User login
The daunting challenge of schizophrenia: Hundreds of biotypes and dozens of theories
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
Islands of knowledge in an ocean of ignorance. That summarizes the advances in unraveling the enigma of schizophrenia, arguably the most complex psychiatric brain disorder. The more breakthroughs are made, the more questions emerge.
Progress is definitely being made and the published literature, replete with new findings, is growing logarithmically. Particularly exciting are the recent advances in the etiology of schizophrenia, both genetic and environmental. Collaboration among geneticists around the world has enabled genome-wide association studies on almost 50,000 DNA samples and has revealed 3 genetic pathways to disrupted brain development, which lead to schizophrenia in early adulthood. Those genetic pathways include:
1. Susceptibility genes—more than 340 of them—are found significantly more often in patients with schizophrenia compared with the general population. These risk genes are scattered across all 23 pairs of chromosomes. They influence neurotransmitter functions, neuroplasticity, and immune regulation. The huge task that lies ahead is identifying what each of the risk genes disrupts in brain structure and/or function.
2. Copy number variants (CNVs), such as deletions (1 allele instead of the normal 2) or duplications (3 alleles), are much more frequent in patients with schizophrenia compared with the general population. That means too little or too much protein is made, which can disrupt the 4 stages of brain development (proliferation, migration, differentiation, and elimination).
3. de novo nonsense mutations, leading to complete absence of protein coding by the affected genes, with adverse ripple effects on brain development.
Approximately 10,000 genes (close to 50% of all 22,000 coding genes in the human genome) are involved in constructing the human brain. The latest estimate is that 79% of the hundreds of biotypes of schizophrenia are genetic in etiology.
In addition, multiple environmental factors can disrupt brain development and lead to schizophrenia. These include older paternal age (>45 years) at the time of conception, pregnancy complications (infections, gestational diabetes, vitamin D deficiency, hypoxia during delivery), childhood maltreatment (sexual or physical abuse or neglect) in the first 5 to 6 years of life, as well as migration and urbanicity (being born and raised in a large metropolitan area).
The bottom line: Schizophrenia is not only very complex, but also an extremely heterogeneous brain syndrome, both biologically and clinically. Psychiatric practitioners are fully cognizant of the extensive clinical variability in patients with schizophrenia, including the presence, absence, or severity of various signs and symptoms, such as insight, delusions, hallucinations, conceptual disorganization, bizarre behaviors, emotional withdrawal, agitation, depression, suicidality, anxiety, substance use, somatic concerns, hostility, idiosyncratic mannerisms, blunted affect, apathy, avolition, self-neglect, poor attention, memory impairment, and problems with decision-making, planning ahead, or organizing one’s life.
In addition, heterogeneity is encountered in such variables as age of onset, minor physical anomalies, soft neurologic signs, naturally occurring movement disorders, premorbid functioning, family history, general medical comorbidities, psychiatry comorbidities, structural brain abnormalities on neuroimaging, neurophysiological deviations (pre-pulse inhibition, p50, p300, N100, mismatch negativity, smooth pursuit eye movements), pituitary volume, rapidity and extent of response to antipsychotics, type and frequency of adverse effects, and functional disability or restoration of vocational functioning.
No wonder, then, given the daunting biologic and clinical heterogeneity of this complex brain syndrome, that myriad hypotheses have been proposed over the past century. The Table lists approximately 50 hypotheses, some discredited but others plausible and still viable. The most absurd hypotheses are the double bind theory of schizophrenia in the 1950s by Gregory Bateson et al, or the latent homosexuality theory of Freud. Some hypotheses may be related to a specific biotype within the schizophrenia syndrome (such as the megavitamin theory) that do not apply to other biotypes. Some of the hypotheses seem to be the product of the rich imagination of an enthusiastic researcher based on limited data.
Another consequence of the extensive heterogeneity of schizophrenia is the large number of “lab tests” that have been reported over the past few decades.1 Those hundreds of biomarkers probably mirror the biologies of the numerous disease subtypes within the schizophrenia syndrome. Some are blood tests, others neurophysiological or neuroimaging, others molecular or genetic, along with many postmortem tissue markers. Obviously, none of these “lab tests” can be used clinically because there would be an unacceptably large number of false positives and false negatives when applied to a heterogeneous sample of patients with schizophrenia.
Heterogeneity also represents a formidable challenge for researchers. Replication of a research finding by investigators across the world can be quite challenging because of the variable composition of biotypes in different countries. This heterogeneity also complicates FDA clinical trials by pharmaceutical companies seeking approval for a new drug to treat schizophrenia. The FDA requires use of DSM diagnostic criteria, which would include patients with similar clinical symptoms, but who can vary widely at the biological level. This results in failed clinical trials where only 20% or 30% of patients with schizophrenia show significant improvement compared with placebo. Given the advances in schizophrenia, a better strategy is to recruit participants who share a specific biomarker to assemble a biologically more homogeneous sample of schizophrenia. If the clinical trial is successful, the same biomarker can then be used by practitioners to predict response to the new drug. That would fulfill the aspirations of applying precision medicine in psychiatric practice.
The famous Eugen Bleuler (whose sister suffered from schizophrenia) was prescient when a century ago he published his classic book titled Dementia Praecox or the Group of Schizophrenias.2 His astute clinical observations led him to recognize the heterogeneity of the syndrome whose name he coined (schizophrenia, or disconnected thoughts). His conceptualization of schizophrenia as a spectrum of disorders of variable outcomes contrasted with that of Emil Kraepelin’s model,3 which regarded dementia praecox as a single, homogeneous, deteriorating disease. But neither Bleuler nor Kraepelin, both of whom relied on clinical observations without any biologic studies, could even imagine the spectacular complexity of the neurobiology of the schizophrenia syndrome and how difficult it is to identify its many biotypes. The monumental advances in neuroscience and neurogenetics, with their sophisticated methodologies, will eventually decipher the mysteries of this neuropsychiatric syndrome, which generates so many aberrations in thought, affect, mood, cognition, and behavior, often leading to severe functional disability among young adults, and untold anguish for their families.
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.
1. Nasrallah HA. Lab tests for psychiatric disorders: Few clinicians are aware of them. Current Psychiatry. 2013;12(2):5-7.
2. Bleuler E. Dementia praecox or the group of schizophrenias. New York, NY: International University Press; 1950.
3. Hippius H, Muller N. The work of Emil Kraepelin and his research group in Munich. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 2):3-11.

A transgender adolescent with chronic pain, depression, and PTSD
X, a 17-year-old Mexican-American transgender male (assigned female at birth) experienced a traumatic brain injury (TBI) 4 years ago and subsequently developed posttraumatic stress disorder (PTSD). I came to treat X at a pediatric outpatient psychiatric clinic after he developed physiologic dysregulation of his nervous system and began to experience panic attacks, major depressive disorder, and auditory hallucinations. X also developed chronic widespread pain during the next few years, including migraines, abdominal pain, and back pain, which significantly impaired his ability to function socially and academically. X was treated by a child and adolescent psychiatrist who used an integrative approach of traditional and complementary medical practices in a pediatric chronic pain clinic.
X’s treatment course at the pediatric psychiatric clinic included 2 years of field capable mental health services. During this time, fluoxetine was started and titrated up to 40 mg/d to target anxiety and depressive symptoms such as pervasive sadness, poor self-esteem, poor concentration, physiologic arousal, and sleep disruption. Risperidone, 2 mg/d, was temporarily added to address residual mood symptoms and the auditory hallucinations X experienced at school. Neuropsychological testing did not indicate that X had cognitive impairments from the TBI. In the pain clinic, X was encouraged to continue with psychotherapy and the selective serotonin reuptake inhibitor. Another recommendation was to seek out acupuncture and yoga. Over the course of 1 year, X’s pain symptoms began to resolve, and his functioning improved significantly. It was during this year that X came out as transgender, first to his friends, and then to his family and his physicians.
The link between PTSD and chronic pain
X’s PTSD presented as nightmares, hypervigilance, and anxiety, especially when he was in school. He would often describe how his chronic pain symptoms prevented him from functioning academically and socially. I wondered if X’s presentation of PTSD indicated a predisposition for chronic widespread pain symptoms or pain syndromes. This theory could be approximated by an association, but research suggests there is a significant temporal relationship between PTSD and widespread pain symptoms, such as in fibromyalgia.
One multicenter study of patients with fibromyalgia found that the prevalence of comorbid PTSD was 45%.1 In two-thirds of patients with fibromyalgia, traumatic life events and PTSD symptoms preceded the onset of chronic widespread pain, while in roughly one-third, traumatic life events and PTSD symptoms followed the onset of chronic widespread pain.1 This study suggests that PTSD could be viewed as a marker of stress vulnerability in which individuals susceptible to stress are more likely to develop chronic widespread pain and other health problems, including fibromyalgia, when a traumatic event occurs.
Benefits of transgender-specific care
During the course of X’s psychiatric treatment, he eventually revealed that he had been experiencing gender dysphoria for many years. His gender transition was occurring during adolescence; during this time, identity formation is a central developmental task.2 X was not comfortable asking others to use his preferred pronouns until he had physiologically transitioned. Any further delay to accessing transgender-specific services would increase the likelihood of a poor prognosis, both behaviorally and medically, because sexual minority adolescents are 3 to 4 times more likely to meet criteria for an internalizing disorder and 2 to 5 times more likely to meet criteria for externalizing disorders.3 My understanding of the minority stress model raised concerns that if X did not get appropriate treatment, the interdependence of stressors of being a sexual minority as well as an ethnic minority would further burden his mental health.
Now that X had access to transgender-specific care, how would management affect his pain symptoms or response to treatment? While some of his pain symptoms began to remit before he came out as transgender, I considered whether hormone therapy might improve his subjective pain. Little research has been conducted in transgender patients to determine whether sex-steroid administration might alter nociception. One study that examined daily fluctuations of sex hormones in 8 women with fibromyalgia found trends suggesting progesterone and testosterone are inversely associated with pain, with peaks of those hormones occurring on days with lower reported pain.4 A small study of female-to-male transgender patients found that administration of sex steroids was associated with relief from chronic painful conditions (headaches, musculoskeletal pain) in 6 of 16 patients who received testosterone injections.5 What little evidence I found in regards to an association between gender-affirming hormone therapy and chronic pain left me feeling optimistic that hormone therapy would not negatively affect the prognosis of X’s chronic pain.
Another consideration in treating X was the practice of chest binding, the compression of chest tissue for masculine gender expression among people who were assigned female sex at birth. One study found that chest binding can improve mood; decrease suicidality, anxiety, and dysphoria; and increase self-esteem.6 However, 97.2% of participants reported at least one negative outcome they attributed to binding. The most common was back pain (53.8%), which X had been experiencing before he began chest binding. I found it notable that X’s primary doctors in the transgender clinic kept this adverse effect in mind when they recommended that he take breaks and limit daily hours of chest binding to minimize the risk of increased chronic back pain.
This particular case spanned several specialized services and required coordination and careful consideration to address X’s developmental and gender-related needs. X experienced significant symptoms incited by a TBI; however, the manifestation of his chronic pain symptoms were more than likely influenced by several overlapping stressors, including belonging to an ethnic minority, transitioning into adulthood, transitioning publicly as a male, and mood symptoms. While it pleased me to see how X responded positively to the integrative and holistic treatment he received, I remain concerned that simply not enough research exists that addresses how transgender individuals are affected, physically and affectively, by chronic levels of stress attributable to their minority status.
1. Häuser W, Galek A, Erbslöh-Möller B, et al. Posttraumatic stress disorder in fibromyalgia syndrome: prevalence, temporal relationship between posttraumatic stress and fibromyalgia symptoms, and impact on clinical outcome. Pain. 2013;154(8):1216-1223.
2. Erikson EH. Identity: Youth and crisis. New York, NY: W.W. Norton & Company; 1968.
3. Fergusson DM, Horwood LJ, Beautrais AL. Is sexual orientation related to mental health problems and suicidality in young people? Arch Gen Psychiatry. 1999;56(10):876-880.
4. Schertzinger M, Wesson-Sides K, Parkitny L, et al. Daily fluctuations of progesterone and testosterone are associated with fibromyalgia pain severity. J Pain. 2018;19(4):410-417.
5. Aloisi AM, Bachiocco V, Costantino A, et al. Cross-sex hormone administration changes pain in transsexual women and men. Pain. 2007;132(suppl 1):S60-S67.
6. Peitzmeier S, Gardner I, Weinand J et al. Health impact of chest binding among transgender adults: a community-engaged, cross-sectional study. Cult Health Sex. 2017;19(1):64-75.

Dr. Anaya is a PGY-5 resident, Department of Psychiatry, Los Angeles County+USC Medical Center, Los Angeles, California.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Anaya is a PGY-5 resident, Department of Psychiatry, Los Angeles County+USC Medical Center, Los Angeles, California.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Anaya is a PGY-5 resident, Department of Psychiatry, Los Angeles County+USC Medical Center, Los Angeles, California.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
X, a 17-year-old Mexican-American transgender male (assigned female at birth) experienced a traumatic brain injury (TBI) 4 years ago and subsequently developed posttraumatic stress disorder (PTSD). I came to treat X at a pediatric outpatient psychiatric clinic after he developed physiologic dysregulation of his nervous system and began to experience panic attacks, major depressive disorder, and auditory hallucinations. X also developed chronic widespread pain during the next few years, including migraines, abdominal pain, and back pain, which significantly impaired his ability to function socially and academically. X was treated by a child and adolescent psychiatrist who used an integrative approach of traditional and complementary medical practices in a pediatric chronic pain clinic.
X’s treatment course at the pediatric psychiatric clinic included 2 years of field capable mental health services. During this time, fluoxetine was started and titrated up to 40 mg/d to target anxiety and depressive symptoms such as pervasive sadness, poor self-esteem, poor concentration, physiologic arousal, and sleep disruption. Risperidone, 2 mg/d, was temporarily added to address residual mood symptoms and the auditory hallucinations X experienced at school. Neuropsychological testing did not indicate that X had cognitive impairments from the TBI. In the pain clinic, X was encouraged to continue with psychotherapy and the selective serotonin reuptake inhibitor. Another recommendation was to seek out acupuncture and yoga. Over the course of 1 year, X’s pain symptoms began to resolve, and his functioning improved significantly. It was during this year that X came out as transgender, first to his friends, and then to his family and his physicians.
The link between PTSD and chronic pain
X’s PTSD presented as nightmares, hypervigilance, and anxiety, especially when he was in school. He would often describe how his chronic pain symptoms prevented him from functioning academically and socially. I wondered if X’s presentation of PTSD indicated a predisposition for chronic widespread pain symptoms or pain syndromes. This theory could be approximated by an association, but research suggests there is a significant temporal relationship between PTSD and widespread pain symptoms, such as in fibromyalgia.
One multicenter study of patients with fibromyalgia found that the prevalence of comorbid PTSD was 45%.1 In two-thirds of patients with fibromyalgia, traumatic life events and PTSD symptoms preceded the onset of chronic widespread pain, while in roughly one-third, traumatic life events and PTSD symptoms followed the onset of chronic widespread pain.1 This study suggests that PTSD could be viewed as a marker of stress vulnerability in which individuals susceptible to stress are more likely to develop chronic widespread pain and other health problems, including fibromyalgia, when a traumatic event occurs.
Benefits of transgender-specific care
During the course of X’s psychiatric treatment, he eventually revealed that he had been experiencing gender dysphoria for many years. His gender transition was occurring during adolescence; during this time, identity formation is a central developmental task.2 X was not comfortable asking others to use his preferred pronouns until he had physiologically transitioned. Any further delay to accessing transgender-specific services would increase the likelihood of a poor prognosis, both behaviorally and medically, because sexual minority adolescents are 3 to 4 times more likely to meet criteria for an internalizing disorder and 2 to 5 times more likely to meet criteria for externalizing disorders.3 My understanding of the minority stress model raised concerns that if X did not get appropriate treatment, the interdependence of stressors of being a sexual minority as well as an ethnic minority would further burden his mental health.
Now that X had access to transgender-specific care, how would management affect his pain symptoms or response to treatment? While some of his pain symptoms began to remit before he came out as transgender, I considered whether hormone therapy might improve his subjective pain. Little research has been conducted in transgender patients to determine whether sex-steroid administration might alter nociception. One study that examined daily fluctuations of sex hormones in 8 women with fibromyalgia found trends suggesting progesterone and testosterone are inversely associated with pain, with peaks of those hormones occurring on days with lower reported pain.4 A small study of female-to-male transgender patients found that administration of sex steroids was associated with relief from chronic painful conditions (headaches, musculoskeletal pain) in 6 of 16 patients who received testosterone injections.5 What little evidence I found in regards to an association between gender-affirming hormone therapy and chronic pain left me feeling optimistic that hormone therapy would not negatively affect the prognosis of X’s chronic pain.
Another consideration in treating X was the practice of chest binding, the compression of chest tissue for masculine gender expression among people who were assigned female sex at birth. One study found that chest binding can improve mood; decrease suicidality, anxiety, and dysphoria; and increase self-esteem.6 However, 97.2% of participants reported at least one negative outcome they attributed to binding. The most common was back pain (53.8%), which X had been experiencing before he began chest binding. I found it notable that X’s primary doctors in the transgender clinic kept this adverse effect in mind when they recommended that he take breaks and limit daily hours of chest binding to minimize the risk of increased chronic back pain.
This particular case spanned several specialized services and required coordination and careful consideration to address X’s developmental and gender-related needs. X experienced significant symptoms incited by a TBI; however, the manifestation of his chronic pain symptoms were more than likely influenced by several overlapping stressors, including belonging to an ethnic minority, transitioning into adulthood, transitioning publicly as a male, and mood symptoms. While it pleased me to see how X responded positively to the integrative and holistic treatment he received, I remain concerned that simply not enough research exists that addresses how transgender individuals are affected, physically and affectively, by chronic levels of stress attributable to their minority status.
X, a 17-year-old Mexican-American transgender male (assigned female at birth) experienced a traumatic brain injury (TBI) 4 years ago and subsequently developed posttraumatic stress disorder (PTSD). I came to treat X at a pediatric outpatient psychiatric clinic after he developed physiologic dysregulation of his nervous system and began to experience panic attacks, major depressive disorder, and auditory hallucinations. X also developed chronic widespread pain during the next few years, including migraines, abdominal pain, and back pain, which significantly impaired his ability to function socially and academically. X was treated by a child and adolescent psychiatrist who used an integrative approach of traditional and complementary medical practices in a pediatric chronic pain clinic.
X’s treatment course at the pediatric psychiatric clinic included 2 years of field capable mental health services. During this time, fluoxetine was started and titrated up to 40 mg/d to target anxiety and depressive symptoms such as pervasive sadness, poor self-esteem, poor concentration, physiologic arousal, and sleep disruption. Risperidone, 2 mg/d, was temporarily added to address residual mood symptoms and the auditory hallucinations X experienced at school. Neuropsychological testing did not indicate that X had cognitive impairments from the TBI. In the pain clinic, X was encouraged to continue with psychotherapy and the selective serotonin reuptake inhibitor. Another recommendation was to seek out acupuncture and yoga. Over the course of 1 year, X’s pain symptoms began to resolve, and his functioning improved significantly. It was during this year that X came out as transgender, first to his friends, and then to his family and his physicians.
The link between PTSD and chronic pain
X’s PTSD presented as nightmares, hypervigilance, and anxiety, especially when he was in school. He would often describe how his chronic pain symptoms prevented him from functioning academically and socially. I wondered if X’s presentation of PTSD indicated a predisposition for chronic widespread pain symptoms or pain syndromes. This theory could be approximated by an association, but research suggests there is a significant temporal relationship between PTSD and widespread pain symptoms, such as in fibromyalgia.
One multicenter study of patients with fibromyalgia found that the prevalence of comorbid PTSD was 45%.1 In two-thirds of patients with fibromyalgia, traumatic life events and PTSD symptoms preceded the onset of chronic widespread pain, while in roughly one-third, traumatic life events and PTSD symptoms followed the onset of chronic widespread pain.1 This study suggests that PTSD could be viewed as a marker of stress vulnerability in which individuals susceptible to stress are more likely to develop chronic widespread pain and other health problems, including fibromyalgia, when a traumatic event occurs.
Benefits of transgender-specific care
During the course of X’s psychiatric treatment, he eventually revealed that he had been experiencing gender dysphoria for many years. His gender transition was occurring during adolescence; during this time, identity formation is a central developmental task.2 X was not comfortable asking others to use his preferred pronouns until he had physiologically transitioned. Any further delay to accessing transgender-specific services would increase the likelihood of a poor prognosis, both behaviorally and medically, because sexual minority adolescents are 3 to 4 times more likely to meet criteria for an internalizing disorder and 2 to 5 times more likely to meet criteria for externalizing disorders.3 My understanding of the minority stress model raised concerns that if X did not get appropriate treatment, the interdependence of stressors of being a sexual minority as well as an ethnic minority would further burden his mental health.
Now that X had access to transgender-specific care, how would management affect his pain symptoms or response to treatment? While some of his pain symptoms began to remit before he came out as transgender, I considered whether hormone therapy might improve his subjective pain. Little research has been conducted in transgender patients to determine whether sex-steroid administration might alter nociception. One study that examined daily fluctuations of sex hormones in 8 women with fibromyalgia found trends suggesting progesterone and testosterone are inversely associated with pain, with peaks of those hormones occurring on days with lower reported pain.4 A small study of female-to-male transgender patients found that administration of sex steroids was associated with relief from chronic painful conditions (headaches, musculoskeletal pain) in 6 of 16 patients who received testosterone injections.5 What little evidence I found in regards to an association between gender-affirming hormone therapy and chronic pain left me feeling optimistic that hormone therapy would not negatively affect the prognosis of X’s chronic pain.
Another consideration in treating X was the practice of chest binding, the compression of chest tissue for masculine gender expression among people who were assigned female sex at birth. One study found that chest binding can improve mood; decrease suicidality, anxiety, and dysphoria; and increase self-esteem.6 However, 97.2% of participants reported at least one negative outcome they attributed to binding. The most common was back pain (53.8%), which X had been experiencing before he began chest binding. I found it notable that X’s primary doctors in the transgender clinic kept this adverse effect in mind when they recommended that he take breaks and limit daily hours of chest binding to minimize the risk of increased chronic back pain.
This particular case spanned several specialized services and required coordination and careful consideration to address X’s developmental and gender-related needs. X experienced significant symptoms incited by a TBI; however, the manifestation of his chronic pain symptoms were more than likely influenced by several overlapping stressors, including belonging to an ethnic minority, transitioning into adulthood, transitioning publicly as a male, and mood symptoms. While it pleased me to see how X responded positively to the integrative and holistic treatment he received, I remain concerned that simply not enough research exists that addresses how transgender individuals are affected, physically and affectively, by chronic levels of stress attributable to their minority status.
1. Häuser W, Galek A, Erbslöh-Möller B, et al. Posttraumatic stress disorder in fibromyalgia syndrome: prevalence, temporal relationship between posttraumatic stress and fibromyalgia symptoms, and impact on clinical outcome. Pain. 2013;154(8):1216-1223.
2. Erikson EH. Identity: Youth and crisis. New York, NY: W.W. Norton & Company; 1968.
3. Fergusson DM, Horwood LJ, Beautrais AL. Is sexual orientation related to mental health problems and suicidality in young people? Arch Gen Psychiatry. 1999;56(10):876-880.
4. Schertzinger M, Wesson-Sides K, Parkitny L, et al. Daily fluctuations of progesterone and testosterone are associated with fibromyalgia pain severity. J Pain. 2018;19(4):410-417.
5. Aloisi AM, Bachiocco V, Costantino A, et al. Cross-sex hormone administration changes pain in transsexual women and men. Pain. 2007;132(suppl 1):S60-S67.
6. Peitzmeier S, Gardner I, Weinand J et al. Health impact of chest binding among transgender adults: a community-engaged, cross-sectional study. Cult Health Sex. 2017;19(1):64-75.
1. Häuser W, Galek A, Erbslöh-Möller B, et al. Posttraumatic stress disorder in fibromyalgia syndrome: prevalence, temporal relationship between posttraumatic stress and fibromyalgia symptoms, and impact on clinical outcome. Pain. 2013;154(8):1216-1223.
2. Erikson EH. Identity: Youth and crisis. New York, NY: W.W. Norton & Company; 1968.
3. Fergusson DM, Horwood LJ, Beautrais AL. Is sexual orientation related to mental health problems and suicidality in young people? Arch Gen Psychiatry. 1999;56(10):876-880.
4. Schertzinger M, Wesson-Sides K, Parkitny L, et al. Daily fluctuations of progesterone and testosterone are associated with fibromyalgia pain severity. J Pain. 2018;19(4):410-417.
5. Aloisi AM, Bachiocco V, Costantino A, et al. Cross-sex hormone administration changes pain in transsexual women and men. Pain. 2007;132(suppl 1):S60-S67.
6. Peitzmeier S, Gardner I, Weinand J et al. Health impact of chest binding among transgender adults: a community-engaged, cross-sectional study. Cult Health Sex. 2017;19(1):64-75.
Resilience: Our only remedy?
Resilience is like patience; we all wish we had more of it, but we hope to avoid getting it the hard way. This wasn’t really an area of interest for me, until it needed to be. When one academic year brings the suicide of one colleague and the murder of another, resilience becomes the only alternative to despair.
I realize that even though the particular pain or trauma we endured may be unique, it’s becoming increasingly common. The alarming studies of resident depression and suicide are too difficult for us to ignore. Now we must look in that evidence-based mirror and decide where we will go from here, as a profession and as trainees. The 2018 American Psychiatric Association annual meeting gave us a rude awakening that we may not have it figured out. Even during a year-long theme on wellness, and several sessions at the meeting focusing on the same, we all found ourselves mourning the loss of 2 colleagues to suicide that very weekend only a few miles away from the gathering of the world’s experts.
It brought an eerie element to the conversation.
The wellness “window dressing” will not get the job done. I recently had a candid discussion with a mentor in administrative leadership, and his words surprised as well as challenged me. He told me that the “system” will not save you. You must save yourself. I have decided to respectfully reject that. I think everyone should be involved, including the “system” that is entrusted with my training, and the least that it ought to ensure is that I get out alive.
Has that really become too much to ask of our profession?
We must hold our system to a higher standard. More mindfulness and better breathing will surely be helpful—but I hope we can begin to admit that this is not the answer. Unfortunately, the culture of “pay your dues” and “you know how much harder it was when I was a resident?” is still the norm. We now receive our training in an environment where the pressure is extraordinarily high, the margin for error very low, and the possibility of support is almost a fantasy. “Sure, you can get the help you need ... but don’t take time off or you will be off cycle and create extra work for all your colleagues, who are also equally stressed and will hate you. In the meantime … enjoy this free ice cream and breathing exercise to mindfully cope with the madness around you.”
The perfectly resilient resident may very well be a mythical figure, a clinical unicorn, that we continue chasing. This is the resident who remarkably discovers posttraumatic growth in every stressor. The vicarious trauma they experience from their patients only bolsters their deep compassion, and they thrive under pressure, so we can continue to pile it on. In our search for this “super resident,” we seem to continue to lose a few ordinary residents along the way.
Are we brave enough as a health care culture to take a closer look at the way we are training the next generation of healers? As I get to the end of this article, I wish I had more answers. I’m just a trainee. What do I know? My fear is that we’ve been avoiding this question altogether and have had our eyes closed to the real problem while pacifying ourselves with one “wellness” activity after another. My sincere hope is that this article will make you angry enough to be driven by a conviction that this is not

Dr. Mirhom is a PGY-3 resident, BronxCare Hospital Center, Mount Sinai Health System, Bronx, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Mirhom is a PGY-3 resident, BronxCare Hospital Center, Mount Sinai Health System, Bronx, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Mirhom is a PGY-3 resident, BronxCare Hospital Center, Mount Sinai Health System, Bronx, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Resilience is like patience; we all wish we had more of it, but we hope to avoid getting it the hard way. This wasn’t really an area of interest for me, until it needed to be. When one academic year brings the suicide of one colleague and the murder of another, resilience becomes the only alternative to despair.
I realize that even though the particular pain or trauma we endured may be unique, it’s becoming increasingly common. The alarming studies of resident depression and suicide are too difficult for us to ignore. Now we must look in that evidence-based mirror and decide where we will go from here, as a profession and as trainees. The 2018 American Psychiatric Association annual meeting gave us a rude awakening that we may not have it figured out. Even during a year-long theme on wellness, and several sessions at the meeting focusing on the same, we all found ourselves mourning the loss of 2 colleagues to suicide that very weekend only a few miles away from the gathering of the world’s experts.
It brought an eerie element to the conversation.
The wellness “window dressing” will not get the job done. I recently had a candid discussion with a mentor in administrative leadership, and his words surprised as well as challenged me. He told me that the “system” will not save you. You must save yourself. I have decided to respectfully reject that. I think everyone should be involved, including the “system” that is entrusted with my training, and the least that it ought to ensure is that I get out alive.
Has that really become too much to ask of our profession?
We must hold our system to a higher standard. More mindfulness and better breathing will surely be helpful—but I hope we can begin to admit that this is not the answer. Unfortunately, the culture of “pay your dues” and “you know how much harder it was when I was a resident?” is still the norm. We now receive our training in an environment where the pressure is extraordinarily high, the margin for error very low, and the possibility of support is almost a fantasy. “Sure, you can get the help you need ... but don’t take time off or you will be off cycle and create extra work for all your colleagues, who are also equally stressed and will hate you. In the meantime … enjoy this free ice cream and breathing exercise to mindfully cope with the madness around you.”
The perfectly resilient resident may very well be a mythical figure, a clinical unicorn, that we continue chasing. This is the resident who remarkably discovers posttraumatic growth in every stressor. The vicarious trauma they experience from their patients only bolsters their deep compassion, and they thrive under pressure, so we can continue to pile it on. In our search for this “super resident,” we seem to continue to lose a few ordinary residents along the way.
Are we brave enough as a health care culture to take a closer look at the way we are training the next generation of healers? As I get to the end of this article, I wish I had more answers. I’m just a trainee. What do I know? My fear is that we’ve been avoiding this question altogether and have had our eyes closed to the real problem while pacifying ourselves with one “wellness” activity after another. My sincere hope is that this article will make you angry enough to be driven by a conviction that this is not
Resilience is like patience; we all wish we had more of it, but we hope to avoid getting it the hard way. This wasn’t really an area of interest for me, until it needed to be. When one academic year brings the suicide of one colleague and the murder of another, resilience becomes the only alternative to despair.
I realize that even though the particular pain or trauma we endured may be unique, it’s becoming increasingly common. The alarming studies of resident depression and suicide are too difficult for us to ignore. Now we must look in that evidence-based mirror and decide where we will go from here, as a profession and as trainees. The 2018 American Psychiatric Association annual meeting gave us a rude awakening that we may not have it figured out. Even during a year-long theme on wellness, and several sessions at the meeting focusing on the same, we all found ourselves mourning the loss of 2 colleagues to suicide that very weekend only a few miles away from the gathering of the world’s experts.
It brought an eerie element to the conversation.
The wellness “window dressing” will not get the job done. I recently had a candid discussion with a mentor in administrative leadership, and his words surprised as well as challenged me. He told me that the “system” will not save you. You must save yourself. I have decided to respectfully reject that. I think everyone should be involved, including the “system” that is entrusted with my training, and the least that it ought to ensure is that I get out alive.
Has that really become too much to ask of our profession?
We must hold our system to a higher standard. More mindfulness and better breathing will surely be helpful—but I hope we can begin to admit that this is not the answer. Unfortunately, the culture of “pay your dues” and “you know how much harder it was when I was a resident?” is still the norm. We now receive our training in an environment where the pressure is extraordinarily high, the margin for error very low, and the possibility of support is almost a fantasy. “Sure, you can get the help you need ... but don’t take time off or you will be off cycle and create extra work for all your colleagues, who are also equally stressed and will hate you. In the meantime … enjoy this free ice cream and breathing exercise to mindfully cope with the madness around you.”
The perfectly resilient resident may very well be a mythical figure, a clinical unicorn, that we continue chasing. This is the resident who remarkably discovers posttraumatic growth in every stressor. The vicarious trauma they experience from their patients only bolsters their deep compassion, and they thrive under pressure, so we can continue to pile it on. In our search for this “super resident,” we seem to continue to lose a few ordinary residents along the way.
Are we brave enough as a health care culture to take a closer look at the way we are training the next generation of healers? As I get to the end of this article, I wish I had more answers. I’m just a trainee. What do I know? My fear is that we’ve been avoiding this question altogether and have had our eyes closed to the real problem while pacifying ourselves with one “wellness” activity after another. My sincere hope is that this article will make you angry enough to be driven by a conviction that this is not
Risperidone extended-release injectable suspension
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3

Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
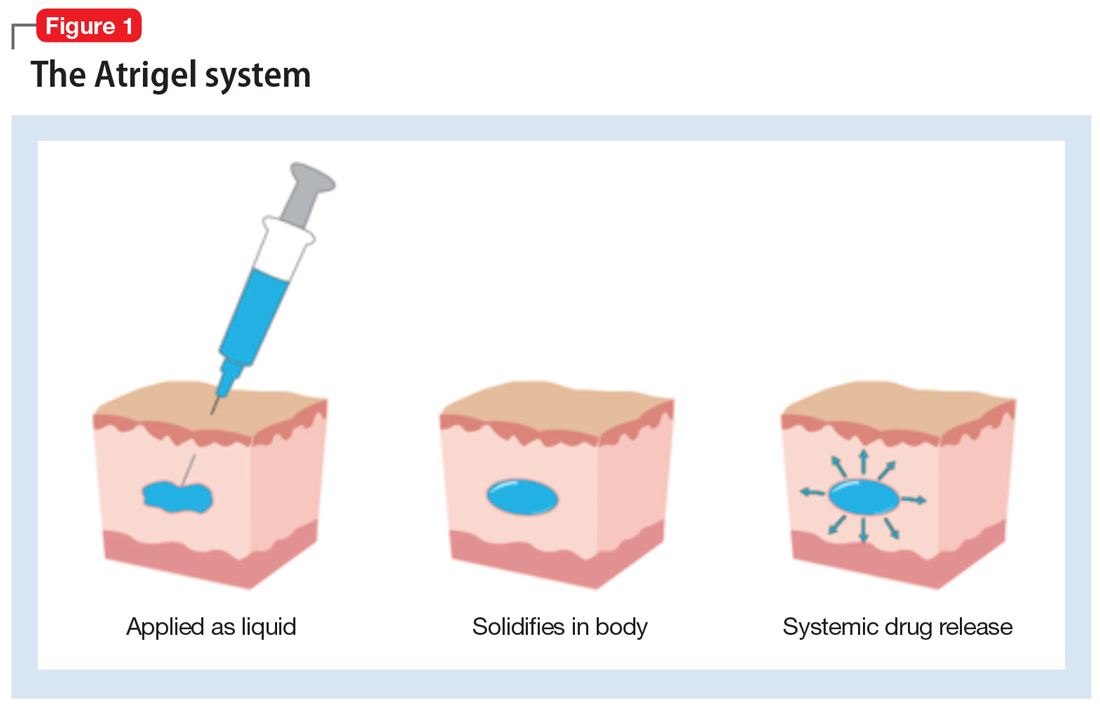
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13

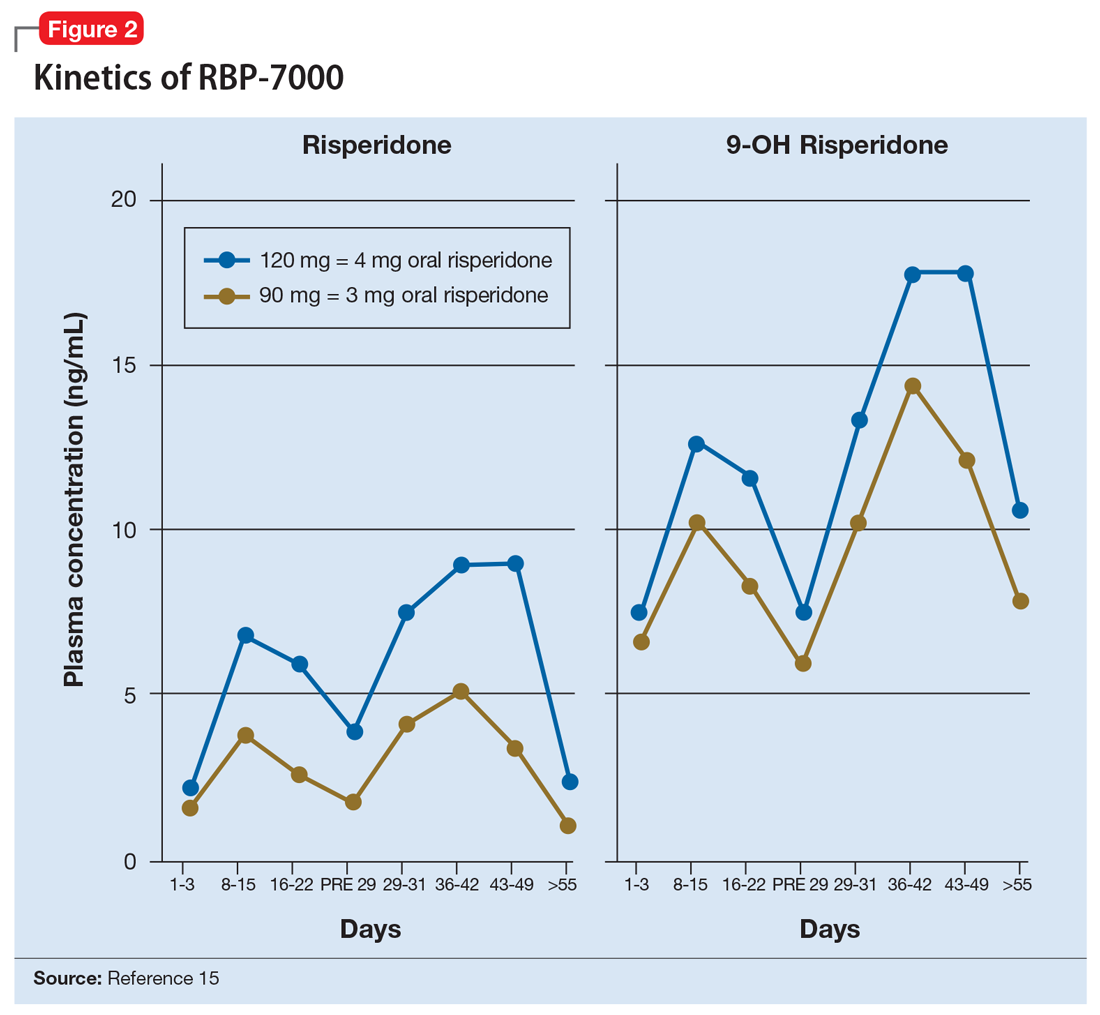
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Catatonia: Recognition, management, and prevention of complications
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous

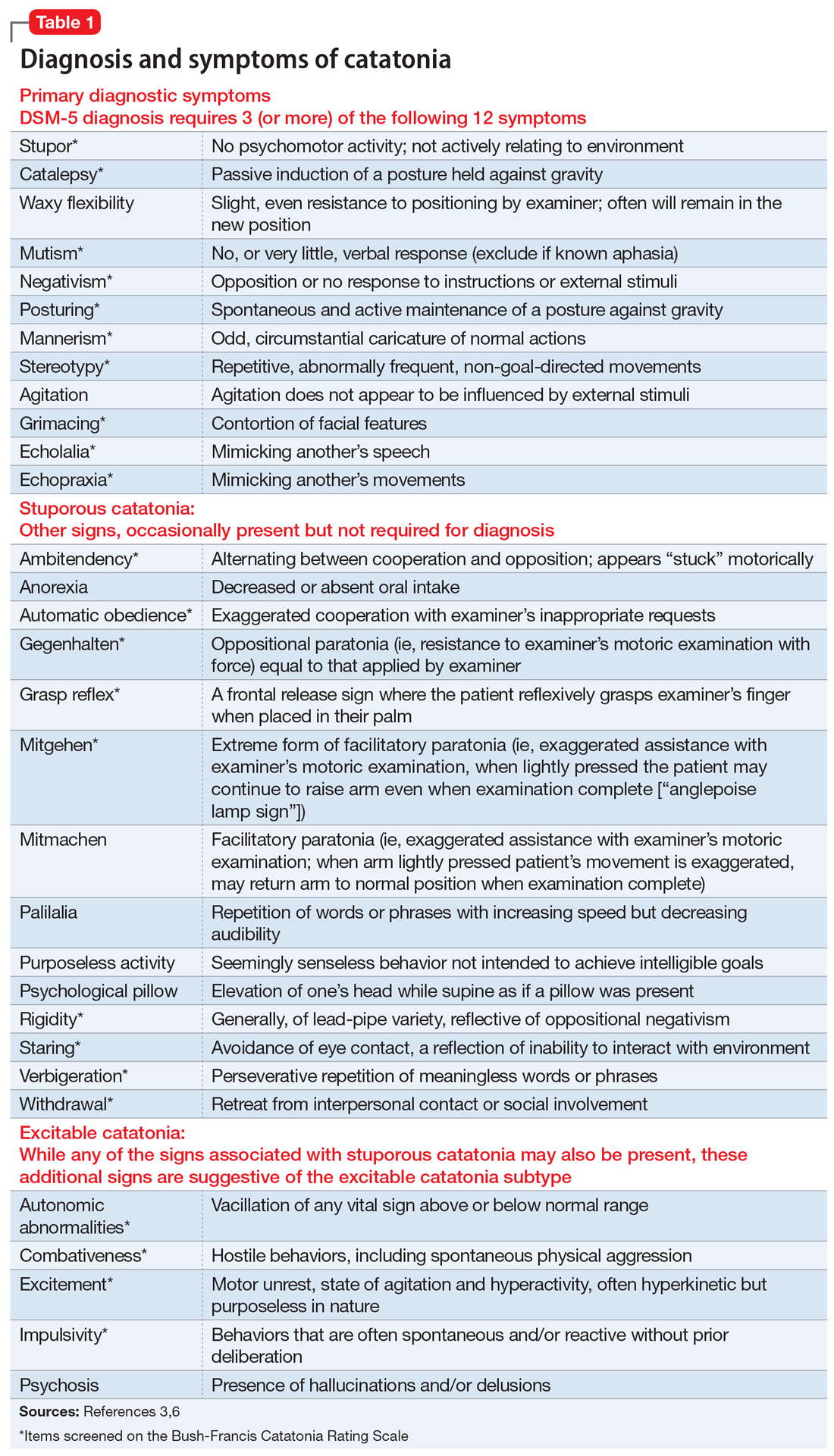
Continue to: Medical complications can be fatal
Medical complications can be fatal

Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Pharmacologic performance enhancement: What to consider before prescribing
Performance enhancement in sports (“doping”) dates back to Ancient Greece. This was an era when Olympic athletes would attempt to improve their physical performance by consuming magic potions, herbal medications, and even exotic meats such as sheep testicles—a delicacy high in testosterone. Advances in medical and pharmaceutical technologies have increased both the range of enhancement agents available and their efficacy, leading to the development of anti-doping agencies and routine screening for doping in athletics. This has led to the renouncement of titles, medals, and financial sponsorship of athletes found to have been using prohibited substances during competition.
While doping in elite athletes often forms the nidus of media attention, the pressure to compete and perform at, or even beyond, one’s potential extends into many facets of today’s achievementfocused society. In the face of these pressures, individuals are increasingly seeking medications to enhance their performance across numerous domains, including cognitive, athletic, and artistic endeavors. Medication classes used to enhance performance include stimulants, which increase attention, executive function, and energy; cholinesterase inhibitors, which may ameliorate age-related memory decline; and beta-blockers, which decrease physiologic symptoms of anxiety and have been demonstrated to be beneficial for musical performance.1 Fifty-three percent of college athletes report using prescription medications to enhance athletic performance,2 and most college students who take stimulants without a prescription use them to study (84%) or stay awake (51%).3
Pharmacologic performance enhancement is the use of medications by healthy individuals to improve function in the absence of mental illness. Psychiatrists are increasingly finding themselves in the controversial position of “gatekeeper” of these medications for enhancement purposes. In this article we:
- outline arguments that support the use of psychopharmacology for performance enhancement, as well as some serious concerns with this practice
- discuss special considerations for pediatric populations and the risk of malpractice when prescribing for performance enhancement
- offer practice guidelines for approaching requests for psychopharmacologic performance enhancement.
Performance enhancement: The wave of the future?
The ethical principle that supports providing medication for performance enhancement is beneficence, the promotion of the patient’s well-being. In other words, it is a physician’s duty to help his or her patient in need. Individuals seeking performance enhancement typically present with suffering, and the principle of beneficence would call upon the psychiatrist to help ameliorate that suffering. Furthermore, patients who seek performance enhancement may present with impairing “subsyndromal” psychiatric symptoms (for example, low-grade attentional difficulty that occurs only in one setting), which, even if they do not rise to the threshold of a DSM diagnosis, may improve with psychiatric medications.
Using medical knowledge and skills beyond the traditional physician duty to diagnose and treat medical conditions is not unprecedented (eg, when surgeons perform cosmetic enhancement). Might elective enhancement of cognition and psychological performance through the judicious use of medication be part of the future of psychiatry? If cognitive and emotional enhancement becomes a more widely accepted standard of care, might this increase both individual and societal innovation and productivity?
Dilemma: Cautions against performance enhancement
One of the major cautions against prescribing psychotropics for the purpose of performance enhancement is the lack of clearly supported efficacy. Psychiatric medications generally are studied in individuals who meet criteria for mental illness, and they are FDA-approved for use in ill persons. It may be erroneous to extrapolate that a medication that improves symptoms in a patient with an illness would achieve the same target effect in a healthy individual. For example, data on whether stimulants provide neurocognitive enhancement in healthy individuals without attention-deficit/hyperactivity disorder is mixed, and these agents may even promote risky behavior in healthy controls.4 Furthermore, dopamine agonism may compress cognitive performance in both directions,5 as it has been observed that methylphenidate improves executive function in healthy controls, but is less beneficial for those with strong executive function at baseline.6
In the face of unclear benefit, it is particularly important to consider the risk of medications used for performance enhancement. Pharmacologic performance enhancement in individuals without psychopathology can be considered an “elective” intervention, for which individuals typically tolerate less risk. Physical risks, including medication-related adverse effects, must be considered, particularly in settings where there may be temptation to use more than prescribed, or to divert medication to others who may use it without medical monitoring. In addition to physical harm, there may be psychological harm associated with prescribing performance enhancers, such as pathologizing variants of “normal,” diminishing one’s sense of self-efficacy, or decreasing one’s ability to bear failure.
Continue to: Finally, there are ethical quandaries
Finally, there are ethical quandaries regarding using medications for performance enhancement. Widespread adoption of pharmacologic performance enhancement may lead to implicit coercion for all individuals to enhance their abilities. As a greater proportion of society receives pharmacologic enhancement, society as a whole faces stronger pressures to seek pharmacologic enhancement, ultimately constricting an individual’s freedom of choice to enhance.6 In this setting, distributive justice would become a consideration, because insurance companies are unlikely to reimburse for medications used for enhancement,7 which would give an advantage to individuals with higher socioeconomic status. Research shows that children from higher socioeconomic communities and from states with higher academic standards are more likely to use stimulants.8
Areas of controversy
Pediatric populations. There are special considerations when prescribing performance-enhancing medications for children and adolescents. First, such prescribing may inhibit normal child development, shifting the focus away from the normative tasks of social and emotional development that occur through leisure and creativity, experimentation, and play to an emphasis on performance and outcomes-based achievement.9 Second, during childhood and adolescence, one develops a sense of his or her identity, morals, and values. Taking a medication during childhood to enhance performance may inhibit the process of learning to tolerate failure, become aware of one’s weaknesses, and value effort in addition to outcome.
Malpractice risk. Practicing medicine beyond the scope of one’s expertise is unethical and unlawful. In the past 30 years, medical malpractice has become one of the most difficult health care issues in the U.S.10 In addition to billions of dollars in legal fees and court costs, medical malpractice premiums in the U.S. total more than $5 billion annually,11 and “defensive medicine”— procedures performed to protect against litigation—is estimated to cost more than $14 billion a year.12
When considering performance-enhancing treatment, it is the physician’s duty to conduct a diagnostic assessment, including noting target symptoms that are interfering with the patient’s function, and to tailor such treatment toward measurable goals and outcomes. Aside from medication, this could include a therapeutic approach to improving performance that might include cognitive-behavioral therapy and promotion of a healthy diet and exercise.
Treatment rises to the level of malpractice when there is a dereliction of duty that directly leads to damages.13 Part of a physician’s duty is to educate patients about the pros and cons of different treatment options. For performance-enhancing medications, the risks of addiction and dependence are adverse effects that require discussion. And for a pediatric patient, this would require the guardian’s engagement and understanding.
Continue to: What to do if you decide to prescribe
What to do if you decide to prescribe
Inevitably, the decision to prescribe psychotropic medications for performance enhancement is a physician-specific one. Certainly, psychiatrists should not feel obligated to prescribe performance enhancers. Given our current state of pharmacology, it is unclear whether medications would be helpful in the absence of psychopathology. When deciding whether to prescribe for performance enhancement in the absence of psychopathology, we suggest first carefully considering how to maintain the ethical value of nonmaleficence by weighing both the potential physical and psychologic harms of prescribing as well as the legal risks and rules of applicable sport governing bodies.
For a psychiatrist who chooses to prescribe for performance enhancement, we recommend conducting a thorough psychiatric assessment to determine whether the patient has a treatable mental illness. If so, then effective treatment of that illness should take priority. Before prescribing, the psychiatrist and patient should discuss the patient’s specific performance goals and how to measure them.
Any prescription for a performance-enhancing medication should be given in conjunction with nonpharmacologic approaches, including optimizing diet, exercise, and sleep. Therapy to address problem-solving techniques and skills to cope with stress may also be appropriate. The patient and psychiatrist should engage in regular follow-up to assess the efficacy of the medication, as well as its safety and tolerability. Finally, if a medication is not efficacious as a performance enhancer, then both the patient and psychiatrist should be open to re-evaluating the treatment plan, and when appropriate, stopping the medication.
1. Brantigan CO, Brantigan TA, Joseph N. Effect of beta blockade and beta stimulation on stage fright. Am J Med. 1982;72(1):88-94.
2. Hoyte CO, Albert D, Heard KJ. The use of energy drinks, dietary supplements, and prescription medications by United States college students to enhance athletic performance. J Community Health. 2013;38(3):575-850.
3. Advokat CD, Guidry D, Martino L. Licit and illicit use of medications for attention-deficit hyperactivity disorder in undergraduate college students. J Am Coll Health. 2008;56(6):601-606.
4. Advokat C, Scheithauer M. Attention-deficit hyperactivity disorder (ADHD) stimulant medications as cognitive enhancers. Front Neurosci. 2013;7:82.
5. Kimberg DY, D’Esposito M, Farah MJ. Effects of bromocriptine on human subjects depend on working memory capacity. Neuroreport. 1997;8(16):3581-3585.
6. Farah MJ, Illes J, Cook-Deegan R, et al. Neurocognitive enhancement: what can we do and what should we do? Nat Rev Neurosci. 2004;5(5):421-425.
7. Larriviere D, Williams MA, Rizzo M, et al; AAN Ethics, Law and Humanities Committee. Responding to requests from adult patients for neuroenhancements: guidance of the Ethics, Law and Humanities Committee. Neurology. 2009;73(17):1406-1412.
8. Colaneri N, Sheldon M, Adesman A. Pharmacological cognitive enhancement in pediatrics. Curr Opin Pediatr. 2018;30(3):430-437.
9. Gaucher N, Payot A, Racine E. Cognitive enhancement in children and adolescents: Is it in their best interests? Acta Paediatr. 2013;102(12):1118-1124.
10. Moore PJ, Adler, NE, Robertson, PA. Medical malpractice; the effect of doctor-patient relations on medical patient perceptions and malpractice intentions. West J Med. 2000;173(4):244-250.
11. Hiatt H. Medical malpractice. Bull N Y Acad Med. 1992;68(2):254-260.
12. Rubin RJ, Mendelson DN. How much does defensive medicine cost? J Am Health Policy. 1994;4(4):7-15.
13. Kloss D. The duty of care: medical negligence. Br Med J (Clin Res Ed). 1984;289(6436):66-68.
Performance enhancement in sports (“doping”) dates back to Ancient Greece. This was an era when Olympic athletes would attempt to improve their physical performance by consuming magic potions, herbal medications, and even exotic meats such as sheep testicles—a delicacy high in testosterone. Advances in medical and pharmaceutical technologies have increased both the range of enhancement agents available and their efficacy, leading to the development of anti-doping agencies and routine screening for doping in athletics. This has led to the renouncement of titles, medals, and financial sponsorship of athletes found to have been using prohibited substances during competition.
While doping in elite athletes often forms the nidus of media attention, the pressure to compete and perform at, or even beyond, one’s potential extends into many facets of today’s achievementfocused society. In the face of these pressures, individuals are increasingly seeking medications to enhance their performance across numerous domains, including cognitive, athletic, and artistic endeavors. Medication classes used to enhance performance include stimulants, which increase attention, executive function, and energy; cholinesterase inhibitors, which may ameliorate age-related memory decline; and beta-blockers, which decrease physiologic symptoms of anxiety and have been demonstrated to be beneficial for musical performance.1 Fifty-three percent of college athletes report using prescription medications to enhance athletic performance,2 and most college students who take stimulants without a prescription use them to study (84%) or stay awake (51%).3
Pharmacologic performance enhancement is the use of medications by healthy individuals to improve function in the absence of mental illness. Psychiatrists are increasingly finding themselves in the controversial position of “gatekeeper” of these medications for enhancement purposes. In this article we:
- outline arguments that support the use of psychopharmacology for performance enhancement, as well as some serious concerns with this practice
- discuss special considerations for pediatric populations and the risk of malpractice when prescribing for performance enhancement
- offer practice guidelines for approaching requests for psychopharmacologic performance enhancement.
Performance enhancement: The wave of the future?
The ethical principle that supports providing medication for performance enhancement is beneficence, the promotion of the patient’s well-being. In other words, it is a physician’s duty to help his or her patient in need. Individuals seeking performance enhancement typically present with suffering, and the principle of beneficence would call upon the psychiatrist to help ameliorate that suffering. Furthermore, patients who seek performance enhancement may present with impairing “subsyndromal” psychiatric symptoms (for example, low-grade attentional difficulty that occurs only in one setting), which, even if they do not rise to the threshold of a DSM diagnosis, may improve with psychiatric medications.
Using medical knowledge and skills beyond the traditional physician duty to diagnose and treat medical conditions is not unprecedented (eg, when surgeons perform cosmetic enhancement). Might elective enhancement of cognition and psychological performance through the judicious use of medication be part of the future of psychiatry? If cognitive and emotional enhancement becomes a more widely accepted standard of care, might this increase both individual and societal innovation and productivity?
Dilemma: Cautions against performance enhancement
One of the major cautions against prescribing psychotropics for the purpose of performance enhancement is the lack of clearly supported efficacy. Psychiatric medications generally are studied in individuals who meet criteria for mental illness, and they are FDA-approved for use in ill persons. It may be erroneous to extrapolate that a medication that improves symptoms in a patient with an illness would achieve the same target effect in a healthy individual. For example, data on whether stimulants provide neurocognitive enhancement in healthy individuals without attention-deficit/hyperactivity disorder is mixed, and these agents may even promote risky behavior in healthy controls.4 Furthermore, dopamine agonism may compress cognitive performance in both directions,5 as it has been observed that methylphenidate improves executive function in healthy controls, but is less beneficial for those with strong executive function at baseline.6
In the face of unclear benefit, it is particularly important to consider the risk of medications used for performance enhancement. Pharmacologic performance enhancement in individuals without psychopathology can be considered an “elective” intervention, for which individuals typically tolerate less risk. Physical risks, including medication-related adverse effects, must be considered, particularly in settings where there may be temptation to use more than prescribed, or to divert medication to others who may use it without medical monitoring. In addition to physical harm, there may be psychological harm associated with prescribing performance enhancers, such as pathologizing variants of “normal,” diminishing one’s sense of self-efficacy, or decreasing one’s ability to bear failure.
Continue to: Finally, there are ethical quandaries
Finally, there are ethical quandaries regarding using medications for performance enhancement. Widespread adoption of pharmacologic performance enhancement may lead to implicit coercion for all individuals to enhance their abilities. As a greater proportion of society receives pharmacologic enhancement, society as a whole faces stronger pressures to seek pharmacologic enhancement, ultimately constricting an individual’s freedom of choice to enhance.6 In this setting, distributive justice would become a consideration, because insurance companies are unlikely to reimburse for medications used for enhancement,7 which would give an advantage to individuals with higher socioeconomic status. Research shows that children from higher socioeconomic communities and from states with higher academic standards are more likely to use stimulants.8
Areas of controversy
Pediatric populations. There are special considerations when prescribing performance-enhancing medications for children and adolescents. First, such prescribing may inhibit normal child development, shifting the focus away from the normative tasks of social and emotional development that occur through leisure and creativity, experimentation, and play to an emphasis on performance and outcomes-based achievement.9 Second, during childhood and adolescence, one develops a sense of his or her identity, morals, and values. Taking a medication during childhood to enhance performance may inhibit the process of learning to tolerate failure, become aware of one’s weaknesses, and value effort in addition to outcome.
Malpractice risk. Practicing medicine beyond the scope of one’s expertise is unethical and unlawful. In the past 30 years, medical malpractice has become one of the most difficult health care issues in the U.S.10 In addition to billions of dollars in legal fees and court costs, medical malpractice premiums in the U.S. total more than $5 billion annually,11 and “defensive medicine”— procedures performed to protect against litigation—is estimated to cost more than $14 billion a year.12
When considering performance-enhancing treatment, it is the physician’s duty to conduct a diagnostic assessment, including noting target symptoms that are interfering with the patient’s function, and to tailor such treatment toward measurable goals and outcomes. Aside from medication, this could include a therapeutic approach to improving performance that might include cognitive-behavioral therapy and promotion of a healthy diet and exercise.
Treatment rises to the level of malpractice when there is a dereliction of duty that directly leads to damages.13 Part of a physician’s duty is to educate patients about the pros and cons of different treatment options. For performance-enhancing medications, the risks of addiction and dependence are adverse effects that require discussion. And for a pediatric patient, this would require the guardian’s engagement and understanding.
Continue to: What to do if you decide to prescribe
What to do if you decide to prescribe
Inevitably, the decision to prescribe psychotropic medications for performance enhancement is a physician-specific one. Certainly, psychiatrists should not feel obligated to prescribe performance enhancers. Given our current state of pharmacology, it is unclear whether medications would be helpful in the absence of psychopathology. When deciding whether to prescribe for performance enhancement in the absence of psychopathology, we suggest first carefully considering how to maintain the ethical value of nonmaleficence by weighing both the potential physical and psychologic harms of prescribing as well as the legal risks and rules of applicable sport governing bodies.
For a psychiatrist who chooses to prescribe for performance enhancement, we recommend conducting a thorough psychiatric assessment to determine whether the patient has a treatable mental illness. If so, then effective treatment of that illness should take priority. Before prescribing, the psychiatrist and patient should discuss the patient’s specific performance goals and how to measure them.
Any prescription for a performance-enhancing medication should be given in conjunction with nonpharmacologic approaches, including optimizing diet, exercise, and sleep. Therapy to address problem-solving techniques and skills to cope with stress may also be appropriate. The patient and psychiatrist should engage in regular follow-up to assess the efficacy of the medication, as well as its safety and tolerability. Finally, if a medication is not efficacious as a performance enhancer, then both the patient and psychiatrist should be open to re-evaluating the treatment plan, and when appropriate, stopping the medication.
Performance enhancement in sports (“doping”) dates back to Ancient Greece. This was an era when Olympic athletes would attempt to improve their physical performance by consuming magic potions, herbal medications, and even exotic meats such as sheep testicles—a delicacy high in testosterone. Advances in medical and pharmaceutical technologies have increased both the range of enhancement agents available and their efficacy, leading to the development of anti-doping agencies and routine screening for doping in athletics. This has led to the renouncement of titles, medals, and financial sponsorship of athletes found to have been using prohibited substances during competition.
While doping in elite athletes often forms the nidus of media attention, the pressure to compete and perform at, or even beyond, one’s potential extends into many facets of today’s achievementfocused society. In the face of these pressures, individuals are increasingly seeking medications to enhance their performance across numerous domains, including cognitive, athletic, and artistic endeavors. Medication classes used to enhance performance include stimulants, which increase attention, executive function, and energy; cholinesterase inhibitors, which may ameliorate age-related memory decline; and beta-blockers, which decrease physiologic symptoms of anxiety and have been demonstrated to be beneficial for musical performance.1 Fifty-three percent of college athletes report using prescription medications to enhance athletic performance,2 and most college students who take stimulants without a prescription use them to study (84%) or stay awake (51%).3
Pharmacologic performance enhancement is the use of medications by healthy individuals to improve function in the absence of mental illness. Psychiatrists are increasingly finding themselves in the controversial position of “gatekeeper” of these medications for enhancement purposes. In this article we:
- outline arguments that support the use of psychopharmacology for performance enhancement, as well as some serious concerns with this practice
- discuss special considerations for pediatric populations and the risk of malpractice when prescribing for performance enhancement
- offer practice guidelines for approaching requests for psychopharmacologic performance enhancement.
Performance enhancement: The wave of the future?
The ethical principle that supports providing medication for performance enhancement is beneficence, the promotion of the patient’s well-being. In other words, it is a physician’s duty to help his or her patient in need. Individuals seeking performance enhancement typically present with suffering, and the principle of beneficence would call upon the psychiatrist to help ameliorate that suffering. Furthermore, patients who seek performance enhancement may present with impairing “subsyndromal” psychiatric symptoms (for example, low-grade attentional difficulty that occurs only in one setting), which, even if they do not rise to the threshold of a DSM diagnosis, may improve with psychiatric medications.
Using medical knowledge and skills beyond the traditional physician duty to diagnose and treat medical conditions is not unprecedented (eg, when surgeons perform cosmetic enhancement). Might elective enhancement of cognition and psychological performance through the judicious use of medication be part of the future of psychiatry? If cognitive and emotional enhancement becomes a more widely accepted standard of care, might this increase both individual and societal innovation and productivity?
Dilemma: Cautions against performance enhancement
One of the major cautions against prescribing psychotropics for the purpose of performance enhancement is the lack of clearly supported efficacy. Psychiatric medications generally are studied in individuals who meet criteria for mental illness, and they are FDA-approved for use in ill persons. It may be erroneous to extrapolate that a medication that improves symptoms in a patient with an illness would achieve the same target effect in a healthy individual. For example, data on whether stimulants provide neurocognitive enhancement in healthy individuals without attention-deficit/hyperactivity disorder is mixed, and these agents may even promote risky behavior in healthy controls.4 Furthermore, dopamine agonism may compress cognitive performance in both directions,5 as it has been observed that methylphenidate improves executive function in healthy controls, but is less beneficial for those with strong executive function at baseline.6
In the face of unclear benefit, it is particularly important to consider the risk of medications used for performance enhancement. Pharmacologic performance enhancement in individuals without psychopathology can be considered an “elective” intervention, for which individuals typically tolerate less risk. Physical risks, including medication-related adverse effects, must be considered, particularly in settings where there may be temptation to use more than prescribed, or to divert medication to others who may use it without medical monitoring. In addition to physical harm, there may be psychological harm associated with prescribing performance enhancers, such as pathologizing variants of “normal,” diminishing one’s sense of self-efficacy, or decreasing one’s ability to bear failure.
Continue to: Finally, there are ethical quandaries
Finally, there are ethical quandaries regarding using medications for performance enhancement. Widespread adoption of pharmacologic performance enhancement may lead to implicit coercion for all individuals to enhance their abilities. As a greater proportion of society receives pharmacologic enhancement, society as a whole faces stronger pressures to seek pharmacologic enhancement, ultimately constricting an individual’s freedom of choice to enhance.6 In this setting, distributive justice would become a consideration, because insurance companies are unlikely to reimburse for medications used for enhancement,7 which would give an advantage to individuals with higher socioeconomic status. Research shows that children from higher socioeconomic communities and from states with higher academic standards are more likely to use stimulants.8
Areas of controversy
Pediatric populations. There are special considerations when prescribing performance-enhancing medications for children and adolescents. First, such prescribing may inhibit normal child development, shifting the focus away from the normative tasks of social and emotional development that occur through leisure and creativity, experimentation, and play to an emphasis on performance and outcomes-based achievement.9 Second, during childhood and adolescence, one develops a sense of his or her identity, morals, and values. Taking a medication during childhood to enhance performance may inhibit the process of learning to tolerate failure, become aware of one’s weaknesses, and value effort in addition to outcome.
Malpractice risk. Practicing medicine beyond the scope of one’s expertise is unethical and unlawful. In the past 30 years, medical malpractice has become one of the most difficult health care issues in the U.S.10 In addition to billions of dollars in legal fees and court costs, medical malpractice premiums in the U.S. total more than $5 billion annually,11 and “defensive medicine”— procedures performed to protect against litigation—is estimated to cost more than $14 billion a year.12
When considering performance-enhancing treatment, it is the physician’s duty to conduct a diagnostic assessment, including noting target symptoms that are interfering with the patient’s function, and to tailor such treatment toward measurable goals and outcomes. Aside from medication, this could include a therapeutic approach to improving performance that might include cognitive-behavioral therapy and promotion of a healthy diet and exercise.
Treatment rises to the level of malpractice when there is a dereliction of duty that directly leads to damages.13 Part of a physician’s duty is to educate patients about the pros and cons of different treatment options. For performance-enhancing medications, the risks of addiction and dependence are adverse effects that require discussion. And for a pediatric patient, this would require the guardian’s engagement and understanding.
Continue to: What to do if you decide to prescribe
What to do if you decide to prescribe
Inevitably, the decision to prescribe psychotropic medications for performance enhancement is a physician-specific one. Certainly, psychiatrists should not feel obligated to prescribe performance enhancers. Given our current state of pharmacology, it is unclear whether medications would be helpful in the absence of psychopathology. When deciding whether to prescribe for performance enhancement in the absence of psychopathology, we suggest first carefully considering how to maintain the ethical value of nonmaleficence by weighing both the potential physical and psychologic harms of prescribing as well as the legal risks and rules of applicable sport governing bodies.
For a psychiatrist who chooses to prescribe for performance enhancement, we recommend conducting a thorough psychiatric assessment to determine whether the patient has a treatable mental illness. If so, then effective treatment of that illness should take priority. Before prescribing, the psychiatrist and patient should discuss the patient’s specific performance goals and how to measure them.
Any prescription for a performance-enhancing medication should be given in conjunction with nonpharmacologic approaches, including optimizing diet, exercise, and sleep. Therapy to address problem-solving techniques and skills to cope with stress may also be appropriate. The patient and psychiatrist should engage in regular follow-up to assess the efficacy of the medication, as well as its safety and tolerability. Finally, if a medication is not efficacious as a performance enhancer, then both the patient and psychiatrist should be open to re-evaluating the treatment plan, and when appropriate, stopping the medication.
1. Brantigan CO, Brantigan TA, Joseph N. Effect of beta blockade and beta stimulation on stage fright. Am J Med. 1982;72(1):88-94.
2. Hoyte CO, Albert D, Heard KJ. The use of energy drinks, dietary supplements, and prescription medications by United States college students to enhance athletic performance. J Community Health. 2013;38(3):575-850.
3. Advokat CD, Guidry D, Martino L. Licit and illicit use of medications for attention-deficit hyperactivity disorder in undergraduate college students. J Am Coll Health. 2008;56(6):601-606.
4. Advokat C, Scheithauer M. Attention-deficit hyperactivity disorder (ADHD) stimulant medications as cognitive enhancers. Front Neurosci. 2013;7:82.
5. Kimberg DY, D’Esposito M, Farah MJ. Effects of bromocriptine on human subjects depend on working memory capacity. Neuroreport. 1997;8(16):3581-3585.
6. Farah MJ, Illes J, Cook-Deegan R, et al. Neurocognitive enhancement: what can we do and what should we do? Nat Rev Neurosci. 2004;5(5):421-425.
7. Larriviere D, Williams MA, Rizzo M, et al; AAN Ethics, Law and Humanities Committee. Responding to requests from adult patients for neuroenhancements: guidance of the Ethics, Law and Humanities Committee. Neurology. 2009;73(17):1406-1412.
8. Colaneri N, Sheldon M, Adesman A. Pharmacological cognitive enhancement in pediatrics. Curr Opin Pediatr. 2018;30(3):430-437.
9. Gaucher N, Payot A, Racine E. Cognitive enhancement in children and adolescents: Is it in their best interests? Acta Paediatr. 2013;102(12):1118-1124.
10. Moore PJ, Adler, NE, Robertson, PA. Medical malpractice; the effect of doctor-patient relations on medical patient perceptions and malpractice intentions. West J Med. 2000;173(4):244-250.
11. Hiatt H. Medical malpractice. Bull N Y Acad Med. 1992;68(2):254-260.
12. Rubin RJ, Mendelson DN. How much does defensive medicine cost? J Am Health Policy. 1994;4(4):7-15.
13. Kloss D. The duty of care: medical negligence. Br Med J (Clin Res Ed). 1984;289(6436):66-68.
1. Brantigan CO, Brantigan TA, Joseph N. Effect of beta blockade and beta stimulation on stage fright. Am J Med. 1982;72(1):88-94.
2. Hoyte CO, Albert D, Heard KJ. The use of energy drinks, dietary supplements, and prescription medications by United States college students to enhance athletic performance. J Community Health. 2013;38(3):575-850.
3. Advokat CD, Guidry D, Martino L. Licit and illicit use of medications for attention-deficit hyperactivity disorder in undergraduate college students. J Am Coll Health. 2008;56(6):601-606.
4. Advokat C, Scheithauer M. Attention-deficit hyperactivity disorder (ADHD) stimulant medications as cognitive enhancers. Front Neurosci. 2013;7:82.
5. Kimberg DY, D’Esposito M, Farah MJ. Effects of bromocriptine on human subjects depend on working memory capacity. Neuroreport. 1997;8(16):3581-3585.
6. Farah MJ, Illes J, Cook-Deegan R, et al. Neurocognitive enhancement: what can we do and what should we do? Nat Rev Neurosci. 2004;5(5):421-425.
7. Larriviere D, Williams MA, Rizzo M, et al; AAN Ethics, Law and Humanities Committee. Responding to requests from adult patients for neuroenhancements: guidance of the Ethics, Law and Humanities Committee. Neurology. 2009;73(17):1406-1412.
8. Colaneri N, Sheldon M, Adesman A. Pharmacological cognitive enhancement in pediatrics. Curr Opin Pediatr. 2018;30(3):430-437.
9. Gaucher N, Payot A, Racine E. Cognitive enhancement in children and adolescents: Is it in their best interests? Acta Paediatr. 2013;102(12):1118-1124.
10. Moore PJ, Adler, NE, Robertson, PA. Medical malpractice; the effect of doctor-patient relations on medical patient perceptions and malpractice intentions. West J Med. 2000;173(4):244-250.
11. Hiatt H. Medical malpractice. Bull N Y Acad Med. 1992;68(2):254-260.
12. Rubin RJ, Mendelson DN. How much does defensive medicine cost? J Am Health Policy. 1994;4(4):7-15.
13. Kloss D. The duty of care: medical negligence. Br Med J (Clin Res Ed). 1984;289(6436):66-68.

A mood disorder complicated by multiple sclerosis
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.

Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
Urine drug screens: Not just for job applicants
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
Although urine drug screens (UDS) are most commonly used to screen job applicants, some clinicians have started to use them as a tool for improving their patients’ clinical outcomes.1 Recently, some clinicians have begun using UDS to help patients who experience chronic pain and dependency (mainly on opioids) and for those who use diverted drugs to relieve these conditions. Many psychiatrists are concerned about the high cost of drug diversion, as well as the possibility of diversion-related patient mortality. Clinicians should therefore consider using UDS as a tool to help address these challenges.
Consider individualized UDS monitoring
The standard 5-substance UDS test panel consists of tetrahydrocannabinol, opiates, amphetamines, cocaine, and phencyclidine. Although this panel was sufficient for an employment screening-related UDS, the American Society of Addiction Medicine (ASAM) has rejected its use for patients with substance abuse. As part of its emphasis on the importance of incorporating preventative procedures, diagnostics, and surveillance protocols, the ASAM advocates using a rotating test panel in conjunction with a patient-specific UDS.2 This type of patient-specific regimen would take into account the dynamic nature of a patient’s health profile factors, including comorbid and psychosocial status, subjective pain features, and diverted drug use. Furthermore, the ASAM recommends evaluating patients for the concurrent use of other substances and agents, such as benzodiazepines, sleep-inducing medications, stimulants, and alcohol, because these can interact with opioids.
Consider extending individualized monitoring by implementing standard “cutoff” values for each drug; patients whose levels of a specific substance are above the established cutoff value are categorized as testing positive for the use of that substance. The Substance Abuse Mental Health Services Administration favors adjusting UDS cutoffs, specifically the use of decreased cutoffs, to improve patient compliance.3 However, standardized drug concentration cutoff values may not be applicable for each patient; therefore, such values may need to be carefully tailored to each patient.
Additional drug monitoring techniques
Existing UDS practices, such as medication adherence and compliance, can be supplemented or alternately used with UDS panels that are modified to account for a patient’s fluctuating clinical conditions and concurrent medications. Point-of-care immunoassays, which provide accurate screening for medication compliance and adherence and possible drug diversion, should be used for routine monitoring. Using DNA-authenticated UDS also adds further control in monitoring a patient’s use of different drugs.4,5
In addition to being helpful for monitoring opioid use, a DNA-verified UDS can be used to evaluate for the presence of synthetic urine substitutes.6-8 Diversion remains a growing epidemiologic concern, and the number of cases is vastly underreported in the literature. The DNA-authenticated UDS can give clinicians greater precision in identifying synthetic and substituted urine among patient-provided samples.4
Using a combination of the methods described here can help expand a clinician’s ability to perform individualized drug monitoring, and verify whether a patient is adhering to his or her treatment regimen.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
1. Choudhry Z, Islam F, Siddiqui W, et al. UDS in mental health: is it time to move forward? J Psychiatry. 2015;18(5): doi: 10.4172/2378-5756.1000319.
2. Drug testing: a white paper of the American Society of Addiction Medicine. Chevy Chase, MD: American Society of Addiction Medicine; https://www.asam.org/docs/default-source/public-policy-statements/drug-testing-a-white-paper-by-asam.pdf. Published October 26, 2013. Accessed November 13, 2018.
3. Substance Abuse Mental Health Services Administration (SAMHSA). Technical Assistance Publication Series, TAP 32. Clinical drug testing in primary care. Rockville, MD: U.S. Department of Health and Human Services; 2012.
4. Genotox Laboratories. DNA Authenticated Drug Screen (ToxProtect). https://genotoxlabs.com/. Accessed October 11, 2018
5. 3RX Holdings Inc. 3RX Toxicology Urinary Drug Testing. http://3rxholdings.com/. Accessed October 11, 2018.
6. Genetic testing to confirm the identity of laboratory specimens. Document No GENE.00041. Medical Policy. Virginia Beach, VA: Amerigroup; 2018.
7. UnitedHealthcare Services. Drug Testing Policy. Reimbursement policy No 2018R6005A. https://www.uhcprovider.com/content/dam/provider/docs/public/policies/comm-reimbursement/COMM-Drug-Testing-Policy.pdf. Accessed October 12, 2018.
8. OzMed Laboratory Services. DNA-Verified Urine Drug Testing. http://www.ozmed.org/. Accessed October 11, 2018.
Study reveals long-term survival in MM patients

A retrospective study suggests one in seven patients with newly diagnosed multiple myeloma (MM) who are eligible for transplant may live at least as long as similar individuals in the general population.
The study included more than 7,000 MM patients, and 14.3% of those patients were able to meet or exceed their expected survival based on data from matched subjects in the general population.
Researchers believe that figure may be even higher today, as more than 90% of patients in this study were treated in the era before novel therapies became available.
Saad Z. Usmani, MD, of the Levine Cancer Institute/Atrium Health in Charlotte, North Carolina, and his colleagues described this study in Blood Cancer Journal.
The researchers studied 7,291 patients with newly diagnosed MM who were up to 75 years old and eligible for treatment with high-dose melphalan and autologous stem cell transplant. The patients were treated on clinical trials in 10 countries.
Factors associated with survival
Patients who had achieved a complete response (CR) 1 year after diagnosis had better median progression-free survival (PFS) than patients who did not achieve a CR—3.3 years and 2.6 years, respectively (P<0.0001).
Patients with a CR also had better median overall survival (OS)—8.5 years and 6.3 years, respectively (P<0.0001).
The identification of early CR as a predictor of PFS and OS “underscores the importance of depth of response as we explore novel regimens for newly diagnosed MM along with MRD [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote.
They did acknowledge, however, that the patients studied were a selected group eligible for transplant and treated on trials.
Dr. Usmani and his colleagues also performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death. The results indicated that patients were less likely to be alive at 10 years if they:
- Were older than 65 (odds ratio [OR]for death, 1.87, P=0.002)
- Had an IgA isotype (OR=1.53; P=0.004)
- Had an albumin level lower than 3.5 g/dL (OR=1.36; P=0.023)
- Had a beta-2 microglobulin level of at least 3.5 mg/dL (OR=1.86; P<0.001)
- Had a serum creatinine level of at least 2 mg/dL (OR=1.77; P=0.005)
- Had a hemoglobin level below 10 g/dL (OR=1.55; P=0.003)
- Had a platelet count below 150,000/μL (OR=2.26; P<0.001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Comparison to general population
Overall, the MM patients had a relative survival of about 0.9 compared with the matched general population. Relative survival was the ratio of observed survival among MM patients to expected survival in a population with comparable characteristics, such as nationality, age, and sex.
With follow-up out to about 20 years, the cure fraction—or the proportion of patients achieving or exceeding expected survival compared with the matched general population—was 14.3%.
The researchers noted that recent therapeutic advances “have re-ignited the debate on possible functional curability of a subset of MM patients. [T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s.”
“This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [a partial response] yet had good long-term survival.”
Dr. Usmani reported relationships with AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, SkylineDx, Array Biopharma, and Pharmacyclics.
A retrospective study suggests one in seven patients with newly diagnosed multiple myeloma (MM) who are eligible for transplant may live at least as long as similar individuals in the general population.
The study included more than 7,000 MM patients, and 14.3% of those patients were able to meet or exceed their expected survival based on data from matched subjects in the general population.
Researchers believe that figure may be even higher today, as more than 90% of patients in this study were treated in the era before novel therapies became available.
Saad Z. Usmani, MD, of the Levine Cancer Institute/Atrium Health in Charlotte, North Carolina, and his colleagues described this study in Blood Cancer Journal.
The researchers studied 7,291 patients with newly diagnosed MM who were up to 75 years old and eligible for treatment with high-dose melphalan and autologous stem cell transplant. The patients were treated on clinical trials in 10 countries.
Factors associated with survival
Patients who had achieved a complete response (CR) 1 year after diagnosis had better median progression-free survival (PFS) than patients who did not achieve a CR—3.3 years and 2.6 years, respectively (P<0.0001).
Patients with a CR also had better median overall survival (OS)—8.5 years and 6.3 years, respectively (P<0.0001).
The identification of early CR as a predictor of PFS and OS “underscores the importance of depth of response as we explore novel regimens for newly diagnosed MM along with MRD [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote.
They did acknowledge, however, that the patients studied were a selected group eligible for transplant and treated on trials.
Dr. Usmani and his colleagues also performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death. The results indicated that patients were less likely to be alive at 10 years if they:
- Were older than 65 (odds ratio [OR]for death, 1.87, P=0.002)
- Had an IgA isotype (OR=1.53; P=0.004)
- Had an albumin level lower than 3.5 g/dL (OR=1.36; P=0.023)
- Had a beta-2 microglobulin level of at least 3.5 mg/dL (OR=1.86; P<0.001)
- Had a serum creatinine level of at least 2 mg/dL (OR=1.77; P=0.005)
- Had a hemoglobin level below 10 g/dL (OR=1.55; P=0.003)
- Had a platelet count below 150,000/μL (OR=2.26; P<0.001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Comparison to general population
Overall, the MM patients had a relative survival of about 0.9 compared with the matched general population. Relative survival was the ratio of observed survival among MM patients to expected survival in a population with comparable characteristics, such as nationality, age, and sex.
With follow-up out to about 20 years, the cure fraction—or the proportion of patients achieving or exceeding expected survival compared with the matched general population—was 14.3%.
The researchers noted that recent therapeutic advances “have re-ignited the debate on possible functional curability of a subset of MM patients. [T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s.”
“This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [a partial response] yet had good long-term survival.”
Dr. Usmani reported relationships with AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, SkylineDx, Array Biopharma, and Pharmacyclics.
A retrospective study suggests one in seven patients with newly diagnosed multiple myeloma (MM) who are eligible for transplant may live at least as long as similar individuals in the general population.
The study included more than 7,000 MM patients, and 14.3% of those patients were able to meet or exceed their expected survival based on data from matched subjects in the general population.
Researchers believe that figure may be even higher today, as more than 90% of patients in this study were treated in the era before novel therapies became available.
Saad Z. Usmani, MD, of the Levine Cancer Institute/Atrium Health in Charlotte, North Carolina, and his colleagues described this study in Blood Cancer Journal.
The researchers studied 7,291 patients with newly diagnosed MM who were up to 75 years old and eligible for treatment with high-dose melphalan and autologous stem cell transplant. The patients were treated on clinical trials in 10 countries.
Factors associated with survival
Patients who had achieved a complete response (CR) 1 year after diagnosis had better median progression-free survival (PFS) than patients who did not achieve a CR—3.3 years and 2.6 years, respectively (P<0.0001).
Patients with a CR also had better median overall survival (OS)—8.5 years and 6.3 years, respectively (P<0.0001).
The identification of early CR as a predictor of PFS and OS “underscores the importance of depth of response as we explore novel regimens for newly diagnosed MM along with MRD [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote.
They did acknowledge, however, that the patients studied were a selected group eligible for transplant and treated on trials.
Dr. Usmani and his colleagues also performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death. The results indicated that patients were less likely to be alive at 10 years if they:
- Were older than 65 (odds ratio [OR]for death, 1.87, P=0.002)
- Had an IgA isotype (OR=1.53; P=0.004)
- Had an albumin level lower than 3.5 g/dL (OR=1.36; P=0.023)
- Had a beta-2 microglobulin level of at least 3.5 mg/dL (OR=1.86; P<0.001)
- Had a serum creatinine level of at least 2 mg/dL (OR=1.77; P=0.005)
- Had a hemoglobin level below 10 g/dL (OR=1.55; P=0.003)
- Had a platelet count below 150,000/μL (OR=2.26; P<0.001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Comparison to general population
Overall, the MM patients had a relative survival of about 0.9 compared with the matched general population. Relative survival was the ratio of observed survival among MM patients to expected survival in a population with comparable characteristics, such as nationality, age, and sex.
With follow-up out to about 20 years, the cure fraction—or the proportion of patients achieving or exceeding expected survival compared with the matched general population—was 14.3%.
The researchers noted that recent therapeutic advances “have re-ignited the debate on possible functional curability of a subset of MM patients. [T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s.”
“This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [a partial response] yet had good long-term survival.”
Dr. Usmani reported relationships with AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, SkylineDx, Array Biopharma, and Pharmacyclics.
The case for longer treatment in MM: Part 1

In Part 1 of this editorial, Katja Weisel, MD, of University Hospital Tubingen in Germany, describes the benefits of longer treatment in patients with multiple myeloma.
Despite recent progress in advancing the care of patients with multiple myeloma (MM), this cancer remains incurable.
Although novel combination regimens have driven major improvements in patient outcomes, most MM patients still experience multiple relapses, even those who respond to treatment initially.1
Historically, MM was treated for a fixed duration, followed by a treatment-free interval and additional treatment at relapse. However, evidence suggests that continuous therapy after an initial response may be a better approach.2,3
Pooled data from three large, phase 3 trials in newly diagnosed MM patients suggest that continuous therapy may lead to an increase in progression-free survival (PFS) and overall survival (OS).2
These results are supported by a meta-analysis, which showed favorable outcomes in PFS and OS with lenalidomide maintenance compared to placebo or observation in newly diagnosed MM patients who had received high-dose therapy and autologous stem cell transplant.3
Given these emerging findings and the availability of effective and tolerable therapies suitable for longer use, there is an opportunity to increase the adoption of this treatment strategy to improve outcomes for MM patients.
The concept of longer treatment for MM is not new. The first clinical trials in which researchers evaluated the efficacy and safety of this approach were conducted 40 years ago in patients initially treated with melphalan and prednisone. However, modest efficacy and substantial toxicity limited longer treatment with those agents.4-7
The intervening years saw the introduction of new agents with different mechanisms of action, such as proteasome inhibitors and immunomodulators. These therapies, commonly used as initial treatment, provided physicians with additional options for treating patients longer.
Research has shown that longer treatment with immunomodulatory agents and proteasome inhibitors can be clinically effective.8
Longer treatment—integrated in the first-line treatment strategy and before a patient relapses—may enhance conventional induction strategies, resulting in better PFS and OS.9,10
Continuous treatment, in which a patient receives treatment beyond a fixed induction period, has demonstrated extended PFS and OS as well.2,3
Data supporting the benefits of prolonged therapy with immunomodulatory drugs has been a key driver behind the shifting paradigm in favor of longer treatment as the standard of care.11,3
Additionally, continuing treatment with a proteasome inhibitor beyond induction therapy is associated with an improvement in the depth of response and prolonged OS.12
Longer treatment with proteasome inhibitors is also associated with deepening response rates and improved PFS following hematopoietic stem cell transplant.13-15
Recent research has also shown that patients may achieve deeper remission with longer treatment,16,17 overturning the long-held belief that longer duration of therapy can only extend a response rather than improve it.
Moreover, treating patients for longer may now be possible because of the favorable toxicity profile of some of the novel therapies currently available, which have fewer cumulative or late-onset toxicities.18
Dr. Weisel has received honoraria and/or consultancy fees from Amgen, BMS, Celgene, Janssen, Juno, Sanofi, and Takeda. She has received research funding from Amgen, Celgene, Sanofi, and Janssen.
The W2O Group provided writing support for this editorial, which was funded by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
1. Lonial S. Hematology Am Soc Hematol Educ Program. 2010; 2010:303-9. doi: 10.1182/asheducation-2010.1.303
2. Palumbo A et al. J Clin Oncol. 2015; 33(30):3459-66. doi: 10.1200/JCO.2014.60.2466
3. McCarthy PL et al. J Clin Oncol. 2017; 35(29):3279-3289. doi: 10.1200/JCO.2017.72.6679
4. Joks M et al. Eur J Haematol. 2015 ;94(2):109-14. doi: 10.1111/ejh.12412
5. Berenson JR et al. Blood. 2002; 99:3163-8. doi: http://www.bloodjournal.org/content/99/9/3163.long
6. Shustik C et al. Br J Haematol. 2007; 126:201-11. doi: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2141.2006.06405.x
7. Fritz E, Ludwig H. Ann Oncol. 2000 Nov;11(11):1427-36
8. Ludwig H et al. Blood. 2012; 119:3003-3015. doi: https://doi.org/10.1182/blood-2011-11-374249
9. Mateos MV et al. Am J Hematol. 2015; 90(4):314-9. doi: 10.1002/ajh.23933
10. Benboubker L et al. N Engl J Med. 2014; 371(10):906-17. doi: 10.1056/NEJMoa1402551
11. Holstein SA et al. Lancet Haematol. 2017; 4(9):e431-e442. doi: 10.1016/S2352-3026(17)30140-0
12. Mateos MV et al. Blood. 2014; 124:1887-1893. doi: https://doi.org/10.1182/blood-2014-05-573733
13. Sonneveld P et al. ASH Annual Meeting Abstracts. Blood. 2010;116. Abstract 40
14. Rosiñol L et al. Blood. 2012; 120(8):1589-96. doi: https://doi.org/10.1182/blood-2012-02-408922
15. Richardson PG et al. N Engl J Med. 2005; 352(24):2487-98. doi: 10.1056/NEJMoa043445
16. de Tute RM et al. ASH Annual Meeting Abstracts. Blood. 2017; 130: 904. Abstract 904
17. Dimopoulos M et al. J Hematol Oncol. 2018;11(1):49. doi: 10.1186/s13045-018-0583-7
18. Lipe B et al. Blood Cancer J. 2016; 6(10): e485. doi: 10.1038/bcj.2016.89
In Part 1 of this editorial, Katja Weisel, MD, of University Hospital Tubingen in Germany, describes the benefits of longer treatment in patients with multiple myeloma.
Despite recent progress in advancing the care of patients with multiple myeloma (MM), this cancer remains incurable.
Although novel combination regimens have driven major improvements in patient outcomes, most MM patients still experience multiple relapses, even those who respond to treatment initially.1
Historically, MM was treated for a fixed duration, followed by a treatment-free interval and additional treatment at relapse. However, evidence suggests that continuous therapy after an initial response may be a better approach.2,3
Pooled data from three large, phase 3 trials in newly diagnosed MM patients suggest that continuous therapy may lead to an increase in progression-free survival (PFS) and overall survival (OS).2
These results are supported by a meta-analysis, which showed favorable outcomes in PFS and OS with lenalidomide maintenance compared to placebo or observation in newly diagnosed MM patients who had received high-dose therapy and autologous stem cell transplant.3
Given these emerging findings and the availability of effective and tolerable therapies suitable for longer use, there is an opportunity to increase the adoption of this treatment strategy to improve outcomes for MM patients.
The concept of longer treatment for MM is not new. The first clinical trials in which researchers evaluated the efficacy and safety of this approach were conducted 40 years ago in patients initially treated with melphalan and prednisone. However, modest efficacy and substantial toxicity limited longer treatment with those agents.4-7
The intervening years saw the introduction of new agents with different mechanisms of action, such as proteasome inhibitors and immunomodulators. These therapies, commonly used as initial treatment, provided physicians with additional options for treating patients longer.
Research has shown that longer treatment with immunomodulatory agents and proteasome inhibitors can be clinically effective.8
Longer treatment—integrated in the first-line treatment strategy and before a patient relapses—may enhance conventional induction strategies, resulting in better PFS and OS.9,10
Continuous treatment, in which a patient receives treatment beyond a fixed induction period, has demonstrated extended PFS and OS as well.2,3
Data supporting the benefits of prolonged therapy with immunomodulatory drugs has been a key driver behind the shifting paradigm in favor of longer treatment as the standard of care.11,3
Additionally, continuing treatment with a proteasome inhibitor beyond induction therapy is associated with an improvement in the depth of response and prolonged OS.12
Longer treatment with proteasome inhibitors is also associated with deepening response rates and improved PFS following hematopoietic stem cell transplant.13-15
Recent research has also shown that patients may achieve deeper remission with longer treatment,16,17 overturning the long-held belief that longer duration of therapy can only extend a response rather than improve it.
Moreover, treating patients for longer may now be possible because of the favorable toxicity profile of some of the novel therapies currently available, which have fewer cumulative or late-onset toxicities.18
Dr. Weisel has received honoraria and/or consultancy fees from Amgen, BMS, Celgene, Janssen, Juno, Sanofi, and Takeda. She has received research funding from Amgen, Celgene, Sanofi, and Janssen.
The W2O Group provided writing support for this editorial, which was funded by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
1. Lonial S. Hematology Am Soc Hematol Educ Program. 2010; 2010:303-9. doi: 10.1182/asheducation-2010.1.303
2. Palumbo A et al. J Clin Oncol. 2015; 33(30):3459-66. doi: 10.1200/JCO.2014.60.2466
3. McCarthy PL et al. J Clin Oncol. 2017; 35(29):3279-3289. doi: 10.1200/JCO.2017.72.6679
4. Joks M et al. Eur J Haematol. 2015 ;94(2):109-14. doi: 10.1111/ejh.12412
5. Berenson JR et al. Blood. 2002; 99:3163-8. doi: http://www.bloodjournal.org/content/99/9/3163.long
6. Shustik C et al. Br J Haematol. 2007; 126:201-11. doi: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2141.2006.06405.x
7. Fritz E, Ludwig H. Ann Oncol. 2000 Nov;11(11):1427-36
8. Ludwig H et al. Blood. 2012; 119:3003-3015. doi: https://doi.org/10.1182/blood-2011-11-374249
9. Mateos MV et al. Am J Hematol. 2015; 90(4):314-9. doi: 10.1002/ajh.23933
10. Benboubker L et al. N Engl J Med. 2014; 371(10):906-17. doi: 10.1056/NEJMoa1402551
11. Holstein SA et al. Lancet Haematol. 2017; 4(9):e431-e442. doi: 10.1016/S2352-3026(17)30140-0
12. Mateos MV et al. Blood. 2014; 124:1887-1893. doi: https://doi.org/10.1182/blood-2014-05-573733
13. Sonneveld P et al. ASH Annual Meeting Abstracts. Blood. 2010;116. Abstract 40
14. Rosiñol L et al. Blood. 2012; 120(8):1589-96. doi: https://doi.org/10.1182/blood-2012-02-408922
15. Richardson PG et al. N Engl J Med. 2005; 352(24):2487-98. doi: 10.1056/NEJMoa043445
16. de Tute RM et al. ASH Annual Meeting Abstracts. Blood. 2017; 130: 904. Abstract 904
17. Dimopoulos M et al. J Hematol Oncol. 2018;11(1):49. doi: 10.1186/s13045-018-0583-7
18. Lipe B et al. Blood Cancer J. 2016; 6(10): e485. doi: 10.1038/bcj.2016.89
In Part 1 of this editorial, Katja Weisel, MD, of University Hospital Tubingen in Germany, describes the benefits of longer treatment in patients with multiple myeloma.
Despite recent progress in advancing the care of patients with multiple myeloma (MM), this cancer remains incurable.
Although novel combination regimens have driven major improvements in patient outcomes, most MM patients still experience multiple relapses, even those who respond to treatment initially.1
Historically, MM was treated for a fixed duration, followed by a treatment-free interval and additional treatment at relapse. However, evidence suggests that continuous therapy after an initial response may be a better approach.2,3
Pooled data from three large, phase 3 trials in newly diagnosed MM patients suggest that continuous therapy may lead to an increase in progression-free survival (PFS) and overall survival (OS).2
These results are supported by a meta-analysis, which showed favorable outcomes in PFS and OS with lenalidomide maintenance compared to placebo or observation in newly diagnosed MM patients who had received high-dose therapy and autologous stem cell transplant.3
Given these emerging findings and the availability of effective and tolerable therapies suitable for longer use, there is an opportunity to increase the adoption of this treatment strategy to improve outcomes for MM patients.
The concept of longer treatment for MM is not new. The first clinical trials in which researchers evaluated the efficacy and safety of this approach were conducted 40 years ago in patients initially treated with melphalan and prednisone. However, modest efficacy and substantial toxicity limited longer treatment with those agents.4-7
The intervening years saw the introduction of new agents with different mechanisms of action, such as proteasome inhibitors and immunomodulators. These therapies, commonly used as initial treatment, provided physicians with additional options for treating patients longer.
Research has shown that longer treatment with immunomodulatory agents and proteasome inhibitors can be clinically effective.8
Longer treatment—integrated in the first-line treatment strategy and before a patient relapses—may enhance conventional induction strategies, resulting in better PFS and OS.9,10
Continuous treatment, in which a patient receives treatment beyond a fixed induction period, has demonstrated extended PFS and OS as well.2,3
Data supporting the benefits of prolonged therapy with immunomodulatory drugs has been a key driver behind the shifting paradigm in favor of longer treatment as the standard of care.11,3
Additionally, continuing treatment with a proteasome inhibitor beyond induction therapy is associated with an improvement in the depth of response and prolonged OS.12
Longer treatment with proteasome inhibitors is also associated with deepening response rates and improved PFS following hematopoietic stem cell transplant.13-15
Recent research has also shown that patients may achieve deeper remission with longer treatment,16,17 overturning the long-held belief that longer duration of therapy can only extend a response rather than improve it.
Moreover, treating patients for longer may now be possible because of the favorable toxicity profile of some of the novel therapies currently available, which have fewer cumulative or late-onset toxicities.18
Dr. Weisel has received honoraria and/or consultancy fees from Amgen, BMS, Celgene, Janssen, Juno, Sanofi, and Takeda. She has received research funding from Amgen, Celgene, Sanofi, and Janssen.
The W2O Group provided writing support for this editorial, which was funded by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
1. Lonial S. Hematology Am Soc Hematol Educ Program. 2010; 2010:303-9. doi: 10.1182/asheducation-2010.1.303
2. Palumbo A et al. J Clin Oncol. 2015; 33(30):3459-66. doi: 10.1200/JCO.2014.60.2466
3. McCarthy PL et al. J Clin Oncol. 2017; 35(29):3279-3289. doi: 10.1200/JCO.2017.72.6679
4. Joks M et al. Eur J Haematol. 2015 ;94(2):109-14. doi: 10.1111/ejh.12412
5. Berenson JR et al. Blood. 2002; 99:3163-8. doi: http://www.bloodjournal.org/content/99/9/3163.long
6. Shustik C et al. Br J Haematol. 2007; 126:201-11. doi: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1365-2141.2006.06405.x
7. Fritz E, Ludwig H. Ann Oncol. 2000 Nov;11(11):1427-36
8. Ludwig H et al. Blood. 2012; 119:3003-3015. doi: https://doi.org/10.1182/blood-2011-11-374249
9. Mateos MV et al. Am J Hematol. 2015; 90(4):314-9. doi: 10.1002/ajh.23933
10. Benboubker L et al. N Engl J Med. 2014; 371(10):906-17. doi: 10.1056/NEJMoa1402551
11. Holstein SA et al. Lancet Haematol. 2017; 4(9):e431-e442. doi: 10.1016/S2352-3026(17)30140-0
12. Mateos MV et al. Blood. 2014; 124:1887-1893. doi: https://doi.org/10.1182/blood-2014-05-573733
13. Sonneveld P et al. ASH Annual Meeting Abstracts. Blood. 2010;116. Abstract 40
14. Rosiñol L et al. Blood. 2012; 120(8):1589-96. doi: https://doi.org/10.1182/blood-2012-02-408922
15. Richardson PG et al. N Engl J Med. 2005; 352(24):2487-98. doi: 10.1056/NEJMoa043445
16. de Tute RM et al. ASH Annual Meeting Abstracts. Blood. 2017; 130: 904. Abstract 904
17. Dimopoulos M et al. J Hematol Oncol. 2018;11(1):49. doi: 10.1186/s13045-018-0583-7
18. Lipe B et al. Blood Cancer J. 2016; 6(10): e485. doi: 10.1038/bcj.2016.89