User login
Comparison of Dermatologist Ratings on Health Care–Specific and General Consumer Websites
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
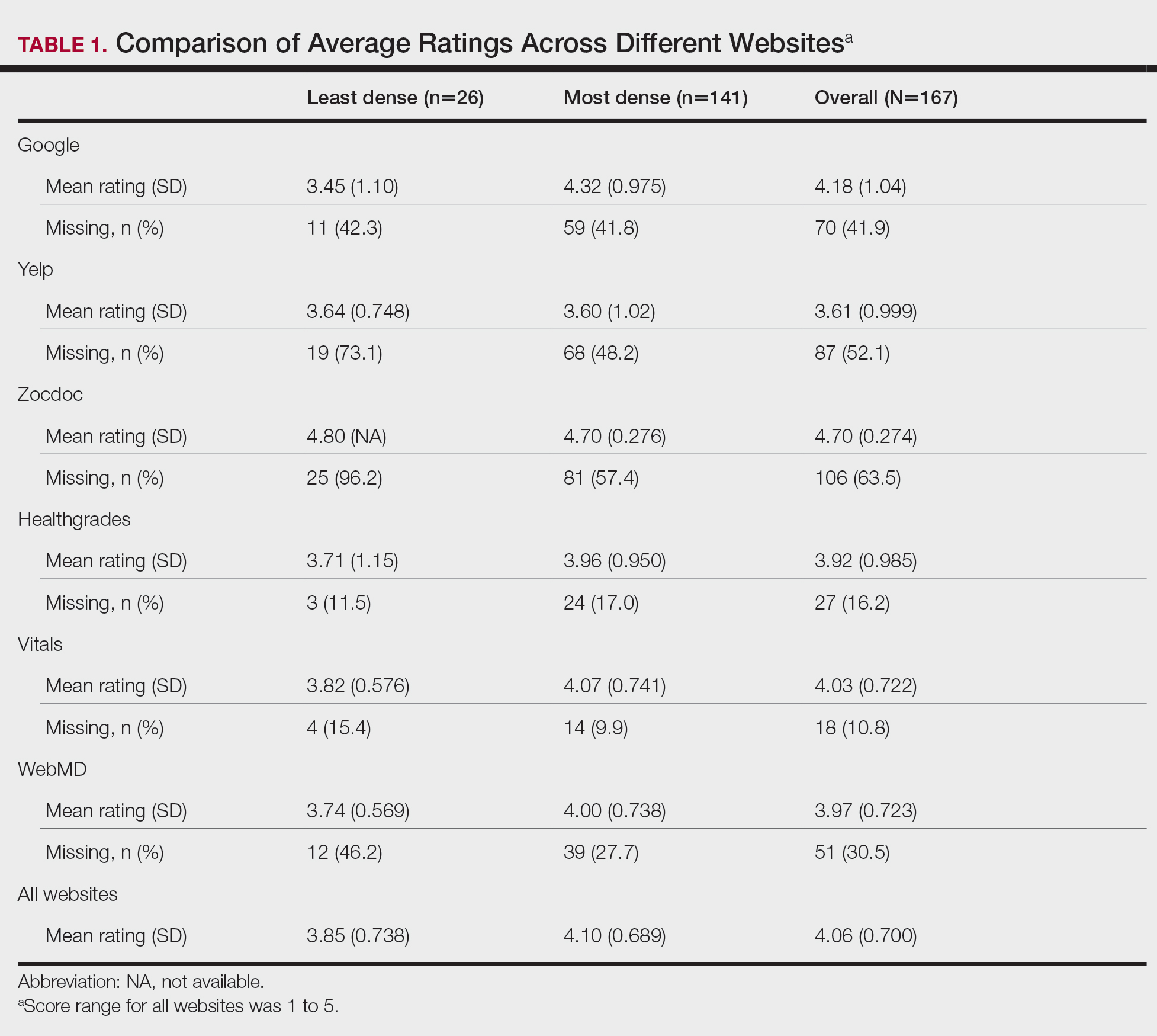
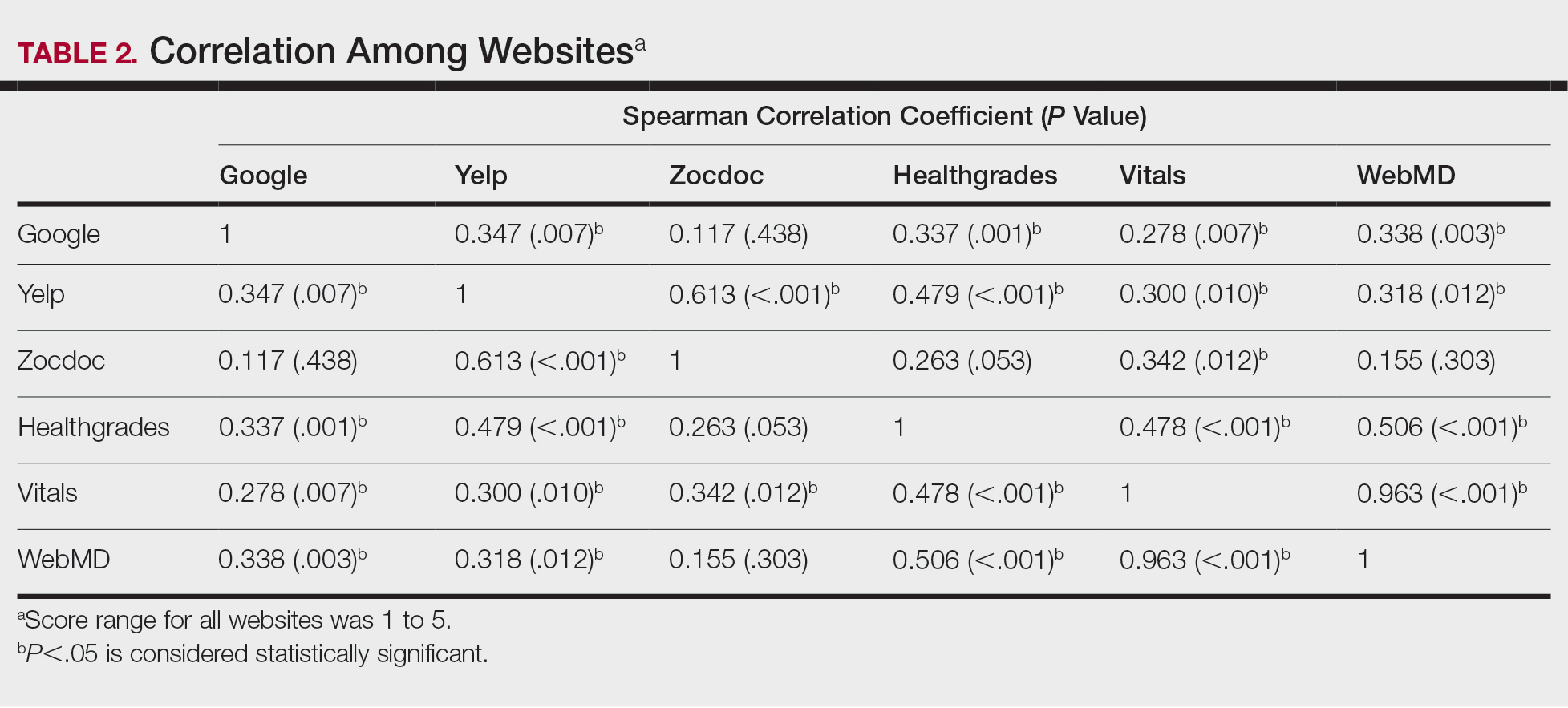
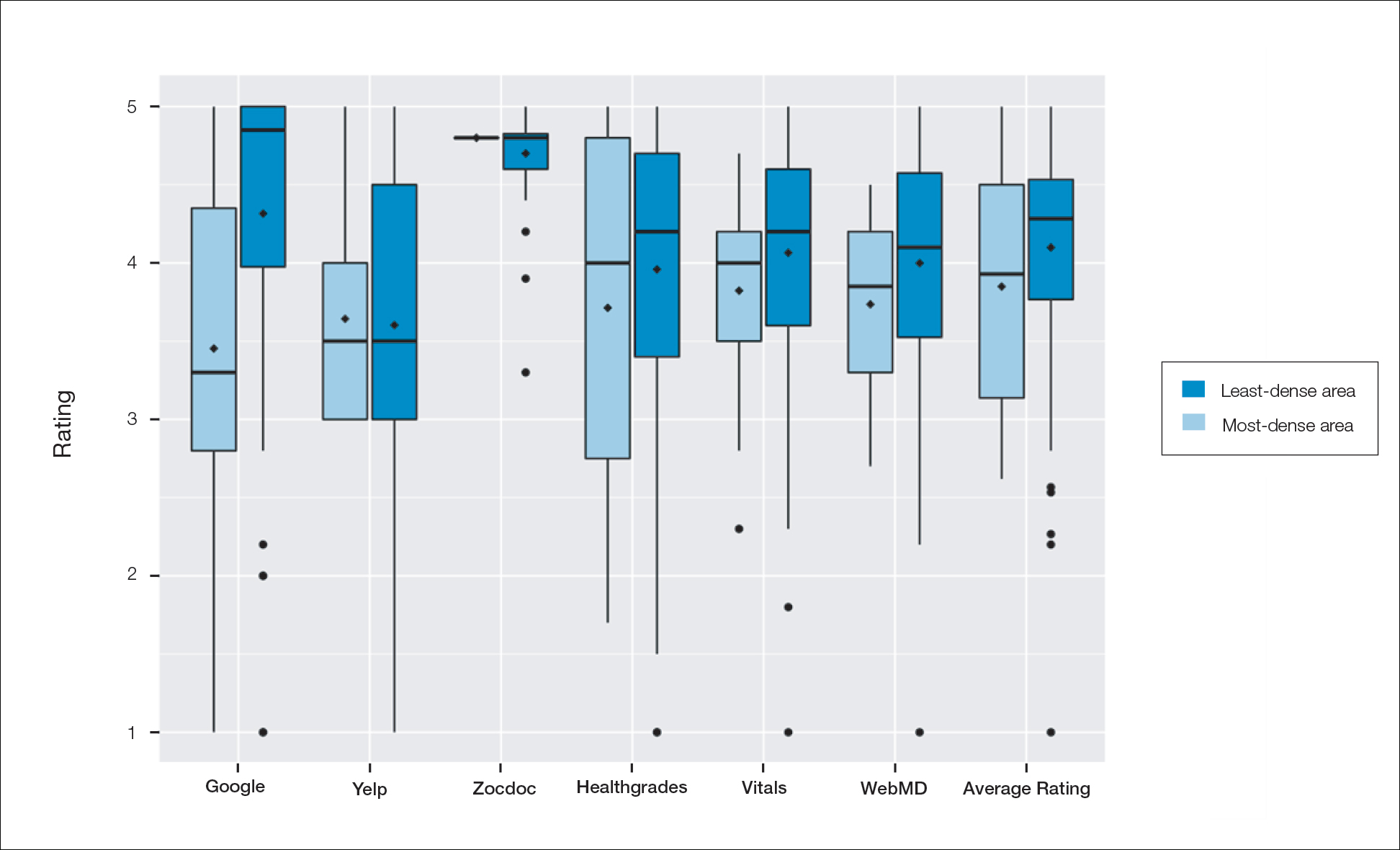
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
Practice Points
- Online physician-rating websites are commonly used by patients to find physicians and review experiences.
- Health care–specific sites may more accurately reflect patient sentiment than general consumer sites.
- Dermatologists can use health care–specific sites to understand patient sentiment and learn how to improve patient experiences.
List of COVID-19 high-risk comorbidities expanded
The list of medical according to the Centers for Disease Control and Prevention.

The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
The list of medical according to the Centers for Disease Control and Prevention.
The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
The list of medical according to the Centers for Disease Control and Prevention.
The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
COVID-19 Monoclonal Antibody Infusions: A Multidisciplinary Initiative to Operationalize EUA Novel Treatment Options
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).

The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.

On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.

Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.

The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.

Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.

Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; kathleen.jodoin@msmc.com.
Financial disclosures: None.
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).
The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.
On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.
Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.
The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.
Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.
Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; kathleen.jodoin@msmc.com.
Financial disclosures: None.
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).
The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.
On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.
Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.
The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.
Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.
Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; kathleen.jodoin@msmc.com.
Financial disclosures: None.
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download

Implementing the AMI READMITS Risk Assessment Score to Increase Referrals Among Patients With Type I Myocardial Infarction
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.

Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.

The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”

Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; nmuganl1@jhmi.edu.
Financial disclosures: None.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.
Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.
The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”
Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; nmuganl1@jhmi.edu.
Financial disclosures: None.
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.
Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.
The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”
Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; nmuganl1@jhmi.edu.
Financial disclosures: None.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
FDA warning letters target OTC cannabidiol product claims for pain relief
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.

In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
Doctors Found Jet Fuel in Veteran’s Lungs. He Can’t Get Full Benefits.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
The lungs Bill Thompson was born with told a gruesome, harrowing and unmistakable tale to Dr. Anthony Szema when he analyzed them and found the black spots, scarring, partially combusted jet fuel and metal inside.
The retired Army staff sergeant had suffered catastrophic lung damage from breathing incinerated waste burned in massive open-air pits and probably other irritants during his tour of duty in Iraq.
“There’s black spots that are burns, particles all over; there’s metal. It was all scarred,” said Szema, a pulmonologist and professor who studies toxic exposures and examined Thompson’s preserved lung tissue. “There was no gas exchange anywhere in that lung.”
Thompson is still alive, surviving on his second transplanted set of lungs. Yet the story burned into the veteran’s internal organs is not one that has been entirely convincing to the U.S. government.
The military has not linked the burn pits to illness. That means many who were exposed to burn pits and are sick do not qualify for benefits under any existing program.
Retirement and health benefits for members of the military depend on factors like length of service, active or reserve status, deployments to combat zones and whether the military considers specific injuries or illnesses to be service-related. Thompson has been able to get care through the Department of Veterans Affairs for his lung disease but has not been able to secure other benefits, like early retirement pay.
“I was denied my Army retirement because if it was not a combat action, then I don’t receive that retirement,” Thompson said at a Senate Veterans’ Affairs Committee hearing last week on service members’ exposures to toxic substances.
Thompson is one of at least 3.5 million veterans since 2001 who have served in war zones where the U.S. military decided to dispose of its trash by burning it, according to VA estimates.
It’s not clear how many people within that population have gotten sick from exposure. Only a small fraction — 234,000 — have enrolled in the VA’s online burn pit registry. Veterans’ advocacy groups have said the majority of claims to the agency stemming from toxic exposures are denied, even as most former service members report contacts with toxins in their deployments.
Soldiers returning from tours in the global war on terror have reported debilitating illnesses almost from its beginning, but got little traction with the military. This year, though, the likelihood of congressional action is high, with Democrats expressing interest and a president who suspects burn pits are to blame for his son’s death.
President Joe Biden’s son Beau died of brain cancer in 2015 at age 46. He had deployed to Iraq in two sites with burn pits — at Baghdad and Balad — around the same time Thompson was at Camp Striker, near the Baghdad airport.
“Because of exposure to burn pits — in my view, I can’t prove it yet — he came back with stage 4 glioblastoma,” Biden said in a 2019 speech.
In testimony at the March 10 hearing, Shane Liermann, who works for the group Disabled American Veterans, told the committee that 78% of burn pit claims are denied. “Part of the problem is VA is not recognizing that exposure as being toxic exposures,” Liermann said.
Aleks Morosky, with the Wounded Warrior Project, said that in his group’s survey of 28,000 veterans last year, 71% said they had “definitely” been exposed to toxic substances or hazardous chemicals, and 18% said they had “probably” been exposed. Half of those people rated their health as poor or fair. Only about 16% of the service members who believed they had suffered exposure said they got treatment from the VA, and 11% said they were denied treatment.
Thompson, who is 49, said care for his lung disease is often slow and sometimes denied. It took the VA three years to approve an air purifier for his home to filter out allergens, and the VA refused to help pay for the removal of dust-trapping carpets, he said.
Thompson’s presence at the hearing, though, was not just meant to put the spotlight on the VA. The military’s entire approach to toxic exposure is a morass that leaves ill soldiers and veterans like Thompson trying to navigate a bureaucracy more labyrinthine than the Pentagon’s corridors.
After Thompson was shipped back to Fort Stewart in Georgia, his medical ordeal was at first addressed within the military system, including a year at Walter Reed National Military Medical Center in Bethesda, Maryland, where doctors found his lungs filled with titanium, magnesium, iron and silica.
Yet he said he didn’t qualify for the Army’s traumatic-injury insurance program, which might have helped him pay to retrofit his home in West Virginia. And he can’t get his military retirement pay until he’s 60.
“I may not live to be age 60. I turn 50 this year,” Thompson said.
Illustrating the problem, several officials at the hearing with the Department of Defense, the Army and the National Guard were unable to explain why Thompson — with 23 years of service between the Guard and Army — might have such a hard time qualifying for retirement benefits when the evidence of his lungs and the findings of the Army’s own doctors are so vivid and extreme.
For advocates who have been working on the problem for decades, it reminds them all too vividly of Agent Orange, which the military is still coming to grips with.
“It’s already been, since the first Persian Gulf [War] — we’re talking 30 years — and since burn pits were again active, since 2001,” said Liermann. “We’re way behind the curve here.”
Although Congress has done relatively little to deal with burn pits, many members seem to at least be thinking along the same lines. The Senate Veterans’ Affairs hearing promised to be something of a kickoff to a year when lawmakers are poised to offer a slew of bills designed to confront the military’s inability to care for service members poisoned during their deployments.
“Make no mistake about it,” said the committee chairman, Sen. Jon Tester (D-Mont.). “We hold these hearings for two reasons: to gather information for the committee members and to help educate the VA that they might take action before Congress does.”
Republicans have also shown growing interest in the problem, offering targeted bills to ensure a handful of toxin-related diseases are covered by the VA.
At the hearing, conservative freshman Sen. Tommy Tuberville (R-Ala.) seemed especially moved.
“We got to do a better job of taking care of our young people,” Tuberville said. “If we’re going to go to war, we got to understand we got to pay the price for it on both ends.”
There is also likely to be high-profile support and attention when revised legislation starts rolling out this spring.
The broadest bill likely to be offered was first introduced by Sen. Kirsten Gillibrand (D-N.Y.) in the Senate and Rep. Raul Ruiz (D-Calif.) in the House in late 2019, with a boost from former “Daily Show” host Jon Stewart and a cadre of 9/11 responders who are turning their attention to toxic exposures.
Indeed, Ruiz and Gillibrand’s legislation is modeled in part on the 9/11 health act that passed in 2015. The burn pit bill would remove the burden of proving a service-related connection.
It would vastly simplify the lives of people like Thompson.
“I am a warrior of the United States of America. I gave my lungs for my country,” Thompson said.
He was cut off before he could finish, but his prepared remarks concluded, “Hopefully, after hearing my story, it will bring awareness for not only me but others who are battling the same or similar injuries related to burn pit exposures from Iraq or Afghanistan.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
USE OUR CONTENT
This story can be republished for free (details).
Subscribe to KHN's free Morning Briefing.
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
A return of holism? It never left osteopathic medicine
I enjoyed Dr. Jonas’s article, “A new model of care to return holism to family medicine” (J Fam Pract. 2020;69:493-498).
However, I wanted to point out that for more than 100 years the concept of the patient-centered medical home, and the outgrowth of that, has been part of osteopathic medical education, founded by A.T. Still, MD, in the 1800s.
Congratulations to the allopathic medicine profession for recognizing its significance.
Steven Shapiro, DO
Fenton, MI
I enjoyed Dr. Jonas’s article, “A new model of care to return holism to family medicine” (J Fam Pract. 2020;69:493-498).
However, I wanted to point out that for more than 100 years the concept of the patient-centered medical home, and the outgrowth of that, has been part of osteopathic medical education, founded by A.T. Still, MD, in the 1800s.
Congratulations to the allopathic medicine profession for recognizing its significance.
Steven Shapiro, DO
Fenton, MI
I enjoyed Dr. Jonas’s article, “A new model of care to return holism to family medicine” (J Fam Pract. 2020;69:493-498).
However, I wanted to point out that for more than 100 years the concept of the patient-centered medical home, and the outgrowth of that, has been part of osteopathic medical education, founded by A.T. Still, MD, in the 1800s.
Congratulations to the allopathic medicine profession for recognizing its significance.
Steven Shapiro, DO
Fenton, MI
Is there liability if you don’t test for BRCA?
CASE Young woman with family history of breast cancer detects lump
Two weeks after noting a lump on her breast when her cat happened to jump on her in that spot, a 28-year-old woman (G0) went to her primary care provider. She was referred to her gynecologist; breast imaging, ultrasonography, and mammography were obtained, with microcalcifications noted. A fine needle aspiration diagnosed intraductal malignancy. The surgical breast tissue specimen was estrogen receptor (ER)- and progestogen receptor (PR)-positive and HER2-negative. Other tumor markers were obtained, including carcinoembryonic antigen, and tissue polypeptide specific antigen, p53, cathepsin D, cyclin E, and nestin, but results were not available.
With regard to family history, the woman’s mother and maternal grandmother had a history of breast cancer. The patient and her family underwent gene testing. The patient was found to be BRCA1- and BRCA2-positive; her mother was BRCA1-positive, an older sister was BRCA2-positive, and her grandmother was not tested.
The question arose in light of her family history as to why she was not tested for BRCA and appropriately counseled by her gynecologist prior to the cancer diagnosis. Litigation was initiated. While the case did not go forward regarding litigation, it is indeed a case in point. (Please note that this is a hypothetical case. It is based on a composite of several cases.)
Medical considerations
Breast cancer is the most common type of cancer affecting women in the Western world.1 Advances in clinical testing for gene mutations have escalated and allowed for identification of patients at increased risk for breast and ovarian cancer. Along with these advances come professional liability risk. After looking at the medical considerations for BRCA1 and 2 testing, we will consider a number of important legal issues. In the view of some commentators, the failure to diagnose genetic mutations in patients predisposed to cancer is “poised to become the next wave of medical professional liability lawsuits.”2
BRCA1 and BRCA2 genes provide tumor suppressor proteins, and assessment for mutations is recommended for individuals at high risk for breast and/or ovarian cancer; mutations in BRCA genes cause DNA damage, which increases the chance of developing cancer. The other way to look at it is, BRCA1 and 2 are tumor suppressor genes that are integrally involved with DNA damage control. Once there is a mutation, it adversely affects the beneficial effects of the gene. Mutations in these genes account for 5% to 10% of all hereditary breast cancers.3 Of note, men with BRCA2 are at increased risk for prostate cancer.
A patient who presents to her gynecologist stating that there is a family history of breast cancer, without knowledge of genetic components, presents a challenge (and a medicolegal risk) for the provider to assess. Prediction models have been used to determine specific patient risk for carrying a genetic mutation with resultant breast cancer development.4 Risk prediction models do not appear to be a good answer to predicting who is more likely to develop breast or ovarian cancer, however. A Mayo model may assist (FIGURE).5 Clinicians should also be aware of other models of risk assessment, including the Gail Model (TABLE 1).6

Continue to: Guidelines for genetic testing...
Guidelines for genetic testing
The American College of Obstetricians and Gynecologists states that patient medical history and family history are paramount in obtaining information regarding risk for breast and ovarian cancer. First- and second-degree relatives are allocated to this category. Information regarding age of diagnosis, maternal and paternal lineage, and ethnic background can imply a need for genetic testing (TABLE 2).7,8 A number of genetics national organizations have participated in recommendations and include the American College of Medical Genetics and Genomics, the National Society for Genetic Counselors, and the Society of Gynecologic Oncology.7

The question always surfaces, could the clinical outcome of the cancer when diagnosed have been changed if screening were undertaken, with earlier diagnosis, or prevented with prophylactic mastectomy, and changed the end result. In addition, it is well known that breast augmentation mammoplasty alters the ability to accurately evaluate mammograms. Patients considering this type of plastic surgery, ideally, should be counselled accordingly.9
Bottom line, we as clinicians must be cognizant of both ACOG and United States Preventive Services Task Force (USPSTF) recommendations regarding screening and gene testing for women considered high risk for breast cancer based on family history.7
Legal considerations
The case presented demonstrates that the discovery of the BRCA1 and BRCA2 genes, and reliable tests for determining the existence of the genes, brought with them legal issues as well as medical advantages. We look at professional liability (malpractice) questions this technology raises, and then consider the outcome of the hypothetical case. (BRCA is used here to apply broadly—not only to BRCA1 and 2 but also to PALB2, CHEK2, and similar genetic abnormalities.)
To date, the most visible BRCA legal issues covered in cases and law reviews have focused more on patent law than malpractice. The most important of these was a decision of the US Supreme Court in Association for Molecular Pathology v Myriad Genetics.10 The US Patent Office was granting patents to companies finding useful, naturally occurring segments of human DNA, and had granted Myriad several patents on BRCA1 and BRCA2 genes. This patent policy had the potential to seriously interfere with broad scientific use of these genes.11 Fortunately, the Supreme Court stepped in and unanimously invalidated such patents. It held that a “naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated.” The Court noted, “Finding the location of the BRCA1 and BRCA2 genes does not render the genes patent eligible ‘new . . . composition[s] of matter.’”8 The Court did allow the patenting of tests for specific gene structures, and artificial changes in naturally occurring genes.
Malpractice and BRCA
While the BRCA patent wars have lingered, the potential for a significant increase in BRCA-related malpractice cases is of increasing concern. Like most malpractice liability, these new claims are based on very old principles of negligence.12 To prevail, the plaintiff (ordinarily, an injured patient) must demonstrate 4 things:
- A duty. That is, the physician owed a duty to the injured party. Usually (but not always) that requires a professional relationship between the physician and the person injured.
- A breach of that duty. Malpractice liability is based on the fact that the physician did something that a reasonably careful physician (generally, of the same specialty) would not have done, or that the physician failed to do something that a reasonable physician would have done. This usually means that the profession itself sees what the physician did (or did not do) as medically inappropriate. In medical malpractice cases, that is ordinarily measured by what the usual or common practice is among prudent physicians. In rare circumstances, courts have found the standard practice of a profession to be negligent. Where, for example, it was custom for a professional not to give an eye pressure test to anyone under age 40, a court found that common standard to be inappropriate.13 In the words of Judge Learned Hand (speaking about a different case), “a whole calling may have unduly lagged in the adoption of new and available devices. It never may set its own tests.”14 Underlying negligence is a cost-benefit analysis (discussed below).
- Damages. There must have been some damage that courts recognize, usually loss of money or opportunity to work, the cost of care, pain and suffering, or loss of enjoyment/quality of life. In malpractice, many states now recognize the “loss of chance” or the “loss of a chance.” That means, if a “physician negligently fails to diagnose a curable disease, and the patient is harmed by the disease, the physician should be liable for causing the ‘loss of a chance of a cure.’”15 (Delay in diagnosis is the most common reason for claims in breast cancer care.)16
- Causation. The breach of duty (negligence) must have caused the damages. The causation must have been reasonably close. If a driver drives through a stop sign, or a physician misreads a test, and someone is injured but there is no connection between the negligence and the injury, there is not tort liability.
The 4 elements of malpractice just described are raised in some way in the possible liability associated with BRCA testing. We next look at the ways in which liability may arise from that testing (or lack of it).
Underlying much of the following discussion is the “cost-benefit” consideration noted above. This concept is that the total cost (financial and health) of testing should be compared with the value of the benefits of testing, taking into account the probabilities that the testing will result in better health outcomes. BRCA testing, for example, is essentially cost-free in terms of physical risk. Its financial cost, while not trivial, is not great, and it is commonly covered by health insurance.17 In terms of benefits, the testing has the potential for providing critical information in making treatment decisions for a meaningful percentage of patients and their families. There are many ways of analyzing the liability risks of genetic malpractice,7,18 and the following is intended to discuss some of the greatest risks related to BRCA testing.
Continue to: Areas of liability...
Areas of liability
The failure to recommend a test. The circumstances in which BRCA testing should be undertaken are set out by professional organizations (noted above). These recommendations are not static, however. They change from time to time. Given the potential harm caused by the failure to test in relevant circumstances, malpractice liability is certainly a possibility when the failure to recommend a test to a patient results in a cancer that might have been prevented had the genetic problem been identified in a timely manner. The circumstances in which testing should be considered continue to change, placing an obligation on clinicians to stay well informed of changing genetic understandings. Another risk is that one specialist may assume that it is the job of another specialist to order the test. Whatever the cause of the failure to test, or unnecessary delay in testing, it appears to be the primary basis for BRCA liability.
The failure to properly interpret a test. Any test that is misinterpreted may lead to harm for the patient. A false negative, of course, may mean that preventive treatment that could have been undertaken will be foregone, as a “loss of a chance.” On the other hand, a false positive can lead to radical, unnecessary surgery or treatment. If a misinterpretation occurred because of carelessness by the testing organization, or confusion by a practitioner, there is a likelihood of negligence.19
A different form of “misinterpretation” could be reasonable—and not negligent. Advances in scientific-medical understanding may result in the outcome of tests being reconsidered and changed. That has been the case with genetic testing and breast cancer. The availability of multiple breast cancer SNPs (single nucleotide polymorphisms), and combining this information with other risk factors for example, results in a polygenic risk score that may be at odds with the level of risk from earlier testing.20,21 This naturally leads to the question of when later, updated testing should be recommended to look for a better current interpretation.22,23
The failure to act on BRCA test results. Testing is of no value, of course, if the results are not used properly. Test results or analyses that are not sent to the proper physicians, or are somehow ignored when properly directed, is a “never” event—it should never happen. It almost always would be considered negligence, and if the patient were injured, could lead to liability. Amazingly, one study found that, in genetic testing liability cases, nearly 20% of the claims arose from failure to return test results to patients.24 In addition, when a patient is found to be BRCA-positive, there is an obligation to discuss the options for dealing with the increased risk associated with the gene mutation(s), as well as to recommend the prudent course of action or to refer the patient to someone who will have that discussion.
Informed consent to the patient. BRCA testing requires informed consent. The physical risks of the testing process are minimal, of course, but it carries a number of other emotional and family risks. The informed consent process is an invitation to an honest discussion between clinicians and patients. It should be an opportunity to discuss what the testing is, and is not, and what the test may mean for treatment. It may also be an opportunity to discuss the implications for other members of the patient’s family (noted below).
One element of informed consent is a discussion of the consequences of failure to consent, or to undertake one of the alternatives. In the case of BRCA testing, this is especially important in cases in which a patient expresses a hesitancy to be tested with an “I’d rather not know philosophy.” Although clinicians should not practice law, some patient concerns about discrimination may be addressed by the protection that the federal Genetic Information Nondiscrimination Act (GINA) and other laws provide (which prohibit insurance and employment discrimination based on genetic information). A good source of information about GINA and related nondiscrimination laws is provided by the National Human Genome Research Institute.25 In addition, the National Institutes of Health has a website that may be helpful to many patients26 (and a much more complex site for health professionals).27 At the same time, courts have resisted plaintiffs/patients who have tried to use informed consent as a way of suing for failure to offer genetic testing.28,29
The failure to refer. In some cases, a patient should be formally referred for genetics consultation. The considerations here are similar to other circumstances in modern, fast developing medical practice that require special sensitivity to those occasions in which a patient will benefit from additional expertise. It is a principle that the AMA Council on Ethical and Judicial Affairs has expressed this way: “In the absence of adequate expertise in pretest and posttest counseling, a physician should refer the patient to an appropriate specialist.”30 The failure to refer, when that deviates from acceptable practice, may result in liability.
Informing others. BRCA testing is an area of medicine in which results may be of great significance not only to the patient but also to the patient’s family.31 Physicians should counsel patients on the importance of informing relatives about relevant results and “should make themselves available to assist patients in communicating with relatives to discuss opportunities for counseling and testing, as appropriate.”30 The question may arise, however, of whether in some circumstances physicians should go a step further in ensuring relatives receive important information regarding their loved one’s health.32 The law has been reluctant to impose liability to “third parties” (someone not a patient). Duties usually arise through the physician-patient relationship. There are exceptions. Perhaps the best known has been the obligation of mental health professionals to take action to protect third parties from patients who have made believable threats against identifiable victims.33 There are indications that some courts could find, in extreme circumstances, a “duty to warn” nonpatients in some instances where it is essential to inform third parties that they should receive a specific form of genetic testing.34,35 Such a duty would, of course, have to protect the privacy rights of the patient to the maximum extent possible. A general duty of this type has not been established widely, but may be part of the future.
Continue to: Was there liability in our example case?...
Was there liability in our example case?
The hypothetical case provided above suggests that there could be liability. Routine medical history by the primary care physician would have produced the fact that the patient’s mother, sister, and maternal grandmother had breast cancer. That would clearly have put her in a category of those who should have received genetic testing. Yet, she was not tested until after her cancer was found. From the limited facts we have, it appears that this timeline of events would have been outside accepted practice—and negligent. The case was not pursued by the patient, however, and this may represent the current state of liability for BRCA issues.
The extent of liability seems to be significant
Our discussion of liability suggests that there is significant potential for BRCA testing negligence within practice, and that the damages in these cases could be substantial. Yet the predicted “tsunami” of malpractice lawsuits related to genetic testing has not appeared.36,37 One study of cases in the United States (through 2016) found a “slowly rising tide” of liability cases instead of a tsunami,24 as the number of claims made was low. On the other hand, the payments where damages were awarded were an order of magnitude larger than other malpractice cases—a mean of $5.3 million and median of $2 million. This is compared with mean values in the range of $275,000 to $600,000 in other areas of malpractice.
The majority of the genetic malpractice cases involve prenatal and newborn testing, and diagnosis/susceptibility/pharmacogenomic accounting for about 25% of cases. In terms of type of errors claimed, approximately 50% were diagnostic-interpretation errors, 30% failure to offer testing, nearly 20% failure to return test results to the patients, and a few remaining cases of failure to properly treat in light of genetic testing.24
Despite a few very large payments, however, the fact remains that there is a surprisingly low number of genetics malpractice cases. Gary Marchant and colleagues suggest that several reasons may account for this:
- the clinical implementation of genetic science has been slower than expected
- the lack of expertise of many physicians in genetic science
- expert witnesses have sometimes been hard to find
- the lack of understanding by plaintiffs’ attorneys of genetic malpractice
- potential plaintiffs’ lack of understanding of the nature of genetic testing and the harms resulting from genetic negligence.17,24,37
The tide is slowly coming in
By all appearances, there is every reason to think that genetic malpractice will be increasing, and that the recent past of much higher damages per claim paid in the genetics area will be part of that tide. The National Human Genome Research LawSeq project has suggested a number of useful ways of dealing with the liability issues.18 In addition to the BRCA issues that we have considered in this article for ObGyns, there are other critical issues of prenatal and newborn genetic testing.38 But those are topics for another day. ●
- Sevilla C, Moatti JP, Reynier CJ, et al. Testing for BRCA1 mutations: a cost-effective analysis. Europ J Human Genetics. 2002;10:599-606.
- Cotton V, Kirkpatrick D. Failure to recommend genetic counseling in breast cancer: is the next wave of medical professional liability lawsuits? Contemp OB/GYN. June 1, 2017.
- Suryavanshi M, Kumar D, Panigrahi M, et al. Detection of false positive mutations in BRCA gene by next generation sequencing. Fam Cancer. 2017;16:311-317.
- Black L, Knoppers B, Avard D, et al. Legal liability and the uncertain nature of risk prediction: the case of breast cancer risk prediction models. Public Health Genomics. 2012;15:335-340.
- McClintock A, Gollab A, Laya M. Breast cancer risk assessment, a step-wise approach for primary care physicians on the front lines of shared decision making. Mayo Clin Proc. 2020;95:1268-1275.
- National Cancer Institute. The Breast Cancer Risk Assessment Tool. https://bcrisktool.cancer.gov/. Accessed February 25, 2021.
- Neff J, Richardson G, Phelps J. Legal liabilities associated with hereditary breast and ovarian cancers. J Reprod Med. 2020;65:227-230.
- American College of Obstetricians and Gynecologists. Practice Bulletin No 182: hereditary breast and ovarian cancer syndrome. Obstet Gynecol. 2017;130:e110-e126.
- Sá dos Reis C, Gremion I, and Meystre NR. Study of breast implants mammography examinations for identification of suitable image quality criteria. Insights Imaging. 2020;11:3.
- Association for Molecular Pathology v Myriad Genetics, 569 U.S. 576 (2013).
- Smith SR. The Supreme Court 2012-2013: dogs, DNA, and DOMA. Register Rep. 2013;39(Fall):26-33.
- Bal BS. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467:339-347.
- Helling v Carey, 83 Wn.2d 514, 519 P.2d 981 (1974).
- The T.J. Hooper, 60 F.2d 737, 740 (2d Cir.1932), cert. denied 287 U.S. 662 (1932).
- Fischer DA. Tort recovery for loss of a chance. Wake Forest L Rev. 2001;36:605-655.
- Murphy BL, Ray-Zack MD, Reddy PN, et al. Breast cancer litigation in the 21st century. Ann Surg Oncol. 2018;25:2939- 2947.
- Prince AE. Prevention for those who can pay: insurance reimbursement of genetic-based preventive interventions in the liminal state between health and disease. J Law Biosci. 2015;2:365-395.
- Marchant G, Barnes M, Evans JP, et al; LawSeq Liability Task Force. From genetics to genomics: facing the liability implications in clinical care. J Law Med Ethics. 2020;48:11-43.
- Complaint, Held v Ambry Genetics Corp., No. 15-CV-8683, 2015 WL 6750024 (S.D.N.Y. Nov. 4, 2015); Order of Dismissal, Held v Ambry Genetics Corp., No. 15-CV-8683, (S.D.N.Y. Dec. 6, 2016).
- Pederson HJ. Breast cancer risk assessment and treatment: current concepts in genetics and genomics. Contemp OB/ GYN. 2017; 62:A1-A4.
- Pederson HJ. Who needs breast cancer genetics testing? OBG Manag. 2018;30:34-39.
- Roberts JL, Foulkes A. Genetic duties. William Mary L Rev. 2020;62:143-212.
- Thorogood A, Cook-Deegan R, Knoppers B. Public variant databases: liability? Genet Med. 2017;19:838–841.
- Marchant G, Lindor R. Genomic malpractice: an emerging tide or gentle ripple? Food Drug Law J. 2018;73:1-37.
- National Human Genome Research Institute. Genetic discrimination. https://www.genome.gov/about-genomics /policy-issues/Genetic-Discrimination. Updated September 16, 2020. Accessed February 25, 2021.
- National Cancer Institute. BRCA mutations: cancer risk and genetic testing. https://www.cancer.gov/about-cancer /causes-prevention/genetics/brca-fact-sheet. Reviewed November 19, 2020. Accessed February 25, 2021.
- National Cancer Institute. Genetics of breast and gynecologic cancers (PDQ®)–Health Professional Version. https://www .cancer.gov/types/breast/hp/breast-ovarian-genetics-pdq. Updated February 12, 2021. Accessed February 25, 2021.
- Reed v Campagnolo, 630 A.2d 1145, 1152–54 (Md. 1993).
- Munro v Regents of Univ. of Cal.,263 Cal. Rptr. 878, 885, 988 (1989).
- AMA Council on Ethical and Judicial Affairs. AMA Code of Medical Ethics’ opinions on genetic testing. Opinion 2.131. 2009;11:683-685. https://journalofethics.ama-assn .org/article/ama-code-medical-ethics-opinions-genetictesting/2009-09.
- Gilbar R, Barnoy S. Disclosing genetic test results to the patient’ relatives: how does the law influence clinical practice? J Law Technol Policy. 2019;125-168.
- Song K. Warning third parties of genetic risks in the era of personalized medicine. U.C. Davis L Rev. 2016;49:1987-2018.
- Tarasoff v Regents of the University of California, 551 P.2d 334, 131 Cal. Rptr. 14 (Cal. 1976).
- Safer v Estate of Pack, 677 A.2d 1188 (N.J. App. 1996), cert. denied, 683 A.2d 1163 (N.J. 1996).
- Pate v Threlkel, 661 So.2d 278 (Fla. 1995).
- Rothstein MA. Liability issues in pharmacogenomics. Louisiana L Rev. 2005;66:117-124.
- Marchant G, Lindor R. Personalized medicine and genetic malpractice. Genet Med. 2013;15:921-922.
- Westbrook M. Transforming the physician’s standard of care in the context of whole genome sequencing technologies: finding guidance in best practice standards. Saint Louis U J Health Law Policy. 2015;9:111-148.

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.

Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.
Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.
Mr. Smith is Professor Emeritus and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
CASE Young woman with family history of breast cancer detects lump
Two weeks after noting a lump on her breast when her cat happened to jump on her in that spot, a 28-year-old woman (G0) went to her primary care provider. She was referred to her gynecologist; breast imaging, ultrasonography, and mammography were obtained, with microcalcifications noted. A fine needle aspiration diagnosed intraductal malignancy. The surgical breast tissue specimen was estrogen receptor (ER)- and progestogen receptor (PR)-positive and HER2-negative. Other tumor markers were obtained, including carcinoembryonic antigen, and tissue polypeptide specific antigen, p53, cathepsin D, cyclin E, and nestin, but results were not available.
With regard to family history, the woman’s mother and maternal grandmother had a history of breast cancer. The patient and her family underwent gene testing. The patient was found to be BRCA1- and BRCA2-positive; her mother was BRCA1-positive, an older sister was BRCA2-positive, and her grandmother was not tested.
The question arose in light of her family history as to why she was not tested for BRCA and appropriately counseled by her gynecologist prior to the cancer diagnosis. Litigation was initiated. While the case did not go forward regarding litigation, it is indeed a case in point. (Please note that this is a hypothetical case. It is based on a composite of several cases.)
Medical considerations
Breast cancer is the most common type of cancer affecting women in the Western world.1 Advances in clinical testing for gene mutations have escalated and allowed for identification of patients at increased risk for breast and ovarian cancer. Along with these advances come professional liability risk. After looking at the medical considerations for BRCA1 and 2 testing, we will consider a number of important legal issues. In the view of some commentators, the failure to diagnose genetic mutations in patients predisposed to cancer is “poised to become the next wave of medical professional liability lawsuits.”2
BRCA1 and BRCA2 genes provide tumor suppressor proteins, and assessment for mutations is recommended for individuals at high risk for breast and/or ovarian cancer; mutations in BRCA genes cause DNA damage, which increases the chance of developing cancer. The other way to look at it is, BRCA1 and 2 are tumor suppressor genes that are integrally involved with DNA damage control. Once there is a mutation, it adversely affects the beneficial effects of the gene. Mutations in these genes account for 5% to 10% of all hereditary breast cancers.3 Of note, men with BRCA2 are at increased risk for prostate cancer.
A patient who presents to her gynecologist stating that there is a family history of breast cancer, without knowledge of genetic components, presents a challenge (and a medicolegal risk) for the provider to assess. Prediction models have been used to determine specific patient risk for carrying a genetic mutation with resultant breast cancer development.4 Risk prediction models do not appear to be a good answer to predicting who is more likely to develop breast or ovarian cancer, however. A Mayo model may assist (FIGURE).5 Clinicians should also be aware of other models of risk assessment, including the Gail Model (TABLE 1).6
Continue to: Guidelines for genetic testing...
Guidelines for genetic testing
The American College of Obstetricians and Gynecologists states that patient medical history and family history are paramount in obtaining information regarding risk for breast and ovarian cancer. First- and second-degree relatives are allocated to this category. Information regarding age of diagnosis, maternal and paternal lineage, and ethnic background can imply a need for genetic testing (TABLE 2).7,8 A number of genetics national organizations have participated in recommendations and include the American College of Medical Genetics and Genomics, the National Society for Genetic Counselors, and the Society of Gynecologic Oncology.7
The question always surfaces, could the clinical outcome of the cancer when diagnosed have been changed if screening were undertaken, with earlier diagnosis, or prevented with prophylactic mastectomy, and changed the end result. In addition, it is well known that breast augmentation mammoplasty alters the ability to accurately evaluate mammograms. Patients considering this type of plastic surgery, ideally, should be counselled accordingly.9
Bottom line, we as clinicians must be cognizant of both ACOG and United States Preventive Services Task Force (USPSTF) recommendations regarding screening and gene testing for women considered high risk for breast cancer based on family history.7
Legal considerations
The case presented demonstrates that the discovery of the BRCA1 and BRCA2 genes, and reliable tests for determining the existence of the genes, brought with them legal issues as well as medical advantages. We look at professional liability (malpractice) questions this technology raises, and then consider the outcome of the hypothetical case. (BRCA is used here to apply broadly—not only to BRCA1 and 2 but also to PALB2, CHEK2, and similar genetic abnormalities.)
To date, the most visible BRCA legal issues covered in cases and law reviews have focused more on patent law than malpractice. The most important of these was a decision of the US Supreme Court in Association for Molecular Pathology v Myriad Genetics.10 The US Patent Office was granting patents to companies finding useful, naturally occurring segments of human DNA, and had granted Myriad several patents on BRCA1 and BRCA2 genes. This patent policy had the potential to seriously interfere with broad scientific use of these genes.11 Fortunately, the Supreme Court stepped in and unanimously invalidated such patents. It held that a “naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated.” The Court noted, “Finding the location of the BRCA1 and BRCA2 genes does not render the genes patent eligible ‘new . . . composition[s] of matter.’”8 The Court did allow the patenting of tests for specific gene structures, and artificial changes in naturally occurring genes.
Malpractice and BRCA
While the BRCA patent wars have lingered, the potential for a significant increase in BRCA-related malpractice cases is of increasing concern. Like most malpractice liability, these new claims are based on very old principles of negligence.12 To prevail, the plaintiff (ordinarily, an injured patient) must demonstrate 4 things:
- A duty. That is, the physician owed a duty to the injured party. Usually (but not always) that requires a professional relationship between the physician and the person injured.
- A breach of that duty. Malpractice liability is based on the fact that the physician did something that a reasonably careful physician (generally, of the same specialty) would not have done, or that the physician failed to do something that a reasonable physician would have done. This usually means that the profession itself sees what the physician did (or did not do) as medically inappropriate. In medical malpractice cases, that is ordinarily measured by what the usual or common practice is among prudent physicians. In rare circumstances, courts have found the standard practice of a profession to be negligent. Where, for example, it was custom for a professional not to give an eye pressure test to anyone under age 40, a court found that common standard to be inappropriate.13 In the words of Judge Learned Hand (speaking about a different case), “a whole calling may have unduly lagged in the adoption of new and available devices. It never may set its own tests.”14 Underlying negligence is a cost-benefit analysis (discussed below).
- Damages. There must have been some damage that courts recognize, usually loss of money or opportunity to work, the cost of care, pain and suffering, or loss of enjoyment/quality of life. In malpractice, many states now recognize the “loss of chance” or the “loss of a chance.” That means, if a “physician negligently fails to diagnose a curable disease, and the patient is harmed by the disease, the physician should be liable for causing the ‘loss of a chance of a cure.’”15 (Delay in diagnosis is the most common reason for claims in breast cancer care.)16
- Causation. The breach of duty (negligence) must have caused the damages. The causation must have been reasonably close. If a driver drives through a stop sign, or a physician misreads a test, and someone is injured but there is no connection between the negligence and the injury, there is not tort liability.
The 4 elements of malpractice just described are raised in some way in the possible liability associated with BRCA testing. We next look at the ways in which liability may arise from that testing (or lack of it).
Underlying much of the following discussion is the “cost-benefit” consideration noted above. This concept is that the total cost (financial and health) of testing should be compared with the value of the benefits of testing, taking into account the probabilities that the testing will result in better health outcomes. BRCA testing, for example, is essentially cost-free in terms of physical risk. Its financial cost, while not trivial, is not great, and it is commonly covered by health insurance.17 In terms of benefits, the testing has the potential for providing critical information in making treatment decisions for a meaningful percentage of patients and their families. There are many ways of analyzing the liability risks of genetic malpractice,7,18 and the following is intended to discuss some of the greatest risks related to BRCA testing.
Continue to: Areas of liability...
Areas of liability
The failure to recommend a test. The circumstances in which BRCA testing should be undertaken are set out by professional organizations (noted above). These recommendations are not static, however. They change from time to time. Given the potential harm caused by the failure to test in relevant circumstances, malpractice liability is certainly a possibility when the failure to recommend a test to a patient results in a cancer that might have been prevented had the genetic problem been identified in a timely manner. The circumstances in which testing should be considered continue to change, placing an obligation on clinicians to stay well informed of changing genetic understandings. Another risk is that one specialist may assume that it is the job of another specialist to order the test. Whatever the cause of the failure to test, or unnecessary delay in testing, it appears to be the primary basis for BRCA liability.
The failure to properly interpret a test. Any test that is misinterpreted may lead to harm for the patient. A false negative, of course, may mean that preventive treatment that could have been undertaken will be foregone, as a “loss of a chance.” On the other hand, a false positive can lead to radical, unnecessary surgery or treatment. If a misinterpretation occurred because of carelessness by the testing organization, or confusion by a practitioner, there is a likelihood of negligence.19
A different form of “misinterpretation” could be reasonable—and not negligent. Advances in scientific-medical understanding may result in the outcome of tests being reconsidered and changed. That has been the case with genetic testing and breast cancer. The availability of multiple breast cancer SNPs (single nucleotide polymorphisms), and combining this information with other risk factors for example, results in a polygenic risk score that may be at odds with the level of risk from earlier testing.20,21 This naturally leads to the question of when later, updated testing should be recommended to look for a better current interpretation.22,23
The failure to act on BRCA test results. Testing is of no value, of course, if the results are not used properly. Test results or analyses that are not sent to the proper physicians, or are somehow ignored when properly directed, is a “never” event—it should never happen. It almost always would be considered negligence, and if the patient were injured, could lead to liability. Amazingly, one study found that, in genetic testing liability cases, nearly 20% of the claims arose from failure to return test results to patients.24 In addition, when a patient is found to be BRCA-positive, there is an obligation to discuss the options for dealing with the increased risk associated with the gene mutation(s), as well as to recommend the prudent course of action or to refer the patient to someone who will have that discussion.
Informed consent to the patient. BRCA testing requires informed consent. The physical risks of the testing process are minimal, of course, but it carries a number of other emotional and family risks. The informed consent process is an invitation to an honest discussion between clinicians and patients. It should be an opportunity to discuss what the testing is, and is not, and what the test may mean for treatment. It may also be an opportunity to discuss the implications for other members of the patient’s family (noted below).
One element of informed consent is a discussion of the consequences of failure to consent, or to undertake one of the alternatives. In the case of BRCA testing, this is especially important in cases in which a patient expresses a hesitancy to be tested with an “I’d rather not know philosophy.” Although clinicians should not practice law, some patient concerns about discrimination may be addressed by the protection that the federal Genetic Information Nondiscrimination Act (GINA) and other laws provide (which prohibit insurance and employment discrimination based on genetic information). A good source of information about GINA and related nondiscrimination laws is provided by the National Human Genome Research Institute.25 In addition, the National Institutes of Health has a website that may be helpful to many patients26 (and a much more complex site for health professionals).27 At the same time, courts have resisted plaintiffs/patients who have tried to use informed consent as a way of suing for failure to offer genetic testing.28,29
The failure to refer. In some cases, a patient should be formally referred for genetics consultation. The considerations here are similar to other circumstances in modern, fast developing medical practice that require special sensitivity to those occasions in which a patient will benefit from additional expertise. It is a principle that the AMA Council on Ethical and Judicial Affairs has expressed this way: “In the absence of adequate expertise in pretest and posttest counseling, a physician should refer the patient to an appropriate specialist.”30 The failure to refer, when that deviates from acceptable practice, may result in liability.
Informing others. BRCA testing is an area of medicine in which results may be of great significance not only to the patient but also to the patient’s family.31 Physicians should counsel patients on the importance of informing relatives about relevant results and “should make themselves available to assist patients in communicating with relatives to discuss opportunities for counseling and testing, as appropriate.”30 The question may arise, however, of whether in some circumstances physicians should go a step further in ensuring relatives receive important information regarding their loved one’s health.32 The law has been reluctant to impose liability to “third parties” (someone not a patient). Duties usually arise through the physician-patient relationship. There are exceptions. Perhaps the best known has been the obligation of mental health professionals to take action to protect third parties from patients who have made believable threats against identifiable victims.33 There are indications that some courts could find, in extreme circumstances, a “duty to warn” nonpatients in some instances where it is essential to inform third parties that they should receive a specific form of genetic testing.34,35 Such a duty would, of course, have to protect the privacy rights of the patient to the maximum extent possible. A general duty of this type has not been established widely, but may be part of the future.
Continue to: Was there liability in our example case?...
Was there liability in our example case?
The hypothetical case provided above suggests that there could be liability. Routine medical history by the primary care physician would have produced the fact that the patient’s mother, sister, and maternal grandmother had breast cancer. That would clearly have put her in a category of those who should have received genetic testing. Yet, she was not tested until after her cancer was found. From the limited facts we have, it appears that this timeline of events would have been outside accepted practice—and negligent. The case was not pursued by the patient, however, and this may represent the current state of liability for BRCA issues.
The extent of liability seems to be significant
Our discussion of liability suggests that there is significant potential for BRCA testing negligence within practice, and that the damages in these cases could be substantial. Yet the predicted “tsunami” of malpractice lawsuits related to genetic testing has not appeared.36,37 One study of cases in the United States (through 2016) found a “slowly rising tide” of liability cases instead of a tsunami,24 as the number of claims made was low. On the other hand, the payments where damages were awarded were an order of magnitude larger than other malpractice cases—a mean of $5.3 million and median of $2 million. This is compared with mean values in the range of $275,000 to $600,000 in other areas of malpractice.
The majority of the genetic malpractice cases involve prenatal and newborn testing, and diagnosis/susceptibility/pharmacogenomic accounting for about 25% of cases. In terms of type of errors claimed, approximately 50% were diagnostic-interpretation errors, 30% failure to offer testing, nearly 20% failure to return test results to the patients, and a few remaining cases of failure to properly treat in light of genetic testing.24
Despite a few very large payments, however, the fact remains that there is a surprisingly low number of genetics malpractice cases. Gary Marchant and colleagues suggest that several reasons may account for this:
- the clinical implementation of genetic science has been slower than expected
- the lack of expertise of many physicians in genetic science
- expert witnesses have sometimes been hard to find
- the lack of understanding by plaintiffs’ attorneys of genetic malpractice
- potential plaintiffs’ lack of understanding of the nature of genetic testing and the harms resulting from genetic negligence.17,24,37
The tide is slowly coming in
By all appearances, there is every reason to think that genetic malpractice will be increasing, and that the recent past of much higher damages per claim paid in the genetics area will be part of that tide. The National Human Genome Research LawSeq project has suggested a number of useful ways of dealing with the liability issues.18 In addition to the BRCA issues that we have considered in this article for ObGyns, there are other critical issues of prenatal and newborn genetic testing.38 But those are topics for another day. ●
CASE Young woman with family history of breast cancer detects lump
Two weeks after noting a lump on her breast when her cat happened to jump on her in that spot, a 28-year-old woman (G0) went to her primary care provider. She was referred to her gynecologist; breast imaging, ultrasonography, and mammography were obtained, with microcalcifications noted. A fine needle aspiration diagnosed intraductal malignancy. The surgical breast tissue specimen was estrogen receptor (ER)- and progestogen receptor (PR)-positive and HER2-negative. Other tumor markers were obtained, including carcinoembryonic antigen, and tissue polypeptide specific antigen, p53, cathepsin D, cyclin E, and nestin, but results were not available.
With regard to family history, the woman’s mother and maternal grandmother had a history of breast cancer. The patient and her family underwent gene testing. The patient was found to be BRCA1- and BRCA2-positive; her mother was BRCA1-positive, an older sister was BRCA2-positive, and her grandmother was not tested.
The question arose in light of her family history as to why she was not tested for BRCA and appropriately counseled by her gynecologist prior to the cancer diagnosis. Litigation was initiated. While the case did not go forward regarding litigation, it is indeed a case in point. (Please note that this is a hypothetical case. It is based on a composite of several cases.)
Medical considerations
Breast cancer is the most common type of cancer affecting women in the Western world.1 Advances in clinical testing for gene mutations have escalated and allowed for identification of patients at increased risk for breast and ovarian cancer. Along with these advances come professional liability risk. After looking at the medical considerations for BRCA1 and 2 testing, we will consider a number of important legal issues. In the view of some commentators, the failure to diagnose genetic mutations in patients predisposed to cancer is “poised to become the next wave of medical professional liability lawsuits.”2
BRCA1 and BRCA2 genes provide tumor suppressor proteins, and assessment for mutations is recommended for individuals at high risk for breast and/or ovarian cancer; mutations in BRCA genes cause DNA damage, which increases the chance of developing cancer. The other way to look at it is, BRCA1 and 2 are tumor suppressor genes that are integrally involved with DNA damage control. Once there is a mutation, it adversely affects the beneficial effects of the gene. Mutations in these genes account for 5% to 10% of all hereditary breast cancers.3 Of note, men with BRCA2 are at increased risk for prostate cancer.
A patient who presents to her gynecologist stating that there is a family history of breast cancer, without knowledge of genetic components, presents a challenge (and a medicolegal risk) for the provider to assess. Prediction models have been used to determine specific patient risk for carrying a genetic mutation with resultant breast cancer development.4 Risk prediction models do not appear to be a good answer to predicting who is more likely to develop breast or ovarian cancer, however. A Mayo model may assist (FIGURE).5 Clinicians should also be aware of other models of risk assessment, including the Gail Model (TABLE 1).6
Continue to: Guidelines for genetic testing...
Guidelines for genetic testing
The American College of Obstetricians and Gynecologists states that patient medical history and family history are paramount in obtaining information regarding risk for breast and ovarian cancer. First- and second-degree relatives are allocated to this category. Information regarding age of diagnosis, maternal and paternal lineage, and ethnic background can imply a need for genetic testing (TABLE 2).7,8 A number of genetics national organizations have participated in recommendations and include the American College of Medical Genetics and Genomics, the National Society for Genetic Counselors, and the Society of Gynecologic Oncology.7
The question always surfaces, could the clinical outcome of the cancer when diagnosed have been changed if screening were undertaken, with earlier diagnosis, or prevented with prophylactic mastectomy, and changed the end result. In addition, it is well known that breast augmentation mammoplasty alters the ability to accurately evaluate mammograms. Patients considering this type of plastic surgery, ideally, should be counselled accordingly.9
Bottom line, we as clinicians must be cognizant of both ACOG and United States Preventive Services Task Force (USPSTF) recommendations regarding screening and gene testing for women considered high risk for breast cancer based on family history.7
Legal considerations
The case presented demonstrates that the discovery of the BRCA1 and BRCA2 genes, and reliable tests for determining the existence of the genes, brought with them legal issues as well as medical advantages. We look at professional liability (malpractice) questions this technology raises, and then consider the outcome of the hypothetical case. (BRCA is used here to apply broadly—not only to BRCA1 and 2 but also to PALB2, CHEK2, and similar genetic abnormalities.)
To date, the most visible BRCA legal issues covered in cases and law reviews have focused more on patent law than malpractice. The most important of these was a decision of the US Supreme Court in Association for Molecular Pathology v Myriad Genetics.10 The US Patent Office was granting patents to companies finding useful, naturally occurring segments of human DNA, and had granted Myriad several patents on BRCA1 and BRCA2 genes. This patent policy had the potential to seriously interfere with broad scientific use of these genes.11 Fortunately, the Supreme Court stepped in and unanimously invalidated such patents. It held that a “naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated.” The Court noted, “Finding the location of the BRCA1 and BRCA2 genes does not render the genes patent eligible ‘new . . . composition[s] of matter.’”8 The Court did allow the patenting of tests for specific gene structures, and artificial changes in naturally occurring genes.
Malpractice and BRCA
While the BRCA patent wars have lingered, the potential for a significant increase in BRCA-related malpractice cases is of increasing concern. Like most malpractice liability, these new claims are based on very old principles of negligence.12 To prevail, the plaintiff (ordinarily, an injured patient) must demonstrate 4 things:
- A duty. That is, the physician owed a duty to the injured party. Usually (but not always) that requires a professional relationship between the physician and the person injured.
- A breach of that duty. Malpractice liability is based on the fact that the physician did something that a reasonably careful physician (generally, of the same specialty) would not have done, or that the physician failed to do something that a reasonable physician would have done. This usually means that the profession itself sees what the physician did (or did not do) as medically inappropriate. In medical malpractice cases, that is ordinarily measured by what the usual or common practice is among prudent physicians. In rare circumstances, courts have found the standard practice of a profession to be negligent. Where, for example, it was custom for a professional not to give an eye pressure test to anyone under age 40, a court found that common standard to be inappropriate.13 In the words of Judge Learned Hand (speaking about a different case), “a whole calling may have unduly lagged in the adoption of new and available devices. It never may set its own tests.”14 Underlying negligence is a cost-benefit analysis (discussed below).
- Damages. There must have been some damage that courts recognize, usually loss of money or opportunity to work, the cost of care, pain and suffering, or loss of enjoyment/quality of life. In malpractice, many states now recognize the “loss of chance” or the “loss of a chance.” That means, if a “physician negligently fails to diagnose a curable disease, and the patient is harmed by the disease, the physician should be liable for causing the ‘loss of a chance of a cure.’”15 (Delay in diagnosis is the most common reason for claims in breast cancer care.)16
- Causation. The breach of duty (negligence) must have caused the damages. The causation must have been reasonably close. If a driver drives through a stop sign, or a physician misreads a test, and someone is injured but there is no connection between the negligence and the injury, there is not tort liability.
The 4 elements of malpractice just described are raised in some way in the possible liability associated with BRCA testing. We next look at the ways in which liability may arise from that testing (or lack of it).
Underlying much of the following discussion is the “cost-benefit” consideration noted above. This concept is that the total cost (financial and health) of testing should be compared with the value of the benefits of testing, taking into account the probabilities that the testing will result in better health outcomes. BRCA testing, for example, is essentially cost-free in terms of physical risk. Its financial cost, while not trivial, is not great, and it is commonly covered by health insurance.17 In terms of benefits, the testing has the potential for providing critical information in making treatment decisions for a meaningful percentage of patients and their families. There are many ways of analyzing the liability risks of genetic malpractice,7,18 and the following is intended to discuss some of the greatest risks related to BRCA testing.
Continue to: Areas of liability...
Areas of liability
The failure to recommend a test. The circumstances in which BRCA testing should be undertaken are set out by professional organizations (noted above). These recommendations are not static, however. They change from time to time. Given the potential harm caused by the failure to test in relevant circumstances, malpractice liability is certainly a possibility when the failure to recommend a test to a patient results in a cancer that might have been prevented had the genetic problem been identified in a timely manner. The circumstances in which testing should be considered continue to change, placing an obligation on clinicians to stay well informed of changing genetic understandings. Another risk is that one specialist may assume that it is the job of another specialist to order the test. Whatever the cause of the failure to test, or unnecessary delay in testing, it appears to be the primary basis for BRCA liability.
The failure to properly interpret a test. Any test that is misinterpreted may lead to harm for the patient. A false negative, of course, may mean that preventive treatment that could have been undertaken will be foregone, as a “loss of a chance.” On the other hand, a false positive can lead to radical, unnecessary surgery or treatment. If a misinterpretation occurred because of carelessness by the testing organization, or confusion by a practitioner, there is a likelihood of negligence.19
A different form of “misinterpretation” could be reasonable—and not negligent. Advances in scientific-medical understanding may result in the outcome of tests being reconsidered and changed. That has been the case with genetic testing and breast cancer. The availability of multiple breast cancer SNPs (single nucleotide polymorphisms), and combining this information with other risk factors for example, results in a polygenic risk score that may be at odds with the level of risk from earlier testing.20,21 This naturally leads to the question of when later, updated testing should be recommended to look for a better current interpretation.22,23
The failure to act on BRCA test results. Testing is of no value, of course, if the results are not used properly. Test results or analyses that are not sent to the proper physicians, or are somehow ignored when properly directed, is a “never” event—it should never happen. It almost always would be considered negligence, and if the patient were injured, could lead to liability. Amazingly, one study found that, in genetic testing liability cases, nearly 20% of the claims arose from failure to return test results to patients.24 In addition, when a patient is found to be BRCA-positive, there is an obligation to discuss the options for dealing with the increased risk associated with the gene mutation(s), as well as to recommend the prudent course of action or to refer the patient to someone who will have that discussion.
Informed consent to the patient. BRCA testing requires informed consent. The physical risks of the testing process are minimal, of course, but it carries a number of other emotional and family risks. The informed consent process is an invitation to an honest discussion between clinicians and patients. It should be an opportunity to discuss what the testing is, and is not, and what the test may mean for treatment. It may also be an opportunity to discuss the implications for other members of the patient’s family (noted below).
One element of informed consent is a discussion of the consequences of failure to consent, or to undertake one of the alternatives. In the case of BRCA testing, this is especially important in cases in which a patient expresses a hesitancy to be tested with an “I’d rather not know philosophy.” Although clinicians should not practice law, some patient concerns about discrimination may be addressed by the protection that the federal Genetic Information Nondiscrimination Act (GINA) and other laws provide (which prohibit insurance and employment discrimination based on genetic information). A good source of information about GINA and related nondiscrimination laws is provided by the National Human Genome Research Institute.25 In addition, the National Institutes of Health has a website that may be helpful to many patients26 (and a much more complex site for health professionals).27 At the same time, courts have resisted plaintiffs/patients who have tried to use informed consent as a way of suing for failure to offer genetic testing.28,29
The failure to refer. In some cases, a patient should be formally referred for genetics consultation. The considerations here are similar to other circumstances in modern, fast developing medical practice that require special sensitivity to those occasions in which a patient will benefit from additional expertise. It is a principle that the AMA Council on Ethical and Judicial Affairs has expressed this way: “In the absence of adequate expertise in pretest and posttest counseling, a physician should refer the patient to an appropriate specialist.”30 The failure to refer, when that deviates from acceptable practice, may result in liability.
Informing others. BRCA testing is an area of medicine in which results may be of great significance not only to the patient but also to the patient’s family.31 Physicians should counsel patients on the importance of informing relatives about relevant results and “should make themselves available to assist patients in communicating with relatives to discuss opportunities for counseling and testing, as appropriate.”30 The question may arise, however, of whether in some circumstances physicians should go a step further in ensuring relatives receive important information regarding their loved one’s health.32 The law has been reluctant to impose liability to “third parties” (someone not a patient). Duties usually arise through the physician-patient relationship. There are exceptions. Perhaps the best known has been the obligation of mental health professionals to take action to protect third parties from patients who have made believable threats against identifiable victims.33 There are indications that some courts could find, in extreme circumstances, a “duty to warn” nonpatients in some instances where it is essential to inform third parties that they should receive a specific form of genetic testing.34,35 Such a duty would, of course, have to protect the privacy rights of the patient to the maximum extent possible. A general duty of this type has not been established widely, but may be part of the future.
Continue to: Was there liability in our example case?...
Was there liability in our example case?
The hypothetical case provided above suggests that there could be liability. Routine medical history by the primary care physician would have produced the fact that the patient’s mother, sister, and maternal grandmother had breast cancer. That would clearly have put her in a category of those who should have received genetic testing. Yet, she was not tested until after her cancer was found. From the limited facts we have, it appears that this timeline of events would have been outside accepted practice—and negligent. The case was not pursued by the patient, however, and this may represent the current state of liability for BRCA issues.
The extent of liability seems to be significant
Our discussion of liability suggests that there is significant potential for BRCA testing negligence within practice, and that the damages in these cases could be substantial. Yet the predicted “tsunami” of malpractice lawsuits related to genetic testing has not appeared.36,37 One study of cases in the United States (through 2016) found a “slowly rising tide” of liability cases instead of a tsunami,24 as the number of claims made was low. On the other hand, the payments where damages were awarded were an order of magnitude larger than other malpractice cases—a mean of $5.3 million and median of $2 million. This is compared with mean values in the range of $275,000 to $600,000 in other areas of malpractice.
The majority of the genetic malpractice cases involve prenatal and newborn testing, and diagnosis/susceptibility/pharmacogenomic accounting for about 25% of cases. In terms of type of errors claimed, approximately 50% were diagnostic-interpretation errors, 30% failure to offer testing, nearly 20% failure to return test results to the patients, and a few remaining cases of failure to properly treat in light of genetic testing.24
Despite a few very large payments, however, the fact remains that there is a surprisingly low number of genetics malpractice cases. Gary Marchant and colleagues suggest that several reasons may account for this:
- the clinical implementation of genetic science has been slower than expected
- the lack of expertise of many physicians in genetic science
- expert witnesses have sometimes been hard to find
- the lack of understanding by plaintiffs’ attorneys of genetic malpractice
- potential plaintiffs’ lack of understanding of the nature of genetic testing and the harms resulting from genetic negligence.17,24,37
The tide is slowly coming in
By all appearances, there is every reason to think that genetic malpractice will be increasing, and that the recent past of much higher damages per claim paid in the genetics area will be part of that tide. The National Human Genome Research LawSeq project has suggested a number of useful ways of dealing with the liability issues.18 In addition to the BRCA issues that we have considered in this article for ObGyns, there are other critical issues of prenatal and newborn genetic testing.38 But those are topics for another day. ●
- Sevilla C, Moatti JP, Reynier CJ, et al. Testing for BRCA1 mutations: a cost-effective analysis. Europ J Human Genetics. 2002;10:599-606.
- Cotton V, Kirkpatrick D. Failure to recommend genetic counseling in breast cancer: is the next wave of medical professional liability lawsuits? Contemp OB/GYN. June 1, 2017.
- Suryavanshi M, Kumar D, Panigrahi M, et al. Detection of false positive mutations in BRCA gene by next generation sequencing. Fam Cancer. 2017;16:311-317.
- Black L, Knoppers B, Avard D, et al. Legal liability and the uncertain nature of risk prediction: the case of breast cancer risk prediction models. Public Health Genomics. 2012;15:335-340.
- McClintock A, Gollab A, Laya M. Breast cancer risk assessment, a step-wise approach for primary care physicians on the front lines of shared decision making. Mayo Clin Proc. 2020;95:1268-1275.
- National Cancer Institute. The Breast Cancer Risk Assessment Tool. https://bcrisktool.cancer.gov/. Accessed February 25, 2021.
- Neff J, Richardson G, Phelps J. Legal liabilities associated with hereditary breast and ovarian cancers. J Reprod Med. 2020;65:227-230.
- American College of Obstetricians and Gynecologists. Practice Bulletin No 182: hereditary breast and ovarian cancer syndrome. Obstet Gynecol. 2017;130:e110-e126.
- Sá dos Reis C, Gremion I, and Meystre NR. Study of breast implants mammography examinations for identification of suitable image quality criteria. Insights Imaging. 2020;11:3.
- Association for Molecular Pathology v Myriad Genetics, 569 U.S. 576 (2013).
- Smith SR. The Supreme Court 2012-2013: dogs, DNA, and DOMA. Register Rep. 2013;39(Fall):26-33.
- Bal BS. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467:339-347.
- Helling v Carey, 83 Wn.2d 514, 519 P.2d 981 (1974).
- The T.J. Hooper, 60 F.2d 737, 740 (2d Cir.1932), cert. denied 287 U.S. 662 (1932).
- Fischer DA. Tort recovery for loss of a chance. Wake Forest L Rev. 2001;36:605-655.
- Murphy BL, Ray-Zack MD, Reddy PN, et al. Breast cancer litigation in the 21st century. Ann Surg Oncol. 2018;25:2939- 2947.
- Prince AE. Prevention for those who can pay: insurance reimbursement of genetic-based preventive interventions in the liminal state between health and disease. J Law Biosci. 2015;2:365-395.
- Marchant G, Barnes M, Evans JP, et al; LawSeq Liability Task Force. From genetics to genomics: facing the liability implications in clinical care. J Law Med Ethics. 2020;48:11-43.
- Complaint, Held v Ambry Genetics Corp., No. 15-CV-8683, 2015 WL 6750024 (S.D.N.Y. Nov. 4, 2015); Order of Dismissal, Held v Ambry Genetics Corp., No. 15-CV-8683, (S.D.N.Y. Dec. 6, 2016).
- Pederson HJ. Breast cancer risk assessment and treatment: current concepts in genetics and genomics. Contemp OB/ GYN. 2017; 62:A1-A4.
- Pederson HJ. Who needs breast cancer genetics testing? OBG Manag. 2018;30:34-39.
- Roberts JL, Foulkes A. Genetic duties. William Mary L Rev. 2020;62:143-212.
- Thorogood A, Cook-Deegan R, Knoppers B. Public variant databases: liability? Genet Med. 2017;19:838–841.
- Marchant G, Lindor R. Genomic malpractice: an emerging tide or gentle ripple? Food Drug Law J. 2018;73:1-37.
- National Human Genome Research Institute. Genetic discrimination. https://www.genome.gov/about-genomics /policy-issues/Genetic-Discrimination. Updated September 16, 2020. Accessed February 25, 2021.
- National Cancer Institute. BRCA mutations: cancer risk and genetic testing. https://www.cancer.gov/about-cancer /causes-prevention/genetics/brca-fact-sheet. Reviewed November 19, 2020. Accessed February 25, 2021.
- National Cancer Institute. Genetics of breast and gynecologic cancers (PDQ®)–Health Professional Version. https://www .cancer.gov/types/breast/hp/breast-ovarian-genetics-pdq. Updated February 12, 2021. Accessed February 25, 2021.
- Reed v Campagnolo, 630 A.2d 1145, 1152–54 (Md. 1993).
- Munro v Regents of Univ. of Cal.,263 Cal. Rptr. 878, 885, 988 (1989).
- AMA Council on Ethical and Judicial Affairs. AMA Code of Medical Ethics’ opinions on genetic testing. Opinion 2.131. 2009;11:683-685. https://journalofethics.ama-assn .org/article/ama-code-medical-ethics-opinions-genetictesting/2009-09.
- Gilbar R, Barnoy S. Disclosing genetic test results to the patient’ relatives: how does the law influence clinical practice? J Law Technol Policy. 2019;125-168.
- Song K. Warning third parties of genetic risks in the era of personalized medicine. U.C. Davis L Rev. 2016;49:1987-2018.
- Tarasoff v Regents of the University of California, 551 P.2d 334, 131 Cal. Rptr. 14 (Cal. 1976).
- Safer v Estate of Pack, 677 A.2d 1188 (N.J. App. 1996), cert. denied, 683 A.2d 1163 (N.J. 1996).
- Pate v Threlkel, 661 So.2d 278 (Fla. 1995).
- Rothstein MA. Liability issues in pharmacogenomics. Louisiana L Rev. 2005;66:117-124.
- Marchant G, Lindor R. Personalized medicine and genetic malpractice. Genet Med. 2013;15:921-922.
- Westbrook M. Transforming the physician’s standard of care in the context of whole genome sequencing technologies: finding guidance in best practice standards. Saint Louis U J Health Law Policy. 2015;9:111-148.
- Sevilla C, Moatti JP, Reynier CJ, et al. Testing for BRCA1 mutations: a cost-effective analysis. Europ J Human Genetics. 2002;10:599-606.
- Cotton V, Kirkpatrick D. Failure to recommend genetic counseling in breast cancer: is the next wave of medical professional liability lawsuits? Contemp OB/GYN. June 1, 2017.
- Suryavanshi M, Kumar D, Panigrahi M, et al. Detection of false positive mutations in BRCA gene by next generation sequencing. Fam Cancer. 2017;16:311-317.
- Black L, Knoppers B, Avard D, et al. Legal liability and the uncertain nature of risk prediction: the case of breast cancer risk prediction models. Public Health Genomics. 2012;15:335-340.
- McClintock A, Gollab A, Laya M. Breast cancer risk assessment, a step-wise approach for primary care physicians on the front lines of shared decision making. Mayo Clin Proc. 2020;95:1268-1275.
- National Cancer Institute. The Breast Cancer Risk Assessment Tool. https://bcrisktool.cancer.gov/. Accessed February 25, 2021.
- Neff J, Richardson G, Phelps J. Legal liabilities associated with hereditary breast and ovarian cancers. J Reprod Med. 2020;65:227-230.
- American College of Obstetricians and Gynecologists. Practice Bulletin No 182: hereditary breast and ovarian cancer syndrome. Obstet Gynecol. 2017;130:e110-e126.
- Sá dos Reis C, Gremion I, and Meystre NR. Study of breast implants mammography examinations for identification of suitable image quality criteria. Insights Imaging. 2020;11:3.
- Association for Molecular Pathology v Myriad Genetics, 569 U.S. 576 (2013).
- Smith SR. The Supreme Court 2012-2013: dogs, DNA, and DOMA. Register Rep. 2013;39(Fall):26-33.
- Bal BS. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467:339-347.
- Helling v Carey, 83 Wn.2d 514, 519 P.2d 981 (1974).
- The T.J. Hooper, 60 F.2d 737, 740 (2d Cir.1932), cert. denied 287 U.S. 662 (1932).
- Fischer DA. Tort recovery for loss of a chance. Wake Forest L Rev. 2001;36:605-655.
- Murphy BL, Ray-Zack MD, Reddy PN, et al. Breast cancer litigation in the 21st century. Ann Surg Oncol. 2018;25:2939- 2947.
- Prince AE. Prevention for those who can pay: insurance reimbursement of genetic-based preventive interventions in the liminal state between health and disease. J Law Biosci. 2015;2:365-395.
- Marchant G, Barnes M, Evans JP, et al; LawSeq Liability Task Force. From genetics to genomics: facing the liability implications in clinical care. J Law Med Ethics. 2020;48:11-43.
- Complaint, Held v Ambry Genetics Corp., No. 15-CV-8683, 2015 WL 6750024 (S.D.N.Y. Nov. 4, 2015); Order of Dismissal, Held v Ambry Genetics Corp., No. 15-CV-8683, (S.D.N.Y. Dec. 6, 2016).
- Pederson HJ. Breast cancer risk assessment and treatment: current concepts in genetics and genomics. Contemp OB/ GYN. 2017; 62:A1-A4.
- Pederson HJ. Who needs breast cancer genetics testing? OBG Manag. 2018;30:34-39.
- Roberts JL, Foulkes A. Genetic duties. William Mary L Rev. 2020;62:143-212.
- Thorogood A, Cook-Deegan R, Knoppers B. Public variant databases: liability? Genet Med. 2017;19:838–841.
- Marchant G, Lindor R. Genomic malpractice: an emerging tide or gentle ripple? Food Drug Law J. 2018;73:1-37.
- National Human Genome Research Institute. Genetic discrimination. https://www.genome.gov/about-genomics /policy-issues/Genetic-Discrimination. Updated September 16, 2020. Accessed February 25, 2021.
- National Cancer Institute. BRCA mutations: cancer risk and genetic testing. https://www.cancer.gov/about-cancer /causes-prevention/genetics/brca-fact-sheet. Reviewed November 19, 2020. Accessed February 25, 2021.
- National Cancer Institute. Genetics of breast and gynecologic cancers (PDQ®)–Health Professional Version. https://www .cancer.gov/types/breast/hp/breast-ovarian-genetics-pdq. Updated February 12, 2021. Accessed February 25, 2021.
- Reed v Campagnolo, 630 A.2d 1145, 1152–54 (Md. 1993).
- Munro v Regents of Univ. of Cal.,263 Cal. Rptr. 878, 885, 988 (1989).
- AMA Council on Ethical and Judicial Affairs. AMA Code of Medical Ethics’ opinions on genetic testing. Opinion 2.131. 2009;11:683-685. https://journalofethics.ama-assn .org/article/ama-code-medical-ethics-opinions-genetictesting/2009-09.
- Gilbar R, Barnoy S. Disclosing genetic test results to the patient’ relatives: how does the law influence clinical practice? J Law Technol Policy. 2019;125-168.
- Song K. Warning third parties of genetic risks in the era of personalized medicine. U.C. Davis L Rev. 2016;49:1987-2018.
- Tarasoff v Regents of the University of California, 551 P.2d 334, 131 Cal. Rptr. 14 (Cal. 1976).
- Safer v Estate of Pack, 677 A.2d 1188 (N.J. App. 1996), cert. denied, 683 A.2d 1163 (N.J. 1996).
- Pate v Threlkel, 661 So.2d 278 (Fla. 1995).
- Rothstein MA. Liability issues in pharmacogenomics. Louisiana L Rev. 2005;66:117-124.
- Marchant G, Lindor R. Personalized medicine and genetic malpractice. Genet Med. 2013;15:921-922.
- Westbrook M. Transforming the physician’s standard of care in the context of whole genome sequencing technologies: finding guidance in best practice standards. Saint Louis U J Health Law Policy. 2015;9:111-148.
FDA authorizes first molecular at-home, OTC COVID-19 test
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.