User login
Caution with IVC filters in elderly
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.

Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.
Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: Acute pulmonary embolism is a common cause of morbidity and mortality in older adults, and IVC filters have historically and frequently been used to prevent subsequent PE. Almost one in six elderly Medicare fee-for-service (FFS) beneficiaries with PE currently receives an IVC filter.
Study design: Retrospective, matched cohort study.
Setting: United States inpatients during 2011-2014.
Synopsis: Of 214,579 Medicare FFS patients aged 65 years or older who were hospitalized for acute PE, 13.4% received an IVC filter. Mortality was higher in those receiving an IVC filter (11.6%), compared with those who did not receive an IVC filter (9.3%), with an adjusted odds ratio of 30-day mortality of 1.02 (95% CI, 0.98-1.06). One-year mortality rates were 20.5% in the IVC filter group and 13.4% in the group with no IVC filter, with an adjusted OR of 1.35 (95% CI, 1.31-1.40).
In the 76,198 Medicare FFS patients hospitalized with acute PE in the matched cohort group, 18.2% received an IVC filter. The IVC-filter group had higher odds for 30-day mortality, compared with the no–IVC filter group (OR, 2.19; 95% CI, 2.06-2.33).
Bottom line: In patients aged 65 years or older, use caution when considering IVC filter placement for prevention of subsequent PE. Future studies across patient subgroups are needed to analyze the safety and value of IVC filters.
Citation: Bikdeli B et al. Association of inferior vena cava filter use with mortality rates in older adults with acute pulmonary embolism. JAMA Intern Med. 2019;179(2):263-5.
Dr. Trammell Velasquez is an associate professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Using reimbursement to drive innovation in tumor biomarker tests
Implementing a tiered, value-based reimbursement policy for tumor biomarker tests could be an important part of improving innovation and the quality of this tests.
“Precision medicine in cancer care depends on on accurate tumor biomarker tests (TBTs) to determine prognosis and guide treatment selection,” wrote Michaela Dinan, PhD, of Duke University, Durham, N.C., and colleagues. Their report is in JCO Precision Oncology.
“The true clinical utility of a TBT is often difficult to determine, initiating a vicious cycle in which TBTs are often undervalued and poorly reimbursed because of a lack of high-level evidence of clinical utility,” they continued. “An inconsistent regulatory and reimbursement environment results in unfavorable return on investment in the research and development (R&D) needed to generate the high levels of evidence (LOEs) necessary to demonstrate the clinical utility of TBTs.”
To that end, the authors have developed a tiered, value-based reimbursement policy to help spur innovation and improve the clinical utility of TBTs.
The tiering involves three categories: Opt-In, Opt-Out, and Opt-Alt, with each of these terms referring to a decision that might be made with the results of the TBT compared with decisions that would be made based on standard of care (SOC) without TBT results.
“Often in oncology, the SOC is to treat all patients in a given context, knowing that all patients will be exposed to potential risks and costs but only a fraction will benefit,” Dr. Dinan and colleagues wrote. “Opt-Out TBTs have value and the potential to be directly cost saving by reducing the use of unnecessary or ineffective yet toxic and costly therapies, either by indicating that the patient does not need additional therapy (prognosis) or that the therapy under consideration is unlikely to work.”
And example of Opt-Out TBTs are tests used to determine whether adjuvant chemotherapy should be given to patients with estrogen receptor–positive, human epidermal growth factor receptor 2–negative, and node-negative breast cancer.
For the Opt-In tier, “the SOC is to not treat, and the TBT leads to treatment,” the authors noted. “In this case, the value of the TBT is generated by turning a minimally effective or unacceptable therapy into one that is indicated, and therefore acceptable, because of an altered risk-benefit that justifies or refines treatment. An Opt-In TBT will typically increase costs, but still has value because it may result in improved long-term clinical outcomes.”
An example of an Opt-In TBT is an analysis of germline susceptibility genes, such as BRCA1 or BRCA2, which could show increased odds of developing and dying as a result of breast and/or ovarian cancer, though the risk can be cut with prophylactic mastectomy and salpingo-oophorectomy, respectively.
“In this case, these otherwise unacceptable interventions become appropriate because of the increased chances of long-term survival as a result of the intervention in a high-risk population,” the authors wrote.
The Opt-Alt TBT is a subcategory of Opt-In, where the SOC is treatment A but a positive TBT results in the selection of a different treatment.
In each teir, the minimum reimbursement of the TBT increases with the analytic validity and clinical utility of the test. For example, the highest minimum reimbursement would come with tests that have been validated as a primary endpoint in prospective randomized trials, while the lowest would be TBTs with ad hoc analyses of convenience cohorts. In the middle would be those validated ad secondary endpoints in prospective trials.
“Our tiered approach provides a clear and achievable opportunity for companies to develop high-quality TBTs with a guarantee for a commensurate return on investment,” the authors state. “Although our system does not reward a TBT for having slightly better performance than another for a similar indication, it does so indirectly. We posit that by ensuring a fertile ground for multiple high-quality competing TBTs, market forces will be allowed to then appropriately drive the use of TBTs that perform best, given current and local practices.”
The authors make no economic estimates related to the implementation of this type of system.
SOURCE: Michaela Dinan et al. JCO Precis Oncol. 2019 Sep 18. doi: 10.1200/PO.19.00210.
Implementing a tiered, value-based reimbursement policy for tumor biomarker tests could be an important part of improving innovation and the quality of this tests.
“Precision medicine in cancer care depends on on accurate tumor biomarker tests (TBTs) to determine prognosis and guide treatment selection,” wrote Michaela Dinan, PhD, of Duke University, Durham, N.C., and colleagues. Their report is in JCO Precision Oncology.
“The true clinical utility of a TBT is often difficult to determine, initiating a vicious cycle in which TBTs are often undervalued and poorly reimbursed because of a lack of high-level evidence of clinical utility,” they continued. “An inconsistent regulatory and reimbursement environment results in unfavorable return on investment in the research and development (R&D) needed to generate the high levels of evidence (LOEs) necessary to demonstrate the clinical utility of TBTs.”
To that end, the authors have developed a tiered, value-based reimbursement policy to help spur innovation and improve the clinical utility of TBTs.
The tiering involves three categories: Opt-In, Opt-Out, and Opt-Alt, with each of these terms referring to a decision that might be made with the results of the TBT compared with decisions that would be made based on standard of care (SOC) without TBT results.
“Often in oncology, the SOC is to treat all patients in a given context, knowing that all patients will be exposed to potential risks and costs but only a fraction will benefit,” Dr. Dinan and colleagues wrote. “Opt-Out TBTs have value and the potential to be directly cost saving by reducing the use of unnecessary or ineffective yet toxic and costly therapies, either by indicating that the patient does not need additional therapy (prognosis) or that the therapy under consideration is unlikely to work.”
And example of Opt-Out TBTs are tests used to determine whether adjuvant chemotherapy should be given to patients with estrogen receptor–positive, human epidermal growth factor receptor 2–negative, and node-negative breast cancer.
For the Opt-In tier, “the SOC is to not treat, and the TBT leads to treatment,” the authors noted. “In this case, the value of the TBT is generated by turning a minimally effective or unacceptable therapy into one that is indicated, and therefore acceptable, because of an altered risk-benefit that justifies or refines treatment. An Opt-In TBT will typically increase costs, but still has value because it may result in improved long-term clinical outcomes.”
An example of an Opt-In TBT is an analysis of germline susceptibility genes, such as BRCA1 or BRCA2, which could show increased odds of developing and dying as a result of breast and/or ovarian cancer, though the risk can be cut with prophylactic mastectomy and salpingo-oophorectomy, respectively.
“In this case, these otherwise unacceptable interventions become appropriate because of the increased chances of long-term survival as a result of the intervention in a high-risk population,” the authors wrote.
The Opt-Alt TBT is a subcategory of Opt-In, where the SOC is treatment A but a positive TBT results in the selection of a different treatment.
In each teir, the minimum reimbursement of the TBT increases with the analytic validity and clinical utility of the test. For example, the highest minimum reimbursement would come with tests that have been validated as a primary endpoint in prospective randomized trials, while the lowest would be TBTs with ad hoc analyses of convenience cohorts. In the middle would be those validated ad secondary endpoints in prospective trials.
“Our tiered approach provides a clear and achievable opportunity for companies to develop high-quality TBTs with a guarantee for a commensurate return on investment,” the authors state. “Although our system does not reward a TBT for having slightly better performance than another for a similar indication, it does so indirectly. We posit that by ensuring a fertile ground for multiple high-quality competing TBTs, market forces will be allowed to then appropriately drive the use of TBTs that perform best, given current and local practices.”
The authors make no economic estimates related to the implementation of this type of system.
SOURCE: Michaela Dinan et al. JCO Precis Oncol. 2019 Sep 18. doi: 10.1200/PO.19.00210.
Implementing a tiered, value-based reimbursement policy for tumor biomarker tests could be an important part of improving innovation and the quality of this tests.
“Precision medicine in cancer care depends on on accurate tumor biomarker tests (TBTs) to determine prognosis and guide treatment selection,” wrote Michaela Dinan, PhD, of Duke University, Durham, N.C., and colleagues. Their report is in JCO Precision Oncology.
“The true clinical utility of a TBT is often difficult to determine, initiating a vicious cycle in which TBTs are often undervalued and poorly reimbursed because of a lack of high-level evidence of clinical utility,” they continued. “An inconsistent regulatory and reimbursement environment results in unfavorable return on investment in the research and development (R&D) needed to generate the high levels of evidence (LOEs) necessary to demonstrate the clinical utility of TBTs.”
To that end, the authors have developed a tiered, value-based reimbursement policy to help spur innovation and improve the clinical utility of TBTs.
The tiering involves three categories: Opt-In, Opt-Out, and Opt-Alt, with each of these terms referring to a decision that might be made with the results of the TBT compared with decisions that would be made based on standard of care (SOC) without TBT results.
“Often in oncology, the SOC is to treat all patients in a given context, knowing that all patients will be exposed to potential risks and costs but only a fraction will benefit,” Dr. Dinan and colleagues wrote. “Opt-Out TBTs have value and the potential to be directly cost saving by reducing the use of unnecessary or ineffective yet toxic and costly therapies, either by indicating that the patient does not need additional therapy (prognosis) or that the therapy under consideration is unlikely to work.”
And example of Opt-Out TBTs are tests used to determine whether adjuvant chemotherapy should be given to patients with estrogen receptor–positive, human epidermal growth factor receptor 2–negative, and node-negative breast cancer.
For the Opt-In tier, “the SOC is to not treat, and the TBT leads to treatment,” the authors noted. “In this case, the value of the TBT is generated by turning a minimally effective or unacceptable therapy into one that is indicated, and therefore acceptable, because of an altered risk-benefit that justifies or refines treatment. An Opt-In TBT will typically increase costs, but still has value because it may result in improved long-term clinical outcomes.”
An example of an Opt-In TBT is an analysis of germline susceptibility genes, such as BRCA1 or BRCA2, which could show increased odds of developing and dying as a result of breast and/or ovarian cancer, though the risk can be cut with prophylactic mastectomy and salpingo-oophorectomy, respectively.
“In this case, these otherwise unacceptable interventions become appropriate because of the increased chances of long-term survival as a result of the intervention in a high-risk population,” the authors wrote.
The Opt-Alt TBT is a subcategory of Opt-In, where the SOC is treatment A but a positive TBT results in the selection of a different treatment.
In each teir, the minimum reimbursement of the TBT increases with the analytic validity and clinical utility of the test. For example, the highest minimum reimbursement would come with tests that have been validated as a primary endpoint in prospective randomized trials, while the lowest would be TBTs with ad hoc analyses of convenience cohorts. In the middle would be those validated ad secondary endpoints in prospective trials.
“Our tiered approach provides a clear and achievable opportunity for companies to develop high-quality TBTs with a guarantee for a commensurate return on investment,” the authors state. “Although our system does not reward a TBT for having slightly better performance than another for a similar indication, it does so indirectly. We posit that by ensuring a fertile ground for multiple high-quality competing TBTs, market forces will be allowed to then appropriately drive the use of TBTs that perform best, given current and local practices.”
The authors make no economic estimates related to the implementation of this type of system.
SOURCE: Michaela Dinan et al. JCO Precis Oncol. 2019 Sep 18. doi: 10.1200/PO.19.00210.
FROM JCO PRECISION ONCOLOGY
Transcervical ablation of symptomatic uterine fibroids under US guidance
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
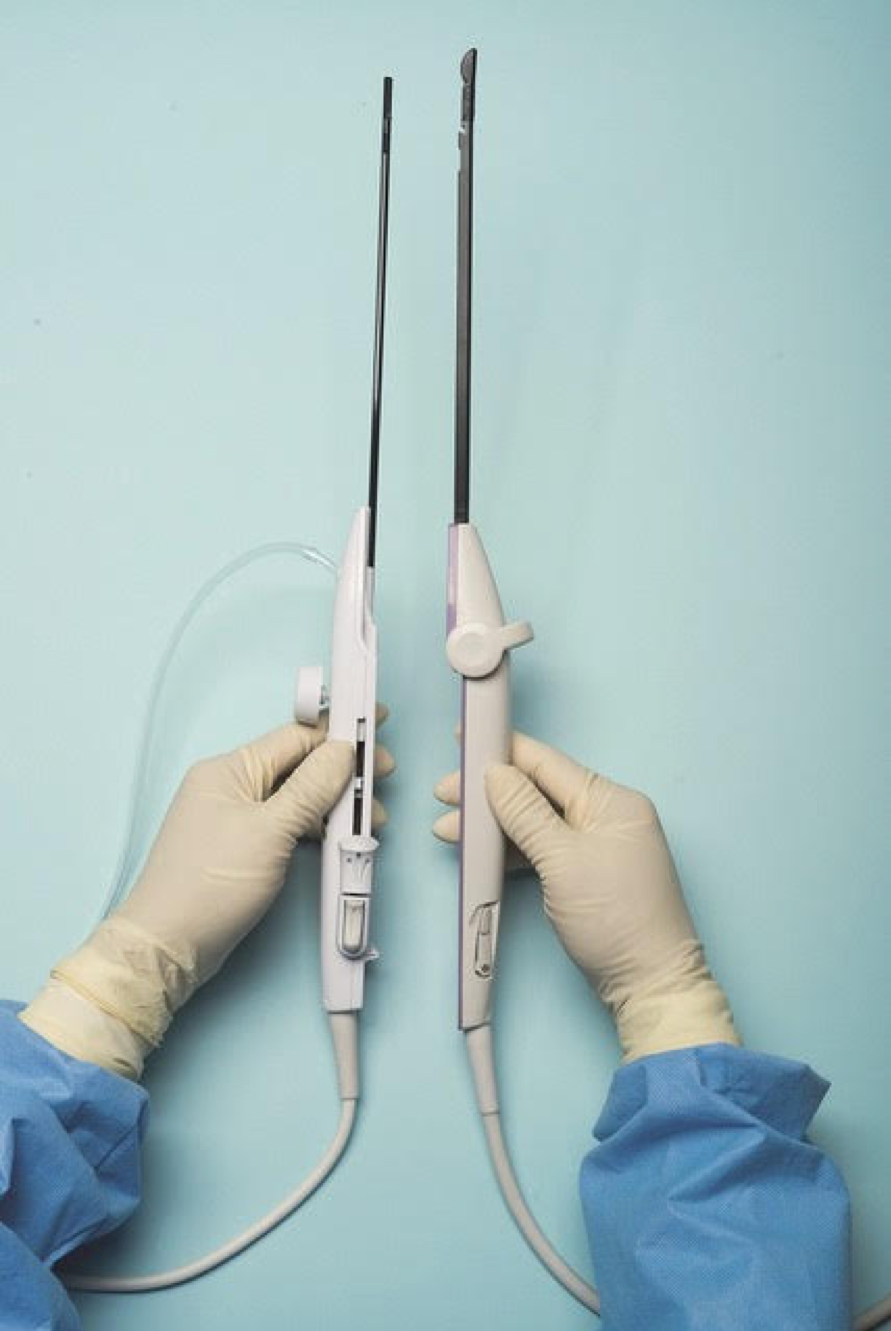
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
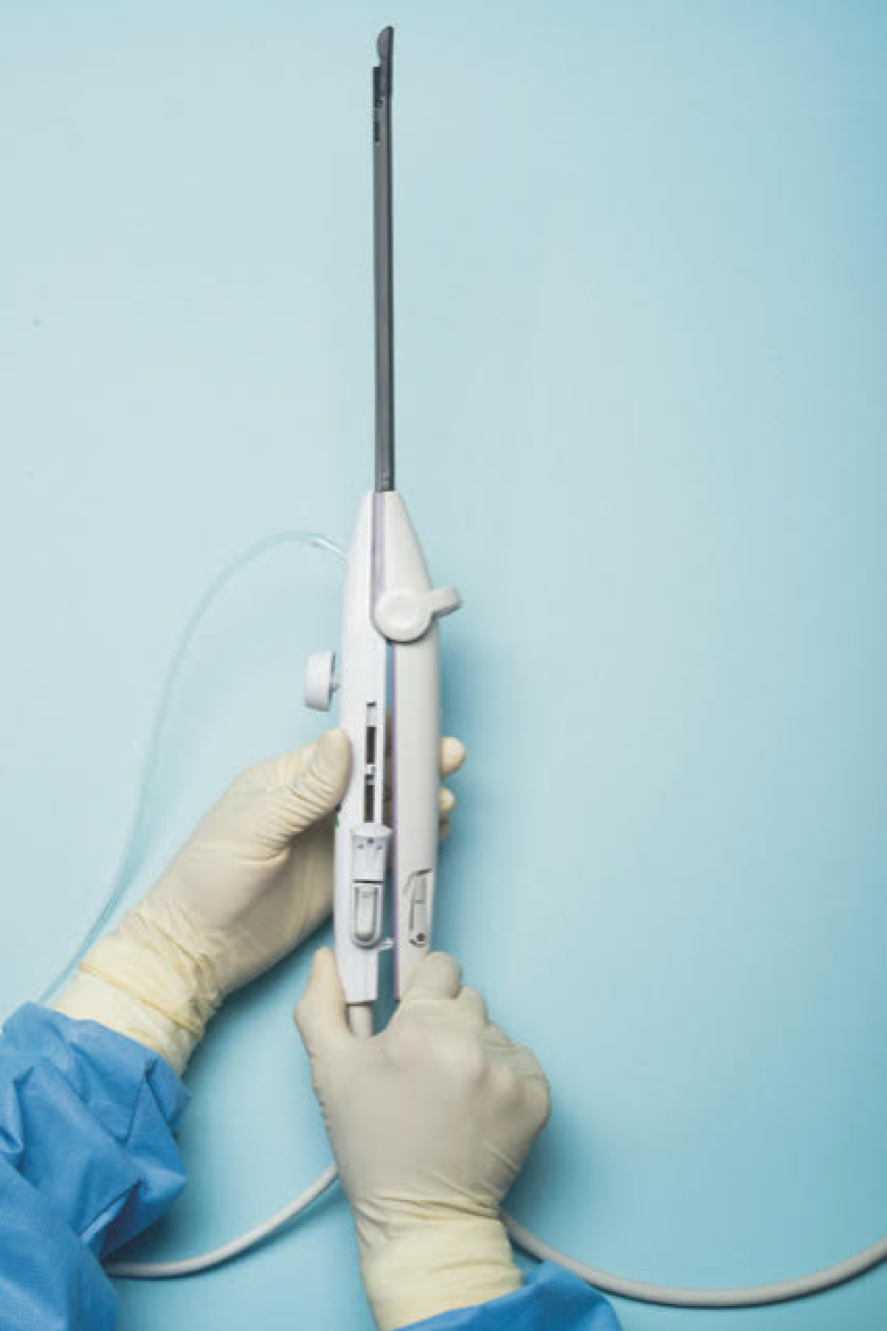
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.
As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
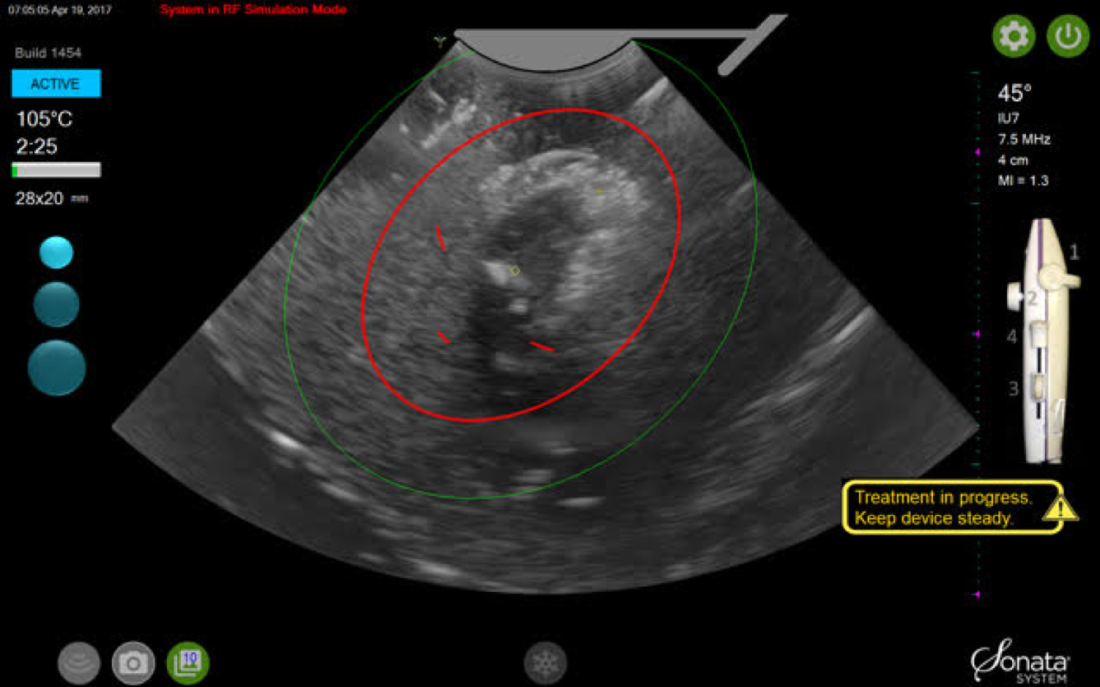
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.
I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.
As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.
I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
On Aug. 29, 2019, the first commercial case utilizing the Sonata system to transcervically ablate symptomatic uterine fibroids under ultrasound guidance was performed at Stamford (Conn.) Hospital. This truly minimally invasive new treatment expands our options in the surgical management of uterine fibroids.
Uterine fibroids are the most common benign tumors of the reproductive tract. It has been estimated that nearly half of the 70%-80% of women who develop fibroids during their reproductive years are symptomatic. Given that some patients present with fertility concerns, it also has been estimated that at least one in three women with fibroids have symptoms such as heavy bleeding (menorrhagia) and bulk symptoms, pain (dyspareunia, dysmenorrhea, noncyclic pain), and increased urinary frequency.
Fibroids are the most common cause of hysterectomy in the United States, with 240,000 (40% of 600,000) performed annually, yet research shows that many women are interested in minimally invasive options and in uterine conservation. In a 2013 national survey published in the American Journal of Obstetrics and Gynecology, 79% of women expressed an interest in minimally invasive approaches for fibroid treatment, and over 50% reported a desire for uterine conservation.1
Both myomectomy and uterine artery embolization are uterine-sparing procedures. However, uterine artery embolization should not be performed in a woman interested in pregnancy. Moreover, there are reports of ovarian reserve issues when the procedure is performed in women in their later reproductive years.
Depending on the technique performed, women undergoing hysteroscopic myomectomy are at risk of fluid overload, hyponatremia, gas-related embolism, and postoperative adhesions. The suture requirements of a laparoscopic myomectomy make this approach an often-difficult one to master, even with robotic assistance. It also requires intubation and potentially places the patient at risk for bleeding and infection. Furthermore, long-term risks include adhesions and the need for C-section with pregnancy.
The impact of uterine fibroids on patients’ lives and their desire for uterine conservation has spurred growing interest in the use of radiofrequency (RF) energy to ablate uterine fibroids. In a 2018 systematic review of nonresective treatments for uterine fibroids published in the International Journal of Hyperthermia, investigators found that the pooled fibroid volume reductions at 6 months after RF ablation and uterine artery embolization were 70% and 54%, respectively.2
The first commercially available system utilizing RF frequency to shrink fibrosis – Acessa – involves laparoscopy, and thus requires abdominal incisions. In August 2018, the Sonata system (Gynesonics: Redwood, Calif.) received Food and Drug Administration clearance after having received European CE-Mark approval in 2010 (for the original device, the VizAblate) and in 2014 (for the next-generation device, the Sonata).
The technology
For a complete description of transcervical, intrauterine sonography–guided radiofrequency ablation of uterine fibroids, one can refer to the excellent outline by David Toub, MD, in Current Obstetrics and Gynecology Reports.3 Basically, the Sonata system allows for real-time, image-guided treatment through the use of a reusable intrauterine ultrasound (IUUS) probe, a single-use RF ablation (RFA) handpiece, and graphical guidance software for diagnosis and targeting.
Initially, the IUUS probe enables identification of fibroids from within the uterine cavity, then guides deployment of an introducer and needle electrode into the targeted fibroid(s). The probe image is curvilinear, penetrates more than 9 cm, and provides a 90-degree field of view.
The RFA handpiece contains the introducer and needle electrode array. It snaps together with the IUUS probe to form and integrate into a single treatment device that contains all controls needed to place and size the ablation. Mechanical stops and lockouts within the RFA handpiece further enhance proper localization and sizing of the ablation.
The system’s graphical guidance software, also known as the SMART Guide, is a real-time graphical overlay on the ultrasound display, which enables one to visually select deployment length, width, and position of the ablation guides. In so doing, the mechanical stops for the introducer and needle electrodes are determined prior to their insertion into the targeted fibroid(s). This was validated in more than 4,000 ablations in bovine muscle and human-extirpated uteri, as well as in vivo at time of laparotomy.
By displaying the ellipsoidal region where the ablation will take place (ablation zone) along with a surrounding ellipsoid (thermal safety border) where tissue temperature will be elevated, the SMART Guide provides a safer and more accurate understanding of the ablation than if it showed only the ablation zone.
As with transabdominal or transvaginal sonography, the serosa will appear hyperechoic at the time of intrauterine ultrasound. By using the SMART Guide, the ablation is sized and positioned to encompass as much of the fibroid as possible while maintaining thermal energy within the uterine serosal margin. Once the desired ablation size has been selected, and safe placement of the needle electrodes is confirmed by rotating the IUUS probe in multiple planes, therapeutic RF energy is delivered to the fibroid; the fixed treatment cycle is dependent on ablation size.
The system will modulate power (up to 150W) to keep temperature at the tips of the needle electrode at 105° C. Moreover, the time of energy delivery at the temperature of 105° – 2-7 minutes – is automatically set based on ablation size, which is a continuum up to 4 cm wide and up to 5 cm long. Multiple ablations may be utilized in a particularly large fibroid.
Unlike hysteroscopic myomectomy, only a small amount of hypotonic solution is instilled within the uterine cavity to enhance acoustic coupling. Furthermore, the treatment device (RFA handpiece and IUUS probe) is only 8.3 mm in diameter. This requires Hegar dilatation of the cervix to 9.
The procedure
After administering anesthesia (regional or sedation), dispersive electrode pads are placed on the anterior thighs. After the cervix is dilated to Hegar dilatation of 9, the treatment device is inserted transcervically into the uterine cavity and the fibroid(s) are identified with the ultrasound probe. The physician plans and optimizes the ablation by sizing and aligning the graphical overlay targeting guide (the SMART Guide) over the live image. Once the size and location of the ablation are set, the trocar-tipped introducer is advanced into the fibroid. After ensuring the guide is within the serosal boundary, the needle electrodes are deployed.
A second visual safety check is completed, and the delivery of RF energy is initiated using a footswitch control. The time of energy delivery is determined based on the size of the desired ablation, up to 7 minutes for the largest ablation size (5 cm x 4 cm). The targeting and treatment steps are repeated as required to treat additional fibroids. Once the treatment is completed, the needle electrodes and introducer are retracted, and the treatment device removed.
Study results and the future
The 12-month safety and effectiveness data for ultrasound-guided transcervical ablation of uterine fibroids were reported in January 2019 in Obstetrics & Gynecology.4 Women enrolled in the prospective, multicenter, single-arm, interventional trial had 1-10 fibroids – the International Federation of Gynecology and Obstetrics (FIGO) types 1, 2, 3, 4, and 2-5 (pedunculated fibroids excluded) – with diameters of 1-5 centimeters. Patients also were required to have at least one fibroid indenting or impinging on the endometrial cavity (FIGO type 1, 2, 3, or 2-5).
Upon study entry, the pictorial assessment blood loss was required to be 150-500 cc. The study included 147 patients. Both coprimary endpoints were satisfied at 12 months; that is, 65% of patients experienced a 50% or greater reduction in menstrual bleeding, and 99% were free from surgical intervention at 1 year.
The mean pictorial blood loss decreased by 39%, 48%, and 51% at 3, 6, and 12 months respectively. Moreover, 95% of the study population experienced some reduction in menstrual bleeding at 12 months. There also were mean improvements in symptom severity and health-related quality-of-life parameters. Mean maximal fibroid volume reduction per patient was 62%.
More than half of the patients returned to normal activity within 1 day, 96% of patients reported symptom improvement at 12 months, and 97% expressed satisfaction with the procedure and results at 12 months. There were no device-related adverse events.
I am the lead author for the 2-year follow-up study utilizing transcervical RFA of symptomatic uterine fibroids, which currently is in press. Suffice it to say, the quality-of-life data, symptom improvement, and lower rate of surgical reintervention all are significant and compelling. Ultimately, I believe Sonata will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.
Dr. Miller is a clinical associate professor at the University of Illinois in Chicago and past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in metropolitan Chicago and the director of minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill. Dr. Miller disclosed that he is a consultant for Gynesonics and holds a stock option agreement with the company.
References
1. Am J Obstet Gynecol. 2013 Oct;209(4):319.e1-319.e20.
2. Int J Hyperthermia. 2019;36(1):295-301.
3. Curr Obstet Gynecol Rep. 2017; 6(1): 67-73.
4. Obstet Gynecol. 2019 Jan;133(1):13-22.
Treating uterine fibroids
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.

The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.
The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Uterine fibroids are the most common benign tumor in women originating from the smooth muscles of the myometrium. While some women are asymptomatic, others experience pelvic pain, pressure, and abnormal uterine bleeding. Uterine fibroids also are associated with gastrointestinal disturbances; urinary problems; infertility; and obstetrical complications including miscarriages, preterm delivery, and cesarean sections.
The first successful abdominal myomectomy was described in 1845 but the procedure quickly fell out of favor because of unacceptably high mortality rates. Myomectomies require special skills and, at times, are associated with bleeding resulting in massive transfusions or sometimes unwanted hysterectomies. In 1922, Victor Bonney developed a uterine artery clamp which significantly decreased bleeding associated with morbidity and mortality.1
The latter part of the 20th century belonged to the minimally invasive surgery (MIS) evolution. Currently, video- or robotic-assisted laparoscopic myomectomies are increasingly employed in fertility-sparing surgery. In 2014, electromechanical morcellators came under scrutiny with concerns about iatrogenic dissemination of both benign and malignant tissues. A media storm ensued, resulting in the 2014 Food and Drug Administration black-box warning, and electromechanical morcellators were pulled from shelves. Data are being collected to quantify and understand these risks more clearly.
While exposing patients to even a small risk of dissemination of an occult uterine malignancy is unwise, MIS should not be abandoned altogether given its advantages to patients.2 Most recently, the American College of Obstetricians and Gynecologists concluded that, although abdominal hysterectomy or myomectomy may reduce the chance of spreading undiagnosed leiomyosarcoma cells, it is associated with increased morbidity, compared with noninvasive approaches, and ob.gyns. should engage in open decision-making processes and explain nonsurgical options with patients.3
The author of this Master Class, Dr. Charles Miller, a world-renowned MIS surgeon, will enlighten readers on the latest development in noninvasive treatment of symptomatic patients. The Sonata system, a promising transcervical (and thus incisionless) treatment modality utilizing intrauterine sonography–guided radiofrequency ablation for uterine fibroids which does not require general anesthesia or hospitalization. He believes that Sonata “will not only be a treatment of choice in the appropriate patient presenting with heavy menstrual flow or bulk symptoms secondary to uterine fibroids, but will prove to be beneficial in women with impinging or deep submucosal fibroids and implantation failure.”
Dr. Miller is on the editorial advisory boards of numerous academic journals and serves as the editor of the award-winning Master Class in Gynecologic Surgery column. For this installment, he has stepped into the role of guest author. Dr. Miller has received numerous awards for his educational contributions and was recently granted the distinct honor of taking the lead in the March 28, 2020 Worldwide EndoMarch–Chicago. It is my pleasure to take part in this introduction.
Dr. Nezhat is director of minimally invasive surgery and robotics as well as the medical director of training and education at Northside Hospital, both in Atlanta. He is fellowship director at Atlanta Center for Special Minimally Invasive Surgery & Reproductive Medicine. Dr. Nezhat also is an adjunct professor of gynecology and obstetrics at Emory University, Atlanta, and is past president of the Society of Reproductive Surgeons and the AAGL. He reported that he has no disclosures relevant to this Master Class. Email him at obnews@mdedge.com.
References
1. BJOG. 2018 Apr;125(5):586.
2. JAMA Oncol. 2015;1(1):78-9.
3. Obstet Gynecol. 2019 Mar;133(3):e238-48.
Cannabis-using MS patients improve cognition with 28 days of abstinence
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.

“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.
“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
STOCKHOLM – The good news about cognitive impairment in patients with multiple sclerosis who’ve been using cannabis heavily for symptom relief – even for many years – is that their memory, executive function, and information processing speed will improve significantly once they’ve been off the drug for just 28 days, according to the results of a randomized trial presented at the annual congress of the European Committee for Treatment and Research in Multiple Sclerosis.
“It’s good for neurologists to know that, if they prescribe cannabis or their patient is self-medicating and chooses to stop, their cognition will improve considerably,” observed Cecilia Meza, a coinvestigator in the study led by Anthony Feinstein, MD, professor of psychiatry at the University of Toronto.
But there’s a surprise twist to this study, she explained in an interview: “We showed patients their results, and they also felt that their cognition was doing a lot better, but despite that, they would rather be using cannabis to feel better than to have their memory intact. The pain was that bad,” said Ms. Meza, a research coordinator at the university’s Sunnybrook Research Institute.
It’s known that cognitive impairment in healthy long-term cannabis users, provided they started as adults, is fully reversed after 28 days of abstinence. But disease-related cognitive dysfunction affects 40%-80% of patients with MS, and cannabis use may compound this impairment.
The study included 40 MS patients with global impairment of cognition, none of whom were cannabis users prior to their diagnosis. They typically started using it for MS symptom relief 2-3 years after receiving their diagnosis. By the time they were approached for study participation, they had been using cannabis four to five times per day or more for an average of 7 years for relief of symptoms, including incontinence, spasticity, poor sleep, headaches, and difficulties in eating.
All participants were willing to try 28 days of abstinence; half were randomized to do so, while the others stayed the course. Study endpoints included change from baseline to day 28 in the Brief Repeatable Neuropsychological Battery, functional MRI done while taking the Symbol Digit Modalities Test, and urine testing to assure compliance with abstinence.
By day 28, the abstinence group – and with one exception, urine testing confirmed they were bona fide cannabis quitters for the study duration – performed significantly better on the neuropsychological test battery than at baseline, with an associated significant increase in brain activation in the bilateral inferior frontal gyri, as well as the caudate and declive cerebellum while executing the Symbol Digit Modalities Test. The control group who kept on using cannabis showed no such improvements.
The full study details were published in conjunction with Ms. Meza’s presentation (Brain. 2019 Sep 1;142[9]:2800-12).
She reported having no financial conflicts regarding the study, funded by the Multiple Sclerosis Society of Canada.
SOURCE: Meza C. ECTRIMS 2019, Abstract P542.
REPORTING FROM ECTRIMS 2019
Preemptive pacifier promotion
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.

Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?
Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.
Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?
Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
I recently encountered an article aimed at parents who were struggling with what to do about their child’s persistent attachment to his pacifier (“How to Ditch the Pacifier,” by Anna Nowogrodski, New York Times, 2019 Sept. 16). For the most part, the author presented a sampling of sound advice from pediatricians and other health experts.
Most children will abandon their pacifiers at a time that is consistent with their developmental stage. Pacifiers seldom do any permanent damage, although they aren’t terribly appealing to look at when hanging out of a toddling toddler’s mouth. Parents were urged to be patient and consistent and were told that allowing the gooey thing to self-destruct often works, as does accelerating the process with a razor blade. Enlisting the aid of the Pacifier Fairy was suggested, but I’m not so sure that would work terribly well.
As I finished perusing the article, I couldn’t help think of how this vexing issue of pacifier removal can be avoided if parents follow a simple rule when they first introduced a pacifier to their child. If experienced parents think back to when they first resorted to using the pacifier, it wasn’t because the plastic and rubber gadget was a family heirloom that had been passed down from generation to generation like an engraved silver spoon. It wasn’t because the dentist told them that children who use pacifiers are less likely to need braces on their teeth. Nor was it a rumor filtered down from speech therapists that pacifiers improve articulation.
Parents reach for a pacifier in hopes that it will help their child will fall asleep. I think most parents of older children agree that at the beginning the pacifier was first and foremost a sleep aid. But here is where the critical oversight occurs: If you give your children pacifiers when you want them to go to sleep, why not simply add the stipulation of where you would like them to go to sleep as well?
Most parents prefer that their children sleep in their own space. We can argue of whether that should be in a side sleeper or their own crib, but most parents don’t want their 3-year-olds sleeping in their bed. Nor do they want their children sleeping on the couch in the living room with them while they watch a movie at 10:30 at night. And as pediatricians, we prefer that children not sleep with their necks flexed in a car seat or baby rocker, particularly if they’re a preemie.
Augmenting the primary association between sleep and the pacifier by adding a place has several important advantages. It gives parents more control of where their children will sleep or, more importantly, where they won’t be sleeping. It helps transitions to nonhome sleeping places like day care and long trips to grandma’s house go more smoothly.
Even more importantly, the crib/pacifier association helps parents who have had trouble reading their children’s cues. If they want a pacifier, it means they are tired and want to go to where the pacifier lives: bed
Finally, maintaining the link between sleeping and the pacifier promotes a more natural weaning process than going cold turkey or hiring the Pacifier Fairy. As naps disappear, the pacifier gradually become a less obvious accessory in the child’s life. However, it may linger in the background as a reminder of when the child needs some restorative sleep.
Of course, helping parents to think clearly enough to create and enforce a simple rule long enough to forge a healthy association when they are sleep deprived themselves is just another one of those challenges we must accept as concerned primary care pediatricians.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Is My Child Overtired? The Sleep Solution for Raising Happier, Healthier Children.” Email him at pdnews@mdedge.com.
FDA approves rituximab to treat children with rare vasculitis
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.

These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
The Food and Drug Administration approved rituximab (Rituxan) by injection to treat granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) in children 2 years of age and older in combination with glucocorticoid treatment, according to an FDA news release.
These rare forms of vasculitis damage small blood vessels through inflammation and can lead to serious organ failure, including lungs and kidneys.
The Genentech drug received priority review and an orphan drug designation based on the results of a pediatric clinical trial of 25 patients aged 6-17 years with active GPA or MPA who were treated with rituximab in an international multicenter, open-label, uncontrolled study. Patients in the trial were also given methylprednisolone prior to starting treatment.
The trial consisted of a 6-month remission induction phase where patients were treated only with rituximab and glucocorticoids. In addition, patients who had not achieved remission could receive additional treatment, including other therapies, at the discretion of the investigator, according to the FDA. By 6 months, 14 of the patients were in remission, and after 18 months, all 25 patients were in remission.
Rituximab contains a boxed warning regarding increased risks of fatal infusion reactions, potentially fatal severe skin and mouth reactions, hepatitis B virus reactivation that may cause serious or lethal liver problems, and progressive multifocal leukoencephalopathy, a rare, potentially lethal brain infection.
The trial was conducted and sponsored by F. Hoffmann-La Roche, which owns Genentech.
Labeling of medication warnings
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.

The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Question: Which one of the following statements regarding medication warnings is incorrect?
A. The drug package “insert” or “label” contains, among other things, a drug’s pharmacology, indications, contraindications, risks and warnings.
B. The Physicians’ Desk Reference (PDR) is an annually updated drug compendium, which can be admitted into evidence as a learned treatise.
C. Drug labeling is a dual responsibility of the manufacturer and the Food and Drug Administration.
D. The FDA is solely responsible for a drug’s warnings and sets the absolute standard of care regarding side effects and complications.
E. State law can impose liability for negligent failure to warn even if the FDA has not included the warning in the drug’s label.
Answer: D. In medical products liability, injured plaintiffs frequently claim a failure to warn of known risks. An example is the cardiovascular deaths caused by Vioxx, a nonsteroidal, anti-inflammatory drug that was withdrawn in 2004. Other examples alleging failure to warn are Actos-associated bladder cancer and Baycol-related rhabdomyolysis. At the time of product approval, the FDA sets out the labeling that goes with each drug, and then makes periodic changes to reflect new indications, warnings and risks. The manufacturer has the prime responsibility for submitting all updated information, especially of augmented risks that come with field experience. In 2012, for example, the FDA mandated the revision of the labeling of Lipitor and other statins to warn of the increased risk of diabetes.
The drug manufacturer stands in the unique position as having the most detailed and up-to-date data and bears a serious responsibility to submit its full findings to the FDA, including its request for label change. Litigation over failure to warn of risks frequently turns on whether the drug manufacturer knew or should have known, had failed to inform the FDA, or whether the FDA itself had declined to make the changes, e.g., because of incomplete or premature data. Notwithstanding the FDA’s overarching federal status, a plaintiff may still attempt to use state tort law to hold a manufacturer liable should the federally approved labeling be silent on the matter.
Two U.S. Supreme Court cases sought to clarify the rules under which a drug manufacturer, when sued for failure to warn, may seek protection under its FDA-approved labeling. The first case involved Diana Levine, a Vermont musician and migraine sufferer, who lost her arm after the drug Phenergan, given by intravenous push, accidentally entered an artery and caused gangrene. Although the intravenous use of Phenergan is approved by the FDA and the risk of such use is clearly stated in the drug’s package insert, the lawsuit alleged that under state law, such a warning was inadequate and should have been strengthened to prohibit this mode of administration. A Vermont jury awarded damages of $6.7 million. On appeal, Wyeth, the defendant pharmaceutical company, maintained that its warning was appropriate, as it had been approved by the federal government through the FDA. It further argued that the drug’s package insert could not be unilaterally altered or modified without running afoul of federal regulations.
In a 6-3 decision,1 the U.S. Supreme Court ruled that the manufacturer was in fact at liberty to issue a more stringent warning, and FDA approval does not bar lawsuits. The Court opined that “Federal law does not pre-empt Levine’s claim that Phenergan’s label did not contain an adequate warning about the IV-push method of administration.” Wyeth had argued that it was impossible for the company to provide additional warnings, since it was the FDA that made the sole determination of the nature and scope of a drug’s label. However, the court held that Wyeth never attempted to change the label to warn of the risk and failed to provide “clear” evidence that the FDA would have prevented it from changing its label. Without defining what constituted “clear” evidence, it rejected Wyeth’s broad assertion that unilaterally changing the Phenergan label would have violated federal law, which was based on the fundamental misunderstanding that the FDA, rather than the manufacturer, bears primary responsibility for drug labeling.
In 2019, the landmark case of Merck Sharp & Dohme Corp v. Albrecht et al.2 reached the U.S. Supreme Court. This class-action suit involved more than 500 individuals who took Fosamax, an effective anti-resorptive drug for treating osteoporosis, and suffered atypical femoral fractures between 1999 and 2010. When the FDA first approved of the manufacture and sale of Fosamax in 1995, the Fosamax label did not warn of the then-speculative risk of atypical femoral fractures. But stronger evidence connecting Fosamax to atypical fractures developed after 1995, prompting the FDA to add a warning in 2011. Merck argued that plaintiffs’ state-law failure-to-warn claims should be dismissed as preempted by federal law. It conceded that the FDA regulations would have permitted Merck to try to change the label to add a warning before 2010 but believed the FDA would have rejected that attempt. In particular, it claimed that the FDA’s rejection of Merck’s 2008 attempt to warn of a risk of “stress fractures” showed that the FDA would also have rejected any attempt by Merck to warn of the risk of atypical femoral fractures. In short, Merck was relying on the legal doctrine of “impossibility preemption,” i.e., it was impossible to comply with both state law (adequate label warning of atypical fractures) and federal law (FDA control of warning labels). The plaintiffs’ position was that Merck’s proposed warning to the FDA had minimized the seriousness of the femoral fracture risk, characterizing them only as “stress fractures.”3
The Court’s earlier Levine decision had held that a state-law failure-to-warn claim is preempted where there is “clear” evidence the FDA would not have approved a label change. In the Albrecht decision, which also sided with the plaintiffs, the court indicated that “Clear evidence is evidence that shows the court that the drug manufacturer fully informed the FDA of the justifications for the warning required by state law and that the FDA, in turn, informed the drug manufacturer that the FDA would not approve a change to the drug’s label to include that warning.” The court also held that issues relating to presumption of impossibility are law-based, and thus it remains for the judge, not the jury, to make that determination.
Issuing timely warnings regarding medical products promotes patient safety, and the law appears to place the major onus on the manufacturer. Still, striking the proper balance is important. During oral arguments in Albrecht, Associate Justice Neil Gorsuch is said to have cautioned against “ ... incentives for companies to submit weakly supported label changes to the agency, knowing that when those label changes are rejected the companies will be free of further liability.” And as pointed out in the earlier cited Johnston article: “ ... a system that creates incentives for manufacturers to over-warn physicians and patients could harm patients by listing the important warnings of adverse effects among numerous less important warnings, which may discourage physicians and patients from choosing potentially useful drugs. On the other hand, a shift of responsibility for labeling to the FDA raises questions about whether the agency, which has resources that are dwarfed by the combined resources of industry, is necessarily capable to serve in this role ...”
Finally, this issue is more complex for devices because of the Medical Device Amendments Act of 1976 (MDA), which may preempt state-based lawsuits. In a claim brought after a Medtronic catheter ruptured in a patient’s coronary artery during heart surgery, the plaintiff alleged that the device was designed, labeled, and manufactured in a manner that violated New York common law. The case was appealed to the U.S. Supreme Court. The court held that the MDA preempted petitioner’s common-law claims challenging the safety or effectiveness of a medical device marketed in a form that received premarket approval from the FDA.4 The court ruled that MDA created a scheme of federal safety oversight for medical devices while sweeping back state oversight schemes.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. This article is meant to be educational and does not constitute medical, ethical, or legal advice. For additional information, readers may contact the author at siang@hawaii.edu.
References
1. Wyeth v. Levine, 555 U.S. 2 (2009).
2. Merck, Sharp & Dohme Corp. v. Albrecht et al., 587 U. S. ____ (2019).
3. Johnston MC et al., A new Supreme Court ruling on drug liability. JAMA 2019;322(7):607-8.
4. Riegel v. Medtronic, 128 S. Ct. 999 (2008).
Apply for the International Scholars Program
If you are a young vascular surgeon from outside North America, consider applying for the International Scholars Program. Recipients of the award will receive a $5,000 stipend, spend two weeks in the U.S, visiting universities and clinics, and attend the 2020 VAM in Toronto. Scholars will work with a mentor to schedule various vascular program visits, including clinical, teaching and research programs. Apply before Oct. 7 to be considered. Learn more here.
If you are a young vascular surgeon from outside North America, consider applying for the International Scholars Program. Recipients of the award will receive a $5,000 stipend, spend two weeks in the U.S, visiting universities and clinics, and attend the 2020 VAM in Toronto. Scholars will work with a mentor to schedule various vascular program visits, including clinical, teaching and research programs. Apply before Oct. 7 to be considered. Learn more here.
If you are a young vascular surgeon from outside North America, consider applying for the International Scholars Program. Recipients of the award will receive a $5,000 stipend, spend two weeks in the U.S, visiting universities and clinics, and attend the 2020 VAM in Toronto. Scholars will work with a mentor to schedule various vascular program visits, including clinical, teaching and research programs. Apply before Oct. 7 to be considered. Learn more here.
FDA adds diabetic kidney disease, heart failure indications to canagliflozin
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.

FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.
FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) for the treatment of diabetic kidney disease and for reduction of the risk of hospitalization for heart failure in patients with type 2 diabetes and diabetic kidney disease, which makes it the first drug indicated for diabetic kidney disease treatment in 20 years.
FDA approval, which was announced in a press release by Janssen, the drug’s manufacturer, is based on results from the phase 3 CREDENCE trial. In that study patients with type 2 diabetes and chronic diabetic kidney disease received either 100 mg canagliflozin or placebo. Patients who received canagliflozin experienced a 30% reduction in the risk of the primary composite endpoint, which included end-stage kidney disease, doubling of serum creatinine, and renal or cardiovascular death. The risk of secondary outcomes were also reduced in patients receiving canagliflozin, including a 39% reduction in the risk of hospitalization for heart failure.
The most common adverse events associated with canagliflozin, according to the label, are female genital mycotic infections, urinary tract infection, and increased urination. Serious adverse events associated with canagliflozin include ketoacidosis, kidney problems, serious urinary tract infections, hypoglycemia, necrotizing fasciitis, serious allergic reaction, and bone fractures.
“The real battle to turn the tide on kidney disease is in early detection and slowing its progression so that patients stay healthier and fewer patients reach kidney failure,” LaVerne A. Burton, president and CEO of the American Kidney Fund, said in the press release. “We are so grateful that advances in kidney disease research are producing treatment options that help to slow the progression of diabetic kidney disease and reduce the risk of hospitalization for heart failure.”
Find the full press release on the Janssen website.