User login
Seasonality of birth and psychiatric illness
“To every thing there is a season, and a time to every purpose under the heaven.”
— Ecclesiastes
The month of birth is not just relevant to one’s astrological sign. It may have medical consequences. An impressive number of published studies have found that the month and season of birth may be related to a higher risk of various medical and psychiatric disorders.
For decades, it has been reported in more than 250 studies1 that a disproportionate number of individuals with schizophrenia are born during the winter months (January/February/March in the Northern Hemisphere and July/August/September in the Southern Hemisphere). This seasonal pattern was eventually linked to the lack of sunlight during winter months and a deficiency of vitamin D, a hormone that is critical for normal brain development. Recent studies have reported that very low serum levels of vitamin D during pregnancy significantly increase the risk of schizophrenia in offspring.2
But the plot thickens. Numerous studies over the past 20 to 30 years have reported an association between month or season of birth with sundry general medical and psychiatric conditions. Even longevity has been reported to vary with season of birth, with a longer life span for people born in autumn (October to December), compared with those born in spring (April to June).3 Of note, a longer life span for an individual born in autumn has been attributed to a higher birth weight during that season compared with those born in other seasons. In addition, the shorter life span of those with spring births has been attributed to factors during fetal life that increase the susceptibility to disease later in life (after age 50).
The following studies have reported an association between month/season of birth and general medical disorders:
- Higher rate of myopia for summer births4
- Tenfold higher risk of respiratory syntactical virus in babies born in January compared with October, and a 2 to 3 times higher risk of hospitalization5
- Higher rates of asthma during childhood for March and April births6
- Lower rate of lung cancer for winter births compared with all other seasons7
- An excess of colon and rectal cancer for people born in September, and the lowest rate for spring births8
- Lowest diabetes risk for summer births9
- For males: Cardiac mortality is 11% less likely for 4th-quarter births compared with 1st-quarter births. For females: Cancer mortality is lowest in 3rd-quarter vs 1st-quarter births10
- The peak risk for both Hodgkin and non-Hodgkin lymphoma is for April births compared with other months11
- A strong trend for malignant neoplasm in males was reported for births during the 1st trimester of the year (January through April) compared with the rest of the year12
- Higher rate of spring births among patients who have insulin-dependent diabetes13
- Breast cancer is 5% higher for June births compared with December births14
- Higher risk of developing an allergy later in life for those born approximately 3 months before the main allergy season.15
The above studies may imply that birth seasonality is medical destiny. However, most such reports need further replication, or may be due to chance findings in various databases. However, they are worth considering as hypothesis-generating signals.
Continue to: And now for the risk of psychiatric disorders...
And now for the risk of psychiatric disorders and month or season of birth. Here, too, there are multiple published reports:
- Higher social anhedonia and schizoid features among persons born in June and July16
- Higher autism rates for children conceived in December to March compared with those conceived during summer months17
- In contrast to the above report, the risk of autism spectrum disorders in the United Kingdom was higher for those born in summer18
- Another study labeled seasonality of birth in autism as “fiction”!19
- Significant spring births for persons with anxiety20
- Highest occurrence of postpartum depression in December21
- High prepartum depression in winter and postpartum depression in fall22
- Lower performance IQ among spring births23
- Disproportionate excess of births in April, May, and June for those who die by suicide24
- Suicide by burning oneself is higher among individuals born in January compared with any other month25
- Relative increase in March and August births among patients with anorexia26
- Season of birth is a predictor of emotional and behavioral regulation27
- Serotonin metabolites show a peak in spring and a trough in fall28
- Increase of spring births in individuals with Down syndrome29
- Excess of spring births among patients with Alzheimer’s disease.30
As with the seasonality of medical illness risk, the association of the month or season of birth with psychiatric disorders may be based on skewed samples or simply a chance finding. However, there may be some seasonal environmental factors that could increase the risk for disorders of the body or the brain/mind. The most plausible factors may be season-related fetal developmental disruptions caused by maternal infection, diet, lack of sunlight, temperature, substance use, or immune dysregulation from comorbid medical conditions during pregnancy. Some researchers have speculated that fluctuations in the availability of various fresh fruits and vegetables during certain seasons of the year may influence fetal development or increase the susceptibility to some medical disorders. This may be at the time of conception or during the 2nd trimester of pregnancy, when the brain develops.
On the other hand, those studies, published in peer-reviewed journals, may constitute a sophisticated form of “psychiatric astrology” whose credibility could be as suspect as the imaginative predictions of one’s horoscope in the daily newspaper…
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Torrey EF, Miller J, Rawlings R, et al. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res. 1997;28(1):1-38.
2. McGrath J, Welham J, Pemberton M. Month of birth, hemisphere of birth and schizophrenia. Br J Psychiatry. 1995;167(6):783-785.
3. Doblhammer G, Vaupel JW. Lifespan depends on month of birth. Proc Natl Acad Sci U S A. 2001;98(5):2934-2939.
4. Mandel Y, Grotto I, El-Yaniv R, et al. Season of birth, natural light, and myopia. Ophthalmology. 2008;115(4):686-692.
5. Lloyd PC, May L, Hoffman D, et al. The effect of birth month on the risk of respiratory syncytial virus hospitalization in the first year of life in the United States. Pediatr Infect Dis J. 2014;33(6):e135-e140.
6. Gazala E, Ron-Feldman V, Alterman M, et al. The association between birth season and future development of childhood asthma. Pediatr Pulmonol. 2006;41(12):1125-1128.
7. Hao Y, Yan L, Ke E, et al. Birth in winter can reduce the risk of lung cancer: A retrospective study of the birth season of patients with lung cancer in Beijing area, China. Chronobiol Int. 2017;34(4):511-518.
8. Francis NK, Curtis NJ, Noble E, et al. Is month of birth a risk factor for colorectal cancer? Gastroenterol Res Pract. 2017;2017:5423765. doi: 10.1155/2017/5423765.
9. Si J, Yu C, Guo Y, et al; China Kadoorie Biobank Collaborative Group. Season of birth and the risk of type 2 diabetes in adulthood: a prospective cohort study of 0.5 million Chinese adults. Diabetologia. 2017;60(5):836-842.
10. Sohn K. The influence of birth season on mortality in the United States. Am J Hum Biol. 2016;28(5):662-670.
11. Crump C, Sundquist J, Sieh W, et al. Season of birth and risk of Hodgkin and non-Hodgkin lymphoma. Int J Cancer. 2014;135(11):2735-2739.
12. Stoupel E, Abramson E, Fenig E. Birth month of patients with malignant neoplasms: links to longevity? J Basic Clin Physiol Pharmacol. 2012;23(2):57-60.
13. Rothwell PM, Gutnikov SA, McKinney PA, et al. Seasonality of birth in children with diabetes in Europe: multicentre cohort study. European Diabetes Study Group. BMJ. 1999;319(7214):887-888.
14. Yuen J, Ekbom A, Trichopoulos D, et al. Season of birth and breast cancer risk in Sweden. Br J Cancer. 1994;70(3):564-568.
15. Aalberse RC, Nieuwenhuys EJ, Hey M, et al. ‘Horoscope effect’ not only for seasonal but also for non-seasonal allergens. Clin Exp Allergy. 1992;22(11):1003-1006.
16. Kirkpatrick B, Messias E, LaPorte D. Schizoid-like features and season of birth in a nonpatient sample. Schizophr Res. 2008;103:151-155.
17. Zerbo O, Iosif AM, Delwiche L, et al. Month of conception and risk of autism. Epidemiology. 2011;22(4):469-475.
18. Hebert KJ, Miller LL, Joinson CJ. Association of autistic spectrum disorder with season of birth and conception in a UK cohort. Autism Res. 2010;3(4):185-190.
19. Landau EC, Cicchetti DV, Klin A, et al. Season of birth in autism: a fiction revisited. J Autism Dev Disord. 1999;29(5):385-393.
20. Parker G, Neilson M. Mental disorder and season of birth--a southern hemisphere study. Br J Psychiatry. 1976;129:355-361.
21. Sit D, Seltman H, Wisner KL. Seasonal effects on depression risk and suicidal symptoms in postpartum women. Depress Anxiety. 2011;28(5):400-405.
22. Chan JE, Samaranayaka A, Paterson H. Seasonal and gestational variation in perinatal depression in a prospective cohort in New Zealand. Aust N Z J Obstet Gynaecol. 2018. [Epub ahead of print]. doi: 10.1111/ajo.12912.
23. Grootendorst-van Mil NH, Steegers-Theunissen RP, Hofman A, et al. Brighter children? The association between seasonality of birth and child IQ in a population-based birth cohort. BMJ Open. 2017;7(2):e012406. doi: 10.1136/bmjopen-2016-012406.
24. Salib E, Cortina-Borja M. Effect of month of birth on the risk of suicide. Br J Psychiatry. 2006;188:416-422.
25. Salib E, Cortina-Borja M. An association between month of birth and method of suicide. Int J Psychiatry Clin Pract. 2010;14(1):8-17.
26. Brewerton TD, Dansky BS, O’Neil PM, et al. Seasonal patterns of birth for subjects with bulimia nervosa, binge eating, and purging: results from the National Women’s Study. Int J Eat Disord. 2012;45(1):131-134.
27. Asano R, Tsuchiya KJ, Harada T, et al; for Hamamatsu Birth Cohort (HBC) Study Team. Season of birth predicts emotional and behavioral regulation in 18-month-old infants: Hamamatsu birth cohort for mothers and children (HBC Study). Front Public Health. 2016;4:152.
28. Luykx JJ, Bakker SC, Lentjes E, et al. Season of sampling and season of birth influence serotonin metabolite levels in human cerebrospinal fluid. PLoS One. 2012;7(2):e30497. doi: 10.1371/journal.pone.0030497.
29. Videbech P, Nielsen J. Chromosome abnormalities and season of birth. Hum Genet. 1984;65(3):221-231.
30. Vézina H, Houde L, Charbonneau H, et al. Season of birth and Alzheimer’s disease: a population-based study in Saguenay-Lac-St-Jean/Québec (IMAGE Project). Psychol Med. 1996;26(1):143-149.
“To every thing there is a season, and a time to every purpose under the heaven.”
— Ecclesiastes
The month of birth is not just relevant to one’s astrological sign. It may have medical consequences. An impressive number of published studies have found that the month and season of birth may be related to a higher risk of various medical and psychiatric disorders.
For decades, it has been reported in more than 250 studies1 that a disproportionate number of individuals with schizophrenia are born during the winter months (January/February/March in the Northern Hemisphere and July/August/September in the Southern Hemisphere). This seasonal pattern was eventually linked to the lack of sunlight during winter months and a deficiency of vitamin D, a hormone that is critical for normal brain development. Recent studies have reported that very low serum levels of vitamin D during pregnancy significantly increase the risk of schizophrenia in offspring.2
But the plot thickens. Numerous studies over the past 20 to 30 years have reported an association between month or season of birth with sundry general medical and psychiatric conditions. Even longevity has been reported to vary with season of birth, with a longer life span for people born in autumn (October to December), compared with those born in spring (April to June).3 Of note, a longer life span for an individual born in autumn has been attributed to a higher birth weight during that season compared with those born in other seasons. In addition, the shorter life span of those with spring births has been attributed to factors during fetal life that increase the susceptibility to disease later in life (after age 50).
The following studies have reported an association between month/season of birth and general medical disorders:
- Higher rate of myopia for summer births4
- Tenfold higher risk of respiratory syntactical virus in babies born in January compared with October, and a 2 to 3 times higher risk of hospitalization5
- Higher rates of asthma during childhood for March and April births6
- Lower rate of lung cancer for winter births compared with all other seasons7
- An excess of colon and rectal cancer for people born in September, and the lowest rate for spring births8
- Lowest diabetes risk for summer births9
- For males: Cardiac mortality is 11% less likely for 4th-quarter births compared with 1st-quarter births. For females: Cancer mortality is lowest in 3rd-quarter vs 1st-quarter births10
- The peak risk for both Hodgkin and non-Hodgkin lymphoma is for April births compared with other months11
- A strong trend for malignant neoplasm in males was reported for births during the 1st trimester of the year (January through April) compared with the rest of the year12
- Higher rate of spring births among patients who have insulin-dependent diabetes13
- Breast cancer is 5% higher for June births compared with December births14
- Higher risk of developing an allergy later in life for those born approximately 3 months before the main allergy season.15
The above studies may imply that birth seasonality is medical destiny. However, most such reports need further replication, or may be due to chance findings in various databases. However, they are worth considering as hypothesis-generating signals.
Continue to: And now for the risk of psychiatric disorders...
And now for the risk of psychiatric disorders and month or season of birth. Here, too, there are multiple published reports:
- Higher social anhedonia and schizoid features among persons born in June and July16
- Higher autism rates for children conceived in December to March compared with those conceived during summer months17
- In contrast to the above report, the risk of autism spectrum disorders in the United Kingdom was higher for those born in summer18
- Another study labeled seasonality of birth in autism as “fiction”!19
- Significant spring births for persons with anxiety20
- Highest occurrence of postpartum depression in December21
- High prepartum depression in winter and postpartum depression in fall22
- Lower performance IQ among spring births23
- Disproportionate excess of births in April, May, and June for those who die by suicide24
- Suicide by burning oneself is higher among individuals born in January compared with any other month25
- Relative increase in March and August births among patients with anorexia26
- Season of birth is a predictor of emotional and behavioral regulation27
- Serotonin metabolites show a peak in spring and a trough in fall28
- Increase of spring births in individuals with Down syndrome29
- Excess of spring births among patients with Alzheimer’s disease.30
As with the seasonality of medical illness risk, the association of the month or season of birth with psychiatric disorders may be based on skewed samples or simply a chance finding. However, there may be some seasonal environmental factors that could increase the risk for disorders of the body or the brain/mind. The most plausible factors may be season-related fetal developmental disruptions caused by maternal infection, diet, lack of sunlight, temperature, substance use, or immune dysregulation from comorbid medical conditions during pregnancy. Some researchers have speculated that fluctuations in the availability of various fresh fruits and vegetables during certain seasons of the year may influence fetal development or increase the susceptibility to some medical disorders. This may be at the time of conception or during the 2nd trimester of pregnancy, when the brain develops.
On the other hand, those studies, published in peer-reviewed journals, may constitute a sophisticated form of “psychiatric astrology” whose credibility could be as suspect as the imaginative predictions of one’s horoscope in the daily newspaper…
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
“To every thing there is a season, and a time to every purpose under the heaven.”
— Ecclesiastes
The month of birth is not just relevant to one’s astrological sign. It may have medical consequences. An impressive number of published studies have found that the month and season of birth may be related to a higher risk of various medical and psychiatric disorders.
For decades, it has been reported in more than 250 studies1 that a disproportionate number of individuals with schizophrenia are born during the winter months (January/February/March in the Northern Hemisphere and July/August/September in the Southern Hemisphere). This seasonal pattern was eventually linked to the lack of sunlight during winter months and a deficiency of vitamin D, a hormone that is critical for normal brain development. Recent studies have reported that very low serum levels of vitamin D during pregnancy significantly increase the risk of schizophrenia in offspring.2
But the plot thickens. Numerous studies over the past 20 to 30 years have reported an association between month or season of birth with sundry general medical and psychiatric conditions. Even longevity has been reported to vary with season of birth, with a longer life span for people born in autumn (October to December), compared with those born in spring (April to June).3 Of note, a longer life span for an individual born in autumn has been attributed to a higher birth weight during that season compared with those born in other seasons. In addition, the shorter life span of those with spring births has been attributed to factors during fetal life that increase the susceptibility to disease later in life (after age 50).
The following studies have reported an association between month/season of birth and general medical disorders:
- Higher rate of myopia for summer births4
- Tenfold higher risk of respiratory syntactical virus in babies born in January compared with October, and a 2 to 3 times higher risk of hospitalization5
- Higher rates of asthma during childhood for March and April births6
- Lower rate of lung cancer for winter births compared with all other seasons7
- An excess of colon and rectal cancer for people born in September, and the lowest rate for spring births8
- Lowest diabetes risk for summer births9
- For males: Cardiac mortality is 11% less likely for 4th-quarter births compared with 1st-quarter births. For females: Cancer mortality is lowest in 3rd-quarter vs 1st-quarter births10
- The peak risk for both Hodgkin and non-Hodgkin lymphoma is for April births compared with other months11
- A strong trend for malignant neoplasm in males was reported for births during the 1st trimester of the year (January through April) compared with the rest of the year12
- Higher rate of spring births among patients who have insulin-dependent diabetes13
- Breast cancer is 5% higher for June births compared with December births14
- Higher risk of developing an allergy later in life for those born approximately 3 months before the main allergy season.15
The above studies may imply that birth seasonality is medical destiny. However, most such reports need further replication, or may be due to chance findings in various databases. However, they are worth considering as hypothesis-generating signals.
Continue to: And now for the risk of psychiatric disorders...
And now for the risk of psychiatric disorders and month or season of birth. Here, too, there are multiple published reports:
- Higher social anhedonia and schizoid features among persons born in June and July16
- Higher autism rates for children conceived in December to March compared with those conceived during summer months17
- In contrast to the above report, the risk of autism spectrum disorders in the United Kingdom was higher for those born in summer18
- Another study labeled seasonality of birth in autism as “fiction”!19
- Significant spring births for persons with anxiety20
- Highest occurrence of postpartum depression in December21
- High prepartum depression in winter and postpartum depression in fall22
- Lower performance IQ among spring births23
- Disproportionate excess of births in April, May, and June for those who die by suicide24
- Suicide by burning oneself is higher among individuals born in January compared with any other month25
- Relative increase in March and August births among patients with anorexia26
- Season of birth is a predictor of emotional and behavioral regulation27
- Serotonin metabolites show a peak in spring and a trough in fall28
- Increase of spring births in individuals with Down syndrome29
- Excess of spring births among patients with Alzheimer’s disease.30
As with the seasonality of medical illness risk, the association of the month or season of birth with psychiatric disorders may be based on skewed samples or simply a chance finding. However, there may be some seasonal environmental factors that could increase the risk for disorders of the body or the brain/mind. The most plausible factors may be season-related fetal developmental disruptions caused by maternal infection, diet, lack of sunlight, temperature, substance use, or immune dysregulation from comorbid medical conditions during pregnancy. Some researchers have speculated that fluctuations in the availability of various fresh fruits and vegetables during certain seasons of the year may influence fetal development or increase the susceptibility to some medical disorders. This may be at the time of conception or during the 2nd trimester of pregnancy, when the brain develops.
On the other hand, those studies, published in peer-reviewed journals, may constitute a sophisticated form of “psychiatric astrology” whose credibility could be as suspect as the imaginative predictions of one’s horoscope in the daily newspaper…
To comment on this editorial or other topics of interest: henry.nasrallah@currentpsychiatry.com.
1. Torrey EF, Miller J, Rawlings R, et al. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res. 1997;28(1):1-38.
2. McGrath J, Welham J, Pemberton M. Month of birth, hemisphere of birth and schizophrenia. Br J Psychiatry. 1995;167(6):783-785.
3. Doblhammer G, Vaupel JW. Lifespan depends on month of birth. Proc Natl Acad Sci U S A. 2001;98(5):2934-2939.
4. Mandel Y, Grotto I, El-Yaniv R, et al. Season of birth, natural light, and myopia. Ophthalmology. 2008;115(4):686-692.
5. Lloyd PC, May L, Hoffman D, et al. The effect of birth month on the risk of respiratory syncytial virus hospitalization in the first year of life in the United States. Pediatr Infect Dis J. 2014;33(6):e135-e140.
6. Gazala E, Ron-Feldman V, Alterman M, et al. The association between birth season and future development of childhood asthma. Pediatr Pulmonol. 2006;41(12):1125-1128.
7. Hao Y, Yan L, Ke E, et al. Birth in winter can reduce the risk of lung cancer: A retrospective study of the birth season of patients with lung cancer in Beijing area, China. Chronobiol Int. 2017;34(4):511-518.
8. Francis NK, Curtis NJ, Noble E, et al. Is month of birth a risk factor for colorectal cancer? Gastroenterol Res Pract. 2017;2017:5423765. doi: 10.1155/2017/5423765.
9. Si J, Yu C, Guo Y, et al; China Kadoorie Biobank Collaborative Group. Season of birth and the risk of type 2 diabetes in adulthood: a prospective cohort study of 0.5 million Chinese adults. Diabetologia. 2017;60(5):836-842.
10. Sohn K. The influence of birth season on mortality in the United States. Am J Hum Biol. 2016;28(5):662-670.
11. Crump C, Sundquist J, Sieh W, et al. Season of birth and risk of Hodgkin and non-Hodgkin lymphoma. Int J Cancer. 2014;135(11):2735-2739.
12. Stoupel E, Abramson E, Fenig E. Birth month of patients with malignant neoplasms: links to longevity? J Basic Clin Physiol Pharmacol. 2012;23(2):57-60.
13. Rothwell PM, Gutnikov SA, McKinney PA, et al. Seasonality of birth in children with diabetes in Europe: multicentre cohort study. European Diabetes Study Group. BMJ. 1999;319(7214):887-888.
14. Yuen J, Ekbom A, Trichopoulos D, et al. Season of birth and breast cancer risk in Sweden. Br J Cancer. 1994;70(3):564-568.
15. Aalberse RC, Nieuwenhuys EJ, Hey M, et al. ‘Horoscope effect’ not only for seasonal but also for non-seasonal allergens. Clin Exp Allergy. 1992;22(11):1003-1006.
16. Kirkpatrick B, Messias E, LaPorte D. Schizoid-like features and season of birth in a nonpatient sample. Schizophr Res. 2008;103:151-155.
17. Zerbo O, Iosif AM, Delwiche L, et al. Month of conception and risk of autism. Epidemiology. 2011;22(4):469-475.
18. Hebert KJ, Miller LL, Joinson CJ. Association of autistic spectrum disorder with season of birth and conception in a UK cohort. Autism Res. 2010;3(4):185-190.
19. Landau EC, Cicchetti DV, Klin A, et al. Season of birth in autism: a fiction revisited. J Autism Dev Disord. 1999;29(5):385-393.
20. Parker G, Neilson M. Mental disorder and season of birth--a southern hemisphere study. Br J Psychiatry. 1976;129:355-361.
21. Sit D, Seltman H, Wisner KL. Seasonal effects on depression risk and suicidal symptoms in postpartum women. Depress Anxiety. 2011;28(5):400-405.
22. Chan JE, Samaranayaka A, Paterson H. Seasonal and gestational variation in perinatal depression in a prospective cohort in New Zealand. Aust N Z J Obstet Gynaecol. 2018. [Epub ahead of print]. doi: 10.1111/ajo.12912.
23. Grootendorst-van Mil NH, Steegers-Theunissen RP, Hofman A, et al. Brighter children? The association between seasonality of birth and child IQ in a population-based birth cohort. BMJ Open. 2017;7(2):e012406. doi: 10.1136/bmjopen-2016-012406.
24. Salib E, Cortina-Borja M. Effect of month of birth on the risk of suicide. Br J Psychiatry. 2006;188:416-422.
25. Salib E, Cortina-Borja M. An association between month of birth and method of suicide. Int J Psychiatry Clin Pract. 2010;14(1):8-17.
26. Brewerton TD, Dansky BS, O’Neil PM, et al. Seasonal patterns of birth for subjects with bulimia nervosa, binge eating, and purging: results from the National Women’s Study. Int J Eat Disord. 2012;45(1):131-134.
27. Asano R, Tsuchiya KJ, Harada T, et al; for Hamamatsu Birth Cohort (HBC) Study Team. Season of birth predicts emotional and behavioral regulation in 18-month-old infants: Hamamatsu birth cohort for mothers and children (HBC Study). Front Public Health. 2016;4:152.
28. Luykx JJ, Bakker SC, Lentjes E, et al. Season of sampling and season of birth influence serotonin metabolite levels in human cerebrospinal fluid. PLoS One. 2012;7(2):e30497. doi: 10.1371/journal.pone.0030497.
29. Videbech P, Nielsen J. Chromosome abnormalities and season of birth. Hum Genet. 1984;65(3):221-231.
30. Vézina H, Houde L, Charbonneau H, et al. Season of birth and Alzheimer’s disease: a population-based study in Saguenay-Lac-St-Jean/Québec (IMAGE Project). Psychol Med. 1996;26(1):143-149.
1. Torrey EF, Miller J, Rawlings R, et al. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr Res. 1997;28(1):1-38.
2. McGrath J, Welham J, Pemberton M. Month of birth, hemisphere of birth and schizophrenia. Br J Psychiatry. 1995;167(6):783-785.
3. Doblhammer G, Vaupel JW. Lifespan depends on month of birth. Proc Natl Acad Sci U S A. 2001;98(5):2934-2939.
4. Mandel Y, Grotto I, El-Yaniv R, et al. Season of birth, natural light, and myopia. Ophthalmology. 2008;115(4):686-692.
5. Lloyd PC, May L, Hoffman D, et al. The effect of birth month on the risk of respiratory syncytial virus hospitalization in the first year of life in the United States. Pediatr Infect Dis J. 2014;33(6):e135-e140.
6. Gazala E, Ron-Feldman V, Alterman M, et al. The association between birth season and future development of childhood asthma. Pediatr Pulmonol. 2006;41(12):1125-1128.
7. Hao Y, Yan L, Ke E, et al. Birth in winter can reduce the risk of lung cancer: A retrospective study of the birth season of patients with lung cancer in Beijing area, China. Chronobiol Int. 2017;34(4):511-518.
8. Francis NK, Curtis NJ, Noble E, et al. Is month of birth a risk factor for colorectal cancer? Gastroenterol Res Pract. 2017;2017:5423765. doi: 10.1155/2017/5423765.
9. Si J, Yu C, Guo Y, et al; China Kadoorie Biobank Collaborative Group. Season of birth and the risk of type 2 diabetes in adulthood: a prospective cohort study of 0.5 million Chinese adults. Diabetologia. 2017;60(5):836-842.
10. Sohn K. The influence of birth season on mortality in the United States. Am J Hum Biol. 2016;28(5):662-670.
11. Crump C, Sundquist J, Sieh W, et al. Season of birth and risk of Hodgkin and non-Hodgkin lymphoma. Int J Cancer. 2014;135(11):2735-2739.
12. Stoupel E, Abramson E, Fenig E. Birth month of patients with malignant neoplasms: links to longevity? J Basic Clin Physiol Pharmacol. 2012;23(2):57-60.
13. Rothwell PM, Gutnikov SA, McKinney PA, et al. Seasonality of birth in children with diabetes in Europe: multicentre cohort study. European Diabetes Study Group. BMJ. 1999;319(7214):887-888.
14. Yuen J, Ekbom A, Trichopoulos D, et al. Season of birth and breast cancer risk in Sweden. Br J Cancer. 1994;70(3):564-568.
15. Aalberse RC, Nieuwenhuys EJ, Hey M, et al. ‘Horoscope effect’ not only for seasonal but also for non-seasonal allergens. Clin Exp Allergy. 1992;22(11):1003-1006.
16. Kirkpatrick B, Messias E, LaPorte D. Schizoid-like features and season of birth in a nonpatient sample. Schizophr Res. 2008;103:151-155.
17. Zerbo O, Iosif AM, Delwiche L, et al. Month of conception and risk of autism. Epidemiology. 2011;22(4):469-475.
18. Hebert KJ, Miller LL, Joinson CJ. Association of autistic spectrum disorder with season of birth and conception in a UK cohort. Autism Res. 2010;3(4):185-190.
19. Landau EC, Cicchetti DV, Klin A, et al. Season of birth in autism: a fiction revisited. J Autism Dev Disord. 1999;29(5):385-393.
20. Parker G, Neilson M. Mental disorder and season of birth--a southern hemisphere study. Br J Psychiatry. 1976;129:355-361.
21. Sit D, Seltman H, Wisner KL. Seasonal effects on depression risk and suicidal symptoms in postpartum women. Depress Anxiety. 2011;28(5):400-405.
22. Chan JE, Samaranayaka A, Paterson H. Seasonal and gestational variation in perinatal depression in a prospective cohort in New Zealand. Aust N Z J Obstet Gynaecol. 2018. [Epub ahead of print]. doi: 10.1111/ajo.12912.
23. Grootendorst-van Mil NH, Steegers-Theunissen RP, Hofman A, et al. Brighter children? The association between seasonality of birth and child IQ in a population-based birth cohort. BMJ Open. 2017;7(2):e012406. doi: 10.1136/bmjopen-2016-012406.
24. Salib E, Cortina-Borja M. Effect of month of birth on the risk of suicide. Br J Psychiatry. 2006;188:416-422.
25. Salib E, Cortina-Borja M. An association between month of birth and method of suicide. Int J Psychiatry Clin Pract. 2010;14(1):8-17.
26. Brewerton TD, Dansky BS, O’Neil PM, et al. Seasonal patterns of birth for subjects with bulimia nervosa, binge eating, and purging: results from the National Women’s Study. Int J Eat Disord. 2012;45(1):131-134.
27. Asano R, Tsuchiya KJ, Harada T, et al; for Hamamatsu Birth Cohort (HBC) Study Team. Season of birth predicts emotional and behavioral regulation in 18-month-old infants: Hamamatsu birth cohort for mothers and children (HBC Study). Front Public Health. 2016;4:152.
28. Luykx JJ, Bakker SC, Lentjes E, et al. Season of sampling and season of birth influence serotonin metabolite levels in human cerebrospinal fluid. PLoS One. 2012;7(2):e30497. doi: 10.1371/journal.pone.0030497.
29. Videbech P, Nielsen J. Chromosome abnormalities and season of birth. Hum Genet. 1984;65(3):221-231.
30. Vézina H, Houde L, Charbonneau H, et al. Season of birth and Alzheimer’s disease: a population-based study in Saguenay-Lac-St-Jean/Québec (IMAGE Project). Psychol Med. 1996;26(1):143-149.

Fulfillment within success: A physician’s dilemma
They say success without fulfillment is of little value in life. Whether this concept is actually driving the spate of depression and substance abuse currently experienced by youth and middle-aged adults in developed countries is rarely discussed and needs to be explored.
We have all reflected on the tragic ends of Anthony Bourdain, Kate Spade, and Robin Williams. Much has been said about the accolades they achieved and the heights they scaled, and just as much about their struggles with substance abuse over the years. Sensational portrayals by the media also encouraged youth to spend time dissecting the details of these high-profile deaths, lending popularity to the notion of suicide contagion. But somewhere in the myriad theories and conclusions, we still seem baffled by the questions of why these suicides occurred, and why no one had seen them coming.
As humans, we are designed to build. For many people, including physicians, the final product is a rewarding career built on years of hard work, or a flourishing family to look back on be proud of. Sometimes, however, these larger ideas barely intersect with our pictures of success.
As physicians and high achievers, we dream of goals and ambitions and set stringent deadlines for achieving them. Falling short sometimes finds us grappling with self-punishment and doubt. When one goal is achieved, another one is automatically created, or the goal post is pushed further. And the cycle continues.
Having said this, I will ask: What are you looking for? What is it that will give you a sense of purpose?
This is not a redundant question, nor is it an easy one. So are you really taking the time to think about it? Does any of this border on self-reflection and self-awareness for you? If it does, then developing that insight into yourself is perhaps a better way of serving your patients.
Peace and gratification often lie in the little things; not everything you do has to be acknowledged with an award. There is a sense of fulfillment that comes from developing others. The key is to realize that there is never a moment to start doing that—it is an ongoing journey. Therefore, give generously, of your time, of your skills, of your knowledge, but above all, of your kindness. Do it because in the end, you will have something to look back on and be proud of. Do it because maybe somewhere you will find meaning in it. And your success may not be bereft of fulfillment.

Dr. Virani is a PGY-3 resident, Department of Psychiatry, Maimonides Medical Center, Brooklyn, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Virani is a PGY-3 resident, Department of Psychiatry, Maimonides Medical Center, Brooklyn, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Virani is a PGY-3 resident, Department of Psychiatry, Maimonides Medical Center, Brooklyn, New York.
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article, or with manufacturers of competing products.
They say success without fulfillment is of little value in life. Whether this concept is actually driving the spate of depression and substance abuse currently experienced by youth and middle-aged adults in developed countries is rarely discussed and needs to be explored.
We have all reflected on the tragic ends of Anthony Bourdain, Kate Spade, and Robin Williams. Much has been said about the accolades they achieved and the heights they scaled, and just as much about their struggles with substance abuse over the years. Sensational portrayals by the media also encouraged youth to spend time dissecting the details of these high-profile deaths, lending popularity to the notion of suicide contagion. But somewhere in the myriad theories and conclusions, we still seem baffled by the questions of why these suicides occurred, and why no one had seen them coming.
As humans, we are designed to build. For many people, including physicians, the final product is a rewarding career built on years of hard work, or a flourishing family to look back on be proud of. Sometimes, however, these larger ideas barely intersect with our pictures of success.
As physicians and high achievers, we dream of goals and ambitions and set stringent deadlines for achieving them. Falling short sometimes finds us grappling with self-punishment and doubt. When one goal is achieved, another one is automatically created, or the goal post is pushed further. And the cycle continues.
Having said this, I will ask: What are you looking for? What is it that will give you a sense of purpose?
This is not a redundant question, nor is it an easy one. So are you really taking the time to think about it? Does any of this border on self-reflection and self-awareness for you? If it does, then developing that insight into yourself is perhaps a better way of serving your patients.
Peace and gratification often lie in the little things; not everything you do has to be acknowledged with an award. There is a sense of fulfillment that comes from developing others. The key is to realize that there is never a moment to start doing that—it is an ongoing journey. Therefore, give generously, of your time, of your skills, of your knowledge, but above all, of your kindness. Do it because in the end, you will have something to look back on and be proud of. Do it because maybe somewhere you will find meaning in it. And your success may not be bereft of fulfillment.
They say success without fulfillment is of little value in life. Whether this concept is actually driving the spate of depression and substance abuse currently experienced by youth and middle-aged adults in developed countries is rarely discussed and needs to be explored.
We have all reflected on the tragic ends of Anthony Bourdain, Kate Spade, and Robin Williams. Much has been said about the accolades they achieved and the heights they scaled, and just as much about their struggles with substance abuse over the years. Sensational portrayals by the media also encouraged youth to spend time dissecting the details of these high-profile deaths, lending popularity to the notion of suicide contagion. But somewhere in the myriad theories and conclusions, we still seem baffled by the questions of why these suicides occurred, and why no one had seen them coming.
As humans, we are designed to build. For many people, including physicians, the final product is a rewarding career built on years of hard work, or a flourishing family to look back on be proud of. Sometimes, however, these larger ideas barely intersect with our pictures of success.
As physicians and high achievers, we dream of goals and ambitions and set stringent deadlines for achieving them. Falling short sometimes finds us grappling with self-punishment and doubt. When one goal is achieved, another one is automatically created, or the goal post is pushed further. And the cycle continues.
Having said this, I will ask: What are you looking for? What is it that will give you a sense of purpose?
This is not a redundant question, nor is it an easy one. So are you really taking the time to think about it? Does any of this border on self-reflection and self-awareness for you? If it does, then developing that insight into yourself is perhaps a better way of serving your patients.
Peace and gratification often lie in the little things; not everything you do has to be acknowledged with an award. There is a sense of fulfillment that comes from developing others. The key is to realize that there is never a moment to start doing that—it is an ongoing journey. Therefore, give generously, of your time, of your skills, of your knowledge, but above all, of your kindness. Do it because in the end, you will have something to look back on and be proud of. Do it because maybe somewhere you will find meaning in it. And your success may not be bereft of fulfillment.
Can lifestyle modifications delay or prevent Alzheimer’s disease?
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4

Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75

On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.

Abuse of psychiatric medications: Not just stimulants and benzodiazepines
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15

Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40

Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73

These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.

With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15
Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40
Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73
These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.
With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
While some classes of medications used to treat psychiatric disorders, such as stimulants and benzodiazepines, are well-recognized as controlled substances and drugs of abuse, clinicians may be less familiar with the potential misuse/abuse of other psychiatric medications. This article reviews the evidence related to the misuse/abuse of anticholinergics, antidepressants, antipsychotics, and gabapentinoids.
The terms “misuse,” “abuse,” and “addiction” are used variably in the literature without standardized definitions. For this review, “misuse/abuse (M/A)” will be used to collectively describe self-administration that is recreational or otherwise inconsistent with legal or medical guidelines, unless a specific distinction is made. Whether or not the medications reviewed are truly “addictive” will be briefly discussed for each drug class, but the focus will be on clinically relevant aspects of M/A, including:
- excessive self-administration
- self-administration by non-oral routes
- co-administration with other drugs of abuse
- malingering of psychiatric symptoms to obtain prescriptions
- diversion for sale to third parties
- toxicity from overdose.
Anticholinergic medications
The first case describing the deliberate M/A of an anticholinergic medication for its euphoric effects was published in 1960.Further reportsfollowed in Europe before the M/A potential of prescription anticholinergic medications among psychiatric patients with an overdose syndrome characterized by atropinism and toxic psychosis was more widely recognized in the United States in the 1970s. Most reported cases of M/A to date have occurred among patients with psychiatric illness because anticholinergic medications, including trihexyphenidyl, benztropine, biperiden, procyclidine, and orphenadrine, were commonly prescribed for the management of first-generation and high dopamine D2-affinity antipsychotic-induced extrapyramidal symptoms (EPS). For example, one study of 234 consecutively hospitalized patients with schizophrenia noted an anticholinergic M/A incidence of 6.5%.1
However, anticholinergic M/A is not limited to individuals with psychotic disorders. A UK study of 154 admissions to an inpatient unit specializing in behavioral disturbances found a 12-month trihexyphenidyl M/A incidence of 17%; the most common diagnosis among abusers was antisocial personality disorder.2 Anticholinergic M/A has also been reported among patients with a primary diagnosis of substance use disorders (SUDs)3 as well as more indiscriminately in prison settings,4 with some inmates exchanging trihexyphenidyl as currency and using it recreationally by crushing it into powder and smoking it with tobacco.5 Others have noted that abusers sometimes take anticholinergics with alcohol in order to “potentiate” the effects of each substance.6,7 Pullen et al8 described individuals with and without psychiatric illness who stole anticholinergic medications, purchased them from other patients, or bought them “on the street.” Malingering EPS in order to obtain anticholinergic medications has also been well documented.9 Clearly, anticholinergic M/A can occur in psychiatric and non-psychiatric populations, both within and outside of clinical settings. Although anticholinergic M/A appears to be less frequent in the United States now that second-generation antipsychotics (SGAs) are more frequently prescribed, M/A remains common in some settings outside of the United States.7
Among the various anticholinergic medications prescribed for EPS, trihexyphenidyl has been reported to have the greatest M/A potential, which has been attributed to its potency,10 its stimulating effects (whereas benztropine is more sedating),11 and its former popularity among prescribers.8 Marken et al11 published a review of 110 reports of M/A occurring in patients receiving anticholinergic medications as part of psychiatric treatment in which 69% of cases involved taking trihexyphenidyl 15 to 60 mg at a time (recommended dosing is 6 to 10 mg/d in divided doses).Most of these patients were prescribed anticholinergic medications for diagnostically appropriate reasons—only 7% were described as “true abusers” with no medical indication. Anticholinergic M/A was typically driven by a desire for euphoric and psychedelic/hallucinogenic effects, although in some cases, anticholinergic M/A was attributed to self-medication of EPS and depressive symptoms. These findings illustrate the blurred distinction between recreational use and perceived subjective benefit, and match those of a subsequent study of 50 psychiatric patients who reported anticholinergic M/A not only to “get high,” but to “decrease depression,” “increase energy,” and decrease antipsychotic adverse effects.12 Once again, trihexyphenidyl was the most frequently misused anticholinergic in this sample.
Table 12,3,7,8,10-15 outlines the subjective effects sought and experienced by anticholinergic abusers as well as potential toxic effects; there is the potential for overlap. Several authors have also described physiologic dependence with long-term trihexyphenidyl use, including tolerance and a withdrawal/abstinence syndrome.7,16 In addition, there have been several reports of coma13 and death in the setting of intended suicide by overdose of anticholinergic medications.14,15
Although anticholinergic M/A in the United States now appears to be less common, clinicians should remain aware of the M/A potential of anticholinergic medications prescribed for EPS. Management of M/A involves:
- detection
- reducing anticholinergic exposure by managing EPS with alternative strategies, such as switching or reducing the dose of the antipsychotic medication
- gradual tapering of anticholinergic medications to minimize withdrawal.11
Continue to: Antidepressants
Antidepressants
Haddad17 published a review of 21 English-language case reports from 1966 to 1998 describing antidepressant use in which individuals met DSM-IV criteria for substance dependence to the medication. An additional 14 cases of antidepressant M/A were excluded based on insufficient details to support a diagnosis of dependence. The 21 reported cases involved:
- tranylcypromine (a monoamine oxidase inhibitor [MAOI])
- amitriptyline (a tricyclic antidepressant [TCA])
- fluoxetine (a selective serotonin reuptake inhibitor [SSRI])
- amineptine (a TCA previously available in France but removed from the market in 1999 in part due to its abuse potential)
- nomifensine (a norepinephrine/dopamine reuptake inhibitor previously available in the United Kingdom but removed in 1986 due to hemolytic anemia).
In 95% of cases, the antidepressants were prescribed for treatment of an affective disorder but were abused for stimulant effects or the perceived ability to lift mood, cause euphoria or a “high,” or to improve functioning. Two-thirds of cases involved patients with preexisting substance misuse. Placing the case reports in the context of the millions of patients prescribed antidepressants during this period, Haddad concluded the “incidence of [antidepressant] addiction [is] so low as to be clinically irrelevant.”17
Despite this conclusion, Haddad singled out amineptine and tranylcypromine as antidepressants with some evidence of true addictive potential.17,18 A more recent case series described 14 patients who met DSM-IV criteria for substance abuse of tertiary amine TCAs (which have strong anticholinergic activity) and concluded that “misuse of [TCAs] is more common than generally appreciated.”19 In keeping with that claim, a study of 54 outpatients taking unspecified antidepressants found that up to 15% met DSM-III-R criteria for substance dependence (for the antidepressant) in the past year, although that rate was much lower than the rate of benzodiazepine dependence (47%) in a comparative sample.20 Finally, a comprehensive review by Evans and Sullivan21 found anecdotal reports published before 2014 that detailed misuse, abuse, and dependence with MAOIs, TCAs, fluoxetine, venlafaxine, bupropion, tianeptine, and amineptine. Taken together, existing evidence indicates that select individuals—typically those with other SUD comorbidity—sometimes misuse antidepressants in a way that suggests addiction.
Still, while it is well known that abrupt cessation of antidepressants can result in a discontinuation syndrome characterized by flu-like symptoms, nausea, and dizziness,22 physiologic withdrawal effects must be distinguished from historical definitions of substance “abuse” and the broader concept of psychological “addiction” or drug dependence18,23 now incorporated into the DSM-5 definition of SUDs.24 Indeed, although withdrawal symptoms were reported by more than half of those who took antidepressants and responded to a recent online survey,25 evidence to support the existence of significant antidepressant tolerance, craving, or compulsive use is lacking.17,18 Antidepressants as a class do not appear to be significantly rewarding or reinforcing and, on the contrary, discontinuation by patients is common in clinical practice.26 The popular claim that some individuals taking antidepressants “can’t quit”27 must also be disentangled from loss of therapeutic effects upon cessation.
Bupropion. A more convincing argument for antidepressant addiction can be made for bupropion, a weak norepinephrine and dopamine reuptake inhibitor with an otherwise unclear mechanism of action.28 In 2002, the first report of recreational bupropion M/A described a 13-year-old girl who took 2,400 mg orally (recommended maximum dose is 450 mg/d in divided doses) after being told it would give her “a better high than amphetamine.”29 This was followed in the same year by the first report of recreational M/A of bupropion via nasal insufflation (snorting), resulting in a seizure,30 and in 2013 by the first published case of M/A by IV self-administration.31
Continue to: The M/A potential of bupropion...
The M/A potential of bupropion, most commonly via intranasal administration, is now broadly recognized based on several case reports describing desired effects that include a euphoric high and a stimulating “buzz” similar to that of cocaine or methamphetamine but less intense.29-36 Among recreational users, bupropion tablets are referred to as “welbys,” “wellies,” “dubs,” or “barnies.”37 Media coverage of a 2013 outbreak of bupropion M/A in Toronto detailed administration by snorting, smoking, and injection, and described bupropion as “poor man’s cocaine.”38 Between 2003 and 2016, 2,232 cases of bupropion misuse/abuse/dependence adverse drug reactions were reported to the European Monitoring Agency.37 A review of intentional bupropion M/A reported to US Poison Control Centers between 2000 to 2013 found 975 such cases, with the yearly number tripling between 2000 and 2012.39 In this sample, nearly half (45%) of the users were age 13 to 19, and 76% of cases involved oral ingestion. In addition to bupropion M/A among younger people, individuals who misuse bupropion often include those with existing SUDs but limited access to illicit stimulants and those trying to evade detection by urine toxicology screening.33 For example, widespread use and diversion has been well documented within correctional settings, and as a result, many facilities have removed bupropion from their formularies.21,28,33,34,40
Beyond desired effects, the most common adverse events associated with bupropion M/A are listed in Table 2,28,30,32-34,36,39 along with their incidence based on cases brought to the attention of US Poison Control Centers.39 With relatively little evidence of a significant bupropion withdrawal syndrome,37 the argument in favor of modeling bupropion as a truly addictive drug is limited to anecdotal reports of cravings and compulsive self-administration35 and pro-dopaminergic activity (reuptake inhibition) that might provide a mechanism for potential rewarding and reinforcing effects.40 While early preclinical studies of bupropion failed to provide evidence of amphetamine-like abuse potential,41,42 non-oral administration in amounts well beyond therapeutic dosing could account for euphoric effects and a greater risk of psychological dependence and addiction.21,28,40
Bupropion also has an FDA indication as an aid to smoking cessation treatment, and the medication demonstrated early promise in the pharmacologic treatment of psychostimulant use disorders, with reported improvements in cravings and other SUD outcomes.43-45 However, subsequent randomized controlled trials (RCTs) failed to demonstrate a clear therapeutic role for bupropion in the treatment of cocaine46,47 and methamphetamine use disorders (although some secondary analyses suggest possible therapeutic effects among non-daily stimulant users who are able to maintain good adherence with bupropion).48-51 Given these overall discouraging results, the additive seizure risk of bupropion use with concomitant psychostimulant use, and the potential for M/A and diversion of bupropion (particularly among those with existing SUDs), the use of bupropion for the off-label treatment of stimulant use disorders is not advised.
Antipsychotics
As dopamine antagonists, antipsychotics are typically considered to have low potential for rewarding or reinforcing effects. Indeed, misuse of antipsychotics was a rarity in the first-generation era, with only a few published reports of haloperidol M/A within a small cluster of naïve young people who developed acute EPS,52 and a report of diversion in a prison with the “sadistic” intent of inflicting dystonic reactions on others.53 A more recent report described 2additional cases of M/A involving haloperidol and trifluoperazine.54 Some authors have described occasional drug-seeking behavior for low-potency D2 blockers such as chlorpromazine, presumably based on their M/A as anticholinergic medications.55
The potential for antipsychotic M/A has gained wider recognition since the advent of the SGAs. Three cases of prescription olanzapine M/A have been published to date. One involved a man who malingered manic symptoms to obtain olanzapine, taking ≥40 mg at a time (beyond his prescribed dose of 20 mg twice daily) to get a “buzz,” and combining it with alcohol and benzodiazepines for additive effects or to “come down” from cocaine.56 This patient noted that olanzapine was “a popular drug at parties” and was bought, sold, or traded among users, and occasionally administered intravenously. Two other cases described women who self-administered olanzapine, 40 to 50 mg/d, for euphoric and anxiolytic effects.57,58 James et al59 detailed a sample of 28 adults who reported “non-medical use” of olanzapine for anxiolytic effects, as a sleep aid, or to “escape from worries.”
Continue to: Quetiapine
Quetiapine. In contrast to some reports of olanzapine M/A in which the line between M/A and “self-medication” was blurred, quetiapine has become a more convincing example of clear recreational antipsychotic M/A. Since the first report of oral and intranasal quetiapine M/A in the Los Angeles County Jail published in 2004,55 subsequent cases have detailed other novel methods of recreational self-administration60-68 (Table 355,60-68), and additional reports have been published in non-English language journals.69,70 Collectively, these case reports have detailed that quetiapine is:
- misused for primary subjective effects as well as to mitigate the unpleasant effects of other drugs60,67
- referred to as “quell,”“Q,” “Susie-Q,” “squirrel,” and “baby heroin”55,71,72
- often obtained by malingering psychiatric symptoms55,61,63,65
- diverted/sold with “street value” both within and outside of psychiatric facilities and correctional settings.55,60-62,67,68,73
These anecdotal accounts of quetiapine M/A have since been corroborated on a larger scale based on several retrospective studies. Although early reports of quetiapine M/A occurring in correctional settings have resulted in formulary removal,71,74 quetiapine M/A is by no means limited to forensic populations and is especially common among those with comorbid SUDs. A survey of 74 patients enrolled in a Canadian methadone program reported that nearly 60% had misused quetiapine at some point.75 Among an Australian sample of 868 individuals with active IV drug abuse, 31% reported having misused quetiapine.76 Finally, within a small sample of patients with SUDs admitted to a detoxification unit in New York City, 17% reported M/A of SGAs.77 In this study, SGAs were often taken in conjunction with other drugs of abuse in order to “recover” from or “enhance” the effects of other drugs or to “experiment.” Quetiapine was by far the most frequently abused SGA, reported in 96% of the sample; the most frequently reported SGA/drug combinations were quetiapine/alcohol/opioids, quetiapine/cocaine, and quetiapine/opioids.
Looking more broadly at poison center data, reports to the US National Poison Data System (NPDS) from 2005 to 2011 included 3,116 cases of quetiapine abuse (37.5%, defined as intentional recreational use in order to obtain a “high”) or misuse (62.5%, defined as improper use or dosing for non-recreational purposes).78 A more recent analysis of NPDS reports from 2003 to 2013 found 2,118 cases of quetiapine abuse, representing 61% of all cases of reported SGA abuse.79 An analysis of the European Medicines Agency Adverse Drug Database yielded 18,112 reports of quetiapine misuse, abuse, dependence, and withdrawal for quetiapine (from 2005 to 2016) compared with 4,178 for olanzapine (from 2004 to 2016).80 These reports identified 368 fatalities associated with quetiapine.
The rate of quetiapine M/A appears to be increasing sharply. Reports of quetiapine M/A to poison centers in Australia increased nearly 7-fold from 2006 to 2016.81 Based on reports to the Drug Abuse Warning System, US emergency department visits for M/A of quetiapine increased from 19,195 in 2005 to 32,024 in 2011 (an average of 27,114 visits/year), with 75% of cases involving quetiapine taken in combination with other prescription drugs, alcohol, or illicit drugs.82 Consistent with poison center data, M/A was reported for other antipsychotics, but none nearly as frequently as for quetiapine.
With increasingly frequent quetiapine M/A, clinicians should be vigilant in monitoring for medical morbidity related to quetiapine and cumulative toxicity with other drugs. The most frequent adverse events associated with quetiapine M/A reported to US Poison Control Centers are presented in Table 4.78,79
Continue to: Unlike bupropion...
Unlike bupropion, quetiapine’s dopamine antagonism makes it unlikely to be a truly addictive drug, although this mechanism of action could mediate an increase in concurrent psychostimulant use.83 A few case reports have described a quetiapine discontinuation syndrome similar to that of antidepressants,60,65,84-88 but withdrawal symptoms suggestive of physiologic dependence may be mediated by non-dopaminergic effects through histamine and serotonin receptors.84,89 Evidence for quetiapine misuse being associated with craving and compulsive use is lacking, and true quetiapine addiction is probably rare.
Similar to bupropion, preliminary findings have suggested promise for quetiapine as a putative therapy for other SUDs.90-93 However, subsequent RCTs have failed to demonstrate a therapeutic effect for alcohol and cocaine use disorders.94-96 Given these negative results and the clear M/A potential of quetiapine, off-label use of quetiapine for the treatment of SUDs and psychiatric symptoms among those with SUDs must be considered judiciously, with an eye towards possible diversion and avoiding the substitution of one drug of abuse for another.
Gabapentinoids
In 1997, the first published case report of gabapentin M/A described a woman who self-administered her husband’s gabapentin to reduce cravings for and withdrawal from cocaine.97 The authors highlighted the possible therapeutic benefit of gabapentin in this regard rather than raising concerns about diversion and M/A. By 2004, however, reports of recreational gabapentin M/A emerged among inmates incarcerated within Florida correctional facilities who self-administered intranasal gabapentin to achieve a “high” that was “reminiscent of prior effects from intranasal ingestion of cocaine powder.”98 In 2007, a single case of gabapentin misuse up to 7,200 mg/d (recommended dosing is ≤3,600 mg/d) was reported, with documentation of both tolerance and withdrawal symptoms.99 As of 2017, a total of 36 cases of gabapentin M/A and 19 cases of pregabalin M/A have been published.100
In the past decade, anecdotal reports have given way to larger-scale epidemiologic data painting a clear picture of the now-widespread M/A of gabapentin and other gabapentinoids. For example, a study of online descriptions of gabapentin and pregabalin M/A from 2008 to 2010 documented:
- oral and IM use (gabapentin)
- IV and rectal (“plugging”) use (pregabalin)
- “parachuting” (emptying the contents of capsules for a larger dose) (pregabalin)
- euphoric, entactogenic, stimulant, calming/anxiolytic, and dissociative subjective effects (gabapentin/pregabalin)
- rapid development of tolerance to euphoric effects leading to self-administration of increasing doses (gabapentin/pregabalin)
- frequent co-administration with other drugs of abuse, including alcohol, benzodiazepines, cannabis, stimulants, opiates, hallucinogens, gamma-hydroxybutyrate, mephedrone, and Salvia divinorum (gabapentin/pregabalin)101
Several systematic reviews of both anecdotal reports and epidemiologic studies published in the past few years provide additional evidence of the above, such as:
- excessive dosing with self-administration
- intranasal and inhaled routes of administration
- diversion and “street value”
- greater M/A potential of pregabalin than gabapentin
- the presence of gabapentinoids in postmortem toxicology analyses, suggesting a role in overdose fatalities when combined with other drugs.100,102,103
Continue to: The European Medicine Agency's EudraVigilance database...
The European Medicine Agency’s EudraVigilance database included 4,301 reports of gabapentin misuse, abuse, or dependence, and 7,639 such reports for pregabalin, from 2006 to 2015 (rising sharply after 2012), with 86 gabapentin-related and 27 pregabalin-related fatalities.104 Data from the Drug Diversion Program of the Researched Abuse, Diversion, and Addiction-Related Surveillance System from 2002 to 2015 have likewise revealed that gabapentin diversion increased significantly in 2013.105
While the prevalence of gabapentinoid M/A is not known, rates appear to be significantly lower than for traditional drugs of abuse such as cannabis, cocaine, 3,4-methylenedioxymethamphetamine (MDMA), and opioids.106,107 However, gabapentin and pregabalin M/A appears to be increasingly common among individuals with SUDs and in particular among those with opioid use disorders (OUDs). For example, a 2015 report indicated that 15% of an adult cohort in Appalachian Kentucky with nonmedical use of diverted prescription opioids reported gabapentin M/A, an increase of nearly 3,000% since 2008.108 Based on data from a US insurance enrollment and claims database, researchers found that the rate of gabapentin overuse among those also overusing opioids was 12% compared with only 2% for those using gabapentin alone.109 It has also been reported that gabapentin is sometimes used as a “cutting agent” for heroin.110
Those who use gabapentinoids together with opioids report that gabapentin and pregabalin potentiate the euphoric effects of methadone111 and endorse specific beliefs that pregabalin increases both the desired effects of heroin as well as negative effects such as “blackouts,” loss of control, and risk of overdose.112 Indeed, sustained M/A of gabapentin and opioids together has been found to increase emergency department utilization, drug-related hospitalization, and respiratory depression.113 Based on a case-control study of opioid users in Canada, co-prescription of gabapentin and opioids was associated with a 50% increase in death from opioid-related causes compared with prescription of opioids alone.114
Case reports documenting tolerance, withdrawal, craving, and loss of control suggest a true addictive potential for gabapentinoids, but Bonnet and Sherbaum100 concluded that while there is robust evidence of abusers “liking” gabapentin and pregabalin (eg, reward), evidence of “wanting” them (eg, psychological dependence) in the absence of other SUDs has been limited to only a few anecdotal reports with pregabalin. Accordingly, the risk of true addiction to gabapentinoids by those without preexisting SUDs appears to be low. Nonetheless, the M/A potential of both gabapentin and pregabalin is clear and in the context of a nationwide opioid epidemic, the increased morbidity/mortality risk related to combined use of gabapentinoids and opioids is both striking and concerning. Consequently, the state of Kentucky recently recognized the M/A potential of gabapentin by designating it a Schedule V controlled substance (pregabalin is already a Schedule V drug according to the US Drug Enforcement Agency),103,113 and several other states now mandate the reporting of gabapentin prescriptions to prescription drug monitoring programs.115
Following a similar pattern to antidepressants and antipsychotics, a potential role for gabapentin in the treatment of cocaine use disorders was supported in preliminary studies,116-118 but not in subsequent RCTs.119-121 However, there is evidence from RCTs to support the use of gabapentin and pregabalin in the treatment of alcohol use disorders.122-124 Gabapentin was also found to significantly reduce cannabis use and withdrawal symptoms in patients compared with placebo in an RCT of individuals with cannabis use disorders.125 The perceived safety of gabapentinoids by clinicians, their subjective desirability by patients with SUDs, and efficacy data supporting a therapeutic role in SUDs must be balanced with recognition that approximately 80% of gabapentin prescriptions are written for off-label indications for which there is little supporting evidence,109 such as low back pain.126 Clinicians considering prescribing gabapentinoids to manage psychiatric symptoms, such as anxiety and insomnia, should carefully consider the risk of M/A and other potential morbidities, especially in the setting of SUDs and OUD in particular.
Continue to: Problematic, even if not addictive
Problematic, even if not addictive
It is sometimes claimed that “addiction” to psychiatric medications is not limited to stimulants and benzodiazepines.27,127 Although anticholinergics, antidepressants, antipsychotics, and gabapentinoids can be drugs of abuse, with some users reporting physiologic withdrawal upon discontinuation, there is only limited evidence that the M/A of these psychiatric medications is associated with the characteristic features of a more complete definition of “addiction,” which may include:
- inability to consistently abstain
- impairment in behavioral control
- diminished recognition of significant problems associated with use
- a dysfunctional emotional response to chronic use.128
Nonetheless, the literature documenting anticholinergic, antidepressant, antipsychotic, and gabapentinoid M/A includes several common features, including:
- initial reports among those with limited access to illicit drugs (eg, young people and incarcerated individuals) and subsequent spread to a wider population with more unconventional routes of administration
- use for recreational purposes and other subjective pseudo-therapeutic effects, often in combination with alcohol and illicit drugs
- greater M/A potential of certain medications within each of these drug classes (eg, trihexyphenidyl, bupropion, quetiapine)
- malingering psychiatric symptoms in order to obtain medications from prescribers and diversion for black market sale
- observations that medications might constitute therapy for SUDs that were not supported in subsequent RCTs (with the exception of gabapentin for alcohol and cannabis use disorders)
- increasing evidence of toxicity related to M/A, which suggests that prescription by clinicians has limited benefit and high risk for patients with SUDs.
Bottom Line
Some psychiatric medications are taken as drugs of abuse. Clinicians should be particularly aware of the misuse/abuse potential of anticholinergics, antidepressants, antipsychotics, and gabapentinoids, and use them cautiously, if at all, when treating patients with existing substance use disorders.
Related Resources
- Substance Abuse and Mental Health Services Administration. Prescription drug misuse and abuse. https://www.samhsa.gov/topics/prescription-drug-misuse-abuse.
- Substance Abuse and Mental Health Services Administration. Types of commonly misused or abused drugs. https://www.samhsa.gov/prescription-drug-misuse-abuse/types.
- National Institute on Drug Abuse. Misuse of prescription drugs. https://www.drugabuse.gov/publications/research-reports/misuse-prescription-drugs/summary.
- National Institute on Drug Abuse. New clinician screening tool available for substance use. https://www.drugabuse.gov/news-events/news-releases/2018/06/newclinician-screening-tool-available-substance-use.
Drug Brand Names
Amitriptyline • Elavil, Endep
Benztropine • Cogentin
Biperiden • Akineton
Bupropion • Wellbutrin, Zyban
Chlorpromazine • Thorzine
Fluoxetine • Prozac
Haloperidol • Haldol
Olanzapine • Zyprexa
Orphenadrine • Disipal, Norflex
Pregabalin • Lyrica, Lyrica CR
Procyclidine • Kemadrin
Quetiapine • Seroquel
Tianeptine • Coaxil, Stablon
Tranylcypromine • Parnate
Trifluoperazine • Stelazine
Trihexyphenidyl • Artane, Tremin
Venlafaxine • Effexor
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.
1. Zemishlany Z, Aizenberg D, Weiner Z, et al. Trihexyphenidyl (Artane) abuse in schizophrenic patients. Int Clin Psychopharmacol. 1996;11(3):199-202.
2. Crawshaw JA, Mullen PE. A study of benzhexol abuse. Brit J Psychiatry. 1984;145:300-303.
3. Woody GE, O’Brien CP. Anticholinergic toxic psychosis in drug abusers treated with benztropine. Comp Psychiatry. 1974;15(5):439-442.
4. Lowry TP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
5. Rouchell AM, Dixon SP. Trihexyphenidyl abuse. Am J Psychiatry. 1977;134(11):1315.
6. Kaminer Y, Munitz H, Wijsenbeek H. Trihexyphenidyl (Artane) abuse: euphoriant and anxiolytic. Brit J Psychiatry. 1982;140(5):473-474.
7. Nappo SA, de Oliviera LG, Sanchez Zv, et al. Trihexyphenidyl (Artane): a Brazilian study of its abuse. Subst Use Misuse. 2005;40(4):473-482.
8. Pullen GP, Best NR, Macguire J. Anticholinergic drug abuse: a common problem? Brit Med J (Clin Res Ed). 1984;289(6445):612-613.
9. Rubinstein JS. Abuse of antiparkinsonian drugs: feigning of extrapyramidal symptoms to obtain trihexyphenidyl. JAMA. 1978;239(22):2365-2366.
10. Mohan D, Mohandas E, Dube S. Trihexyphenidyl abuse. Brit J Addiction. 1981:76(2);195-197.
11. Marken PA, Stoner SC, Bunker MT. Anticholinergic drug abuse and misuse. CNS Drugs. 1996;5(3):190-199.
12. Buhrich N, Weller A, Kevans P. Misuse of anticholinergic drugs by people with serious mental illness. Psychiatric Serv. 2000;51(7):928-929.
13. Goldstein MR, Kasper R. Hyperpyrexia and coma due to overdose of benztropine. South Med J. 1968;61(9):984.
14. Petkovi
15. McIntyre IM, Mallett P, Burton CG, et al. Acute benztropine intoxication and fatality. J Forensic Sci. 2014;59(6):1675-1678.
16. Dilsaver SC. Antimuscarinic agents as substances of abuse: A review. J Clin Psychopharmacol. 1988:8(1):14-22.
17. Haddad P. Do antidepressants have any potential to cause addiction? J Psychopharmacol. 1999;13(3):300-307.
18. Haddad PM. Do antidepressants cause dependence? Epidemiol Psichiatr Soc. 2005;14(2):58-62.
19. Shenouda R, Desan PH. Abuse of tricyclic antidepressant drugs: a case series. J Clin Psychopharmacol. 2013;33(3):440-442.
20. van Broekhoven F, Kan CC, Zitman FG. Dependence potential of antidepressants compared to benzodiazepines. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):939-943.
21. Evans EA, Sullivan MA. Abuse and misuse of antidepressants. Subst Abuse Rehabil. 2014;5:107-120.
22. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449-456.
23. Lichtigfeld FJ, Gillman MA. Antidepressants are not drugs of abuse or dependence. Postgrad Med J. 1998;74(875):529-532.
24. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
25. Read J, Cartwright C, Gibson K. Adverse emotional and interpersonal effects reported by 1829 New Zealanders while taking antidepressants. Psychiatry Res. 2014;216(1):67-73.
26. Haddad P, Anderson I. Antidepressants aren’t addictive: clinicians have depended on them for years. J Psychopharmacol. 1999;13(3):291-292.
27. Carey B, Gebeloff R. Many people taking antidepressants discover they cannot quit. New York Times. https://www.nytimes.com/2018/04/07/health/antidepressants-withdrawal-prozac-cymbalta.html. Published April 7, 2018. Accessed December 11, 2018.
28. Kim D, Steinhart B. Seizures induced by recreational abuse of bupropion tablets via nasal insufflation. CJEM. 2010;12(2):158-161.
29. McCormick J. Recreational bupropion in a teenager. Br J Clin Pharmacol. 2002;53(2):214.
30. Welsh C, Doyon S. Seizure induced by insufflation of bupropion. N Engl J Med. 2002; 347(2):951.
31. Baribeau D, Araki KF. Intravenous bupropion: A previously undocumented method of abuse of a commonly prescribed antidepressant agent. J Addict Med. 2013;7(3):216-217.
32. Hill SH, Sikand H, Lee J. A case report of seizure induced by bupropion nasal insufflation. Prim Care Companion J Clin Psych. 2007;9(1):67-69.
33. Yoon G, Westermeyer J. Intranasal bupropion abuse. Am J Addict. 2013;22(2):180.
34. Reeves RR, Ladner ME. Additional evidence of the abuse potential of bupropion. J Clin Psychopharmacol. 2013;33(4):584-585.
35. Oppek K, Koller G, Zwergal A, et al. Intravenous administration and abuse of bupropion: a case report and a review of the literature. J Addict Med. 2014;8(4):290-293.
36. Strike M, Hatcher S. Bupropion injection resulting in tissue necrosis and psychosis: previously undocumented complications of intravenous bupropion use disorder. J Addict Med. 2015;9(3):246-250.
37. Schifano F, Chiappini S. Is there a potential of misuse for venlafaxine and bupropion? Front Pharmacol. 2018;9:239.
38. Tryon J, Logan N. Antidepressant Wellbutrin becomes ‘poor man’s cocaine’ on Toronto streets. Global News. https://globalnews.ca/news/846576/antidepressant-wellbutrin-becomes-poor-mans-cocaine-on-toronto-streets/. Published September 18, 2013. Accessed December 11, 2018.
39. Stassinos GL, Klein-Schwartz W. Bupropion “abuse” reported to US Poison Centers. J Addict Med. 2016;10(5):357-362.
40. Hilliard WT, Barloon L, Farley P, et al. Bupropion diversion and misuse in the correctional facility. J Correct Health Care. 2013;19(3):211-217.
41. Griffith JD, Carranza J, Griffith C, et al. Bupropion clinical assay for amphetamine-like abuse potential. J Clin Psychiatry.1983;44(5 Pt 2):206-208.
42. Miller L, Griffith J. A comparison of bupropion, dextroamphetamine, and placebo in mixed-substance abusers. Psychopharmacol (Berl). 1983;80(3):199-205.
43. Berigan TR, Russell ML. Treatment of methamphetamine cravings with bupropion: A case report. Prim Care Companion J Clin Psychiatry. 2001;3(6):267-268.
44. Tardieu T, Poirier Y, Micallef J, et al. Amphetamine-like stimulant cessation in an abusing patient treated with bupropion. Acta Psychiatr Scand. 2004;109(1):75-78.
45. Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced cravings. Neuropsychopharmacology. 2006;31(7):1537-1544.
46. Margolin A, Kosten TR, Avants SK, et al. A multicenter trial for cocaine dependence in methadone-maintained patients. Drug Alcohol Depend. 1995;40(2):125-131.
47. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Bupropion hydrochloride versus placebo, in combination with cognitive behavioral therapy, for the treatment of cocaine abuse/dependence. J Addict Dis. 2008;27(1):13-23.
48. Anderson AL, Li S, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non-daily users: a randomized, double-blind placebo-controlled trial. Drug Alcohol Depend. 2015;150:170-174.
49. Shoptaw S, Heinzerling KG, Rotheram-Fuller E, et al. Randomized, placebo-controlled trial of bupropion for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2008;96(3):222-232.
50. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
51. Heinzerling KG, Swanson A, Hall TM, et al. Randomized, placebo-controlled trial of bupropion in methamphetamine-dependent participants with less than daily methamphetamine use. Addiction. 2014;109(11):1878-1886.
52. Doenecke AL, Heuerman RC. Treatment of haloperidol abuse with diphenhydramine. Am J Psychiatry. 1980;137(4):487-488.
53. Weddington WW, Leventhal BL. Sadistic abuse of haloperidol. Am J Psychiatry. 1982;139:132-133.
54. Basu D, Marudkar M, Khurana H. Abuse of neuroleptic drugs by psychiatric patients. Indian J Med Sci. 2000;54(2):59-62.
55. Pierre JM, Shnayder I, Wirshing DA, et al. Intranasal quetiapine abuse. Am J Psychiatry 2004;161(9):1718.
56. Reeves RR. Abuse of olanzapine by substance abusers. J Psychoactive Drugs. 2007;39(3):297-299.
57. Kumsar NA, Erol A. Olanzapine abuse. Subst Abus. 2013;34(1):73-74.
58. Lai C. Olanzapine abuse was relieved after switching to aripiprazole in a patient with psychotic depression. Prog Neuropsychpharmacol Biol Psychiatry. 2010;34(7):1363-1364.
59. James PD, Fida AS, Konovalov P, et al. Non-medical use of olanzapine by people on methadone treatment. BJPsych Bull. 2016;40(6):314-317.
60. Reeves RR, Brister JC. Additional evidence of the abuse potential of quetiapine. South Med J. 2007;100(8):834-836.
61. Murphy D, Bailey K, Stone M, et al. Addictive potential of quetiapine. Am J Psychiatry. 2008;165(7):918.
62. Paparrigopoulos T, Karaiskos D, Liappas J. Quetiapine: another drug with potential for misuse? J Clin Psychiatry. 2008;69(1):162-163.
63. Reeves RR, Burke RS. Abuse of the combination of gabapentin and quetiapine. Prim Care Companion CNS Disord. 2014;16(5): doi: 10.4088/PCC.14l01660.
64. Morin AK. Possible intranasal quetiapine misuse. Am J Health Syst Pharm. 2007;64(7):723-725.
65. Caniato RN, Gundabawady A, Baune BT, et al. Malingered psychotic symptoms and quetiapine abuse in a forensic setting. J Forens Psychiatr Psychol. 2009;20(6):928-935.
66. Hussain MZ, Waheed W, Hussain S. Intravenous quetiapine abuse. Am J Psychiatry. 2005; 162(9):1755-1756.
67. Waters BM, Joshi KG. Intravenous quetiapine-cocaine use (“Q-ball”). Am J Psychiatry. 2007;164(1):173-174.
68. Haridas A, Kushon D, Gurmu S, et al. Smoking quetiapine: a “Maq ball?” Prim Psychiatry. 2010;17:38-39.
69. Cubala WJ, Springer J. Quetiapine abuse and dependence in psychiatric patients: a systematic review of 25 case reports in the literature. J Subs Use. 2014;19(5):388-393.
70. Piróg-Balcerzak A, Habrat B, Mierzejewski P. Misuse and abuse of quetiapine [in Polish]. Psychiatr Pol. 2015;49(1):81-93.
71. Pinta ER, Taylor RE. Quetiapine addiction? Am J Psychiatry. 2007;164(1):174.
72. Tamburello AC, Lieberman JA, Baum RM, et al. Successful removal of quetiapine from a correctional formulary. J Amer Acad Psychiatr Law. 2012;40(4):502-508.
73. Tarasoff G, Osti K. Black-market value of antipsychotics, antidepressants, and hypnotics in Las Vegas, Nevada. Am J Psychiatry. 2007;164(2):350.
74. Reccoppa L. Less abuse potential with XR formulation of quetiapine. Am J Addiction. 2010;20(2):178.
75. McLarnon ME, Fulton HG, MacIsaac C, et al. Characteristics of quetiapine misuse among clients of a community-based methadone maintenance program. J Clin Psychopharmacol. 2012;32(5):721-723.
76. Reddel SE, Bruno R, Burns L, et al. Prevalence and associations of quetiapine fumarate misuse among an Australian national city sample of people who regularly inject drugs. Addiction. 2013;109(2):295-302.
77. Malekshahi T, Tioleco N, Ahmed N, et al. Misuse of atypical antipsychotics in conjunction with alcohol and other drugs of abuse. J Subs Abuse Treat. 2015;48(1):8-12.
78. Klein-Schwartz W, Schwartz EK, Anderson BD. Evaluation of quetiapine abuse and misuse reported to poison centers. J Addict Med. 2014;8(3):195-198.
79. Klein L, Bangh S, Cole JB. Intentional recreational abuse of quetiapine compared to other second-generation antipsychotics. West J Emerg Med. 2017;18(2):243-250.
80. Chiappini S, Schifano F. Is there a potential of misuse for quetiapine?: Literature review and analysis of the European Medicines Agency/European Medicines Agency Adverse Drug Reactions’ Database. J Clin Psychopharmacol. 2018;38(1):72-79.
81. Lee J, Pilgrim J, Gerostamoulos D, et al. Increasing rates of quetiapine overdose, misuse, and mortality in Victoria, Australia. Drug Alcohol Depend. 2018;187:95-99.
82. Mattson ME, Albright VA, Yoon J, et al. Emergency department visits involving misuse and abuse of the antipsychotic quetiapine: Results from the
83. Brutcher RE, Nader SH, Nader MA. Evaluation of the reinforcing effect of quetiapine, alone and in combination with cocaine, in rhesus monkeys. J Pharmacol Exp Ther. 2016;356(2):244-250.
84. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry. 2005;162(5):1020.
85. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry. 2000;61(8):602-603.
86. Kohen I, Kremen N. A case report of quetiapine withdrawal syndrome in a geriatric patient. World J Biol Psychiatry. 2009;10(4 pt 3):985-986.
87. Yargic I, Caferov C. Quetiapine dependence and withdrawal: a case report. Subst Abus. 2011;32(3):168-169.
88. Koch HJ. Severe quetiapine withdrawal syndrome with nausea and vomiting in a 65-year-old patient with psychotic depression. Therapie. 2015;70(6):537-538.
89. Fischer BA, Boggs DL. The role of antihistaminic effects in the misuse of quetiapine: a case report and review of the literature. Neurosci Biobehav Rev. 2010;34(4):555-558.
90. Longoria J, Brown ES, Perantie DC, et al. Quetiapine for alcohol use and craving in bipolar disorder. J Clin Psychopharmacol. 2004;24(1):101-102.
91. Monnelly EP, Ciraulo DA, Knapp C, et al. Quetiapine for treatment of alcohol dependence. J Clin Psychopharmacol. 2004;24(5):532-535.
92. Kennedy A, Wood AE, Saxon AJ, et al. Quetiapine for the treatment of cocaine dependence: an open-label trial. J Clin Psychopharmacol. 2008;28(2):221-224.
93. Mariani JJ, Pavlicova M, Mamczur A, et al. Open-label pilot study of quetiapine treatment for cannabis dependence. Am J Drug Alcohol Abuse. 2014;40(4):280-284.
94. Guardia J, Roncero C, Galan J, et al. A double-blind, placebo-controlled, randomized pilot study comparing quetiapine with placebo, associated to naltrexone, in the treatment of alcohol-dependent patients. Addict Behav. 2011;36(3):265-269.
95. Litten RZ, Fertig JB, Falk DE, et al; NCIG 001 Study Group. A double-blind, placebo-controlled trial to assess the efficacy of quetiapine fumarate XR in very heavy-drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012;36(3):406-416.
96. Tapp A, Wood AE, Kennedy A, et al. Quetiapine for the treatment of cocaine use disorder. Drug Alcohol Depend. 2015;149:18-24.
97. Markowitz JS, Finkenbine R, Myrick H, et al. Gabapentin abuse in a cocaine user: Implications for treatment. J Clin Psychopharmacol. 1997;17(5):423-424.
98. Reccoppa L, Malcolm R, Ware M. Gabapentin abuse in inmates with prior history of cocaine dependence. Am J Addict. 2004;13(3):321-323.
99. Victorri-Vigneau C, Guelais M, Jolliet P. Abuse, dependency and withdrawal with gabapentin: a first case report. Pharmacopsychiatry. 2007;40(1):43-44.
100. Bonnet U, Sherbaum N. How addictive are gabapentin and pregabalin? A systematic review. Eur Neuropsychopharmacol. 2017;27(12):1185-1215.
101. Schifano F, D’Offizi S, Piccione M, et al. Is there a recreational misuse potential for pregabalin? Analysis of anecdotal online reports in comparison with related gabapentin and clonazepam data. Psychother Psychosom. 2011;80(2):118-122.
102. Evoy KE, Morrison MD, Saklad SR. Abuse and misuse of pregabalin and gabapentin. Drugs. 2017;77(4):403-426.
103. Smith RV, Havens JR, Walsh SL. Gabapentin misuse, abuse and diversion: a systematic review. Addiction. 2016;111(7):1160-1174.
104. Chiappini S, Shifano F. A decade of gabapentinoid misuse: an analysis of the European Medicines Agency’s ‘suspected adverse drug reactions’ database. CNS Drugs. 2016;30(7):647-654.
105. Buttram ME, Kurtz SP, Dart R, et al. Law enforcement-derived data on gabapentin diversion and misuse, 2002-2015: diversion rates and qualitative research findings. Pharmacoepidemiol Drug Saf. 2017;26(9):1083-1086.
106. Kapil V, Green JL, Le Lait M, et al. Misuse of the y-aminobutyric acid analogues baclofen, gabapentin and pregabalin in the UK. Br J Clin Pharmacol. 2013;78(1):190-191.
107. Peckham AM, Fairman KA, Sclar DA. Prevalence of gabapentin abuse: comparison with agents with known abuse potential in a commercially insured US population. Clin Drug Invest. 2017;37(8):763-773.
108. Smith RV, Lofwall MR, Havens JR. Abuse and diversion of gabapentin among nonmedical prescription opioid users in Appalachian Kentucky. Am J Psychiatry. 2015;172(5):487-488.
109. Peckham AM, Evoy KE, Covvey JR, et al. Predictors of gabapentin overuse with or without concomitant opioids in a commercially insured U.S. population. Pharmacotherapy. 2018;38(4):436-443.
110. Smith BH, Higgins C, Baldacchino A, et al. Substance misuse of gabapentin. Br J Gen Pract. 2012;62(601):401-407.
111. Baird CRW, Fox P, Colvin LA. Gabapentinoid abuse in order to potentiate the effect of methadone: a survey among substance misusers. Eur Addict Res. 2014;20(3):115-118.
112. Lyndon A, Audrey S, Wells C, et al. Risk to heroin users of polydrug use of pregabalin or gabapentin. Addiction. 2017;112(9):1580-1589.
113. Peckham AM, Fairman KA, Sclar DA. All-cause and drug-related medical events associated with overuse of gabapentin and/or opioid medications: a retrospective cohort analysis of a commercially insured US population. Drug Saf. 2018;41(2):213-228.
114. Gomes T, Juurlink DN, Antoniou T, et al. Gabapentin, opioids, and the risk of opioid-related death: a population-based nested case-control study. PLoS Med. 2017;14(10):e10022396. doi: 10.1371/journal.pmed.1002396.
115. Peckham AM, Fairman K, Sclar DA. Policies to mitigate nonmedical use of prescription medications: how should emerging evidence of gabapentin misuse be addressed? Exp Opin Drug Saf. 2018;17(5):519-523.
116. Raby WN. Gabapentin for cocaine cravings. Am J Psychiatry. 2000;157(12):2058-2059.
117. Myrick H, Henderson S, Brady KT, et al. Gabapentin in the treatment of cocaine dependence: a case series. J CLin Psychiatry. 2001;62(1):19-23.
118. Raby WN, Coomaraswamy S. Gabapentin reduces cocaine use among addicts from a community clinic sample. J Clin Psychiatry. 2004;65(1):84-86.
119. Hart CL, Ward AS, Collins ED, et al. Gabapentin maintenance decreases smoked cocaine-related subjective effects, but not self-administration by humans. Drug Alcohol Depend. 2004;73(3):279-287.
120. Bisaga A, Aharonovich E, Garawi F, et al. A randomized placebo-controlled trial of gabapentin for cocaine dependence. Drug Alc Depend. 2006;81(3):267-274.
121. Hart CL, Haney M, Collins ED, et al. Smoked cocaine self-administration by humans is not reduced by large gabapentin maintenance doses. Drug Alcohol Depend. 2007;86(2-3):274-277.
122. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
123. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
124. Martinotti G, Di Nicola M, Tedeschi D, et al. Pregabalin versus naltrexone in alcohol dependence: a randomised, double-blind, comparison trial. J Psychopharmacol. 2010;24(9):1367-1374.
125. Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychpharmacology. 2012;27(7):1689-1698.
126. Enke O, New HA, New CH, et al. Anticonvulsants in the treatment of low back pain and lumbar radicular pain: a systematic review and meta-analysis. CMAJ. 2018;190(26):E786-E793.
127. Cartwright C, Gibson K, Read J, et al. Long-term antidepressant use: patient perspectives of benefits and adverse effects. Patient Prefer Adherence. 2016;10:1401-1407.
128. American Society of Addiction Medicine. Public policy statement: definition of addiction. https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf?sfvrsn=a8f64512_4. Published August 15, 2011. Accessed July 23, 2018.

What is your diagnosis? - January 2019
Primary intestinal lymphangiectasia
Histologic examination shows chronic inflammation of the ileum characterized by increased lymphoplasma cell infiltration of lamina propria without malignancy. Moreover, marked dilatation of lymphatic ducts that involved the mucosa was identified (Figure F, arrows; stain: hematoxylin and eosin; original magnification, ×100). On the basis of pathologic examinations, a diagnosis of primary intestinal lymphangiectasia (PIL) was made.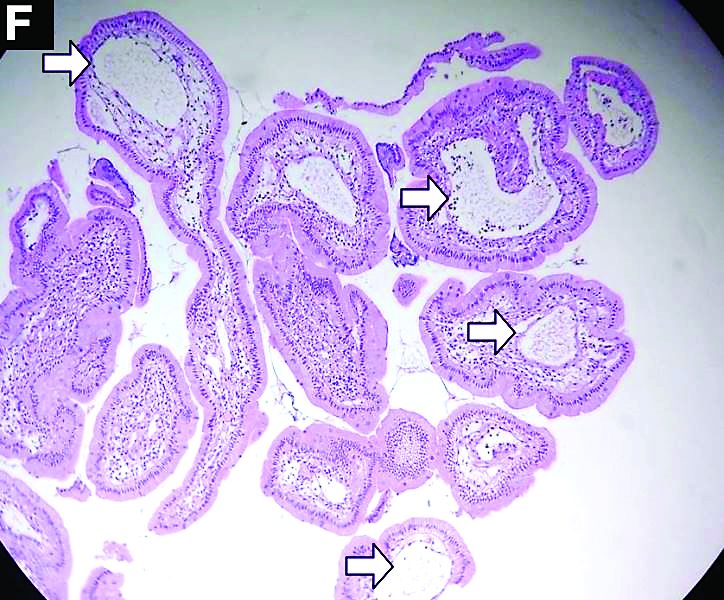
PIL is an extremely rare cause of protein-losing enteropathy characterized by the presence of dilated lymphatic channels in the mucosa, submucosa, or subserosa leading to protein-losing enteropathy.1 The true incidence and prevalence of this disease remains unclear. The disease affects males and females equally, and usually occurs in children and young adults. To date, less than 200 cases of PIL have been reported in the literature. The clinical manifestations of PIL may be asymptomatic or symptomatic such as abdominal pain, edema, diarrhea, and dyspnea. The diagnosis is based on the typical endoscopic findings of diffuse scattered mucosal white blebs with characteristic histologic findings of abnormal lymphatic dilatation. Double-balloon enteroscopy and capsule endoscopy are powerful modalities to evaluate the entire affected area of PIL.2 Although diet modification is a major treatment of PIL, several medicines have been reported to be useful such as corticosteroids, octreotide, and antiplasmin.3 Moreover, in patients with segmental lesions, surgery with local bowel resection is a useful treatment.3 In addition, PIL had a 5% risk of malignant transformation into lymphoma.3
References
1. Waldmann TA, Steinfeld JL, Dutcher TF, et al. The role of the gastrointestinal system in “idiopathic hypoproteinemia.” Gastroenterology. 1961;41:197-207.
2. Oh TG, Chung JW, Kim HM, et al. Primary intestinal lymphangiectasia diagnosed by capsule endoscopy and double balloon enteroscopy. World J Gastrointest Endosc. 2011;3:235-40.
3. Wen J, Tang Q, Wu, J. Primary intestinal lymphangiectasia: four case reports and a review of the literature. Dig Dis Sci. 2010;55:3466-72.
Primary intestinal lymphangiectasia
Histologic examination shows chronic inflammation of the ileum characterized by increased lymphoplasma cell infiltration of lamina propria without malignancy. Moreover, marked dilatation of lymphatic ducts that involved the mucosa was identified (Figure F, arrows; stain: hematoxylin and eosin; original magnification, ×100). On the basis of pathologic examinations, a diagnosis of primary intestinal lymphangiectasia (PIL) was made.
PIL is an extremely rare cause of protein-losing enteropathy characterized by the presence of dilated lymphatic channels in the mucosa, submucosa, or subserosa leading to protein-losing enteropathy.1 The true incidence and prevalence of this disease remains unclear. The disease affects males and females equally, and usually occurs in children and young adults. To date, less than 200 cases of PIL have been reported in the literature. The clinical manifestations of PIL may be asymptomatic or symptomatic such as abdominal pain, edema, diarrhea, and dyspnea. The diagnosis is based on the typical endoscopic findings of diffuse scattered mucosal white blebs with characteristic histologic findings of abnormal lymphatic dilatation. Double-balloon enteroscopy and capsule endoscopy are powerful modalities to evaluate the entire affected area of PIL.2 Although diet modification is a major treatment of PIL, several medicines have been reported to be useful such as corticosteroids, octreotide, and antiplasmin.3 Moreover, in patients with segmental lesions, surgery with local bowel resection is a useful treatment.3 In addition, PIL had a 5% risk of malignant transformation into lymphoma.3
References
1. Waldmann TA, Steinfeld JL, Dutcher TF, et al. The role of the gastrointestinal system in “idiopathic hypoproteinemia.” Gastroenterology. 1961;41:197-207.
2. Oh TG, Chung JW, Kim HM, et al. Primary intestinal lymphangiectasia diagnosed by capsule endoscopy and double balloon enteroscopy. World J Gastrointest Endosc. 2011;3:235-40.
3. Wen J, Tang Q, Wu, J. Primary intestinal lymphangiectasia: four case reports and a review of the literature. Dig Dis Sci. 2010;55:3466-72.
Primary intestinal lymphangiectasia
Histologic examination shows chronic inflammation of the ileum characterized by increased lymphoplasma cell infiltration of lamina propria without malignancy. Moreover, marked dilatation of lymphatic ducts that involved the mucosa was identified (Figure F, arrows; stain: hematoxylin and eosin; original magnification, ×100). On the basis of pathologic examinations, a diagnosis of primary intestinal lymphangiectasia (PIL) was made.
PIL is an extremely rare cause of protein-losing enteropathy characterized by the presence of dilated lymphatic channels in the mucosa, submucosa, or subserosa leading to protein-losing enteropathy.1 The true incidence and prevalence of this disease remains unclear. The disease affects males and females equally, and usually occurs in children and young adults. To date, less than 200 cases of PIL have been reported in the literature. The clinical manifestations of PIL may be asymptomatic or symptomatic such as abdominal pain, edema, diarrhea, and dyspnea. The diagnosis is based on the typical endoscopic findings of diffuse scattered mucosal white blebs with characteristic histologic findings of abnormal lymphatic dilatation. Double-balloon enteroscopy and capsule endoscopy are powerful modalities to evaluate the entire affected area of PIL.2 Although diet modification is a major treatment of PIL, several medicines have been reported to be useful such as corticosteroids, octreotide, and antiplasmin.3 Moreover, in patients with segmental lesions, surgery with local bowel resection is a useful treatment.3 In addition, PIL had a 5% risk of malignant transformation into lymphoma.3
References
1. Waldmann TA, Steinfeld JL, Dutcher TF, et al. The role of the gastrointestinal system in “idiopathic hypoproteinemia.” Gastroenterology. 1961;41:197-207.
2. Oh TG, Chung JW, Kim HM, et al. Primary intestinal lymphangiectasia diagnosed by capsule endoscopy and double balloon enteroscopy. World J Gastrointest Endosc. 2011;3:235-40.
3. Wen J, Tang Q, Wu, J. Primary intestinal lymphangiectasia: four case reports and a review of the literature. Dig Dis Sci. 2010;55:3466-72.
A 19-year-old boy presented to our hospital because of a 6-month history of progressive dyspnea and generalized edema. He developed cough, abdominal fullness, diarrhea, and leg edema 5 years ago. 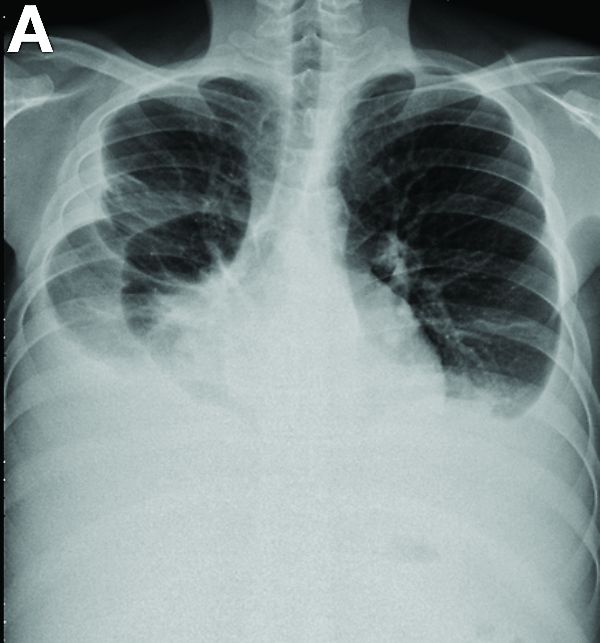
Liver cirrhosis was suspected at that time. However, he seemed to have a poor response to medical treatment. Physical examination showed decreased breathing sounds and rales of the bilateral lower chest area, a distended abdomen with multiple purple striae, and edema of bilateral lower legs. 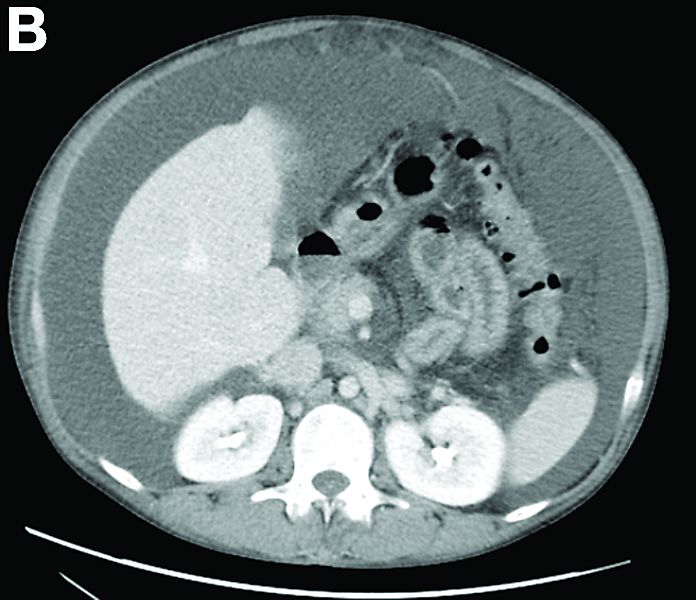
Laboratory tests showed a low serum total protein of 3.8 g/dL (normal range, 5.5–8), albumin of 2.0 g/dL (normal range, 3.8–5.4), total calcium of 7 mg/dL (normal range, 8.4–10.8), C-reactive protein of 11.02 mg/dL (normal, below 0.8). His hemogram showed a white blood cell count of 13,310 × 109/L (normal range, 3.5–11 × 109/L) with lymphocytopenia (9.8%). 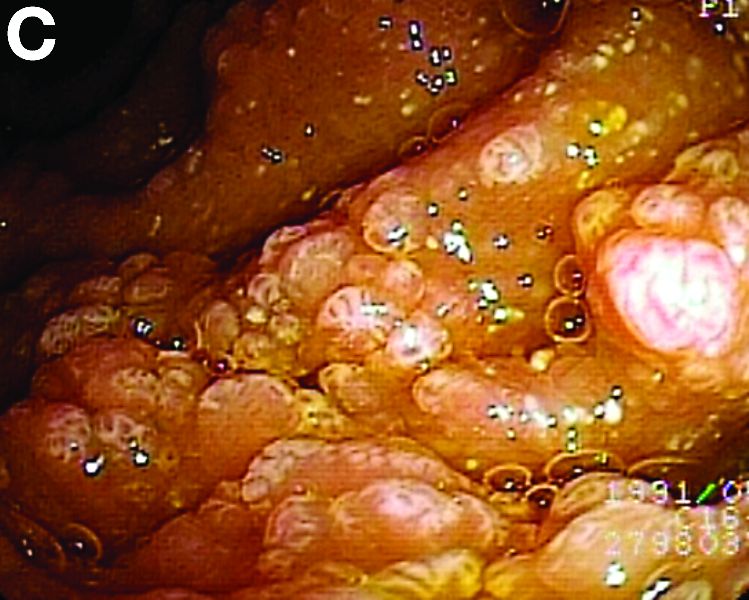
Other blood tests were within normal limits. The urinalysis and stool analysis were normal. Chest radiography showed bilateral pleural effusions (Figure A). Abdominal computed tomography demonstrated large ascites (Figure B). Paracentesis showed his serum ascites albumin gradient was 1.9 g/dL. 
Subsequently, antegrade double-balloon enteroscopy (Fujinon EN-450T5; Fujinon, Saitama, Japan) demonstrated nodular mucosal lesions with a milk-like surface in the duodenum (Figure C).
Moreover, a snowflake appearance of mucosa was found in the jejunum and proximal ileum (Figure D). However, normal appearance of mucosa was identified in the middle ileum (Figure E). Biopsy specimens from these abnormal mucosal lesions were taken for pathology.
What is the diagnosis?
Identifying Melanoma With Dermoscopy
Identifying Melanoma With Dermoscopy: 7- Point Checklist
Clinical Guidance: Thiopurine agents for the treatment of IBD
A new clinical practice update recommends combination therapy with tumor necrosis factor (TNF) inhibitors and thiopurines, as opposed to either therapy alone, for the treatment of ulcerative colitis (UC) and Crohn’s disease (CD). The commentary was published in Gastroenterology.

Clinicians should also note that while several clinical trials use weight-based dosing to monitor clinical response following thiopurine therapy, 6-thioguanine levels have inevitably shown to better predict prognosis, wrote Stephen B. Hanauer, MD, AGAF, of Northwestern University in Chicago and his colleagues.
The thiopurine drug class is composed of many different agents, including thioguanine, azathioprine, and mercaptopurine. Methotrexate, a folate antagonist affecting thymidylate production, is commonly used alongside thiopurines as steroid-sparing agents for patients with UC and CD. Among these therapies, various different dosing strategies and routes of administration are used to manage active disease.
Initially, thiopurines were studied exclusively as monotherapy for the treatment of patients with steroid-intractable CD; however, results showed only marginal benefit when using these agents alone. As a result, combination trials were performed subsequently, and these revealed modest efficacy for use as maintenance therapies in both UC and CD. Further studies reported that methotrexate is beneficial only as a maintenance therapy for CD given that trial evidence confirmed treatment limitations in patients with UC.
“Thiopurines also have the potential to reduce postoperative recurrence of Crohn’s disease, in particular when administered with imidazole antibiotics,” the experts wrote. “There is currently no controlled data regarding the efficacy of methotrexate as maintenance therapy in ulcerative colitis,” they added.
Despite its limitations in UC, 25 mg of methotrexate administered intramuscularly once weekly in combination with oral steroids has shown benefits for inducing disease remission and limiting steroid use in the management of active CD. Comparatively, other trials have failed to show the same benefits with oral methotrexate. In addition, a number of clinical case series have reported benefit for use of methotrexate as a maintenance therapy for CD in patients who initially responded to methotrexate induction therapy.
Consequently, Dr. Hanauer and his colleagues recommended that methotrexate only be given in combination with biologics if being used for the treatment of UC.
“Thiopurines and methotrexate can be used in combination with anti-TNF biologics, in particular infliximab, to reduce immunogenicity and increase blood levels,” they stated.
One agent in particular, thioguanine, exhibits unique therapeutic efficacy in patients allergic to azathioprine or mercaptopurine. Despite this benefit, thioguanine use has been linked with an increased risk of developing hepatic nodular regenerative hyperplasia, as well as venoocclusive disease. Given these limitations, long-term use of thioguanine was not recommended by the authors.
With respect to safety, routine laboratory monitoring for both liver and hematologic adverse effects is recommended. In rare cases, patients may develop secondary lymphomas in response to thiopurine treatment. Moreover, regular follow-up is essential because of the higher prevalence of nonmelanoma skin cancers seen with thiopurines use.
“Patients using thiopurines for the treatment of IBD, particularly Caucasian patients, should avoid excessive sun exposure and use high-strength sun block,” the experts wrote. “Health care deliverers should ensure patients undergo appropriate dermatologic evaluations and investigate suspicious skin lesions in these patients,” they further reported.
Another important monitoring consideration is ongoing infection risk, in particular with opportunistic and viral pathogens. Because of the immunosuppressive effects of therapy, both methotrexate and thiopurine use are linked with a greater chance of developing these infections. Accordingly, Dr. Hanauer and his colleagues recommended that, before initiation of these therapies, applicable preventative measures should be taken, including administration of influenza, human papillomavirus, varicella zoster virus, pneumococcus, and hepatitis B vaccines.
“Live vaccines are contraindicated once therapy has begun; however, zoster vaccination can be given while patients are receiving azathioprine at less than 2 mg/kg,” they stated.
The experts went on to report that withdrawal of thiopurine agents, when used in combination therapy, has the potential to reduce therapeutic levels of infliximab and promote development of antidrug antibodies. However, the experts did not suggest a method to manage these complications. Further studies are needed to answer these and other remaining questions regarding thiopurine use in the setting of IBD.
SOURCE: Hanauer SB et al. Gastroenterology. 2018 Sep 6. doi: 10.1053/j.gastro.2018.08.043.
*This story was updated on January 4, 2019.
A new clinical practice update recommends combination therapy with tumor necrosis factor (TNF) inhibitors and thiopurines, as opposed to either therapy alone, for the treatment of ulcerative colitis (UC) and Crohn’s disease (CD). The commentary was published in Gastroenterology.
Clinicians should also note that while several clinical trials use weight-based dosing to monitor clinical response following thiopurine therapy, 6-thioguanine levels have inevitably shown to better predict prognosis, wrote Stephen B. Hanauer, MD, AGAF, of Northwestern University in Chicago and his colleagues.
The thiopurine drug class is composed of many different agents, including thioguanine, azathioprine, and mercaptopurine. Methotrexate, a folate antagonist affecting thymidylate production, is commonly used alongside thiopurines as steroid-sparing agents for patients with UC and CD. Among these therapies, various different dosing strategies and routes of administration are used to manage active disease.
Initially, thiopurines were studied exclusively as monotherapy for the treatment of patients with steroid-intractable CD; however, results showed only marginal benefit when using these agents alone. As a result, combination trials were performed subsequently, and these revealed modest efficacy for use as maintenance therapies in both UC and CD. Further studies reported that methotrexate is beneficial only as a maintenance therapy for CD given that trial evidence confirmed treatment limitations in patients with UC.
“Thiopurines also have the potential to reduce postoperative recurrence of Crohn’s disease, in particular when administered with imidazole antibiotics,” the experts wrote. “There is currently no controlled data regarding the efficacy of methotrexate as maintenance therapy in ulcerative colitis,” they added.
Despite its limitations in UC, 25 mg of methotrexate administered intramuscularly once weekly in combination with oral steroids has shown benefits for inducing disease remission and limiting steroid use in the management of active CD. Comparatively, other trials have failed to show the same benefits with oral methotrexate. In addition, a number of clinical case series have reported benefit for use of methotrexate as a maintenance therapy for CD in patients who initially responded to methotrexate induction therapy.
Consequently, Dr. Hanauer and his colleagues recommended that methotrexate only be given in combination with biologics if being used for the treatment of UC.
“Thiopurines and methotrexate can be used in combination with anti-TNF biologics, in particular infliximab, to reduce immunogenicity and increase blood levels,” they stated.
One agent in particular, thioguanine, exhibits unique therapeutic efficacy in patients allergic to azathioprine or mercaptopurine. Despite this benefit, thioguanine use has been linked with an increased risk of developing hepatic nodular regenerative hyperplasia, as well as venoocclusive disease. Given these limitations, long-term use of thioguanine was not recommended by the authors.
With respect to safety, routine laboratory monitoring for both liver and hematologic adverse effects is recommended. In rare cases, patients may develop secondary lymphomas in response to thiopurine treatment. Moreover, regular follow-up is essential because of the higher prevalence of nonmelanoma skin cancers seen with thiopurines use.
“Patients using thiopurines for the treatment of IBD, particularly Caucasian patients, should avoid excessive sun exposure and use high-strength sun block,” the experts wrote. “Health care deliverers should ensure patients undergo appropriate dermatologic evaluations and investigate suspicious skin lesions in these patients,” they further reported.
Another important monitoring consideration is ongoing infection risk, in particular with opportunistic and viral pathogens. Because of the immunosuppressive effects of therapy, both methotrexate and thiopurine use are linked with a greater chance of developing these infections. Accordingly, Dr. Hanauer and his colleagues recommended that, before initiation of these therapies, applicable preventative measures should be taken, including administration of influenza, human papillomavirus, varicella zoster virus, pneumococcus, and hepatitis B vaccines.
“Live vaccines are contraindicated once therapy has begun; however, zoster vaccination can be given while patients are receiving azathioprine at less than 2 mg/kg,” they stated.
The experts went on to report that withdrawal of thiopurine agents, when used in combination therapy, has the potential to reduce therapeutic levels of infliximab and promote development of antidrug antibodies. However, the experts did not suggest a method to manage these complications. Further studies are needed to answer these and other remaining questions regarding thiopurine use in the setting of IBD.
SOURCE: Hanauer SB et al. Gastroenterology. 2018 Sep 6. doi: 10.1053/j.gastro.2018.08.043.
*This story was updated on January 4, 2019.
A new clinical practice update recommends combination therapy with tumor necrosis factor (TNF) inhibitors and thiopurines, as opposed to either therapy alone, for the treatment of ulcerative colitis (UC) and Crohn’s disease (CD). The commentary was published in Gastroenterology.
Clinicians should also note that while several clinical trials use weight-based dosing to monitor clinical response following thiopurine therapy, 6-thioguanine levels have inevitably shown to better predict prognosis, wrote Stephen B. Hanauer, MD, AGAF, of Northwestern University in Chicago and his colleagues.
The thiopurine drug class is composed of many different agents, including thioguanine, azathioprine, and mercaptopurine. Methotrexate, a folate antagonist affecting thymidylate production, is commonly used alongside thiopurines as steroid-sparing agents for patients with UC and CD. Among these therapies, various different dosing strategies and routes of administration are used to manage active disease.
Initially, thiopurines were studied exclusively as monotherapy for the treatment of patients with steroid-intractable CD; however, results showed only marginal benefit when using these agents alone. As a result, combination trials were performed subsequently, and these revealed modest efficacy for use as maintenance therapies in both UC and CD. Further studies reported that methotrexate is beneficial only as a maintenance therapy for CD given that trial evidence confirmed treatment limitations in patients with UC.
“Thiopurines also have the potential to reduce postoperative recurrence of Crohn’s disease, in particular when administered with imidazole antibiotics,” the experts wrote. “There is currently no controlled data regarding the efficacy of methotrexate as maintenance therapy in ulcerative colitis,” they added.
Despite its limitations in UC, 25 mg of methotrexate administered intramuscularly once weekly in combination with oral steroids has shown benefits for inducing disease remission and limiting steroid use in the management of active CD. Comparatively, other trials have failed to show the same benefits with oral methotrexate. In addition, a number of clinical case series have reported benefit for use of methotrexate as a maintenance therapy for CD in patients who initially responded to methotrexate induction therapy.
Consequently, Dr. Hanauer and his colleagues recommended that methotrexate only be given in combination with biologics if being used for the treatment of UC.
“Thiopurines and methotrexate can be used in combination with anti-TNF biologics, in particular infliximab, to reduce immunogenicity and increase blood levels,” they stated.
One agent in particular, thioguanine, exhibits unique therapeutic efficacy in patients allergic to azathioprine or mercaptopurine. Despite this benefit, thioguanine use has been linked with an increased risk of developing hepatic nodular regenerative hyperplasia, as well as venoocclusive disease. Given these limitations, long-term use of thioguanine was not recommended by the authors.
With respect to safety, routine laboratory monitoring for both liver and hematologic adverse effects is recommended. In rare cases, patients may develop secondary lymphomas in response to thiopurine treatment. Moreover, regular follow-up is essential because of the higher prevalence of nonmelanoma skin cancers seen with thiopurines use.
“Patients using thiopurines for the treatment of IBD, particularly Caucasian patients, should avoid excessive sun exposure and use high-strength sun block,” the experts wrote. “Health care deliverers should ensure patients undergo appropriate dermatologic evaluations and investigate suspicious skin lesions in these patients,” they further reported.
Another important monitoring consideration is ongoing infection risk, in particular with opportunistic and viral pathogens. Because of the immunosuppressive effects of therapy, both methotrexate and thiopurine use are linked with a greater chance of developing these infections. Accordingly, Dr. Hanauer and his colleagues recommended that, before initiation of these therapies, applicable preventative measures should be taken, including administration of influenza, human papillomavirus, varicella zoster virus, pneumococcus, and hepatitis B vaccines.
“Live vaccines are contraindicated once therapy has begun; however, zoster vaccination can be given while patients are receiving azathioprine at less than 2 mg/kg,” they stated.
The experts went on to report that withdrawal of thiopurine agents, when used in combination therapy, has the potential to reduce therapeutic levels of infliximab and promote development of antidrug antibodies. However, the experts did not suggest a method to manage these complications. Further studies are needed to answer these and other remaining questions regarding thiopurine use in the setting of IBD.
SOURCE: Hanauer SB et al. Gastroenterology. 2018 Sep 6. doi: 10.1053/j.gastro.2018.08.043.
*This story was updated on January 4, 2019.
FROM GASTROENTEROLOGY
Key clinical point: Best clinical practices surrounding the use of thiopurines in patients with inflammatory bowel disease (IBD) were summarized by a group of experts.
Major finding:
Study details: Expert opinion consensus–based review of current evidence surrounding thiopurine therapy for IBD, without complete systematic review of the literature.
Disclosures: The authors reported no conflicts of interest.
Source: Hanauer SB et al. Gastroenterology. 2018 Sep 6. doi: 10.1053/j.gastro.2018.08.043.
AGA Clinical Practice Update: Endoscopic submucosal dissection
The surgical technique published in Clinical Gastroenterology and Hepatology.
Clinicians should recognize ESD as one of the main treatment modalities for GI cancer enclosed within the superficial esophageal mucosa, which includes squamous cell dysplasia, wrote Peter V. Draganov, MD, of the University of Florida in Gainesville with his fellow experts.
Endoscopic resection is a surgical method used to treat both malignant and nonmalignant GI lesions. Over the past several years, the technique has advanced significantly, progressing from snare polypectomy to endoscopic mucosal resection, with current practice now ESD. The minimally invasive technique is considered first-line therapy in patients with colorectal lesions lacking invasive cancer.
While the technique is widely used in Asian countries, and as practice continues to rise throughout Europe, uptake in the United States has been slow. Several factors may be responsible for this delay, including a lack of ESD experts and training centers, underestimation of the benefits associated with ESD, and a likely bias of American oncologists toward treatment with surgical resection. In recent years, extensive improvements have occurred in ESD technique, such as incorporation of pocket and tunnel strategies, which have significantly contributed to the overall safety and efficacy of the procedure.
“With low thresholds for performing endoscopy for upper GI symptoms and the promotion of screening colonoscopy for colon cancer prevention, more precancerous lesions and early cancers are being detected that may be amenable to endoscopic resection by ESD,” the experts wrote.
For mucosal lesions too large to be removed by standard endoscopic resection, or lesions at high risk of being deemed malignant, the guidelines recommend using ESD to remove these lesions. Dr. Draganov and his colleagues acknowledged that the probability of lymph node metastasis is marginally higher when the procedure is used for these widened indications; however, the risk of metastasis remains sufficiently low. Along those lines, several additional recommendations were made related to the expanded indications for ESD, including use in certain patients with Barrett’s esophagus, colorectal neoplasia, and other forms of superficial gastric cancer.
“Expanded indications for gastric ESD include moderately and well-differentiated superficial cancers that are [more than] 2 cm, lesions [up to] 3 cm with ulceration or that contain early submucosal invasion, and poorly differentiated superficial cancers [up to] 2 cm in size,” the experts stated.
With respect to cost, endoscopic resection was found to provide significant savings in comparison to surgical techniques for the removal of colorectal lesions. The economic analysis revealed that using a lesion-specific ESD model for high-risk patients could allow for notable cost reductions.
“Although some insurers have begun preapproving and covering their members who might benefit from ESD, the hurdles preventing other patients from being covered for this innovative and potentially cost-saving procedure should be removed,” they added.
Other recommendations were made in regards to effective implementation of a stepwise ESD educational model to train American endoscopists on how to properly perform the procedure. The proposed strategy involves completion of a formal training program, independent study, self-practice using animal models, and live viewing of cases by ESD experts. In addition, they recommend that newly trained endoscopists complete their first procedures on patients with absolute indications for ESD.
“At present, there is no standardized approach for ESD training in the United States,” the experts wrote. They further explained that “the usual starting point is to attend an ESD course or series of courses that provide increasingly more in-depth exposure.” And they concluded, “a guiding principle should be that our patients’ interests and welfare stand above all else and that patients must not be used as an opportunity for practice or skills acquisition.”
The practice update also recommends that endoscopists avoid the use of techniques that have the ability to produce submucosal fibrosis. Dr. Draganov and his colleagues warn that these practices, such as “tattooing in close proximity to or beneath a lesion for marking” and “partial snare resection of a portion of a lesion for histopathology,” can impede subsequent endoscopic procedures.
Dr. Draganov and several coauthors disclosed financial affiliations with AbbVie, Boston Scientific Corporation, Cook Medical, Olympus America, and others.
SOURCE: Draganov PV et al. Clin Gastroenterol Hepatol. 2018 Aug 2. doi: 10.1016/j.cgh.2018.07.041.
The surgical technique published in Clinical Gastroenterology and Hepatology.
Clinicians should recognize ESD as one of the main treatment modalities for GI cancer enclosed within the superficial esophageal mucosa, which includes squamous cell dysplasia, wrote Peter V. Draganov, MD, of the University of Florida in Gainesville with his fellow experts.
Endoscopic resection is a surgical method used to treat both malignant and nonmalignant GI lesions. Over the past several years, the technique has advanced significantly, progressing from snare polypectomy to endoscopic mucosal resection, with current practice now ESD. The minimally invasive technique is considered first-line therapy in patients with colorectal lesions lacking invasive cancer.
While the technique is widely used in Asian countries, and as practice continues to rise throughout Europe, uptake in the United States has been slow. Several factors may be responsible for this delay, including a lack of ESD experts and training centers, underestimation of the benefits associated with ESD, and a likely bias of American oncologists toward treatment with surgical resection. In recent years, extensive improvements have occurred in ESD technique, such as incorporation of pocket and tunnel strategies, which have significantly contributed to the overall safety and efficacy of the procedure.
“With low thresholds for performing endoscopy for upper GI symptoms and the promotion of screening colonoscopy for colon cancer prevention, more precancerous lesions and early cancers are being detected that may be amenable to endoscopic resection by ESD,” the experts wrote.
For mucosal lesions too large to be removed by standard endoscopic resection, or lesions at high risk of being deemed malignant, the guidelines recommend using ESD to remove these lesions. Dr. Draganov and his colleagues acknowledged that the probability of lymph node metastasis is marginally higher when the procedure is used for these widened indications; however, the risk of metastasis remains sufficiently low. Along those lines, several additional recommendations were made related to the expanded indications for ESD, including use in certain patients with Barrett’s esophagus, colorectal neoplasia, and other forms of superficial gastric cancer.
“Expanded indications for gastric ESD include moderately and well-differentiated superficial cancers that are [more than] 2 cm, lesions [up to] 3 cm with ulceration or that contain early submucosal invasion, and poorly differentiated superficial cancers [up to] 2 cm in size,” the experts stated.
With respect to cost, endoscopic resection was found to provide significant savings in comparison to surgical techniques for the removal of colorectal lesions. The economic analysis revealed that using a lesion-specific ESD model for high-risk patients could allow for notable cost reductions.
“Although some insurers have begun preapproving and covering their members who might benefit from ESD, the hurdles preventing other patients from being covered for this innovative and potentially cost-saving procedure should be removed,” they added.
Other recommendations were made in regards to effective implementation of a stepwise ESD educational model to train American endoscopists on how to properly perform the procedure. The proposed strategy involves completion of a formal training program, independent study, self-practice using animal models, and live viewing of cases by ESD experts. In addition, they recommend that newly trained endoscopists complete their first procedures on patients with absolute indications for ESD.
“At present, there is no standardized approach for ESD training in the United States,” the experts wrote. They further explained that “the usual starting point is to attend an ESD course or series of courses that provide increasingly more in-depth exposure.” And they concluded, “a guiding principle should be that our patients’ interests and welfare stand above all else and that patients must not be used as an opportunity for practice or skills acquisition.”
The practice update also recommends that endoscopists avoid the use of techniques that have the ability to produce submucosal fibrosis. Dr. Draganov and his colleagues warn that these practices, such as “tattooing in close proximity to or beneath a lesion for marking” and “partial snare resection of a portion of a lesion for histopathology,” can impede subsequent endoscopic procedures.
Dr. Draganov and several coauthors disclosed financial affiliations with AbbVie, Boston Scientific Corporation, Cook Medical, Olympus America, and others.
SOURCE: Draganov PV et al. Clin Gastroenterol Hepatol. 2018 Aug 2. doi: 10.1016/j.cgh.2018.07.041.
The surgical technique published in Clinical Gastroenterology and Hepatology.
Clinicians should recognize ESD as one of the main treatment modalities for GI cancer enclosed within the superficial esophageal mucosa, which includes squamous cell dysplasia, wrote Peter V. Draganov, MD, of the University of Florida in Gainesville with his fellow experts.
Endoscopic resection is a surgical method used to treat both malignant and nonmalignant GI lesions. Over the past several years, the technique has advanced significantly, progressing from snare polypectomy to endoscopic mucosal resection, with current practice now ESD. The minimally invasive technique is considered first-line therapy in patients with colorectal lesions lacking invasive cancer.
While the technique is widely used in Asian countries, and as practice continues to rise throughout Europe, uptake in the United States has been slow. Several factors may be responsible for this delay, including a lack of ESD experts and training centers, underestimation of the benefits associated with ESD, and a likely bias of American oncologists toward treatment with surgical resection. In recent years, extensive improvements have occurred in ESD technique, such as incorporation of pocket and tunnel strategies, which have significantly contributed to the overall safety and efficacy of the procedure.
“With low thresholds for performing endoscopy for upper GI symptoms and the promotion of screening colonoscopy for colon cancer prevention, more precancerous lesions and early cancers are being detected that may be amenable to endoscopic resection by ESD,” the experts wrote.
For mucosal lesions too large to be removed by standard endoscopic resection, or lesions at high risk of being deemed malignant, the guidelines recommend using ESD to remove these lesions. Dr. Draganov and his colleagues acknowledged that the probability of lymph node metastasis is marginally higher when the procedure is used for these widened indications; however, the risk of metastasis remains sufficiently low. Along those lines, several additional recommendations were made related to the expanded indications for ESD, including use in certain patients with Barrett’s esophagus, colorectal neoplasia, and other forms of superficial gastric cancer.
“Expanded indications for gastric ESD include moderately and well-differentiated superficial cancers that are [more than] 2 cm, lesions [up to] 3 cm with ulceration or that contain early submucosal invasion, and poorly differentiated superficial cancers [up to] 2 cm in size,” the experts stated.
With respect to cost, endoscopic resection was found to provide significant savings in comparison to surgical techniques for the removal of colorectal lesions. The economic analysis revealed that using a lesion-specific ESD model for high-risk patients could allow for notable cost reductions.
“Although some insurers have begun preapproving and covering their members who might benefit from ESD, the hurdles preventing other patients from being covered for this innovative and potentially cost-saving procedure should be removed,” they added.
Other recommendations were made in regards to effective implementation of a stepwise ESD educational model to train American endoscopists on how to properly perform the procedure. The proposed strategy involves completion of a formal training program, independent study, self-practice using animal models, and live viewing of cases by ESD experts. In addition, they recommend that newly trained endoscopists complete their first procedures on patients with absolute indications for ESD.
“At present, there is no standardized approach for ESD training in the United States,” the experts wrote. They further explained that “the usual starting point is to attend an ESD course or series of courses that provide increasingly more in-depth exposure.” And they concluded, “a guiding principle should be that our patients’ interests and welfare stand above all else and that patients must not be used as an opportunity for practice or skills acquisition.”
The practice update also recommends that endoscopists avoid the use of techniques that have the ability to produce submucosal fibrosis. Dr. Draganov and his colleagues warn that these practices, such as “tattooing in close proximity to or beneath a lesion for marking” and “partial snare resection of a portion of a lesion for histopathology,” can impede subsequent endoscopic procedures.
Dr. Draganov and several coauthors disclosed financial affiliations with AbbVie, Boston Scientific Corporation, Cook Medical, Olympus America, and others.
SOURCE: Draganov PV et al. Clin Gastroenterol Hepatol. 2018 Aug 2. doi: 10.1016/j.cgh.2018.07.041.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: The American Gastroenterological Association (AGA) has released clinical guidance regarding the use of endoscopic submucosal dissection (ESD).
Major finding: ESD should be established as an endoscopic technique that allows for total removal of malignant lesions that could otherwise lead to future complications for patients.
Study details: Expert review focused on the current and upcoming role of ESD in clinical gastroenterology practice in the United States.
Disclosures: Dr. Draganov and several coauthors disclosed financial affiliations with AbbVie, Boston Scientific, Cook Medical, Olympus America, and others.
Source: Draganov PV et al. Clin Gastroenterol Hepatol. 2018 Aug 2. doi: 10.1016/j.cgh.2018.07.041.
Fungal failure
Two months ago I met Ed, still working at age 71. “My life’s ambition,” he said, “has been to help high school science teachers do their jobs better.”
“How’s it going?” I asked.
Ed sighed. “I’m still at it,” he said. “Let’s just say we’re not there yet.”
I too, dear colleagues, have had a life’s ambition, secret until right now:
Alas, like Ed’s, my work is not yet done.
I get reminders of this all the time, but last week the evidence got so overwhelming that I had to take a breath to settle down. And a nip. Ten cases. In 24 hours.
1. A 66-year-old woman energetically smeared econazole cream twice daily for weeks for an itchy, lichenified rash on both dorsal feet and ankles. Switched to betamethasone. Cleared in 5 days.
2. A 48-year-old woman with scaly patches on both legs. No response to terbinafine cream, then to ketoconazole cream, then to oral fluconazole. Cleared promptly on triamcinolone.
3. A 26-year-old with an erosive vulvar rash lasting month, unresponsive to Nystatin. After 5 days on a steroid, it was gone.
4. A 45-year-old man with lots of dermatoheliosis and idiopathic guttate hypomelanosis on arms and legs. No luck with topical selenium sulfide for tinea versicolor.
5. A 42-year-old nurse treated for weeks with topical antifungals. She came in with globs of fungus cream sealed in with Tegaderm (to prevent spread). Her roommates wanted to cancel her lease. Cleared of both rash and Tegaderm in 1 week. Now allowed to touch doorknobs.
6. A 27-year-old man with 8 weeks of lichenified patches all over his torso. Antifungal creams not working. Steroids do!
7. A 25-year-old recent émigré from India, where he was treated for his itchy groin rash with a succession of antifungal creams. He cannot sleep. (Imagine the plane trip from Delhi!) Has lichenified inguinal folds and scrotum. Cleared in 1 week with a topical steroid.
8. A 22-year-old woman with widespread atopic dermatitis. No response to antifungals. She had a rash at age 2 that was called “allergy to shampoo.” Clears promptly on a steroid.
9. A 22-year-old man being treated for a scaly, bilateral periocular rash with oral cephalexin. Clears promptly on a weak topical steroid.
10. A 29-year-old woman who has been suffering for months with “sensitivity” of her vulvar skin that has been diagnosed and treated as “a yeast infection,” in the absence of any rash or discharge. Her only visible finding is inverse psoriasis in the gluteal cleft. Guess what clears her up?
And so it goes, and so it has gone, week after week, year after year, decade after decade. Medicine scales Olympus: genomics, immunotherapy, stereotactic surgery. Meantime, the it’s-not-a-fungus problem seems impervious to both education and even to daily observation as obvious as it is ineffective: If a supposed fungus does not respond to antifungal treatment, then it must be a very bad fungus. If it doesn’t respond to yet another antifungal cream, then it must be terrible fungus. Reconsidering that it may not be a fungus at all seems to demand a mental paradigm shift whose achievement will have to await a more discerning generation.
In the meantime, patients not only don’t get better, but they feel defiled and dirty. They avoid human contact, intimate and otherwise, and do a lot of superfluous and expensive cleaning of house and wardrobe. If you doubt this, ask them. If you think I overstate, spend a day with me.
Early in my career I inherited the once-yearly dermatology slot at Medical Grand Rounds at the local community hospital. I spoke about cutaneous fungus, with emphasis on the fact that lots of round rashes are nummular eczema rather than fungus, as well as what it means to patients to be told they are “fungal.”
I didn’t get much direct feedback, but the chief of medicine sprang into action. He canceled the dermatology slot. Not medical enough, I guess.
Ed tells me that many high school science teachers don’t know much science. They teach it because they thought they might like to, or because there was an opening. After Ed hangs up his cleats, there will be plenty of his work left to be done.
But then, there always is.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Two months ago I met Ed, still working at age 71. “My life’s ambition,” he said, “has been to help high school science teachers do their jobs better.”
“How’s it going?” I asked.
Ed sighed. “I’m still at it,” he said. “Let’s just say we’re not there yet.”
I too, dear colleagues, have had a life’s ambition, secret until right now:
Alas, like Ed’s, my work is not yet done.
I get reminders of this all the time, but last week the evidence got so overwhelming that I had to take a breath to settle down. And a nip. Ten cases. In 24 hours.
1. A 66-year-old woman energetically smeared econazole cream twice daily for weeks for an itchy, lichenified rash on both dorsal feet and ankles. Switched to betamethasone. Cleared in 5 days.
2. A 48-year-old woman with scaly patches on both legs. No response to terbinafine cream, then to ketoconazole cream, then to oral fluconazole. Cleared promptly on triamcinolone.
3. A 26-year-old with an erosive vulvar rash lasting month, unresponsive to Nystatin. After 5 days on a steroid, it was gone.
4. A 45-year-old man with lots of dermatoheliosis and idiopathic guttate hypomelanosis on arms and legs. No luck with topical selenium sulfide for tinea versicolor.
5. A 42-year-old nurse treated for weeks with topical antifungals. She came in with globs of fungus cream sealed in with Tegaderm (to prevent spread). Her roommates wanted to cancel her lease. Cleared of both rash and Tegaderm in 1 week. Now allowed to touch doorknobs.
6. A 27-year-old man with 8 weeks of lichenified patches all over his torso. Antifungal creams not working. Steroids do!
7. A 25-year-old recent émigré from India, where he was treated for his itchy groin rash with a succession of antifungal creams. He cannot sleep. (Imagine the plane trip from Delhi!) Has lichenified inguinal folds and scrotum. Cleared in 1 week with a topical steroid.
8. A 22-year-old woman with widespread atopic dermatitis. No response to antifungals. She had a rash at age 2 that was called “allergy to shampoo.” Clears promptly on a steroid.
9. A 22-year-old man being treated for a scaly, bilateral periocular rash with oral cephalexin. Clears promptly on a weak topical steroid.
10. A 29-year-old woman who has been suffering for months with “sensitivity” of her vulvar skin that has been diagnosed and treated as “a yeast infection,” in the absence of any rash or discharge. Her only visible finding is inverse psoriasis in the gluteal cleft. Guess what clears her up?
And so it goes, and so it has gone, week after week, year after year, decade after decade. Medicine scales Olympus: genomics, immunotherapy, stereotactic surgery. Meantime, the it’s-not-a-fungus problem seems impervious to both education and even to daily observation as obvious as it is ineffective: If a supposed fungus does not respond to antifungal treatment, then it must be a very bad fungus. If it doesn’t respond to yet another antifungal cream, then it must be terrible fungus. Reconsidering that it may not be a fungus at all seems to demand a mental paradigm shift whose achievement will have to await a more discerning generation.
In the meantime, patients not only don’t get better, but they feel defiled and dirty. They avoid human contact, intimate and otherwise, and do a lot of superfluous and expensive cleaning of house and wardrobe. If you doubt this, ask them. If you think I overstate, spend a day with me.
Early in my career I inherited the once-yearly dermatology slot at Medical Grand Rounds at the local community hospital. I spoke about cutaneous fungus, with emphasis on the fact that lots of round rashes are nummular eczema rather than fungus, as well as what it means to patients to be told they are “fungal.”
I didn’t get much direct feedback, but the chief of medicine sprang into action. He canceled the dermatology slot. Not medical enough, I guess.
Ed tells me that many high school science teachers don’t know much science. They teach it because they thought they might like to, or because there was an opening. After Ed hangs up his cleats, there will be plenty of his work left to be done.
But then, there always is.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.
Two months ago I met Ed, still working at age 71. “My life’s ambition,” he said, “has been to help high school science teachers do their jobs better.”
“How’s it going?” I asked.
Ed sighed. “I’m still at it,” he said. “Let’s just say we’re not there yet.”
I too, dear colleagues, have had a life’s ambition, secret until right now:
Alas, like Ed’s, my work is not yet done.
I get reminders of this all the time, but last week the evidence got so overwhelming that I had to take a breath to settle down. And a nip. Ten cases. In 24 hours.
1. A 66-year-old woman energetically smeared econazole cream twice daily for weeks for an itchy, lichenified rash on both dorsal feet and ankles. Switched to betamethasone. Cleared in 5 days.
2. A 48-year-old woman with scaly patches on both legs. No response to terbinafine cream, then to ketoconazole cream, then to oral fluconazole. Cleared promptly on triamcinolone.
3. A 26-year-old with an erosive vulvar rash lasting month, unresponsive to Nystatin. After 5 days on a steroid, it was gone.
4. A 45-year-old man with lots of dermatoheliosis and idiopathic guttate hypomelanosis on arms and legs. No luck with topical selenium sulfide for tinea versicolor.
5. A 42-year-old nurse treated for weeks with topical antifungals. She came in with globs of fungus cream sealed in with Tegaderm (to prevent spread). Her roommates wanted to cancel her lease. Cleared of both rash and Tegaderm in 1 week. Now allowed to touch doorknobs.
6. A 27-year-old man with 8 weeks of lichenified patches all over his torso. Antifungal creams not working. Steroids do!
7. A 25-year-old recent émigré from India, where he was treated for his itchy groin rash with a succession of antifungal creams. He cannot sleep. (Imagine the plane trip from Delhi!) Has lichenified inguinal folds and scrotum. Cleared in 1 week with a topical steroid.
8. A 22-year-old woman with widespread atopic dermatitis. No response to antifungals. She had a rash at age 2 that was called “allergy to shampoo.” Clears promptly on a steroid.
9. A 22-year-old man being treated for a scaly, bilateral periocular rash with oral cephalexin. Clears promptly on a weak topical steroid.
10. A 29-year-old woman who has been suffering for months with “sensitivity” of her vulvar skin that has been diagnosed and treated as “a yeast infection,” in the absence of any rash or discharge. Her only visible finding is inverse psoriasis in the gluteal cleft. Guess what clears her up?
And so it goes, and so it has gone, week after week, year after year, decade after decade. Medicine scales Olympus: genomics, immunotherapy, stereotactic surgery. Meantime, the it’s-not-a-fungus problem seems impervious to both education and even to daily observation as obvious as it is ineffective: If a supposed fungus does not respond to antifungal treatment, then it must be a very bad fungus. If it doesn’t respond to yet another antifungal cream, then it must be terrible fungus. Reconsidering that it may not be a fungus at all seems to demand a mental paradigm shift whose achievement will have to await a more discerning generation.
In the meantime, patients not only don’t get better, but they feel defiled and dirty. They avoid human contact, intimate and otherwise, and do a lot of superfluous and expensive cleaning of house and wardrobe. If you doubt this, ask them. If you think I overstate, spend a day with me.
Early in my career I inherited the once-yearly dermatology slot at Medical Grand Rounds at the local community hospital. I spoke about cutaneous fungus, with emphasis on the fact that lots of round rashes are nummular eczema rather than fungus, as well as what it means to patients to be told they are “fungal.”
I didn’t get much direct feedback, but the chief of medicine sprang into action. He canceled the dermatology slot. Not medical enough, I guess.
Ed tells me that many high school science teachers don’t know much science. They teach it because they thought they might like to, or because there was an opening. After Ed hangs up his cleats, there will be plenty of his work left to be done.
But then, there always is.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@mdedge.com.