenia and heparin-induced thrombocytopenia-thrombosis.
Other ASH awardees include William Eaton, MD, PhD, and Richard A. Larson, MD, who will receive the 2019 Henry M. Stratton Medal “for their seminal contributions to basic and clinical/translational hematology research, respectively.”
Dr. Eaton, of the National Institute of Diabetes and Digestive and Kidney Diseases conducts research on sickle cell disease, and his work contributed to the development of hydroxyurea. Dr. Larson, of the University of Chicago is the namesake of the Larson regimen (CALGB 8811) for acute lymphoblastic leukemia, and he played a key role in research that led to the U.S. approval of midostaurin.
Philip Greenberg, MD, will deliver the 2019 E. Donnall Thomas Lecture in recognition of “his outstanding contributions to the field of immunotherapy.” Dr. Greenberg, of theFred Hutchinson Cancer Research Center and the University of Washington in Seattle, is known for contributing to the development of T-cell adoptive immune therapy. His lecture will focus on the engineering of T cells to target acute myeloid leukemia and other malignancies.
Sriram Krishnaswamy, PhD, and Jeffrey I. Weitz, MD, will deliver the 2019 Ernest Beutler Lecture in recognition of “their significant research contributions to the understanding and treatment of blood clots.” The researchers will each deliver one part of the lecture at the meeting, and both will discuss research related to novel anticoagulants.
Dr. Krishnaswamy, of the University of Pennsylvania and Children’s Hospital of Philadelphia, is considered an authority on the function of surface-dependent coagulation complexes. Dr. Weitz, of McMaster University in Hamilton, Ont., has conducted research that led to the development of novel anticoagulants.
Emmanuelle Passegué, PhD, will receive the 2019 William Dameshek Prize “for her outstanding contributions to the understanding of hematopoietic stem cells.” Dr. Passegué, of Columbia University Irving Medical Center in New York, conducts research focused on changes to hematopoietic stem cells in the contexts of myeloid malignancies and physiological aging.
Griffin Rodgers, MD, will receive the ASH Award for Leadership in Promoting Diversity “for his extraordinary commitment to diversity and inclusion in hematology.” As director of the National Institute of Diabetes and Digestive and Kidney Diseases, Dr. Rodgers has worked to promote diversity in the scientific workforce and in clinical trials.
Leonard Zon, MD, and Michael R. DeBaun, MD, will receive the 2019 ASH Mentor Award “for their sustained, outstanding commitment to the training and career development of early career hematologists.”
Dr. DeBaun, of Vanderbilt University School of Medicine in Nashville, Tenn., has mentees ranging from high school students with sickle cell disease to tenured faculty members at medical schools. Dr. Zon, of Harvard University and Boston Children’s Hospital, organizes mentoring events for postdocs, graduate students, and technicians.
The American Society of Hematology (ASH) plans to honor 10 researchers with awards and lectures at this year’s annual meeting, scheduled to take place Dec. 7-10 in Orlando.
Richard Aster, MD, will receive the 2019 Wallace H. Coulter Award for Lifetime Achievement in Hematology. Dr. Aster, of the Medical College of Wisconsin and Versiti Blood Center of Wisconsin in Milwaukee, “will be honored for his significant contributions to hematology through research, mentorship, and education throughout his 62-year career,” according to ASH.
Dr. Aster is known for his research on immune diseases that affect blood cells, particularly platelets. His work has led to improvements in platelet preparation, storage, and matching. In addition, he and his team developed the standard techniques for diagnosing immune thrombocytopenia and heparin-induced thrombocytopenia-thrombosis.
Other ASH awardees include William Eaton, MD, PhD, and Richard A. Larson, MD, who will receive the 2019 Henry M. Stratton Medal “for their seminal contributions to basic and clinical/translational hematology research, respectively.”
Dr. Eaton, of the National Institute of Diabetes and Digestive and Kidney Diseases conducts research on sickle cell disease, and his work contributed to the development of hydroxyurea. Dr. Larson, of the University of Chicago is the namesake of the Larson regimen (CALGB 8811) for acute lymphoblastic leukemia, and he played a key role in research that led to the U.S. approval of midostaurin.
Philip Greenberg, MD, will deliver the 2019 E. Donnall Thomas Lecture in recognition of “his outstanding contributions to the field of immunotherapy.” Dr. Greenberg, of theFred Hutchinson Cancer Research Center and the University of Washington in Seattle, is known for contributing to the development of T-cell adoptive immune therapy. His lecture will focus on the engineering of T cells to target acute myeloid leukemia and other malignancies.
Sriram Krishnaswamy, PhD, and Jeffrey I. Weitz, MD, will deliver the 2019 Ernest Beutler Lecture in recognition of “their significant research contributions to the understanding and treatment of blood clots.” The researchers will each deliver one part of the lecture at the meeting, and both will discuss research related to novel anticoagulants.
Dr. Krishnaswamy, of the University of Pennsylvania and Children’s Hospital of Philadelphia, is considered an authority on the function of surface-dependent coagulation complexes. Dr. Weitz, of McMaster University in Hamilton, Ont., has conducted research that led to the development of novel anticoagulants.
Emmanuelle Passegué, PhD, will receive the 2019 William Dameshek Prize “for her outstanding contributions to the understanding of hematopoietic stem cells.” Dr. Passegué, of Columbia University Irving Medical Center in New York, conducts research focused on changes to hematopoietic stem cells in the contexts of myeloid malignancies and physiological aging.
Griffin Rodgers, MD, will receive the ASH Award for Leadership in Promoting Diversity “for his extraordinary commitment to diversity and inclusion in hematology.” As director of the National Institute of Diabetes and Digestive and Kidney Diseases, Dr. Rodgers has worked to promote diversity in the scientific workforce and in clinical trials.
Leonard Zon, MD, and Michael R. DeBaun, MD, will receive the 2019 ASH Mentor Award “for their sustained, outstanding commitment to the training and career development of early career hematologists.”
Dr. DeBaun, of Vanderbilt University School of Medicine in Nashville, Tenn., has mentees ranging from high school students with sickle cell disease to tenured faculty members at medical schools. Dr. Zon, of Harvard University and Boston Children’s Hospital, organizes mentoring events for postdocs, graduate students, and technicians.
Presence
The plan was in motion before I got on the plane.

When your leukemia came back suddenly 3 years after your stem cell transplant, it was devastating. But we had a plan. Your cancer developed a new mutation we could target with a chemotherapy drug. If we got you into a second remission, we could consolidate it by infusing more of your donor’s stem cells.
We met in the hospital, but I was adamant to “keep” you as my patient when you got to the clinic. I made swaps to see you, to get the continuity I value so much but often lose as a fellow, rotating from clinic to hospital to clinic.
I was grateful to see how well you dealt with the chemotherapy. Typically it’s a tough regimen, but you hardly had side effects. Between visits, I would check your blood counts on my phone, watching your blast count fall and your normal blood cells rise. Watching the cancer disappear.
Your lumbar punctures were negative, negative, negative – my favorite word, I told you. I was involved in long email threads coordinating the timing of your stem cell infusion with the remission we were achieving.
We were on our way.
One day, your lumbar puncture came back with a few “atypical” cells. I called the pathologist, and upon further review they were convinced the cells were reactive, not cancer. The next lumbar puncture was normal, but it was hard to ignore.
“Are you worried?” I asked my attending in clinic.
“I’m always worried,” she said. Neither of us truly believed the leukemia was back, but with the odds against us, we pored over every detail, always on the alert for a clue to an outcome we feared.
By now, the stem cell infusion was all set up. The donor was ready; so was the medical team; so were you. It was exciting. I thought of how a different attending described his interest in leukemia: There’s a subset you get to cure. Yes, you were going to be one of them.
Your big day coincided with a vacation I had scheduled months before. I was sorry I would be missing the actual moment, but happy I would come back to good news.
I left my coat and badge at the hospital, packed my bags, and got on the plane. I refrained from immediately checking your blood counts on my phone as soon as we landed. That night, jet lagged, I let myself look before I go to sleep. Relief. Your numbers still looked good.
Every day, I explored. My Internet was spotty during my travels, and when I would I finally get service I would peek at your latest blood tests.
Day 1. Cooled lava canyons. Black sand beaches. Circulating blast count: 0%.
Day 2: Glacier tour. A national park. Geysers. Blast count: 2%.
Day 3: We drive along the shore to see a famous waterfall, where you can climb a set of winding stairs to the top.
I check my phone before we start the climb. No service.
And so we begin. The wind cuts as I count steps. 403, 404, 405 … and 406. We are there. The air is thin, the world quiet. My nose is running from the cold.
We hike a bit, and I glance down again. Still no signal. It’s probably for the best. The scenery is spectacular.
Two miles later, I get service. I open the blood work first. Circulating blast count: 5%. But the other counts are okay. It could still be reactive, I say to myself, though on a deeper level I think of my attending’s words: I’m always worried. The stem cell infusion is scheduled for tomorrow.
I hear the rush of the water hitting the rocks below. Icicles form to our left. Sheep graze on our right. I appreciate the feeling of my muscles aching as we climb, higher and higher, a reminder of where I am and my place in it.
At the very top, we pause to take photos. And I get a signal again. I open the bone marrow biopsy report and skim the pathologist’s words. My eyes glue on the summary: 80% blasts, compatible with relapsed leukemia.
I let out an audible gasp.
Do you know? How will they tell you? I am painfully aware of the distance between us, in so many ways.
I want to be present. And soon I will be back, and I will be visiting in the hospital, and we will be having hard conversations and thinking about hard decisions.
But I’m not there right now. Someone else is. Here, now, I realize what I cannot do. The best way I can be present for you later is to be present where I am now.
I stuff my phone in my backpack and zip it closed. I step carefully forward on the rocks, slippery from the rain. My nose is running again, but not from the cold.
“What do you think?” my partner asks.
“The views are incredible,” I say.
Minor details of this story were changed to protect privacy.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
The plan was in motion before I got on the plane.
When your leukemia came back suddenly 3 years after your stem cell transplant, it was devastating. But we had a plan. Your cancer developed a new mutation we could target with a chemotherapy drug. If we got you into a second remission, we could consolidate it by infusing more of your donor’s stem cells.
We met in the hospital, but I was adamant to “keep” you as my patient when you got to the clinic. I made swaps to see you, to get the continuity I value so much but often lose as a fellow, rotating from clinic to hospital to clinic.
I was grateful to see how well you dealt with the chemotherapy. Typically it’s a tough regimen, but you hardly had side effects. Between visits, I would check your blood counts on my phone, watching your blast count fall and your normal blood cells rise. Watching the cancer disappear.
Your lumbar punctures were negative, negative, negative – my favorite word, I told you. I was involved in long email threads coordinating the timing of your stem cell infusion with the remission we were achieving.
We were on our way.
One day, your lumbar puncture came back with a few “atypical” cells. I called the pathologist, and upon further review they were convinced the cells were reactive, not cancer. The next lumbar puncture was normal, but it was hard to ignore.
“Are you worried?” I asked my attending in clinic.
“I’m always worried,” she said. Neither of us truly believed the leukemia was back, but with the odds against us, we pored over every detail, always on the alert for a clue to an outcome we feared.
By now, the stem cell infusion was all set up. The donor was ready; so was the medical team; so were you. It was exciting. I thought of how a different attending described his interest in leukemia: There’s a subset you get to cure. Yes, you were going to be one of them.
Your big day coincided with a vacation I had scheduled months before. I was sorry I would be missing the actual moment, but happy I would come back to good news.
I left my coat and badge at the hospital, packed my bags, and got on the plane. I refrained from immediately checking your blood counts on my phone as soon as we landed. That night, jet lagged, I let myself look before I go to sleep. Relief. Your numbers still looked good.
Every day, I explored. My Internet was spotty during my travels, and when I would I finally get service I would peek at your latest blood tests.
Day 1. Cooled lava canyons. Black sand beaches. Circulating blast count: 0%.
Day 2: Glacier tour. A national park. Geysers. Blast count: 2%.
Day 3: We drive along the shore to see a famous waterfall, where you can climb a set of winding stairs to the top.
I check my phone before we start the climb. No service.
And so we begin. The wind cuts as I count steps. 403, 404, 405 … and 406. We are there. The air is thin, the world quiet. My nose is running from the cold.
We hike a bit, and I glance down again. Still no signal. It’s probably for the best. The scenery is spectacular.
Two miles later, I get service. I open the blood work first. Circulating blast count: 5%. But the other counts are okay. It could still be reactive, I say to myself, though on a deeper level I think of my attending’s words: I’m always worried. The stem cell infusion is scheduled for tomorrow.
I hear the rush of the water hitting the rocks below. Icicles form to our left. Sheep graze on our right. I appreciate the feeling of my muscles aching as we climb, higher and higher, a reminder of where I am and my place in it.
At the very top, we pause to take photos. And I get a signal again. I open the bone marrow biopsy report and skim the pathologist’s words. My eyes glue on the summary: 80% blasts, compatible with relapsed leukemia.
I let out an audible gasp.
Do you know? How will they tell you? I am painfully aware of the distance between us, in so many ways.
I want to be present. And soon I will be back, and I will be visiting in the hospital, and we will be having hard conversations and thinking about hard decisions.
But I’m not there right now. Someone else is. Here, now, I realize what I cannot do. The best way I can be present for you later is to be present where I am now.
I stuff my phone in my backpack and zip it closed. I step carefully forward on the rocks, slippery from the rain. My nose is running again, but not from the cold.
“What do you think?” my partner asks.
“The views are incredible,” I say.
Minor details of this story were changed to protect privacy.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
The plan was in motion before I got on the plane.
When your leukemia came back suddenly 3 years after your stem cell transplant, it was devastating. But we had a plan. Your cancer developed a new mutation we could target with a chemotherapy drug. If we got you into a second remission, we could consolidate it by infusing more of your donor’s stem cells.
We met in the hospital, but I was adamant to “keep” you as my patient when you got to the clinic. I made swaps to see you, to get the continuity I value so much but often lose as a fellow, rotating from clinic to hospital to clinic.
I was grateful to see how well you dealt with the chemotherapy. Typically it’s a tough regimen, but you hardly had side effects. Between visits, I would check your blood counts on my phone, watching your blast count fall and your normal blood cells rise. Watching the cancer disappear.
Your lumbar punctures were negative, negative, negative – my favorite word, I told you. I was involved in long email threads coordinating the timing of your stem cell infusion with the remission we were achieving.
We were on our way.
One day, your lumbar puncture came back with a few “atypical” cells. I called the pathologist, and upon further review they were convinced the cells were reactive, not cancer. The next lumbar puncture was normal, but it was hard to ignore.
“Are you worried?” I asked my attending in clinic.
“I’m always worried,” she said. Neither of us truly believed the leukemia was back, but with the odds against us, we pored over every detail, always on the alert for a clue to an outcome we feared.
By now, the stem cell infusion was all set up. The donor was ready; so was the medical team; so were you. It was exciting. I thought of how a different attending described his interest in leukemia: There’s a subset you get to cure. Yes, you were going to be one of them.
Your big day coincided with a vacation I had scheduled months before. I was sorry I would be missing the actual moment, but happy I would come back to good news.
I left my coat and badge at the hospital, packed my bags, and got on the plane. I refrained from immediately checking your blood counts on my phone as soon as we landed. That night, jet lagged, I let myself look before I go to sleep. Relief. Your numbers still looked good.
Every day, I explored. My Internet was spotty during my travels, and when I would I finally get service I would peek at your latest blood tests.
Day 1. Cooled lava canyons. Black sand beaches. Circulating blast count: 0%.
Day 2: Glacier tour. A national park. Geysers. Blast count: 2%.
Day 3: We drive along the shore to see a famous waterfall, where you can climb a set of winding stairs to the top.
I check my phone before we start the climb. No service.
And so we begin. The wind cuts as I count steps. 403, 404, 405 … and 406. We are there. The air is thin, the world quiet. My nose is running from the cold.
We hike a bit, and I glance down again. Still no signal. It’s probably for the best. The scenery is spectacular.
Two miles later, I get service. I open the blood work first. Circulating blast count: 5%. But the other counts are okay. It could still be reactive, I say to myself, though on a deeper level I think of my attending’s words: I’m always worried. The stem cell infusion is scheduled for tomorrow.
I hear the rush of the water hitting the rocks below. Icicles form to our left. Sheep graze on our right. I appreciate the feeling of my muscles aching as we climb, higher and higher, a reminder of where I am and my place in it.
At the very top, we pause to take photos. And I get a signal again. I open the bone marrow biopsy report and skim the pathologist’s words. My eyes glue on the summary: 80% blasts, compatible with relapsed leukemia.
I let out an audible gasp.
Do you know? How will they tell you? I am painfully aware of the distance between us, in so many ways.
I want to be present. And soon I will be back, and I will be visiting in the hospital, and we will be having hard conversations and thinking about hard decisions.
But I’m not there right now. Someone else is. Here, now, I realize what I cannot do. The best way I can be present for you later is to be present where I am now.
I stuff my phone in my backpack and zip it closed. I step carefully forward on the rocks, slippery from the rain. My nose is running again, but not from the cold.
“What do you think?” my partner asks.
“The views are incredible,” I say.
Minor details of this story were changed to protect privacy.
Dr. Yurkiewicz is a fellow in hematology and oncology at Stanford (Calif.) University. Follow her on Twitter @ilanayurkiewicz and listen to her each week on the Blood & Cancer podcast.
Functional heartburn: An underrecognized cause of PPI-refractory symptoms
A 44-year-old woman presents with an 8-year history of intermittent heartburn, and in the past year she has been experiencing her symptoms daily. She says the heartburn is constant and is worse immediately after eating spicy or acidic foods. She says she has had no dysphagia, weight loss, or vomiting. Her symptoms have persisted despite taking a histamine (H)2-receptor antagonist twice daily plus a proton pump inhibitor (PPI) before breakfast and dinner for more than 3 months.
She has undergone upper endoscopy 3 times in the past 8 years. Each time, the esophagus was normal with a regular Z-line and normal biopsy results from the proximal and distal esophagus.
The patient believes she has severe gastroesophageal reflux disease (GERD) and asks if she is a candidate for fundoplication surgery.
HEARTBURN IS A SYMPTOM; GERD IS A CONDITION
A distinction should be made between heartburn—the symptom of persistent retrosternal burning and discomfort—and gastroesophageal reflux disease—the condition in which reflux of stomach contents causes troublesome symptoms or complications.1 While many clinicians initially diagnose patients who have heartburn as having GERD, there are many other potential causes of their symptoms.
For patients with persistent heartburn, an empiric trial of a once-daily PPI is usually effective, but one-third of patients continue to have heartburn.2,3 The most common cause of this PPI-refractory heartburn is functional heartburn, a functional or hypersensitivity disorder of the esophagus.4
PATHOPHYSIOLOGY IS POORLY UNDERSTOOD

DIAGNOSTIC EVALUATION

Clinicians have several tests available for diagnosing these conditions.
Upper endoscopy
Upper endoscopy is recommended for patients with heartburn that does not respond to a 3-month trial of a PPI.9 Endoscopy is also indicated in any patient who has any of the following “alarm symptoms” that could be due to malignancy or peptic ulcer:
- Dysphagia
- Odynophagia
- Vomiting
- Unexplained weight loss or anemia
- Signs of gastrointestinal bleeding
- Anorexia
- New onset of dyspepsia in a patient over age 60.
During upper endoscopy, the esophagus is evaluated for reflux esophagitis, Barrett esophagus, and other inflammatory disorders such as infectious esophagitis. But even if the esophageal mucosa appears normal, the proximal and distal esophagus should be biopsied to rule out an inflammatory disorder such as eosinophilic or lymphocytic esophagitis.
Esophageal manometry
If endoscopic and esophageal biopsy results are inconclusive, a workup for an esophageal motility disorder is the next step. Dysphagia is the most common symptom of these disorders, although the initial presenting symptom may be heartburn or regurgitation that persists despite PPI therapy.
Manometry is used to test for motility disorders such as achalasia and esophageal spasm.10 After applying a local anesthetic inside the nares, the clinician inserts a flexible catheter (about 4 mm in diameter) with 36 pressure sensors spaced at 1-cm intervals into the nares and passes it through the esophagus and lower esophageal sphincter. The patient then swallows liquid, and the sensors relay the esophageal response, creating a topographic plot that shows esophageal peristalsis and lower esophageal sphincter relaxation.
Achalasia is identified by incomplete lower esophageal sphincter relaxation combined with 100% failed peristalsis in the body of the esophagus. Esophageal spasms are identified by a shortened distal latency, which corresponds to premature contraction of the esophagus during peristalsis.11
Esophageal pH testing
Measuring esophageal pH levels is an important step to quantify gastroesophageal reflux and determine if symptoms occur during reflux events. According to the updated Porto GERD consensus group recommendations,12 a pH test is positive if the acid exposure time is greater than 6% of the testing period. Testing the pH differentiates between GERD (abnormal acid exposure), reflux hypersensitivity (normal acid exposure, strong correlation between symptoms and reflux events), and functional heartburn (normal acid exposure, negative correlation between reflux events and symptoms).5 For this test, a pH probe is placed in the esophagus transnasally or endoscopically. The probe records esophageal pH levels for 24 to 96 hours in an outpatient setting. Antisecretory therapy needs to be withheld for 7 to 10 days before the test.
Transnasal pH probe. For this approach, a thin catheter is inserted through the nares and advanced until the tip is 5 cm proximal to the lower esophageal sphincter. (The placement is guided by the results of esophageal manometry, which is done immediately before pH catheter placement.) The tube is secured with clear tape on the side of the patient’s face, and the end is connected to a portable recorder that compiles the data. The patient pushes a button on the recorder when experiencing heartburn symptoms. (A nurse instructs the patient on proper procedure.) After 24 hours, the patient either removes the catheter or has the clinic remove it. The pH and symptom data are downloaded and analyzed.
Transnasal pH testing can be combined with impedance measurement, which can detect nonacid reflux or weakly acid reflux. However, the clinical significance of this measurement is unclear, as multiple studies have found total acid exposure time to be a better predictor of response to therapy than weakly acid or nonacid reflux.12
Wireless pH probe. This method uses a disposable, catheter-free, capsule device to measure esophageal pH. The capsule, about the size of a gel capsule or pencil eraser, is attached to the patient’s esophageal lining, usually during upper endoscopy. The capsule records pH levels in the lower esophagus for 48 to 96 hours and transmits the data wirelessly to a receiver the patient wears. The patient pushes buttons on the receiver to record symptom-specific data when experiencing heartburn, chest pain, regurgitation, or cough. The capsule detaches from the esophagus spontaneously, generally within 7 days, and is passed out of the body through a bowel movement.
Diagnosing functional heartburn

CASE CONTINUED: NORMAL RESULTS ON TESTING
 for most of the test.")
Based on these results, her condition is diagnosed as functional heartburn, consistent with the Rome IV criteria.5
TREATMENT
Patient education is key
Patient education about the pathogenesis, natural history, and treatment options is the most important aspect of treating any functional gastrointestinal disorder. This includes the “brain-gut connection” and potential mechanisms of dysregulation. Patient education along with assessment of symptoms should be part of every visit, especially before discussing treatment options.
Patients whose condition is diagnosed as functional heartburn need reassurance that the condition is benign and, in particular, that the risk of progression to esophageal adenocarcinoma is minimal in the absence of Barrett esophagus.13 Also important to point out is that the disorder may spontaneously resolve: resolution rates of up to 40% have been reported for other functional gastrointestinal disorders.14
Antisecretory medications may work for some
A PPI or H2-receptor antagonist is the most common first-line treatment for heartburn symptoms. Although most patients with functional heartburn experience no improvement in symptoms with an antisecretory agent, a small number report some relief, which suggests that acid-suppression therapy may have an indirect impact on pain modulation in the esophagus.15 In patients who report symptom relief with an antisecretory agent, we suggest continuing the medication tapered to the lowest effective dose, with repeated reassurance that the medication can be discontinued safely at any time.
Antireflux surgery should be avoided
Antireflux surgery should be avoided in patients with normal pH testing and no objective finding of reflux, as this is associated with worse subjective outcomes than in patients with abnormal pH test results.16
Neuromodulators

It is important to discuss with patients the concept of neuromodulation, including the fact that antidepressants are often used because of their effects on serotonin and norepinephrine, which decrease visceral hypersensitivity.
The selective serotonin reuptake inhibitor citalopram has been shown to reduce esophageal hypersensitivity,17 and a tricyclic antidepressant has been shown to improve quality of life.18 These results have led experts to recommend a trial of a low dose of either type of medication.19 The dose of tricyclic antidepressant often needs to be increased sequentially every 2 to 4 weeks.
Interestingly, melatonin 6 mg at bedtime has also shown efficacy for functional heartburn, potentially due to its antinociceptive properties.20
Alternative and complementary therapies
Many esophageal centers use cognitive behavioral therapy and hypnotherapy as first-line treatment for functional esophageal disorders. Here again, it is important for the patient to understand the rationale of therapy for functional gastrointestinal disorders, given the stigma in the general population regarding psychotherapy.
Cognitive behavioral therapy has been used for functional gastrointestinal disorders for many years, as it has been shown to modulate visceral perception.21 Although published studies are limited, research regarding other functional esophageal disorders suggests that patients who commit to long-term behavioral therapy have had a significant improvement in symptoms.22
The goal of esophageal-directed behavioral therapy is to promote focused relaxation using deep breathing techniques, which can help patients manage esophageal hypervigilance, especially if symptoms continue despite neuromodulator therapy. Specifically, hypnotherapy has been shown to modulate functional chest pain through the visceral sensory pathway and also to suppress gastric acid secretion.21,23 A study of a 7-week hypnotherapy program reported significant benefits in heartburn relief and improved quality of life in patients with functional heartburn.24 The data support the use of behavioral therapies as first-line therapy or as adjunctive therapy for patients already taking a neuromodulator.
CASE FOLLOW-UP: IMPROVEMENT WITH TREATMENT
During a follow-up visit, the patient is given several printed resources, including the Rome Foundation article on functional heartburn.5 We again emphasize the benign nature of functional heartburn, noting the minimal risk of progression to esophageal adenocarcinoma, as she had no evidence of Barrett esophagus on endoscopy. And we discuss the natural course of functional heartburn, including the spontaneous resolution rate of about 40%.
For treatment, we present her the rationale for using neuromodulators and reassure her that these medications are for treatment of visceral hypersensitivity, not for anxiety or depression. After the discussion, the patient opts to start amitriptyline therapy at 10 mg every night at bedtime, increasing the dose by 10 mg every 2 weeks until symptoms improve, up to 75–100 mg every day.
After 3 months, the patient reports a 90% improvement in symptoms while on amitriptyline 30 mg every night. She is also able to taper her antisecretory medications once symptoms are controlled. We plan to continue amitriptyline at the current dose for 6 to 12 months, then discuss a slow taper to see if her symptoms spontaneously resolve.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101(8):1900–1920.
- Dean BB, Gano AD Jr, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2(8):656–664. pmid:15290657
- Hachem C, Shaheen NJ. Diagnosis and management of functional heartburn. Am J Gastroenterol 2016; 111(1):53–61. doi:10.1038/ajg.2015.376
- Fass R, Sifrim D. Management of heartburn not responding to proton pump inhibitors. Gut 2009; 58(2):295–309. doi:10.1136/gut.2007.145581
- Aziz Q, Fass R, Gyawali CP, Miwa H, Pandolfino JE, Zerbib F. Esophageal disorders. Gastroenterology 2016; 150(6):1368-1379. doi:10.1053/j.gastro.2016.02.012
- Kondo T, Miwa H. The role of esophageal hypersensitivity in functional heartburn. J Clin Gastroenterol 2017; 51(7):571–578. doi:10.1097/MCG.0000000000000885
- Farmer AD, Ruffle JK, Aziz Q. The role of esophageal hypersensitivity in functional esophageal disorders. J Clin Gastroenterol 2017; 51(2):91–99. doi:10.1097/MCG.0000000000000757
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55(10):1398–1402. doi:10.1136/gut.2005.087668
- Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013; 108(3):308–328. doi:10.1038/ajg.2012.444
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27(2):160–174. doi:10.1111/nmo.12477
- Kichler AJ, Gabbard S. A man with progressive dysphagia. Cleve Clin J Med 2017; 84(6):443–449. doi:10.3949/ccjm.84a.16055
- Roman S, Gyawali CP, Savarino E, et al; GERD consensus group. Ambulatory reflux monitoring for diagnosis of gastro-esophageal reflux disease: update of the Porto consensus and recommendations from an international consensus group. Neurogastroenterol Motil 2017; 29(10):1–15. doi:10.1111/nmo.13067
- Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG clinical guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016; 111(1):30–50. doi:10.1038/ajg.2015.322
- Halder SL, Locke GR 3rd, Schleck CD, Zinsmeister AR, Melton LJ 3rd, Talley NJ. Natural history of functional gastrointestinal disorders: a 12-year longitudinal population-based study. Gastroenterology 2007; 133(3):799–807. doi:10.1053/j.gastro.2007.06.010
- Park EY, Choi MG, Baeg M, et al. The value of early wireless esophageal pH monitoring in diagnosing functional heartburn in refractory gastroesophageal reflux disease. Dig Dis Sci 2013; 58(10):2933–2939. doi:10.1007/s10620-013-2728-4
- Khajanchee YS, Hong D, Hansen PD, Swanström LL. Outcomes of antireflux surgery in patients with normal preoperative 24-hour pH test results. Am J Surg 2004; 187(5):599–603. doi:10.1016/j.amjsurg.2004.01.010
- Viazis N, Keyoglou A, Kanellopoulos AK, et al. Selective serotonin reuptake inhibitors for the treatment of hypersensitive esophagus: a randomized, double-blind, placebo-controlled study. Am J Gastroenterol 2012; 107(11):1662–1667. doi:10.1038/ajg.2011.179
- Limsrivilai J, Charatcharoenwitthaya P, Pausawasdi N, Leelakusolvong S. Imipramine for treatment of esophageal hypersensitivity and functional heartburn: a randomized placebo-controlled trial. Am J Gastroenterol 2016; 111(2):217–224. doi:10.1038/ajg.2015.413
- Keefer L, Kahrilas PJ. Low-dose tricyclics for esophageal hypersensitivity: is it all placebo effect? Am J Gastroenterol 2016; 111(2):225–227. doi:10.1038/ajg.2016.13
- Basu PP, Hempole H, Krishnaswamy N, Shah NJ, Aloysius, M. The effect of melatonin in functional heartburn: a randomized, placebo-controlled clinical trial. Open J Gastroenterol 2014; 4(2):56–61. doi:10.4236/ojgas.2014.42010
- Watanabe S, Hattori T, Kanazawa M, Kano M, Fukudo S. Role of histaminergic neurons in hypnotic modulation of brain processing of visceral perception. Neurogastroenterol Motil 2007; 19(10):831–838. doi:10.1111/j.1365-2982.2007.00959.x
- Riehl ME, Kinsinger S, Kahrilas PJ, Pandolfino JE, Keefer L. Role of a health psychologist in the management of functional esophageal complaints. Dis Esophagus 2015; 28(5):428–436. doi:10.1111/dote.12219
- Klein KB, Spiegel D. Modulation of gastric acid secretion by hypnosis. Gastroenterology 1989; 96(6):1383–1387. pmid:2714570
- Riehl ME, Pandolfino JE, Palsson OS, Keefer L. Feasibility and acceptability of esophageal-directed hypnotherapy for functional heartburn. Dis Esophagus 2016; 29(5):490–496. doi:10.1111/dote.12353
A 44-year-old woman presents with an 8-year history of intermittent heartburn, and in the past year she has been experiencing her symptoms daily. She says the heartburn is constant and is worse immediately after eating spicy or acidic foods. She says she has had no dysphagia, weight loss, or vomiting. Her symptoms have persisted despite taking a histamine (H)2-receptor antagonist twice daily plus a proton pump inhibitor (PPI) before breakfast and dinner for more than 3 months.
She has undergone upper endoscopy 3 times in the past 8 years. Each time, the esophagus was normal with a regular Z-line and normal biopsy results from the proximal and distal esophagus.
The patient believes she has severe gastroesophageal reflux disease (GERD) and asks if she is a candidate for fundoplication surgery.
HEARTBURN IS A SYMPTOM; GERD IS A CONDITION
A distinction should be made between heartburn—the symptom of persistent retrosternal burning and discomfort—and gastroesophageal reflux disease—the condition in which reflux of stomach contents causes troublesome symptoms or complications.1 While many clinicians initially diagnose patients who have heartburn as having GERD, there are many other potential causes of their symptoms.
For patients with persistent heartburn, an empiric trial of a once-daily PPI is usually effective, but one-third of patients continue to have heartburn.2,3 The most common cause of this PPI-refractory heartburn is functional heartburn, a functional or hypersensitivity disorder of the esophagus.4
PATHOPHYSIOLOGY IS POORLY UNDERSTOOD
DIAGNOSTIC EVALUATION
Clinicians have several tests available for diagnosing these conditions.
Upper endoscopy
Upper endoscopy is recommended for patients with heartburn that does not respond to a 3-month trial of a PPI.9 Endoscopy is also indicated in any patient who has any of the following “alarm symptoms” that could be due to malignancy or peptic ulcer:
- Dysphagia
- Odynophagia
- Vomiting
- Unexplained weight loss or anemia
- Signs of gastrointestinal bleeding
- Anorexia
- New onset of dyspepsia in a patient over age 60.
During upper endoscopy, the esophagus is evaluated for reflux esophagitis, Barrett esophagus, and other inflammatory disorders such as infectious esophagitis. But even if the esophageal mucosa appears normal, the proximal and distal esophagus should be biopsied to rule out an inflammatory disorder such as eosinophilic or lymphocytic esophagitis.
Esophageal manometry
If endoscopic and esophageal biopsy results are inconclusive, a workup for an esophageal motility disorder is the next step. Dysphagia is the most common symptom of these disorders, although the initial presenting symptom may be heartburn or regurgitation that persists despite PPI therapy.
Manometry is used to test for motility disorders such as achalasia and esophageal spasm.10 After applying a local anesthetic inside the nares, the clinician inserts a flexible catheter (about 4 mm in diameter) with 36 pressure sensors spaced at 1-cm intervals into the nares and passes it through the esophagus and lower esophageal sphincter. The patient then swallows liquid, and the sensors relay the esophageal response, creating a topographic plot that shows esophageal peristalsis and lower esophageal sphincter relaxation.
Achalasia is identified by incomplete lower esophageal sphincter relaxation combined with 100% failed peristalsis in the body of the esophagus. Esophageal spasms are identified by a shortened distal latency, which corresponds to premature contraction of the esophagus during peristalsis.11
Esophageal pH testing
Measuring esophageal pH levels is an important step to quantify gastroesophageal reflux and determine if symptoms occur during reflux events. According to the updated Porto GERD consensus group recommendations,12 a pH test is positive if the acid exposure time is greater than 6% of the testing period. Testing the pH differentiates between GERD (abnormal acid exposure), reflux hypersensitivity (normal acid exposure, strong correlation between symptoms and reflux events), and functional heartburn (normal acid exposure, negative correlation between reflux events and symptoms).5 For this test, a pH probe is placed in the esophagus transnasally or endoscopically. The probe records esophageal pH levels for 24 to 96 hours in an outpatient setting. Antisecretory therapy needs to be withheld for 7 to 10 days before the test.
Transnasal pH probe. For this approach, a thin catheter is inserted through the nares and advanced until the tip is 5 cm proximal to the lower esophageal sphincter. (The placement is guided by the results of esophageal manometry, which is done immediately before pH catheter placement.) The tube is secured with clear tape on the side of the patient’s face, and the end is connected to a portable recorder that compiles the data. The patient pushes a button on the recorder when experiencing heartburn symptoms. (A nurse instructs the patient on proper procedure.) After 24 hours, the patient either removes the catheter or has the clinic remove it. The pH and symptom data are downloaded and analyzed.
Transnasal pH testing can be combined with impedance measurement, which can detect nonacid reflux or weakly acid reflux. However, the clinical significance of this measurement is unclear, as multiple studies have found total acid exposure time to be a better predictor of response to therapy than weakly acid or nonacid reflux.12
Wireless pH probe. This method uses a disposable, catheter-free, capsule device to measure esophageal pH. The capsule, about the size of a gel capsule or pencil eraser, is attached to the patient’s esophageal lining, usually during upper endoscopy. The capsule records pH levels in the lower esophagus for 48 to 96 hours and transmits the data wirelessly to a receiver the patient wears. The patient pushes buttons on the receiver to record symptom-specific data when experiencing heartburn, chest pain, regurgitation, or cough. The capsule detaches from the esophagus spontaneously, generally within 7 days, and is passed out of the body through a bowel movement.
Diagnosing functional heartburn
CASE CONTINUED: NORMAL RESULTS ON TESTING
Based on these results, her condition is diagnosed as functional heartburn, consistent with the Rome IV criteria.5
TREATMENT
Patient education is key
Patient education about the pathogenesis, natural history, and treatment options is the most important aspect of treating any functional gastrointestinal disorder. This includes the “brain-gut connection” and potential mechanisms of dysregulation. Patient education along with assessment of symptoms should be part of every visit, especially before discussing treatment options.
Patients whose condition is diagnosed as functional heartburn need reassurance that the condition is benign and, in particular, that the risk of progression to esophageal adenocarcinoma is minimal in the absence of Barrett esophagus.13 Also important to point out is that the disorder may spontaneously resolve: resolution rates of up to 40% have been reported for other functional gastrointestinal disorders.14
Antisecretory medications may work for some
A PPI or H2-receptor antagonist is the most common first-line treatment for heartburn symptoms. Although most patients with functional heartburn experience no improvement in symptoms with an antisecretory agent, a small number report some relief, which suggests that acid-suppression therapy may have an indirect impact on pain modulation in the esophagus.15 In patients who report symptom relief with an antisecretory agent, we suggest continuing the medication tapered to the lowest effective dose, with repeated reassurance that the medication can be discontinued safely at any time.
Antireflux surgery should be avoided
Antireflux surgery should be avoided in patients with normal pH testing and no objective finding of reflux, as this is associated with worse subjective outcomes than in patients with abnormal pH test results.16
Neuromodulators
It is important to discuss with patients the concept of neuromodulation, including the fact that antidepressants are often used because of their effects on serotonin and norepinephrine, which decrease visceral hypersensitivity.
The selective serotonin reuptake inhibitor citalopram has been shown to reduce esophageal hypersensitivity,17 and a tricyclic antidepressant has been shown to improve quality of life.18 These results have led experts to recommend a trial of a low dose of either type of medication.19 The dose of tricyclic antidepressant often needs to be increased sequentially every 2 to 4 weeks.
Interestingly, melatonin 6 mg at bedtime has also shown efficacy for functional heartburn, potentially due to its antinociceptive properties.20
Alternative and complementary therapies
Many esophageal centers use cognitive behavioral therapy and hypnotherapy as first-line treatment for functional esophageal disorders. Here again, it is important for the patient to understand the rationale of therapy for functional gastrointestinal disorders, given the stigma in the general population regarding psychotherapy.
Cognitive behavioral therapy has been used for functional gastrointestinal disorders for many years, as it has been shown to modulate visceral perception.21 Although published studies are limited, research regarding other functional esophageal disorders suggests that patients who commit to long-term behavioral therapy have had a significant improvement in symptoms.22
The goal of esophageal-directed behavioral therapy is to promote focused relaxation using deep breathing techniques, which can help patients manage esophageal hypervigilance, especially if symptoms continue despite neuromodulator therapy. Specifically, hypnotherapy has been shown to modulate functional chest pain through the visceral sensory pathway and also to suppress gastric acid secretion.21,23 A study of a 7-week hypnotherapy program reported significant benefits in heartburn relief and improved quality of life in patients with functional heartburn.24 The data support the use of behavioral therapies as first-line therapy or as adjunctive therapy for patients already taking a neuromodulator.
CASE FOLLOW-UP: IMPROVEMENT WITH TREATMENT
During a follow-up visit, the patient is given several printed resources, including the Rome Foundation article on functional heartburn.5 We again emphasize the benign nature of functional heartburn, noting the minimal risk of progression to esophageal adenocarcinoma, as she had no evidence of Barrett esophagus on endoscopy. And we discuss the natural course of functional heartburn, including the spontaneous resolution rate of about 40%.
For treatment, we present her the rationale for using neuromodulators and reassure her that these medications are for treatment of visceral hypersensitivity, not for anxiety or depression. After the discussion, the patient opts to start amitriptyline therapy at 10 mg every night at bedtime, increasing the dose by 10 mg every 2 weeks until symptoms improve, up to 75–100 mg every day.
After 3 months, the patient reports a 90% improvement in symptoms while on amitriptyline 30 mg every night. She is also able to taper her antisecretory medications once symptoms are controlled. We plan to continue amitriptyline at the current dose for 6 to 12 months, then discuss a slow taper to see if her symptoms spontaneously resolve.
A 44-year-old woman presents with an 8-year history of intermittent heartburn, and in the past year she has been experiencing her symptoms daily. She says the heartburn is constant and is worse immediately after eating spicy or acidic foods. She says she has had no dysphagia, weight loss, or vomiting. Her symptoms have persisted despite taking a histamine (H)2-receptor antagonist twice daily plus a proton pump inhibitor (PPI) before breakfast and dinner for more than 3 months.
She has undergone upper endoscopy 3 times in the past 8 years. Each time, the esophagus was normal with a regular Z-line and normal biopsy results from the proximal and distal esophagus.
The patient believes she has severe gastroesophageal reflux disease (GERD) and asks if she is a candidate for fundoplication surgery.
HEARTBURN IS A SYMPTOM; GERD IS A CONDITION
A distinction should be made between heartburn—the symptom of persistent retrosternal burning and discomfort—and gastroesophageal reflux disease—the condition in which reflux of stomach contents causes troublesome symptoms or complications.1 While many clinicians initially diagnose patients who have heartburn as having GERD, there are many other potential causes of their symptoms.
For patients with persistent heartburn, an empiric trial of a once-daily PPI is usually effective, but one-third of patients continue to have heartburn.2,3 The most common cause of this PPI-refractory heartburn is functional heartburn, a functional or hypersensitivity disorder of the esophagus.4
PATHOPHYSIOLOGY IS POORLY UNDERSTOOD
DIAGNOSTIC EVALUATION
Clinicians have several tests available for diagnosing these conditions.
Upper endoscopy
Upper endoscopy is recommended for patients with heartburn that does not respond to a 3-month trial of a PPI.9 Endoscopy is also indicated in any patient who has any of the following “alarm symptoms” that could be due to malignancy or peptic ulcer:
- Dysphagia
- Odynophagia
- Vomiting
- Unexplained weight loss or anemia
- Signs of gastrointestinal bleeding
- Anorexia
- New onset of dyspepsia in a patient over age 60.
During upper endoscopy, the esophagus is evaluated for reflux esophagitis, Barrett esophagus, and other inflammatory disorders such as infectious esophagitis. But even if the esophageal mucosa appears normal, the proximal and distal esophagus should be biopsied to rule out an inflammatory disorder such as eosinophilic or lymphocytic esophagitis.
Esophageal manometry
If endoscopic and esophageal biopsy results are inconclusive, a workup for an esophageal motility disorder is the next step. Dysphagia is the most common symptom of these disorders, although the initial presenting symptom may be heartburn or regurgitation that persists despite PPI therapy.
Manometry is used to test for motility disorders such as achalasia and esophageal spasm.10 After applying a local anesthetic inside the nares, the clinician inserts a flexible catheter (about 4 mm in diameter) with 36 pressure sensors spaced at 1-cm intervals into the nares and passes it through the esophagus and lower esophageal sphincter. The patient then swallows liquid, and the sensors relay the esophageal response, creating a topographic plot that shows esophageal peristalsis and lower esophageal sphincter relaxation.
Achalasia is identified by incomplete lower esophageal sphincter relaxation combined with 100% failed peristalsis in the body of the esophagus. Esophageal spasms are identified by a shortened distal latency, which corresponds to premature contraction of the esophagus during peristalsis.11
Esophageal pH testing
Measuring esophageal pH levels is an important step to quantify gastroesophageal reflux and determine if symptoms occur during reflux events. According to the updated Porto GERD consensus group recommendations,12 a pH test is positive if the acid exposure time is greater than 6% of the testing period. Testing the pH differentiates between GERD (abnormal acid exposure), reflux hypersensitivity (normal acid exposure, strong correlation between symptoms and reflux events), and functional heartburn (normal acid exposure, negative correlation between reflux events and symptoms).5 For this test, a pH probe is placed in the esophagus transnasally or endoscopically. The probe records esophageal pH levels for 24 to 96 hours in an outpatient setting. Antisecretory therapy needs to be withheld for 7 to 10 days before the test.
Transnasal pH probe. For this approach, a thin catheter is inserted through the nares and advanced until the tip is 5 cm proximal to the lower esophageal sphincter. (The placement is guided by the results of esophageal manometry, which is done immediately before pH catheter placement.) The tube is secured with clear tape on the side of the patient’s face, and the end is connected to a portable recorder that compiles the data. The patient pushes a button on the recorder when experiencing heartburn symptoms. (A nurse instructs the patient on proper procedure.) After 24 hours, the patient either removes the catheter or has the clinic remove it. The pH and symptom data are downloaded and analyzed.
Transnasal pH testing can be combined with impedance measurement, which can detect nonacid reflux or weakly acid reflux. However, the clinical significance of this measurement is unclear, as multiple studies have found total acid exposure time to be a better predictor of response to therapy than weakly acid or nonacid reflux.12
Wireless pH probe. This method uses a disposable, catheter-free, capsule device to measure esophageal pH. The capsule, about the size of a gel capsule or pencil eraser, is attached to the patient’s esophageal lining, usually during upper endoscopy. The capsule records pH levels in the lower esophagus for 48 to 96 hours and transmits the data wirelessly to a receiver the patient wears. The patient pushes buttons on the receiver to record symptom-specific data when experiencing heartburn, chest pain, regurgitation, or cough. The capsule detaches from the esophagus spontaneously, generally within 7 days, and is passed out of the body through a bowel movement.
Diagnosing functional heartburn
CASE CONTINUED: NORMAL RESULTS ON TESTING
Based on these results, her condition is diagnosed as functional heartburn, consistent with the Rome IV criteria.5
TREATMENT
Patient education is key
Patient education about the pathogenesis, natural history, and treatment options is the most important aspect of treating any functional gastrointestinal disorder. This includes the “brain-gut connection” and potential mechanisms of dysregulation. Patient education along with assessment of symptoms should be part of every visit, especially before discussing treatment options.
Patients whose condition is diagnosed as functional heartburn need reassurance that the condition is benign and, in particular, that the risk of progression to esophageal adenocarcinoma is minimal in the absence of Barrett esophagus.13 Also important to point out is that the disorder may spontaneously resolve: resolution rates of up to 40% have been reported for other functional gastrointestinal disorders.14
Antisecretory medications may work for some
A PPI or H2-receptor antagonist is the most common first-line treatment for heartburn symptoms. Although most patients with functional heartburn experience no improvement in symptoms with an antisecretory agent, a small number report some relief, which suggests that acid-suppression therapy may have an indirect impact on pain modulation in the esophagus.15 In patients who report symptom relief with an antisecretory agent, we suggest continuing the medication tapered to the lowest effective dose, with repeated reassurance that the medication can be discontinued safely at any time.
Antireflux surgery should be avoided
Antireflux surgery should be avoided in patients with normal pH testing and no objective finding of reflux, as this is associated with worse subjective outcomes than in patients with abnormal pH test results.16
Neuromodulators
It is important to discuss with patients the concept of neuromodulation, including the fact that antidepressants are often used because of their effects on serotonin and norepinephrine, which decrease visceral hypersensitivity.
The selective serotonin reuptake inhibitor citalopram has been shown to reduce esophageal hypersensitivity,17 and a tricyclic antidepressant has been shown to improve quality of life.18 These results have led experts to recommend a trial of a low dose of either type of medication.19 The dose of tricyclic antidepressant often needs to be increased sequentially every 2 to 4 weeks.
Interestingly, melatonin 6 mg at bedtime has also shown efficacy for functional heartburn, potentially due to its antinociceptive properties.20
Alternative and complementary therapies
Many esophageal centers use cognitive behavioral therapy and hypnotherapy as first-line treatment for functional esophageal disorders. Here again, it is important for the patient to understand the rationale of therapy for functional gastrointestinal disorders, given the stigma in the general population regarding psychotherapy.
Cognitive behavioral therapy has been used for functional gastrointestinal disorders for many years, as it has been shown to modulate visceral perception.21 Although published studies are limited, research regarding other functional esophageal disorders suggests that patients who commit to long-term behavioral therapy have had a significant improvement in symptoms.22
The goal of esophageal-directed behavioral therapy is to promote focused relaxation using deep breathing techniques, which can help patients manage esophageal hypervigilance, especially if symptoms continue despite neuromodulator therapy. Specifically, hypnotherapy has been shown to modulate functional chest pain through the visceral sensory pathway and also to suppress gastric acid secretion.21,23 A study of a 7-week hypnotherapy program reported significant benefits in heartburn relief and improved quality of life in patients with functional heartburn.24 The data support the use of behavioral therapies as first-line therapy or as adjunctive therapy for patients already taking a neuromodulator.
CASE FOLLOW-UP: IMPROVEMENT WITH TREATMENT
During a follow-up visit, the patient is given several printed resources, including the Rome Foundation article on functional heartburn.5 We again emphasize the benign nature of functional heartburn, noting the minimal risk of progression to esophageal adenocarcinoma, as she had no evidence of Barrett esophagus on endoscopy. And we discuss the natural course of functional heartburn, including the spontaneous resolution rate of about 40%.
For treatment, we present her the rationale for using neuromodulators and reassure her that these medications are for treatment of visceral hypersensitivity, not for anxiety or depression. After the discussion, the patient opts to start amitriptyline therapy at 10 mg every night at bedtime, increasing the dose by 10 mg every 2 weeks until symptoms improve, up to 75–100 mg every day.
After 3 months, the patient reports a 90% improvement in symptoms while on amitriptyline 30 mg every night. She is also able to taper her antisecretory medications once symptoms are controlled. We plan to continue amitriptyline at the current dose for 6 to 12 months, then discuss a slow taper to see if her symptoms spontaneously resolve.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101(8):1900–1920.
- Dean BB, Gano AD Jr, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2(8):656–664. pmid:15290657
- Hachem C, Shaheen NJ. Diagnosis and management of functional heartburn. Am J Gastroenterol 2016; 111(1):53–61. doi:10.1038/ajg.2015.376
- Fass R, Sifrim D. Management of heartburn not responding to proton pump inhibitors. Gut 2009; 58(2):295–309. doi:10.1136/gut.2007.145581
- Aziz Q, Fass R, Gyawali CP, Miwa H, Pandolfino JE, Zerbib F. Esophageal disorders. Gastroenterology 2016; 150(6):1368-1379. doi:10.1053/j.gastro.2016.02.012
- Kondo T, Miwa H. The role of esophageal hypersensitivity in functional heartburn. J Clin Gastroenterol 2017; 51(7):571–578. doi:10.1097/MCG.0000000000000885
- Farmer AD, Ruffle JK, Aziz Q. The role of esophageal hypersensitivity in functional esophageal disorders. J Clin Gastroenterol 2017; 51(2):91–99. doi:10.1097/MCG.0000000000000757
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55(10):1398–1402. doi:10.1136/gut.2005.087668
- Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013; 108(3):308–328. doi:10.1038/ajg.2012.444
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27(2):160–174. doi:10.1111/nmo.12477
- Kichler AJ, Gabbard S. A man with progressive dysphagia. Cleve Clin J Med 2017; 84(6):443–449. doi:10.3949/ccjm.84a.16055
- Roman S, Gyawali CP, Savarino E, et al; GERD consensus group. Ambulatory reflux monitoring for diagnosis of gastro-esophageal reflux disease: update of the Porto consensus and recommendations from an international consensus group. Neurogastroenterol Motil 2017; 29(10):1–15. doi:10.1111/nmo.13067
- Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG clinical guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016; 111(1):30–50. doi:10.1038/ajg.2015.322
- Halder SL, Locke GR 3rd, Schleck CD, Zinsmeister AR, Melton LJ 3rd, Talley NJ. Natural history of functional gastrointestinal disorders: a 12-year longitudinal population-based study. Gastroenterology 2007; 133(3):799–807. doi:10.1053/j.gastro.2007.06.010
- Park EY, Choi MG, Baeg M, et al. The value of early wireless esophageal pH monitoring in diagnosing functional heartburn in refractory gastroesophageal reflux disease. Dig Dis Sci 2013; 58(10):2933–2939. doi:10.1007/s10620-013-2728-4
- Khajanchee YS, Hong D, Hansen PD, Swanström LL. Outcomes of antireflux surgery in patients with normal preoperative 24-hour pH test results. Am J Surg 2004; 187(5):599–603. doi:10.1016/j.amjsurg.2004.01.010
- Viazis N, Keyoglou A, Kanellopoulos AK, et al. Selective serotonin reuptake inhibitors for the treatment of hypersensitive esophagus: a randomized, double-blind, placebo-controlled study. Am J Gastroenterol 2012; 107(11):1662–1667. doi:10.1038/ajg.2011.179
- Limsrivilai J, Charatcharoenwitthaya P, Pausawasdi N, Leelakusolvong S. Imipramine for treatment of esophageal hypersensitivity and functional heartburn: a randomized placebo-controlled trial. Am J Gastroenterol 2016; 111(2):217–224. doi:10.1038/ajg.2015.413
- Keefer L, Kahrilas PJ. Low-dose tricyclics for esophageal hypersensitivity: is it all placebo effect? Am J Gastroenterol 2016; 111(2):225–227. doi:10.1038/ajg.2016.13
- Basu PP, Hempole H, Krishnaswamy N, Shah NJ, Aloysius, M. The effect of melatonin in functional heartburn: a randomized, placebo-controlled clinical trial. Open J Gastroenterol 2014; 4(2):56–61. doi:10.4236/ojgas.2014.42010
- Watanabe S, Hattori T, Kanazawa M, Kano M, Fukudo S. Role of histaminergic neurons in hypnotic modulation of brain processing of visceral perception. Neurogastroenterol Motil 2007; 19(10):831–838. doi:10.1111/j.1365-2982.2007.00959.x
- Riehl ME, Kinsinger S, Kahrilas PJ, Pandolfino JE, Keefer L. Role of a health psychologist in the management of functional esophageal complaints. Dis Esophagus 2015; 28(5):428–436. doi:10.1111/dote.12219
- Klein KB, Spiegel D. Modulation of gastric acid secretion by hypnosis. Gastroenterology 1989; 96(6):1383–1387. pmid:2714570
- Riehl ME, Pandolfino JE, Palsson OS, Keefer L. Feasibility and acceptability of esophageal-directed hypnotherapy for functional heartburn. Dis Esophagus 2016; 29(5):490–496. doi:10.1111/dote.12353
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101(8):1900–1920.
- Dean BB, Gano AD Jr, Knight K, Ofman JJ, Fass R. Effectiveness of proton pump inhibitors in nonerosive reflux disease. Clin Gastroenterol Hepatol 2004; 2(8):656–664. pmid:15290657
- Hachem C, Shaheen NJ. Diagnosis and management of functional heartburn. Am J Gastroenterol 2016; 111(1):53–61. doi:10.1038/ajg.2015.376
- Fass R, Sifrim D. Management of heartburn not responding to proton pump inhibitors. Gut 2009; 58(2):295–309. doi:10.1136/gut.2007.145581
- Aziz Q, Fass R, Gyawali CP, Miwa H, Pandolfino JE, Zerbib F. Esophageal disorders. Gastroenterology 2016; 150(6):1368-1379. doi:10.1053/j.gastro.2016.02.012
- Kondo T, Miwa H. The role of esophageal hypersensitivity in functional heartburn. J Clin Gastroenterol 2017; 51(7):571–578. doi:10.1097/MCG.0000000000000885
- Farmer AD, Ruffle JK, Aziz Q. The role of esophageal hypersensitivity in functional esophageal disorders. J Clin Gastroenterol 2017; 51(2):91–99. doi:10.1097/MCG.0000000000000757
- Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006; 55(10):1398–1402. doi:10.1136/gut.2005.087668
- Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2013; 108(3):308–328. doi:10.1038/ajg.2012.444
- Kahrilas PJ, Bredenoord AJ, Fox M, et al; International High Resolution Manometry Working Group. The Chicago classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil 2015; 27(2):160–174. doi:10.1111/nmo.12477
- Kichler AJ, Gabbard S. A man with progressive dysphagia. Cleve Clin J Med 2017; 84(6):443–449. doi:10.3949/ccjm.84a.16055
- Roman S, Gyawali CP, Savarino E, et al; GERD consensus group. Ambulatory reflux monitoring for diagnosis of gastro-esophageal reflux disease: update of the Porto consensus and recommendations from an international consensus group. Neurogastroenterol Motil 2017; 29(10):1–15. doi:10.1111/nmo.13067
- Shaheen NJ, Falk GW, Iyer PG, Gerson LB; American College of Gastroenterology. ACG clinical guideline: diagnosis and management of Barrett’s esophagus. Am J Gastroenterol 2016; 111(1):30–50. doi:10.1038/ajg.2015.322
- Halder SL, Locke GR 3rd, Schleck CD, Zinsmeister AR, Melton LJ 3rd, Talley NJ. Natural history of functional gastrointestinal disorders: a 12-year longitudinal population-based study. Gastroenterology 2007; 133(3):799–807. doi:10.1053/j.gastro.2007.06.010
- Park EY, Choi MG, Baeg M, et al. The value of early wireless esophageal pH monitoring in diagnosing functional heartburn in refractory gastroesophageal reflux disease. Dig Dis Sci 2013; 58(10):2933–2939. doi:10.1007/s10620-013-2728-4
- Khajanchee YS, Hong D, Hansen PD, Swanström LL. Outcomes of antireflux surgery in patients with normal preoperative 24-hour pH test results. Am J Surg 2004; 187(5):599–603. doi:10.1016/j.amjsurg.2004.01.010
- Viazis N, Keyoglou A, Kanellopoulos AK, et al. Selective serotonin reuptake inhibitors for the treatment of hypersensitive esophagus: a randomized, double-blind, placebo-controlled study. Am J Gastroenterol 2012; 107(11):1662–1667. doi:10.1038/ajg.2011.179
- Limsrivilai J, Charatcharoenwitthaya P, Pausawasdi N, Leelakusolvong S. Imipramine for treatment of esophageal hypersensitivity and functional heartburn: a randomized placebo-controlled trial. Am J Gastroenterol 2016; 111(2):217–224. doi:10.1038/ajg.2015.413
- Keefer L, Kahrilas PJ. Low-dose tricyclics for esophageal hypersensitivity: is it all placebo effect? Am J Gastroenterol 2016; 111(2):225–227. doi:10.1038/ajg.2016.13
- Basu PP, Hempole H, Krishnaswamy N, Shah NJ, Aloysius, M. The effect of melatonin in functional heartburn: a randomized, placebo-controlled clinical trial. Open J Gastroenterol 2014; 4(2):56–61. doi:10.4236/ojgas.2014.42010
- Watanabe S, Hattori T, Kanazawa M, Kano M, Fukudo S. Role of histaminergic neurons in hypnotic modulation of brain processing of visceral perception. Neurogastroenterol Motil 2007; 19(10):831–838. doi:10.1111/j.1365-2982.2007.00959.x
- Riehl ME, Kinsinger S, Kahrilas PJ, Pandolfino JE, Keefer L. Role of a health psychologist in the management of functional esophageal complaints. Dis Esophagus 2015; 28(5):428–436. doi:10.1111/dote.12219
- Klein KB, Spiegel D. Modulation of gastric acid secretion by hypnosis. Gastroenterology 1989; 96(6):1383–1387. pmid:2714570
- Riehl ME, Pandolfino JE, Palsson OS, Keefer L. Feasibility and acceptability of esophageal-directed hypnotherapy for functional heartburn. Dis Esophagus 2016; 29(5):490–496. doi:10.1111/dote.12353
KEY POINTS
- Functional heartburn accounts for more than half of all referrals for PPI-refractory GERD.
- Diagnostic criteria require at least 3 months of symptoms in the 6 months before presentation.
- Results of upper endoscopy with biopsy, esophageal manometry, and esophageal pH monitoring must be normal.
- Patient education is key, with reassurance that the risk of progression to malignancy is low in the absence of Barrett esophagus, and that the condition remits spontaneously in up to 40% of cases.
- Neuromodulators to reduce pain perception are the mainstay of treatment for functional gastrointestinal disorders such as functional heartburn. Cognitive behavioral therapy and hypnotherapy are also used as first-line treatment.

Beyond depression: Other uses for tricyclic antidepressants
Most tricyclic antidepressants (TCAs) have US Food and Drug Administration approval for treatment of depression and anxiety disorders, but they are also a viable off-label option that should be considered by clinicians in specialties beyond psychiatry, especially for treating pain syndromes. Given the ongoing epidemic of opioid use disorder, increasing attention has been drawn to alternative strategies for chronic pain management, renewing an interest in the use of TCAs.
This review summarizes the pharmacologic properties of TCAs, their potential indications in conditions other than depression, and safety considerations.
BRIEF HISTORY OF TRICYCLICS
TCAs were originally designed in the 1950s and marketed later for treating depression. Due to their adverse effects and lethality in overdose quantities, over time they have been largely replaced by selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) in depression management. However, TCAs have been applied to conditions other than depression with varying degrees of efficacy and safety.
TCA PHARMACOLOGY

TCAs are absorbed in the small intestine and undergo first-pass metabolism in the liver. They bind extensively to proteins, leading to interactions with other protein-bound drugs. They are widely distributed throughout the systemic circulation because they are highly lipophilic, resulting in systemic effects including central nervous system manifestations.
Peak plasma concentration is at about 2 to 6 hours, and elimination half-life is around 24 hours for most agents, providing a long duration of action. Clearance depends on cytochrome P450 oxidative enzymes.1
MECHANISMS OF ACTION
TCAs inhibit reuptake of norepinephrine and serotonin, resulting in accumulation of these neurotransmitters in the presynaptic cleft. They also block postsynaptic histamine, alpha-adrenergic, and muscarinic-acetylcholine receptors, causing a variety of adverse effects, including dry mouth, confusion, cognitive impairment, hypotension, orthostasis, blurred vision, urinary retention, drowsiness, and sedation.1
Research suggests that TCAs relieve pain centrally through a descending pathway that inhibits transmission of pain signals in the spinal cord, as well as peripherally through complex anti-neuroimmune actions.2 Norepinephrine appears to play a more important role in this process than serotonin, although both are deemed necessary for the “dual action” often cited in pain management,1 which is also the rationale for widespread use of SNRIs to control pain.
Table 1 compares neurotransmitter reuptake mechanisms, adverse effect profiles, and typical dosages for depression for commonly prescribed TCAs.
POTENTIAL USES
Headache and migraine
TCAs have been shown to be effective for managing and preventing chronic headache syndromes.3,4 Amitriptyline has been the most studied of the TCAs for both chronic daily and episodic migraine headache, showing the most efficacy among diverse drug classes (angiotensin II receptor blockers, anticonvulsants, beta-blockers, SSRIs) compared with placebo. However, in head-to-head trials, amitriptyline was no more effective than SSRIs, venlafaxine, topiramate, or propranolol.4 Jackson et al4 suggested that prophylactic medication choices should be tailored to patient characteristics and expected adverse effects, and specifically recommended that TCAs—particularly amitriptyline—be reserved for patients who have both migraine and depression.
Neuropathic pain
Neuropathic pain is defined as pain secondary to a lesion or disease of the somatosensory nervous system5 and is the pathomechanistic component of a number of conditions, including postherpetic neuralgia,6 diabetic and nondiabetic painful polyneuropathy,7 posttraumatic or postsurgical neuropathic pain8 (including plexus avulsion and complex regional pain syndrome9), central poststroke pain,10 spinal cord injury pain,11 and multiple sclerosis-associated pain.12
As a group, TCAs appear to have a role as first-line agents for managing these varied neuropathic pain syndromes. In a recent meta-analysis,13 16 (89%) of 18 placebo-controlled trials of TCAs (mainly amitriptyline at 25–150 mg/day) for these pain conditions were positive, with a combined number needed to treat of 3.6, suggesting a role for TCAs in these conditions. Of note, the TCAs desipramine14 and nortriptyline15 have demonstrated little evidence of efficacy in neuropathic pain syndromes.
Chronic low back pain
Chronic low back pain is a leading cause of loss of work, excessive healthcare expenditure, and disability in the United States. It can be due to numerous spinal conditions, including degenerative disk disease, spinal stenosis, lumbar spondylosis, and spinal arthropathy.
TCAs have been used to treat chronic low back pain for decades and have been repeatedly shown to be more effective than placebo in reducing pain severity.16,17 A double-blind controlled trial18 from 1999 compared the effects of the TCA maprotiline (up to 150 mg daily), the SSRI paroxetine (up to 30 mg daily), and placebo and found a statistically significant reduction in back pain with maprotiline compared with paroxetine and placebo. However, a 2008 meta-analysis suggested little evidence that TCAs were superior to placebo.19
Evidence of TCA efficacy for back pain was reported in 2018 with a well-designed 6-month double-blind randomized controlled trial20 comparing low-dose amitriptyline (25 mg) with an active comparator (benztropine 1 mg). The authors reported that amitriptyline was effective in reducing pain and pain-related disability without incurring serious adverse effects. They suggested continued use of TCAs for chronic low back pain if complicated with pain-related disability, insomnia, depression, or other comorbidity, although they called for further large-scale studies. They also cautioned that patients started the trial with symptoms similar to the adverse effects of TCAs themselves; this has implications for monitoring of symptoms as well as TCA adverse effects while using these drugs.
Fibromyalgia and chronic widespread pain
Fibromyalgia is a common, frustrating, noninflammatory pain syndrome characterized by diffuse hyperalgesia and multiple comorbidities.21 Although sleep hygiene, exercise, cognitive-behavioral therapy, some gabapentinoids (pregabalin), and a combination of these therapies have demonstrated efficacy, TCAs also offer robust benefits.
A meta-analysis of 9 placebo-controlled TCA trials showed large effect sizes for pain reduction, fatigue reduction, improved sleep quality, and reduced stiffness and tenderness, with the most significant of these improvements being for sleep.22 A separate meta-analysis calculated that the number needed to treat with amitriptyline for a positive outcome is 4.9.23 Recent systematic reviews have supported these findings, listing TCAs as second-line agents after pregabalin, duloxetine, and milnacipran.24
Of note, TCA monotherapy rarely produces a complete response in patients with moderate to severe fibromyalgia, chronic widespread pain, or significant comorbidities (depression, anxiety). Supplementation with cognitive-behavioral therapy, physical therapy, functional restoration, and other modalities is strongly recommended.
Abdominal and gastrointestinal pain
TCAs have been applied to a number of gastrointestinal syndromes with or without pain. Patients with irritable bowel syndrome have long been known to benefit from TCAs; the number needed to treat for symptomatic benefit over placebo is 3.5.25,26
Although there is no substantial evidence that TCAs are useful in reducing active inflammation in inflammatory bowel disease, a study involving 81 patients found that residual noninflammatory gastrointestinal symptoms (such as diarrhea and pain) responded to TCAs, including nortriptyline and amitriptyline, with greater benefit for ulcerative colitis than for Crohn disease.27
TCAs have also shown prophylactic benefit in cyclic vomiting syndrome, with a clinical response in over 75% of patients in controlled cohort studies.28
The efficacy of TCAs in other abdominal or gastrointestinal syndromes is unclear or modest at best.29 However, few alternative treatments exist for these conditions. Amitriptyline may help symptoms of functional dyspepsia,30 but nortriptyline has proven ineffective in gastroparesis.31 Nonetheless, some authors29 suggest considering TCAs on an individualized basis, with proper monitoring, in many if not most functional gastrointestinal disorders, especially when paired with behavioral therapies.
Pelvic and urogynecologic symptoms
Chronic pelvic pain affects up to 24% of women32 and 5% to 10% of men.33 TCAs have shown efficacy in treating chronic pelvic pain with or without comorbid depression.34 Amitriptyline and to a lesser extent nortriptyline are the TCAs most often prescribed. Pain relief appears to be independent of antidepressant effects and may be achieved at low doses; initial dosing ranges from 10 to 25 mg at bedtime, which may be increased to 100 mg as tolerated.34
Based on a randomized, double-blind trial,35 amitriptyline was recommended as a treatment option for interstitial cystitis or bladder pain, with the greatest symptom improvement in patients tolerating a daily dose of 50 mg.
Another study36 randomized 56 women with chronic pelvic pain to amitriptyline or gabapentin, or a combination of the drugs for 24 months. Although each regimen resulted in significant reduction in pain, fewer adverse effects occurred with gabapentin than amitriptyline. Poor compliance and early discontinuation of amitriptyline were common due to anticholinergic effects.
In small uncontrolled studies,37 about half of women with chronic pelvic pain became pain-free after 8 weeks of treatment with nortriptyline and imipramine.
Randomized controlled studies are needed to confirm potential benefits of TCAs in chronic urologic and pelvic pain.
Insomnia
Insomnia affects 23% to 56% of people in the United States, Europe, and Asia38 and is the reason for more than 5.5 million primary care visits annually.39 TCAs (especially doxepin, maprotiline, and amitriptyline40) have been shown to be an effective treatment, with an 82% increase in somnolence compared with placebo, as well as measurably improved total sleep time, enhanced sleep efficiency, reduced latency to persistent sleep, and decreased wake times after sleep onset.38
Dosing should be kept at a minimum to minimize harsh anticholinergic effects and avoid daytime sedation. Patients should be advised to take new doses or dose escalations earlier in the night to ensure less hangover sedation the next morning.
For patients with insomnia and comorbid depression, the American Academy of Sleep Medicine suggests the addition of a low dose (eg, 10–25 mg) of a TCA at nighttime to complement preexisting, full-dose, non-TCA antidepressants, while monitoring for serotonin syndrome and other potential but exceedingly rare drug-drug interactions.41
Psychiatric indications other than depression
Beyond the known benefits in major depressive disorder, TCAs have been shown to be effective for obsessive-compulsive disorder, panic disorder, posttraumatic stress disorder, bulimia nervosa, and childhood enuresis.42 Given the shortage of mental health clinicians and the high prevalence of these conditions, nonpsychiatrist physicians should be familiar with the therapeutic potential of TCAs for these indications.
ADVERSE EFFECTS
Adverse effects vary among TCAs. Common ones include blurred vision, dry mouth, constipation, urinary retention, hypotension, tachycardia, tremor, weight gain, and sexual dysfunction.43 Tertiary amines are generally more sedating than secondary amines and cause more anticholinergic effects (Table 1).

Despite widespread perceptions that TCAs are less tolerable than newer antidepressants, studies repeatedly suggest that they have an adverse-effect burden similar to that of SSRIs and SNRIs, although SSRIs have a greater tendency to produce nausea, whereas TCAs are more likely to cause constipation.44
Discontinuation syndrome
Abrupt discontinuation or unintentionally missed doses of TCAs have been associated with a discontinuation syndrome in about 40% of users.45 Patients should be warned about this possibility and the syndrome’s potential effects: dizziness, insomnia, headaches, nausea, vomiting, flulike achiness, and restlessness. Rebound depression, anxiety, panic, or other psychiatric symptoms may also occur. Symptoms generally present within 2 to 5 days after dose discontinuation and last 7 to 14 days.45
However, all TCAs have a long half-life, allowing for sufficient coverage with once-daily dosing and thus carry a lower risk of discontinuation syndrome than many other antidepressants (78% with venlafaxine; 55% with paroxetine).45
To discontinue therapy safely, the dosage should be reduced gradually. As is pharmacologically expected, the greatest likelihood of discontinuation syndrome is associated with longer duration of continuous treatment.
CONTRAINDICATIONS
Cardiac conduction abnormalities
TCAs should not be prescribed to patients who have right bundle branch block, a severe electrolyte disturbance, or other cardiac conduction deficit or arrhythmia that can prolong the QTc interval and elevate the risk of lethal arrhythmia.46,47 Cardiac effects from TCAs are largely dose-dependent. Nevertheless, a baseline electrocardiogram can be obtained to assess cardiac risk, and dose escalation can proceed if results are normal (eg, appropriate conduction intervals, QTc ≤ 450 ms).
Advanced age
For elderly patients, TCAs should be prescribed with caution and sometimes not at all,48 because anticholinergic effects may worsen preexisting urinary retention (including benign prostatic hyperplasia), narrow-angle glaucoma, imbalance and gait issues, and cognitive impairment and dementia. Dehydration and orthostatic hypotension are contraindications for TCAs, as they may precipitate falls or hypotensive shock.
Epilepsy
TCAs should also be used with caution in patients with epilepsy, as they lower the seizure threshold.
Concomitant monoamine oxidase inhibitor treatment
Giving TCAs together with monoamine oxidase inhibitor antidepressants should be avoided, given the risk of hypertensive crisis.
Suicide risk
TCAs are dangerous and potentially lethal in overdose and so should not be prescribed to suicidal or otherwise impulsive patients.
Pregnancy
TCAs are in pregnancy risk category C (animal studies show adverse effects on fetus; no adequate or well-controlled studies in humans; potential benefits may warrant use despite risks). Using TCAs during pregnancy has very rarely led to neonatal withdrawal such as irritability, jitteriness, and convulsions, as well as fetal QTc interval prolongation.49
The American College of Obstetricians and Gynecologists recommends that therapy for depression during pregnancy be individualized, incorporating the expertise of the patient’s mental health clinician, obstetrician, primary healthcare provider, and pediatrician. In general, they recommend that TCAs should be avoided if possible and that alternatives such as SSRIs or SNRIs should be considered.50
TCAs are excreted in breast milk, but they have not been detected in the serum of nursing infants, and no adverse events have been reported.
OVERDOSE IS HIGHLY DANGEROUS
Severe morbidity and death are associated with TCA overdose, characterized by convulsions, cardiac arrest, and coma (the “3 Cs”). These dangers occur at much higher rates with TCAs than with other antidepressants.43 Signs and symptoms of toxicity develop rapidly, usually within the first hour of overdose. Manifestations of overdose include prolonged QTc, cardiac arrhythmias, tachycardia, hypertension, severe hypotension, agitation, seizures, central nervous system depression, hallucinations, seizures, and coma.
Overdose management includes activated charcoal, seizure control, cardioversion, hydration, electrolyte stabilization, and other intensive care.
OFF-LABEL TCA MANAGEMENT
Dosing recommendations for off-label use of TCAs vary based on the condition, the medication, and the suggestions of individual authors and researchers. In general, dosing ranges for pain and other nondepression indications may be lower than for severe depression (Table 2).1
As with any pharmacologic titration, monitoring for rate-limiting adverse effects is recommended. We suggest caution, tailoring the approach to the patient, and routinely assessing for adverse effects and other safety considerations.
In addition, we strongly recommend supplementing TCA therapy with nonpharmacologic strategies such as lifestyle changes, dietary modifications, exercise, physical therapy, and mental health optimization.
- Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci 2017; 18(11). doi:10.3390/ijms18112483
- Kremer M, Yalcin I, Goumon Y, et al. A dual noradrenergic mechanism for the relief of neuropathic allodynia by the antidepressant drugs duloxetine and amitriptyline. J Neurosci 2018; 38(46):9934–9954. doi:10.1523/JNEUROSCI.1004-18.2018
- Tomkins GE, Jackson JL, O’Malley PG, Balden E, Santoro JE. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med 2001; 111(1):54–63. doi:10.1016/s0002-9343(01)00762-8
- Jackson JL, Cogbill E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PLoS One 2015; 10(7):e0130733. doi:10.1371/journal.pone.0130733
- International Association for the Study of Pain (IASP). IASP Terminology. www.iasp-pain.org/Education/Content.aspx?ItemNumber=1698&navItemNumber=576. Accessed November 20, 2019.
- Feller L, Khammissa RAG, Fourie J, Bouckaert M, Lemmer J. Postherpectic neuralgia and trigeminal neuralgia. Pain Res Treat 2017; 2017:1681765. doi:10.1155/2017/1681765
- Shillo P, Sloan G, Greig M, et al. Painful and painless diabetic neuropathies: what is the difference? Curr Diab Rep 2019; 19(6):32. doi:10.1007/s11892-019-1150-5
- Schwartzman RJ, Maleki J. Postinjury neuropathic pain syndromes. Med Clin North Am 1999; 83(3):597–626. doi:10.1016/s0025-7125(05)70126-7
- Oaklander AL, Horowitz SH. The complex regional pain syndrome. Handb Clin Neurol 2015; 131:481–503. doi:10.1016/B978-0-444-62627-1.00026-3
- Akyuz G, Kuru P. Systematic review of central post stroke pain: what is happening in the central nervous system? Am J Phys Med Rehabil 2016; 95(8):618–627. doi:10.1097/PHM.0000000000000542
- Shiao R, Lee-Kubli CA. Neuropathic pain after spinal cord injury: challenges and research perspectives. Neurotherapeutics 2018; 15(3):635–653. doi:10.1007/s13311-018-0633-4
- Ceruti S. What role does multiple sclerosis play in the development of untreatable painful conditions? Pain Manag 2018; 8(1):37–44. doi:10.2217/pmt-2017-0038
- Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015; 14(2):162–173. doi:10.1016/S1474-4422(14)70251-0
- Hearn L, Moore RA, Derry S, Wiffen PJ, Phillips T. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev 2014; (9):CD011003. doi:10.1002/14651858.CD011003.pub2
- Derry S, Whiffen PJ, Aldington D, Moore RA. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev 2015; 1:CD011209. doi:10.1002/14651858.CD011209.pub2
- Salerno SM, Browning R, Jackson JL. The effect of antidepressant treatment on chronic back pain: a meta-analysis. Arch Intern Med 2002; 162(1):19–24. doi:10.1001/archinte.162.1.19
- Staiger TO, Gaster B, Sullivan MD, Deyo RA. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976) 2003; 28(22):2540–2545. doi:10.1097/01.BRS.0000092372.73527.BA
- Atkinson JH, Slater MA, Wahlgren DR, et al. Effects of noradrenergic and serotonergic antidepressants on chronic low back pain intensity. Pain 1999; 83(2):137–145. doi:10.1016/s0304-3959(99)00082-2
- Urquhart DM, Hoving JL, Assendelft WW, Roland M, van Tulder MW. Antidepressants for non-specific back pain. Cochrane Database Syst Rev 2008; (1):CD001703. doi:10.1002/14651858.CD001703.pub3
- Urquhart DM, Wluka AE, van Tulder M, et al. Efficacy of low-dose amitriptyline for chronic low back pain: a randomized clinical trial. JAMA Intern Med 2018; 178(11):1474–1481. doi:10.1001/jamainternmed.2018.4222
- Clauw DJ. Fibromyalgia: a clinical review. JAMA 2014; 311(15):1547–1555. doi:10.1001/jama.2014.3266
- Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41(2):104–113. pmid:10749947
- Hauser W, Wolfe F, Tolle T, Uceyler N, Sommer C. The role of antidepressants in the management of fibromyalgia syndrome: a systematic review and meta-analysis. CNS Drugs 2012; 26(4):297–307. doi:10.2165/11598970-000000000-00000
- Calandre EP, Rico-Villademoros F, Slim M. An update on pharmacotherapy for the treatment of fibromyalgia. Expert Opin Pharmacother 2015; 16(9):1347–1368. doi:10.1517/14656566.2015.1047343
- Jackson JL, O’Malley PG, Tomkins G, Balden E, Santoro J, Kroenke K. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med 2000; 108(1):65–72. doi:10.1016/s0002-9343(99)00299-5
- Rahimi R, Nikfar S, Rezaie A, Abdollahi M. Efficacy of tricyclic antidepressants in irritable bowel syndrome: a meta-analysis. World J Gastroenterol 2009; 15(13):1548–1553. doi:10.3748/wjg.15.1548
- Iskandar HN, Cassell B, Kanuri N, et al. Tricyclic antidepressants for management of residual symptoms in inflammatory bowel disease. J Clin Gastroenterol 2014; 48(5):423–429. doi:10.1097/MCG.0000000000000049
- Lee LY, Abbott L, Mahlangu B, Moodie SJ, Anderson S. The management of cyclic vomiting syndrome: a systematic review. Eur J Gastroenterol Hepatol 2012; 24(9):1001–1006. doi:10.1097/MEG.0b013e328355638f
- Thorkelson G, Bielefeldt K, Szigethy E. Empirically supported use of psychiatric medications in adolescents and adults with IBD. Inflamm Bowel Dis 2016; 22(6):1509–1522. doi:10.1097/MIB.0000000000000734
- Braak B, Klooker TK, Wouters MM, et al. Randomised clinical trial: the effects of amitriptyline on drinking capacity and symptoms in patients with functional dyspepsia, a double-blind placebo-controlled study. Aliment Pharmacol Ther 2011; 34(6):638–648. doi:10.1111/j.1365-2036.2011.04775.x
- Parkman HP, Van Natta ML, Abell TL, et al. Effect of nortriptyline on symptoms of idiopathic gastroparesis: the NORIG randomized clinical trial. JAMA 2013; 310(24):2640–2649. doi:10.1001/jama.2013.282833
- Latthe P, Latthe M, Say L, Gulmezoglu M, Khan KS. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health 2006; 6:177. doi:10.1186/1471-2458-6-177
- Moise G, Capodice J, Winfree CJ. Treatment of chronic pelvic pain in men and women. Expert Rev Neurother 2007; 7(5):507–520. doi:10.1586/14737175.7.5.507
- Lai HH. Management of interstitial cystitis/bladder pain syndrome with tricyclic antidepressants. In: Moldwin RM, ed. Urological and Gynaecological Chronic Pelvic Pain. Cham, Switzerland: Springer; 2017:107–118.
- American Urological Association. Diagnosis and treatment interstitial cystitis/bladder pain syndrome (2014). www.auanet.org/guidelines/interstitial-cystitis/bladder-pain-syndrome-(2011-amended-2014). Accessed November 19, 2019.
- Carey ET, As-Sanie S. New developments in the pharmacotherapy of neuropathic chronic pelvic pain. Future Sci OA 2016; 2(4):FSO148. doi:10.4155/fsoa-2016-0048
- Papandreou C, Skapinakis P, Giannakis D, Sofikitis N, Mavreas V. Antidepressant drugs for chronic urological pelvic pain: an evidence-based review. Adv Urol 2009; 2009:797031. doi:10.1155/2009/797031
- Liu Y, Xu X, Dong M, Jia S, Wei Y. Treatment of insomnia with tricyclic antidepressants: a meta-analysis of polysomnographic randomized controlled trials. Sleep Med 2017; 34:126–133. doi:10.1016/j.sleep.2017.03.007
- Matheson E, Hainer BL. Insomnia: pharmacologic therapy. Am Fam Physician 2017; 96(1):29–35. pmid:28671376
- McCall C, McCall WV. What is the role of sedating antidepressants, antipsychotics, and anticonvulsants in the management of insomnia? Curr Psychiatry Rep 2012; 14(5):494–502. doi:10.1007/s11920-012-0302-y
- Clark MS, Smith PO, Jamieson B. FPIN’s clinical inquiries: antidepressants for the treatment of insomnia in patients with depression. Am Fam Physician 2011; 84(9):1–2. pmid:22164891
- Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s Synopsis of Psychiatry. New York, NY: Lippincott Williams & Wilkins; 2014.
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J 2018; 54(2):101–112. doi:10.4068/cmj.2018.54.2.101.
- Trindade E, Menon D, Topfer LA, Coloma C. Adverse effects associated with selective serotonin reuptake inhibitors and tricyclic antidepressants: a meta-analysis. CMAJ 1998; 159(10):1245–1252. pmid:9861221
- Fava M. Prospective studies of adverse events related to antidepressant discontinuation. J Clin Psychiatry 2006; 67(suppl 4):14–21. pmid:16683858
- Gintant G. An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 2011; 129(2):109–119. doi:10.1016/j.pharmthera.2010.08.008.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54(1):1–13. doi:10.1016/j.psym.2012.11.001
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63(11):2227–2246. doi:10.1111/jgs.13702
- Fukushima N, Nanao K, Fukushima H, Namera A, Miura M. A neonatal prolonged QT syndrome due to maternal use of oral tricyclic antidepressants. Eur J Pediatr 2016; 175(8):1129–1132. doi:10.1007/s00431-016-2722-x
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111(4):1001–1020. doi:10.1097/AOG.0b013e31816fd910
Most tricyclic antidepressants (TCAs) have US Food and Drug Administration approval for treatment of depression and anxiety disorders, but they are also a viable off-label option that should be considered by clinicians in specialties beyond psychiatry, especially for treating pain syndromes. Given the ongoing epidemic of opioid use disorder, increasing attention has been drawn to alternative strategies for chronic pain management, renewing an interest in the use of TCAs.
This review summarizes the pharmacologic properties of TCAs, their potential indications in conditions other than depression, and safety considerations.
BRIEF HISTORY OF TRICYCLICS
TCAs were originally designed in the 1950s and marketed later for treating depression. Due to their adverse effects and lethality in overdose quantities, over time they have been largely replaced by selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) in depression management. However, TCAs have been applied to conditions other than depression with varying degrees of efficacy and safety.
TCA PHARMACOLOGY
TCAs are absorbed in the small intestine and undergo first-pass metabolism in the liver. They bind extensively to proteins, leading to interactions with other protein-bound drugs. They are widely distributed throughout the systemic circulation because they are highly lipophilic, resulting in systemic effects including central nervous system manifestations.
Peak plasma concentration is at about 2 to 6 hours, and elimination half-life is around 24 hours for most agents, providing a long duration of action. Clearance depends on cytochrome P450 oxidative enzymes.1
MECHANISMS OF ACTION
TCAs inhibit reuptake of norepinephrine and serotonin, resulting in accumulation of these neurotransmitters in the presynaptic cleft. They also block postsynaptic histamine, alpha-adrenergic, and muscarinic-acetylcholine receptors, causing a variety of adverse effects, including dry mouth, confusion, cognitive impairment, hypotension, orthostasis, blurred vision, urinary retention, drowsiness, and sedation.1
Research suggests that TCAs relieve pain centrally through a descending pathway that inhibits transmission of pain signals in the spinal cord, as well as peripherally through complex anti-neuroimmune actions.2 Norepinephrine appears to play a more important role in this process than serotonin, although both are deemed necessary for the “dual action” often cited in pain management,1 which is also the rationale for widespread use of SNRIs to control pain.
Table 1 compares neurotransmitter reuptake mechanisms, adverse effect profiles, and typical dosages for depression for commonly prescribed TCAs.
POTENTIAL USES
Headache and migraine
TCAs have been shown to be effective for managing and preventing chronic headache syndromes.3,4 Amitriptyline has been the most studied of the TCAs for both chronic daily and episodic migraine headache, showing the most efficacy among diverse drug classes (angiotensin II receptor blockers, anticonvulsants, beta-blockers, SSRIs) compared with placebo. However, in head-to-head trials, amitriptyline was no more effective than SSRIs, venlafaxine, topiramate, or propranolol.4 Jackson et al4 suggested that prophylactic medication choices should be tailored to patient characteristics and expected adverse effects, and specifically recommended that TCAs—particularly amitriptyline—be reserved for patients who have both migraine and depression.
Neuropathic pain
Neuropathic pain is defined as pain secondary to a lesion or disease of the somatosensory nervous system5 and is the pathomechanistic component of a number of conditions, including postherpetic neuralgia,6 diabetic and nondiabetic painful polyneuropathy,7 posttraumatic or postsurgical neuropathic pain8 (including plexus avulsion and complex regional pain syndrome9), central poststroke pain,10 spinal cord injury pain,11 and multiple sclerosis-associated pain.12
As a group, TCAs appear to have a role as first-line agents for managing these varied neuropathic pain syndromes. In a recent meta-analysis,13 16 (89%) of 18 placebo-controlled trials of TCAs (mainly amitriptyline at 25–150 mg/day) for these pain conditions were positive, with a combined number needed to treat of 3.6, suggesting a role for TCAs in these conditions. Of note, the TCAs desipramine14 and nortriptyline15 have demonstrated little evidence of efficacy in neuropathic pain syndromes.
Chronic low back pain
Chronic low back pain is a leading cause of loss of work, excessive healthcare expenditure, and disability in the United States. It can be due to numerous spinal conditions, including degenerative disk disease, spinal stenosis, lumbar spondylosis, and spinal arthropathy.
TCAs have been used to treat chronic low back pain for decades and have been repeatedly shown to be more effective than placebo in reducing pain severity.16,17 A double-blind controlled trial18 from 1999 compared the effects of the TCA maprotiline (up to 150 mg daily), the SSRI paroxetine (up to 30 mg daily), and placebo and found a statistically significant reduction in back pain with maprotiline compared with paroxetine and placebo. However, a 2008 meta-analysis suggested little evidence that TCAs were superior to placebo.19
Evidence of TCA efficacy for back pain was reported in 2018 with a well-designed 6-month double-blind randomized controlled trial20 comparing low-dose amitriptyline (25 mg) with an active comparator (benztropine 1 mg). The authors reported that amitriptyline was effective in reducing pain and pain-related disability without incurring serious adverse effects. They suggested continued use of TCAs for chronic low back pain if complicated with pain-related disability, insomnia, depression, or other comorbidity, although they called for further large-scale studies. They also cautioned that patients started the trial with symptoms similar to the adverse effects of TCAs themselves; this has implications for monitoring of symptoms as well as TCA adverse effects while using these drugs.
Fibromyalgia and chronic widespread pain
Fibromyalgia is a common, frustrating, noninflammatory pain syndrome characterized by diffuse hyperalgesia and multiple comorbidities.21 Although sleep hygiene, exercise, cognitive-behavioral therapy, some gabapentinoids (pregabalin), and a combination of these therapies have demonstrated efficacy, TCAs also offer robust benefits.
A meta-analysis of 9 placebo-controlled TCA trials showed large effect sizes for pain reduction, fatigue reduction, improved sleep quality, and reduced stiffness and tenderness, with the most significant of these improvements being for sleep.22 A separate meta-analysis calculated that the number needed to treat with amitriptyline for a positive outcome is 4.9.23 Recent systematic reviews have supported these findings, listing TCAs as second-line agents after pregabalin, duloxetine, and milnacipran.24
Of note, TCA monotherapy rarely produces a complete response in patients with moderate to severe fibromyalgia, chronic widespread pain, or significant comorbidities (depression, anxiety). Supplementation with cognitive-behavioral therapy, physical therapy, functional restoration, and other modalities is strongly recommended.
Abdominal and gastrointestinal pain
TCAs have been applied to a number of gastrointestinal syndromes with or without pain. Patients with irritable bowel syndrome have long been known to benefit from TCAs; the number needed to treat for symptomatic benefit over placebo is 3.5.25,26
Although there is no substantial evidence that TCAs are useful in reducing active inflammation in inflammatory bowel disease, a study involving 81 patients found that residual noninflammatory gastrointestinal symptoms (such as diarrhea and pain) responded to TCAs, including nortriptyline and amitriptyline, with greater benefit for ulcerative colitis than for Crohn disease.27
TCAs have also shown prophylactic benefit in cyclic vomiting syndrome, with a clinical response in over 75% of patients in controlled cohort studies.28
The efficacy of TCAs in other abdominal or gastrointestinal syndromes is unclear or modest at best.29 However, few alternative treatments exist for these conditions. Amitriptyline may help symptoms of functional dyspepsia,30 but nortriptyline has proven ineffective in gastroparesis.31 Nonetheless, some authors29 suggest considering TCAs on an individualized basis, with proper monitoring, in many if not most functional gastrointestinal disorders, especially when paired with behavioral therapies.
Pelvic and urogynecologic symptoms
Chronic pelvic pain affects up to 24% of women32 and 5% to 10% of men.33 TCAs have shown efficacy in treating chronic pelvic pain with or without comorbid depression.34 Amitriptyline and to a lesser extent nortriptyline are the TCAs most often prescribed. Pain relief appears to be independent of antidepressant effects and may be achieved at low doses; initial dosing ranges from 10 to 25 mg at bedtime, which may be increased to 100 mg as tolerated.34
Based on a randomized, double-blind trial,35 amitriptyline was recommended as a treatment option for interstitial cystitis or bladder pain, with the greatest symptom improvement in patients tolerating a daily dose of 50 mg.
Another study36 randomized 56 women with chronic pelvic pain to amitriptyline or gabapentin, or a combination of the drugs for 24 months. Although each regimen resulted in significant reduction in pain, fewer adverse effects occurred with gabapentin than amitriptyline. Poor compliance and early discontinuation of amitriptyline were common due to anticholinergic effects.
In small uncontrolled studies,37 about half of women with chronic pelvic pain became pain-free after 8 weeks of treatment with nortriptyline and imipramine.
Randomized controlled studies are needed to confirm potential benefits of TCAs in chronic urologic and pelvic pain.
Insomnia
Insomnia affects 23% to 56% of people in the United States, Europe, and Asia38 and is the reason for more than 5.5 million primary care visits annually.39 TCAs (especially doxepin, maprotiline, and amitriptyline40) have been shown to be an effective treatment, with an 82% increase in somnolence compared with placebo, as well as measurably improved total sleep time, enhanced sleep efficiency, reduced latency to persistent sleep, and decreased wake times after sleep onset.38
Dosing should be kept at a minimum to minimize harsh anticholinergic effects and avoid daytime sedation. Patients should be advised to take new doses or dose escalations earlier in the night to ensure less hangover sedation the next morning.
For patients with insomnia and comorbid depression, the American Academy of Sleep Medicine suggests the addition of a low dose (eg, 10–25 mg) of a TCA at nighttime to complement preexisting, full-dose, non-TCA antidepressants, while monitoring for serotonin syndrome and other potential but exceedingly rare drug-drug interactions.41
Psychiatric indications other than depression
Beyond the known benefits in major depressive disorder, TCAs have been shown to be effective for obsessive-compulsive disorder, panic disorder, posttraumatic stress disorder, bulimia nervosa, and childhood enuresis.42 Given the shortage of mental health clinicians and the high prevalence of these conditions, nonpsychiatrist physicians should be familiar with the therapeutic potential of TCAs for these indications.
ADVERSE EFFECTS
Adverse effects vary among TCAs. Common ones include blurred vision, dry mouth, constipation, urinary retention, hypotension, tachycardia, tremor, weight gain, and sexual dysfunction.43 Tertiary amines are generally more sedating than secondary amines and cause more anticholinergic effects (Table 1).
Despite widespread perceptions that TCAs are less tolerable than newer antidepressants, studies repeatedly suggest that they have an adverse-effect burden similar to that of SSRIs and SNRIs, although SSRIs have a greater tendency to produce nausea, whereas TCAs are more likely to cause constipation.44
Discontinuation syndrome
Abrupt discontinuation or unintentionally missed doses of TCAs have been associated with a discontinuation syndrome in about 40% of users.45 Patients should be warned about this possibility and the syndrome’s potential effects: dizziness, insomnia, headaches, nausea, vomiting, flulike achiness, and restlessness. Rebound depression, anxiety, panic, or other psychiatric symptoms may also occur. Symptoms generally present within 2 to 5 days after dose discontinuation and last 7 to 14 days.45
However, all TCAs have a long half-life, allowing for sufficient coverage with once-daily dosing and thus carry a lower risk of discontinuation syndrome than many other antidepressants (78% with venlafaxine; 55% with paroxetine).45
To discontinue therapy safely, the dosage should be reduced gradually. As is pharmacologically expected, the greatest likelihood of discontinuation syndrome is associated with longer duration of continuous treatment.
CONTRAINDICATIONS
Cardiac conduction abnormalities
TCAs should not be prescribed to patients who have right bundle branch block, a severe electrolyte disturbance, or other cardiac conduction deficit or arrhythmia that can prolong the QTc interval and elevate the risk of lethal arrhythmia.46,47 Cardiac effects from TCAs are largely dose-dependent. Nevertheless, a baseline electrocardiogram can be obtained to assess cardiac risk, and dose escalation can proceed if results are normal (eg, appropriate conduction intervals, QTc ≤ 450 ms).
Advanced age
For elderly patients, TCAs should be prescribed with caution and sometimes not at all,48 because anticholinergic effects may worsen preexisting urinary retention (including benign prostatic hyperplasia), narrow-angle glaucoma, imbalance and gait issues, and cognitive impairment and dementia. Dehydration and orthostatic hypotension are contraindications for TCAs, as they may precipitate falls or hypotensive shock.
Epilepsy
TCAs should also be used with caution in patients with epilepsy, as they lower the seizure threshold.
Concomitant monoamine oxidase inhibitor treatment
Giving TCAs together with monoamine oxidase inhibitor antidepressants should be avoided, given the risk of hypertensive crisis.
Suicide risk
TCAs are dangerous and potentially lethal in overdose and so should not be prescribed to suicidal or otherwise impulsive patients.
Pregnancy
TCAs are in pregnancy risk category C (animal studies show adverse effects on fetus; no adequate or well-controlled studies in humans; potential benefits may warrant use despite risks). Using TCAs during pregnancy has very rarely led to neonatal withdrawal such as irritability, jitteriness, and convulsions, as well as fetal QTc interval prolongation.49
The American College of Obstetricians and Gynecologists recommends that therapy for depression during pregnancy be individualized, incorporating the expertise of the patient’s mental health clinician, obstetrician, primary healthcare provider, and pediatrician. In general, they recommend that TCAs should be avoided if possible and that alternatives such as SSRIs or SNRIs should be considered.50
TCAs are excreted in breast milk, but they have not been detected in the serum of nursing infants, and no adverse events have been reported.
OVERDOSE IS HIGHLY DANGEROUS
Severe morbidity and death are associated with TCA overdose, characterized by convulsions, cardiac arrest, and coma (the “3 Cs”). These dangers occur at much higher rates with TCAs than with other antidepressants.43 Signs and symptoms of toxicity develop rapidly, usually within the first hour of overdose. Manifestations of overdose include prolonged QTc, cardiac arrhythmias, tachycardia, hypertension, severe hypotension, agitation, seizures, central nervous system depression, hallucinations, seizures, and coma.
Overdose management includes activated charcoal, seizure control, cardioversion, hydration, electrolyte stabilization, and other intensive care.
OFF-LABEL TCA MANAGEMENT
Dosing recommendations for off-label use of TCAs vary based on the condition, the medication, and the suggestions of individual authors and researchers. In general, dosing ranges for pain and other nondepression indications may be lower than for severe depression (Table 2).1
As with any pharmacologic titration, monitoring for rate-limiting adverse effects is recommended. We suggest caution, tailoring the approach to the patient, and routinely assessing for adverse effects and other safety considerations.
In addition, we strongly recommend supplementing TCA therapy with nonpharmacologic strategies such as lifestyle changes, dietary modifications, exercise, physical therapy, and mental health optimization.
Most tricyclic antidepressants (TCAs) have US Food and Drug Administration approval for treatment of depression and anxiety disorders, but they are also a viable off-label option that should be considered by clinicians in specialties beyond psychiatry, especially for treating pain syndromes. Given the ongoing epidemic of opioid use disorder, increasing attention has been drawn to alternative strategies for chronic pain management, renewing an interest in the use of TCAs.
This review summarizes the pharmacologic properties of TCAs, their potential indications in conditions other than depression, and safety considerations.
BRIEF HISTORY OF TRICYCLICS
TCAs were originally designed in the 1950s and marketed later for treating depression. Due to their adverse effects and lethality in overdose quantities, over time they have been largely replaced by selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) in depression management. However, TCAs have been applied to conditions other than depression with varying degrees of efficacy and safety.
TCA PHARMACOLOGY
TCAs are absorbed in the small intestine and undergo first-pass metabolism in the liver. They bind extensively to proteins, leading to interactions with other protein-bound drugs. They are widely distributed throughout the systemic circulation because they are highly lipophilic, resulting in systemic effects including central nervous system manifestations.
Peak plasma concentration is at about 2 to 6 hours, and elimination half-life is around 24 hours for most agents, providing a long duration of action. Clearance depends on cytochrome P450 oxidative enzymes.1
MECHANISMS OF ACTION
TCAs inhibit reuptake of norepinephrine and serotonin, resulting in accumulation of these neurotransmitters in the presynaptic cleft. They also block postsynaptic histamine, alpha-adrenergic, and muscarinic-acetylcholine receptors, causing a variety of adverse effects, including dry mouth, confusion, cognitive impairment, hypotension, orthostasis, blurred vision, urinary retention, drowsiness, and sedation.1
Research suggests that TCAs relieve pain centrally through a descending pathway that inhibits transmission of pain signals in the spinal cord, as well as peripherally through complex anti-neuroimmune actions.2 Norepinephrine appears to play a more important role in this process than serotonin, although both are deemed necessary for the “dual action” often cited in pain management,1 which is also the rationale for widespread use of SNRIs to control pain.
Table 1 compares neurotransmitter reuptake mechanisms, adverse effect profiles, and typical dosages for depression for commonly prescribed TCAs.
POTENTIAL USES
Headache and migraine
TCAs have been shown to be effective for managing and preventing chronic headache syndromes.3,4 Amitriptyline has been the most studied of the TCAs for both chronic daily and episodic migraine headache, showing the most efficacy among diverse drug classes (angiotensin II receptor blockers, anticonvulsants, beta-blockers, SSRIs) compared with placebo. However, in head-to-head trials, amitriptyline was no more effective than SSRIs, venlafaxine, topiramate, or propranolol.4 Jackson et al4 suggested that prophylactic medication choices should be tailored to patient characteristics and expected adverse effects, and specifically recommended that TCAs—particularly amitriptyline—be reserved for patients who have both migraine and depression.
Neuropathic pain
Neuropathic pain is defined as pain secondary to a lesion or disease of the somatosensory nervous system5 and is the pathomechanistic component of a number of conditions, including postherpetic neuralgia,6 diabetic and nondiabetic painful polyneuropathy,7 posttraumatic or postsurgical neuropathic pain8 (including plexus avulsion and complex regional pain syndrome9), central poststroke pain,10 spinal cord injury pain,11 and multiple sclerosis-associated pain.12
As a group, TCAs appear to have a role as first-line agents for managing these varied neuropathic pain syndromes. In a recent meta-analysis,13 16 (89%) of 18 placebo-controlled trials of TCAs (mainly amitriptyline at 25–150 mg/day) for these pain conditions were positive, with a combined number needed to treat of 3.6, suggesting a role for TCAs in these conditions. Of note, the TCAs desipramine14 and nortriptyline15 have demonstrated little evidence of efficacy in neuropathic pain syndromes.
Chronic low back pain
Chronic low back pain is a leading cause of loss of work, excessive healthcare expenditure, and disability in the United States. It can be due to numerous spinal conditions, including degenerative disk disease, spinal stenosis, lumbar spondylosis, and spinal arthropathy.
TCAs have been used to treat chronic low back pain for decades and have been repeatedly shown to be more effective than placebo in reducing pain severity.16,17 A double-blind controlled trial18 from 1999 compared the effects of the TCA maprotiline (up to 150 mg daily), the SSRI paroxetine (up to 30 mg daily), and placebo and found a statistically significant reduction in back pain with maprotiline compared with paroxetine and placebo. However, a 2008 meta-analysis suggested little evidence that TCAs were superior to placebo.19
Evidence of TCA efficacy for back pain was reported in 2018 with a well-designed 6-month double-blind randomized controlled trial20 comparing low-dose amitriptyline (25 mg) with an active comparator (benztropine 1 mg). The authors reported that amitriptyline was effective in reducing pain and pain-related disability without incurring serious adverse effects. They suggested continued use of TCAs for chronic low back pain if complicated with pain-related disability, insomnia, depression, or other comorbidity, although they called for further large-scale studies. They also cautioned that patients started the trial with symptoms similar to the adverse effects of TCAs themselves; this has implications for monitoring of symptoms as well as TCA adverse effects while using these drugs.
Fibromyalgia and chronic widespread pain
Fibromyalgia is a common, frustrating, noninflammatory pain syndrome characterized by diffuse hyperalgesia and multiple comorbidities.21 Although sleep hygiene, exercise, cognitive-behavioral therapy, some gabapentinoids (pregabalin), and a combination of these therapies have demonstrated efficacy, TCAs also offer robust benefits.
A meta-analysis of 9 placebo-controlled TCA trials showed large effect sizes for pain reduction, fatigue reduction, improved sleep quality, and reduced stiffness and tenderness, with the most significant of these improvements being for sleep.22 A separate meta-analysis calculated that the number needed to treat with amitriptyline for a positive outcome is 4.9.23 Recent systematic reviews have supported these findings, listing TCAs as second-line agents after pregabalin, duloxetine, and milnacipran.24
Of note, TCA monotherapy rarely produces a complete response in patients with moderate to severe fibromyalgia, chronic widespread pain, or significant comorbidities (depression, anxiety). Supplementation with cognitive-behavioral therapy, physical therapy, functional restoration, and other modalities is strongly recommended.
Abdominal and gastrointestinal pain
TCAs have been applied to a number of gastrointestinal syndromes with or without pain. Patients with irritable bowel syndrome have long been known to benefit from TCAs; the number needed to treat for symptomatic benefit over placebo is 3.5.25,26
Although there is no substantial evidence that TCAs are useful in reducing active inflammation in inflammatory bowel disease, a study involving 81 patients found that residual noninflammatory gastrointestinal symptoms (such as diarrhea and pain) responded to TCAs, including nortriptyline and amitriptyline, with greater benefit for ulcerative colitis than for Crohn disease.27
TCAs have also shown prophylactic benefit in cyclic vomiting syndrome, with a clinical response in over 75% of patients in controlled cohort studies.28
The efficacy of TCAs in other abdominal or gastrointestinal syndromes is unclear or modest at best.29 However, few alternative treatments exist for these conditions. Amitriptyline may help symptoms of functional dyspepsia,30 but nortriptyline has proven ineffective in gastroparesis.31 Nonetheless, some authors29 suggest considering TCAs on an individualized basis, with proper monitoring, in many if not most functional gastrointestinal disorders, especially when paired with behavioral therapies.
Pelvic and urogynecologic symptoms
Chronic pelvic pain affects up to 24% of women32 and 5% to 10% of men.33 TCAs have shown efficacy in treating chronic pelvic pain with or without comorbid depression.34 Amitriptyline and to a lesser extent nortriptyline are the TCAs most often prescribed. Pain relief appears to be independent of antidepressant effects and may be achieved at low doses; initial dosing ranges from 10 to 25 mg at bedtime, which may be increased to 100 mg as tolerated.34
Based on a randomized, double-blind trial,35 amitriptyline was recommended as a treatment option for interstitial cystitis or bladder pain, with the greatest symptom improvement in patients tolerating a daily dose of 50 mg.
Another study36 randomized 56 women with chronic pelvic pain to amitriptyline or gabapentin, or a combination of the drugs for 24 months. Although each regimen resulted in significant reduction in pain, fewer adverse effects occurred with gabapentin than amitriptyline. Poor compliance and early discontinuation of amitriptyline were common due to anticholinergic effects.
In small uncontrolled studies,37 about half of women with chronic pelvic pain became pain-free after 8 weeks of treatment with nortriptyline and imipramine.
Randomized controlled studies are needed to confirm potential benefits of TCAs in chronic urologic and pelvic pain.
Insomnia
Insomnia affects 23% to 56% of people in the United States, Europe, and Asia38 and is the reason for more than 5.5 million primary care visits annually.39 TCAs (especially doxepin, maprotiline, and amitriptyline40) have been shown to be an effective treatment, with an 82% increase in somnolence compared with placebo, as well as measurably improved total sleep time, enhanced sleep efficiency, reduced latency to persistent sleep, and decreased wake times after sleep onset.38
Dosing should be kept at a minimum to minimize harsh anticholinergic effects and avoid daytime sedation. Patients should be advised to take new doses or dose escalations earlier in the night to ensure less hangover sedation the next morning.
For patients with insomnia and comorbid depression, the American Academy of Sleep Medicine suggests the addition of a low dose (eg, 10–25 mg) of a TCA at nighttime to complement preexisting, full-dose, non-TCA antidepressants, while monitoring for serotonin syndrome and other potential but exceedingly rare drug-drug interactions.41
Psychiatric indications other than depression
Beyond the known benefits in major depressive disorder, TCAs have been shown to be effective for obsessive-compulsive disorder, panic disorder, posttraumatic stress disorder, bulimia nervosa, and childhood enuresis.42 Given the shortage of mental health clinicians and the high prevalence of these conditions, nonpsychiatrist physicians should be familiar with the therapeutic potential of TCAs for these indications.
ADVERSE EFFECTS
Adverse effects vary among TCAs. Common ones include blurred vision, dry mouth, constipation, urinary retention, hypotension, tachycardia, tremor, weight gain, and sexual dysfunction.43 Tertiary amines are generally more sedating than secondary amines and cause more anticholinergic effects (Table 1).
Despite widespread perceptions that TCAs are less tolerable than newer antidepressants, studies repeatedly suggest that they have an adverse-effect burden similar to that of SSRIs and SNRIs, although SSRIs have a greater tendency to produce nausea, whereas TCAs are more likely to cause constipation.44
Discontinuation syndrome
Abrupt discontinuation or unintentionally missed doses of TCAs have been associated with a discontinuation syndrome in about 40% of users.45 Patients should be warned about this possibility and the syndrome’s potential effects: dizziness, insomnia, headaches, nausea, vomiting, flulike achiness, and restlessness. Rebound depression, anxiety, panic, or other psychiatric symptoms may also occur. Symptoms generally present within 2 to 5 days after dose discontinuation and last 7 to 14 days.45
However, all TCAs have a long half-life, allowing for sufficient coverage with once-daily dosing and thus carry a lower risk of discontinuation syndrome than many other antidepressants (78% with venlafaxine; 55% with paroxetine).45
To discontinue therapy safely, the dosage should be reduced gradually. As is pharmacologically expected, the greatest likelihood of discontinuation syndrome is associated with longer duration of continuous treatment.
CONTRAINDICATIONS
Cardiac conduction abnormalities
TCAs should not be prescribed to patients who have right bundle branch block, a severe electrolyte disturbance, or other cardiac conduction deficit or arrhythmia that can prolong the QTc interval and elevate the risk of lethal arrhythmia.46,47 Cardiac effects from TCAs are largely dose-dependent. Nevertheless, a baseline electrocardiogram can be obtained to assess cardiac risk, and dose escalation can proceed if results are normal (eg, appropriate conduction intervals, QTc ≤ 450 ms).
Advanced age
For elderly patients, TCAs should be prescribed with caution and sometimes not at all,48 because anticholinergic effects may worsen preexisting urinary retention (including benign prostatic hyperplasia), narrow-angle glaucoma, imbalance and gait issues, and cognitive impairment and dementia. Dehydration and orthostatic hypotension are contraindications for TCAs, as they may precipitate falls or hypotensive shock.
Epilepsy
TCAs should also be used with caution in patients with epilepsy, as they lower the seizure threshold.
Concomitant monoamine oxidase inhibitor treatment
Giving TCAs together with monoamine oxidase inhibitor antidepressants should be avoided, given the risk of hypertensive crisis.
Suicide risk
TCAs are dangerous and potentially lethal in overdose and so should not be prescribed to suicidal or otherwise impulsive patients.
Pregnancy
TCAs are in pregnancy risk category C (animal studies show adverse effects on fetus; no adequate or well-controlled studies in humans; potential benefits may warrant use despite risks). Using TCAs during pregnancy has very rarely led to neonatal withdrawal such as irritability, jitteriness, and convulsions, as well as fetal QTc interval prolongation.49
The American College of Obstetricians and Gynecologists recommends that therapy for depression during pregnancy be individualized, incorporating the expertise of the patient’s mental health clinician, obstetrician, primary healthcare provider, and pediatrician. In general, they recommend that TCAs should be avoided if possible and that alternatives such as SSRIs or SNRIs should be considered.50
TCAs are excreted in breast milk, but they have not been detected in the serum of nursing infants, and no adverse events have been reported.
OVERDOSE IS HIGHLY DANGEROUS
Severe morbidity and death are associated with TCA overdose, characterized by convulsions, cardiac arrest, and coma (the “3 Cs”). These dangers occur at much higher rates with TCAs than with other antidepressants.43 Signs and symptoms of toxicity develop rapidly, usually within the first hour of overdose. Manifestations of overdose include prolonged QTc, cardiac arrhythmias, tachycardia, hypertension, severe hypotension, agitation, seizures, central nervous system depression, hallucinations, seizures, and coma.
Overdose management includes activated charcoal, seizure control, cardioversion, hydration, electrolyte stabilization, and other intensive care.
OFF-LABEL TCA MANAGEMENT
Dosing recommendations for off-label use of TCAs vary based on the condition, the medication, and the suggestions of individual authors and researchers. In general, dosing ranges for pain and other nondepression indications may be lower than for severe depression (Table 2).1
As with any pharmacologic titration, monitoring for rate-limiting adverse effects is recommended. We suggest caution, tailoring the approach to the patient, and routinely assessing for adverse effects and other safety considerations.
In addition, we strongly recommend supplementing TCA therapy with nonpharmacologic strategies such as lifestyle changes, dietary modifications, exercise, physical therapy, and mental health optimization.
- Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci 2017; 18(11). doi:10.3390/ijms18112483
- Kremer M, Yalcin I, Goumon Y, et al. A dual noradrenergic mechanism for the relief of neuropathic allodynia by the antidepressant drugs duloxetine and amitriptyline. J Neurosci 2018; 38(46):9934–9954. doi:10.1523/JNEUROSCI.1004-18.2018
- Tomkins GE, Jackson JL, O’Malley PG, Balden E, Santoro JE. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med 2001; 111(1):54–63. doi:10.1016/s0002-9343(01)00762-8
- Jackson JL, Cogbill E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PLoS One 2015; 10(7):e0130733. doi:10.1371/journal.pone.0130733
- International Association for the Study of Pain (IASP). IASP Terminology. www.iasp-pain.org/Education/Content.aspx?ItemNumber=1698&navItemNumber=576. Accessed November 20, 2019.
- Feller L, Khammissa RAG, Fourie J, Bouckaert M, Lemmer J. Postherpectic neuralgia and trigeminal neuralgia. Pain Res Treat 2017; 2017:1681765. doi:10.1155/2017/1681765
- Shillo P, Sloan G, Greig M, et al. Painful and painless diabetic neuropathies: what is the difference? Curr Diab Rep 2019; 19(6):32. doi:10.1007/s11892-019-1150-5
- Schwartzman RJ, Maleki J. Postinjury neuropathic pain syndromes. Med Clin North Am 1999; 83(3):597–626. doi:10.1016/s0025-7125(05)70126-7
- Oaklander AL, Horowitz SH. The complex regional pain syndrome. Handb Clin Neurol 2015; 131:481–503. doi:10.1016/B978-0-444-62627-1.00026-3
- Akyuz G, Kuru P. Systematic review of central post stroke pain: what is happening in the central nervous system? Am J Phys Med Rehabil 2016; 95(8):618–627. doi:10.1097/PHM.0000000000000542
- Shiao R, Lee-Kubli CA. Neuropathic pain after spinal cord injury: challenges and research perspectives. Neurotherapeutics 2018; 15(3):635–653. doi:10.1007/s13311-018-0633-4
- Ceruti S. What role does multiple sclerosis play in the development of untreatable painful conditions? Pain Manag 2018; 8(1):37–44. doi:10.2217/pmt-2017-0038
- Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015; 14(2):162–173. doi:10.1016/S1474-4422(14)70251-0
- Hearn L, Moore RA, Derry S, Wiffen PJ, Phillips T. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev 2014; (9):CD011003. doi:10.1002/14651858.CD011003.pub2
- Derry S, Whiffen PJ, Aldington D, Moore RA. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev 2015; 1:CD011209. doi:10.1002/14651858.CD011209.pub2
- Salerno SM, Browning R, Jackson JL. The effect of antidepressant treatment on chronic back pain: a meta-analysis. Arch Intern Med 2002; 162(1):19–24. doi:10.1001/archinte.162.1.19
- Staiger TO, Gaster B, Sullivan MD, Deyo RA. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976) 2003; 28(22):2540–2545. doi:10.1097/01.BRS.0000092372.73527.BA
- Atkinson JH, Slater MA, Wahlgren DR, et al. Effects of noradrenergic and serotonergic antidepressants on chronic low back pain intensity. Pain 1999; 83(2):137–145. doi:10.1016/s0304-3959(99)00082-2
- Urquhart DM, Hoving JL, Assendelft WW, Roland M, van Tulder MW. Antidepressants for non-specific back pain. Cochrane Database Syst Rev 2008; (1):CD001703. doi:10.1002/14651858.CD001703.pub3
- Urquhart DM, Wluka AE, van Tulder M, et al. Efficacy of low-dose amitriptyline for chronic low back pain: a randomized clinical trial. JAMA Intern Med 2018; 178(11):1474–1481. doi:10.1001/jamainternmed.2018.4222
- Clauw DJ. Fibromyalgia: a clinical review. JAMA 2014; 311(15):1547–1555. doi:10.1001/jama.2014.3266
- Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41(2):104–113. pmid:10749947
- Hauser W, Wolfe F, Tolle T, Uceyler N, Sommer C. The role of antidepressants in the management of fibromyalgia syndrome: a systematic review and meta-analysis. CNS Drugs 2012; 26(4):297–307. doi:10.2165/11598970-000000000-00000
- Calandre EP, Rico-Villademoros F, Slim M. An update on pharmacotherapy for the treatment of fibromyalgia. Expert Opin Pharmacother 2015; 16(9):1347–1368. doi:10.1517/14656566.2015.1047343
- Jackson JL, O’Malley PG, Tomkins G, Balden E, Santoro J, Kroenke K. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med 2000; 108(1):65–72. doi:10.1016/s0002-9343(99)00299-5
- Rahimi R, Nikfar S, Rezaie A, Abdollahi M. Efficacy of tricyclic antidepressants in irritable bowel syndrome: a meta-analysis. World J Gastroenterol 2009; 15(13):1548–1553. doi:10.3748/wjg.15.1548
- Iskandar HN, Cassell B, Kanuri N, et al. Tricyclic antidepressants for management of residual symptoms in inflammatory bowel disease. J Clin Gastroenterol 2014; 48(5):423–429. doi:10.1097/MCG.0000000000000049
- Lee LY, Abbott L, Mahlangu B, Moodie SJ, Anderson S. The management of cyclic vomiting syndrome: a systematic review. Eur J Gastroenterol Hepatol 2012; 24(9):1001–1006. doi:10.1097/MEG.0b013e328355638f
- Thorkelson G, Bielefeldt K, Szigethy E. Empirically supported use of psychiatric medications in adolescents and adults with IBD. Inflamm Bowel Dis 2016; 22(6):1509–1522. doi:10.1097/MIB.0000000000000734
- Braak B, Klooker TK, Wouters MM, et al. Randomised clinical trial: the effects of amitriptyline on drinking capacity and symptoms in patients with functional dyspepsia, a double-blind placebo-controlled study. Aliment Pharmacol Ther 2011; 34(6):638–648. doi:10.1111/j.1365-2036.2011.04775.x
- Parkman HP, Van Natta ML, Abell TL, et al. Effect of nortriptyline on symptoms of idiopathic gastroparesis: the NORIG randomized clinical trial. JAMA 2013; 310(24):2640–2649. doi:10.1001/jama.2013.282833
- Latthe P, Latthe M, Say L, Gulmezoglu M, Khan KS. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health 2006; 6:177. doi:10.1186/1471-2458-6-177
- Moise G, Capodice J, Winfree CJ. Treatment of chronic pelvic pain in men and women. Expert Rev Neurother 2007; 7(5):507–520. doi:10.1586/14737175.7.5.507
- Lai HH. Management of interstitial cystitis/bladder pain syndrome with tricyclic antidepressants. In: Moldwin RM, ed. Urological and Gynaecological Chronic Pelvic Pain. Cham, Switzerland: Springer; 2017:107–118.
- American Urological Association. Diagnosis and treatment interstitial cystitis/bladder pain syndrome (2014). www.auanet.org/guidelines/interstitial-cystitis/bladder-pain-syndrome-(2011-amended-2014). Accessed November 19, 2019.
- Carey ET, As-Sanie S. New developments in the pharmacotherapy of neuropathic chronic pelvic pain. Future Sci OA 2016; 2(4):FSO148. doi:10.4155/fsoa-2016-0048
- Papandreou C, Skapinakis P, Giannakis D, Sofikitis N, Mavreas V. Antidepressant drugs for chronic urological pelvic pain: an evidence-based review. Adv Urol 2009; 2009:797031. doi:10.1155/2009/797031
- Liu Y, Xu X, Dong M, Jia S, Wei Y. Treatment of insomnia with tricyclic antidepressants: a meta-analysis of polysomnographic randomized controlled trials. Sleep Med 2017; 34:126–133. doi:10.1016/j.sleep.2017.03.007
- Matheson E, Hainer BL. Insomnia: pharmacologic therapy. Am Fam Physician 2017; 96(1):29–35. pmid:28671376
- McCall C, McCall WV. What is the role of sedating antidepressants, antipsychotics, and anticonvulsants in the management of insomnia? Curr Psychiatry Rep 2012; 14(5):494–502. doi:10.1007/s11920-012-0302-y
- Clark MS, Smith PO, Jamieson B. FPIN’s clinical inquiries: antidepressants for the treatment of insomnia in patients with depression. Am Fam Physician 2011; 84(9):1–2. pmid:22164891
- Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s Synopsis of Psychiatry. New York, NY: Lippincott Williams & Wilkins; 2014.
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J 2018; 54(2):101–112. doi:10.4068/cmj.2018.54.2.101.
- Trindade E, Menon D, Topfer LA, Coloma C. Adverse effects associated with selective serotonin reuptake inhibitors and tricyclic antidepressants: a meta-analysis. CMAJ 1998; 159(10):1245–1252. pmid:9861221
- Fava M. Prospective studies of adverse events related to antidepressant discontinuation. J Clin Psychiatry 2006; 67(suppl 4):14–21. pmid:16683858
- Gintant G. An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 2011; 129(2):109–119. doi:10.1016/j.pharmthera.2010.08.008.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54(1):1–13. doi:10.1016/j.psym.2012.11.001
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63(11):2227–2246. doi:10.1111/jgs.13702
- Fukushima N, Nanao K, Fukushima H, Namera A, Miura M. A neonatal prolonged QT syndrome due to maternal use of oral tricyclic antidepressants. Eur J Pediatr 2016; 175(8):1129–1132. doi:10.1007/s00431-016-2722-x
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111(4):1001–1020. doi:10.1097/AOG.0b013e31816fd910
- Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci 2017; 18(11). doi:10.3390/ijms18112483
- Kremer M, Yalcin I, Goumon Y, et al. A dual noradrenergic mechanism for the relief of neuropathic allodynia by the antidepressant drugs duloxetine and amitriptyline. J Neurosci 2018; 38(46):9934–9954. doi:10.1523/JNEUROSCI.1004-18.2018
- Tomkins GE, Jackson JL, O’Malley PG, Balden E, Santoro JE. Treatment of chronic headache with antidepressants: a meta-analysis. Am J Med 2001; 111(1):54–63. doi:10.1016/s0002-9343(01)00762-8
- Jackson JL, Cogbill E, Santana-Davila R, et al. A comparative effectiveness meta-analysis of drugs for the prophylaxis of migraine headache. PLoS One 2015; 10(7):e0130733. doi:10.1371/journal.pone.0130733
- International Association for the Study of Pain (IASP). IASP Terminology. www.iasp-pain.org/Education/Content.aspx?ItemNumber=1698&navItemNumber=576. Accessed November 20, 2019.
- Feller L, Khammissa RAG, Fourie J, Bouckaert M, Lemmer J. Postherpectic neuralgia and trigeminal neuralgia. Pain Res Treat 2017; 2017:1681765. doi:10.1155/2017/1681765
- Shillo P, Sloan G, Greig M, et al. Painful and painless diabetic neuropathies: what is the difference? Curr Diab Rep 2019; 19(6):32. doi:10.1007/s11892-019-1150-5
- Schwartzman RJ, Maleki J. Postinjury neuropathic pain syndromes. Med Clin North Am 1999; 83(3):597–626. doi:10.1016/s0025-7125(05)70126-7
- Oaklander AL, Horowitz SH. The complex regional pain syndrome. Handb Clin Neurol 2015; 131:481–503. doi:10.1016/B978-0-444-62627-1.00026-3
- Akyuz G, Kuru P. Systematic review of central post stroke pain: what is happening in the central nervous system? Am J Phys Med Rehabil 2016; 95(8):618–627. doi:10.1097/PHM.0000000000000542
- Shiao R, Lee-Kubli CA. Neuropathic pain after spinal cord injury: challenges and research perspectives. Neurotherapeutics 2018; 15(3):635–653. doi:10.1007/s13311-018-0633-4
- Ceruti S. What role does multiple sclerosis play in the development of untreatable painful conditions? Pain Manag 2018; 8(1):37–44. doi:10.2217/pmt-2017-0038
- Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015; 14(2):162–173. doi:10.1016/S1474-4422(14)70251-0
- Hearn L, Moore RA, Derry S, Wiffen PJ, Phillips T. Desipramine for neuropathic pain in adults. Cochrane Database Syst Rev 2014; (9):CD011003. doi:10.1002/14651858.CD011003.pub2
- Derry S, Whiffen PJ, Aldington D, Moore RA. Nortriptyline for neuropathic pain in adults. Cochrane Database Syst Rev 2015; 1:CD011209. doi:10.1002/14651858.CD011209.pub2
- Salerno SM, Browning R, Jackson JL. The effect of antidepressant treatment on chronic back pain: a meta-analysis. Arch Intern Med 2002; 162(1):19–24. doi:10.1001/archinte.162.1.19
- Staiger TO, Gaster B, Sullivan MD, Deyo RA. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976) 2003; 28(22):2540–2545. doi:10.1097/01.BRS.0000092372.73527.BA
- Atkinson JH, Slater MA, Wahlgren DR, et al. Effects of noradrenergic and serotonergic antidepressants on chronic low back pain intensity. Pain 1999; 83(2):137–145. doi:10.1016/s0304-3959(99)00082-2
- Urquhart DM, Hoving JL, Assendelft WW, Roland M, van Tulder MW. Antidepressants for non-specific back pain. Cochrane Database Syst Rev 2008; (1):CD001703. doi:10.1002/14651858.CD001703.pub3
- Urquhart DM, Wluka AE, van Tulder M, et al. Efficacy of low-dose amitriptyline for chronic low back pain: a randomized clinical trial. JAMA Intern Med 2018; 178(11):1474–1481. doi:10.1001/jamainternmed.2018.4222
- Clauw DJ. Fibromyalgia: a clinical review. JAMA 2014; 311(15):1547–1555. doi:10.1001/jama.2014.3266
- Arnold LM, Keck PE Jr, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41(2):104–113. pmid:10749947
- Hauser W, Wolfe F, Tolle T, Uceyler N, Sommer C. The role of antidepressants in the management of fibromyalgia syndrome: a systematic review and meta-analysis. CNS Drugs 2012; 26(4):297–307. doi:10.2165/11598970-000000000-00000
- Calandre EP, Rico-Villademoros F, Slim M. An update on pharmacotherapy for the treatment of fibromyalgia. Expert Opin Pharmacother 2015; 16(9):1347–1368. doi:10.1517/14656566.2015.1047343
- Jackson JL, O’Malley PG, Tomkins G, Balden E, Santoro J, Kroenke K. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta-analysis. Am J Med 2000; 108(1):65–72. doi:10.1016/s0002-9343(99)00299-5
- Rahimi R, Nikfar S, Rezaie A, Abdollahi M. Efficacy of tricyclic antidepressants in irritable bowel syndrome: a meta-analysis. World J Gastroenterol 2009; 15(13):1548–1553. doi:10.3748/wjg.15.1548
- Iskandar HN, Cassell B, Kanuri N, et al. Tricyclic antidepressants for management of residual symptoms in inflammatory bowel disease. J Clin Gastroenterol 2014; 48(5):423–429. doi:10.1097/MCG.0000000000000049
- Lee LY, Abbott L, Mahlangu B, Moodie SJ, Anderson S. The management of cyclic vomiting syndrome: a systematic review. Eur J Gastroenterol Hepatol 2012; 24(9):1001–1006. doi:10.1097/MEG.0b013e328355638f
- Thorkelson G, Bielefeldt K, Szigethy E. Empirically supported use of psychiatric medications in adolescents and adults with IBD. Inflamm Bowel Dis 2016; 22(6):1509–1522. doi:10.1097/MIB.0000000000000734
- Braak B, Klooker TK, Wouters MM, et al. Randomised clinical trial: the effects of amitriptyline on drinking capacity and symptoms in patients with functional dyspepsia, a double-blind placebo-controlled study. Aliment Pharmacol Ther 2011; 34(6):638–648. doi:10.1111/j.1365-2036.2011.04775.x
- Parkman HP, Van Natta ML, Abell TL, et al. Effect of nortriptyline on symptoms of idiopathic gastroparesis: the NORIG randomized clinical trial. JAMA 2013; 310(24):2640–2649. doi:10.1001/jama.2013.282833
- Latthe P, Latthe M, Say L, Gulmezoglu M, Khan KS. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health 2006; 6:177. doi:10.1186/1471-2458-6-177
- Moise G, Capodice J, Winfree CJ. Treatment of chronic pelvic pain in men and women. Expert Rev Neurother 2007; 7(5):507–520. doi:10.1586/14737175.7.5.507
- Lai HH. Management of interstitial cystitis/bladder pain syndrome with tricyclic antidepressants. In: Moldwin RM, ed. Urological and Gynaecological Chronic Pelvic Pain. Cham, Switzerland: Springer; 2017:107–118.
- American Urological Association. Diagnosis and treatment interstitial cystitis/bladder pain syndrome (2014). www.auanet.org/guidelines/interstitial-cystitis/bladder-pain-syndrome-(2011-amended-2014). Accessed November 19, 2019.
- Carey ET, As-Sanie S. New developments in the pharmacotherapy of neuropathic chronic pelvic pain. Future Sci OA 2016; 2(4):FSO148. doi:10.4155/fsoa-2016-0048
- Papandreou C, Skapinakis P, Giannakis D, Sofikitis N, Mavreas V. Antidepressant drugs for chronic urological pelvic pain: an evidence-based review. Adv Urol 2009; 2009:797031. doi:10.1155/2009/797031
- Liu Y, Xu X, Dong M, Jia S, Wei Y. Treatment of insomnia with tricyclic antidepressants: a meta-analysis of polysomnographic randomized controlled trials. Sleep Med 2017; 34:126–133. doi:10.1016/j.sleep.2017.03.007
- Matheson E, Hainer BL. Insomnia: pharmacologic therapy. Am Fam Physician 2017; 96(1):29–35. pmid:28671376
- McCall C, McCall WV. What is the role of sedating antidepressants, antipsychotics, and anticonvulsants in the management of insomnia? Curr Psychiatry Rep 2012; 14(5):494–502. doi:10.1007/s11920-012-0302-y
- Clark MS, Smith PO, Jamieson B. FPIN’s clinical inquiries: antidepressants for the treatment of insomnia in patients with depression. Am Fam Physician 2011; 84(9):1–2. pmid:22164891
- Sadock BJ, Sadock VA, Ruiz P. Kaplan and Sadock’s Synopsis of Psychiatry. New York, NY: Lippincott Williams & Wilkins; 2014.
- Wang SM, Han C, Bahk WM, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J 2018; 54(2):101–112. doi:10.4068/cmj.2018.54.2.101.
- Trindade E, Menon D, Topfer LA, Coloma C. Adverse effects associated with selective serotonin reuptake inhibitors and tricyclic antidepressants: a meta-analysis. CMAJ 1998; 159(10):1245–1252. pmid:9861221
- Fava M. Prospective studies of adverse events related to antidepressant discontinuation. J Clin Psychiatry 2006; 67(suppl 4):14–21. pmid:16683858
- Gintant G. An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 2011; 129(2):109–119. doi:10.1016/j.pharmthera.2010.08.008.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54(1):1–13. doi:10.1016/j.psym.2012.11.001
- American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2015; 63(11):2227–2246. doi:10.1111/jgs.13702
- Fukushima N, Nanao K, Fukushima H, Namera A, Miura M. A neonatal prolonged QT syndrome due to maternal use of oral tricyclic antidepressants. Eur J Pediatr 2016; 175(8):1129–1132. doi:10.1007/s00431-016-2722-x
- ACOG Committee on Practice Bulletins—Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol 2008; 111(4):1001–1020. doi:10.1097/AOG.0b013e31816fd910
KEY POINTS
- Amitriptyline is the most useful TCA for many painful conditions.
- TCAs can be especially helpful for patients with a pain syndrome or insomnia with comorbid depression, although their benefits appear to be independent of antidepressant effects.
- TCAs have long half-lives and so can be taken once a day.
- Effective dosages for symptom control in many conditions are lower than for severe depression; dosage should start low and be gradually increased while monitoring efficacy and adverse effects.
- TCAs should not be used concurrently with a monoamine oxidase inhibitor and by certain patient groups: the elderly, pregnant women, and patients with certain cardiac conduction abnormalities, epilepsy, or risk of suicide.
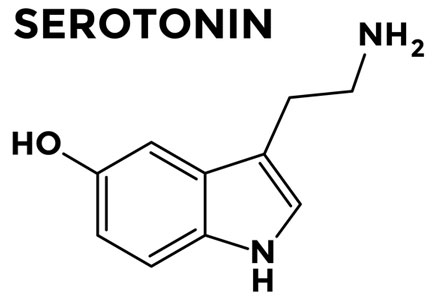
Gabapentin for alcohol use disorder: A good option, or cause for concern?
Perceptions regarding the use of gabapentin for alcohol use disorder (AUD) have shifted over time.1–4 Early on, the drug was deemed to be benign and effective.4–6 But more and more, concerns are being raised about its recreational use to achieve euphoria,7 and the drug is often misused by vulnerable populations, particularly those with opioid use disorder.7–9
Given the large number of gabapentin prescriptions written off-label for AUD, it is incumbent on providers to understand how to prescribe it responsibly.7–9 To that end, this article focuses on the benefits—and concerns—of this treatment option. We describe the effects of gabapentin on the central nervous system and how it may mitigate alcohol withdrawal and increase the likelihood of abstinence. In addition, we review clinical trials that evaluated potential roles of gabapentin in AUD, discuss the drug’s misuse potential, and suggest a framework for its appropriate use in AUD management.
ALCOHOL USE DISORDER IS COMMON AND SERIOUS
AUD affects about 14% of US adults and represents a significant health burden,1 often with severe clinical and social implications. It manifests as compulsive drinking and loss of control despite adverse consequences on various life domains.10 It is generally associated with cravings, tolerance, and withdrawal symptoms upon cessation. Alcohol withdrawal is characterized by tremors, anxiety, sweating, nausea, and tachycardia, and in severe cases, may involve hallucinations, seizures, and delirium tremens. Untreated, alcohol withdrawal can be fatal.10

Even though psychosocial treatments for AUD by themselves are associated with high relapse rates, pharmacotherapy is underutilized. Three drugs approved by the US Food and Drug Administration (FDA) are available to treat it, but they are often poorly accepted and have limited efficacy. For these reasons, there is considerable interest in finding alternatives. Gabapentin is one of several agents that have been studied (Table 1). The topic has been reviewed in depth by Soyka and Müller.11
GABAPENTIN REDUCES EXCITATION
The anticonvulsant gabapentin is FDA-approved for treating epilepsy, postherpetic neuralgia, and restless leg syndrome.8,12–14 It binds and selectively impedes voltage-sensitive calcium channels, the pores in cell membrane that permit calcium to enter a neuron in response to changes in electrical currents.15
Gabapentin is believed to decrease excitation of the central nervous system in multiple ways:
- It reduces the release of glutamate, a key component of the excitatory system16
- It increases the concentration of gamma-aminobutyric acid (GABA), the main inhibitory neurotransmitter in the brain7
- By binding the alpha-2-delta type 1 subunit of voltage-sensitive calcium channels,8,15–17 it inhibits excitatory synapse formation independent of calcium channel activity16
- By blocking excitatory neurotransmission, it also may indirectly increase the concentration of GABA in the central nervous system16,17
- It modulates action of glutamic acid decarboxylase (involved in the synthesis of GABA) and glutamate synthesizing enzyme to increase GABA and decrease glutamate.17
ALCOHOL’S ACTIONS
The actions of alcohol on the brain are also complex.18 Alpha-2-delta type 1 subunits of calcium channels are upregulated in the reward centers of the brain by addictive substances, including alcohol.16 Alcohol interacts with corticotropin-releasing factor and several neurotransmitters,18 and specifically affects neuropathways involving norepinephrine, GABA, and glutamate.19 Alcohol has reinforcing effects mediated by the release of dopamine in the nucleus accumbens.20
Acutely, alcohol promotes GABA release and may also reduce GABA degradation, producing sedative and anxiolytic effects.21 Chronic alcohol use leads to a decrease in the number of GABAA receptors. Clinically, this downregulation manifests as tolerance to alcohol’s sedating effects.21
Alcohol affects the signaling of glutamatergic interaction with the N-methyl-d-aspartate (NMDA) receptor.22 Glutamate activates this receptor as well as the voltage-gated ion channels, modifying calcium influx and increasing neuronal excitability.22,23 Acutely, alcohol has an antagonistic effect on the NMDA receptor, while chronic drinking upregulates (increases) the number of NMDA receptors and voltage-gated calcium channels.22,23
Alcohol withdrawal increases excitatory effects
Patients experiencing alcohol withdrawal have decreased GABA-ergic functioning and increased glutamatergic action throughout the central nervous system.19,24
Withdrawal can be subdivided into an acute phase (lasting up to about 5 days) and a protracted phase (of undetermined duration). During withdrawal, the brain activates its “stress system,” leading to overexpression of corticotropin-releasing factor in the amygdala. Protracted withdrawal dysregulates the prefrontal cortex, increasing cravings and worsening negative emotional states and sleep.16
GABAPENTIN FOR ALCOHOL WITHDRAWAL
Benzodiazepines are the standard treatment for alcohol withdrawal.3,24 They relieve symptoms and can prevent seizures and delirium tremens,24 but they are sedating and cause psychomotor impairments.3 Because of the potential for addiction, benzodiazepine use is limited to acute alcohol withdrawal.3
Gabapentin shows promise as an agent that can be used in withdrawal and continued through early abstinence without the highly addictive potential of benzodiazepines.16 It is thought to affect drinking behaviors during early abstinence by normalizing GABA and glutamate activity.2,16
Early preclinical studies in mouse models found that gabapentin decreases anxiogenic and epileptic effects of alcohol withdrawal. Compared with other antidrinking medications, gabapentin has the benefits of lacking elimination via hepatic metabolism, few pharmacokinetic interactions, and good reported tolerability in this population.
Inpatient trials show no benefit over standard treatments
Bonnet et al25 conducted a double-blind placebo-controlled trial in Germany in inpatients experiencing acute alcohol withdrawal to determine whether gabapentin might be an effective adjunct to clomethiazole, a GABAA modulator commonly used in Europe for alcohol withdrawal. Participants (N = 61) were randomized to receive placebo or gabapentin (400 mg every 6 hours) for 72 hours, with tapering over the next 3 days. All patients could receive rescue doses of clomethiazole, using a symptom-triggered protocol.
The study revealed no differences in the amount of clomethiazole administered between the 2 groups, suggesting that gabapentin had no adjunctive effect. Side effects (vertigo, nausea, dizziness, and ataxia) were mild and comparable between groups.
Nichols et al26 conducted a retrospective cohort study in a South Carolina academic psychiatric hospital to assess the adjunctive effect of gabapentin on the as-needed use of benzodiazepines for alcohol withdrawal. The active group (n = 40) received gabapentin as well as a symptom-triggered alcohol withdrawal protocol of benzodiazepine. The control group (n = 43) received only the symptom-triggered alcohol withdrawal protocol without gabapentin.
No effect was found of gabapentin use for benzodiazepine treatment of alcohol withdrawal. It is notable that Bonnet et al and Nichols et al had similar findings despite their studies being conducted in different countries using distinct comparators and methods.
Bonnet et al,27 in another study, tried a different design to investigate a possible role for gabapentin in inpatient alcohol withdrawal. The study included 37 patients with severe alcohol withdrawal (Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised [CIWA-Ar] > 15).
All participants received gabapentin 800 mg. Those whose CIWA-Ar score improved within 2 hours were considered “early responders” (n = 27) and next received 2 days of gabapentin 600 mg 4 times a day before starting a taper. The nonresponders whose CIWA-Ar score worsened (associated with greater anxiety and depressive symptoms; n = 10) were switched to standard treatment with clomethiazole (n = 4) or clonazepam (n = 6). Scores of 3 early responders subsequently worsened; 2 of these participants developed seizures and were switched to standard treatment.
The authors concluded that gabapentin in a dose of 3,200 mg in the first 24 hours is useful only for milder forms of alcohol withdrawal. Hence, subsequent efforts on the use of gabapentin for alcohol withdrawal have focused on outpatients.
Outpatient trials reveal benefits over benzodiazepines
Myrick et al3 compared gabapentin vs lorazepam in 100 outpatients seeking treatment for alcohol withdrawal. Participants were randomized to 1 of 4 groups: gabapentin 600 mg, 900 mg, or 1,200 mg, or lorazepam 6 mg, each tapering over 4 days. Alcohol withdrawal was measured by the CIWA-Ar score. Only 68 patients completed all follow-up appointments to day 12.
Gabapentin 600 mg was discontinued because of seizures in 2 patients, but it was generally well tolerated and was associated with diminished symptoms of alcohol withdrawal, especially at the 1,200 mg dose. The gabapentin groups experienced less anxiety and sedation and fewer cravings than the lorazepam group. Those treated with lorazepam fared worse for achieving early abstinence and were more likely to return to drinking when the intervention was discontinued. However, significant relapse by day 12 occurred in both groups.
The authors concluded that gabapentin was at least as effective as lorazepam in the outpatient treatment of alcohol withdrawal, with the 1,200-mg gabapentin dosage being more effective than 900 mg. At 1,200 mg, gabapentin was associated with better sleep, less anxiety, and better self-reported ability to work than lorazepam, and at the 900-mg dose it was associated with less depression than lorazepam.
Stock et al28 conducted a randomized, double-blind study of gabapentin in acute alcohol withdrawal in 26 military veterans in an outpatient setting. Patients were randomized to one of the following:
- Gabapentin 1,200 mg orally for 3 days, followed by 900 mg, 600 mg, and 300 mg for 1 day each (n = 17)
- Chlordiazepoxide 100 mg orally for 3 days, followed by 75 mg, 50 mg, and 25 mg for 1 day each (n = 9).
Withdrawal scores improved similarly in both groups. Early on (days 1–4), neither cravings nor sleep differed significantly between groups; but later (days 5–7), the gabapentin group had superior scores for these measures. Gabapentin was also associated with significantly less sedation than chlordiazepoxide and trended to less alcohol craving.
Bottom line: Gabapentin is useful for mild withdrawal
Data suggest that gabapentin offers benefits for managing mild alcohol withdrawal. Improved residual craving and sleep measures are clinically important because they are risk factors for relapse. Mood and anxiety also improve with gabapentin, further indicating a therapeutic effect.
Gabapentin’s benefits for moderate and severe alcohol withdrawal have not been established. Seizures occurred during withdrawal despite gabapentin treatment, but whether from an insufficient dose, patient susceptibility, or lack of gabapentin efficacy is not clear. Best results occurred at the 1,200-mg daily dose, but benefits may not apply to patients with severe withdrawal. In addition, many studies were small, limiting the strength of conclusions.
Across most studies of gabapentin for alcohol withdrawal, advantages included a smoother transition into early abstinence due to improved sleep, mood, and anxiety, alleviating common triggers for a return to drinking. Gabapentin also carries less reinforcing potential than benzodiazepines. These qualities fueled interest in trying gabapentin to improve long-term abstinence.
GABAPENTIN FOR RELAPSE PREVENTION
Although naltrexone and acamprosate are the first-line treatments for relapse prevention, they do not help all patients and are more effective when combined with cognitive behavioral therapy.1,29,30 For patients in whom standard treatments are not effective or tolerated, gabapentin may provide a reasonable alternative, and several randomized controlled trials have examined its use for this role.
Gabapentin alone is better than placebo
Furieri and Nakamura-Palacios4 assessed the use of gabapentin for relapse prevention in Brazilian outpatients (N = 60) who had averaged 27 years of drinking and consumed 17 drinks daily for the 90 days before baseline. After detoxification with diazepam and vitamins, patients were randomized to either gabapentin 300 mg twice daily or placebo for 4 weeks.
Compared with placebo, gabapentin significantly reduced cravings and lowered the percentage of heavy drinking days and the number of drinks per day, with a significant increase in the percentage of abstinent days. These self-reported measures correlated with decreases in gamma-glutamyl transferase, a biological marker for heavy drinking.
Brower et al31 investigated the use of gabapentin in 21 outpatients with AUD and insomnia who desired to remain abstinent. They were randomized to gabapentin (up to 1,500 mg at night) or placebo for 6 weeks. Just 14 participants completed the study; all but 2 were followed without treatment until week 12.
Gabapentin was associated with significantly lower relapse rates at 6 weeks (3 of 10 in the gabapentin group vs 9 of 11 in the placebo group) and at 12 weeks (6 of 10 in the gabapentin group vs 11 of 11 in the placebo group, assuming the 2 patients lost to follow-up relapsed). No difference between groups was detected for sleep measures in this small study. However, other studies have found that gabapentin for AUD improves measures of insomnia and daytime drowsiness—predictors of relapse—compared with other medications.16
High-dose gabapentin is better
Mason et al2 randomized 150 outpatients with alcohol dependence to 12 weeks of daily treatment with either gabapentin (900 mg or 1,800 mg) or placebo after at least 3 days of abstinence. All participants received counseling. Drinking quantity and frequency were assessed by gamma-glutamyl transferase testing.
Patients taking gabapentin had better rates of abstinence and cessation of heavy drinking than those taking placebo. During the 12-week study, the 1,800-mg daily dose showed a substantially higher abstinence rate (17%) than either 900 mg (11%) or placebo (4%). Significant dose-related improvements were also found for heavy drinking days, total drinking quantity, and frequency of alcohol withdrawal symptoms that predispose to early relapse, such as poor sleep, cravings, and poor mood. There were also significant linear dose effects on rates of abstinence and nondrinking days at the 24-week posttreatment follow-up.
Gabapentin plus naltrexone is better than naltrexone alone
Anton et al5 examined the efficacy of gabapentin combined with naltrexone during early abstinence. The study randomly assigned 150 people with AUD to one of the following groups:
- 16 weeks of naltrexone (50 mg/day) alone
- 6 weeks of naltrexone (50 mg/day) plus gabapentin (up to 1,200 mg/day), followed by 10 weeks of naltrexone alone
- Placebo.
All participants received medical management.
Over the first 6 weeks, those receiving naltrexone plus gabapentin had a longer interval to heavy drinking than those taking only naltrexone. By week 6, about half of those taking placebo or naltrexone alone had a heavy drinking day, compared with about 35% of those taking naltrexone plus gabapentin. Those receiving the combination also had fewer days of heavy drinking, fewer drinks per drinking day, and better sleep than the other groups. Participants in the naltrexone-alone group were more likely to drink heavily during periods in which they reported poor sleep. No significant group differences were found in measures of mood.
Gabapentin enacarbil is no better than placebo
Falk et al,32 in a 2019 preliminary analysis, examined data from a trial of gabapentin enacarbil, a prodrug formulation of gabapentin. In this 6-month double-blind study, 346 people with moderate AUD at 10 sites were randomized to gabapentin enacarbil extended-release 600 mg twice a day or placebo. All subjects received a computerized behavioral intervention.
No significant differences between groups were found in drinking measures or alcohol cravings, sleep problems, depression, or anxiety symptoms. However, a dose-response analysis found significantly less drinking for higher doses of the drug.
Bottom line: Evidence of benefits mixed but risk low
The efficacy of gabapentin as a treatment for AUD has varied across studies as a function of dosing and formulation. Daily doses have ranged from 600 mg to 1,800 mg, with the highest dose showing advantages in one study for cravings, insomnia, anxiety, dysphoria, and relapse.2 Thus far, gabapentin immediate-release has performed better than gabapentin enacarbil extended-release. All forms of gabapentin have been well-tolerated in AUD trials.
The 2018 American Psychiatric Association guidelines stated that gabapentin had a small positive effect on drinking outcomes, but the harm of treatment was deemed minimal, especially relative to the harms of chronic drinking.33 The guidelines endorse the use of gabapentin in patients with moderate to severe AUD who select gabapentin from the available options, or for those who are nonresponsive to or cannot tolerate naltrexone or acamprosate, as long as no contraindications exist. It was also noted that even small effects may be clinically important, considering the significant morbidity associated with AUD.
POTENTIAL FOR MISUSE
The use of gabapentin has become controversial owing to the growing recognition that it may not be as benign as initially thought.7–9,34 A review of US legislative actions reflects concerns about its misuse.35 In July 2017, Kentucky classified it as a schedule V controlled substance with prescription drug monitoring,35 as did Tennessee in 201836 and Michigan in January 2019.37 Currently, 8 other states (Massachusetts, Minnesota, Nebraska, North Dakota, Ohio, Virginia, Wyoming, and West Virginia) require prescription drug monitoring of gabapentin, and other states are considering it.35
Efforts to understand gabapentin misuse derive largely from people with drug use disorders. A review of postmortem toxicology reports in fatal drug overdoses found gabapentin present in 22%.38 Although it was not necessarily a cause of death, its high rate of detection suggests wide misuse among drug users.
Among a cohort of 503 prescription opioid misusers in Appalachian Kentucky, 15% reported using gabapentin “to get high.” Those who reported misusing gabapentin were 6 times more likely than nonusers to be abusing opioids and benzodiazepines. The main sources of gabapentin were doctors (52%) and dealers (36%). The average cost of gabapentin on the street was less than $1.00 per pill.39
Gabapentin misuse by methadone clinic patients is also reported. Baird et al40 surveyed patients in 6 addiction clinics in the United Kingdom for gabapentin and pregabalin abuse and found that 22% disclosed misusing these medications. Of these, 38% said they did so to enhance the methadone high.
In a review article, Quintero41 also cited enhancement of methadone euphoria and treatment of opioid withdrawal as motivations for misuse. Opioid-dependent gabapentin misusers consumed doses of gabapentin 3 to 20 times higher than clinically recommended and in combination with multiple drugs.4 Such use can cause dissociative and psychedelic effects.
Gabapentin also potentiates the sedative effects of opioids, thus increasing the risk of falls, accidents, and other adverse events.34,35 Risk of opioid-related deaths was increased with coprescription of gabapentin and with moderate to high gabapentin doses.34
Are people with AUD at higher risk of gabapentin abuse?
Despite concerns, patients in clinical trials of gabapentin treatment for AUD were not identified as at high risk for misuse of the drug.2,4,5,16 Further, no such trials reported serious drug-related adverse events resulting in gabapentin discontinuation or side effects that differed from placebo in frequency or severity.2,4,5,16
Clinical laboratory studies also have found no significant interactions between alcohol and gabapentin.42,43 In fact, they showed no influence of gabapentin on the pharmacokinetics of alcohol or on alcohol’s subjective effects. Relative to placebo, gabapentin did not affect blood alcohol levels, the degree of intoxication, sedation, craving, or alcohol self-administration.
Smith et al9 reported estimates that only 1% of the general population misuse gabapentin. Another review concluded that gabapentin is seldom a drug of choice.17 Most patients prescribed gabapentin do not experience cravings or loss of control, which are hallmarks of addiction. Hence, with adequate precautions, the off-label use of gabapentin for AUD is reasonable.
CLINICAL IMPLICATIONS OF GABAPENTIN PRESCRIBING
Overall, evidence for the benefit of gabapentin in AUD is mixed. Subgroups of alcoholic patients, such as those who do not respond to or tolerate standard therapies, may particularly benefit, as may those with comorbid insomnia or neuropathic pain.44 Clinicians should prescribe gabapentin only when it is likely to be helpful and should carefully document its efficacy.2,45
At each visit, an open and honest assessment of the benefits and risks serves to promote shared decision-making regarding initiating, continuing, or discontinuing gabapentin.
For alcohol withdrawal
Before gabapentin is prescribed for alcohol withdrawal, potential benefits (reduction of withdrawal symptoms), side effects (sedation, fatigue), and risks (falls) should be discussed with the patient.46 Patients should also be informed that benzodiazepines are the gold standard for alcohol withdrawal and that gabapentin is not effective for severe withdrawal.46
For relapse prevention
When initiating treatment for relapse prevention, the patient and the prescriber should agree on specific goals (eg, reduction of drinking, anxiety, and insomnia).2,16 Ongoing monitoring is essential and includes assessing and documenting improvement with respect to these goals.
In the AUD studies, gabapentin was well tolerated.16 Frequently observed side effects including headache, insomnia, fatigue, muscle aches, and gastrointestinal distress did not occur at a statistically different rate from placebo. However, patients in studies are selected samples, and their experience may not be generalizable to clinical practice. Thus, it is necessary to exercise caution and check for comorbidities that may put patients at risk of complications.47 Older patients and those on hemodialysis are more susceptible to gabapentin side effects such as sedation, dizziness, ataxia, and mental status changes,34 and prescribers should be alert for signs of toxicity (eg, ataxia, mental status changes).47,48
Gabapentin misuse was not observed in AUD studies,2,4,5,16 but evidence indicates that patients with opioid use disorder, prisoners, and polydrug users are at high risk for gabapentin misuse.39–41 In all cases, clinicians should monitor for red flags that may indicate abuse, such as missed appointments, early refill requests, demands for increased dosage, and simultaneous opiate and benzodiazepine use.49
Acknowledgment: The authors wish to thank Nick Mulligan for his invaluable assistance with formatting and grammar.
- Kranzler HR, Soyka M. Diagnosis and pharmacotherapy of alcohol use disorder: a review. JAMA 2018; 320(8):815–824. doi:10.1001/jama.2018.11406
- Mason BJ, Quello S, Goodell V, Shadan F, Kyle M, Begovic A. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med 2014; 174(1):70–77. doi:10.1001/jamainternmed.2013.11950
- Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res 2009; 33(9):1582–1588. doi:10.1111/j.1530-0277.2009.00986.x
- Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007; 68(11):1691–1700. pmid:18052562
- Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry 2011; 168(7):709–717. doi:10.1176/appi.ajp.2011.10101436
- Mack A. Examination of the evidence for off-label use of gabapentin. J Manag Care Pharm 2003; 9(6):559–568. doi:10.18553/jmcp.2003.9.6.559
- Schifano F. Misuse and abuse of pregabalin and gabapentin: cause for concern? CNS Drugs 2014; 28(6):491–496. doi:10.1007/s40263-014-0164-4
- Goodman CW, Brett AS. Gabapentin and pregabalin for pain—is increased prescribing a cause for concern? N Engl J Med 2017; 377(5):411–414. doi:10.1056/NEJMp1704633
- Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction 2016; 111(7):1160–1174. doi:10.1111/add.13324
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- Soyka M, Müller CA. Pharmacotherapy of alcoholism—an update on approved and off-label medications. Expert Opin Pharmacother 2017; 18(12):1187-1199. doi:10.1080/14656566.2017.1349098
- Zhang M, Gao CX, Ma KT, et al. A meta-analysis of therapeutic efficacy and safety of gabapentin i n the treatment of postherpetic neuralgia from randomized controlled trials. Biomed Res Int 2018; 2018:7474207. doi:10.1155/2018/7474207
- Winkelmann J, Allen RP, Högl B, et al. Treatment of restless legs syndrome: evidence-based review and implications for clinical practice (Revised 2017). Mov Disord 2018; 33(7):1077–1091. doi:10.1002/mds.27260
- Honarmand A, Safavi M, Zare M. Gabapentin: an update of its pharmacological properties and therapeutic use in epilepsy. J Res Med Sci 2011; 16(8):1062–1069. pmid:22279483
- van Hooft JA, Dougherty JJ, Endeman D, Nichols RA, Wadman WJ. Gabapentin inhibits presynaptic Ca(2+) influx and synaptic transmission in rat hippocampus and neocortex. Eur J Pharmacol 2002; 449(3):221–228. doi:10.1016/s0014-2999(02)02044-7
- Mason BJ, Quello S, Shadan F. Gabapentin for the treatment of alcohol use disorder. Expert Opin Investig Drugs 2018; 27(1):113–124. doi:10.1080/13543784.2018.1417383
- Taylor CP. Mechanisms of action of gabapentin. Rev Neurol (Paris) 1997; 153(suppl 1):S39–S45. pmid:9686247
- Agoglia AE, Herman MA. The center of the emotional universe: alcohol, stress, and CRF1 amygdala circuitry. Alcohol 2018; 72:61–73. doi:10.1016/j.alcohol.2018.03.009
- Nevo I, Hamon M. Neurotransmitter and neuromodulatory mechanisms involved in alcohol abuse and alcoholism. Neurochem Int 1995; 26(4):305–336. pmid:7633325
- You C, Vandegrift B, Brodie MS. Ethanol actions on the ventral tegmental area: novel potential targets on reward pathway neurons. Psychopharmacology (Berl) 2018; 235(6):1711–1726. doi:10.1007/s00213-018-4875-y
- Lovinger DM. Presynaptic ethanol actions: potential roles in ethanol seeking. Handb Exp Pharmacol 2018; 248:29–54. doi:10.1007/164_2017_76
- Williams SB, Yorgason JT, Nelson AC, et al. Glutamate transmission to ventral tegmental area GABA neurons is altered by acute and chronic ethanol. Alcohol Clin Exp Res 2018; 42(11):2186–2195. doi:10.1111/acer.13883
- N’Gouemo P. Voltage-sensitive calcium channels in the brain: relevance to alcohol intoxication and withdrawal. Handb Exp Pharmacol 2018; 248:263–280. doi:10.1007/164_2018_93
- Modesto-Lowe V, Huard J, Conrad C. Alcohol withdrawal kindling: is there a role for anticonvulsants? Psychiatry (Edgmont) 2005; 2(5):25–31. pmid:21152146
- Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol 2003; 23(5):514–519. doi:10.1097/01.jcp.0000088905.24613.ad
- Nichols TA, Robert S, Taber DJ, Cluver J. Alcohol withdrawal-related outcomes associated with gabapentin use in an inpatient psychiatric facility. Ment Health Clin 2019 ; 9(1):1–5. doi:10.9740/mhc.2019.01.001
- Bonnet U, Hamzavi-Abedi R, Specka M, Wiltfang J, Lieb B, Scherbaum N. An open trial of gabapentin in acute alcohol withdrawal using an oral loading protocol. Alcohol Alcohol 2010; 45(2):143–145. doi:10.1093/alcalc/agp085
- Stock CJ, Carpenter L, Ying J, Greene T. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother 2013; 47(7–8):961–969. doi:10.1345/aph.1R751
- Blanco-Gandía MC, Rodríguez-Arias M. Pharmacological treatments for opiate and alcohol addiction: a historical perspective of the last 50 years. Eur J Pharmacol 2018; 836:89–101. doi:10.1016/j.ejphar.2018.08.007
- Anton RF, Moak DH, Latham P, et al. Naltrexone combined with either cognitive behavioral or motivational enhancement therapy for alcohol dependence. J Clin Psychopharmacol 2005; 25(4):349–357. pmid:16012278
- Brower KJ, Myra Kim H, Strobbe S, Karam-Hage MA, Consens F, Zucker RA. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res 2008; 32(8):1429–1438. doi:10.1111/j.1530-0277.2008.00706.x
- Falk DE, Ryan ML, Fertig JB, et al; National Institute on Alcohol Abuse and Alcoholism Clinical Investigations Group (NCIG) Study Group. Gabapentin enacarbil extended-release for alcohol use disorder: a randomized, double-blind, placebo-controlled, multisite trial assessing efficacy and safety. Alcohol Clin Exp Res 2019; 43(1):158–169. doi:10.1111/acer.13917
- The American Psychiatric Association. Practice Guideline for the Pharmacological Treatment of Patients with Alcohol Use Disorder. https://psychiatryonline.org/doi/pdf/10.1176/appi.books.9781615371969. Accessed October 10, 2019.
- Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med 2017; 14(10):e1002396. doi:10.1371/journal.pmed.1002396
- Peckham AM, Ananickal MJ, Sclar DA. Gabapentin use, abuse, and the US opioid epidemic: the case for reclassification as a controlled substance and the need for pharmacovigilance. Risk Manag Healthc Policy 2018; 11:109–116. doi:10.2147/RMHP.S168504
- Tennessee Pharmacists Association. Advocacy alert: end of session summary. www.tnpharm.org/news/news-posts-pages/advocacy-alert-4-30-18/? Accessed October 10, 2019.
- Michigan.gov. Gabapentin scheduled as controlled substance to help with state’s opioid epidemic. www.michigan.gov/som/0,4669,7-192-47796-487050--,00.html. Accessed October 10, 2019.
- Slavova S, Miller A, Bunn TL, et al. Prevalence of gabapentin in drug overdose postmortem toxicology testing results. Drug Alcohol Depend 2018; 186:80–85. doi:10.1016/j.drugalcdep.2018.01.018
- Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry 2015; 172(5):487–488. doi:10.1176/appi.ajp.2014.14101272
- Baird CR, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res 2014; 20(3):115–118. doi:10.1159/000355268
- Quintero GC. Review about gabapentin misuse, interactions, contraindications and side effects. J Exp Pharmacol 2017; 9:13–21. doi:10.2147/JEP.S124391
- Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend 2006; 83(1):25–32. doi:10.1016/j.drugalcdep.2005.10.008
- Myrick H, Anton R, Voronin K, Wang W, Henderson S. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res 2007; 31(2):221–227. doi:10.1111/j.1530-0277.2006.00299.x
- Tzellos TG, Papazisis G, Toulis KA, Sardeli CH, Kouvelas D. A2delta ligands gabapentin and pregabalin: future implications in daily clinical practice. Hippokratia 2010; 14(2):71–75. pmid:20596259
- Morrison EE, Sandilands EA, Webb DJ. Gabapentin and pregabalin: do the benefits outweigh the harms? J R Coll Physicians Edinb 2017; 47(4):310–313. doi:10.4997/JRCPE.2017.402
- Leung JG, Rakocevic DB, Allen ND, et al. Use of a gabapentin protocol for the management of alcohol withdrawal: a preliminary experience expanding from the consultation-liaison psychiatry service. Psychosomatics 2018; 59(5):496–505. doi:10.1016/j.psym.2018.03.002
- Fleet JL, Dixon SN, Kuwornu PJ, et al. Gabapentin dose and the 30-day risk of altered mental status in older adults: a retrospective population-based study. PLoS One 2018; 13(3):e0193134. doi:10.1371/journal.pone.0193134
- Chiappini S, Schifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs 2016; 30(7):647–654. doi:10.1007/s40263-016-0359-y
- Modesto-Lowe V, Chaplin M, Sinha S, Woodard K. Universal precautions to reduce stimulant misuse in treating adult ADHD. Cleve Clin J Med 2015; 82(8):506–512. doi:10.3949/ccjm.82a.14131
Perceptions regarding the use of gabapentin for alcohol use disorder (AUD) have shifted over time.1–4 Early on, the drug was deemed to be benign and effective.4–6 But more and more, concerns are being raised about its recreational use to achieve euphoria,7 and the drug is often misused by vulnerable populations, particularly those with opioid use disorder.7–9
Given the large number of gabapentin prescriptions written off-label for AUD, it is incumbent on providers to understand how to prescribe it responsibly.7–9 To that end, this article focuses on the benefits—and concerns—of this treatment option. We describe the effects of gabapentin on the central nervous system and how it may mitigate alcohol withdrawal and increase the likelihood of abstinence. In addition, we review clinical trials that evaluated potential roles of gabapentin in AUD, discuss the drug’s misuse potential, and suggest a framework for its appropriate use in AUD management.
ALCOHOL USE DISORDER IS COMMON AND SERIOUS
AUD affects about 14% of US adults and represents a significant health burden,1 often with severe clinical and social implications. It manifests as compulsive drinking and loss of control despite adverse consequences on various life domains.10 It is generally associated with cravings, tolerance, and withdrawal symptoms upon cessation. Alcohol withdrawal is characterized by tremors, anxiety, sweating, nausea, and tachycardia, and in severe cases, may involve hallucinations, seizures, and delirium tremens. Untreated, alcohol withdrawal can be fatal.10
Even though psychosocial treatments for AUD by themselves are associated with high relapse rates, pharmacotherapy is underutilized. Three drugs approved by the US Food and Drug Administration (FDA) are available to treat it, but they are often poorly accepted and have limited efficacy. For these reasons, there is considerable interest in finding alternatives. Gabapentin is one of several agents that have been studied (Table 1). The topic has been reviewed in depth by Soyka and Müller.11
GABAPENTIN REDUCES EXCITATION
The anticonvulsant gabapentin is FDA-approved for treating epilepsy, postherpetic neuralgia, and restless leg syndrome.8,12–14 It binds and selectively impedes voltage-sensitive calcium channels, the pores in cell membrane that permit calcium to enter a neuron in response to changes in electrical currents.15
Gabapentin is believed to decrease excitation of the central nervous system in multiple ways:
- It reduces the release of glutamate, a key component of the excitatory system16
- It increases the concentration of gamma-aminobutyric acid (GABA), the main inhibitory neurotransmitter in the brain7
- By binding the alpha-2-delta type 1 subunit of voltage-sensitive calcium channels,8,15–17 it inhibits excitatory synapse formation independent of calcium channel activity16
- By blocking excitatory neurotransmission, it also may indirectly increase the concentration of GABA in the central nervous system16,17
- It modulates action of glutamic acid decarboxylase (involved in the synthesis of GABA) and glutamate synthesizing enzyme to increase GABA and decrease glutamate.17
ALCOHOL’S ACTIONS
The actions of alcohol on the brain are also complex.18 Alpha-2-delta type 1 subunits of calcium channels are upregulated in the reward centers of the brain by addictive substances, including alcohol.16 Alcohol interacts with corticotropin-releasing factor and several neurotransmitters,18 and specifically affects neuropathways involving norepinephrine, GABA, and glutamate.19 Alcohol has reinforcing effects mediated by the release of dopamine in the nucleus accumbens.20
Acutely, alcohol promotes GABA release and may also reduce GABA degradation, producing sedative and anxiolytic effects.21 Chronic alcohol use leads to a decrease in the number of GABAA receptors. Clinically, this downregulation manifests as tolerance to alcohol’s sedating effects.21
Alcohol affects the signaling of glutamatergic interaction with the N-methyl-d-aspartate (NMDA) receptor.22 Glutamate activates this receptor as well as the voltage-gated ion channels, modifying calcium influx and increasing neuronal excitability.22,23 Acutely, alcohol has an antagonistic effect on the NMDA receptor, while chronic drinking upregulates (increases) the number of NMDA receptors and voltage-gated calcium channels.22,23
Alcohol withdrawal increases excitatory effects
Patients experiencing alcohol withdrawal have decreased GABA-ergic functioning and increased glutamatergic action throughout the central nervous system.19,24
Withdrawal can be subdivided into an acute phase (lasting up to about 5 days) and a protracted phase (of undetermined duration). During withdrawal, the brain activates its “stress system,” leading to overexpression of corticotropin-releasing factor in the amygdala. Protracted withdrawal dysregulates the prefrontal cortex, increasing cravings and worsening negative emotional states and sleep.16
GABAPENTIN FOR ALCOHOL WITHDRAWAL
Benzodiazepines are the standard treatment for alcohol withdrawal.3,24 They relieve symptoms and can prevent seizures and delirium tremens,24 but they are sedating and cause psychomotor impairments.3 Because of the potential for addiction, benzodiazepine use is limited to acute alcohol withdrawal.3
Gabapentin shows promise as an agent that can be used in withdrawal and continued through early abstinence without the highly addictive potential of benzodiazepines.16 It is thought to affect drinking behaviors during early abstinence by normalizing GABA and glutamate activity.2,16
Early preclinical studies in mouse models found that gabapentin decreases anxiogenic and epileptic effects of alcohol withdrawal. Compared with other antidrinking medications, gabapentin has the benefits of lacking elimination via hepatic metabolism, few pharmacokinetic interactions, and good reported tolerability in this population.
Inpatient trials show no benefit over standard treatments
Bonnet et al25 conducted a double-blind placebo-controlled trial in Germany in inpatients experiencing acute alcohol withdrawal to determine whether gabapentin might be an effective adjunct to clomethiazole, a GABAA modulator commonly used in Europe for alcohol withdrawal. Participants (N = 61) were randomized to receive placebo or gabapentin (400 mg every 6 hours) for 72 hours, with tapering over the next 3 days. All patients could receive rescue doses of clomethiazole, using a symptom-triggered protocol.
The study revealed no differences in the amount of clomethiazole administered between the 2 groups, suggesting that gabapentin had no adjunctive effect. Side effects (vertigo, nausea, dizziness, and ataxia) were mild and comparable between groups.
Nichols et al26 conducted a retrospective cohort study in a South Carolina academic psychiatric hospital to assess the adjunctive effect of gabapentin on the as-needed use of benzodiazepines for alcohol withdrawal. The active group (n = 40) received gabapentin as well as a symptom-triggered alcohol withdrawal protocol of benzodiazepine. The control group (n = 43) received only the symptom-triggered alcohol withdrawal protocol without gabapentin.
No effect was found of gabapentin use for benzodiazepine treatment of alcohol withdrawal. It is notable that Bonnet et al and Nichols et al had similar findings despite their studies being conducted in different countries using distinct comparators and methods.
Bonnet et al,27 in another study, tried a different design to investigate a possible role for gabapentin in inpatient alcohol withdrawal. The study included 37 patients with severe alcohol withdrawal (Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised [CIWA-Ar] > 15).
All participants received gabapentin 800 mg. Those whose CIWA-Ar score improved within 2 hours were considered “early responders” (n = 27) and next received 2 days of gabapentin 600 mg 4 times a day before starting a taper. The nonresponders whose CIWA-Ar score worsened (associated with greater anxiety and depressive symptoms; n = 10) were switched to standard treatment with clomethiazole (n = 4) or clonazepam (n = 6). Scores of 3 early responders subsequently worsened; 2 of these participants developed seizures and were switched to standard treatment.
The authors concluded that gabapentin in a dose of 3,200 mg in the first 24 hours is useful only for milder forms of alcohol withdrawal. Hence, subsequent efforts on the use of gabapentin for alcohol withdrawal have focused on outpatients.
Outpatient trials reveal benefits over benzodiazepines
Myrick et al3 compared gabapentin vs lorazepam in 100 outpatients seeking treatment for alcohol withdrawal. Participants were randomized to 1 of 4 groups: gabapentin 600 mg, 900 mg, or 1,200 mg, or lorazepam 6 mg, each tapering over 4 days. Alcohol withdrawal was measured by the CIWA-Ar score. Only 68 patients completed all follow-up appointments to day 12.
Gabapentin 600 mg was discontinued because of seizures in 2 patients, but it was generally well tolerated and was associated with diminished symptoms of alcohol withdrawal, especially at the 1,200 mg dose. The gabapentin groups experienced less anxiety and sedation and fewer cravings than the lorazepam group. Those treated with lorazepam fared worse for achieving early abstinence and were more likely to return to drinking when the intervention was discontinued. However, significant relapse by day 12 occurred in both groups.
The authors concluded that gabapentin was at least as effective as lorazepam in the outpatient treatment of alcohol withdrawal, with the 1,200-mg gabapentin dosage being more effective than 900 mg. At 1,200 mg, gabapentin was associated with better sleep, less anxiety, and better self-reported ability to work than lorazepam, and at the 900-mg dose it was associated with less depression than lorazepam.
Stock et al28 conducted a randomized, double-blind study of gabapentin in acute alcohol withdrawal in 26 military veterans in an outpatient setting. Patients were randomized to one of the following:
- Gabapentin 1,200 mg orally for 3 days, followed by 900 mg, 600 mg, and 300 mg for 1 day each (n = 17)
- Chlordiazepoxide 100 mg orally for 3 days, followed by 75 mg, 50 mg, and 25 mg for 1 day each (n = 9).
Withdrawal scores improved similarly in both groups. Early on (days 1–4), neither cravings nor sleep differed significantly between groups; but later (days 5–7), the gabapentin group had superior scores for these measures. Gabapentin was also associated with significantly less sedation than chlordiazepoxide and trended to less alcohol craving.
Bottom line: Gabapentin is useful for mild withdrawal
Data suggest that gabapentin offers benefits for managing mild alcohol withdrawal. Improved residual craving and sleep measures are clinically important because they are risk factors for relapse. Mood and anxiety also improve with gabapentin, further indicating a therapeutic effect.
Gabapentin’s benefits for moderate and severe alcohol withdrawal have not been established. Seizures occurred during withdrawal despite gabapentin treatment, but whether from an insufficient dose, patient susceptibility, or lack of gabapentin efficacy is not clear. Best results occurred at the 1,200-mg daily dose, but benefits may not apply to patients with severe withdrawal. In addition, many studies were small, limiting the strength of conclusions.
Across most studies of gabapentin for alcohol withdrawal, advantages included a smoother transition into early abstinence due to improved sleep, mood, and anxiety, alleviating common triggers for a return to drinking. Gabapentin also carries less reinforcing potential than benzodiazepines. These qualities fueled interest in trying gabapentin to improve long-term abstinence.
GABAPENTIN FOR RELAPSE PREVENTION
Although naltrexone and acamprosate are the first-line treatments for relapse prevention, they do not help all patients and are more effective when combined with cognitive behavioral therapy.1,29,30 For patients in whom standard treatments are not effective or tolerated, gabapentin may provide a reasonable alternative, and several randomized controlled trials have examined its use for this role.
Gabapentin alone is better than placebo
Furieri and Nakamura-Palacios4 assessed the use of gabapentin for relapse prevention in Brazilian outpatients (N = 60) who had averaged 27 years of drinking and consumed 17 drinks daily for the 90 days before baseline. After detoxification with diazepam and vitamins, patients were randomized to either gabapentin 300 mg twice daily or placebo for 4 weeks.
Compared with placebo, gabapentin significantly reduced cravings and lowered the percentage of heavy drinking days and the number of drinks per day, with a significant increase in the percentage of abstinent days. These self-reported measures correlated with decreases in gamma-glutamyl transferase, a biological marker for heavy drinking.
Brower et al31 investigated the use of gabapentin in 21 outpatients with AUD and insomnia who desired to remain abstinent. They were randomized to gabapentin (up to 1,500 mg at night) or placebo for 6 weeks. Just 14 participants completed the study; all but 2 were followed without treatment until week 12.
Gabapentin was associated with significantly lower relapse rates at 6 weeks (3 of 10 in the gabapentin group vs 9 of 11 in the placebo group) and at 12 weeks (6 of 10 in the gabapentin group vs 11 of 11 in the placebo group, assuming the 2 patients lost to follow-up relapsed). No difference between groups was detected for sleep measures in this small study. However, other studies have found that gabapentin for AUD improves measures of insomnia and daytime drowsiness—predictors of relapse—compared with other medications.16
High-dose gabapentin is better
Mason et al2 randomized 150 outpatients with alcohol dependence to 12 weeks of daily treatment with either gabapentin (900 mg or 1,800 mg) or placebo after at least 3 days of abstinence. All participants received counseling. Drinking quantity and frequency were assessed by gamma-glutamyl transferase testing.
Patients taking gabapentin had better rates of abstinence and cessation of heavy drinking than those taking placebo. During the 12-week study, the 1,800-mg daily dose showed a substantially higher abstinence rate (17%) than either 900 mg (11%) or placebo (4%). Significant dose-related improvements were also found for heavy drinking days, total drinking quantity, and frequency of alcohol withdrawal symptoms that predispose to early relapse, such as poor sleep, cravings, and poor mood. There were also significant linear dose effects on rates of abstinence and nondrinking days at the 24-week posttreatment follow-up.
Gabapentin plus naltrexone is better than naltrexone alone
Anton et al5 examined the efficacy of gabapentin combined with naltrexone during early abstinence. The study randomly assigned 150 people with AUD to one of the following groups:
- 16 weeks of naltrexone (50 mg/day) alone
- 6 weeks of naltrexone (50 mg/day) plus gabapentin (up to 1,200 mg/day), followed by 10 weeks of naltrexone alone
- Placebo.
All participants received medical management.
Over the first 6 weeks, those receiving naltrexone plus gabapentin had a longer interval to heavy drinking than those taking only naltrexone. By week 6, about half of those taking placebo or naltrexone alone had a heavy drinking day, compared with about 35% of those taking naltrexone plus gabapentin. Those receiving the combination also had fewer days of heavy drinking, fewer drinks per drinking day, and better sleep than the other groups. Participants in the naltrexone-alone group were more likely to drink heavily during periods in which they reported poor sleep. No significant group differences were found in measures of mood.
Gabapentin enacarbil is no better than placebo
Falk et al,32 in a 2019 preliminary analysis, examined data from a trial of gabapentin enacarbil, a prodrug formulation of gabapentin. In this 6-month double-blind study, 346 people with moderate AUD at 10 sites were randomized to gabapentin enacarbil extended-release 600 mg twice a day or placebo. All subjects received a computerized behavioral intervention.
No significant differences between groups were found in drinking measures or alcohol cravings, sleep problems, depression, or anxiety symptoms. However, a dose-response analysis found significantly less drinking for higher doses of the drug.
Bottom line: Evidence of benefits mixed but risk low
The efficacy of gabapentin as a treatment for AUD has varied across studies as a function of dosing and formulation. Daily doses have ranged from 600 mg to 1,800 mg, with the highest dose showing advantages in one study for cravings, insomnia, anxiety, dysphoria, and relapse.2 Thus far, gabapentin immediate-release has performed better than gabapentin enacarbil extended-release. All forms of gabapentin have been well-tolerated in AUD trials.
The 2018 American Psychiatric Association guidelines stated that gabapentin had a small positive effect on drinking outcomes, but the harm of treatment was deemed minimal, especially relative to the harms of chronic drinking.33 The guidelines endorse the use of gabapentin in patients with moderate to severe AUD who select gabapentin from the available options, or for those who are nonresponsive to or cannot tolerate naltrexone or acamprosate, as long as no contraindications exist. It was also noted that even small effects may be clinically important, considering the significant morbidity associated with AUD.
POTENTIAL FOR MISUSE
The use of gabapentin has become controversial owing to the growing recognition that it may not be as benign as initially thought.7–9,34 A review of US legislative actions reflects concerns about its misuse.35 In July 2017, Kentucky classified it as a schedule V controlled substance with prescription drug monitoring,35 as did Tennessee in 201836 and Michigan in January 2019.37 Currently, 8 other states (Massachusetts, Minnesota, Nebraska, North Dakota, Ohio, Virginia, Wyoming, and West Virginia) require prescription drug monitoring of gabapentin, and other states are considering it.35
Efforts to understand gabapentin misuse derive largely from people with drug use disorders. A review of postmortem toxicology reports in fatal drug overdoses found gabapentin present in 22%.38 Although it was not necessarily a cause of death, its high rate of detection suggests wide misuse among drug users.
Among a cohort of 503 prescription opioid misusers in Appalachian Kentucky, 15% reported using gabapentin “to get high.” Those who reported misusing gabapentin were 6 times more likely than nonusers to be abusing opioids and benzodiazepines. The main sources of gabapentin were doctors (52%) and dealers (36%). The average cost of gabapentin on the street was less than $1.00 per pill.39
Gabapentin misuse by methadone clinic patients is also reported. Baird et al40 surveyed patients in 6 addiction clinics in the United Kingdom for gabapentin and pregabalin abuse and found that 22% disclosed misusing these medications. Of these, 38% said they did so to enhance the methadone high.
In a review article, Quintero41 also cited enhancement of methadone euphoria and treatment of opioid withdrawal as motivations for misuse. Opioid-dependent gabapentin misusers consumed doses of gabapentin 3 to 20 times higher than clinically recommended and in combination with multiple drugs.4 Such use can cause dissociative and psychedelic effects.
Gabapentin also potentiates the sedative effects of opioids, thus increasing the risk of falls, accidents, and other adverse events.34,35 Risk of opioid-related deaths was increased with coprescription of gabapentin and with moderate to high gabapentin doses.34
Are people with AUD at higher risk of gabapentin abuse?
Despite concerns, patients in clinical trials of gabapentin treatment for AUD were not identified as at high risk for misuse of the drug.2,4,5,16 Further, no such trials reported serious drug-related adverse events resulting in gabapentin discontinuation or side effects that differed from placebo in frequency or severity.2,4,5,16
Clinical laboratory studies also have found no significant interactions between alcohol and gabapentin.42,43 In fact, they showed no influence of gabapentin on the pharmacokinetics of alcohol or on alcohol’s subjective effects. Relative to placebo, gabapentin did not affect blood alcohol levels, the degree of intoxication, sedation, craving, or alcohol self-administration.
Smith et al9 reported estimates that only 1% of the general population misuse gabapentin. Another review concluded that gabapentin is seldom a drug of choice.17 Most patients prescribed gabapentin do not experience cravings or loss of control, which are hallmarks of addiction. Hence, with adequate precautions, the off-label use of gabapentin for AUD is reasonable.
CLINICAL IMPLICATIONS OF GABAPENTIN PRESCRIBING
Overall, evidence for the benefit of gabapentin in AUD is mixed. Subgroups of alcoholic patients, such as those who do not respond to or tolerate standard therapies, may particularly benefit, as may those with comorbid insomnia or neuropathic pain.44 Clinicians should prescribe gabapentin only when it is likely to be helpful and should carefully document its efficacy.2,45
At each visit, an open and honest assessment of the benefits and risks serves to promote shared decision-making regarding initiating, continuing, or discontinuing gabapentin.
For alcohol withdrawal
Before gabapentin is prescribed for alcohol withdrawal, potential benefits (reduction of withdrawal symptoms), side effects (sedation, fatigue), and risks (falls) should be discussed with the patient.46 Patients should also be informed that benzodiazepines are the gold standard for alcohol withdrawal and that gabapentin is not effective for severe withdrawal.46
For relapse prevention
When initiating treatment for relapse prevention, the patient and the prescriber should agree on specific goals (eg, reduction of drinking, anxiety, and insomnia).2,16 Ongoing monitoring is essential and includes assessing and documenting improvement with respect to these goals.
In the AUD studies, gabapentin was well tolerated.16 Frequently observed side effects including headache, insomnia, fatigue, muscle aches, and gastrointestinal distress did not occur at a statistically different rate from placebo. However, patients in studies are selected samples, and their experience may not be generalizable to clinical practice. Thus, it is necessary to exercise caution and check for comorbidities that may put patients at risk of complications.47 Older patients and those on hemodialysis are more susceptible to gabapentin side effects such as sedation, dizziness, ataxia, and mental status changes,34 and prescribers should be alert for signs of toxicity (eg, ataxia, mental status changes).47,48
Gabapentin misuse was not observed in AUD studies,2,4,5,16 but evidence indicates that patients with opioid use disorder, prisoners, and polydrug users are at high risk for gabapentin misuse.39–41 In all cases, clinicians should monitor for red flags that may indicate abuse, such as missed appointments, early refill requests, demands for increased dosage, and simultaneous opiate and benzodiazepine use.49
Acknowledgment: The authors wish to thank Nick Mulligan for his invaluable assistance with formatting and grammar.
Perceptions regarding the use of gabapentin for alcohol use disorder (AUD) have shifted over time.1–4 Early on, the drug was deemed to be benign and effective.4–6 But more and more, concerns are being raised about its recreational use to achieve euphoria,7 and the drug is often misused by vulnerable populations, particularly those with opioid use disorder.7–9
Given the large number of gabapentin prescriptions written off-label for AUD, it is incumbent on providers to understand how to prescribe it responsibly.7–9 To that end, this article focuses on the benefits—and concerns—of this treatment option. We describe the effects of gabapentin on the central nervous system and how it may mitigate alcohol withdrawal and increase the likelihood of abstinence. In addition, we review clinical trials that evaluated potential roles of gabapentin in AUD, discuss the drug’s misuse potential, and suggest a framework for its appropriate use in AUD management.
ALCOHOL USE DISORDER IS COMMON AND SERIOUS
AUD affects about 14% of US adults and represents a significant health burden,1 often with severe clinical and social implications. It manifests as compulsive drinking and loss of control despite adverse consequences on various life domains.10 It is generally associated with cravings, tolerance, and withdrawal symptoms upon cessation. Alcohol withdrawal is characterized by tremors, anxiety, sweating, nausea, and tachycardia, and in severe cases, may involve hallucinations, seizures, and delirium tremens. Untreated, alcohol withdrawal can be fatal.10
Even though psychosocial treatments for AUD by themselves are associated with high relapse rates, pharmacotherapy is underutilized. Three drugs approved by the US Food and Drug Administration (FDA) are available to treat it, but they are often poorly accepted and have limited efficacy. For these reasons, there is considerable interest in finding alternatives. Gabapentin is one of several agents that have been studied (Table 1). The topic has been reviewed in depth by Soyka and Müller.11
GABAPENTIN REDUCES EXCITATION
The anticonvulsant gabapentin is FDA-approved for treating epilepsy, postherpetic neuralgia, and restless leg syndrome.8,12–14 It binds and selectively impedes voltage-sensitive calcium channels, the pores in cell membrane that permit calcium to enter a neuron in response to changes in electrical currents.15
Gabapentin is believed to decrease excitation of the central nervous system in multiple ways:
- It reduces the release of glutamate, a key component of the excitatory system16
- It increases the concentration of gamma-aminobutyric acid (GABA), the main inhibitory neurotransmitter in the brain7
- By binding the alpha-2-delta type 1 subunit of voltage-sensitive calcium channels,8,15–17 it inhibits excitatory synapse formation independent of calcium channel activity16
- By blocking excitatory neurotransmission, it also may indirectly increase the concentration of GABA in the central nervous system16,17
- It modulates action of glutamic acid decarboxylase (involved in the synthesis of GABA) and glutamate synthesizing enzyme to increase GABA and decrease glutamate.17
ALCOHOL’S ACTIONS
The actions of alcohol on the brain are also complex.18 Alpha-2-delta type 1 subunits of calcium channels are upregulated in the reward centers of the brain by addictive substances, including alcohol.16 Alcohol interacts with corticotropin-releasing factor and several neurotransmitters,18 and specifically affects neuropathways involving norepinephrine, GABA, and glutamate.19 Alcohol has reinforcing effects mediated by the release of dopamine in the nucleus accumbens.20
Acutely, alcohol promotes GABA release and may also reduce GABA degradation, producing sedative and anxiolytic effects.21 Chronic alcohol use leads to a decrease in the number of GABAA receptors. Clinically, this downregulation manifests as tolerance to alcohol’s sedating effects.21
Alcohol affects the signaling of glutamatergic interaction with the N-methyl-d-aspartate (NMDA) receptor.22 Glutamate activates this receptor as well as the voltage-gated ion channels, modifying calcium influx and increasing neuronal excitability.22,23 Acutely, alcohol has an antagonistic effect on the NMDA receptor, while chronic drinking upregulates (increases) the number of NMDA receptors and voltage-gated calcium channels.22,23
Alcohol withdrawal increases excitatory effects
Patients experiencing alcohol withdrawal have decreased GABA-ergic functioning and increased glutamatergic action throughout the central nervous system.19,24
Withdrawal can be subdivided into an acute phase (lasting up to about 5 days) and a protracted phase (of undetermined duration). During withdrawal, the brain activates its “stress system,” leading to overexpression of corticotropin-releasing factor in the amygdala. Protracted withdrawal dysregulates the prefrontal cortex, increasing cravings and worsening negative emotional states and sleep.16
GABAPENTIN FOR ALCOHOL WITHDRAWAL
Benzodiazepines are the standard treatment for alcohol withdrawal.3,24 They relieve symptoms and can prevent seizures and delirium tremens,24 but they are sedating and cause psychomotor impairments.3 Because of the potential for addiction, benzodiazepine use is limited to acute alcohol withdrawal.3
Gabapentin shows promise as an agent that can be used in withdrawal and continued through early abstinence without the highly addictive potential of benzodiazepines.16 It is thought to affect drinking behaviors during early abstinence by normalizing GABA and glutamate activity.2,16
Early preclinical studies in mouse models found that gabapentin decreases anxiogenic and epileptic effects of alcohol withdrawal. Compared with other antidrinking medications, gabapentin has the benefits of lacking elimination via hepatic metabolism, few pharmacokinetic interactions, and good reported tolerability in this population.
Inpatient trials show no benefit over standard treatments
Bonnet et al25 conducted a double-blind placebo-controlled trial in Germany in inpatients experiencing acute alcohol withdrawal to determine whether gabapentin might be an effective adjunct to clomethiazole, a GABAA modulator commonly used in Europe for alcohol withdrawal. Participants (N = 61) were randomized to receive placebo or gabapentin (400 mg every 6 hours) for 72 hours, with tapering over the next 3 days. All patients could receive rescue doses of clomethiazole, using a symptom-triggered protocol.
The study revealed no differences in the amount of clomethiazole administered between the 2 groups, suggesting that gabapentin had no adjunctive effect. Side effects (vertigo, nausea, dizziness, and ataxia) were mild and comparable between groups.
Nichols et al26 conducted a retrospective cohort study in a South Carolina academic psychiatric hospital to assess the adjunctive effect of gabapentin on the as-needed use of benzodiazepines for alcohol withdrawal. The active group (n = 40) received gabapentin as well as a symptom-triggered alcohol withdrawal protocol of benzodiazepine. The control group (n = 43) received only the symptom-triggered alcohol withdrawal protocol without gabapentin.
No effect was found of gabapentin use for benzodiazepine treatment of alcohol withdrawal. It is notable that Bonnet et al and Nichols et al had similar findings despite their studies being conducted in different countries using distinct comparators and methods.
Bonnet et al,27 in another study, tried a different design to investigate a possible role for gabapentin in inpatient alcohol withdrawal. The study included 37 patients with severe alcohol withdrawal (Clinical Institute Withdrawal Assessment of Alcohol Scale, Revised [CIWA-Ar] > 15).
All participants received gabapentin 800 mg. Those whose CIWA-Ar score improved within 2 hours were considered “early responders” (n = 27) and next received 2 days of gabapentin 600 mg 4 times a day before starting a taper. The nonresponders whose CIWA-Ar score worsened (associated with greater anxiety and depressive symptoms; n = 10) were switched to standard treatment with clomethiazole (n = 4) or clonazepam (n = 6). Scores of 3 early responders subsequently worsened; 2 of these participants developed seizures and were switched to standard treatment.
The authors concluded that gabapentin in a dose of 3,200 mg in the first 24 hours is useful only for milder forms of alcohol withdrawal. Hence, subsequent efforts on the use of gabapentin for alcohol withdrawal have focused on outpatients.
Outpatient trials reveal benefits over benzodiazepines
Myrick et al3 compared gabapentin vs lorazepam in 100 outpatients seeking treatment for alcohol withdrawal. Participants were randomized to 1 of 4 groups: gabapentin 600 mg, 900 mg, or 1,200 mg, or lorazepam 6 mg, each tapering over 4 days. Alcohol withdrawal was measured by the CIWA-Ar score. Only 68 patients completed all follow-up appointments to day 12.
Gabapentin 600 mg was discontinued because of seizures in 2 patients, but it was generally well tolerated and was associated with diminished symptoms of alcohol withdrawal, especially at the 1,200 mg dose. The gabapentin groups experienced less anxiety and sedation and fewer cravings than the lorazepam group. Those treated with lorazepam fared worse for achieving early abstinence and were more likely to return to drinking when the intervention was discontinued. However, significant relapse by day 12 occurred in both groups.
The authors concluded that gabapentin was at least as effective as lorazepam in the outpatient treatment of alcohol withdrawal, with the 1,200-mg gabapentin dosage being more effective than 900 mg. At 1,200 mg, gabapentin was associated with better sleep, less anxiety, and better self-reported ability to work than lorazepam, and at the 900-mg dose it was associated with less depression than lorazepam.
Stock et al28 conducted a randomized, double-blind study of gabapentin in acute alcohol withdrawal in 26 military veterans in an outpatient setting. Patients were randomized to one of the following:
- Gabapentin 1,200 mg orally for 3 days, followed by 900 mg, 600 mg, and 300 mg for 1 day each (n = 17)
- Chlordiazepoxide 100 mg orally for 3 days, followed by 75 mg, 50 mg, and 25 mg for 1 day each (n = 9).
Withdrawal scores improved similarly in both groups. Early on (days 1–4), neither cravings nor sleep differed significantly between groups; but later (days 5–7), the gabapentin group had superior scores for these measures. Gabapentin was also associated with significantly less sedation than chlordiazepoxide and trended to less alcohol craving.
Bottom line: Gabapentin is useful for mild withdrawal
Data suggest that gabapentin offers benefits for managing mild alcohol withdrawal. Improved residual craving and sleep measures are clinically important because they are risk factors for relapse. Mood and anxiety also improve with gabapentin, further indicating a therapeutic effect.
Gabapentin’s benefits for moderate and severe alcohol withdrawal have not been established. Seizures occurred during withdrawal despite gabapentin treatment, but whether from an insufficient dose, patient susceptibility, or lack of gabapentin efficacy is not clear. Best results occurred at the 1,200-mg daily dose, but benefits may not apply to patients with severe withdrawal. In addition, many studies were small, limiting the strength of conclusions.
Across most studies of gabapentin for alcohol withdrawal, advantages included a smoother transition into early abstinence due to improved sleep, mood, and anxiety, alleviating common triggers for a return to drinking. Gabapentin also carries less reinforcing potential than benzodiazepines. These qualities fueled interest in trying gabapentin to improve long-term abstinence.
GABAPENTIN FOR RELAPSE PREVENTION
Although naltrexone and acamprosate are the first-line treatments for relapse prevention, they do not help all patients and are more effective when combined with cognitive behavioral therapy.1,29,30 For patients in whom standard treatments are not effective or tolerated, gabapentin may provide a reasonable alternative, and several randomized controlled trials have examined its use for this role.
Gabapentin alone is better than placebo
Furieri and Nakamura-Palacios4 assessed the use of gabapentin for relapse prevention in Brazilian outpatients (N = 60) who had averaged 27 years of drinking and consumed 17 drinks daily for the 90 days before baseline. After detoxification with diazepam and vitamins, patients were randomized to either gabapentin 300 mg twice daily or placebo for 4 weeks.
Compared with placebo, gabapentin significantly reduced cravings and lowered the percentage of heavy drinking days and the number of drinks per day, with a significant increase in the percentage of abstinent days. These self-reported measures correlated with decreases in gamma-glutamyl transferase, a biological marker for heavy drinking.
Brower et al31 investigated the use of gabapentin in 21 outpatients with AUD and insomnia who desired to remain abstinent. They were randomized to gabapentin (up to 1,500 mg at night) or placebo for 6 weeks. Just 14 participants completed the study; all but 2 were followed without treatment until week 12.
Gabapentin was associated with significantly lower relapse rates at 6 weeks (3 of 10 in the gabapentin group vs 9 of 11 in the placebo group) and at 12 weeks (6 of 10 in the gabapentin group vs 11 of 11 in the placebo group, assuming the 2 patients lost to follow-up relapsed). No difference between groups was detected for sleep measures in this small study. However, other studies have found that gabapentin for AUD improves measures of insomnia and daytime drowsiness—predictors of relapse—compared with other medications.16
High-dose gabapentin is better
Mason et al2 randomized 150 outpatients with alcohol dependence to 12 weeks of daily treatment with either gabapentin (900 mg or 1,800 mg) or placebo after at least 3 days of abstinence. All participants received counseling. Drinking quantity and frequency were assessed by gamma-glutamyl transferase testing.
Patients taking gabapentin had better rates of abstinence and cessation of heavy drinking than those taking placebo. During the 12-week study, the 1,800-mg daily dose showed a substantially higher abstinence rate (17%) than either 900 mg (11%) or placebo (4%). Significant dose-related improvements were also found for heavy drinking days, total drinking quantity, and frequency of alcohol withdrawal symptoms that predispose to early relapse, such as poor sleep, cravings, and poor mood. There were also significant linear dose effects on rates of abstinence and nondrinking days at the 24-week posttreatment follow-up.
Gabapentin plus naltrexone is better than naltrexone alone
Anton et al5 examined the efficacy of gabapentin combined with naltrexone during early abstinence. The study randomly assigned 150 people with AUD to one of the following groups:
- 16 weeks of naltrexone (50 mg/day) alone
- 6 weeks of naltrexone (50 mg/day) plus gabapentin (up to 1,200 mg/day), followed by 10 weeks of naltrexone alone
- Placebo.
All participants received medical management.
Over the first 6 weeks, those receiving naltrexone plus gabapentin had a longer interval to heavy drinking than those taking only naltrexone. By week 6, about half of those taking placebo or naltrexone alone had a heavy drinking day, compared with about 35% of those taking naltrexone plus gabapentin. Those receiving the combination also had fewer days of heavy drinking, fewer drinks per drinking day, and better sleep than the other groups. Participants in the naltrexone-alone group were more likely to drink heavily during periods in which they reported poor sleep. No significant group differences were found in measures of mood.
Gabapentin enacarbil is no better than placebo
Falk et al,32 in a 2019 preliminary analysis, examined data from a trial of gabapentin enacarbil, a prodrug formulation of gabapentin. In this 6-month double-blind study, 346 people with moderate AUD at 10 sites were randomized to gabapentin enacarbil extended-release 600 mg twice a day or placebo. All subjects received a computerized behavioral intervention.
No significant differences between groups were found in drinking measures or alcohol cravings, sleep problems, depression, or anxiety symptoms. However, a dose-response analysis found significantly less drinking for higher doses of the drug.
Bottom line: Evidence of benefits mixed but risk low
The efficacy of gabapentin as a treatment for AUD has varied across studies as a function of dosing and formulation. Daily doses have ranged from 600 mg to 1,800 mg, with the highest dose showing advantages in one study for cravings, insomnia, anxiety, dysphoria, and relapse.2 Thus far, gabapentin immediate-release has performed better than gabapentin enacarbil extended-release. All forms of gabapentin have been well-tolerated in AUD trials.
The 2018 American Psychiatric Association guidelines stated that gabapentin had a small positive effect on drinking outcomes, but the harm of treatment was deemed minimal, especially relative to the harms of chronic drinking.33 The guidelines endorse the use of gabapentin in patients with moderate to severe AUD who select gabapentin from the available options, or for those who are nonresponsive to or cannot tolerate naltrexone or acamprosate, as long as no contraindications exist. It was also noted that even small effects may be clinically important, considering the significant morbidity associated with AUD.
POTENTIAL FOR MISUSE
The use of gabapentin has become controversial owing to the growing recognition that it may not be as benign as initially thought.7–9,34 A review of US legislative actions reflects concerns about its misuse.35 In July 2017, Kentucky classified it as a schedule V controlled substance with prescription drug monitoring,35 as did Tennessee in 201836 and Michigan in January 2019.37 Currently, 8 other states (Massachusetts, Minnesota, Nebraska, North Dakota, Ohio, Virginia, Wyoming, and West Virginia) require prescription drug monitoring of gabapentin, and other states are considering it.35
Efforts to understand gabapentin misuse derive largely from people with drug use disorders. A review of postmortem toxicology reports in fatal drug overdoses found gabapentin present in 22%.38 Although it was not necessarily a cause of death, its high rate of detection suggests wide misuse among drug users.
Among a cohort of 503 prescription opioid misusers in Appalachian Kentucky, 15% reported using gabapentin “to get high.” Those who reported misusing gabapentin were 6 times more likely than nonusers to be abusing opioids and benzodiazepines. The main sources of gabapentin were doctors (52%) and dealers (36%). The average cost of gabapentin on the street was less than $1.00 per pill.39
Gabapentin misuse by methadone clinic patients is also reported. Baird et al40 surveyed patients in 6 addiction clinics in the United Kingdom for gabapentin and pregabalin abuse and found that 22% disclosed misusing these medications. Of these, 38% said they did so to enhance the methadone high.
In a review article, Quintero41 also cited enhancement of methadone euphoria and treatment of opioid withdrawal as motivations for misuse. Opioid-dependent gabapentin misusers consumed doses of gabapentin 3 to 20 times higher than clinically recommended and in combination with multiple drugs.4 Such use can cause dissociative and psychedelic effects.
Gabapentin also potentiates the sedative effects of opioids, thus increasing the risk of falls, accidents, and other adverse events.34,35 Risk of opioid-related deaths was increased with coprescription of gabapentin and with moderate to high gabapentin doses.34
Are people with AUD at higher risk of gabapentin abuse?
Despite concerns, patients in clinical trials of gabapentin treatment for AUD were not identified as at high risk for misuse of the drug.2,4,5,16 Further, no such trials reported serious drug-related adverse events resulting in gabapentin discontinuation or side effects that differed from placebo in frequency or severity.2,4,5,16
Clinical laboratory studies also have found no significant interactions between alcohol and gabapentin.42,43 In fact, they showed no influence of gabapentin on the pharmacokinetics of alcohol or on alcohol’s subjective effects. Relative to placebo, gabapentin did not affect blood alcohol levels, the degree of intoxication, sedation, craving, or alcohol self-administration.
Smith et al9 reported estimates that only 1% of the general population misuse gabapentin. Another review concluded that gabapentin is seldom a drug of choice.17 Most patients prescribed gabapentin do not experience cravings or loss of control, which are hallmarks of addiction. Hence, with adequate precautions, the off-label use of gabapentin for AUD is reasonable.
CLINICAL IMPLICATIONS OF GABAPENTIN PRESCRIBING
Overall, evidence for the benefit of gabapentin in AUD is mixed. Subgroups of alcoholic patients, such as those who do not respond to or tolerate standard therapies, may particularly benefit, as may those with comorbid insomnia or neuropathic pain.44 Clinicians should prescribe gabapentin only when it is likely to be helpful and should carefully document its efficacy.2,45
At each visit, an open and honest assessment of the benefits and risks serves to promote shared decision-making regarding initiating, continuing, or discontinuing gabapentin.
For alcohol withdrawal
Before gabapentin is prescribed for alcohol withdrawal, potential benefits (reduction of withdrawal symptoms), side effects (sedation, fatigue), and risks (falls) should be discussed with the patient.46 Patients should also be informed that benzodiazepines are the gold standard for alcohol withdrawal and that gabapentin is not effective for severe withdrawal.46
For relapse prevention
When initiating treatment for relapse prevention, the patient and the prescriber should agree on specific goals (eg, reduction of drinking, anxiety, and insomnia).2,16 Ongoing monitoring is essential and includes assessing and documenting improvement with respect to these goals.
In the AUD studies, gabapentin was well tolerated.16 Frequently observed side effects including headache, insomnia, fatigue, muscle aches, and gastrointestinal distress did not occur at a statistically different rate from placebo. However, patients in studies are selected samples, and their experience may not be generalizable to clinical practice. Thus, it is necessary to exercise caution and check for comorbidities that may put patients at risk of complications.47 Older patients and those on hemodialysis are more susceptible to gabapentin side effects such as sedation, dizziness, ataxia, and mental status changes,34 and prescribers should be alert for signs of toxicity (eg, ataxia, mental status changes).47,48
Gabapentin misuse was not observed in AUD studies,2,4,5,16 but evidence indicates that patients with opioid use disorder, prisoners, and polydrug users are at high risk for gabapentin misuse.39–41 In all cases, clinicians should monitor for red flags that may indicate abuse, such as missed appointments, early refill requests, demands for increased dosage, and simultaneous opiate and benzodiazepine use.49
Acknowledgment: The authors wish to thank Nick Mulligan for his invaluable assistance with formatting and grammar.
- Kranzler HR, Soyka M. Diagnosis and pharmacotherapy of alcohol use disorder: a review. JAMA 2018; 320(8):815–824. doi:10.1001/jama.2018.11406
- Mason BJ, Quello S, Goodell V, Shadan F, Kyle M, Begovic A. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med 2014; 174(1):70–77. doi:10.1001/jamainternmed.2013.11950
- Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res 2009; 33(9):1582–1588. doi:10.1111/j.1530-0277.2009.00986.x
- Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007; 68(11):1691–1700. pmid:18052562
- Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry 2011; 168(7):709–717. doi:10.1176/appi.ajp.2011.10101436
- Mack A. Examination of the evidence for off-label use of gabapentin. J Manag Care Pharm 2003; 9(6):559–568. doi:10.18553/jmcp.2003.9.6.559
- Schifano F. Misuse and abuse of pregabalin and gabapentin: cause for concern? CNS Drugs 2014; 28(6):491–496. doi:10.1007/s40263-014-0164-4
- Goodman CW, Brett AS. Gabapentin and pregabalin for pain—is increased prescribing a cause for concern? N Engl J Med 2017; 377(5):411–414. doi:10.1056/NEJMp1704633
- Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction 2016; 111(7):1160–1174. doi:10.1111/add.13324
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- Soyka M, Müller CA. Pharmacotherapy of alcoholism—an update on approved and off-label medications. Expert Opin Pharmacother 2017; 18(12):1187-1199. doi:10.1080/14656566.2017.1349098
- Zhang M, Gao CX, Ma KT, et al. A meta-analysis of therapeutic efficacy and safety of gabapentin i n the treatment of postherpetic neuralgia from randomized controlled trials. Biomed Res Int 2018; 2018:7474207. doi:10.1155/2018/7474207
- Winkelmann J, Allen RP, Högl B, et al. Treatment of restless legs syndrome: evidence-based review and implications for clinical practice (Revised 2017). Mov Disord 2018; 33(7):1077–1091. doi:10.1002/mds.27260
- Honarmand A, Safavi M, Zare M. Gabapentin: an update of its pharmacological properties and therapeutic use in epilepsy. J Res Med Sci 2011; 16(8):1062–1069. pmid:22279483
- van Hooft JA, Dougherty JJ, Endeman D, Nichols RA, Wadman WJ. Gabapentin inhibits presynaptic Ca(2+) influx and synaptic transmission in rat hippocampus and neocortex. Eur J Pharmacol 2002; 449(3):221–228. doi:10.1016/s0014-2999(02)02044-7
- Mason BJ, Quello S, Shadan F. Gabapentin for the treatment of alcohol use disorder. Expert Opin Investig Drugs 2018; 27(1):113–124. doi:10.1080/13543784.2018.1417383
- Taylor CP. Mechanisms of action of gabapentin. Rev Neurol (Paris) 1997; 153(suppl 1):S39–S45. pmid:9686247
- Agoglia AE, Herman MA. The center of the emotional universe: alcohol, stress, and CRF1 amygdala circuitry. Alcohol 2018; 72:61–73. doi:10.1016/j.alcohol.2018.03.009
- Nevo I, Hamon M. Neurotransmitter and neuromodulatory mechanisms involved in alcohol abuse and alcoholism. Neurochem Int 1995; 26(4):305–336. pmid:7633325
- You C, Vandegrift B, Brodie MS. Ethanol actions on the ventral tegmental area: novel potential targets on reward pathway neurons. Psychopharmacology (Berl) 2018; 235(6):1711–1726. doi:10.1007/s00213-018-4875-y
- Lovinger DM. Presynaptic ethanol actions: potential roles in ethanol seeking. Handb Exp Pharmacol 2018; 248:29–54. doi:10.1007/164_2017_76
- Williams SB, Yorgason JT, Nelson AC, et al. Glutamate transmission to ventral tegmental area GABA neurons is altered by acute and chronic ethanol. Alcohol Clin Exp Res 2018; 42(11):2186–2195. doi:10.1111/acer.13883
- N’Gouemo P. Voltage-sensitive calcium channels in the brain: relevance to alcohol intoxication and withdrawal. Handb Exp Pharmacol 2018; 248:263–280. doi:10.1007/164_2018_93
- Modesto-Lowe V, Huard J, Conrad C. Alcohol withdrawal kindling: is there a role for anticonvulsants? Psychiatry (Edgmont) 2005; 2(5):25–31. pmid:21152146
- Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol 2003; 23(5):514–519. doi:10.1097/01.jcp.0000088905.24613.ad
- Nichols TA, Robert S, Taber DJ, Cluver J. Alcohol withdrawal-related outcomes associated with gabapentin use in an inpatient psychiatric facility. Ment Health Clin 2019 ; 9(1):1–5. doi:10.9740/mhc.2019.01.001
- Bonnet U, Hamzavi-Abedi R, Specka M, Wiltfang J, Lieb B, Scherbaum N. An open trial of gabapentin in acute alcohol withdrawal using an oral loading protocol. Alcohol Alcohol 2010; 45(2):143–145. doi:10.1093/alcalc/agp085
- Stock CJ, Carpenter L, Ying J, Greene T. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother 2013; 47(7–8):961–969. doi:10.1345/aph.1R751
- Blanco-Gandía MC, Rodríguez-Arias M. Pharmacological treatments for opiate and alcohol addiction: a historical perspective of the last 50 years. Eur J Pharmacol 2018; 836:89–101. doi:10.1016/j.ejphar.2018.08.007
- Anton RF, Moak DH, Latham P, et al. Naltrexone combined with either cognitive behavioral or motivational enhancement therapy for alcohol dependence. J Clin Psychopharmacol 2005; 25(4):349–357. pmid:16012278
- Brower KJ, Myra Kim H, Strobbe S, Karam-Hage MA, Consens F, Zucker RA. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res 2008; 32(8):1429–1438. doi:10.1111/j.1530-0277.2008.00706.x
- Falk DE, Ryan ML, Fertig JB, et al; National Institute on Alcohol Abuse and Alcoholism Clinical Investigations Group (NCIG) Study Group. Gabapentin enacarbil extended-release for alcohol use disorder: a randomized, double-blind, placebo-controlled, multisite trial assessing efficacy and safety. Alcohol Clin Exp Res 2019; 43(1):158–169. doi:10.1111/acer.13917
- The American Psychiatric Association. Practice Guideline for the Pharmacological Treatment of Patients with Alcohol Use Disorder. https://psychiatryonline.org/doi/pdf/10.1176/appi.books.9781615371969. Accessed October 10, 2019.
- Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med 2017; 14(10):e1002396. doi:10.1371/journal.pmed.1002396
- Peckham AM, Ananickal MJ, Sclar DA. Gabapentin use, abuse, and the US opioid epidemic: the case for reclassification as a controlled substance and the need for pharmacovigilance. Risk Manag Healthc Policy 2018; 11:109–116. doi:10.2147/RMHP.S168504
- Tennessee Pharmacists Association. Advocacy alert: end of session summary. www.tnpharm.org/news/news-posts-pages/advocacy-alert-4-30-18/? Accessed October 10, 2019.
- Michigan.gov. Gabapentin scheduled as controlled substance to help with state’s opioid epidemic. www.michigan.gov/som/0,4669,7-192-47796-487050--,00.html. Accessed October 10, 2019.
- Slavova S, Miller A, Bunn TL, et al. Prevalence of gabapentin in drug overdose postmortem toxicology testing results. Drug Alcohol Depend 2018; 186:80–85. doi:10.1016/j.drugalcdep.2018.01.018
- Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry 2015; 172(5):487–488. doi:10.1176/appi.ajp.2014.14101272
- Baird CR, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res 2014; 20(3):115–118. doi:10.1159/000355268
- Quintero GC. Review about gabapentin misuse, interactions, contraindications and side effects. J Exp Pharmacol 2017; 9:13–21. doi:10.2147/JEP.S124391
- Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend 2006; 83(1):25–32. doi:10.1016/j.drugalcdep.2005.10.008
- Myrick H, Anton R, Voronin K, Wang W, Henderson S. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res 2007; 31(2):221–227. doi:10.1111/j.1530-0277.2006.00299.x
- Tzellos TG, Papazisis G, Toulis KA, Sardeli CH, Kouvelas D. A2delta ligands gabapentin and pregabalin: future implications in daily clinical practice. Hippokratia 2010; 14(2):71–75. pmid:20596259
- Morrison EE, Sandilands EA, Webb DJ. Gabapentin and pregabalin: do the benefits outweigh the harms? J R Coll Physicians Edinb 2017; 47(4):310–313. doi:10.4997/JRCPE.2017.402
- Leung JG, Rakocevic DB, Allen ND, et al. Use of a gabapentin protocol for the management of alcohol withdrawal: a preliminary experience expanding from the consultation-liaison psychiatry service. Psychosomatics 2018; 59(5):496–505. doi:10.1016/j.psym.2018.03.002
- Fleet JL, Dixon SN, Kuwornu PJ, et al. Gabapentin dose and the 30-day risk of altered mental status in older adults: a retrospective population-based study. PLoS One 2018; 13(3):e0193134. doi:10.1371/journal.pone.0193134
- Chiappini S, Schifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs 2016; 30(7):647–654. doi:10.1007/s40263-016-0359-y
- Modesto-Lowe V, Chaplin M, Sinha S, Woodard K. Universal precautions to reduce stimulant misuse in treating adult ADHD. Cleve Clin J Med 2015; 82(8):506–512. doi:10.3949/ccjm.82a.14131
- Kranzler HR, Soyka M. Diagnosis and pharmacotherapy of alcohol use disorder: a review. JAMA 2018; 320(8):815–824. doi:10.1001/jama.2018.11406
- Mason BJ, Quello S, Goodell V, Shadan F, Kyle M, Begovic A. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med 2014; 174(1):70–77. doi:10.1001/jamainternmed.2013.11950
- Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res 2009; 33(9):1582–1588. doi:10.1111/j.1530-0277.2009.00986.x
- Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007; 68(11):1691–1700. pmid:18052562
- Anton RF, Myrick H, Wright TM, et al. Gabapentin combined with naltrexone for the treatment of alcohol dependence. Am J Psychiatry 2011; 168(7):709–717. doi:10.1176/appi.ajp.2011.10101436
- Mack A. Examination of the evidence for off-label use of gabapentin. J Manag Care Pharm 2003; 9(6):559–568. doi:10.18553/jmcp.2003.9.6.559
- Schifano F. Misuse and abuse of pregabalin and gabapentin: cause for concern? CNS Drugs 2014; 28(6):491–496. doi:10.1007/s40263-014-0164-4
- Goodman CW, Brett AS. Gabapentin and pregabalin for pain—is increased prescribing a cause for concern? N Engl J Med 2017; 377(5):411–414. doi:10.1056/NEJMp1704633
- Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction 2016; 111(7):1160–1174. doi:10.1111/add.13324
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
- Soyka M, Müller CA. Pharmacotherapy of alcoholism—an update on approved and off-label medications. Expert Opin Pharmacother 2017; 18(12):1187-1199. doi:10.1080/14656566.2017.1349098
- Zhang M, Gao CX, Ma KT, et al. A meta-analysis of therapeutic efficacy and safety of gabapentin i n the treatment of postherpetic neuralgia from randomized controlled trials. Biomed Res Int 2018; 2018:7474207. doi:10.1155/2018/7474207
- Winkelmann J, Allen RP, Högl B, et al. Treatment of restless legs syndrome: evidence-based review and implications for clinical practice (Revised 2017). Mov Disord 2018; 33(7):1077–1091. doi:10.1002/mds.27260
- Honarmand A, Safavi M, Zare M. Gabapentin: an update of its pharmacological properties and therapeutic use in epilepsy. J Res Med Sci 2011; 16(8):1062–1069. pmid:22279483
- van Hooft JA, Dougherty JJ, Endeman D, Nichols RA, Wadman WJ. Gabapentin inhibits presynaptic Ca(2+) influx and synaptic transmission in rat hippocampus and neocortex. Eur J Pharmacol 2002; 449(3):221–228. doi:10.1016/s0014-2999(02)02044-7
- Mason BJ, Quello S, Shadan F. Gabapentin for the treatment of alcohol use disorder. Expert Opin Investig Drugs 2018; 27(1):113–124. doi:10.1080/13543784.2018.1417383
- Taylor CP. Mechanisms of action of gabapentin. Rev Neurol (Paris) 1997; 153(suppl 1):S39–S45. pmid:9686247
- Agoglia AE, Herman MA. The center of the emotional universe: alcohol, stress, and CRF1 amygdala circuitry. Alcohol 2018; 72:61–73. doi:10.1016/j.alcohol.2018.03.009
- Nevo I, Hamon M. Neurotransmitter and neuromodulatory mechanisms involved in alcohol abuse and alcoholism. Neurochem Int 1995; 26(4):305–336. pmid:7633325
- You C, Vandegrift B, Brodie MS. Ethanol actions on the ventral tegmental area: novel potential targets on reward pathway neurons. Psychopharmacology (Berl) 2018; 235(6):1711–1726. doi:10.1007/s00213-018-4875-y
- Lovinger DM. Presynaptic ethanol actions: potential roles in ethanol seeking. Handb Exp Pharmacol 2018; 248:29–54. doi:10.1007/164_2017_76
- Williams SB, Yorgason JT, Nelson AC, et al. Glutamate transmission to ventral tegmental area GABA neurons is altered by acute and chronic ethanol. Alcohol Clin Exp Res 2018; 42(11):2186–2195. doi:10.1111/acer.13883
- N’Gouemo P. Voltage-sensitive calcium channels in the brain: relevance to alcohol intoxication and withdrawal. Handb Exp Pharmacol 2018; 248:263–280. doi:10.1007/164_2018_93
- Modesto-Lowe V, Huard J, Conrad C. Alcohol withdrawal kindling: is there a role for anticonvulsants? Psychiatry (Edgmont) 2005; 2(5):25–31. pmid:21152146
- Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol 2003; 23(5):514–519. doi:10.1097/01.jcp.0000088905.24613.ad
- Nichols TA, Robert S, Taber DJ, Cluver J. Alcohol withdrawal-related outcomes associated with gabapentin use in an inpatient psychiatric facility. Ment Health Clin 2019 ; 9(1):1–5. doi:10.9740/mhc.2019.01.001
- Bonnet U, Hamzavi-Abedi R, Specka M, Wiltfang J, Lieb B, Scherbaum N. An open trial of gabapentin in acute alcohol withdrawal using an oral loading protocol. Alcohol Alcohol 2010; 45(2):143–145. doi:10.1093/alcalc/agp085
- Stock CJ, Carpenter L, Ying J, Greene T. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother 2013; 47(7–8):961–969. doi:10.1345/aph.1R751
- Blanco-Gandía MC, Rodríguez-Arias M. Pharmacological treatments for opiate and alcohol addiction: a historical perspective of the last 50 years. Eur J Pharmacol 2018; 836:89–101. doi:10.1016/j.ejphar.2018.08.007
- Anton RF, Moak DH, Latham P, et al. Naltrexone combined with either cognitive behavioral or motivational enhancement therapy for alcohol dependence. J Clin Psychopharmacol 2005; 25(4):349–357. pmid:16012278
- Brower KJ, Myra Kim H, Strobbe S, Karam-Hage MA, Consens F, Zucker RA. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res 2008; 32(8):1429–1438. doi:10.1111/j.1530-0277.2008.00706.x
- Falk DE, Ryan ML, Fertig JB, et al; National Institute on Alcohol Abuse and Alcoholism Clinical Investigations Group (NCIG) Study Group. Gabapentin enacarbil extended-release for alcohol use disorder: a randomized, double-blind, placebo-controlled, multisite trial assessing efficacy and safety. Alcohol Clin Exp Res 2019; 43(1):158–169. doi:10.1111/acer.13917
- The American Psychiatric Association. Practice Guideline for the Pharmacological Treatment of Patients with Alcohol Use Disorder. https://psychiatryonline.org/doi/pdf/10.1176/appi.books.9781615371969. Accessed October 10, 2019.
- Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med 2017; 14(10):e1002396. doi:10.1371/journal.pmed.1002396
- Peckham AM, Ananickal MJ, Sclar DA. Gabapentin use, abuse, and the US opioid epidemic: the case for reclassification as a controlled substance and the need for pharmacovigilance. Risk Manag Healthc Policy 2018; 11:109–116. doi:10.2147/RMHP.S168504
- Tennessee Pharmacists Association. Advocacy alert: end of session summary. www.tnpharm.org/news/news-posts-pages/advocacy-alert-4-30-18/? Accessed October 10, 2019.
- Michigan.gov. Gabapentin scheduled as controlled substance to help with state’s opioid epidemic. www.michigan.gov/som/0,4669,7-192-47796-487050--,00.html. Accessed October 10, 2019.
- Slavova S, Miller A, Bunn TL, et al. Prevalence of gabapentin in drug overdose postmortem toxicology testing results. Drug Alcohol Depend 2018; 186:80–85. doi:10.1016/j.drugalcdep.2018.01.018
- Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry 2015; 172(5):487–488. doi:10.1176/appi.ajp.2014.14101272
- Baird CR, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res 2014; 20(3):115–118. doi:10.1159/000355268
- Quintero GC. Review about gabapentin misuse, interactions, contraindications and side effects. J Exp Pharmacol 2017; 9:13–21. doi:10.2147/JEP.S124391
- Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend 2006; 83(1):25–32. doi:10.1016/j.drugalcdep.2005.10.008
- Myrick H, Anton R, Voronin K, Wang W, Henderson S. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res 2007; 31(2):221–227. doi:10.1111/j.1530-0277.2006.00299.x
- Tzellos TG, Papazisis G, Toulis KA, Sardeli CH, Kouvelas D. A2delta ligands gabapentin and pregabalin: future implications in daily clinical practice. Hippokratia 2010; 14(2):71–75. pmid:20596259
- Morrison EE, Sandilands EA, Webb DJ. Gabapentin and pregabalin: do the benefits outweigh the harms? J R Coll Physicians Edinb 2017; 47(4):310–313. doi:10.4997/JRCPE.2017.402
- Leung JG, Rakocevic DB, Allen ND, et al. Use of a gabapentin protocol for the management of alcohol withdrawal: a preliminary experience expanding from the consultation-liaison psychiatry service. Psychosomatics 2018; 59(5):496–505. doi:10.1016/j.psym.2018.03.002
- Fleet JL, Dixon SN, Kuwornu PJ, et al. Gabapentin dose and the 30-day risk of altered mental status in older adults: a retrospective population-based study. PLoS One 2018; 13(3):e0193134. doi:10.1371/journal.pone.0193134
- Chiappini S, Schifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs 2016; 30(7):647–654. doi:10.1007/s40263-016-0359-y
- Modesto-Lowe V, Chaplin M, Sinha S, Woodard K. Universal precautions to reduce stimulant misuse in treating adult ADHD. Cleve Clin J Med 2015; 82(8):506–512. doi:10.3949/ccjm.82a.14131
KEY POINTS
- Gabapentin has been shown to be safe and effective for mild alcohol withdrawal but is not appropriate as monotherapy for severe withdrawal owing to risk of seizures.
- During early abstinence, gabapentin may improve sleep, cravings, and mood—factors associated with relapse.
- Gabapentin is being used recreationally to achieve or enhance euphoria, but its misuse potential appears to be low when taken at therapeutic doses by patients without a history of drug abuse.

Off-label and oft-prescribed

In no way do I minimize the value of the imprimatur of FDA approval stating a drug, after appropriate preclinical and clinical studies, is deemed safe and effective. Whatever the agency’s shortcomings, the story of thalidomide (a drug never approved by the FDA) gives credence to the value of having a robust approval process. Arguments will likely continue forever as to whether the agency errs on the side of being too permissive or too restrictive in its approval process.
Nonetheless, I believe there are valid clinical reasons why we should continue to prescribe FDA-approved medications for nonapproved indications. In my practice, I treat some conditions that are sufficiently uncommon or heterogeneous in expression that large-scale clinical trials are logistically hard to carry out or deemed financially unviable by the corporate sponsor, even though clinical experience has informed us of a reasonable likelihood of efficacy. Sometimes drugs have “failed” in clinical trials, but experience and post hoc subset analysis of data have indicated a likely positive response in certain patients.
Although a drug that has been FDA approved has passed significant safety testing, the patients exposed to the drug when it was evaluated for treating a certain disease may be strikingly different from patients who have a different disease—the age, sex, comorbidities, and coprescribed medications may all differ significantly in the population of patients with the “off-label” disorder. Hence, appropriate caution is warranted, and if relevant, this should be explained to patients before giving them the medication.
In this issue of the Journal, 2 papers address the use of medications in an “off-label” manner. Schneider and colleagues discuss several frequent clinical uses of tricyclic antidepressants for reasons other than depression, and Modesto-Lowe and colleagues review the more controversial use of gabapentin in patients with alcohol use disorder. The hoped-for benefits in both circumstances are symptomatic, and both benefits and side effects are dose-related in ways not necessarily coinciding with those in the FDA-labeled indications.
My experience in using tricyclics as adjunctive treatment for fibromyalgia is that patients are quite sensitive to some of the side effects of the drugs (eg, oral dryness and fatigue), even in low doses. Moreover, we should expect only modest benefits, which should be explicitly described to the patient: improved quality of sleep with resultant decreased fatigue (while we watch closely for worsened fatigue from too-high dosing) and a modest reduction in pain over time as part of a multimodality treatment plan. I often find that practitioners who are less familiar with the use of these medications in this setting tend to start at lowish (but higher than often tolerated) doses, have patients take the medication too close to bedtime (resulting in some morning hangover sensation), fail to discuss the timing and degree of expected pain relief, don’t titrate the dose over time, and are not aware of the different responses that patients may experience with different medications within the same class. As with all prescribed medications, the benefits and ill effects must be frequently assessed, and particularly with these medications, one must be willing to discontinue them if appropriate outcomes are not achieved.
“Off-label” should not imply off the table as a therapeutic option. But it is incumbent on us to devote sufficient time to explain to each patient the anticipated side effects and hoped-for benefits, particularly since in most cases, we and our patients cannot refer to the results of definitive phase 3 clinical trials or patient online information sites that are totally relevant, reliable, data supported, and FDA reviewed.
In no way do I minimize the value of the imprimatur of FDA approval stating a drug, after appropriate preclinical and clinical studies, is deemed safe and effective. Whatever the agency’s shortcomings, the story of thalidomide (a drug never approved by the FDA) gives credence to the value of having a robust approval process. Arguments will likely continue forever as to whether the agency errs on the side of being too permissive or too restrictive in its approval process.
Nonetheless, I believe there are valid clinical reasons why we should continue to prescribe FDA-approved medications for nonapproved indications. In my practice, I treat some conditions that are sufficiently uncommon or heterogeneous in expression that large-scale clinical trials are logistically hard to carry out or deemed financially unviable by the corporate sponsor, even though clinical experience has informed us of a reasonable likelihood of efficacy. Sometimes drugs have “failed” in clinical trials, but experience and post hoc subset analysis of data have indicated a likely positive response in certain patients.
Although a drug that has been FDA approved has passed significant safety testing, the patients exposed to the drug when it was evaluated for treating a certain disease may be strikingly different from patients who have a different disease—the age, sex, comorbidities, and coprescribed medications may all differ significantly in the population of patients with the “off-label” disorder. Hence, appropriate caution is warranted, and if relevant, this should be explained to patients before giving them the medication.
In this issue of the Journal, 2 papers address the use of medications in an “off-label” manner. Schneider and colleagues discuss several frequent clinical uses of tricyclic antidepressants for reasons other than depression, and Modesto-Lowe and colleagues review the more controversial use of gabapentin in patients with alcohol use disorder. The hoped-for benefits in both circumstances are symptomatic, and both benefits and side effects are dose-related in ways not necessarily coinciding with those in the FDA-labeled indications.
My experience in using tricyclics as adjunctive treatment for fibromyalgia is that patients are quite sensitive to some of the side effects of the drugs (eg, oral dryness and fatigue), even in low doses. Moreover, we should expect only modest benefits, which should be explicitly described to the patient: improved quality of sleep with resultant decreased fatigue (while we watch closely for worsened fatigue from too-high dosing) and a modest reduction in pain over time as part of a multimodality treatment plan. I often find that practitioners who are less familiar with the use of these medications in this setting tend to start at lowish (but higher than often tolerated) doses, have patients take the medication too close to bedtime (resulting in some morning hangover sensation), fail to discuss the timing and degree of expected pain relief, don’t titrate the dose over time, and are not aware of the different responses that patients may experience with different medications within the same class. As with all prescribed medications, the benefits and ill effects must be frequently assessed, and particularly with these medications, one must be willing to discontinue them if appropriate outcomes are not achieved.
“Off-label” should not imply off the table as a therapeutic option. But it is incumbent on us to devote sufficient time to explain to each patient the anticipated side effects and hoped-for benefits, particularly since in most cases, we and our patients cannot refer to the results of definitive phase 3 clinical trials or patient online information sites that are totally relevant, reliable, data supported, and FDA reviewed.
In no way do I minimize the value of the imprimatur of FDA approval stating a drug, after appropriate preclinical and clinical studies, is deemed safe and effective. Whatever the agency’s shortcomings, the story of thalidomide (a drug never approved by the FDA) gives credence to the value of having a robust approval process. Arguments will likely continue forever as to whether the agency errs on the side of being too permissive or too restrictive in its approval process.
Nonetheless, I believe there are valid clinical reasons why we should continue to prescribe FDA-approved medications for nonapproved indications. In my practice, I treat some conditions that are sufficiently uncommon or heterogeneous in expression that large-scale clinical trials are logistically hard to carry out or deemed financially unviable by the corporate sponsor, even though clinical experience has informed us of a reasonable likelihood of efficacy. Sometimes drugs have “failed” in clinical trials, but experience and post hoc subset analysis of data have indicated a likely positive response in certain patients.
Although a drug that has been FDA approved has passed significant safety testing, the patients exposed to the drug when it was evaluated for treating a certain disease may be strikingly different from patients who have a different disease—the age, sex, comorbidities, and coprescribed medications may all differ significantly in the population of patients with the “off-label” disorder. Hence, appropriate caution is warranted, and if relevant, this should be explained to patients before giving them the medication.
In this issue of the Journal, 2 papers address the use of medications in an “off-label” manner. Schneider and colleagues discuss several frequent clinical uses of tricyclic antidepressants for reasons other than depression, and Modesto-Lowe and colleagues review the more controversial use of gabapentin in patients with alcohol use disorder. The hoped-for benefits in both circumstances are symptomatic, and both benefits and side effects are dose-related in ways not necessarily coinciding with those in the FDA-labeled indications.
My experience in using tricyclics as adjunctive treatment for fibromyalgia is that patients are quite sensitive to some of the side effects of the drugs (eg, oral dryness and fatigue), even in low doses. Moreover, we should expect only modest benefits, which should be explicitly described to the patient: improved quality of sleep with resultant decreased fatigue (while we watch closely for worsened fatigue from too-high dosing) and a modest reduction in pain over time as part of a multimodality treatment plan. I often find that practitioners who are less familiar with the use of these medications in this setting tend to start at lowish (but higher than often tolerated) doses, have patients take the medication too close to bedtime (resulting in some morning hangover sensation), fail to discuss the timing and degree of expected pain relief, don’t titrate the dose over time, and are not aware of the different responses that patients may experience with different medications within the same class. As with all prescribed medications, the benefits and ill effects must be frequently assessed, and particularly with these medications, one must be willing to discontinue them if appropriate outcomes are not achieved.
“Off-label” should not imply off the table as a therapeutic option. But it is incumbent on us to devote sufficient time to explain to each patient the anticipated side effects and hoped-for benefits, particularly since in most cases, we and our patients cannot refer to the results of definitive phase 3 clinical trials or patient online information sites that are totally relevant, reliable, data supported, and FDA reviewed.
Desquamating pustular rash
A 66-year-old man presented with burning pain and erythema over the left axilla, and pustules that had ruptured and crusted over. The rash also involved the right axilla, trunk, abdomen, and face.
He said the symptoms had developed 3 days after starting to use ciprofloxacin eye drops for eye redness and purulent discharge that had been diagnosed as bacterial conjunctivitis. He was taking no other new medications. He was afebrile.


The ciprofloxacin drops were stopped. The skin lesions were treated with emollients, topical steroids, and topical mupirocin. Improvement was noted 3 days into the hospitalization, as the lesions started to crust over and dry up and no new lesions were forming. The conjunctivitis improved with topical bacitracin ointment and prednisolone drops.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
The differential diagnosis of drug-related acute generalized exanthematous pustulosis includes Stevens-Johnson syndrome, pustular psoriasis, folliculitis, and varicella infection. The characteristic features of the rash and lesions and the temporal relationship between the start of ciprofloxacin eye drops and the development of symptoms, combined with rapid resolution of symptoms within days after discontinuing the drops, accompanied by skin biopsy study showing diffuse spongiosis with scattered eosinophils and a subcorneal pustule, confirmed the diagnosis of AGEP.
Key features of AGEP include numerous small, sterile, nonfollicular pustules on an erythematous background with associated fever and sometimes neutrophilia and eosinophilia.1 It usually begins on the face or in the intertriginous areas and then spreads to the trunk and lower limbs with rare mucosal involvement.1 It can be associated with viral infections, but most reported cases are related to drug reactions.2
Our patient’s case was unusual because AGEP triggered by topical medications is rarely reported, especially with ophthalmic medications.3 Drugs most commonly implicated are antibiotics including penicillins, sulfonamides, and quinolones, but other drugs such as terbinafine, diltiazem, and hydroxychloroquine have also been associated.2
AGEP may present with extensive skin desquamation, as in our patient, sometimes with bullae formation and skin sloughing manifesting as AGEP with overlapping toxic epidermal necrolysis.4
Diagnosis entails a careful review of medications, attention to lesion morphology, compatible disease course, and a high index of suspicion. Treatment is supportive and consists of stopping the offending agent, wound care, and antipyretics. Evidence for the use of steroids is weak.5
TAKE-HOME POINTS
AGEP should be considered in sudden-onset pustular desquamating erythematous rash related to use of a new medication. It is important to be aware that topical and ophthalmic medications are possible triggers. A thorough medication review should be done. Antibiotics are the most commonly implicated medications. An alternative medication should be tried. Treatment is supportive, as the condition is usually self-limiting once the offending medication is discontinued. Rarely, extensive desquamation and bullae formation may occur, which may be a manifestation of overlap features with toxic epidermal necrolysis.
- Speeckaert MM, Speeckaert R, Lambert J, Brochez L. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts. Eur J Dermatol 2010; 20(4):425–433. doi:10.1684/ejd.2010.0932
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case–control study (EuroSCAR). Br J Dermatol 2007; 157(5):989–996. doi:10.1111/j.1365-2133.2007.08156.x
- Beltran C, Vergier B, Doutre MS, Beylot C, Beylot-Barry M. Acute generalized exanthematous pustulosis induced by topical application of Algipan. Ann Dermatol Venereol 2009; 136(10):709–712. French. doi:10.1016/j.annder.2008.10.042
- Peermohamed S, Haber RM. Acute generalized exanthematous pustulosis simulating toxic epidermal necrolysis: a case report and review of the literature. Arch Dermatol 2011; 147(6):697–701. doi:10.1001/archdermatol.2011.147
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci 2016; 17(8). pii:E1214. doi:10.3390/ijms17081214
A 66-year-old man presented with burning pain and erythema over the left axilla, and pustules that had ruptured and crusted over. The rash also involved the right axilla, trunk, abdomen, and face.
He said the symptoms had developed 3 days after starting to use ciprofloxacin eye drops for eye redness and purulent discharge that had been diagnosed as bacterial conjunctivitis. He was taking no other new medications. He was afebrile.
The ciprofloxacin drops were stopped. The skin lesions were treated with emollients, topical steroids, and topical mupirocin. Improvement was noted 3 days into the hospitalization, as the lesions started to crust over and dry up and no new lesions were forming. The conjunctivitis improved with topical bacitracin ointment and prednisolone drops.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
The differential diagnosis of drug-related acute generalized exanthematous pustulosis includes Stevens-Johnson syndrome, pustular psoriasis, folliculitis, and varicella infection. The characteristic features of the rash and lesions and the temporal relationship between the start of ciprofloxacin eye drops and the development of symptoms, combined with rapid resolution of symptoms within days after discontinuing the drops, accompanied by skin biopsy study showing diffuse spongiosis with scattered eosinophils and a subcorneal pustule, confirmed the diagnosis of AGEP.
Key features of AGEP include numerous small, sterile, nonfollicular pustules on an erythematous background with associated fever and sometimes neutrophilia and eosinophilia.1 It usually begins on the face or in the intertriginous areas and then spreads to the trunk and lower limbs with rare mucosal involvement.1 It can be associated with viral infections, but most reported cases are related to drug reactions.2
Our patient’s case was unusual because AGEP triggered by topical medications is rarely reported, especially with ophthalmic medications.3 Drugs most commonly implicated are antibiotics including penicillins, sulfonamides, and quinolones, but other drugs such as terbinafine, diltiazem, and hydroxychloroquine have also been associated.2
AGEP may present with extensive skin desquamation, as in our patient, sometimes with bullae formation and skin sloughing manifesting as AGEP with overlapping toxic epidermal necrolysis.4
Diagnosis entails a careful review of medications, attention to lesion morphology, compatible disease course, and a high index of suspicion. Treatment is supportive and consists of stopping the offending agent, wound care, and antipyretics. Evidence for the use of steroids is weak.5
TAKE-HOME POINTS
AGEP should be considered in sudden-onset pustular desquamating erythematous rash related to use of a new medication. It is important to be aware that topical and ophthalmic medications are possible triggers. A thorough medication review should be done. Antibiotics are the most commonly implicated medications. An alternative medication should be tried. Treatment is supportive, as the condition is usually self-limiting once the offending medication is discontinued. Rarely, extensive desquamation and bullae formation may occur, which may be a manifestation of overlap features with toxic epidermal necrolysis.
A 66-year-old man presented with burning pain and erythema over the left axilla, and pustules that had ruptured and crusted over. The rash also involved the right axilla, trunk, abdomen, and face.
He said the symptoms had developed 3 days after starting to use ciprofloxacin eye drops for eye redness and purulent discharge that had been diagnosed as bacterial conjunctivitis. He was taking no other new medications. He was afebrile.
The ciprofloxacin drops were stopped. The skin lesions were treated with emollients, topical steroids, and topical mupirocin. Improvement was noted 3 days into the hospitalization, as the lesions started to crust over and dry up and no new lesions were forming. The conjunctivitis improved with topical bacitracin ointment and prednisolone drops.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
The differential diagnosis of drug-related acute generalized exanthematous pustulosis includes Stevens-Johnson syndrome, pustular psoriasis, folliculitis, and varicella infection. The characteristic features of the rash and lesions and the temporal relationship between the start of ciprofloxacin eye drops and the development of symptoms, combined with rapid resolution of symptoms within days after discontinuing the drops, accompanied by skin biopsy study showing diffuse spongiosis with scattered eosinophils and a subcorneal pustule, confirmed the diagnosis of AGEP.
Key features of AGEP include numerous small, sterile, nonfollicular pustules on an erythematous background with associated fever and sometimes neutrophilia and eosinophilia.1 It usually begins on the face or in the intertriginous areas and then spreads to the trunk and lower limbs with rare mucosal involvement.1 It can be associated with viral infections, but most reported cases are related to drug reactions.2
Our patient’s case was unusual because AGEP triggered by topical medications is rarely reported, especially with ophthalmic medications.3 Drugs most commonly implicated are antibiotics including penicillins, sulfonamides, and quinolones, but other drugs such as terbinafine, diltiazem, and hydroxychloroquine have also been associated.2
AGEP may present with extensive skin desquamation, as in our patient, sometimes with bullae formation and skin sloughing manifesting as AGEP with overlapping toxic epidermal necrolysis.4
Diagnosis entails a careful review of medications, attention to lesion morphology, compatible disease course, and a high index of suspicion. Treatment is supportive and consists of stopping the offending agent, wound care, and antipyretics. Evidence for the use of steroids is weak.5
TAKE-HOME POINTS
AGEP should be considered in sudden-onset pustular desquamating erythematous rash related to use of a new medication. It is important to be aware that topical and ophthalmic medications are possible triggers. A thorough medication review should be done. Antibiotics are the most commonly implicated medications. An alternative medication should be tried. Treatment is supportive, as the condition is usually self-limiting once the offending medication is discontinued. Rarely, extensive desquamation and bullae formation may occur, which may be a manifestation of overlap features with toxic epidermal necrolysis.
- Speeckaert MM, Speeckaert R, Lambert J, Brochez L. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts. Eur J Dermatol 2010; 20(4):425–433. doi:10.1684/ejd.2010.0932
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case–control study (EuroSCAR). Br J Dermatol 2007; 157(5):989–996. doi:10.1111/j.1365-2133.2007.08156.x
- Beltran C, Vergier B, Doutre MS, Beylot C, Beylot-Barry M. Acute generalized exanthematous pustulosis induced by topical application of Algipan. Ann Dermatol Venereol 2009; 136(10):709–712. French. doi:10.1016/j.annder.2008.10.042
- Peermohamed S, Haber RM. Acute generalized exanthematous pustulosis simulating toxic epidermal necrolysis: a case report and review of the literature. Arch Dermatol 2011; 147(6):697–701. doi:10.1001/archdermatol.2011.147
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci 2016; 17(8). pii:E1214. doi:10.3390/ijms17081214
- Speeckaert MM, Speeckaert R, Lambert J, Brochez L. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts. Eur J Dermatol 2010; 20(4):425–433. doi:10.1684/ejd.2010.0932
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case–control study (EuroSCAR). Br J Dermatol 2007; 157(5):989–996. doi:10.1111/j.1365-2133.2007.08156.x
- Beltran C, Vergier B, Doutre MS, Beylot C, Beylot-Barry M. Acute generalized exanthematous pustulosis induced by topical application of Algipan. Ann Dermatol Venereol 2009; 136(10):709–712. French. doi:10.1016/j.annder.2008.10.042
- Peermohamed S, Haber RM. Acute generalized exanthematous pustulosis simulating toxic epidermal necrolysis: a case report and review of the literature. Arch Dermatol 2011; 147(6):697–701. doi:10.1001/archdermatol.2011.147
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci 2016; 17(8). pii:E1214. doi:10.3390/ijms17081214

How to respond to flu vaccine doubters
The benefits of influenza vaccination are clear to those in the medical community. Yet misinformation and unfounded fears continue to discourage some people from getting a flu shot. During the 2018–2019 influenza season, only 45% of US adults and 63% of children were vaccinated.1

‘IT DOESN’T WORK FOR MANY PEOPLE’
Multiple studies have shown that the flu vaccine prevents millions of flu cases and flu-related doctor’s visits each year. During the 2016–2017 flu season, flu vaccine prevented an estimated 5.3 million influenza cases, 2.6 million influenza-associated medical visits, and 85,000 influenza-associated hospitalizations.2
Several viral and host factors affect vaccine effectiveness. In seasons when the vaccine viruses have matched circulating strains, flu vaccine has been shown to reduce the following:
- The risk of having to go to the doctor with flu by 40% to 60%
- Children’s risk of flu-related death and intensive care unit (ICU) admission by 74%
- The risk in adults of flu-associated hospitalizations by 40% and ICU admission by 82%
- The rate of cardiac events in people with heart disease
- Hospitalizations in people with diabetes or underlying chronic lung disease.3
In people hospitalized with influenza despite receiving the flu vaccine for the season, studies have shown that receiving the flu vaccine shortens the average duration of hospitalization, reduces the chance of ICU admission by 59%, shortens the duration of ICU stay by 4 days, and reduces deaths.3

‘IT TARGETS THE WRONG VIRUS’
Selecting an effective influenza vaccine is a challenge. Every year, the World Health Organization and the CDC decide on the influenza strains expected to circulate in the upcoming flu season in the Northern Hemisphere, based on data for circulating strains in the Southern Hemisphere. This decision takes place about 7 months before the expected onset of the flu season. Flu viruses may mutate between the time the decision is made and the time the vaccine is administered (as well as after the flu season starts). Also, vaccine production in eggs needs time, which is why this decision must be made several months ahead of the flu season.
Vaccine effectiveness varies by virus serotype. Vaccines are typically less effective against influenza A H3N2 viruses than against influenza A H1N1 and influenza B viruses. Effectiveness also varies from season to season depending on how close the vaccine serotypes match the circulating serotypes, but some effectiveness is retained even in seasons when some of the serotypes don’t match circulating viruses. For example, in the 2017–2018 season, when the influenza A H3N2 vaccine serotype did not match the circulating serotype, the overall effectiveness in preventing medically attended, laboratory-confirmed influenza virus infection was 36%.5
A universal flu vaccine that does not need to be updated annually is the ultimate solution, but according to the National Institute of Allergy and Infectious Diseases, such a vaccine is likely several years away.6
‘IT MAKES PEOPLE SICK’
Pain at the injection site of a flu shot occurs in 10% to 65% of people, lasts less than 2 days, and does not usually interfere with daily activities.7
Systemic symptoms such as fever, malaise, and myalgia may occur in people who have had no previous exposure to the influenza virus antigens in the vaccine, particularly in children. In adults, the frequency of systemic symptoms after the flu shot is similar to that with placebo.
The Vaccine Adverse Event Reporting System, which has been capturing data since 1990, shows that the influenza vaccine accounted for 5.7% of people who developed malaise after receiving any vaccine.8
The injectable inactivated influenza vaccine cannot biologically cause an influenza virus-related illness, since the inactivated vaccine viruses can elicit a protective immune response but cannot replicate. The nasal live-attenuated flu vaccine can in theory cause acute illness in the person receiving it, but because it is cold-adapted, it multiplies only in the colder environment of the nasal epithelium, not in the lower airways where the temperature is higher. Consequently, the vaccine virus triggers immunity by multiplying in the nose, but doesn’t infect the lungs.
From 10% to 50% of people who receive the nasal live-attenuated vaccine develop runny nose, wheezing, headache, vomiting, muscle aches, fever, sore throat, or cough shortly after receiving the vaccine, but these symptoms are usually mild and short-lived.
The most common reactions people have to flu vaccines are considerably less severe than the symptoms caused by actual flu illness.
While influenza illness results in natural immunity to the specific viral serotype causing it, this illness results in hospitalization in 2% and is fatal in 0.16% of people. Influenza vaccine results in immunity to the serotypes included in the vaccine, and multiple studies have not found a causal relationship between vaccination and death.9
‘IT CAUSES GUILLAIN-BARRÉ SYNDROME’
In the United States, 3,000 to 6,000 people per year develop Guillain-Barré syndrome, or 1 to 2 of every 100,000, which translates to 80 to 160 cases per week.10 While the exact cause of Guillain-Barré syndrome is unknown, about two-thirds of people have an acute diarrheal or respiratory illness within 3 months before the onset of symptoms. In 1976, the estimated attributable risk of influenza vaccine-related Guillain-Barré syndrome in the US adult population was 1 case per 100,000 in the 6 weeks after vaccination.11 Studies in subsequent influenza seasons have not shown similar findings.12 In fact, one study showed that the risk of developing Guillain-Barré syndrome was 15 times higher after influenza illness than after influenza vaccination.13
Since 5% to 15% of the US population develop symptomatic influenza annually,14 the decision to vaccinate with respect to the risk of Guillain-Barré syndrome should be obvious: vaccinate. The correct question to ask before influenza vaccination should be, “Have you previously developed Guillain-Barré syndrome within 6 weeks after receiving the flu vaccine?” If the answer is yes, the CDC considers this a caution, not a contraindication against receiving the influenza vaccine, since the benefit may still outweigh the risk.
‘I GOT THE FLU SHOT AND STILL GOT SICK’
The flu vaccine does not prevent illnesses caused by other viruses or bacteria that can make people sick during flu season. Influenza, the common cold, and streptococcal pharyngitis can have similar symptoms that make it difficult for patients—and, frequently, even healthcare providers—to distinguish between these illnesses with certainty.
One study suggested that influenza vaccine recipients had an increased risk of virologically confirmed noninfluenza respiratory viral infections,15 citing the phenomenon of virus interference that was described in the 1940s16 as a potential explanation. In essence, people protected against influenza by the vaccine may lack temporary nonspecific immunity against other respiratory viruses. However, these findings have not been replicated in subsequent studies.17
Viral gastroenteritis, mistakenly called “stomach flu,” is also not prevented by influenza vaccination.
‘I’M ALLERGIC TO EGGS’
The prevalence of egg allergy in US children is 0.5% to 2.5%.18 Most outgrow it by school age, but in one-third, the allergy persists into adulthood.
In general, people who can eat lightly cooked eggs (eg, scrambled eggs) without a reaction are unlikely to be allergic. On the other hand, the fact that egg-allergic people may tolerate egg included in baked products does not exclude the possibility of egg allergy. Egg allergy can be confirmed by a consistent medical history of adverse reaction to eggs and egg-containing foods, in addition to skin or blood testing for immunoglobulin E directed against egg proteins.19
Most currently available influenza vaccines are prepared by propagation of virus in embryonated eggs and so may contain trace amounts of egg proteins such as ovalbumin, with the exception of the inactivated quadrivalent recombinant influenza vaccine (Flublok) and the inactivated quadrivalent cell culture-based vaccine (Flucelvax).
The ACIP recommends that persons with a history of urticaria (hives) after exposure to eggs should receive any licensed, recommended influenza vaccine that is otherwise appropriate for their age and health status. Persons who report having angioedema, respiratory distress, lightheadedness, or recurrent vomiting, or who required epinephrine or another emergency medical intervention after exposure to eggs, should receive the influenza vaccine in an inpatient or outpatient medical setting under the supervision of a healthcare provider who is able to recognize and manage severe allergic reactions.
A history of severe allergic reaction such as anaphylaxis to a previous dose of any influenza vaccine, regardless of the vaccine component (including eggs) suspected of being responsible for the reaction, is a contraindication to influenza vaccination. The ACIP recommends that vaccine providers consider observing patients for 15 minutes after administration of any vaccine (regardless of history of egg allergy) to decrease the risk of injury should syncope occur.20
‘I DON’T WANT TO PUT POISONOUS MERCURY IN MY BODY’

A process of biomagnification of methylmercury occurs when humans eat large fish that have eaten smaller fish. Thus, larger fish such as shark can be hazardous for women who are or may become pregnant, for nursing mothers, and for young children, while smaller fish such as herring are relatively safe.
As a precautionary measure, thimerosal was taken out of childhood vaccines in the United States in 2001. Thimerosal-free influenza vaccine formulations include the nasal live-attenuated flu vaccine, the inactivated quadrivalent recombinant influenza vaccine, and the inactivated quadrivalent cell culture-based vaccine.
‘I DON’T LIKE NEEDLES’
At least 10% of US adults have aichmophobia, the fear of sharp objects including needles.22 Vasovagal syncope is the most common manifestation. Behavioral therapy, topical anesthetics, and systemic anxiolytics have variable efficacy in treating needle phobia. For those who are absolutely averse to needles, the nasal flu vaccine is an appropriate alternative.
‘I DON’T WANT TO TAKE ANYTHING THAT CAN MESS WITH MY OTHER MEDICATIONS’
Some immunosuppressive medications may decrease influenza vaccine immunogenicity. Concomitant administration of the inactivated influenza vaccine with other vaccines is safe and does not alter immunogenicity of other vaccines.1 The live-attenuated influenza vaccine is contraindicated in children and adolescents taking aspirin or other salicylates due to the risk of Reye syndrome.
‘I’M AFRAID IT WILL TRIGGER AN IMMUNE RESPONSE THAT WILL MAKE MY ASTHMA WORSE’
A recent systematic review and meta-analysis showed that the inactivated influenza vaccine is not associated with asthma exacerbation.23 However, the nasal live-attenuated influenza vaccine is contraindicated in children 2 to 4 years old who have asthma and should be used with caution in persons with asthma 5 years old and older. In the systematic review, influenza vaccine prevented 59% to 78% of asthma attacks leading to emergency visits or hospitalization.23 In other immune-mediated diseases such as rheumatoid arthritis, influenza vaccine does not precipitate exacerbations.24
‘I HAD AN ORGAN TRANSPLANT, AND I’M AFRAID THE FLU SHOT WILL CAUSE ORGAN REJECTION’
A study of 51,730 kidney transplant recipients found that receipt of the inactivated influenza vaccine in the first year after transplant was associated with a lower risk of subsequent allograft loss (adjusted hazard ratio 0.77; 95% confidence interval 0.69–0.85; P < .001) and death (adjusted hazard ratio 0.82; 95% confidence interval 0.76–0.89; P < .001).25 In the same study, although acute rejection in the first year was not associated with influenza vaccination, influenza infection in the first year was associated with rejection (odds ratio 1.58; 95% confidence interval 1.10–2.26; P < 0.001), but not with graft loss or death. Solid organ transplant recipients should receive the inactivated influenza vaccine starting 3 months after transplant.26
Influenza vaccination has not been shown to precipitate graft-vs-host disease in hematopoietic stem cell transplant recipients. These patients should also receive the inactivated influenza vaccine starting 3 to 6 months after transplant.27
The nasal live-attenuated influenza vaccine is contraindicated in these immunocompromised patients.
‘I’M PREGNANT, AND I DON’T WANT TO EXPOSE MY UNBORN BABY TO ANYTHING POTENTIALLY HARMFUL’
The morbidity and mortality risk from influenza is high in children under 2 years old because of low immunogenicity to flu vaccine. This is particularly true in children younger than 6 months, but the vaccine is not recommended in this population. The best way to protect infants is for all household members to be vaccinated against the flu.
Equally important, morbidity and mortality risk from influenza is much higher in pregnant women than in the general population. Many studies have shown the value of influenza vaccination during pregnancy for both mothers and their infants. A recently published study showed that 18% of infants who developed influenza required hospitalization.28 In that study, prenatal and postpartum maternal influenza vaccination decreased the odds of influenza in infants by 61% and 53%, respectively. Another study showed that vaccine effectiveness did not vary by gestational age at vaccination.29 A post hoc analysis of an influenza vaccination study in pregnant women suggested that the vaccine was also associated with decreased rates of pertussis in these women.30
Healthcare providers should try to understand the public’s misconceptions31 about seasonal influenza and influenza vaccines in order to best address them.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2018–19 influenza season. www.cdc.gov/flu/fluvaxview/coverage-1819estimates.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Immunogenicity, efficacy, and effectiveness of influenza vaccines. www.cdc.gov/flu/professionals/acip/immunogenicity.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). What are the benefits of flu vaccination? www.cdc.gov/flu/prevent/vaccine-benefits.htm. Accessed November 13, 2019.
- Grohskopf LA, Alyanak E, Broder KR, Walter EB, Fry AM, Jernigan DB. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices—United States, 2019–20 influenza season. MMWR Recomm Rep 2019; 68(3):1–21. doi:10.15585/mmwr.rr6803a1
- Flannery B, Chung JR, Belongia EA, et al. Interim estimates of 2017–18 seasonal influenza vaccine effectiveness—United States, February 2018. MMWR Morb Mortal Wkly Rep 2018; 67(6):180–185. doi:10.15585/mmwr.mm6706a2
- Erbelding EJ, Post DJ, Stemmy EJ, et al. A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 2018; 218(3):347–354. doi:10.1093/infdis/jiy103
- Centers for Disease Control and Prevention (CDC). Seasonal influenza vaccine safety: a summary for clinicians. www.cdc.gov/flu/professionals/vaccination/vaccine_safety.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). About the Vaccine Adverse Event Reporting System (VAERS). https://wonder.cdc.gov/vaers.html. Accessed November 13, 2019.
- Miller ER, Moro PL, Cano M, Shimabukuro TT. Deaths following vaccination: what does the evidence show? Vaccine 2015; 33(29):3288–3292. doi:10.1016/j.vaccine.2015.05.023
- Centers for Disease Control and Prevention (CDC). Guillain-Barré syndrome and flu vaccine. www.cdc.gov/flu/prevent/guillainbarre.htm. Accessed November 13, 2019.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barre syndrome following vaccination in the national influenza immunization program, United States, 1976–1977. Am J Epidemiol 1979; 110(2):105–123. doi:10.1093/oxfordjournals.aje.a112795
- Baxter R, Bakshi N, Fireman B, et al. Lack of association of Guillain-Barré syndrome with vaccinations. Clin Infect Dis 2013; 57(2):197–204. doi:10.1093/cid/cit222
- Kwong JC, Vasa PP, Campitelli MA, et al. Risk of Guillain-Barré syndrome after seasonal influenza vaccination and influenza health-care encounters: a self-controlled study. Lancet Infect Dis 2013; 13(9):769–776. doi:10.1016/S1473-3099(13)70104-X
- Centers for Disease Control and Prevention (CDC). Disease burden of influenza. www.cdc.gov/flu/about/burden/index.html. Accessed November 13, 2019.
- Cowling BJ, Fang VJ, Nishiura H, et al. Increased risk of noninfluenza respiratory virus infections associated with receipt of inactivated influenza vaccine. Clin Infect Dis 2012; 54(12):1778–1783. doi:10.1093/cid/cis307
- Henle W, Henle G. Interference of inactive virus with the propagation of virus of influenza. Science 1943; 98(2534):87–89. doi:10.1126/science.98.2534.87
- Sundaram ME, McClure DL, VanWormer JJ, Friedrich TC, Meece JK, Belongia EA. Influenza vaccination is not associated with detection of noninfluenza respiratory viruses in seasonal studies of influenza vaccine effectiveness. Clin Infect Dis 2013; 57(6):789–793. doi:10.1093/cid/cit379
- Caubet JC, Wang J. Current understanding of egg allergy. Pediatr Clin North Am 2011; 58(2):427–443. doi:10.1016/j.pcl.2011.02.014
- Erlewyn-Lajeunesse M, Brathwaite N, Lucas JS, Warner JO. Recommendations for the administration of influenza vaccine in children allergic to egg. BMJ 2009; 339:b3680. doi:10.1136/bmj.b3680
- Ezeanolue E, Harriman K, Hunter P, Kroger A, Pellegrini C. General Best Practice Guidelines for Immunization. Best Practices Guidance of the Advisory Committee on Immunization Practices (ACIP). https://www.cdc.gov/vaccines/hcp/acip-recs/general-recs/downloads/general-recs.pdf. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Thimerosal in vaccines. www.cdc.gov/vaccinesafety/concerns/thimerosal/index.html. Accessed November 13, 2019.
- Hamilton JG. Needle phobia: a neglected diagnosis. J Fam Pract 1995; 41(2):169–175. pmid:7636457
- Vasileiou E, Sheikh A, Butler C, et al. Effectiveness of influenza vaccines in asthma: a systematic review and meta-analysis. Clin Infect Dis 2017; 65(8):1388–1395. doi:10.1093/cid/cix524
- Fomin I, Caspi D, Levy V, et al. Vaccination against influenza in rheumatoid arthritis: the effect of disease modifying drugs, including TNF alpha blockers. Ann Rheum Dis 2006; 65(2):191–194. doi:10.1136/ard.2005.036434
- Hurst FP, Lee JJ, Jindal RM, Agodoa LY, Abbott KC. Outcomes associated with influenza vaccination in the first year after kidney transplantation. Clin J Am Soc Nephrol 2011; 6(5):1192–1197. doi:10.2215/CJN.05430610
- Chong PP, Handler L, Weber DJ. A systematic review of safety and immunogenicity of influenza vaccination strategies in solid organ transplant recipients. Clin Infect Dis 2018; 66(11):1802–1811. doi:10.1093/cid/cix1081
- Ljungman P, Avetisyan G. Influenza vaccination in hematopoietic SCT recipients. Bone Marrow Transplant 2008; 42(10):637–641. doi:10.1038/bmt.2008.264
- Ohfuji S, Deguchi M, Tachibana D, et al; Osaka Pregnant Women Influenza Study Group. Protective effect of maternal influenza vaccination on influenza in their infants: a prospective cohort study. J Infect Dis 2018; 217(6):878–886. doi:10.1093/infdis/jix629
- Katz J, Englund JA, Steinhoff MC, et al. Impact of timing of influenza vaccination in pregnancy on transplacental antibody transfer, influenza incidence, and birth outcomes: a randomized trial in rural Nepal. Clin Infect Dis 2018; 67(3):334–340. doi:10.1093/cid/ciy090
- Nunes MC, Cutland CL, Madhi SA. Influenza vaccination during pregnancy and protection against pertussis. N Engl J Med 2018; 378(13):1257–1258. doi:10.1056/NEJMc1705208
- Centers for Disease Control and Prevention (CDC). Misconceptions about seasonal flu and flu vaccines. www.cdc.gov/flu/prevent/misconceptions.htm. Accessed November 13, 2019.
The benefits of influenza vaccination are clear to those in the medical community. Yet misinformation and unfounded fears continue to discourage some people from getting a flu shot. During the 2018–2019 influenza season, only 45% of US adults and 63% of children were vaccinated.1
‘IT DOESN’T WORK FOR MANY PEOPLE’
Multiple studies have shown that the flu vaccine prevents millions of flu cases and flu-related doctor’s visits each year. During the 2016–2017 flu season, flu vaccine prevented an estimated 5.3 million influenza cases, 2.6 million influenza-associated medical visits, and 85,000 influenza-associated hospitalizations.2
Several viral and host factors affect vaccine effectiveness. In seasons when the vaccine viruses have matched circulating strains, flu vaccine has been shown to reduce the following:
- The risk of having to go to the doctor with flu by 40% to 60%
- Children’s risk of flu-related death and intensive care unit (ICU) admission by 74%
- The risk in adults of flu-associated hospitalizations by 40% and ICU admission by 82%
- The rate of cardiac events in people with heart disease
- Hospitalizations in people with diabetes or underlying chronic lung disease.3
In people hospitalized with influenza despite receiving the flu vaccine for the season, studies have shown that receiving the flu vaccine shortens the average duration of hospitalization, reduces the chance of ICU admission by 59%, shortens the duration of ICU stay by 4 days, and reduces deaths.3
‘IT TARGETS THE WRONG VIRUS’
Selecting an effective influenza vaccine is a challenge. Every year, the World Health Organization and the CDC decide on the influenza strains expected to circulate in the upcoming flu season in the Northern Hemisphere, based on data for circulating strains in the Southern Hemisphere. This decision takes place about 7 months before the expected onset of the flu season. Flu viruses may mutate between the time the decision is made and the time the vaccine is administered (as well as after the flu season starts). Also, vaccine production in eggs needs time, which is why this decision must be made several months ahead of the flu season.
Vaccine effectiveness varies by virus serotype. Vaccines are typically less effective against influenza A H3N2 viruses than against influenza A H1N1 and influenza B viruses. Effectiveness also varies from season to season depending on how close the vaccine serotypes match the circulating serotypes, but some effectiveness is retained even in seasons when some of the serotypes don’t match circulating viruses. For example, in the 2017–2018 season, when the influenza A H3N2 vaccine serotype did not match the circulating serotype, the overall effectiveness in preventing medically attended, laboratory-confirmed influenza virus infection was 36%.5
A universal flu vaccine that does not need to be updated annually is the ultimate solution, but according to the National Institute of Allergy and Infectious Diseases, such a vaccine is likely several years away.6
‘IT MAKES PEOPLE SICK’
Pain at the injection site of a flu shot occurs in 10% to 65% of people, lasts less than 2 days, and does not usually interfere with daily activities.7
Systemic symptoms such as fever, malaise, and myalgia may occur in people who have had no previous exposure to the influenza virus antigens in the vaccine, particularly in children. In adults, the frequency of systemic symptoms after the flu shot is similar to that with placebo.
The Vaccine Adverse Event Reporting System, which has been capturing data since 1990, shows that the influenza vaccine accounted for 5.7% of people who developed malaise after receiving any vaccine.8
The injectable inactivated influenza vaccine cannot biologically cause an influenza virus-related illness, since the inactivated vaccine viruses can elicit a protective immune response but cannot replicate. The nasal live-attenuated flu vaccine can in theory cause acute illness in the person receiving it, but because it is cold-adapted, it multiplies only in the colder environment of the nasal epithelium, not in the lower airways where the temperature is higher. Consequently, the vaccine virus triggers immunity by multiplying in the nose, but doesn’t infect the lungs.
From 10% to 50% of people who receive the nasal live-attenuated vaccine develop runny nose, wheezing, headache, vomiting, muscle aches, fever, sore throat, or cough shortly after receiving the vaccine, but these symptoms are usually mild and short-lived.
The most common reactions people have to flu vaccines are considerably less severe than the symptoms caused by actual flu illness.
While influenza illness results in natural immunity to the specific viral serotype causing it, this illness results in hospitalization in 2% and is fatal in 0.16% of people. Influenza vaccine results in immunity to the serotypes included in the vaccine, and multiple studies have not found a causal relationship between vaccination and death.9
‘IT CAUSES GUILLAIN-BARRÉ SYNDROME’
In the United States, 3,000 to 6,000 people per year develop Guillain-Barré syndrome, or 1 to 2 of every 100,000, which translates to 80 to 160 cases per week.10 While the exact cause of Guillain-Barré syndrome is unknown, about two-thirds of people have an acute diarrheal or respiratory illness within 3 months before the onset of symptoms. In 1976, the estimated attributable risk of influenza vaccine-related Guillain-Barré syndrome in the US adult population was 1 case per 100,000 in the 6 weeks after vaccination.11 Studies in subsequent influenza seasons have not shown similar findings.12 In fact, one study showed that the risk of developing Guillain-Barré syndrome was 15 times higher after influenza illness than after influenza vaccination.13
Since 5% to 15% of the US population develop symptomatic influenza annually,14 the decision to vaccinate with respect to the risk of Guillain-Barré syndrome should be obvious: vaccinate. The correct question to ask before influenza vaccination should be, “Have you previously developed Guillain-Barré syndrome within 6 weeks after receiving the flu vaccine?” If the answer is yes, the CDC considers this a caution, not a contraindication against receiving the influenza vaccine, since the benefit may still outweigh the risk.
‘I GOT THE FLU SHOT AND STILL GOT SICK’
The flu vaccine does not prevent illnesses caused by other viruses or bacteria that can make people sick during flu season. Influenza, the common cold, and streptococcal pharyngitis can have similar symptoms that make it difficult for patients—and, frequently, even healthcare providers—to distinguish between these illnesses with certainty.
One study suggested that influenza vaccine recipients had an increased risk of virologically confirmed noninfluenza respiratory viral infections,15 citing the phenomenon of virus interference that was described in the 1940s16 as a potential explanation. In essence, people protected against influenza by the vaccine may lack temporary nonspecific immunity against other respiratory viruses. However, these findings have not been replicated in subsequent studies.17
Viral gastroenteritis, mistakenly called “stomach flu,” is also not prevented by influenza vaccination.
‘I’M ALLERGIC TO EGGS’
The prevalence of egg allergy in US children is 0.5% to 2.5%.18 Most outgrow it by school age, but in one-third, the allergy persists into adulthood.
In general, people who can eat lightly cooked eggs (eg, scrambled eggs) without a reaction are unlikely to be allergic. On the other hand, the fact that egg-allergic people may tolerate egg included in baked products does not exclude the possibility of egg allergy. Egg allergy can be confirmed by a consistent medical history of adverse reaction to eggs and egg-containing foods, in addition to skin or blood testing for immunoglobulin E directed against egg proteins.19
Most currently available influenza vaccines are prepared by propagation of virus in embryonated eggs and so may contain trace amounts of egg proteins such as ovalbumin, with the exception of the inactivated quadrivalent recombinant influenza vaccine (Flublok) and the inactivated quadrivalent cell culture-based vaccine (Flucelvax).
The ACIP recommends that persons with a history of urticaria (hives) after exposure to eggs should receive any licensed, recommended influenza vaccine that is otherwise appropriate for their age and health status. Persons who report having angioedema, respiratory distress, lightheadedness, or recurrent vomiting, or who required epinephrine or another emergency medical intervention after exposure to eggs, should receive the influenza vaccine in an inpatient or outpatient medical setting under the supervision of a healthcare provider who is able to recognize and manage severe allergic reactions.
A history of severe allergic reaction such as anaphylaxis to a previous dose of any influenza vaccine, regardless of the vaccine component (including eggs) suspected of being responsible for the reaction, is a contraindication to influenza vaccination. The ACIP recommends that vaccine providers consider observing patients for 15 minutes after administration of any vaccine (regardless of history of egg allergy) to decrease the risk of injury should syncope occur.20
‘I DON’T WANT TO PUT POISONOUS MERCURY IN MY BODY’
A process of biomagnification of methylmercury occurs when humans eat large fish that have eaten smaller fish. Thus, larger fish such as shark can be hazardous for women who are or may become pregnant, for nursing mothers, and for young children, while smaller fish such as herring are relatively safe.
As a precautionary measure, thimerosal was taken out of childhood vaccines in the United States in 2001. Thimerosal-free influenza vaccine formulations include the nasal live-attenuated flu vaccine, the inactivated quadrivalent recombinant influenza vaccine, and the inactivated quadrivalent cell culture-based vaccine.
‘I DON’T LIKE NEEDLES’
At least 10% of US adults have aichmophobia, the fear of sharp objects including needles.22 Vasovagal syncope is the most common manifestation. Behavioral therapy, topical anesthetics, and systemic anxiolytics have variable efficacy in treating needle phobia. For those who are absolutely averse to needles, the nasal flu vaccine is an appropriate alternative.
‘I DON’T WANT TO TAKE ANYTHING THAT CAN MESS WITH MY OTHER MEDICATIONS’
Some immunosuppressive medications may decrease influenza vaccine immunogenicity. Concomitant administration of the inactivated influenza vaccine with other vaccines is safe and does not alter immunogenicity of other vaccines.1 The live-attenuated influenza vaccine is contraindicated in children and adolescents taking aspirin or other salicylates due to the risk of Reye syndrome.
‘I’M AFRAID IT WILL TRIGGER AN IMMUNE RESPONSE THAT WILL MAKE MY ASTHMA WORSE’
A recent systematic review and meta-analysis showed that the inactivated influenza vaccine is not associated with asthma exacerbation.23 However, the nasal live-attenuated influenza vaccine is contraindicated in children 2 to 4 years old who have asthma and should be used with caution in persons with asthma 5 years old and older. In the systematic review, influenza vaccine prevented 59% to 78% of asthma attacks leading to emergency visits or hospitalization.23 In other immune-mediated diseases such as rheumatoid arthritis, influenza vaccine does not precipitate exacerbations.24
‘I HAD AN ORGAN TRANSPLANT, AND I’M AFRAID THE FLU SHOT WILL CAUSE ORGAN REJECTION’
A study of 51,730 kidney transplant recipients found that receipt of the inactivated influenza vaccine in the first year after transplant was associated with a lower risk of subsequent allograft loss (adjusted hazard ratio 0.77; 95% confidence interval 0.69–0.85; P < .001) and death (adjusted hazard ratio 0.82; 95% confidence interval 0.76–0.89; P < .001).25 In the same study, although acute rejection in the first year was not associated with influenza vaccination, influenza infection in the first year was associated with rejection (odds ratio 1.58; 95% confidence interval 1.10–2.26; P < 0.001), but not with graft loss or death. Solid organ transplant recipients should receive the inactivated influenza vaccine starting 3 months after transplant.26
Influenza vaccination has not been shown to precipitate graft-vs-host disease in hematopoietic stem cell transplant recipients. These patients should also receive the inactivated influenza vaccine starting 3 to 6 months after transplant.27
The nasal live-attenuated influenza vaccine is contraindicated in these immunocompromised patients.
‘I’M PREGNANT, AND I DON’T WANT TO EXPOSE MY UNBORN BABY TO ANYTHING POTENTIALLY HARMFUL’
The morbidity and mortality risk from influenza is high in children under 2 years old because of low immunogenicity to flu vaccine. This is particularly true in children younger than 6 months, but the vaccine is not recommended in this population. The best way to protect infants is for all household members to be vaccinated against the flu.
Equally important, morbidity and mortality risk from influenza is much higher in pregnant women than in the general population. Many studies have shown the value of influenza vaccination during pregnancy for both mothers and their infants. A recently published study showed that 18% of infants who developed influenza required hospitalization.28 In that study, prenatal and postpartum maternal influenza vaccination decreased the odds of influenza in infants by 61% and 53%, respectively. Another study showed that vaccine effectiveness did not vary by gestational age at vaccination.29 A post hoc analysis of an influenza vaccination study in pregnant women suggested that the vaccine was also associated with decreased rates of pertussis in these women.30
Healthcare providers should try to understand the public’s misconceptions31 about seasonal influenza and influenza vaccines in order to best address them.
The benefits of influenza vaccination are clear to those in the medical community. Yet misinformation and unfounded fears continue to discourage some people from getting a flu shot. During the 2018–2019 influenza season, only 45% of US adults and 63% of children were vaccinated.1
‘IT DOESN’T WORK FOR MANY PEOPLE’
Multiple studies have shown that the flu vaccine prevents millions of flu cases and flu-related doctor’s visits each year. During the 2016–2017 flu season, flu vaccine prevented an estimated 5.3 million influenza cases, 2.6 million influenza-associated medical visits, and 85,000 influenza-associated hospitalizations.2
Several viral and host factors affect vaccine effectiveness. In seasons when the vaccine viruses have matched circulating strains, flu vaccine has been shown to reduce the following:
- The risk of having to go to the doctor with flu by 40% to 60%
- Children’s risk of flu-related death and intensive care unit (ICU) admission by 74%
- The risk in adults of flu-associated hospitalizations by 40% and ICU admission by 82%
- The rate of cardiac events in people with heart disease
- Hospitalizations in people with diabetes or underlying chronic lung disease.3
In people hospitalized with influenza despite receiving the flu vaccine for the season, studies have shown that receiving the flu vaccine shortens the average duration of hospitalization, reduces the chance of ICU admission by 59%, shortens the duration of ICU stay by 4 days, and reduces deaths.3
‘IT TARGETS THE WRONG VIRUS’
Selecting an effective influenza vaccine is a challenge. Every year, the World Health Organization and the CDC decide on the influenza strains expected to circulate in the upcoming flu season in the Northern Hemisphere, based on data for circulating strains in the Southern Hemisphere. This decision takes place about 7 months before the expected onset of the flu season. Flu viruses may mutate between the time the decision is made and the time the vaccine is administered (as well as after the flu season starts). Also, vaccine production in eggs needs time, which is why this decision must be made several months ahead of the flu season.
Vaccine effectiveness varies by virus serotype. Vaccines are typically less effective against influenza A H3N2 viruses than against influenza A H1N1 and influenza B viruses. Effectiveness also varies from season to season depending on how close the vaccine serotypes match the circulating serotypes, but some effectiveness is retained even in seasons when some of the serotypes don’t match circulating viruses. For example, in the 2017–2018 season, when the influenza A H3N2 vaccine serotype did not match the circulating serotype, the overall effectiveness in preventing medically attended, laboratory-confirmed influenza virus infection was 36%.5
A universal flu vaccine that does not need to be updated annually is the ultimate solution, but according to the National Institute of Allergy and Infectious Diseases, such a vaccine is likely several years away.6
‘IT MAKES PEOPLE SICK’
Pain at the injection site of a flu shot occurs in 10% to 65% of people, lasts less than 2 days, and does not usually interfere with daily activities.7
Systemic symptoms such as fever, malaise, and myalgia may occur in people who have had no previous exposure to the influenza virus antigens in the vaccine, particularly in children. In adults, the frequency of systemic symptoms after the flu shot is similar to that with placebo.
The Vaccine Adverse Event Reporting System, which has been capturing data since 1990, shows that the influenza vaccine accounted for 5.7% of people who developed malaise after receiving any vaccine.8
The injectable inactivated influenza vaccine cannot biologically cause an influenza virus-related illness, since the inactivated vaccine viruses can elicit a protective immune response but cannot replicate. The nasal live-attenuated flu vaccine can in theory cause acute illness in the person receiving it, but because it is cold-adapted, it multiplies only in the colder environment of the nasal epithelium, not in the lower airways where the temperature is higher. Consequently, the vaccine virus triggers immunity by multiplying in the nose, but doesn’t infect the lungs.
From 10% to 50% of people who receive the nasal live-attenuated vaccine develop runny nose, wheezing, headache, vomiting, muscle aches, fever, sore throat, or cough shortly after receiving the vaccine, but these symptoms are usually mild and short-lived.
The most common reactions people have to flu vaccines are considerably less severe than the symptoms caused by actual flu illness.
While influenza illness results in natural immunity to the specific viral serotype causing it, this illness results in hospitalization in 2% and is fatal in 0.16% of people. Influenza vaccine results in immunity to the serotypes included in the vaccine, and multiple studies have not found a causal relationship between vaccination and death.9
‘IT CAUSES GUILLAIN-BARRÉ SYNDROME’
In the United States, 3,000 to 6,000 people per year develop Guillain-Barré syndrome, or 1 to 2 of every 100,000, which translates to 80 to 160 cases per week.10 While the exact cause of Guillain-Barré syndrome is unknown, about two-thirds of people have an acute diarrheal or respiratory illness within 3 months before the onset of symptoms. In 1976, the estimated attributable risk of influenza vaccine-related Guillain-Barré syndrome in the US adult population was 1 case per 100,000 in the 6 weeks after vaccination.11 Studies in subsequent influenza seasons have not shown similar findings.12 In fact, one study showed that the risk of developing Guillain-Barré syndrome was 15 times higher after influenza illness than after influenza vaccination.13
Since 5% to 15% of the US population develop symptomatic influenza annually,14 the decision to vaccinate with respect to the risk of Guillain-Barré syndrome should be obvious: vaccinate. The correct question to ask before influenza vaccination should be, “Have you previously developed Guillain-Barré syndrome within 6 weeks after receiving the flu vaccine?” If the answer is yes, the CDC considers this a caution, not a contraindication against receiving the influenza vaccine, since the benefit may still outweigh the risk.
‘I GOT THE FLU SHOT AND STILL GOT SICK’
The flu vaccine does not prevent illnesses caused by other viruses or bacteria that can make people sick during flu season. Influenza, the common cold, and streptococcal pharyngitis can have similar symptoms that make it difficult for patients—and, frequently, even healthcare providers—to distinguish between these illnesses with certainty.
One study suggested that influenza vaccine recipients had an increased risk of virologically confirmed noninfluenza respiratory viral infections,15 citing the phenomenon of virus interference that was described in the 1940s16 as a potential explanation. In essence, people protected against influenza by the vaccine may lack temporary nonspecific immunity against other respiratory viruses. However, these findings have not been replicated in subsequent studies.17
Viral gastroenteritis, mistakenly called “stomach flu,” is also not prevented by influenza vaccination.
‘I’M ALLERGIC TO EGGS’
The prevalence of egg allergy in US children is 0.5% to 2.5%.18 Most outgrow it by school age, but in one-third, the allergy persists into adulthood.
In general, people who can eat lightly cooked eggs (eg, scrambled eggs) without a reaction are unlikely to be allergic. On the other hand, the fact that egg-allergic people may tolerate egg included in baked products does not exclude the possibility of egg allergy. Egg allergy can be confirmed by a consistent medical history of adverse reaction to eggs and egg-containing foods, in addition to skin or blood testing for immunoglobulin E directed against egg proteins.19
Most currently available influenza vaccines are prepared by propagation of virus in embryonated eggs and so may contain trace amounts of egg proteins such as ovalbumin, with the exception of the inactivated quadrivalent recombinant influenza vaccine (Flublok) and the inactivated quadrivalent cell culture-based vaccine (Flucelvax).
The ACIP recommends that persons with a history of urticaria (hives) after exposure to eggs should receive any licensed, recommended influenza vaccine that is otherwise appropriate for their age and health status. Persons who report having angioedema, respiratory distress, lightheadedness, or recurrent vomiting, or who required epinephrine or another emergency medical intervention after exposure to eggs, should receive the influenza vaccine in an inpatient or outpatient medical setting under the supervision of a healthcare provider who is able to recognize and manage severe allergic reactions.
A history of severe allergic reaction such as anaphylaxis to a previous dose of any influenza vaccine, regardless of the vaccine component (including eggs) suspected of being responsible for the reaction, is a contraindication to influenza vaccination. The ACIP recommends that vaccine providers consider observing patients for 15 minutes after administration of any vaccine (regardless of history of egg allergy) to decrease the risk of injury should syncope occur.20
‘I DON’T WANT TO PUT POISONOUS MERCURY IN MY BODY’
A process of biomagnification of methylmercury occurs when humans eat large fish that have eaten smaller fish. Thus, larger fish such as shark can be hazardous for women who are or may become pregnant, for nursing mothers, and for young children, while smaller fish such as herring are relatively safe.
As a precautionary measure, thimerosal was taken out of childhood vaccines in the United States in 2001. Thimerosal-free influenza vaccine formulations include the nasal live-attenuated flu vaccine, the inactivated quadrivalent recombinant influenza vaccine, and the inactivated quadrivalent cell culture-based vaccine.
‘I DON’T LIKE NEEDLES’
At least 10% of US adults have aichmophobia, the fear of sharp objects including needles.22 Vasovagal syncope is the most common manifestation. Behavioral therapy, topical anesthetics, and systemic anxiolytics have variable efficacy in treating needle phobia. For those who are absolutely averse to needles, the nasal flu vaccine is an appropriate alternative.
‘I DON’T WANT TO TAKE ANYTHING THAT CAN MESS WITH MY OTHER MEDICATIONS’
Some immunosuppressive medications may decrease influenza vaccine immunogenicity. Concomitant administration of the inactivated influenza vaccine with other vaccines is safe and does not alter immunogenicity of other vaccines.1 The live-attenuated influenza vaccine is contraindicated in children and adolescents taking aspirin or other salicylates due to the risk of Reye syndrome.
‘I’M AFRAID IT WILL TRIGGER AN IMMUNE RESPONSE THAT WILL MAKE MY ASTHMA WORSE’
A recent systematic review and meta-analysis showed that the inactivated influenza vaccine is not associated with asthma exacerbation.23 However, the nasal live-attenuated influenza vaccine is contraindicated in children 2 to 4 years old who have asthma and should be used with caution in persons with asthma 5 years old and older. In the systematic review, influenza vaccine prevented 59% to 78% of asthma attacks leading to emergency visits or hospitalization.23 In other immune-mediated diseases such as rheumatoid arthritis, influenza vaccine does not precipitate exacerbations.24
‘I HAD AN ORGAN TRANSPLANT, AND I’M AFRAID THE FLU SHOT WILL CAUSE ORGAN REJECTION’
A study of 51,730 kidney transplant recipients found that receipt of the inactivated influenza vaccine in the first year after transplant was associated with a lower risk of subsequent allograft loss (adjusted hazard ratio 0.77; 95% confidence interval 0.69–0.85; P < .001) and death (adjusted hazard ratio 0.82; 95% confidence interval 0.76–0.89; P < .001).25 In the same study, although acute rejection in the first year was not associated with influenza vaccination, influenza infection in the first year was associated with rejection (odds ratio 1.58; 95% confidence interval 1.10–2.26; P < 0.001), but not with graft loss or death. Solid organ transplant recipients should receive the inactivated influenza vaccine starting 3 months after transplant.26
Influenza vaccination has not been shown to precipitate graft-vs-host disease in hematopoietic stem cell transplant recipients. These patients should also receive the inactivated influenza vaccine starting 3 to 6 months after transplant.27
The nasal live-attenuated influenza vaccine is contraindicated in these immunocompromised patients.
‘I’M PREGNANT, AND I DON’T WANT TO EXPOSE MY UNBORN BABY TO ANYTHING POTENTIALLY HARMFUL’
The morbidity and mortality risk from influenza is high in children under 2 years old because of low immunogenicity to flu vaccine. This is particularly true in children younger than 6 months, but the vaccine is not recommended in this population. The best way to protect infants is for all household members to be vaccinated against the flu.
Equally important, morbidity and mortality risk from influenza is much higher in pregnant women than in the general population. Many studies have shown the value of influenza vaccination during pregnancy for both mothers and their infants. A recently published study showed that 18% of infants who developed influenza required hospitalization.28 In that study, prenatal and postpartum maternal influenza vaccination decreased the odds of influenza in infants by 61% and 53%, respectively. Another study showed that vaccine effectiveness did not vary by gestational age at vaccination.29 A post hoc analysis of an influenza vaccination study in pregnant women suggested that the vaccine was also associated with decreased rates of pertussis in these women.30
Healthcare providers should try to understand the public’s misconceptions31 about seasonal influenza and influenza vaccines in order to best address them.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2018–19 influenza season. www.cdc.gov/flu/fluvaxview/coverage-1819estimates.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Immunogenicity, efficacy, and effectiveness of influenza vaccines. www.cdc.gov/flu/professionals/acip/immunogenicity.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). What are the benefits of flu vaccination? www.cdc.gov/flu/prevent/vaccine-benefits.htm. Accessed November 13, 2019.
- Grohskopf LA, Alyanak E, Broder KR, Walter EB, Fry AM, Jernigan DB. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices—United States, 2019–20 influenza season. MMWR Recomm Rep 2019; 68(3):1–21. doi:10.15585/mmwr.rr6803a1
- Flannery B, Chung JR, Belongia EA, et al. Interim estimates of 2017–18 seasonal influenza vaccine effectiveness—United States, February 2018. MMWR Morb Mortal Wkly Rep 2018; 67(6):180–185. doi:10.15585/mmwr.mm6706a2
- Erbelding EJ, Post DJ, Stemmy EJ, et al. A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 2018; 218(3):347–354. doi:10.1093/infdis/jiy103
- Centers for Disease Control and Prevention (CDC). Seasonal influenza vaccine safety: a summary for clinicians. www.cdc.gov/flu/professionals/vaccination/vaccine_safety.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). About the Vaccine Adverse Event Reporting System (VAERS). https://wonder.cdc.gov/vaers.html. Accessed November 13, 2019.
- Miller ER, Moro PL, Cano M, Shimabukuro TT. Deaths following vaccination: what does the evidence show? Vaccine 2015; 33(29):3288–3292. doi:10.1016/j.vaccine.2015.05.023
- Centers for Disease Control and Prevention (CDC). Guillain-Barré syndrome and flu vaccine. www.cdc.gov/flu/prevent/guillainbarre.htm. Accessed November 13, 2019.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barre syndrome following vaccination in the national influenza immunization program, United States, 1976–1977. Am J Epidemiol 1979; 110(2):105–123. doi:10.1093/oxfordjournals.aje.a112795
- Baxter R, Bakshi N, Fireman B, et al. Lack of association of Guillain-Barré syndrome with vaccinations. Clin Infect Dis 2013; 57(2):197–204. doi:10.1093/cid/cit222
- Kwong JC, Vasa PP, Campitelli MA, et al. Risk of Guillain-Barré syndrome after seasonal influenza vaccination and influenza health-care encounters: a self-controlled study. Lancet Infect Dis 2013; 13(9):769–776. doi:10.1016/S1473-3099(13)70104-X
- Centers for Disease Control and Prevention (CDC). Disease burden of influenza. www.cdc.gov/flu/about/burden/index.html. Accessed November 13, 2019.
- Cowling BJ, Fang VJ, Nishiura H, et al. Increased risk of noninfluenza respiratory virus infections associated with receipt of inactivated influenza vaccine. Clin Infect Dis 2012; 54(12):1778–1783. doi:10.1093/cid/cis307
- Henle W, Henle G. Interference of inactive virus with the propagation of virus of influenza. Science 1943; 98(2534):87–89. doi:10.1126/science.98.2534.87
- Sundaram ME, McClure DL, VanWormer JJ, Friedrich TC, Meece JK, Belongia EA. Influenza vaccination is not associated with detection of noninfluenza respiratory viruses in seasonal studies of influenza vaccine effectiveness. Clin Infect Dis 2013; 57(6):789–793. doi:10.1093/cid/cit379
- Caubet JC, Wang J. Current understanding of egg allergy. Pediatr Clin North Am 2011; 58(2):427–443. doi:10.1016/j.pcl.2011.02.014
- Erlewyn-Lajeunesse M, Brathwaite N, Lucas JS, Warner JO. Recommendations for the administration of influenza vaccine in children allergic to egg. BMJ 2009; 339:b3680. doi:10.1136/bmj.b3680
- Ezeanolue E, Harriman K, Hunter P, Kroger A, Pellegrini C. General Best Practice Guidelines for Immunization. Best Practices Guidance of the Advisory Committee on Immunization Practices (ACIP). https://www.cdc.gov/vaccines/hcp/acip-recs/general-recs/downloads/general-recs.pdf. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Thimerosal in vaccines. www.cdc.gov/vaccinesafety/concerns/thimerosal/index.html. Accessed November 13, 2019.
- Hamilton JG. Needle phobia: a neglected diagnosis. J Fam Pract 1995; 41(2):169–175. pmid:7636457
- Vasileiou E, Sheikh A, Butler C, et al. Effectiveness of influenza vaccines in asthma: a systematic review and meta-analysis. Clin Infect Dis 2017; 65(8):1388–1395. doi:10.1093/cid/cix524
- Fomin I, Caspi D, Levy V, et al. Vaccination against influenza in rheumatoid arthritis: the effect of disease modifying drugs, including TNF alpha blockers. Ann Rheum Dis 2006; 65(2):191–194. doi:10.1136/ard.2005.036434
- Hurst FP, Lee JJ, Jindal RM, Agodoa LY, Abbott KC. Outcomes associated with influenza vaccination in the first year after kidney transplantation. Clin J Am Soc Nephrol 2011; 6(5):1192–1197. doi:10.2215/CJN.05430610
- Chong PP, Handler L, Weber DJ. A systematic review of safety and immunogenicity of influenza vaccination strategies in solid organ transplant recipients. Clin Infect Dis 2018; 66(11):1802–1811. doi:10.1093/cid/cix1081
- Ljungman P, Avetisyan G. Influenza vaccination in hematopoietic SCT recipients. Bone Marrow Transplant 2008; 42(10):637–641. doi:10.1038/bmt.2008.264
- Ohfuji S, Deguchi M, Tachibana D, et al; Osaka Pregnant Women Influenza Study Group. Protective effect of maternal influenza vaccination on influenza in their infants: a prospective cohort study. J Infect Dis 2018; 217(6):878–886. doi:10.1093/infdis/jix629
- Katz J, Englund JA, Steinhoff MC, et al. Impact of timing of influenza vaccination in pregnancy on transplacental antibody transfer, influenza incidence, and birth outcomes: a randomized trial in rural Nepal. Clin Infect Dis 2018; 67(3):334–340. doi:10.1093/cid/ciy090
- Nunes MC, Cutland CL, Madhi SA. Influenza vaccination during pregnancy and protection against pertussis. N Engl J Med 2018; 378(13):1257–1258. doi:10.1056/NEJMc1705208
- Centers for Disease Control and Prevention (CDC). Misconceptions about seasonal flu and flu vaccines. www.cdc.gov/flu/prevent/misconceptions.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Flu vaccination coverage, United States, 2018–19 influenza season. www.cdc.gov/flu/fluvaxview/coverage-1819estimates.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Immunogenicity, efficacy, and effectiveness of influenza vaccines. www.cdc.gov/flu/professionals/acip/immunogenicity.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). What are the benefits of flu vaccination? www.cdc.gov/flu/prevent/vaccine-benefits.htm. Accessed November 13, 2019.
- Grohskopf LA, Alyanak E, Broder KR, Walter EB, Fry AM, Jernigan DB. Prevention and control of seasonal influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices—United States, 2019–20 influenza season. MMWR Recomm Rep 2019; 68(3):1–21. doi:10.15585/mmwr.rr6803a1
- Flannery B, Chung JR, Belongia EA, et al. Interim estimates of 2017–18 seasonal influenza vaccine effectiveness—United States, February 2018. MMWR Morb Mortal Wkly Rep 2018; 67(6):180–185. doi:10.15585/mmwr.mm6706a2
- Erbelding EJ, Post DJ, Stemmy EJ, et al. A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 2018; 218(3):347–354. doi:10.1093/infdis/jiy103
- Centers for Disease Control and Prevention (CDC). Seasonal influenza vaccine safety: a summary for clinicians. www.cdc.gov/flu/professionals/vaccination/vaccine_safety.htm. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). About the Vaccine Adverse Event Reporting System (VAERS). https://wonder.cdc.gov/vaers.html. Accessed November 13, 2019.
- Miller ER, Moro PL, Cano M, Shimabukuro TT. Deaths following vaccination: what does the evidence show? Vaccine 2015; 33(29):3288–3292. doi:10.1016/j.vaccine.2015.05.023
- Centers for Disease Control and Prevention (CDC). Guillain-Barré syndrome and flu vaccine. www.cdc.gov/flu/prevent/guillainbarre.htm. Accessed November 13, 2019.
- Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, et al. Guillain-Barre syndrome following vaccination in the national influenza immunization program, United States, 1976–1977. Am J Epidemiol 1979; 110(2):105–123. doi:10.1093/oxfordjournals.aje.a112795
- Baxter R, Bakshi N, Fireman B, et al. Lack of association of Guillain-Barré syndrome with vaccinations. Clin Infect Dis 2013; 57(2):197–204. doi:10.1093/cid/cit222
- Kwong JC, Vasa PP, Campitelli MA, et al. Risk of Guillain-Barré syndrome after seasonal influenza vaccination and influenza health-care encounters: a self-controlled study. Lancet Infect Dis 2013; 13(9):769–776. doi:10.1016/S1473-3099(13)70104-X
- Centers for Disease Control and Prevention (CDC). Disease burden of influenza. www.cdc.gov/flu/about/burden/index.html. Accessed November 13, 2019.
- Cowling BJ, Fang VJ, Nishiura H, et al. Increased risk of noninfluenza respiratory virus infections associated with receipt of inactivated influenza vaccine. Clin Infect Dis 2012; 54(12):1778–1783. doi:10.1093/cid/cis307
- Henle W, Henle G. Interference of inactive virus with the propagation of virus of influenza. Science 1943; 98(2534):87–89. doi:10.1126/science.98.2534.87
- Sundaram ME, McClure DL, VanWormer JJ, Friedrich TC, Meece JK, Belongia EA. Influenza vaccination is not associated with detection of noninfluenza respiratory viruses in seasonal studies of influenza vaccine effectiveness. Clin Infect Dis 2013; 57(6):789–793. doi:10.1093/cid/cit379
- Caubet JC, Wang J. Current understanding of egg allergy. Pediatr Clin North Am 2011; 58(2):427–443. doi:10.1016/j.pcl.2011.02.014
- Erlewyn-Lajeunesse M, Brathwaite N, Lucas JS, Warner JO. Recommendations for the administration of influenza vaccine in children allergic to egg. BMJ 2009; 339:b3680. doi:10.1136/bmj.b3680
- Ezeanolue E, Harriman K, Hunter P, Kroger A, Pellegrini C. General Best Practice Guidelines for Immunization. Best Practices Guidance of the Advisory Committee on Immunization Practices (ACIP). https://www.cdc.gov/vaccines/hcp/acip-recs/general-recs/downloads/general-recs.pdf. Accessed November 13, 2019.
- Centers for Disease Control and Prevention (CDC). Thimerosal in vaccines. www.cdc.gov/vaccinesafety/concerns/thimerosal/index.html. Accessed November 13, 2019.
- Hamilton JG. Needle phobia: a neglected diagnosis. J Fam Pract 1995; 41(2):169–175. pmid:7636457
- Vasileiou E, Sheikh A, Butler C, et al. Effectiveness of influenza vaccines in asthma: a systematic review and meta-analysis. Clin Infect Dis 2017; 65(8):1388–1395. doi:10.1093/cid/cix524
- Fomin I, Caspi D, Levy V, et al. Vaccination against influenza in rheumatoid arthritis: the effect of disease modifying drugs, including TNF alpha blockers. Ann Rheum Dis 2006; 65(2):191–194. doi:10.1136/ard.2005.036434
- Hurst FP, Lee JJ, Jindal RM, Agodoa LY, Abbott KC. Outcomes associated with influenza vaccination in the first year after kidney transplantation. Clin J Am Soc Nephrol 2011; 6(5):1192–1197. doi:10.2215/CJN.05430610
- Chong PP, Handler L, Weber DJ. A systematic review of safety and immunogenicity of influenza vaccination strategies in solid organ transplant recipients. Clin Infect Dis 2018; 66(11):1802–1811. doi:10.1093/cid/cix1081
- Ljungman P, Avetisyan G. Influenza vaccination in hematopoietic SCT recipients. Bone Marrow Transplant 2008; 42(10):637–641. doi:10.1038/bmt.2008.264
- Ohfuji S, Deguchi M, Tachibana D, et al; Osaka Pregnant Women Influenza Study Group. Protective effect of maternal influenza vaccination on influenza in their infants: a prospective cohort study. J Infect Dis 2018; 217(6):878–886. doi:10.1093/infdis/jix629
- Katz J, Englund JA, Steinhoff MC, et al. Impact of timing of influenza vaccination in pregnancy on transplacental antibody transfer, influenza incidence, and birth outcomes: a randomized trial in rural Nepal. Clin Infect Dis 2018; 67(3):334–340. doi:10.1093/cid/ciy090
- Nunes MC, Cutland CL, Madhi SA. Influenza vaccination during pregnancy and protection against pertussis. N Engl J Med 2018; 378(13):1257–1258. doi:10.1056/NEJMc1705208
- Centers for Disease Control and Prevention (CDC). Misconceptions about seasonal flu and flu vaccines. www.cdc.gov/flu/prevent/misconceptions.htm. Accessed November 13, 2019.
Colorectal cancer screening: Folic acid supplementation alters the equation
To the Editor: In their paper on colorectal cancer screening, Mankaney and colleagues noted the increasing rates of colorectal cancer in young adults in the United States.1 Recent epidemiologic data demonstrate an increasing incidence of the disease in people ages 40 through 49 since the mid-1990s.2 Even though screening starting at age 45 is not uniformly accepted,1 there is evidence supporting earlier screening.
During the mid-1990s, the US government mandated that all enriched flour and uncooked cereal grains were to be fortified with folic acid in order to prevent births complicated by neural tube defects.3 Subsequently, there was a 2-fold increase in plasma folate concentrations and, disturbingly, a temporally associated significant increase in the incidence of colorectal cancer.3
Notably, a US trial4 testing the efficacy of folic acid 1 mg taken daily for 6 years to prevent colorectal adenomas in those with a history of colorectal adenomas failed to show a reduction in adenoma risk. Instead, participants randomized to folic acid exhibited a significantly increased risk of an advanced adenoma. Another trial,5 conducted in the Netherlands, where there is no mandatory folic acid fortification, investigated folic acid 400 µg and vitamin B12 500 µg daily over 2 to 3 years for the prevention of osteoporotic fractures. The group randomized to the vitamins had a nearly 2-fold increase in the risk of colorectal cancer.
Folic acid can be a double-edged sword.3,5 Although folic acid intake may protect against carcinogenesis through increased genetic stability, if precancerous or neoplastic cells are present, excess folic acid may promote cancer by increasing DNA synthesis and cell proliferation. Cancer cells have folic acid receptors.
Since screening colonoscopy is typically done in individuals over 50, advanced adenomas from folic acid exposure in people younger than 50 likely go undiagnosed. Therefore, colorectal cancer screening should start at a younger age in countries where folic acid fortification is mandatory.
- Mankaney G, Sutton RA, Burke CA. Colorectal cancer screening: choosing the right test. Cleve Clin J Med 2019; 86(6):385–392. doi:10.3949/ccjm.86a.17125
- Meester RGS, Mannalithara A, Lansdorp-Vogelaar I, Ladabaum U. Trends in incidence and stage at diagnosis of colorectal cancer in adults aged 40 through 49 years, 1975–2015. JAMA 2019; 321(19):1933–1934. doi:10.1001/jama.2019.3076
- Mason JB, Dickstein A, Jacques PF, et al. A temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: a hypothesis. Cancer Epidemiol Biomarkers Prev 2007;16(7):1325–1329. doi:10.1158/1055-9965.EPI-07-0329
- Cole BF, Baron JA, Sandler RS, et al; POlyp Prevention Study Group. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA 2007; 297(21):2351–2359. doi:10.1001/jama.297.21.2351
- Oliai Araghi S, Kiefte-de Jong JC, van Dijk SC, et al. Folic acid and vitamin B12 supplementation and the risk of cancer: long-term follow-up of the B vitamins for the prevention of osteoporotic fractures (B-PROOF) Trial. Cancer Epidemiol Biomarkers Prev 2019; 28(2):275–282. doi:10.1158/1055-9965.EPI-17-1198
To the Editor: In their paper on colorectal cancer screening, Mankaney and colleagues noted the increasing rates of colorectal cancer in young adults in the United States.1 Recent epidemiologic data demonstrate an increasing incidence of the disease in people ages 40 through 49 since the mid-1990s.2 Even though screening starting at age 45 is not uniformly accepted,1 there is evidence supporting earlier screening.
During the mid-1990s, the US government mandated that all enriched flour and uncooked cereal grains were to be fortified with folic acid in order to prevent births complicated by neural tube defects.3 Subsequently, there was a 2-fold increase in plasma folate concentrations and, disturbingly, a temporally associated significant increase in the incidence of colorectal cancer.3
Notably, a US trial4 testing the efficacy of folic acid 1 mg taken daily for 6 years to prevent colorectal adenomas in those with a history of colorectal adenomas failed to show a reduction in adenoma risk. Instead, participants randomized to folic acid exhibited a significantly increased risk of an advanced adenoma. Another trial,5 conducted in the Netherlands, where there is no mandatory folic acid fortification, investigated folic acid 400 µg and vitamin B12 500 µg daily over 2 to 3 years for the prevention of osteoporotic fractures. The group randomized to the vitamins had a nearly 2-fold increase in the risk of colorectal cancer.
Folic acid can be a double-edged sword.3,5 Although folic acid intake may protect against carcinogenesis through increased genetic stability, if precancerous or neoplastic cells are present, excess folic acid may promote cancer by increasing DNA synthesis and cell proliferation. Cancer cells have folic acid receptors.
Since screening colonoscopy is typically done in individuals over 50, advanced adenomas from folic acid exposure in people younger than 50 likely go undiagnosed. Therefore, colorectal cancer screening should start at a younger age in countries where folic acid fortification is mandatory.
To the Editor: In their paper on colorectal cancer screening, Mankaney and colleagues noted the increasing rates of colorectal cancer in young adults in the United States.1 Recent epidemiologic data demonstrate an increasing incidence of the disease in people ages 40 through 49 since the mid-1990s.2 Even though screening starting at age 45 is not uniformly accepted,1 there is evidence supporting earlier screening.
During the mid-1990s, the US government mandated that all enriched flour and uncooked cereal grains were to be fortified with folic acid in order to prevent births complicated by neural tube defects.3 Subsequently, there was a 2-fold increase in plasma folate concentrations and, disturbingly, a temporally associated significant increase in the incidence of colorectal cancer.3
Notably, a US trial4 testing the efficacy of folic acid 1 mg taken daily for 6 years to prevent colorectal adenomas in those with a history of colorectal adenomas failed to show a reduction in adenoma risk. Instead, participants randomized to folic acid exhibited a significantly increased risk of an advanced adenoma. Another trial,5 conducted in the Netherlands, where there is no mandatory folic acid fortification, investigated folic acid 400 µg and vitamin B12 500 µg daily over 2 to 3 years for the prevention of osteoporotic fractures. The group randomized to the vitamins had a nearly 2-fold increase in the risk of colorectal cancer.
Folic acid can be a double-edged sword.3,5 Although folic acid intake may protect against carcinogenesis through increased genetic stability, if precancerous or neoplastic cells are present, excess folic acid may promote cancer by increasing DNA synthesis and cell proliferation. Cancer cells have folic acid receptors.
Since screening colonoscopy is typically done in individuals over 50, advanced adenomas from folic acid exposure in people younger than 50 likely go undiagnosed. Therefore, colorectal cancer screening should start at a younger age in countries where folic acid fortification is mandatory.
- Mankaney G, Sutton RA, Burke CA. Colorectal cancer screening: choosing the right test. Cleve Clin J Med 2019; 86(6):385–392. doi:10.3949/ccjm.86a.17125
- Meester RGS, Mannalithara A, Lansdorp-Vogelaar I, Ladabaum U. Trends in incidence and stage at diagnosis of colorectal cancer in adults aged 40 through 49 years, 1975–2015. JAMA 2019; 321(19):1933–1934. doi:10.1001/jama.2019.3076
- Mason JB, Dickstein A, Jacques PF, et al. A temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: a hypothesis. Cancer Epidemiol Biomarkers Prev 2007;16(7):1325–1329. doi:10.1158/1055-9965.EPI-07-0329
- Cole BF, Baron JA, Sandler RS, et al; POlyp Prevention Study Group. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA 2007; 297(21):2351–2359. doi:10.1001/jama.297.21.2351
- Oliai Araghi S, Kiefte-de Jong JC, van Dijk SC, et al. Folic acid and vitamin B12 supplementation and the risk of cancer: long-term follow-up of the B vitamins for the prevention of osteoporotic fractures (B-PROOF) Trial. Cancer Epidemiol Biomarkers Prev 2019; 28(2):275–282. doi:10.1158/1055-9965.EPI-17-1198
- Mankaney G, Sutton RA, Burke CA. Colorectal cancer screening: choosing the right test. Cleve Clin J Med 2019; 86(6):385–392. doi:10.3949/ccjm.86a.17125
- Meester RGS, Mannalithara A, Lansdorp-Vogelaar I, Ladabaum U. Trends in incidence and stage at diagnosis of colorectal cancer in adults aged 40 through 49 years, 1975–2015. JAMA 2019; 321(19):1933–1934. doi:10.1001/jama.2019.3076
- Mason JB, Dickstein A, Jacques PF, et al. A temporal association between folic acid fortification and an increase in colorectal cancer rates may be illuminating important biological principles: a hypothesis. Cancer Epidemiol Biomarkers Prev 2007;16(7):1325–1329. doi:10.1158/1055-9965.EPI-07-0329
- Cole BF, Baron JA, Sandler RS, et al; POlyp Prevention Study Group. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA 2007; 297(21):2351–2359. doi:10.1001/jama.297.21.2351
- Oliai Araghi S, Kiefte-de Jong JC, van Dijk SC, et al. Folic acid and vitamin B12 supplementation and the risk of cancer: long-term follow-up of the B vitamins for the prevention of osteoporotic fractures (B-PROOF) Trial. Cancer Epidemiol Biomarkers Prev 2019; 28(2):275–282. doi:10.1158/1055-9965.EPI-17-1198
Colorectal cancer screening: Colonoscopy has disadvantages
To the Editor: In the article, “Colorectal cancer screening: Choosing the right test,” the authors offer an excellent review, but restrict the discussion to just 2 of the many options. Screening compliance improves when clinicians and patients can select their preferred screening approach, and other noninvasive or minimally invasive approaches also deserve consideration and may well be superior. It is important that both the patient and the healthcare provider be fully aware of the advantages and disadvantages of each method.
The article is overly generous in its description of the accuracy and sensitivity of optical colonoscopy. The statement that colonoscopy visualizes the entire colon in more than 98% of cases is not supported by the biomedical literature or clinical experience. The measure of colonoscopy accuracy is best quantified by a review of more than 15,000 tandem colonoscopies that showed an average polyp miss rate of 22% using standard colonoscopes, and a 69% polyp miss rate compared with full-spectrum colonoscopes with greater fields of view.1–3 Between 5% and 10% of colonoscopies are technically incomplete and do not reach the cecum. Only 35% of colonoscopy bowel preps are excellent, and 21% are so poor that the procedure cannot be completed.4–8 Colorectal cancers are frequently missed at colonoscopy, with a rate of 7% quoted in the literature for interval cancer development.9–16 Studies of computed tomography colonography (virtual colonoscopy) have confirmed that between 10% and 20% of the colonic mucosa is hidden from view on optical colonoscopy by tall haustral mucosal folds.17,18 The operator variation measured by adenoma detection rates can exceed a 10-fold differential.
Colonoscopy is an important and valuable diagnostic and therapeutic tool. The disadvantages include significant cancer and polyp miss rates, high discomfort, high expense, potentially life-threatening complications, time- and resource-intensive utilization, high loss of patient work productivity, challenging and frequently inadequate preparation, higher risk of metachronous cancer and polyp spread, and high operator variability of quality.19–24 Unfortunately, while colonoscopy is an important tool, it does not come anywhere close to a score of 98% and should not be considered the gold standard for colorectal cancer screening.25
- Zhao S, Wang S, Pan P, et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: a systemic review and meta-analysis. Gastroenterology 2019; 156(6):1661–1674. doi:10.1053/j.gastro.2019.01.260
- van Rijn JC, Reitsma JB, Stoker J, Bossuyt PM, van Deventer SJ, Dekker E. Polyp miss rate determined by tandem colonoscopy: a systematic review. Am J Gastroenterol 2006; 101(2):343–350. doi:10.1111/j.1572-0241.2006.00390.x
- Gralnek IM, Siersema PD, Halpern Z, et al. Standard forward-viewing colonoscopy versus full-spectrum endoscopy: an international, multicenter, randomised, tandem colonoscopy trial. Lancet Oncol 2014; 15(3):353–360. doi:10.1016/S1470-2045(14)70020-8
- Ness RM, Manam R, Hoen H, Chalasani N. Predictors of inadequate bowel preparation for colonoscopy. Am J Gastroenterol 2001; 96(6):1797–1802. doi:10.1111/j.1572-0241.2001.03874.x
- Kluge M, Williams J, Wu C, et al. Inadequate Boston Bowel Preparation Scale scores predict the risk of missed neoplasia on the next colonoscopy. Gastrointest Endosc 2018; 87(3):744–751. doi:10.1016/j.gie.2017.06.012
- Gagneja H, Parekh P, Burleson D, et al. HyGIeaCare® preparation for colonoscopy – a technical update for success. J Gastrointest Dig Syst 2016; 6:4. doi:10.4172/2161-069X.1000458
- Das A, Parekh P, Bekal P, et al. Bowel preparation for colonoscopy: a comparative cost-effective analysis of traditional per os purgatory prep versus a novel method using high-volume colonic water irrigation. Gastroenterol Hepatol Int J 2017; 2(4):000132.
- D’Souza SM, Parekh PJ, Johnson DA. The dirty side of colonoscopy: predictors of poor bowel preparation and novel approaches to overcome the shortcomings. Br J Gastroenterol 2019: 1:1. https://hygieacare.com/wp-content/uploads/2019/06/The-Dirty-Side-of-Colonoscopy-PDF.pdf. Accessed October 23, 2019.
- Mouchli M, Ouk L, Scheitel M. Colonoscopy surveillance for high risk polyps does not always prevent colorectal cancer. World J Gastroenterol 2018; 24(8):905–916. doi:10.3748/wjg.v24.i8.905
- Adler J, Robertson DJ. Interval colorectal cancer after colonoscopy: exploring explanations and solutions. Am J Gastroenterol 2015; 110(12):1657–1664. doi:10.1038/ajg.2015.365
- Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multi-cohort analysis. Gut 2014; 63(6):949–956. doi:10.1136/gutjnl-2012-303796
- Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 2011; 154(1):22–30. doi:10.7326/0003-4819-154-1-201101040-00004
- Brenner H, Chang-Claude J, Seiler CM, Hoffmeister M. Long-term risk of colorectal cancer after negative colonoscopy. J Clin Oncol 2011; 29(28):3761–3767. doi:10.1200/JCO.2011.35.9307
- Pohl H, Robertson DJ. Colorectal cancers detected after colonoscopy frequently result from missed lesions. Clin Gastroenterol Hepatol 2010; 8(10):858–864. doi:10.1016/j.cgh.2010.06.028
- Singh H, Nugent Z, Demers AA, Bernstein CN. Rate and predictors of early/missed colorectal cancers after colonoscopy in Manitoba: a population-based study. Am J Gastroenterol 2010; 105(12):2588–2596. doi:10.1038/ajg.2010.390
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013; 369(12):1095–1105. doi:10.1056/NEJMoa1301969
- Thompson A, Jones R, Pou P, et al. Taller haustral folds in the proximal colon: a potential factor contributing to interval colorectal cancer. J Colon Rectal Cancer 2016; 1(1):45–54. doi:10.14302/issn.2471-7061.jcrc-15-899
- Zhu H, Barish M, Pickhardt P, et al. Haustral fold segmentation with curvature-guided level set evolution. IEEE Trans Biomed Eng 2013; 60(2):321–331. doi:10.1109/TBME.2012.2226242
- Chukmaitov A, Bradley CJ, Dahman B, Siangphoe U, Warren JL, Klabunde CN. Association of polypectomy techniques, endoscopist volume, and facility type with colonoscopy complications. Gastrointest Endosc 2013; 77(3):436–446. doi:10.1016/j.gie.2012.11.012
- Reumkens A, Rondagh EJ, Bakker CM, et al. Post-colonoscopy complications: a systematic review, time trends, and meta-analysis of population-based studies. Am J Gastroenterol 2016; 111(8):1092–1101. doi:10.1038/ajg.2016.234
- ASGE Standards of Practice Committee, Fisher DA, Maple JT, Ben-Menachem T, et al. Complications of colonoscopy. Gastrointest Endosc 2011; 74(4):745–752. doi:10.1016/j.gie.2011.07.025
- Warren JL, Klabunde CN, Mariotto AB, et al. Adverse events after outpatient colonoscopy in the Medicare population. Ann Intern Med 2009; 150(12):849–857. doi:10.7326/0003-4819-150-12-200906160-00008
- Whitlock EP, Lin JS, Liles E, et al. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2008; 149(9):638–658. doi:10.7326/0003-4819-149-9-200811040-00245
- Backes Y, Seerden T, van Gestel R, et al. Tumor seeding during colonoscopy as a possible cause for metachronous colorectal cancer. Gastroenterology 2019; Aug 13. pii: S0016-5085(19)41229-8. [Epub ahead of print] doi:10.1053/j.gastro.2019.07.062
- Lin JS, Piper MA, Perdue LA, et al. Screening for colorectal cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315(23):2576–2594. doi:10.1001/jama.2016.3332
To the Editor: In the article, “Colorectal cancer screening: Choosing the right test,” the authors offer an excellent review, but restrict the discussion to just 2 of the many options. Screening compliance improves when clinicians and patients can select their preferred screening approach, and other noninvasive or minimally invasive approaches also deserve consideration and may well be superior. It is important that both the patient and the healthcare provider be fully aware of the advantages and disadvantages of each method.
The article is overly generous in its description of the accuracy and sensitivity of optical colonoscopy. The statement that colonoscopy visualizes the entire colon in more than 98% of cases is not supported by the biomedical literature or clinical experience. The measure of colonoscopy accuracy is best quantified by a review of more than 15,000 tandem colonoscopies that showed an average polyp miss rate of 22% using standard colonoscopes, and a 69% polyp miss rate compared with full-spectrum colonoscopes with greater fields of view.1–3 Between 5% and 10% of colonoscopies are technically incomplete and do not reach the cecum. Only 35% of colonoscopy bowel preps are excellent, and 21% are so poor that the procedure cannot be completed.4–8 Colorectal cancers are frequently missed at colonoscopy, with a rate of 7% quoted in the literature for interval cancer development.9–16 Studies of computed tomography colonography (virtual colonoscopy) have confirmed that between 10% and 20% of the colonic mucosa is hidden from view on optical colonoscopy by tall haustral mucosal folds.17,18 The operator variation measured by adenoma detection rates can exceed a 10-fold differential.
Colonoscopy is an important and valuable diagnostic and therapeutic tool. The disadvantages include significant cancer and polyp miss rates, high discomfort, high expense, potentially life-threatening complications, time- and resource-intensive utilization, high loss of patient work productivity, challenging and frequently inadequate preparation, higher risk of metachronous cancer and polyp spread, and high operator variability of quality.19–24 Unfortunately, while colonoscopy is an important tool, it does not come anywhere close to a score of 98% and should not be considered the gold standard for colorectal cancer screening.25
To the Editor: In the article, “Colorectal cancer screening: Choosing the right test,” the authors offer an excellent review, but restrict the discussion to just 2 of the many options. Screening compliance improves when clinicians and patients can select their preferred screening approach, and other noninvasive or minimally invasive approaches also deserve consideration and may well be superior. It is important that both the patient and the healthcare provider be fully aware of the advantages and disadvantages of each method.
The article is overly generous in its description of the accuracy and sensitivity of optical colonoscopy. The statement that colonoscopy visualizes the entire colon in more than 98% of cases is not supported by the biomedical literature or clinical experience. The measure of colonoscopy accuracy is best quantified by a review of more than 15,000 tandem colonoscopies that showed an average polyp miss rate of 22% using standard colonoscopes, and a 69% polyp miss rate compared with full-spectrum colonoscopes with greater fields of view.1–3 Between 5% and 10% of colonoscopies are technically incomplete and do not reach the cecum. Only 35% of colonoscopy bowel preps are excellent, and 21% are so poor that the procedure cannot be completed.4–8 Colorectal cancers are frequently missed at colonoscopy, with a rate of 7% quoted in the literature for interval cancer development.9–16 Studies of computed tomography colonography (virtual colonoscopy) have confirmed that between 10% and 20% of the colonic mucosa is hidden from view on optical colonoscopy by tall haustral mucosal folds.17,18 The operator variation measured by adenoma detection rates can exceed a 10-fold differential.
Colonoscopy is an important and valuable diagnostic and therapeutic tool. The disadvantages include significant cancer and polyp miss rates, high discomfort, high expense, potentially life-threatening complications, time- and resource-intensive utilization, high loss of patient work productivity, challenging and frequently inadequate preparation, higher risk of metachronous cancer and polyp spread, and high operator variability of quality.19–24 Unfortunately, while colonoscopy is an important tool, it does not come anywhere close to a score of 98% and should not be considered the gold standard for colorectal cancer screening.25
- Zhao S, Wang S, Pan P, et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: a systemic review and meta-analysis. Gastroenterology 2019; 156(6):1661–1674. doi:10.1053/j.gastro.2019.01.260
- van Rijn JC, Reitsma JB, Stoker J, Bossuyt PM, van Deventer SJ, Dekker E. Polyp miss rate determined by tandem colonoscopy: a systematic review. Am J Gastroenterol 2006; 101(2):343–350. doi:10.1111/j.1572-0241.2006.00390.x
- Gralnek IM, Siersema PD, Halpern Z, et al. Standard forward-viewing colonoscopy versus full-spectrum endoscopy: an international, multicenter, randomised, tandem colonoscopy trial. Lancet Oncol 2014; 15(3):353–360. doi:10.1016/S1470-2045(14)70020-8
- Ness RM, Manam R, Hoen H, Chalasani N. Predictors of inadequate bowel preparation for colonoscopy. Am J Gastroenterol 2001; 96(6):1797–1802. doi:10.1111/j.1572-0241.2001.03874.x
- Kluge M, Williams J, Wu C, et al. Inadequate Boston Bowel Preparation Scale scores predict the risk of missed neoplasia on the next colonoscopy. Gastrointest Endosc 2018; 87(3):744–751. doi:10.1016/j.gie.2017.06.012
- Gagneja H, Parekh P, Burleson D, et al. HyGIeaCare® preparation for colonoscopy – a technical update for success. J Gastrointest Dig Syst 2016; 6:4. doi:10.4172/2161-069X.1000458
- Das A, Parekh P, Bekal P, et al. Bowel preparation for colonoscopy: a comparative cost-effective analysis of traditional per os purgatory prep versus a novel method using high-volume colonic water irrigation. Gastroenterol Hepatol Int J 2017; 2(4):000132.
- D’Souza SM, Parekh PJ, Johnson DA. The dirty side of colonoscopy: predictors of poor bowel preparation and novel approaches to overcome the shortcomings. Br J Gastroenterol 2019: 1:1. https://hygieacare.com/wp-content/uploads/2019/06/The-Dirty-Side-of-Colonoscopy-PDF.pdf. Accessed October 23, 2019.
- Mouchli M, Ouk L, Scheitel M. Colonoscopy surveillance for high risk polyps does not always prevent colorectal cancer. World J Gastroenterol 2018; 24(8):905–916. doi:10.3748/wjg.v24.i8.905
- Adler J, Robertson DJ. Interval colorectal cancer after colonoscopy: exploring explanations and solutions. Am J Gastroenterol 2015; 110(12):1657–1664. doi:10.1038/ajg.2015.365
- Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multi-cohort analysis. Gut 2014; 63(6):949–956. doi:10.1136/gutjnl-2012-303796
- Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 2011; 154(1):22–30. doi:10.7326/0003-4819-154-1-201101040-00004
- Brenner H, Chang-Claude J, Seiler CM, Hoffmeister M. Long-term risk of colorectal cancer after negative colonoscopy. J Clin Oncol 2011; 29(28):3761–3767. doi:10.1200/JCO.2011.35.9307
- Pohl H, Robertson DJ. Colorectal cancers detected after colonoscopy frequently result from missed lesions. Clin Gastroenterol Hepatol 2010; 8(10):858–864. doi:10.1016/j.cgh.2010.06.028
- Singh H, Nugent Z, Demers AA, Bernstein CN. Rate and predictors of early/missed colorectal cancers after colonoscopy in Manitoba: a population-based study. Am J Gastroenterol 2010; 105(12):2588–2596. doi:10.1038/ajg.2010.390
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013; 369(12):1095–1105. doi:10.1056/NEJMoa1301969
- Thompson A, Jones R, Pou P, et al. Taller haustral folds in the proximal colon: a potential factor contributing to interval colorectal cancer. J Colon Rectal Cancer 2016; 1(1):45–54. doi:10.14302/issn.2471-7061.jcrc-15-899
- Zhu H, Barish M, Pickhardt P, et al. Haustral fold segmentation with curvature-guided level set evolution. IEEE Trans Biomed Eng 2013; 60(2):321–331. doi:10.1109/TBME.2012.2226242
- Chukmaitov A, Bradley CJ, Dahman B, Siangphoe U, Warren JL, Klabunde CN. Association of polypectomy techniques, endoscopist volume, and facility type with colonoscopy complications. Gastrointest Endosc 2013; 77(3):436–446. doi:10.1016/j.gie.2012.11.012
- Reumkens A, Rondagh EJ, Bakker CM, et al. Post-colonoscopy complications: a systematic review, time trends, and meta-analysis of population-based studies. Am J Gastroenterol 2016; 111(8):1092–1101. doi:10.1038/ajg.2016.234
- ASGE Standards of Practice Committee, Fisher DA, Maple JT, Ben-Menachem T, et al. Complications of colonoscopy. Gastrointest Endosc 2011; 74(4):745–752. doi:10.1016/j.gie.2011.07.025
- Warren JL, Klabunde CN, Mariotto AB, et al. Adverse events after outpatient colonoscopy in the Medicare population. Ann Intern Med 2009; 150(12):849–857. doi:10.7326/0003-4819-150-12-200906160-00008
- Whitlock EP, Lin JS, Liles E, et al. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2008; 149(9):638–658. doi:10.7326/0003-4819-149-9-200811040-00245
- Backes Y, Seerden T, van Gestel R, et al. Tumor seeding during colonoscopy as a possible cause for metachronous colorectal cancer. Gastroenterology 2019; Aug 13. pii: S0016-5085(19)41229-8. [Epub ahead of print] doi:10.1053/j.gastro.2019.07.062
- Lin JS, Piper MA, Perdue LA, et al. Screening for colorectal cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315(23):2576–2594. doi:10.1001/jama.2016.3332
- Zhao S, Wang S, Pan P, et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: a systemic review and meta-analysis. Gastroenterology 2019; 156(6):1661–1674. doi:10.1053/j.gastro.2019.01.260
- van Rijn JC, Reitsma JB, Stoker J, Bossuyt PM, van Deventer SJ, Dekker E. Polyp miss rate determined by tandem colonoscopy: a systematic review. Am J Gastroenterol 2006; 101(2):343–350. doi:10.1111/j.1572-0241.2006.00390.x
- Gralnek IM, Siersema PD, Halpern Z, et al. Standard forward-viewing colonoscopy versus full-spectrum endoscopy: an international, multicenter, randomised, tandem colonoscopy trial. Lancet Oncol 2014; 15(3):353–360. doi:10.1016/S1470-2045(14)70020-8
- Ness RM, Manam R, Hoen H, Chalasani N. Predictors of inadequate bowel preparation for colonoscopy. Am J Gastroenterol 2001; 96(6):1797–1802. doi:10.1111/j.1572-0241.2001.03874.x
- Kluge M, Williams J, Wu C, et al. Inadequate Boston Bowel Preparation Scale scores predict the risk of missed neoplasia on the next colonoscopy. Gastrointest Endosc 2018; 87(3):744–751. doi:10.1016/j.gie.2017.06.012
- Gagneja H, Parekh P, Burleson D, et al. HyGIeaCare® preparation for colonoscopy – a technical update for success. J Gastrointest Dig Syst 2016; 6:4. doi:10.4172/2161-069X.1000458
- Das A, Parekh P, Bekal P, et al. Bowel preparation for colonoscopy: a comparative cost-effective analysis of traditional per os purgatory prep versus a novel method using high-volume colonic water irrigation. Gastroenterol Hepatol Int J 2017; 2(4):000132.
- D’Souza SM, Parekh PJ, Johnson DA. The dirty side of colonoscopy: predictors of poor bowel preparation and novel approaches to overcome the shortcomings. Br J Gastroenterol 2019: 1:1. https://hygieacare.com/wp-content/uploads/2019/06/The-Dirty-Side-of-Colonoscopy-PDF.pdf. Accessed October 23, 2019.
- Mouchli M, Ouk L, Scheitel M. Colonoscopy surveillance for high risk polyps does not always prevent colorectal cancer. World J Gastroenterol 2018; 24(8):905–916. doi:10.3748/wjg.v24.i8.905
- Adler J, Robertson DJ. Interval colorectal cancer after colonoscopy: exploring explanations and solutions. Am J Gastroenterol 2015; 110(12):1657–1664. doi:10.1038/ajg.2015.365
- Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multi-cohort analysis. Gut 2014; 63(6):949–956. doi:10.1136/gutjnl-2012-303796
- Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 2011; 154(1):22–30. doi:10.7326/0003-4819-154-1-201101040-00004
- Brenner H, Chang-Claude J, Seiler CM, Hoffmeister M. Long-term risk of colorectal cancer after negative colonoscopy. J Clin Oncol 2011; 29(28):3761–3767. doi:10.1200/JCO.2011.35.9307
- Pohl H, Robertson DJ. Colorectal cancers detected after colonoscopy frequently result from missed lesions. Clin Gastroenterol Hepatol 2010; 8(10):858–864. doi:10.1016/j.cgh.2010.06.028
- Singh H, Nugent Z, Demers AA, Bernstein CN. Rate and predictors of early/missed colorectal cancers after colonoscopy in Manitoba: a population-based study. Am J Gastroenterol 2010; 105(12):2588–2596. doi:10.1038/ajg.2010.390
- Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013; 369(12):1095–1105. doi:10.1056/NEJMoa1301969
- Thompson A, Jones R, Pou P, et al. Taller haustral folds in the proximal colon: a potential factor contributing to interval colorectal cancer. J Colon Rectal Cancer 2016; 1(1):45–54. doi:10.14302/issn.2471-7061.jcrc-15-899
- Zhu H, Barish M, Pickhardt P, et al. Haustral fold segmentation with curvature-guided level set evolution. IEEE Trans Biomed Eng 2013; 60(2):321–331. doi:10.1109/TBME.2012.2226242
- Chukmaitov A, Bradley CJ, Dahman B, Siangphoe U, Warren JL, Klabunde CN. Association of polypectomy techniques, endoscopist volume, and facility type with colonoscopy complications. Gastrointest Endosc 2013; 77(3):436–446. doi:10.1016/j.gie.2012.11.012
- Reumkens A, Rondagh EJ, Bakker CM, et al. Post-colonoscopy complications: a systematic review, time trends, and meta-analysis of population-based studies. Am J Gastroenterol 2016; 111(8):1092–1101. doi:10.1038/ajg.2016.234
- ASGE Standards of Practice Committee, Fisher DA, Maple JT, Ben-Menachem T, et al. Complications of colonoscopy. Gastrointest Endosc 2011; 74(4):745–752. doi:10.1016/j.gie.2011.07.025
- Warren JL, Klabunde CN, Mariotto AB, et al. Adverse events after outpatient colonoscopy in the Medicare population. Ann Intern Med 2009; 150(12):849–857. doi:10.7326/0003-4819-150-12-200906160-00008
- Whitlock EP, Lin JS, Liles E, et al. Screening for colorectal cancer: a targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2008; 149(9):638–658. doi:10.7326/0003-4819-149-9-200811040-00245
- Backes Y, Seerden T, van Gestel R, et al. Tumor seeding during colonoscopy as a possible cause for metachronous colorectal cancer. Gastroenterology 2019; Aug 13. pii: S0016-5085(19)41229-8. [Epub ahead of print] doi:10.1053/j.gastro.2019.07.062
- Lin JS, Piper MA, Perdue LA, et al. Screening for colorectal cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 315(23):2576–2594. doi:10.1001/jama.2016.3332