User login
New postmenopausal osteoporosis guidelines emphasize patient priorities
NEW ORLEANS – In new guidelines for the pharmacologic management of osteoporosis, bisphosphonates have been identified as the first-line therapy with denosumab (Prolia) listed as an acceptable alternative that is particularly well suited for high-risk patients, according to a presentation at the annual meeting of the Endocrine Society.

“We hope our guideline will not only improve patient care but provide confidence in treatment,” reported guideline writing committee member Clifford J. Rosen, MD, director of the Center for Clinical and Translational Research at Maine Medical Center Research Institute, Scarborough.
The new guidelines are evidence based, relying on randomized, controlled trials to evaluate the data quality of treatment options with GRADE methodology, but Dr. Rosen said that the guideline writing committee also considered patient preferences because of concerns about the abundant evidence that adherence to pharmacologic therapies for osteoporosis is poor.
“There is a considerable gap in the treatment of osteoporosis. Most women will not take anti-osteoporosis therapies despite their efficacy, and those who do often stop,” Dr. Rosen observed. He said it was the intention of the writing committee to provide acceptable recommendations with a clear outline of benefits and risks in order to enlist patients more successfully in understanding and participating in fracture prevention.
The Endocrine Society guidelines, which are available online and will soon appear in print (J Clin Endocrinol Metab. 2019;104:1-28), are focused on pharmacologic management and therefore differ from guidelines on diagnosis and treatment published previously by the American Association of Clinical Endocrinologists (AACE) (Endocr Pract. 2016;22[Suppl 4]:1-42).
The AACE guidelines, which devote considerable space to prevention, indicated that bisphosphonates should “be generally considered as initial options for most patients who are candidates for treatment.” The AACE guidelines identify denosumab as the “treatment of choice” in patients with renal insufficiency (although not in those on dialysis or with end-stage renal disease).
In outlining some of the key features of the new guidelines at ENDO 2019, Dr. Rosen drew attention to a call for reevaluation of the need for bisphosphonates after patients have been on this therapy for 3 or more years. For those found at this time to be at low or moderate risk of fracture, a drug holiday is recommended based on guideline-cited evidence that bisphosphonates offer a residual therapeutic effect after stopping.
However, stopping is not recommended in those who remain at high risk. In these patients, bone density should be monitored at regular intervals for the goal of switching or intensifying therapy if needed. This includes use of teriparatide (Forteo) or abaloparatide (Tymlos) for periods of up to 2 years in patients with a history of severe or multiple fractures. These and other choices are included in a detailed algorithm covering both low- and high-risk patients.
Although many postmenopausal women hope to avoid pharmacologic therapy with high dietary intake of calcium and vitamin D, Dr. Rosen stressed the limited benefit of these nutrients in preventing fracture for those with established osteoporosis. While acknowledging that calcium and vitamin D enhance mineralization and maintenance of bone mass, he characterized them as “supplements” once pharmacologic therapies are indicated.
Unsurprisingly, patients prefer oral therapies that are effective but with a low burden of adverse events, according to a review of evidence undertaken by the guideline committee. Cost was a less important consideration. Dr. Rosen indicated that recognizing patient goals and priorities while explaining relative risks might engage patients in selecting a therapy to which they are willing to adhere.
He reported having no relevant financial relationships.
NEW ORLEANS – In new guidelines for the pharmacologic management of osteoporosis, bisphosphonates have been identified as the first-line therapy with denosumab (Prolia) listed as an acceptable alternative that is particularly well suited for high-risk patients, according to a presentation at the annual meeting of the Endocrine Society.
“We hope our guideline will not only improve patient care but provide confidence in treatment,” reported guideline writing committee member Clifford J. Rosen, MD, director of the Center for Clinical and Translational Research at Maine Medical Center Research Institute, Scarborough.
The new guidelines are evidence based, relying on randomized, controlled trials to evaluate the data quality of treatment options with GRADE methodology, but Dr. Rosen said that the guideline writing committee also considered patient preferences because of concerns about the abundant evidence that adherence to pharmacologic therapies for osteoporosis is poor.
“There is a considerable gap in the treatment of osteoporosis. Most women will not take anti-osteoporosis therapies despite their efficacy, and those who do often stop,” Dr. Rosen observed. He said it was the intention of the writing committee to provide acceptable recommendations with a clear outline of benefits and risks in order to enlist patients more successfully in understanding and participating in fracture prevention.
The Endocrine Society guidelines, which are available online and will soon appear in print (J Clin Endocrinol Metab. 2019;104:1-28), are focused on pharmacologic management and therefore differ from guidelines on diagnosis and treatment published previously by the American Association of Clinical Endocrinologists (AACE) (Endocr Pract. 2016;22[Suppl 4]:1-42).
The AACE guidelines, which devote considerable space to prevention, indicated that bisphosphonates should “be generally considered as initial options for most patients who are candidates for treatment.” The AACE guidelines identify denosumab as the “treatment of choice” in patients with renal insufficiency (although not in those on dialysis or with end-stage renal disease).
In outlining some of the key features of the new guidelines at ENDO 2019, Dr. Rosen drew attention to a call for reevaluation of the need for bisphosphonates after patients have been on this therapy for 3 or more years. For those found at this time to be at low or moderate risk of fracture, a drug holiday is recommended based on guideline-cited evidence that bisphosphonates offer a residual therapeutic effect after stopping.
However, stopping is not recommended in those who remain at high risk. In these patients, bone density should be monitored at regular intervals for the goal of switching or intensifying therapy if needed. This includes use of teriparatide (Forteo) or abaloparatide (Tymlos) for periods of up to 2 years in patients with a history of severe or multiple fractures. These and other choices are included in a detailed algorithm covering both low- and high-risk patients.
Although many postmenopausal women hope to avoid pharmacologic therapy with high dietary intake of calcium and vitamin D, Dr. Rosen stressed the limited benefit of these nutrients in preventing fracture for those with established osteoporosis. While acknowledging that calcium and vitamin D enhance mineralization and maintenance of bone mass, he characterized them as “supplements” once pharmacologic therapies are indicated.
Unsurprisingly, patients prefer oral therapies that are effective but with a low burden of adverse events, according to a review of evidence undertaken by the guideline committee. Cost was a less important consideration. Dr. Rosen indicated that recognizing patient goals and priorities while explaining relative risks might engage patients in selecting a therapy to which they are willing to adhere.
He reported having no relevant financial relationships.
NEW ORLEANS – In new guidelines for the pharmacologic management of osteoporosis, bisphosphonates have been identified as the first-line therapy with denosumab (Prolia) listed as an acceptable alternative that is particularly well suited for high-risk patients, according to a presentation at the annual meeting of the Endocrine Society.
“We hope our guideline will not only improve patient care but provide confidence in treatment,” reported guideline writing committee member Clifford J. Rosen, MD, director of the Center for Clinical and Translational Research at Maine Medical Center Research Institute, Scarborough.
The new guidelines are evidence based, relying on randomized, controlled trials to evaluate the data quality of treatment options with GRADE methodology, but Dr. Rosen said that the guideline writing committee also considered patient preferences because of concerns about the abundant evidence that adherence to pharmacologic therapies for osteoporosis is poor.
“There is a considerable gap in the treatment of osteoporosis. Most women will not take anti-osteoporosis therapies despite their efficacy, and those who do often stop,” Dr. Rosen observed. He said it was the intention of the writing committee to provide acceptable recommendations with a clear outline of benefits and risks in order to enlist patients more successfully in understanding and participating in fracture prevention.
The Endocrine Society guidelines, which are available online and will soon appear in print (J Clin Endocrinol Metab. 2019;104:1-28), are focused on pharmacologic management and therefore differ from guidelines on diagnosis and treatment published previously by the American Association of Clinical Endocrinologists (AACE) (Endocr Pract. 2016;22[Suppl 4]:1-42).
The AACE guidelines, which devote considerable space to prevention, indicated that bisphosphonates should “be generally considered as initial options for most patients who are candidates for treatment.” The AACE guidelines identify denosumab as the “treatment of choice” in patients with renal insufficiency (although not in those on dialysis or with end-stage renal disease).
In outlining some of the key features of the new guidelines at ENDO 2019, Dr. Rosen drew attention to a call for reevaluation of the need for bisphosphonates after patients have been on this therapy for 3 or more years. For those found at this time to be at low or moderate risk of fracture, a drug holiday is recommended based on guideline-cited evidence that bisphosphonates offer a residual therapeutic effect after stopping.
However, stopping is not recommended in those who remain at high risk. In these patients, bone density should be monitored at regular intervals for the goal of switching or intensifying therapy if needed. This includes use of teriparatide (Forteo) or abaloparatide (Tymlos) for periods of up to 2 years in patients with a history of severe or multiple fractures. These and other choices are included in a detailed algorithm covering both low- and high-risk patients.
Although many postmenopausal women hope to avoid pharmacologic therapy with high dietary intake of calcium and vitamin D, Dr. Rosen stressed the limited benefit of these nutrients in preventing fracture for those with established osteoporosis. While acknowledging that calcium and vitamin D enhance mineralization and maintenance of bone mass, he characterized them as “supplements” once pharmacologic therapies are indicated.
Unsurprisingly, patients prefer oral therapies that are effective but with a low burden of adverse events, according to a review of evidence undertaken by the guideline committee. Cost was a less important consideration. Dr. Rosen indicated that recognizing patient goals and priorities while explaining relative risks might engage patients in selecting a therapy to which they are willing to adhere.
He reported having no relevant financial relationships.
REPORTING FROM ENDO 2019
Some cardiac devices vulnerable to cybersecurity threats
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.

The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.
The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
Implantable cardiac devices made by Medtronic have cybersecurity vulnerabilities, according to a safety communication from the Food and Drug Administration. That said, so far the FDA is unaware of any reports of harm related to these vulnerabilities, and the agency still advises doctors and patients to continue using the devices as intended and in accordance with device labeling.
The Conexus wireless telemetry protocol used with Medtronic’s implantable cardioverter defibrillators and cardiac resynchronization therapy defibrillators, as well as with certain models of Medtronic’s CareLink Programmer and the MyCareLink Monitor, lacks encryption, authentication, or authorization, which leaves the devices open to exploitation. Such exploitation “could allow unauthorized individuals ... to access and potentially manipulate an implantable device, home monitor, or clinic programmer,” the agency said in its safety communication.
The FDA provides several recommendations in the safety communication, including obtaining these devices “directly from the manufacturer to ensure integrity of the system” and operating “the programmers within well-managed networks.”
FDA panel calls for changes to breast implant rupture screening
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
AT AN FDA ADVISORY PANEL MEETING
2018 FDA-approved new drugs
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.

There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.
If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at obnews@mdedge.com.
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.
There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.
If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at obnews@mdedge.com.
In 2018, the Food and Drug Administration approved a record 58 new drugs for humans. One of these agents, Annovera (segesterone acetate and ethinyl estradiol), is a vaginal ring to prevent pregnancy and is not relevant in this article. A second drug, Asparlas (calaspargase pegol-mknl), indicated to treat acute lymphoblastic leukemia, has not yet been released by its manufacturer. The agents with molecular weights (MW) less than 1,000 probably cross the placenta, but nearly all, regardless of MW, will cross in the second half of pregnancy.
There is no human pregnancy data for these agents, but there are five drugs included in pregnancy registries. However, it will take some time before the outcomes of these drugs are published. The routine absence of pregnancy data for most drugs was pointed out in a reference that I coauthored (“Should pregnant women be included in phase 4 clinical drug trials?” Am J Obstet Gynecol. 2015 Dec;213[6]:810-5). The article makes a strong argument for including some drugs in these trials.
Amyloidosis
Onpattro (patisiran) is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. The drug caused embryo-fetal death and reduced fetal body weight in rabbits at doses also associated with maternal toxicity. No developmental toxicity was observed in rats.
Anti-infectives
Aemcolo (rifamycin), which has a MW of 720, is indicated for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli. No adverse fetal effects were observed in rats and rabbits that received close to human doses.
Krintafel (tafenoquine) is an antimalarial agent that is used to prevent relapse in patients who are receiving appropriate antimalarial therapy for Plasmodium vivax infection. The drug may cause hemolytic anemia in a fetus deficient in glucose-6-phosphate dehydrogenase. In rabbits, the drug caused dose-related abortions and maternal toxicity was observed in rabbits and rats. Treatment with this drug in pregnancy is not recommended, according to the manufacturer.
Tpoxx (tecovirimat monohydrate), which has a MW of about 394, is indicated for the treatment of smallpox disease. The drug did not cause embryo-fetal toxicity in pregnant mice and rabbits, but the maximum exposure in rabbits was only 0.4 times the human exposure.
Xofluza (baloxavir marboxil), which has a MW of about 572, is a prodrug that is converted by hydrolysis to baloxavir. It is indicated for the treatment of acute uncomplicated influenza. No adverse developmental effects were observed in rats and rabbits.
Zemdri (plazomicin), which has a MW of about 593, is an aminoglycoside indicated for the treatment of complicated urinary tract infections including pyelonephritis. The drug did not cause fetal harm in rats and rabbits at doses that did not cause maternal toxicity; however, prolonged use of an aminoglycoside (such as streptomycin) has caused irreversible, bilateral congenital deafness in children exposed in utero to prolonged use and is a potential complication.
Three new drugs in 2018 are indicated for treating HIV-1:
Biktarvy is a three-drug combination that includes bictegravir, emtricitabine, and tenofovir. The latter two drugs are included in the 11th edition of my book (“Drugs in Pregnancy and Lactation,” 11th ed. [Riverwoods, Ill.: Wolters Kluwer, 2017) and are not included here. Both are classified as compatible in pregnancy. Bictegravir has a MW of about 471. No adverse embryo-fetal effects in rats and rabbits were observed with this agent.
Trogarzo (ibalizumab-uiyk), which has a MW of about 150,000, is a monoclonal antibody antiretroviral agent used in combination with other antiretrovirals. There are no animal data. Although the MW is very high, monoclonal antibodies are transported across the placenta as pregnancy progresses.
Pifeltro (doravirine), which has a MW of about 426, is a nonnucleoside reverse transcriptase inhibitor used in combination with other antiretroviral agents for the treatment of HIV-1. The drug caused no significant toxicologic effects on embryo-fetal rats and rabbits.
If Biktarvy, Pifeltro, or Trogarzo are used in pregnancy, health care providers are encouraged to register the patient in the Antiretroviral Pregnancy Registry by calling 1-800-258-4263.
There are three new agents in the tetracycline class.
Nuzyra (omadacycline), which has a MW of about 729, is for community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections.
Seysara (sarecycline), which has a MW of about 524, is for inflammatory lesions of nonnodular, moderate to severe acne vulgaris.
Xerava (eravacycline), which has a MW of about 632, is for complicated intra-abdominal infection.
The various dose-related toxicities observed with the three drugs in rats and rabbits included maternal deaths; increased postimplantation loss; reduced fetal body weights; delays in skeletal ossification; and fetal malformations of the skeleton, heart, and lung. Use of these drugs in the last half of pregnancy may cause permanent discoloration of the teeth and enamel hypoplasia, as well as inhibition of bone growth.
Antilipemic agents
Crysvita (burosumab-twza), which has a MW of about 147,000, is a fibroblast growth factor–blocking antibody indicated for the treatment of X-linked hypophosphatemia. In pregnant cynomolgus monkeys, doses slightly higher than the human dose were not teratogenic. The drug was detected in fetal serum indicating that it crossed the monkey placenta.
Tegsedi (inotersen), which has a MW of about 7,601, is an amyloidosis inhibitor used for polyneuropathy of hereditary transthyretin-mediated amyloidosis. It is available only through a restricted program. The drug was not teratogenic in mice and rabbits; however, it does decrease vitamin A levels, so supplementation with the vitamin is recommended.
Antineoplastics
The manufacturers recommend avoiding these drugs during pregnancy. Effective contraception should be used.
Daurismo (glasdegib), which has a MW of about 491, is a hedgehog pathway inhibitor indicated in combination with low-dose cytarabine for newly diagnosed acute myeloid leukemia (AML). The drug caused embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits at doses less than the human dose.
Erleada (apalutamide), which has a MW of about 477, is an androgen receptor inhibitor indicated for nonmetastatic, castration-resistant prostate cancer. Animal studies were not conducted because the drug should not be used in females.
Elzonris (tagraxofusp-erzs), which has a MW of 57,695, is a cytotoxin indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm. Animal studies have not been conducted.
Lumoxiti (moxetumomab pasudotox–tdfk) which as a MW of about 63,000, is indicated for relapsed or refractory hairy cell leukemia. Studies have not been conducted in pregnant animals. Two life-threatening outcomes have occurred with the drug: capillary leak syndrome and hemolytic uremic syndrome. The drug should be discontinued if either occurs.
Lutathera (lutetium Lu 177 dotatate), which has a MW of about 1,610, is a radiolabeled somatostatin analogue given as a single intravenous dose every 8 weeks for four doses for the treatment of gastroenteropancreatic neuroendocrine tumors. Reproductive studies in animals have not been conducted. However, all radiopharmaceuticals have the potential to cause embryo-fetal harm. They also can cause infertility in males and females.
Talzenna (talazoparib), which has a MW of about 553, is a poly (ADP-ribose) polymerase inhibitor indicated for the treatment of certain types of breast cancer. At doses much less then the human dose, the drug caused fetal malformations and embryo-fetal death in rats.
Tibsovo (ivosidenib), which has a MW of 583, is an isocitrate dehydrogenase 1 inhibitor used for patients with relapsed or refractory AML. The drug caused embryo-fetal toxicity in rats and rabbits at doses slighter higher than the human dose.
There are seven new kinase inhibitors.
Braftovi (encorafenib), which has a MW of 540, is indicated in combination with Mektovi for patients with a specific type of metastatic melanoma. The drug caused embryo-fetal toxicity in rats and rabbits.
Copiktra (duvelisib), which has a MW of about 435, is indicated for treatment of chronic lymphocytic leukemia and follicular lymphoma. In rats and rabbits, the drug caused embryo-fetal death, lower fetal weights, and malformations.
Lorbrena (lorlatinib), which has a MW of about 406, is given for the treatment of metastatic non–small cell lung cancer. In rats and rabbits, the drug caused abortions, decreased fetal body weight, and major malformations.
Mektovi (binimetinib), which has a MW of about 441, is used in combination with Braftovi for patients with a specific type of melanoma. The drug was embryotoxic and abortifacient in rabbits.
Vitrakvi (larotrectinib), which has a MW of about 527, is used for patients with solid tumors. Studies in rats revealed fetal anasarca (extreme generalized edema) and omphalocele in rabbits.
Vizimpro (dacomitinib), which has a MW of about 488, is indicated for metastatic non–small cell lung cancer. The drug caused embryo-fetal toxicity in rats and mice.
Xospata (gilteritinib), which has a MW of about 1,222, is indicated for relapsed or refractory AML. In rats, the drug caused embryo-fetal death, suppressed fetal growth, and caused multiple malformations.
Three drugs are classified as monoclonal antibodies.
Gamifant (emapalumab), which has a MW of about 148,000, is indicated for primary hemophagocytic lymphohistiocytosis. A murine surrogate antimouse antibody was given to pregnant mice throughout gestation and no fetal harm was observed.
Libtayo (cemiplimab-rwlc), which has a MW of 146,000, is indicated for patients with metastatic or locally advanced cutaneous squamous cell carcinoma. Animal reproduction studies have not been conducted; however, based on its mechanism, increased rates of abortion or stillbirth may occur if the drug is used in human pregnancy.
Poteligeo (mogamulizumab-kpkc), which has a MW of about 149,000, is given for relapsed/refractory mycosis fungoides or Sézary syndrome. In pregnant monkeys, there was no embryo-fetal lethality, teratogenicity, fetal growth restriction, spontaneous abortion, or increased fetal death.
Central nervous system
There are three antimigraine agents that are monoclonal antibodies given as a subcutaneous injection.
Aimovig (erenumab-aooe), which has a MW of about 150,000, caused no adverse effects in monkey offspring.
Ajovy (fremanezumab-vfrm), which has a MW of about 148,000, had no adverse effect in rat and rabbit offspring.
Emgality (galcanezumab-gnlm), which has a MW of about 147,000, produced no adverse effects in rat and rabbit offspring.
Diacomit (stiripentol), which has a MW of about 234, is an anticonvulsant used to treat seizures associated with Dravet syndrome. The drug caused severe embryo-fetal toxicity in mice, rabbits, and rats. The drug is included in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. Patients can enroll themselves by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Epidiolex (cannabidiol), which has a MW of about 314, is an anticonvulsant indicated for the treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome. In pregnant rats, doses up to about 16 times the recommended human dose (RHD) caused no embryo-fetal adverse effects. The drug caused decreased fetal body weights, increased fetal structural variations, and maternal toxicity when the drug was given to pregnant rabbits throughout organogenesis. The no-effect dose for embryo-fetal toxicity was less than the human dose. Patients can enroll themselves in the NAAED Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://aedpregnancyregistry.org/.
Firdapse (amifampridine), a potassium channel blocker with a MW of about 201, is used for the treatment of Lambert-Eaton myasthenic syndrome. No adverse effects on embryo-fetal development were observed in rats and rabbits given the drug throughout organogenesis. However, in rats given the drug throughout pregnancy and lactation, there was an increase in stillbirths and pup deaths, reduced pup weight, and delayed sexual development in female pups.
Lucemyra (lofexidine), which has a MW of about 296, is used to mitigate opioid withdrawal symptoms to facilitate abrupt opioid discontinuation in adults. The drug caused severe toxicity in the fetuses of rats and rabbits.
Olumiant (baricitinib), which has a MW of about 371, is a Janus kinase inhibitor indicated for the treatment of rheumatoid arthritis. The drug was teratogenic in pregnant rats given doses about 20 times greater than the maximum RHD based on area under the curve. In rabbits, embryo death and rib anomalies were observed with doses 84 times greater than the maximum RHD, but no developmental toxicity was seen with doses 12 times greater than the maximum RHD.
Orilissa (elagolix), which has a MW of about 654, is a gonadotropin-releasing hormone receptor antagonist indicated for the management of pain associated with endometriosis. The drug caused abortions in rats and rabbits. Because the drug may increase the risk of early pregnancy loss, the manufacturer classifies it as contraindicated in pregnancy.
Dermatologic agents
Ilumya (tildrakizumab), which has a MW of about 147,000, is given by subcutaneous injection for the treatment of moderate to severe plaque psoriasis. When given during organogenesis in monkeys, no maternal or embryo-fetal toxicities were observed. However, when given throughout pregnancy a few neonatal deaths occurred, but the clinical significance of these nonclinical findings were unknown.
Fabry disease
Galafold (migalastat), which has a MW of about 200, is an alpha-galactosidase A pharmacologic chaperone indicated for the treatment of Fabry disease. Three pregnant women with Fabry disease were exposed to the drug in clinical studies but no information was provided on the pregnancy outcomes. No adverse developmental effects were observed in pregnant rats and rabbits.
Gastrointestinal agents
Akynzeo (netupitant or fosnetupitant palonosetron), which have MWs of about 579, 333, and 762, respectively, is available as an oral capsule (netupitant + palonosetron) and as an intravenous formulation (fosnetupitant + palonosetron). They are indicated, in combination with dexamethasone, for the prevention of nausea and vomiting related to cancer chemotherapy. Netupitant and fosnetupitant produced no embryo-fetal adverse effects in rats but were toxic to rabbit embryos. Palonosetron caused no embryo-fetal adverse effects in rats and rabbits.
Motegrity (prucalopride), which has a MW of about 486, is indicated for chronic idiopathic constipation. No adverse embryo-fetal developmental effects were observed in rats and rabbits.
Hematologic agents
Doptelet (avatrombopag), which has a MW of about 766, is indicated for the treatment of thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. No embryo-fetal effects were observed in rats, but in rabbits the drug was associated with spontaneous abortions.
Lokelma (sodium zirconium cyclosilicate) is a nonabsorbed zirconium silicate that exchanges potassium for hydrogen and sodium. Animal studies have not been conducted. Because it is not absorbed, it is not expected to result in fetal exposure to the drug.
Mulpleta (lusutrombopag), which has a MW of about 592, a thrombopoietin receptor agonist, is indicated for the treatment of thrombocytopenia in patients with chronic liver disease. High levels of the drug in pregnant rats were associated with adverse developmental outcomes. No adverse embryo-fetal effects were seen in pregnant rabbits.
Palynziq (pegvaliase-pqpz), which has a MW of about 1,000,000, is a phenylalanine-metabolizing enzyme indicated to reduce blood phenylalanine concentrations in patients with phenylketonuria. In pregnant rats, the drug caused an increase in skeletal variations. In rabbits, the drug caused a high incidence of multiple malformations.
Takhzyro (lanadelumab-flyo), which has a MW of about 49,000, is a monoclonal antibody indicated for prophylaxis to prevent attacks of hereditary angioedema. The drug caused no fetal harm in monkeys.
Tavalisse (fostamatinib disodium hexahydrate), which has a MW of about 733, is a kinase inhibitor used for the treatment of thrombocytopenia. In pregnant rats and rabbits, the drug caused adverse developmental outcomes including embryo-fetal mortality, lower fetal weights, and structural anomalies.
Ultomiris (ravulizumab), which has a MW of about 148,000, is a humanized monoclonal antibody indicated for adult patients with paroxysmal nocturnal hemoglobinuria. In mice, the drug was associated with increased rates of developmental abnormalities and an increased rate of dead and moribund offspring.
Immunologic agent
Revcovi (elapegademase-lvlr), which has a MW of about 113,000, is a recombinant adenosine deaminase indicated for the treatment of adenosine deaminase severe combined immune deficiency. Animal studies in pregnancy have not been conducted.
Nutrient/Nutritional supplement
Fish oil is indicated as a source of calories and fatty acids in pediatric patients with parenteral nutrition-associated cholestasis. Animal reproduction studies have not been conducted. It is doubtful if this product will be used in pregnancy.
Ophthalmic – nerve growth factor
Oxervate (cenegermin-bkbj), which has a MW of 13,266, is a solution that contains 118 amino acids. It is a recombinant human nerve growth factor indicated for neurotrophic keratitis. In rats and rabbits given the drug during organogenesis, there was a slight increase in postimplantation loss at doses greater than or equal to 267 times the human dose.
Respiratory drugs
Symdeko (tezacaftor + ivacaftor), which have MWs of about 521 and 392, is indicated for the treatment of patients with cystic fibrosis who are homozygous for the F508del mutation or who have at least one mutation in the cystic fibrosis transmembrane conductance regulator gene. There were no adverse developmental effects in pregnant rats and rabbits when the drugs were used separately or combined.
Yupelri (revefenacin), which has a MW of about 598, is an anticholinergic drug. It is an inhaled solution for the maintenance treatment of chronic obstructive pulmonary disease. In rats and rabbits, doses that were about 209 times the RHD produced no evidence of fetal harm.
Mr. Briggs is clinical professor of pharmacy at the University of California, San Francisco, and adjunct professor of pharmacy at the University of Southern California, Los Angeles, as well as at Washington State University, Spokane. Mr. Briggs reported no relevant financial disclosures. Email him at obnews@mdedge.com.
Postpartum hypertension
When managing our pregnant patients, we often might be tempted to view the delivery of the baby as the conclusion of prenatal care. For many women, the baby’s birth coincides with a resolution of health conditions that they may have experienced during pregnancy, including edema, gestational diabetes, and hypertensive disorders. However, the postpartum period remains a critical time in the health of the mother. Indeed, the weeks immediately following parturition often are colloquially referred to as the fourth trimester, further emphasizing the importance of appropriate patient management and care during this time.

One of the key health conditions we must monitor in the immediate postpartum period is hypertension. According to a 2018 report compiling data from nine of the Centers for Disease Control and Prevention’s Maternal Mortality Review Committees, hypertensive disorders accounted for approximately 9.3% of pregnancy-related maternal deaths within 42 days after delivery (http://reviewtoaction.org/Report_from_Nine_MMRCs). Although women who have hypertensive disorders during pregnancy are at risk for complications after giving birth, women without gestational hypertension, preeclampsia, or eclampsia can experience these conditions post partum at a rate between 0.3% and 27.5% (Am J Obstet Gynecol 2012 Jun;206[6]:470-5). Therefore, we cannot assume that a patient with an uncomplicated pregnancy is completely “in the clear” after delivery.
Despite these somewhat grim statistics, With vigilant monitoring and strong communication with our patients, ob.gyns. can reduce the risks of these complications from occurring, more quickly resolve symptoms as they might arise, and significantly improve the health and well-being of new mothers in the fourth trimester.
The importance of caring for all of our patients along the continuum of pregnancy, especially as it pertains to monitoring and preventing postpartum hypertension, is the focus of the third and final installment of this Master Class series on hypertension in pregnancy authored by Dr. Baha Sibai, professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
When managing our pregnant patients, we often might be tempted to view the delivery of the baby as the conclusion of prenatal care. For many women, the baby’s birth coincides with a resolution of health conditions that they may have experienced during pregnancy, including edema, gestational diabetes, and hypertensive disorders. However, the postpartum period remains a critical time in the health of the mother. Indeed, the weeks immediately following parturition often are colloquially referred to as the fourth trimester, further emphasizing the importance of appropriate patient management and care during this time.
One of the key health conditions we must monitor in the immediate postpartum period is hypertension. According to a 2018 report compiling data from nine of the Centers for Disease Control and Prevention’s Maternal Mortality Review Committees, hypertensive disorders accounted for approximately 9.3% of pregnancy-related maternal deaths within 42 days after delivery (http://reviewtoaction.org/Report_from_Nine_MMRCs). Although women who have hypertensive disorders during pregnancy are at risk for complications after giving birth, women without gestational hypertension, preeclampsia, or eclampsia can experience these conditions post partum at a rate between 0.3% and 27.5% (Am J Obstet Gynecol 2012 Jun;206[6]:470-5). Therefore, we cannot assume that a patient with an uncomplicated pregnancy is completely “in the clear” after delivery.
Despite these somewhat grim statistics, With vigilant monitoring and strong communication with our patients, ob.gyns. can reduce the risks of these complications from occurring, more quickly resolve symptoms as they might arise, and significantly improve the health and well-being of new mothers in the fourth trimester.
The importance of caring for all of our patients along the continuum of pregnancy, especially as it pertains to monitoring and preventing postpartum hypertension, is the focus of the third and final installment of this Master Class series on hypertension in pregnancy authored by Dr. Baha Sibai, professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
When managing our pregnant patients, we often might be tempted to view the delivery of the baby as the conclusion of prenatal care. For many women, the baby’s birth coincides with a resolution of health conditions that they may have experienced during pregnancy, including edema, gestational diabetes, and hypertensive disorders. However, the postpartum period remains a critical time in the health of the mother. Indeed, the weeks immediately following parturition often are colloquially referred to as the fourth trimester, further emphasizing the importance of appropriate patient management and care during this time.
One of the key health conditions we must monitor in the immediate postpartum period is hypertension. According to a 2018 report compiling data from nine of the Centers for Disease Control and Prevention’s Maternal Mortality Review Committees, hypertensive disorders accounted for approximately 9.3% of pregnancy-related maternal deaths within 42 days after delivery (http://reviewtoaction.org/Report_from_Nine_MMRCs). Although women who have hypertensive disorders during pregnancy are at risk for complications after giving birth, women without gestational hypertension, preeclampsia, or eclampsia can experience these conditions post partum at a rate between 0.3% and 27.5% (Am J Obstet Gynecol 2012 Jun;206[6]:470-5). Therefore, we cannot assume that a patient with an uncomplicated pregnancy is completely “in the clear” after delivery.
Despite these somewhat grim statistics, With vigilant monitoring and strong communication with our patients, ob.gyns. can reduce the risks of these complications from occurring, more quickly resolve symptoms as they might arise, and significantly improve the health and well-being of new mothers in the fourth trimester.
The importance of caring for all of our patients along the continuum of pregnancy, especially as it pertains to monitoring and preventing postpartum hypertension, is the focus of the third and final installment of this Master Class series on hypertension in pregnancy authored by Dr. Baha Sibai, professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston.
Dr. Reece, who specializes in maternal-fetal medicine, is executive vice president for medical affairs at the University of Maryland, Baltimore, as well as the John Z. and Akiko K. Bowers Distinguished Professor and dean of the school of medicine. He is the medical editor of this column. He said he had no relevant financial disclosures. Contact him at obnews@mdedge.com.
Recognition, evaluation, and management of postpartum hypertension
Postpartum hypertension has a host of potential causes, some of which may be benign (such as the persistence of mild gestational hypertension or mild chronic hypertension) whereas others (such as severe de novo preeclampsia-eclampsia and HELLP syndrome [a complication of pregnancy characterized by hemolysis, elevated liver enzymes, and a low platelet count]) can be life threatening.
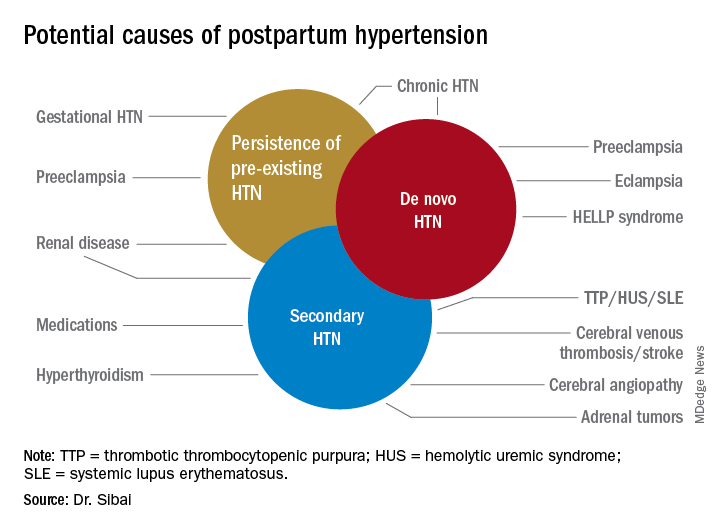
Postpartum hypertension may occur secondary to lupus, hyperthyroidism, hemolytic uremic syndrome, stroke, and other conditions, which means that we must have a high index of suspicion for secondary dangerous causes of hypertension when evaluating such women.
With monitoring, reporting, and prompt evaluation of symptoms in the postpartum period – and with patient education on signs and symptoms of severe hypertension and preeclampsia (PE) – we can expect to avoid a range of potential maternal complications, from hypertensive encephalopathy, liver hemorrhage, renal failure, and the development of eclampsia, ischemic stroke/cerebral hemorrhage, pulmonary edema, and cardiomyopathy.
Most women with gestational hypertension (GHTN) become normotensive during the first week post partum, but in women who develop PE during pregnancy, hypertension often takes longer to resolve. Some of these women may have an initial decrease in blood pressure immediately post partum followed by development of hypertension again between days 3 and 6. Therefore, This can be achieved either in-hospital, through home BP monitoring, or with in-office visits.
In addition, all women – including those who did not have hypertension during their pregnancies – should be educated about the signs and symptoms of severe hypertension or PE and instructed to report these to a medical provider in a timely fashion. Severe hypertension or PE with severe features may develop for the first time during the postpartum period either before or after hospital discharge. It is important to appreciate, moreover, that approximately 25%-40% of cases of eclampsia develop in the postpartum period with onset ranging from 2 days to 6 weeks after delivery. Moreover, almost one-third of women who develop the HELLP syndrome do so during the postpartum period.
Management of persistent hypertension
The most common causes for persistent hypertension beyond 48 hours after delivery are GHTN, PE, or chronic hypertension. Initial management will depend on history, clinical findings, presence or absence of associated symptoms, results of laboratory findings (urine protein, platelet count, liver enzymes, serum creatinine, and electrolytes), and response to prior treatment of hypertension.
Certain medications that frequently are prescribed in the postpartum period, such as ergonovine and decongestants, should be discontinued if they are being used. These agents can aggravate preexisting hypertension or result in new-onset hypertension if used in large or frequent doses. Their use also may be associated with cerebral symptoms, nausea, and vomiting.
Subsequent management includes close observation until resolution of hypertension and associated symptoms. If the patient has hypertension only with no symptoms, no proteinuria, and normal laboratory findings, BP control is the focus; antihypertensives are used if systolic BP remains persistently greater than or equal to 150 mm Hg and/or if diastolic BP persists at greater than or equal to 100 mm Hg. Intravenous boluses of either labetalol or hydralazine or oral rapid-acting nifedipine are used initially if systolic BP is greater than or equal to 160 mm Hg or diastolic BP greater than or equal to 110 mm Hg persists for at least 30 minutes. This is followed by oral medication to keep systolic BP less than 150 mg Hg and diastolic BP less than 100 mm Hg.
For patients with persistent hypertension after GHTN or PE, I recommend oral long-acting nifedipine XL (30 mg every 12 hours) or oral labetalol (200 mg every 8-12 hours). Compared with labetalol, oral nifedipine is associated with improved renal blood flow with resultant diuresis, which makes it the drug of choice in women with volume overload. In some, it is necessary to switch to a new agent such as an angiotensin-converting enzyme (ACE) inhibitor; an ACE inhibitor is the drug of choice in those with pregestational diabetes mellitus, renal disease, or cardiomyopathy. In addition, thiazide or loop diuretics may be needed in women with circulatory overload and in those with pulmonary edema. Antihypertensives such as nifedipine, labetalol, furosemide, captopril, and enalapril are compatible with breastfeeding.
If the BP remains less than 150 mm Hg (systolic) and/or less than 100 mm Hg (diastolic) for 24 hours, and there are no maternal symptoms, the patient may be discharged home with instructions for daily BP measurements (self or by a visiting nurse) and the reporting of symptoms until her next visit in 1 week. Antihypertensives then are discontinued if the BP remains below the hypertensive levels for at least 48 hours. This may take 1 or several weeks to achieve.
Women with PE with severe features should receive close monitoring of BP and of symptoms during the immediate postpartum period, as well as accurate measurements of fluid intake, urinary output, and weight gain. These women often have received large amounts of IV fluids during labor as a result of prehydration before epidural analgesia, as well as IV fluids administered during the use of oxytocin and magnesium sulfate in labor and post partum. Mobilization of extracellular fluid also leads to increased intravascular volume. As a result, women who have PE with severe features – particularly those with abnormal renal function, capillary leak, or early-onset disease – are at increased risk for pulmonary edema and exacerbation of severe hypertension.
Careful evaluation of the volume of IV fluids, oral intake, blood products, urine output, respiratory symptoms, and vital signs is advised. Patients who develop tachycardia or respiratory symptoms such as dry cough, shortness of breath, or orthopnea also should be monitored with pulse oximetry and frequent chest auscultation, as well as chest x-ray.
New-onset severe symptoms
Because severe hypertension or PE with severe features may develop for the first time during the postpartum period, postpartum women – and the medical providers and personnel who respond to patient phone calls – should be well educated about the signs and symptoms of severe hypertension or PE. These include new-onset severe headaches that do not respond to maximum doses of analgesics, persistent severe visual changes, and new-onset epigastric pain with nausea and vomiting, dyspnea, orthopnea, shortness of breath, or palpitations. These women are at increased risk for eclampsia, pulmonary edema, stroke, and thromboembolism; these women require careful evaluation and potential hospitalization.
Severe new onset of persistent headaches and/or visual symptoms. Women with hypertension in association with new-onset persistent headaches and/or visual changes should be suspected to have severe PE. Patients who have hypertension with seizure should be initially treated as having eclampsia and should receive brain imaging to rule out other etiologies. Magnesium sulfate therapy must be initiated promptly for seizure prophylaxis and/or treatment. In addition, intravenous antihypertensive medications are recommended to lower BP to the desired goal while considering an alternative cause for the cerebral symptoms.
Women presenting with hypertension in association with refractory and/or thunderclap headaches, visual disturbances, or neurologic deficits should be evaluated for possible cerebrovascular complications such as reversible cerebral vasoconstriction syndrome (RCVS), cerebral venous thrombosis, or stroke. These women will require selective diagnostic neuroimaging and consultation with neurology and/or neurosurgery. Such an evaluation may include CT scan for hemorrhage, MRI for detection of vasogenic edema and/or ischemia or infarction, cerebral angiography for diagnosis of RCVS, and cerebral venography for detection of cerebral venous thrombosis. Subsequent treatment will depend on the etiology.
Severe new-onset epigastric/right upper quadrant pain with nausea and vomiting. Women with persistent nausea, vomiting, or epigastric pain should be evaluated for HELLP syndrome because up to 30% who develop the syndrome do so post partum. The time of onset of clinical and laboratory findings ranges from 1 to 7 days post partum. Women are managed as they are before delivery, with the use of magnesium sulfate, antihypertensives, and close monitoring of vital signs and laboratory values.
In general, patients with HELLP syndrome will demonstrate an improvement in clinical and laboratory findings within 72 hours after treatment. If there is either no improvement or a deterioration in these findings, then it is important to consult with appropriate specialists for evaluation and subsequent management of possible rare syndromes such as acute fatty liver, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, or exacerbation of lupus.
Severe new-onset shortness of breath, dyspnea, orthopnea, or palpitations. Women with these symptoms in the postpartum period should be evaluated for possible pulmonary edema, pulmonary embolism, or peripartum cardiomyopathy. Women with postpartum hypertension are at risk for pulmonary edema with onset at 3-6 days after delivery. Diagnosis is confirmed by physical exam (tachycardia, tachypnea), presence of rales on lung exam, pulse oximetry (oxygen saturation less than 93%), and chest x-ray, and echocardiography to exclude other etiologies. Treatment of pulmonary edema includes oxygen supplementation, 40 mg IV furosemide, control of severe hypertension, fluid restriction, and supportive care.
Pulmonary embolism usually is confirmed by chest CT angiography and managed with therapeutic anticoagulation. Peripartum cardiomyopathy is diagnosed by echocardiography revealing left ventricular systolic dysfunction (ejection fraction less than 45%, dilated left ventricle). Treatment includes IV furosemide, use of a vasodilator, and ACE inhibitor therapy.
Remote prognosis
Recent research suggests that women who develop PE may be at increased risk for future cardiovascular disease such as heart failure, coronary artery disease, and stroke later in life. Indeed, many of the risk factors and pathophysiologic abnormalities of PE are similar to those of coronary artery disease.
The American College of Obstetricians and Gynecologists and the American Heart Association recommend that women with PE receive close observation in the postpartum period and careful evaluation in the first year after delivery to identify those who could benefit from early intervention to prevent subsequent cardiovascular disease. In general, when pregnancies are complicated by PE, there are opportunities for lifestyle and risk factor modification.
Dr. Sibai is professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston
Postpartum hypertension has a host of potential causes, some of which may be benign (such as the persistence of mild gestational hypertension or mild chronic hypertension) whereas others (such as severe de novo preeclampsia-eclampsia and HELLP syndrome [a complication of pregnancy characterized by hemolysis, elevated liver enzymes, and a low platelet count]) can be life threatening.
Postpartum hypertension may occur secondary to lupus, hyperthyroidism, hemolytic uremic syndrome, stroke, and other conditions, which means that we must have a high index of suspicion for secondary dangerous causes of hypertension when evaluating such women.
With monitoring, reporting, and prompt evaluation of symptoms in the postpartum period – and with patient education on signs and symptoms of severe hypertension and preeclampsia (PE) – we can expect to avoid a range of potential maternal complications, from hypertensive encephalopathy, liver hemorrhage, renal failure, and the development of eclampsia, ischemic stroke/cerebral hemorrhage, pulmonary edema, and cardiomyopathy.
Most women with gestational hypertension (GHTN) become normotensive during the first week post partum, but in women who develop PE during pregnancy, hypertension often takes longer to resolve. Some of these women may have an initial decrease in blood pressure immediately post partum followed by development of hypertension again between days 3 and 6. Therefore, This can be achieved either in-hospital, through home BP monitoring, or with in-office visits.
In addition, all women – including those who did not have hypertension during their pregnancies – should be educated about the signs and symptoms of severe hypertension or PE and instructed to report these to a medical provider in a timely fashion. Severe hypertension or PE with severe features may develop for the first time during the postpartum period either before or after hospital discharge. It is important to appreciate, moreover, that approximately 25%-40% of cases of eclampsia develop in the postpartum period with onset ranging from 2 days to 6 weeks after delivery. Moreover, almost one-third of women who develop the HELLP syndrome do so during the postpartum period.
Management of persistent hypertension
The most common causes for persistent hypertension beyond 48 hours after delivery are GHTN, PE, or chronic hypertension. Initial management will depend on history, clinical findings, presence or absence of associated symptoms, results of laboratory findings (urine protein, platelet count, liver enzymes, serum creatinine, and electrolytes), and response to prior treatment of hypertension.
Certain medications that frequently are prescribed in the postpartum period, such as ergonovine and decongestants, should be discontinued if they are being used. These agents can aggravate preexisting hypertension or result in new-onset hypertension if used in large or frequent doses. Their use also may be associated with cerebral symptoms, nausea, and vomiting.
Subsequent management includes close observation until resolution of hypertension and associated symptoms. If the patient has hypertension only with no symptoms, no proteinuria, and normal laboratory findings, BP control is the focus; antihypertensives are used if systolic BP remains persistently greater than or equal to 150 mm Hg and/or if diastolic BP persists at greater than or equal to 100 mm Hg. Intravenous boluses of either labetalol or hydralazine or oral rapid-acting nifedipine are used initially if systolic BP is greater than or equal to 160 mm Hg or diastolic BP greater than or equal to 110 mm Hg persists for at least 30 minutes. This is followed by oral medication to keep systolic BP less than 150 mg Hg and diastolic BP less than 100 mm Hg.
For patients with persistent hypertension after GHTN or PE, I recommend oral long-acting nifedipine XL (30 mg every 12 hours) or oral labetalol (200 mg every 8-12 hours). Compared with labetalol, oral nifedipine is associated with improved renal blood flow with resultant diuresis, which makes it the drug of choice in women with volume overload. In some, it is necessary to switch to a new agent such as an angiotensin-converting enzyme (ACE) inhibitor; an ACE inhibitor is the drug of choice in those with pregestational diabetes mellitus, renal disease, or cardiomyopathy. In addition, thiazide or loop diuretics may be needed in women with circulatory overload and in those with pulmonary edema. Antihypertensives such as nifedipine, labetalol, furosemide, captopril, and enalapril are compatible with breastfeeding.
If the BP remains less than 150 mm Hg (systolic) and/or less than 100 mm Hg (diastolic) for 24 hours, and there are no maternal symptoms, the patient may be discharged home with instructions for daily BP measurements (self or by a visiting nurse) and the reporting of symptoms until her next visit in 1 week. Antihypertensives then are discontinued if the BP remains below the hypertensive levels for at least 48 hours. This may take 1 or several weeks to achieve.
Women with PE with severe features should receive close monitoring of BP and of symptoms during the immediate postpartum period, as well as accurate measurements of fluid intake, urinary output, and weight gain. These women often have received large amounts of IV fluids during labor as a result of prehydration before epidural analgesia, as well as IV fluids administered during the use of oxytocin and magnesium sulfate in labor and post partum. Mobilization of extracellular fluid also leads to increased intravascular volume. As a result, women who have PE with severe features – particularly those with abnormal renal function, capillary leak, or early-onset disease – are at increased risk for pulmonary edema and exacerbation of severe hypertension.
Careful evaluation of the volume of IV fluids, oral intake, blood products, urine output, respiratory symptoms, and vital signs is advised. Patients who develop tachycardia or respiratory symptoms such as dry cough, shortness of breath, or orthopnea also should be monitored with pulse oximetry and frequent chest auscultation, as well as chest x-ray.
New-onset severe symptoms
Because severe hypertension or PE with severe features may develop for the first time during the postpartum period, postpartum women – and the medical providers and personnel who respond to patient phone calls – should be well educated about the signs and symptoms of severe hypertension or PE. These include new-onset severe headaches that do not respond to maximum doses of analgesics, persistent severe visual changes, and new-onset epigastric pain with nausea and vomiting, dyspnea, orthopnea, shortness of breath, or palpitations. These women are at increased risk for eclampsia, pulmonary edema, stroke, and thromboembolism; these women require careful evaluation and potential hospitalization.
Severe new onset of persistent headaches and/or visual symptoms. Women with hypertension in association with new-onset persistent headaches and/or visual changes should be suspected to have severe PE. Patients who have hypertension with seizure should be initially treated as having eclampsia and should receive brain imaging to rule out other etiologies. Magnesium sulfate therapy must be initiated promptly for seizure prophylaxis and/or treatment. In addition, intravenous antihypertensive medications are recommended to lower BP to the desired goal while considering an alternative cause for the cerebral symptoms.
Women presenting with hypertension in association with refractory and/or thunderclap headaches, visual disturbances, or neurologic deficits should be evaluated for possible cerebrovascular complications such as reversible cerebral vasoconstriction syndrome (RCVS), cerebral venous thrombosis, or stroke. These women will require selective diagnostic neuroimaging and consultation with neurology and/or neurosurgery. Such an evaluation may include CT scan for hemorrhage, MRI for detection of vasogenic edema and/or ischemia or infarction, cerebral angiography for diagnosis of RCVS, and cerebral venography for detection of cerebral venous thrombosis. Subsequent treatment will depend on the etiology.
Severe new-onset epigastric/right upper quadrant pain with nausea and vomiting. Women with persistent nausea, vomiting, or epigastric pain should be evaluated for HELLP syndrome because up to 30% who develop the syndrome do so post partum. The time of onset of clinical and laboratory findings ranges from 1 to 7 days post partum. Women are managed as they are before delivery, with the use of magnesium sulfate, antihypertensives, and close monitoring of vital signs and laboratory values.
In general, patients with HELLP syndrome will demonstrate an improvement in clinical and laboratory findings within 72 hours after treatment. If there is either no improvement or a deterioration in these findings, then it is important to consult with appropriate specialists for evaluation and subsequent management of possible rare syndromes such as acute fatty liver, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, or exacerbation of lupus.
Severe new-onset shortness of breath, dyspnea, orthopnea, or palpitations. Women with these symptoms in the postpartum period should be evaluated for possible pulmonary edema, pulmonary embolism, or peripartum cardiomyopathy. Women with postpartum hypertension are at risk for pulmonary edema with onset at 3-6 days after delivery. Diagnosis is confirmed by physical exam (tachycardia, tachypnea), presence of rales on lung exam, pulse oximetry (oxygen saturation less than 93%), and chest x-ray, and echocardiography to exclude other etiologies. Treatment of pulmonary edema includes oxygen supplementation, 40 mg IV furosemide, control of severe hypertension, fluid restriction, and supportive care.
Pulmonary embolism usually is confirmed by chest CT angiography and managed with therapeutic anticoagulation. Peripartum cardiomyopathy is diagnosed by echocardiography revealing left ventricular systolic dysfunction (ejection fraction less than 45%, dilated left ventricle). Treatment includes IV furosemide, use of a vasodilator, and ACE inhibitor therapy.
Remote prognosis
Recent research suggests that women who develop PE may be at increased risk for future cardiovascular disease such as heart failure, coronary artery disease, and stroke later in life. Indeed, many of the risk factors and pathophysiologic abnormalities of PE are similar to those of coronary artery disease.
The American College of Obstetricians and Gynecologists and the American Heart Association recommend that women with PE receive close observation in the postpartum period and careful evaluation in the first year after delivery to identify those who could benefit from early intervention to prevent subsequent cardiovascular disease. In general, when pregnancies are complicated by PE, there are opportunities for lifestyle and risk factor modification.
Dr. Sibai is professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston
Postpartum hypertension has a host of potential causes, some of which may be benign (such as the persistence of mild gestational hypertension or mild chronic hypertension) whereas others (such as severe de novo preeclampsia-eclampsia and HELLP syndrome [a complication of pregnancy characterized by hemolysis, elevated liver enzymes, and a low platelet count]) can be life threatening.
Postpartum hypertension may occur secondary to lupus, hyperthyroidism, hemolytic uremic syndrome, stroke, and other conditions, which means that we must have a high index of suspicion for secondary dangerous causes of hypertension when evaluating such women.
With monitoring, reporting, and prompt evaluation of symptoms in the postpartum period – and with patient education on signs and symptoms of severe hypertension and preeclampsia (PE) – we can expect to avoid a range of potential maternal complications, from hypertensive encephalopathy, liver hemorrhage, renal failure, and the development of eclampsia, ischemic stroke/cerebral hemorrhage, pulmonary edema, and cardiomyopathy.
Most women with gestational hypertension (GHTN) become normotensive during the first week post partum, but in women who develop PE during pregnancy, hypertension often takes longer to resolve. Some of these women may have an initial decrease in blood pressure immediately post partum followed by development of hypertension again between days 3 and 6. Therefore, This can be achieved either in-hospital, through home BP monitoring, or with in-office visits.
In addition, all women – including those who did not have hypertension during their pregnancies – should be educated about the signs and symptoms of severe hypertension or PE and instructed to report these to a medical provider in a timely fashion. Severe hypertension or PE with severe features may develop for the first time during the postpartum period either before or after hospital discharge. It is important to appreciate, moreover, that approximately 25%-40% of cases of eclampsia develop in the postpartum period with onset ranging from 2 days to 6 weeks after delivery. Moreover, almost one-third of women who develop the HELLP syndrome do so during the postpartum period.
Management of persistent hypertension
The most common causes for persistent hypertension beyond 48 hours after delivery are GHTN, PE, or chronic hypertension. Initial management will depend on history, clinical findings, presence or absence of associated symptoms, results of laboratory findings (urine protein, platelet count, liver enzymes, serum creatinine, and electrolytes), and response to prior treatment of hypertension.
Certain medications that frequently are prescribed in the postpartum period, such as ergonovine and decongestants, should be discontinued if they are being used. These agents can aggravate preexisting hypertension or result in new-onset hypertension if used in large or frequent doses. Their use also may be associated with cerebral symptoms, nausea, and vomiting.
Subsequent management includes close observation until resolution of hypertension and associated symptoms. If the patient has hypertension only with no symptoms, no proteinuria, and normal laboratory findings, BP control is the focus; antihypertensives are used if systolic BP remains persistently greater than or equal to 150 mm Hg and/or if diastolic BP persists at greater than or equal to 100 mm Hg. Intravenous boluses of either labetalol or hydralazine or oral rapid-acting nifedipine are used initially if systolic BP is greater than or equal to 160 mm Hg or diastolic BP greater than or equal to 110 mm Hg persists for at least 30 minutes. This is followed by oral medication to keep systolic BP less than 150 mg Hg and diastolic BP less than 100 mm Hg.
For patients with persistent hypertension after GHTN or PE, I recommend oral long-acting nifedipine XL (30 mg every 12 hours) or oral labetalol (200 mg every 8-12 hours). Compared with labetalol, oral nifedipine is associated with improved renal blood flow with resultant diuresis, which makes it the drug of choice in women with volume overload. In some, it is necessary to switch to a new agent such as an angiotensin-converting enzyme (ACE) inhibitor; an ACE inhibitor is the drug of choice in those with pregestational diabetes mellitus, renal disease, or cardiomyopathy. In addition, thiazide or loop diuretics may be needed in women with circulatory overload and in those with pulmonary edema. Antihypertensives such as nifedipine, labetalol, furosemide, captopril, and enalapril are compatible with breastfeeding.
If the BP remains less than 150 mm Hg (systolic) and/or less than 100 mm Hg (diastolic) for 24 hours, and there are no maternal symptoms, the patient may be discharged home with instructions for daily BP measurements (self or by a visiting nurse) and the reporting of symptoms until her next visit in 1 week. Antihypertensives then are discontinued if the BP remains below the hypertensive levels for at least 48 hours. This may take 1 or several weeks to achieve.
Women with PE with severe features should receive close monitoring of BP and of symptoms during the immediate postpartum period, as well as accurate measurements of fluid intake, urinary output, and weight gain. These women often have received large amounts of IV fluids during labor as a result of prehydration before epidural analgesia, as well as IV fluids administered during the use of oxytocin and magnesium sulfate in labor and post partum. Mobilization of extracellular fluid also leads to increased intravascular volume. As a result, women who have PE with severe features – particularly those with abnormal renal function, capillary leak, or early-onset disease – are at increased risk for pulmonary edema and exacerbation of severe hypertension.
Careful evaluation of the volume of IV fluids, oral intake, blood products, urine output, respiratory symptoms, and vital signs is advised. Patients who develop tachycardia or respiratory symptoms such as dry cough, shortness of breath, or orthopnea also should be monitored with pulse oximetry and frequent chest auscultation, as well as chest x-ray.
New-onset severe symptoms
Because severe hypertension or PE with severe features may develop for the first time during the postpartum period, postpartum women – and the medical providers and personnel who respond to patient phone calls – should be well educated about the signs and symptoms of severe hypertension or PE. These include new-onset severe headaches that do not respond to maximum doses of analgesics, persistent severe visual changes, and new-onset epigastric pain with nausea and vomiting, dyspnea, orthopnea, shortness of breath, or palpitations. These women are at increased risk for eclampsia, pulmonary edema, stroke, and thromboembolism; these women require careful evaluation and potential hospitalization.
Severe new onset of persistent headaches and/or visual symptoms. Women with hypertension in association with new-onset persistent headaches and/or visual changes should be suspected to have severe PE. Patients who have hypertension with seizure should be initially treated as having eclampsia and should receive brain imaging to rule out other etiologies. Magnesium sulfate therapy must be initiated promptly for seizure prophylaxis and/or treatment. In addition, intravenous antihypertensive medications are recommended to lower BP to the desired goal while considering an alternative cause for the cerebral symptoms.
Women presenting with hypertension in association with refractory and/or thunderclap headaches, visual disturbances, or neurologic deficits should be evaluated for possible cerebrovascular complications such as reversible cerebral vasoconstriction syndrome (RCVS), cerebral venous thrombosis, or stroke. These women will require selective diagnostic neuroimaging and consultation with neurology and/or neurosurgery. Such an evaluation may include CT scan for hemorrhage, MRI for detection of vasogenic edema and/or ischemia or infarction, cerebral angiography for diagnosis of RCVS, and cerebral venography for detection of cerebral venous thrombosis. Subsequent treatment will depend on the etiology.
Severe new-onset epigastric/right upper quadrant pain with nausea and vomiting. Women with persistent nausea, vomiting, or epigastric pain should be evaluated for HELLP syndrome because up to 30% who develop the syndrome do so post partum. The time of onset of clinical and laboratory findings ranges from 1 to 7 days post partum. Women are managed as they are before delivery, with the use of magnesium sulfate, antihypertensives, and close monitoring of vital signs and laboratory values.
In general, patients with HELLP syndrome will demonstrate an improvement in clinical and laboratory findings within 72 hours after treatment. If there is either no improvement or a deterioration in these findings, then it is important to consult with appropriate specialists for evaluation and subsequent management of possible rare syndromes such as acute fatty liver, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, or exacerbation of lupus.
Severe new-onset shortness of breath, dyspnea, orthopnea, or palpitations. Women with these symptoms in the postpartum period should be evaluated for possible pulmonary edema, pulmonary embolism, or peripartum cardiomyopathy. Women with postpartum hypertension are at risk for pulmonary edema with onset at 3-6 days after delivery. Diagnosis is confirmed by physical exam (tachycardia, tachypnea), presence of rales on lung exam, pulse oximetry (oxygen saturation less than 93%), and chest x-ray, and echocardiography to exclude other etiologies. Treatment of pulmonary edema includes oxygen supplementation, 40 mg IV furosemide, control of severe hypertension, fluid restriction, and supportive care.
Pulmonary embolism usually is confirmed by chest CT angiography and managed with therapeutic anticoagulation. Peripartum cardiomyopathy is diagnosed by echocardiography revealing left ventricular systolic dysfunction (ejection fraction less than 45%, dilated left ventricle). Treatment includes IV furosemide, use of a vasodilator, and ACE inhibitor therapy.
Remote prognosis
Recent research suggests that women who develop PE may be at increased risk for future cardiovascular disease such as heart failure, coronary artery disease, and stroke later in life. Indeed, many of the risk factors and pathophysiologic abnormalities of PE are similar to those of coronary artery disease.
The American College of Obstetricians and Gynecologists and the American Heart Association recommend that women with PE receive close observation in the postpartum period and careful evaluation in the first year after delivery to identify those who could benefit from early intervention to prevent subsequent cardiovascular disease. In general, when pregnancies are complicated by PE, there are opportunities for lifestyle and risk factor modification.
Dr. Sibai is professor of obstetrics, gynecology, and reproductive sciences at the University of Texas McGovern Medical School, Houston
When Diet Is an Emergency
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.
At age 35, a woman underwent Roux-en-Y gastric bypass surgery. About 1 month later, she began vomiting and became unable to keep down any food or liquids. She was admitted to the hospital.
Two days after her admission, a dietitian evaluated the patient and recommended that she receive total parenteral nutrition (TPN). However, the attending physician did not order TPN during the patient’s 12-day hospital stay. As a result, the patient experienced vitamin deficiencies, including low thiamine. The patient developed symptoms of neurologic complications but was discharged.
Within 1 week, she was readmitted with the same symptoms, as well as signs of delirium and reduced level of consciousness. Her mental state continued to decline, and she became comatose for a period of time.
The patient now has Wernicke encephalopathy, which she alleged was caused by a lack of thiamine. She has no short-term memory, is wheelchair bound, and lives in a nursing home.
VERDICT
The jury found in favor of the plaintiff, awarding her $14,285,505.86 in damages, including $133,202 for loss of past earning capacity, $888,429 for loss of earning capacity, and $13,263,874.86 for medical care expenses.
COMMENTARY
It is foolish to think of diet as ancillary to medicine. While we often consider the long-term health implications of diet—obesity, atherosclerosis—we may overlook the urgent and emergent conditions that can result from a patient’s diet.
A familiar example is hypoglycemia. We associate it with agents used to treat diabetes. But it also can occur in the context of renal failure, tumor, severe infection, alcohol, or starvation. Similarly, thiamine deficiency would be an obvious consideration in a patient who presented in a coma or with altered mental status. But, as this case shows, thiamine deficiency can sneak up on you.
Continue to: In this case...
In this case, the patient’s altered structural anatomy rendered her more susceptible to thiamine deficiency, which was ultimately found to be causally related to the physician’s failure to order TPN. This raises an important issue in the management of patients who have had bariatric procedures.
This plaintiff had Roux-en-Y gastric bypass, a significant procedure that short circuits a sizable portion of the stomach and about 75 to 100 cm of the small intestine. This surgery carries long-term risks including bowel obstruction, hernias, ulcers, dumping syndrome, low blood sugar, and malnutrition. The last of these can manifest as low levels of B12, folate, thiamine, iron, calcium, and vitamin D.
Roux-en-Y bypass requires adherence to dietary recommendations, lifelong vitamin/mineral supplementation, and follow-up compliance. Patients who have had bariatric procedures are at increased risk for complications—which also raise malpractice risks. Clinicians must be aware that patients who have had a bariatric procedure have altered anatomy. We must take steps to understand the nature of those alterations and how they impact the present clinical picture.
In this case, the altered anatomy in combination with the failure to order TPN resulted in Wernicke encephalopathy—a condition caused by a biochemical lesion that occurs after stores of B vitamin are exhausted. Classic Wernicke encephalopathy is advertised as a triad of ophthalmoplegia, ataxia, and confusion, but only 10% of patients will demonstrate a true triad.
Wernicke encephalopathy typically occurs in the setting of alcoholism. However, certain other conditions can cause it, including recurrent dialysis, uremia, hyperemesis, thyrotoxicosis, cancer, AIDS, and starvation. It may be caused by surgical GI changes (eg, gastric bypass and banding) and nonsurgical GI causes (eg, pancreatitis, liver dysfunction, chronic diarrhea, celiac disease, and Crohn disease).
Continue to: Prognosis depends on...
Prognosis depends on how quickly the condition is recognized. Prompt treatment can lead to cure. But treatment delays (or lack of treatment) give the disorder the opportunity to progress, and memory and learning impairment may not completely resolve. Mortality in those untreated is 10% to 20%. And 80% of untreated or undertreated patients will develop Korsakoff psychosis, with severe retrograde and anterograde amnesia, disorientation, and emotional changes.
These factors make Wernicke encephalopathy a medical malpractice risk. If the diagnosis is missed, the damage is clear, substantial, and irreversible—as seen in this unfortunate case. This plaintiff’s attorney had a dream of a closing argument to make: Her client will suffer the effects of brain damage, robbing her of her life, her memory, and her very personality. This damage could have been prevented—not through use of an experimental new procedure or an investigational drug, but through use of a simple and readily available vitamin.
Worse still, cases such as this can involve a punitive element. Jurors would be invited to conclude that treating clinicians ignored the patient and left her to starve in her own bed. Always act in the patient’s best interest—and be attuned to situations that may evolve into claims that the patient was abandoned, neglected, or ignored.
Finally, you must address anything in the patient’s written record that is contrary to your plan. The fact pattern makes clear that a nutritional evaluation was obtained 2 days after the plaintiff’s admission. The dietitian recommended TPN. The record is not clear on why the physician did not order it. If you plan to take an action at odds with a prior observation or recommendation, be sure to clearly explain the rationale supporting your course of treatment. If you perform a risk-benefit analysis that leads you to a different conclusion, document that in the record—preferably with a second opinion from another clinician who supports your decision to deviate from the recommendation.
IN SUMMARY
Nutrition can be critically important. Make sure you consider both short- and long-term consequences of nutritional deficiencies. Bariatric surgery patients have altered anatomy, so be cautious with them. Consider the possibility of thiamine encephalopathy—which can be devastating—when the setting is suggestive. And make sure that all recommendations from other clinicians recorded in the patient’s chart are acted on. If you select a course of treatment that departs from prior recommendation, make clear your risk-benefit analysis and consider obtaining a second opinion in support of your decision.

FDA approves siponimod for relapsing forms of MS
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.

Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
Pigmented Fungiform Papillae of the Tongue in an Indian Male
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
To the Editor:
The tongue is composed of 4 different types of papillae: fungiform, foliate, circumvallate, and filiform. Fungiform papillae, primarily located on the tip and sides of the tongue, are mushroom-shaped epithelial elevations composed of taste buds at the upper surface overlying a core of connective tissue.1 Foliate and circumvallate papillae are likewise associated with taste buds, while the filiform papillae are hypothesized to exclusively provide a frictional surface for proper food manipulation. Pigmented fungiform papillae of the tongue (PFPT) was first reported by Leonard2 in 1905, who described discrete hyperpigmentation present only on the surface of fungiform papillae, mainly in black patients. Although they have been primarily described in black individuals, PFPT also has been occasionally reported in Asian and Middle Eastern individuals as well as Indian women.3-6
A 36-year-old Indian man initially presented to his primary care provider with brown discoloration of the dorsolateral aspects of the tongue that had been present since childhood. His primary care provider was concerned about a potential syndrome or systemic illness and referred the patient to dermatology for further evaluation. The patient denied any oral mucosal bleeding or discomfort, and a review of systems was unremarkable. His medical and family history were otherwise noncontributory, and he denied a history of tobacco use.
Physical examination of the tongue and oral mucosa revealed numerous 0.5- to 1.0-mm brown papillae in a symmetric distribution, primarily located on the tip and lateral aspects of the tongue (Figure). No hyperpigmentation was present on the posterior aspect of the tongue or on any other mucosal surface. Routine laboratory values were notable for mild elevations in aspartate aminotransferase and alanine aminotransferase (47 U/L [reference range, 10–30 U/L] and 64 U/L [reference range, 10–40 U/L], respectively) and mild hyperbilirubinemia (total bilirubin, 1.8 mg/dL [reference range, 0.3–1.2 mg/dL]). A complete blood cell count and electrolytes were within reference range. Based on the clinical appearance of the lesions and their presence since childhood, the patient was diagnosed with PFPT. No intervention was undertaken, and the patient was reassured of the benign nature of the lesions.
Pigmented fungiform papillae of the tongue presents in 3 variants. The first variant involves hyperpigmentation of all fungiform papillae located on the lateral and frontal aspects of the tongue and is the most common manifestation of PFPT.3 Our patient falls into this category. The second and third variants involve the dorsal surface, with the former involving only a few fungiform papillae on the dorsal aspect of the tongue and the latter variant involving all papillae.3 In 1974, Holzwanger et al3 conducted a survey of 300 random individuals, finding that 30% of black women and 25% of black men had some hyperpigmentation of the tongue, while only 1 white individual demonstrated lingual pigmentation. The physiology of PFPT remains largely unknown. Dermoscopic evaluation often demonstrates elevations with pigmented borders in a rose petal shape.7 Histopathologic evaluation reveals melanophages without inflammation that are positive for melanin on Fontana-Masson silver staining but negative for iron on Prussian blue staining.8
Despite the fact that PFPT is not a rare condition, the diagnosis remains notably missing from many standard dermatology textbooks and online dermatology resources, making it a potentially overlooked clinical entity.4-6 The tongue has a number of normal variations that are unlikely to be fully appreciated or acknowledged by dermatologists on routine physical examination but may cause distress to patients and raise concerns from primary care providers. Given that PFPT are benign, physicians should be aware of this diagnosis so as to provide reassurance to patients and avoid unnecessary testing. However, because the tongue can represent a harbinger of systemic disease, the differential diagnosis for the hyperpigmented lesions must always be considered, including Peutz-Jeghers syndrome, hemochromatosis, Addison disease, and Laugier-Hunziker syndrome (a rarer condition causing pigmented lesions on the lips, palate, and tongue), particularly if the hyperpigmented lesions extend beyond the fungiform papillae and do not fit into the 3 categories of PFPT.9
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
- Ross MH, Pawlina W. Digestive system I: oral cavity and associated structures. In: Ross MH, Pawlina W. Histology: A Text and Atlas, With Correlated Cell and Molecular Biology. 6th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2010:526-567.
- Leonard TMR. Ankylostomiasis or uncinariasis. JAMA. 1905;45:588-594.
- Holzwanger JM, Rudolph RI, Heaton CL. Pigmented fungiform papillae of the tongue: a common variant of oral pigmentation. Int J Dermatol. 1974;13:403-408.
- Tan C, Liu Y, Min ZS, et al. A clinical analysis of 58 Chinese cases of pigmented fungiform papillae of the tongue. J Eur Acad Dermatol Venereol. 2014;28:242-245.
- Romiti R, Molina De Medeiros L. Pigmented fungiform papillae of the tongue. Pediatr Dermatol. 2010;27:398-399.
- Millington GW, Shah SN. A case of pigmented fungiform lingual papillae in an Indian woman. J Eur Acad Dermatol Venereol. 2007;21:705.
- Mukamal LV, Ormiga P, Ramos ESM. Dermoscopy of the pigmented fungiform papillae of the tongue. J Dermatol. 2012;39:397-399.
- Werchniak AE, Storm CA, Dinulos JG. Hyperpigmented patches on the tongue of a young girl. Pigmented fungiform papillae of the tongue. Arch Dermatol. 2004;140:1275-1280.
- Urbina F, Sudy E. Pigmented fungiform papillae of the tongue in Laugier disease or Laugier-Hunziker syndrome. Actas Dermosifiliogr. 2013;104:173-174.
Practice Points
- Pigmented fungiform papillae of the tongue are common lingual hyperpigmented macules in patients with skin of color.
- It is important to be aware of this benign entity to provide reassurance to patients and avoid unnecessary testing.

AAP updates 2019-2020 flu vaccine recommendations to include nasal spray
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.
Although the American Academy of Pediatrics had cited a preference for injected flu vaccines for children during the 2018-2019 flu season, this year’s recommendations say either that or the nasal spray formulation are acceptable, according to a press release. The Centers for Disease Control and Prevention has given similar guidance.
Because the spray did not work as well against A/H1N1 as the injected vaccine had during the 2013-2014 and 2014-2015 seasons, the AAP did not recommend the spray during the 2015-2016 and 2016-2017 seasons. However, in 2017 the spray’s manufacturer included a new strain of A/H1N1, and new data has supported the spray’s effectiveness against some strains.
according to the CDC. That said, the spray is especially appropriate for patients who refuse to receive the injected form, so the choice of formulation is at the pediatrician’s discretion, according to the AAP release.