User login
Pembrolizumab/lenvatinib active against urothelial carcinoma
SAN FRANCISCO – A combination of a targeted therapy and an immune checkpoint inhibitor showed promising activity against advanced urothelial cancer in early data from a phase 1b/2 study.

In a cohort of 20 patients with urothelial carcinoma who were enrolled in a larger clinical trial testing the combination of the tyrosine kinase inhibitor (TKI) lenvatinib (Lenvima) and the checkpoint inhibitor pembrolizumab (Keytruda) against urinary tract and other solid malignancies, 5 had an objective response to the combination, including one complete and four partial responses, for an objective response rate of 25%, reported Nicholas J. Vogelzang, MD, from Comprehensive Cancers Centers of Nevada in Las Vegas.
“This response rate warrants further investigation. The lenvatinib plus pembrolizumab combination will be studied in a phase 3 trial in urothelial carcinoma,” he said at the American Society of Clinical Oncology (ASCO) - Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
Dr. Vogelzang noted that urothelial carcinomas account for more than 90% of all bladder cancers. Pembrolizumab monotherapy is approved for treatment of patients with urothelial carcinoma who are ineligible for cisplatin and whose tumors have a combined positive score (CPS) for programmed death-ligand 1 (PD-L1) of 10 or greater or who are ineligible for platinum-based chemotherapy regimens and, in the second line, for advanced or metastatic urothelial carcinoma.
Lenvatinib, a multikinase inhibitor, is approved as monotherapy for radioiodine-refractory differentiated thyroid cancer, unresectable hepatocellular carcinoma, and in combination with everolimus for advanced renal cell carcinoma (RCC) after one year of antiangiogenic therapy.
Dr. Vogelzang reported results of the urothelial cancer cohort from a multicohort study testing the combination.
Twenty patients with histologically confirmed metastatic urothelial cancer were enrolled. The patients all had no more than two prior systemic regimens, good performance status, and a life expectancy of at least 12 weeks. The patients received oral lenvatinib 20 mg daily and pembrolizumab 200 mg intravenously every 21 days. The median patient age was 72 years. The cohort included 14 men and six women.
The objective response rate (ORR) at 24 weeks, the primary endpoint, was 25%, comprising one complete and four partial responses. Nine patients had stable disease, two had disease progression, and four were not evaluable for efficacy. The results were identical according to immune-related Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and modified RECIST version 1.1. Of the 16 evaluable patients, 12 experienced tumor-size reductions from baseline.
“Although there were five objective responses, there were an additional seven patients or more who had minor regressions of disease – clearly an active regimen,” Dr. Vogelzang said. Four of the patients, including one with a PD-L1–positive tumor and three with PD-L1–negative tumors were still alive, with the longest survival past 80 weeks since the start of therapy. The majority of patients, however, had no objective response or disease progression within about 20 weeks.
After a median follow-up of 11.7 months, the median progression-free survival (PFS) was 5.4 months, and the 12-month PFS rate was 26%.
In all, 18 of the 20 patients (90%) experienced a treatment-related adverse event of any grade, 10 had grade 3 or 4 events, and 6 had serious adverse events including one death from gastrointestinal hemorrhage that Dr. Vogelzang said appeared to be related to lenvatinib. A total of four patients (20%) had a treatment-related event leading to withdrawal or discontinuation, seven had a dose reduction, and 12 had an interruption in therapy, primarily of lenvatinib. The most common toxicities were proteinuria, diarrhea, hypertension, fatigue, hypothyroidism, decreased appetite with nausea, pancreatitis with increased lipase, skin rash, vomiting, and dry mouth.
In addition to the planned phase 3 trial of the combination in urothelial carcinoma, lenvatinib/pembrolizumab is also being studied for the treatment of RCC.
The study was supported by Eisai and Merck Sharp & Dohme. Dr. Vogelzang disclosed financial relationships with Caris Life Sciences, Pfizer, Up to Date, AstraZeneca, MedImmune, and other companies. Five coauthors are employees of Merck or Esai.
SOURCE: Vogelzang NJ et al. ASCO-SITC, Abstract 11.
SAN FRANCISCO – A combination of a targeted therapy and an immune checkpoint inhibitor showed promising activity against advanced urothelial cancer in early data from a phase 1b/2 study.
In a cohort of 20 patients with urothelial carcinoma who were enrolled in a larger clinical trial testing the combination of the tyrosine kinase inhibitor (TKI) lenvatinib (Lenvima) and the checkpoint inhibitor pembrolizumab (Keytruda) against urinary tract and other solid malignancies, 5 had an objective response to the combination, including one complete and four partial responses, for an objective response rate of 25%, reported Nicholas J. Vogelzang, MD, from Comprehensive Cancers Centers of Nevada in Las Vegas.
“This response rate warrants further investigation. The lenvatinib plus pembrolizumab combination will be studied in a phase 3 trial in urothelial carcinoma,” he said at the American Society of Clinical Oncology (ASCO) - Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
Dr. Vogelzang noted that urothelial carcinomas account for more than 90% of all bladder cancers. Pembrolizumab monotherapy is approved for treatment of patients with urothelial carcinoma who are ineligible for cisplatin and whose tumors have a combined positive score (CPS) for programmed death-ligand 1 (PD-L1) of 10 or greater or who are ineligible for platinum-based chemotherapy regimens and, in the second line, for advanced or metastatic urothelial carcinoma.
Lenvatinib, a multikinase inhibitor, is approved as monotherapy for radioiodine-refractory differentiated thyroid cancer, unresectable hepatocellular carcinoma, and in combination with everolimus for advanced renal cell carcinoma (RCC) after one year of antiangiogenic therapy.
Dr. Vogelzang reported results of the urothelial cancer cohort from a multicohort study testing the combination.
Twenty patients with histologically confirmed metastatic urothelial cancer were enrolled. The patients all had no more than two prior systemic regimens, good performance status, and a life expectancy of at least 12 weeks. The patients received oral lenvatinib 20 mg daily and pembrolizumab 200 mg intravenously every 21 days. The median patient age was 72 years. The cohort included 14 men and six women.
The objective response rate (ORR) at 24 weeks, the primary endpoint, was 25%, comprising one complete and four partial responses. Nine patients had stable disease, two had disease progression, and four were not evaluable for efficacy. The results were identical according to immune-related Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and modified RECIST version 1.1. Of the 16 evaluable patients, 12 experienced tumor-size reductions from baseline.
“Although there were five objective responses, there were an additional seven patients or more who had minor regressions of disease – clearly an active regimen,” Dr. Vogelzang said. Four of the patients, including one with a PD-L1–positive tumor and three with PD-L1–negative tumors were still alive, with the longest survival past 80 weeks since the start of therapy. The majority of patients, however, had no objective response or disease progression within about 20 weeks.
After a median follow-up of 11.7 months, the median progression-free survival (PFS) was 5.4 months, and the 12-month PFS rate was 26%.
In all, 18 of the 20 patients (90%) experienced a treatment-related adverse event of any grade, 10 had grade 3 or 4 events, and 6 had serious adverse events including one death from gastrointestinal hemorrhage that Dr. Vogelzang said appeared to be related to lenvatinib. A total of four patients (20%) had a treatment-related event leading to withdrawal or discontinuation, seven had a dose reduction, and 12 had an interruption in therapy, primarily of lenvatinib. The most common toxicities were proteinuria, diarrhea, hypertension, fatigue, hypothyroidism, decreased appetite with nausea, pancreatitis with increased lipase, skin rash, vomiting, and dry mouth.
In addition to the planned phase 3 trial of the combination in urothelial carcinoma, lenvatinib/pembrolizumab is also being studied for the treatment of RCC.
The study was supported by Eisai and Merck Sharp & Dohme. Dr. Vogelzang disclosed financial relationships with Caris Life Sciences, Pfizer, Up to Date, AstraZeneca, MedImmune, and other companies. Five coauthors are employees of Merck or Esai.
SOURCE: Vogelzang NJ et al. ASCO-SITC, Abstract 11.
SAN FRANCISCO – A combination of a targeted therapy and an immune checkpoint inhibitor showed promising activity against advanced urothelial cancer in early data from a phase 1b/2 study.
In a cohort of 20 patients with urothelial carcinoma who were enrolled in a larger clinical trial testing the combination of the tyrosine kinase inhibitor (TKI) lenvatinib (Lenvima) and the checkpoint inhibitor pembrolizumab (Keytruda) against urinary tract and other solid malignancies, 5 had an objective response to the combination, including one complete and four partial responses, for an objective response rate of 25%, reported Nicholas J. Vogelzang, MD, from Comprehensive Cancers Centers of Nevada in Las Vegas.
“This response rate warrants further investigation. The lenvatinib plus pembrolizumab combination will be studied in a phase 3 trial in urothelial carcinoma,” he said at the American Society of Clinical Oncology (ASCO) - Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
Dr. Vogelzang noted that urothelial carcinomas account for more than 90% of all bladder cancers. Pembrolizumab monotherapy is approved for treatment of patients with urothelial carcinoma who are ineligible for cisplatin and whose tumors have a combined positive score (CPS) for programmed death-ligand 1 (PD-L1) of 10 or greater or who are ineligible for platinum-based chemotherapy regimens and, in the second line, for advanced or metastatic urothelial carcinoma.
Lenvatinib, a multikinase inhibitor, is approved as monotherapy for radioiodine-refractory differentiated thyroid cancer, unresectable hepatocellular carcinoma, and in combination with everolimus for advanced renal cell carcinoma (RCC) after one year of antiangiogenic therapy.
Dr. Vogelzang reported results of the urothelial cancer cohort from a multicohort study testing the combination.
Twenty patients with histologically confirmed metastatic urothelial cancer were enrolled. The patients all had no more than two prior systemic regimens, good performance status, and a life expectancy of at least 12 weeks. The patients received oral lenvatinib 20 mg daily and pembrolizumab 200 mg intravenously every 21 days. The median patient age was 72 years. The cohort included 14 men and six women.
The objective response rate (ORR) at 24 weeks, the primary endpoint, was 25%, comprising one complete and four partial responses. Nine patients had stable disease, two had disease progression, and four were not evaluable for efficacy. The results were identical according to immune-related Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and modified RECIST version 1.1. Of the 16 evaluable patients, 12 experienced tumor-size reductions from baseline.
“Although there were five objective responses, there were an additional seven patients or more who had minor regressions of disease – clearly an active regimen,” Dr. Vogelzang said. Four of the patients, including one with a PD-L1–positive tumor and three with PD-L1–negative tumors were still alive, with the longest survival past 80 weeks since the start of therapy. The majority of patients, however, had no objective response or disease progression within about 20 weeks.
After a median follow-up of 11.7 months, the median progression-free survival (PFS) was 5.4 months, and the 12-month PFS rate was 26%.
In all, 18 of the 20 patients (90%) experienced a treatment-related adverse event of any grade, 10 had grade 3 or 4 events, and 6 had serious adverse events including one death from gastrointestinal hemorrhage that Dr. Vogelzang said appeared to be related to lenvatinib. A total of four patients (20%) had a treatment-related event leading to withdrawal or discontinuation, seven had a dose reduction, and 12 had an interruption in therapy, primarily of lenvatinib. The most common toxicities were proteinuria, diarrhea, hypertension, fatigue, hypothyroidism, decreased appetite with nausea, pancreatitis with increased lipase, skin rash, vomiting, and dry mouth.
In addition to the planned phase 3 trial of the combination in urothelial carcinoma, lenvatinib/pembrolizumab is also being studied for the treatment of RCC.
The study was supported by Eisai and Merck Sharp & Dohme. Dr. Vogelzang disclosed financial relationships with Caris Life Sciences, Pfizer, Up to Date, AstraZeneca, MedImmune, and other companies. Five coauthors are employees of Merck or Esai.
SOURCE: Vogelzang NJ et al. ASCO-SITC, Abstract 11.
REPORTING FROM ASCO-SITC
Distinct features found in young-onset CRC
Young-onset colorectal cancer (CRC) has distinct clinical and molecular features, compared with disease diagnosed later in life, according to investigators who conducted a review that included more than 36,000 patients.
investigators said. Conversely, those younger patients were less likely to have BRAF V600 mutations than were patients 50 years old and older, the investigators reported in the journal Cancer.
Very young patients were more likely to have signet ring histology and less likely to have adenomatous polyposis coli (APC) mutations, according to senior study author Jonathan M. Loree, MD, and his coinvestigators at The University of Texas MD Anderson Cancer Center, Houston.
“We need to appreciate that there are unique biologic subtypes within young patients that may affect how their cancers behave and may require a personalized approach to treatment,” Dr. Loree said in a press statement. “Going forward, special clinical consideration should be given to, and further scientific investigations should be performed for, both very young patients with colorectal cancer and those with predisposing medical conditions.”
The incidence of young-onset CRC has increased 1%-3% annually in recent years, Dr. Loree and colleagues wrote in their report.
Although smaller studies have characterized molecular features of CRC in younger patients, there has so far been no comprehensive molecular characterization of these patients, they added. Accordingly, the investigators conducted a retrospective analysis that included more than 36,000 patients in four different patient cohorts.
Patients under 50 years more likely had synchronous metastases (P = .009), more likely had primary tumors in the distal colon and rectum (P less than .0001), and were more likely to be MSI-high (P = .038), compared with their older counterparts, Dr. Loree and coauthors reported.
BRAF V600 mutations were infrequent in patients under 30 years of age, at a prevalence of 4% or less, increasing to a high of 13% in patients aged 70 years or older (P less than 0.001), investigators also reported.
Very young patients, or those aged 18-29 years, had a higher prevalence of signet ring histology, compared with the other age groups (P = .0003), and they had a nearly fivefold increased odds of signet ring histology compared with patients in the 30-49 year age range, investigators wrote.
There were also considerably fewer APC mutations in the patients younger than 30 years, compared with older patients in the young-onset CRC group, with an odds ratio of 0.56 (95% confidence interval, 0.35-0.90; P = .015).
Hispanic patients were significantly overrepresented in the under-30-years age group (P = .0015), according to the report.
For patients under 50 years who also had inflammatory bowel disease, odds of mucinous or signet ring histology were higher (OR, 5.54; 95% CI, 2.24-13.74; P = .0004), and odds of APC mutations were lower (OR, 0.24; 95% CI, 0.07-0.75; P = .019), compared with younger patients with no such predisposing conditions.
“These notable differences in very young patients with CRC and those with predisposing conditions highlight that early-onset CRC has unique subsets within the population of patients younger than 50 years,” Dr. Loree and coauthors concluded.
Support to investigators in the study came from the National Cancer Institute, National Institutes of Health, and the MD Anderson Colorectal Cancer Moon Shot Program. One coinvestigator reported disclosures related to Roche, Genentech, EMD Serono, Merck, Karyopharm, Amal, Navire, Symphogen, Holystone, Biocartis, Amgen, and Novartis.
SOURCE: Willauer AN et al. Cancer. 2019 Mar 11. doi: 10.1002/cncr.31994
Young-onset colorectal cancer (CRC) has distinct clinical and molecular features, compared with disease diagnosed later in life, according to investigators who conducted a review that included more than 36,000 patients.
investigators said. Conversely, those younger patients were less likely to have BRAF V600 mutations than were patients 50 years old and older, the investigators reported in the journal Cancer.
Very young patients were more likely to have signet ring histology and less likely to have adenomatous polyposis coli (APC) mutations, according to senior study author Jonathan M. Loree, MD, and his coinvestigators at The University of Texas MD Anderson Cancer Center, Houston.
“We need to appreciate that there are unique biologic subtypes within young patients that may affect how their cancers behave and may require a personalized approach to treatment,” Dr. Loree said in a press statement. “Going forward, special clinical consideration should be given to, and further scientific investigations should be performed for, both very young patients with colorectal cancer and those with predisposing medical conditions.”
The incidence of young-onset CRC has increased 1%-3% annually in recent years, Dr. Loree and colleagues wrote in their report.
Although smaller studies have characterized molecular features of CRC in younger patients, there has so far been no comprehensive molecular characterization of these patients, they added. Accordingly, the investigators conducted a retrospective analysis that included more than 36,000 patients in four different patient cohorts.
Patients under 50 years more likely had synchronous metastases (P = .009), more likely had primary tumors in the distal colon and rectum (P less than .0001), and were more likely to be MSI-high (P = .038), compared with their older counterparts, Dr. Loree and coauthors reported.
BRAF V600 mutations were infrequent in patients under 30 years of age, at a prevalence of 4% or less, increasing to a high of 13% in patients aged 70 years or older (P less than 0.001), investigators also reported.
Very young patients, or those aged 18-29 years, had a higher prevalence of signet ring histology, compared with the other age groups (P = .0003), and they had a nearly fivefold increased odds of signet ring histology compared with patients in the 30-49 year age range, investigators wrote.
There were also considerably fewer APC mutations in the patients younger than 30 years, compared with older patients in the young-onset CRC group, with an odds ratio of 0.56 (95% confidence interval, 0.35-0.90; P = .015).
Hispanic patients were significantly overrepresented in the under-30-years age group (P = .0015), according to the report.
For patients under 50 years who also had inflammatory bowel disease, odds of mucinous or signet ring histology were higher (OR, 5.54; 95% CI, 2.24-13.74; P = .0004), and odds of APC mutations were lower (OR, 0.24; 95% CI, 0.07-0.75; P = .019), compared with younger patients with no such predisposing conditions.
“These notable differences in very young patients with CRC and those with predisposing conditions highlight that early-onset CRC has unique subsets within the population of patients younger than 50 years,” Dr. Loree and coauthors concluded.
Support to investigators in the study came from the National Cancer Institute, National Institutes of Health, and the MD Anderson Colorectal Cancer Moon Shot Program. One coinvestigator reported disclosures related to Roche, Genentech, EMD Serono, Merck, Karyopharm, Amal, Navire, Symphogen, Holystone, Biocartis, Amgen, and Novartis.
SOURCE: Willauer AN et al. Cancer. 2019 Mar 11. doi: 10.1002/cncr.31994
Young-onset colorectal cancer (CRC) has distinct clinical and molecular features, compared with disease diagnosed later in life, according to investigators who conducted a review that included more than 36,000 patients.
investigators said. Conversely, those younger patients were less likely to have BRAF V600 mutations than were patients 50 years old and older, the investigators reported in the journal Cancer.
Very young patients were more likely to have signet ring histology and less likely to have adenomatous polyposis coli (APC) mutations, according to senior study author Jonathan M. Loree, MD, and his coinvestigators at The University of Texas MD Anderson Cancer Center, Houston.
“We need to appreciate that there are unique biologic subtypes within young patients that may affect how their cancers behave and may require a personalized approach to treatment,” Dr. Loree said in a press statement. “Going forward, special clinical consideration should be given to, and further scientific investigations should be performed for, both very young patients with colorectal cancer and those with predisposing medical conditions.”
The incidence of young-onset CRC has increased 1%-3% annually in recent years, Dr. Loree and colleagues wrote in their report.
Although smaller studies have characterized molecular features of CRC in younger patients, there has so far been no comprehensive molecular characterization of these patients, they added. Accordingly, the investigators conducted a retrospective analysis that included more than 36,000 patients in four different patient cohorts.
Patients under 50 years more likely had synchronous metastases (P = .009), more likely had primary tumors in the distal colon and rectum (P less than .0001), and were more likely to be MSI-high (P = .038), compared with their older counterparts, Dr. Loree and coauthors reported.
BRAF V600 mutations were infrequent in patients under 30 years of age, at a prevalence of 4% or less, increasing to a high of 13% in patients aged 70 years or older (P less than 0.001), investigators also reported.
Very young patients, or those aged 18-29 years, had a higher prevalence of signet ring histology, compared with the other age groups (P = .0003), and they had a nearly fivefold increased odds of signet ring histology compared with patients in the 30-49 year age range, investigators wrote.
There were also considerably fewer APC mutations in the patients younger than 30 years, compared with older patients in the young-onset CRC group, with an odds ratio of 0.56 (95% confidence interval, 0.35-0.90; P = .015).
Hispanic patients were significantly overrepresented in the under-30-years age group (P = .0015), according to the report.
For patients under 50 years who also had inflammatory bowel disease, odds of mucinous or signet ring histology were higher (OR, 5.54; 95% CI, 2.24-13.74; P = .0004), and odds of APC mutations were lower (OR, 0.24; 95% CI, 0.07-0.75; P = .019), compared with younger patients with no such predisposing conditions.
“These notable differences in very young patients with CRC and those with predisposing conditions highlight that early-onset CRC has unique subsets within the population of patients younger than 50 years,” Dr. Loree and coauthors concluded.
Support to investigators in the study came from the National Cancer Institute, National Institutes of Health, and the MD Anderson Colorectal Cancer Moon Shot Program. One coinvestigator reported disclosures related to Roche, Genentech, EMD Serono, Merck, Karyopharm, Amal, Navire, Symphogen, Holystone, Biocartis, Amgen, and Novartis.
SOURCE: Willauer AN et al. Cancer. 2019 Mar 11. doi: 10.1002/cncr.31994
FROM CANCER
Antenatal steroids for preterm birth is cost effective
Administering antenatal corticosteroids to pregnant women at high risk for preterm birth was a cost-effective intervention that improved infant respiratory outcomes, according to a new study.
“This intervention has a potential cost saving in the United States of approximately $100 million dollars annually from the benefit in the immediate neonatal outcome alone,” Cynthia Gyamfi-Bannerman, MD, of Columbia University, New York, and her associates reported in JAMA Pediatrics. “Because late preterm birth comprises a large proportion of all preterm births, our findings have the potential for a large influence on public health.”
The researchers conducted a retrospective secondary analysis of the randomized Antenatal Late Preterm Steroids (ALPS) clinical trial October 2010 to February 2015. The trial enrolled randomly assigned antenatal administration of betamethasone or placebo to women pregnant with a singleton and at high risk for preterm birth while between 34 weeks, 6 days, and 36 weeks, 0 days, of gestation.
Antenatal corticosteroid administration was regarded as effective if a newborn did not require treatment in the first 72 hours for respiratory distress or illness. Treatment could include “continuous positive airway pressure or high-flow nasal cannula for 2 hours or more, supplemental oxygen with a fraction of inspired oxygen of 30% or more for 4 hours or more, and extracorporeal membrane oxygenation or mechanical ventilation,” Dr. Gyamfi-Bannerman and her associates wrote.
To tally the costs, the researchers used Medicaid rates to estimate the total in 2015 U.S. dollars for betamethasone, outpatient visits or inpatient stays to administer it, and all direct newborn care costs, including neonatal ICU daily costs stratified by respiratory illness severity. Betamethasone administration included an initial 12-mg intramuscular dose followed by another after 24 hours if the infant had not been delivered.
“Because therapy often persists for longer than this 72-hour duration, we measured costs through hospital discharge,” the authors wrote. “The analysis took the perspective of a third-party payer in which we included direct medical costs and associated overhead accruing to hospitals and medical payers for the care of enrolled patients and their infants.”
Among 2,821 mothers not lost to follow-up during the secondary analysis, 1,426 received betamethasone and 1,395 received placebo. For mothers who received betamethasone antenatally, the total mean cost was $4,681 per mother-infant pair. Total mean cost for those in the placebo group was $5,379 per pair, resulting in a significant mean $698 savings (P = .02). Respiratory morbidity was 2.9% lower in infants whose mothers received antenatal corticosteroid treatment.
“Thus, because the treated group had lower costs and this strategy was more effective, administration of betamethasone to women at risk for late preterm birth was judged to be a dominant strategy, which is defined as one in which costs are lower and effectiveness is higher than a comparator (incremental cost-effectiveness ratio [ICER], −23 986),” Dr. Gyamfi-Bannerman and her associates reported. ICER is defined as the difference in mean total cost per patient in the betamethasone and placebo arms divided by the difference in the effectiveness.
Study limitations were an inability to estimate costs according to quality-adjusted life years or to include families’/caregivers’ costs.
SOURCE: Gyamfi-Bannerman C. JAMA Pediatr. 2019 Mar 11. doi: 10.1001/jamapediatrics.2019.0032.
Administering antenatal corticosteroids to pregnant women at high risk for preterm birth was a cost-effective intervention that improved infant respiratory outcomes, according to a new study.
“This intervention has a potential cost saving in the United States of approximately $100 million dollars annually from the benefit in the immediate neonatal outcome alone,” Cynthia Gyamfi-Bannerman, MD, of Columbia University, New York, and her associates reported in JAMA Pediatrics. “Because late preterm birth comprises a large proportion of all preterm births, our findings have the potential for a large influence on public health.”
The researchers conducted a retrospective secondary analysis of the randomized Antenatal Late Preterm Steroids (ALPS) clinical trial October 2010 to February 2015. The trial enrolled randomly assigned antenatal administration of betamethasone or placebo to women pregnant with a singleton and at high risk for preterm birth while between 34 weeks, 6 days, and 36 weeks, 0 days, of gestation.
Antenatal corticosteroid administration was regarded as effective if a newborn did not require treatment in the first 72 hours for respiratory distress or illness. Treatment could include “continuous positive airway pressure or high-flow nasal cannula for 2 hours or more, supplemental oxygen with a fraction of inspired oxygen of 30% or more for 4 hours or more, and extracorporeal membrane oxygenation or mechanical ventilation,” Dr. Gyamfi-Bannerman and her associates wrote.
To tally the costs, the researchers used Medicaid rates to estimate the total in 2015 U.S. dollars for betamethasone, outpatient visits or inpatient stays to administer it, and all direct newborn care costs, including neonatal ICU daily costs stratified by respiratory illness severity. Betamethasone administration included an initial 12-mg intramuscular dose followed by another after 24 hours if the infant had not been delivered.
“Because therapy often persists for longer than this 72-hour duration, we measured costs through hospital discharge,” the authors wrote. “The analysis took the perspective of a third-party payer in which we included direct medical costs and associated overhead accruing to hospitals and medical payers for the care of enrolled patients and their infants.”
Among 2,821 mothers not lost to follow-up during the secondary analysis, 1,426 received betamethasone and 1,395 received placebo. For mothers who received betamethasone antenatally, the total mean cost was $4,681 per mother-infant pair. Total mean cost for those in the placebo group was $5,379 per pair, resulting in a significant mean $698 savings (P = .02). Respiratory morbidity was 2.9% lower in infants whose mothers received antenatal corticosteroid treatment.
“Thus, because the treated group had lower costs and this strategy was more effective, administration of betamethasone to women at risk for late preterm birth was judged to be a dominant strategy, which is defined as one in which costs are lower and effectiveness is higher than a comparator (incremental cost-effectiveness ratio [ICER], −23 986),” Dr. Gyamfi-Bannerman and her associates reported. ICER is defined as the difference in mean total cost per patient in the betamethasone and placebo arms divided by the difference in the effectiveness.
Study limitations were an inability to estimate costs according to quality-adjusted life years or to include families’/caregivers’ costs.
SOURCE: Gyamfi-Bannerman C. JAMA Pediatr. 2019 Mar 11. doi: 10.1001/jamapediatrics.2019.0032.
Administering antenatal corticosteroids to pregnant women at high risk for preterm birth was a cost-effective intervention that improved infant respiratory outcomes, according to a new study.
“This intervention has a potential cost saving in the United States of approximately $100 million dollars annually from the benefit in the immediate neonatal outcome alone,” Cynthia Gyamfi-Bannerman, MD, of Columbia University, New York, and her associates reported in JAMA Pediatrics. “Because late preterm birth comprises a large proportion of all preterm births, our findings have the potential for a large influence on public health.”
The researchers conducted a retrospective secondary analysis of the randomized Antenatal Late Preterm Steroids (ALPS) clinical trial October 2010 to February 2015. The trial enrolled randomly assigned antenatal administration of betamethasone or placebo to women pregnant with a singleton and at high risk for preterm birth while between 34 weeks, 6 days, and 36 weeks, 0 days, of gestation.
Antenatal corticosteroid administration was regarded as effective if a newborn did not require treatment in the first 72 hours for respiratory distress or illness. Treatment could include “continuous positive airway pressure or high-flow nasal cannula for 2 hours or more, supplemental oxygen with a fraction of inspired oxygen of 30% or more for 4 hours or more, and extracorporeal membrane oxygenation or mechanical ventilation,” Dr. Gyamfi-Bannerman and her associates wrote.
To tally the costs, the researchers used Medicaid rates to estimate the total in 2015 U.S. dollars for betamethasone, outpatient visits or inpatient stays to administer it, and all direct newborn care costs, including neonatal ICU daily costs stratified by respiratory illness severity. Betamethasone administration included an initial 12-mg intramuscular dose followed by another after 24 hours if the infant had not been delivered.
“Because therapy often persists for longer than this 72-hour duration, we measured costs through hospital discharge,” the authors wrote. “The analysis took the perspective of a third-party payer in which we included direct medical costs and associated overhead accruing to hospitals and medical payers for the care of enrolled patients and their infants.”
Among 2,821 mothers not lost to follow-up during the secondary analysis, 1,426 received betamethasone and 1,395 received placebo. For mothers who received betamethasone antenatally, the total mean cost was $4,681 per mother-infant pair. Total mean cost for those in the placebo group was $5,379 per pair, resulting in a significant mean $698 savings (P = .02). Respiratory morbidity was 2.9% lower in infants whose mothers received antenatal corticosteroid treatment.
“Thus, because the treated group had lower costs and this strategy was more effective, administration of betamethasone to women at risk for late preterm birth was judged to be a dominant strategy, which is defined as one in which costs are lower and effectiveness is higher than a comparator (incremental cost-effectiveness ratio [ICER], −23 986),” Dr. Gyamfi-Bannerman and her associates reported. ICER is defined as the difference in mean total cost per patient in the betamethasone and placebo arms divided by the difference in the effectiveness.
Study limitations were an inability to estimate costs according to quality-adjusted life years or to include families’/caregivers’ costs.
SOURCE: Gyamfi-Bannerman C. JAMA Pediatr. 2019 Mar 11. doi: 10.1001/jamapediatrics.2019.0032.
FROM JAMA PEDIATRICS
Daily aspirin associated with lower risk of COPD flareup
Daily aspirin use could reduce the risk of acute exacerbations of chronic obstructive pulmonary disease, new data suggest.
Researchers reported the outcomes of an observational cohort study of 1,698 individuals with COPD, 45% of whom said they were taking daily aspirin at baseline. Their findings were published in Chest.
After a median follow up of 2.7 years, aspirin users had an overall 22% lower incidence of acute COPD exacerbations compared with nonusers. This was largely accounted for by a 25% reduction in moderate exacerbations, but there was no significant difference between aspirin users and nonusers in severe exacerbations.
A similar pattern was seen after just 1 year of follow-up, with an overall 30% reduction in the incidence of exacerbations, a 37% reduction in moderate exacerbations, but no significant reduction in severe exacerbations.
“Though aspirin use has previously been linked with reduced mortality risk in patients with COPD, to our knowledge, this is the first study to investigate the association of daily aspirin use with respiratory morbidity in COPD,” wrote Ashraf Fawzy, MD, of the division of pulmonary and critical care medicine at Johns Hopkins University, Baltimore, and his coauthors.
The association between aspirin use and reduced incidence of exacerbations was stronger among individuals with chronic bronchitis, which prompted the authors to suggest that future studies of aspirin in COPD should focus on participants with chronic bronchitis.
However, the association was not affected by COPD severity, emphysema presence or severity, or cardiometabolic phenotype.
Aspirin users reported better respiratory-specific quality of life than that of nonusers, including 34% lower odds of reporting moderate to severe dyspnea, and better baseline COPD health status.
“Findings of this study add to the existing literature by highlighting that aspirin use is also associated with reduced respiratory morbidity across several domains – including exacerbation risk, quality of life, and dyspnea – factors related to patient well-being and healthcare utilization,” the authors wrote.
Aspirin users were more likely to be white, male, and obese, and less likely to be smokers. They had better lung function but more cardiovascular comorbidities at baseline, although the aspirin users and nonusers were matched on baseline characteristics.
Speculating on the mechanisms by which aspirin might impact COPD exacerbations, the authors noted that the drug has both systemic and local pulmonary mechanisms of action.
For example, a pathway that results in elevated levels of a urinary metabolite in patients with COPD is irreversibly blocked by aspirin. Aspirin also attenuates the elevation of inflammatory markers interleukin-6 and C-reactive protein, which are part of the inflammatory phenotype of COPD. Aspirin has been shown to reduce proinflammatory cytokines in the lung.
The authors did note that aspirin use was self-reported, so they did not have data on dosage or duration of use.
The National Institutes of Health funded the study. Six authors declared advisory board positions, research support, and other funding from the pharmaceutical sector. One author was also a founder of a company commercializing lung image analysis software. No other conflicts of interest were declared.
SOURCE: Fawzy A et al. Chest. 2019 Mar;155(3): 519-27. doi: 10.1016/j.chest.2018.11.028.
Daily aspirin use could reduce the risk of acute exacerbations of chronic obstructive pulmonary disease, new data suggest.
Researchers reported the outcomes of an observational cohort study of 1,698 individuals with COPD, 45% of whom said they were taking daily aspirin at baseline. Their findings were published in Chest.
After a median follow up of 2.7 years, aspirin users had an overall 22% lower incidence of acute COPD exacerbations compared with nonusers. This was largely accounted for by a 25% reduction in moderate exacerbations, but there was no significant difference between aspirin users and nonusers in severe exacerbations.
A similar pattern was seen after just 1 year of follow-up, with an overall 30% reduction in the incidence of exacerbations, a 37% reduction in moderate exacerbations, but no significant reduction in severe exacerbations.
“Though aspirin use has previously been linked with reduced mortality risk in patients with COPD, to our knowledge, this is the first study to investigate the association of daily aspirin use with respiratory morbidity in COPD,” wrote Ashraf Fawzy, MD, of the division of pulmonary and critical care medicine at Johns Hopkins University, Baltimore, and his coauthors.
The association between aspirin use and reduced incidence of exacerbations was stronger among individuals with chronic bronchitis, which prompted the authors to suggest that future studies of aspirin in COPD should focus on participants with chronic bronchitis.
However, the association was not affected by COPD severity, emphysema presence or severity, or cardiometabolic phenotype.
Aspirin users reported better respiratory-specific quality of life than that of nonusers, including 34% lower odds of reporting moderate to severe dyspnea, and better baseline COPD health status.
“Findings of this study add to the existing literature by highlighting that aspirin use is also associated with reduced respiratory morbidity across several domains – including exacerbation risk, quality of life, and dyspnea – factors related to patient well-being and healthcare utilization,” the authors wrote.
Aspirin users were more likely to be white, male, and obese, and less likely to be smokers. They had better lung function but more cardiovascular comorbidities at baseline, although the aspirin users and nonusers were matched on baseline characteristics.
Speculating on the mechanisms by which aspirin might impact COPD exacerbations, the authors noted that the drug has both systemic and local pulmonary mechanisms of action.
For example, a pathway that results in elevated levels of a urinary metabolite in patients with COPD is irreversibly blocked by aspirin. Aspirin also attenuates the elevation of inflammatory markers interleukin-6 and C-reactive protein, which are part of the inflammatory phenotype of COPD. Aspirin has been shown to reduce proinflammatory cytokines in the lung.
The authors did note that aspirin use was self-reported, so they did not have data on dosage or duration of use.
The National Institutes of Health funded the study. Six authors declared advisory board positions, research support, and other funding from the pharmaceutical sector. One author was also a founder of a company commercializing lung image analysis software. No other conflicts of interest were declared.
SOURCE: Fawzy A et al. Chest. 2019 Mar;155(3): 519-27. doi: 10.1016/j.chest.2018.11.028.
Daily aspirin use could reduce the risk of acute exacerbations of chronic obstructive pulmonary disease, new data suggest.
Researchers reported the outcomes of an observational cohort study of 1,698 individuals with COPD, 45% of whom said they were taking daily aspirin at baseline. Their findings were published in Chest.
After a median follow up of 2.7 years, aspirin users had an overall 22% lower incidence of acute COPD exacerbations compared with nonusers. This was largely accounted for by a 25% reduction in moderate exacerbations, but there was no significant difference between aspirin users and nonusers in severe exacerbations.
A similar pattern was seen after just 1 year of follow-up, with an overall 30% reduction in the incidence of exacerbations, a 37% reduction in moderate exacerbations, but no significant reduction in severe exacerbations.
“Though aspirin use has previously been linked with reduced mortality risk in patients with COPD, to our knowledge, this is the first study to investigate the association of daily aspirin use with respiratory morbidity in COPD,” wrote Ashraf Fawzy, MD, of the division of pulmonary and critical care medicine at Johns Hopkins University, Baltimore, and his coauthors.
The association between aspirin use and reduced incidence of exacerbations was stronger among individuals with chronic bronchitis, which prompted the authors to suggest that future studies of aspirin in COPD should focus on participants with chronic bronchitis.
However, the association was not affected by COPD severity, emphysema presence or severity, or cardiometabolic phenotype.
Aspirin users reported better respiratory-specific quality of life than that of nonusers, including 34% lower odds of reporting moderate to severe dyspnea, and better baseline COPD health status.
“Findings of this study add to the existing literature by highlighting that aspirin use is also associated with reduced respiratory morbidity across several domains – including exacerbation risk, quality of life, and dyspnea – factors related to patient well-being and healthcare utilization,” the authors wrote.
Aspirin users were more likely to be white, male, and obese, and less likely to be smokers. They had better lung function but more cardiovascular comorbidities at baseline, although the aspirin users and nonusers were matched on baseline characteristics.
Speculating on the mechanisms by which aspirin might impact COPD exacerbations, the authors noted that the drug has both systemic and local pulmonary mechanisms of action.
For example, a pathway that results in elevated levels of a urinary metabolite in patients with COPD is irreversibly blocked by aspirin. Aspirin also attenuates the elevation of inflammatory markers interleukin-6 and C-reactive protein, which are part of the inflammatory phenotype of COPD. Aspirin has been shown to reduce proinflammatory cytokines in the lung.
The authors did note that aspirin use was self-reported, so they did not have data on dosage or duration of use.
The National Institutes of Health funded the study. Six authors declared advisory board positions, research support, and other funding from the pharmaceutical sector. One author was also a founder of a company commercializing lung image analysis software. No other conflicts of interest were declared.
SOURCE: Fawzy A et al. Chest. 2019 Mar;155(3): 519-27. doi: 10.1016/j.chest.2018.11.028.
FROM CHEST
Bariatric surgery leads to less improvement in black patients
“Per this analysis, there are significant racial disparities in perioperative outcomes, weight loss, and quality of life after bariatric surgery,” wrote lead author Michael H. Wood, MD, of Wayne State University, Detroit, and his coauthors, adding that, “while biological differences may explain some of the disparity in outcomes, environmental, social, and behavioral factors likely play a role.” The study was published online in JAMA Surgery.
This study reviewed data from 14,210 participants in the Michigan Bariatric Surgery Collaborative (MBSC), a state-wide consortium and clinical registry of bariatric surgery patients. Matching cohorts were established for black (n = 7,105) and white (n = 7,105) patients who underwent a primary bariatric operation (Roux-en-Y gastric bypass, sleeve gastrectomy, or adjustable gastric banding) between June 2006 and January 2017. The only significant differences between cohorts – clarified as “never more than 1 or 2 percentage points” – were in regard to income brackets and procedure type.
At 30-day follow-up, the rate of overall complications was higher in black patients (628, 8.8%) than in white patients (481, 6.8%; adjusted odds ratio, 1.33; 95% confidence interval, 1.17-1.51; P = .02), as was the length of stay (mean, 2.2 days vs. 1.9 days; aOR, 0.30; 95% CI, 0.20-0.40; P less than .001). Black patients also had a higher rate of both ED visits (541 [11.6%] vs. 826 [7.6%]; aOR, 1.60; 95% CI, 1.43-1.79; P less than .001) and readmissions (414 [5.8%] vs. 245 [3.5%]; aOR, 1.73; 95% CI, 1.47-2.03; P less than .001).
In addition, at 1-year follow-up, black patients had a lower mean weight loss (32.0 kg vs. 38.3 kg; P less than .001) and percentage of total weight loss (26% vs. 29%; P less than .001) compared with white patients. And though black patients were more likely than white patients to report a high quality of life before surgery (2,672 [49.5%] vs. 2,354 [41.4%]; P less than .001), they were less likely to do so 1 year afterward (1,379 [87.2%] vs. 2,133 [90.4%]; P = .002).
The coauthors acknowledged the limitations of their study, including potential unmeasured factors between cohorts such as disease duration or severity. They also noted that a wider time horizon than 30 days post surgery could have altered the results, although “serious adverse events and resource use tend to be highest within the first month after surgery, and we anticipate that this effect would have been negligible.”
The study was funded by Blue Cross Blue Shield Michigan/Blue Care Network. Dr. Wood reported no conflicts of interest. Three of his coauthors reported receiving salary support from Blue Cross Blue Shield Michigan/Blue Care Network for their work with the MBSC, and one other coauthor reported receiving an honorarium for being the MBSC’s executive committee chair.
SOURCE: Wood MH et al. JAMA Surg. 2019 Mar 6. doi: 10.1001/jamasurg.2019.0029.
The well-documented disparities between black and white patients after bariatric surgery are brought back to the forefront via to this study from Wood et al., according to Brian Hodgens, MD, and Kenric M. Murayama, MD, of the University of Hawaii, Honolulu.
Some of the findings hint at the cultural differences that permeate the time before and after a surgery like this: In particular, they highlighted how black patients were more likely to report good or very good quality of life before surgery but less likely after. This could be related to a “difference in perceptions of obesity by black patients,” where they are more hesitant to pursue the surgery than their white counterparts, Dr. Hodgens and Dr. Murayama wrote.
More work is needed, they added, but “this study and others like it can better equip practicing bariatric surgeons to educate themselves and patients on expectations before and after bariatric surgery.”
These comments are adapted from an accompanying editorial ( JAMA Surg. 2019 Mar 6. doi: 1 0.1001/jamasurg.2019.0067 ). Dr. Murayama reported receiving personal fees from Medtronic outside the submitted work.
The well-documented disparities between black and white patients after bariatric surgery are brought back to the forefront via to this study from Wood et al., according to Brian Hodgens, MD, and Kenric M. Murayama, MD, of the University of Hawaii, Honolulu.
Some of the findings hint at the cultural differences that permeate the time before and after a surgery like this: In particular, they highlighted how black patients were more likely to report good or very good quality of life before surgery but less likely after. This could be related to a “difference in perceptions of obesity by black patients,” where they are more hesitant to pursue the surgery than their white counterparts, Dr. Hodgens and Dr. Murayama wrote.
More work is needed, they added, but “this study and others like it can better equip practicing bariatric surgeons to educate themselves and patients on expectations before and after bariatric surgery.”
These comments are adapted from an accompanying editorial ( JAMA Surg. 2019 Mar 6. doi: 1 0.1001/jamasurg.2019.0067 ). Dr. Murayama reported receiving personal fees from Medtronic outside the submitted work.
The well-documented disparities between black and white patients after bariatric surgery are brought back to the forefront via to this study from Wood et al., according to Brian Hodgens, MD, and Kenric M. Murayama, MD, of the University of Hawaii, Honolulu.
Some of the findings hint at the cultural differences that permeate the time before and after a surgery like this: In particular, they highlighted how black patients were more likely to report good or very good quality of life before surgery but less likely after. This could be related to a “difference in perceptions of obesity by black patients,” where they are more hesitant to pursue the surgery than their white counterparts, Dr. Hodgens and Dr. Murayama wrote.
More work is needed, they added, but “this study and others like it can better equip practicing bariatric surgeons to educate themselves and patients on expectations before and after bariatric surgery.”
These comments are adapted from an accompanying editorial ( JAMA Surg. 2019 Mar 6. doi: 1 0.1001/jamasurg.2019.0067 ). Dr. Murayama reported receiving personal fees from Medtronic outside the submitted work.
“Per this analysis, there are significant racial disparities in perioperative outcomes, weight loss, and quality of life after bariatric surgery,” wrote lead author Michael H. Wood, MD, of Wayne State University, Detroit, and his coauthors, adding that, “while biological differences may explain some of the disparity in outcomes, environmental, social, and behavioral factors likely play a role.” The study was published online in JAMA Surgery.
This study reviewed data from 14,210 participants in the Michigan Bariatric Surgery Collaborative (MBSC), a state-wide consortium and clinical registry of bariatric surgery patients. Matching cohorts were established for black (n = 7,105) and white (n = 7,105) patients who underwent a primary bariatric operation (Roux-en-Y gastric bypass, sleeve gastrectomy, or adjustable gastric banding) between June 2006 and January 2017. The only significant differences between cohorts – clarified as “never more than 1 or 2 percentage points” – were in regard to income brackets and procedure type.
At 30-day follow-up, the rate of overall complications was higher in black patients (628, 8.8%) than in white patients (481, 6.8%; adjusted odds ratio, 1.33; 95% confidence interval, 1.17-1.51; P = .02), as was the length of stay (mean, 2.2 days vs. 1.9 days; aOR, 0.30; 95% CI, 0.20-0.40; P less than .001). Black patients also had a higher rate of both ED visits (541 [11.6%] vs. 826 [7.6%]; aOR, 1.60; 95% CI, 1.43-1.79; P less than .001) and readmissions (414 [5.8%] vs. 245 [3.5%]; aOR, 1.73; 95% CI, 1.47-2.03; P less than .001).
In addition, at 1-year follow-up, black patients had a lower mean weight loss (32.0 kg vs. 38.3 kg; P less than .001) and percentage of total weight loss (26% vs. 29%; P less than .001) compared with white patients. And though black patients were more likely than white patients to report a high quality of life before surgery (2,672 [49.5%] vs. 2,354 [41.4%]; P less than .001), they were less likely to do so 1 year afterward (1,379 [87.2%] vs. 2,133 [90.4%]; P = .002).
The coauthors acknowledged the limitations of their study, including potential unmeasured factors between cohorts such as disease duration or severity. They also noted that a wider time horizon than 30 days post surgery could have altered the results, although “serious adverse events and resource use tend to be highest within the first month after surgery, and we anticipate that this effect would have been negligible.”
The study was funded by Blue Cross Blue Shield Michigan/Blue Care Network. Dr. Wood reported no conflicts of interest. Three of his coauthors reported receiving salary support from Blue Cross Blue Shield Michigan/Blue Care Network for their work with the MBSC, and one other coauthor reported receiving an honorarium for being the MBSC’s executive committee chair.
SOURCE: Wood MH et al. JAMA Surg. 2019 Mar 6. doi: 10.1001/jamasurg.2019.0029.
“Per this analysis, there are significant racial disparities in perioperative outcomes, weight loss, and quality of life after bariatric surgery,” wrote lead author Michael H. Wood, MD, of Wayne State University, Detroit, and his coauthors, adding that, “while biological differences may explain some of the disparity in outcomes, environmental, social, and behavioral factors likely play a role.” The study was published online in JAMA Surgery.
This study reviewed data from 14,210 participants in the Michigan Bariatric Surgery Collaborative (MBSC), a state-wide consortium and clinical registry of bariatric surgery patients. Matching cohorts were established for black (n = 7,105) and white (n = 7,105) patients who underwent a primary bariatric operation (Roux-en-Y gastric bypass, sleeve gastrectomy, or adjustable gastric banding) between June 2006 and January 2017. The only significant differences between cohorts – clarified as “never more than 1 or 2 percentage points” – were in regard to income brackets and procedure type.
At 30-day follow-up, the rate of overall complications was higher in black patients (628, 8.8%) than in white patients (481, 6.8%; adjusted odds ratio, 1.33; 95% confidence interval, 1.17-1.51; P = .02), as was the length of stay (mean, 2.2 days vs. 1.9 days; aOR, 0.30; 95% CI, 0.20-0.40; P less than .001). Black patients also had a higher rate of both ED visits (541 [11.6%] vs. 826 [7.6%]; aOR, 1.60; 95% CI, 1.43-1.79; P less than .001) and readmissions (414 [5.8%] vs. 245 [3.5%]; aOR, 1.73; 95% CI, 1.47-2.03; P less than .001).
In addition, at 1-year follow-up, black patients had a lower mean weight loss (32.0 kg vs. 38.3 kg; P less than .001) and percentage of total weight loss (26% vs. 29%; P less than .001) compared with white patients. And though black patients were more likely than white patients to report a high quality of life before surgery (2,672 [49.5%] vs. 2,354 [41.4%]; P less than .001), they were less likely to do so 1 year afterward (1,379 [87.2%] vs. 2,133 [90.4%]; P = .002).
The coauthors acknowledged the limitations of their study, including potential unmeasured factors between cohorts such as disease duration or severity. They also noted that a wider time horizon than 30 days post surgery could have altered the results, although “serious adverse events and resource use tend to be highest within the first month after surgery, and we anticipate that this effect would have been negligible.”
The study was funded by Blue Cross Blue Shield Michigan/Blue Care Network. Dr. Wood reported no conflicts of interest. Three of his coauthors reported receiving salary support from Blue Cross Blue Shield Michigan/Blue Care Network for their work with the MBSC, and one other coauthor reported receiving an honorarium for being the MBSC’s executive committee chair.
SOURCE: Wood MH et al. JAMA Surg. 2019 Mar 6. doi: 10.1001/jamasurg.2019.0029.
FROM JAMA SURGERY
Peer-Review Transparency
Federal health care providers live under a microscope, so it seems only fair that we at Fed Pract honor that reality and open ourselves up to scrutiny as well.1 We hope that by shedding light on our peer-review process and manuscript acceptance rate, we will not only highlight our accomplishments, but identify areas for improvement.
Free access to Fed Pract content has always been our priority. While many journals charge authors or readers, Fed Pract has been and will remain free for readers and authors.2 Advertising enables the journal to support this free model of publishing, but we take care to ensure that advertisements do not influence content in any way. Our advertising policy can be found at www.mdedge.com/fedprac/page/advertising.
In January 2019, Fed Pract placed > 400 peer-reviewed articles published since January 2015 in the PubMed Central (PMC) database (ncbi.nlm.nih.gov/pmc). The full text of these and all future Fed Pract peer-reviewed articles will be available at PMC (no registration required), and the citations also will be included in PubMed. We hope that this process will make it even easier for anyone to access our authors’ works.
In 2018 about 36,000 federal health care providers (HCPs) received hard copies of this journal. The print journal is free, but circulation is limited to HCPs who work at the US Department of Veterans Affairs (VA), US Department of Defense (DoD), and the US Public Health Service (PHS). The mdedge.com/fedprac website, which includes every article published since 2003, had 1.4 million page views in 2018. After reading 3 online articles, readers in the US are asked to complete a simple registration form to help us better customize the reader experience. In some cases, international readers may be asked to pay for access to articles online; however, any VA, DoD, or PHS officer stationed overseas can contact the editorial staff (fedprac@mdedge.com) to ensure that they can access the articles for free.
In 2018 the journal received 164 manuscripts and published 94 articles written by 357 different federal HCPs. The 164 manuscript submissions represented a 45% growth over previous years. Not surprisingly, the increased rate of submissions began shortly after the May 2018 announcement that journal articles would be included in PMC. Most of those articles (83%) were submitted unsolicited.
Fed Pract has always prided itself on being an early promoter of interdisciplinary health care professional publications. Nearly half of its listed authors were physicians (48%), while pharmacists made up the next largest cohort (18%). There were smaller numbers of PhDs, nurses, social workers, and physical therapists. The majority were written by HCPs affiliated with the VA (95% of articles and 93% of authors), and no articles in 2018 were written by PHS officers. Physicians comprise about two-thirds of the audience, while pharmacists make up 17% and nurses 9%. PHS and DoD HCPs make up 19% of the Fed Pract audience, suggesting that the journal needs to do more work to encourage these HCPs to contribute articles to the journal.3
Articles published in 2018 covered a broad range of topics from “Anesthesia Care Practice Models in the VHA” and “Army Behavioral Health System” to “Vitreous Hemorrhage in the Setting of a Vascular Loop” and “A Workforce Assessment of VA Home-Based Primary Care Pharmacists.” Categorizing the articles is a challenge. Few health care topics fit neatly into a single topic or specialty. This is especially true in federal health care where much of the care is delivered by multidisciplinary patient-centered medical homes or patient aligned care teams. Nevertheless, a few broad outlines can be discerned. Articles were roughly split between primary care and hospital-based and/or specialty care topics; one-quarter of the articles were case studies or case series articles, and about 20% were editorials or opinion columns. Nineteen articles dealt explicitly with chronic conditions, and 10 articles focused on mental health care.
Peer reviewers are an essential part of the process. Reviewers are blinded to the identityof the authors, ensuring fairness and reducing potential conflicts of interest. We are extremely grateful to each and every reviewer for the time and energy they contribute to the journal. Peer reviewers do not get nearly enough recognition for their important work. In 2018 Fed Pract invited 1,205 reviewers for 164 manuscript submissions and 94 manuscript revisions. More than 200 different reviewers submitted 487 reviews with a median (SD) of 2 reviews (1.8) and a range of 1 to 10. The top 20 reviewers completed 134 reviews with a median (SD) of 6 reviews (1.2). The results stand in contrast to some journals that must offer many invitations per review and depend on a small number of reviewers.1,4-6
The reviewers recommended to reject 14% and to revise 26% of the articles, which is a much lower rejection rate than many other journals (Table).4 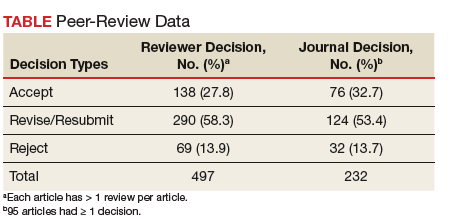
These data suggest that Fed Pract and its peer-review process is on a sound foundation but needs to make improvements. Moving into 2019, the journal expects that an increasing number of submissions will require a higher rejection rate. Moreover, we will need to do a better job reaching out to underrepresented portions of our audience. To decrease the time to publication for accepted manuscripts, in 2019 we will publish more articles online ahead of the print publication as we strive to improve the experience for authors, reviewers, readers, and the entire Fed Pract audience.
None of this work can be done without our small and dedicated staff. I would like to thank Managing Editor Joyce Brody who sent out each and every one of those reviewer invitations, Deputy Editor Robert Fee, who manages the special issues, Web Editor Teraya Smith, who runs our entire digital operation, and of course, Editor in Chief Cynthia Geppert, who oversees it all. Finally, it is important that you let us know how we are doing and whether we are meeting your needs. Visit mdedge.com/fedprac to take the readership survey or reach out to me at rpaul@mdedge.com.
1. Geppert CMA. Caring under a microscope. Fed Pract. 2018;35(7):6-7.
2. Smith R. Peer review: a flawed process at the heart of science and journals. J R Soc Med. 2006;99(4):178-182.
3. BPA Worldwide. Federal Practitioner brand report for the 6 month period ending June 2018. https://www.frontlinemedcom.com/wp-content/uploads/FEDPRAC_BPA.pdf. Updated June 2018. Accessed March 5, 2019.
4. Fontanarosa PB, Bauchner H, Golub RM. Thank you to JAMA authors, peer reviewers, and readers. JAMA. 2017;317(8):812-813.
5. Publons, Clarivate Analytics. 2018 global state of peer review. https://publons.com/static/Publons-Global-State-Of-Peer-Review-2018.pdf. Published September 2018. Accessed March 5, 2019.
6. Malcom D. It’s time we fix the peer review system. Am J Pharm Educ. 2018;82(5):7144.
Federal health care providers live under a microscope, so it seems only fair that we at Fed Pract honor that reality and open ourselves up to scrutiny as well.1 We hope that by shedding light on our peer-review process and manuscript acceptance rate, we will not only highlight our accomplishments, but identify areas for improvement.
Free access to Fed Pract content has always been our priority. While many journals charge authors or readers, Fed Pract has been and will remain free for readers and authors.2 Advertising enables the journal to support this free model of publishing, but we take care to ensure that advertisements do not influence content in any way. Our advertising policy can be found at www.mdedge.com/fedprac/page/advertising.
In January 2019, Fed Pract placed > 400 peer-reviewed articles published since January 2015 in the PubMed Central (PMC) database (ncbi.nlm.nih.gov/pmc). The full text of these and all future Fed Pract peer-reviewed articles will be available at PMC (no registration required), and the citations also will be included in PubMed. We hope that this process will make it even easier for anyone to access our authors’ works.
In 2018 about 36,000 federal health care providers (HCPs) received hard copies of this journal. The print journal is free, but circulation is limited to HCPs who work at the US Department of Veterans Affairs (VA), US Department of Defense (DoD), and the US Public Health Service (PHS). The mdedge.com/fedprac website, which includes every article published since 2003, had 1.4 million page views in 2018. After reading 3 online articles, readers in the US are asked to complete a simple registration form to help us better customize the reader experience. In some cases, international readers may be asked to pay for access to articles online; however, any VA, DoD, or PHS officer stationed overseas can contact the editorial staff (fedprac@mdedge.com) to ensure that they can access the articles for free.
In 2018 the journal received 164 manuscripts and published 94 articles written by 357 different federal HCPs. The 164 manuscript submissions represented a 45% growth over previous years. Not surprisingly, the increased rate of submissions began shortly after the May 2018 announcement that journal articles would be included in PMC. Most of those articles (83%) were submitted unsolicited.
Fed Pract has always prided itself on being an early promoter of interdisciplinary health care professional publications. Nearly half of its listed authors were physicians (48%), while pharmacists made up the next largest cohort (18%). There were smaller numbers of PhDs, nurses, social workers, and physical therapists. The majority were written by HCPs affiliated with the VA (95% of articles and 93% of authors), and no articles in 2018 were written by PHS officers. Physicians comprise about two-thirds of the audience, while pharmacists make up 17% and nurses 9%. PHS and DoD HCPs make up 19% of the Fed Pract audience, suggesting that the journal needs to do more work to encourage these HCPs to contribute articles to the journal.3
Articles published in 2018 covered a broad range of topics from “Anesthesia Care Practice Models in the VHA” and “Army Behavioral Health System” to “Vitreous Hemorrhage in the Setting of a Vascular Loop” and “A Workforce Assessment of VA Home-Based Primary Care Pharmacists.” Categorizing the articles is a challenge. Few health care topics fit neatly into a single topic or specialty. This is especially true in federal health care where much of the care is delivered by multidisciplinary patient-centered medical homes or patient aligned care teams. Nevertheless, a few broad outlines can be discerned. Articles were roughly split between primary care and hospital-based and/or specialty care topics; one-quarter of the articles were case studies or case series articles, and about 20% were editorials or opinion columns. Nineteen articles dealt explicitly with chronic conditions, and 10 articles focused on mental health care.
Peer reviewers are an essential part of the process. Reviewers are blinded to the identityof the authors, ensuring fairness and reducing potential conflicts of interest. We are extremely grateful to each and every reviewer for the time and energy they contribute to the journal. Peer reviewers do not get nearly enough recognition for their important work. In 2018 Fed Pract invited 1,205 reviewers for 164 manuscript submissions and 94 manuscript revisions. More than 200 different reviewers submitted 487 reviews with a median (SD) of 2 reviews (1.8) and a range of 1 to 10. The top 20 reviewers completed 134 reviews with a median (SD) of 6 reviews (1.2). The results stand in contrast to some journals that must offer many invitations per review and depend on a small number of reviewers.1,4-6
The reviewers recommended to reject 14% and to revise 26% of the articles, which is a much lower rejection rate than many other journals (Table).4
These data suggest that Fed Pract and its peer-review process is on a sound foundation but needs to make improvements. Moving into 2019, the journal expects that an increasing number of submissions will require a higher rejection rate. Moreover, we will need to do a better job reaching out to underrepresented portions of our audience. To decrease the time to publication for accepted manuscripts, in 2019 we will publish more articles online ahead of the print publication as we strive to improve the experience for authors, reviewers, readers, and the entire Fed Pract audience.
None of this work can be done without our small and dedicated staff. I would like to thank Managing Editor Joyce Brody who sent out each and every one of those reviewer invitations, Deputy Editor Robert Fee, who manages the special issues, Web Editor Teraya Smith, who runs our entire digital operation, and of course, Editor in Chief Cynthia Geppert, who oversees it all. Finally, it is important that you let us know how we are doing and whether we are meeting your needs. Visit mdedge.com/fedprac to take the readership survey or reach out to me at rpaul@mdedge.com.
Federal health care providers live under a microscope, so it seems only fair that we at Fed Pract honor that reality and open ourselves up to scrutiny as well.1 We hope that by shedding light on our peer-review process and manuscript acceptance rate, we will not only highlight our accomplishments, but identify areas for improvement.
Free access to Fed Pract content has always been our priority. While many journals charge authors or readers, Fed Pract has been and will remain free for readers and authors.2 Advertising enables the journal to support this free model of publishing, but we take care to ensure that advertisements do not influence content in any way. Our advertising policy can be found at www.mdedge.com/fedprac/page/advertising.
In January 2019, Fed Pract placed > 400 peer-reviewed articles published since January 2015 in the PubMed Central (PMC) database (ncbi.nlm.nih.gov/pmc). The full text of these and all future Fed Pract peer-reviewed articles will be available at PMC (no registration required), and the citations also will be included in PubMed. We hope that this process will make it even easier for anyone to access our authors’ works.
In 2018 about 36,000 federal health care providers (HCPs) received hard copies of this journal. The print journal is free, but circulation is limited to HCPs who work at the US Department of Veterans Affairs (VA), US Department of Defense (DoD), and the US Public Health Service (PHS). The mdedge.com/fedprac website, which includes every article published since 2003, had 1.4 million page views in 2018. After reading 3 online articles, readers in the US are asked to complete a simple registration form to help us better customize the reader experience. In some cases, international readers may be asked to pay for access to articles online; however, any VA, DoD, or PHS officer stationed overseas can contact the editorial staff (fedprac@mdedge.com) to ensure that they can access the articles for free.
In 2018 the journal received 164 manuscripts and published 94 articles written by 357 different federal HCPs. The 164 manuscript submissions represented a 45% growth over previous years. Not surprisingly, the increased rate of submissions began shortly after the May 2018 announcement that journal articles would be included in PMC. Most of those articles (83%) were submitted unsolicited.
Fed Pract has always prided itself on being an early promoter of interdisciplinary health care professional publications. Nearly half of its listed authors were physicians (48%), while pharmacists made up the next largest cohort (18%). There were smaller numbers of PhDs, nurses, social workers, and physical therapists. The majority were written by HCPs affiliated with the VA (95% of articles and 93% of authors), and no articles in 2018 were written by PHS officers. Physicians comprise about two-thirds of the audience, while pharmacists make up 17% and nurses 9%. PHS and DoD HCPs make up 19% of the Fed Pract audience, suggesting that the journal needs to do more work to encourage these HCPs to contribute articles to the journal.3
Articles published in 2018 covered a broad range of topics from “Anesthesia Care Practice Models in the VHA” and “Army Behavioral Health System” to “Vitreous Hemorrhage in the Setting of a Vascular Loop” and “A Workforce Assessment of VA Home-Based Primary Care Pharmacists.” Categorizing the articles is a challenge. Few health care topics fit neatly into a single topic or specialty. This is especially true in federal health care where much of the care is delivered by multidisciplinary patient-centered medical homes or patient aligned care teams. Nevertheless, a few broad outlines can be discerned. Articles were roughly split between primary care and hospital-based and/or specialty care topics; one-quarter of the articles were case studies or case series articles, and about 20% were editorials or opinion columns. Nineteen articles dealt explicitly with chronic conditions, and 10 articles focused on mental health care.
Peer reviewers are an essential part of the process. Reviewers are blinded to the identityof the authors, ensuring fairness and reducing potential conflicts of interest. We are extremely grateful to each and every reviewer for the time and energy they contribute to the journal. Peer reviewers do not get nearly enough recognition for their important work. In 2018 Fed Pract invited 1,205 reviewers for 164 manuscript submissions and 94 manuscript revisions. More than 200 different reviewers submitted 487 reviews with a median (SD) of 2 reviews (1.8) and a range of 1 to 10. The top 20 reviewers completed 134 reviews with a median (SD) of 6 reviews (1.2). The results stand in contrast to some journals that must offer many invitations per review and depend on a small number of reviewers.1,4-6
The reviewers recommended to reject 14% and to revise 26% of the articles, which is a much lower rejection rate than many other journals (Table).4
These data suggest that Fed Pract and its peer-review process is on a sound foundation but needs to make improvements. Moving into 2019, the journal expects that an increasing number of submissions will require a higher rejection rate. Moreover, we will need to do a better job reaching out to underrepresented portions of our audience. To decrease the time to publication for accepted manuscripts, in 2019 we will publish more articles online ahead of the print publication as we strive to improve the experience for authors, reviewers, readers, and the entire Fed Pract audience.
None of this work can be done without our small and dedicated staff. I would like to thank Managing Editor Joyce Brody who sent out each and every one of those reviewer invitations, Deputy Editor Robert Fee, who manages the special issues, Web Editor Teraya Smith, who runs our entire digital operation, and of course, Editor in Chief Cynthia Geppert, who oversees it all. Finally, it is important that you let us know how we are doing and whether we are meeting your needs. Visit mdedge.com/fedprac to take the readership survey or reach out to me at rpaul@mdedge.com.
1. Geppert CMA. Caring under a microscope. Fed Pract. 2018;35(7):6-7.
2. Smith R. Peer review: a flawed process at the heart of science and journals. J R Soc Med. 2006;99(4):178-182.
3. BPA Worldwide. Federal Practitioner brand report for the 6 month period ending June 2018. https://www.frontlinemedcom.com/wp-content/uploads/FEDPRAC_BPA.pdf. Updated June 2018. Accessed March 5, 2019.
4. Fontanarosa PB, Bauchner H, Golub RM. Thank you to JAMA authors, peer reviewers, and readers. JAMA. 2017;317(8):812-813.
5. Publons, Clarivate Analytics. 2018 global state of peer review. https://publons.com/static/Publons-Global-State-Of-Peer-Review-2018.pdf. Published September 2018. Accessed March 5, 2019.
6. Malcom D. It’s time we fix the peer review system. Am J Pharm Educ. 2018;82(5):7144.
1. Geppert CMA. Caring under a microscope. Fed Pract. 2018;35(7):6-7.
2. Smith R. Peer review: a flawed process at the heart of science and journals. J R Soc Med. 2006;99(4):178-182.
3. BPA Worldwide. Federal Practitioner brand report for the 6 month period ending June 2018. https://www.frontlinemedcom.com/wp-content/uploads/FEDPRAC_BPA.pdf. Updated June 2018. Accessed March 5, 2019.
4. Fontanarosa PB, Bauchner H, Golub RM. Thank you to JAMA authors, peer reviewers, and readers. JAMA. 2017;317(8):812-813.
5. Publons, Clarivate Analytics. 2018 global state of peer review. https://publons.com/static/Publons-Global-State-Of-Peer-Review-2018.pdf. Published September 2018. Accessed March 5, 2019.
6. Malcom D. It’s time we fix the peer review system. Am J Pharm Educ. 2018;82(5):7144.
Juvenile idiopathic arthritis: Old disease, new tactics
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.

SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27

Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.
SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27
Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
Juvenile idiopathic arthritis (JIA) is a clinically heterogeneous group of arthritides that are characterized by onset before 16 years of age and defined in part as lasting ≥6 weeks.1 Significantly, the etiology of JIA is unknown, making it a diagnosis of exclusion.2
The most common autoimmune condition of childhood, JIA has a prevalence of 3.8 to 400 affected children for every 100,000 people.3,4 As the leading cause of musculoskeletal disability in children,5 and comprising 7 categories of disease, JIA must be managed with appropriate initial and ongoing intervention.
The amalgam of care that a JIA patient requires—medical, social, physical, psychological—calls for a primary care physician’s expert ability to collaborate and coordinate with medical specialists and subspecialists, including rheumatology, ophthalmology, social work, physical and occupational therapy, and psychology. The goal? As this article describes, the goal is to provide prompt diagnosis, suitable and effective intervention, and continuity of care. (JIA is a lifelong disease, in many cases.)
How JIA is classifiedfor diagnosis and treatment
JIA comprises 7 categories, or classes.6 The scheme devised by the International League of Associations for Rheumatology (ILAR), now widely accepted, classifies JIA on the basis of clinical and biochemical markers that aid detection and treatment of the disorder, as well as research. (See “How efforts to classify JIA have caused confusion.”7-10) The ILAR classes (TABLE11) are:
- enthesitis-related arthritis (ERA)
- extended oligo-articular JIA (eoJIA), which involves ≤4 joints
- juvenile psoriatic arthritis (jPsA)
- rheumatoid factor (RF)-positive polyarticular JIA (RF+ pJIA)
- RF-negative polyarticular JIA (RF– pJIA)
- systemic-onset JIA (sJIA)
- undifferentiated JIA, which, generally, involves ≥4 joints.
SIDEBAR
How efforts to classiy JIA have caused confusion7-10
Various classifications of juvenile arthritis have been proposed and used over the past 3 decades. First was the American College of Rheumatology’s 1972 criteria for juvenile rheumatoid arthritis7; next came the European League against Rheumatism (EULAR) criteria for juvenile chronic arthritis, developed in 1977.8 Being contemporaneous, the 2 classifications led to a complicated, dichotomous definition of JIA among clinicians and researchers.
As a result of this disarray, the 1997 Durban, South Africa, meeting of the Pediatric Standing Committee of the International League of Associations for Rheumatology (ILAR)9 proposed that juvenile idiopathic arthritis be adopted as the umbrella term for the misunderstood terms juvenile rheumatoid arthritis and juvenile chronic arthritis. The intent of including “idiopathic” in the term was to acknowledge that the cause of these diseases was (and is still) unknown.
The novel classification proposed by the Pediatric Standing Committee was followed, in 2001, by an ILAR task force meeting in Edmonton, Alberta, Canada, on the classification of childhood arthritis. The outcome was a recommendation to add exclusion and inclusion criteria, to make all classes of JIA mutually exclusive.10 Most recently, as discussed in the body of this article, updated ILAR guidelines on JIA classification emphasize 1) heterogeneity among the 7 disease subtypes and 2) the fact that overlapping and exclusive features exist from class to class.
Updated guidelines regarding the 7 ILAR classes of JIA emphasize heterogeneity among disease subtypes, with overlapping and exclusive features noted from class to class.11
Extended oligo-articular JIA (27%-56%), pJIA (13%-35%), sJIA (4%-17%), and ERA,(3%-11%) are the most common JIA subtypes,12 with age of onset and sex predilection differing according to JIA class.11 The disease occurs more often in girls than in boys,11 and the predisposition is higher among Whites and Asians. The incidence of JIA (all classes taken together, for every 100,000 people) is: in Japan, 10 to 15 cases13; in Turkey, 64 cases14; in Norway, 65 cases15; and in the United States and Canada, taken together, 10 to 15 cases.16
What causes JIA?
The etiology of JIA remains unclear. It is known that the disease involves inflammation of the synovium and destruction of hard and soft tissues in joints.17 It has been postulated, therefore, that a combination of genetic, environmental, and immunogenic mechanisms might be responsible for JIA.
Continue to: For example, there is an increased...
For example, there is an increased frequency of autoimmune diseases among JIA patients.18 There are also reports documenting an increased rate of infection, including with enteric pathogens, parvovirus B,19 rubella, mumps, hepatitis B, Epstein-Barr virus, mycoplasma, and chlamydia.19 Stress and trauma have also been implicated.12
The T-lymphocyte percentage is increased in the synovial fluid of JIA patients, although that percentage varies from subtype to subtype.20 This elevation results in an increase in the number of macrophages, which are induced by secreted cytokines to produce interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-a). This activity of cellular immunity leads to joint destruction.21
Clinical features
The most common signs and symptoms of JIA are arthralgias (39%), arthritis (25%), fever (18%), limping (9%), rash (8%), abdominal pain (1.3%), and uveitis (1.3%).15 Forty percent of JIA patients are reported to have temporomandibular joint involvement at some point in their life; mandibular asymmetry secondary to condylar resorption and remodeling17 is the most common presenting complaint—not arthralgia or pain, as would be expected.
Most JIA patients (52%) first present to the emergency department; another 42% present to the office of a general medical practitioner.15 On average, 3 visits to a physician, over the course of approximately 3 months, are made before a definitive diagnosis (usually by a pediatric rheumatologist) is made.15
Pertinent questions to ask a patient who has a confirmed diagnosis of JIA include the nature, severity, and duration of morning stiffness and pain, as well as any encumbering factors to regular functioning at home or school.22 Different scoring charts can be used to determine the extent of pain and disability, including the Juvenile Arthritis Disease Activity Score (JADAS)23 and the clinical JADAS (cJADAS),24 which measure minimal disease activity25 and clinically inactive disease26 cutoffs.
Continue to: Macrophage-activating syndrome increases risk of morbidity, mortality
Macrophage-activating syndrome increases risk of morbidity, mortality
An overactivation and expansion of T lymphocytes and macrophagic histiocytes with hemophagocytic activity, macrophage-activating syndrome (MAS) occurs in approximately 10% of JIA patients,27 increasing their risk of morbidity and mortality. The syndrome, which typically presents as fever, seizures, hypotension, purpura, hepatitis, splenomegaly, and occasionally, multisystem organ failure, is seen in 30% to 40% of sJIA patients; approximately 11% of them experience sudden death as a consequence.28
The clinical setting of MAS includes presenting symptoms of fever and a salmon-pink macular rash (FIGURE). For many sJIA patients with MAS, the diagnosis is made when laboratory results show hyperferritinemia, thrombocytopenia, anemia, leukopenia, coagulopathy, and elevated levels of C-reactive protein and D-dimer.27
Different classes, different features
The following clinical profiles have been documented in different classes of JIA:
Systemic JIA presents with intermittent fever of at least 2 weeks’ duration, arthritis, and occasionally, a rash.
Extended oligo-articular JIA involves pain, in a mono-articular lower-extremity joint, that can develop suddenly or insidiously, and is characterized by early-morning stiffness and uveitis (especially in early-onset, antinuclear antibody-positive JIA patients).
Continue to: Poly-articular JIA
Poly-articular JIA patients present with mild fever, weight loss, and anemia.
Enthesis-related arthritis patients have findings of enthesopathy; asymmetric arthritis of the lower extremities, particularly the Achilles tendon29; and recurrent acute, symptomatic iridocyclitis.30
Juvenile psoriatic arthritis can involve any joint but is readily differentiated from pJIA by involvement of distal interphalangeal joints and psoriatic skin and nail changes.29
Investigations
Imaging
Radiography is still the most widely used imaging tool for making the diagnosis of JIA. Plain films demonstrate structural joint damage and disturbances of growth and maturation in bones. Radiography has poor sensitivity for detecting acute synovitis and limited utility in visualizing erosion changes early in the course of disease, however, which has led to increased use of ultrasonography (US) and contrast-enhanced magnetic resonance imaging (MRI) to diagnose JIA.30
Contrast-enhanced MRI is superior to US for detecting early inflammation and monitoring subsequent joint disease. Of course, MRI is more expensive than US, and less widely available. Other imaging options are computed tomography and positron emission tomography, but these scans are not as sensitive as contrast-enhanced MRI and have the disadvantage of radiation exposure (in the former) and cost (in the latter).
Continue to: Laboratory testing
Laboratory testing
No diagnostic tests for JIA exist. Assays of acute-phase reactants, including C-reactive protein, the erythrocyte sedimentation rate, and serum amyloid-A proteins, can be utilized to demonstrate inflammation but not to confirm the diagnosis. For some classes of JIA, various tests, including rheumatoid factor, antinuclear antibody, human leukocyte antigen B-27, and cyclic citrullated peptide antibodies, can be used to confirm a specific class but, again, are not recommended for confirming JIA.6
The complete blood count, blood cultures, and tests of uric acid and lactate dehydrogenase can be ordered during treatment to monitor for complications, such as malignancy, infection, MAS, and sepsis.
Treatment is based on disease class
Nonsteroidal anti-inflammatory drugs (NSAIDs) and intra-articular steroids are used in all JIA classes, as an adjunct to class-specific treatment, or as induction agents.31 These therapies, although they alleviate acute signs and symptoms, such as pain, inflammation, swelling and joint contractures, are not useful for long-term treatment of JIA because they do not halt disease progression.
Systemic steroids can be utilized in exceptional cases, including chronic uveitis with arthritis or in patients with destructive arthritis and poor prognostic features, including cyclic citrullated peptide antibodies, positive RF, erosions, and joint-space narrowing.32
Other drugs. Options include traditional disease-modifying anti-rheumatic drugs (csDMARDs), such as methotrexate and leflunomide; biologic agents, such as TNF-a inhibitors (eg, etanercept, adalimumab, and infliximab); and anti-IL monoclonal antibody drugs (eg, the IL-6 inhibitor tocilizumab and IL-1 inhibitors anakinra, and canakinumab).31 Indications by class include:
- csDMARDs as first-line therapy in persistent eoJIA and pJIA;
- TNF-Symbolα inhibitors for refractory eoJIA and for pJIA episodes31;
- tocilizumab, recommended for sJIA patients who have persistent systemic signs; and
- anakinra and canakinumab for refractory SJIA patients.32
Continue to: Failure
Failure
When treatment of JIA fails with a given drug, options include increasing the dosage; switching to another agent in the same drug class; switching to a different class; and combining an NSAID with a csDMARD or a biologic agent.32 In class-specific JIA cases, a change in a drug regimen is warranted on the basis of the evidence-based historical clinical response rate.32
What is the prognosis?
Treatment of JIA with novel agents, such as biologics, has opened up the possibility that JIA patients can live not just with suppressed symptoms but immunologically inactive disease. This is the result of better understanding of the pathogenesis of JIA and the mechanism of action of targeted drugs, and identification of biomarkers that are helpful in predicting prognosis, adverse effects, and response to treatment.
JIA is often a lifelong disease; one-third of patients continue to exhibit symptoms into adulthood.4 If their disease is properly managed, however, these patients do not develop typical features of rheumatoid arthritis, including hand, limb, and spine deformities. Last, patients with JIA who have only intermittent disease tend to do better over the long term than those whose disease is continual.32
The mortality rate of JIA has dropped: from 1% to 4% in the mid-1970s to 0.3% to 1% today4—an improvement in life expectancy that is echoed in enhanced quality of life for patients. According to the 4-level Steinbrocker functional classification scale33 (used to rate the extent of physical disability), 15% of JIA patients were Class III (limited to few or no activities of the patient’s usual occupation) or Class IV (bedridden with little or no self-care) in the period from 1976 to 1994—a percentage that had declined to 5% by 2002.34
The family physician plays pivotal role in JIA care
For the family physician, appropriate initial intervention in the management of JIA is imperative. This includes ordering imaging (whether plain films or MRI), laboratory tests as described earlier (although not to make the diagnosis), and the use of NSAIDs, intra-articular steroids, and other induction agents. Once the diagnosis is made, and a drug regimen is put in place, you will need to monitor for adverse effects. This monitoring will need to occur when a patient is escalated to csDMARDs, biological agents, or systemic steroids; is maintained on an NSAID; or is placed on a combination regimen.
Continue to: Before beginning therapy with a biologic agent...
Before beginning therapy with a biologic agent, it’s important to screen for hepatitis B, hepatitis C, human immunodeficiency virus infection, tuberculosis, and fungal infection (eg, Histoplasma capsulatum, Coccidioides immitis32). Be sure to make a timely referral to the ophthalmology service for a bi-annual eye exam and, in the event that surgery is necessary, conduct a preoperative evaluation, with the knowledge of how long before surgery a biologic agent must be withheld (duration varies by drug).32
CORRESPONDENCE
Tobe Momah, MD, Department of Family Medicine, Clinical Science Building, 4th Floor, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216; tmomah@umc.edu.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
1. Adriano LS, de França Fonteles MM, de Fátima Menezes Azevedo M, et al. Medication adherence in patients with juvenile idiopathic arthritis. Rev Bras Reumatol Engl Ed. 2017;57:23-29.
2. Akioka S. A better understanding of juvenile idiopathic arthritis with classification criteria. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39:513-521.
3. Thierry S, Fautrel B, Lemelle I, Guillemin F. Prevalence and incidence of juvenile idiopathic arthritis: a systematic review. Joint Bone Spine. 2014;81:112-117.
4. Petty RE, Laxer RM, Lindsley CB, et al. Pediatric Rheumatology. Philadelphia, PA: Elsevier; 2016:188-201.e6.
5. Scott C, Brice N. Juvenile idiopathic arthritis–an update on its diagnosis and management. S Afr Med J. 2015;105:1077.
6. Giancane G, Consolaro A, Lanni S, et al. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther. 2016;3:187-207.
7. Criteria for the classification of juvenile rheumatoid arthritis. Bull Rheum Dis. 1972;23:712-719.
8. Wood PHN: Special meeting on nomenclature and classification of arthritis in children. In: Munthe E, ed. The Care of Rheumatic Children. Basel, Switzerland: EULAR Publishers; 1978:47-50.
9. Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991-1994.
10. Petty RE, Southwood TR, Manners P, et al; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390-392.
11. Basra HAS, Humphries PD. Juvenile idiopathic arthritis: what is the utility of ultrasound? Br J Radiol. 2017;90:20160920.
12. Weiss J, Ilowite NT. Juvenile idiopathic arthritis. Pediatr Clin North Am. 2005;52:413-442, vi.
13. Fujikawa S, Okuni M. A nationwide surveillance study of rheumatic diseases among Japanese children. Acta Pediatric Jpn. 1997:39:242-244.
14. Ozen S, Karaaslan Y, Ozdemir O, et al. Prevalence of juvenile chronic arthritis and familial Mediterranean fever in Turkey: a field study. J Rheumatol. 1998;25:2445-2449.
15. Aoust L, Rossi-Semerano L, Koné-PauL I, et al. Time to diagnosis in juvenile idiopathic arthritis: a French perspective. Orphanet J Rare Dis. 2017;12:43.
16. Moe N, Rygg M. Epidemiology of juvenile chronic arthritis in northern Norway; a ten-year retrospective study. Clin Exp Rheumatol. 1998;16:99-101.
17. Abramowicz S, Kim S, Prahalad S, et al. Juvenile arthritis: current concepts in terminology, etiopathogenesis, diagnosis, and management. Int J Oral Maxillofac Surg. 2016;45:801-812.
18. Prahalad S, Shear ES, Thompson SD, et al. Increased prevalence of familial autoimmunity in simplex and multiplex families with juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1851-1856.
19. Gonzalez B, Larrañaga C, León O, et al. Parvovirus B19 may have a role in the pathogenesis of juvenile idiopathic arthritis. J Rheumatol. 2007;34:1336-1340.
20. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138-2149.
21. Zhou J, Ding Y, Zhang Y, et al. CD3+CD56+ natural killer T cell activity in children with different forms of juvenile idiopathic arthritis and the influence of etanercept treatment on polyarticular subgroup. Clin Immunol. 2016;176:1-11.
22. Shoop-Worrall SJW, Verstappen SMM, Baildam E, et al. How common is clinically inactive disease in a prospective cohort of patients with juvenile idiopathic arthritis? The importance of definition. Ann Rheum Dis. 2017;0:1-8.
23. Nordal EB, Zak M, Berntson L, et al. Juvenile Arthritis Disease Activity Score (JADAS) based on CRP; validity and predictive ability in a Nordic population-based setting. Pediatr Rheumatol Online J. 2011;9(suppl 1):155.
24. Swart JF, Dijkhuizen EHP, Wulffraat NM, et al. Clinical Juvenile Arthritis Disease Activity Score proves to be a useful tool in treat-to-target therapy in juvenile idiopathic arthritis. Ann Rheum Dis. 2018;77:336-342.
25. Horneff G, Klein A, Ganser G, et al. Protocols on classification, monitoring and therapy in children’s rheumatology (PRO-KIND): results of the working group polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2017;15:78.
26. Shoop-Worrall SJW, Verstappen SMM, McDonagh JE, et al. Long‐term outcomes following achievement of clinically inactive disease in juvenile idiopathic arthritis. Arthritis Rheumatol. 2018;70:1519-1529.
27. Ahn SS, Yoo BW, Jung SM, et al. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Sem Arthritis Rheum. 2017;.47:216-221.
28. Yokota S, Mori M, Imagawa T, et al. Proposal for juvenile idiopathic arthritis guidance on diagnosis and treatment for primary care pediatricians and nonpediatric rheumatologists (2007). Mod Rheumatol. 2007;17:353-363.
29. Barut K, Adrovic A, Şahin S, et al. Juvenile idiopathic arthritis. Balkan Med J. 2017;34:90-101.
30. Colebatch-Bourn AN, Edwards CJ, et al. EULAR-PReS points to consider for the use of imaging in the diagnosis and management of juvenile idiopathic arthritis in clinical practice. Ann Rheum Dis. 2015;74:1946-1957.
31. Blazina Š, Markelj G, AvramoviČ MZ, et al. Management of juvenile idiopathic arthritis: a clinical guide. Pediatr Drugs. 2016;18:397-412.
32. Santos MJ, Conde M, Mourão AF, et al. 2016 update of the Portuguese recommendations for the use of biologic therapies in children and adolescents with juvenile idiopathic arthritis. Acta Rheumatol Port. 2016;41:194-212.
33. Steinbrocker 0, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. JAMA. 1949;140:659-662.
34. Oen K, Malleson PN, Cabral D, et al. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29:1989-1999.
PRACTICE RECOMMENDATIONS
› Pair the findings of your clinical exam with the results of imaging and laboratory testing to make the diagnosis of juvenile idiopathic arthritis (JIA), as it is a diagnosis of exclusion. B
› Individualize treatment based on where the patient falls in the JIA disease spectrum to increase the likelihood that medical therapy will be effective. A
› Consider treating diagnosed JIA with an available biologic agent, which can provide a long asymptomatic period. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Patient, heal thyself!
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”

Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”
Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Octavio has prostate cancer. His prostate growth is large but localized.
“What do your doctors suggest?” I asked him.
“They sent me to two specialists at the medical center,” he said. “One does robotic surgery, the other does radiation. Each one told me why they recommend their technique.”
“How will you decide?”
“I’ll do some reading,” he said.
“What about the doctor who sent you to them?”
“He hasn’t discussed the choice with me, just sent me to get opinions. I have to make up own mind.”
Out of training for some time, I gather from students and family medical interactions that patient autonomy is now a reigning principle. Here is one definition:
. Patient autonomy does allow for health care providers to educate the patient but does not allow the health care provider to make the decision for the patient.
This sounds sensible, even admirable: no more paternalistic physicians talking down to patients and ordering them around. Yet a closer look shows a contradiction:
1. The second sentence says that patient autonomy “does not allow the health care provider to make the decision for the patient.”
2. But the first one says that patients should decide, “without their health care provider trying to influence the decision.”
Is “trying to influence” the same as making the decision for the patient?
Some would argue that it is: The power discrepancy between the parties makes a doctor’s attempt to influence amount to coercion.
Do you agree, esteemed colleagues, those of you who, like me, treat patients all day? If the choice is between freezing an actinic keratosis, burning it, or using topical chemotherapy, do you just lay all three options out there and ask the patient to pick one? What if your patient works in public and doesn’t have 2 weeks to wait while the reaction to topical 5-fluorouracil that makes his skin look like raw lobster subsides? Can you point that out? Or would that be “trying to influence” and thus not allowed?
You and I can think of many other examples, about medical choices large and small, where we could pose similar questions. This is not abstract philosophy; it is what we do all day.
Look up robot-assisted surgery and radiation for prostate cancer. You will find proponents of both, each making claims concerning survival, recurrence, discomfort, complications. Which is more important – a 15% greater chance of living 2 years longer or a 22% lower risk of incontinence? Will reading such statistics make your choice easier? What if other studies show different numbers?
Octavio chose surgery. I asked him how he decided.
“I talked with an internist I know socially,” he said. “He shared his experience with patients he’s referred for my problem and advised surgery as the better choice. I also saw a story online about a lawyer who chose one method, then 5 years later had to do the other.”
Octavio is sophisticated and well read. He lives near Boston, the self-described hub of medical expertise and academic excellence. Yet he makes up his mind the way everybody does: by asking a trusted adviser, by hearing an arresting anecdote. It’s not science. It’s how people think.
You don’t have to be a behavioral psychologist to know how hard it is for patients, especially frightened ones, to interpret statistical variances or compare disparate categories. Which is better – shorter life with less pain or longer life with more? How much less? How much more? There are ways to address such questions, but having an expert, trusted, and sympathetic adviser is a pretty good way to start. Only an abstract ethicist with no practical exposure to (or sympathy with) actual existing patients and their actual existing providers could possibly think otherwise.
“Let’s freeze those actinics off,” I suggest to a patient. “That won’t scar, you won’t need a dozen shots of lidocaine, and you won’t have to hide for 3 weeks.”
Did I influence her health care decision? Sure. Guilty as charged, with no apologies. When I am a patient, I want nothing less for myself: sympathetic, experienced guidance, shared by someone who knows me and appears to care one way or the other how I do.
Lord preserve us, doctors and patients both, from dogmatists who would demand otherwise.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.

Neurologists grappling with patients who embrace ‘stem cell tourism’
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.

In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.
In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.
In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
REPORTING FROM ACTRIMS FORUM 2019
Understanding AD as immune-driven disease has opened the door to new therapies
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.

“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.
“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.
“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
EXPERT ANALYSIS FROM AAD 19