User login
Half of psychiatry, psychology trial abstracts contain spin
About half of papers in a sample of psychiatry and psychology journals have evidence of “spin” in the abstract, according to an analysis published in BMJ Evidence-Based Medicine.
Samuel Jellison, a third-year medical student at Oklahoma State University, Tulsa, and coauthors wrote that the results of randomized, controlled trials should be reported objectively because of their importance for psychiatry clinical practice. However, given that researchers were allowed more latitude in the abstract of a paper to highlight certain results or conclusions, the abstract might not accurately represent the findings of the study.
To evaluate this, the authors investigated the use of spin in abstracts, which they defined as “‘use of specific reporting strategies, from whatever motive, to highlight that the experimental treatment is beneficial, despite a statistically nonsignificant difference for the primary outcome, or to distract the reader from statistically nonsignificant results.”
They analyzed 116 randomized, controlled trials of interventions where there was a nonsignificant primary endpoint and found that 56% of those contained spin in the abstract. Spin was evident in 2% of publication titles, 21% of abstract results sections, and 49.1% of abstract conclusions sections.
Spin was more common in trials of pharmacologic treatments, compared with nonpharmacologic treatments. However, the study did not find a higher level of spin in industry-funded studies, and in fact, spin was more common in publicly funded research.
The most common form of spin was focusing on a statistically significant primary or secondary endpoint while omitting one or more nonsignificant primary endpoints. Other spin strategies included claiming noninferiority or equivalence for a statistically nonsignificant endpoint; using phrases such as “trend towards significance”; or focusing on statistically significant subgroup analyses, such as per protocol instead of intention to treat.
The authors observed that most physicians only read article abstracts, and up to one-quarter of editorial decisions are based on the abstract alone.
“Adding spin to the abstract of an article may mislead physicians who are attempting to draw conclusions about a treatment for patients,” they wrote, while calling for efforts to discourage spin in abstracts.
In an interview, Paul S. Nestadt, MD, said the findings were not surprising.
“The proportion [56%] of psychiatry and psychology abstracts which Jellison et al. found to contain spin is similar to that found in broader studies of all biomedical literature in previous reviews,” said Dr. Nestadt, assistant professor in the department of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore. “It is disheartening that attempts to mislead have become ‘standard of care’ in medical literature, but it is a predictable outcome of increasing competition for shrinking grant funding, awarded partly on the basis of publication history in leading journals that maintain clear publication biases toward positive results.
“As the authors point out, we all share a responsibility to call out spin when we see it, whether in our role as reviewer, editor, coauthor, or as the writers themselves.”
Neither Mr. Jellison nor his coauthors reported funding or conflicts of interest.
SOURCE: Jellison S et al. BMJ Evid Based Med. 2019 Aug 5. doi: 10.1136/bmjebm-2019-111176.
About half of papers in a sample of psychiatry and psychology journals have evidence of “spin” in the abstract, according to an analysis published in BMJ Evidence-Based Medicine.
Samuel Jellison, a third-year medical student at Oklahoma State University, Tulsa, and coauthors wrote that the results of randomized, controlled trials should be reported objectively because of their importance for psychiatry clinical practice. However, given that researchers were allowed more latitude in the abstract of a paper to highlight certain results or conclusions, the abstract might not accurately represent the findings of the study.
To evaluate this, the authors investigated the use of spin in abstracts, which they defined as “‘use of specific reporting strategies, from whatever motive, to highlight that the experimental treatment is beneficial, despite a statistically nonsignificant difference for the primary outcome, or to distract the reader from statistically nonsignificant results.”
They analyzed 116 randomized, controlled trials of interventions where there was a nonsignificant primary endpoint and found that 56% of those contained spin in the abstract. Spin was evident in 2% of publication titles, 21% of abstract results sections, and 49.1% of abstract conclusions sections.
Spin was more common in trials of pharmacologic treatments, compared with nonpharmacologic treatments. However, the study did not find a higher level of spin in industry-funded studies, and in fact, spin was more common in publicly funded research.
The most common form of spin was focusing on a statistically significant primary or secondary endpoint while omitting one or more nonsignificant primary endpoints. Other spin strategies included claiming noninferiority or equivalence for a statistically nonsignificant endpoint; using phrases such as “trend towards significance”; or focusing on statistically significant subgroup analyses, such as per protocol instead of intention to treat.
The authors observed that most physicians only read article abstracts, and up to one-quarter of editorial decisions are based on the abstract alone.
“Adding spin to the abstract of an article may mislead physicians who are attempting to draw conclusions about a treatment for patients,” they wrote, while calling for efforts to discourage spin in abstracts.
In an interview, Paul S. Nestadt, MD, said the findings were not surprising.
“The proportion [56%] of psychiatry and psychology abstracts which Jellison et al. found to contain spin is similar to that found in broader studies of all biomedical literature in previous reviews,” said Dr. Nestadt, assistant professor in the department of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore. “It is disheartening that attempts to mislead have become ‘standard of care’ in medical literature, but it is a predictable outcome of increasing competition for shrinking grant funding, awarded partly on the basis of publication history in leading journals that maintain clear publication biases toward positive results.
“As the authors point out, we all share a responsibility to call out spin when we see it, whether in our role as reviewer, editor, coauthor, or as the writers themselves.”
Neither Mr. Jellison nor his coauthors reported funding or conflicts of interest.
SOURCE: Jellison S et al. BMJ Evid Based Med. 2019 Aug 5. doi: 10.1136/bmjebm-2019-111176.
About half of papers in a sample of psychiatry and psychology journals have evidence of “spin” in the abstract, according to an analysis published in BMJ Evidence-Based Medicine.
Samuel Jellison, a third-year medical student at Oklahoma State University, Tulsa, and coauthors wrote that the results of randomized, controlled trials should be reported objectively because of their importance for psychiatry clinical practice. However, given that researchers were allowed more latitude in the abstract of a paper to highlight certain results or conclusions, the abstract might not accurately represent the findings of the study.
To evaluate this, the authors investigated the use of spin in abstracts, which they defined as “‘use of specific reporting strategies, from whatever motive, to highlight that the experimental treatment is beneficial, despite a statistically nonsignificant difference for the primary outcome, or to distract the reader from statistically nonsignificant results.”
They analyzed 116 randomized, controlled trials of interventions where there was a nonsignificant primary endpoint and found that 56% of those contained spin in the abstract. Spin was evident in 2% of publication titles, 21% of abstract results sections, and 49.1% of abstract conclusions sections.
Spin was more common in trials of pharmacologic treatments, compared with nonpharmacologic treatments. However, the study did not find a higher level of spin in industry-funded studies, and in fact, spin was more common in publicly funded research.
The most common form of spin was focusing on a statistically significant primary or secondary endpoint while omitting one or more nonsignificant primary endpoints. Other spin strategies included claiming noninferiority or equivalence for a statistically nonsignificant endpoint; using phrases such as “trend towards significance”; or focusing on statistically significant subgroup analyses, such as per protocol instead of intention to treat.
The authors observed that most physicians only read article abstracts, and up to one-quarter of editorial decisions are based on the abstract alone.
“Adding spin to the abstract of an article may mislead physicians who are attempting to draw conclusions about a treatment for patients,” they wrote, while calling for efforts to discourage spin in abstracts.
In an interview, Paul S. Nestadt, MD, said the findings were not surprising.
“The proportion [56%] of psychiatry and psychology abstracts which Jellison et al. found to contain spin is similar to that found in broader studies of all biomedical literature in previous reviews,” said Dr. Nestadt, assistant professor in the department of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore. “It is disheartening that attempts to mislead have become ‘standard of care’ in medical literature, but it is a predictable outcome of increasing competition for shrinking grant funding, awarded partly on the basis of publication history in leading journals that maintain clear publication biases toward positive results.
“As the authors point out, we all share a responsibility to call out spin when we see it, whether in our role as reviewer, editor, coauthor, or as the writers themselves.”
Neither Mr. Jellison nor his coauthors reported funding or conflicts of interest.
SOURCE: Jellison S et al. BMJ Evid Based Med. 2019 Aug 5. doi: 10.1136/bmjebm-2019-111176.
FROM BMJ EVIDENCE-BASED MEDICINE
Key clinical point. Spin is common in abstracts of psychiatry and psychology clinical trial papers.
Major finding: Spin was found in 56% of abstracts for psychiatry and psychology trials with a nonsignificant primary outcome.
Study details: An analysis of 116 randomized, controlled trials of interventions where there was a nonsignificant primary outcome.
Disclosures: No funding or conflicts of interest were reported.
Source: Jellison S et al. BMJ Evid Based Med. 2019 Aug 5. doi: 10.1136/bmjebm-2019-111176.
Lynch syndrome screening shows low efficiency in elderly
The need for universal screening for Lynch syndrome in elderly patients with newly diagnosed colorectal cancer (CRC) has been questioned, according to results from a retrospective cohort study.
In addition, discontinuing reflex CRC screening for Lynch syndrome in patients over age 80 years could be feasible because of very low efficiency.
“The universal strategy advocates screening all patients with newly diagnosed CRC for Lynch syndrome and has been shown to be the most sensitive method,” wrote Dan Li, MD, of Kaiser Permanente Northern California, Santa Clara, and colleagues. The findings were published in Annals of Internal Medicine.
The researchers studied 3,891 patients with newly diagnosed CRC who were screened for Lynch syndrome from 2011 to 2016. Data were collected from a population-based screening program at Kaiser Permanente Northern California.
“The system provides comprehensive medical care for more than 4 million members across 21 medical centers covering urban, suburban, and semirural areas,” Dr. Li and his colleagues wrote.
To compare universal and age-restricted screening, the team obtained surgical samples of all newly diagnosed CRC tumors and tested them for reflex mismatch repair protein expression using immunohistochemistry.
Subsequently, the age-restricted screening groups were divided into several age categories, ranging from age 50 to 85 years.
The diagnostic yield, defined as the “percentage of patients with pathogenic reflex mismatch repair gene variants among all patients with CRC screened with immunohistochemistry,” was measured and compared with the universal screening technique.
“We calculated the number of patients with CRC who needed to be screened in each age group to identify one case of Lynch syndrome by dividing the number of patients screened in each age group by the number of Lynch syndrome cases diagnosed in that group,” they explained.
After analysis, the researchers detected a total of 63 cases of Lynch syndrome (diagnostic yield, 1.62%) with universal screening, among which 5 (7.9%) were over age 70 years and 1 (1.6%) was over age 80 years.
When patients with CRC who were universally screened were used as the denominator, 58 cases (diagnostic yield, 1.49%) were detected in those with CRC diagnosed at or prior to age 70 years.
In addition, in patients diagnosed at or before age 75 and 80 years, 60 and 62 cases of Lynch syndrome (diagnostic yield, 1.54% and 1.59%) were detected, respectively.
“The incremental diagnostic yield decreased substantially after age 70 to 75 years,” they wrote.
With these findings, Dr. Li and his colleagues suggested that cessation of screening for Lynch syndrome post age 80 years may be acceptable, especially in resource-limited environments.
“Using age as the primary criterion is a simple method of selecting patients for Lynch syndrome screening in clinical practice,” they added.
In accordance with previous studies, a major reduction in Lynch syndrome incidence has been noted among elderly populations.
There remains a need for additional studies exploring the effects of diagnosing Lynch syndrome in elderly patients on family members.
The researchers acknowledged a key limitation of the study was that patients who did not finish germline analysis but were eligible for it were excluded from certain measurements. To reduce potential bias, the team conducted a sensitivity analysis, and the findings were negligible with respect to main results.
“Given the geographic variation in the reported prevalence of Lynch syndrome, the diagnostic efficiency of Lynch syndrome screening among elderly populations should be further investigated in other populations,” they concluded.
The study was funded by Kaiser Permanente Northern California. The authors reported financial affiliations with Bayer, Clinical Genomics, Covidien, Exact Sciences, Motus GI, Quorum, Universal DX, and the National Cancer Institute.
SOURCE: Li D et al. Ann Intern Med. 2019 Jun 11. doi: 10.7326/M18-3316.
The need for universal screening for Lynch syndrome in elderly patients with newly diagnosed colorectal cancer (CRC) has been questioned, according to results from a retrospective cohort study.
In addition, discontinuing reflex CRC screening for Lynch syndrome in patients over age 80 years could be feasible because of very low efficiency.
“The universal strategy advocates screening all patients with newly diagnosed CRC for Lynch syndrome and has been shown to be the most sensitive method,” wrote Dan Li, MD, of Kaiser Permanente Northern California, Santa Clara, and colleagues. The findings were published in Annals of Internal Medicine.
The researchers studied 3,891 patients with newly diagnosed CRC who were screened for Lynch syndrome from 2011 to 2016. Data were collected from a population-based screening program at Kaiser Permanente Northern California.
“The system provides comprehensive medical care for more than 4 million members across 21 medical centers covering urban, suburban, and semirural areas,” Dr. Li and his colleagues wrote.
To compare universal and age-restricted screening, the team obtained surgical samples of all newly diagnosed CRC tumors and tested them for reflex mismatch repair protein expression using immunohistochemistry.
Subsequently, the age-restricted screening groups were divided into several age categories, ranging from age 50 to 85 years.
The diagnostic yield, defined as the “percentage of patients with pathogenic reflex mismatch repair gene variants among all patients with CRC screened with immunohistochemistry,” was measured and compared with the universal screening technique.
“We calculated the number of patients with CRC who needed to be screened in each age group to identify one case of Lynch syndrome by dividing the number of patients screened in each age group by the number of Lynch syndrome cases diagnosed in that group,” they explained.
After analysis, the researchers detected a total of 63 cases of Lynch syndrome (diagnostic yield, 1.62%) with universal screening, among which 5 (7.9%) were over age 70 years and 1 (1.6%) was over age 80 years.
When patients with CRC who were universally screened were used as the denominator, 58 cases (diagnostic yield, 1.49%) were detected in those with CRC diagnosed at or prior to age 70 years.
In addition, in patients diagnosed at or before age 75 and 80 years, 60 and 62 cases of Lynch syndrome (diagnostic yield, 1.54% and 1.59%) were detected, respectively.
“The incremental diagnostic yield decreased substantially after age 70 to 75 years,” they wrote.
With these findings, Dr. Li and his colleagues suggested that cessation of screening for Lynch syndrome post age 80 years may be acceptable, especially in resource-limited environments.
“Using age as the primary criterion is a simple method of selecting patients for Lynch syndrome screening in clinical practice,” they added.
In accordance with previous studies, a major reduction in Lynch syndrome incidence has been noted among elderly populations.
There remains a need for additional studies exploring the effects of diagnosing Lynch syndrome in elderly patients on family members.
The researchers acknowledged a key limitation of the study was that patients who did not finish germline analysis but were eligible for it were excluded from certain measurements. To reduce potential bias, the team conducted a sensitivity analysis, and the findings were negligible with respect to main results.
“Given the geographic variation in the reported prevalence of Lynch syndrome, the diagnostic efficiency of Lynch syndrome screening among elderly populations should be further investigated in other populations,” they concluded.
The study was funded by Kaiser Permanente Northern California. The authors reported financial affiliations with Bayer, Clinical Genomics, Covidien, Exact Sciences, Motus GI, Quorum, Universal DX, and the National Cancer Institute.
SOURCE: Li D et al. Ann Intern Med. 2019 Jun 11. doi: 10.7326/M18-3316.
The need for universal screening for Lynch syndrome in elderly patients with newly diagnosed colorectal cancer (CRC) has been questioned, according to results from a retrospective cohort study.
In addition, discontinuing reflex CRC screening for Lynch syndrome in patients over age 80 years could be feasible because of very low efficiency.
“The universal strategy advocates screening all patients with newly diagnosed CRC for Lynch syndrome and has been shown to be the most sensitive method,” wrote Dan Li, MD, of Kaiser Permanente Northern California, Santa Clara, and colleagues. The findings were published in Annals of Internal Medicine.
The researchers studied 3,891 patients with newly diagnosed CRC who were screened for Lynch syndrome from 2011 to 2016. Data were collected from a population-based screening program at Kaiser Permanente Northern California.
“The system provides comprehensive medical care for more than 4 million members across 21 medical centers covering urban, suburban, and semirural areas,” Dr. Li and his colleagues wrote.
To compare universal and age-restricted screening, the team obtained surgical samples of all newly diagnosed CRC tumors and tested them for reflex mismatch repair protein expression using immunohistochemistry.
Subsequently, the age-restricted screening groups were divided into several age categories, ranging from age 50 to 85 years.
The diagnostic yield, defined as the “percentage of patients with pathogenic reflex mismatch repair gene variants among all patients with CRC screened with immunohistochemistry,” was measured and compared with the universal screening technique.
“We calculated the number of patients with CRC who needed to be screened in each age group to identify one case of Lynch syndrome by dividing the number of patients screened in each age group by the number of Lynch syndrome cases diagnosed in that group,” they explained.
After analysis, the researchers detected a total of 63 cases of Lynch syndrome (diagnostic yield, 1.62%) with universal screening, among which 5 (7.9%) were over age 70 years and 1 (1.6%) was over age 80 years.
When patients with CRC who were universally screened were used as the denominator, 58 cases (diagnostic yield, 1.49%) were detected in those with CRC diagnosed at or prior to age 70 years.
In addition, in patients diagnosed at or before age 75 and 80 years, 60 and 62 cases of Lynch syndrome (diagnostic yield, 1.54% and 1.59%) were detected, respectively.
“The incremental diagnostic yield decreased substantially after age 70 to 75 years,” they wrote.
With these findings, Dr. Li and his colleagues suggested that cessation of screening for Lynch syndrome post age 80 years may be acceptable, especially in resource-limited environments.
“Using age as the primary criterion is a simple method of selecting patients for Lynch syndrome screening in clinical practice,” they added.
In accordance with previous studies, a major reduction in Lynch syndrome incidence has been noted among elderly populations.
There remains a need for additional studies exploring the effects of diagnosing Lynch syndrome in elderly patients on family members.
The researchers acknowledged a key limitation of the study was that patients who did not finish germline analysis but were eligible for it were excluded from certain measurements. To reduce potential bias, the team conducted a sensitivity analysis, and the findings were negligible with respect to main results.
“Given the geographic variation in the reported prevalence of Lynch syndrome, the diagnostic efficiency of Lynch syndrome screening among elderly populations should be further investigated in other populations,” they concluded.
The study was funded by Kaiser Permanente Northern California. The authors reported financial affiliations with Bayer, Clinical Genomics, Covidien, Exact Sciences, Motus GI, Quorum, Universal DX, and the National Cancer Institute.
SOURCE: Li D et al. Ann Intern Med. 2019 Jun 11. doi: 10.7326/M18-3316.
FROM ANNALS OF INTERNAL MEDICINE
CVD risk after breast cancer: Adipose distribution trumps BMI
When it comes to cardiovascular disease (CVD) risk after a breast cancer diagnosis, it’s not so much a matter of the amount of body fat carried but rather where it is located, results of a retrospective cohort study of nearly 3,000 survivors suggest.
“It is well known that higher body mass index (BMI) is associated with CVD mortality in the general population. However, BMI is not always an accurate proxy for individual-level adiposity and does not describe adipose tissue distribution,” note lead investigator Elizabeth M. Cespedes Feliciano, ScD, of Kaiser Permanente Northern California, Oakland, Calif., and coinvestigators.
The investigators studied 2,943 survivors of nonmetastatic breast cancer having a mean age of 56 years who were initially CVD free, using clinically acquired CT scans obtained near diagnosis to measure adiposity in three compartments: visceral, subcutaneous, and intramuscular.
The cohort experienced 328 CVD events (nonfatal stroke, myocardial infarction, heart failure, or CVD death) during a median follow-up of 6 years, the investigators reported in Journal of Clinical Oncology. The 10-year cumulative incidence was 15%.
In analyses that were adjusted for potential confounders and took into account competing risks, survivors’ CVD risk increased significantly with each standard deviation (SD) increase in visceral adiposity (hazard ratio, 1.15; 95% confidence interval, 1.03-1.29) and each SD increase in intramuscular adiposity (HR, 1.21; 95% CI, 1.06-1.37). The association for subcutaneous adiposity was not significant.
Findings were similar across all BMI categories. Of particular note, among survivors having a normal BMI, risk of CVD events increased by 70% with each SD greater visceral adiposity (HR, 1.70; 95% CI, 1.10-2.62).
Risk also rose with BMI exceeding the normal range, but the association became significant only in survivors with a BMI placing them in obesity class II (35 kg/m2 or greater) (HR, 1.70; 95% CI, 1.20-2.42).
“Although it has been assumed that excess adiposity increases the risk of CVD after breast cancer, this first-of-its-kind study demonstrates that adipose tissue distribution best identifies patients with breast cancer with higher CVD risk after diagnosis, including those with normal BMI,” Dr. Cespedes Feliciano and coinvestigators note.
“Software is now available that automatically measures body composition from clinically acquired CT scans, facilitating clinical integration,” they note. “Measures of adipose tissue distribution from CT or anthropometry (e.g., waist circumference) may help identify individuals with high CVD risk and tailor prevention efforts to patients’ body composition.”
Dr. Cespedes Feliciano disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Cespedes Feliciano EM et al. J Clin Oncol. 2019 Aug 1. doi: 10.1200/JCO.19.00286.
When it comes to cardiovascular disease (CVD) risk after a breast cancer diagnosis, it’s not so much a matter of the amount of body fat carried but rather where it is located, results of a retrospective cohort study of nearly 3,000 survivors suggest.
“It is well known that higher body mass index (BMI) is associated with CVD mortality in the general population. However, BMI is not always an accurate proxy for individual-level adiposity and does not describe adipose tissue distribution,” note lead investigator Elizabeth M. Cespedes Feliciano, ScD, of Kaiser Permanente Northern California, Oakland, Calif., and coinvestigators.
The investigators studied 2,943 survivors of nonmetastatic breast cancer having a mean age of 56 years who were initially CVD free, using clinically acquired CT scans obtained near diagnosis to measure adiposity in three compartments: visceral, subcutaneous, and intramuscular.
The cohort experienced 328 CVD events (nonfatal stroke, myocardial infarction, heart failure, or CVD death) during a median follow-up of 6 years, the investigators reported in Journal of Clinical Oncology. The 10-year cumulative incidence was 15%.
In analyses that were adjusted for potential confounders and took into account competing risks, survivors’ CVD risk increased significantly with each standard deviation (SD) increase in visceral adiposity (hazard ratio, 1.15; 95% confidence interval, 1.03-1.29) and each SD increase in intramuscular adiposity (HR, 1.21; 95% CI, 1.06-1.37). The association for subcutaneous adiposity was not significant.
Findings were similar across all BMI categories. Of particular note, among survivors having a normal BMI, risk of CVD events increased by 70% with each SD greater visceral adiposity (HR, 1.70; 95% CI, 1.10-2.62).
Risk also rose with BMI exceeding the normal range, but the association became significant only in survivors with a BMI placing them in obesity class II (35 kg/m2 or greater) (HR, 1.70; 95% CI, 1.20-2.42).
“Although it has been assumed that excess adiposity increases the risk of CVD after breast cancer, this first-of-its-kind study demonstrates that adipose tissue distribution best identifies patients with breast cancer with higher CVD risk after diagnosis, including those with normal BMI,” Dr. Cespedes Feliciano and coinvestigators note.
“Software is now available that automatically measures body composition from clinically acquired CT scans, facilitating clinical integration,” they note. “Measures of adipose tissue distribution from CT or anthropometry (e.g., waist circumference) may help identify individuals with high CVD risk and tailor prevention efforts to patients’ body composition.”
Dr. Cespedes Feliciano disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Cespedes Feliciano EM et al. J Clin Oncol. 2019 Aug 1. doi: 10.1200/JCO.19.00286.
When it comes to cardiovascular disease (CVD) risk after a breast cancer diagnosis, it’s not so much a matter of the amount of body fat carried but rather where it is located, results of a retrospective cohort study of nearly 3,000 survivors suggest.
“It is well known that higher body mass index (BMI) is associated with CVD mortality in the general population. However, BMI is not always an accurate proxy for individual-level adiposity and does not describe adipose tissue distribution,” note lead investigator Elizabeth M. Cespedes Feliciano, ScD, of Kaiser Permanente Northern California, Oakland, Calif., and coinvestigators.
The investigators studied 2,943 survivors of nonmetastatic breast cancer having a mean age of 56 years who were initially CVD free, using clinically acquired CT scans obtained near diagnosis to measure adiposity in three compartments: visceral, subcutaneous, and intramuscular.
The cohort experienced 328 CVD events (nonfatal stroke, myocardial infarction, heart failure, or CVD death) during a median follow-up of 6 years, the investigators reported in Journal of Clinical Oncology. The 10-year cumulative incidence was 15%.
In analyses that were adjusted for potential confounders and took into account competing risks, survivors’ CVD risk increased significantly with each standard deviation (SD) increase in visceral adiposity (hazard ratio, 1.15; 95% confidence interval, 1.03-1.29) and each SD increase in intramuscular adiposity (HR, 1.21; 95% CI, 1.06-1.37). The association for subcutaneous adiposity was not significant.
Findings were similar across all BMI categories. Of particular note, among survivors having a normal BMI, risk of CVD events increased by 70% with each SD greater visceral adiposity (HR, 1.70; 95% CI, 1.10-2.62).
Risk also rose with BMI exceeding the normal range, but the association became significant only in survivors with a BMI placing them in obesity class II (35 kg/m2 or greater) (HR, 1.70; 95% CI, 1.20-2.42).
“Although it has been assumed that excess adiposity increases the risk of CVD after breast cancer, this first-of-its-kind study demonstrates that adipose tissue distribution best identifies patients with breast cancer with higher CVD risk after diagnosis, including those with normal BMI,” Dr. Cespedes Feliciano and coinvestigators note.
“Software is now available that automatically measures body composition from clinically acquired CT scans, facilitating clinical integration,” they note. “Measures of adipose tissue distribution from CT or anthropometry (e.g., waist circumference) may help identify individuals with high CVD risk and tailor prevention efforts to patients’ body composition.”
Dr. Cespedes Feliciano disclosed no relevant conflicts of interest. The study did not receive any specific funding.
SOURCE: Cespedes Feliciano EM et al. J Clin Oncol. 2019 Aug 1. doi: 10.1200/JCO.19.00286.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
International lupus community sets out top barriers to improving lupus outcomes
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.

A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.
A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
The heterogeneity of lupus and the subsequent lack of a clear disease definition have been identified by an international group of experts as the primary barriers hindering timely diagnosis, improved treatment options, and appropriate access to care.
A report published in Lupus Science & Medicine titled “Global Consensus Building and Prioritization of Fundamental Lupus Challenges: The ALPHA Project” describes the results of a first-ever global consensus on key barriers to advances in lupus care, including a lack of validated biomarkers and flawed clinical trial design.
A lack of access to medical professionals familiar with lupus, challenges in managing lupus because of social determinants, and lack of treatment adherence were also considered to be barriers to improving the outcomes of people living with lupus.
First author Susan Manzi, MD, codirector of the Lupus Center of Excellence at Allegheny Health Network, Pittsburgh, and her colleagues said that, in contrast to other autoimmune diseases such as rheumatoid arthritis and psoriasis, the field of lupus has struggled with establishing a clear pathway for lupus drug development because of “persistent challenges in understanding the biology of the disease, defining clinical trial entry criteria and end points, developing instruments to measure changes in clinical activity, and controlling background medications.”
The authors noted that the intention of the Addressing Lupus Pillars for Health Advancement (ALPHA) Project was to build on the work of other initiatives, including some that were international in scope or were still ongoing.
“The ALPHA project was founded as the first step in an ongoing commitment to identify, prioritize, and implement strategies to address the most pressing challenges that limit progress in lupus across the continuum,” they wrote. In a joint initiative, the Lupus Foundation of America (LFA) and the Tufts Center for the Study of Drug Development (Tufts CSDD) set up a Global Advisory Committee (GAC) that included 13 lupus experts from the United States, Australia, United Kingdom, Germany, and South Korea to guide and oversee the study. Members had extensive knowledge of the disease, with specific expertise in rheumatology, dermatology, immunology, nephrology, and pediatrics.
Next, in-depth interviews were conducted with 17 experts who were well respected in the lupus scientific and care communities and represented all stakeholders. Using information garnered from these interviews, the LFA, Tufts CSDD, and GAC collaborated to develop a survey that included 23 questions addressing attitudes and perceptions about lupus as well as the prioritization of the most pressing challenges to improving diagnosis, care, treatment, and research.
The online survey was sent to 366 candidates, from whom the researchers received 127 completed responses. Of these, 82 (65%) were clinician-researcher-scientists and 14 (11%) worked in industry/biotechnology, 13 (10%) were researcher-scientists, and 12 (9%) were clinicians; 5% marked “other.”
The research team used a weighting system to prioritize barriers ranked by respondents, whereby higher ratings represented the challenges of highest impact (a score of 9 was highest rating, with 1 the lowest).
Survey respondents ranked the following as the top barriers to improving outcomes in lupus:
- A lack of diagnostic, predictive, and prognostic biomarkers for lupus (weighted prioritization score of 7.294) and lack of biomarkers to predict drug response in clinical trials (weighted prioritization score of 6.614).
- Flawed clinical trial design (weighted prioritization score of 6.370).
- Lack of access to clinicians familiar with lupus (weighted prioritization score of 6.873), and limited awareness of lupus among nonexpert medical professionals (weighted prioritization score of 5.800).
- Barriers to effective management of lupus because of social determinants of care in predominantly lower socioeconomic status areas (weighted prioritization score of 6.937).
- A lack of treatment adherence (weighted prioritization score of 6.717).
“A strong consensus built throughout the study, as themes and insights gathered from the in-depth interviews were highly consistent with those collected in the survey,” the researchers noted.
They said it was not surprising that the development of biomarkers had received a high ranking, as advances in this area would help accelerate drug development and precision medicine as well as more practical aspects of clinical care.
The research team acknowledged that substantial funds would be needed to address the top priorities identified in the study, and some of the issues may be more easily addressed than others.
“In the past decade, the overall funding landscape for lupus has been on a decline, particularly through the National Institutes of Health – the largest public funder of lupus research in the world – during a time in which arguably, lupus research has been prolific,” they wrote.
They concluded that comprehensive measures were needed to transform the lupus research and health care landscape.
“Lupus experts must convene to determine feasible and coordinated approaches for addressing long-standing barriers across the global lupus community,” they stressed.
The next part of the project will involve an international stakeholder meeting to develop a global road map of specific recommendations to address identified barriers, which “may include multipronged strategies using regulatory and advocacy approaches, scientific consensus building, communication efforts, among other possible tactics,” they added.
The ALPHA Project was launched in partnership with founding partner EMD Serono Research & Development (a business of Merck KGaA) and through additional support by GlaxoSmithKline. Many authors of the report had financial connections to the pharmaceutical industry.
SOURCE: Manzi S et al. Lupus Sci Med. 2019;6:e000342. doi: 10.1136/lupus-2019-000342.
REPORTING FROM LUPUS SCIENCE & MEDICINE
Why do so many women aged 65 years and older die of cervical cancer?
Surprisingly, the cervical cancer death rate is greater among women aged >65 years than among younger women1,2 (FIGURE). Paradoxically, most of our screening programs focus on women <65 years of age. A nationwide study from Denmark estimated that the cervical cancer death rate per 100,000 women at ages 40 to 44 and 65 to 69 was 3.8 and 9.0, respectively.1 In other words, the cervical cancer death rate at age 65 to 69 years was 2.36 times higher than at age 40 to 44 years.1
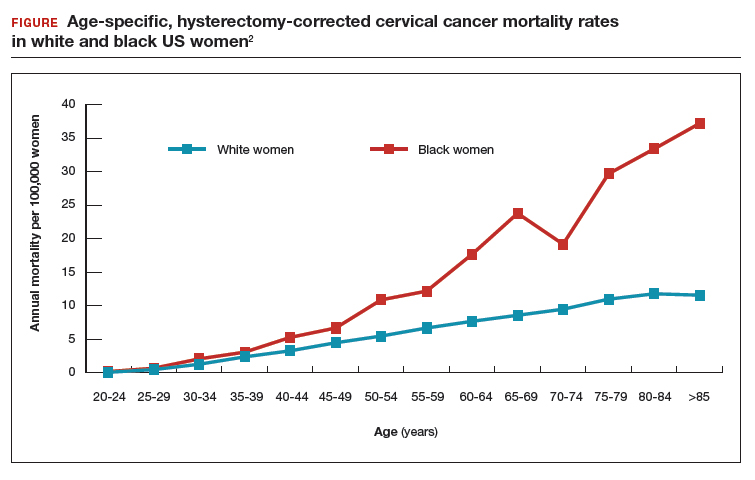
A study from the United States estimated that the cervical cancer death rate per 100,000 white women at ages 40 to 44 and 65 to 69 was 3.3 and 8.6, respectively,2 very similar to the findings from Denmark. The same US study estimated that the cervical cancer death rate per 100,000 black women at ages 40 to 44 and 65 to 69 was 5.3 and 23.8, highlighting the fact that, in the United States, cervical cancer disease burden is disproportionately greater among black than among white women.2 In addition, the cervical cancer death rate among black women at age 65 to 69 was 4.49 times higher than at age 40 to 44 years.2
Given the high death rate from cervical cancer in women >65 years of age, it is paradoxical that most professional society guidelines recommend discontinuing cervical cancer screening at 65 years of age, if previous cervical cancer screening is normal.3,4 Is the problem due to an inability to implement the current guidelines? Or is the problem that the guidelines are not optimally designed to reduce cervical cancer risk in women >65 years of age?
The American College of Obstetricians and Gynecologists (ACOG) and the US Preventive Services Task Force (USPSTF) recommend against cervical cancer screening in women >65 years of age who have had adequate prior screening and are not otherwise at high risk for cervical cancer. However, ACOG and the USPSTF caution that there are many groups of women that may benefit from continued screening after 65 years of age, including women with HIV infection, a compromised immune system, or previous high-grade precancerous lesion or cervicalcancer; women with limited access to care; women from racial/ethnic minority groups; and migrant women.4 Many clinicians remember the guidance, “discontinue cervical cancer screening at 65 years” but do not recall all the clinical factors that might warrant continued screening past age 65. Of special concern is that black,2 Hispanic,5 and migrant women6 are at much higher risk for invasive cervical cancer than white or US-born women.
The optimal implementation of the ACOG and USPSTF guidelines are undermined by a fractured health care system, where key pieces of information may be unavailable to the clinician tasked with making a decision about discontinuing cervical cancer screening. Imagine the case in which a 65-year-old woman pre‑sents to her primary care physician for cervical cancer screening. The clinician performs a cervical cytology test and obtains a report of “no intraepithelial lesion or malignancy.” The clinician then recommends that the patient discontinue cervical cancer screening. Unbeknownst to the clinician, the patient had a positive HPV 16/18/45 test within the past 10 years in another health system. In this case, it would be inappropriate to terminate the patient from cervical cancer screening.
Continue to: Testing for hrHPV is superior to cervical cytology in women >65 years...
Testing for hrHPV is superior to cervical cytology in women >65 years
In Sweden, about 30% of cervical cancer cases occur in women aged >60 years.7 To assess the prevalence of oncogenic high-risk HPV (hrHPV), women at ages 60, 65, 70, and 75 years were invited to send sequential self-collected vaginal samples for nucleic acid testing for hrHPV. The prevalence of hrHPV was found to be 4.4%. Women with a second positive, self-collected, hrHPV test were invited for colposcopy, cervical biopsy, and cytology testing. Among the women with two positive hrHPV tests, cervical biopsy revealed 7 cases of cervical intraepithelial neoplasia grade 2 (CIN2), 6 cases of CIN1, and 4 biopsies without CIN. In these women 94% of the cervical cytology samples returned, “no intraepithelial lesion or malignancy” and 6% revealed atypical squamous cells of undetermined significance. This study suggests that, in women aged >65 years, cervical cytology may have a high rate of false-negative results, possibly due to epithelial atrophy. An evolving clinical pearl is that, when using the current cervical cancer screening guidelines, the final screen for cervical cancer must include a nucleic acid test for hrHPV.
In women 65 to 90 years, the prevalence of hrHPV is approximately 5%
In a study of 40,382 women aged 14 to 95 years, the prevalence of hrHPV was 46% in 20- to 23-year-old women and 5.7% in women older than 65 years of age.8 In a study of more than 108,000 women aged 69 to >89 years the prevalence of hrHPV was 4.3%, and similar prevalence rates were seen across all ages from 69 to >89 years.9 The carcinogenic role of persistent hrHPV infection in women >65 years is an important area for future research.
Latent HPV virus infection
Following a primary varicella-zoster infection (chickenpox), the virus may remain in a latent state in sensory ganglia, reactivating later in life to cause shingles. Thirty percent of people who have a primary chickenpox infection eventually will develop a case of shingles. Immunocompromised populations are at an increased risk of developing shingles because of reduced T-cell mediated immunity.
A recent hypothesis is that in immunocompromised and older women, latent HPV can reactivate and cause clinically significant infection.10 Following renal transplantation investigators have reported a significant increase in the prevalence of genital HPV, without a change in sexual behavior.11 In cervical tissue from women with no evidence of active HPV infection, highly sensitive PCR-based assays detected HPV16 virus in a latent state in some women, possibly due to disruption of the viral E2 gene.12 If latent HPV infection is a valid biological concept, it suggests that there is no “safe age” at which to discontinue screening for HPV infection because the virus cannot be detected in screening samples while it is latent.
Options for cervical cancer screening in women >65 years
Three options might reduce the morbidity and mortality associated with cervical cancer in women >65 years.
Option 1: Double-down on trying to effectively implement current guidelines. The high rate of cervical cancer mortality in women >65 years of age indicates that the current guidelines, as implemented in real clinical practice, are not working. A problem with the current screening guidelines is that clinicians are expected to be capable of finding all relevant cervical cancer test results and properly interpreting the results. Clinicians are over-taxed and fallible, and the current approach is not likely to be successful unless additional information technology solutions are implemented.
Continue to: Health systems could use information...
Health systems could use information technology to mitigate these problems. For example, health systems could deploy software to assemble every cervical screening result on each woman and pre‑sent those results to clinicians in a single integrated view in the electronic record. Additionally, once all lifetime screening results are consolidated in one view, artificial intelligence systems could be used to analyze the totality of results and identify women who would benefit by continued screening past age 65 and women who could safely discontinue screening.
Option 2: Adopt the Australian approach to cervical cancer screening. The current Australian approach to cervical cancer screening is built on 3 pillars: 1) school-based vaccination of all children against hrHPV, 2) screening all women from 25 to 74 years of age every 5 years using nucleic acid testing for hrHPV, and 3) providing a system for the testing of samples self-collected by women who are reluctant to visit a clinician for screening.13 Australia has one of the lowest cervical cancer death rates in the world.
Option 3: Continue screening most women past age 65. Women >65 years of age are known to be infected with hrHPV genotypes. hrHPV infection causes cervical cancer. Cervical cancer causes many deaths in women aged >65 years. There is no strong rationale for ignoring these three facts. hrHPV screening every 5 years as long as the woman is healthy and has a reasonable life expectancy is an option that could be evaluated in randomized studies.
Given the high rate of cervical cancer death in women >65 years of age, I plan to be very cautious about discontinuing cervical cancer screening until I can personally ensure that my patient has no evidence of hrHPV infection.
In 2008, Harald zur Hausen, MD, received the Nobel Prize in Physiology or Medicine for discovering that human papilloma virus (HPV) caused cervical cancer. In a recent study, 74% of cervical cancers were associated with HPV 16 or 18 infections. A total of 89% of the cancers were associated with one of the high-risk HPV genotypes, including HPV 16/18/31/33/45/52/58.1
Recently, HPV has been shown to be a major cause of oropharyngeal cancer. The Centers for Disease Control and Prevention calculated that in CY2015 in the United States there were 18,917 cases of HPV-associated oropharyngeal squamous cell cancer and 11,788 cases of cervical cancer.2 Most cases of HPV-associated oropharyngeal cancer occur in men, and HPV vaccination of boys may help to prevent this cancer type. Oncogenic HPV produce two proteins (E6 and E7) that promote viral replication and squamous cell growth by inhibiting the function of p53 and retinoblastoma protein. The immortalized HeLa cell line, derived from Ms. Henrietta Lack's cervical cancer, contains integrated HPV18 nucleic acid sequences.3,4
The discovery that HPV causes cancer catalyzed the development of nucleic acid tests to identify high-risk oncogenic HPV and vaccines against high-risk oncogenic HPV genotypes that prevent cervical cancer. From a public health perspective, it is more effective to vaccinate the population against oncogenic HPV genotypes than to screen and treat cancer. In the United States, vaccination rates range from a high of 92% (District of Columbia) and 89% (Rhode Island) to a low of 47% (Wyoming) and 50% (Kentucky and Mississippi).5 To reduce HPV-associated cancer mortality, the gap in vaccination compliance must be closed.
References
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Van Dyne EA, Henley SJ, Saraiya M, et al. Trends in human papillomavirus-associated cancers - United States, 1999-2015. MMWR Morb Mortal Wkly Rep. 2018;67:918-924.
- Rosl F, Westphal EM, zur Hausen H. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol Carcinog. 1989;2:72-80.
- Adey A, Burton JN, Kitzman, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207-211.
- Walker TY, Elam-Evans LD, Singleton JA, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13-17 years - United States, 2016. MMWR Morb Mortal Wkly Rep. 2017;66:874-882.
- Hammer A, Kahlert J, Gravitt PE, et al. Hysterectomy-corrected cervical cancer mortality rates in Denmark during 2002-2015: a registry-based cohort study. Acta Obstet Gynecol Scand. 2019;98:1063-1069.
- Beavis AL, Gravitt PE, Rositch AF. Hysterectomy-corrected cervical cancer mortality rates reveal a larger racial disparity in the United States. Cancer. 2017;123:1044-1050.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. Practice Bulletin No. 168: cervical cancer screening and prevention. Obstet Gynecol. 2016;128:e111-30.
- Curry SJ, Krist AH, Owens DK, et al; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;320:674-686.
- Stang A, Hawk H, Knowlton R, et al. Hysterectomy-corrected incidence rates of cervical and uterine cancers in Massachusetts, 1995-2010. Ann Epidemiol. 2014;24:849-854.
- Hallowell BD, Endeshaw M, McKenna MT, et al. Cervical cancer death rates among U.S.- and foreign-born women: U.S., 2005-2014. Am J Prev Med. 2019;56:869-874.
- Lindström AK, Hermansson RS, Gustavsson I, et al. Cervical dysplasia in elderly women performing repeated self-sampling for HPV testing. PLoS One. 2018;13:e0207714.
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Andersen B, Christensen BS, Christensen J, et al. HPV-prevalence in elderly women in Denmark. Gynecol Oncol. 2019;154:118-123.
- Gravitt PE, Winer RL. Natural history of HPV infection across the lifespan: role of viral latency. Viruses. 2017;9:E267.
- Hinten F, Hilbrands LB, Meeuwis KAP, et al. Reactivation of latent HPV infections after renal transplantation. Am J Transplant. 2017;17:1563-1573.
- Leonard SM, Pereira M, Roberts S, et al. Evidence of disrupted high-risk human papillomavirus DNA in morphologically normal cervices of older women. Sci Rep. 2016;6:20847.
- Cervical cancer screening. Cancer Council website. https://www.cancer.org.au/about-cancer/early-detection/screening-programs/cervical-cancer-screening.html. Updated March 15, 2019. Accessed July 23, 2019.

Robert L. Barbieri, MD
Editor in Chief, OBG MANAGEMENT
Chair, Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG MANAGEMENT
Chair, Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Dr. Barbieri reports no financial relationships relevant to this article.
Robert L. Barbieri, MD
Editor in Chief, OBG MANAGEMENT
Chair, Obstetrics and Gynecology
Brigham and Women’s Hospital
Kate Macy Ladd Professor of Obstetrics,
Gynecology and Reproductive Biology
Harvard Medical School
Dr. Barbieri reports no financial relationships relevant to this article.
Surprisingly, the cervical cancer death rate is greater among women aged >65 years than among younger women1,2 (FIGURE). Paradoxically, most of our screening programs focus on women <65 years of age. A nationwide study from Denmark estimated that the cervical cancer death rate per 100,000 women at ages 40 to 44 and 65 to 69 was 3.8 and 9.0, respectively.1 In other words, the cervical cancer death rate at age 65 to 69 years was 2.36 times higher than at age 40 to 44 years.1
A study from the United States estimated that the cervical cancer death rate per 100,000 white women at ages 40 to 44 and 65 to 69 was 3.3 and 8.6, respectively,2 very similar to the findings from Denmark. The same US study estimated that the cervical cancer death rate per 100,000 black women at ages 40 to 44 and 65 to 69 was 5.3 and 23.8, highlighting the fact that, in the United States, cervical cancer disease burden is disproportionately greater among black than among white women.2 In addition, the cervical cancer death rate among black women at age 65 to 69 was 4.49 times higher than at age 40 to 44 years.2
Given the high death rate from cervical cancer in women >65 years of age, it is paradoxical that most professional society guidelines recommend discontinuing cervical cancer screening at 65 years of age, if previous cervical cancer screening is normal.3,4 Is the problem due to an inability to implement the current guidelines? Or is the problem that the guidelines are not optimally designed to reduce cervical cancer risk in women >65 years of age?
The American College of Obstetricians and Gynecologists (ACOG) and the US Preventive Services Task Force (USPSTF) recommend against cervical cancer screening in women >65 years of age who have had adequate prior screening and are not otherwise at high risk for cervical cancer. However, ACOG and the USPSTF caution that there are many groups of women that may benefit from continued screening after 65 years of age, including women with HIV infection, a compromised immune system, or previous high-grade precancerous lesion or cervicalcancer; women with limited access to care; women from racial/ethnic minority groups; and migrant women.4 Many clinicians remember the guidance, “discontinue cervical cancer screening at 65 years” but do not recall all the clinical factors that might warrant continued screening past age 65. Of special concern is that black,2 Hispanic,5 and migrant women6 are at much higher risk for invasive cervical cancer than white or US-born women.
The optimal implementation of the ACOG and USPSTF guidelines are undermined by a fractured health care system, where key pieces of information may be unavailable to the clinician tasked with making a decision about discontinuing cervical cancer screening. Imagine the case in which a 65-year-old woman pre‑sents to her primary care physician for cervical cancer screening. The clinician performs a cervical cytology test and obtains a report of “no intraepithelial lesion or malignancy.” The clinician then recommends that the patient discontinue cervical cancer screening. Unbeknownst to the clinician, the patient had a positive HPV 16/18/45 test within the past 10 years in another health system. In this case, it would be inappropriate to terminate the patient from cervical cancer screening.
Continue to: Testing for hrHPV is superior to cervical cytology in women >65 years...
Testing for hrHPV is superior to cervical cytology in women >65 years
In Sweden, about 30% of cervical cancer cases occur in women aged >60 years.7 To assess the prevalence of oncogenic high-risk HPV (hrHPV), women at ages 60, 65, 70, and 75 years were invited to send sequential self-collected vaginal samples for nucleic acid testing for hrHPV. The prevalence of hrHPV was found to be 4.4%. Women with a second positive, self-collected, hrHPV test were invited for colposcopy, cervical biopsy, and cytology testing. Among the women with two positive hrHPV tests, cervical biopsy revealed 7 cases of cervical intraepithelial neoplasia grade 2 (CIN2), 6 cases of CIN1, and 4 biopsies without CIN. In these women 94% of the cervical cytology samples returned, “no intraepithelial lesion or malignancy” and 6% revealed atypical squamous cells of undetermined significance. This study suggests that, in women aged >65 years, cervical cytology may have a high rate of false-negative results, possibly due to epithelial atrophy. An evolving clinical pearl is that, when using the current cervical cancer screening guidelines, the final screen for cervical cancer must include a nucleic acid test for hrHPV.
In women 65 to 90 years, the prevalence of hrHPV is approximately 5%
In a study of 40,382 women aged 14 to 95 years, the prevalence of hrHPV was 46% in 20- to 23-year-old women and 5.7% in women older than 65 years of age.8 In a study of more than 108,000 women aged 69 to >89 years the prevalence of hrHPV was 4.3%, and similar prevalence rates were seen across all ages from 69 to >89 years.9 The carcinogenic role of persistent hrHPV infection in women >65 years is an important area for future research.
Latent HPV virus infection
Following a primary varicella-zoster infection (chickenpox), the virus may remain in a latent state in sensory ganglia, reactivating later in life to cause shingles. Thirty percent of people who have a primary chickenpox infection eventually will develop a case of shingles. Immunocompromised populations are at an increased risk of developing shingles because of reduced T-cell mediated immunity.
A recent hypothesis is that in immunocompromised and older women, latent HPV can reactivate and cause clinically significant infection.10 Following renal transplantation investigators have reported a significant increase in the prevalence of genital HPV, without a change in sexual behavior.11 In cervical tissue from women with no evidence of active HPV infection, highly sensitive PCR-based assays detected HPV16 virus in a latent state in some women, possibly due to disruption of the viral E2 gene.12 If latent HPV infection is a valid biological concept, it suggests that there is no “safe age” at which to discontinue screening for HPV infection because the virus cannot be detected in screening samples while it is latent.
Options for cervical cancer screening in women >65 years
Three options might reduce the morbidity and mortality associated with cervical cancer in women >65 years.
Option 1: Double-down on trying to effectively implement current guidelines. The high rate of cervical cancer mortality in women >65 years of age indicates that the current guidelines, as implemented in real clinical practice, are not working. A problem with the current screening guidelines is that clinicians are expected to be capable of finding all relevant cervical cancer test results and properly interpreting the results. Clinicians are over-taxed and fallible, and the current approach is not likely to be successful unless additional information technology solutions are implemented.
Continue to: Health systems could use information...
Health systems could use information technology to mitigate these problems. For example, health systems could deploy software to assemble every cervical screening result on each woman and pre‑sent those results to clinicians in a single integrated view in the electronic record. Additionally, once all lifetime screening results are consolidated in one view, artificial intelligence systems could be used to analyze the totality of results and identify women who would benefit by continued screening past age 65 and women who could safely discontinue screening.
Option 2: Adopt the Australian approach to cervical cancer screening. The current Australian approach to cervical cancer screening is built on 3 pillars: 1) school-based vaccination of all children against hrHPV, 2) screening all women from 25 to 74 years of age every 5 years using nucleic acid testing for hrHPV, and 3) providing a system for the testing of samples self-collected by women who are reluctant to visit a clinician for screening.13 Australia has one of the lowest cervical cancer death rates in the world.
Option 3: Continue screening most women past age 65. Women >65 years of age are known to be infected with hrHPV genotypes. hrHPV infection causes cervical cancer. Cervical cancer causes many deaths in women aged >65 years. There is no strong rationale for ignoring these three facts. hrHPV screening every 5 years as long as the woman is healthy and has a reasonable life expectancy is an option that could be evaluated in randomized studies.
Given the high rate of cervical cancer death in women >65 years of age, I plan to be very cautious about discontinuing cervical cancer screening until I can personally ensure that my patient has no evidence of hrHPV infection.
In 2008, Harald zur Hausen, MD, received the Nobel Prize in Physiology or Medicine for discovering that human papilloma virus (HPV) caused cervical cancer. In a recent study, 74% of cervical cancers were associated with HPV 16 or 18 infections. A total of 89% of the cancers were associated with one of the high-risk HPV genotypes, including HPV 16/18/31/33/45/52/58.1
Recently, HPV has been shown to be a major cause of oropharyngeal cancer. The Centers for Disease Control and Prevention calculated that in CY2015 in the United States there were 18,917 cases of HPV-associated oropharyngeal squamous cell cancer and 11,788 cases of cervical cancer.2 Most cases of HPV-associated oropharyngeal cancer occur in men, and HPV vaccination of boys may help to prevent this cancer type. Oncogenic HPV produce two proteins (E6 and E7) that promote viral replication and squamous cell growth by inhibiting the function of p53 and retinoblastoma protein. The immortalized HeLa cell line, derived from Ms. Henrietta Lack's cervical cancer, contains integrated HPV18 nucleic acid sequences.3,4
The discovery that HPV causes cancer catalyzed the development of nucleic acid tests to identify high-risk oncogenic HPV and vaccines against high-risk oncogenic HPV genotypes that prevent cervical cancer. From a public health perspective, it is more effective to vaccinate the population against oncogenic HPV genotypes than to screen and treat cancer. In the United States, vaccination rates range from a high of 92% (District of Columbia) and 89% (Rhode Island) to a low of 47% (Wyoming) and 50% (Kentucky and Mississippi).5 To reduce HPV-associated cancer mortality, the gap in vaccination compliance must be closed.
References
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Van Dyne EA, Henley SJ, Saraiya M, et al. Trends in human papillomavirus-associated cancers - United States, 1999-2015. MMWR Morb Mortal Wkly Rep. 2018;67:918-924.
- Rosl F, Westphal EM, zur Hausen H. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol Carcinog. 1989;2:72-80.
- Adey A, Burton JN, Kitzman, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207-211.
- Walker TY, Elam-Evans LD, Singleton JA, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13-17 years - United States, 2016. MMWR Morb Mortal Wkly Rep. 2017;66:874-882.
Surprisingly, the cervical cancer death rate is greater among women aged >65 years than among younger women1,2 (FIGURE). Paradoxically, most of our screening programs focus on women <65 years of age. A nationwide study from Denmark estimated that the cervical cancer death rate per 100,000 women at ages 40 to 44 and 65 to 69 was 3.8 and 9.0, respectively.1 In other words, the cervical cancer death rate at age 65 to 69 years was 2.36 times higher than at age 40 to 44 years.1
A study from the United States estimated that the cervical cancer death rate per 100,000 white women at ages 40 to 44 and 65 to 69 was 3.3 and 8.6, respectively,2 very similar to the findings from Denmark. The same US study estimated that the cervical cancer death rate per 100,000 black women at ages 40 to 44 and 65 to 69 was 5.3 and 23.8, highlighting the fact that, in the United States, cervical cancer disease burden is disproportionately greater among black than among white women.2 In addition, the cervical cancer death rate among black women at age 65 to 69 was 4.49 times higher than at age 40 to 44 years.2
Given the high death rate from cervical cancer in women >65 years of age, it is paradoxical that most professional society guidelines recommend discontinuing cervical cancer screening at 65 years of age, if previous cervical cancer screening is normal.3,4 Is the problem due to an inability to implement the current guidelines? Or is the problem that the guidelines are not optimally designed to reduce cervical cancer risk in women >65 years of age?
The American College of Obstetricians and Gynecologists (ACOG) and the US Preventive Services Task Force (USPSTF) recommend against cervical cancer screening in women >65 years of age who have had adequate prior screening and are not otherwise at high risk for cervical cancer. However, ACOG and the USPSTF caution that there are many groups of women that may benefit from continued screening after 65 years of age, including women with HIV infection, a compromised immune system, or previous high-grade precancerous lesion or cervicalcancer; women with limited access to care; women from racial/ethnic minority groups; and migrant women.4 Many clinicians remember the guidance, “discontinue cervical cancer screening at 65 years” but do not recall all the clinical factors that might warrant continued screening past age 65. Of special concern is that black,2 Hispanic,5 and migrant women6 are at much higher risk for invasive cervical cancer than white or US-born women.
The optimal implementation of the ACOG and USPSTF guidelines are undermined by a fractured health care system, where key pieces of information may be unavailable to the clinician tasked with making a decision about discontinuing cervical cancer screening. Imagine the case in which a 65-year-old woman pre‑sents to her primary care physician for cervical cancer screening. The clinician performs a cervical cytology test and obtains a report of “no intraepithelial lesion or malignancy.” The clinician then recommends that the patient discontinue cervical cancer screening. Unbeknownst to the clinician, the patient had a positive HPV 16/18/45 test within the past 10 years in another health system. In this case, it would be inappropriate to terminate the patient from cervical cancer screening.
Continue to: Testing for hrHPV is superior to cervical cytology in women >65 years...
Testing for hrHPV is superior to cervical cytology in women >65 years
In Sweden, about 30% of cervical cancer cases occur in women aged >60 years.7 To assess the prevalence of oncogenic high-risk HPV (hrHPV), women at ages 60, 65, 70, and 75 years were invited to send sequential self-collected vaginal samples for nucleic acid testing for hrHPV. The prevalence of hrHPV was found to be 4.4%. Women with a second positive, self-collected, hrHPV test were invited for colposcopy, cervical biopsy, and cytology testing. Among the women with two positive hrHPV tests, cervical biopsy revealed 7 cases of cervical intraepithelial neoplasia grade 2 (CIN2), 6 cases of CIN1, and 4 biopsies without CIN. In these women 94% of the cervical cytology samples returned, “no intraepithelial lesion or malignancy” and 6% revealed atypical squamous cells of undetermined significance. This study suggests that, in women aged >65 years, cervical cytology may have a high rate of false-negative results, possibly due to epithelial atrophy. An evolving clinical pearl is that, when using the current cervical cancer screening guidelines, the final screen for cervical cancer must include a nucleic acid test for hrHPV.
In women 65 to 90 years, the prevalence of hrHPV is approximately 5%
In a study of 40,382 women aged 14 to 95 years, the prevalence of hrHPV was 46% in 20- to 23-year-old women and 5.7% in women older than 65 years of age.8 In a study of more than 108,000 women aged 69 to >89 years the prevalence of hrHPV was 4.3%, and similar prevalence rates were seen across all ages from 69 to >89 years.9 The carcinogenic role of persistent hrHPV infection in women >65 years is an important area for future research.
Latent HPV virus infection
Following a primary varicella-zoster infection (chickenpox), the virus may remain in a latent state in sensory ganglia, reactivating later in life to cause shingles. Thirty percent of people who have a primary chickenpox infection eventually will develop a case of shingles. Immunocompromised populations are at an increased risk of developing shingles because of reduced T-cell mediated immunity.
A recent hypothesis is that in immunocompromised and older women, latent HPV can reactivate and cause clinically significant infection.10 Following renal transplantation investigators have reported a significant increase in the prevalence of genital HPV, without a change in sexual behavior.11 In cervical tissue from women with no evidence of active HPV infection, highly sensitive PCR-based assays detected HPV16 virus in a latent state in some women, possibly due to disruption of the viral E2 gene.12 If latent HPV infection is a valid biological concept, it suggests that there is no “safe age” at which to discontinue screening for HPV infection because the virus cannot be detected in screening samples while it is latent.
Options for cervical cancer screening in women >65 years
Three options might reduce the morbidity and mortality associated with cervical cancer in women >65 years.
Option 1: Double-down on trying to effectively implement current guidelines. The high rate of cervical cancer mortality in women >65 years of age indicates that the current guidelines, as implemented in real clinical practice, are not working. A problem with the current screening guidelines is that clinicians are expected to be capable of finding all relevant cervical cancer test results and properly interpreting the results. Clinicians are over-taxed and fallible, and the current approach is not likely to be successful unless additional information technology solutions are implemented.
Continue to: Health systems could use information...
Health systems could use information technology to mitigate these problems. For example, health systems could deploy software to assemble every cervical screening result on each woman and pre‑sent those results to clinicians in a single integrated view in the electronic record. Additionally, once all lifetime screening results are consolidated in one view, artificial intelligence systems could be used to analyze the totality of results and identify women who would benefit by continued screening past age 65 and women who could safely discontinue screening.
Option 2: Adopt the Australian approach to cervical cancer screening. The current Australian approach to cervical cancer screening is built on 3 pillars: 1) school-based vaccination of all children against hrHPV, 2) screening all women from 25 to 74 years of age every 5 years using nucleic acid testing for hrHPV, and 3) providing a system for the testing of samples self-collected by women who are reluctant to visit a clinician for screening.13 Australia has one of the lowest cervical cancer death rates in the world.
Option 3: Continue screening most women past age 65. Women >65 years of age are known to be infected with hrHPV genotypes. hrHPV infection causes cervical cancer. Cervical cancer causes many deaths in women aged >65 years. There is no strong rationale for ignoring these three facts. hrHPV screening every 5 years as long as the woman is healthy and has a reasonable life expectancy is an option that could be evaluated in randomized studies.
Given the high rate of cervical cancer death in women >65 years of age, I plan to be very cautious about discontinuing cervical cancer screening until I can personally ensure that my patient has no evidence of hrHPV infection.
In 2008, Harald zur Hausen, MD, received the Nobel Prize in Physiology or Medicine for discovering that human papilloma virus (HPV) caused cervical cancer. In a recent study, 74% of cervical cancers were associated with HPV 16 or 18 infections. A total of 89% of the cancers were associated with one of the high-risk HPV genotypes, including HPV 16/18/31/33/45/52/58.1
Recently, HPV has been shown to be a major cause of oropharyngeal cancer. The Centers for Disease Control and Prevention calculated that in CY2015 in the United States there were 18,917 cases of HPV-associated oropharyngeal squamous cell cancer and 11,788 cases of cervical cancer.2 Most cases of HPV-associated oropharyngeal cancer occur in men, and HPV vaccination of boys may help to prevent this cancer type. Oncogenic HPV produce two proteins (E6 and E7) that promote viral replication and squamous cell growth by inhibiting the function of p53 and retinoblastoma protein. The immortalized HeLa cell line, derived from Ms. Henrietta Lack's cervical cancer, contains integrated HPV18 nucleic acid sequences.3,4
The discovery that HPV causes cancer catalyzed the development of nucleic acid tests to identify high-risk oncogenic HPV and vaccines against high-risk oncogenic HPV genotypes that prevent cervical cancer. From a public health perspective, it is more effective to vaccinate the population against oncogenic HPV genotypes than to screen and treat cancer. In the United States, vaccination rates range from a high of 92% (District of Columbia) and 89% (Rhode Island) to a low of 47% (Wyoming) and 50% (Kentucky and Mississippi).5 To reduce HPV-associated cancer mortality, the gap in vaccination compliance must be closed.
References
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Van Dyne EA, Henley SJ, Saraiya M, et al. Trends in human papillomavirus-associated cancers - United States, 1999-2015. MMWR Morb Mortal Wkly Rep. 2018;67:918-924.
- Rosl F, Westphal EM, zur Hausen H. Chromatin structure and transcriptional regulation of human papillomavirus type 18 DNA in HeLa cells. Mol Carcinog. 1989;2:72-80.
- Adey A, Burton JN, Kitzman, et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207-211.
- Walker TY, Elam-Evans LD, Singleton JA, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13-17 years - United States, 2016. MMWR Morb Mortal Wkly Rep. 2017;66:874-882.
- Hammer A, Kahlert J, Gravitt PE, et al. Hysterectomy-corrected cervical cancer mortality rates in Denmark during 2002-2015: a registry-based cohort study. Acta Obstet Gynecol Scand. 2019;98:1063-1069.
- Beavis AL, Gravitt PE, Rositch AF. Hysterectomy-corrected cervical cancer mortality rates reveal a larger racial disparity in the United States. Cancer. 2017;123:1044-1050.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. Practice Bulletin No. 168: cervical cancer screening and prevention. Obstet Gynecol. 2016;128:e111-30.
- Curry SJ, Krist AH, Owens DK, et al; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;320:674-686.
- Stang A, Hawk H, Knowlton R, et al. Hysterectomy-corrected incidence rates of cervical and uterine cancers in Massachusetts, 1995-2010. Ann Epidemiol. 2014;24:849-854.
- Hallowell BD, Endeshaw M, McKenna MT, et al. Cervical cancer death rates among U.S.- and foreign-born women: U.S., 2005-2014. Am J Prev Med. 2019;56:869-874.
- Lindström AK, Hermansson RS, Gustavsson I, et al. Cervical dysplasia in elderly women performing repeated self-sampling for HPV testing. PLoS One. 2018;13:e0207714.
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Andersen B, Christensen BS, Christensen J, et al. HPV-prevalence in elderly women in Denmark. Gynecol Oncol. 2019;154:118-123.
- Gravitt PE, Winer RL. Natural history of HPV infection across the lifespan: role of viral latency. Viruses. 2017;9:E267.
- Hinten F, Hilbrands LB, Meeuwis KAP, et al. Reactivation of latent HPV infections after renal transplantation. Am J Transplant. 2017;17:1563-1573.
- Leonard SM, Pereira M, Roberts S, et al. Evidence of disrupted high-risk human papillomavirus DNA in morphologically normal cervices of older women. Sci Rep. 2016;6:20847.
- Cervical cancer screening. Cancer Council website. https://www.cancer.org.au/about-cancer/early-detection/screening-programs/cervical-cancer-screening.html. Updated March 15, 2019. Accessed July 23, 2019.
- Hammer A, Kahlert J, Gravitt PE, et al. Hysterectomy-corrected cervical cancer mortality rates in Denmark during 2002-2015: a registry-based cohort study. Acta Obstet Gynecol Scand. 2019;98:1063-1069.
- Beavis AL, Gravitt PE, Rositch AF. Hysterectomy-corrected cervical cancer mortality rates reveal a larger racial disparity in the United States. Cancer. 2017;123:1044-1050.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins--Gynecology. Practice Bulletin No. 168: cervical cancer screening and prevention. Obstet Gynecol. 2016;128:e111-30.
- Curry SJ, Krist AH, Owens DK, et al; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;320:674-686.
- Stang A, Hawk H, Knowlton R, et al. Hysterectomy-corrected incidence rates of cervical and uterine cancers in Massachusetts, 1995-2010. Ann Epidemiol. 2014;24:849-854.
- Hallowell BD, Endeshaw M, McKenna MT, et al. Cervical cancer death rates among U.S.- and foreign-born women: U.S., 2005-2014. Am J Prev Med. 2019;56:869-874.
- Lindström AK, Hermansson RS, Gustavsson I, et al. Cervical dysplasia in elderly women performing repeated self-sampling for HPV testing. PLoS One. 2018;13:e0207714.
- Kjaer SK, Munk C, Junge J, et al. Carcinogenic HPV prevalence and age-specific type distribution in 40,382 women with normal cervical cytology, ACSUC/LSIL, HSIL, or cervical cancer: what is the potential for prevention? Cancer Causes Control. 2014;25:179-189.
- Andersen B, Christensen BS, Christensen J, et al. HPV-prevalence in elderly women in Denmark. Gynecol Oncol. 2019;154:118-123.
- Gravitt PE, Winer RL. Natural history of HPV infection across the lifespan: role of viral latency. Viruses. 2017;9:E267.
- Hinten F, Hilbrands LB, Meeuwis KAP, et al. Reactivation of latent HPV infections after renal transplantation. Am J Transplant. 2017;17:1563-1573.
- Leonard SM, Pereira M, Roberts S, et al. Evidence of disrupted high-risk human papillomavirus DNA in morphologically normal cervices of older women. Sci Rep. 2016;6:20847.
- Cervical cancer screening. Cancer Council website. https://www.cancer.org.au/about-cancer/early-detection/screening-programs/cervical-cancer-screening.html. Updated March 15, 2019. Accessed July 23, 2019.
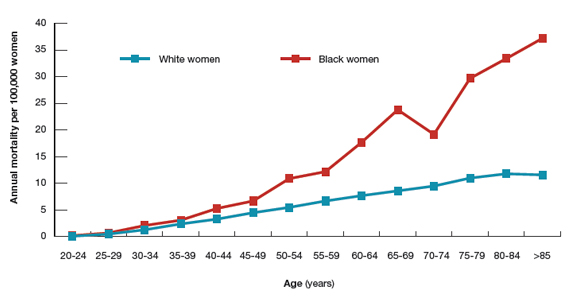
August 2019 - Quick Quiz Question 2
Q2. Correct answer: E
Rationale:
Radiographic evaluation is commonly employed in the diagnosis and management of patients with lower GI bleeding. CT scans, tagged red blood cell scintigraphy, and angiography all have roles in the care of these patients. Though tagged red blood cell scintigraphy is the most sensitive modality at detecting active bleeding, requiring rates from 0.05-0.1 cc/min, it is relatively poor at localizing the bleeding, accurately predicting the location in only 60%-70% of cases. CT scans have the advantage of being quickly performed and are widely available. If extravasation is seen, its location is also accurately determined. It is not as sensitive as red blood cell scintigraphy, however, and requires bleeding rates of 0.3-0.5 cc/min to be positive. Angiography has the advantage of being both diagnostic and potentially therapeutic. It is best performed in sicker patients with hypotension and high transfusion demands as it is higher yield in these situations. Angiography is the least sensitive of these modalities, requiring bleeding rates between 0.5 and 1 cc/min to be positive.
Reference:
1. Strate LL, Naumann CR. The role of colonoscopy and radiological procedures in the management of acute lower intestinal bleeding. Clin Gastroenterol Hepatol. 2010 Apr;8(4):333-43.
ginews@gastro.org
Q2. Correct answer: E
Rationale:
Radiographic evaluation is commonly employed in the diagnosis and management of patients with lower GI bleeding. CT scans, tagged red blood cell scintigraphy, and angiography all have roles in the care of these patients. Though tagged red blood cell scintigraphy is the most sensitive modality at detecting active bleeding, requiring rates from 0.05-0.1 cc/min, it is relatively poor at localizing the bleeding, accurately predicting the location in only 60%-70% of cases. CT scans have the advantage of being quickly performed and are widely available. If extravasation is seen, its location is also accurately determined. It is not as sensitive as red blood cell scintigraphy, however, and requires bleeding rates of 0.3-0.5 cc/min to be positive. Angiography has the advantage of being both diagnostic and potentially therapeutic. It is best performed in sicker patients with hypotension and high transfusion demands as it is higher yield in these situations. Angiography is the least sensitive of these modalities, requiring bleeding rates between 0.5 and 1 cc/min to be positive.
Reference:
1. Strate LL, Naumann CR. The role of colonoscopy and radiological procedures in the management of acute lower intestinal bleeding. Clin Gastroenterol Hepatol. 2010 Apr;8(4):333-43.
ginews@gastro.org
Q2. Correct answer: E
Rationale:
Radiographic evaluation is commonly employed in the diagnosis and management of patients with lower GI bleeding. CT scans, tagged red blood cell scintigraphy, and angiography all have roles in the care of these patients. Though tagged red blood cell scintigraphy is the most sensitive modality at detecting active bleeding, requiring rates from 0.05-0.1 cc/min, it is relatively poor at localizing the bleeding, accurately predicting the location in only 60%-70% of cases. CT scans have the advantage of being quickly performed and are widely available. If extravasation is seen, its location is also accurately determined. It is not as sensitive as red blood cell scintigraphy, however, and requires bleeding rates of 0.3-0.5 cc/min to be positive. Angiography has the advantage of being both diagnostic and potentially therapeutic. It is best performed in sicker patients with hypotension and high transfusion demands as it is higher yield in these situations. Angiography is the least sensitive of these modalities, requiring bleeding rates between 0.5 and 1 cc/min to be positive.
Reference:
1. Strate LL, Naumann CR. The role of colonoscopy and radiological procedures in the management of acute lower intestinal bleeding. Clin Gastroenterol Hepatol. 2010 Apr;8(4):333-43.
ginews@gastro.org
Q2:
August 2019 - Question 1
Q1. Correct answer: C
Rationale:
The two standard treatment regimens for AIH include corticosteroids (prednisone or prednisolone) alone, or corticosteroids combined with azathioprine. The combination regimen allows for a lower dose of steroids and fewer side effects with the same therapeutic efficacy. This patient appears to have developed azathioprine-induced pancreatitis, which is a rare complication more often seen in patients with Crohn's disease treated with azathioprine. In patients who are intolerant of azathioprine, mycophenolate mofetil and calcineurin inhibitors have been used with success.
There are data supporting the use of budesonide in place of prednisone, but this regimen is not as effective in patients with cirrhosis or advanced fibrosis, so it is reserved for patients with lesser degrees of liver fibrosis. The TNF-alpha inhibitors are not used to treat AIH, nor is the IL-1 inhibitor anakinra.
References:
1. Czaja AJ. Diagnosis and management of autoimmune hepatitis: Current status and future directions. Gut Liver. 2016;10:177-203.
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune Hepatitis. J Hepatol. 2015:63:971-1004.
3. Manns MP, et al. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:1-31.
Q1. Correct answer: C
Rationale:
The two standard treatment regimens for AIH include corticosteroids (prednisone or prednisolone) alone, or corticosteroids combined with azathioprine. The combination regimen allows for a lower dose of steroids and fewer side effects with the same therapeutic efficacy. This patient appears to have developed azathioprine-induced pancreatitis, which is a rare complication more often seen in patients with Crohn's disease treated with azathioprine. In patients who are intolerant of azathioprine, mycophenolate mofetil and calcineurin inhibitors have been used with success.
There are data supporting the use of budesonide in place of prednisone, but this regimen is not as effective in patients with cirrhosis or advanced fibrosis, so it is reserved for patients with lesser degrees of liver fibrosis. The TNF-alpha inhibitors are not used to treat AIH, nor is the IL-1 inhibitor anakinra.
References:
1. Czaja AJ. Diagnosis and management of autoimmune hepatitis: Current status and future directions. Gut Liver. 2016;10:177-203.
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune Hepatitis. J Hepatol. 2015:63:971-1004.
3. Manns MP, et al. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:1-31.
Q1. Correct answer: C
Rationale:
The two standard treatment regimens for AIH include corticosteroids (prednisone or prednisolone) alone, or corticosteroids combined with azathioprine. The combination regimen allows for a lower dose of steroids and fewer side effects with the same therapeutic efficacy. This patient appears to have developed azathioprine-induced pancreatitis, which is a rare complication more often seen in patients with Crohn's disease treated with azathioprine. In patients who are intolerant of azathioprine, mycophenolate mofetil and calcineurin inhibitors have been used with success.
There are data supporting the use of budesonide in place of prednisone, but this regimen is not as effective in patients with cirrhosis or advanced fibrosis, so it is reserved for patients with lesser degrees of liver fibrosis. The TNF-alpha inhibitors are not used to treat AIH, nor is the IL-1 inhibitor anakinra.
References:
1. Czaja AJ. Diagnosis and management of autoimmune hepatitis: Current status and future directions. Gut Liver. 2016;10:177-203.
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune Hepatitis. J Hepatol. 2015:63:971-1004.
3. Manns MP, et al. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51:1-31.
A 21-year-old woman is diagnosed with autoimmune hepatitis and is started on prednisone and azathioprine. Within a week, she develops mid-abdominal pain, radiating to the back, and her lipase level is 537 U/L.
How do new BP guidelines affect identifying risk for hypertensive disorders of pregnancy?
Hauspurg A, Parry S, Mercer BM, et al. Blood pressure trajectory and category and risk of hypertensive disorders of pregnancy in nulliparous women. Am J Obstet Gynecol. 2019. pii: S0002-9378(19)30807-5. doi: 10.1016/j.ajog.2019.06.031.
EXPERT COMMENTARY
Hauspurg and colleagues set out to determine whether redefined BP category (normal, < 120/80 mm Hg) and trajectory (a difference of ≥ 5 mm Hg systolic, diastolic, or mean arterial pressure between the first and second prenatal visit) helps to identify women at increased risk for developing hypertensive disorders of pregnancy or preeclampsia.
With respect to the former variable, such an association was demonstrated in the first National Institutes of Health–funded preeclampsia prevention trial published in 1993, which used low-dose aspirin.1 In that trial, low-dose aspirin was not found to be effective in preventing preeclampsia in young, healthy nulliparous women. Interestingly, the 2 factors most associated with developing preeclampsia were an initial systolic BP of 120 to 134 mm Hg and an initial weight of >60 kg. For most clinicians, these findings would not be helpful in trying to better identify a high-risk group.
Details of the study
The idea of BP “trajectory” is interesting in the Hauspurg and colleagues’ study. The authors analyzed data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be (nuMoM2b), a prospective cohort study, and included a very large population of almost 9,000 women in the analysis. Participants were classified according to their BP measurement at the first study visit, with BP categories based on updated American College of Cardiology/American Heart Association guidelines. The primary outcome was the risk of hypertensive disorders of pregnancy, including gestational hypertension and preeclampsia.
The data analysis found that elevated BP was associated with an adjusted risk ratio (aRR) of 1.54 (95% confidence interval [CI], 1.18–2.02). Stage 1 hypertension was associated with an aRR of 2.16 (95% CI, 1.31–3.57). Compared with women whose BP had a downward systolic trajectory, women with normal BP and an upward systolic trajectory had a 41% increased risk of any hypertensive disorder of pregnancy (aRR, 1.41; 95% CI, 1.20–1.65).
Study strengths and limitations
While the large study population is a strength of this study, there are a number of limitations, such as the use of BP measurements during pregnancy only, without having pre-pregnancy measurements available. Further, a single BP measurement during each visit is also a drawback, although the standardized measurement by study staff is a strength.
Anticlimactic conclusions. The conclusions of the study, however, are either not surprising, not clinically meaningful, or of little value to clinicians at present, at least with respect to patient management.
Continue to: Conclusions that were not surprising included...
Conclusions that were not surprising included a statistically lower chance of indicated preterm delivery in the normal BP group than in the elevated BP or stage 1 hypertension groups. Conclusions that were not meaningful included a statistically significant lower birthweight in the elevated BP group (3,269 g) and in the stage 1 hypertension group (3,258 g) compared with the normal BP group (3,279 g), but the clinical significance of these differences is arguable.
Lastly is the issue of what these data mean for clinical practice. The idea of identifying high-risk groups is attractive, provided that there are effective intervention strategies available. If one follows the United States Preventive Services Task Force (USPSTF) recommendations for preeclampsia prevention,2 then virtually every nulliparous woman is a candidate for low-dose aspirin for preeclampsia prophylaxis. Beyond that, the current data do not support any change in the standard clinical practice of managing these “now identified” high-risk women. Increasing prenatal visits, using biomarkers to further delineate risk, and using uterine artery Doppler studies are all strategies that have been or are being investigated, but as yet they are not supported by conclusive data documenting improved outcomes—a sentiment supported by both the USPSTF3 and the authors of the study.
Until further data are available, my advice to clinicians is to pay close attention to all risk factors for any of the hypertensive disorders of pregnancy. Initial BP and BP trajectory are important but probably something that sound clinical judgment would identify anyway. My recommendation is to continue to use those methods of prophylaxis, fetal surveillance, and indications for delivery that are supported by current data and await the additional investigations that Hauspurg and colleagues suggest need to be done before altering your management of women at increased risk for any of the hypertensive disorders of pregnancy.
JOHN T. REPKE, MD
- Sibai BM, Caritis SN, Thom E, et al; National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine. Prevention of preeclampsia with low-dose aspirin in healthy nulliparous pregnant women. N Engl J Med. 1993;329:1213-1218.
- United States Preventive Services Task Force. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. September 2014. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed July 30, 2019.
- United States Preventive Service Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA. 2017;387:1661-1667.
Hauspurg A, Parry S, Mercer BM, et al. Blood pressure trajectory and category and risk of hypertensive disorders of pregnancy in nulliparous women. Am J Obstet Gynecol. 2019. pii: S0002-9378(19)30807-5. doi: 10.1016/j.ajog.2019.06.031.
EXPERT COMMENTARY
Hauspurg and colleagues set out to determine whether redefined BP category (normal, < 120/80 mm Hg) and trajectory (a difference of ≥ 5 mm Hg systolic, diastolic, or mean arterial pressure between the first and second prenatal visit) helps to identify women at increased risk for developing hypertensive disorders of pregnancy or preeclampsia.
With respect to the former variable, such an association was demonstrated in the first National Institutes of Health–funded preeclampsia prevention trial published in 1993, which used low-dose aspirin.1 In that trial, low-dose aspirin was not found to be effective in preventing preeclampsia in young, healthy nulliparous women. Interestingly, the 2 factors most associated with developing preeclampsia were an initial systolic BP of 120 to 134 mm Hg and an initial weight of >60 kg. For most clinicians, these findings would not be helpful in trying to better identify a high-risk group.
Details of the study
The idea of BP “trajectory” is interesting in the Hauspurg and colleagues’ study. The authors analyzed data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be (nuMoM2b), a prospective cohort study, and included a very large population of almost 9,000 women in the analysis. Participants were classified according to their BP measurement at the first study visit, with BP categories based on updated American College of Cardiology/American Heart Association guidelines. The primary outcome was the risk of hypertensive disorders of pregnancy, including gestational hypertension and preeclampsia.
The data analysis found that elevated BP was associated with an adjusted risk ratio (aRR) of 1.54 (95% confidence interval [CI], 1.18–2.02). Stage 1 hypertension was associated with an aRR of 2.16 (95% CI, 1.31–3.57). Compared with women whose BP had a downward systolic trajectory, women with normal BP and an upward systolic trajectory had a 41% increased risk of any hypertensive disorder of pregnancy (aRR, 1.41; 95% CI, 1.20–1.65).
Study strengths and limitations
While the large study population is a strength of this study, there are a number of limitations, such as the use of BP measurements during pregnancy only, without having pre-pregnancy measurements available. Further, a single BP measurement during each visit is also a drawback, although the standardized measurement by study staff is a strength.
Anticlimactic conclusions. The conclusions of the study, however, are either not surprising, not clinically meaningful, or of little value to clinicians at present, at least with respect to patient management.
Continue to: Conclusions that were not surprising included...
Conclusions that were not surprising included a statistically lower chance of indicated preterm delivery in the normal BP group than in the elevated BP or stage 1 hypertension groups. Conclusions that were not meaningful included a statistically significant lower birthweight in the elevated BP group (3,269 g) and in the stage 1 hypertension group (3,258 g) compared with the normal BP group (3,279 g), but the clinical significance of these differences is arguable.
Lastly is the issue of what these data mean for clinical practice. The idea of identifying high-risk groups is attractive, provided that there are effective intervention strategies available. If one follows the United States Preventive Services Task Force (USPSTF) recommendations for preeclampsia prevention,2 then virtually every nulliparous woman is a candidate for low-dose aspirin for preeclampsia prophylaxis. Beyond that, the current data do not support any change in the standard clinical practice of managing these “now identified” high-risk women. Increasing prenatal visits, using biomarkers to further delineate risk, and using uterine artery Doppler studies are all strategies that have been or are being investigated, but as yet they are not supported by conclusive data documenting improved outcomes—a sentiment supported by both the USPSTF3 and the authors of the study.
Until further data are available, my advice to clinicians is to pay close attention to all risk factors for any of the hypertensive disorders of pregnancy. Initial BP and BP trajectory are important but probably something that sound clinical judgment would identify anyway. My recommendation is to continue to use those methods of prophylaxis, fetal surveillance, and indications for delivery that are supported by current data and await the additional investigations that Hauspurg and colleagues suggest need to be done before altering your management of women at increased risk for any of the hypertensive disorders of pregnancy.
JOHN T. REPKE, MD
Hauspurg A, Parry S, Mercer BM, et al. Blood pressure trajectory and category and risk of hypertensive disorders of pregnancy in nulliparous women. Am J Obstet Gynecol. 2019. pii: S0002-9378(19)30807-5. doi: 10.1016/j.ajog.2019.06.031.
EXPERT COMMENTARY
Hauspurg and colleagues set out to determine whether redefined BP category (normal, < 120/80 mm Hg) and trajectory (a difference of ≥ 5 mm Hg systolic, diastolic, or mean arterial pressure between the first and second prenatal visit) helps to identify women at increased risk for developing hypertensive disorders of pregnancy or preeclampsia.
With respect to the former variable, such an association was demonstrated in the first National Institutes of Health–funded preeclampsia prevention trial published in 1993, which used low-dose aspirin.1 In that trial, low-dose aspirin was not found to be effective in preventing preeclampsia in young, healthy nulliparous women. Interestingly, the 2 factors most associated with developing preeclampsia were an initial systolic BP of 120 to 134 mm Hg and an initial weight of >60 kg. For most clinicians, these findings would not be helpful in trying to better identify a high-risk group.
Details of the study
The idea of BP “trajectory” is interesting in the Hauspurg and colleagues’ study. The authors analyzed data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be (nuMoM2b), a prospective cohort study, and included a very large population of almost 9,000 women in the analysis. Participants were classified according to their BP measurement at the first study visit, with BP categories based on updated American College of Cardiology/American Heart Association guidelines. The primary outcome was the risk of hypertensive disorders of pregnancy, including gestational hypertension and preeclampsia.
The data analysis found that elevated BP was associated with an adjusted risk ratio (aRR) of 1.54 (95% confidence interval [CI], 1.18–2.02). Stage 1 hypertension was associated with an aRR of 2.16 (95% CI, 1.31–3.57). Compared with women whose BP had a downward systolic trajectory, women with normal BP and an upward systolic trajectory had a 41% increased risk of any hypertensive disorder of pregnancy (aRR, 1.41; 95% CI, 1.20–1.65).
Study strengths and limitations
While the large study population is a strength of this study, there are a number of limitations, such as the use of BP measurements during pregnancy only, without having pre-pregnancy measurements available. Further, a single BP measurement during each visit is also a drawback, although the standardized measurement by study staff is a strength.
Anticlimactic conclusions. The conclusions of the study, however, are either not surprising, not clinically meaningful, or of little value to clinicians at present, at least with respect to patient management.
Continue to: Conclusions that were not surprising included...
Conclusions that were not surprising included a statistically lower chance of indicated preterm delivery in the normal BP group than in the elevated BP or stage 1 hypertension groups. Conclusions that were not meaningful included a statistically significant lower birthweight in the elevated BP group (3,269 g) and in the stage 1 hypertension group (3,258 g) compared with the normal BP group (3,279 g), but the clinical significance of these differences is arguable.
Lastly is the issue of what these data mean for clinical practice. The idea of identifying high-risk groups is attractive, provided that there are effective intervention strategies available. If one follows the United States Preventive Services Task Force (USPSTF) recommendations for preeclampsia prevention,2 then virtually every nulliparous woman is a candidate for low-dose aspirin for preeclampsia prophylaxis. Beyond that, the current data do not support any change in the standard clinical practice of managing these “now identified” high-risk women. Increasing prenatal visits, using biomarkers to further delineate risk, and using uterine artery Doppler studies are all strategies that have been or are being investigated, but as yet they are not supported by conclusive data documenting improved outcomes—a sentiment supported by both the USPSTF3 and the authors of the study.
Until further data are available, my advice to clinicians is to pay close attention to all risk factors for any of the hypertensive disorders of pregnancy. Initial BP and BP trajectory are important but probably something that sound clinical judgment would identify anyway. My recommendation is to continue to use those methods of prophylaxis, fetal surveillance, and indications for delivery that are supported by current data and await the additional investigations that Hauspurg and colleagues suggest need to be done before altering your management of women at increased risk for any of the hypertensive disorders of pregnancy.
JOHN T. REPKE, MD
- Sibai BM, Caritis SN, Thom E, et al; National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine. Prevention of preeclampsia with low-dose aspirin in healthy nulliparous pregnant women. N Engl J Med. 1993;329:1213-1218.
- United States Preventive Services Task Force. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. September 2014. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed July 30, 2019.
- United States Preventive Service Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA. 2017;387:1661-1667.
- Sibai BM, Caritis SN, Thom E, et al; National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine. Prevention of preeclampsia with low-dose aspirin in healthy nulliparous pregnant women. N Engl J Med. 1993;329:1213-1218.
- United States Preventive Services Task Force. Low-dose aspirin use for the prevention of morbidity and mortality from preeclampsia: preventive medication. September 2014. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/low-dose-aspirin-use-for-the-prevention-of-morbidity-and-mortality-from-preeclampsia-preventive-medication. Accessed July 30, 2019.
- United States Preventive Service Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA. 2017;387:1661-1667.

Gastrointestinal Stromal Tumors: Management of Localized Disease
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.

When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Efficacy of erenumab is sustained over more than 4 years of treatment
PHILADELPHIA – “Erenumab was well tolerated and safe, with no safety signals detected over this period,” said Messoud Ashina, MD, PhD, professor of neurology at the University of Copenhagen. Dr. Ashina presented the interim data from a 5-year, open-label extension study of erenumab at the annual meeting of the American Headache Society.

Erenumab is a monoclonal antibody that targets and blocks the calcitonin gene-related peptide (CGRP) receptor. In May 2018, the Food and Drug Administration approved erenumab for the preventive treatment of migraine in adults. The treatment, marketed as Aimovig, is administered once monthly by self-injection.
During the open-label study, patients initially received 70 mg of erenumab monthly. After approximately 2 years, patients switched to 140 mg of erenumab monthly. The researchers’ interim efficacy analysis included all patients on 140 mg of erenumab with data about monthly migraine days after more than 4 years of treatment. The safety analysis included all patients who enrolled in the open-label treatment period and received at least one dose of erenumab.
Of 250 patients who increased the erenumab dose from 70 mg to 140 mg, a total of 221 (88%) completed the open-label treatment period or remained on 140 mg after more than 4 years. Patients’ average number of monthly migraine days at study baseline was 8.7, and the average change from baseline to the most recent month in the interim analysis was –5.8.
During the most recent month of assessment, 77% of patients had at least a 50% reduction in monthly migraine days from baseline, 56% had at least a 75% reduction, and 33% had a 100% reduction.
Mean change from baseline in acute migraine‐specific medication treatment days was –4.6, from a baseline of 6.1.
Among the 383 patients who entered the open-label treatment period and received at least one dose of erenumab (mean age, 41.3; 79% female), the median erenumab exposure was 58.5 months. The exposure‐adjusted incidence of adverse events per 100 patient‐years was 124.9, and the three most frequent adverse events (per 100 patient-years) were nasopharyngitis (10.9), upper respiratory tract infection (6.8), and influenza (4.7). The exposure‐adjusted incidence rate per 100 patient‐years for constipation was 1.3 (9/383) for 70-mg erenumab and 2.6 (15/250) for 140-mg erenumab.
“The exposure‐adjusted incidence rate per 100 patient‐years of serious adverse events was 3.8, similar to the rate observed for erenumab and placebo during the placebo‐controlled periods of studies,” the researchers said.
The study was sponsored by Amgen, and several study authors are employees of Amgen or Novartis, the companies that market erenumab. Dr. Ashina is a consultant for Amgen, Novartis, and other companies.
SOURCE: Ashina M et al. AHS 2019, Abstract IOR10.
PHILADELPHIA – “Erenumab was well tolerated and safe, with no safety signals detected over this period,” said Messoud Ashina, MD, PhD, professor of neurology at the University of Copenhagen. Dr. Ashina presented the interim data from a 5-year, open-label extension study of erenumab at the annual meeting of the American Headache Society.
Erenumab is a monoclonal antibody that targets and blocks the calcitonin gene-related peptide (CGRP) receptor. In May 2018, the Food and Drug Administration approved erenumab for the preventive treatment of migraine in adults. The treatment, marketed as Aimovig, is administered once monthly by self-injection.
During the open-label study, patients initially received 70 mg of erenumab monthly. After approximately 2 years, patients switched to 140 mg of erenumab monthly. The researchers’ interim efficacy analysis included all patients on 140 mg of erenumab with data about monthly migraine days after more than 4 years of treatment. The safety analysis included all patients who enrolled in the open-label treatment period and received at least one dose of erenumab.
Of 250 patients who increased the erenumab dose from 70 mg to 140 mg, a total of 221 (88%) completed the open-label treatment period or remained on 140 mg after more than 4 years. Patients’ average number of monthly migraine days at study baseline was 8.7, and the average change from baseline to the most recent month in the interim analysis was –5.8.
During the most recent month of assessment, 77% of patients had at least a 50% reduction in monthly migraine days from baseline, 56% had at least a 75% reduction, and 33% had a 100% reduction.
Mean change from baseline in acute migraine‐specific medication treatment days was –4.6, from a baseline of 6.1.
Among the 383 patients who entered the open-label treatment period and received at least one dose of erenumab (mean age, 41.3; 79% female), the median erenumab exposure was 58.5 months. The exposure‐adjusted incidence of adverse events per 100 patient‐years was 124.9, and the three most frequent adverse events (per 100 patient-years) were nasopharyngitis (10.9), upper respiratory tract infection (6.8), and influenza (4.7). The exposure‐adjusted incidence rate per 100 patient‐years for constipation was 1.3 (9/383) for 70-mg erenumab and 2.6 (15/250) for 140-mg erenumab.
“The exposure‐adjusted incidence rate per 100 patient‐years of serious adverse events was 3.8, similar to the rate observed for erenumab and placebo during the placebo‐controlled periods of studies,” the researchers said.
The study was sponsored by Amgen, and several study authors are employees of Amgen or Novartis, the companies that market erenumab. Dr. Ashina is a consultant for Amgen, Novartis, and other companies.
SOURCE: Ashina M et al. AHS 2019, Abstract IOR10.
PHILADELPHIA – “Erenumab was well tolerated and safe, with no safety signals detected over this period,” said Messoud Ashina, MD, PhD, professor of neurology at the University of Copenhagen. Dr. Ashina presented the interim data from a 5-year, open-label extension study of erenumab at the annual meeting of the American Headache Society.
Erenumab is a monoclonal antibody that targets and blocks the calcitonin gene-related peptide (CGRP) receptor. In May 2018, the Food and Drug Administration approved erenumab for the preventive treatment of migraine in adults. The treatment, marketed as Aimovig, is administered once monthly by self-injection.
During the open-label study, patients initially received 70 mg of erenumab monthly. After approximately 2 years, patients switched to 140 mg of erenumab monthly. The researchers’ interim efficacy analysis included all patients on 140 mg of erenumab with data about monthly migraine days after more than 4 years of treatment. The safety analysis included all patients who enrolled in the open-label treatment period and received at least one dose of erenumab.
Of 250 patients who increased the erenumab dose from 70 mg to 140 mg, a total of 221 (88%) completed the open-label treatment period or remained on 140 mg after more than 4 years. Patients’ average number of monthly migraine days at study baseline was 8.7, and the average change from baseline to the most recent month in the interim analysis was –5.8.
During the most recent month of assessment, 77% of patients had at least a 50% reduction in monthly migraine days from baseline, 56% had at least a 75% reduction, and 33% had a 100% reduction.
Mean change from baseline in acute migraine‐specific medication treatment days was –4.6, from a baseline of 6.1.
Among the 383 patients who entered the open-label treatment period and received at least one dose of erenumab (mean age, 41.3; 79% female), the median erenumab exposure was 58.5 months. The exposure‐adjusted incidence of adverse events per 100 patient‐years was 124.9, and the three most frequent adverse events (per 100 patient-years) were nasopharyngitis (10.9), upper respiratory tract infection (6.8), and influenza (4.7). The exposure‐adjusted incidence rate per 100 patient‐years for constipation was 1.3 (9/383) for 70-mg erenumab and 2.6 (15/250) for 140-mg erenumab.
“The exposure‐adjusted incidence rate per 100 patient‐years of serious adverse events was 3.8, similar to the rate observed for erenumab and placebo during the placebo‐controlled periods of studies,” the researchers said.
The study was sponsored by Amgen, and several study authors are employees of Amgen or Novartis, the companies that market erenumab. Dr. Ashina is a consultant for Amgen, Novartis, and other companies.
SOURCE: Ashina M et al. AHS 2019, Abstract IOR10.
REPORTING FROM AHS 2019