User login
Use of genetic testing for congenital heart defect management
The average student in America learns that genes form the building blocks of what makes us human by the time they receive their high school diploma. Indeed, the completion of the Human Genome Project in 2003 paved the way for our genetic makeup, much like our medical history, to become a routine part of our health care. For example, our faculty at the University of Maryland School of Medicine discovered an important gene – CYP2C19 – which is involved in the metabolism of the antiplatelet medicine clopidogrel (Plavix). Although most people have this gene, some don’t. Therefore, when we manage a patient with coronary disease, we use a genetic screen to determine whether that patient has CYP2C19 and then modify therapy based on these results.

Our genes also have become commodities – from companies willing to analyze our genes to determine our racial and ethnic ancestry or propensity for certain diseases to those that can sequence the family dog’s genes.
Advances in genomics similarly have impacted ob.gyn. practice. Because of rapidly evolving gene analysis tools, we can now, for example, noninvasively test a developing fetus’s risk for chromosomal abnormalities and determine a baby’s sex by merely examining fetal DNA in a pregnant woman’s bloodstream. Although not diagnostic, these gene-based prenatal screening tests have reduced the need for unnecessary, costly, and highly invasive procedures for many of our patients.
Importantly, our recognition that certain genes can confer a higher risk of disease has meant that performing a prenatal genetic evaluation can greatly inform the mother and her care team about potential problems her baby may have that may require additional management. For babies who have congenital heart defects, a genetic evaluation performed in addition to sonographic examination can provide ob.gyns. with crucial details to enhance pregnancy management and postnatal care decisions.
The importance of genetic testing and analysis in the detection, treatment, and prevention of congenital heart defects is the topic of part two of this two-part Master Class series authored by Shifa Turan, MD, associate professor of obstetrics, gynecology, and reproductive sciences at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. By using a combination of three- and four-dimensional ultrasound with gene assays, Dr. Turan and her colleagues can greatly enhance and personalize the care they deliver to their patients.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland School of Medicine, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
The average student in America learns that genes form the building blocks of what makes us human by the time they receive their high school diploma. Indeed, the completion of the Human Genome Project in 2003 paved the way for our genetic makeup, much like our medical history, to become a routine part of our health care. For example, our faculty at the University of Maryland School of Medicine discovered an important gene – CYP2C19 – which is involved in the metabolism of the antiplatelet medicine clopidogrel (Plavix). Although most people have this gene, some don’t. Therefore, when we manage a patient with coronary disease, we use a genetic screen to determine whether that patient has CYP2C19 and then modify therapy based on these results.
Our genes also have become commodities – from companies willing to analyze our genes to determine our racial and ethnic ancestry or propensity for certain diseases to those that can sequence the family dog’s genes.
Advances in genomics similarly have impacted ob.gyn. practice. Because of rapidly evolving gene analysis tools, we can now, for example, noninvasively test a developing fetus’s risk for chromosomal abnormalities and determine a baby’s sex by merely examining fetal DNA in a pregnant woman’s bloodstream. Although not diagnostic, these gene-based prenatal screening tests have reduced the need for unnecessary, costly, and highly invasive procedures for many of our patients.
Importantly, our recognition that certain genes can confer a higher risk of disease has meant that performing a prenatal genetic evaluation can greatly inform the mother and her care team about potential problems her baby may have that may require additional management. For babies who have congenital heart defects, a genetic evaluation performed in addition to sonographic examination can provide ob.gyns. with crucial details to enhance pregnancy management and postnatal care decisions.
The importance of genetic testing and analysis in the detection, treatment, and prevention of congenital heart defects is the topic of part two of this two-part Master Class series authored by Shifa Turan, MD, associate professor of obstetrics, gynecology, and reproductive sciences at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. By using a combination of three- and four-dimensional ultrasound with gene assays, Dr. Turan and her colleagues can greatly enhance and personalize the care they deliver to their patients.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland School of Medicine, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
The average student in America learns that genes form the building blocks of what makes us human by the time they receive their high school diploma. Indeed, the completion of the Human Genome Project in 2003 paved the way for our genetic makeup, much like our medical history, to become a routine part of our health care. For example, our faculty at the University of Maryland School of Medicine discovered an important gene – CYP2C19 – which is involved in the metabolism of the antiplatelet medicine clopidogrel (Plavix). Although most people have this gene, some don’t. Therefore, when we manage a patient with coronary disease, we use a genetic screen to determine whether that patient has CYP2C19 and then modify therapy based on these results.
Our genes also have become commodities – from companies willing to analyze our genes to determine our racial and ethnic ancestry or propensity for certain diseases to those that can sequence the family dog’s genes.
Advances in genomics similarly have impacted ob.gyn. practice. Because of rapidly evolving gene analysis tools, we can now, for example, noninvasively test a developing fetus’s risk for chromosomal abnormalities and determine a baby’s sex by merely examining fetal DNA in a pregnant woman’s bloodstream. Although not diagnostic, these gene-based prenatal screening tests have reduced the need for unnecessary, costly, and highly invasive procedures for many of our patients.
Importantly, our recognition that certain genes can confer a higher risk of disease has meant that performing a prenatal genetic evaluation can greatly inform the mother and her care team about potential problems her baby may have that may require additional management. For babies who have congenital heart defects, a genetic evaluation performed in addition to sonographic examination can provide ob.gyns. with crucial details to enhance pregnancy management and postnatal care decisions.
The importance of genetic testing and analysis in the detection, treatment, and prevention of congenital heart defects is the topic of part two of this two-part Master Class series authored by Shifa Turan, MD, associate professor of obstetrics, gynecology, and reproductive sciences at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. By using a combination of three- and four-dimensional ultrasound with gene assays, Dr. Turan and her colleagues can greatly enhance and personalize the care they deliver to their patients.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland School of Medicine, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
Genetic assessment for CHD: Case-specific, stepwise
Congenital heart defects (CHDs) are etiologically heterogeneous, but in recent years it has become clear that genetics plays a larger role in the development of CHDs than was previously thought. Research has been shifting from a focus on risk – estimating the magnitude of increased risk, for instance, based on maternal or familial risk factors – to a focus on the etiology of cardiac defects.
In practice, advances in genetic testing technologies have made the underlying causes of CHDs increasingly detectable. Chromosomal microarray analysis (CMA) – technology that detects significantly more and smaller changes in the amount of chromosomal material than traditional karyotype – has been proven to increase the diagnostic yield in cases of isolated CHDs and CHDs with extracardiac anomalies. Targeted next-generation sequencing also is now available as an additional approach in selective cases, and a clinically viable option for whole-exome sequencing is fast approaching.
For researchers, genetic evaluation carries the potential to unravel remaining mysteries about underlying causes of CHDs – to provide pathological insights and identify potential therapeutic targets. Currently, about 6 % of the total pie of presumed genetic determinants of CHDs is attributed to chromosomal anomalies, 10% to copy number variants, and 12% to single-gene defects. The remaining 72% of etiology, approximately, is undetermined.
As Helen Taussig, MD, (known as the founder of pediatric cardiology) once said, common cardiac malformations occurring in otherwise “normal” individuals “must be genetic in origin.”1 Greater use of genetic testing – and in particular, of whole-exome sequencing – will drive down this “undetermined” piece of the genetics pie.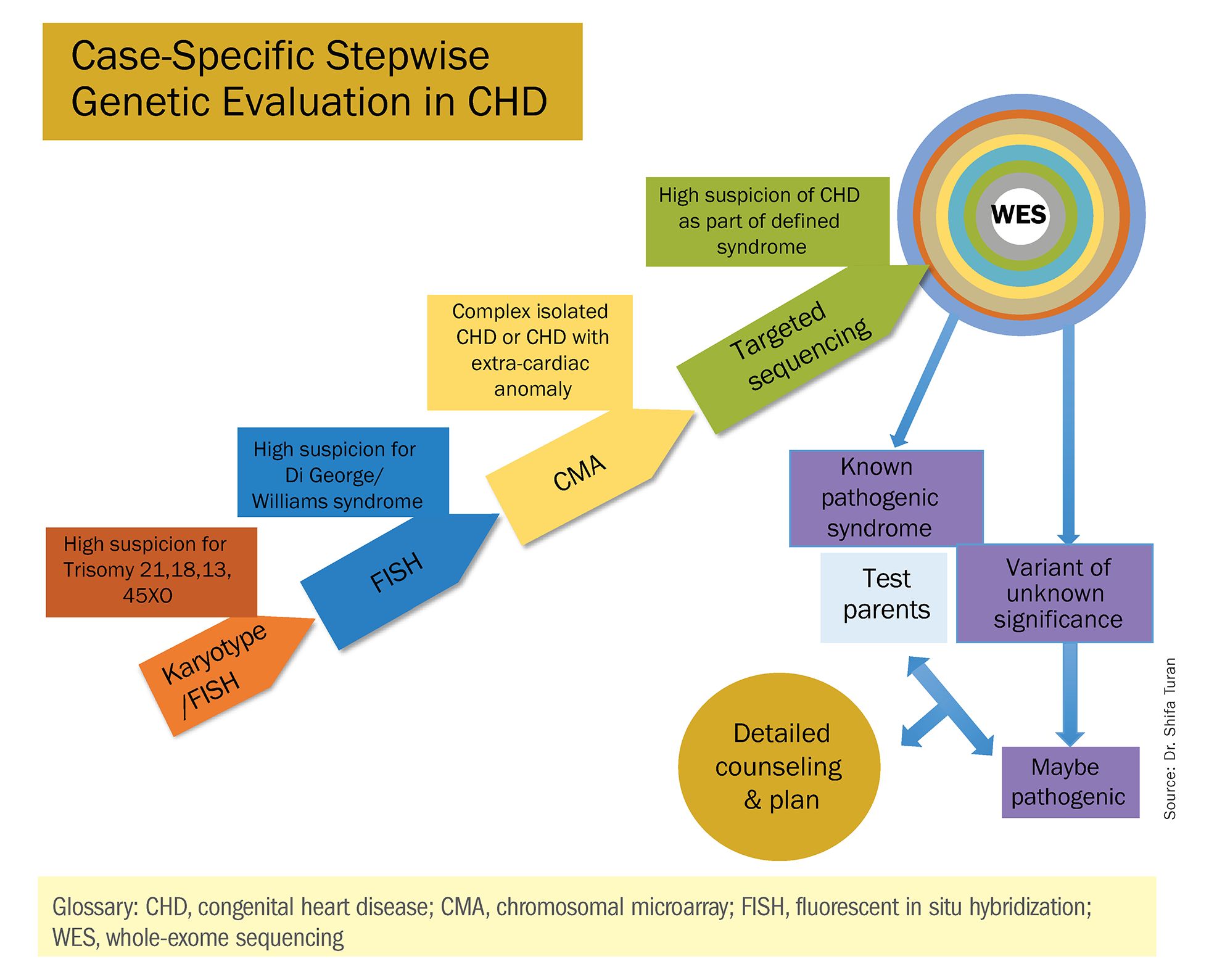
For clinicians and patients, prenatal genetic evaluation can inform clinical management, guiding decisions on the mode, timing, and location of delivery. Genetic assessments help guide the neonatal health care team in taking optimal care of the infant, and the surgeon in preparing for neonatal surgeries and postsurgical complications.
In a recent analysis of the Society of Thoracic Surgeons Congenital Heart Surgery Database, prenatal diagnosis was associated with a lower overall prevalence of major preoperative risk factors for cardiac surgery.2 Surgical outcomes themselves also have been shown to be better after the prenatal diagnosis of complex CHDs, mainly because of improvements in perioperative care.3
When genetic etiology is elucidated, the cardiologist also is better able to counsel patients about anticipated challenges – such as the propensity, with certain genetic variants of CHD, to develop neurodevelopmental delays or other cardiac complications – and to target patient follow-up. Patients also can make informed decisions about termination or continuation of a current pregnancy and about family planning in the future.
Fortunately, advances in genetics technology have paralleled technological advancements in ultrasound. As I discussed in part one of this two-part Master Class series, it is now possible to detect many major CHDs well before 16 weeks’ gestation. Checking the structure of the fetal heart at the first-trimester screening and sonography (11-14 weeks of gestation) offers the opportunity for early genetic assessment, counseling, and planning when anomalies are detected.
A personalized approach
There has been growing interest in recent years in CMA for the prenatal genetic workup of CHDs. Microarray targets chromosomal regions at a much higher resolution than traditional karyotype. Traditional karyotype assesses both changes in chromosome number as well as more subtle structural changes such as chromosomal deletions and duplications. CMA finds what traditional karyotype identifies, but in addition, it identifies much smaller, clinically relevant chromosomal deletions and duplications that are not detected by karyotype performed with or without fluorescence in-situ hybridization (FISH). FISH uses DNA probes that carry fluorescent tags to detect chromosomal DNA.
At our center, we studied the prenatal genetic test results of 145 fetuses diagnosed with CHDs. Each case involved FISH for aneuploidy/karyotype, followed by CMA in cases of a negative karyotype result. CMA increased the diagnostic yield in cases of CHD by 19.8% overall – 17.4% in cases of isolated CHD and 24.5% in cases of CHD plus extracardiac anomalies.4
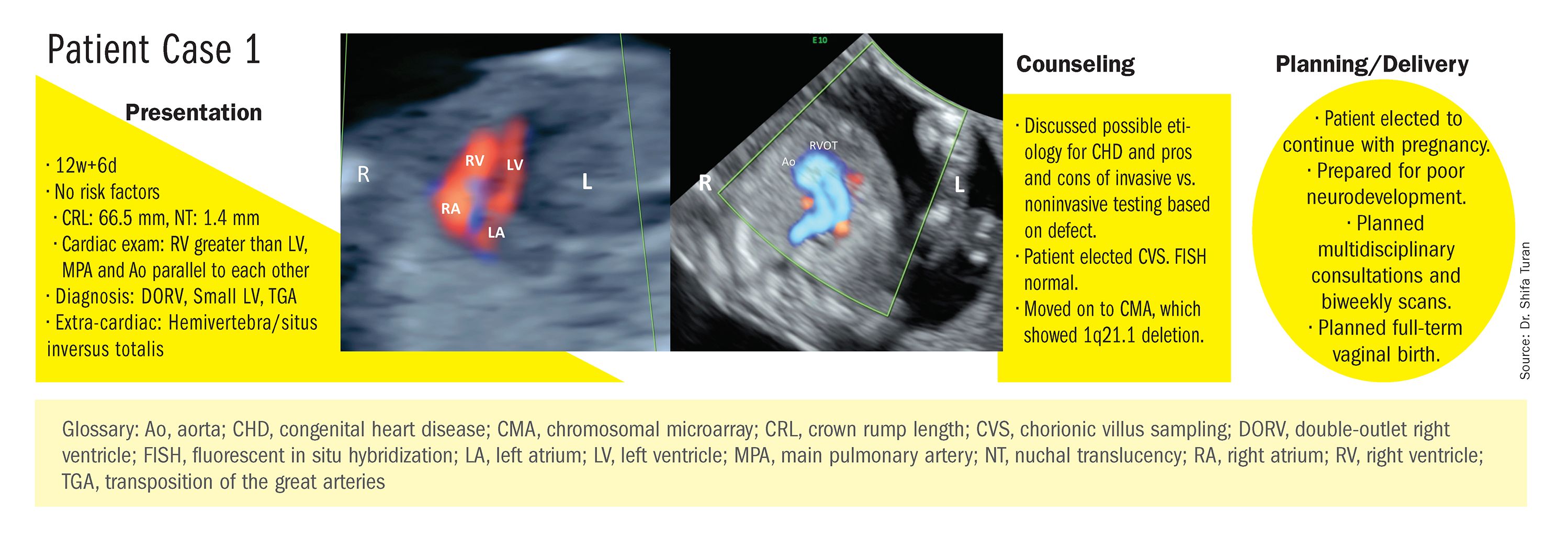
Indeed, although a microarray costs more and takes an additional 2 weeks to run, CMA should be strongly considered as first-line testing for the prenatal genetic evaluation of fetuses with major structural cardiac abnormalities detected by ultrasound. However, there still are cases in which a karyotype might be sufficient. For instance, if I see that a fetus has an atrial-ventricular septal defect on a prenatal ultrasound, and there are markers for trisomy 21, 13, or 18, or Turner’s syndrome (45 XO), I usually recommend a karyotype or FISH rather than an initial CMA. If the karyotype is abnormal – which is likely in such a scenario – there isn’t a need for more extensive testing.
Similarly, when there is high suspicion for DiGeorge syndrome (the 22q11.2 deletion, which often includes cleft palate and aortic arch abnormalities), usually it is most appropriate to perform a FISH test.
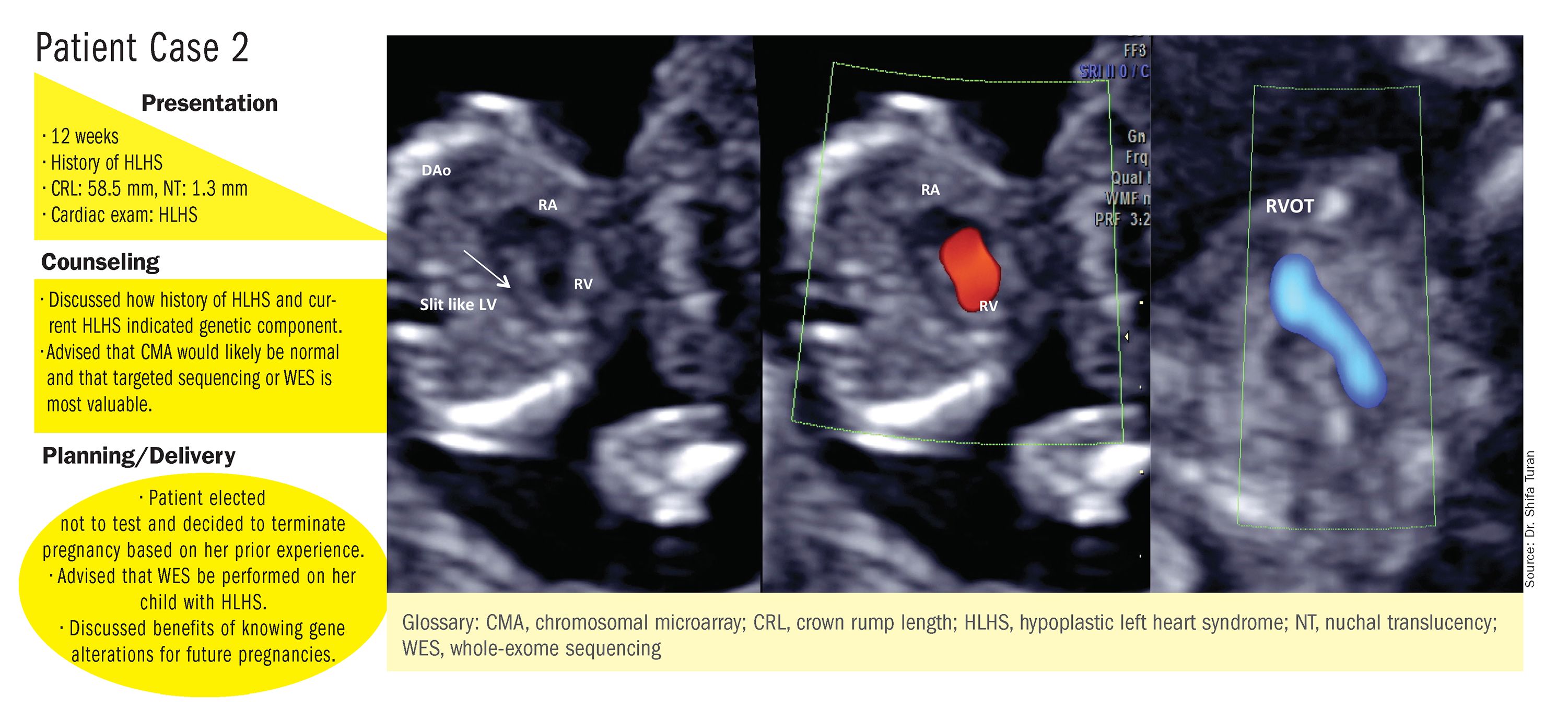
CMA is the preferred first modality, however, when prenatal imaging suggests severe CHD – for instance, when there are signs of hypoplastic left heart syndrome or tetralogy of Fallot (a conotruncal defect) – or complex CHD with extracardiac anomalies. In these cases, there is a high likelihood of detecting a small deletion or duplication that would be missed with karyotype.
In the past decade, karyotype and CMA have become the major methods used in our practice. However, targeted next‐generation sequencing and whole‐exome sequencing may become more widely used because these technologies enable rapid analysis of a large number of gene sequences and facilitate discovery of novel causative genes in many genetic diseases that cause CHDs.
Currently, targeted next-generation sequencing has mainly been used in the postnatal setting, and there are limited data available on its prenatal use. Compared with whole-exome sequencing, which sequences all of the protein-coding regions of the genome, targeted next-generation sequencing panels select regions of genes that are known to be associated with diseases of interest.
For CHDs, some perinatal centers have begun using a customized gene panel that targets 77 CHD-associated genes. This particular panel has been shown to be useful in addition to current methods and is an effective tool for prenatal genetic diagnosis.5
Whole-exome sequencing is currently expensive and time consuming. While sometimes it is used in the postnatal context, it is not yet part of routine practice as a prenatal diagnostic tool. As technology advances this will change – early in the next decade, I believe. For now, whole-exome sequencing may be an option for some patients who want to know more when severe CHD is evident on ultrasound and there are negative results from CMA or targeted sequencing. We have diagnosed some rare genetic syndromes using whole-exome sequencing; these diagnoses helped us to better manage the pregnancies.
These choices are part of the case-specific, stepwise approach to genetic evaluation that we take in our fetal heart program. Our goal is to pursue information that will be accurate and valuable for the patient and clinicians, in the most cost-effective and timely manner.
Limitations of noninvasive screening
In our fetal heart program we see increasing numbers of referred patients who have chosen noninvasive cell-free fetal DNA screening (cfDNA) after a cardiac anomaly is detected on ultrasound examination, and who believe that their “low risk” results demonstrate very little or no risk of CHD. Many of these patients express a belief that noninvasive testing is highly sensitive and accurate for fetal anomalies, including CHDs, and are not easily convinced of the value of other genetic tests.
We recently conducted a retrospective chart analysis (unpublished) in which we found that 41% of cases of CHD with abnormal genetics results were not detectable by cfDNA screening.
In the case of atrial-ventricular septal defects and conotruncal abnormalities that often are more associated with common aneuploidies (trisomy 21, 18, 13, and 45 XO), a “high-risk” result from cfDNA screening may offer the family and cardiology/neonatal team some guidance, but a “low-risk” result does not eliminate the risk of a microarray abnormality and thus may provide false reassurance.
Other research has shown that noninvasive screening will miss up to 7.3% of karyotype abnormalities in pregnancies at high risk for common aneuploidies.6
While invasive testing poses a very small risk of miscarriage, it is hard without such testing to elucidate the potential genetic etiologies of CHDs and truly understand the problems. We must take time to thoughtfully counsel patients who decline invasive testing about the limitations of cfDNA screening for CHDs and other anomalies.
Dr. Turan is an associate professor of obstetrics, gynecology, and reproductive sciences, and director of the fetal heart program at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. Dr. Turan reported that she has no disclosures relevant to this Master Class. Email her at obnews@mdedge.com.
References
1. J Am Coll Cardiol. 1988 Oct;12(4):1079-86.
2. Pediatr Cardiol. 2019 Mar;40(3):489-96.
3. Ann Pediatr Cardiol. 2017 May-Aug;10(2):126-30.
4. Eur J Obstet Gynecol Reprod Biol 2018;221:172-76.
5. Ultrasound Obstet Gynecol. 2018 Aug;52(2):205-11.
6. PLoS One. 2016 Jan 15;11(1):e0146794.
Congenital heart defects (CHDs) are etiologically heterogeneous, but in recent years it has become clear that genetics plays a larger role in the development of CHDs than was previously thought. Research has been shifting from a focus on risk – estimating the magnitude of increased risk, for instance, based on maternal or familial risk factors – to a focus on the etiology of cardiac defects.
In practice, advances in genetic testing technologies have made the underlying causes of CHDs increasingly detectable. Chromosomal microarray analysis (CMA) – technology that detects significantly more and smaller changes in the amount of chromosomal material than traditional karyotype – has been proven to increase the diagnostic yield in cases of isolated CHDs and CHDs with extracardiac anomalies. Targeted next-generation sequencing also is now available as an additional approach in selective cases, and a clinically viable option for whole-exome sequencing is fast approaching.
For researchers, genetic evaluation carries the potential to unravel remaining mysteries about underlying causes of CHDs – to provide pathological insights and identify potential therapeutic targets. Currently, about 6 % of the total pie of presumed genetic determinants of CHDs is attributed to chromosomal anomalies, 10% to copy number variants, and 12% to single-gene defects. The remaining 72% of etiology, approximately, is undetermined.
As Helen Taussig, MD, (known as the founder of pediatric cardiology) once said, common cardiac malformations occurring in otherwise “normal” individuals “must be genetic in origin.”1 Greater use of genetic testing – and in particular, of whole-exome sequencing – will drive down this “undetermined” piece of the genetics pie.
For clinicians and patients, prenatal genetic evaluation can inform clinical management, guiding decisions on the mode, timing, and location of delivery. Genetic assessments help guide the neonatal health care team in taking optimal care of the infant, and the surgeon in preparing for neonatal surgeries and postsurgical complications.
In a recent analysis of the Society of Thoracic Surgeons Congenital Heart Surgery Database, prenatal diagnosis was associated with a lower overall prevalence of major preoperative risk factors for cardiac surgery.2 Surgical outcomes themselves also have been shown to be better after the prenatal diagnosis of complex CHDs, mainly because of improvements in perioperative care.3
When genetic etiology is elucidated, the cardiologist also is better able to counsel patients about anticipated challenges – such as the propensity, with certain genetic variants of CHD, to develop neurodevelopmental delays or other cardiac complications – and to target patient follow-up. Patients also can make informed decisions about termination or continuation of a current pregnancy and about family planning in the future.
Fortunately, advances in genetics technology have paralleled technological advancements in ultrasound. As I discussed in part one of this two-part Master Class series, it is now possible to detect many major CHDs well before 16 weeks’ gestation. Checking the structure of the fetal heart at the first-trimester screening and sonography (11-14 weeks of gestation) offers the opportunity for early genetic assessment, counseling, and planning when anomalies are detected.
A personalized approach
There has been growing interest in recent years in CMA for the prenatal genetic workup of CHDs. Microarray targets chromosomal regions at a much higher resolution than traditional karyotype. Traditional karyotype assesses both changes in chromosome number as well as more subtle structural changes such as chromosomal deletions and duplications. CMA finds what traditional karyotype identifies, but in addition, it identifies much smaller, clinically relevant chromosomal deletions and duplications that are not detected by karyotype performed with or without fluorescence in-situ hybridization (FISH). FISH uses DNA probes that carry fluorescent tags to detect chromosomal DNA.
At our center, we studied the prenatal genetic test results of 145 fetuses diagnosed with CHDs. Each case involved FISH for aneuploidy/karyotype, followed by CMA in cases of a negative karyotype result. CMA increased the diagnostic yield in cases of CHD by 19.8% overall – 17.4% in cases of isolated CHD and 24.5% in cases of CHD plus extracardiac anomalies.4
Indeed, although a microarray costs more and takes an additional 2 weeks to run, CMA should be strongly considered as first-line testing for the prenatal genetic evaluation of fetuses with major structural cardiac abnormalities detected by ultrasound. However, there still are cases in which a karyotype might be sufficient. For instance, if I see that a fetus has an atrial-ventricular septal defect on a prenatal ultrasound, and there are markers for trisomy 21, 13, or 18, or Turner’s syndrome (45 XO), I usually recommend a karyotype or FISH rather than an initial CMA. If the karyotype is abnormal – which is likely in such a scenario – there isn’t a need for more extensive testing.
Similarly, when there is high suspicion for DiGeorge syndrome (the 22q11.2 deletion, which often includes cleft palate and aortic arch abnormalities), usually it is most appropriate to perform a FISH test.
CMA is the preferred first modality, however, when prenatal imaging suggests severe CHD – for instance, when there are signs of hypoplastic left heart syndrome or tetralogy of Fallot (a conotruncal defect) – or complex CHD with extracardiac anomalies. In these cases, there is a high likelihood of detecting a small deletion or duplication that would be missed with karyotype.
In the past decade, karyotype and CMA have become the major methods used in our practice. However, targeted next‐generation sequencing and whole‐exome sequencing may become more widely used because these technologies enable rapid analysis of a large number of gene sequences and facilitate discovery of novel causative genes in many genetic diseases that cause CHDs.
Currently, targeted next-generation sequencing has mainly been used in the postnatal setting, and there are limited data available on its prenatal use. Compared with whole-exome sequencing, which sequences all of the protein-coding regions of the genome, targeted next-generation sequencing panels select regions of genes that are known to be associated with diseases of interest.
For CHDs, some perinatal centers have begun using a customized gene panel that targets 77 CHD-associated genes. This particular panel has been shown to be useful in addition to current methods and is an effective tool for prenatal genetic diagnosis.5
Whole-exome sequencing is currently expensive and time consuming. While sometimes it is used in the postnatal context, it is not yet part of routine practice as a prenatal diagnostic tool. As technology advances this will change – early in the next decade, I believe. For now, whole-exome sequencing may be an option for some patients who want to know more when severe CHD is evident on ultrasound and there are negative results from CMA or targeted sequencing. We have diagnosed some rare genetic syndromes using whole-exome sequencing; these diagnoses helped us to better manage the pregnancies.
These choices are part of the case-specific, stepwise approach to genetic evaluation that we take in our fetal heart program. Our goal is to pursue information that will be accurate and valuable for the patient and clinicians, in the most cost-effective and timely manner.
Limitations of noninvasive screening
In our fetal heart program we see increasing numbers of referred patients who have chosen noninvasive cell-free fetal DNA screening (cfDNA) after a cardiac anomaly is detected on ultrasound examination, and who believe that their “low risk” results demonstrate very little or no risk of CHD. Many of these patients express a belief that noninvasive testing is highly sensitive and accurate for fetal anomalies, including CHDs, and are not easily convinced of the value of other genetic tests.
We recently conducted a retrospective chart analysis (unpublished) in which we found that 41% of cases of CHD with abnormal genetics results were not detectable by cfDNA screening.
In the case of atrial-ventricular septal defects and conotruncal abnormalities that often are more associated with common aneuploidies (trisomy 21, 18, 13, and 45 XO), a “high-risk” result from cfDNA screening may offer the family and cardiology/neonatal team some guidance, but a “low-risk” result does not eliminate the risk of a microarray abnormality and thus may provide false reassurance.
Other research has shown that noninvasive screening will miss up to 7.3% of karyotype abnormalities in pregnancies at high risk for common aneuploidies.6
While invasive testing poses a very small risk of miscarriage, it is hard without such testing to elucidate the potential genetic etiologies of CHDs and truly understand the problems. We must take time to thoughtfully counsel patients who decline invasive testing about the limitations of cfDNA screening for CHDs and other anomalies.
Dr. Turan is an associate professor of obstetrics, gynecology, and reproductive sciences, and director of the fetal heart program at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. Dr. Turan reported that she has no disclosures relevant to this Master Class. Email her at obnews@mdedge.com.
References
1. J Am Coll Cardiol. 1988 Oct;12(4):1079-86.
2. Pediatr Cardiol. 2019 Mar;40(3):489-96.
3. Ann Pediatr Cardiol. 2017 May-Aug;10(2):126-30.
4. Eur J Obstet Gynecol Reprod Biol 2018;221:172-76.
5. Ultrasound Obstet Gynecol. 2018 Aug;52(2):205-11.
6. PLoS One. 2016 Jan 15;11(1):e0146794.
Congenital heart defects (CHDs) are etiologically heterogeneous, but in recent years it has become clear that genetics plays a larger role in the development of CHDs than was previously thought. Research has been shifting from a focus on risk – estimating the magnitude of increased risk, for instance, based on maternal or familial risk factors – to a focus on the etiology of cardiac defects.
In practice, advances in genetic testing technologies have made the underlying causes of CHDs increasingly detectable. Chromosomal microarray analysis (CMA) – technology that detects significantly more and smaller changes in the amount of chromosomal material than traditional karyotype – has been proven to increase the diagnostic yield in cases of isolated CHDs and CHDs with extracardiac anomalies. Targeted next-generation sequencing also is now available as an additional approach in selective cases, and a clinically viable option for whole-exome sequencing is fast approaching.
For researchers, genetic evaluation carries the potential to unravel remaining mysteries about underlying causes of CHDs – to provide pathological insights and identify potential therapeutic targets. Currently, about 6 % of the total pie of presumed genetic determinants of CHDs is attributed to chromosomal anomalies, 10% to copy number variants, and 12% to single-gene defects. The remaining 72% of etiology, approximately, is undetermined.
As Helen Taussig, MD, (known as the founder of pediatric cardiology) once said, common cardiac malformations occurring in otherwise “normal” individuals “must be genetic in origin.”1 Greater use of genetic testing – and in particular, of whole-exome sequencing – will drive down this “undetermined” piece of the genetics pie.
For clinicians and patients, prenatal genetic evaluation can inform clinical management, guiding decisions on the mode, timing, and location of delivery. Genetic assessments help guide the neonatal health care team in taking optimal care of the infant, and the surgeon in preparing for neonatal surgeries and postsurgical complications.
In a recent analysis of the Society of Thoracic Surgeons Congenital Heart Surgery Database, prenatal diagnosis was associated with a lower overall prevalence of major preoperative risk factors for cardiac surgery.2 Surgical outcomes themselves also have been shown to be better after the prenatal diagnosis of complex CHDs, mainly because of improvements in perioperative care.3
When genetic etiology is elucidated, the cardiologist also is better able to counsel patients about anticipated challenges – such as the propensity, with certain genetic variants of CHD, to develop neurodevelopmental delays or other cardiac complications – and to target patient follow-up. Patients also can make informed decisions about termination or continuation of a current pregnancy and about family planning in the future.
Fortunately, advances in genetics technology have paralleled technological advancements in ultrasound. As I discussed in part one of this two-part Master Class series, it is now possible to detect many major CHDs well before 16 weeks’ gestation. Checking the structure of the fetal heart at the first-trimester screening and sonography (11-14 weeks of gestation) offers the opportunity for early genetic assessment, counseling, and planning when anomalies are detected.
A personalized approach
There has been growing interest in recent years in CMA for the prenatal genetic workup of CHDs. Microarray targets chromosomal regions at a much higher resolution than traditional karyotype. Traditional karyotype assesses both changes in chromosome number as well as more subtle structural changes such as chromosomal deletions and duplications. CMA finds what traditional karyotype identifies, but in addition, it identifies much smaller, clinically relevant chromosomal deletions and duplications that are not detected by karyotype performed with or without fluorescence in-situ hybridization (FISH). FISH uses DNA probes that carry fluorescent tags to detect chromosomal DNA.
At our center, we studied the prenatal genetic test results of 145 fetuses diagnosed with CHDs. Each case involved FISH for aneuploidy/karyotype, followed by CMA in cases of a negative karyotype result. CMA increased the diagnostic yield in cases of CHD by 19.8% overall – 17.4% in cases of isolated CHD and 24.5% in cases of CHD plus extracardiac anomalies.4
Indeed, although a microarray costs more and takes an additional 2 weeks to run, CMA should be strongly considered as first-line testing for the prenatal genetic evaluation of fetuses with major structural cardiac abnormalities detected by ultrasound. However, there still are cases in which a karyotype might be sufficient. For instance, if I see that a fetus has an atrial-ventricular septal defect on a prenatal ultrasound, and there are markers for trisomy 21, 13, or 18, or Turner’s syndrome (45 XO), I usually recommend a karyotype or FISH rather than an initial CMA. If the karyotype is abnormal – which is likely in such a scenario – there isn’t a need for more extensive testing.
Similarly, when there is high suspicion for DiGeorge syndrome (the 22q11.2 deletion, which often includes cleft palate and aortic arch abnormalities), usually it is most appropriate to perform a FISH test.
CMA is the preferred first modality, however, when prenatal imaging suggests severe CHD – for instance, when there are signs of hypoplastic left heart syndrome or tetralogy of Fallot (a conotruncal defect) – or complex CHD with extracardiac anomalies. In these cases, there is a high likelihood of detecting a small deletion or duplication that would be missed with karyotype.
In the past decade, karyotype and CMA have become the major methods used in our practice. However, targeted next‐generation sequencing and whole‐exome sequencing may become more widely used because these technologies enable rapid analysis of a large number of gene sequences and facilitate discovery of novel causative genes in many genetic diseases that cause CHDs.
Currently, targeted next-generation sequencing has mainly been used in the postnatal setting, and there are limited data available on its prenatal use. Compared with whole-exome sequencing, which sequences all of the protein-coding regions of the genome, targeted next-generation sequencing panels select regions of genes that are known to be associated with diseases of interest.
For CHDs, some perinatal centers have begun using a customized gene panel that targets 77 CHD-associated genes. This particular panel has been shown to be useful in addition to current methods and is an effective tool for prenatal genetic diagnosis.5
Whole-exome sequencing is currently expensive and time consuming. While sometimes it is used in the postnatal context, it is not yet part of routine practice as a prenatal diagnostic tool. As technology advances this will change – early in the next decade, I believe. For now, whole-exome sequencing may be an option for some patients who want to know more when severe CHD is evident on ultrasound and there are negative results from CMA or targeted sequencing. We have diagnosed some rare genetic syndromes using whole-exome sequencing; these diagnoses helped us to better manage the pregnancies.
These choices are part of the case-specific, stepwise approach to genetic evaluation that we take in our fetal heart program. Our goal is to pursue information that will be accurate and valuable for the patient and clinicians, in the most cost-effective and timely manner.
Limitations of noninvasive screening
In our fetal heart program we see increasing numbers of referred patients who have chosen noninvasive cell-free fetal DNA screening (cfDNA) after a cardiac anomaly is detected on ultrasound examination, and who believe that their “low risk” results demonstrate very little or no risk of CHD. Many of these patients express a belief that noninvasive testing is highly sensitive and accurate for fetal anomalies, including CHDs, and are not easily convinced of the value of other genetic tests.
We recently conducted a retrospective chart analysis (unpublished) in which we found that 41% of cases of CHD with abnormal genetics results were not detectable by cfDNA screening.
In the case of atrial-ventricular septal defects and conotruncal abnormalities that often are more associated with common aneuploidies (trisomy 21, 18, 13, and 45 XO), a “high-risk” result from cfDNA screening may offer the family and cardiology/neonatal team some guidance, but a “low-risk” result does not eliminate the risk of a microarray abnormality and thus may provide false reassurance.
Other research has shown that noninvasive screening will miss up to 7.3% of karyotype abnormalities in pregnancies at high risk for common aneuploidies.6
While invasive testing poses a very small risk of miscarriage, it is hard without such testing to elucidate the potential genetic etiologies of CHDs and truly understand the problems. We must take time to thoughtfully counsel patients who decline invasive testing about the limitations of cfDNA screening for CHDs and other anomalies.
Dr. Turan is an associate professor of obstetrics, gynecology, and reproductive sciences, and director of the fetal heart program at the University of Maryland School of Medicine and director of the Fetal Heart Program at the University of Maryland Medical Center. Dr. Turan reported that she has no disclosures relevant to this Master Class. Email her at obnews@mdedge.com.
References
1. J Am Coll Cardiol. 1988 Oct;12(4):1079-86.
2. Pediatr Cardiol. 2019 Mar;40(3):489-96.
3. Ann Pediatr Cardiol. 2017 May-Aug;10(2):126-30.
4. Eur J Obstet Gynecol Reprod Biol 2018;221:172-76.
5. Ultrasound Obstet Gynecol. 2018 Aug;52(2):205-11.
6. PLoS One. 2016 Jan 15;11(1):e0146794.
Consider triple therapy for the management of COPD
Background: The Global Initiative for Obstructive Lung Disease (GOLD) recommends triple therapy with inhaled corticosteroids, long-acting beta2-adrenoceptor agonists (LABA), and long-acting muscarinic receptor antagonists (LAMA) for patients with severe COPD who have frequent exacerbations despite treatment with a LABA and LAMA. Triple therapy has been shown to improve forced expiratory volume in 1 second (FEV1), but its effect on preventing exacerbations has not been well documented in previous meta-analyses.

Study design: Meta-analysis.
Setting: Studies published on PubMed, Embase, Cochrane Library website, Cochrane Central Register of Controlled Trials (CENTRAL), and ClinicalTrials.gov databases.
Synopsis: 21 randomized, controlled trials of triple therapy in stable cases of moderate to very severe COPD were included in this meta-analysis. Triple therapy was associated with a significantly greater reduction in the rate of COPD exacerbations, compared with dual therapy of LAMA and LABA (rate ratio, 0.78; 95% confidence interval, 0.70-0.88), inhaled corticosteroid and LABA (rate ratio, 0.77; 95% CI, 0.66-0.91), or LAMA monotherapy (rate ratio, 0.71; 95% CI, 0.60-0.85). Triple therapy was also associated with greater improvement in FEV1.
There was a significantly higher incidence of pneumonia in patients using triple therapy, compared with those using dual therapy (LAMA and LABA), and there also was a trend toward increased pneumonia incidence with triple therapy, compared with LAMA monotherapy. Triple therapy was not shown to improve survival; however, most trials lasted less than 6 months, which limits their analysis of survival outcomes.
Bottom line: In patients with advanced COPD, triple therapy is associated with lower rates of COPD exacerbations and improved lung function, compared with dual therapy or monotherapy.
Citation: Zheng Y et al. Triple therapy in the management of chronic obstructive pulmonary disease: Systemic review and meta-analysis. BMJ. 2018;363:k4388.
Dr. Chace is an associate physician in the division of hospital medicine at the University of California, San Diego.
Background: The Global Initiative for Obstructive Lung Disease (GOLD) recommends triple therapy with inhaled corticosteroids, long-acting beta2-adrenoceptor agonists (LABA), and long-acting muscarinic receptor antagonists (LAMA) for patients with severe COPD who have frequent exacerbations despite treatment with a LABA and LAMA. Triple therapy has been shown to improve forced expiratory volume in 1 second (FEV1), but its effect on preventing exacerbations has not been well documented in previous meta-analyses.
Study design: Meta-analysis.
Setting: Studies published on PubMed, Embase, Cochrane Library website, Cochrane Central Register of Controlled Trials (CENTRAL), and ClinicalTrials.gov databases.
Synopsis: 21 randomized, controlled trials of triple therapy in stable cases of moderate to very severe COPD were included in this meta-analysis. Triple therapy was associated with a significantly greater reduction in the rate of COPD exacerbations, compared with dual therapy of LAMA and LABA (rate ratio, 0.78; 95% confidence interval, 0.70-0.88), inhaled corticosteroid and LABA (rate ratio, 0.77; 95% CI, 0.66-0.91), or LAMA monotherapy (rate ratio, 0.71; 95% CI, 0.60-0.85). Triple therapy was also associated with greater improvement in FEV1.
There was a significantly higher incidence of pneumonia in patients using triple therapy, compared with those using dual therapy (LAMA and LABA), and there also was a trend toward increased pneumonia incidence with triple therapy, compared with LAMA monotherapy. Triple therapy was not shown to improve survival; however, most trials lasted less than 6 months, which limits their analysis of survival outcomes.
Bottom line: In patients with advanced COPD, triple therapy is associated with lower rates of COPD exacerbations and improved lung function, compared with dual therapy or monotherapy.
Citation: Zheng Y et al. Triple therapy in the management of chronic obstructive pulmonary disease: Systemic review and meta-analysis. BMJ. 2018;363:k4388.
Dr. Chace is an associate physician in the division of hospital medicine at the University of California, San Diego.
Background: The Global Initiative for Obstructive Lung Disease (GOLD) recommends triple therapy with inhaled corticosteroids, long-acting beta2-adrenoceptor agonists (LABA), and long-acting muscarinic receptor antagonists (LAMA) for patients with severe COPD who have frequent exacerbations despite treatment with a LABA and LAMA. Triple therapy has been shown to improve forced expiratory volume in 1 second (FEV1), but its effect on preventing exacerbations has not been well documented in previous meta-analyses.
Study design: Meta-analysis.
Setting: Studies published on PubMed, Embase, Cochrane Library website, Cochrane Central Register of Controlled Trials (CENTRAL), and ClinicalTrials.gov databases.
Synopsis: 21 randomized, controlled trials of triple therapy in stable cases of moderate to very severe COPD were included in this meta-analysis. Triple therapy was associated with a significantly greater reduction in the rate of COPD exacerbations, compared with dual therapy of LAMA and LABA (rate ratio, 0.78; 95% confidence interval, 0.70-0.88), inhaled corticosteroid and LABA (rate ratio, 0.77; 95% CI, 0.66-0.91), or LAMA monotherapy (rate ratio, 0.71; 95% CI, 0.60-0.85). Triple therapy was also associated with greater improvement in FEV1.
There was a significantly higher incidence of pneumonia in patients using triple therapy, compared with those using dual therapy (LAMA and LABA), and there also was a trend toward increased pneumonia incidence with triple therapy, compared with LAMA monotherapy. Triple therapy was not shown to improve survival; however, most trials lasted less than 6 months, which limits their analysis of survival outcomes.
Bottom line: In patients with advanced COPD, triple therapy is associated with lower rates of COPD exacerbations and improved lung function, compared with dual therapy or monotherapy.
Citation: Zheng Y et al. Triple therapy in the management of chronic obstructive pulmonary disease: Systemic review and meta-analysis. BMJ. 2018;363:k4388.
Dr. Chace is an associate physician in the division of hospital medicine at the University of California, San Diego.
Try testosterone for some women with sexual dysfunction, but not others
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
A new international position statement on testosterone therapy for women concludes that a trial of testosterone is appropriate for postmenopausal women with hypoactive sexual desire dysfunction (HSDD) and that its use for any other condition, symptom, or reason is not supported by available evidence.
The seven-page position statement, developed by an international task force of experts from the Endocrine Society, the American College of Gynecologists and Obstetricians, and multiple other medical societies, also emphasized that blood concentrations of testosterone should approximate premenopausal physiological conditions.
“When testosterone therapy is given, the resultant blood levels should not be above those seen in healthy young women,” said lead author Susan Ruth Davis, PhD, MBBS, of Monash University in Melbourne, Australia, in a press release issued by the Endocrine Society. Dr. Davis is president of the International Menopause Society, which coordinated the panel.
The statement was published in the Journal of Clinical Endocrinology & Metabolism and three other medical journals.
Margaret E. Wierman, MD, who represented the Endocrine Society on the task force, said in an interview that there has been “growing concern about testosterone being prescribed for a variety of signs and symptoms without data to support” such use. At the same time, there is significant concern about the ongoing lack of approved formulations licensed specifically for women, she said.
In part, the statement is about a renewed “call to industry to make some [female-specific] formulations so that we can examine other potential roles of testosterone in women,” said Dr. Wierman, professor of medicine and physiology at the University of Colorado at Denver, Aurora, and chief of endocrinology at the Rocky Mountain Regional Veterans Affairs Medical Center in Aurora.
“Testosterone may be useful [for indications other than HSDD], but we don’t know. There may be no [breast or cardiovascular disease risk], but we don’t know,” she said. “And without a formulation to study potential benefits and risks, it’s good to be cautious. It’s good to really outline where we have data and where we don’t.”
The Endocrine Society’s 2014 clinical practice guideline on androgen therapy in women, for which Dr. Wierman was the lead author, also recommended against the off-label use of testosterone for sexual dysfunction other than HSDD or for any other reason, such as cognitive, cardiovascular, metabolic, or bone health. As with the new statement, the society’s position statement was guided by an international, multisociety task force, albeit a smaller one.
For the new global position statement, the task force’s review of evidence includes a recently published systematic review and meta-analysis of randomized controlled trial data – of at least 12 weeks’ duration – on the use of testosterone for sexual function, cardiometabolic variables, cognitive measures, and musculoskeletal health. Some of the data from the randomized controlled trials were unpublished.
The meta-analysis, led by Dr. Davis and published in July in the Lancet Diabetes & Endocrinology, found that, compared with placebo or a comparator (such as estrogen, with or without progesterone), testosterone in either oral or transdermal form significantly improved sexual function in postmenopausal women. However, data about the effects of testosterone for other indications, its long-term safety, and its use in premenopausal women, were insufficient for drawing any conclusions (Lancet Diabetes Endocrinol. 2019 Jul 25. doi: 10.1016/S2213-8587[19]30189-5).
In addition, testosterone administered orally – but not nonorally (patch or cream) – was associated with adverse lipid profiles, Dr. Davis and her colleagues reported.
Another systematic review and meta-analysis, published in Fertility and Sterility in 2017 and included in the task force’s evidence review, focused specifically on transdermal testosterone for menopausal women with HSDD, with or without estrogen and progestin therapy. It also showed short-term efficacy in terms of improvement in sexual function, as well as short-term safety (Fertil Steril. 2017;107(2):475-82).
The new position statement warns about the lack of long-term safety data, stating that “safety data for testosterone in physiologic doses are not available beyond 24 months of treatment.”
In the short term, testosterone therapy for postmenopausal women (in doses approximating testosterone concentrations for premenopausal women), is associated with mild increases in acne and body/facial hair growth in some women, but not with alopecia, clitoromegaly, or voice change. Short-term transdermal therapy also does not seem to affect breast cancer risk or have any significant effects on lipid profiles, the statement says.
The panel points out, however, that randomized controlled trials with testosterone therapy have excluded women who are at high risk of cardiometabolic disease, and that women with a previous diagnosis of breast cancer have also been excluded from randomized trials of testosterone in women with HSDD. This is a “big issue,” said Dr. Wierman, and means that recommendations regarding the effect of testosterone in postmenopausal women with HSDD may not be generalizable to possible at-risk subpopulations.
The panel endorsed testosterone therapy specifically for women with HSDD because most of the studies reporting on sexual function have recruited women with diagnosed HSDD. Demonstrated benefits of testosterone in these cases include improved sexual desire, arousal, orgasm, and pleasure, and reduced concerns and distress about sex. HSDD should be diagnosed after formal biopsychosocial assessment, the statement notes.
“We don’t completely understand the control of sexual function in women, but it’s very dependent on estrogen status. And it’s also dependent on psychosocial factors, emotional health, relationship issues, and physical issues,” Dr. Wierman said in the interview.
“In practice, we look at all these issues, and we first optimize estrogen status. Once that’s done, and we’ve looked at all the other components of sexual function, then we can consider off-label use of testosterone,” she said. “If there’s no response in 3-6 months, we stop it.”
Testosterone levels do not correlate with sexual dysfunction, Dr. Wierman emphasized, and direct assays for the measurement of total and free testosterone are unreliable. The statement acknowledges that but still recommends measurement of testosterone using direct assays, in cases in which liquid/gas chromatography and tandem mass spectrometry assay (which has “high accuracy and reproducibility”) are not available. This is “to exclude high baseline concentrations and also to exclude supraphysiological concentrations during treatment,” the panel said.
Most endocrinologists and other experts who prescribe testosterone therapy for women use an approved male formulation off label and adjust it – an approach that the panel says is reasonable as long as hormone concentrations are “maintained in the physiologic female range.”
Compounded “bioidentical” testosterone therapy “cannot be recommended for the treatment of HSDD because of the lack of evidence for safety and efficacy,” the statement says.
“A big concern of many endocrinologists,” Dr. Wierman added, “is the recent explosion of using pharmacological levels of both estrogen and testosterone in either [injections] or pellets.” The Endocrine Society and other societies have alerted the Food and Drug Administration to “this new cottage industry, which may have significant side effects and risks for our patients,” she said.
Dr. Wierman reported received funding from Corcept Therapeutics, Novartis, and the Cancer League of Colorado, and honoraria or consultation fees from Pfizer to review ASPIRE grant applications for studies of acromegaly as well as Endocrine Society honorarium for teaching in the Endocrine Board Review and Clinical Endocrine Update. Dr. Davis reported receiving funding from a National Health and Medical Research Council Project Grant, a National Breast Foundation accelerator grant, and the Grollo-Ruzenne Foundation, as well as honoraria from Besins and Pfizer Australia. She has been a consultant to Besins Healthcare, Mayne Pharmaceuticals, Lawley Pharmaceuticals, and Que Oncology. Disclosures for other authors of the position statement are listed with the statement.
SOURCE: Davis SR et al. J Clin Endocrinol Metab. 2019 Sep 2. doi: 10.1210/jc.2019-01603.
FROM JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM
Solitary Papule on the Leg
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
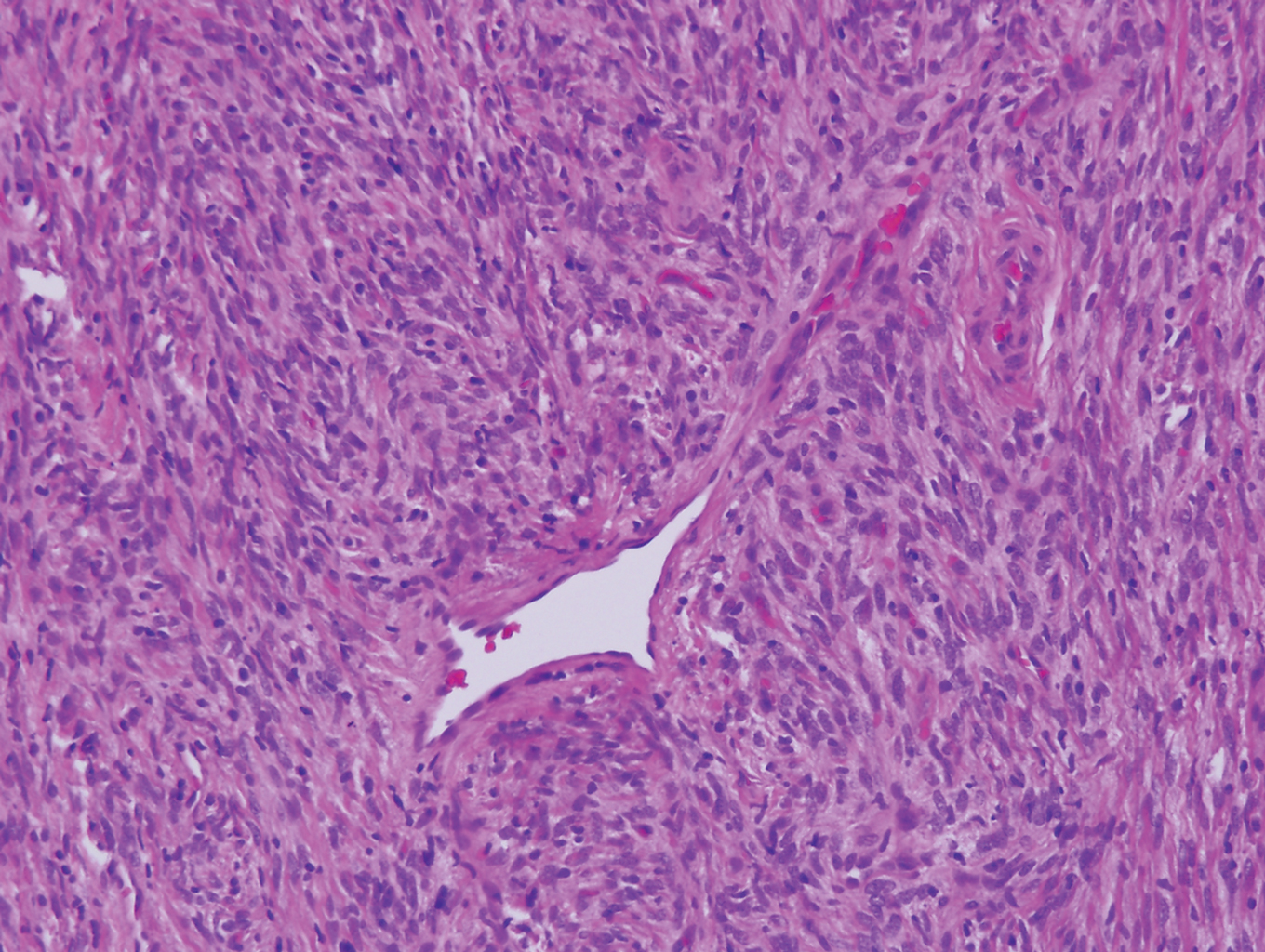
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
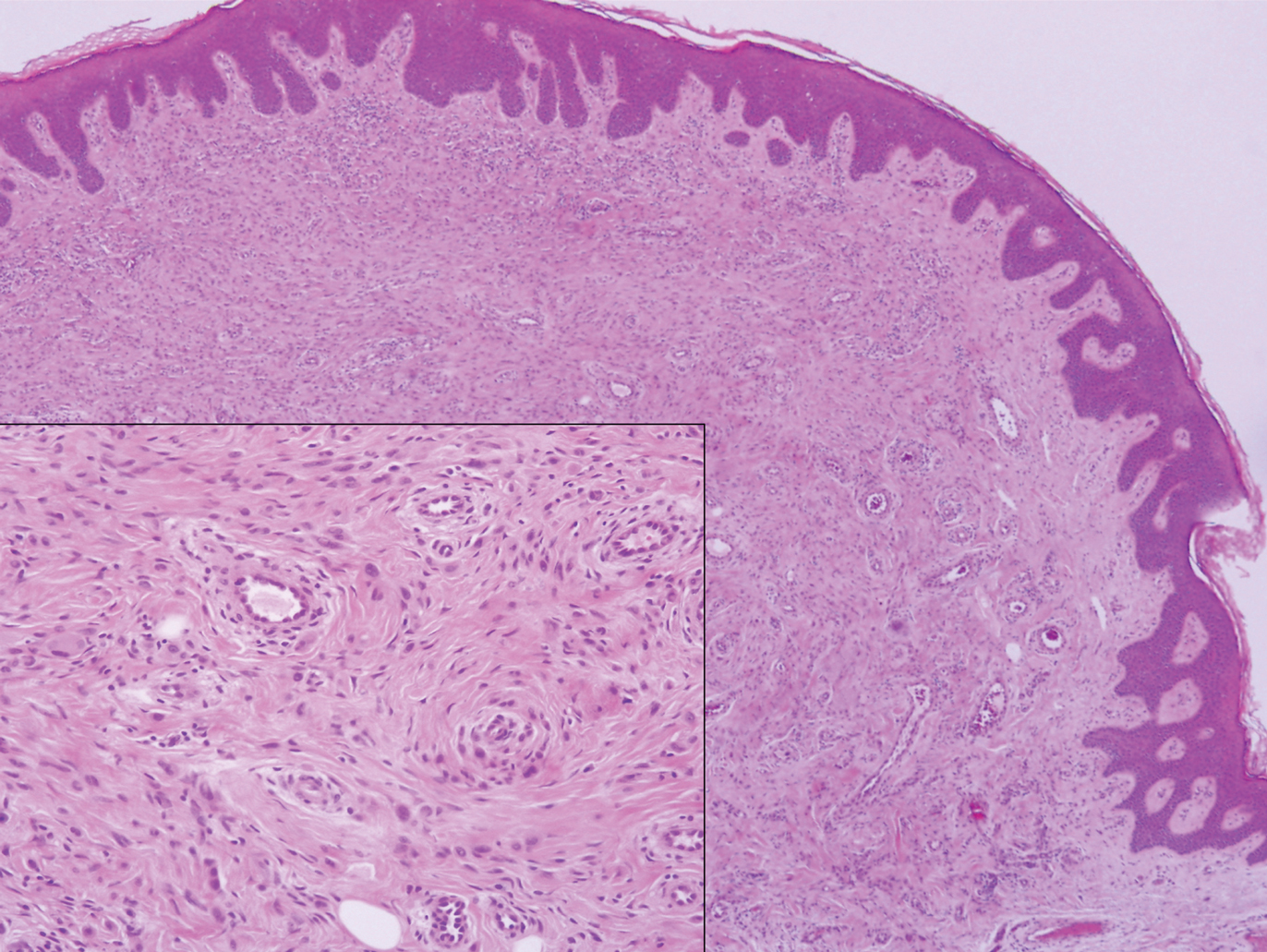
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18

Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22

Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
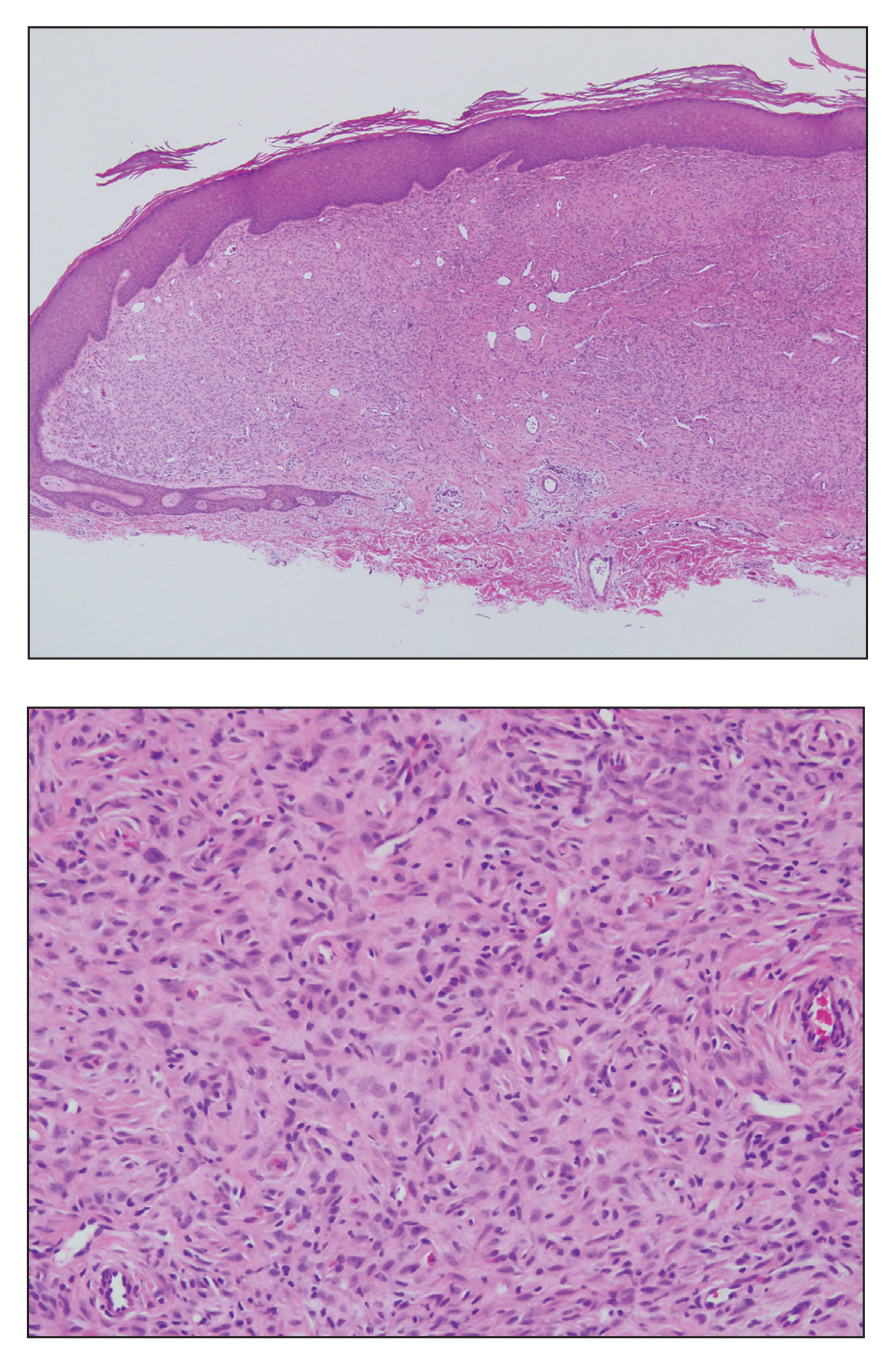
A 28-year-old man presented with a growing asymptomatic papule on the right leg.
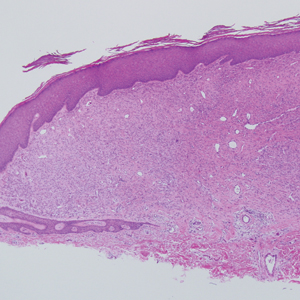
Ankylosing spondylitis severity, comorbidities higher in blacks than in whites
While ankylosing spondylitis may be more common among whites, black patients have more comorbidities and express higher disease activity, Dilpreet Kaur Singh, MD, and Marina Magrey, MBBS, reported in The Journal of Rheumatology.
A retrospective review of a large U.S. medical database conducted by Dr. Singh and Dr. Magrey of MetroHealth Medical Center, Cleveland, found that black patients had higher erythrocyte sedimentation rates and C-reactive protein, and a higher prevalence of anterior uveitis, hypertension, diabetes, and depression, compared with white patients. (Dr. Singh, who was a rheumatology fellow at MetroHealth at the time of the study, is now a practicing rheumatologist in Springfield, Mass.)
Disease severity in AS is “thought to be genetically mediated but cultural, social, or economic factors may also be contributing to this racial disparity,” the investigators wrote. “Further research is needed to determine the role of factors other than genetic factors like HLA-B27 positivity that contribute to worsening disease severity.”
The authors extracted data recorded during 1999-2017 from the Explorys platform, a clinical research informatics tool with data from more than 50 million patients in 26 major integrated health care systems in the United States.
The current study comprised 10,990 AS patients with at least two visits to a rheumatologist. Most (84%) were white; 8% were black. Sex was equally distributed in both groups. Positivity for HLA-B27 was similar among whites (26%) and blacks (20%). A majority of patients smoked (65%), and smoking was more common among whites than among blacks (67% vs. 59%).
Disease characteristics suggested that AS was more severe among blacks. Significantly greater proportions of black patients had elevated erythrocyte sedimentation rate (62% vs. 48% of whites) and C-reactive protein (68% vs. 54%). Blacks also experienced significantly greater rates of anterior uveitis (8% vs. 4%), hypertension (29% vs. 22%), diabetes (27% vs. 17%), and depression (36% vs. 32%).
Blacks experienced higher rates of peripheral arthritis, enthesopathy, dactylitis, and inflammatory bowel disease, although these differences were not statistically significant when compared with whites.
Whites, however, had significantly higher rates of psoriasis (10% vs. 6.5%).
Most of the cohort (87%) received NSAIDs; 39% used tumor necrosis factor inhibitors. There were no significant between-group treatment differences.
The authors reported no potential conflicts of interest and no source of financial support.
SOURCE: Singh DK et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.181019.
While ankylosing spondylitis may be more common among whites, black patients have more comorbidities and express higher disease activity, Dilpreet Kaur Singh, MD, and Marina Magrey, MBBS, reported in The Journal of Rheumatology.
A retrospective review of a large U.S. medical database conducted by Dr. Singh and Dr. Magrey of MetroHealth Medical Center, Cleveland, found that black patients had higher erythrocyte sedimentation rates and C-reactive protein, and a higher prevalence of anterior uveitis, hypertension, diabetes, and depression, compared with white patients. (Dr. Singh, who was a rheumatology fellow at MetroHealth at the time of the study, is now a practicing rheumatologist in Springfield, Mass.)
Disease severity in AS is “thought to be genetically mediated but cultural, social, or economic factors may also be contributing to this racial disparity,” the investigators wrote. “Further research is needed to determine the role of factors other than genetic factors like HLA-B27 positivity that contribute to worsening disease severity.”
The authors extracted data recorded during 1999-2017 from the Explorys platform, a clinical research informatics tool with data from more than 50 million patients in 26 major integrated health care systems in the United States.
The current study comprised 10,990 AS patients with at least two visits to a rheumatologist. Most (84%) were white; 8% were black. Sex was equally distributed in both groups. Positivity for HLA-B27 was similar among whites (26%) and blacks (20%). A majority of patients smoked (65%), and smoking was more common among whites than among blacks (67% vs. 59%).
Disease characteristics suggested that AS was more severe among blacks. Significantly greater proportions of black patients had elevated erythrocyte sedimentation rate (62% vs. 48% of whites) and C-reactive protein (68% vs. 54%). Blacks also experienced significantly greater rates of anterior uveitis (8% vs. 4%), hypertension (29% vs. 22%), diabetes (27% vs. 17%), and depression (36% vs. 32%).
Blacks experienced higher rates of peripheral arthritis, enthesopathy, dactylitis, and inflammatory bowel disease, although these differences were not statistically significant when compared with whites.
Whites, however, had significantly higher rates of psoriasis (10% vs. 6.5%).
Most of the cohort (87%) received NSAIDs; 39% used tumor necrosis factor inhibitors. There were no significant between-group treatment differences.
The authors reported no potential conflicts of interest and no source of financial support.
SOURCE: Singh DK et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.181019.
While ankylosing spondylitis may be more common among whites, black patients have more comorbidities and express higher disease activity, Dilpreet Kaur Singh, MD, and Marina Magrey, MBBS, reported in The Journal of Rheumatology.
A retrospective review of a large U.S. medical database conducted by Dr. Singh and Dr. Magrey of MetroHealth Medical Center, Cleveland, found that black patients had higher erythrocyte sedimentation rates and C-reactive protein, and a higher prevalence of anterior uveitis, hypertension, diabetes, and depression, compared with white patients. (Dr. Singh, who was a rheumatology fellow at MetroHealth at the time of the study, is now a practicing rheumatologist in Springfield, Mass.)
Disease severity in AS is “thought to be genetically mediated but cultural, social, or economic factors may also be contributing to this racial disparity,” the investigators wrote. “Further research is needed to determine the role of factors other than genetic factors like HLA-B27 positivity that contribute to worsening disease severity.”
The authors extracted data recorded during 1999-2017 from the Explorys platform, a clinical research informatics tool with data from more than 50 million patients in 26 major integrated health care systems in the United States.
The current study comprised 10,990 AS patients with at least two visits to a rheumatologist. Most (84%) were white; 8% were black. Sex was equally distributed in both groups. Positivity for HLA-B27 was similar among whites (26%) and blacks (20%). A majority of patients smoked (65%), and smoking was more common among whites than among blacks (67% vs. 59%).
Disease characteristics suggested that AS was more severe among blacks. Significantly greater proportions of black patients had elevated erythrocyte sedimentation rate (62% vs. 48% of whites) and C-reactive protein (68% vs. 54%). Blacks also experienced significantly greater rates of anterior uveitis (8% vs. 4%), hypertension (29% vs. 22%), diabetes (27% vs. 17%), and depression (36% vs. 32%).
Blacks experienced higher rates of peripheral arthritis, enthesopathy, dactylitis, and inflammatory bowel disease, although these differences were not statistically significant when compared with whites.
Whites, however, had significantly higher rates of psoriasis (10% vs. 6.5%).
Most of the cohort (87%) received NSAIDs; 39% used tumor necrosis factor inhibitors. There were no significant between-group treatment differences.
The authors reported no potential conflicts of interest and no source of financial support.
SOURCE: Singh DK et al. J Rheumatol. 2019 Sep 1. doi: 10.3899/jrheum.181019.
FROM THE JOURNAL OF RHEUMATOLOGY
For trans children, early gender ID conversion efforts damage lifelong mental health
Gender identity conversion efforts during early childhood quadruple lifetime risk of suicidal behavior for transgender people, a study of almost 28,000 adults has determined.
The findings prompted the authors to issue a blanket warning against the controversial treatment, which already has been decried by several medical associations.
“Our results support policy positions … which state that gender identity–conversion therapy should not be conducted for transgender patients at any age, Jack L. Turban, MD, and colleagues wrote in JAMA Psychiatry.
The American Academy of Child and Adolescent Psychiatry, the American Psychiatric Association, the American Academy of Pediatrics, and the American Medical Association all strongly warn against any kind of gender conversion efforts.
The significantly increased risk of suicidal behavior was similar whether the gender identity–conversion effort (GICE) was administered by a clinician or a cleric. “This suggests that any process of intervening to alter gender identity is associated with poorer mental health regardless of whether the intervention occurred within a secular or religious framework,” wrote Dr. Turban of Massachusetts General Hospital, Boston, and coauthors.
The study – the largest of its kind thus far – comprised 27,715 transgender adults included in the 2015 U.S. Transgender Survey, conducted by the National Center for Transgender Equality.
In the current study, investigators focused on the question: “Did any professional (such as a psychologist, counselor, or religious advisor) try to make you identify only with your sex assigned at birth (in other words, try to stop you being trans)?”
They compared responses among subjects who reported exposure to GICE before age 10 years with those who did not.
Subjects were aged a mean of 31 years when they participated in the survey. Slightly less than half (42.8%) were assigned male sex at birth. Most (19,751) had discussed their gender identity with a professional. Nearly 20% (3,869) reported some exposure to GICE; 35% said a religious adviser had conducted the effort.
In this group, exposure had significant negative lifetime effects on mental health. These subjects were at a 56% increased risk of psychological distress within the month before taking the survey (odds ratio, 1.56), and more than twice as likely to have tried at least once to end their lives by suicide (OR, 2.27).
But the negative effects were even more pronounced among the small group of 206 who reported exposure to GICE before they were 10 years old. In a multivariate analysis adjusted for demographics, GICE before age 10 more than quadrupled the risk of lifetime suicide attempts (OR, 4.15). Again, it didn’t matter whether a medical or religious professional administered GICE.
“A plausible association of these practices with poor mental health outcomes can be conceptualized through the minority stress framework; that is, elevated stigma-related stress from exposure to GICE may increase general emotion dysregulation, interpersonal dysfunction, and maladaptive cognitions,” the investigators wrote. “Although this study suggests that exposure to GICE is associated with increased odds of suicide attempts, GICE are not the only way in which minority group stress manifests, and thus, other factors are also likely to be associated with suicidality among gender-diverse people.”
“One potential explanation for this is that, compared with persons in the sexual-minority group, many persons in the gender-minority group must interact with clinical professionals to be medically and surgically affirmed in their identities. This higher prevalence of interactions with clinical professionals among people in the gender minority group may lead to greater risk of experiencing conversion effort.”
The authors cited the sample size of the study as one of its many strengths. One limitation includes the study’s cross-sectional design.
Dr. Turban reported collecting royalties from Springer for an upcoming textbook about pediatric gender identity. The other coauthors reported no disclosures.
SOURCE: Turban JL et al. JAMA Psychiatry. 2019 Sep 11. doi: 10.1001/jamapsychiatry.2019.2285.
Gender identity conversion efforts during early childhood quadruple lifetime risk of suicidal behavior for transgender people, a study of almost 28,000 adults has determined.
The findings prompted the authors to issue a blanket warning against the controversial treatment, which already has been decried by several medical associations.
“Our results support policy positions … which state that gender identity–conversion therapy should not be conducted for transgender patients at any age, Jack L. Turban, MD, and colleagues wrote in JAMA Psychiatry.
The American Academy of Child and Adolescent Psychiatry, the American Psychiatric Association, the American Academy of Pediatrics, and the American Medical Association all strongly warn against any kind of gender conversion efforts.
The significantly increased risk of suicidal behavior was similar whether the gender identity–conversion effort (GICE) was administered by a clinician or a cleric. “This suggests that any process of intervening to alter gender identity is associated with poorer mental health regardless of whether the intervention occurred within a secular or religious framework,” wrote Dr. Turban of Massachusetts General Hospital, Boston, and coauthors.
The study – the largest of its kind thus far – comprised 27,715 transgender adults included in the 2015 U.S. Transgender Survey, conducted by the National Center for Transgender Equality.
In the current study, investigators focused on the question: “Did any professional (such as a psychologist, counselor, or religious advisor) try to make you identify only with your sex assigned at birth (in other words, try to stop you being trans)?”
They compared responses among subjects who reported exposure to GICE before age 10 years with those who did not.
Subjects were aged a mean of 31 years when they participated in the survey. Slightly less than half (42.8%) were assigned male sex at birth. Most (19,751) had discussed their gender identity with a professional. Nearly 20% (3,869) reported some exposure to GICE; 35% said a religious adviser had conducted the effort.
In this group, exposure had significant negative lifetime effects on mental health. These subjects were at a 56% increased risk of psychological distress within the month before taking the survey (odds ratio, 1.56), and more than twice as likely to have tried at least once to end their lives by suicide (OR, 2.27).
But the negative effects were even more pronounced among the small group of 206 who reported exposure to GICE before they were 10 years old. In a multivariate analysis adjusted for demographics, GICE before age 10 more than quadrupled the risk of lifetime suicide attempts (OR, 4.15). Again, it didn’t matter whether a medical or religious professional administered GICE.
“A plausible association of these practices with poor mental health outcomes can be conceptualized through the minority stress framework; that is, elevated stigma-related stress from exposure to GICE may increase general emotion dysregulation, interpersonal dysfunction, and maladaptive cognitions,” the investigators wrote. “Although this study suggests that exposure to GICE is associated with increased odds of suicide attempts, GICE are not the only way in which minority group stress manifests, and thus, other factors are also likely to be associated with suicidality among gender-diverse people.”
“One potential explanation for this is that, compared with persons in the sexual-minority group, many persons in the gender-minority group must interact with clinical professionals to be medically and surgically affirmed in their identities. This higher prevalence of interactions with clinical professionals among people in the gender minority group may lead to greater risk of experiencing conversion effort.”
The authors cited the sample size of the study as one of its many strengths. One limitation includes the study’s cross-sectional design.
Dr. Turban reported collecting royalties from Springer for an upcoming textbook about pediatric gender identity. The other coauthors reported no disclosures.
SOURCE: Turban JL et al. JAMA Psychiatry. 2019 Sep 11. doi: 10.1001/jamapsychiatry.2019.2285.
Gender identity conversion efforts during early childhood quadruple lifetime risk of suicidal behavior for transgender people, a study of almost 28,000 adults has determined.
The findings prompted the authors to issue a blanket warning against the controversial treatment, which already has been decried by several medical associations.
“Our results support policy positions … which state that gender identity–conversion therapy should not be conducted for transgender patients at any age, Jack L. Turban, MD, and colleagues wrote in JAMA Psychiatry.
The American Academy of Child and Adolescent Psychiatry, the American Psychiatric Association, the American Academy of Pediatrics, and the American Medical Association all strongly warn against any kind of gender conversion efforts.
The significantly increased risk of suicidal behavior was similar whether the gender identity–conversion effort (GICE) was administered by a clinician or a cleric. “This suggests that any process of intervening to alter gender identity is associated with poorer mental health regardless of whether the intervention occurred within a secular or religious framework,” wrote Dr. Turban of Massachusetts General Hospital, Boston, and coauthors.
The study – the largest of its kind thus far – comprised 27,715 transgender adults included in the 2015 U.S. Transgender Survey, conducted by the National Center for Transgender Equality.
In the current study, investigators focused on the question: “Did any professional (such as a psychologist, counselor, or religious advisor) try to make you identify only with your sex assigned at birth (in other words, try to stop you being trans)?”
They compared responses among subjects who reported exposure to GICE before age 10 years with those who did not.
Subjects were aged a mean of 31 years when they participated in the survey. Slightly less than half (42.8%) were assigned male sex at birth. Most (19,751) had discussed their gender identity with a professional. Nearly 20% (3,869) reported some exposure to GICE; 35% said a religious adviser had conducted the effort.
In this group, exposure had significant negative lifetime effects on mental health. These subjects were at a 56% increased risk of psychological distress within the month before taking the survey (odds ratio, 1.56), and more than twice as likely to have tried at least once to end their lives by suicide (OR, 2.27).
But the negative effects were even more pronounced among the small group of 206 who reported exposure to GICE before they were 10 years old. In a multivariate analysis adjusted for demographics, GICE before age 10 more than quadrupled the risk of lifetime suicide attempts (OR, 4.15). Again, it didn’t matter whether a medical or religious professional administered GICE.
“A plausible association of these practices with poor mental health outcomes can be conceptualized through the minority stress framework; that is, elevated stigma-related stress from exposure to GICE may increase general emotion dysregulation, interpersonal dysfunction, and maladaptive cognitions,” the investigators wrote. “Although this study suggests that exposure to GICE is associated with increased odds of suicide attempts, GICE are not the only way in which minority group stress manifests, and thus, other factors are also likely to be associated with suicidality among gender-diverse people.”
“One potential explanation for this is that, compared with persons in the sexual-minority group, many persons in the gender-minority group must interact with clinical professionals to be medically and surgically affirmed in their identities. This higher prevalence of interactions with clinical professionals among people in the gender minority group may lead to greater risk of experiencing conversion effort.”
The authors cited the sample size of the study as one of its many strengths. One limitation includes the study’s cross-sectional design.
Dr. Turban reported collecting royalties from Springer for an upcoming textbook about pediatric gender identity. The other coauthors reported no disclosures.
SOURCE: Turban JL et al. JAMA Psychiatry. 2019 Sep 11. doi: 10.1001/jamapsychiatry.2019.2285.
FROM JAMA PSYCHIATRY
Key clinical point: Gender identity–conversion efforts exert lifelong negative mental health impacts on transgender children.
Major finding: Exposure before age 10 years more than quadrupled the risk of lifetime suicidal behavior.
Study details: The cohort comprised 27,715 transgender adults.
Disclosures: Dr. Turban reported collecting royalties from Springer for an upcoming textbook about pediatric gender identity. No other financial disclosures were reported.
Source: Turban JL et al. JAMA Psychiatry. 2019 Sep 11. doi: 10.1001/jamapsychiatry.2019.2285.
Breaking a 10-year streak, the number of uninsured Americans rises
For the first time in a decade, the number of Americans without health insurance has risen – by about 2 million people in 2018 – according to the annual U.S. Census Bureau report released Sept. 10, 2019.
The Census found that 8.5% of the U.S. population went without medical insurance for all of 2018, up from 7.9% in 2017. By contrast, in 2013, before the Affordable Care Act took full effect, 13.3% were uninsured. It was the first year-to-year increase since 2008-09, Census officials said, adding that most of the drop in health coverage was related to a 0.7% decline in Medicaid participants. The number of people with private insurance remained steady and there was a 0.4% increase in those on Medicare.
Many of those losing coverage were noncitizens, a possible fallout from the Trump administration’s tough immigration policies and rhetoric. About 574,000 noncitizens lost coverage in 2018, a drop of about 2.3%, the report found.
“Uninsured noncitizens account for almost a third of the increase in uninsured, which may reflect the administration’s more aggressive stance on immigration,” said Joseph Antos, a health economist at the American Enterprise Institute.
The increase in the number of uninsured people in 2018 was remarkable because uninsured rates typically fall or hold steady when unemployment rates drop. The U.S. unemployment rate fell slightly from about 4.3% in 2017 to 4% in 2018.
The uninsured rate continued to vary by poverty status and whether a state expanded its Medicaid program under Obamacare. Texas (17.7%), Oklahoma (14.2%), Georgia (13.7%), and Florida (13%) had the highest uninsured rates in 2018, according to the report. None of those states have expanded Medicaid under Obamacare.
The percentage of uninsured children aged under 19 years increased by 0.6 percentage points from 2017 to 2018, to 5.5%.
“The Census data are clear – the uninsured rate for kids is up sharply and it’s due to a loss of public coverage – mostly Medicaid,” Joan Alker, executive director of Georgetown University Center for Children and Families, said in a statement.
“These children are not getting private coverage as the Trump administration has suggested but rather becoming uninsured,” she said. “This serious erosion of children’s health coverage is due in large part to the Trump administration’s actions that have made health care harder to access and have deterred families from enrolling their children.”
The share of Americans without medical insurance fell steadily since 2014 but then leveled off in 2017, the year Mr. Trump became president.
Health care advocates have complained that efforts by the Trump administration and Congress are jeopardizing insurance enrollment. They point to cuts in outreach programs that aim to tell consumers about their health care options under Obamacare and the elimination of the ACA’s tax penalty for people who don’t have health coverage.
Ms. Alker complained that the administration’s policies are causing the loss of children’s coverage. “In a period of continued economic and job growth, we shouldn’t be going backwards on health coverage,” said Judy Solomon, a senior fellow for the Center on Budget and Policy Priorities, a left-leaning think tank. “This backsliding almost certainly reflects, at least in part, Trump administration policies to weaken public health coverage.”
She attributed the drop to the Trump administration making it harder for families to enroll for coverage in Medicaid by curtailing outreach efforts, allowing states to ask for more paperwork and proposing a so-called public charge rule that would make it harder for legal immigrants to get permanent resident status if they have received certain kinds of public assistance – including Medicaid.
Tom Miller, a resident fellow at the American Enterprise Institute, a conservative think tank, said the drop in Medicaid coverage “is a positive.”
“When the economy grows Medicaid eventually drops,” he said.
One reason for the drop in health coverage is that middle-income families can’t afford the rising cost of insurance in the individual market, particularly if they don’t qualify for government subsidies, he added.
“On balance, this is some short-term noise,” he said of the uptick in the uninsured rate. “I would put more stake in it if happens for several years.”
Chris Pope, a senior fellow with the conservative Manhattan Institute, also said he considered the change “fairly small” and likely caused by increasing wages “pushing people above the income eligibility cutoff in Medicaid expansion states.”
But he suggested that next year would be a better indicator of how changes in the ACA are playing out. “I expect that the mandate repeal will make next year’s increase in the uninsured more significant.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
For the first time in a decade, the number of Americans without health insurance has risen – by about 2 million people in 2018 – according to the annual U.S. Census Bureau report released Sept. 10, 2019.
The Census found that 8.5% of the U.S. population went without medical insurance for all of 2018, up from 7.9% in 2017. By contrast, in 2013, before the Affordable Care Act took full effect, 13.3% were uninsured. It was the first year-to-year increase since 2008-09, Census officials said, adding that most of the drop in health coverage was related to a 0.7% decline in Medicaid participants. The number of people with private insurance remained steady and there was a 0.4% increase in those on Medicare.
Many of those losing coverage were noncitizens, a possible fallout from the Trump administration’s tough immigration policies and rhetoric. About 574,000 noncitizens lost coverage in 2018, a drop of about 2.3%, the report found.
“Uninsured noncitizens account for almost a third of the increase in uninsured, which may reflect the administration’s more aggressive stance on immigration,” said Joseph Antos, a health economist at the American Enterprise Institute.
The increase in the number of uninsured people in 2018 was remarkable because uninsured rates typically fall or hold steady when unemployment rates drop. The U.S. unemployment rate fell slightly from about 4.3% in 2017 to 4% in 2018.
The uninsured rate continued to vary by poverty status and whether a state expanded its Medicaid program under Obamacare. Texas (17.7%), Oklahoma (14.2%), Georgia (13.7%), and Florida (13%) had the highest uninsured rates in 2018, according to the report. None of those states have expanded Medicaid under Obamacare.
The percentage of uninsured children aged under 19 years increased by 0.6 percentage points from 2017 to 2018, to 5.5%.
“The Census data are clear – the uninsured rate for kids is up sharply and it’s due to a loss of public coverage – mostly Medicaid,” Joan Alker, executive director of Georgetown University Center for Children and Families, said in a statement.
“These children are not getting private coverage as the Trump administration has suggested but rather becoming uninsured,” she said. “This serious erosion of children’s health coverage is due in large part to the Trump administration’s actions that have made health care harder to access and have deterred families from enrolling their children.”
The share of Americans without medical insurance fell steadily since 2014 but then leveled off in 2017, the year Mr. Trump became president.
Health care advocates have complained that efforts by the Trump administration and Congress are jeopardizing insurance enrollment. They point to cuts in outreach programs that aim to tell consumers about their health care options under Obamacare and the elimination of the ACA’s tax penalty for people who don’t have health coverage.
Ms. Alker complained that the administration’s policies are causing the loss of children’s coverage. “In a period of continued economic and job growth, we shouldn’t be going backwards on health coverage,” said Judy Solomon, a senior fellow for the Center on Budget and Policy Priorities, a left-leaning think tank. “This backsliding almost certainly reflects, at least in part, Trump administration policies to weaken public health coverage.”
She attributed the drop to the Trump administration making it harder for families to enroll for coverage in Medicaid by curtailing outreach efforts, allowing states to ask for more paperwork and proposing a so-called public charge rule that would make it harder for legal immigrants to get permanent resident status if they have received certain kinds of public assistance – including Medicaid.
Tom Miller, a resident fellow at the American Enterprise Institute, a conservative think tank, said the drop in Medicaid coverage “is a positive.”
“When the economy grows Medicaid eventually drops,” he said.
One reason for the drop in health coverage is that middle-income families can’t afford the rising cost of insurance in the individual market, particularly if they don’t qualify for government subsidies, he added.
“On balance, this is some short-term noise,” he said of the uptick in the uninsured rate. “I would put more stake in it if happens for several years.”
Chris Pope, a senior fellow with the conservative Manhattan Institute, also said he considered the change “fairly small” and likely caused by increasing wages “pushing people above the income eligibility cutoff in Medicaid expansion states.”
But he suggested that next year would be a better indicator of how changes in the ACA are playing out. “I expect that the mandate repeal will make next year’s increase in the uninsured more significant.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
For the first time in a decade, the number of Americans without health insurance has risen – by about 2 million people in 2018 – according to the annual U.S. Census Bureau report released Sept. 10, 2019.
The Census found that 8.5% of the U.S. population went without medical insurance for all of 2018, up from 7.9% in 2017. By contrast, in 2013, before the Affordable Care Act took full effect, 13.3% were uninsured. It was the first year-to-year increase since 2008-09, Census officials said, adding that most of the drop in health coverage was related to a 0.7% decline in Medicaid participants. The number of people with private insurance remained steady and there was a 0.4% increase in those on Medicare.
Many of those losing coverage were noncitizens, a possible fallout from the Trump administration’s tough immigration policies and rhetoric. About 574,000 noncitizens lost coverage in 2018, a drop of about 2.3%, the report found.
“Uninsured noncitizens account for almost a third of the increase in uninsured, which may reflect the administration’s more aggressive stance on immigration,” said Joseph Antos, a health economist at the American Enterprise Institute.
The increase in the number of uninsured people in 2018 was remarkable because uninsured rates typically fall or hold steady when unemployment rates drop. The U.S. unemployment rate fell slightly from about 4.3% in 2017 to 4% in 2018.
The uninsured rate continued to vary by poverty status and whether a state expanded its Medicaid program under Obamacare. Texas (17.7%), Oklahoma (14.2%), Georgia (13.7%), and Florida (13%) had the highest uninsured rates in 2018, according to the report. None of those states have expanded Medicaid under Obamacare.
The percentage of uninsured children aged under 19 years increased by 0.6 percentage points from 2017 to 2018, to 5.5%.
“The Census data are clear – the uninsured rate for kids is up sharply and it’s due to a loss of public coverage – mostly Medicaid,” Joan Alker, executive director of Georgetown University Center for Children and Families, said in a statement.
“These children are not getting private coverage as the Trump administration has suggested but rather becoming uninsured,” she said. “This serious erosion of children’s health coverage is due in large part to the Trump administration’s actions that have made health care harder to access and have deterred families from enrolling their children.”
The share of Americans without medical insurance fell steadily since 2014 but then leveled off in 2017, the year Mr. Trump became president.
Health care advocates have complained that efforts by the Trump administration and Congress are jeopardizing insurance enrollment. They point to cuts in outreach programs that aim to tell consumers about their health care options under Obamacare and the elimination of the ACA’s tax penalty for people who don’t have health coverage.
Ms. Alker complained that the administration’s policies are causing the loss of children’s coverage. “In a period of continued economic and job growth, we shouldn’t be going backwards on health coverage,” said Judy Solomon, a senior fellow for the Center on Budget and Policy Priorities, a left-leaning think tank. “This backsliding almost certainly reflects, at least in part, Trump administration policies to weaken public health coverage.”
She attributed the drop to the Trump administration making it harder for families to enroll for coverage in Medicaid by curtailing outreach efforts, allowing states to ask for more paperwork and proposing a so-called public charge rule that would make it harder for legal immigrants to get permanent resident status if they have received certain kinds of public assistance – including Medicaid.
Tom Miller, a resident fellow at the American Enterprise Institute, a conservative think tank, said the drop in Medicaid coverage “is a positive.”
“When the economy grows Medicaid eventually drops,” he said.
One reason for the drop in health coverage is that middle-income families can’t afford the rising cost of insurance in the individual market, particularly if they don’t qualify for government subsidies, he added.
“On balance, this is some short-term noise,” he said of the uptick in the uninsured rate. “I would put more stake in it if happens for several years.”
Chris Pope, a senior fellow with the conservative Manhattan Institute, also said he considered the change “fairly small” and likely caused by increasing wages “pushing people above the income eligibility cutoff in Medicaid expansion states.”
But he suggested that next year would be a better indicator of how changes in the ACA are playing out. “I expect that the mandate repeal will make next year’s increase in the uninsured more significant.”
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Long-term advantages may not endure after early tight control in psoriatic arthritis
, based on data from 110 patients in the TIght COntrol of inflammation in early Psoriatic Arthritis (TICOPA) study.

In the original study, significantly more adults with psoriatic arthritis (PsA) who were randomized to a tight control, treat-to-target group achieved minimal disease activity criteria after 48 weeks, compared with a standard care group (40% vs. 25%).
“Following exit from this study, we hypothesized that this advantage would translate to a clinical advantage in the medium term,” wrote Laura C. Coates, MBChB, PhD, of the University of Oxford (England) and colleagues.
In a study published in Rheumatology, the researchers examined data from 54 patients in the tight control arm of TICOPA and 56 patients in the standard care arm.
At 5 years after the completion of the TICOPA study, 69% of patients in the tight control group and 76% of patients in the standard care group were considered to be in low disease activity. In addition, methotrexate use after 5 years was similar between the tight control and standard care groups (44% and 54%, respectively) and both groups had reduced methotrexate use since the study’s end.
Overall use of biologic drugs was similar between the tight control and standard care groups after 5 years (54% and 52%, respectively), although overall use of biologics was higher in the tight control group at the end of the original study, compared with the standard care group (33% vs. 9%).
The findings were limited by several factors, notably the lack of intention to continue treatment or observations beyond the end of the original TICOPA study, and the patients’ status at 5 years was based on routine clinician notes with no formal assessment of minimal disease activity or objective measure of disease status, the researchers noted.
However, “this result reflects clinical practice in routine rheumatology care,” and any benefit of early tight control on later disease activity could not be determined, they added.
The current study was funded by the National Institute for Health Research infrastructure at Leeds and Oxford (England). The original TICOPA study was funded by Arthritis Research UK (now called Versus Arthritis) and Pfizer. Some of the investigators disclosed financial relationships with companies that market drugs for PsA.
SOURCE: Coates LC et al. Rheumatology. 2019 Aug 31. doi: 10.1093/rheumatology/kez369.
, based on data from 110 patients in the TIght COntrol of inflammation in early Psoriatic Arthritis (TICOPA) study.
In the original study, significantly more adults with psoriatic arthritis (PsA) who were randomized to a tight control, treat-to-target group achieved minimal disease activity criteria after 48 weeks, compared with a standard care group (40% vs. 25%).
“Following exit from this study, we hypothesized that this advantage would translate to a clinical advantage in the medium term,” wrote Laura C. Coates, MBChB, PhD, of the University of Oxford (England) and colleagues.
In a study published in Rheumatology, the researchers examined data from 54 patients in the tight control arm of TICOPA and 56 patients in the standard care arm.
At 5 years after the completion of the TICOPA study, 69% of patients in the tight control group and 76% of patients in the standard care group were considered to be in low disease activity. In addition, methotrexate use after 5 years was similar between the tight control and standard care groups (44% and 54%, respectively) and both groups had reduced methotrexate use since the study’s end.
Overall use of biologic drugs was similar between the tight control and standard care groups after 5 years (54% and 52%, respectively), although overall use of biologics was higher in the tight control group at the end of the original study, compared with the standard care group (33% vs. 9%).
The findings were limited by several factors, notably the lack of intention to continue treatment or observations beyond the end of the original TICOPA study, and the patients’ status at 5 years was based on routine clinician notes with no formal assessment of minimal disease activity or objective measure of disease status, the researchers noted.
However, “this result reflects clinical practice in routine rheumatology care,” and any benefit of early tight control on later disease activity could not be determined, they added.
The current study was funded by the National Institute for Health Research infrastructure at Leeds and Oxford (England). The original TICOPA study was funded by Arthritis Research UK (now called Versus Arthritis) and Pfizer. Some of the investigators disclosed financial relationships with companies that market drugs for PsA.
SOURCE: Coates LC et al. Rheumatology. 2019 Aug 31. doi: 10.1093/rheumatology/kez369.
, based on data from 110 patients in the TIght COntrol of inflammation in early Psoriatic Arthritis (TICOPA) study.
In the original study, significantly more adults with psoriatic arthritis (PsA) who were randomized to a tight control, treat-to-target group achieved minimal disease activity criteria after 48 weeks, compared with a standard care group (40% vs. 25%).
“Following exit from this study, we hypothesized that this advantage would translate to a clinical advantage in the medium term,” wrote Laura C. Coates, MBChB, PhD, of the University of Oxford (England) and colleagues.
In a study published in Rheumatology, the researchers examined data from 54 patients in the tight control arm of TICOPA and 56 patients in the standard care arm.
At 5 years after the completion of the TICOPA study, 69% of patients in the tight control group and 76% of patients in the standard care group were considered to be in low disease activity. In addition, methotrexate use after 5 years was similar between the tight control and standard care groups (44% and 54%, respectively) and both groups had reduced methotrexate use since the study’s end.
Overall use of biologic drugs was similar between the tight control and standard care groups after 5 years (54% and 52%, respectively), although overall use of biologics was higher in the tight control group at the end of the original study, compared with the standard care group (33% vs. 9%).
The findings were limited by several factors, notably the lack of intention to continue treatment or observations beyond the end of the original TICOPA study, and the patients’ status at 5 years was based on routine clinician notes with no formal assessment of minimal disease activity or objective measure of disease status, the researchers noted.
However, “this result reflects clinical practice in routine rheumatology care,” and any benefit of early tight control on later disease activity could not be determined, they added.
The current study was funded by the National Institute for Health Research infrastructure at Leeds and Oxford (England). The original TICOPA study was funded by Arthritis Research UK (now called Versus Arthritis) and Pfizer. Some of the investigators disclosed financial relationships with companies that market drugs for PsA.
SOURCE: Coates LC et al. Rheumatology. 2019 Aug 31. doi: 10.1093/rheumatology/kez369.
FROM RHEUMATOLOGY
Key clinical point: Patients in a psoriatic arthritis study comparing tight control and standard care showed no significant difference in disease activity 5 years later.
Major finding: At 5 years after the end of the TICOPA trial, 69% in the tight control group vs. 76% in the standard care group were considered to be in low disease activity.
Study details: The data come from a follow-up of 110 patients from the TIght COntrol of inflammation in early Psoriatic Arthritis (TICOPA) study.
Disclosures: The current study was funded by the National Institute for Health Research infrastructure at Leeds and Oxford (England). The original TICOPA study was funded by Arthritis Research UK (now called Versus Arthritis) and Pfizer. Some of the investigators disclosed financial relationships with companies that market drugs for PsA.
Source: Coates LC et al. Rheumatology. 2019 Aug 31. doi: 10.1093/rheumatology/kez369.
Dr. John Mann discusses suicide prevention

