User login
ISAR-REACT 5: Prasugrel superior to ticagrelor in ACS
PARIS – Prasugrel proved superior to ticagrelor in patients with acute coronary syndrome (ACS) in what was hailed as “a landmark study” presented at the annual congress of the European Society of Cardiology.

The results of ISAR-REACT 5 (Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment 5) were unequivocal: “In ACS patients with or without ST-segment elevation, treatment with prasugrel as compared with ticagrelor significantly reduced the composite rate of death, myocardial infarction, or stroke at 1 year without an increase in major bleeding,” declared first author Stephanie Schuepke, MD, a cardiologist at the German Heart Center in Munich.
The study outcome was totally unexpected. Indeed, the result didn’t merely turn heads, it no doubt caused numerous wrenched necks caused by strenuous double-takes on the part of interventional cardiologists at the congress. That’s because, even though both ticagrelor and prasugrel enjoy a class I recommendation for use in ACS in the ESC guidelines, it has been widely assumed – based on previous evidence plus the fact that ticagrelor is the more potent platelet inhibitor – that ticagrelor is the superior drug in this setting. It turns out, however, that those earlier studies weren’t germane to the issue directly addressed in ISAR-REACT 5, the first-ever direct head-to-head comparison of the two potent P2Y12 inhibitors in the setting of ACS with a planned invasive strategy.
“Obviously, very surprising results,” commented Roxana Mehran, MD, professor of medicine and director of interventional cardiology research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York, who cochaired a press conference where Dr. Schuepke shared the study findings.
“We were surprised as well,” confessed Dr. Schuepke. “We assumed that ticagrelor is superior to prasugrel in terms of clinical outcomes in patients with ACS with a planned invasive strategy. But the results show us that the opposite is true.”
ISAR-REACT 5 was an open-label, investigator-initiated randomized trial conducted at 23 centers in Germany and Italy. It included 4,018 participants with ST-elevation segment MI (STEMI), without STEMI, or with unstable angina, all with scheduled coronary angiography. Participants were randomized to ticagrelor or prasugrel and were expected to remain on their assigned potent antiplatelet agent plus aspirin for 1 year of dual-antiplatelet therapy.
The primary outcome was the composite of death, MI, or stroke at 1 year of follow-up. This endpoint occurred in 9.3% of the ticagrelor group and 6.9% of patients in the prasugrel group, for a highly significant 36% increased relative risk in the ticagrelor-treated patients. Prasugrel had a numeric advantage in each of the individual components of the endpoint: the 1-year rate of all-cause mortality was 4.5% with ticagrelor versus 3.7% with prasugrel; for MI, the incidence was 4.8% with ticagrelor and 3.0% with prasugrel, a statistically significant difference; and for stroke, 1.1% versus 1.0%.
Major bleeding as defined by the Bleeding Academic Research Consortium scale occurred in 5.4% of patients in the ticagrelor arm, and was similar at 4.8% in the prasugrel group, Dr. Schuepke continued.
Definite or probable stent thrombosis occurred in 1.3% of the ticagrelor group and 1.0% of patients assigned to prasugrel.
The mechanism for prasugrel’s superior results is unclear, she said. Possibilities include the fact that it’s a once-daily drug, compared with twice-daily ticagrelor, which could affect adherence; a differential profile in terms of drug interactions; and prasugrel’s reversibility of action and capacity for step-down dosing in patients at high bleeding risk.
Discussant Gilles Montalescot, MD, PhD, called ISAR-REACT 5 a “fascinating” study. He elaborated: “It is a pragmatic study to answer a pragmatic question. It’s not a drug trial; really, it’s more a strategy trial, with a comparison of two drugs and two strategies.”

In ISAR-REACT 5, ticagrelor was utilized in standard fashion: Patients, regardless of their ACS type, received a 180-mg loading dose as soon as possible after randomization, typically about 3 hours after presentation. That was also the protocol in the prasugrel arm, but only in patients with STEMI, who quickly got a 60-mg loading dose. In contrast, patients without STEMI or with unstable angina didn’t get a loading dose of prasugrel until they’d undergone coronary angiography and their coronary anatomy was known. That was the ISAR-REACT 5 strategy in light of an earlier study, which concluded that giving prasugrel prior to angiography in such patients led to increased bleeding without any improvement in clinical outcomes.
The essence of ISAR-REACT 5 lies in that difference in treatment strategies and the impact it had on outcomes. “The one-size-fits-all strategy – here, with ticagrelor – was inferior to an individualized strategy, here with prasugrel,” observed Dr. Montalescot, professor of cardiology at the University of Paris VI and director of the cardiac care unit at Paris-Salpetriere Hospital.
The study results were notably consistent, favoring prasugrel over ticagrelor numerically across the board regardless of whether a patient presented with STEMI, without STEMI, or with unstable angina. Particularly noteworthy was the finding that this was also true in terms of the individual components of the 1-year composite endpoint. Dr. Montalescot drew special attention to the large subset of patients who had presented with STEMI and thus received the same treatment strategy involving a loading dose of their assigned P2Y12 inhibitor prior to angiography. This allowed for a direct head-to-head comparison of the clinical efficacy of the two antiplatelet agents in STEMI patients. The clear winner here was prasugrel, as the composite event rate was 10.1% in the ticagrelor group, compared with 7.9% in the prasugrel group.
“ISAR-REACT 5 is a landmark study which is going to impact our practice and the next set of guidelines to come in 2020,” the interventional cardiologist predicted.
Dr. Schuepke reported having no financial conflicts regarding the ISAR-REACT 5 study, sponsored by the German Heart Center and the German Center for Cardiovascular Research.
Simultaneous with her presentation of ISAR-REACT 5, the study results were published online (N Engl J Med. 2019 Sep 1. doi: 10.1056/NEJMoa1908973).
PARIS – Prasugrel proved superior to ticagrelor in patients with acute coronary syndrome (ACS) in what was hailed as “a landmark study” presented at the annual congress of the European Society of Cardiology.
The results of ISAR-REACT 5 (Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment 5) were unequivocal: “In ACS patients with or without ST-segment elevation, treatment with prasugrel as compared with ticagrelor significantly reduced the composite rate of death, myocardial infarction, or stroke at 1 year without an increase in major bleeding,” declared first author Stephanie Schuepke, MD, a cardiologist at the German Heart Center in Munich.
The study outcome was totally unexpected. Indeed, the result didn’t merely turn heads, it no doubt caused numerous wrenched necks caused by strenuous double-takes on the part of interventional cardiologists at the congress. That’s because, even though both ticagrelor and prasugrel enjoy a class I recommendation for use in ACS in the ESC guidelines, it has been widely assumed – based on previous evidence plus the fact that ticagrelor is the more potent platelet inhibitor – that ticagrelor is the superior drug in this setting. It turns out, however, that those earlier studies weren’t germane to the issue directly addressed in ISAR-REACT 5, the first-ever direct head-to-head comparison of the two potent P2Y12 inhibitors in the setting of ACS with a planned invasive strategy.
“Obviously, very surprising results,” commented Roxana Mehran, MD, professor of medicine and director of interventional cardiology research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York, who cochaired a press conference where Dr. Schuepke shared the study findings.
“We were surprised as well,” confessed Dr. Schuepke. “We assumed that ticagrelor is superior to prasugrel in terms of clinical outcomes in patients with ACS with a planned invasive strategy. But the results show us that the opposite is true.”
ISAR-REACT 5 was an open-label, investigator-initiated randomized trial conducted at 23 centers in Germany and Italy. It included 4,018 participants with ST-elevation segment MI (STEMI), without STEMI, or with unstable angina, all with scheduled coronary angiography. Participants were randomized to ticagrelor or prasugrel and were expected to remain on their assigned potent antiplatelet agent plus aspirin for 1 year of dual-antiplatelet therapy.
The primary outcome was the composite of death, MI, or stroke at 1 year of follow-up. This endpoint occurred in 9.3% of the ticagrelor group and 6.9% of patients in the prasugrel group, for a highly significant 36% increased relative risk in the ticagrelor-treated patients. Prasugrel had a numeric advantage in each of the individual components of the endpoint: the 1-year rate of all-cause mortality was 4.5% with ticagrelor versus 3.7% with prasugrel; for MI, the incidence was 4.8% with ticagrelor and 3.0% with prasugrel, a statistically significant difference; and for stroke, 1.1% versus 1.0%.
Major bleeding as defined by the Bleeding Academic Research Consortium scale occurred in 5.4% of patients in the ticagrelor arm, and was similar at 4.8% in the prasugrel group, Dr. Schuepke continued.
Definite or probable stent thrombosis occurred in 1.3% of the ticagrelor group and 1.0% of patients assigned to prasugrel.
The mechanism for prasugrel’s superior results is unclear, she said. Possibilities include the fact that it’s a once-daily drug, compared with twice-daily ticagrelor, which could affect adherence; a differential profile in terms of drug interactions; and prasugrel’s reversibility of action and capacity for step-down dosing in patients at high bleeding risk.
Discussant Gilles Montalescot, MD, PhD, called ISAR-REACT 5 a “fascinating” study. He elaborated: “It is a pragmatic study to answer a pragmatic question. It’s not a drug trial; really, it’s more a strategy trial, with a comparison of two drugs and two strategies.”
In ISAR-REACT 5, ticagrelor was utilized in standard fashion: Patients, regardless of their ACS type, received a 180-mg loading dose as soon as possible after randomization, typically about 3 hours after presentation. That was also the protocol in the prasugrel arm, but only in patients with STEMI, who quickly got a 60-mg loading dose. In contrast, patients without STEMI or with unstable angina didn’t get a loading dose of prasugrel until they’d undergone coronary angiography and their coronary anatomy was known. That was the ISAR-REACT 5 strategy in light of an earlier study, which concluded that giving prasugrel prior to angiography in such patients led to increased bleeding without any improvement in clinical outcomes.
The essence of ISAR-REACT 5 lies in that difference in treatment strategies and the impact it had on outcomes. “The one-size-fits-all strategy – here, with ticagrelor – was inferior to an individualized strategy, here with prasugrel,” observed Dr. Montalescot, professor of cardiology at the University of Paris VI and director of the cardiac care unit at Paris-Salpetriere Hospital.
The study results were notably consistent, favoring prasugrel over ticagrelor numerically across the board regardless of whether a patient presented with STEMI, without STEMI, or with unstable angina. Particularly noteworthy was the finding that this was also true in terms of the individual components of the 1-year composite endpoint. Dr. Montalescot drew special attention to the large subset of patients who had presented with STEMI and thus received the same treatment strategy involving a loading dose of their assigned P2Y12 inhibitor prior to angiography. This allowed for a direct head-to-head comparison of the clinical efficacy of the two antiplatelet agents in STEMI patients. The clear winner here was prasugrel, as the composite event rate was 10.1% in the ticagrelor group, compared with 7.9% in the prasugrel group.
“ISAR-REACT 5 is a landmark study which is going to impact our practice and the next set of guidelines to come in 2020,” the interventional cardiologist predicted.
Dr. Schuepke reported having no financial conflicts regarding the ISAR-REACT 5 study, sponsored by the German Heart Center and the German Center for Cardiovascular Research.
Simultaneous with her presentation of ISAR-REACT 5, the study results were published online (N Engl J Med. 2019 Sep 1. doi: 10.1056/NEJMoa1908973).
PARIS – Prasugrel proved superior to ticagrelor in patients with acute coronary syndrome (ACS) in what was hailed as “a landmark study” presented at the annual congress of the European Society of Cardiology.
The results of ISAR-REACT 5 (Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment 5) were unequivocal: “In ACS patients with or without ST-segment elevation, treatment with prasugrel as compared with ticagrelor significantly reduced the composite rate of death, myocardial infarction, or stroke at 1 year without an increase in major bleeding,” declared first author Stephanie Schuepke, MD, a cardiologist at the German Heart Center in Munich.
The study outcome was totally unexpected. Indeed, the result didn’t merely turn heads, it no doubt caused numerous wrenched necks caused by strenuous double-takes on the part of interventional cardiologists at the congress. That’s because, even though both ticagrelor and prasugrel enjoy a class I recommendation for use in ACS in the ESC guidelines, it has been widely assumed – based on previous evidence plus the fact that ticagrelor is the more potent platelet inhibitor – that ticagrelor is the superior drug in this setting. It turns out, however, that those earlier studies weren’t germane to the issue directly addressed in ISAR-REACT 5, the first-ever direct head-to-head comparison of the two potent P2Y12 inhibitors in the setting of ACS with a planned invasive strategy.
“Obviously, very surprising results,” commented Roxana Mehran, MD, professor of medicine and director of interventional cardiology research and clinical trials at the Icahn School of Medicine at Mount Sinai, New York, who cochaired a press conference where Dr. Schuepke shared the study findings.
“We were surprised as well,” confessed Dr. Schuepke. “We assumed that ticagrelor is superior to prasugrel in terms of clinical outcomes in patients with ACS with a planned invasive strategy. But the results show us that the opposite is true.”
ISAR-REACT 5 was an open-label, investigator-initiated randomized trial conducted at 23 centers in Germany and Italy. It included 4,018 participants with ST-elevation segment MI (STEMI), without STEMI, or with unstable angina, all with scheduled coronary angiography. Participants were randomized to ticagrelor or prasugrel and were expected to remain on their assigned potent antiplatelet agent plus aspirin for 1 year of dual-antiplatelet therapy.
The primary outcome was the composite of death, MI, or stroke at 1 year of follow-up. This endpoint occurred in 9.3% of the ticagrelor group and 6.9% of patients in the prasugrel group, for a highly significant 36% increased relative risk in the ticagrelor-treated patients. Prasugrel had a numeric advantage in each of the individual components of the endpoint: the 1-year rate of all-cause mortality was 4.5% with ticagrelor versus 3.7% with prasugrel; for MI, the incidence was 4.8% with ticagrelor and 3.0% with prasugrel, a statistically significant difference; and for stroke, 1.1% versus 1.0%.
Major bleeding as defined by the Bleeding Academic Research Consortium scale occurred in 5.4% of patients in the ticagrelor arm, and was similar at 4.8% in the prasugrel group, Dr. Schuepke continued.
Definite or probable stent thrombosis occurred in 1.3% of the ticagrelor group and 1.0% of patients assigned to prasugrel.
The mechanism for prasugrel’s superior results is unclear, she said. Possibilities include the fact that it’s a once-daily drug, compared with twice-daily ticagrelor, which could affect adherence; a differential profile in terms of drug interactions; and prasugrel’s reversibility of action and capacity for step-down dosing in patients at high bleeding risk.
Discussant Gilles Montalescot, MD, PhD, called ISAR-REACT 5 a “fascinating” study. He elaborated: “It is a pragmatic study to answer a pragmatic question. It’s not a drug trial; really, it’s more a strategy trial, with a comparison of two drugs and two strategies.”
In ISAR-REACT 5, ticagrelor was utilized in standard fashion: Patients, regardless of their ACS type, received a 180-mg loading dose as soon as possible after randomization, typically about 3 hours after presentation. That was also the protocol in the prasugrel arm, but only in patients with STEMI, who quickly got a 60-mg loading dose. In contrast, patients without STEMI or with unstable angina didn’t get a loading dose of prasugrel until they’d undergone coronary angiography and their coronary anatomy was known. That was the ISAR-REACT 5 strategy in light of an earlier study, which concluded that giving prasugrel prior to angiography in such patients led to increased bleeding without any improvement in clinical outcomes.
The essence of ISAR-REACT 5 lies in that difference in treatment strategies and the impact it had on outcomes. “The one-size-fits-all strategy – here, with ticagrelor – was inferior to an individualized strategy, here with prasugrel,” observed Dr. Montalescot, professor of cardiology at the University of Paris VI and director of the cardiac care unit at Paris-Salpetriere Hospital.
The study results were notably consistent, favoring prasugrel over ticagrelor numerically across the board regardless of whether a patient presented with STEMI, without STEMI, or with unstable angina. Particularly noteworthy was the finding that this was also true in terms of the individual components of the 1-year composite endpoint. Dr. Montalescot drew special attention to the large subset of patients who had presented with STEMI and thus received the same treatment strategy involving a loading dose of their assigned P2Y12 inhibitor prior to angiography. This allowed for a direct head-to-head comparison of the clinical efficacy of the two antiplatelet agents in STEMI patients. The clear winner here was prasugrel, as the composite event rate was 10.1% in the ticagrelor group, compared with 7.9% in the prasugrel group.
“ISAR-REACT 5 is a landmark study which is going to impact our practice and the next set of guidelines to come in 2020,” the interventional cardiologist predicted.
Dr. Schuepke reported having no financial conflicts regarding the ISAR-REACT 5 study, sponsored by the German Heart Center and the German Center for Cardiovascular Research.
Simultaneous with her presentation of ISAR-REACT 5, the study results were published online (N Engl J Med. 2019 Sep 1. doi: 10.1056/NEJMoa1908973).
REPORTING FROM THE ESC CONGRESS 2019
Adolescent and young adult (AYA) survival trends
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
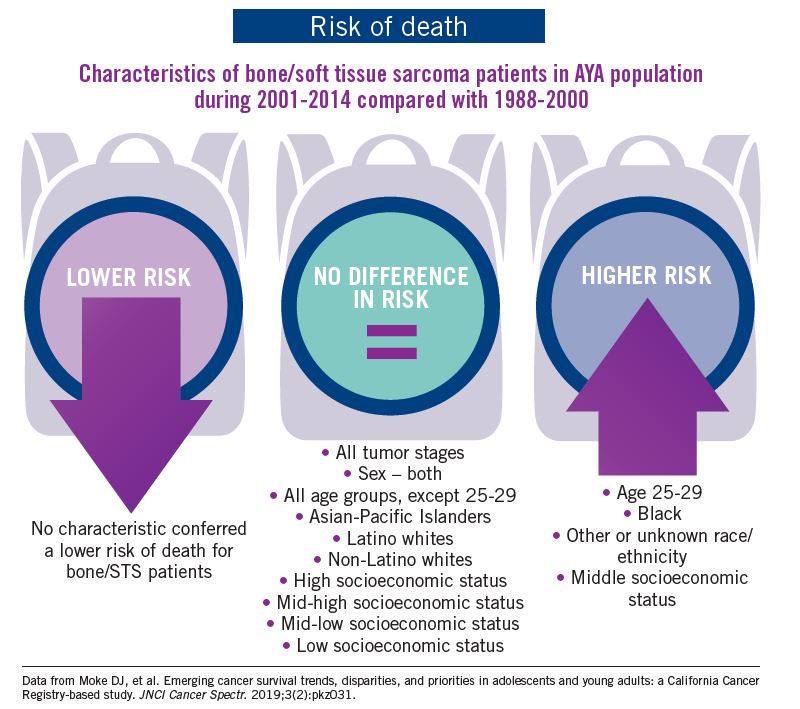
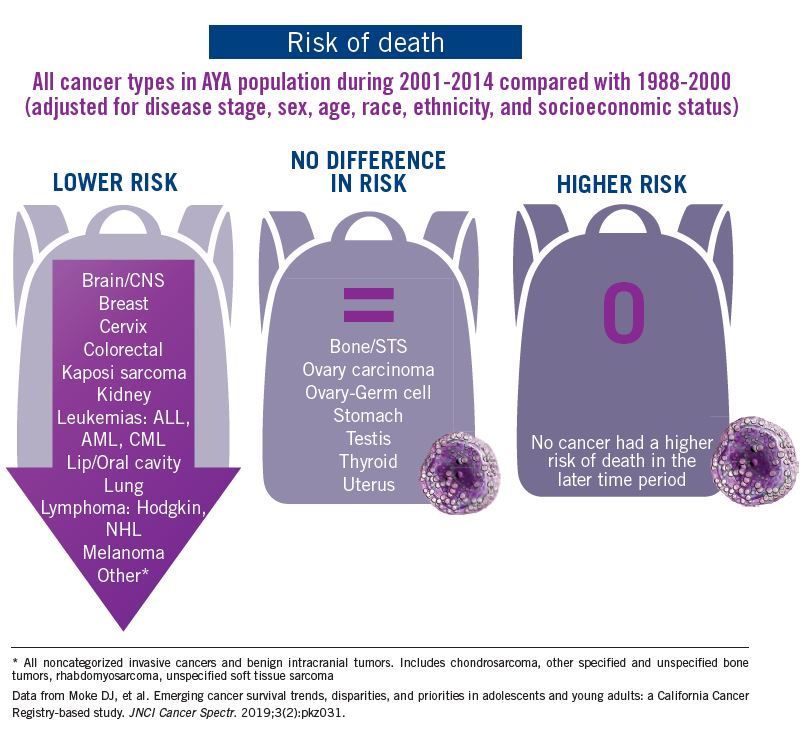
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
Observation versus inpatient status
A dilemma for hospitalists and patients
A federal effort to reduce health care expenditures has left many older Medicare recipients experiencing the sticker shock of “observation status.” Patients who are not sick enough to meet inpatient admission criteria, however, still require hospitalization, and may be placed under Medicare observation care.

Seniors can get frustrated, confused, and anxious as their status can be changed while they are in the hospital, and they may receive large medical bills after they are discharged. The Centers for Medicare & Medicaid Services’ “3-day rule” mandates that Medicare will not pay for skilled nursing facility care unless the patient is admitted as an “inpatient” for at least 3 days. Observation days do not count towards this 3-day hospital stay.
There has been an increase in outpatient services over the years since 2006. The 2018 State of Hospital Medicine Report (SoHM) highlights the percentage of discharges based on hospitalists’ billed Current Procedural Terminology codes. Codes 99217 (observation discharge) and 99238-99239 (inpatient discharge) were used to calculate the percentages. 80.7% of adult medicine hospitalist discharges were coded using inpatient discharge codes, while 19.3% of patients were discharged with observation discharge codes.
In the 2016 SoHM report, the ratio was 76.0% inpatient and 21.1% observation codes and in the 2014 report we saw 80.3% inpatient and 16.1% observation discharges (see table 1). But in both of those surveys, same-day admission/discharge codes were also separately reported, which did not occur in 2018. That makes year-over-year comparison of the data challenging.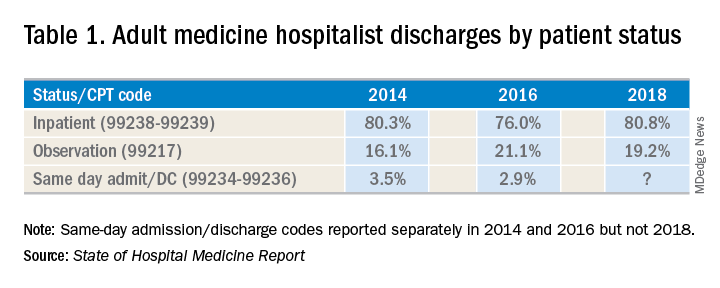
Interestingly, the 2017 CMS data on Evaluation and Management Codes by Specialty for the first time included separate data for hospitalists, based on hospitalists who credentialed with Medicare using the new C6 specialty code. Based on that data, when looking only at inpatient (99238-99239) and observation (99217) codes, 83% of the discharges were inpatient and 17% were observation.
Physicians feel the pressure of strained patient-physician relationships as a consequence of patients feeling the brunt of the financing gap related to observation status. Patients often feel they were not warned adequately about the financial ramifications of observation status. Even if Medicare beneficiaries have received the Medicare Outpatient Observation Notice, outlined by the Notice of Observation Treatment and Implication for Care Eligibility Act, they have no rights to appeal.
Currently Medicare beneficiaries admitted as inpatients only incur a Part A deductible; they are not liable for tests, procedures, and nursing care. On the other hand, in observation status all services are billed separately. For Medicare Part B services (which covers observation care) patients must pay 20% of services after the Part B deductible, which could result in a huge financial burden. Costs for skilled nursing facilities, when they are not covered by Medicare Part A, because of the 3-day rule, can easily go up to $20,000 or more. Medicare beneficiaries have no cap on costs for an observation stay. In some cases, hospitals have to apply a condition code 44 and retroactively change the stay to observation status.
I attended the 2019 Society of Hospital Medicine Annual Conference in Washington. Hospitalists from all parts of the country advocated on Capitol Hill against the “observation bill,” and “meet and greets” with congressional representatives increased their opposition to the bill. These efforts may work in favor of protecting patients from surprise medical bills. Hospital medicine physicians are on the front lines for providing health care in the hospital setting; they have demanded a fix to this legislative loophole which brings high out of pocket costs to our nation’s most vulnerable seniors. The observation status “2-midnight rule” utilized by CMS has increased financial barriers and decreased access to postacute care, affecting the provision of high-quality care for patients.
My hospital has a utilization review committee which reviews all cases to determine the appropriateness of an inpatient versus an observation designation. (An interesting question is whether the financial resources used to support this additional staff could be better assigned to provide high-quality care.) Distribution of these patients is determined on very specific criteria as outlined by Medicare. Observation is basically considered a billing method implemented by payers to decrease dollars paid to acute care hospitals for inpatient care. It pertains to admission status, not to the level of care provided in the hospital. Unfortunately, it is felt that no two payers define observation the same way. A few examples of common observation diagnoses are chest pain, abdominal pain, syncope, and migraine headache; in other words, patients with diagnoses where it is suspected that a less than 24-hour stay in the hospital could be sufficient.
Observation care is increasing and can sometimes contribute to work flow impediments and frustrations in hospitalists; thus, hospitalists are demanding reform. It has been proposed that observation could be eliminated altogether by creating a payment blend of inpatient/outpatient rates. Another option could be to assign lower Diagnosis Related Group coding to lower acuity disease processes, instead of separate observation reimbursement.
Patients and doctors lament that “Once you are in the hospital, you are admitted!” I don’t know the right answer that would solve the observation versus inpatient dilemma, but it is intriguing to consider changes in policy that might focus on the complete elimination of observation status.
Dr. Puri is a hospitalist at Lahey Hospital and Medical Center in Burlington, Mass.
A dilemma for hospitalists and patients
A dilemma for hospitalists and patients
A federal effort to reduce health care expenditures has left many older Medicare recipients experiencing the sticker shock of “observation status.” Patients who are not sick enough to meet inpatient admission criteria, however, still require hospitalization, and may be placed under Medicare observation care.
Seniors can get frustrated, confused, and anxious as their status can be changed while they are in the hospital, and they may receive large medical bills after they are discharged. The Centers for Medicare & Medicaid Services’ “3-day rule” mandates that Medicare will not pay for skilled nursing facility care unless the patient is admitted as an “inpatient” for at least 3 days. Observation days do not count towards this 3-day hospital stay.
There has been an increase in outpatient services over the years since 2006. The 2018 State of Hospital Medicine Report (SoHM) highlights the percentage of discharges based on hospitalists’ billed Current Procedural Terminology codes. Codes 99217 (observation discharge) and 99238-99239 (inpatient discharge) were used to calculate the percentages. 80.7% of adult medicine hospitalist discharges were coded using inpatient discharge codes, while 19.3% of patients were discharged with observation discharge codes.
In the 2016 SoHM report, the ratio was 76.0% inpatient and 21.1% observation codes and in the 2014 report we saw 80.3% inpatient and 16.1% observation discharges (see table 1). But in both of those surveys, same-day admission/discharge codes were also separately reported, which did not occur in 2018. That makes year-over-year comparison of the data challenging.
Interestingly, the 2017 CMS data on Evaluation and Management Codes by Specialty for the first time included separate data for hospitalists, based on hospitalists who credentialed with Medicare using the new C6 specialty code. Based on that data, when looking only at inpatient (99238-99239) and observation (99217) codes, 83% of the discharges were inpatient and 17% were observation.
Physicians feel the pressure of strained patient-physician relationships as a consequence of patients feeling the brunt of the financing gap related to observation status. Patients often feel they were not warned adequately about the financial ramifications of observation status. Even if Medicare beneficiaries have received the Medicare Outpatient Observation Notice, outlined by the Notice of Observation Treatment and Implication for Care Eligibility Act, they have no rights to appeal.
Currently Medicare beneficiaries admitted as inpatients only incur a Part A deductible; they are not liable for tests, procedures, and nursing care. On the other hand, in observation status all services are billed separately. For Medicare Part B services (which covers observation care) patients must pay 20% of services after the Part B deductible, which could result in a huge financial burden. Costs for skilled nursing facilities, when they are not covered by Medicare Part A, because of the 3-day rule, can easily go up to $20,000 or more. Medicare beneficiaries have no cap on costs for an observation stay. In some cases, hospitals have to apply a condition code 44 and retroactively change the stay to observation status.
I attended the 2019 Society of Hospital Medicine Annual Conference in Washington. Hospitalists from all parts of the country advocated on Capitol Hill against the “observation bill,” and “meet and greets” with congressional representatives increased their opposition to the bill. These efforts may work in favor of protecting patients from surprise medical bills. Hospital medicine physicians are on the front lines for providing health care in the hospital setting; they have demanded a fix to this legislative loophole which brings high out of pocket costs to our nation’s most vulnerable seniors. The observation status “2-midnight rule” utilized by CMS has increased financial barriers and decreased access to postacute care, affecting the provision of high-quality care for patients.
My hospital has a utilization review committee which reviews all cases to determine the appropriateness of an inpatient versus an observation designation. (An interesting question is whether the financial resources used to support this additional staff could be better assigned to provide high-quality care.) Distribution of these patients is determined on very specific criteria as outlined by Medicare. Observation is basically considered a billing method implemented by payers to decrease dollars paid to acute care hospitals for inpatient care. It pertains to admission status, not to the level of care provided in the hospital. Unfortunately, it is felt that no two payers define observation the same way. A few examples of common observation diagnoses are chest pain, abdominal pain, syncope, and migraine headache; in other words, patients with diagnoses where it is suspected that a less than 24-hour stay in the hospital could be sufficient.
Observation care is increasing and can sometimes contribute to work flow impediments and frustrations in hospitalists; thus, hospitalists are demanding reform. It has been proposed that observation could be eliminated altogether by creating a payment blend of inpatient/outpatient rates. Another option could be to assign lower Diagnosis Related Group coding to lower acuity disease processes, instead of separate observation reimbursement.
Patients and doctors lament that “Once you are in the hospital, you are admitted!” I don’t know the right answer that would solve the observation versus inpatient dilemma, but it is intriguing to consider changes in policy that might focus on the complete elimination of observation status.
Dr. Puri is a hospitalist at Lahey Hospital and Medical Center in Burlington, Mass.
A federal effort to reduce health care expenditures has left many older Medicare recipients experiencing the sticker shock of “observation status.” Patients who are not sick enough to meet inpatient admission criteria, however, still require hospitalization, and may be placed under Medicare observation care.
Seniors can get frustrated, confused, and anxious as their status can be changed while they are in the hospital, and they may receive large medical bills after they are discharged. The Centers for Medicare & Medicaid Services’ “3-day rule” mandates that Medicare will not pay for skilled nursing facility care unless the patient is admitted as an “inpatient” for at least 3 days. Observation days do not count towards this 3-day hospital stay.
There has been an increase in outpatient services over the years since 2006. The 2018 State of Hospital Medicine Report (SoHM) highlights the percentage of discharges based on hospitalists’ billed Current Procedural Terminology codes. Codes 99217 (observation discharge) and 99238-99239 (inpatient discharge) were used to calculate the percentages. 80.7% of adult medicine hospitalist discharges were coded using inpatient discharge codes, while 19.3% of patients were discharged with observation discharge codes.
In the 2016 SoHM report, the ratio was 76.0% inpatient and 21.1% observation codes and in the 2014 report we saw 80.3% inpatient and 16.1% observation discharges (see table 1). But in both of those surveys, same-day admission/discharge codes were also separately reported, which did not occur in 2018. That makes year-over-year comparison of the data challenging.
Interestingly, the 2017 CMS data on Evaluation and Management Codes by Specialty for the first time included separate data for hospitalists, based on hospitalists who credentialed with Medicare using the new C6 specialty code. Based on that data, when looking only at inpatient (99238-99239) and observation (99217) codes, 83% of the discharges were inpatient and 17% were observation.
Physicians feel the pressure of strained patient-physician relationships as a consequence of patients feeling the brunt of the financing gap related to observation status. Patients often feel they were not warned adequately about the financial ramifications of observation status. Even if Medicare beneficiaries have received the Medicare Outpatient Observation Notice, outlined by the Notice of Observation Treatment and Implication for Care Eligibility Act, they have no rights to appeal.
Currently Medicare beneficiaries admitted as inpatients only incur a Part A deductible; they are not liable for tests, procedures, and nursing care. On the other hand, in observation status all services are billed separately. For Medicare Part B services (which covers observation care) patients must pay 20% of services after the Part B deductible, which could result in a huge financial burden. Costs for skilled nursing facilities, when they are not covered by Medicare Part A, because of the 3-day rule, can easily go up to $20,000 or more. Medicare beneficiaries have no cap on costs for an observation stay. In some cases, hospitals have to apply a condition code 44 and retroactively change the stay to observation status.
I attended the 2019 Society of Hospital Medicine Annual Conference in Washington. Hospitalists from all parts of the country advocated on Capitol Hill against the “observation bill,” and “meet and greets” with congressional representatives increased their opposition to the bill. These efforts may work in favor of protecting patients from surprise medical bills. Hospital medicine physicians are on the front lines for providing health care in the hospital setting; they have demanded a fix to this legislative loophole which brings high out of pocket costs to our nation’s most vulnerable seniors. The observation status “2-midnight rule” utilized by CMS has increased financial barriers and decreased access to postacute care, affecting the provision of high-quality care for patients.
My hospital has a utilization review committee which reviews all cases to determine the appropriateness of an inpatient versus an observation designation. (An interesting question is whether the financial resources used to support this additional staff could be better assigned to provide high-quality care.) Distribution of these patients is determined on very specific criteria as outlined by Medicare. Observation is basically considered a billing method implemented by payers to decrease dollars paid to acute care hospitals for inpatient care. It pertains to admission status, not to the level of care provided in the hospital. Unfortunately, it is felt that no two payers define observation the same way. A few examples of common observation diagnoses are chest pain, abdominal pain, syncope, and migraine headache; in other words, patients with diagnoses where it is suspected that a less than 24-hour stay in the hospital could be sufficient.
Observation care is increasing and can sometimes contribute to work flow impediments and frustrations in hospitalists; thus, hospitalists are demanding reform. It has been proposed that observation could be eliminated altogether by creating a payment blend of inpatient/outpatient rates. Another option could be to assign lower Diagnosis Related Group coding to lower acuity disease processes, instead of separate observation reimbursement.
Patients and doctors lament that “Once you are in the hospital, you are admitted!” I don’t know the right answer that would solve the observation versus inpatient dilemma, but it is intriguing to consider changes in policy that might focus on the complete elimination of observation status.
Dr. Puri is a hospitalist at Lahey Hospital and Medical Center in Burlington, Mass.
My patient tells me that they are transgender – now what?
I am privileged to work in a university hospital system where I have access to colleagues with expertise in LGBT health; however, medical providers in the community may not enjoy such resources. Many transgender and gender-diverse (TGD) youth now are seeking help from their community primary care providers to affirm their gender identity, but many community primary care providers do not have the luxury of referring these patients to an expert in gender-affirming care when their TGD patients express the desire to affirm their gender through medical and surgical means. This is even more difficult if the nearest referral center is hundreds of miles away. Nevertheless, 
If a TGD youth discloses their gender identity to you, it is critical that you make the patient feel safe and supported. Showing support is important in maintaining rapport between you and the patient. Furthermore, you may be one of the very few adults in the child’s life whom they can trust.
First of all, thank them. For many TGD patients, disclosing their gender identity to a health care provider can be a herculean task. They may have spent many hours trying to find the right words to say to disclose an important aspect of themselves to you. They also probably spent a fair amount of time worrying about whether or not you would react positively to this disclosure. This fear is reasonable. About one-fifth of transgender people have reported being kicked out of a medical practice because of their disclosure of their gender identity.1
Secondly, assure the TGD patient that your treatment would be no different from the care provided for other patients. Discrimination from a health care provider has frequently been reported by TGD patients1 and is expected from this population.2 By emphasizing this, you have signaled to them that you are committed to treating them with dignity and respect. Furthermore, signal your commitment to this treatment by making the clinic a safe and welcoming place for LGBT youth. Several resources exist that can help with this. The American Medical Association provides a good example on how to draft a nondiscrimination statement that can be posted in waiting areas;3 the Fenway Institute has a good example of an intake form that is LGBT friendly.4
In addition, a good way to help affirm their gender identity is to tell them that being transgender or gender-diverse is normal and healthy. Many times, TGD youth will hear narratives that gender diversity is pathological or aberrant; however, hearing that they are healthy and normal, especially from a health care provider, can make a powerful impact on feeling supported and affirmed.
Furthermore, inform your TGD youth of their right to confidentiality. Many TGD youth may not be out to their parents, and you may be the first person to whom they disclosed their gender identity. This is especially helpful if you describe their right to and the limits of confidentiality (e.g., suicidality) at the beginning of the visit. Assurance of confidentiality is a vital reason adolescents and young adults seek health care from a medical provider,5 and the same can be said of TGD youth; however, keep in mind that if they do desire to transition using cross-sex hormones or surgery, parental permission is required.
If they are not out to their parents and they are planning to come out to their parents, offer to be there when they do. Having someone to support the child – someone who is a medical provider – can add to the sense of legitimacy that what the child is experiencing is normal and healthy. Providing guidance on how parents can support their TGD child is essential for successful affirmation, and some suggestions can be found in an LGBT Youth Consult column I wrote titled, “Guidance for parents of LGBT youth.”
If you practice in a location where the nearest expert in gender-affirming care can be hundreds of miles away, educate yourself on gender-affirming care. Several guidelines are available. The World Professional Society for Transgender Standards of Care (SOC) focuses on the mental health aspects of gender-affirming care. The SOC recommends, but no longer requires, letters from a mental health therapist to start gender-affirming medical treatments and does allow for a discussion between you and the patient on the risks, benefits, alternatives, unknowns, limitations of treatment, and risks of not treating (i.e., obtaining informed consent) as the threshold for hormone therapy.6 This approach, known as the “informed consent model,” can be helpful in expanding health care access for TGD youth. Furthermore, there’s the Endocrine Society Clinical Practice Guidelines7 and the University of California, San Francisco, Guidelines,8 which focus on the medical aspects of gender-affirming care, such as when to start pubertal blockers and dosing for cross-sex hormones. Finally, there are resources that allow providers to consult an expert remotely for more complicated cases. Transline is a transgender medical consultation service staffed by medical providers with expertise in gender-affirming care. Providers can learn more about this valuable service on the website: http://project-health.org/transline/.
Working in a major medical center is not necessary in providing gender-affirming care to TGD youth. Being respectful, supportive, and having the willingness to learn are the minimal requirements. Resources are available to help guide you on the more technical aspects of gender-affirming care. Maintaining a supportive environment and using these resources will help you expand health care access for this population.
Dr. Montano is an assistant professor of pediatrics at the University of Pittsburgh and an adolescent medicine physician at Children’s Hospital of Pittsburgh of UPMC. Email him at pdnews@mdedge.com.
References
1. Injustice at every turn: A report of the national transgender discrimination survey (National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011).
2. Psychol Bull. 2003 Sep;129(5):674-97.
3. “Creating an LGBTQ-friendly practice,” American Medical Association.
4. Fenway Health Client Registration Form.
5. JAMA. 1993 Mar 17;269(11):1404-7.
6. Int J Transgenderism 2012;13:165-232.
7. J Clin Endocrinol Metab. 2017 Nov 1;102(11):3869-903.
8. “Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People,” 2nd edition (San Francisco, CA: University of California, San Francisco, June 17, 2016).
I am privileged to work in a university hospital system where I have access to colleagues with expertise in LGBT health; however, medical providers in the community may not enjoy such resources. Many transgender and gender-diverse (TGD) youth now are seeking help from their community primary care providers to affirm their gender identity, but many community primary care providers do not have the luxury of referring these patients to an expert in gender-affirming care when their TGD patients express the desire to affirm their gender through medical and surgical means. This is even more difficult if the nearest referral center is hundreds of miles away. Nevertheless,
If a TGD youth discloses their gender identity to you, it is critical that you make the patient feel safe and supported. Showing support is important in maintaining rapport between you and the patient. Furthermore, you may be one of the very few adults in the child’s life whom they can trust.
First of all, thank them. For many TGD patients, disclosing their gender identity to a health care provider can be a herculean task. They may have spent many hours trying to find the right words to say to disclose an important aspect of themselves to you. They also probably spent a fair amount of time worrying about whether or not you would react positively to this disclosure. This fear is reasonable. About one-fifth of transgender people have reported being kicked out of a medical practice because of their disclosure of their gender identity.1
Secondly, assure the TGD patient that your treatment would be no different from the care provided for other patients. Discrimination from a health care provider has frequently been reported by TGD patients1 and is expected from this population.2 By emphasizing this, you have signaled to them that you are committed to treating them with dignity and respect. Furthermore, signal your commitment to this treatment by making the clinic a safe and welcoming place for LGBT youth. Several resources exist that can help with this. The American Medical Association provides a good example on how to draft a nondiscrimination statement that can be posted in waiting areas;3 the Fenway Institute has a good example of an intake form that is LGBT friendly.4
In addition, a good way to help affirm their gender identity is to tell them that being transgender or gender-diverse is normal and healthy. Many times, TGD youth will hear narratives that gender diversity is pathological or aberrant; however, hearing that they are healthy and normal, especially from a health care provider, can make a powerful impact on feeling supported and affirmed.
Furthermore, inform your TGD youth of their right to confidentiality. Many TGD youth may not be out to their parents, and you may be the first person to whom they disclosed their gender identity. This is especially helpful if you describe their right to and the limits of confidentiality (e.g., suicidality) at the beginning of the visit. Assurance of confidentiality is a vital reason adolescents and young adults seek health care from a medical provider,5 and the same can be said of TGD youth; however, keep in mind that if they do desire to transition using cross-sex hormones or surgery, parental permission is required.
If they are not out to their parents and they are planning to come out to their parents, offer to be there when they do. Having someone to support the child – someone who is a medical provider – can add to the sense of legitimacy that what the child is experiencing is normal and healthy. Providing guidance on how parents can support their TGD child is essential for successful affirmation, and some suggestions can be found in an LGBT Youth Consult column I wrote titled, “Guidance for parents of LGBT youth.”
If you practice in a location where the nearest expert in gender-affirming care can be hundreds of miles away, educate yourself on gender-affirming care. Several guidelines are available. The World Professional Society for Transgender Standards of Care (SOC) focuses on the mental health aspects of gender-affirming care. The SOC recommends, but no longer requires, letters from a mental health therapist to start gender-affirming medical treatments and does allow for a discussion between you and the patient on the risks, benefits, alternatives, unknowns, limitations of treatment, and risks of not treating (i.e., obtaining informed consent) as the threshold for hormone therapy.6 This approach, known as the “informed consent model,” can be helpful in expanding health care access for TGD youth. Furthermore, there’s the Endocrine Society Clinical Practice Guidelines7 and the University of California, San Francisco, Guidelines,8 which focus on the medical aspects of gender-affirming care, such as when to start pubertal blockers and dosing for cross-sex hormones. Finally, there are resources that allow providers to consult an expert remotely for more complicated cases. Transline is a transgender medical consultation service staffed by medical providers with expertise in gender-affirming care. Providers can learn more about this valuable service on the website: http://project-health.org/transline/.
Working in a major medical center is not necessary in providing gender-affirming care to TGD youth. Being respectful, supportive, and having the willingness to learn are the minimal requirements. Resources are available to help guide you on the more technical aspects of gender-affirming care. Maintaining a supportive environment and using these resources will help you expand health care access for this population.
Dr. Montano is an assistant professor of pediatrics at the University of Pittsburgh and an adolescent medicine physician at Children’s Hospital of Pittsburgh of UPMC. Email him at pdnews@mdedge.com.
References
1. Injustice at every turn: A report of the national transgender discrimination survey (National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011).
2. Psychol Bull. 2003 Sep;129(5):674-97.
3. “Creating an LGBTQ-friendly practice,” American Medical Association.
4. Fenway Health Client Registration Form.
5. JAMA. 1993 Mar 17;269(11):1404-7.
6. Int J Transgenderism 2012;13:165-232.
7. J Clin Endocrinol Metab. 2017 Nov 1;102(11):3869-903.
8. “Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People,” 2nd edition (San Francisco, CA: University of California, San Francisco, June 17, 2016).
I am privileged to work in a university hospital system where I have access to colleagues with expertise in LGBT health; however, medical providers in the community may not enjoy such resources. Many transgender and gender-diverse (TGD) youth now are seeking help from their community primary care providers to affirm their gender identity, but many community primary care providers do not have the luxury of referring these patients to an expert in gender-affirming care when their TGD patients express the desire to affirm their gender through medical and surgical means. This is even more difficult if the nearest referral center is hundreds of miles away. Nevertheless,
If a TGD youth discloses their gender identity to you, it is critical that you make the patient feel safe and supported. Showing support is important in maintaining rapport between you and the patient. Furthermore, you may be one of the very few adults in the child’s life whom they can trust.
First of all, thank them. For many TGD patients, disclosing their gender identity to a health care provider can be a herculean task. They may have spent many hours trying to find the right words to say to disclose an important aspect of themselves to you. They also probably spent a fair amount of time worrying about whether or not you would react positively to this disclosure. This fear is reasonable. About one-fifth of transgender people have reported being kicked out of a medical practice because of their disclosure of their gender identity.1
Secondly, assure the TGD patient that your treatment would be no different from the care provided for other patients. Discrimination from a health care provider has frequently been reported by TGD patients1 and is expected from this population.2 By emphasizing this, you have signaled to them that you are committed to treating them with dignity and respect. Furthermore, signal your commitment to this treatment by making the clinic a safe and welcoming place for LGBT youth. Several resources exist that can help with this. The American Medical Association provides a good example on how to draft a nondiscrimination statement that can be posted in waiting areas;3 the Fenway Institute has a good example of an intake form that is LGBT friendly.4
In addition, a good way to help affirm their gender identity is to tell them that being transgender or gender-diverse is normal and healthy. Many times, TGD youth will hear narratives that gender diversity is pathological or aberrant; however, hearing that they are healthy and normal, especially from a health care provider, can make a powerful impact on feeling supported and affirmed.
Furthermore, inform your TGD youth of their right to confidentiality. Many TGD youth may not be out to their parents, and you may be the first person to whom they disclosed their gender identity. This is especially helpful if you describe their right to and the limits of confidentiality (e.g., suicidality) at the beginning of the visit. Assurance of confidentiality is a vital reason adolescents and young adults seek health care from a medical provider,5 and the same can be said of TGD youth; however, keep in mind that if they do desire to transition using cross-sex hormones or surgery, parental permission is required.
If they are not out to their parents and they are planning to come out to their parents, offer to be there when they do. Having someone to support the child – someone who is a medical provider – can add to the sense of legitimacy that what the child is experiencing is normal and healthy. Providing guidance on how parents can support their TGD child is essential for successful affirmation, and some suggestions can be found in an LGBT Youth Consult column I wrote titled, “Guidance for parents of LGBT youth.”
If you practice in a location where the nearest expert in gender-affirming care can be hundreds of miles away, educate yourself on gender-affirming care. Several guidelines are available. The World Professional Society for Transgender Standards of Care (SOC) focuses on the mental health aspects of gender-affirming care. The SOC recommends, but no longer requires, letters from a mental health therapist to start gender-affirming medical treatments and does allow for a discussion between you and the patient on the risks, benefits, alternatives, unknowns, limitations of treatment, and risks of not treating (i.e., obtaining informed consent) as the threshold for hormone therapy.6 This approach, known as the “informed consent model,” can be helpful in expanding health care access for TGD youth. Furthermore, there’s the Endocrine Society Clinical Practice Guidelines7 and the University of California, San Francisco, Guidelines,8 which focus on the medical aspects of gender-affirming care, such as when to start pubertal blockers and dosing for cross-sex hormones. Finally, there are resources that allow providers to consult an expert remotely for more complicated cases. Transline is a transgender medical consultation service staffed by medical providers with expertise in gender-affirming care. Providers can learn more about this valuable service on the website: http://project-health.org/transline/.
Working in a major medical center is not necessary in providing gender-affirming care to TGD youth. Being respectful, supportive, and having the willingness to learn are the minimal requirements. Resources are available to help guide you on the more technical aspects of gender-affirming care. Maintaining a supportive environment and using these resources will help you expand health care access for this population.
Dr. Montano is an assistant professor of pediatrics at the University of Pittsburgh and an adolescent medicine physician at Children’s Hospital of Pittsburgh of UPMC. Email him at pdnews@mdedge.com.
References
1. Injustice at every turn: A report of the national transgender discrimination survey (National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011).
2. Psychol Bull. 2003 Sep;129(5):674-97.
3. “Creating an LGBTQ-friendly practice,” American Medical Association.
4. Fenway Health Client Registration Form.
5. JAMA. 1993 Mar 17;269(11):1404-7.
6. Int J Transgenderism 2012;13:165-232.
7. J Clin Endocrinol Metab. 2017 Nov 1;102(11):3869-903.
8. “Guidelines for the Primary and Gender-Affirming Care of Transgender and Gender Nonbinary People,” 2nd edition (San Francisco, CA: University of California, San Francisco, June 17, 2016).

Wildfire smoke has acute cardiorespiratory impact, but long-term effects still under study
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.
John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires

Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]). 
Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.

Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).

“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”
Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.
John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires
Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]).
Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.
Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).
“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”
Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
The 2019 wildfire season is underway in many locales across the United States, exposing millions of individuals to smoky conditions that will have health consequences ranging from stinging eyes to scratchy throats to a trip to the ED for asthma or chronic obstructive pulmonary disease (COPD) exacerbation. Questions about long-term health impacts are on the minds of many, including physicians and their patients who live with cardiorespiratory conditions.
John R. Balmes, MD, a pulmonologist at the University of California, San Francisco, and an expert on the respiratory and cardiovascular effects of air pollutants, suggested that the best available published literature points to “pretty strong evidence for acute effects of wildfire smoke on respiratory health, meaning people with preexisting asthma and COPD are at risk for exacerbations, and probably for respiratory tract infections as well.” He said, “It’s a little less clear, but there’s good biological plausibility for increased risk of respiratory tract infections because when your alveolar macrophages are overloaded with carbon particles that are toxic to those cells, they don’t function as well as a first line of defense against bacterial infection, for example.”
The new normal of wildfires
Warmer, drier summers in recent years in the western United States and many other regions, attributed by climate experts to global climate change, have produced catastrophic wildfires (PNAS;2016 Oct 18;113[42]11770-5; Science 2006 Aug 18;313:940-3). The Camp Fire in Northern California broke out in November 2018, took the lives of at least 85 people, and cost more than $16 billion in damage. Smoke from that blaze reached hazardous levels in San Francisco, Sacramento, Fresno, and many other smaller towns. Other forest fires in that year caused heavy smoke conditions in Portland, Seattle, Vancouver, and Anchorage. Such events are expected to be repeated often in the coming years (Int J Environ Res Public Health. 2019 Jul 6;16[13]).
Wildfire smoke can contain a wide range of substances, chemicals, and gases with known and unknown cardiorespiratory implications. “Smoke is composed primarily of carbon dioxide, water vapor, carbon monoxide, particulate matter, hydrocarbons and other organic chemicals, nitrogen oxides, trace minerals and several thousand other compounds,” according to the U.S. Environmental Protection Agency (Wildfire smoke: A guide for public health officials 2019. Washington, D.C.: EPA, 2019). The EPA report noted, “Particles with diameters less than 10 mcm (particulate matter, or PM10) can be inhaled into the lungs and affect the lungs, heart, and blood vessels. The smallest particles, those less than 2.5 mcm in diameter (PM2.5), are the greatest risk to public health because they can reach deep into the lungs and may even make it into the bloodstream.”
Research on health impact
In early June of 2008, Wayne Cascio, MD, awoke in his Greenville, N.C., home to the stench of smoke emanating from a large peat fire burning some 65 miles away. By the time he reached the parking lot at East Carolina University in Greenville to begin his workday as chief of cardiology, the haze of smoke had thickened to the point where he could only see a few feet in front of him.
Over the next several weeks, the fire scorched 41,000 acres and produced haze and air pollution that far exceeded National Ambient Air Quality Standards for particulate matter and blanketed rural communities in the state’s eastern region. The price tag for management of the blaze reached $20 million. Because of his interest in the health effects of wildfire smoke and because of his relationship with investigators at the EPA, Dr. Cascio initiated an epidemiology study to investigate the effects of exposure on cardiorespiratory outcomes in the population affected by the fire (Environ Health Perspect. 2011 Oct;119[10]:1415-20).
By combining satellite data with syndromic surveillance drawn from hospital records in 41 counties contained in the North Carolina Disease Event Tracking and Epidemiologic Collection Tool, he and his colleagues found that exposure to the peat wildfire smoke led to increases in the cumulative risk ratio for asthma (relative risk, 1.65), chronic obstructive pulmonary disease (RR, 1.73), and pneumonia and acute bronchitis (RR, 1.59). ED visits related to cardiopulmonary symptoms and heart failure also were significantly increased (RR, 1.23 and 1.37, respectively). “That was really the first study to strongly identify a cardiac endpoint related to wildfire smoke exposure,” said Dr. Cascio, who now directs the EPA’s National Health and Environmental Effects Research Laboratory. “It really pointed out how little we knew about the health effects of wildfire up until that time.”
Those early findings have been replicated in subsequent research about the acute health effects of exposure to wildfire smoke, which contains PM2.5 and other toxic substances from structures, electronic devices, and automobiles destroyed in the path of flames, including heavy metals and asbestos. Most of the work has focused on smoke-related cardiovascular and respiratory ED visits and hospitalizations.
A study of the 2008 California wildfire impact on ED visits accounted for ozone levels in addition to PM2.5 in the smoke. During the active fire periods, PM2.5 was significantly associated with exacerbations of asthma and COPD and these effects remained after controlling for ozone levels. PM2.5 inhalation during the wildfires was associated with increased risk of an ED visit for asthma (RR, 1.112; 95% confidence interval, 1.087-1.138) for a 10 mcg/m3 increase in PM2.5 and COPD (RR, 1.05; 95% CI, 1.019-1.0825), as well as for combined respiratory visits (RR, 1.035; 95% CI, 1.023-1.046) (Environ Int. 2109 Aug;129:291-8).
Researchers who evaluated the health impacts of wildfires in California during the 2015 fire season found an increase in all-cause cardiovascular and respiratory ED visits, especially among those aged 65 years and older during smoke days. The population-based study included 1,196,233 ED visits during May 1–Sept. 30 that year. PM2.5 concentrations were categorized as light, medium, or dense. Relative risk rose with the amount of smoke in the air. Rates of all-cause cardiovascular ED visits were elevated across levels of smoke density, with the greatest increase on dense smoke days and among those aged 65 years or older (RR,1.15; 95% CI, 1.09-1.22). All-cause cerebrovascular visits were associated with dense smoke days, especially among those aged 65 years and older (RR, 1.22; 95% CI, 1.00-1.49). Respiratory conditions also were increased on dense smoke days (RR, 1.18; 95% CI, 1.08-1.28) (J Am Heart Assoc. 2018 Apr 11;7:e007492. doi: 10.1161/JAHA.117.007492).
Long-term effects unknown
When it comes to the long-term effects of wildfire smoke on human health outcomes, much less is known. In a recent literature review, Colleen E. Reid, PhD, and Melissa May Maestas, PhD, found only one study that investigated long-term respiratory health impacts of wildfire smoke, and only a few studies that have estimated future health impacts of wildfires under likely climate change scenarios (Curr Opin Pulm Med. 2019 Mar;25:179-87).
“We know that there are immediate respiratory health effects from wildfire smoke,” said Dr. Reid of the department of geography at the University of Colorado Boulder. “What’s less known is everything else. That’s challenging, because people want to know about the long-term health effects.”
Evidence from the scientific literature suggests that exposure to air pollution adversely affects cardiovascular health, but whether exposure to wildfire smoke confers a similar risk is less clear. “Until just a few years ago we haven’t been able to study wildfire exposure measures on a large scale,” said EPA scientist Ana G. Rappold, PhD, a statistician there in the environmental public health division of the National Health and Environmental Effects Research Laboratory. “It’s also hard to predict wildfires, so it’s hard to plan for an epidemiologic study if you don’t know where they’re going to occur.”
Dr. Rappold and colleagues examined cardiopulmonary hospitalizations among adults aged 65 years and older in 692 U.S. counties within 200 km of 123 large wildfires during 2008-2010 (Environ Health Perspect. 2019;127[3]:37006. doi: 10.1289/EHP3860). They observed that an increased risk of PM2.5-related cardiopulmonary hospitalizations was similar on smoke and nonsmoke days across multiple lags and exposure metrics, while risk for asthma-related hospitalizations was higher during smoke days. “One hypothesis is that this was an older study population, so naturally if you’re inhaling smoke, the first organ that’s impacted in an older population is the lungs,” Dr. Rappold said. “If you go to the hospital for asthma, wheezing, or bronchitis, you are taken out of the risk pool for cardiovascular and other diseases. That could explain why in other studies we don’t see a clear cardiovascular signal as we have for air pollution studies in general. Another aspect to this study is, the exposure metric was PM2.5, but smoke contains many other components, particularly gases, which are respiratory irritants. It could be that this triggers a higher risk for respiratory [effects] than regular episodes of high PM2.5 exposure, just because of the additional gases that people are exposed to.”
Another complicating factor is the paucity of data about solutions to long-term exposure to wildfire smoke. “If you’re impacted by high-exposure levels for 60 days, that is not something we have experienced before,” Dr. Rappold noted. “What are the solutions for that community? What works? Can we show that by implementing community-level resilience plans with HEPA [high-efficiency particulate air] filters or other interventions, do the overall outcomes improve? Doctors are the first ones to talk with their patients about their symptoms and about how to take care of their conditions. They can clearly make a difference in emphasizing reducing exposures in a way that fits their patients individually, either reducing the amount of time spent outside, the duration of exposure, and the level of exposure. Maybe change activities based on the intensity of exposure. Don’t go for a run outside when it’s smoky, because your ventilation rate is higher and you will breathe in more smoke. Become aware of those things.”
Advising vulnerable patients
While research in this field advances, the unforgiving wildfire season looms, assuring more destruction of property and threats to cardiorespiratory health. “There are a lot of questions that research will have an opportunity to address as we go forward, including the utility and the benefit of N95 masks, the utility of HEPA filters used in the house, and even with HVAC [heating, ventilation, and air conditioning] systems,” Dr. Cascio said. “Can we really clean up the indoor air well enough to protect us from wildfire smoke?”
The way he sees it, the time is ripe for clinicians and officials in public and private practice settings to refine how they distribute information to people living in areas affected by wildfire smoke. “We can’t force people do anything, but at least if they’re informed, then they understand they can make an informed decision about how they might want to affect what they do that would limit their exposure,” he said. “As a patient, my health care system sends text and email messages to me. So, why couldn’t the hospital send out a text message or an email to all of the patients with COPD, coronary disease, and heart failure when an area is impacted by smoke, saying, ‘Check your air quality and take action if air quality is poor?’ Physicians don’t have time to do this kind of education in the office for all of their patients. I know that from experience. But if one were to only focus on those at highest risk, and encourage them to follow our guidelines, which might include doing HEPA filter treatment in the home, we probably would reduce the number of clinical events in a cost-effective way.”
Study confirms prognostic impact of MYC partner gene in DLBCL
MYC rearrangement (MYC-R) has negative prognostic significance in patients with diffuse large B-cell lymphoma (DLBCL) in relation to the MYC partner gene, according to a retrospective study.
![Micrograph of a 'diffuse large B cell lymphoma. Lymph node FNA specimen. Field stain.(http://www.gnu.org/copyleft/fdl.html)], via Wikimedia Commons](https://cdn.mdedge.com/files/s3fs-public/Image/June-2017/DLBCL_web.jpg)
The negative prognostic effect of MYC-R was largely limited to patients with MYC–double hit/MYC–triple hit status and an immunoglobulin (IG) partner within 24 months following diagnosis.
“The primary objective of the study was to validate the prognostic relevance of [MYC–single hit] and [MYC–double hit/MYC–triple hit] status within the context of the MYC translocation partner (MYC-IG v. MYC-non-IG) in patients with DLBCL morphology,” wrote Andreas Rosenwald, MD, of the University of Würzburg (Germany) and colleagues in the Journal of Clinical Oncology.
The researchers identified 5,117 patients who all received R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) or R-CHOP–like immunochemotherapy, 2,383 of whom were evaluable for MYC-R and had complete clinical data. The cohort consisted of patients enrolled in various population-based registries and prospective clinical studies throughout North America and Europe.
The team used fluorescence in situ hybridization testing to identify MYC-R, in addition to BCL2 and/or BCL6 translocations if MYC-R was detected. Subsequently, these data were correlated with clinical endpoints.
After analysis, the researchers found that MYC-R was detected in 11% of patients. The presence of MYC-R was associated with significantly reduced survival, particularly within the initial 24 months following diagnosis.
Adverse prognostic implications were largely apparent in patients with accompanying rearrangement of BCL2 and/or BCL6 translocations (MYC–double-hit/MYC–triple hit status) and an immunoglobulin (IG) partner (hazard ratio, 2.4; 95% confidence interval, 1.6-3.6; P less than .001).
“These data suggest that little justification exists for altering initial therapeutic approaches in patients with DLBCL whose tumors carry an MYC translocation alone [MYC single hit],” they wrote. “However, for [MYC double hit/MYC triple hit] DLBCL, the major negative prognostic impact and 2-year effect support the practice of optimizing first-line treatment approaches to achieve maximum complete response rates because salvage treatment at relapse is not effective.”
Dr. Rosenwald and colleagues suggested that, in the future, diagnostic approaches should be implemented to detect patients in this high-risk group and that risk-modified treatment strategies should be further developed.
The study was supported by unrestricted grants to the Lunenburg Lymphoma Biomarker Consortium from several pharmaceutical companies and Bloodwise. Dr. Rosenwald reported having no conflicts of interest, but several coauthors reported relationships with industry.
SOURCE: : Rosenwald A et al. J Clin Oncol. 2019 Sep 9. doi: 10.1200/JCO.19.00743.
MYC rearrangement (MYC-R) has negative prognostic significance in patients with diffuse large B-cell lymphoma (DLBCL) in relation to the MYC partner gene, according to a retrospective study.
The negative prognostic effect of MYC-R was largely limited to patients with MYC–double hit/MYC–triple hit status and an immunoglobulin (IG) partner within 24 months following diagnosis.
“The primary objective of the study was to validate the prognostic relevance of [MYC–single hit] and [MYC–double hit/MYC–triple hit] status within the context of the MYC translocation partner (MYC-IG v. MYC-non-IG) in patients with DLBCL morphology,” wrote Andreas Rosenwald, MD, of the University of Würzburg (Germany) and colleagues in the Journal of Clinical Oncology.
The researchers identified 5,117 patients who all received R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) or R-CHOP–like immunochemotherapy, 2,383 of whom were evaluable for MYC-R and had complete clinical data. The cohort consisted of patients enrolled in various population-based registries and prospective clinical studies throughout North America and Europe.
The team used fluorescence in situ hybridization testing to identify MYC-R, in addition to BCL2 and/or BCL6 translocations if MYC-R was detected. Subsequently, these data were correlated with clinical endpoints.
After analysis, the researchers found that MYC-R was detected in 11% of patients. The presence of MYC-R was associated with significantly reduced survival, particularly within the initial 24 months following diagnosis.
Adverse prognostic implications were largely apparent in patients with accompanying rearrangement of BCL2 and/or BCL6 translocations (MYC–double-hit/MYC–triple hit status) and an immunoglobulin (IG) partner (hazard ratio, 2.4; 95% confidence interval, 1.6-3.6; P less than .001).
“These data suggest that little justification exists for altering initial therapeutic approaches in patients with DLBCL whose tumors carry an MYC translocation alone [MYC single hit],” they wrote. “However, for [MYC double hit/MYC triple hit] DLBCL, the major negative prognostic impact and 2-year effect support the practice of optimizing first-line treatment approaches to achieve maximum complete response rates because salvage treatment at relapse is not effective.”
Dr. Rosenwald and colleagues suggested that, in the future, diagnostic approaches should be implemented to detect patients in this high-risk group and that risk-modified treatment strategies should be further developed.
The study was supported by unrestricted grants to the Lunenburg Lymphoma Biomarker Consortium from several pharmaceutical companies and Bloodwise. Dr. Rosenwald reported having no conflicts of interest, but several coauthors reported relationships with industry.
SOURCE: : Rosenwald A et al. J Clin Oncol. 2019 Sep 9. doi: 10.1200/JCO.19.00743.
MYC rearrangement (MYC-R) has negative prognostic significance in patients with diffuse large B-cell lymphoma (DLBCL) in relation to the MYC partner gene, according to a retrospective study.
The negative prognostic effect of MYC-R was largely limited to patients with MYC–double hit/MYC–triple hit status and an immunoglobulin (IG) partner within 24 months following diagnosis.
“The primary objective of the study was to validate the prognostic relevance of [MYC–single hit] and [MYC–double hit/MYC–triple hit] status within the context of the MYC translocation partner (MYC-IG v. MYC-non-IG) in patients with DLBCL morphology,” wrote Andreas Rosenwald, MD, of the University of Würzburg (Germany) and colleagues in the Journal of Clinical Oncology.
The researchers identified 5,117 patients who all received R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) or R-CHOP–like immunochemotherapy, 2,383 of whom were evaluable for MYC-R and had complete clinical data. The cohort consisted of patients enrolled in various population-based registries and prospective clinical studies throughout North America and Europe.
The team used fluorescence in situ hybridization testing to identify MYC-R, in addition to BCL2 and/or BCL6 translocations if MYC-R was detected. Subsequently, these data were correlated with clinical endpoints.
After analysis, the researchers found that MYC-R was detected in 11% of patients. The presence of MYC-R was associated with significantly reduced survival, particularly within the initial 24 months following diagnosis.
Adverse prognostic implications were largely apparent in patients with accompanying rearrangement of BCL2 and/or BCL6 translocations (MYC–double-hit/MYC–triple hit status) and an immunoglobulin (IG) partner (hazard ratio, 2.4; 95% confidence interval, 1.6-3.6; P less than .001).
“These data suggest that little justification exists for altering initial therapeutic approaches in patients with DLBCL whose tumors carry an MYC translocation alone [MYC single hit],” they wrote. “However, for [MYC double hit/MYC triple hit] DLBCL, the major negative prognostic impact and 2-year effect support the practice of optimizing first-line treatment approaches to achieve maximum complete response rates because salvage treatment at relapse is not effective.”
Dr. Rosenwald and colleagues suggested that, in the future, diagnostic approaches should be implemented to detect patients in this high-risk group and that risk-modified treatment strategies should be further developed.
The study was supported by unrestricted grants to the Lunenburg Lymphoma Biomarker Consortium from several pharmaceutical companies and Bloodwise. Dr. Rosenwald reported having no conflicts of interest, but several coauthors reported relationships with industry.
SOURCE: : Rosenwald A et al. J Clin Oncol. 2019 Sep 9. doi: 10.1200/JCO.19.00743.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Flesh-Colored Papules on the Scrotum
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
A 44-year-old black man presented with "bumps on the scrotum" of approximately 4 months' duration. They were asymptomatic and untreated. The patient denied extramarital sexual contacts or a history of any sexually transmitted infection. His medical history was notable for sarcoidosis diagnosed 4 years prior to presentation when hilar lymphadenopathy was incidentally found on routine screening. His condition was managed with regular follow-up without treatment. He also had a positive tuberculosis skin test in the past without radiologic evidence of active pulmonary disease. Physical examination revealed multiple 2- to 5-mm, flesh-colored, shiny, polygonal, flat-topped papules spread diffusely over the scrotum. A 1-cm, barely palpable, nonscaly, violaceous to brown plaque also was seen on the tip of the nose. A punch biopsy was taken from a lesion on the scrotum.
Clinical research in private practice? It can be done, and here’s how
Not every physician coming out of medical school wants to go down the path of conducting clinical research. But for those who do, the decision is a rewarding one that can make a real difference in patients’ lives.

I took a nontraditional route to get to my current role as the director of clinical research and education at the largest gastroenterology practice in the United States. I had a business degree coming out of college and worked for years in the business world prior to going to medical school. After graduation, I got an offer to join the pharmaceutical industry in medical affairs. There, I focused on inflammatory bowel disease (IBD), where my role was to share high-level scientific data and liaise with IBD disease state experts. Part of the role was working internally with clinical operations and externally with sites conducting research throughout the United States. In the next 6 years, I had the opportunity to observe how research was run in both the academic and community practice settings, noting those characteristics that allowed some to succeed and far more to stagnate or fail.
In 2014, I joined Texas Digestive Disease Consultants with the goal to ramp up the practice’s clinical research arm and create a department expressly for that purpose. To date, the department has become incredibly successful and ultimately sustainable. If you’re considering joining a practice based on its clinical research program or starting one in your current practice, here is what I’ve learned along the way:
Know the benefit to patients
You have to decide to conduct clinical research because of its benefit to the patient, not because you see it as an alternative revenue stream. It will never be sustainable if viewed as a profit center. Clinical research offers therapeutic advancements that will not be available to most for the next 5-10 years. Additionally, patients do not need insurance in order to participate in a study. The sponsor covers all study visits, procedures, and therapeutics.
Know the value to the practice
Having a clinical trials program brings both direct and indirect enterprise value to the practice. It may come in the form of referrals from other practices that don’t have the same capabilities. Patients may view your practice differently, knowing that it has the added value of research. There is a halo effect from having clinical trials capabilities in how you are viewed by patients and other physicians in the community.
Get the right people in place
First, you will need an enthusiastic primary investigator who can take a bit of time from practice to conduct clinical trials. But just as importantly, you need a knowledgeable clinical research coordinator. Without an effective coordinator, the program is doomed. A good coordinator should be well rounded in all aspects of research (e.g., regulatory, patient recruitment, quality assurance, contracts, budgets, running labs, conducting patient visits) and able to deal with the day-to-day intricacies of running clinical trials, which will allow a doctor to take care of the existing practice. They should also be versed in the requisite equipment (e.g., locking refrigerator, freezer, and ambient cabinet, temperature monitoring devices, EKG machine, and centrifuge) necessary to run a clinical study.
Know how to recruit patients
The database at Texas Digestive Disease Consultants/GI Alliance (TDDC/GIA) is vast, which makes identification of patients who may qualify for a trial an easier task. Many practices will have an electronic medical record that will aid in identification, but if that is not the case at your practice, there are a number of ways you can go about this. First, talk to physicians in your practice and in other practices – internists, family physicians, and other gastroenterologists come to mind – about what you can offer. Send a letter or an email out to physicians in the community with the inclusion/exclusion criteria for your study, and always direct them to https://clinicaltrials.gov/ for additional information. Patient advocacy organizations are another good source of referrals for clinical trials. And of course, pharmaceutical companies have recruitment services that they use and can help steer patients your way.
Understand the ethics
There are numerous ethical and legal considerations when it comes to running clinical trials. The principles of Good Clinical Practice (GCP) and Human Subject Protection (HSP) guide the conduct of clinical trials in the United States. Understanding the concepts and regulations around caring for patients in trials is not only “good practice,” it’s a necessity. There are free Good Clinical Practice training courses available, which are required every few years for everyone conducting clinical research. These courses are quite lengthy (3-6 hours), but provide a great overview of the Food and Drug Administration regulations, ethical considerations, and other advice on successfully operating a clinical trials program. Again, a capable clinical research coordinator will help guide you here.
Adopt standard operating procedures
Every clinical trials sponsor will require standard operating procedures for such facets of research as informed consent, reporting requirements, and safety monitoring. Here again, a seasoned clinical trials coordinator can put you on the right path. You may choose to purchase standardized templates or work with another group that already has them and would be willing to share.
Be smart about spending
While this may depend on the scale you want to grow your research, at TDDC/GIA we knew we wanted to build a sustainable program. We invested a lot of time and money into our infrastructure, as well as adopting a robust Clinical Trials Management System (CTMS). A CTMS system will provide the ability to measure productivity, finances, schedule patient visits, and pay patient stipends. Some systems even automate the regulatory process (E-Regulatory) or source process (E-source), which can cut down on the paper burden on a site.
Seek mentors
Look for someone to emulate who is already established in operating a clinical research program in private practice. Reach out to the practices or physicians that are already doing this work and doing it well. For physicians who are just starting out or still in training, look for opportunities to publish or be involved with the clinical trials program at your institution.
Contacts in the pharmaceutical industry are very important to getting a new program off the ground. Ask the pharma sales representatives who visit your clinic to put you in touch with their medical science liaison (MSL). The MSL can get you information about the clinical trials their company is currently running and those they have in the pipeline.
Dr. Chris Fourment, director of clinical research and education, Texas Digestive Disease Consultants/GI Alliance.
Not every physician coming out of medical school wants to go down the path of conducting clinical research. But for those who do, the decision is a rewarding one that can make a real difference in patients’ lives.
I took a nontraditional route to get to my current role as the director of clinical research and education at the largest gastroenterology practice in the United States. I had a business degree coming out of college and worked for years in the business world prior to going to medical school. After graduation, I got an offer to join the pharmaceutical industry in medical affairs. There, I focused on inflammatory bowel disease (IBD), where my role was to share high-level scientific data and liaise with IBD disease state experts. Part of the role was working internally with clinical operations and externally with sites conducting research throughout the United States. In the next 6 years, I had the opportunity to observe how research was run in both the academic and community practice settings, noting those characteristics that allowed some to succeed and far more to stagnate or fail.
In 2014, I joined Texas Digestive Disease Consultants with the goal to ramp up the practice’s clinical research arm and create a department expressly for that purpose. To date, the department has become incredibly successful and ultimately sustainable. If you’re considering joining a practice based on its clinical research program or starting one in your current practice, here is what I’ve learned along the way:
Know the benefit to patients
You have to decide to conduct clinical research because of its benefit to the patient, not because you see it as an alternative revenue stream. It will never be sustainable if viewed as a profit center. Clinical research offers therapeutic advancements that will not be available to most for the next 5-10 years. Additionally, patients do not need insurance in order to participate in a study. The sponsor covers all study visits, procedures, and therapeutics.
Know the value to the practice
Having a clinical trials program brings both direct and indirect enterprise value to the practice. It may come in the form of referrals from other practices that don’t have the same capabilities. Patients may view your practice differently, knowing that it has the added value of research. There is a halo effect from having clinical trials capabilities in how you are viewed by patients and other physicians in the community.
Get the right people in place
First, you will need an enthusiastic primary investigator who can take a bit of time from practice to conduct clinical trials. But just as importantly, you need a knowledgeable clinical research coordinator. Without an effective coordinator, the program is doomed. A good coordinator should be well rounded in all aspects of research (e.g., regulatory, patient recruitment, quality assurance, contracts, budgets, running labs, conducting patient visits) and able to deal with the day-to-day intricacies of running clinical trials, which will allow a doctor to take care of the existing practice. They should also be versed in the requisite equipment (e.g., locking refrigerator, freezer, and ambient cabinet, temperature monitoring devices, EKG machine, and centrifuge) necessary to run a clinical study.
Know how to recruit patients
The database at Texas Digestive Disease Consultants/GI Alliance (TDDC/GIA) is vast, which makes identification of patients who may qualify for a trial an easier task. Many practices will have an electronic medical record that will aid in identification, but if that is not the case at your practice, there are a number of ways you can go about this. First, talk to physicians in your practice and in other practices – internists, family physicians, and other gastroenterologists come to mind – about what you can offer. Send a letter or an email out to physicians in the community with the inclusion/exclusion criteria for your study, and always direct them to https://clinicaltrials.gov/ for additional information. Patient advocacy organizations are another good source of referrals for clinical trials. And of course, pharmaceutical companies have recruitment services that they use and can help steer patients your way.
Understand the ethics
There are numerous ethical and legal considerations when it comes to running clinical trials. The principles of Good Clinical Practice (GCP) and Human Subject Protection (HSP) guide the conduct of clinical trials in the United States. Understanding the concepts and regulations around caring for patients in trials is not only “good practice,” it’s a necessity. There are free Good Clinical Practice training courses available, which are required every few years for everyone conducting clinical research. These courses are quite lengthy (3-6 hours), but provide a great overview of the Food and Drug Administration regulations, ethical considerations, and other advice on successfully operating a clinical trials program. Again, a capable clinical research coordinator will help guide you here.
Adopt standard operating procedures
Every clinical trials sponsor will require standard operating procedures for such facets of research as informed consent, reporting requirements, and safety monitoring. Here again, a seasoned clinical trials coordinator can put you on the right path. You may choose to purchase standardized templates or work with another group that already has them and would be willing to share.
Be smart about spending
While this may depend on the scale you want to grow your research, at TDDC/GIA we knew we wanted to build a sustainable program. We invested a lot of time and money into our infrastructure, as well as adopting a robust Clinical Trials Management System (CTMS). A CTMS system will provide the ability to measure productivity, finances, schedule patient visits, and pay patient stipends. Some systems even automate the regulatory process (E-Regulatory) or source process (E-source), which can cut down on the paper burden on a site.
Seek mentors
Look for someone to emulate who is already established in operating a clinical research program in private practice. Reach out to the practices or physicians that are already doing this work and doing it well. For physicians who are just starting out or still in training, look for opportunities to publish or be involved with the clinical trials program at your institution.
Contacts in the pharmaceutical industry are very important to getting a new program off the ground. Ask the pharma sales representatives who visit your clinic to put you in touch with their medical science liaison (MSL). The MSL can get you information about the clinical trials their company is currently running and those they have in the pipeline.
Dr. Chris Fourment, director of clinical research and education, Texas Digestive Disease Consultants/GI Alliance.
Not every physician coming out of medical school wants to go down the path of conducting clinical research. But for those who do, the decision is a rewarding one that can make a real difference in patients’ lives.
I took a nontraditional route to get to my current role as the director of clinical research and education at the largest gastroenterology practice in the United States. I had a business degree coming out of college and worked for years in the business world prior to going to medical school. After graduation, I got an offer to join the pharmaceutical industry in medical affairs. There, I focused on inflammatory bowel disease (IBD), where my role was to share high-level scientific data and liaise with IBD disease state experts. Part of the role was working internally with clinical operations and externally with sites conducting research throughout the United States. In the next 6 years, I had the opportunity to observe how research was run in both the academic and community practice settings, noting those characteristics that allowed some to succeed and far more to stagnate or fail.
In 2014, I joined Texas Digestive Disease Consultants with the goal to ramp up the practice’s clinical research arm and create a department expressly for that purpose. To date, the department has become incredibly successful and ultimately sustainable. If you’re considering joining a practice based on its clinical research program or starting one in your current practice, here is what I’ve learned along the way:
Know the benefit to patients
You have to decide to conduct clinical research because of its benefit to the patient, not because you see it as an alternative revenue stream. It will never be sustainable if viewed as a profit center. Clinical research offers therapeutic advancements that will not be available to most for the next 5-10 years. Additionally, patients do not need insurance in order to participate in a study. The sponsor covers all study visits, procedures, and therapeutics.
Know the value to the practice
Having a clinical trials program brings both direct and indirect enterprise value to the practice. It may come in the form of referrals from other practices that don’t have the same capabilities. Patients may view your practice differently, knowing that it has the added value of research. There is a halo effect from having clinical trials capabilities in how you are viewed by patients and other physicians in the community.
Get the right people in place
First, you will need an enthusiastic primary investigator who can take a bit of time from practice to conduct clinical trials. But just as importantly, you need a knowledgeable clinical research coordinator. Without an effective coordinator, the program is doomed. A good coordinator should be well rounded in all aspects of research (e.g., regulatory, patient recruitment, quality assurance, contracts, budgets, running labs, conducting patient visits) and able to deal with the day-to-day intricacies of running clinical trials, which will allow a doctor to take care of the existing practice. They should also be versed in the requisite equipment (e.g., locking refrigerator, freezer, and ambient cabinet, temperature monitoring devices, EKG machine, and centrifuge) necessary to run a clinical study.
Know how to recruit patients
The database at Texas Digestive Disease Consultants/GI Alliance (TDDC/GIA) is vast, which makes identification of patients who may qualify for a trial an easier task. Many practices will have an electronic medical record that will aid in identification, but if that is not the case at your practice, there are a number of ways you can go about this. First, talk to physicians in your practice and in other practices – internists, family physicians, and other gastroenterologists come to mind – about what you can offer. Send a letter or an email out to physicians in the community with the inclusion/exclusion criteria for your study, and always direct them to https://clinicaltrials.gov/ for additional information. Patient advocacy organizations are another good source of referrals for clinical trials. And of course, pharmaceutical companies have recruitment services that they use and can help steer patients your way.
Understand the ethics
There are numerous ethical and legal considerations when it comes to running clinical trials. The principles of Good Clinical Practice (GCP) and Human Subject Protection (HSP) guide the conduct of clinical trials in the United States. Understanding the concepts and regulations around caring for patients in trials is not only “good practice,” it’s a necessity. There are free Good Clinical Practice training courses available, which are required every few years for everyone conducting clinical research. These courses are quite lengthy (3-6 hours), but provide a great overview of the Food and Drug Administration regulations, ethical considerations, and other advice on successfully operating a clinical trials program. Again, a capable clinical research coordinator will help guide you here.
Adopt standard operating procedures
Every clinical trials sponsor will require standard operating procedures for such facets of research as informed consent, reporting requirements, and safety monitoring. Here again, a seasoned clinical trials coordinator can put you on the right path. You may choose to purchase standardized templates or work with another group that already has them and would be willing to share.
Be smart about spending
While this may depend on the scale you want to grow your research, at TDDC/GIA we knew we wanted to build a sustainable program. We invested a lot of time and money into our infrastructure, as well as adopting a robust Clinical Trials Management System (CTMS). A CTMS system will provide the ability to measure productivity, finances, schedule patient visits, and pay patient stipends. Some systems even automate the regulatory process (E-Regulatory) or source process (E-source), which can cut down on the paper burden on a site.
Seek mentors
Look for someone to emulate who is already established in operating a clinical research program in private practice. Reach out to the practices or physicians that are already doing this work and doing it well. For physicians who are just starting out or still in training, look for opportunities to publish or be involved with the clinical trials program at your institution.
Contacts in the pharmaceutical industry are very important to getting a new program off the ground. Ask the pharma sales representatives who visit your clinic to put you in touch with their medical science liaison (MSL). The MSL can get you information about the clinical trials their company is currently running and those they have in the pipeline.
Dr. Chris Fourment, director of clinical research and education, Texas Digestive Disease Consultants/GI Alliance.
VRD pretransplant induction deepens responses in myeloma
Pretransplant induction therapy with subcutaneous bortezomib, lenalidomide, and dexamethasone (VRD) deepened responses in patients with newly diagnosed multiple myeloma, according to an interim analysis of a phase 3 study.
Overall, the regimen was well tolerated, with a minimal number of patients discontinuing treatment because of treatment-emergent adverse events.
The ongoing, open-label, randomized, phase 3 study is designed to compare two transplant-conditioning regimens – intravenous busulfan plus melphalan versus melphalan – in patients who received VRD induction and consolidation, wrote Laura Rosiñol, MD, PhD, of the August Pi i Sunyer Biomedical Research Institute in Barcelona, and colleagues. The findings were published in Blood.
The PETHEMA/GEM2012 study included 458 patients with newly diagnosed multiple myeloma who were eligible for autologous stem cell transplantation. Study patients were previously untreated and aged younger than 65 years.
All patients received VRD induction, which consisted of subcutaneous bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 of each cycle; lenalidomide 25 mg/day on days 1-21; and dexamethasone 40 mg on days 1-4 and 9-12 at 4-week intervals for six cycles. Posttransplant consolidation consisted of two cycles of VRD.
The researchers conducted a grouped-response analysis of three different treatment phases: induction, transplant, and consolidation.
After analysis, the researchers found that responses deepened over the duration of treatment. In patients who started the sixth induction cycle, the response rates were 55.6%, 63.8%, 68.3%, and 70.4% after cycles 3, 4, 5, and post induction, respectively.
After six cycles of induction, the complete response rate was 33.4%, with a rate of undetectable minimal residual disease of 28.8%, which further increased at transplant (42.1%), and consolidation (45.2%).
With respect to safety, the most frequently reported grade 3 or higher treatment-emergent adverse events were neutropenia (12.9%) and infection (9.2%). The rate of grade 2 or higher peripheral neuropathy throughout induction was 17.0%, with lower rates of grade 3 (3.7%) and 4 (0.2%) toxicities.
“The regimen [used in the present study] has the highest lenalidomide and dexamethasone dose intensity per cycle and a lower bortezomib dose intensity per cycle than the 21-day regimens, which may offer high activity with low levels of toxicity, thereby enabling delivery of all planned induction cycles,” the researchers wrote, adding that “these results confirm that VRD is an effective pretransplant induction regimen and may be considered a new standard of care.”
The study was supported by Celgene, Janssen, Pierre Fabré, and the Instituto de Salud Carlos III. The authors reported financial affiliations with Celgene, Janssen, and several other companies.
SOURCE: Rosiñol L et al. Blood. 2019 Sep 4. doi: 10.1182/blood.2019000241.
Pretransplant induction therapy with subcutaneous bortezomib, lenalidomide, and dexamethasone (VRD) deepened responses in patients with newly diagnosed multiple myeloma, according to an interim analysis of a phase 3 study.
Overall, the regimen was well tolerated, with a minimal number of patients discontinuing treatment because of treatment-emergent adverse events.
The ongoing, open-label, randomized, phase 3 study is designed to compare two transplant-conditioning regimens – intravenous busulfan plus melphalan versus melphalan – in patients who received VRD induction and consolidation, wrote Laura Rosiñol, MD, PhD, of the August Pi i Sunyer Biomedical Research Institute in Barcelona, and colleagues. The findings were published in Blood.
The PETHEMA/GEM2012 study included 458 patients with newly diagnosed multiple myeloma who were eligible for autologous stem cell transplantation. Study patients were previously untreated and aged younger than 65 years.
All patients received VRD induction, which consisted of subcutaneous bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 of each cycle; lenalidomide 25 mg/day on days 1-21; and dexamethasone 40 mg on days 1-4 and 9-12 at 4-week intervals for six cycles. Posttransplant consolidation consisted of two cycles of VRD.
The researchers conducted a grouped-response analysis of three different treatment phases: induction, transplant, and consolidation.
After analysis, the researchers found that responses deepened over the duration of treatment. In patients who started the sixth induction cycle, the response rates were 55.6%, 63.8%, 68.3%, and 70.4% after cycles 3, 4, 5, and post induction, respectively.
After six cycles of induction, the complete response rate was 33.4%, with a rate of undetectable minimal residual disease of 28.8%, which further increased at transplant (42.1%), and consolidation (45.2%).
With respect to safety, the most frequently reported grade 3 or higher treatment-emergent adverse events were neutropenia (12.9%) and infection (9.2%). The rate of grade 2 or higher peripheral neuropathy throughout induction was 17.0%, with lower rates of grade 3 (3.7%) and 4 (0.2%) toxicities.
“The regimen [used in the present study] has the highest lenalidomide and dexamethasone dose intensity per cycle and a lower bortezomib dose intensity per cycle than the 21-day regimens, which may offer high activity with low levels of toxicity, thereby enabling delivery of all planned induction cycles,” the researchers wrote, adding that “these results confirm that VRD is an effective pretransplant induction regimen and may be considered a new standard of care.”
The study was supported by Celgene, Janssen, Pierre Fabré, and the Instituto de Salud Carlos III. The authors reported financial affiliations with Celgene, Janssen, and several other companies.
SOURCE: Rosiñol L et al. Blood. 2019 Sep 4. doi: 10.1182/blood.2019000241.
Pretransplant induction therapy with subcutaneous bortezomib, lenalidomide, and dexamethasone (VRD) deepened responses in patients with newly diagnosed multiple myeloma, according to an interim analysis of a phase 3 study.
Overall, the regimen was well tolerated, with a minimal number of patients discontinuing treatment because of treatment-emergent adverse events.
The ongoing, open-label, randomized, phase 3 study is designed to compare two transplant-conditioning regimens – intravenous busulfan plus melphalan versus melphalan – in patients who received VRD induction and consolidation, wrote Laura Rosiñol, MD, PhD, of the August Pi i Sunyer Biomedical Research Institute in Barcelona, and colleagues. The findings were published in Blood.
The PETHEMA/GEM2012 study included 458 patients with newly diagnosed multiple myeloma who were eligible for autologous stem cell transplantation. Study patients were previously untreated and aged younger than 65 years.
All patients received VRD induction, which consisted of subcutaneous bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 of each cycle; lenalidomide 25 mg/day on days 1-21; and dexamethasone 40 mg on days 1-4 and 9-12 at 4-week intervals for six cycles. Posttransplant consolidation consisted of two cycles of VRD.
The researchers conducted a grouped-response analysis of three different treatment phases: induction, transplant, and consolidation.
After analysis, the researchers found that responses deepened over the duration of treatment. In patients who started the sixth induction cycle, the response rates were 55.6%, 63.8%, 68.3%, and 70.4% after cycles 3, 4, 5, and post induction, respectively.
After six cycles of induction, the complete response rate was 33.4%, with a rate of undetectable minimal residual disease of 28.8%, which further increased at transplant (42.1%), and consolidation (45.2%).
With respect to safety, the most frequently reported grade 3 or higher treatment-emergent adverse events were neutropenia (12.9%) and infection (9.2%). The rate of grade 2 or higher peripheral neuropathy throughout induction was 17.0%, with lower rates of grade 3 (3.7%) and 4 (0.2%) toxicities.
“The regimen [used in the present study] has the highest lenalidomide and dexamethasone dose intensity per cycle and a lower bortezomib dose intensity per cycle than the 21-day regimens, which may offer high activity with low levels of toxicity, thereby enabling delivery of all planned induction cycles,” the researchers wrote, adding that “these results confirm that VRD is an effective pretransplant induction regimen and may be considered a new standard of care.”
The study was supported by Celgene, Janssen, Pierre Fabré, and the Instituto de Salud Carlos III. The authors reported financial affiliations with Celgene, Janssen, and several other companies.
SOURCE: Rosiñol L et al. Blood. 2019 Sep 4. doi: 10.1182/blood.2019000241.
FROM BLOOD
Conference Coverage: ASCO 2019
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.