User login
Why STEMI patients benefit from PCI of nonculprit lesions
PHILADELPHIA – Nearly half of patients with ST-elevation MI and multivessel coronary artery disease in the landmark COMPLETE trial had an obstructive coronary lesion with vulnerable plaque morphology in a segment far from the culprit lesion, Natalia Pinilla-Echeverri, MD, reported at the American Heart Association scientific sessions.

This novel finding from an optical coherence tomography (OCT) substudy of COMPLETE provides a likely mechanistic explanation for the major clinical benefits documented in the full COMPLETE trial, noted Dr. Pinilla-Echeverri, a cardiologist at the Population Health Research Institute at McMaster University, Hamilton, Ont.
COMPLETE was a multinational trial which randomized 4,041 ST-elevation MI (STEMI) patients with multivessel disease to culprit lesion–only percutaneous coronary intervention (PCI) or additional routine angiography–guided staged PCI of nonculprit obstructive lesions with at least 70% stenosis. As previously reported, the risk of the coprimary composite endpoint comprising cardiovascular death, new MI, or ischemia-driven revascularization was reduced by 49% over 3 years of follow-up in the group with staged PCI of nonculprit lesions, with an impressive number needed to treat of just 13 (N Engl J Med. 2019 Oct 10;381[15]:1411-21).
Dr. Pinella-Echeverri reported on the 93 patients who participated in the OCT substudy, the purpose of which was to determine the prevalence of high-risk, vulnerable plaque in obstructive and nonobstructive nonculprit lesions. For this purpose, vulnerable plaque was defined as thin-cap fibroatheroma (TCFA), a coronary lesion known to pose high risk of worsening stenosis, plaque rupture, and cardiovascular events.
Of note, these 93 patients had a total of 425 diseased segments: 150 obstructive and 275 nonobstructive.
“This is reassuring that the concept of acute coronary syndrome implies a diffuse pathophysiology of affecting not only the culprit segment but the coronary vasculature as a whole,” Dr. Pinella-Echeverri observed.
The main study finding, however, was that TCFA was significantly more prevalent in obstructive, compared with nonobstructive, nonculprit lesions by a margin of 35% to 23%. The obstructive and nonobstructive TCFA lesions had a similar lipid-rich composition; however, the obstructive ones were significantly longer and had a smaller mean lumen area.
When breaking down the prevalence of TCFA per patient, 47% of patients had a nonculprit obstructive lesion with vulnerable plaque morphology. Another 20% had nonobstructive TCFA lesions. And only 32% of the STEMI patients had no TCFA in their obstructive or nonobstructive segments.
Discussant Frans Van de Werf, MD, PhD, commented: “This [OCT substudy result] immediately explains the clinical benefit observed with preventive PCI in STEMI patients with obstructive multivessel disease.”
The finding that 20% of the STEMI patients had nonobstructive lesions with vulnerable plaque morphology by OCT provides powerful support for the current guideline-recommended strategy of immediately starting STEMI patients on intensive lipid-lowering therapy, added Dr. Van de Werf, professor of medicine at the Catholic University of Leuven (Belgium).
He argued that the decision to revascularize nonculprit lesions by means of PCI versus the more complete revascularization achieved via coronary artery bypass graft surgery shouldn’t be made during the initial primary PCI, citing evidence that when the decision gets made at that time, coronary artery bypass grafting (CABG) is less likely to be chosen.
“I believe that OCT and [fractional flow reserve] should not be performed during the index primary PCI, not only for the comfort of the patient, but also for the better selection of complete revascularization. Interventional cardiologists should not forget that CABG might be a better revascularization treatment in some cases, such as left main disease and diabetes mellitus,” the cardiologist cautioned.
The COMPLETE OCT Substudy was supported by Abbott Vascular, the Population Health Research Institute, Hamilton Health Sciences, and the Canadian Institutes of Health Research.
PHILADELPHIA – Nearly half of patients with ST-elevation MI and multivessel coronary artery disease in the landmark COMPLETE trial had an obstructive coronary lesion with vulnerable plaque morphology in a segment far from the culprit lesion, Natalia Pinilla-Echeverri, MD, reported at the American Heart Association scientific sessions.
This novel finding from an optical coherence tomography (OCT) substudy of COMPLETE provides a likely mechanistic explanation for the major clinical benefits documented in the full COMPLETE trial, noted Dr. Pinilla-Echeverri, a cardiologist at the Population Health Research Institute at McMaster University, Hamilton, Ont.
COMPLETE was a multinational trial which randomized 4,041 ST-elevation MI (STEMI) patients with multivessel disease to culprit lesion–only percutaneous coronary intervention (PCI) or additional routine angiography–guided staged PCI of nonculprit obstructive lesions with at least 70% stenosis. As previously reported, the risk of the coprimary composite endpoint comprising cardiovascular death, new MI, or ischemia-driven revascularization was reduced by 49% over 3 years of follow-up in the group with staged PCI of nonculprit lesions, with an impressive number needed to treat of just 13 (N Engl J Med. 2019 Oct 10;381[15]:1411-21).
Dr. Pinella-Echeverri reported on the 93 patients who participated in the OCT substudy, the purpose of which was to determine the prevalence of high-risk, vulnerable plaque in obstructive and nonobstructive nonculprit lesions. For this purpose, vulnerable plaque was defined as thin-cap fibroatheroma (TCFA), a coronary lesion known to pose high risk of worsening stenosis, plaque rupture, and cardiovascular events.
Of note, these 93 patients had a total of 425 diseased segments: 150 obstructive and 275 nonobstructive.
“This is reassuring that the concept of acute coronary syndrome implies a diffuse pathophysiology of affecting not only the culprit segment but the coronary vasculature as a whole,” Dr. Pinella-Echeverri observed.
The main study finding, however, was that TCFA was significantly more prevalent in obstructive, compared with nonobstructive, nonculprit lesions by a margin of 35% to 23%. The obstructive and nonobstructive TCFA lesions had a similar lipid-rich composition; however, the obstructive ones were significantly longer and had a smaller mean lumen area.
When breaking down the prevalence of TCFA per patient, 47% of patients had a nonculprit obstructive lesion with vulnerable plaque morphology. Another 20% had nonobstructive TCFA lesions. And only 32% of the STEMI patients had no TCFA in their obstructive or nonobstructive segments.
Discussant Frans Van de Werf, MD, PhD, commented: “This [OCT substudy result] immediately explains the clinical benefit observed with preventive PCI in STEMI patients with obstructive multivessel disease.”
The finding that 20% of the STEMI patients had nonobstructive lesions with vulnerable plaque morphology by OCT provides powerful support for the current guideline-recommended strategy of immediately starting STEMI patients on intensive lipid-lowering therapy, added Dr. Van de Werf, professor of medicine at the Catholic University of Leuven (Belgium).
He argued that the decision to revascularize nonculprit lesions by means of PCI versus the more complete revascularization achieved via coronary artery bypass graft surgery shouldn’t be made during the initial primary PCI, citing evidence that when the decision gets made at that time, coronary artery bypass grafting (CABG) is less likely to be chosen.
“I believe that OCT and [fractional flow reserve] should not be performed during the index primary PCI, not only for the comfort of the patient, but also for the better selection of complete revascularization. Interventional cardiologists should not forget that CABG might be a better revascularization treatment in some cases, such as left main disease and diabetes mellitus,” the cardiologist cautioned.
The COMPLETE OCT Substudy was supported by Abbott Vascular, the Population Health Research Institute, Hamilton Health Sciences, and the Canadian Institutes of Health Research.
PHILADELPHIA – Nearly half of patients with ST-elevation MI and multivessel coronary artery disease in the landmark COMPLETE trial had an obstructive coronary lesion with vulnerable plaque morphology in a segment far from the culprit lesion, Natalia Pinilla-Echeverri, MD, reported at the American Heart Association scientific sessions.
This novel finding from an optical coherence tomography (OCT) substudy of COMPLETE provides a likely mechanistic explanation for the major clinical benefits documented in the full COMPLETE trial, noted Dr. Pinilla-Echeverri, a cardiologist at the Population Health Research Institute at McMaster University, Hamilton, Ont.
COMPLETE was a multinational trial which randomized 4,041 ST-elevation MI (STEMI) patients with multivessel disease to culprit lesion–only percutaneous coronary intervention (PCI) or additional routine angiography–guided staged PCI of nonculprit obstructive lesions with at least 70% stenosis. As previously reported, the risk of the coprimary composite endpoint comprising cardiovascular death, new MI, or ischemia-driven revascularization was reduced by 49% over 3 years of follow-up in the group with staged PCI of nonculprit lesions, with an impressive number needed to treat of just 13 (N Engl J Med. 2019 Oct 10;381[15]:1411-21).
Dr. Pinella-Echeverri reported on the 93 patients who participated in the OCT substudy, the purpose of which was to determine the prevalence of high-risk, vulnerable plaque in obstructive and nonobstructive nonculprit lesions. For this purpose, vulnerable plaque was defined as thin-cap fibroatheroma (TCFA), a coronary lesion known to pose high risk of worsening stenosis, plaque rupture, and cardiovascular events.
Of note, these 93 patients had a total of 425 diseased segments: 150 obstructive and 275 nonobstructive.
“This is reassuring that the concept of acute coronary syndrome implies a diffuse pathophysiology of affecting not only the culprit segment but the coronary vasculature as a whole,” Dr. Pinella-Echeverri observed.
The main study finding, however, was that TCFA was significantly more prevalent in obstructive, compared with nonobstructive, nonculprit lesions by a margin of 35% to 23%. The obstructive and nonobstructive TCFA lesions had a similar lipid-rich composition; however, the obstructive ones were significantly longer and had a smaller mean lumen area.
When breaking down the prevalence of TCFA per patient, 47% of patients had a nonculprit obstructive lesion with vulnerable plaque morphology. Another 20% had nonobstructive TCFA lesions. And only 32% of the STEMI patients had no TCFA in their obstructive or nonobstructive segments.
Discussant Frans Van de Werf, MD, PhD, commented: “This [OCT substudy result] immediately explains the clinical benefit observed with preventive PCI in STEMI patients with obstructive multivessel disease.”
The finding that 20% of the STEMI patients had nonobstructive lesions with vulnerable plaque morphology by OCT provides powerful support for the current guideline-recommended strategy of immediately starting STEMI patients on intensive lipid-lowering therapy, added Dr. Van de Werf, professor of medicine at the Catholic University of Leuven (Belgium).
He argued that the decision to revascularize nonculprit lesions by means of PCI versus the more complete revascularization achieved via coronary artery bypass graft surgery shouldn’t be made during the initial primary PCI, citing evidence that when the decision gets made at that time, coronary artery bypass grafting (CABG) is less likely to be chosen.
“I believe that OCT and [fractional flow reserve] should not be performed during the index primary PCI, not only for the comfort of the patient, but also for the better selection of complete revascularization. Interventional cardiologists should not forget that CABG might be a better revascularization treatment in some cases, such as left main disease and diabetes mellitus,” the cardiologist cautioned.
The COMPLETE OCT Substudy was supported by Abbott Vascular, the Population Health Research Institute, Hamilton Health Sciences, and the Canadian Institutes of Health Research.
REPORTING FROM AHA 2019
Cardiac biomarkers refine antihypertensive drug initiation decisions
PHILADELPHIA – Incorporation of cardiac biomarkers into current guideline-based decision-making regarding initiation of antihypertensive medication in patients with previously untreated mild or moderate high blood pressure leads to more appropriate and selective matching of intensive blood pressure control with true patient risk, Ambarish Pandey, MD, reported at the American Heart Association scientific sessions.

That’s because the 2017 American College of Cardiology/AHA blood pressure guidelines recommend incorporating the ACC/AHA 10-Year Atherosclerotic Cardiovascular Disease (ASCVD) Risk Calculator into decision making as to whether to start antihypertensive drug therapy in patients with stage 1 hypertension (130-139/80-89 mm Hg), but the risk calculator doesn’t account for the risk of heart failure.
Yet by far the greatest benefit of intensive BP lowering is in reducing the risk of developing heart failure, as demonstrated in the landmark SPRINT trial, which showed that intensive BP lowering achieved much greater risk reduction in new-onset heart failure than in atherosclerotic cardiovascular events.
Thus, there’s a need for better strategies to guide antihypertensive therapy. And therein lies the rationale for incorporating into the risk assessment an individual’s values for N-terminal pro–brain natriuretic peptide (NT-proBNP), which reflects chronic myocardial stress, and high-sensitivity cardiac troponin T (hs-cTnT), which when elevated signals myocardial injury.
“Cardiac biomarkers are intermediate phenotypes from hypertension to future cardiovascular events. They can identify individuals at increased risk for atherosclerotic events, and at even higher risk for heart failure events,” explained Dr. Pandey, a cardiologist at the University of Texas Southwestern Medical Center, Dallas.
He presented a study of 12,987 participants in three major U.S. cohort studies: the Atherosclerosis Risk In Communities (ARIC) study, the Multi-Ethnic Study of Atherosclerosis (MESA), and the Dallas Heart Study. At baseline, none of the participants were on antihypertensive therapy or had known cardiovascular disease. During 10 years of prospective follow-up, 825 of them experienced a first cardiovascular disease event: 251 developed heart failure and 574 had an MI, stroke, or cardiovascular death. Dr. Pandey and his coworkers calculated the cardiovascular event incidence rate and number-needed-to-treat with intensive antihypertensive drug therapy to prevent a first cardiovascular disease event on the basis of whether patients in the various BP categories were positive or negative for one or more biomarkers.
The results
Fifty-four percent of subjects had normal BP, defined in the guidelines as less than 120/80 mm Hg. Another 3% had BP in excess of 160/100 mm Hg. No controversy exists regarding pharmacotherapy in either of these groups: It’s not warranted in the former, essential in the latter.
Another 3,000 individuals had what the ACC/AHA guidelines define as elevated BP, meaning 120-129/<80 mm Hg, or low-risk stage 1 hypertension of 130-139/80-89 mm Hg and a 10-year ASCVD risk score of less than 10%. Initiation of antihypertensive medication in these groups is not recommended in the guidelines. Yet 36% of these individuals had at least one positive cardiac biomarker. And here’s the eye-opening finding: Notably, the 10-year cardiovascular event incidence rate in this biomarker group not currently recommended for antihypertensive pharmacotherapy was 11%, more than double the 4.6% rate in the biomarker-negative group, which in turn was comparable to the 3.8% in the normal BP participants.
Antihypertensive therapy was recommended according to the guidelines in 20% of the total study population, comprising patients with stage 1 hypertension who had an ASCVD risk score of 10% or more as well as those with stage 2 hypertension, defined as BP greater than 140/90 mm Hg but less than 160/100 mm Hg. Forty-eight percent of these subjects were positive for at least one biomarker. Their cardiovascular incidence rate was 15.1%, compared to the 7.9% rate in biomarker-negative individuals.
The estimated number-needed-to-treat (NNT) with intensive blood pressure–lowering therapy to a target systolic BP of less than 120 mm Hg, as in SPRINT, to prevent one cardiovascular event in individuals not currently guideline-recommended for antihypertensive medications was 86 in those who were biomarker-negative. The NNT dropped to 36 in the biomarker-positive subgroup, a far more attractive figure that suggests a reasonable likelihood of benefit from intensive blood pressure control, in Dr. Pandey’s view.
Similarly, among individuals currently recommended for pharmacotherapy initiation, the NNTs were 49 if biomarker-negative, improving to 26 in those positive for one or both biomarkers, which was comparable to the NNT of 22 in the group with blood pressures greater than 160/100 mm Hg. The NNT of 49 in the biomarker-negative subgroup is in a borderline gray zone warranting individualized shared decision-making regarding pharmacotherapy, Dr. Pandey said.
In this study, an elevated hs-cTnT was defined as 6 ng/L or more, while an elevated NT-proBNP was considered to be at least 100 pg/mL.
“It’s noteworthy that the degree of elevation in hs-cTnT and NT-proBNP which were observed in our study were pretty subtle and much below the threshold used for diagnosis of ischemic events or heart failure. Thus, these elevations were largely representative of subtle chronic injury and not acute events,” according to the cardiologist.
One audience member asked if the elevated biomarkers could simply be a surrogate for longer duration of exposure of the heart to high BP. Sure, Dr. Pandey replied, pointing to the 6-year greater average age of the biomarker-positive participants.
“It is likely that biomarker-positive status is capturing the culmination of longstanding exposure. But the thing about hypertension is there are no symptoms that can signal to the patient or the doctor that they have this disease, so testing for the biomarkers can actually capture the high-risk group that may have had hypertension for a long duration but now needs to be treated in order to prevent the advance of downstream adverse events,” he said.
Dr. Pandey reported having no financial conflicts of interest regarding his study, conducted free of commercial support.
SOURCE: Pandey A. AHA 2019 Abstract EP.AOS.521.141
PHILADELPHIA – Incorporation of cardiac biomarkers into current guideline-based decision-making regarding initiation of antihypertensive medication in patients with previously untreated mild or moderate high blood pressure leads to more appropriate and selective matching of intensive blood pressure control with true patient risk, Ambarish Pandey, MD, reported at the American Heart Association scientific sessions.
That’s because the 2017 American College of Cardiology/AHA blood pressure guidelines recommend incorporating the ACC/AHA 10-Year Atherosclerotic Cardiovascular Disease (ASCVD) Risk Calculator into decision making as to whether to start antihypertensive drug therapy in patients with stage 1 hypertension (130-139/80-89 mm Hg), but the risk calculator doesn’t account for the risk of heart failure.
Yet by far the greatest benefit of intensive BP lowering is in reducing the risk of developing heart failure, as demonstrated in the landmark SPRINT trial, which showed that intensive BP lowering achieved much greater risk reduction in new-onset heart failure than in atherosclerotic cardiovascular events.
Thus, there’s a need for better strategies to guide antihypertensive therapy. And therein lies the rationale for incorporating into the risk assessment an individual’s values for N-terminal pro–brain natriuretic peptide (NT-proBNP), which reflects chronic myocardial stress, and high-sensitivity cardiac troponin T (hs-cTnT), which when elevated signals myocardial injury.
“Cardiac biomarkers are intermediate phenotypes from hypertension to future cardiovascular events. They can identify individuals at increased risk for atherosclerotic events, and at even higher risk for heart failure events,” explained Dr. Pandey, a cardiologist at the University of Texas Southwestern Medical Center, Dallas.
He presented a study of 12,987 participants in three major U.S. cohort studies: the Atherosclerosis Risk In Communities (ARIC) study, the Multi-Ethnic Study of Atherosclerosis (MESA), and the Dallas Heart Study. At baseline, none of the participants were on antihypertensive therapy or had known cardiovascular disease. During 10 years of prospective follow-up, 825 of them experienced a first cardiovascular disease event: 251 developed heart failure and 574 had an MI, stroke, or cardiovascular death. Dr. Pandey and his coworkers calculated the cardiovascular event incidence rate and number-needed-to-treat with intensive antihypertensive drug therapy to prevent a first cardiovascular disease event on the basis of whether patients in the various BP categories were positive or negative for one or more biomarkers.
The results
Fifty-four percent of subjects had normal BP, defined in the guidelines as less than 120/80 mm Hg. Another 3% had BP in excess of 160/100 mm Hg. No controversy exists regarding pharmacotherapy in either of these groups: It’s not warranted in the former, essential in the latter.
Another 3,000 individuals had what the ACC/AHA guidelines define as elevated BP, meaning 120-129/<80 mm Hg, or low-risk stage 1 hypertension of 130-139/80-89 mm Hg and a 10-year ASCVD risk score of less than 10%. Initiation of antihypertensive medication in these groups is not recommended in the guidelines. Yet 36% of these individuals had at least one positive cardiac biomarker. And here’s the eye-opening finding: Notably, the 10-year cardiovascular event incidence rate in this biomarker group not currently recommended for antihypertensive pharmacotherapy was 11%, more than double the 4.6% rate in the biomarker-negative group, which in turn was comparable to the 3.8% in the normal BP participants.
Antihypertensive therapy was recommended according to the guidelines in 20% of the total study population, comprising patients with stage 1 hypertension who had an ASCVD risk score of 10% or more as well as those with stage 2 hypertension, defined as BP greater than 140/90 mm Hg but less than 160/100 mm Hg. Forty-eight percent of these subjects were positive for at least one biomarker. Their cardiovascular incidence rate was 15.1%, compared to the 7.9% rate in biomarker-negative individuals.
The estimated number-needed-to-treat (NNT) with intensive blood pressure–lowering therapy to a target systolic BP of less than 120 mm Hg, as in SPRINT, to prevent one cardiovascular event in individuals not currently guideline-recommended for antihypertensive medications was 86 in those who were biomarker-negative. The NNT dropped to 36 in the biomarker-positive subgroup, a far more attractive figure that suggests a reasonable likelihood of benefit from intensive blood pressure control, in Dr. Pandey’s view.
Similarly, among individuals currently recommended for pharmacotherapy initiation, the NNTs were 49 if biomarker-negative, improving to 26 in those positive for one or both biomarkers, which was comparable to the NNT of 22 in the group with blood pressures greater than 160/100 mm Hg. The NNT of 49 in the biomarker-negative subgroup is in a borderline gray zone warranting individualized shared decision-making regarding pharmacotherapy, Dr. Pandey said.
In this study, an elevated hs-cTnT was defined as 6 ng/L or more, while an elevated NT-proBNP was considered to be at least 100 pg/mL.
“It’s noteworthy that the degree of elevation in hs-cTnT and NT-proBNP which were observed in our study were pretty subtle and much below the threshold used for diagnosis of ischemic events or heart failure. Thus, these elevations were largely representative of subtle chronic injury and not acute events,” according to the cardiologist.
One audience member asked if the elevated biomarkers could simply be a surrogate for longer duration of exposure of the heart to high BP. Sure, Dr. Pandey replied, pointing to the 6-year greater average age of the biomarker-positive participants.
“It is likely that biomarker-positive status is capturing the culmination of longstanding exposure. But the thing about hypertension is there are no symptoms that can signal to the patient or the doctor that they have this disease, so testing for the biomarkers can actually capture the high-risk group that may have had hypertension for a long duration but now needs to be treated in order to prevent the advance of downstream adverse events,” he said.
Dr. Pandey reported having no financial conflicts of interest regarding his study, conducted free of commercial support.
SOURCE: Pandey A. AHA 2019 Abstract EP.AOS.521.141
PHILADELPHIA – Incorporation of cardiac biomarkers into current guideline-based decision-making regarding initiation of antihypertensive medication in patients with previously untreated mild or moderate high blood pressure leads to more appropriate and selective matching of intensive blood pressure control with true patient risk, Ambarish Pandey, MD, reported at the American Heart Association scientific sessions.
That’s because the 2017 American College of Cardiology/AHA blood pressure guidelines recommend incorporating the ACC/AHA 10-Year Atherosclerotic Cardiovascular Disease (ASCVD) Risk Calculator into decision making as to whether to start antihypertensive drug therapy in patients with stage 1 hypertension (130-139/80-89 mm Hg), but the risk calculator doesn’t account for the risk of heart failure.
Yet by far the greatest benefit of intensive BP lowering is in reducing the risk of developing heart failure, as demonstrated in the landmark SPRINT trial, which showed that intensive BP lowering achieved much greater risk reduction in new-onset heart failure than in atherosclerotic cardiovascular events.
Thus, there’s a need for better strategies to guide antihypertensive therapy. And therein lies the rationale for incorporating into the risk assessment an individual’s values for N-terminal pro–brain natriuretic peptide (NT-proBNP), which reflects chronic myocardial stress, and high-sensitivity cardiac troponin T (hs-cTnT), which when elevated signals myocardial injury.
“Cardiac biomarkers are intermediate phenotypes from hypertension to future cardiovascular events. They can identify individuals at increased risk for atherosclerotic events, and at even higher risk for heart failure events,” explained Dr. Pandey, a cardiologist at the University of Texas Southwestern Medical Center, Dallas.
He presented a study of 12,987 participants in three major U.S. cohort studies: the Atherosclerosis Risk In Communities (ARIC) study, the Multi-Ethnic Study of Atherosclerosis (MESA), and the Dallas Heart Study. At baseline, none of the participants were on antihypertensive therapy or had known cardiovascular disease. During 10 years of prospective follow-up, 825 of them experienced a first cardiovascular disease event: 251 developed heart failure and 574 had an MI, stroke, or cardiovascular death. Dr. Pandey and his coworkers calculated the cardiovascular event incidence rate and number-needed-to-treat with intensive antihypertensive drug therapy to prevent a first cardiovascular disease event on the basis of whether patients in the various BP categories were positive or negative for one or more biomarkers.
The results
Fifty-four percent of subjects had normal BP, defined in the guidelines as less than 120/80 mm Hg. Another 3% had BP in excess of 160/100 mm Hg. No controversy exists regarding pharmacotherapy in either of these groups: It’s not warranted in the former, essential in the latter.
Another 3,000 individuals had what the ACC/AHA guidelines define as elevated BP, meaning 120-129/<80 mm Hg, or low-risk stage 1 hypertension of 130-139/80-89 mm Hg and a 10-year ASCVD risk score of less than 10%. Initiation of antihypertensive medication in these groups is not recommended in the guidelines. Yet 36% of these individuals had at least one positive cardiac biomarker. And here’s the eye-opening finding: Notably, the 10-year cardiovascular event incidence rate in this biomarker group not currently recommended for antihypertensive pharmacotherapy was 11%, more than double the 4.6% rate in the biomarker-negative group, which in turn was comparable to the 3.8% in the normal BP participants.
Antihypertensive therapy was recommended according to the guidelines in 20% of the total study population, comprising patients with stage 1 hypertension who had an ASCVD risk score of 10% or more as well as those with stage 2 hypertension, defined as BP greater than 140/90 mm Hg but less than 160/100 mm Hg. Forty-eight percent of these subjects were positive for at least one biomarker. Their cardiovascular incidence rate was 15.1%, compared to the 7.9% rate in biomarker-negative individuals.
The estimated number-needed-to-treat (NNT) with intensive blood pressure–lowering therapy to a target systolic BP of less than 120 mm Hg, as in SPRINT, to prevent one cardiovascular event in individuals not currently guideline-recommended for antihypertensive medications was 86 in those who were biomarker-negative. The NNT dropped to 36 in the biomarker-positive subgroup, a far more attractive figure that suggests a reasonable likelihood of benefit from intensive blood pressure control, in Dr. Pandey’s view.
Similarly, among individuals currently recommended for pharmacotherapy initiation, the NNTs were 49 if biomarker-negative, improving to 26 in those positive for one or both biomarkers, which was comparable to the NNT of 22 in the group with blood pressures greater than 160/100 mm Hg. The NNT of 49 in the biomarker-negative subgroup is in a borderline gray zone warranting individualized shared decision-making regarding pharmacotherapy, Dr. Pandey said.
In this study, an elevated hs-cTnT was defined as 6 ng/L or more, while an elevated NT-proBNP was considered to be at least 100 pg/mL.
“It’s noteworthy that the degree of elevation in hs-cTnT and NT-proBNP which were observed in our study were pretty subtle and much below the threshold used for diagnosis of ischemic events or heart failure. Thus, these elevations were largely representative of subtle chronic injury and not acute events,” according to the cardiologist.
One audience member asked if the elevated biomarkers could simply be a surrogate for longer duration of exposure of the heart to high BP. Sure, Dr. Pandey replied, pointing to the 6-year greater average age of the biomarker-positive participants.
“It is likely that biomarker-positive status is capturing the culmination of longstanding exposure. But the thing about hypertension is there are no symptoms that can signal to the patient or the doctor that they have this disease, so testing for the biomarkers can actually capture the high-risk group that may have had hypertension for a long duration but now needs to be treated in order to prevent the advance of downstream adverse events,” he said.
Dr. Pandey reported having no financial conflicts of interest regarding his study, conducted free of commercial support.
SOURCE: Pandey A. AHA 2019 Abstract EP.AOS.521.141
REPORTING FROM AHA 2019
Ofatumumab works safely for elderly patients with CLL, comorbidities
For elderly patients with chronic lymphocytic leukemia (CLL) and comorbidities, the anti-CD20 monoclonal antibody ofatumumab may be a safe and effective treatment option, according to a recent phase 2 trial.
Among 32 patients with a median age of 73 years, the overall response rate was 72%, and no grade 4 adverse events occurred, reported lead author Candida Vitale, MD, PhD, of the University of Torino (Italy) and colleagues.
These findings help fill in a knowledge gap created by clinical trial exclusions, which currently make treatment planning “a significant challenge,” the investigators wrote in Journal of Geriatric Oncology.
The study, which was conducted at MD Anderson Cancer Center in Houston, enrolled 34 treatment-naive patients with CLL who were 65 years or older. All patients had an Eastern Cooperative Oncology Group performance status of 2 or 3, or a Charlson comorbidity index of at least 2. Patients with other serious medical conditions and/or primary malignancies were eligible, given that they were not already receiving anticancer therapy.
More than half of the patients (53%) had advanced-stage disease and almost one-third (29%) had at least one other primary cancer diagnosis. Many patients also had high-risk disease characteristics, including a complex karyotype involving three or more chromosomal abnormalities (15%), and/or unmutated immunoglobulin heavy chain variable region (IGHV, 59%).
Among 32 patients eligible for efficacy analysis, the overall response rate was 72%, of which 53% were partial and 19% were complete. Six percent (6%) of patients achieved minimal residual disease negativity. The benefits of ofatumumab extended to patients with high-risk disease characteristics, including unmutated IGHV (65% response rate) and/or a complex karyotype (60% response rate).
Ofatumumab also demonstrated a favorable safety profile, according to the investigators.
With all 34 patients evaluable for safety data, 19 (56%) experienced a grade 1 or 2 infusion-related reaction, and 1 (3%) experienced a grade 3 infusion-related reaction. Twenty-one grade 2 infections were reported, and one grade 3 infection occurred. Other grade 3 treatment-related adverse events included gastrointestinal disturbances, pulmonary embolism, allergic reaction, and hyperglycemia, each of which occurred in one patient. No grade 4 adverse events, or grade 2 or higher hematologic toxicities occurred.
“Our findings show that older patients with poor performance status and comorbidities can safely undergo treatment with ofatumumab,” the investigators concluded. “[The results] also support the possibility of enrolling these patients in clinical trials, so that a larger number of patients will be included and their characteristics will more closely mirror those of typical patients seen in the community.”
The study was funded by Novartis, which markets the antibody. The investigators reported additional relationships with AbbVie, Roche, Celgene, and others.
SOURCE: Vitale et al. J Geriatr Oncol. 2019 Apr 18. doi: 10.1016/j.jgo.2019.04.002.
For elderly patients with chronic lymphocytic leukemia (CLL) and comorbidities, the anti-CD20 monoclonal antibody ofatumumab may be a safe and effective treatment option, according to a recent phase 2 trial.
Among 32 patients with a median age of 73 years, the overall response rate was 72%, and no grade 4 adverse events occurred, reported lead author Candida Vitale, MD, PhD, of the University of Torino (Italy) and colleagues.
These findings help fill in a knowledge gap created by clinical trial exclusions, which currently make treatment planning “a significant challenge,” the investigators wrote in Journal of Geriatric Oncology.
The study, which was conducted at MD Anderson Cancer Center in Houston, enrolled 34 treatment-naive patients with CLL who were 65 years or older. All patients had an Eastern Cooperative Oncology Group performance status of 2 or 3, or a Charlson comorbidity index of at least 2. Patients with other serious medical conditions and/or primary malignancies were eligible, given that they were not already receiving anticancer therapy.
More than half of the patients (53%) had advanced-stage disease and almost one-third (29%) had at least one other primary cancer diagnosis. Many patients also had high-risk disease characteristics, including a complex karyotype involving three or more chromosomal abnormalities (15%), and/or unmutated immunoglobulin heavy chain variable region (IGHV, 59%).
Among 32 patients eligible for efficacy analysis, the overall response rate was 72%, of which 53% were partial and 19% were complete. Six percent (6%) of patients achieved minimal residual disease negativity. The benefits of ofatumumab extended to patients with high-risk disease characteristics, including unmutated IGHV (65% response rate) and/or a complex karyotype (60% response rate).
Ofatumumab also demonstrated a favorable safety profile, according to the investigators.
With all 34 patients evaluable for safety data, 19 (56%) experienced a grade 1 or 2 infusion-related reaction, and 1 (3%) experienced a grade 3 infusion-related reaction. Twenty-one grade 2 infections were reported, and one grade 3 infection occurred. Other grade 3 treatment-related adverse events included gastrointestinal disturbances, pulmonary embolism, allergic reaction, and hyperglycemia, each of which occurred in one patient. No grade 4 adverse events, or grade 2 or higher hematologic toxicities occurred.
“Our findings show that older patients with poor performance status and comorbidities can safely undergo treatment with ofatumumab,” the investigators concluded. “[The results] also support the possibility of enrolling these patients in clinical trials, so that a larger number of patients will be included and their characteristics will more closely mirror those of typical patients seen in the community.”
The study was funded by Novartis, which markets the antibody. The investigators reported additional relationships with AbbVie, Roche, Celgene, and others.
SOURCE: Vitale et al. J Geriatr Oncol. 2019 Apr 18. doi: 10.1016/j.jgo.2019.04.002.
For elderly patients with chronic lymphocytic leukemia (CLL) and comorbidities, the anti-CD20 monoclonal antibody ofatumumab may be a safe and effective treatment option, according to a recent phase 2 trial.
Among 32 patients with a median age of 73 years, the overall response rate was 72%, and no grade 4 adverse events occurred, reported lead author Candida Vitale, MD, PhD, of the University of Torino (Italy) and colleagues.
These findings help fill in a knowledge gap created by clinical trial exclusions, which currently make treatment planning “a significant challenge,” the investigators wrote in Journal of Geriatric Oncology.
The study, which was conducted at MD Anderson Cancer Center in Houston, enrolled 34 treatment-naive patients with CLL who were 65 years or older. All patients had an Eastern Cooperative Oncology Group performance status of 2 or 3, or a Charlson comorbidity index of at least 2. Patients with other serious medical conditions and/or primary malignancies were eligible, given that they were not already receiving anticancer therapy.
More than half of the patients (53%) had advanced-stage disease and almost one-third (29%) had at least one other primary cancer diagnosis. Many patients also had high-risk disease characteristics, including a complex karyotype involving three or more chromosomal abnormalities (15%), and/or unmutated immunoglobulin heavy chain variable region (IGHV, 59%).
Among 32 patients eligible for efficacy analysis, the overall response rate was 72%, of which 53% were partial and 19% were complete. Six percent (6%) of patients achieved minimal residual disease negativity. The benefits of ofatumumab extended to patients with high-risk disease characteristics, including unmutated IGHV (65% response rate) and/or a complex karyotype (60% response rate).
Ofatumumab also demonstrated a favorable safety profile, according to the investigators.
With all 34 patients evaluable for safety data, 19 (56%) experienced a grade 1 or 2 infusion-related reaction, and 1 (3%) experienced a grade 3 infusion-related reaction. Twenty-one grade 2 infections were reported, and one grade 3 infection occurred. Other grade 3 treatment-related adverse events included gastrointestinal disturbances, pulmonary embolism, allergic reaction, and hyperglycemia, each of which occurred in one patient. No grade 4 adverse events, or grade 2 or higher hematologic toxicities occurred.
“Our findings show that older patients with poor performance status and comorbidities can safely undergo treatment with ofatumumab,” the investigators concluded. “[The results] also support the possibility of enrolling these patients in clinical trials, so that a larger number of patients will be included and their characteristics will more closely mirror those of typical patients seen in the community.”
The study was funded by Novartis, which markets the antibody. The investigators reported additional relationships with AbbVie, Roche, Celgene, and others.
SOURCE: Vitale et al. J Geriatr Oncol. 2019 Apr 18. doi: 10.1016/j.jgo.2019.04.002.
FROM THE JOURNAL OF GERIATRIC ONCOLOGY
ATLAS Opens New Telehealth Site With Walmart
Groceries, maybe a new shirt, and now some veterans can fit in some shopping at their next health care visit. In a pilot project, the US Department of Veterans Affairs (VA) is partnering with Walmart to offer veterans easy access to health care at 5 sites.
The VA-led ATLAS (Accessing telehealth through local area stations) program is part of the VA Anywhere to Anywhere telehealth initiative, which aims to provide care to veterans no matter where they live. Other telehealth pilot sites are in Wisconsin, Michigan, and Iowa. In addition to Walmart, ATLAS sites are located at American Legion posts and Veterans of Foreign Wars (VFW) posts.
The local VA facility associated with the ATLAS site determines which clinical services the site offers. The health care services do not require hands-on exams. Clinical services may include, for instance, primary care, mental health counseling, clinical pharmacy, nutrition services, and social work. On-site attendants provide information, help the veterans get started, troubleshoot technical issues, and clean the space between appointments. Walmart donated equipment and space, where veterans can meet with a VA provider in a private room via video technology.
Last year, nearly 500,000 veterans logged > 1.3 million VA video telehealth encounters. It is the “way of the future,” says VA Secretary Robert Wilkie. “Veterans need the expansion of choice, and this partnership is vital to affording them convenient access to VA health care services where they live.”
Daryl Risinger, Chief Growth Officer for Walmart US Health and Wellness, is a veteran of the Air Force, and has a son and son-in-law serving. He says, “I know firsthand how important support and access is for our military, especially when it comes to health care. …This is another way we are helping our communities live better.”
For a veteran to attend an appointment at an ATLAS site, the site must be associated with the VA Medical Center where the veteran is enrolled. Family members who receive care through the VA can also visit ATLAS sites for select appointments.
Groceries, maybe a new shirt, and now some veterans can fit in some shopping at their next health care visit. In a pilot project, the US Department of Veterans Affairs (VA) is partnering with Walmart to offer veterans easy access to health care at 5 sites.
The VA-led ATLAS (Accessing telehealth through local area stations) program is part of the VA Anywhere to Anywhere telehealth initiative, which aims to provide care to veterans no matter where they live. Other telehealth pilot sites are in Wisconsin, Michigan, and Iowa. In addition to Walmart, ATLAS sites are located at American Legion posts and Veterans of Foreign Wars (VFW) posts.
The local VA facility associated with the ATLAS site determines which clinical services the site offers. The health care services do not require hands-on exams. Clinical services may include, for instance, primary care, mental health counseling, clinical pharmacy, nutrition services, and social work. On-site attendants provide information, help the veterans get started, troubleshoot technical issues, and clean the space between appointments. Walmart donated equipment and space, where veterans can meet with a VA provider in a private room via video technology.
Last year, nearly 500,000 veterans logged > 1.3 million VA video telehealth encounters. It is the “way of the future,” says VA Secretary Robert Wilkie. “Veterans need the expansion of choice, and this partnership is vital to affording them convenient access to VA health care services where they live.”
Daryl Risinger, Chief Growth Officer for Walmart US Health and Wellness, is a veteran of the Air Force, and has a son and son-in-law serving. He says, “I know firsthand how important support and access is for our military, especially when it comes to health care. …This is another way we are helping our communities live better.”
For a veteran to attend an appointment at an ATLAS site, the site must be associated with the VA Medical Center where the veteran is enrolled. Family members who receive care through the VA can also visit ATLAS sites for select appointments.
Groceries, maybe a new shirt, and now some veterans can fit in some shopping at their next health care visit. In a pilot project, the US Department of Veterans Affairs (VA) is partnering with Walmart to offer veterans easy access to health care at 5 sites.
The VA-led ATLAS (Accessing telehealth through local area stations) program is part of the VA Anywhere to Anywhere telehealth initiative, which aims to provide care to veterans no matter where they live. Other telehealth pilot sites are in Wisconsin, Michigan, and Iowa. In addition to Walmart, ATLAS sites are located at American Legion posts and Veterans of Foreign Wars (VFW) posts.
The local VA facility associated with the ATLAS site determines which clinical services the site offers. The health care services do not require hands-on exams. Clinical services may include, for instance, primary care, mental health counseling, clinical pharmacy, nutrition services, and social work. On-site attendants provide information, help the veterans get started, troubleshoot technical issues, and clean the space between appointments. Walmart donated equipment and space, where veterans can meet with a VA provider in a private room via video technology.
Last year, nearly 500,000 veterans logged > 1.3 million VA video telehealth encounters. It is the “way of the future,” says VA Secretary Robert Wilkie. “Veterans need the expansion of choice, and this partnership is vital to affording them convenient access to VA health care services where they live.”
Daryl Risinger, Chief Growth Officer for Walmart US Health and Wellness, is a veteran of the Air Force, and has a son and son-in-law serving. He says, “I know firsthand how important support and access is for our military, especially when it comes to health care. …This is another way we are helping our communities live better.”
For a veteran to attend an appointment at an ATLAS site, the site must be associated with the VA Medical Center where the veteran is enrolled. Family members who receive care through the VA can also visit ATLAS sites for select appointments.
Teprotumumab gets FDA go-ahead for thyroid eye disease
according to a press release.

Thyroid eye disease is a rare, progressive, autoimmune condition that causes the eyes to bulge (proptosis) and can lead to blindness. Until now, treatment has focused on managing its symptoms – which can include eye pain, double vision, or sensitivity to light – with steroids, and in some cases, multiple invasive surgeries.
The human monoclonal antibody and a targeted inhibitor of the insulinlike growth factor-1 receptor is administered to patients once every 3 weeks, for a total of eight infusions, according to a statement from Horizon Therapeutics, which manufactures the drug.
The approval was based on the findings from two similarly designed, parallel-group studies (Studies 1 and 2) involving 170 patients with thyroid eye disease who were randomized to receive either teprotumumab or placebo. Of those receiving the study drug, 71% in Study 1 and 83% in Study 2 had a reduction of more than 2 mm in eye protrusion, compared with 20% and 10%, respectively, among the placebo participants.
The most common adverse reactions in patients receiving teprotumumab were muscle spasm, nausea, alopecia, diarrhea, fatigue, and hyperglycemia. The treatment is contraindicated for pregnancy.
according to a press release.
Thyroid eye disease is a rare, progressive, autoimmune condition that causes the eyes to bulge (proptosis) and can lead to blindness. Until now, treatment has focused on managing its symptoms – which can include eye pain, double vision, or sensitivity to light – with steroids, and in some cases, multiple invasive surgeries.
The human monoclonal antibody and a targeted inhibitor of the insulinlike growth factor-1 receptor is administered to patients once every 3 weeks, for a total of eight infusions, according to a statement from Horizon Therapeutics, which manufactures the drug.
The approval was based on the findings from two similarly designed, parallel-group studies (Studies 1 and 2) involving 170 patients with thyroid eye disease who were randomized to receive either teprotumumab or placebo. Of those receiving the study drug, 71% in Study 1 and 83% in Study 2 had a reduction of more than 2 mm in eye protrusion, compared with 20% and 10%, respectively, among the placebo participants.
The most common adverse reactions in patients receiving teprotumumab were muscle spasm, nausea, alopecia, diarrhea, fatigue, and hyperglycemia. The treatment is contraindicated for pregnancy.
according to a press release.
Thyroid eye disease is a rare, progressive, autoimmune condition that causes the eyes to bulge (proptosis) and can lead to blindness. Until now, treatment has focused on managing its symptoms – which can include eye pain, double vision, or sensitivity to light – with steroids, and in some cases, multiple invasive surgeries.
The human monoclonal antibody and a targeted inhibitor of the insulinlike growth factor-1 receptor is administered to patients once every 3 weeks, for a total of eight infusions, according to a statement from Horizon Therapeutics, which manufactures the drug.
The approval was based on the findings from two similarly designed, parallel-group studies (Studies 1 and 2) involving 170 patients with thyroid eye disease who were randomized to receive either teprotumumab or placebo. Of those receiving the study drug, 71% in Study 1 and 83% in Study 2 had a reduction of more than 2 mm in eye protrusion, compared with 20% and 10%, respectively, among the placebo participants.
The most common adverse reactions in patients receiving teprotumumab were muscle spasm, nausea, alopecia, diarrhea, fatigue, and hyperglycemia. The treatment is contraindicated for pregnancy.
FROM THE FDA
Washington state patient is first U.S. case of novel coronavirus
The first case of the novel coronavirus, named 2019-nCoV, in the United States has been diagnosed in a traveler from China who came through Seattle-Tacoma International Airport on Jan 15, the Centers for Disease Control and Prevention announced today at a press briefing.
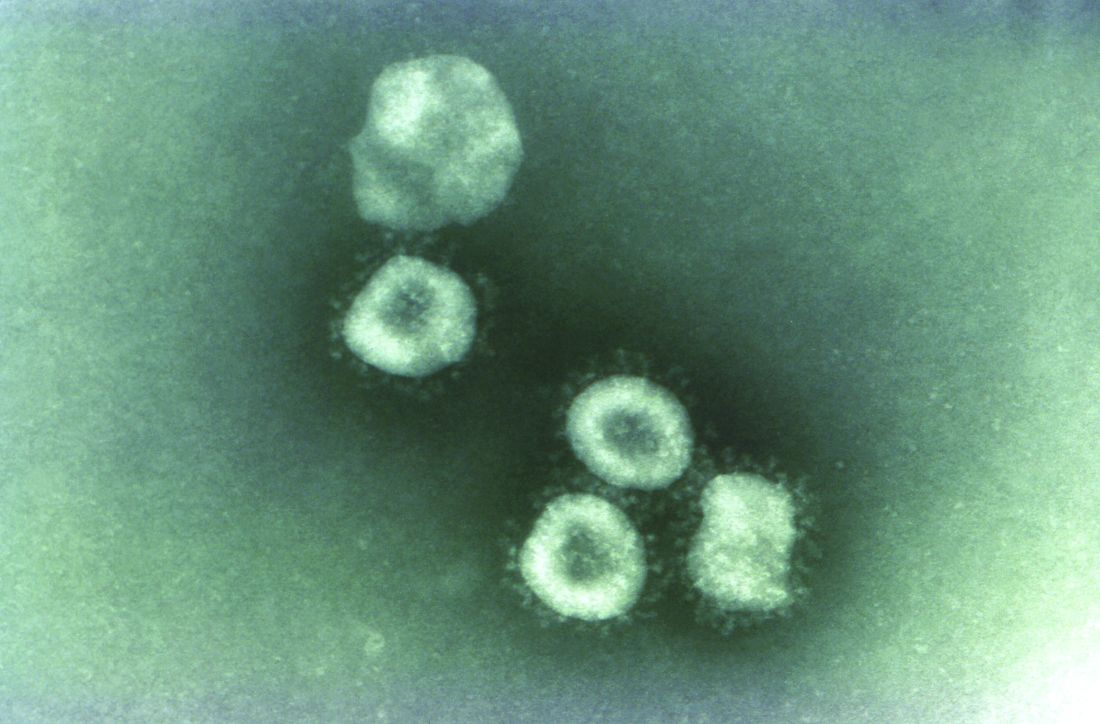
The outbreak began at a animal and meat market in China and now has spread to at least three other countries, including Thailand, Japan and South Korea. While originally thought to be spreading from animal to person, it appears that limited person-to-person transmission is occurring, although it is currently unknown how easily this virus spreads between people.
More than 300 cases have been reported and six deaths have occurred. Fourteen health care workers have been infected.
Scott Lindquist, MD, MPH, Washington state epidemiologist, said at the briefing that the patient, a man who had been in Wuhan, arrived at Sea-Tac on Jan. 15, 2 days before airport screening had been initiated. He was symptom free at the time of his arrival and probably would not have been identified as infected with 2019-nCoV. The patient had been aware of the public health and news media coverage of 2019-nCoV and, after developing symptoms, contacted his health care provider on Jan. 19. The patient did not fly directly from Wuhan, but Dr. Lindquist said that he has been fully cooperative and has been helpful to authorities in tracing his route and contacts. The man is being treated at Providence Regional Medical Center, Everett, Wash.
The CDC obtained a specimen from the patient immediately and identified the 2019-nCoV within 24 hours.
Screening at airports is part of a multipart strategy to address this type of infection that includes public health information dissemination, patient education, as well as hospital preparation and training exercises. Currently, a strategy referred to as “funneling” is being implemented wherein travelers from China are rerouted and reticketed to one of the five airports conducting screening. At present, JFK in New York, San Francisco International, Los Angeles International, Hartsfield-Jackson Atlanta International Airport, and Chicago O’Hare International Airport are conducting inbound traveler screening.
The CDC is working in close cooperation with the Department of Homeland Security and the Federal Aviation Administration to coordinate travel screenings and reroutings. In addition, the CDC is working with the World Health Organization and the international global health community to share information about this outbreak. The CDC also has staff on site in Wuhan and is communicating with local health authorities. The CDC has activated its Emergency Operations Center to better provide ongoing support to the 2019-nCoV response. Currently, the focus is on tracing contacts and the means of transmission of this virus.
Updates on the outbreak will be posted on the CDC coronavirus website.
CORRECTION: 1/21/2020: The name of the medical center where the 2019-nCoV patient is being treated was corrected.
The first case of the novel coronavirus, named 2019-nCoV, in the United States has been diagnosed in a traveler from China who came through Seattle-Tacoma International Airport on Jan 15, the Centers for Disease Control and Prevention announced today at a press briefing.
The outbreak began at a animal and meat market in China and now has spread to at least three other countries, including Thailand, Japan and South Korea. While originally thought to be spreading from animal to person, it appears that limited person-to-person transmission is occurring, although it is currently unknown how easily this virus spreads between people.
More than 300 cases have been reported and six deaths have occurred. Fourteen health care workers have been infected.
Scott Lindquist, MD, MPH, Washington state epidemiologist, said at the briefing that the patient, a man who had been in Wuhan, arrived at Sea-Tac on Jan. 15, 2 days before airport screening had been initiated. He was symptom free at the time of his arrival and probably would not have been identified as infected with 2019-nCoV. The patient had been aware of the public health and news media coverage of 2019-nCoV and, after developing symptoms, contacted his health care provider on Jan. 19. The patient did not fly directly from Wuhan, but Dr. Lindquist said that he has been fully cooperative and has been helpful to authorities in tracing his route and contacts. The man is being treated at Providence Regional Medical Center, Everett, Wash.
The CDC obtained a specimen from the patient immediately and identified the 2019-nCoV within 24 hours.
Screening at airports is part of a multipart strategy to address this type of infection that includes public health information dissemination, patient education, as well as hospital preparation and training exercises. Currently, a strategy referred to as “funneling” is being implemented wherein travelers from China are rerouted and reticketed to one of the five airports conducting screening. At present, JFK in New York, San Francisco International, Los Angeles International, Hartsfield-Jackson Atlanta International Airport, and Chicago O’Hare International Airport are conducting inbound traveler screening.
The CDC is working in close cooperation with the Department of Homeland Security and the Federal Aviation Administration to coordinate travel screenings and reroutings. In addition, the CDC is working with the World Health Organization and the international global health community to share information about this outbreak. The CDC also has staff on site in Wuhan and is communicating with local health authorities. The CDC has activated its Emergency Operations Center to better provide ongoing support to the 2019-nCoV response. Currently, the focus is on tracing contacts and the means of transmission of this virus.
Updates on the outbreak will be posted on the CDC coronavirus website.
CORRECTION: 1/21/2020: The name of the medical center where the 2019-nCoV patient is being treated was corrected.
The first case of the novel coronavirus, named 2019-nCoV, in the United States has been diagnosed in a traveler from China who came through Seattle-Tacoma International Airport on Jan 15, the Centers for Disease Control and Prevention announced today at a press briefing.
The outbreak began at a animal and meat market in China and now has spread to at least three other countries, including Thailand, Japan and South Korea. While originally thought to be spreading from animal to person, it appears that limited person-to-person transmission is occurring, although it is currently unknown how easily this virus spreads between people.
More than 300 cases have been reported and six deaths have occurred. Fourteen health care workers have been infected.
Scott Lindquist, MD, MPH, Washington state epidemiologist, said at the briefing that the patient, a man who had been in Wuhan, arrived at Sea-Tac on Jan. 15, 2 days before airport screening had been initiated. He was symptom free at the time of his arrival and probably would not have been identified as infected with 2019-nCoV. The patient had been aware of the public health and news media coverage of 2019-nCoV and, after developing symptoms, contacted his health care provider on Jan. 19. The patient did not fly directly from Wuhan, but Dr. Lindquist said that he has been fully cooperative and has been helpful to authorities in tracing his route and contacts. The man is being treated at Providence Regional Medical Center, Everett, Wash.
The CDC obtained a specimen from the patient immediately and identified the 2019-nCoV within 24 hours.
Screening at airports is part of a multipart strategy to address this type of infection that includes public health information dissemination, patient education, as well as hospital preparation and training exercises. Currently, a strategy referred to as “funneling” is being implemented wherein travelers from China are rerouted and reticketed to one of the five airports conducting screening. At present, JFK in New York, San Francisco International, Los Angeles International, Hartsfield-Jackson Atlanta International Airport, and Chicago O’Hare International Airport are conducting inbound traveler screening.
The CDC is working in close cooperation with the Department of Homeland Security and the Federal Aviation Administration to coordinate travel screenings and reroutings. In addition, the CDC is working with the World Health Organization and the international global health community to share information about this outbreak. The CDC also has staff on site in Wuhan and is communicating with local health authorities. The CDC has activated its Emergency Operations Center to better provide ongoing support to the 2019-nCoV response. Currently, the focus is on tracing contacts and the means of transmission of this virus.
Updates on the outbreak will be posted on the CDC coronavirus website.
CORRECTION: 1/21/2020: The name of the medical center where the 2019-nCoV patient is being treated was corrected.
REPORTING FROM CDC
Dermatology News welcomes new advisory board member Dr. Jonathan I. Silverberg
Dermatology News welcomes Jonathan I. Silverberg, MD, PhD, MPH, to its editorial advisory board. Dr. Silverberg is an associate professor of dermatology at George Washington University in Washington, where he is also director of clinical research and contact dermatitis.

Dr. Silverberg’s clinical subspecialty is inflammatory skin disease, particularly atopic dermatitis and contact dermatitis. His research interests include comorbidities and burden of itch and inflammatory skin disease, evidence-based dermatology, patient-reported outcomes, and dermatoepidemiology, as well as health services research, drug development, clinical trial design, and biomarkers. He has published more than 600 peer-reviewed articles, abstracts, and book chapters, and has appeared frequently on the pages of Dermatology News.
‑Elizabeth Mechcatie, editor
Dermatology News welcomes Jonathan I. Silverberg, MD, PhD, MPH, to its editorial advisory board. Dr. Silverberg is an associate professor of dermatology at George Washington University in Washington, where he is also director of clinical research and contact dermatitis.
Dr. Silverberg’s clinical subspecialty is inflammatory skin disease, particularly atopic dermatitis and contact dermatitis. His research interests include comorbidities and burden of itch and inflammatory skin disease, evidence-based dermatology, patient-reported outcomes, and dermatoepidemiology, as well as health services research, drug development, clinical trial design, and biomarkers. He has published more than 600 peer-reviewed articles, abstracts, and book chapters, and has appeared frequently on the pages of Dermatology News.
‑Elizabeth Mechcatie, editor
Dermatology News welcomes Jonathan I. Silverberg, MD, PhD, MPH, to its editorial advisory board. Dr. Silverberg is an associate professor of dermatology at George Washington University in Washington, where he is also director of clinical research and contact dermatitis.
Dr. Silverberg’s clinical subspecialty is inflammatory skin disease, particularly atopic dermatitis and contact dermatitis. His research interests include comorbidities and burden of itch and inflammatory skin disease, evidence-based dermatology, patient-reported outcomes, and dermatoepidemiology, as well as health services research, drug development, clinical trial design, and biomarkers. He has published more than 600 peer-reviewed articles, abstracts, and book chapters, and has appeared frequently on the pages of Dermatology News.
‑Elizabeth Mechcatie, editor
FDA advisers set high bar for new opioids
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?

On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?
On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
During an opioid-addiction epidemic, can any new opioid pain drug meet prevailing safety demands to gain regulatory approval?
On Jan. 14 and 15, a Food and Drug Administration advisory committee voted virtually unanimously against two new opioid formulations and evenly split for and against a third; the 2 days of data and discussion showed how high a bar new opioids face these days for getting onto the U.S. market.
The bar’s height is very understandable given how many Americans have become addicted to opioids over the past decade, more often than not by accident while using pain medications as they believed they had been directed, said experts during the sessions held on the FDA’s campus in White Oak, Md.
Among the many upshots of the opioid crisis, the meetings held to discuss these three contender opioids highlighted the bitter irony confronting attempts to bring new, safer opioids to the U.S. market: While less abusable pain-relief medications that still harness the potent analgesic power of mu opioid receptor agonists are desperately desired, new agents in this space now receive withering scrutiny over their safeguards against misuse and abuse, and over whether they add anything meaningfully new to what’s already available. While these demands seem reasonable, perhaps even essential, it’s unclear whether any new opioid-based pain drugs will ever fully meet the safety that researchers, clinicians, and the public now seek.
A special FDA advisory committee that combined the Anesthetic and Analgesic Drug Products Advisory Committee with members of the Drug Safety and Risk Management Advisory Committee considered the application for three different opioid drugs from three separate companies. None received a clear endorsement. Oxycodegol, a new type of orally delivered opioid molecule engineered to slow brain entry and thereby delay an abuser’s high, got voted down without any votes in favor and 27 votes against agency approval. Aximris XR, an extended-release oxycodone formulation that successfully deterred intravenous abuse but had no deterrence efficacy for intranasal or oral abuse failed by a 2-24 vote against. The third agent, CTC, a novel formulation of the schedule IV opioid tramadol with the NSAID celecoxib designed to be analgesic but with limited opioid-abuse appeal, came the closest to meaningful support with a tied 13-13 vote from advisory committee members for and against agency approval. FDA staff takes advisory committee opinions and votes into account when making their final decisions about drug marketing approvals.
In each case, the committee members, mostly the same roster assembled for each of the three agents, identified specific concerns with the data purported to show each drug’s safety and efficacy. But the gathered experts and consumer representatives also consistently cited holistic challenges to approving new opioids and the stiffer criteria these agents face amid a continuing wave of opioid misuse and abuse.
“In the context of the public health issues, we don’t want to be perceived in any way of taking shortcuts,” said Linda S. Tyler, PharmD,, an advisory committee member and professor of pharmacy and chief pharmacy officer at the University of Utah in Salt Lake City. “There is no question that for a new product to come to market in this space it needs to add to what’s on the market, meet a high bar, and provide advantages compared with what’s already on the market,” she said.
Tramadol plus celecoxib gains some support
The proposed combined formulation of tramadol and celecoxib came closest to meeting that bar, as far as the advisory committee was concerned, coming away with 13 votes favoring approval to match 13 votes against. The premise behind this agent, know as CTC (cocrystal of tramadol and celecoxib), was that it combined a modest dose (44 mg) of the schedule IV opioid tramadol with a 56-mg dose of celecoxib in a twice-daily pill. Eugene R. Viscusi, MD, professor of anesthesiology and director of acute pain management at Thomas Jefferson University in Philadelphia and a speaker at the session on behalf of the applicant company, spelled out the rationale behind CTC: “We are caught in a dilemma. We need to reduce opioid use, but we also need to treat pain. We have an urgent need to have pain treatment options that are effective but have low potential for abuse and dependence. We are looking at multimodal analgesia, that uses combination of agents, recognizing that postoperative pain is a mixed pain syndrome. Multimodal pain treatments are now considered standard care. We want to minimize opioids to the lowest dose possible to produce safe analgesia. Tramadol is the least-preferred opioid for abuse,” and is rated as schedule IV, the U.S. designation for drugs considered to have a low level of potential for causing abuse or dependence. “Opioids used as stand-alone agents have contributed to the current opioid crisis,” Dr. Viscusi told the committee.
In contrast to tramadol’s schedule IV status, the mainstays of recent opioid pain therapy have been hydrocodone and oxycodone, schedule II opioids rated as having a “high potential for abuse.”
Several advisory committee members agreed that CTC minimized patient exposure to an opioid. “This drug isn’t even tramadol; it’s tramadol light. It has about as low a dose [of an opioid] as you can have and still have a drug,” said member Lee A. Hoffer, PhD, a medical anthropologist at Case Western Reserve University, Cleveland, who studies substance use disorders. “All opioids are dangerous, even at a low dose, but there is a linear relationship based on potency, so if we want to have an opioid for acute pain, I’d like it to have the lowest morphine milligram equivalent possible. The ideal is no opioids, but that is not what happens,” he said. The CTC formulation delivers 17.6 morphine milligram equivalents (MME) per pill, the manufacturer’s representatives said. The Centers for Disease Control and Prevention defines a “relatively low” daily opioid dose as 20-50 MME.
Some committee members hailed the CTC formulation as a meaningful step toward cutting opioid consumption.
“We may be very nervous about abuse of scheduled opioids, but a schedule IV opioid in an opioid-sparing formulation is as good as it gets in 2020,” said committee member Kevin L. Zacharoff, MD, a pain medicine specialist at the State University of New York at Stony Brook. “Any opioid has potential for abuse, but this is a safer alternative to the schedule II drugs. There is less public health risk with this,” said committee member Sherif Zaafran, MD, a Houston anesthesiologist. “This represents an incremental but important approach to addressing the opioid crisis, especially if used to replace schedule II opioids,” said Brandon D.L. Marshall, PhD, an epidemiologist and substance abuse researcher at Brown University in Providence, R.I.
But despite agreement that CTC represented a new low in the MME of an opioid given to patients, several committee members still saw the formulation as problematic by introducing any opioid, no matter how small the dose.
“The landscape of tramadol use and prescribing is evolving. There’s been an exponential upturn in tramadol prescribing. It’s perceived [as] safer, but it’s not completely safe. Will this change tramadol abuse and open the door to abuse of other opioids? This is what got us into trouble with opioids in the first place. Patients start with a prescription opioid that they perceive is safe. Patients don’t start with oxycodone or heroin. They start with drugs that are believed to be safe. I feel this combination has less risk for abuse, but I’m worried that it would produce a false sense of security for tolerability and safety,” said committee member Maryann E. Amirshahi, MD, a medical toxicologist at Georgetown University and MedStar Health in Washington.
Several other committee members returned to this point throughout the 2 days of discussions: The majority of Americans who have become hooked on opioids reached that point by taking an opioid pain medication for a legitimate medical reason and using the drug the way they had understood they should.
“I’m most concerned about unintentional misuse leading to addiction and abuse. Most people with an opioid addiction got it inadvertently, misusing it by mistake,” said committee member Suzanne B. Robotti, a consumer representative and executive director of DES Action USA. “I’m concerned about approving an opioid, even an opioid with a low abuse history, without a clearer picture of the human abuse potential data and what would happen if this drug were abused,” she added, referring to the proposed CTC formulation.
“All the patients I work with started [their opioid addiction] as pain patients,” Dr. Hoffer said.
“The most common use and abuse of opioids is orally. We need to avoid having patients who use the drug as prescribed and still end up addicted,” said committee member Friedhelm Sandbrink, MD, a neurologist and director of pain management at the Veterans Affairs (VA) Medical Center in Washington.
What this means, said several panelists, is functionally clamping down a class-wide lid on new opioids. “The way to reduce deaths from abuse is to reduce addiction, and to have an impact you need to reduce opioid exposure.” said committee member Sonia Hernandez-Diaz, MD, professor of epidemiology at the Harvard School of Public Health in Boston.
“In this opioid crisis, we ask for data that we wouldn’t ordinarily ask for. I feel there are unanswered questions about the abuse potential [of CTC]. We have seen a recent reduction in oxycodone use, which is great, but also an increase in tramadol use. We should not be fooled. Tramadol is an opioid, even if it’s schedule IV,” Dr. Tyler said.
Two other opioids faced greater opposition
The other two agents that the committee considered received much less support and sharper skepticism. The application for Aximris XR, an extended release form of oxycodone with a purported abuse-deterrent formulation (ADF) that relies on being difficult to extract for intravenous use as well as possibly having effective deterrence mechanisms for other forms of abuse. But FDA staffers reported that the only effective deterrence they could document was against manipulation for intravenous use, making Aximris XR the first opioid seeking ADF labeling based on deterrence to a single delivery route. This led several committee members, as well as the FDA, to comment on the clinical meaningfulness of ADF for one route. So far, the FDA approved ADF labeling for seven opioids, most notably OxyContin, an extended-release oxycodone with the biggest share of the U.S. market for opioids with ADF labeling.
“For ADF, we label based on what we expect from the premarket data. We don’t really know how that translates into what happens once the drug is on the market. Every company with an ADF in their label is required to do postmarketing studies on the abuse routes that are supposed to be deterred. We see shifts to other routes. Assessment of ADF is incredibly challenging, both scientifically and logistically, because there has not been a lot of uptake of these products, for a variety of reasons,” said Judy Staffa, PhD, associate director for Public Health Initiatives in the Office of Surveillance & Epidemiology in the FDA’s Center for Drug Evaluation and Research. The company that markets OxyContin has been the first to submit to the FDA all of its required postmarketing data on ADF efficacy, and the agency is now reviewing this filing, Dr. Staffa said.
The data presented for Aximris XR appeared to generally fail to convince committee members that it provided a meaningful addition to the range of opioids with ADF designations already available, which meant that their decision mostly came down to whether they felt it made sense to bring a me-too opioid to the U.S. market. Their answer was mostly no.
“In the end, it’s another opioid, and I’m not sure we need another opioid,” said committee member Lonnie K. Zeltzer, MD, professor of pediatrics, anesthesiology, psychiatry, and biobehavioral sciences and director of pediatric pain at the University of California, Los Angeles “There are so many options for patients and for people who abuse these drug. I don’t see this formulation as having a profound impact, but I’m very concerned about adding more prescription opioids,” said Martin Garcia-Bunuel, MD, deputy chief of staff for the VA Maryland Health Care System in Baltimore. Another concern of some committee members was that ADF remains a designation with an uncertain meaning, pending the FDA’s analysis of the OxyContin data.
“At the end of the day, we don’t know whether any of the [ADF] stuff makes a difference,” noted Steve B. Meisel, PharmD, system director of medication safety for M Health Fairview in Minneapolis and a committee member,
The third agent, oxycodegol, a molecule designed to pass more slowly across the blood-brain barrier because of an attached polyethylene glycol chain that’s supposed to prevent a rapid high after ingestion and hence cut abuse potential. It received unanimous committee rejection, primarily because its safety and efficacy evidence had so many holes, but the shadow of opioid abuse permeated the committee’s discussion.
“One dogma in the abuse world is that slowing entry into the brain reduces abuse potential, but the opioid crisis showed that this is not the only factor. Some people have become addicted to slow-acting drugs. The abuse potential of this drug, oxycodegol, needs to be considered given where we’ve been with the opioid crisis,” said Jane B. Acri, PhD, chief of the Medications Discovery and Toxicology Branch of the National Institute on Drug Abuse.
“During the opioid epidemic, do we want to approve more opioids? If the [pain] efficacy is about the same as oxycodone, is better safety or abuse potential a reason to approve it? We need guidance [from the FDA] about what is ‘better enough.’ No opioid will ever be perfect; there will always be abuse and misuse. But what is good enough to justify bringing another opioid onto the market? What is a good enough improvement? I don’t have an answer,” Dr. Hernandez-Diaz said.
Adviser comments showed that the continued threat of widespread opioid addiction has cooled prospects for new opioid approvals by making FDA advisers skittish over how to properly score the incremental value of a new opioid.
“Do we need to go back to the drawing board on how we make decisions on exposing the American public to these kinds of agents?” Dr. Garcia-Bunuel asked. “I don’t think we have the tools to make these decisions.”
Renal denervation rebounds
PHILADELPHIA – Enthusiasm for catheter-based renal denervation as a potential nondrug treatment for hypertension is once again on the rise, Michael Bohm, MD, observed at the American Heart Association scientific sessions.

The field experienced “a big depression” in 2014 with the publication of the unexpectedly negative results of the Symplicity HTN-3 trial (N Engl J Med. 2014;370:1393-401), he said. But post hoc analysis of the trial revealed significant shortcomings in design and execution.
“All of the flaws of this trial have been eliminated and now there is a very tightly controlled program to show whether renal denervation will work or not,” according to Dr. Bohm, director of the department of internal medicine and professor of cardiology at Saarland University in Homburg, Germany.
Indeed, three randomized, double-blind, sham-controlled, proof-of-concept clinical trials – all with strongly positive results – were published in Lancet in 2017 and 2018: SPYRAL HTN-OFF (2017 Nov 11;390:2160-70), RADIANCE SOLO (2018 Jun 9;391:2335-45), and SPYRAL HTN-ON (2018 May 23;391:2346-55). Based on the encouraging findings, four large pivotal trials of renal denervation (RDN) for hypertension are ongoing: RADIANCE HTN, REQUIRE, RADIANCE II, and SPYRAL HTN-ON MED. In addition, the SPYRAL HTN-OFF MED pivotal trial has been completed and will be presented soon, Dr. Bohm said.
Defining who’s most likely to benefit
Treatment response has been quite variable within the various RDN trials. A reliable predictor of response would be an important advance because it would enable physicians to select the best candidates for treatment while sparing others from an invasive procedure – albeit a relatively safe one – that they may not benefit from. On this front, Dr. Bohm and colleagues have recently reported that a baseline 24-hour heart rate above the median value of 73.5 bpm in the SPYRAL HTN-OFF MED trial – a marker for sympathetic overdrive – was associated with a 10.7/7.5 mm Hg greater reduction in average ambulatory blood pressures post-RDN than with a sham procedure. In contrast, blood pressure changes in RDN recipients with a below-median baseline 24-hour heart rate weren’t significant (Eur Heart J. 2019 Mar 1;40:743-51).
“Although this is a little bit rough, there is no other really true and reliable marker,” the cardiologist observed.
A pressing need exists for a reliable intraprocedural indicator of success. Dr. Bohm noted that Australian investigators are pursuing a promising approach in animal studies: intraprocedural transvascular high-frequency pacing of the aorticorenal ganglia. Abolition of the pacing-induced increase in blood pressure may be an indicator of complete RDN (JACC Cardiovasc Interv. 2019 Jun 24;12:1109-20).
Applications other than hypertension
Renal denervation is under early-stage investigation for a range of other cardiovascular diseases in which sympathetic overdrive figures prominently.
“The truly interesting things in renal denervation are what happens beyond hypertension. There are a lot of potential applications,” according to Dr. Bohm.
For example, when RDN was performed alongside pulmonary vein isolation for treatment of paroxysmal atrial fibrillation in hypertensive patients, the arrhythmia recurrence rate was significantly reduced during 1 year of follow-up, compared with AF ablation alone, in the randomized, multicenter, 302-patient ERADICATE-AF trial, presented at the most recent meeting of the Heart Rhythm Society.
Also, a small, uncontrolled registry study of RDN in patients with cardiomyopathy and electrical storm suggests the procedure may have an immediate anti–ventricular arrhythmia effect.
Meanwhile, Dr. Bohm is pressing the German government to sponsor an independent randomized controlled trial of RDN for heart failure. He and others have shown in small pilot studies a promising signal that the treatment may improve myocardial function and the signs and symptoms of heart failure in both patients with reduced and preserved left ventricular ejection fraction – and without reducing their blood pressure, which is often already low.
Dr. Bohm and others have also been exploring the impact of RDN in patients with metabolic syndrome. The treatment has a sound pathophysiologic rationale because insulin resistance is dependent upon sympathetic nervous system activation. Preliminary reports show improved insulin sensitivity in response to RDN. Patients also report better quality of life, presumably because of the reduction in sympathetic overactivity.
A couple of small Chinese studies suggest denervating the pulmonary vein in patients with pulmonary hypertension leads to a salutary reduction in pulmonary blood pressures.
“We haven’t done that yet. There is no properly designed catheter. They’ve used a Spyra unipolar catheter. It could work, but it hasn’t been rigorously investigated,” the cardiologist said.
Dr. Bohm reported serving as a scientific adviser to Abbott, AstraZeneca, BMS, Boehringer Ingelheim, and Servier.
PHILADELPHIA – Enthusiasm for catheter-based renal denervation as a potential nondrug treatment for hypertension is once again on the rise, Michael Bohm, MD, observed at the American Heart Association scientific sessions.
The field experienced “a big depression” in 2014 with the publication of the unexpectedly negative results of the Symplicity HTN-3 trial (N Engl J Med. 2014;370:1393-401), he said. But post hoc analysis of the trial revealed significant shortcomings in design and execution.
“All of the flaws of this trial have been eliminated and now there is a very tightly controlled program to show whether renal denervation will work or not,” according to Dr. Bohm, director of the department of internal medicine and professor of cardiology at Saarland University in Homburg, Germany.
Indeed, three randomized, double-blind, sham-controlled, proof-of-concept clinical trials – all with strongly positive results – were published in Lancet in 2017 and 2018: SPYRAL HTN-OFF (2017 Nov 11;390:2160-70), RADIANCE SOLO (2018 Jun 9;391:2335-45), and SPYRAL HTN-ON (2018 May 23;391:2346-55). Based on the encouraging findings, four large pivotal trials of renal denervation (RDN) for hypertension are ongoing: RADIANCE HTN, REQUIRE, RADIANCE II, and SPYRAL HTN-ON MED. In addition, the SPYRAL HTN-OFF MED pivotal trial has been completed and will be presented soon, Dr. Bohm said.
Defining who’s most likely to benefit
Treatment response has been quite variable within the various RDN trials. A reliable predictor of response would be an important advance because it would enable physicians to select the best candidates for treatment while sparing others from an invasive procedure – albeit a relatively safe one – that they may not benefit from. On this front, Dr. Bohm and colleagues have recently reported that a baseline 24-hour heart rate above the median value of 73.5 bpm in the SPYRAL HTN-OFF MED trial – a marker for sympathetic overdrive – was associated with a 10.7/7.5 mm Hg greater reduction in average ambulatory blood pressures post-RDN than with a sham procedure. In contrast, blood pressure changes in RDN recipients with a below-median baseline 24-hour heart rate weren’t significant (Eur Heart J. 2019 Mar 1;40:743-51).
“Although this is a little bit rough, there is no other really true and reliable marker,” the cardiologist observed.
A pressing need exists for a reliable intraprocedural indicator of success. Dr. Bohm noted that Australian investigators are pursuing a promising approach in animal studies: intraprocedural transvascular high-frequency pacing of the aorticorenal ganglia. Abolition of the pacing-induced increase in blood pressure may be an indicator of complete RDN (JACC Cardiovasc Interv. 2019 Jun 24;12:1109-20).
Applications other than hypertension
Renal denervation is under early-stage investigation for a range of other cardiovascular diseases in which sympathetic overdrive figures prominently.
“The truly interesting things in renal denervation are what happens beyond hypertension. There are a lot of potential applications,” according to Dr. Bohm.
For example, when RDN was performed alongside pulmonary vein isolation for treatment of paroxysmal atrial fibrillation in hypertensive patients, the arrhythmia recurrence rate was significantly reduced during 1 year of follow-up, compared with AF ablation alone, in the randomized, multicenter, 302-patient ERADICATE-AF trial, presented at the most recent meeting of the Heart Rhythm Society.
Also, a small, uncontrolled registry study of RDN in patients with cardiomyopathy and electrical storm suggests the procedure may have an immediate anti–ventricular arrhythmia effect.
Meanwhile, Dr. Bohm is pressing the German government to sponsor an independent randomized controlled trial of RDN for heart failure. He and others have shown in small pilot studies a promising signal that the treatment may improve myocardial function and the signs and symptoms of heart failure in both patients with reduced and preserved left ventricular ejection fraction – and without reducing their blood pressure, which is often already low.
Dr. Bohm and others have also been exploring the impact of RDN in patients with metabolic syndrome. The treatment has a sound pathophysiologic rationale because insulin resistance is dependent upon sympathetic nervous system activation. Preliminary reports show improved insulin sensitivity in response to RDN. Patients also report better quality of life, presumably because of the reduction in sympathetic overactivity.
A couple of small Chinese studies suggest denervating the pulmonary vein in patients with pulmonary hypertension leads to a salutary reduction in pulmonary blood pressures.
“We haven’t done that yet. There is no properly designed catheter. They’ve used a Spyra unipolar catheter. It could work, but it hasn’t been rigorously investigated,” the cardiologist said.
Dr. Bohm reported serving as a scientific adviser to Abbott, AstraZeneca, BMS, Boehringer Ingelheim, and Servier.
PHILADELPHIA – Enthusiasm for catheter-based renal denervation as a potential nondrug treatment for hypertension is once again on the rise, Michael Bohm, MD, observed at the American Heart Association scientific sessions.
The field experienced “a big depression” in 2014 with the publication of the unexpectedly negative results of the Symplicity HTN-3 trial (N Engl J Med. 2014;370:1393-401), he said. But post hoc analysis of the trial revealed significant shortcomings in design and execution.
“All of the flaws of this trial have been eliminated and now there is a very tightly controlled program to show whether renal denervation will work or not,” according to Dr. Bohm, director of the department of internal medicine and professor of cardiology at Saarland University in Homburg, Germany.
Indeed, three randomized, double-blind, sham-controlled, proof-of-concept clinical trials – all with strongly positive results – were published in Lancet in 2017 and 2018: SPYRAL HTN-OFF (2017 Nov 11;390:2160-70), RADIANCE SOLO (2018 Jun 9;391:2335-45), and SPYRAL HTN-ON (2018 May 23;391:2346-55). Based on the encouraging findings, four large pivotal trials of renal denervation (RDN) for hypertension are ongoing: RADIANCE HTN, REQUIRE, RADIANCE II, and SPYRAL HTN-ON MED. In addition, the SPYRAL HTN-OFF MED pivotal trial has been completed and will be presented soon, Dr. Bohm said.
Defining who’s most likely to benefit
Treatment response has been quite variable within the various RDN trials. A reliable predictor of response would be an important advance because it would enable physicians to select the best candidates for treatment while sparing others from an invasive procedure – albeit a relatively safe one – that they may not benefit from. On this front, Dr. Bohm and colleagues have recently reported that a baseline 24-hour heart rate above the median value of 73.5 bpm in the SPYRAL HTN-OFF MED trial – a marker for sympathetic overdrive – was associated with a 10.7/7.5 mm Hg greater reduction in average ambulatory blood pressures post-RDN than with a sham procedure. In contrast, blood pressure changes in RDN recipients with a below-median baseline 24-hour heart rate weren’t significant (Eur Heart J. 2019 Mar 1;40:743-51).
“Although this is a little bit rough, there is no other really true and reliable marker,” the cardiologist observed.
A pressing need exists for a reliable intraprocedural indicator of success. Dr. Bohm noted that Australian investigators are pursuing a promising approach in animal studies: intraprocedural transvascular high-frequency pacing of the aorticorenal ganglia. Abolition of the pacing-induced increase in blood pressure may be an indicator of complete RDN (JACC Cardiovasc Interv. 2019 Jun 24;12:1109-20).
Applications other than hypertension
Renal denervation is under early-stage investigation for a range of other cardiovascular diseases in which sympathetic overdrive figures prominently.
“The truly interesting things in renal denervation are what happens beyond hypertension. There are a lot of potential applications,” according to Dr. Bohm.
For example, when RDN was performed alongside pulmonary vein isolation for treatment of paroxysmal atrial fibrillation in hypertensive patients, the arrhythmia recurrence rate was significantly reduced during 1 year of follow-up, compared with AF ablation alone, in the randomized, multicenter, 302-patient ERADICATE-AF trial, presented at the most recent meeting of the Heart Rhythm Society.
Also, a small, uncontrolled registry study of RDN in patients with cardiomyopathy and electrical storm suggests the procedure may have an immediate anti–ventricular arrhythmia effect.
Meanwhile, Dr. Bohm is pressing the German government to sponsor an independent randomized controlled trial of RDN for heart failure. He and others have shown in small pilot studies a promising signal that the treatment may improve myocardial function and the signs and symptoms of heart failure in both patients with reduced and preserved left ventricular ejection fraction – and without reducing their blood pressure, which is often already low.
Dr. Bohm and others have also been exploring the impact of RDN in patients with metabolic syndrome. The treatment has a sound pathophysiologic rationale because insulin resistance is dependent upon sympathetic nervous system activation. Preliminary reports show improved insulin sensitivity in response to RDN. Patients also report better quality of life, presumably because of the reduction in sympathetic overactivity.
A couple of small Chinese studies suggest denervating the pulmonary vein in patients with pulmonary hypertension leads to a salutary reduction in pulmonary blood pressures.
“We haven’t done that yet. There is no properly designed catheter. They’ve used a Spyra unipolar catheter. It could work, but it hasn’t been rigorously investigated,” the cardiologist said.
Dr. Bohm reported serving as a scientific adviser to Abbott, AstraZeneca, BMS, Boehringer Ingelheim, and Servier.
EXPERT ANALYSIS FROM AHA 2019
Start of myeloma therapy may be delayed for women, minorities
Women and racial minorities with multiple myeloma may be at increased risk of delayed treatment, a situation that should be addressed urgently, according to authors of a recent analysis of a clinical oncology database.
By contrast, patients receiving myeloma treatment sooner after diagnosis included patients who were over 80 years of age, had multiple comorbidities, were treated at specialized cancer programs or in areas other than the Northeast, and had Medicaid or did not have private insurance, the authors reported.
Contrary to what was expected, levels of education and income did not significantly affect the timeliness of treatment in this analysis by Vivek Kumar, MD, of Dana-Farber Cancer Institute in Boston and coinvestigators.
While results of studies to date are “conflicting” as to whether timeliness of myeloma therapy will affect patient outcomes, recent studies in breast cancer and other tumor types suggest earlier treatment intervention may reduce morbidity, improve quality of life, and possibly prolong survival, according to Dr. Kumar and colleagues.
Moreover, the focus of myeloma treatment has shifted toward earlier treatment in light of the superiority of today’s treatment options, which was demonstrated in the 2014 update of the International Myeloma Working Group (IMWG) diagnostic criteria, according to the investigators.
“The definition of active MM [multiple myeloma] has been updated so that patients who may have been considered to have smoldering MM previously are now treated sooner to prevent end-organ damage whenever possible,” said Dr. Kumar and coauthors in their report in JCO Oncology Practice.
The analysis of timely myeloma treatment was based on for 74,722 patients in the National Cancer Database who received a diagnosis of multiple myeloma between 2004 and 2015 and went on to receive systemic treatment within the first year of diagnosis.
Delay in treatment, defined as receiving antimyeloma therapy 40 or more days after diagnosis, occurred in 18,375 of those patients, or about one-quarter of the study cohort. The mean time from diagnosis to start of treatment in that group was 63 days.
Compared with patients who received treatment within 7 days of diagnosis, patients with delays in treatment were more likely to be women (odds ratio, 1.15; 95% confidence interval, 1.1-1.2) and more likely to be non-Hispanic black (OR, 1.21; 95% CI, 1.14-1.28), the investigators reported.
A previous analysis of the SEER-Medicare database suggested that certain antimyeloma agents are used later in racial and ethnic minorities, including Hispanic patients, who had the highest median time to first dose of bortezomib, Dr. Kumar and colleagues noted.
However, no report before the present one had looked at the time to overall initial treatment in racial and ethnic minorities, they added.
Patients diagnosed in more recent years had higher odds of treatment delay, though this could have been caused by an increase in the number of patients diagnosed early; prior to the 2014 IMWG diagnostic criteria revision, many would have been offered therapy only when signs of end-organ damage were present, while patients without end-organ damage would have been said to have smoldering disease, authors said.
Patients 80 years of age and older and those with a higher Charlson comorbidity score had a lower likelihood of treatment delay in this analysis, possibly reflecting the frailty of those patients and an urgent need for treatment, according to investigators.
Uninsured patients and those with Medicaid were less likely than insured patients to experience treatment delay, according to the report.
“This may be associated with the fact that, for these insurances, prior authorization is typically not required before initiating treatment,” said Dr. Kumar and colleagues. “However, this could also depend on several other possible factors, including availability of caregiver support and seeking medical care later.”
Dr. Kumar reported no conflicts of interest related to the analysis. Coauthors reported disclosures with Takeda, Guardant Health, and other pharmaceutical companies.
SOURCE: Kumar V et al. JCO Oncology Practice. 2020 Jan 21. doi: 10.1200/JOP.19.00309.
Women and racial minorities with multiple myeloma may be at increased risk of delayed treatment, a situation that should be addressed urgently, according to authors of a recent analysis of a clinical oncology database.
By contrast, patients receiving myeloma treatment sooner after diagnosis included patients who were over 80 years of age, had multiple comorbidities, were treated at specialized cancer programs or in areas other than the Northeast, and had Medicaid or did not have private insurance, the authors reported.
Contrary to what was expected, levels of education and income did not significantly affect the timeliness of treatment in this analysis by Vivek Kumar, MD, of Dana-Farber Cancer Institute in Boston and coinvestigators.
While results of studies to date are “conflicting” as to whether timeliness of myeloma therapy will affect patient outcomes, recent studies in breast cancer and other tumor types suggest earlier treatment intervention may reduce morbidity, improve quality of life, and possibly prolong survival, according to Dr. Kumar and colleagues.
Moreover, the focus of myeloma treatment has shifted toward earlier treatment in light of the superiority of today’s treatment options, which was demonstrated in the 2014 update of the International Myeloma Working Group (IMWG) diagnostic criteria, according to the investigators.
“The definition of active MM [multiple myeloma] has been updated so that patients who may have been considered to have smoldering MM previously are now treated sooner to prevent end-organ damage whenever possible,” said Dr. Kumar and coauthors in their report in JCO Oncology Practice.
The analysis of timely myeloma treatment was based on for 74,722 patients in the National Cancer Database who received a diagnosis of multiple myeloma between 2004 and 2015 and went on to receive systemic treatment within the first year of diagnosis.
Delay in treatment, defined as receiving antimyeloma therapy 40 or more days after diagnosis, occurred in 18,375 of those patients, or about one-quarter of the study cohort. The mean time from diagnosis to start of treatment in that group was 63 days.
Compared with patients who received treatment within 7 days of diagnosis, patients with delays in treatment were more likely to be women (odds ratio, 1.15; 95% confidence interval, 1.1-1.2) and more likely to be non-Hispanic black (OR, 1.21; 95% CI, 1.14-1.28), the investigators reported.
A previous analysis of the SEER-Medicare database suggested that certain antimyeloma agents are used later in racial and ethnic minorities, including Hispanic patients, who had the highest median time to first dose of bortezomib, Dr. Kumar and colleagues noted.
However, no report before the present one had looked at the time to overall initial treatment in racial and ethnic minorities, they added.
Patients diagnosed in more recent years had higher odds of treatment delay, though this could have been caused by an increase in the number of patients diagnosed early; prior to the 2014 IMWG diagnostic criteria revision, many would have been offered therapy only when signs of end-organ damage were present, while patients without end-organ damage would have been said to have smoldering disease, authors said.
Patients 80 years of age and older and those with a higher Charlson comorbidity score had a lower likelihood of treatment delay in this analysis, possibly reflecting the frailty of those patients and an urgent need for treatment, according to investigators.
Uninsured patients and those with Medicaid were less likely than insured patients to experience treatment delay, according to the report.
“This may be associated with the fact that, for these insurances, prior authorization is typically not required before initiating treatment,” said Dr. Kumar and colleagues. “However, this could also depend on several other possible factors, including availability of caregiver support and seeking medical care later.”
Dr. Kumar reported no conflicts of interest related to the analysis. Coauthors reported disclosures with Takeda, Guardant Health, and other pharmaceutical companies.
SOURCE: Kumar V et al. JCO Oncology Practice. 2020 Jan 21. doi: 10.1200/JOP.19.00309.
Women and racial minorities with multiple myeloma may be at increased risk of delayed treatment, a situation that should be addressed urgently, according to authors of a recent analysis of a clinical oncology database.
By contrast, patients receiving myeloma treatment sooner after diagnosis included patients who were over 80 years of age, had multiple comorbidities, were treated at specialized cancer programs or in areas other than the Northeast, and had Medicaid or did not have private insurance, the authors reported.
Contrary to what was expected, levels of education and income did not significantly affect the timeliness of treatment in this analysis by Vivek Kumar, MD, of Dana-Farber Cancer Institute in Boston and coinvestigators.
While results of studies to date are “conflicting” as to whether timeliness of myeloma therapy will affect patient outcomes, recent studies in breast cancer and other tumor types suggest earlier treatment intervention may reduce morbidity, improve quality of life, and possibly prolong survival, according to Dr. Kumar and colleagues.
Moreover, the focus of myeloma treatment has shifted toward earlier treatment in light of the superiority of today’s treatment options, which was demonstrated in the 2014 update of the International Myeloma Working Group (IMWG) diagnostic criteria, according to the investigators.
“The definition of active MM [multiple myeloma] has been updated so that patients who may have been considered to have smoldering MM previously are now treated sooner to prevent end-organ damage whenever possible,” said Dr. Kumar and coauthors in their report in JCO Oncology Practice.
The analysis of timely myeloma treatment was based on for 74,722 patients in the National Cancer Database who received a diagnosis of multiple myeloma between 2004 and 2015 and went on to receive systemic treatment within the first year of diagnosis.
Delay in treatment, defined as receiving antimyeloma therapy 40 or more days after diagnosis, occurred in 18,375 of those patients, or about one-quarter of the study cohort. The mean time from diagnosis to start of treatment in that group was 63 days.
Compared with patients who received treatment within 7 days of diagnosis, patients with delays in treatment were more likely to be women (odds ratio, 1.15; 95% confidence interval, 1.1-1.2) and more likely to be non-Hispanic black (OR, 1.21; 95% CI, 1.14-1.28), the investigators reported.
A previous analysis of the SEER-Medicare database suggested that certain antimyeloma agents are used later in racial and ethnic minorities, including Hispanic patients, who had the highest median time to first dose of bortezomib, Dr. Kumar and colleagues noted.
However, no report before the present one had looked at the time to overall initial treatment in racial and ethnic minorities, they added.
Patients diagnosed in more recent years had higher odds of treatment delay, though this could have been caused by an increase in the number of patients diagnosed early; prior to the 2014 IMWG diagnostic criteria revision, many would have been offered therapy only when signs of end-organ damage were present, while patients without end-organ damage would have been said to have smoldering disease, authors said.
Patients 80 years of age and older and those with a higher Charlson comorbidity score had a lower likelihood of treatment delay in this analysis, possibly reflecting the frailty of those patients and an urgent need for treatment, according to investigators.
Uninsured patients and those with Medicaid were less likely than insured patients to experience treatment delay, according to the report.
“This may be associated with the fact that, for these insurances, prior authorization is typically not required before initiating treatment,” said Dr. Kumar and colleagues. “However, this could also depend on several other possible factors, including availability of caregiver support and seeking medical care later.”
Dr. Kumar reported no conflicts of interest related to the analysis. Coauthors reported disclosures with Takeda, Guardant Health, and other pharmaceutical companies.
SOURCE: Kumar V et al. JCO Oncology Practice. 2020 Jan 21. doi: 10.1200/JOP.19.00309.
FROM JCO ONCOLOGY PRACTICE
Key clinical point:
Major finding: Patients with delays in treatment were more likely to be women (odds ratio, 1.15) and more likely to be non-Hispanic blacks (OR, 1.21).
Study details: Retrospective analysis of 74,722 patients in the National Cancer Database diagnosed with multiple myeloma between 2004 and 2015.
Disclosures: Dr. Kumar reported no conflicts of interest related to the analysis. Coauthors reported disclosures with Takeda, Guardant Health, and other pharmaceutical companies.
Source: Kumar V et al. JCO Oncology Practice. 2020 Jan 21. doi: 10.1200/JOP.19.00309.