User login
More evidence that statins reduce HCC risk
SAN ANTONIO – The evidence that statin therapy reduces the risk of developing hepatocellular carcinoma, while not rising to the highest-level 1A strata, is nonetheless sufficiently persuasive at this point that consideration should be given to prescribing a statin in all patients with risk factors for the malignancy, regardless of their cardiovascular risk profile, Muhammad Talal Sarmini, MD, asserted at the annual meeting of the American College of Gastroenterology.

This includes individuals with hepatitis B or C virus infection as well as those with cirrhosis. The jury is still out as to whether nonalcoholic steatohepatitis is a risk factor for hepatocellular carcinoma (HCC), observed Dr. Sarmini of the Cleveland Clinic.
He presented a new meta-analysis, which concluded that patients on statin therapy had a 43% lower risk of new-onset HCC than persons not taking a statin. This meta-analysis – the largest ever addressing the issue – included 20 studies totaling more than 2.6 million patients and 24,341 cases of new-onset HCC. There were 11 retrospective case-control studies, 6 cohort studies, and 3 randomized trials. Five studies were from the United States, nine from Asia, and six were European.
In subgroup analyses aimed at assessing the consistency of the study results across various domains, there was a 45% reduction in the risk of HCC in association with statin therapy in the three studies of patients with hepatitis B virus, and significant reductions as well in Asia, Europe, and the United States when those participants were evaluated separately. The reduction was significant in both the case-control and cohort studies, but not when the three randomized, controlled trials (RCTs) were analyzed collectively. However, Dr. Sarmini shrugged off the neutral RCT results.
“It’s worth noting that the RCTs reported data from patients who were on statins with 4-5 years of follow-up. They were not at high risk for HCC. Given the nature of the disease and the relatively short period of follow-up, these studies only reported 81 cases of HCC. So they were very limited,” he said.
Audience members were eager to learn if Dr. Sarmini had found a differential preventive effect for lipophilic statins, such as atorvastatin or simvastatin, versus hydrophilic statins. He replied that, unfortunately, the published study results don’t allow for such an analysis. However, a large, propensity-matched cohort study published too recently for inclusion in his meta-analysis shed light on this matter. This Swedish national registry study included 16,668 propensity score–matched adults with chronic hepatitis B or C infection, of whom 6,554 initiated lipophilic statin therapy, 1,780 began treatment with a hydrophilic statin, and the rest were statin nonusers. The lipophilic statin users had an adjusted 44% reduction in 10-year HCC risk, compared with nonusers, while hydrophilic statins weren’t associated with a significant preventive effect (Ann Intern Med. 2019 Sep 3;171[5]:318-27).
Dr. Sarmini said that the meta-analysis results, together with the Swedish registry findings, highlight the need for additional well-designed cohort studies and RCTs of statins in populations at high risk for HCC in order to verify the existence of an HCC preventive effect and pinpoint which statins are effective at what dosages.
HCC is the fourth-leading cause of cancer-related mortality globally, accounting for 800,000 deaths annually. And the incidence is rising on a year-by-year basis.
Dr. Sarmini reported having no financial conflicts regarding his study, which was conducted free of commercial support.
SAN ANTONIO – The evidence that statin therapy reduces the risk of developing hepatocellular carcinoma, while not rising to the highest-level 1A strata, is nonetheless sufficiently persuasive at this point that consideration should be given to prescribing a statin in all patients with risk factors for the malignancy, regardless of their cardiovascular risk profile, Muhammad Talal Sarmini, MD, asserted at the annual meeting of the American College of Gastroenterology.
This includes individuals with hepatitis B or C virus infection as well as those with cirrhosis. The jury is still out as to whether nonalcoholic steatohepatitis is a risk factor for hepatocellular carcinoma (HCC), observed Dr. Sarmini of the Cleveland Clinic.
He presented a new meta-analysis, which concluded that patients on statin therapy had a 43% lower risk of new-onset HCC than persons not taking a statin. This meta-analysis – the largest ever addressing the issue – included 20 studies totaling more than 2.6 million patients and 24,341 cases of new-onset HCC. There were 11 retrospective case-control studies, 6 cohort studies, and 3 randomized trials. Five studies were from the United States, nine from Asia, and six were European.
In subgroup analyses aimed at assessing the consistency of the study results across various domains, there was a 45% reduction in the risk of HCC in association with statin therapy in the three studies of patients with hepatitis B virus, and significant reductions as well in Asia, Europe, and the United States when those participants were evaluated separately. The reduction was significant in both the case-control and cohort studies, but not when the three randomized, controlled trials (RCTs) were analyzed collectively. However, Dr. Sarmini shrugged off the neutral RCT results.
“It’s worth noting that the RCTs reported data from patients who were on statins with 4-5 years of follow-up. They were not at high risk for HCC. Given the nature of the disease and the relatively short period of follow-up, these studies only reported 81 cases of HCC. So they were very limited,” he said.
Audience members were eager to learn if Dr. Sarmini had found a differential preventive effect for lipophilic statins, such as atorvastatin or simvastatin, versus hydrophilic statins. He replied that, unfortunately, the published study results don’t allow for such an analysis. However, a large, propensity-matched cohort study published too recently for inclusion in his meta-analysis shed light on this matter. This Swedish national registry study included 16,668 propensity score–matched adults with chronic hepatitis B or C infection, of whom 6,554 initiated lipophilic statin therapy, 1,780 began treatment with a hydrophilic statin, and the rest were statin nonusers. The lipophilic statin users had an adjusted 44% reduction in 10-year HCC risk, compared with nonusers, while hydrophilic statins weren’t associated with a significant preventive effect (Ann Intern Med. 2019 Sep 3;171[5]:318-27).
Dr. Sarmini said that the meta-analysis results, together with the Swedish registry findings, highlight the need for additional well-designed cohort studies and RCTs of statins in populations at high risk for HCC in order to verify the existence of an HCC preventive effect and pinpoint which statins are effective at what dosages.
HCC is the fourth-leading cause of cancer-related mortality globally, accounting for 800,000 deaths annually. And the incidence is rising on a year-by-year basis.
Dr. Sarmini reported having no financial conflicts regarding his study, which was conducted free of commercial support.
SAN ANTONIO – The evidence that statin therapy reduces the risk of developing hepatocellular carcinoma, while not rising to the highest-level 1A strata, is nonetheless sufficiently persuasive at this point that consideration should be given to prescribing a statin in all patients with risk factors for the malignancy, regardless of their cardiovascular risk profile, Muhammad Talal Sarmini, MD, asserted at the annual meeting of the American College of Gastroenterology.
This includes individuals with hepatitis B or C virus infection as well as those with cirrhosis. The jury is still out as to whether nonalcoholic steatohepatitis is a risk factor for hepatocellular carcinoma (HCC), observed Dr. Sarmini of the Cleveland Clinic.
He presented a new meta-analysis, which concluded that patients on statin therapy had a 43% lower risk of new-onset HCC than persons not taking a statin. This meta-analysis – the largest ever addressing the issue – included 20 studies totaling more than 2.6 million patients and 24,341 cases of new-onset HCC. There were 11 retrospective case-control studies, 6 cohort studies, and 3 randomized trials. Five studies were from the United States, nine from Asia, and six were European.
In subgroup analyses aimed at assessing the consistency of the study results across various domains, there was a 45% reduction in the risk of HCC in association with statin therapy in the three studies of patients with hepatitis B virus, and significant reductions as well in Asia, Europe, and the United States when those participants were evaluated separately. The reduction was significant in both the case-control and cohort studies, but not when the three randomized, controlled trials (RCTs) were analyzed collectively. However, Dr. Sarmini shrugged off the neutral RCT results.
“It’s worth noting that the RCTs reported data from patients who were on statins with 4-5 years of follow-up. They were not at high risk for HCC. Given the nature of the disease and the relatively short period of follow-up, these studies only reported 81 cases of HCC. So they were very limited,” he said.
Audience members were eager to learn if Dr. Sarmini had found a differential preventive effect for lipophilic statins, such as atorvastatin or simvastatin, versus hydrophilic statins. He replied that, unfortunately, the published study results don’t allow for such an analysis. However, a large, propensity-matched cohort study published too recently for inclusion in his meta-analysis shed light on this matter. This Swedish national registry study included 16,668 propensity score–matched adults with chronic hepatitis B or C infection, of whom 6,554 initiated lipophilic statin therapy, 1,780 began treatment with a hydrophilic statin, and the rest were statin nonusers. The lipophilic statin users had an adjusted 44% reduction in 10-year HCC risk, compared with nonusers, while hydrophilic statins weren’t associated with a significant preventive effect (Ann Intern Med. 2019 Sep 3;171[5]:318-27).
Dr. Sarmini said that the meta-analysis results, together with the Swedish registry findings, highlight the need for additional well-designed cohort studies and RCTs of statins in populations at high risk for HCC in order to verify the existence of an HCC preventive effect and pinpoint which statins are effective at what dosages.
HCC is the fourth-leading cause of cancer-related mortality globally, accounting for 800,000 deaths annually. And the incidence is rising on a year-by-year basis.
Dr. Sarmini reported having no financial conflicts regarding his study, which was conducted free of commercial support.
REPORTING FROM ACG 2019
Oral antibiotics as effective as IV for stable endocarditis patients
Background: Patients with left-sided infective endocarditis often are treated with prolonged courses of intravenous (IV) antibiotics. The safety of switching from IV to oral antibiotics is unknown.

Study design: Randomized, multicenter, noninferiority study.
Setting: Cardiac centers in Denmark during July 2011–August 2017.
Synopsis: The study enrolled 400 patients with left-sided infective endocarditis and positive blood cultures from Streptococcus, Enterococcus, Staphylococcus aureus, or coagulase-negative staph (non–methicillin-resistant Staphylococcus aureus), without evidence of valvular abscess. Following at least 7 days (for those who required surgical intervention) or 10 days (for those who did not require surgical intervention) of IV antibiotics, patients with ongoing fever, leukocytosis, elevated C-reactive protein, or concurrent infections were excluded from the study. Patients were randomized to receive continued IV antibiotic treatment or switch to oral antibiotic treatment. The IV treatment group received a median of 19 additional days of therapy, compared with 17 days in the oral group. The primary composite outcome of death, unplanned cardiac surgery, embolic event, and relapse of bacteremia occurred in 12.1% in the IV therapy group and 9% in the oral therapy group (difference of 3.1%; 95% confidence interval, –3.4 to 9.6; P = .40), meeting the studies prespecified noninferiority criteria. Poor representation of women, obese patients, and patients who use IV drugs may limit the study’s generalizability. An accompanying editorial advocated for additional research before widespread change to current treatment recommendations are made.
Bottom line: For patients with left-sided infective endocarditis who have been stabilized on IV antibiotic treatment, transitioning to an oral antibiotic regimen may be a noninferior approach.
Citation: Iverson K et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Eng J Med. 2019 Jan 31;380(5):415-24.
Dr. Phillips is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine at Harvard Medical School.
Background: Patients with left-sided infective endocarditis often are treated with prolonged courses of intravenous (IV) antibiotics. The safety of switching from IV to oral antibiotics is unknown.
Study design: Randomized, multicenter, noninferiority study.
Setting: Cardiac centers in Denmark during July 2011–August 2017.
Synopsis: The study enrolled 400 patients with left-sided infective endocarditis and positive blood cultures from Streptococcus, Enterococcus, Staphylococcus aureus, or coagulase-negative staph (non–methicillin-resistant Staphylococcus aureus), without evidence of valvular abscess. Following at least 7 days (for those who required surgical intervention) or 10 days (for those who did not require surgical intervention) of IV antibiotics, patients with ongoing fever, leukocytosis, elevated C-reactive protein, or concurrent infections were excluded from the study. Patients were randomized to receive continued IV antibiotic treatment or switch to oral antibiotic treatment. The IV treatment group received a median of 19 additional days of therapy, compared with 17 days in the oral group. The primary composite outcome of death, unplanned cardiac surgery, embolic event, and relapse of bacteremia occurred in 12.1% in the IV therapy group and 9% in the oral therapy group (difference of 3.1%; 95% confidence interval, –3.4 to 9.6; P = .40), meeting the studies prespecified noninferiority criteria. Poor representation of women, obese patients, and patients who use IV drugs may limit the study’s generalizability. An accompanying editorial advocated for additional research before widespread change to current treatment recommendations are made.
Bottom line: For patients with left-sided infective endocarditis who have been stabilized on IV antibiotic treatment, transitioning to an oral antibiotic regimen may be a noninferior approach.
Citation: Iverson K et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Eng J Med. 2019 Jan 31;380(5):415-24.
Dr. Phillips is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine at Harvard Medical School.
Background: Patients with left-sided infective endocarditis often are treated with prolonged courses of intravenous (IV) antibiotics. The safety of switching from IV to oral antibiotics is unknown.
Study design: Randomized, multicenter, noninferiority study.
Setting: Cardiac centers in Denmark during July 2011–August 2017.
Synopsis: The study enrolled 400 patients with left-sided infective endocarditis and positive blood cultures from Streptococcus, Enterococcus, Staphylococcus aureus, or coagulase-negative staph (non–methicillin-resistant Staphylococcus aureus), without evidence of valvular abscess. Following at least 7 days (for those who required surgical intervention) or 10 days (for those who did not require surgical intervention) of IV antibiotics, patients with ongoing fever, leukocytosis, elevated C-reactive protein, or concurrent infections were excluded from the study. Patients were randomized to receive continued IV antibiotic treatment or switch to oral antibiotic treatment. The IV treatment group received a median of 19 additional days of therapy, compared with 17 days in the oral group. The primary composite outcome of death, unplanned cardiac surgery, embolic event, and relapse of bacteremia occurred in 12.1% in the IV therapy group and 9% in the oral therapy group (difference of 3.1%; 95% confidence interval, –3.4 to 9.6; P = .40), meeting the studies prespecified noninferiority criteria. Poor representation of women, obese patients, and patients who use IV drugs may limit the study’s generalizability. An accompanying editorial advocated for additional research before widespread change to current treatment recommendations are made.
Bottom line: For patients with left-sided infective endocarditis who have been stabilized on IV antibiotic treatment, transitioning to an oral antibiotic regimen may be a noninferior approach.
Citation: Iverson K et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Eng J Med. 2019 Jan 31;380(5):415-24.
Dr. Phillips is a hospitalist at Beth Israel Deaconess Medical Center and instructor in medicine at Harvard Medical School.
Probiotics with Lactobacillus reduce loss in spine BMD for postmenopausal women
, according to recent research published in The Lancet Rheumatology.

“The menopausal and early postmenopausal lumbar spine bone loss is substantial in women, and by using a prevention therapy with bacteria naturally occurring in the human gut microbiota we observed a close to complete protection against lumbar spine bone loss in healthy postmenopausal women,” Per-Anders Jansson, MD, chief physician at the University of Gothenburg (Sweden), and colleagues wrote in their study.
Dr. Jansson and colleagues performed a double-blind trial at four centers in Sweden in which 249 postmenopausal women were randomized during April-November 2016 to receive probiotics consisting of three Lactobacillus strains or placebo once per day for 12 months. Participants were healthy women, neither underweight nor overweight, and were postmenopausal, which was defined as being 2-12 years or less from last menstruation. The Lactobacillus strains, L. paracasei 8700:2 (DSM 13434), L. plantarum Heal 9 (DSM 15312), and L. plantarum Heal 19 (DSM 15313), were equally represented in a capsule at a dose of 1 x 1010 colony-forming unit per capsule. The researchers measured the lumbar spine bone mineral density (LS-BMD) at baseline and at 12 months, and also evaluated the safety profile of participants in both the probiotic and placebo groups.
Overall, 234 participants (94%) had data available for analysis at the end of the study. There was a significant reduction in LS-BMD loss for participants who received the probiotic treatment, compared with women in the control group (mean difference, 0.71%; 95% confidence interval, 0.06%-1.35%), while there was a significant loss in LS-BMD for participants in the placebo group (percentage change, –0.72%; 95% CI, –1.22% to –0.22%) compared with loss in the probiotic group (percentage change, –0.01%; 95% CI, –0.50% to 0.48%). Using analysis of covariance, the researchers found the probiotic group had reduced LS-BMD loss after adjustment for factors such as study site, age at baseline, BMD at baseline, and number of years from menopause (mean difference, 7.44 mg/cm2; 95% CI, 0.38 to 14.50).
In a subgroup analysis of women above and below the median time since menopause at baseline (6 years), participants in the probiotic group who were below the median time saw a significant protective effect of Lactobacillus treatment (mean difference, 1.08%; 95% CI, 0.20%-1.96%), compared with women above the median time (mean difference, 0.31%; 95% CI, –0.62% to 1.23%).
Researchers also examined the effects of probiotic treatment on total hip and femoral neck BMD as secondary endpoints. Lactobacillus treatment did not appear to affect total hip (–1.01%; 95% CI, –1.65% to –0.37%) or trochanter BMD (–1.13%; 95% CI, –2.27% to 0.20%), but femoral neck BMD was reduced in the probiotic group (–1.34%; 95% CI, –2.09% to –0.58%), compared with the placebo group (–0.88%; 95% CI, –1.64% to –0.13%).
Limitations of the study included examining only one dose of Lactobacillus treatment and no analysis of the effect of short-chain fatty acids on LS-BMD. The researchers noted that “recent studies have shown that short-chain fatty acids, which are generated by fermentation of complex carbohydrates by the gut microbiota, are important regulators of both bone formation and resorption.”
The researchers also acknowledged that the LS-BMD effect size for the probiotic treatment over the 12 months was a lower magnitude, compared with first-line treatments for osteoporosis in postmenopausal women using bisphosphonates. “Further long-term studies should be done to evaluate if the bone-protective effect becomes more pronounced with prolonged treatment with the Lactobacillus strains used in the present study,” they said.
In a related editorial, Shivani Sahni, PhD, of Harvard Medical School, Boston, and Connie M. Weaver, PhD, of Purdue University, West Lafayette, Ind., reiterated that the effect size of probiotics is “of far less magnitude” than such treatments as bisphosphonates and expressed concern about the reduction of femoral neck BMD in the probiotic group, which was not explained in the study (Lancet Rheumatol. 2019 Nov;1[3]:e135-e137. doi: 10.1016/S2665-9913(19)30073-6). There is a need to learn the optimum dose of probiotics as well as which Lactobacillus strains should be used in future studies, as the strains chosen by Jansson et al. were based on results in mice.
In the meantime, patients might be better off choosing dietary interventions with proven bone protection and no documented negative effects on the hip, such as prebiotics like soluble corn fiber and dried prunes, in tandem with drug therapies, Dr. Sahni and Dr. Weaver said.
“Although Jansson and colleagues’ results are important, more work is needed before such probiotics are ready for consumers,” they concluded.
This study was funded by Probi, which employs two of the study’s authors. Three authors reported being coinventors of a patent involving the effects of probiotics in osteoporosis treatment, and one author is listed as an inventor on a pending patent application on probiotic compositions and uses. Dr. Sahni reported receiving grants from Dairy Management. Dr. Weaver reported no relevant conflicts of interest.
SOURCE: Jansson P-A et al. Lancet Rheumatol. 2019 Nov;1(3):e154-e162. doi: 10.1016/S2665-9913(19)30068-2
, according to recent research published in The Lancet Rheumatology.
“The menopausal and early postmenopausal lumbar spine bone loss is substantial in women, and by using a prevention therapy with bacteria naturally occurring in the human gut microbiota we observed a close to complete protection against lumbar spine bone loss in healthy postmenopausal women,” Per-Anders Jansson, MD, chief physician at the University of Gothenburg (Sweden), and colleagues wrote in their study.
Dr. Jansson and colleagues performed a double-blind trial at four centers in Sweden in which 249 postmenopausal women were randomized during April-November 2016 to receive probiotics consisting of three Lactobacillus strains or placebo once per day for 12 months. Participants were healthy women, neither underweight nor overweight, and were postmenopausal, which was defined as being 2-12 years or less from last menstruation. The Lactobacillus strains, L. paracasei 8700:2 (DSM 13434), L. plantarum Heal 9 (DSM 15312), and L. plantarum Heal 19 (DSM 15313), were equally represented in a capsule at a dose of 1 x 1010 colony-forming unit per capsule. The researchers measured the lumbar spine bone mineral density (LS-BMD) at baseline and at 12 months, and also evaluated the safety profile of participants in both the probiotic and placebo groups.
Overall, 234 participants (94%) had data available for analysis at the end of the study. There was a significant reduction in LS-BMD loss for participants who received the probiotic treatment, compared with women in the control group (mean difference, 0.71%; 95% confidence interval, 0.06%-1.35%), while there was a significant loss in LS-BMD for participants in the placebo group (percentage change, –0.72%; 95% CI, –1.22% to –0.22%) compared with loss in the probiotic group (percentage change, –0.01%; 95% CI, –0.50% to 0.48%). Using analysis of covariance, the researchers found the probiotic group had reduced LS-BMD loss after adjustment for factors such as study site, age at baseline, BMD at baseline, and number of years from menopause (mean difference, 7.44 mg/cm2; 95% CI, 0.38 to 14.50).
In a subgroup analysis of women above and below the median time since menopause at baseline (6 years), participants in the probiotic group who were below the median time saw a significant protective effect of Lactobacillus treatment (mean difference, 1.08%; 95% CI, 0.20%-1.96%), compared with women above the median time (mean difference, 0.31%; 95% CI, –0.62% to 1.23%).
Researchers also examined the effects of probiotic treatment on total hip and femoral neck BMD as secondary endpoints. Lactobacillus treatment did not appear to affect total hip (–1.01%; 95% CI, –1.65% to –0.37%) or trochanter BMD (–1.13%; 95% CI, –2.27% to 0.20%), but femoral neck BMD was reduced in the probiotic group (–1.34%; 95% CI, –2.09% to –0.58%), compared with the placebo group (–0.88%; 95% CI, –1.64% to –0.13%).
Limitations of the study included examining only one dose of Lactobacillus treatment and no analysis of the effect of short-chain fatty acids on LS-BMD. The researchers noted that “recent studies have shown that short-chain fatty acids, which are generated by fermentation of complex carbohydrates by the gut microbiota, are important regulators of both bone formation and resorption.”
The researchers also acknowledged that the LS-BMD effect size for the probiotic treatment over the 12 months was a lower magnitude, compared with first-line treatments for osteoporosis in postmenopausal women using bisphosphonates. “Further long-term studies should be done to evaluate if the bone-protective effect becomes more pronounced with prolonged treatment with the Lactobacillus strains used in the present study,” they said.
In a related editorial, Shivani Sahni, PhD, of Harvard Medical School, Boston, and Connie M. Weaver, PhD, of Purdue University, West Lafayette, Ind., reiterated that the effect size of probiotics is “of far less magnitude” than such treatments as bisphosphonates and expressed concern about the reduction of femoral neck BMD in the probiotic group, which was not explained in the study (Lancet Rheumatol. 2019 Nov;1[3]:e135-e137. doi: 10.1016/S2665-9913(19)30073-6). There is a need to learn the optimum dose of probiotics as well as which Lactobacillus strains should be used in future studies, as the strains chosen by Jansson et al. were based on results in mice.
In the meantime, patients might be better off choosing dietary interventions with proven bone protection and no documented negative effects on the hip, such as prebiotics like soluble corn fiber and dried prunes, in tandem with drug therapies, Dr. Sahni and Dr. Weaver said.
“Although Jansson and colleagues’ results are important, more work is needed before such probiotics are ready for consumers,” they concluded.
This study was funded by Probi, which employs two of the study’s authors. Three authors reported being coinventors of a patent involving the effects of probiotics in osteoporosis treatment, and one author is listed as an inventor on a pending patent application on probiotic compositions and uses. Dr. Sahni reported receiving grants from Dairy Management. Dr. Weaver reported no relevant conflicts of interest.
SOURCE: Jansson P-A et al. Lancet Rheumatol. 2019 Nov;1(3):e154-e162. doi: 10.1016/S2665-9913(19)30068-2
, according to recent research published in The Lancet Rheumatology.
“The menopausal and early postmenopausal lumbar spine bone loss is substantial in women, and by using a prevention therapy with bacteria naturally occurring in the human gut microbiota we observed a close to complete protection against lumbar spine bone loss in healthy postmenopausal women,” Per-Anders Jansson, MD, chief physician at the University of Gothenburg (Sweden), and colleagues wrote in their study.
Dr. Jansson and colleagues performed a double-blind trial at four centers in Sweden in which 249 postmenopausal women were randomized during April-November 2016 to receive probiotics consisting of three Lactobacillus strains or placebo once per day for 12 months. Participants were healthy women, neither underweight nor overweight, and were postmenopausal, which was defined as being 2-12 years or less from last menstruation. The Lactobacillus strains, L. paracasei 8700:2 (DSM 13434), L. plantarum Heal 9 (DSM 15312), and L. plantarum Heal 19 (DSM 15313), were equally represented in a capsule at a dose of 1 x 1010 colony-forming unit per capsule. The researchers measured the lumbar spine bone mineral density (LS-BMD) at baseline and at 12 months, and also evaluated the safety profile of participants in both the probiotic and placebo groups.
Overall, 234 participants (94%) had data available for analysis at the end of the study. There was a significant reduction in LS-BMD loss for participants who received the probiotic treatment, compared with women in the control group (mean difference, 0.71%; 95% confidence interval, 0.06%-1.35%), while there was a significant loss in LS-BMD for participants in the placebo group (percentage change, –0.72%; 95% CI, –1.22% to –0.22%) compared with loss in the probiotic group (percentage change, –0.01%; 95% CI, –0.50% to 0.48%). Using analysis of covariance, the researchers found the probiotic group had reduced LS-BMD loss after adjustment for factors such as study site, age at baseline, BMD at baseline, and number of years from menopause (mean difference, 7.44 mg/cm2; 95% CI, 0.38 to 14.50).
In a subgroup analysis of women above and below the median time since menopause at baseline (6 years), participants in the probiotic group who were below the median time saw a significant protective effect of Lactobacillus treatment (mean difference, 1.08%; 95% CI, 0.20%-1.96%), compared with women above the median time (mean difference, 0.31%; 95% CI, –0.62% to 1.23%).
Researchers also examined the effects of probiotic treatment on total hip and femoral neck BMD as secondary endpoints. Lactobacillus treatment did not appear to affect total hip (–1.01%; 95% CI, –1.65% to –0.37%) or trochanter BMD (–1.13%; 95% CI, –2.27% to 0.20%), but femoral neck BMD was reduced in the probiotic group (–1.34%; 95% CI, –2.09% to –0.58%), compared with the placebo group (–0.88%; 95% CI, –1.64% to –0.13%).
Limitations of the study included examining only one dose of Lactobacillus treatment and no analysis of the effect of short-chain fatty acids on LS-BMD. The researchers noted that “recent studies have shown that short-chain fatty acids, which are generated by fermentation of complex carbohydrates by the gut microbiota, are important regulators of both bone formation and resorption.”
The researchers also acknowledged that the LS-BMD effect size for the probiotic treatment over the 12 months was a lower magnitude, compared with first-line treatments for osteoporosis in postmenopausal women using bisphosphonates. “Further long-term studies should be done to evaluate if the bone-protective effect becomes more pronounced with prolonged treatment with the Lactobacillus strains used in the present study,” they said.
In a related editorial, Shivani Sahni, PhD, of Harvard Medical School, Boston, and Connie M. Weaver, PhD, of Purdue University, West Lafayette, Ind., reiterated that the effect size of probiotics is “of far less magnitude” than such treatments as bisphosphonates and expressed concern about the reduction of femoral neck BMD in the probiotic group, which was not explained in the study (Lancet Rheumatol. 2019 Nov;1[3]:e135-e137. doi: 10.1016/S2665-9913(19)30073-6). There is a need to learn the optimum dose of probiotics as well as which Lactobacillus strains should be used in future studies, as the strains chosen by Jansson et al. were based on results in mice.
In the meantime, patients might be better off choosing dietary interventions with proven bone protection and no documented negative effects on the hip, such as prebiotics like soluble corn fiber and dried prunes, in tandem with drug therapies, Dr. Sahni and Dr. Weaver said.
“Although Jansson and colleagues’ results are important, more work is needed before such probiotics are ready for consumers,” they concluded.
This study was funded by Probi, which employs two of the study’s authors. Three authors reported being coinventors of a patent involving the effects of probiotics in osteoporosis treatment, and one author is listed as an inventor on a pending patent application on probiotic compositions and uses. Dr. Sahni reported receiving grants from Dairy Management. Dr. Weaver reported no relevant conflicts of interest.
SOURCE: Jansson P-A et al. Lancet Rheumatol. 2019 Nov;1(3):e154-e162. doi: 10.1016/S2665-9913(19)30068-2
FROM THE LANCET RHEUMATOLOGY
Spinal progression found more often in men with ankylosing spondylitis
Patients with ankylosing spondylitis who are male, have evidence of spinal damage, or have higher levels of inflammatory markers may be at higher risk of disease progression, a study has found.
“Assessment of AS-related structural changes longitudinally is essential for understanding the natural course of progression and its underlying factors,” Ismail Sari, MD, of the University of Toronto and coauthors wrote in Arthritis Care & Research. “This could help identify the mechanisms responsible for progression and thereby personalizing treatment.”
The researchers found that nearly one-quarter (24.3%) of 350 individuals with ankylosing spondylitis in a longitudinal cohort study showed radiographic evidence of progression, defined as a change of 2 units on the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) in 2 years. Overall, 76% of the group were males, and the group had a mean age of about 38 years with a mean symptom duration of nearly 15 years.
Over the 6-year follow-up, the mean mSASSS increased from 9.3 units at baseline to 17.7 units, with more progression seen in the cervical spine than the lumbar segments. During the first 2 years, the total mSASSS increased by a mean of 1.23 units; in years 2-4, it increased by a mean of 1.47 units, and from 4 to 6 years, it increased by a mean of 1.52 units.
Male sex was associated with more than double the risk of radiographic progression (hazard ratio, 2.46; 95% confidence interval, 1.05-5.76), while individuals with radiographic evidence of spinal damage at baseline had a nearly eightfold higher risk of progression (HR, 7.98; 95% CI, 3.98-16). The risk for disease progression also increased with higher levels of C-reactive protein.
The investigators also found that patients who had used tumor necrosis factor inhibitor therapy for at least 1 year had an 18% reduction in the rate of spinal progression.
However, other factors including symptom duration, presence of HLA-B27, smoking status, presence of radiographic hip disease, or use of disease-modifying antirheumatic drugs or NSAIDs did not appear to influence the risk of disease progression.
No funding or conflicts of interest were declared.
SOURCE: Sari I et al. Arthritis Care Res. 2019 Nov 1. doi: 10.1002/acr.24104.
Patients with ankylosing spondylitis who are male, have evidence of spinal damage, or have higher levels of inflammatory markers may be at higher risk of disease progression, a study has found.
“Assessment of AS-related structural changes longitudinally is essential for understanding the natural course of progression and its underlying factors,” Ismail Sari, MD, of the University of Toronto and coauthors wrote in Arthritis Care & Research. “This could help identify the mechanisms responsible for progression and thereby personalizing treatment.”
The researchers found that nearly one-quarter (24.3%) of 350 individuals with ankylosing spondylitis in a longitudinal cohort study showed radiographic evidence of progression, defined as a change of 2 units on the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) in 2 years. Overall, 76% of the group were males, and the group had a mean age of about 38 years with a mean symptom duration of nearly 15 years.
Over the 6-year follow-up, the mean mSASSS increased from 9.3 units at baseline to 17.7 units, with more progression seen in the cervical spine than the lumbar segments. During the first 2 years, the total mSASSS increased by a mean of 1.23 units; in years 2-4, it increased by a mean of 1.47 units, and from 4 to 6 years, it increased by a mean of 1.52 units.
Male sex was associated with more than double the risk of radiographic progression (hazard ratio, 2.46; 95% confidence interval, 1.05-5.76), while individuals with radiographic evidence of spinal damage at baseline had a nearly eightfold higher risk of progression (HR, 7.98; 95% CI, 3.98-16). The risk for disease progression also increased with higher levels of C-reactive protein.
The investigators also found that patients who had used tumor necrosis factor inhibitor therapy for at least 1 year had an 18% reduction in the rate of spinal progression.
However, other factors including symptom duration, presence of HLA-B27, smoking status, presence of radiographic hip disease, or use of disease-modifying antirheumatic drugs or NSAIDs did not appear to influence the risk of disease progression.
No funding or conflicts of interest were declared.
SOURCE: Sari I et al. Arthritis Care Res. 2019 Nov 1. doi: 10.1002/acr.24104.
Patients with ankylosing spondylitis who are male, have evidence of spinal damage, or have higher levels of inflammatory markers may be at higher risk of disease progression, a study has found.
“Assessment of AS-related structural changes longitudinally is essential for understanding the natural course of progression and its underlying factors,” Ismail Sari, MD, of the University of Toronto and coauthors wrote in Arthritis Care & Research. “This could help identify the mechanisms responsible for progression and thereby personalizing treatment.”
The researchers found that nearly one-quarter (24.3%) of 350 individuals with ankylosing spondylitis in a longitudinal cohort study showed radiographic evidence of progression, defined as a change of 2 units on the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) in 2 years. Overall, 76% of the group were males, and the group had a mean age of about 38 years with a mean symptom duration of nearly 15 years.
Over the 6-year follow-up, the mean mSASSS increased from 9.3 units at baseline to 17.7 units, with more progression seen in the cervical spine than the lumbar segments. During the first 2 years, the total mSASSS increased by a mean of 1.23 units; in years 2-4, it increased by a mean of 1.47 units, and from 4 to 6 years, it increased by a mean of 1.52 units.
Male sex was associated with more than double the risk of radiographic progression (hazard ratio, 2.46; 95% confidence interval, 1.05-5.76), while individuals with radiographic evidence of spinal damage at baseline had a nearly eightfold higher risk of progression (HR, 7.98; 95% CI, 3.98-16). The risk for disease progression also increased with higher levels of C-reactive protein.
The investigators also found that patients who had used tumor necrosis factor inhibitor therapy for at least 1 year had an 18% reduction in the rate of spinal progression.
However, other factors including symptom duration, presence of HLA-B27, smoking status, presence of radiographic hip disease, or use of disease-modifying antirheumatic drugs or NSAIDs did not appear to influence the risk of disease progression.
No funding or conflicts of interest were declared.
SOURCE: Sari I et al. Arthritis Care Res. 2019 Nov 1. doi: 10.1002/acr.24104.
FROM ARTHRITIS CARE & RESEARCH
Psoriatic Arthritis Types and Disease Pattern
Blocking TLR9 may halt brain edema in acute liver failure
A toll-like receptor 9 (TLR9) antagonist may eventually be used to combat brain edema in acute liver failure, according to investigators.
This prediction is based on results of a recent study involving mouse models, which showed that ODN2088, a TLR9 antagonist, could stop ammonia-induced colocalization of DNA with TLR9 in innate immune cells, thereby blocking cytokine production and ensuant brain edema, reported lead author Godhev Kumar Manakkat Vijay of King’s College London and colleagues.
“Ammonia plays a pivotal role in the development of hepatic encephalopathy and brain edema in acute liver failure,” the investigators explained in Cellular and Molecular Gastroenterology and Hepatology. “A robust systemic inflammatory response and susceptibility to developing infection are common in acute liver failure, exacerbate the development of ammonia-induced brain edema and are major prognosticators. Experimental models have unequivocally associated ammonia exposure with astrocyte swelling and brain edema, potentiated by proinflammatory cytokines.”
The investigators added that, “although the evidence base supporting the relationship between ammonia, inflammation, and brain edema is robust in acute liver failure, there is a paucity of data characterizing the specific pathogenic mechanisms entailed.” Previous research suggested that TLR9 plays a key role in acetaminophen-induced liver inflammation, they noted, and that ammonia, in combination with DNA, triggers TLR9 expression in neutrophils, which brought TLR9 into focus for the present study.
Along with wild-type mice, the investigators relied upon two knockout models: TLR9–/– mice, in which TLR9 is entirely absent, and LysM-Cre TLR9fl/fl mice, in which TLR9 is absent from lysozyme-expressing cells (predominantly neutrophils and macrophages). Comparing against controls, the investigators assessed cytokine production and brain edema in each type of mouse when intraperitoneally injected with ammonium acetate (4 mmol/kg). Specifically, 6 hours after injection, they measured intracellular cytokines in splenic macrophages, CD8+ T cells, and CD4+ T cells. In addition, they recorded total plasma DNA and brain water, a measure of brain edema.
Following ammonium acetate injection, wild-type mice developed brain edema and liver enlargement, while TLR9–/– mice and control-injected mice did not. After injection, total plasma DNA levels rose by comparable magnitudes in both wild-type mice and TLR9–/– mice, but did not change in control-injected mice, suggesting that ammonium-acetate injection was causing a release of DNA, which was binding with TLR9, resulting in activation of the innate immune system.
This hypothesis was supported by measurements of cytokines in T cells and splenic macrophages, which showed that wild-type mice had elevations of cytokines, whereas knockout mice did not. Further experiments showed that LysM-Cre TLR9fl/fl mice had similar outcomes as TLR9–/– mice, highlighting that macrophages and neutrophils are the key immune cells linking TLR9 activation with cytokine release, and therefore brain edema.
To ensure that brain edema was not directly caused by the acetate component of ammonium acetate, or acetate’s potential to increase pH, a different set of wild-type mice were injected with sodium acetate adjusted to the same pH as ammonium acetate. This had no impact on cytokine production, brain-water content, or liver-to-body weight ratio, confirming that acetate was not responsible for brain edema while providing further support for the role of TLR9.
Finally, the investigators treated wild-type mice immediately after ammonium acetate injection with the TLR9 antagonist ODN2088 (50 mcg/mouse). This treatment halted cytokine production, inflammation, and brain edema, strongly supporting the link between these ammonia-induced processes and TLR9 activation.
“These data are well supported by the findings of Imaeda et al. (J Clin Invest. 2009 Feb 2. doi: 10.1172/JCI35958), who in an acetaminophen-induced hepatotoxicity model established that inhibition of TLR9 using ODN2088 and IRS954, a TLR7/9 antagonist, down-regulated proinflammatory cytokine release and reduced mortality,” the investigators wrote. “The amelioration of brain edema and cytokine production by ODN2088 supports exploration of TLR9 antagonism as a therapeutic modality in early acute liver failure to prevent the development of brain edema and intracranial hypertension.”
The study was funded by the U.K. Institute of Liver Studies Charitable Fund and the National Institutes of Health. The investigators reported no conflicts of interest.
SOURCE: Vijay GKM et al. Cell Mol Gastroenterol Hepatol. 2019 Aug 8. doi: 10.1016/j.jcmgh.2019.08.002.
Acute liver failure is a devastating disease, which has a high mortality burden and often requires liver transplant. One of the major complications is cerebral edema that leads to encephalopathy and could be fatal. These brain changes are accompanied by inflammation, immune activation, and hyperammonemia, but further mechanistic approaches are needed.

This data adds to the growing literature about the interaction between immune dysfunction and brain diseases such as schizophrenia, autism, depression, and multiple sclerosis. However, further studies in models of brain edema with concomitant liver failure, which are closer to the human disease process, are needed. This exciting investigation of neuroimmune regulation of brain edema could set the basis for new therapeutic options for the prevention and treatment of this feared complication of acute liver failure.
Jasmohan S. Bajaj, MD, AGAF, is professor in the division of gastroenterology, hepatology, and nutrition at Virginia Commonwealth University, Richmond. He reported no conflicts of interest.
Acute liver failure is a devastating disease, which has a high mortality burden and often requires liver transplant. One of the major complications is cerebral edema that leads to encephalopathy and could be fatal. These brain changes are accompanied by inflammation, immune activation, and hyperammonemia, but further mechanistic approaches are needed.
This data adds to the growing literature about the interaction between immune dysfunction and brain diseases such as schizophrenia, autism, depression, and multiple sclerosis. However, further studies in models of brain edema with concomitant liver failure, which are closer to the human disease process, are needed. This exciting investigation of neuroimmune regulation of brain edema could set the basis for new therapeutic options for the prevention and treatment of this feared complication of acute liver failure.
Jasmohan S. Bajaj, MD, AGAF, is professor in the division of gastroenterology, hepatology, and nutrition at Virginia Commonwealth University, Richmond. He reported no conflicts of interest.
Acute liver failure is a devastating disease, which has a high mortality burden and often requires liver transplant. One of the major complications is cerebral edema that leads to encephalopathy and could be fatal. These brain changes are accompanied by inflammation, immune activation, and hyperammonemia, but further mechanistic approaches are needed.
This data adds to the growing literature about the interaction between immune dysfunction and brain diseases such as schizophrenia, autism, depression, and multiple sclerosis. However, further studies in models of brain edema with concomitant liver failure, which are closer to the human disease process, are needed. This exciting investigation of neuroimmune regulation of brain edema could set the basis for new therapeutic options for the prevention and treatment of this feared complication of acute liver failure.
Jasmohan S. Bajaj, MD, AGAF, is professor in the division of gastroenterology, hepatology, and nutrition at Virginia Commonwealth University, Richmond. He reported no conflicts of interest.
A toll-like receptor 9 (TLR9) antagonist may eventually be used to combat brain edema in acute liver failure, according to investigators.
This prediction is based on results of a recent study involving mouse models, which showed that ODN2088, a TLR9 antagonist, could stop ammonia-induced colocalization of DNA with TLR9 in innate immune cells, thereby blocking cytokine production and ensuant brain edema, reported lead author Godhev Kumar Manakkat Vijay of King’s College London and colleagues.
“Ammonia plays a pivotal role in the development of hepatic encephalopathy and brain edema in acute liver failure,” the investigators explained in Cellular and Molecular Gastroenterology and Hepatology. “A robust systemic inflammatory response and susceptibility to developing infection are common in acute liver failure, exacerbate the development of ammonia-induced brain edema and are major prognosticators. Experimental models have unequivocally associated ammonia exposure with astrocyte swelling and brain edema, potentiated by proinflammatory cytokines.”
The investigators added that, “although the evidence base supporting the relationship between ammonia, inflammation, and brain edema is robust in acute liver failure, there is a paucity of data characterizing the specific pathogenic mechanisms entailed.” Previous research suggested that TLR9 plays a key role in acetaminophen-induced liver inflammation, they noted, and that ammonia, in combination with DNA, triggers TLR9 expression in neutrophils, which brought TLR9 into focus for the present study.
Along with wild-type mice, the investigators relied upon two knockout models: TLR9–/– mice, in which TLR9 is entirely absent, and LysM-Cre TLR9fl/fl mice, in which TLR9 is absent from lysozyme-expressing cells (predominantly neutrophils and macrophages). Comparing against controls, the investigators assessed cytokine production and brain edema in each type of mouse when intraperitoneally injected with ammonium acetate (4 mmol/kg). Specifically, 6 hours after injection, they measured intracellular cytokines in splenic macrophages, CD8+ T cells, and CD4+ T cells. In addition, they recorded total plasma DNA and brain water, a measure of brain edema.
Following ammonium acetate injection, wild-type mice developed brain edema and liver enlargement, while TLR9–/– mice and control-injected mice did not. After injection, total plasma DNA levels rose by comparable magnitudes in both wild-type mice and TLR9–/– mice, but did not change in control-injected mice, suggesting that ammonium-acetate injection was causing a release of DNA, which was binding with TLR9, resulting in activation of the innate immune system.
This hypothesis was supported by measurements of cytokines in T cells and splenic macrophages, which showed that wild-type mice had elevations of cytokines, whereas knockout mice did not. Further experiments showed that LysM-Cre TLR9fl/fl mice had similar outcomes as TLR9–/– mice, highlighting that macrophages and neutrophils are the key immune cells linking TLR9 activation with cytokine release, and therefore brain edema.
To ensure that brain edema was not directly caused by the acetate component of ammonium acetate, or acetate’s potential to increase pH, a different set of wild-type mice were injected with sodium acetate adjusted to the same pH as ammonium acetate. This had no impact on cytokine production, brain-water content, or liver-to-body weight ratio, confirming that acetate was not responsible for brain edema while providing further support for the role of TLR9.
Finally, the investigators treated wild-type mice immediately after ammonium acetate injection with the TLR9 antagonist ODN2088 (50 mcg/mouse). This treatment halted cytokine production, inflammation, and brain edema, strongly supporting the link between these ammonia-induced processes and TLR9 activation.
“These data are well supported by the findings of Imaeda et al. (J Clin Invest. 2009 Feb 2. doi: 10.1172/JCI35958), who in an acetaminophen-induced hepatotoxicity model established that inhibition of TLR9 using ODN2088 and IRS954, a TLR7/9 antagonist, down-regulated proinflammatory cytokine release and reduced mortality,” the investigators wrote. “The amelioration of brain edema and cytokine production by ODN2088 supports exploration of TLR9 antagonism as a therapeutic modality in early acute liver failure to prevent the development of brain edema and intracranial hypertension.”
The study was funded by the U.K. Institute of Liver Studies Charitable Fund and the National Institutes of Health. The investigators reported no conflicts of interest.
SOURCE: Vijay GKM et al. Cell Mol Gastroenterol Hepatol. 2019 Aug 8. doi: 10.1016/j.jcmgh.2019.08.002.
A toll-like receptor 9 (TLR9) antagonist may eventually be used to combat brain edema in acute liver failure, according to investigators.
This prediction is based on results of a recent study involving mouse models, which showed that ODN2088, a TLR9 antagonist, could stop ammonia-induced colocalization of DNA with TLR9 in innate immune cells, thereby blocking cytokine production and ensuant brain edema, reported lead author Godhev Kumar Manakkat Vijay of King’s College London and colleagues.
“Ammonia plays a pivotal role in the development of hepatic encephalopathy and brain edema in acute liver failure,” the investigators explained in Cellular and Molecular Gastroenterology and Hepatology. “A robust systemic inflammatory response and susceptibility to developing infection are common in acute liver failure, exacerbate the development of ammonia-induced brain edema and are major prognosticators. Experimental models have unequivocally associated ammonia exposure with astrocyte swelling and brain edema, potentiated by proinflammatory cytokines.”
The investigators added that, “although the evidence base supporting the relationship between ammonia, inflammation, and brain edema is robust in acute liver failure, there is a paucity of data characterizing the specific pathogenic mechanisms entailed.” Previous research suggested that TLR9 plays a key role in acetaminophen-induced liver inflammation, they noted, and that ammonia, in combination with DNA, triggers TLR9 expression in neutrophils, which brought TLR9 into focus for the present study.
Along with wild-type mice, the investigators relied upon two knockout models: TLR9–/– mice, in which TLR9 is entirely absent, and LysM-Cre TLR9fl/fl mice, in which TLR9 is absent from lysozyme-expressing cells (predominantly neutrophils and macrophages). Comparing against controls, the investigators assessed cytokine production and brain edema in each type of mouse when intraperitoneally injected with ammonium acetate (4 mmol/kg). Specifically, 6 hours after injection, they measured intracellular cytokines in splenic macrophages, CD8+ T cells, and CD4+ T cells. In addition, they recorded total plasma DNA and brain water, a measure of brain edema.
Following ammonium acetate injection, wild-type mice developed brain edema and liver enlargement, while TLR9–/– mice and control-injected mice did not. After injection, total plasma DNA levels rose by comparable magnitudes in both wild-type mice and TLR9–/– mice, but did not change in control-injected mice, suggesting that ammonium-acetate injection was causing a release of DNA, which was binding with TLR9, resulting in activation of the innate immune system.
This hypothesis was supported by measurements of cytokines in T cells and splenic macrophages, which showed that wild-type mice had elevations of cytokines, whereas knockout mice did not. Further experiments showed that LysM-Cre TLR9fl/fl mice had similar outcomes as TLR9–/– mice, highlighting that macrophages and neutrophils are the key immune cells linking TLR9 activation with cytokine release, and therefore brain edema.
To ensure that brain edema was not directly caused by the acetate component of ammonium acetate, or acetate’s potential to increase pH, a different set of wild-type mice were injected with sodium acetate adjusted to the same pH as ammonium acetate. This had no impact on cytokine production, brain-water content, or liver-to-body weight ratio, confirming that acetate was not responsible for brain edema while providing further support for the role of TLR9.
Finally, the investigators treated wild-type mice immediately after ammonium acetate injection with the TLR9 antagonist ODN2088 (50 mcg/mouse). This treatment halted cytokine production, inflammation, and brain edema, strongly supporting the link between these ammonia-induced processes and TLR9 activation.
“These data are well supported by the findings of Imaeda et al. (J Clin Invest. 2009 Feb 2. doi: 10.1172/JCI35958), who in an acetaminophen-induced hepatotoxicity model established that inhibition of TLR9 using ODN2088 and IRS954, a TLR7/9 antagonist, down-regulated proinflammatory cytokine release and reduced mortality,” the investigators wrote. “The amelioration of brain edema and cytokine production by ODN2088 supports exploration of TLR9 antagonism as a therapeutic modality in early acute liver failure to prevent the development of brain edema and intracranial hypertension.”
The study was funded by the U.K. Institute of Liver Studies Charitable Fund and the National Institutes of Health. The investigators reported no conflicts of interest.
SOURCE: Vijay GKM et al. Cell Mol Gastroenterol Hepatol. 2019 Aug 8. doi: 10.1016/j.jcmgh.2019.08.002.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
How to use type 2 diabetes meds to lower CV disease risk
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4

Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).

Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32

See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.

ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; Robert.gotfried@utoledo.edu.
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4
Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).
Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32
See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.
ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; Robert.gotfried@utoledo.edu.
The association between type 2 diabetes (T2D) and cardiovascular (CV) disease is well-established:
- Type 2 diabetes approximately doubles the risk of coronary artery disease, stroke, and peripheral arterial disease, independent of conventional risk factors1
- CV disease is the leading cause of morbidity and mortality in patients with T2D
- CV disease is the largest contributor to direct and indirect costs of the health care of patients who have T2D.2
In recent years, new classes of agents for treating T2D have been introduced (TABLE 1). Prior to 2008, the US Food and Drug Administration (FDA) approved drugs in those new classes based simply on their effectiveness in reducing the blood glucose level. Concerns about the CV safety of specific drugs (eg, rosiglitazone, muraglitazar) emerged from a number of trials, suggesting that these agents might increase the risk of CV events.3,4
Consequently, in 2008, the FDA issued guidance to the pharmaceutical industry: Preapproval and postapproval trials of all new antidiabetic drugs must now assess potential excess CV risk.5 CV outcomes trials (CVOTs), performed in accordance with FDA guidelines, have therefore become the focus of evaluating novel treatment options. In most CVOTs, combined primary CV endpoints have included CV mortality, nonfatal myocardial infarction (MI), and nonfatal stroke—taken together, what is known as the composite of these 3 major adverse CV events, or MACE-3.
To date, 15 CVOTs have been completed, assessing 3 novel classes of antihyperglycemic agents:
- dipeptidyl peptidase-4 (DPP-4) inhibitors
- glucagon-like peptide-1 (GLP-1) receptor agonists
- sodium–glucose cotransporter-2 (SGLT-2) inhibitors.
None of these trials identified any increased incidence of MACE; 7 found CV benefit. This review summarizes what the CVOTs revealed about these antihyperglycemic agents and their ability to yield a reduction in MACE and a decrease in all-cause mortality in patients with T2D and elevated CV disease risk. Armed with this information, you will have the tools you need to offer patients with T2D CV benefit while managing their primary disease.
Cardiovascular outcomes trials: DPP-4 inhibitors
Four trials. Trials of DPP-4 inhibitors that have been completed and reported are of saxagliptin (SAVOR-TIMI 536), alogliptin (EXAMINE7), sitagliptin (TECOS8), and linagliptin (CARMELINA9); others are in progress. In general, researchers enrolled patients at high risk of CV events, although inclusion criteria varied substantially. Consistently, these studies demonstrated that DPP-4 inhibition neither increased nor decreased (ie, were noninferior) the 3-point MACE (SAVOR-TIMI 53 noninferiority, P < .001; EXAMINE, P < .001; TECOS, P < .001).
Continue to: Rather than improve...
Rather than improve CV outcomes, there was some evidence that DPP-4 inhibitors might be associated with an increased risk of hospitalization for heart failure (HHF). In the SAVOR-TIMI 53 trial, patients randomized to saxagliptin had a 0.7% absolute increase in risk of HHF (P = .98).6 In the EXAMINE trial, patients treated with alogliptin showed a nonsignificant trend for HHF.10 In both the TECOS and CARMELINA trials, no difference was recorded in the rate of HHF.8,9,11 Subsequent meta-analysis that summarized the risk of HHF in CVOTs with DPP-4 inhibitors indicated a nonsignificant trend to increased risk.12
From these trials alone, it appears that DPP-4 inhibitors are unlikely to provide CV benefit. Data from additional trials are needed to evaluate the possible association between these medications and heart failure (HF). However, largely as a result of the findings from SAVOR-TIMI 53 and EXAMINE, the FDA issued a Drug Safety Communication in April 2016, adding warnings about HF to the labeling of saxagliptin and alogliptin.13
CARMELINA was designed to also evaluate kidney outcomes in patients with T2D. As with other DPP-4 inhibitor trials, the primary aim was to establish noninferiority, compared with placebo, for time to MACE-3 (P < .001). Secondary outcomes were defined as time to first occurrence of end-stage renal disease, death due to renal failure, and sustained decrease from baseline of ≥ 40% in the estimated glomerular filtration rate. The incidence of the secondary kidney composite results was not significantly different between groups randomized to linagliptin or placebo.9
Cardiovascular outcomes trials: GLP-1 receptor agonists
ELIXA. The CV safety of GLP-1 receptor agonists has been evaluated in several randomized clinical trials. The Evaluation of Lixisenatide in Acute Coronary Syndrome (ELIXA) trial was the first14: Lixisenatide was studied in 6068 patients with recent hospitalization for acute coronary syndrome. Lixisenatide therapy was neutral with regard to CV outcomes, which met the primary endpoint: noninferiority to placebo (P < .001). There was no increase in either HF or HHF.
Continue to: LEADER
LEADER. The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results trial (LEADER) evaluated long-term effects of liraglutide, compared to placebo, on CV events in patients with T2D.15 It was a multicenter, double-blind, placebocontrolled study that followed 9340 participants, most (81%) of whom had established CV disease, over 5 years. LEADER is considered a landmark study because it was the first large CVOT to show significant benefit for a GLP-1 receptor agonist.
Liraglutide demonstrated reductions in first occurrence of death from CV causes, nonfatal MI or nonfatal stroke, overall CV mortality, and all-cause mortality. The composite MACE-3 showed a relative risk reduction (RRR) of 13%, equivalent to an absolute risk reduction (ARR) of 1.9% (noninferiority, P < .001; superiority, P < .01). The RRR was 22% for death from CV causes, with an ARR of 1.3% (P = .007); the RRR for death from any cause was 15%, with an ARR of 1.4% (P = .02).
In addition, there was a lower rate of nephropathy (1.5 events for every 100 patient–years in the liraglutide group [P = .003], compared with 1.9 events every 100 patient–years in the placebo group).15
Results clearly demonstrated benefit. No significant difference was seen in the liraglutide rate of HHF, compared to the rate in the placebo group.
SUSTAIN-6. Evidence for the CV benefit of GLP-1 receptor agonists was also demonstrated in the phase 3 Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes (SUSTAIN-6).16 This was a study of 3297 patients with T2D at high risk of CV disease and with a mean hemoglobin A1c (HbA1c) value of 8.7%, 83% of whom had established CV disease. Patients were randomized to semaglutide or placebo. Note: SUSTAIN-6 was a noninferiority safety study; as such, it was not actually designed to assess or establish superiority.
Continue to: The incidence of MACE-3...
The incidence of MACE-3 was significantly reduced among patients treated with semaglutide (P = .02) after median followup of 2.1 years. The expanded composite outcome (death from CV causes, nonfatal MI, nonfatal stroke, coronary revascularization, or hospitalization for unstable angina or HF), also showed a significant reduction with semaglutide (P = .002), compared with placebo. There was no difference in the overall hospitalization rate or rate of death from any cause.
EXSCEL. The Exenatide Study of Cardiovascular Event Lowering trial (EXSCEL)17,18 was a phase III/IV, double-blind, pragmatic placebo-controlled study of 14,752 patients at any level of CV risk, for a median 3.2 years. The study population was intentionally more diverse than in earlier GLP-1 receptor agonist studies. The researchers hypothesized that patients at increased risk of MACE would experience a comparatively greater relative treatment benefit with exenatide than those at lower risk. That did not prove to be the case.
EXSCEL did confirm noninferiority compared with placebo (P < .001), but once-weekly exenatide resulted in a nonsignificant reduction in major adverse CV events, and a trend for RRR in all-cause mortality (RRR = 14%; ARR = 1% [P = .06]).
HARMONY OUTCOMES. The Albiglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes and Cardiovascular Disease study (HARMONY OUTCOMES)19 was a double-blind, randomized, placebocontrolled trial conducted at 610 sites across 28 countries. The study investigated albiglutide, 30 to 50 mg once weekly, compared with placebo. It included 9463 patients ages ≥ 40 years with T2D who had an HbA1c > 7% (median value, 8.7%) and established CV disease. Patients were evaluated for a median 1.6 years.
Albiglutide reduced the risk of CV causes of death, nonfatal MI, and nonfatal stroke by an RRR of 22%, (ARR, 2%) (noninferiority, P < .0001; superiority, P < .0006).
Continue to: REWIND
REWIND. The Researching Cardiovascular Events with a Weekly INcretin in Diabetes trial (REWIND),20 the most recently completed GLP-1 receptor agonist CVOT (presented at the 2019 American Diabetes Association [ADA] Conference in June and published simultaneously in The Lancet), was a multicenter, randomized, double-blind placebo-controlled trial designed to assess the effect of weekly dulaglutide, 1.5 mg, compared with placebo, in 9901 participants enrolled at 371 sites in 24 countries. Mean patient age was 66.2 years, with women constituting 4589 (46.3%) of participants.
REWIND was distinct from other CVOTs in several ways:
- Other CVOTs were designed to show noninferiority compared with placebo regarding CV events; REWIND was designed to establish superiority
- In contrast to trials of other GLP-1 receptor agonists, in which most patients had established CV disease, only 31% of REWIND participants had a history of CV disease or a prior CV event (although 69% did have CV risk factors without underlying disease)
- REWIND was much longer (median follow-up, 5.4 years) than other GLP-1 receptor agonist trials (median follow-up, 1.5 to 3.8 years).
In REWIND, the primary composite outcome of MACE-3 occurred in 12% of participants assigned to dulaglutide, compared with 13.1% assigned to placebo (P = .026). This equated to 2.4 events for every 100 person– years on dulaglutide, compared with 2.7 events for every 100 person–years on placebo. There was a consistent effect on all MACE-3 components, although the greatest reductions were observed in nonfatal stroke (P = .017). Overall risk reduction was the same for primary and secondary prevention cohorts (P = .97), as well as in patients with either an HbA1c value < 7.2% or ≥ 7.2% (P = .75). Risk reduction was consistent across age, sex, duration of T2D, and body mass index.
Dulaglutide did not significantly affect the incidence of all-cause mortality, heart failure, revascularization, or hospital admission. Forty-seven percent of patients taking dulaglutide reported gastrointestinal adverse effects (P = .0001).
In a separate analysis of secondary outcomes, 21 dulaglutide reduced the composite renal outcomes of new-onset macroalbuminuria (P = .0001); decline of ≥ 30% in the estimated glomerular filtration rate (P = .066); and chronic renal replacement therapy (P = .39). Investigators estimated that 1 composite renal outcome event would be prevented for every 31 patients treated with dulaglutide for a median 5.4 years.
Continue to: Cardiovascular outcomes trials...
Cardiovascular outcomes trials: SGLT-2 inhibitors
EMPA-REG OUTCOME. The Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes trial (EMPA-REG OUTCOME) was also a landmark study because it was the first dedicated CVOT to show that an antihyperglycemic agent 1) decreased CV mortality and all-cause mortality, and 2) reduced HHF in patients with T2D and established CV disease.22 In this trial, 7020 patients with T2D who were at high risk of CV events were randomized and treated with empagliflozin, 10 or 25 mg, or placebo, in addition to standard care, and were followed for a median 2.6 years.
Compared with placebo, empagliflozin resulted in an RRR of 14% (ARR, 1.6%) in the primary endpoint of CV death, nonfatal MI, and stroke, confirming study drug superiority (P = .04). When compared with placebo, the empagliflozin group had an RRR of 38% in CV mortality, (ARR < 2.2%) (P < .001); an RRR of 35% in HHF (ARR, 1.4%) (P = .002); and an RRR of 32% (ARR, 2.6%) in death from any cause (P < .001).
CANVAS. The Canagliflozin Cardiovascular Assessment Study (CANVAS) integrated 2 multicenter, placebo-controlled, randomized trials with 10,142 participants and a mean follow-up of 3.6 years.23 Patients were randomized to receive canagliflozin (100-300 mg/d) or placebo. Approximately two-thirds of patients had a history of CV disease (therefore representing secondary prevention); one-third had CV risk factors only (primary prevention).
In CANVAS, patients receiving canagliflozin had a risk reduction in MACE-3, establishing superiority compared with placebo (P < .001). There was also a significant reduction in progression of albuminuria (P < .05). Superiority was not shown for the secondary outcome of death from any cause. Canagliflozin had no effect on the primary endpoint (MACE-3) in the subgroup of participants who did not have a history of CV disease. Similar to what was found with empagliflozin in EMPA-REG OUTCOME, CANVAS participants had a reduced risk of HHF.
Continue to: Patients on canagliflozin...
Patients on canagliflozin unexpectedly had an increased incidence of amputations (6.3 participants, compared with 3.4 participants, for every 1000 patient–years). This finding led to a black box warning for canagliflozin about the risk of lower-limb amputation.
DECLARE-TIMI 58. The Dapagliflozin Effect of Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 trial (DECLARETIMI 58) was the largest SGLT-2 inhibitor outcomes trial to date, enrolling 17,160 patients with T2D who also had established CV disease or multiple risk factors for atherosclerotic CV disease. The trial compared dapagliflozin, 10 mg/d, and placebo, following patients for a median 4.2 years.24 Unlike CANVAS and EMPA-REG OUTCOME, DECLARE-TIMI 58 included CV death and HHF as primary outcomes, in addition to MACE-3.
Dapagliflozin was noninferior to placebo with regard to MACE-3. However, its use did result in a lower rate of CV death and HHF by an RRR of 17% (ARR, 1.9%). Risk reduction was greatest in patients with HF who had a reduced ejection fraction (ARR = 9.2%).25
In October, the FDA approved dapagliflozin to reduce the risk of HHF in adults with T2D and established CV disease or multiple CV risk factors. Before initiating the drug, physicians should evaluate the patient's renal function and monitor periodically.
Meta-analyses of SGLT-2 inhibitors
Systematic review. Usman et al released a meta-analysis in 2018 that included 35 randomized, placebo-controlled trials (including EMPA-REG OUTCOME, CANVAS, and DECLARE-TIMI 58) that had assessed the use of SGLT-2 inhibitors in nearly 35,000 patients with T2D.26 This review concluded that, as a class, SGLT-2 inhibitors reduce all-cause mortality, major adverse cardiac events, nonfatal MI, and HF and HHF, compared with placebo.
Continue to: CVD-REAL
CVD-REAL. A separate study, Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors (CVD-REAL), of 154,528 patients who were treated with canagliflozin, dapagliflozin, or empagliflozin, showed that initiation of SGLT-2 inhibitors, compared with other glucose- lowering therapies, was associated with a 39% reduction in HHF; a 51% reduction in death from any cause; and a 46% reduction in the composite of HHF or death (P < .001).27
CVD-REAL was unique because it was the largest real-world study to assess the effectiveness of SGLT-2 inhibitors on HHF and mortality. The study utilized data from patients in the United States, Norway, Denmark, Sweden, Germany, and the United Kingdom, based on information obtained from medical claims, primary care and hospital records, and national registries that compared patients who were either newly started on an SGLT-2 inhibitor or another glucose-lowering drug. The drug used by most patients in the trial was canagliflozin (53%), followed by dapagliflozin (42%), and empagliflozin (5%).
In this meta-analysis, similar therapeutic effects were seen across countries, regardless of geographic differences, in the use of specific SGLT-2 inhibitors, suggesting a class effect. Of particular significance was that most (87%) patients enrolled in CVD-REAL did not have prior CV disease. Despite this, results for examined outcomes in CVD-REAL were similar to what was seen in other SGLT-2 inhibitor trials that were designed to study patients with established CV disease.
Risk of adverse effects of newer antidiabetic agents
DPP-4 inhibitors. Alogliptin and sitagliptin carry a black-box warning about potential risk of HF. In SAVOR-TIMI, a 27% increase was detected in the rate of HHF after approximately 2 years of saxagliptin therapy.6 Although HF should not be considered a class effect for DPP-4 inhibitors, patients who have risk factors for HF should be monitored for signs and symptoms of HF.
Continue to: Cases of acute pancreatitis...
Cases of acute pancreatitis have been reported in association with all DPP-4 inhibitors available in the United States. A combined analysis of DDP-4 inhibitor trials suggested an increased relative risk of 79% and an absolute risk of 0.13%, which translates to 1 or 2 additional cases of acute pancreatitis for every 1000 patients treated for 2 years.28
There have been numerous postmarketing reports of severe joint pain in patients taking a DPP-4 inhibitor. Most recently, cases of bullous pemphigoid have been reported after initiation of DPP-4 inhibitor therapy.29
GLP-1 receptor agonists carry a black box warning for medullary thyroid (C-cell) tumor risk. GLP-1 receptor agonists are contraindicated in patients with a personal or family history of this cancer, although this FDA warning is based solely on observations from animal models.
In addition, GLP-1 receptor agonists can increase the risk of cholecystitis and pancreatitis. Not uncommonly, they cause gastrointestinal symptoms when first started and when the dosage is titrated upward. Most GLP-1 receptor agonists can be used in patients with renal impairment, although data regarding their use in Stages 4 and 5 chronic kidney disease are limited.30 Semaglutide was found, in the SUSTAIN-6 trial, to be associated with an increased rate of complications of retinopathy, including vitreous hemorrhage and blindness (P = .02)31
SGLT-2 inhibitors are associated with an increased incidence of genitourinary infection, bone fracture (canagliflozin), amputation (canagliflozin), and euglycemic diabetic ketoacidosis. Agents in this class should be avoided in patients with moderate or severe renal impairment, primarily due to a lack of efficacy. They are contraindicated in patients with an estimated glomerular filtration rate (eGFR) < 30 mL/min/1.73 m2. (Dapagliflozin is not recommended when eGFR is < 45 mL/min/ 1.73 m2.) These agents carry an FDA warning about the risk of acute kidney injury.30
Continue to: Summing up
Summing up
All glucose-lowering medications used to treat T2D are not equally effective in reducing CV complications. Recent CVOTs have uncovered evidence that certain antidiabetic agents might confer CV and all-cause mortality benefits (TABLE 26,7,9,11,14-17,19-24).
Discussion of proposed mechanisms for CV outcome superiority of these agents is beyond the scope of this review. It is generally believed that benefits result from mechanisms other than a reduction in the serum glucose level, given the relatively short time frame of the studies and the magnitude of the CV benefit. It is almost certain that mechanisms of CV benefit in the 2 landmark studies—LEADER and EMPA-REG OUTCOME—are distinct from each other.32
See “When planning T2D pharmacotherapy, include newer agents that offer CV benefit,” 33-38 for a stepwise approach to treating T2D, including the role of agents that have efficacy in modifying the risk of CV disease.
SIDEBAR
When planning T2D pharmacotherapy, include newer agents that offer CV benefit33-38
First-line management. The 2019 Standards of Medical Care in Diabetes Guidelines established by the American Diabetes Association (ADA) recommend metformin as first-line pharmacotherapy for type 2 diabetes (T2D).33 This recommendation is based on metformin’s efficacy in reducing the blood glucose level and hemoglobin A1C (HbA1C); safety; tolerability; extensive clinical experience; and findings from the UK Prospective Diabetes Study demonstrating a substantial beneficial effect of metformin on cardiovascular (CV) disease.34 Additional benefits of metformin include a decrease in body weight, low-density lipoprotein level, and the need for insulin.
Second-line additive benefit. In addition, ADA guidelines make a highest level (Level-A) recommendation that patients with T2D and established atherosclerotic CV disease be treated with one of the sodium–glucose cotransporter-2 (SGLT-2) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists that have demonstrated efficacy in CV disease risk reduction as part of an antihyperglycemic regimen.35 Seven agents described in this article from these 2 unique classes of medications meet the CV disease benefit criterion: liraglutide, semaglutide, albiglutide, dulaglutide, empagliflozin, canagliflozin, and dapagliflozin. Only empagliflozin and liraglutide have received a US Food and Drug Administration indication for risk reduction in major CV events in adults with T2D and established CV disease.
Regarding dulaglutide, although the findings of REWIND are encouraging, results were not robust; further analysis is necessary to make a recommendation for treating patients who do not have a history of established CV disease with this medication.
Individualized decision-making. From a clinical perspective, patient-specific considerations and shared decision-making should be incorporated into T2D treatment decisions:
- For patients with T2D and established atherosclerotic CV disease, SGLT-2 inhibitors and GLP-1 receptor agonists are recommended agents after metformin.
- SGLT-2 inhibitors are preferred in T2D patients with established CV disease and a history of heart failure.
- GLP-1 receptor agonists with proven CV disease benefit are preferred in patients with established CV disease and chronic kidney disease.
Add-on Tx. In ADA guidelines, dipeptidyl peptidase-4 (DDP-4) inhibitors are recommended as an optional add-on for patients without clinical atherosclerotic CV disease who are unable to reach their HbA1C goal after taking metformin for 3 months.33 Furthermore, the American Association of Clinical Endocrinologists lists DPP-4 inhibitors as alternatives for patients with an HbA1C < 7.5% in whom metformin is contraindicated.36 DPP-4 inhibitors are not an ideal choice as a second agent when the patient has a history of heart failure, and should not be recommended over GLP-1 receptor agonists or SGLT-2 inhibitors as second-line agents in patients with T2D and CV disease.
Individualizing management. The current algorithm for T2D management,37 based primarily on HbA1C reduction, is shifting toward concurrent attention to reduction of CV risk (FIGURE38). Our challenge, as physicians, is to translate the results of recent CV outcomes trials into a more targeted management strategy that focuses on eligible populations.
ACKNOWLEDGMENTS
Linda Speer, MD, Kevin Phelps, DO, and Jay Shubrook, DO, provided support and editorial assistance.
CORRESPONDENCE
Robert Gotfried, DO, FAAFP, Department of Family Medicine, University of Toledo College of Medicine, 3333 Glendale Avenue, Toledo, OH 43614; Robert.gotfried@utoledo.edu.
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
1. Emerging Risk Factors Collaboration; Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215-2222.
2. Chamberlain JJ, Johnson EL, Leal S, et al. Cardiovascular disease and risk management: review of the American Diabetes Association Standards of Medical Care in Diabetes 2018. Ann Intern Med. 2018;168:640-650.
3. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581-2586.
4. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-2471.
5. Center for Drug Evaluation and Research, US Food and Drug Administration. Guidance document: Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/drugs/guidance
complianceregulatoryinformation/guidances/ucm071627.pdf. Published December 2008. Accessed October 4, 2019.
6. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patient with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
7. White WB, Canon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327-1335.
8. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
9. Rosenstock J, Perkovic V, Johansen OE, et al; CARMELINA Investigators. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA. 2019;321:69-79.
10. Zannad F, Cannon CP, Cushman WC, et al. EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
11. McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes with Sitagliptin Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2016;1:126-135.
12. Toh S, Hampp C, Reichman ME, et al. Risk for hospitalized heart failure among new users of saxagliptin, sitagliptin, and other antihyperglycemic drugs: a retrospective cohort study. Ann Intern Med. 2016;164:705-714.
13. US Food and Drug Administration. FDA drug safety communication: FDA adds warning about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. www.fda.gov/Drugs/DrugSafety/ucm486096.htm. Updated April 5, 2016. Accessed October 4, 2019.
14. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patient with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247-2257.
15. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
16. Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834-1844.
17. Mentz RJ, Bethel MA, Merrill P, et al; EXSCEL Study Group. Effect of once-weekly exenatide on clinical outcomes according to baseline risk in patients with type 2 diabetes mellitus: insights from the EXSCEL Trial. J Am Heart Assoc. 2018;7:e009304.
18. Holman RR, Bethel MA, George J, et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am Heart J. 2016;174:103-110.
19. Hernandez AF, Green JB, Janmohamed S, et al; Harmony Outcomes committees and investigators. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. 2018;392:1519-1529.
20. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121-130.
21. Gerstein HC, Colhoun HM, Dagenais GR, et al; REWIND Investigators. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomized, placebo-controlled trial. Lancet. 2019;394:131-138.
22. Zinman B, Wanner C, Lachin JM, et al; EMPA-REG OUTCOME Investigators. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
23. Neal B, Perkovic V, Mahaffey KW, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657.
24. Wiviott SD, Raz I, Bonaca MP, et al; DECLARE–TIMI 58 Investigators. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347-357.
25. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536.
26. Usman MS, Siddiqi TJ, Memon MM, et al. Sodium-glucose cotransporter 2 inhibitors and cardiovascular outcomes: a systematic review and meta-analysis. Eur J Prev Cardiol. 2018;25:495-502.
27. Kosiborod M, Cavender MA, Fu AZ, et al; CVD-REAL Investigators and Study Group. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation. 2017;136:249-259.
28. Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284-286.
29. Schaffer C, Buclin T, Jornayvaz FR, et al. Use of dipeptidyl-peptidase IV inhibitors and bullous pemphigoid. Dermatology. 2017;233:401-403.
30. Madievsky R. Spotlight on antidiabetic agents with cardiovascular or renoprotective benefits. Perm J. 2018;22:18-034.
31. Vilsbøll T, Bain SC, Leiter LA, et al. Semaglutide, reduction in glycated hemoglobin and the risk of diabetic retinopathy. Diabetes Obes Metab. 2018;20:889-897.
32. Kosiborod M. Following the LEADER–why this and other recent trials signal a major paradigm shift in the management of type 2 diabetes. J Diabetes Complications. 2017;31:517-519.
33. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S90-S102.
34. Holman R. Metformin as first choice in oral diabetes treatment: the UKPDS experience. Journ Annu Diabetol Hotel Dieu. 2007:13-20.
35. American Diabetes Association. 10. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2019. Diabetes Care. 2019;42(Suppl 1):S103-S123.
36. Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2018 executive summary. Endocr Pract. 2018;24:91-120.
37. Inzucci SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140-149.
38. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669-2701.
PRACTICE RECOMMENDATIONS
› Consider American Diabetes Association (ADA) guidance and prescribe a sodium–glucose cotransporter-2 (SGLT-2) inhibitor or glucagon-like peptide- 1 (GLP-1) receptor agonist that has demonstrated cardiovascular (CV) disease benefit for your patients who have type 2 diabetes (T2D) and established atherosclerotic CV disease. A
› Consider ADA’s recommendation for preferred therapy and prescribe an SGLT-2 inhibitor for your patients with T2D who have atherosclerotic CV disease and are at high risk of heart failure or in whom heart failure coexists. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Ketoacidosis is on the rise in children with type 1 diabetes
BARCELONA – As many as 40%-60% of children have diabetic ketoacidosis (DKA) at the time of being diagnosed with type 1 diabetes, according to data from two U.S. analyses – and the figures have been rising for the past 10 years.
Between 2010 and 2017, the prevalence of DKA at diagnosis in children who were followed up at the Barbara Davies Cancer Center in Denver (n = 2,429) went from 41% to 59%, with a 7% annual rise, Arleta Rewers, MD, PhD, of Children’s Hospital Colorado, Denver, reported at the annual meeting of the European Association for the Study of Diabetes.
Meanwhile, in another analysis that included multiple U.S. centers and about 7,600 cases of youth-onset type 1 diabetes, the overall prevalence of DKA at diagnosis was 38.5% between 2010 and 2016. However, the prevalence had increased from 35% in 2010 to 40.6% in 2016, according to Elizabeth T. Jensen, MPH, PhD, of Wake Forest University, Winston-Salem, N.C. The annual increase in prevalence of DKA at diagnosis of type 1 disease was 2%, adjusted for sociodemographic factors.
Rising prevalence
“DKA occurs most commonly at the time of type 1 diabetes diagnosis,” observed Dr. Jensen, who noted that “in the United States, among children, it’s younger children, uninsured or underinsured children, and children from minority racial or ethnic groups, who are at greatest risk.”

Dr. Jensen and colleagues had previously shown that the prevalence of DKA at diagnosis was around 30% between 2002 and 2010, with no significant change in its prevalence. However, more recent reports from referral-based, single-center studies had suggested there was an increase, and that led her and her colleagues to take a closer look at the data.
To characterize the risk factors for DKA and the prevalence of DKA over time, Dr. Jensen and her team used the SEARCH for Diabetes in Youth database, which, she said, was “uniquely suited” for this purpose. SEARCH is a population-based, multicenter study conducted in centers in five U.S. states: South Carolina, Ohio, Colorado, California, and Washington.
A diagnosis of DKA was based on blood bicarbonate levels of less than 15 mmol/L, a venous pH of less than 7.25 or arterial or capillary pH of less than 7.3, or if there was any documentation of a DKA diagnosis.
As expected, the prevalence of DKA was highest in the youngest age group (0-4 years), Dr. Jensen said, but the increase in prevalence in that group was no different from the increases seen over time in the other age groups (5-9 years, 10-14 years, and 15 years or older).
There were no differences in the prevalence of DKA between the sexes, although there was a general increase over time. Similar trends were seen in DKA prevalence by race or ethnicity and by season, or time of year.
Of note, higher rates of DKA were seen in children who were covered by public health insurance, than in those covered by private insurance, although there was no difference in the rate of increase in DKA prevalence between the two groups. Dr. Jensen noted that only 64% of this study population had private insurance.
She said that future research in this area would need to look at the economic drivers and the “changing landscape of health insurance coverage in the United States.”
Expansion in health coverage
In presenting the findings of a study showing an increase in the prevalence of DKA at diagnosis of type 1 diabetes in children in Colorado from 2010 to 2017, Dr. Rewers said that the increase “paradoxically occurred” at a time of increasing health insurance coverage, a reference to the expansion of Medicaid during 2008-2012 and implementation in 2013 of the Affordable Care Act.
“Our group in Colorado has followed the frequency of DKA for almost 2 decades,” Dr. Rewers said. It’s important to study DKA as it is linked to worse glycemic control – with children with DKA having an HbA1c level of around 1% higher than those without DKA – and the potential for future, long-term complications.
Dr. Rewers noted that the increase in DKA at diagnosis of type 1 diabetes was more rapid in the children who had private rather than public health insurance. Of 1,187 patients with DKA, 57% had private health insurance, and 37% had public insurance, compared with 66% and 28%, respectively, in those without DKA. In 2010, the prevalence of DKA at diagnosis was 35.3% in those who were privately insured and 52.2% of those with public health insurance, but by 2017, a similar percentage of DKA was seen in the privately and publicly insured children (59.6% and 58.5%, respectively).
She said one possible explanation for that might be that “increased enrollment in high-deductible insurance plans could discourage families with private insurance from seeking timely care.”
Another explanation is that there is a low awareness of type 1 diabetes in the general population, she added. “Educational campaigns and autoimmunity screening have been shown to reduce DKA at diabetes diagnosis, but unfortunately they are not used widely at this point.”
Identifying at-risk children
“Diabetic ketoacidosis is a serious complication of diabetes [and] is difficult to diagnose because of the variability of the symptoms, said Angela Ibald-Mulli, PhD, who presented the findings of a retrospective cohort study in which she and her colleagues used a “discovery algorithm” called Q-Finder to identify the predictive factors for DKA in youth with type 1 diabetes, based on data from the Diabetes Prospective Follow-up Registry (DPV).

“The better we know the risk factors, the better we can care for our patients,” she emphasized.
The investigators obtained data on 108,223 patients with a diagnosis of type 1 disease and with more than two visits related to diabetes. The prevalence of DKA – defined as a pH of less than 7.3 during hospitalization occurring at least 10 days after the onset of type 1 diabetes – was 5.2%, said Dr. Ibald-Mulli, head of Medical Evidence Generation Primary Care at Sanofi, Paris.
A total of 129 different features were considered for their association with DKA – including comorbidities, sociodemographic factors, laboratory values, and concomitant medications – and were then used to identify, test, and the validate likely risk profiles.
After comparing the characteristics of patients with and without DKA, eight significant factors, all of which have been reported previously in the DPV cohort, were seen: younger age, lower body weight, higher HbA1c, younger age at onset of T1D; shorter disease duration; having a migration background; being less active; and having had more medical visits.
The investigators used the algorithm, and found 11 distinct profiles associated with DKA: an HbA1c higher than 8.87%; being aged 6-10 years; being aged 11-15 years; a diagnosis of nephropathy; DKA being present at onset; a prevalence of hypoglycemia with coma; a diagnosis of thyroiditis; a standardized body mass index lower than 16.9; not using short-acting insulin; younger than age 15 years; and not using continuous glucose monitoring.
Almost two-thirds of patients (64.7%) belonged to at least one of these risk profiles, Dr. Ibald-Mulli observed, with 7.1% of them having DKA, compared with 1.6% who belonged to none of the profiles.
Dr. Ibald-Mulli said it was important to note that the DKA risk profiles could overlap. “The more profiles a patient belongs to, the higher is the risk of having DKA,” she emphasized, adding that most patients (88.8%) with DKA belonged to just one profile, and fewer than 5% belonged to three or more profiles.
“Overall, the results of the algorithm confirmed known risk-factor profiles that had been previously identified by conventional statistical methods,” she concluded. It also provided “additional insights that can be further explored.”
SEARCH is funded by the Centers for Disease and Prevention and the National Institute of Diabetes and Digestive and Kidney Diseases. The DPV Registry is funded by multiple sponsors, including the European Federation for the Study of Diabetes and other academic institutions with the support of several commercial partners. Sanofi sponsored the study presented by Dr. Ibald-Mulli. Dr. Rewers made no disclosures, and Dr. Jensen did not have any conflicts of interest to declare. Dr. Ibald-Mulli is an employee of Sanofi.
SOURCE: Rewers A et al. EASD 2019, Abstract 115; Jensen E et al. EASD 2019, Abstract 116; Ibald-Mulli A et al. EASD 2019, Abstract 117.
BARCELONA – As many as 40%-60% of children have diabetic ketoacidosis (DKA) at the time of being diagnosed with type 1 diabetes, according to data from two U.S. analyses – and the figures have been rising for the past 10 years.
Between 2010 and 2017, the prevalence of DKA at diagnosis in children who were followed up at the Barbara Davies Cancer Center in Denver (n = 2,429) went from 41% to 59%, with a 7% annual rise, Arleta Rewers, MD, PhD, of Children’s Hospital Colorado, Denver, reported at the annual meeting of the European Association for the Study of Diabetes.
Meanwhile, in another analysis that included multiple U.S. centers and about 7,600 cases of youth-onset type 1 diabetes, the overall prevalence of DKA at diagnosis was 38.5% between 2010 and 2016. However, the prevalence had increased from 35% in 2010 to 40.6% in 2016, according to Elizabeth T. Jensen, MPH, PhD, of Wake Forest University, Winston-Salem, N.C. The annual increase in prevalence of DKA at diagnosis of type 1 disease was 2%, adjusted for sociodemographic factors.
Rising prevalence
“DKA occurs most commonly at the time of type 1 diabetes diagnosis,” observed Dr. Jensen, who noted that “in the United States, among children, it’s younger children, uninsured or underinsured children, and children from minority racial or ethnic groups, who are at greatest risk.”
Dr. Jensen and colleagues had previously shown that the prevalence of DKA at diagnosis was around 30% between 2002 and 2010, with no significant change in its prevalence. However, more recent reports from referral-based, single-center studies had suggested there was an increase, and that led her and her colleagues to take a closer look at the data.
To characterize the risk factors for DKA and the prevalence of DKA over time, Dr. Jensen and her team used the SEARCH for Diabetes in Youth database, which, she said, was “uniquely suited” for this purpose. SEARCH is a population-based, multicenter study conducted in centers in five U.S. states: South Carolina, Ohio, Colorado, California, and Washington.
A diagnosis of DKA was based on blood bicarbonate levels of less than 15 mmol/L, a venous pH of less than 7.25 or arterial or capillary pH of less than 7.3, or if there was any documentation of a DKA diagnosis.
As expected, the prevalence of DKA was highest in the youngest age group (0-4 years), Dr. Jensen said, but the increase in prevalence in that group was no different from the increases seen over time in the other age groups (5-9 years, 10-14 years, and 15 years or older).
There were no differences in the prevalence of DKA between the sexes, although there was a general increase over time. Similar trends were seen in DKA prevalence by race or ethnicity and by season, or time of year.
Of note, higher rates of DKA were seen in children who were covered by public health insurance, than in those covered by private insurance, although there was no difference in the rate of increase in DKA prevalence between the two groups. Dr. Jensen noted that only 64% of this study population had private insurance.
She said that future research in this area would need to look at the economic drivers and the “changing landscape of health insurance coverage in the United States.”
Expansion in health coverage
In presenting the findings of a study showing an increase in the prevalence of DKA at diagnosis of type 1 diabetes in children in Colorado from 2010 to 2017, Dr. Rewers said that the increase “paradoxically occurred” at a time of increasing health insurance coverage, a reference to the expansion of Medicaid during 2008-2012 and implementation in 2013 of the Affordable Care Act.
“Our group in Colorado has followed the frequency of DKA for almost 2 decades,” Dr. Rewers said. It’s important to study DKA as it is linked to worse glycemic control – with children with DKA having an HbA1c level of around 1% higher than those without DKA – and the potential for future, long-term complications.
Dr. Rewers noted that the increase in DKA at diagnosis of type 1 diabetes was more rapid in the children who had private rather than public health insurance. Of 1,187 patients with DKA, 57% had private health insurance, and 37% had public insurance, compared with 66% and 28%, respectively, in those without DKA. In 2010, the prevalence of DKA at diagnosis was 35.3% in those who were privately insured and 52.2% of those with public health insurance, but by 2017, a similar percentage of DKA was seen in the privately and publicly insured children (59.6% and 58.5%, respectively).
She said one possible explanation for that might be that “increased enrollment in high-deductible insurance plans could discourage families with private insurance from seeking timely care.”
Another explanation is that there is a low awareness of type 1 diabetes in the general population, she added. “Educational campaigns and autoimmunity screening have been shown to reduce DKA at diabetes diagnosis, but unfortunately they are not used widely at this point.”
Identifying at-risk children
“Diabetic ketoacidosis is a serious complication of diabetes [and] is difficult to diagnose because of the variability of the symptoms, said Angela Ibald-Mulli, PhD, who presented the findings of a retrospective cohort study in which she and her colleagues used a “discovery algorithm” called Q-Finder to identify the predictive factors for DKA in youth with type 1 diabetes, based on data from the Diabetes Prospective Follow-up Registry (DPV).
“The better we know the risk factors, the better we can care for our patients,” she emphasized.
The investigators obtained data on 108,223 patients with a diagnosis of type 1 disease and with more than two visits related to diabetes. The prevalence of DKA – defined as a pH of less than 7.3 during hospitalization occurring at least 10 days after the onset of type 1 diabetes – was 5.2%, said Dr. Ibald-Mulli, head of Medical Evidence Generation Primary Care at Sanofi, Paris.
A total of 129 different features were considered for their association with DKA – including comorbidities, sociodemographic factors, laboratory values, and concomitant medications – and were then used to identify, test, and the validate likely risk profiles.
After comparing the characteristics of patients with and without DKA, eight significant factors, all of which have been reported previously in the DPV cohort, were seen: younger age, lower body weight, higher HbA1c, younger age at onset of T1D; shorter disease duration; having a migration background; being less active; and having had more medical visits.
The investigators used the algorithm, and found 11 distinct profiles associated with DKA: an HbA1c higher than 8.87%; being aged 6-10 years; being aged 11-15 years; a diagnosis of nephropathy; DKA being present at onset; a prevalence of hypoglycemia with coma; a diagnosis of thyroiditis; a standardized body mass index lower than 16.9; not using short-acting insulin; younger than age 15 years; and not using continuous glucose monitoring.
Almost two-thirds of patients (64.7%) belonged to at least one of these risk profiles, Dr. Ibald-Mulli observed, with 7.1% of them having DKA, compared with 1.6% who belonged to none of the profiles.
Dr. Ibald-Mulli said it was important to note that the DKA risk profiles could overlap. “The more profiles a patient belongs to, the higher is the risk of having DKA,” she emphasized, adding that most patients (88.8%) with DKA belonged to just one profile, and fewer than 5% belonged to three or more profiles.
“Overall, the results of the algorithm confirmed known risk-factor profiles that had been previously identified by conventional statistical methods,” she concluded. It also provided “additional insights that can be further explored.”
SEARCH is funded by the Centers for Disease and Prevention and the National Institute of Diabetes and Digestive and Kidney Diseases. The DPV Registry is funded by multiple sponsors, including the European Federation for the Study of Diabetes and other academic institutions with the support of several commercial partners. Sanofi sponsored the study presented by Dr. Ibald-Mulli. Dr. Rewers made no disclosures, and Dr. Jensen did not have any conflicts of interest to declare. Dr. Ibald-Mulli is an employee of Sanofi.
SOURCE: Rewers A et al. EASD 2019, Abstract 115; Jensen E et al. EASD 2019, Abstract 116; Ibald-Mulli A et al. EASD 2019, Abstract 117.
BARCELONA – As many as 40%-60% of children have diabetic ketoacidosis (DKA) at the time of being diagnosed with type 1 diabetes, according to data from two U.S. analyses – and the figures have been rising for the past 10 years.
Between 2010 and 2017, the prevalence of DKA at diagnosis in children who were followed up at the Barbara Davies Cancer Center in Denver (n = 2,429) went from 41% to 59%, with a 7% annual rise, Arleta Rewers, MD, PhD, of Children’s Hospital Colorado, Denver, reported at the annual meeting of the European Association for the Study of Diabetes.
Meanwhile, in another analysis that included multiple U.S. centers and about 7,600 cases of youth-onset type 1 diabetes, the overall prevalence of DKA at diagnosis was 38.5% between 2010 and 2016. However, the prevalence had increased from 35% in 2010 to 40.6% in 2016, according to Elizabeth T. Jensen, MPH, PhD, of Wake Forest University, Winston-Salem, N.C. The annual increase in prevalence of DKA at diagnosis of type 1 disease was 2%, adjusted for sociodemographic factors.
Rising prevalence
“DKA occurs most commonly at the time of type 1 diabetes diagnosis,” observed Dr. Jensen, who noted that “in the United States, among children, it’s younger children, uninsured or underinsured children, and children from minority racial or ethnic groups, who are at greatest risk.”
Dr. Jensen and colleagues had previously shown that the prevalence of DKA at diagnosis was around 30% between 2002 and 2010, with no significant change in its prevalence. However, more recent reports from referral-based, single-center studies had suggested there was an increase, and that led her and her colleagues to take a closer look at the data.
To characterize the risk factors for DKA and the prevalence of DKA over time, Dr. Jensen and her team used the SEARCH for Diabetes in Youth database, which, she said, was “uniquely suited” for this purpose. SEARCH is a population-based, multicenter study conducted in centers in five U.S. states: South Carolina, Ohio, Colorado, California, and Washington.
A diagnosis of DKA was based on blood bicarbonate levels of less than 15 mmol/L, a venous pH of less than 7.25 or arterial or capillary pH of less than 7.3, or if there was any documentation of a DKA diagnosis.
As expected, the prevalence of DKA was highest in the youngest age group (0-4 years), Dr. Jensen said, but the increase in prevalence in that group was no different from the increases seen over time in the other age groups (5-9 years, 10-14 years, and 15 years or older).
There were no differences in the prevalence of DKA between the sexes, although there was a general increase over time. Similar trends were seen in DKA prevalence by race or ethnicity and by season, or time of year.
Of note, higher rates of DKA were seen in children who were covered by public health insurance, than in those covered by private insurance, although there was no difference in the rate of increase in DKA prevalence between the two groups. Dr. Jensen noted that only 64% of this study population had private insurance.
She said that future research in this area would need to look at the economic drivers and the “changing landscape of health insurance coverage in the United States.”
Expansion in health coverage
In presenting the findings of a study showing an increase in the prevalence of DKA at diagnosis of type 1 diabetes in children in Colorado from 2010 to 2017, Dr. Rewers said that the increase “paradoxically occurred” at a time of increasing health insurance coverage, a reference to the expansion of Medicaid during 2008-2012 and implementation in 2013 of the Affordable Care Act.
“Our group in Colorado has followed the frequency of DKA for almost 2 decades,” Dr. Rewers said. It’s important to study DKA as it is linked to worse glycemic control – with children with DKA having an HbA1c level of around 1% higher than those without DKA – and the potential for future, long-term complications.
Dr. Rewers noted that the increase in DKA at diagnosis of type 1 diabetes was more rapid in the children who had private rather than public health insurance. Of 1,187 patients with DKA, 57% had private health insurance, and 37% had public insurance, compared with 66% and 28%, respectively, in those without DKA. In 2010, the prevalence of DKA at diagnosis was 35.3% in those who were privately insured and 52.2% of those with public health insurance, but by 2017, a similar percentage of DKA was seen in the privately and publicly insured children (59.6% and 58.5%, respectively).
She said one possible explanation for that might be that “increased enrollment in high-deductible insurance plans could discourage families with private insurance from seeking timely care.”
Another explanation is that there is a low awareness of type 1 diabetes in the general population, she added. “Educational campaigns and autoimmunity screening have been shown to reduce DKA at diabetes diagnosis, but unfortunately they are not used widely at this point.”
Identifying at-risk children
“Diabetic ketoacidosis is a serious complication of diabetes [and] is difficult to diagnose because of the variability of the symptoms, said Angela Ibald-Mulli, PhD, who presented the findings of a retrospective cohort study in which she and her colleagues used a “discovery algorithm” called Q-Finder to identify the predictive factors for DKA in youth with type 1 diabetes, based on data from the Diabetes Prospective Follow-up Registry (DPV).
“The better we know the risk factors, the better we can care for our patients,” she emphasized.
The investigators obtained data on 108,223 patients with a diagnosis of type 1 disease and with more than two visits related to diabetes. The prevalence of DKA – defined as a pH of less than 7.3 during hospitalization occurring at least 10 days after the onset of type 1 diabetes – was 5.2%, said Dr. Ibald-Mulli, head of Medical Evidence Generation Primary Care at Sanofi, Paris.
A total of 129 different features were considered for their association with DKA – including comorbidities, sociodemographic factors, laboratory values, and concomitant medications – and were then used to identify, test, and the validate likely risk profiles.
After comparing the characteristics of patients with and without DKA, eight significant factors, all of which have been reported previously in the DPV cohort, were seen: younger age, lower body weight, higher HbA1c, younger age at onset of T1D; shorter disease duration; having a migration background; being less active; and having had more medical visits.
The investigators used the algorithm, and found 11 distinct profiles associated with DKA: an HbA1c higher than 8.87%; being aged 6-10 years; being aged 11-15 years; a diagnosis of nephropathy; DKA being present at onset; a prevalence of hypoglycemia with coma; a diagnosis of thyroiditis; a standardized body mass index lower than 16.9; not using short-acting insulin; younger than age 15 years; and not using continuous glucose monitoring.
Almost two-thirds of patients (64.7%) belonged to at least one of these risk profiles, Dr. Ibald-Mulli observed, with 7.1% of them having DKA, compared with 1.6% who belonged to none of the profiles.
Dr. Ibald-Mulli said it was important to note that the DKA risk profiles could overlap. “The more profiles a patient belongs to, the higher is the risk of having DKA,” she emphasized, adding that most patients (88.8%) with DKA belonged to just one profile, and fewer than 5% belonged to three or more profiles.
“Overall, the results of the algorithm confirmed known risk-factor profiles that had been previously identified by conventional statistical methods,” she concluded. It also provided “additional insights that can be further explored.”
SEARCH is funded by the Centers for Disease and Prevention and the National Institute of Diabetes and Digestive and Kidney Diseases. The DPV Registry is funded by multiple sponsors, including the European Federation for the Study of Diabetes and other academic institutions with the support of several commercial partners. Sanofi sponsored the study presented by Dr. Ibald-Mulli. Dr. Rewers made no disclosures, and Dr. Jensen did not have any conflicts of interest to declare. Dr. Ibald-Mulli is an employee of Sanofi.
SOURCE: Rewers A et al. EASD 2019, Abstract 115; Jensen E et al. EASD 2019, Abstract 116; Ibald-Mulli A et al. EASD 2019, Abstract 117.
REPORTING FROM EASD 2019
Lipoprotein(a) Elevation: A New Diagnostic Code with Relevance to Service Members and Veterans (FULL)
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
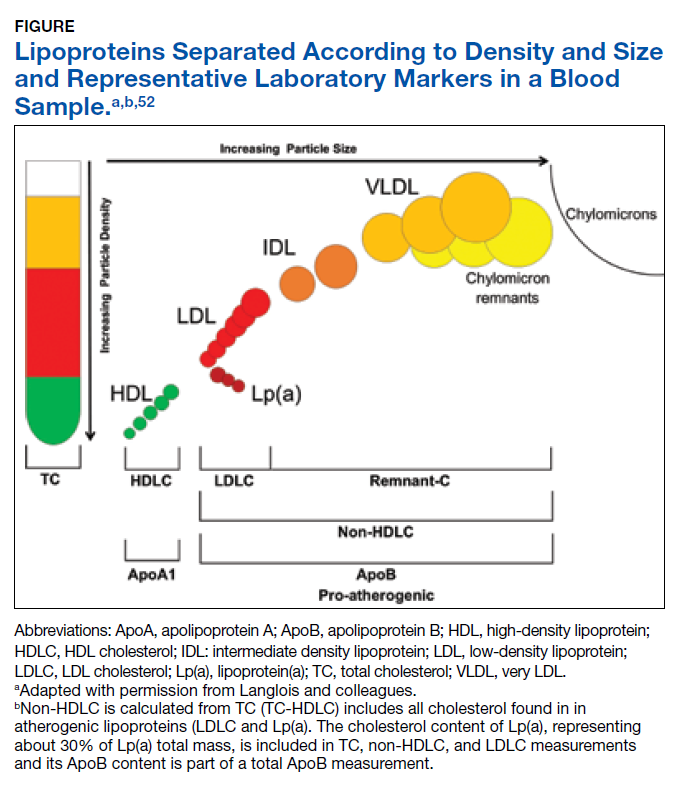
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
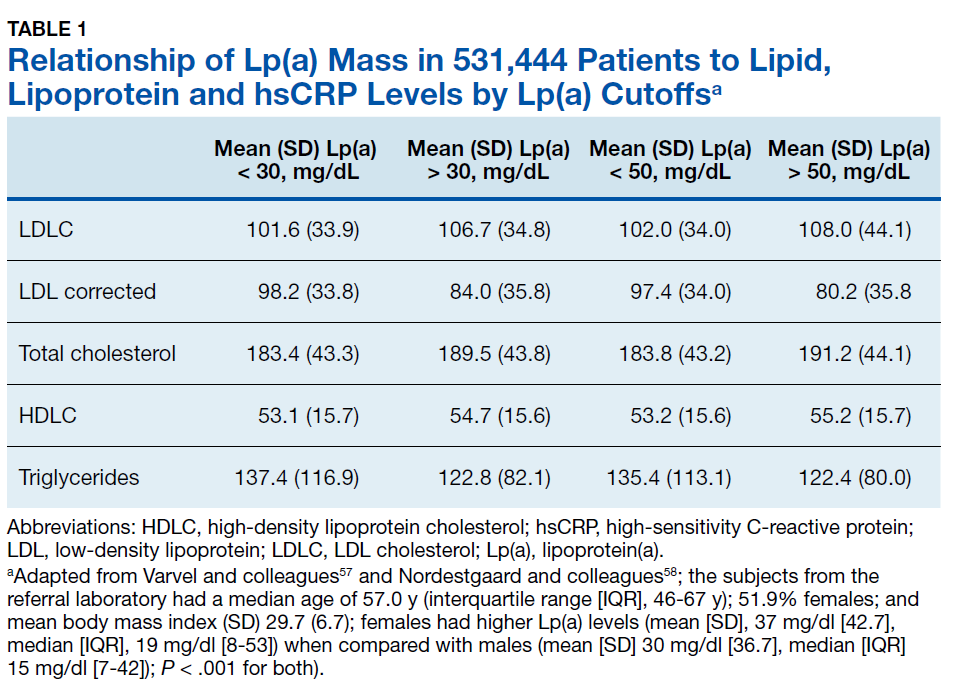
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
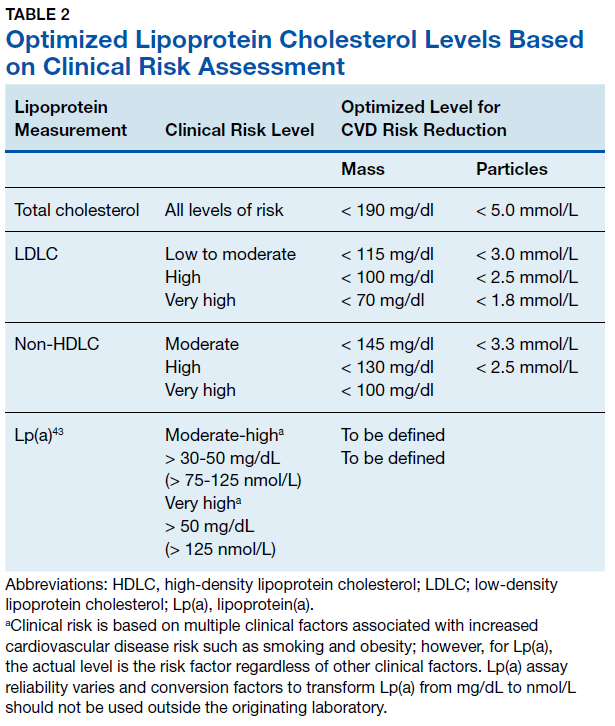
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
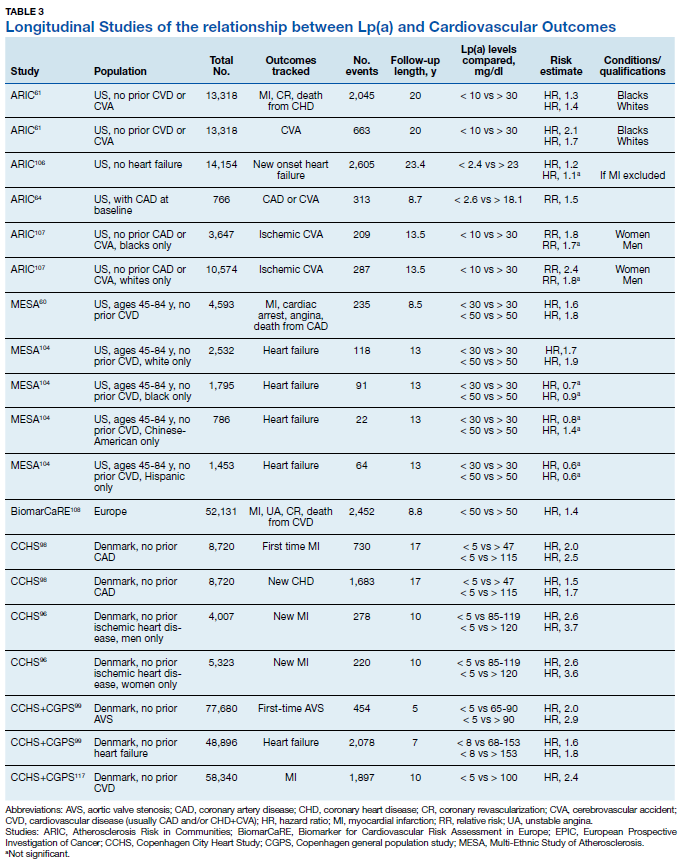
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
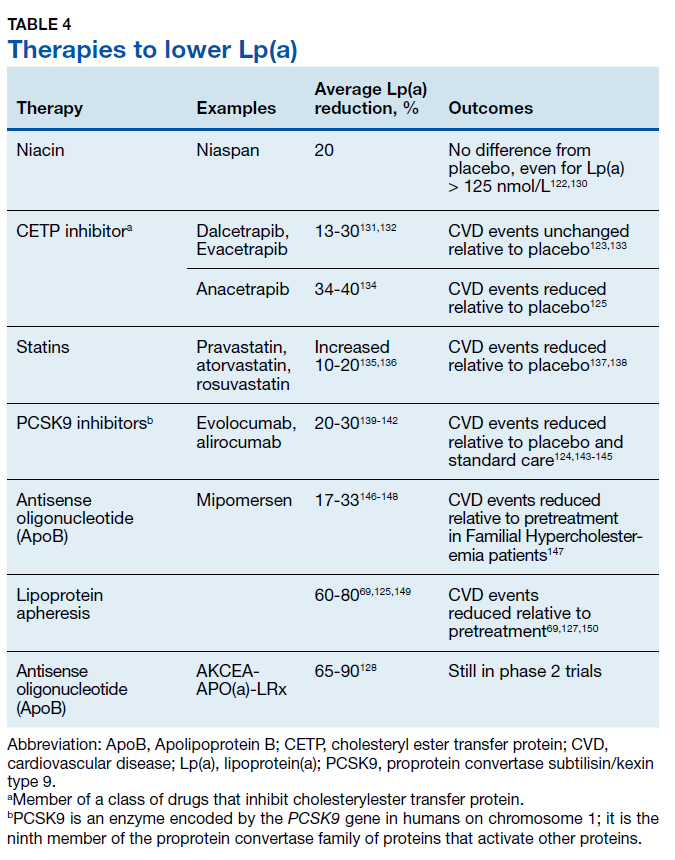
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
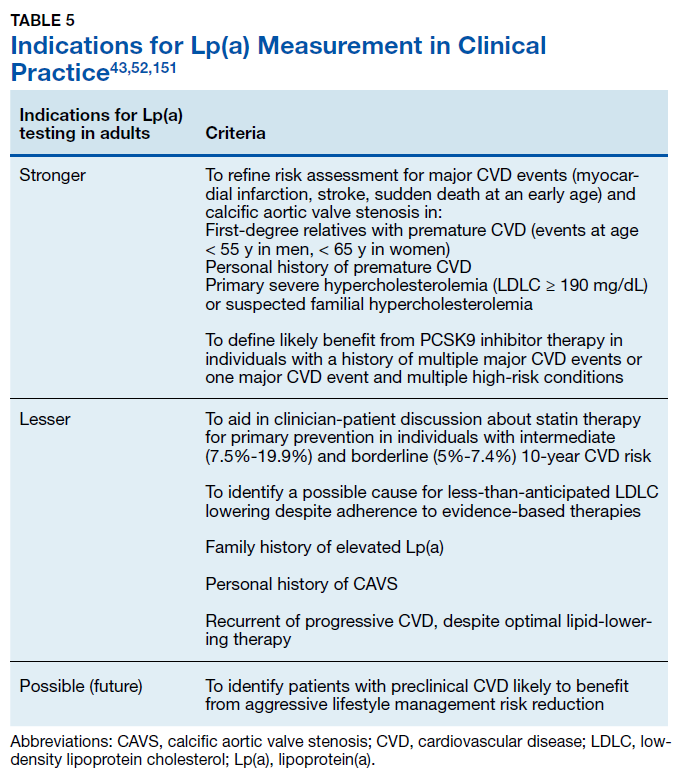
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
Cardiovascular disease (CVD) remains the leading cause of global mortality. In 2015, 41.5% of the US population had at least 1 form of CVD and CVD accounted for nearly 18 million deaths worldwide.1,2 The major disease categories represented include myocardial infarction (MI), sudden death, strokes, calcific aortic valve stenosis (CAVS), and peripheral vascular disease.1,2 In terms of health care costs, quality of life, and caregiver burden, the overall impact of disease prevalence continues to rise.1,3-6 There is an urgent need for more precise and earlier CVD risk assessment to guide lifestyle and therapeutic interventions for prevention of disease progression as well as potential reversal of preclinical disease. Even at a young age, visible coronary atherosclerosis has been found in up to 11% of “healthy” active individuals during autopsies for trauma fatalities.7,8
The impact of CVD on the US and global populations is profound. In 2011, CVD prevalence was predicted to reach 40% by 2030.9 That estimate was exceeded in 2015, and it is now predicted that by 2035, 45% of the US population will suffer from some form of clinical or preclinical CVD. In 2015, the decadeslong decline in CVD mortality was reversed for the first time since 1969, showing a 1% increase in deaths from CVD.1 Nearly 300,000 of those using US Department of Veterans Affairs (VA) services were hospitalized for CVD between 2010 and 2014.10 The annual direct and indirect costs related to CVD in the US are estimated at $329.7 billion, and these costs are predicted to top $1 trillion by 2035.1 Heart attack, coronary atherosclerosis, and stroke accounted for 3 of the 10 most expensive conditions treated in US hospitals in 2013.11 Globally, the estimate for CVD-related direct and indirect costs was $863 billion in 2010 and may exceed $1 trillion by 2030.12
The nature of military service adds additional risk factors, such as posttraumatic stress disorder, depression, sleep disorders and physical trauma which increase CVD morbidity/ mortality in service members, veterans, and their families.13-16 In addition, living in lowerincome areas (countries or neighborhoods) can increase the risk of both CVD incidence and fatalities, particularly in younger individuals.17-20 The Military Health System (MHS) and VA are responsible for the care of those individuals who have voluntarily taken on these additional risks through their time in service. This responsibility calls for rapid translation to practice tools and resources that can support interventions to minimize as many modifiable risk factors as possible and improve longterm health. This strategy aligns with the World Health Organization’s (WHO) focus on prevention of disease progression through interventions targeting modifiable risk.3-6,21-23 The driving force behind the launch of the US Department of Health and Human Services (HHS) Million Hearts program was the goal of preventing 1 million heart attacks and strokes by 2017 with risk reduction through aspirin, blood pressure control, cholesterol management, smoking cessation, sodium reduction, and physical activity.24,25 While some reductions in CVD events have been documented, the outcomes fell short of the goals set, highlighting both the need and value of continued and expanded efforts for CVD risk reduction.26
More precise assessment of risk factors during preventative care, as well as after a diagnosis of CVD, may improve the timeliness and precision of earlier interventions (both lifestyle and therapeutic) that reduce CVD morbidity and mortality.27 Personalized or precision medicine approaches take into account differences in socioeconomic, environmental, and lifestyle factors that are potentially reversible, as well as gender, race, and ethnicity.28-31 Current methods of predicting CVD risk have considerable room for improvement.27 About 40% of patients with newly diagnosed CVD have normal traditional cholesterol profiles, including those whose first cardiac event proves fatal.29-33 Currently available risk scores (hundreds have been described in the literature) mischaracterize risk in minority populations and women, and have shown deficiencies in identifying preclinical atherosclerosis.34,35 The failure to recognize preclinical CVD in military personnel during their active duty life cycle results in missed opportunities for improved health and readiness sustainment.
Most CVD risk prediction models incorporate some form of blood lipids. Total cholesterol (TC) is most commonly used in clinical practice, along with high-density lipoprotein (HDLC), low-density lipoprotein (LDLC), and triglycerides (TG).23,27,36 High LDLC and/or TC are well established as lipid-related CVD risk factors and are incorporated into many CVD risk scoring systems/models described in the literature.27 LDLC reduction is commonly recommended as CVD prevention, but even with optimal statin treatment, there is still considerable residual risk for new and recurrent CVD events.28,32,34,35,37-42
Incorporating novel biomarkers and alternative lipid measurements may improve risk prediction and aid targeted treatment, ultimately reducing CVD events.27 Apolipoprotein B (ApoB) is a major atherogenic component embedded in LDL and VLDL correlating to non-HDLC and may be useful in the setting of triglycerides ≥ 200 mg/d as levels > 130 mg/ dL appear to be risk-enhancing, but measurements may be unreliable.43 According to the 2018 Cholesterol Guidelines, lipoprotein(a) [Lp(a)] elevation also is recognized as a risk-enhancing factor that is particularly implicated when there is a strong family history of premature atherosclerotic CVD or personal history of CVD not explained by major risk factors.43
Lp(a) elevation is a largely underrecognized category of lipid disorder that impacts up to 20% to 30% of the population globally and within the US, although there is considerable variability by geographic location and ethnicity.44 Globally, Lp(a) elevation places > 1 billion people at moderate to high risk for CVD.44 Lp(a) has a strong genetic component and is recognized as a distinct and independent risk factor for MI, sudden death, strokes and CAVS. Lp(a) has an extensive body of evidence to support its distinct role both as a causal factor in CVD and as an augmentation to traditional risk factors.44-48
Lipoproteni(a) Elevation Use For Diagnosis
The importance of Lp(a) elevation as a clinical diagnosis rather than a laboratory abnormality alone was brought forward by the Lipoprotein(a) Foundation. Its founder, Sandra Tremulis, is a survivor of an acute coronary event that occurred when she was 39-years old, despite running marathons and having none of the traditional CVD lifestyle risk factors.49 This experience inspired her to create the Lipoprotein(a) Foundation to give a voice to families living with or at risk for CVD due to Lp(a) elevation.
As often happens in the progress of medicine, patients and their families drive change based on their personal experiences with the gaps in standard clinical practice. It was this foundation—not a member of the medical establishment—that submitted the formal request for the addition of new ICD-10-CM diagnostic and family history codes for Lp(a) elevation during the Centers for Disease Control and Prevention (CDC) September 2017 ICD-10-CM Coordination and Maintenance Committee meeting.50 In June 2018, the final ICD-10-CM code addenda for 2019 was released and included the new codes E78.41 (Elevated Lp[a]) and Z83.430 (Family history of elevated Lp[a]).52 After the new codes were approved, both the American Heart Association and the National Lipid Association added recommendations regarding Lp(a) testing to their clinical practice guidelines.43,52
Practically, these codes standardize billing and payment for legitimate clinical work and laboratory testing. Prior to the addition of Lp(a) elevation as a clinical diagnosis, testing and treatment of Lp(a) elevation was considered experimental and not medically necessary until after a cardiovascular event had already occurred. Services for Lp(a) elevation were therefore not reimbursed by many healthcare organizations and insurance companies. The new ICD-10-CM codes encourage the assessment of Lp(a) both in individuals with early onset major CVD events and in presumably fit, healthy individuals, particularly when there is a family history of Lp(a) elevation. Given that Lp(a) levels do not change significantly over time, the current understanding is that only a single measurement is needed to define the individual risk over a lifetime.41,42,44,45 As therapies targeting Lp(a) levels evolve, repeated measurements may be indicated to monitor response and direct changes in management. “Elevated Lipoprotein(a)” is the first laboratory testing abnormality that has achieved the status of a clinical diagnosis.
Lp(a) Measurements
There is considerable complexity to the measurement of lipoproteins in blood samples due to heterogeneity in both density and size of particles as illustrated in the Figure.53
For traditional lipids measured in clinical practice, the size and density ranges from small high-density lipoprotein (HDL) through LDLC and intermediate- density lipoprotein (IDL) to the largest least dense particles in the very low-density lipoprotein (VLDL) and chylomicron remnant fractions. Standard lipid profiles consist of mass concentration measurements (mg/dL) of TC, TG, HDLC, and LDLC.53 Non-HDLC (calculated as: TC−HDLC) consists of all cholesterol found in atherogenic lipoproteins, including remnant-C and Lp(a). Until recently, the cholesterol content of Lp(a), corresponding to about 30% of Lp(a) total mass, was included in the TC, non-HDLC and LDLC measurements with no separate reporting by the majority of clinical laboratories.
After > 50 years of research on the structure and biochemistry of Lp(a), the physiology and biological functions of these complex and polymorphic lipoprotein particles are not fully understood. Lp(a) is composed of a lipoprotein particle similar in composition to LDL (protein and lipid), containing 1 molecule of ApoB wrapped around a core of cholesteryl ester and triglyceride with phospholipids and unesterified cholesterol at its surface.48 The presence of a unique hydrophilic, highly glycosylated protein referred to as apolopoprotienA (apo[a]), covalently attached to ApoB-100 by a single disulfide bridge, differentiates Lp(a) from LDL.48 Cholesterol rich ApoB is an important component within many lipoproteins pathogenic for atherosclerosis and CVD.45,47,53
The apo(a) contributes to the increased density of Lp(a) compared to LDLC with associated reduced binding affinity to the LDL receptor. This reduced receptor binding affinity is a presumed mechanism for the lack of Lp(a) plasma level response to statin therapies, which increase hepatic LDL receptor activity.47 Apo(a) evolved from the plasminogen gene through duplication and remodeling and demonstrates extensive heterogeneity in protein size, with > 40 different apo(a) isoforms resulting in > 40 different Lp(a) particle sizes. Size of the apo(a) particle is determined by the number of pleated structures known as kringles. Most people (> 80%) carry 2 different-sized apo(a) isoforms. Plasma Lp(a) level is determined by the net production of apo(a) in each isoform, and the smaller apo(a) isoforms are associated with higher plasma levels of Lp(a).45
Given the heterogeneity in Lp(a) molecular weight, which can vary even within individuals, recommendations have been made for reporting results as particle numbers or concentrations (nmol/L or mmol/L) rather than as mass concentration (mg/dL).55 However, the majority of the large CVD morbidity and mortality outcomes studies used Lp(a) mass concentration levels in mg/ dL to characterize risk levels.56,57 There is no standardized method to convert Lp(a) measurements from mg/dL to nmol/L.55 Current assays using WHO standardized reagents and controls are reliable for categorizing risk levels.58
The European Atherosclerosis Society consensus panel recommended that desirable Lp(a) levels should be below the 80th percentile (< 50 mg/dL or < 125 nmol/L) in patients with intermediate or high CVD risk.59 Subsequent epidemiological and Mendelian randomization studies have been performed in general populations with no history of CVD and demonstrated that increased CVD risk can be detected with Lp(a) levels as low as 25 to 30 mg/dL.56,60-63 In secondary prevention populations with prior CVD and optimal treatment (statins, antiplatelet drugs), recurrent event risk was also increased with elevated Lp(a).63-66
Using immunoturbidometric assays, Varvel and colleagues reported the prevalence of elevated Lp(a) mass concentration levels (mg/dL) in > 500,000 US patients undergoing clinical evaluations based on data from a referral laboratory of patients.58 The mean Lp(a) levels were 34.0 mg/dL with median (interquartile range [IQR]) levels at 17 (7-47) mg/dL and overall range of 0 to 907 mg/dL.58 Females had higher Lp(a) levels compared to males but no ethnic or racial breakdown was provided. Lp(a) levels > 30 mg/dL and > 50 mg/dL were present in 35% and 24% of subjects, respectively. Table 1 displays the relationship between various Lp(a) level cut-offs to mean levels of LDLC, estimated LDLC corrected for Lp(a), TC, HDLC, and TG.58 The data demonstrate that Lp(a) elevation cannot be inferred from LDLC levels nor from any of the other traditional lipoprotein measures. Patients with high risk Lp(a) levels may have normal LDLC. While Lp(a) thresholds have been identified for stratification of CVD risk, the target levels for risk reduction have not been specifically defined, particularly since therapies are not widely available for reduction of Lp(a). Table 2 provides an overview of clinical lipoprotein measurements that may be reasonable targets for therapeutic interventions and reduction of CVD risk.44,53,55 In general, existing studies suggest that radical reduction (> 80%) is required to impact long-term outcomes, particularly in individuals with severe disease.68,69
LDLC reduction alone leaves a residual CVD risk that is greater than the risk reduced.40 In addition, the autoimmune inflammation and lipid specific autoantibodies play an important role in increased CVD morbidity and mortality risk.70,71 The presence of autoantibodies such as antiphospholipid antibodies (without a specific autoimmune disease diagnosis) increases the risk of subclinical atherosclerosis.72,73 Certain autoimmune diseases such as systemic lupus erythematosus are recognized as independent risk factors for CVD.74,75 Autoantibodies appear to mediate CVD events and mortality risk, independent of traditional therapies for risk reduction.73 Further research is needed to clarify the role of autoantibodies as markers of increased or decreased CVD risk and their mechanism of action.
Autoantibodies directed at new antigens in lipoproteins within atherosclerotic lesions can modulate the impact of atherosclerosis via activation of the innate and adaptive immune system.76 The lipid-associated neopeptides are recognized as damage-associated or danger- associated molecular patterns (DAMPs), also known as alarmins, which signal molecules that can trigger and perpetuate noninfectious inflammatory responses.77-79 Plasma autoantibodies (immunoglobulin M and G [IgM, IgG]) modify proinflammatory oxidation-specific epitopes on oxidized phospholipids (oxPL) within lipoproteins and are linked with markers of inflammation and CVD events.80-82 Modified LDLC and ApoB-100 immune complexes with specific autoantibodies in the IgG class are associated with increased CVD.76 These and other risk-modulating autoantibodies may explain some of the variability in CVD outcomes by ethnicity and between individuals.
Some antibodies to oxidized LDL (ox-LDL) may have a protective role in the development of atherosclerosis.83,84 In a cohort of > 500 women, the number of carotid atherosclerotic plaques and total carotid plaque area were inversely correlated with a specific IgM autoantibody (MDA-p210).84 High concentrations of Lp(a)- containing circulating immune complexes and Lp(a)-specific IgM and IgG have been described in patients with coronary heart disease (CHD).85 Like ox-LDL, oxidized Lp(a) [ox-Lp(a)] is more potent than native Lp(a) in increasing atherosclerosis risk and is increased in patients with CHD compared to healthy controls.86-88 Ox-Lp(a) levels may represent an even stronger risk marker for CVD than ox-LDL.85
Possible Mechanisms of Pathogenesis
While the precise quantification of Lp(a) in human plasma (or serum) has been challenging, current clinical laboratories use standardized international reference reagents and controls in their assays. Most current Lp(a) assays are based on immunological methods (eg, immunonephelometry, immunoturbidimetry, or enzyme linked immunosorbent assay [ELISA]) using antibodies against apo(a).89 Apo(a) contains 10 subtypes of kringle IV and 1 copy of kringle V. Some assays use antibodies against kringle-IV type 2; however, it has been recommended that newer methods should use antibodies against the specific bridging kringle-IV Type 9 domain, which has a more stable bond and is present as a single copy.48,89 Other approaches to Lp(a) measurement include ultraperformance liquid chromatography/mass spectrometry that can determine both the concentration and particle size of apo(a).48,90 For routine clinical care, currently available assays reporting in mg/dL can be considered fairly accurate for separating low-risk from moderate-to-high-risk patients.45
The physiologic role of Lp(a) in humans remains to be fully defined and individuals with extremely low plasma Lp(a) levels present no disease or deficiency syndromes.91 Lp(a) accumulates in endothelial injuries and binds to components of the vessel wall and subendothelial matrix, presumably due to the strong lysine binding site in apo(a).46 Mediated by apo(a), the binding stimulates chemotactic activation of monocytes/macrophages and thereby modulating angiogenesis and inflammation.89 Lp(a) may contribute to CVD and CAVS via its LDL-like component, with proinflammatory effects of oxidized phospholipids (OxPL) on both ApoB and apo(a) and antifibrinolytic/prothrombotic effects of apo(a).92 In Vitro studies have demonstrated that apo(a) modifies cellular function of cultured vascular endothelial cells (promoting stress fiber formation, endothelial contraction and vascular permeability), smooth muscles, and monocytes/ macrophages (promoting differentiation of proinflammatory M1-1 type macrophages) via complex mechanisms of cell signaling and cytokine production.89 Lp(a) is the only monogenetic risk factor for aortic valve calcification and stenosis93 and is strongly linked specifically with the single nucleotide polymorphism (SNP) rs10455872 in the gene LPA encoding for apo(a).94
CVD Risk Predictive Value
There are a large number of studies demonstrating that Lp(a) elevations are an independent predictor of adverse cardiovascular outcomes including MI, sudden death, strokes, calcific aortic valve stenosis and peripheral vascular disease (Table 3). The Copenhagen City Heart Study and Copenhagen General Population Study are well known prospective population- based cohort studies that track outcomes through national patient registries.95 These studies demonstrate increased risk for MI, CHD, CAVS, and heart failure when subjects with very high Lp(a) levels (50-115 mg/dL) are compared with subjects with very low Lp(a) levels (< 5 mg/dL).96-100 Subjects with less extreme Lp(a) elevations (> 30 mg/dL) also show increased risk of CVD when they have comorbid LDLC elevations.101 However, the Copenhagen studies are composed exclusively of white subjects and the effects of Lp(a) are known to vary with race or ethnicity.
The Multi-Ethnic Study of Atherosclerosis (MESA) recruited an ethnically diverse sample of > 6,000 Americans, aged 45 to 84 years, without CVD, into an ongoing prospective cohort study. Research using subjects from this study has found consistently increased risk of CHD, heart failure, subclinical aortic valve calcification, and more severe CAVS in white subjects with elevated Lp(a).60,102,103 Black subjects with elevated Lp(a) had increased risk of CHD and more severe CAVS and Hispanic subjects with Lp(a) elevation were at higher risk for CHD.60,102 So far, no studies of MESA subjects have identified a relationship between Lp(a) elevation and CVD events for Asian-Americans subjects (predominantly of Chinese descent). There is a need for ongoing research to more precisely define relevant cut-off levels by race, ethnicity and sex.
The Atherosclerosis Risk in Communities (ARIC) Study was a prospective multiethnic cohort study including > 15,000 US adults, aged 45 to 64 years.103 Lp(a) elevations in this cohort were associated with greater risks for first CVD events, heart failure, and recurrent CVD events.61,64,105 The risk of stroke for subjects with elevated Lp(a) was greater for black and white women, and for black men.61,106 However, a meta-analysis of case-control studies showed increased ischemic stroke risk in both men and women with elevated Lp(a).57
A recent European meta-analysis collected blood samples and outcome data from > 50,000 subjects in 7 prospective cohort studies. Using a central laboratory to standardize Lp(a) measurements, researchers found increased risk of major coronary events and new CVD in subjects with Lp(a) > 50 mg/dL compared to those below that threshold.107
Although many of these studies show modest increases in risk of CVD events with Lp(a) elevation, it should be noted that other studies do not demonstrate such consistent associations. This is particularly true in studies of women and nonwhite ethnic groups.103,108-112 The variability of study results may be due to other confounding factors such as autoantibodies that either upregulate or downregulate atherogenicity of LDLC and potentially other lipoproteins. This is particularly relevant to women who have an increased risk for autoimmune disease.
Lp(a) has significant genetic heritability—75% in Europeans and 85% in African Americans.113 In whites, the LPA gene on chromosome 6p26- 27 with the polymorphism genetic variants rs10455872 and rs3798220 is consistently associated with elevated Lp(a) levels.63,100,113 However, the degree of Lp(a) elevation associated with these specific genetic variants varies by ethnicity.78,113,115
Lifestyle and Cardiovascular Health
It is noteworthy that the Lp(a) genetic risks can also be modified by lifestyle risk reduction even in the absence of significant blood level reductions. For example, Khera and colleagues constructed a genetic risk profile for CVD that included genes related to Lp(a).116 Subjects with high genetic risk were more likely to experience CVD events compared with subjects with low genetic risk. However, risks for CVD were attenuated by 4 healthy lifestyle factors: current nonsmoker, body mass index < 30, at least weekly physical activity, and a healthy diet. Subjects with high genetic risk and an unhealthy lifestyle (0 or 1 of the 4 healthy lifestyle factors) were the most likely to develop CVD (Hazard ratio [HR], 3.5), but that risk was lower for subjects with healthy (3 or 4 of the 4 healthy lifestyle factors) and intermediate lifestyles (2 of the 4 healthy lifestyle factors) (HR, 1.9 and 2.2, respectively), despite despite high genetic risk for CVD.
While the independent CVD risk associated with elevated Lp(a) does not appear to be responsive to lifestyle risk reduction alone, certainly elevated LDLC and traditional risk factors can increase the overall CVD risk and are worthy of preventive interventions. In particular, inflammation from any source exacerbates CVD risk. Proatherogenic diet, insufficient sleep, lack of exercise, and maladaptive stress responses are other targets for personalized CVD risk reduction. 28,117 Studies of dietary modifications and other lifestyle factors have shown reduced risk of CVD events, despite lack of reduction in Lp(a) levels.119,120 It is noteworthy that statin therapy (with or without ezetimibe) fails to impact CAVS progression, likely because statins either raise or have no effect on Lp(a) levels.92,119
Until recently, there has been no evidence supporting any therapeutic intervention causing clinically meaningful reductions in Lp(a). Table 4 lists major drug classes and their effects on Lp(a) and CVD outcomes; however, a detailed discussion of each of these therapies is beyond the scope of this review. Drugs that reduce Lp(a) by 20-30% have varying effects on CVD outcomes, from no effect122,123 to a 10% to 20% decrease in CVD events when compared with a placebo.124,125 Because these drugs also produce substantial reductions in LDLC, it is not possible to determine how much of the beneficial effects are due to reductions in Lp(a).
Lipoprotein apheresis produces profound reductions in Lp(a) of 60 to 80% in very highrisk populations.69,126 Within-subjects comparisons show up to 80% reductions in CVD events, relative to event rates prior to treatment initiation.69,127 Early trials of antisense oligonucleotide against apo(a) therapies show potential to produce similar outcomes.128,129 These treatments may be particularly effective in patients with isolated Lp(a) elevations.
Summary
Lp(a) elevation is a major contributor to cardiovascular disease risk and has been recognized as an ICD-10-CM coded clinical diagnosis, the first laboratory abnormality to be defined a clinical disease in the asymptomatic healthy young individuals. This change addresses currently under- diagnosed CVD risk independent of LDLC reduction strategies. A brief overview of recent guidelines for the clinical use of Lp(a) testing from the American Heart Association43,151 and the National Lipid Association52 can be found in Table 5. Although drug therapies for lowering Lp(a) levels remain limited, new treatment options are actively being developed.
Many Americans with high Lp(a) have not yet been identified. Expanded one-time screening can inform these patients of their cardiovascular risk and increase their access to early, aggressive lifestyle modification and optimal lipid-lowering therapy. Given the further increased CVD risk factors for military service members and veterans, a case can be made for broader screening and enhanced surveillance of elevated Lp(a) in these presumably healthy and fit individuals as well as management focused on modifiable risk factors.
Acknowledgements
This program initiative was conducted by the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. as part of the Integrative Cardiac Health Project at Walter Reed National Military Medical Center (WRNMMC), and is made possible by a cooperative agreement that was awarded and administered by the US Army Medical Research & Materiel Command (USAMRMC), at Fort Detrick under Contract Number: W81XWH-16-2-0007. It reflects literature review preparatory work for a research protocol but does not involve an actual research project. The work in this manuscript was supported by the staff of the Integrative Cardiac Health Project (ICHP) with special thanks to Claire Fuller, Elaine Walizer, Dr. Mariam Kashani and the entire health coaching team.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
1. American Heart Association. Cardiovascular disease: a costly burden for America, projections through 2035. http://www.heart.org/idc/groups /heart-public/@wcm/@adv/documents/downloadable /ucm_491543.pdf. Accessed October 10, 2019.
2. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492.
3. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1-25.
4. Thrift AG, Cadilhac DA, Thayabaranathan T, et al. Global stroke statistics. Int J Stroke. 2014;9(1):6-18.
5. Murray CJ, Barber RM, Foreman KJ, et al; GBD 2013 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990-2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145-2191.
6. Mukherjee D, Patil CG. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 suppl):S85-S90.
7. Joseph A, Ackerman D, Talley JD, Johnstone J, Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims—an autopsy study. J Am Coll Cardiol. 1993;22(2):459-467.
8. Webber BJ, Seguin PG, Burnett DG, Clark LL, Otto JL. Prevalence of and risk factors for autopsy-determined atherosclerosis among US service members, 2001-2011. JAMA. 2012;308(24):2577-2583.
9. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933-944.
10. Krishnamurthi N, Francis J, Fihn SD, Meyer CS, Whooley MA. Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One. 2018;13(3):e0193996.
11. Torio CM, Moore BJ. National inpatient hospital costs: the most expensive conditions by payer. Agency for Healthcare Research and Quality Statistical Brief No. 204. http:// www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most -Expensive-Hospital-Conditions.pdf. Published May 2016. Accessed October 10, 2019.
12. Bloom DE, Cafiero ET, Jané-Llopis E, et al. The global economic burden of noncommunicable diseases. https:// www.weforum.org/reports/global-economic-burden-non -communicable-diseases. Published September 18, 2011. Accessed October 10, 2019.
13. Crum-Cianflone NF, Bagnell ME, Schaller E, et al. Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation. 2014;129(18):1813-1820.
14. Fryar CD, Herrick K, Afful J, Ogden CL. Cardiovascular disease risk factors among male veterans, U.S., 2009- 2012. Am J Prev Med. 2016;50(1):101-105.
15. Ulmer CS, Bosworth HB, Germain A, et al; VA Mid-Atlantic Mental Illness Research Education and Clinical Center Registry Workgroup. Associations between sleep difficulties and risk factors for cardiovascular disease in veterans and active duty military personnel of the Iraq and Afghanistan conflicts. J Behav Med. 2015;38(3):544-555.
16. Lutwak N, Dill C. Military sexual trauma increases risk of post-traumatic stress disorder and depression thereby amplifying the possibility of suicidal ideation and cardiovascular disease. Mil Med. 2013;178(4):359-361.
17. Bowry ADK, Lewey J, Dugani SB, Choudhry NK. The burden of cardiovascular disease in low- and middle-income countries: epidemiology and management. Can J Cardiol. 2015;31(9):1151-1159.
18. Reinier K, Stecker EC, Vickers C, Gunson K, Jui J, Chugh SS. Incidence of sudden cardiac arrest is higher in areas of low socioeconomic status: a prospective two year study in a large United States community. Resuscitation. 2006;70(2):186-192.
19. Reinier K, Thomas E, Andrusiek DL, et al; Resuscitation Outcomes Consortium Investigators. Socioeconomic status and incidence of sudden cardiac arrest. CMAJ. 2011;183(15):1705-1712.
20. Yusuf S, Rangarajan S, Teo K, et al; PURE Investigators. Cardiovascular risk and events in 17 low-, middle-, and high-income countries. N Engl J Med. 2014;371(9):818-827.
21. World Health Organization. Health topics: cardiovascular disease. http://www.who.int/cardiovascular_diseases/en/. Updated 2019. Accessed October 10, 2019.
22. Berkowitz AL. Stroke and the noncommunicable diseases: A global burden in need of global advocacy. Neurology. 2015;84(21):2183-2184.
23. Holt T. Predicting cardiovascular disease. BMJ. 2016;353:i2621.
24. Centers for Disease Control and Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors—United States, 2011. MMWR Morb Mortal Wkly Rep. 2011;60(36):1248-1251.
25. Fryar CD, Chen TC, Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: United States, 1999- 2010. NCHS Data Brief. 2012;103:1-8.
26. Ritchey MD, Loustalot F, Wall HK, et al. Million Hearts: description of the national surveillance and modeling methodology used to monitor the number of cardiovascular events prevented during 2012-2016. J Am Heart Assoc. 2017;6(5):pii:e00602.
27. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/ AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2935-2959.
28. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): casecontrol study. Lancet. 2004;364(9438):937-952.
29. Eckel RH, Jakicic JM, Ard JD, et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2960-2984.
30. Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201(suppl 1):S1-S7.
31. Havranek EP, Mujahid MS, Barr DA, et al. Social determinants of risk and outcomes for cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2015;132(9):873-898.
32. Miedema MD, Garberich RF, Schnaidt LJ, et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6(4): pii: e005333.
33. Noheria A, Teodorescu C, Uy-Evanado A, et al. Distinctive profile of sudden cardiac arrest in middle-aged vs. older adults: a community-based study. Int J Cardiol. 2013;168(4):3495-3499.
34. Lieb W, Enserro DM, Larson MG, Vasan RS. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. 2018;5(1):e000722.
35. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157(1):111-117.e2.
36. Damen JA, Hooft L, Schuit E, et al. Prediction models for cardiovascular disease risk in the general population: systematic review. BMJ. 2016;353:i2416.
37. Fulcher J, O’Connell R, Voysey M, et al; Cholesterol Treatment Trialists (CTT) Collaboration. Efficacy and safety of LDL-lowering therapy among men and women: metaanalysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397-1405.
38. Perrone V, Sangiorgi D, Buda S, Degli Esposti L. Residual cardiovascular risk in patients who received lipid-lowering treatment in a real-life setting: retrospective study. Clinicoecon Outcomes Res. 2016;8:649-655.
39. Sirimarco G, Labreuche J, Bruckert E, et al; PERFORM and SPARCL Investigators. Atherogenic dyslipidemia and residual cardiovascular risk in statin-treated patients. Stroke. 2014;45(5):1429-1436.
40. Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617-670.
41. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887-1906.
42. Downs JR, O’Malley PG. Management of dyslipidemia for cardiovascular disease risk reduction: synopsis of the 2014 U.S. Department of Veterans Affairs and U.S. Department of Defense clinical practice guideline. Ann Intern Med. 2015;163(4):291-297.
43. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/ AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/ NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73(24):3168-3209.
44. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177-192.
45. Tsimikas S. A test in context: Lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711.
46. Ellis KL, Boffa MB, Sahebkar A, Koschinsky ML, Watts GF. The renaissance of lipoprotein(a): brave new world for preventive cardiology? Prog Lipid Res. 2017;68:57-82.
47. Thompson GR, Seed M. Lipoprotein(a): the underestimated cardiovascular risk factor. Heart. 2014;100(7):534-535.
48. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526-537.
49. Tremulis SR. Founder’s Story: Lipoprotein(a) Foundation. https://www.lipoproteinafoundation.org/page /Sandrastory. Accessed October 10, 2019.
50. Centers for Disease Control and Prevention. ICD-10 Coordination and Maintenance Committee meeting, September 12-13, 2017 diagnosis agenda. https://www.cdc. gov/nchs/data/icd/Topic_Packet_Sept_2017.pdf. Accessed October 10, 2019.
51. Centers for Medicare & Medicaid Services. 2019 ICD- 10-CM codes descriptions in tabular order. https://www. cms.gov/Medicare/Coding/ICD10/2019-ICD-10-CM.html. Accessed October 10, 2019.
52. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374-392.
53. Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of ldl cholesterol. A consensus statement from EAS and EFLM. Clin Chem. 2018;64(7):1006-1033.
54. Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res. 2017;6:134.
55. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323.
56. Erqou S, Kaptoge S, Perry PL, et al; Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423.
57. Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496-503.
58. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239-2245.
59. Nordestgaard BG, Chapman MJ, Ray K, et al; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853.
60. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996-1001.
61. Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241-249.
62. Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56(12):946-955.
63. Clarke R, Peden JF, Hopewell JC, et al; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New Eng J Med. 2009;361(26):2518-2528.
64. Wattanakit K, Folsom AR, Chambless LE, Nieto FJ. Risk factors for cardiovascular event recurrence in the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2005;149(4):606-612.
65. Ruotolo G, Lincoff MA, Menon V, et al. Lipoprotein(a) is a determinant of residual cardiovascular risk in the setting of optimal LDL-C in statin-treated patients with atherosclerotic cardiovascular disease [Abstract 17400]. Circulation. 2018;136(suppl 1):A17400.
66. Suwa S, Ogita M, Miyauchi K, et al. Impact of lipoprotein (a) on long-term outcomes in patients with coronary artery disease treated with statin after a first percutaneous coronary intervention. J Atheroscler Thromb. 2017;24(11):1125-1131.
67. Nestel PJ, Barnes EH, Tonkin AM, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(12):2902-2908.
68. Burgess S, Ference BA, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627.
69. Roeseler E, Julius U, Heigl F, et al; Pro(a)LiFe-Study Group. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of followup and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019-2027.
70. Matsuura E, Atzeni F, Sarzi-Puttini P, Turiel M, Lopez LR, Nurmohamed MT. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47.
71. Ahearn J, Shields KJ, Liu CC, Manzi S. Cardiovascular disease biomarkers across autoimmune diseases. Clin Immunol. 2015;161(1):59-63.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
72. Di Minno MND, Emmi G, Ambrosino P, et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: a cross-sectional study. Int J Cardiol. 2019;274:1-6.
73. Iseme RA, McEvoy M, Kelly B, et al. A role for autoantibodies in atherogenesis. Cardiovasc Res. 2017;113(10):1102-1112.
74. Sinicato NA, da Silva Cardoso PA, Appenzeller S. Risk factors in cardiovascular disease in systemic lupus erythematosus. Curr Cardiol Rev. 2013;9(1):15-19.
75. Sciatti E, Cavazzana I, Vizzardi E, et al. Systemic lupus erythematosus and endothelial dysfunction: a close relationship. Curr Rheumatol Rev. 2018;15(3):177-188.
76. Prasad A, Clopton P, Ayers C, et al. Relationship of autoantibodies to MDA-LDL and ApoB-Immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol. 2017;37(6):1213-1221.
77. Miller YI, Choi SH, Wiesner P, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108(2):235-248.
78. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092-1104.
79. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485-497.
80. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. 2016;46(8):1818-1825.
81. Ravandi A, Boekholdt SM, Mallat Z, et al. Relationship of oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. 2011;52(10):1829-1836.
82. Tsimikas S, Willeit P, Willeit J, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60(21):2218-2229.
83. Cinoku I, Mavragani CP, Tellis CC, Nezos A, Tselepis AD, Moutsopoulos HM. Autoantibodies to ox-LDL in Sjogren’s syndrome: are they atheroprotective? Clin Exp Rheumatol. 2018;36 Suppl 112(3):61-67.
84. Fagerberg B, Prahl Gullberg U, Alm R, Nilsson J, Fredrikson GN. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS One. 2015;10(3):e0120744.
85. Klesareva EA, Afanas’eva OI, Donskikh VV, Adamova IY, Pokrovskii SN. Characteristics of lipoprotein(a)-containing circulating immune complexes as markers of coronary heart disease. Bull Exp Biol Med. 2016;162(2):231-236.
86. Morishita R, Ishii J, Kusumi Y, et al. Association of serum oxidized lipoprotein(a) concentration with coronary artery disease: potential role of oxidized lipoprotein(a) in the vasucular wall. J Atheroscler Thromb. 2009;16(4):410-418.
87. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1-2):73-78.
88. Wang JJ, Han AZ, Meng Y, et al. Measurement of oxidized lipoprotein (a) in patients with acute coronary syndromes and stable coronary artery disease by 2 ELISAs: using different capture antibody against oxidized lipoprotein (a) or oxidized LDL. Clin Biochem. 2010;43(6):571-575.
89. Orso E, Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12(Suppl 1):31-37.
90. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/ mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101-1106.
91. Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93(2):75-84.
92. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624.
93. Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31(4):440-450.
94. Thanassoulis G, Campbell CY, Owens DS, et al; CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512.
95. Aguib Y, Al Suwaidi J. The Copenhagen City Heart Study (Osterbroundersogelsen). Glob Cardiol Sci Pract. 2015;2015(3):33.
96. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184.
97. Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339.
98. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol.2013;61(11):1146-1156.
99. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477.
100. Kamstrup PR, Nordestgaard BG. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of heart failure in the general population. JACC Heart Fail.2016;4(1):78-87.
101. Verbeek R, Hoogeveen RM, Langsted A, et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J. 2018;39(27):2589-2596.
102. Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(5):1003-1009.
103. Steffen BT, Duprez D, Bertoni AG, Guan W, Tsai M. Lp(a) [lipoprotein(a)]-related risk of heart failure is evident in whites but not in other racial/ethnic groups.Arterioscler Thromb Vasc Biol. 2018;38(10):2498-2504.
104. ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129(4):687-702.
105. Agarwala A, Pokharel Y, Saeed A, et al. The association of lipoprotein(a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis. 2017;262:131-137.
106. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407-1412.
107. Waldeyer C, Makarova N, Zeller T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017;38(32):2490-2498.
108. Cook NR, Mora S, Ridker PM. Lipoprotein(a) and cardiovascular risk prediction among women. J Am Coll Cardiol. 2018;72(3):287-296.
109. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363-1370.
110. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52(2):124-131.
111. Chien KL, Hsu HC, Su TC, Sung FC, Chen MF, Lee YT. Lipoprotein(a) and cardiovascular disease in ethnic Chinese: the Chin-Shan Community Cardiovascular Cohort Study. Clin Chem. 2008;54(2):285-291.
112. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation.2017;135(3):251-263.
113. Zekavat SM, Ruotsalainen S, Handsaker RE, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun.2018;9(1):2606.
114. Zewinger S, Kleber ME, Tragante V, et al. Relations between lipoprotein(a) concentrations, LPA genetic variants, and the risk of mortality in patients with established coronary heart disease: a molecular and genetic association study. Lancet Diabetes Endocrinol. 2017;5(7):534-543.
115. Li J, Lange LA, Sabourin J, et al. Genome- and exomewide association study of serum lipoprotein (a) in the Jackson Heart Study. J Hum Genet. 2015;60(12):755-761.
116. Khera AV, Emdin CA, Drake I, et al, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med.2016;375(24):2349-2358.
117. Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res.2016;57(11):1953-1975.
118. Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a metaanalysis. Eur J Prev Cardiol.2014;21(1):57-64.
119. Estruch R, Ros E, Salas-Salvado J, et al. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med.2018;378(25):e34.
120. Perrot N, Verbeek R, Sandhu M, et al. Ideal cardiovascular health influences cardiovascular disease risk associated with high lipoprotein(a) levels and genotype: The EPICNorfolk prospective population study. Atherosclerosis. 2017;256:47-52.
121. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebocontrolled clinical trials on 2344 patients. Can J Cardiol. 2011;27(6):800-808.
122. Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62(17):1575-1579.
123. Lincoff AM, Nicholls SJ, Riesmeyer JS, et al; ACCELERATE Investigators. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942.
124. Schmidt AF, Pearce LS, Wilkins JT, Overington JP, Hingorani AD, Casas JP. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev.2017;4:CD011748.
125. Bowman L, Hopewell JC, Chen F, et al; PHS3/TIM155-REVEAL Collaborative Group. Effects of anacetrapib in patients with atherosclerotic vascular disease.
126. Leebmann J, Roeseler E, Julius U, et al; Pro(a)LiFe Study Group. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation. 2013;128(24):2567-2576.
127. Heigl F, Hettich R, Lotz N, et al. Efficacy, safety, and tolerability of long-term lipoprotein apheresis in patients with LDL- or Lp(a) hyperlipoproteinemia: Findings gathered from more than 36,000 treatments at one center in Germany. Atheroscler Suppl. 2015;18:154-162.
128. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253.
129. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340-351.
130. Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
131. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519-527.e4.
132. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol.2018;3(2):164-168.
133. Schwartz GG, Olsson AG, Abt M, et al; dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med.2012;367(22):2089-2099.
134. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol.2017;37(9):1770-1775.
135. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635-642.
136. Yeang C, Hung MY, Byun YS, et al. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J Clin Lipidol. 2016;10(3):594-603.
137. Zhou Z, Rahme E, Pilote L. Are statins created equal? Evidence from randomized trials of pravastatin, simvastatin, and atorvastatin for cardiovascular disease prevention.Am Heart J. 2006;151(2):273-281.
138. Ridker PM, MacFadyen JG, Fonseca FA, et al; JUPITER Study Group. Number needed to treat with rosuvastatin to prevent first cardiovascular events and death among men and women with low low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes. 2009;2(6):616-623.
139. Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol.2014;63(13):1278-1288.
140. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509.
141. Koren MJ, Sabatine MS, Giugliano RP, et al. Long-term low-density lipoprotein cholesterol-lowering efficacy, persistence, and safety of evolocumab in treatment of hypercholesterolemia: results up to 4 years from the open-label OSLER-1 extension study. JAMA Cardiol.2017;2(6):598-607.
142. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation.2013;128(9):962-969.
143. Schwartz GG, Steg PG, Szarek M, et al; ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome.N Engl J Med. 2018;379(22):2097-2107.
144. Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular Disease.N Engl J Med. 2017;376(18):1713-1722.
145. Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: A meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6(12):e006910.
146. Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension.Eur Heart J. 2015;36(9):566-575.
147. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011-1021.
148. McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One.2012;7(11):e49006.
149. Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med.2009;6(3):229-239.
150. Rosada A, Kassner U, Vogt A, Willhauck M, Parhofer K, Steinhagen-Thiessen E. Does regular lipid apheresis in Does regular lipid apheresis in patients with isolated elevated lipoprotein(a) levels reduce the incidence of cardiovascular events? Artif Organs. 2014;38(2):135-141.
151. Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646.
HCV testing/awareness successful as part of HIV integrated care
(PWID), according to researchers reporting on a multisite randomized trial of nearly 12,000 HIV-infected individuals in India.
HCV antibody prevalence at these sites ranged from 7.2%-76.6%. Across six integrated care centers (ICCs), 5,263 clients underwent HCV testing, of whom 2,278 were newly diagnosed. At evaluation, PWID in ICC clusters were nearly four times more likely to report being tested for HCV than those in usual care clusters (adjusted prevalence ratio [aPR]: 3.69), according to the report by Sunil Suhas Solomon, MD, of Johns Hopkins University School of Medicine, Baltimore, and colleagues.
PWID in ICC clusters were also seven times more likely to be aware of their HCV status (aPR: 7.11; 95% confidence interval: 1.14, 44.3) and significantly more likely to initiate treatment, (aPR: 9.86; 95% CI: 1.52, 63.8), than individuals in usual care, the authors stated in their report published online ahead of press in the Journal of Hepatology.
“These data provide among the first empirical support of the benefits of integrating HCV testing with HIV prevention and treatment services for PWID. Over a short duration, we observed significant impact on community-level HCV testing and awareness of HCV status among PWID. While additional strategies might be required to improve population awareness levels, integration of HCV testing with HIV programs for PWID particularly given the high burden of HIV/HCV coinfection represents a critical first step,” the researchers concluded.
The study was funded by the National Institutes of Health and the Elton John AIDS Foundation. The authors reported that they had no relevant disclosures.
SOURCE: Solomon, SS et al. J Hepatol. 2019. doi.org/10.1016/j.jhep.2019.09.022.
(PWID), according to researchers reporting on a multisite randomized trial of nearly 12,000 HIV-infected individuals in India.
HCV antibody prevalence at these sites ranged from 7.2%-76.6%. Across six integrated care centers (ICCs), 5,263 clients underwent HCV testing, of whom 2,278 were newly diagnosed. At evaluation, PWID in ICC clusters were nearly four times more likely to report being tested for HCV than those in usual care clusters (adjusted prevalence ratio [aPR]: 3.69), according to the report by Sunil Suhas Solomon, MD, of Johns Hopkins University School of Medicine, Baltimore, and colleagues.
PWID in ICC clusters were also seven times more likely to be aware of their HCV status (aPR: 7.11; 95% confidence interval: 1.14, 44.3) and significantly more likely to initiate treatment, (aPR: 9.86; 95% CI: 1.52, 63.8), than individuals in usual care, the authors stated in their report published online ahead of press in the Journal of Hepatology.
“These data provide among the first empirical support of the benefits of integrating HCV testing with HIV prevention and treatment services for PWID. Over a short duration, we observed significant impact on community-level HCV testing and awareness of HCV status among PWID. While additional strategies might be required to improve population awareness levels, integration of HCV testing with HIV programs for PWID particularly given the high burden of HIV/HCV coinfection represents a critical first step,” the researchers concluded.
The study was funded by the National Institutes of Health and the Elton John AIDS Foundation. The authors reported that they had no relevant disclosures.
SOURCE: Solomon, SS et al. J Hepatol. 2019. doi.org/10.1016/j.jhep.2019.09.022.
(PWID), according to researchers reporting on a multisite randomized trial of nearly 12,000 HIV-infected individuals in India.
HCV antibody prevalence at these sites ranged from 7.2%-76.6%. Across six integrated care centers (ICCs), 5,263 clients underwent HCV testing, of whom 2,278 were newly diagnosed. At evaluation, PWID in ICC clusters were nearly four times more likely to report being tested for HCV than those in usual care clusters (adjusted prevalence ratio [aPR]: 3.69), according to the report by Sunil Suhas Solomon, MD, of Johns Hopkins University School of Medicine, Baltimore, and colleagues.
PWID in ICC clusters were also seven times more likely to be aware of their HCV status (aPR: 7.11; 95% confidence interval: 1.14, 44.3) and significantly more likely to initiate treatment, (aPR: 9.86; 95% CI: 1.52, 63.8), than individuals in usual care, the authors stated in their report published online ahead of press in the Journal of Hepatology.
“These data provide among the first empirical support of the benefits of integrating HCV testing with HIV prevention and treatment services for PWID. Over a short duration, we observed significant impact on community-level HCV testing and awareness of HCV status among PWID. While additional strategies might be required to improve population awareness levels, integration of HCV testing with HIV programs for PWID particularly given the high burden of HIV/HCV coinfection represents a critical first step,” the researchers concluded.
The study was funded by the National Institutes of Health and the Elton John AIDS Foundation. The authors reported that they had no relevant disclosures.
SOURCE: Solomon, SS et al. J Hepatol. 2019. doi.org/10.1016/j.jhep.2019.09.022.
FROM THE JOURNAL OF HEPATOLOGY