User login
mIDH1 inhibitor ivosidenib improves progression-free survival in advanced cholangiocarcinoma
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
BARCELONA – Ivosidenib, a first-in-class, oral, small-molecule inhibitor of the mutant isocitrate dehydrogenase 1 (mIDH1) protein, significantly improved progression-free survival, compared with placebo, for the treatment of advanced cholangiocarcinoma in the global, randomized, phase 3 ClarIDHy trial.
A trend toward favorable overall survival was also seen in the pivotal double-blind trial, Ghassan K. Abou-Alfa, MD, of Memorial Sloan-Kettering Cancer Center, New York, reported at the European Society for Medical Oncology Congress.
Median progression-free survival (PFS) in 124 patients randomized to receive 500 mg of ivosidenib (IVO) once daily was 2.7 months, compared with 1.4 months in 61 patients who received placebo (hazard ratio, 0.37).
The primary study endpoint – PFS by central review – was reached, Dr. Abou-Alfa said, noting that the PFS rates at 6 and 12 months were 32% and 22% in the IVO arm, whereas none of the patients in the placebo arm were progression-free for 6 or more months.
Although the 1.3-month difference in PFS between the treatment and placebo arms “may seem short and some people may question whether this is clinically meaningful,” this outcome actually represents an important breakthrough for patients with a disease that has few treatment options, Angela Lamarca, MD, PhD, of the Christie NHS Foundation Trust, Manchester, England, explained in a press release about the study.
“A treatment that increases the chance of being free from progression by 32% at 6 months after starting treatment and that prolongs survival from 6 months with placebo to 10.8 months with ivosidenib, after adjusting for crossover, is definitely meaningful for our patients with cholangiocarcinoma and their families,” said Dr. Lamarca, representing the ESMO Press & Media Affairs Committee.
Indeed, the overall response rate in the IVO arm was 2.4%, representing three partial responses, and the stable disease rate was 50.8%. The overall response rate and stable disease rates in the placebo arm were 0% and 28%, Dr. Abou-Alfa said.
Median overall survival in the intent-to-treat population, including the 57% of placebo arm patients who crossed over to the treatment arm at the time of radiographic progression as allowed by study protocol, was 10.8 months, compared with 9.7 months in the placebo arm, respectively (HR, 0.69), Dr. Abou-Alfa said.
Crossover-adjusted overall survival was 6 months for the placebo arm (HR, 0.46) as assessed using rank-preserving structural failure time analysis, which suggested that the overall survival benefit would have been statistically significant had there been no crossover.
Study subjects had unresectable or metastatic mIDH1 cholangiocarcinoma, good performance status, and measurable disease. They had a median age of 62 years, and 68 were men. Most had intrahepatic disease (91%) and metastatic disease (92%), and 43% had two prior therapies, he noted.
They were randomized 2:1 to the treatment and placebo arms, respectively.
Since IVO targets mIDH1, which occurs in about 20% of patients with cholangiocarcinoma and results in production of the oncogenesis-promoting oncometabolite D-2-hydroxyglutarate, and since the IVO has shown encouraging activity in smaller prior studies, ClarIDHy was designed to evaluate it in the advanced mIDH1 cholangiocarcinoma setting because patients with this aggressive disease have generally poor prognosis and few treatment options beyond chemotherapy, he explained.
In addition to providing significant, clinically meaningful survival benefit, the treatment also was generally well tolerated, he noted.
Treatment-emergent adverse events occurring in more than 15% of patients in the IVO arm included nausea (32.1%), diarrhea (28.8%), fatigue (23.7%), cough (19.2%), abdominal pain (18.6%), ascites (18.6%), decreased appetite (17.3%), anemia (16.0%), and vomiting (16.0%). Grade 3 or higher adverse events were reported in 46% and 36% of the IVO and placebo patients, respectively.
The findings are notable, in part because of the unmet need and also because this is the first pivotal study demonstrating the clinical benefit of targeting mIDH1 in patients with advanced mIDH1 cholangiocarcinoma, he said, concluding that “these pivotal data demonstrate the clinical relevance and benefit of ivosidenib in mIDH1 cholangiocarcinoma ... [and] establish the role for genomic testing in this rare cancer with a high unmet need.”
He also said studies should investigate IVO in the first-line setting for IDH1-mutated cholangiocarcinoma, in addition to its use in combination therapy and as adjuvant therapy.
In the ESMO press release, Chris Verslype, MD, of University Hospital Leuven (Belgium) called the findings of this study unprecedented given the lack of treatment options in those who fail systemic therapy, which has led to very limited survival.
The findings are “very likely to change clinical practice” and “will, for sure, drive the further development of targeted therapy for the disease,” he said.
Despite being limited by the requirement that patients have good performance status after prior chemotherapy (which means the findings may not be representative of all patients), ClarIDHy is “still a strong study because of the randomization to placebo.”
“It showed a real effect,” he said.
ClarIDHy was funded by Agios Pharmaceuticals. Dr. Abou-Alfa reported both personal and institutional relationships with industry. These include advisory /consulting roles and research grants/funding from numerous pharmaceutical companies.
SOURCE: Abou-Alfa GK et al. ESMO 2019, Abstract LBA10-PR.
REPORTING FROM ESMO 2019
Decoding biosimilar approvals
SAN FRANCISCO – Several factors must be considered when extrapolating biosimilar results, according to a speaker at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.

In this context, “extrapolation” means expanding the use of an approved biosimilar from one indication to another, based on efficacy and safety data from the first indication, Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, explained at the meeting.
To determine if extrapolation is appropriate, regulatory agencies consider the biosimilar’s mechanism of action in each indication; pharmacokinetics, pharmacodynamics, and immunogenicity in the different patient populations; differences in expected toxicities for each condition and population; and any other factor that may affect safety or efficacy.
To illustrate the process, Dr. Zelenetz explained how results with a rituximab biosimilar in rheumatoid arthritis (RA) cannot be extrapolated to B‐cell non‐Hodgkin lymphoma (NHL), but results with that same biosimilar in follicular lymphoma can be extrapolated to other types of B-cell NHL.
The biosimilar is rituximab-abbs (CT‐P10, Truxima). In a phase 1 trial of patients with RA, rituximab-abbs demonstrated biosimilarity to the reference product (Ann Rheum Dis. 2017;76[3]:566‐70).
The RA results cannot be extrapolated to B-cell NHL for a few reasons, according to Dr. Zelenetz. He noted that rituximab’s mechanism of action is antibody-dependent cell‐mediated cytotoxicity in both RA and NHL. However, the target in RA is the normal B cell, and the target in NHL is the malignant B cell.
In addition, the pharmacokinetics of rituximab are “drastically different” in RA and NHL, Dr. Zelenetz said. Differences in pharmacokinetics support different dosing approaches in the two diseases.
Another big difference is immunogenicity. Anti‐CD20 antibodies develop in 15%-17% of RA patients, Dr. Zelenetz said, but the risk of antibody development is less than 1% in lymphoma.
Though extrapolation from RA to B‐cell NHL was not possible, it was possible to extrapolate results with rituximab-abbs in follicular lymphoma to other B-cell NHLs.
The study used was a phase 3 trial comparing rituximab-abbs to rituximab – both in combination with cyclophosphamide, vincristine, and prednisone – in patients with newly diagnosed, advanced stage follicular lymphoma.
This study showed no difference in pharmacokinetics or pharmacodynamics between rituximab-abbs and rituximab. The two agents also had comparable safety profiles and produced similar response rates (Lancet Haematol. 2017 Jul 13;4:e362‐73).
Rituximab‐abbs was approved in the United States based on these data, and results from this trial were extrapolated to other types of B-cell NHL. The results were extrapolated because the mechanism of action, pharmacokinetics, pharmacodynamics, and immunogenicity of rituximab are the same across B-cell NHLs, Dr. Zelenetz noted.
“Extrapolation is a critical part of biosimilarity development,” he said. “As long as scientific justification for extrapolation exists, I believe that extrapolation makes good sense.”
Dr. Zelenetz reported relationships with AbbVie, Adaptive Biotechnologies, Amgen, AstraZeneca, BeiGene, Celgene, Genentech, Gilead Sciences, Janssen, MEI Pharma, MorphoSys AG, Novartis, Pharmacyclics, and Roche.
SAN FRANCISCO – Several factors must be considered when extrapolating biosimilar results, according to a speaker at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
In this context, “extrapolation” means expanding the use of an approved biosimilar from one indication to another, based on efficacy and safety data from the first indication, Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, explained at the meeting.
To determine if extrapolation is appropriate, regulatory agencies consider the biosimilar’s mechanism of action in each indication; pharmacokinetics, pharmacodynamics, and immunogenicity in the different patient populations; differences in expected toxicities for each condition and population; and any other factor that may affect safety or efficacy.
To illustrate the process, Dr. Zelenetz explained how results with a rituximab biosimilar in rheumatoid arthritis (RA) cannot be extrapolated to B‐cell non‐Hodgkin lymphoma (NHL), but results with that same biosimilar in follicular lymphoma can be extrapolated to other types of B-cell NHL.
The biosimilar is rituximab-abbs (CT‐P10, Truxima). In a phase 1 trial of patients with RA, rituximab-abbs demonstrated biosimilarity to the reference product (Ann Rheum Dis. 2017;76[3]:566‐70).
The RA results cannot be extrapolated to B-cell NHL for a few reasons, according to Dr. Zelenetz. He noted that rituximab’s mechanism of action is antibody-dependent cell‐mediated cytotoxicity in both RA and NHL. However, the target in RA is the normal B cell, and the target in NHL is the malignant B cell.
In addition, the pharmacokinetics of rituximab are “drastically different” in RA and NHL, Dr. Zelenetz said. Differences in pharmacokinetics support different dosing approaches in the two diseases.
Another big difference is immunogenicity. Anti‐CD20 antibodies develop in 15%-17% of RA patients, Dr. Zelenetz said, but the risk of antibody development is less than 1% in lymphoma.
Though extrapolation from RA to B‐cell NHL was not possible, it was possible to extrapolate results with rituximab-abbs in follicular lymphoma to other B-cell NHLs.
The study used was a phase 3 trial comparing rituximab-abbs to rituximab – both in combination with cyclophosphamide, vincristine, and prednisone – in patients with newly diagnosed, advanced stage follicular lymphoma.
This study showed no difference in pharmacokinetics or pharmacodynamics between rituximab-abbs and rituximab. The two agents also had comparable safety profiles and produced similar response rates (Lancet Haematol. 2017 Jul 13;4:e362‐73).
Rituximab‐abbs was approved in the United States based on these data, and results from this trial were extrapolated to other types of B-cell NHL. The results were extrapolated because the mechanism of action, pharmacokinetics, pharmacodynamics, and immunogenicity of rituximab are the same across B-cell NHLs, Dr. Zelenetz noted.
“Extrapolation is a critical part of biosimilarity development,” he said. “As long as scientific justification for extrapolation exists, I believe that extrapolation makes good sense.”
Dr. Zelenetz reported relationships with AbbVie, Adaptive Biotechnologies, Amgen, AstraZeneca, BeiGene, Celgene, Genentech, Gilead Sciences, Janssen, MEI Pharma, MorphoSys AG, Novartis, Pharmacyclics, and Roche.
SAN FRANCISCO – Several factors must be considered when extrapolating biosimilar results, according to a speaker at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
In this context, “extrapolation” means expanding the use of an approved biosimilar from one indication to another, based on efficacy and safety data from the first indication, Andrew D. Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York, explained at the meeting.
To determine if extrapolation is appropriate, regulatory agencies consider the biosimilar’s mechanism of action in each indication; pharmacokinetics, pharmacodynamics, and immunogenicity in the different patient populations; differences in expected toxicities for each condition and population; and any other factor that may affect safety or efficacy.
To illustrate the process, Dr. Zelenetz explained how results with a rituximab biosimilar in rheumatoid arthritis (RA) cannot be extrapolated to B‐cell non‐Hodgkin lymphoma (NHL), but results with that same biosimilar in follicular lymphoma can be extrapolated to other types of B-cell NHL.
The biosimilar is rituximab-abbs (CT‐P10, Truxima). In a phase 1 trial of patients with RA, rituximab-abbs demonstrated biosimilarity to the reference product (Ann Rheum Dis. 2017;76[3]:566‐70).
The RA results cannot be extrapolated to B-cell NHL for a few reasons, according to Dr. Zelenetz. He noted that rituximab’s mechanism of action is antibody-dependent cell‐mediated cytotoxicity in both RA and NHL. However, the target in RA is the normal B cell, and the target in NHL is the malignant B cell.
In addition, the pharmacokinetics of rituximab are “drastically different” in RA and NHL, Dr. Zelenetz said. Differences in pharmacokinetics support different dosing approaches in the two diseases.
Another big difference is immunogenicity. Anti‐CD20 antibodies develop in 15%-17% of RA patients, Dr. Zelenetz said, but the risk of antibody development is less than 1% in lymphoma.
Though extrapolation from RA to B‐cell NHL was not possible, it was possible to extrapolate results with rituximab-abbs in follicular lymphoma to other B-cell NHLs.
The study used was a phase 3 trial comparing rituximab-abbs to rituximab – both in combination with cyclophosphamide, vincristine, and prednisone – in patients with newly diagnosed, advanced stage follicular lymphoma.
This study showed no difference in pharmacokinetics or pharmacodynamics between rituximab-abbs and rituximab. The two agents also had comparable safety profiles and produced similar response rates (Lancet Haematol. 2017 Jul 13;4:e362‐73).
Rituximab‐abbs was approved in the United States based on these data, and results from this trial were extrapolated to other types of B-cell NHL. The results were extrapolated because the mechanism of action, pharmacokinetics, pharmacodynamics, and immunogenicity of rituximab are the same across B-cell NHLs, Dr. Zelenetz noted.
“Extrapolation is a critical part of biosimilarity development,” he said. “As long as scientific justification for extrapolation exists, I believe that extrapolation makes good sense.”
Dr. Zelenetz reported relationships with AbbVie, Adaptive Biotechnologies, Amgen, AstraZeneca, BeiGene, Celgene, Genentech, Gilead Sciences, Janssen, MEI Pharma, MorphoSys AG, Novartis, Pharmacyclics, and Roche.
REPORTING FROM NCCN HEMATOLOGIC MALIGNANCIES
Study finds no standard for treatment discontinuation in myeloma
BOSTON — There is “no standard of care and no clear pattern” for discontinuing treatment in multiple myeloma, according to a speaker at the International Myeloma Workshop.

Data from a large, observational study revealed that a wide range of treatment regimens are used for first-, second-, and third-line therapy in multiple myeloma. The duration of therapy and time to next treatment were shorter in this real-world study than in prior clinical trials, and reasons for treatment discontinuation varied by regimen and line of therapy.
Katja Weisel, MD, of University Medical Center Hamburg-Eppendorf in Germany, presented these findings at the workshop, held by the International Myeloma Society.
The study, INSIGHT MM, is the largest global, prospective, observational study of multiple myeloma to date, according to Dr. Weisel. The study, which began July 1, 2016, has enrolled patients in the United States (n = 1,004), Europe (n = 1,612), Latin America (n = 367), and Asia (n = 218).
Dr. Weisel and her colleagues evaluated duration of therapy, reasons for treatment discontinuation, and subsequent therapies in a subset of patients on INSIGHT MM. The researchers’ analysis revealed “broad heterogeneity” across lines of therapy, Dr. Weisel said, adding that patients are receiving multiple regimens in addition to the most commonly prescribed regimens in myeloma.
First-line therapy
“In first-line treatment, we see predominantly bortezomib-based triplets ... regardless of transplant-eligible or transplant-ineligible patients,” Dr. Weisel said. “This is followed by doublets and other more rarely [applied] regimens.”
First-line therapies in 1,175 patients included:
- Bortezomib, cyclophosphamide, and dexamethasone (VCd) – 323 patients.
- Bortezomib, lenalidomide, and dexamethasone (VRd) – 321 patients.
- Bortezomib, thalidomide, and dexamethasone (VTd) – 200 patients.
- Bortezomib and dexamethasone (Vd) – 102 patients.
- Lenalidomide and dexamethasone (Rd) – 90 patients.
- Bortezomib, melphalan, and prednisone (VMP) – 53 patients.
- Carfilzomib, lenalidomide, and dexamethasone (KRd) – 47 patients.
- Daratumumab-based regimens (Dara) – 32 patients.
- Carfilzomib and dexamethasone (Kd) – 5 patients.
- Ixazomib, lenalidomide, and dexamethasone (IRd) – 2 patients.
Of the 1,175 newly diagnosed patients, 894 did not proceed to transplant after first-line therapy, but 281 did. Most of the patients who went on to transplant had received VRd (n = 82), VTd (n = 76), or VCd (n = 75).
Second- and third-line therapies
“In second-line treatment, we have still a dominance of the len-dex regimen all over the world,” Dr. Weisel said. “There is an emerging use of daratumumab in various combinations, and then you see the whole spectrum of approved triplet and doublet regimens.”
In the third line, the most commonly used regimens are daratumumab-based combinations and Rd.
There were 548 patients who received second-line treatment and 332 who received third-line therapy. The regimens used were:
- Rd – 130 patients second line, 71 third line.
- Dara – 121 patients second line, 105 third line.
- KRd – 61 patients second line, 17 third line.
- VCd – 57 patients second line, 19 third line.
- Vd – 48 patients second line, 29 third line.
- VRd – 36 patients second line, 8 third line.
- Kd – 33 patients for both second and third line.
- IRd – 29 patients second line, 43 third line.
- VTd – 25 patients second line, 4 third line.
- VMP – 8 patients second line, 3 third line.
Duration of therapy
Most transplant-eligible patients received any first-line therapy (VRd, VTd, or VCd) for longer than 12 months. Among transplant-ineligible patients, Rd was the first-line therapy most likely to be given for 12 months or more.
None of the second-line regimens lasted longer than 12 months in a majority of patients, but daratumumab-based regimens and IRd were the therapies most likely to exceed 12 months’ duration in both second- and third-line treatment.
Time to next treatment
“The vast majority of [transplant-eligible] patients, close to 90% ... do not need a second-line treatment during the first year of treatment,” Dr. Weisel said. “However, for transplant-ineligible patients, this accounts only for the most effective regimens, VMP and Rd.”
For second- and third-line therapies, a 12-month or longer time to next treatment was most likely among patients who received IRd or daratumumab-based regimens.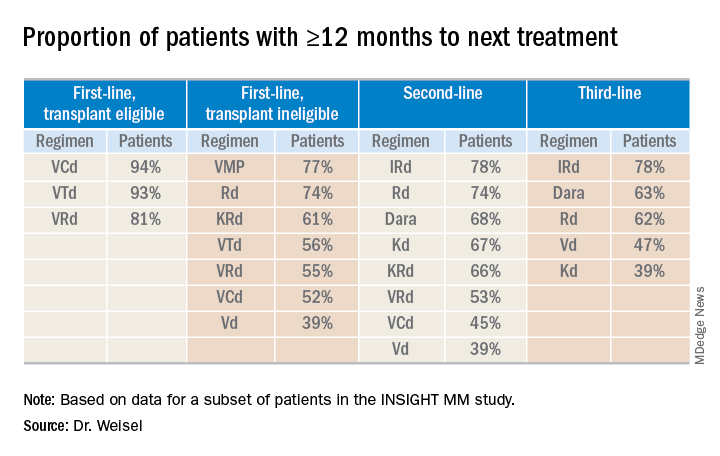
Reasons for discontinuation
“Planned end of therapy only accounts for a small proportion of treatment discontinuations, especially in the relapsed setting,” Dr. Weisel said. “Patients are discontinuing treatment due to reasons other than relapse, ultimately receiving fixed-duration therapy.”
The most common reasons for discontinuation of first-line therapy were:
- Relapse for VCd.
- Planned end of therapy for VRd.
- Adverse events (AEs) for VD and VTd.
- AEs and “other reasons” for Rd.
The most common reasons for discontinuation of second-line therapy were:
- Planned end of therapy for VCd.
- AEs, relapse, and other reasons for VRd.
- Relapse for VD, KRd, and Dara.
- AEs for Rd and IRd.
- AEs and other reasons for Kd.
The most common reasons for discontinuation of third-line therapy were:
- AEs for VCd, Vd, and KRd.
- Relapse for Kd, IRd, and Dara.
- Relapse and other reasons for VRd.
- AEs and other reasons for Rd.
The most common AE leading to discontinuation, across all treatment regimens, was peripheral neuropathy. This suggests peripheral neuropathy is still the “biggest impediment for continuous treatment,” Dr. Weisel said.
INSIGHT MM is sponsored by Takeda. Dr. Weisel reported relationships with Takeda and several other companies.
SOURCE: Weisel K et al. IMW 2019, Abstract OAB-005.
BOSTON — There is “no standard of care and no clear pattern” for discontinuing treatment in multiple myeloma, according to a speaker at the International Myeloma Workshop.
Data from a large, observational study revealed that a wide range of treatment regimens are used for first-, second-, and third-line therapy in multiple myeloma. The duration of therapy and time to next treatment were shorter in this real-world study than in prior clinical trials, and reasons for treatment discontinuation varied by regimen and line of therapy.
Katja Weisel, MD, of University Medical Center Hamburg-Eppendorf in Germany, presented these findings at the workshop, held by the International Myeloma Society.
The study, INSIGHT MM, is the largest global, prospective, observational study of multiple myeloma to date, according to Dr. Weisel. The study, which began July 1, 2016, has enrolled patients in the United States (n = 1,004), Europe (n = 1,612), Latin America (n = 367), and Asia (n = 218).
Dr. Weisel and her colleagues evaluated duration of therapy, reasons for treatment discontinuation, and subsequent therapies in a subset of patients on INSIGHT MM. The researchers’ analysis revealed “broad heterogeneity” across lines of therapy, Dr. Weisel said, adding that patients are receiving multiple regimens in addition to the most commonly prescribed regimens in myeloma.
First-line therapy
“In first-line treatment, we see predominantly bortezomib-based triplets ... regardless of transplant-eligible or transplant-ineligible patients,” Dr. Weisel said. “This is followed by doublets and other more rarely [applied] regimens.”
First-line therapies in 1,175 patients included:
- Bortezomib, cyclophosphamide, and dexamethasone (VCd) – 323 patients.
- Bortezomib, lenalidomide, and dexamethasone (VRd) – 321 patients.
- Bortezomib, thalidomide, and dexamethasone (VTd) – 200 patients.
- Bortezomib and dexamethasone (Vd) – 102 patients.
- Lenalidomide and dexamethasone (Rd) – 90 patients.
- Bortezomib, melphalan, and prednisone (VMP) – 53 patients.
- Carfilzomib, lenalidomide, and dexamethasone (KRd) – 47 patients.
- Daratumumab-based regimens (Dara) – 32 patients.
- Carfilzomib and dexamethasone (Kd) – 5 patients.
- Ixazomib, lenalidomide, and dexamethasone (IRd) – 2 patients.
Of the 1,175 newly diagnosed patients, 894 did not proceed to transplant after first-line therapy, but 281 did. Most of the patients who went on to transplant had received VRd (n = 82), VTd (n = 76), or VCd (n = 75).
Second- and third-line therapies
“In second-line treatment, we have still a dominance of the len-dex regimen all over the world,” Dr. Weisel said. “There is an emerging use of daratumumab in various combinations, and then you see the whole spectrum of approved triplet and doublet regimens.”
In the third line, the most commonly used regimens are daratumumab-based combinations and Rd.
There were 548 patients who received second-line treatment and 332 who received third-line therapy. The regimens used were:
- Rd – 130 patients second line, 71 third line.
- Dara – 121 patients second line, 105 third line.
- KRd – 61 patients second line, 17 third line.
- VCd – 57 patients second line, 19 third line.
- Vd – 48 patients second line, 29 third line.
- VRd – 36 patients second line, 8 third line.
- Kd – 33 patients for both second and third line.
- IRd – 29 patients second line, 43 third line.
- VTd – 25 patients second line, 4 third line.
- VMP – 8 patients second line, 3 third line.
Duration of therapy
Most transplant-eligible patients received any first-line therapy (VRd, VTd, or VCd) for longer than 12 months. Among transplant-ineligible patients, Rd was the first-line therapy most likely to be given for 12 months or more.
None of the second-line regimens lasted longer than 12 months in a majority of patients, but daratumumab-based regimens and IRd were the therapies most likely to exceed 12 months’ duration in both second- and third-line treatment.
Time to next treatment
“The vast majority of [transplant-eligible] patients, close to 90% ... do not need a second-line treatment during the first year of treatment,” Dr. Weisel said. “However, for transplant-ineligible patients, this accounts only for the most effective regimens, VMP and Rd.”
For second- and third-line therapies, a 12-month or longer time to next treatment was most likely among patients who received IRd or daratumumab-based regimens.
Reasons for discontinuation
“Planned end of therapy only accounts for a small proportion of treatment discontinuations, especially in the relapsed setting,” Dr. Weisel said. “Patients are discontinuing treatment due to reasons other than relapse, ultimately receiving fixed-duration therapy.”
The most common reasons for discontinuation of first-line therapy were:
- Relapse for VCd.
- Planned end of therapy for VRd.
- Adverse events (AEs) for VD and VTd.
- AEs and “other reasons” for Rd.
The most common reasons for discontinuation of second-line therapy were:
- Planned end of therapy for VCd.
- AEs, relapse, and other reasons for VRd.
- Relapse for VD, KRd, and Dara.
- AEs for Rd and IRd.
- AEs and other reasons for Kd.
The most common reasons for discontinuation of third-line therapy were:
- AEs for VCd, Vd, and KRd.
- Relapse for Kd, IRd, and Dara.
- Relapse and other reasons for VRd.
- AEs and other reasons for Rd.
The most common AE leading to discontinuation, across all treatment regimens, was peripheral neuropathy. This suggests peripheral neuropathy is still the “biggest impediment for continuous treatment,” Dr. Weisel said.
INSIGHT MM is sponsored by Takeda. Dr. Weisel reported relationships with Takeda and several other companies.
SOURCE: Weisel K et al. IMW 2019, Abstract OAB-005.
BOSTON — There is “no standard of care and no clear pattern” for discontinuing treatment in multiple myeloma, according to a speaker at the International Myeloma Workshop.
Data from a large, observational study revealed that a wide range of treatment regimens are used for first-, second-, and third-line therapy in multiple myeloma. The duration of therapy and time to next treatment were shorter in this real-world study than in prior clinical trials, and reasons for treatment discontinuation varied by regimen and line of therapy.
Katja Weisel, MD, of University Medical Center Hamburg-Eppendorf in Germany, presented these findings at the workshop, held by the International Myeloma Society.
The study, INSIGHT MM, is the largest global, prospective, observational study of multiple myeloma to date, according to Dr. Weisel. The study, which began July 1, 2016, has enrolled patients in the United States (n = 1,004), Europe (n = 1,612), Latin America (n = 367), and Asia (n = 218).
Dr. Weisel and her colleagues evaluated duration of therapy, reasons for treatment discontinuation, and subsequent therapies in a subset of patients on INSIGHT MM. The researchers’ analysis revealed “broad heterogeneity” across lines of therapy, Dr. Weisel said, adding that patients are receiving multiple regimens in addition to the most commonly prescribed regimens in myeloma.
First-line therapy
“In first-line treatment, we see predominantly bortezomib-based triplets ... regardless of transplant-eligible or transplant-ineligible patients,” Dr. Weisel said. “This is followed by doublets and other more rarely [applied] regimens.”
First-line therapies in 1,175 patients included:
- Bortezomib, cyclophosphamide, and dexamethasone (VCd) – 323 patients.
- Bortezomib, lenalidomide, and dexamethasone (VRd) – 321 patients.
- Bortezomib, thalidomide, and dexamethasone (VTd) – 200 patients.
- Bortezomib and dexamethasone (Vd) – 102 patients.
- Lenalidomide and dexamethasone (Rd) – 90 patients.
- Bortezomib, melphalan, and prednisone (VMP) – 53 patients.
- Carfilzomib, lenalidomide, and dexamethasone (KRd) – 47 patients.
- Daratumumab-based regimens (Dara) – 32 patients.
- Carfilzomib and dexamethasone (Kd) – 5 patients.
- Ixazomib, lenalidomide, and dexamethasone (IRd) – 2 patients.
Of the 1,175 newly diagnosed patients, 894 did not proceed to transplant after first-line therapy, but 281 did. Most of the patients who went on to transplant had received VRd (n = 82), VTd (n = 76), or VCd (n = 75).
Second- and third-line therapies
“In second-line treatment, we have still a dominance of the len-dex regimen all over the world,” Dr. Weisel said. “There is an emerging use of daratumumab in various combinations, and then you see the whole spectrum of approved triplet and doublet regimens.”
In the third line, the most commonly used regimens are daratumumab-based combinations and Rd.
There were 548 patients who received second-line treatment and 332 who received third-line therapy. The regimens used were:
- Rd – 130 patients second line, 71 third line.
- Dara – 121 patients second line, 105 third line.
- KRd – 61 patients second line, 17 third line.
- VCd – 57 patients second line, 19 third line.
- Vd – 48 patients second line, 29 third line.
- VRd – 36 patients second line, 8 third line.
- Kd – 33 patients for both second and third line.
- IRd – 29 patients second line, 43 third line.
- VTd – 25 patients second line, 4 third line.
- VMP – 8 patients second line, 3 third line.
Duration of therapy
Most transplant-eligible patients received any first-line therapy (VRd, VTd, or VCd) for longer than 12 months. Among transplant-ineligible patients, Rd was the first-line therapy most likely to be given for 12 months or more.
None of the second-line regimens lasted longer than 12 months in a majority of patients, but daratumumab-based regimens and IRd were the therapies most likely to exceed 12 months’ duration in both second- and third-line treatment.
Time to next treatment
“The vast majority of [transplant-eligible] patients, close to 90% ... do not need a second-line treatment during the first year of treatment,” Dr. Weisel said. “However, for transplant-ineligible patients, this accounts only for the most effective regimens, VMP and Rd.”
For second- and third-line therapies, a 12-month or longer time to next treatment was most likely among patients who received IRd or daratumumab-based regimens.
Reasons for discontinuation
“Planned end of therapy only accounts for a small proportion of treatment discontinuations, especially in the relapsed setting,” Dr. Weisel said. “Patients are discontinuing treatment due to reasons other than relapse, ultimately receiving fixed-duration therapy.”
The most common reasons for discontinuation of first-line therapy were:
- Relapse for VCd.
- Planned end of therapy for VRd.
- Adverse events (AEs) for VD and VTd.
- AEs and “other reasons” for Rd.
The most common reasons for discontinuation of second-line therapy were:
- Planned end of therapy for VCd.
- AEs, relapse, and other reasons for VRd.
- Relapse for VD, KRd, and Dara.
- AEs for Rd and IRd.
- AEs and other reasons for Kd.
The most common reasons for discontinuation of third-line therapy were:
- AEs for VCd, Vd, and KRd.
- Relapse for Kd, IRd, and Dara.
- Relapse and other reasons for VRd.
- AEs and other reasons for Rd.
The most common AE leading to discontinuation, across all treatment regimens, was peripheral neuropathy. This suggests peripheral neuropathy is still the “biggest impediment for continuous treatment,” Dr. Weisel said.
INSIGHT MM is sponsored by Takeda. Dr. Weisel reported relationships with Takeda and several other companies.
SOURCE: Weisel K et al. IMW 2019, Abstract OAB-005.
REPORTING FROM IMW 2019
Lefamulin found noninferior to moxifloxacin for bacterial pneumonia
Persistent high rates of bacterial resistance to current treatments have created the need for more options, especially for the treatment of community-acquired bacterial pneumonia (CABP), which remains a leading cause of hospitalization and death in the United States, wrote Elizabeth Alexander, MD, of Nabriva Therapeutics in King of Prussia, Penn., and colleagues. Lefamulin, “the first pleuromutilin antibiotic approved for intravenous and oral use in humans,” has demonstrated activity against many CABP-causing pathogens, including some not susceptible to other classes of antimicrobials, they noted.
Findings of Lefamulin Evaluation Against Pneumonia 2 (LEAP2) were published in JAMA. In this study, the researchers randomized 370 patients to 600 mg of oral lefamulin every 12 hours for 5 days and 368 patients to 400 mg of oral moxifloxacin every 24 hours for 7 days.
Early clinical response rates at 96 hours were 90.8% for both medications (difference of 0.1%). In addition, the rates of clinical response success were similar between the groups in both the modified intent-to-treat population (87.5% with lefamulin and 89.1% with moxifloxacin) and the clinically evaluable population (89.7% with lefamulin and 93.6% with moxifloxacin).
Gastrointestinal issues of diarrhea and nausea were the two most frequently reported treatment-emergent adverse events in both groups. Both conditions occurred more often in the lefamulin group, compared with the moxifloxacin group, but the differences were not significant (12.2% vs. 1.1% and 5.2% vs. 1.9%, respectively).
The study findings were limited by several factors including strict exclusion criteria that may limit the generalizability of the results, as well as a lack of testing for viral copathogens, low recovery of resistant pathogens, and possible misclassification of patient ethnicity, the researchers noted.
However, the results were strengthened by the randomized design, inclusion of patients with more severe CABP, and low rate of discontinuation, they said. The data support previous studies of lefamulin. Its lack of cross-resistance to other drug classes, coverage of typical and atypical CABP pathogens, and options for both oral and intravenous use suggest that it “may provide an alternative approach for the treatment of vulnerable patients,” the researchers said.
The study was supported by Nabriva Therapeutics. Dr. Alexander and several coauthors are employees of Nabriva Therapeutics and own stock in the company.
SOURCE: Alexander E et al. JAMA. 2019 Sep 27. doi:10.1001/jama.2019.15468.
“The development and approval of a new antibiotic is a rare occurrence and a reason to celebrate” given the scientific, regulatory, and economic challenges to antibiotic development, wrote Preeti N. Malani, MD, in an accompanying editorial. Lefamulin in both oral and intravenous forms was approved by the Food and Drug Administration in August 2019 for the treatment of community-acquired bacterial pneumonia, Dr. Malani said.
Lefamulin will likely be an expensive option. According to a manufacturer press release, lefamulin may cost $205/day for intravenous treatment and $275/day for oral treatment. “This is severalfold more than moxifloxacin or levofloxacin, which are the most commonly prescribed fluoroquinolones for CABP [community-acquired bacterial pneumonia],” said Dr. Malani. However, the addition of lefamulin to the array of antibiotics is important because of the persistent burden of bacterial pneumonia as an indication for antibiotic use, Dr. Malani emphasized.
Dr. Malani is affiliated with the University of Michigan, Ann Arbor, and serves as an associate editor of JAMA, but had no financial conflicts to disclose. These remarks were taken from an accompanying editorial (JAMA. 2019 Sep 27. doi:10.1001/jama.2019.16215).
“The development and approval of a new antibiotic is a rare occurrence and a reason to celebrate” given the scientific, regulatory, and economic challenges to antibiotic development, wrote Preeti N. Malani, MD, in an accompanying editorial. Lefamulin in both oral and intravenous forms was approved by the Food and Drug Administration in August 2019 for the treatment of community-acquired bacterial pneumonia, Dr. Malani said.
Lefamulin will likely be an expensive option. According to a manufacturer press release, lefamulin may cost $205/day for intravenous treatment and $275/day for oral treatment. “This is severalfold more than moxifloxacin or levofloxacin, which are the most commonly prescribed fluoroquinolones for CABP [community-acquired bacterial pneumonia],” said Dr. Malani. However, the addition of lefamulin to the array of antibiotics is important because of the persistent burden of bacterial pneumonia as an indication for antibiotic use, Dr. Malani emphasized.
Dr. Malani is affiliated with the University of Michigan, Ann Arbor, and serves as an associate editor of JAMA, but had no financial conflicts to disclose. These remarks were taken from an accompanying editorial (JAMA. 2019 Sep 27. doi:10.1001/jama.2019.16215).
“The development and approval of a new antibiotic is a rare occurrence and a reason to celebrate” given the scientific, regulatory, and economic challenges to antibiotic development, wrote Preeti N. Malani, MD, in an accompanying editorial. Lefamulin in both oral and intravenous forms was approved by the Food and Drug Administration in August 2019 for the treatment of community-acquired bacterial pneumonia, Dr. Malani said.
Lefamulin will likely be an expensive option. According to a manufacturer press release, lefamulin may cost $205/day for intravenous treatment and $275/day for oral treatment. “This is severalfold more than moxifloxacin or levofloxacin, which are the most commonly prescribed fluoroquinolones for CABP [community-acquired bacterial pneumonia],” said Dr. Malani. However, the addition of lefamulin to the array of antibiotics is important because of the persistent burden of bacterial pneumonia as an indication for antibiotic use, Dr. Malani emphasized.
Dr. Malani is affiliated with the University of Michigan, Ann Arbor, and serves as an associate editor of JAMA, but had no financial conflicts to disclose. These remarks were taken from an accompanying editorial (JAMA. 2019 Sep 27. doi:10.1001/jama.2019.16215).
Persistent high rates of bacterial resistance to current treatments have created the need for more options, especially for the treatment of community-acquired bacterial pneumonia (CABP), which remains a leading cause of hospitalization and death in the United States, wrote Elizabeth Alexander, MD, of Nabriva Therapeutics in King of Prussia, Penn., and colleagues. Lefamulin, “the first pleuromutilin antibiotic approved for intravenous and oral use in humans,” has demonstrated activity against many CABP-causing pathogens, including some not susceptible to other classes of antimicrobials, they noted.
Findings of Lefamulin Evaluation Against Pneumonia 2 (LEAP2) were published in JAMA. In this study, the researchers randomized 370 patients to 600 mg of oral lefamulin every 12 hours for 5 days and 368 patients to 400 mg of oral moxifloxacin every 24 hours for 7 days.
Early clinical response rates at 96 hours were 90.8% for both medications (difference of 0.1%). In addition, the rates of clinical response success were similar between the groups in both the modified intent-to-treat population (87.5% with lefamulin and 89.1% with moxifloxacin) and the clinically evaluable population (89.7% with lefamulin and 93.6% with moxifloxacin).
Gastrointestinal issues of diarrhea and nausea were the two most frequently reported treatment-emergent adverse events in both groups. Both conditions occurred more often in the lefamulin group, compared with the moxifloxacin group, but the differences were not significant (12.2% vs. 1.1% and 5.2% vs. 1.9%, respectively).
The study findings were limited by several factors including strict exclusion criteria that may limit the generalizability of the results, as well as a lack of testing for viral copathogens, low recovery of resistant pathogens, and possible misclassification of patient ethnicity, the researchers noted.
However, the results were strengthened by the randomized design, inclusion of patients with more severe CABP, and low rate of discontinuation, they said. The data support previous studies of lefamulin. Its lack of cross-resistance to other drug classes, coverage of typical and atypical CABP pathogens, and options for both oral and intravenous use suggest that it “may provide an alternative approach for the treatment of vulnerable patients,” the researchers said.
The study was supported by Nabriva Therapeutics. Dr. Alexander and several coauthors are employees of Nabriva Therapeutics and own stock in the company.
SOURCE: Alexander E et al. JAMA. 2019 Sep 27. doi:10.1001/jama.2019.15468.
Persistent high rates of bacterial resistance to current treatments have created the need for more options, especially for the treatment of community-acquired bacterial pneumonia (CABP), which remains a leading cause of hospitalization and death in the United States, wrote Elizabeth Alexander, MD, of Nabriva Therapeutics in King of Prussia, Penn., and colleagues. Lefamulin, “the first pleuromutilin antibiotic approved for intravenous and oral use in humans,” has demonstrated activity against many CABP-causing pathogens, including some not susceptible to other classes of antimicrobials, they noted.
Findings of Lefamulin Evaluation Against Pneumonia 2 (LEAP2) were published in JAMA. In this study, the researchers randomized 370 patients to 600 mg of oral lefamulin every 12 hours for 5 days and 368 patients to 400 mg of oral moxifloxacin every 24 hours for 7 days.
Early clinical response rates at 96 hours were 90.8% for both medications (difference of 0.1%). In addition, the rates of clinical response success were similar between the groups in both the modified intent-to-treat population (87.5% with lefamulin and 89.1% with moxifloxacin) and the clinically evaluable population (89.7% with lefamulin and 93.6% with moxifloxacin).
Gastrointestinal issues of diarrhea and nausea were the two most frequently reported treatment-emergent adverse events in both groups. Both conditions occurred more often in the lefamulin group, compared with the moxifloxacin group, but the differences were not significant (12.2% vs. 1.1% and 5.2% vs. 1.9%, respectively).
The study findings were limited by several factors including strict exclusion criteria that may limit the generalizability of the results, as well as a lack of testing for viral copathogens, low recovery of resistant pathogens, and possible misclassification of patient ethnicity, the researchers noted.
However, the results were strengthened by the randomized design, inclusion of patients with more severe CABP, and low rate of discontinuation, they said. The data support previous studies of lefamulin. Its lack of cross-resistance to other drug classes, coverage of typical and atypical CABP pathogens, and options for both oral and intravenous use suggest that it “may provide an alternative approach for the treatment of vulnerable patients,” the researchers said.
The study was supported by Nabriva Therapeutics. Dr. Alexander and several coauthors are employees of Nabriva Therapeutics and own stock in the company.
SOURCE: Alexander E et al. JAMA. 2019 Sep 27. doi:10.1001/jama.2019.15468.
FROM JAMA
Smoking, inactivity most powerful post-MI lifestyle risk factors
PARIS – All lifestyle-related cardiovascular risk factors aren’t equal in power when it comes to secondary prevention after a first acute MI, according to a massive Swedish registry study.

Insufficient physical activity and current smoking were consistently the strongest risk factors for all-cause mortality, major adverse cardiovascular events, and other key adverse outcomes in an analysis from the SWEDEHEART registry. The study included 65,002 patients discharged after a first MI and 325,010 age- and sex-matched controls with no prior MI followed for a median of 5.5 years and maximum of 12, Emil Hagstrom, MD, PhD, reported at the annual congress of the European Society of Cardiology.
Strongest lifestyle risk factors
The study examined the long-term relative importance of control of six major lifestyle risk factors for secondary cardiovascular prevention: current smoking, insufficient physical activity, blood pressure of 140/90 mm Hg or more, obesity, a fasting blood glucose of at least 126 mg/dL, and an LDL cholesterol of 70 mg/dL or more. Notably, two risk factors that physicians often emphasize in working with their patients with known coronary heart disease – an elevated LDL cholesterol and obesity – barely moved the needle. Out of the six risk factors scrutinized, those two consistently showed the weakest association with long-term risk of adverse outcomes. Occupying the middle ground in terms of predictive strength were hypertension and elevated blood glucose, according to Dr. Hagstrom, a cardiologist at Uppsala (Sweden) University.
Risk factor status was assessed 6-10 weeks post MI. Insufficient physical activity was defined as not engaging in at least 30 minutes of moderate-intensity exercise on at least 5 days per week. And when Dr. Hagstrom recalculated the risk of adverse outcomes using an LDL cholesterol threshold of 55 mg/dL rather than using 70 mg/dL, as recommended in new ESC secondary prevention guidelines released during the congress, the study results remained unchanged.
Cumulative effects
A key SWEDEHEART finding underscoring the importance of lifestyle in secondary prevention was that a linear stepwise relationship existed between the number of risk factors at target levels and the risk of all of the various adverse outcomes assessed, including stroke and heart failure hospitalization as well as all-cause mortality, cardiovascular mortality, and major bleeding.
Moreover, patients with none of the six risk factors outside of target when assessed after their MI had the same risks of all-cause mortality, cardiovascular mortality, and stroke as the matched controls.
For example, in an analysis adjusted for comorbid cancer, chronic obstructive pulmonary disease, and dementia, post-MI patients with zero risk factors had the same long-term risk of cardiovascular mortality as controls without a history of MI at baseline. With one risk factor not at target, a patient had a 41% increased risk compared with controls, a statistically significant difference. With two out-of-whack risk factors, the risk climbed to 102%. With three, 185%. With four risk factors not at target, the all-cause mortality risk jumped to 291%. And patients with more than four of the six risk factors not at target had a 409% greater risk of all-cause mortality than controls who had never had a heart attack.
When Dr. Hagstrom stratified subjects by age at baseline – up to 55, 56-64, 65-70, and 70-75 years – he discovered that, regardless of age, patients with zero risk factors had the same risk of all-cause mortality and other adverse outcomes as controls. However, when risk factors were present, younger patients consistently had a higher risk of all adverse outcomes than older patients with the same number of risk factors. When asked for an explanation of this phenomenon, Dr. Hagstrom noted that younger patients with multiple risk factors have a longer time to be exposed to and accumulate risk.
Follow-up of the study cohort will continue for years to come, the cardiologist promised.
At an ESC congress highlights session that closed out the meeting, Eva Prescott, MD, put the SWEDEHEART study at the top of her list of important developments in preventive cardiology arising from the congress.
“This is an excellent national registry I think we’re all envious of,” commented Dr. Prescott, a cardiologist at Copenhagen University. “The conclusion of this registry-based data, I think, is that lifestyle really remains at the core of prevention of cardiovascular events still today.”
The SWEDEHEART study analysis was funded free of commercial support. Dr. Hagstrom reported serving as a consultant to or receiving speakers’ fees from Amgen, AstraZeneca, Bayer, Novo Nordisk, and Sanofi.
PARIS – All lifestyle-related cardiovascular risk factors aren’t equal in power when it comes to secondary prevention after a first acute MI, according to a massive Swedish registry study.
Insufficient physical activity and current smoking were consistently the strongest risk factors for all-cause mortality, major adverse cardiovascular events, and other key adverse outcomes in an analysis from the SWEDEHEART registry. The study included 65,002 patients discharged after a first MI and 325,010 age- and sex-matched controls with no prior MI followed for a median of 5.5 years and maximum of 12, Emil Hagstrom, MD, PhD, reported at the annual congress of the European Society of Cardiology.
Strongest lifestyle risk factors
The study examined the long-term relative importance of control of six major lifestyle risk factors for secondary cardiovascular prevention: current smoking, insufficient physical activity, blood pressure of 140/90 mm Hg or more, obesity, a fasting blood glucose of at least 126 mg/dL, and an LDL cholesterol of 70 mg/dL or more. Notably, two risk factors that physicians often emphasize in working with their patients with known coronary heart disease – an elevated LDL cholesterol and obesity – barely moved the needle. Out of the six risk factors scrutinized, those two consistently showed the weakest association with long-term risk of adverse outcomes. Occupying the middle ground in terms of predictive strength were hypertension and elevated blood glucose, according to Dr. Hagstrom, a cardiologist at Uppsala (Sweden) University.
Risk factor status was assessed 6-10 weeks post MI. Insufficient physical activity was defined as not engaging in at least 30 minutes of moderate-intensity exercise on at least 5 days per week. And when Dr. Hagstrom recalculated the risk of adverse outcomes using an LDL cholesterol threshold of 55 mg/dL rather than using 70 mg/dL, as recommended in new ESC secondary prevention guidelines released during the congress, the study results remained unchanged.
Cumulative effects
A key SWEDEHEART finding underscoring the importance of lifestyle in secondary prevention was that a linear stepwise relationship existed between the number of risk factors at target levels and the risk of all of the various adverse outcomes assessed, including stroke and heart failure hospitalization as well as all-cause mortality, cardiovascular mortality, and major bleeding.
Moreover, patients with none of the six risk factors outside of target when assessed after their MI had the same risks of all-cause mortality, cardiovascular mortality, and stroke as the matched controls.
For example, in an analysis adjusted for comorbid cancer, chronic obstructive pulmonary disease, and dementia, post-MI patients with zero risk factors had the same long-term risk of cardiovascular mortality as controls without a history of MI at baseline. With one risk factor not at target, a patient had a 41% increased risk compared with controls, a statistically significant difference. With two out-of-whack risk factors, the risk climbed to 102%. With three, 185%. With four risk factors not at target, the all-cause mortality risk jumped to 291%. And patients with more than four of the six risk factors not at target had a 409% greater risk of all-cause mortality than controls who had never had a heart attack.
When Dr. Hagstrom stratified subjects by age at baseline – up to 55, 56-64, 65-70, and 70-75 years – he discovered that, regardless of age, patients with zero risk factors had the same risk of all-cause mortality and other adverse outcomes as controls. However, when risk factors were present, younger patients consistently had a higher risk of all adverse outcomes than older patients with the same number of risk factors. When asked for an explanation of this phenomenon, Dr. Hagstrom noted that younger patients with multiple risk factors have a longer time to be exposed to and accumulate risk.
Follow-up of the study cohort will continue for years to come, the cardiologist promised.
At an ESC congress highlights session that closed out the meeting, Eva Prescott, MD, put the SWEDEHEART study at the top of her list of important developments in preventive cardiology arising from the congress.
“This is an excellent national registry I think we’re all envious of,” commented Dr. Prescott, a cardiologist at Copenhagen University. “The conclusion of this registry-based data, I think, is that lifestyle really remains at the core of prevention of cardiovascular events still today.”
The SWEDEHEART study analysis was funded free of commercial support. Dr. Hagstrom reported serving as a consultant to or receiving speakers’ fees from Amgen, AstraZeneca, Bayer, Novo Nordisk, and Sanofi.
PARIS – All lifestyle-related cardiovascular risk factors aren’t equal in power when it comes to secondary prevention after a first acute MI, according to a massive Swedish registry study.
Insufficient physical activity and current smoking were consistently the strongest risk factors for all-cause mortality, major adverse cardiovascular events, and other key adverse outcomes in an analysis from the SWEDEHEART registry. The study included 65,002 patients discharged after a first MI and 325,010 age- and sex-matched controls with no prior MI followed for a median of 5.5 years and maximum of 12, Emil Hagstrom, MD, PhD, reported at the annual congress of the European Society of Cardiology.
Strongest lifestyle risk factors
The study examined the long-term relative importance of control of six major lifestyle risk factors for secondary cardiovascular prevention: current smoking, insufficient physical activity, blood pressure of 140/90 mm Hg or more, obesity, a fasting blood glucose of at least 126 mg/dL, and an LDL cholesterol of 70 mg/dL or more. Notably, two risk factors that physicians often emphasize in working with their patients with known coronary heart disease – an elevated LDL cholesterol and obesity – barely moved the needle. Out of the six risk factors scrutinized, those two consistently showed the weakest association with long-term risk of adverse outcomes. Occupying the middle ground in terms of predictive strength were hypertension and elevated blood glucose, according to Dr. Hagstrom, a cardiologist at Uppsala (Sweden) University.
Risk factor status was assessed 6-10 weeks post MI. Insufficient physical activity was defined as not engaging in at least 30 minutes of moderate-intensity exercise on at least 5 days per week. And when Dr. Hagstrom recalculated the risk of adverse outcomes using an LDL cholesterol threshold of 55 mg/dL rather than using 70 mg/dL, as recommended in new ESC secondary prevention guidelines released during the congress, the study results remained unchanged.
Cumulative effects
A key SWEDEHEART finding underscoring the importance of lifestyle in secondary prevention was that a linear stepwise relationship existed between the number of risk factors at target levels and the risk of all of the various adverse outcomes assessed, including stroke and heart failure hospitalization as well as all-cause mortality, cardiovascular mortality, and major bleeding.
Moreover, patients with none of the six risk factors outside of target when assessed after their MI had the same risks of all-cause mortality, cardiovascular mortality, and stroke as the matched controls.
For example, in an analysis adjusted for comorbid cancer, chronic obstructive pulmonary disease, and dementia, post-MI patients with zero risk factors had the same long-term risk of cardiovascular mortality as controls without a history of MI at baseline. With one risk factor not at target, a patient had a 41% increased risk compared with controls, a statistically significant difference. With two out-of-whack risk factors, the risk climbed to 102%. With three, 185%. With four risk factors not at target, the all-cause mortality risk jumped to 291%. And patients with more than four of the six risk factors not at target had a 409% greater risk of all-cause mortality than controls who had never had a heart attack.
When Dr. Hagstrom stratified subjects by age at baseline – up to 55, 56-64, 65-70, and 70-75 years – he discovered that, regardless of age, patients with zero risk factors had the same risk of all-cause mortality and other adverse outcomes as controls. However, when risk factors were present, younger patients consistently had a higher risk of all adverse outcomes than older patients with the same number of risk factors. When asked for an explanation of this phenomenon, Dr. Hagstrom noted that younger patients with multiple risk factors have a longer time to be exposed to and accumulate risk.
Follow-up of the study cohort will continue for years to come, the cardiologist promised.
At an ESC congress highlights session that closed out the meeting, Eva Prescott, MD, put the SWEDEHEART study at the top of her list of important developments in preventive cardiology arising from the congress.
“This is an excellent national registry I think we’re all envious of,” commented Dr. Prescott, a cardiologist at Copenhagen University. “The conclusion of this registry-based data, I think, is that lifestyle really remains at the core of prevention of cardiovascular events still today.”
The SWEDEHEART study analysis was funded free of commercial support. Dr. Hagstrom reported serving as a consultant to or receiving speakers’ fees from Amgen, AstraZeneca, Bayer, Novo Nordisk, and Sanofi.
REPORTING FROM THE ESC CONGRESS 2019
PARP inhibitor prolongs PFS in mCRPC
BARCELONA – PARP inhibitors may be able to do for men with advanced castration-resistant prostate cancer what they currently do for women with breast or ovarian cancers linked to BRCA mutations, investigators report.

Among men with metastatic castration-resistant prostate cancer (mCRPC) that had progressed on prior therapy with either abiraterone (Zytiga) or enzalutamide (Xtandi) and that bore DNA-repair mutations (BRCA1, BRCA2, or ATM), those who were randomized in the PROfound trial to receive olaparib (Lynparza) had significant improvement in radiographic progression-free survival (rPFS) compared with patients assigned to the physician’s choice of a new hormonal agent, reported Maha Hussain, MD, of the Robert H. Lurie Comprehensive Cancer Center at Northwestern University in Chicago.
“PROfound is the first positive biomarker-selected phase 3 study evaluating a molecularly-targeted therapy in men with metastatic castration-resistant prostate cancer, highlighting the importance of genomic testing in this population, and also importantly highlighting the feasibility of precision-medicine trials in this disease,” she said at the European Society for Medical Oncology (ESMO) Congress.
Approximately 25% of men with mCRPC have loss-of-function mutations or alterations in homologous recombinant repair (HRR) genes, especially BRCA1, BRCA2, and ATM. Breast and ovarian cancers bearing these mutations are known to be sensitive to PARP (poly ADP ribose polymerase) inhibitors such as olaparib.
To see whether men with mCRPC could derive a similar benefit, the investigators enrolled patients who had experienced disease progression on abiraterone or enzalutamiude and whose tumors had one or more alterations in any qualifying gene with direct or indirect role in homologous recombinant DNA repair.
Patients were stratified by previous taxane use and measurable disease, and then two cohorts were enrolled. Cohort A included 245 men with BRCA1, BRCA2, or ATM mutations, and cohort B included 142 men with other alterations (in BARD1, BIRP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD15B, RAD15C, RAD15D, or RAD54L).
The median age in cohort A was 68, and in cohort B it was 67, with men 86 years of age at the upper end of the range.
“I think it’s remarkable that men in their late 80s and even early 90s were eligible, were able to be enter this study and be treated on it, and I think that’s an important point here,” Dr. Hussain said.
Nearly one-fourth of patients in the trial had metastatic disease at their initial diagnosis, she noted.
Patients in each cohort were randomized on a 2:1 basis to receive either open-label olaparib 300 mg twice daily, or the treating physician’s choice of abireraterone or enzalutamide, plus predisone.
Upon blinded independent central review (BICR) showing disease progression, patients were allowed to cross over to the olaparib arm, which more than 80% of patients eventually did.
Radiographic PFS in the BRCA1, BRCA2 and ATM cohort according to BICR, the primary endpoint, was a median of 7.39 months with olaparib, compared with 3.55 months with the other therapies, for a hazard ratio (HR) for progression on olaparib of 0.34 (P less than .0001).
A somewhat smaller but still significant benefit was seen for olaparib in the overall population (both cohorts), with a median rPFS of 5.82 months vs. 3.52 months, respectively (HR 0.49, P less than .0001).
Among patients in cohort A, the objective response rate was 33.3% with olaparib, compared with 2.3% for the other agents, resulting in an odds ratio for response of 20.86 (P less than .0001)
Olaparib was also associated with longer time to pain progression in patients in cohort A, with the median not reached compared with 9.92 months with the hormonal agents (HR 0.44, P = .0192).
Among patients in the physician’s choice arm who had disease progression, 80.6% in cohort A and 84.6% in cohort B were crossed over to olaparib.
At this interim analysis, median overall survival was 18.5 months with olaparib, compared with 15.11 months with the other agents, but this difference was not statistically significant. Further follow-up will be needed before a difference in overall survival becomes evident, Dr. Hussain said.
“I think this study has demonstrated that now prostate cancer can be treated with a targeted therapy approach,” said Ignacio Duran, MD, of Hospital Universitario Marques de Valdecilla in Santander, Spain. He was the invited discussant and moderator of a briefing where Dr. Hussain outlined the study details prior to presentation in a symposium.
“Not all prostate cancer patients have the same tumors, and this is the first time we’ve been able to identify that we can more precisely characterize the molecular biology, the genetic background of these tumors, and that is going to determine how we treat them,” he said.
The PROfound trial made “a double hit: superiority in terms of efficacy, and it has proved a new concept that in prostate cancer has not been proved before,” he added.
“This is a truly practice-changing study, not just for our practice and our patients, but also for the study design,” said Eleni Efstathiou, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, who was the invited discussant at the symposium where Dr. Hussain presented the full trial data.
She lauded the use of a validated genomic testing tissue-based assay (FoundationOne CDx next-generation sequencing test) to identify patients who might benefit from olaparib.
She said that the PARP inhibition-specific strategy of the trial appears to have paid off, with statistically significant, clinically meaningful improvement in outcomes and an acceptable safety profile.
“And when it comes to a prostate cancer therapy strategry? Well, we’re starting to enter into the targeted therapy era,” she said.
The PROfound trial was sponsored by AstraZeneca and is part of an alliance between AstraZeneca and Merck. Dr. Hussain disclosed travel and/or accommodation support, honoraria, consulting/advisory fees and research support from AstraZeneca and others. Dr. Duran disclosed advisory board fees from Roche and BMS, and speaker honoraria from Roche, Bristol-Myers Squibb, and Merck. Dr. Efstathiou disclosed research support and honoraria from various companies, not including AstraZeneca or Merck.
SOURCE: Hussain M et al. ESMO 2019, Abstract LBA-12.
BARCELONA – PARP inhibitors may be able to do for men with advanced castration-resistant prostate cancer what they currently do for women with breast or ovarian cancers linked to BRCA mutations, investigators report.
Among men with metastatic castration-resistant prostate cancer (mCRPC) that had progressed on prior therapy with either abiraterone (Zytiga) or enzalutamide (Xtandi) and that bore DNA-repair mutations (BRCA1, BRCA2, or ATM), those who were randomized in the PROfound trial to receive olaparib (Lynparza) had significant improvement in radiographic progression-free survival (rPFS) compared with patients assigned to the physician’s choice of a new hormonal agent, reported Maha Hussain, MD, of the Robert H. Lurie Comprehensive Cancer Center at Northwestern University in Chicago.
“PROfound is the first positive biomarker-selected phase 3 study evaluating a molecularly-targeted therapy in men with metastatic castration-resistant prostate cancer, highlighting the importance of genomic testing in this population, and also importantly highlighting the feasibility of precision-medicine trials in this disease,” she said at the European Society for Medical Oncology (ESMO) Congress.
Approximately 25% of men with mCRPC have loss-of-function mutations or alterations in homologous recombinant repair (HRR) genes, especially BRCA1, BRCA2, and ATM. Breast and ovarian cancers bearing these mutations are known to be sensitive to PARP (poly ADP ribose polymerase) inhibitors such as olaparib.
To see whether men with mCRPC could derive a similar benefit, the investigators enrolled patients who had experienced disease progression on abiraterone or enzalutamiude and whose tumors had one or more alterations in any qualifying gene with direct or indirect role in homologous recombinant DNA repair.
Patients were stratified by previous taxane use and measurable disease, and then two cohorts were enrolled. Cohort A included 245 men with BRCA1, BRCA2, or ATM mutations, and cohort B included 142 men with other alterations (in BARD1, BIRP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD15B, RAD15C, RAD15D, or RAD54L).
The median age in cohort A was 68, and in cohort B it was 67, with men 86 years of age at the upper end of the range.
“I think it’s remarkable that men in their late 80s and even early 90s were eligible, were able to be enter this study and be treated on it, and I think that’s an important point here,” Dr. Hussain said.
Nearly one-fourth of patients in the trial had metastatic disease at their initial diagnosis, she noted.
Patients in each cohort were randomized on a 2:1 basis to receive either open-label olaparib 300 mg twice daily, or the treating physician’s choice of abireraterone or enzalutamide, plus predisone.
Upon blinded independent central review (BICR) showing disease progression, patients were allowed to cross over to the olaparib arm, which more than 80% of patients eventually did.
Radiographic PFS in the BRCA1, BRCA2 and ATM cohort according to BICR, the primary endpoint, was a median of 7.39 months with olaparib, compared with 3.55 months with the other therapies, for a hazard ratio (HR) for progression on olaparib of 0.34 (P less than .0001).
A somewhat smaller but still significant benefit was seen for olaparib in the overall population (both cohorts), with a median rPFS of 5.82 months vs. 3.52 months, respectively (HR 0.49, P less than .0001).
Among patients in cohort A, the objective response rate was 33.3% with olaparib, compared with 2.3% for the other agents, resulting in an odds ratio for response of 20.86 (P less than .0001)
Olaparib was also associated with longer time to pain progression in patients in cohort A, with the median not reached compared with 9.92 months with the hormonal agents (HR 0.44, P = .0192).
Among patients in the physician’s choice arm who had disease progression, 80.6% in cohort A and 84.6% in cohort B were crossed over to olaparib.
At this interim analysis, median overall survival was 18.5 months with olaparib, compared with 15.11 months with the other agents, but this difference was not statistically significant. Further follow-up will be needed before a difference in overall survival becomes evident, Dr. Hussain said.
“I think this study has demonstrated that now prostate cancer can be treated with a targeted therapy approach,” said Ignacio Duran, MD, of Hospital Universitario Marques de Valdecilla in Santander, Spain. He was the invited discussant and moderator of a briefing where Dr. Hussain outlined the study details prior to presentation in a symposium.
“Not all prostate cancer patients have the same tumors, and this is the first time we’ve been able to identify that we can more precisely characterize the molecular biology, the genetic background of these tumors, and that is going to determine how we treat them,” he said.
The PROfound trial made “a double hit: superiority in terms of efficacy, and it has proved a new concept that in prostate cancer has not been proved before,” he added.
“This is a truly practice-changing study, not just for our practice and our patients, but also for the study design,” said Eleni Efstathiou, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, who was the invited discussant at the symposium where Dr. Hussain presented the full trial data.
She lauded the use of a validated genomic testing tissue-based assay (FoundationOne CDx next-generation sequencing test) to identify patients who might benefit from olaparib.
She said that the PARP inhibition-specific strategy of the trial appears to have paid off, with statistically significant, clinically meaningful improvement in outcomes and an acceptable safety profile.
“And when it comes to a prostate cancer therapy strategry? Well, we’re starting to enter into the targeted therapy era,” she said.
The PROfound trial was sponsored by AstraZeneca and is part of an alliance between AstraZeneca and Merck. Dr. Hussain disclosed travel and/or accommodation support, honoraria, consulting/advisory fees and research support from AstraZeneca and others. Dr. Duran disclosed advisory board fees from Roche and BMS, and speaker honoraria from Roche, Bristol-Myers Squibb, and Merck. Dr. Efstathiou disclosed research support and honoraria from various companies, not including AstraZeneca or Merck.
SOURCE: Hussain M et al. ESMO 2019, Abstract LBA-12.
BARCELONA – PARP inhibitors may be able to do for men with advanced castration-resistant prostate cancer what they currently do for women with breast or ovarian cancers linked to BRCA mutations, investigators report.
Among men with metastatic castration-resistant prostate cancer (mCRPC) that had progressed on prior therapy with either abiraterone (Zytiga) or enzalutamide (Xtandi) and that bore DNA-repair mutations (BRCA1, BRCA2, or ATM), those who were randomized in the PROfound trial to receive olaparib (Lynparza) had significant improvement in radiographic progression-free survival (rPFS) compared with patients assigned to the physician’s choice of a new hormonal agent, reported Maha Hussain, MD, of the Robert H. Lurie Comprehensive Cancer Center at Northwestern University in Chicago.
“PROfound is the first positive biomarker-selected phase 3 study evaluating a molecularly-targeted therapy in men with metastatic castration-resistant prostate cancer, highlighting the importance of genomic testing in this population, and also importantly highlighting the feasibility of precision-medicine trials in this disease,” she said at the European Society for Medical Oncology (ESMO) Congress.
Approximately 25% of men with mCRPC have loss-of-function mutations or alterations in homologous recombinant repair (HRR) genes, especially BRCA1, BRCA2, and ATM. Breast and ovarian cancers bearing these mutations are known to be sensitive to PARP (poly ADP ribose polymerase) inhibitors such as olaparib.
To see whether men with mCRPC could derive a similar benefit, the investigators enrolled patients who had experienced disease progression on abiraterone or enzalutamiude and whose tumors had one or more alterations in any qualifying gene with direct or indirect role in homologous recombinant DNA repair.
Patients were stratified by previous taxane use and measurable disease, and then two cohorts were enrolled. Cohort A included 245 men with BRCA1, BRCA2, or ATM mutations, and cohort B included 142 men with other alterations (in BARD1, BIRP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD15B, RAD15C, RAD15D, or RAD54L).
The median age in cohort A was 68, and in cohort B it was 67, with men 86 years of age at the upper end of the range.
“I think it’s remarkable that men in their late 80s and even early 90s were eligible, were able to be enter this study and be treated on it, and I think that’s an important point here,” Dr. Hussain said.
Nearly one-fourth of patients in the trial had metastatic disease at their initial diagnosis, she noted.
Patients in each cohort were randomized on a 2:1 basis to receive either open-label olaparib 300 mg twice daily, or the treating physician’s choice of abireraterone or enzalutamide, plus predisone.
Upon blinded independent central review (BICR) showing disease progression, patients were allowed to cross over to the olaparib arm, which more than 80% of patients eventually did.
Radiographic PFS in the BRCA1, BRCA2 and ATM cohort according to BICR, the primary endpoint, was a median of 7.39 months with olaparib, compared with 3.55 months with the other therapies, for a hazard ratio (HR) for progression on olaparib of 0.34 (P less than .0001).
A somewhat smaller but still significant benefit was seen for olaparib in the overall population (both cohorts), with a median rPFS of 5.82 months vs. 3.52 months, respectively (HR 0.49, P less than .0001).
Among patients in cohort A, the objective response rate was 33.3% with olaparib, compared with 2.3% for the other agents, resulting in an odds ratio for response of 20.86 (P less than .0001)
Olaparib was also associated with longer time to pain progression in patients in cohort A, with the median not reached compared with 9.92 months with the hormonal agents (HR 0.44, P = .0192).
Among patients in the physician’s choice arm who had disease progression, 80.6% in cohort A and 84.6% in cohort B were crossed over to olaparib.
At this interim analysis, median overall survival was 18.5 months with olaparib, compared with 15.11 months with the other agents, but this difference was not statistically significant. Further follow-up will be needed before a difference in overall survival becomes evident, Dr. Hussain said.
“I think this study has demonstrated that now prostate cancer can be treated with a targeted therapy approach,” said Ignacio Duran, MD, of Hospital Universitario Marques de Valdecilla in Santander, Spain. He was the invited discussant and moderator of a briefing where Dr. Hussain outlined the study details prior to presentation in a symposium.
“Not all prostate cancer patients have the same tumors, and this is the first time we’ve been able to identify that we can more precisely characterize the molecular biology, the genetic background of these tumors, and that is going to determine how we treat them,” he said.
The PROfound trial made “a double hit: superiority in terms of efficacy, and it has proved a new concept that in prostate cancer has not been proved before,” he added.
“This is a truly practice-changing study, not just for our practice and our patients, but also for the study design,” said Eleni Efstathiou, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, who was the invited discussant at the symposium where Dr. Hussain presented the full trial data.
She lauded the use of a validated genomic testing tissue-based assay (FoundationOne CDx next-generation sequencing test) to identify patients who might benefit from olaparib.
She said that the PARP inhibition-specific strategy of the trial appears to have paid off, with statistically significant, clinically meaningful improvement in outcomes and an acceptable safety profile.
“And when it comes to a prostate cancer therapy strategry? Well, we’re starting to enter into the targeted therapy era,” she said.
The PROfound trial was sponsored by AstraZeneca and is part of an alliance between AstraZeneca and Merck. Dr. Hussain disclosed travel and/or accommodation support, honoraria, consulting/advisory fees and research support from AstraZeneca and others. Dr. Duran disclosed advisory board fees from Roche and BMS, and speaker honoraria from Roche, Bristol-Myers Squibb, and Merck. Dr. Efstathiou disclosed research support and honoraria from various companies, not including AstraZeneca or Merck.
SOURCE: Hussain M et al. ESMO 2019, Abstract LBA-12.
REPORTING FROM ESMO 2019
Heparin Drug Shortage Conservation Strategies
Heparin is the anticoagulant of choice when a rapid anticoagulant is indicated: Onset of action is immediate when administered IV as a bolus.1 The major anticoagulant effect of heparin is mediated by heparin/antithrombin (AT) interaction. Heparin/AT inactivates factor IIa (thrombin) and factors Xa, IXa, XIa, and XIIa. Heparin is approved for multiple indications, such as venous thromboembolism (VTE) treatment and prophylaxis of medical and surgical patients; stroke prevention in atrial fibrillation (AF); acute coronary syndrome (ACS); vascular and cardiac surgeries; and various interventional procedures (eg, diagnostic angiography and percutaneous coronary intervention [PCI]). It also is used as an anticoagulant in blood transfusions, extracorporeal circulation, and for maintaining patency of central vascular access devices (CVADs).
About 60% of the crude heparin used to manufacture heparin in the US originates in China, derived from porcine mucosa. African swine fever, a contagious virus with no cure, has eliminated about 25% to 35% of China’s pig population, or about 150 million pigs. In July 2019, members of the US House of Representatives Committee on Energy and Commerce sent a letter to the US Food and Drug Administration asking for details on the potential impact of African swine fever on the supply of heparin.2
The US Department of Veterans Affairs (VA) heath care system is currently experiencing a shortage of heparin vials and syringes. It is unclear when resolution of this shortage will occur as it could resolve within several weeks or as late as January 2020.3 Although vials and syringes are the current products that are affected, it is possible the shortage may eventually include IV heparin bags as well.
Since the foremost objective of VA health care providers is to provide timely access to medications for veterans, strategies to conserve unfractionated heparin (UfH) must be used since it is a first-line therapy where few evidence-based alternatives exist. Conservation strategies may include drug rationing, therapeutic substitution, and compounding of needed products using the limited stock available in the pharmacy.4 It is important that all staff are educated on facility strategies in order to be familiar with alternatives and limit the potential for near misses, adverse events, and provider frustration.
In shortage situations, the VA-Pharmacy Benefits Management (PBM) defers decisions regarding drug preservation, processes to shift to viable alternatives, and the best practice for safe transitions to local facilities and their subject matter experts.5 At the VA Tennessee Valley Healthcare System, a 1A, tertiary, dual campus health care system, a pharmacy task force has formed to track drug shortages impacting the facility’s efficiencies and budgets. This group communicates with the Pharmacy and Therapeutics committee about potential risks to patient care and develops shortage briefs (following an SBAR [situation, background, assessment, recommendation] design) generally authored and championed by at least 1 clinical pharmacy specialist and supervising physicians who are field experts. Prior to dissemination, the SBAR undergoes a rapid peer-review process.

To date, VA PBM has not issued specific guidance on how pharmacists should proceed in case of a shortage. However, we recommend strategies that may be considered for implementation during a potential UfH shortage. For example, pharmacists can use therapeutic alternatives for which best available evidence suggests no disadvantage.4 The Table lists alternative agents according to indication and patient-specific considerations that may preclude use. Existing UfH products may also be used for drug compounding (eg, use current stock to provide an indicated aliquot) to meet the need of prioritized patients.4 In addition, we suggest prioritizing current UfH/heparinized saline for use for the following groups of patients4:
- Emergent/urgent cardiac surgery1,6;
- Hemodialysis patients1,7-9 for which the low-molecular-weight heparin (LMWH) dalteparin is deemed inappropriate or the patient is not monitored in the intensive care unit for regional citrate administration;
- VTE prophylaxis for patients with epidurals or chest tubes for which urgent invasive management may occur, recent cardiac or neurosurgery, or for patients with a creatine clearance < 15 mL/min or receiving hemodialysis10-12;
- Vascular surgery (eg, limb ischemia) and interventions (eg, carotid stenting, endarterectomy)13,14;
- Mesenteric ischemia (venous thrombosis) with a potential to proceed to laparotomy15;
- Critically ill patients with arterial lines for which normal saline is deemed inappropriate for line flushing16;
- Electrophysiology procedures (eg, AF ablation)17; and
- Contraindication to use of a long-acting alternative listed in the table or a medical necessity exists for using a rapidly reversible agent. Examples for this category include but are not limited to recent gastrointestinal bleeding, central nervous system lesion, and select neurologic diagnoses (eg, cerebral venous sinus thrombosis with hemorrhage, thrombus in vertebral basilar system or anterior circulation, intraparenchymal hemorrhage plus mechanical valve, medium to large cardioembolic stroke with intracardiac thrombus).
Conclusion
The UfH drug shortage represents a significant threat to public health and is a major challenge for US health care systems, including the Veterans Health Administration. Overreliance on a predominant source of crude heparin has affected multiple UfH manufacturers and products. Current alternatives to UfH include low-molecular-weight heparins, IV direct thrombin inhibitors, and SC fondaparinux, with selection supported by guidelines or evolving literature. However, the shortage has the potential to expand to other injectables, such as dalteparin and enoxaparin, and severely limit care for veterans. It is vital that clinicians rapidly address the current shortage by creating a plan to develop efficient and equitable access to UfH, continue to assess supply and update stakeholders, and select evidence-based alternatives while maintaining focus on efficacy and safety.
Acknowledgments
The authors thank Ashley Yost, PharmD, for her coordination of the multidisciplinary task force assigned to efficiently manage the heparin drug shortage. This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville, Tennessee.
1. Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119(1):64S-94S.
2. Bipartisan E&C leaders request FDA briefing on threat to U.S. heparin supply [press release]. Washington, DC: House Committee on Energy and Commerce; July 30, 2019. https://energycommerce.house.gov/newsroom/press-releases/bipartisan-ec-leaders-request-fda-briefing-on-threat-to-us-heparin-supply. Accessed September 19, 2019.
3. American Society of Health-System Pharmacists. Drug Shortages. Heparin injection. https://www.ashp.org/Drug-Shortages/Current-Shortages/Drug-Shortages-List?page=CurrentShortages. Accessed September 19, 2019.
4. Reed BN, Fox ER, Konig M, et al. The impact of drug shortages on patients with cardiovascular disease: causes, consequences, and a call to action. Am Heart J. 2016;175:130-141.
5. US Department of Veterans Affairs. Pharmacy Benefits Management Services, Medical Advisory Panel, VISN Pharmacist Executives, The Center For Medication Safety. Heparin supply status: frequently asked questions. PBM-2018-02. https://www.pbm.va.gov/PBM/vacenterformedicationsafety/HeparinandSalineSyringeRecallDuetoContamination_NationalPBMPati.pdf. Published May 3, 2018. Accessed September 11, 2019.
6. Shore-Lesserson I, Baker RA, Ferraris VA, et al. The Society of Thoracic Surgeons, The Society of Cardiovascular Anesthesiologists, and the American Society of ExtraCorporeal Technology: Clinical Practice Guidelines-anticoagulation during cardiopulmonary bypass. Ann Thorac Surg. 2018;105(2):650-662.
7. Soroka S, Agharazii M, Donnelly S, et al. An adjustable dalteparin sodium dose regimen for the prevention of clotting in the extracorporeal circuit in hemodialysis: a clinical trial of safety and efficacy (the PARROT Study). Can J Kidney Health Dis. 2018;5:1-12.
8. Shantha GPS, Kumar AA, Sethi M, Khanna RC, Pancholy SB. Efficacy and safety of low molecular weight heparin compared to unfractionated heparin for chronic outpatient hemodialysis in end stage renal disease: systematic review and meta-analysis. Peer J. 2015;3:e835.
9. Kessler M, Moureau F, and Nguyen P. Anticoagulation in chronic hemodialysis: progress toward an optimal approach. Semin Dial. 2015;28(5):474-489.
10. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e227s-e277S.
11. Kaye AD, Brunk AJ, Kaye AJ, et al. Regional anesthesia in patients on anticoagulation therapies—evidence-based recommendations. Curr Pain Headache Rep. 2019;23(9):67.
12. Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e195S-e226S.
13. Naylor AR, Ricco JB, de Borst GJ, et al. Management of atherosclerotic carotid and vertebral artery disease: 2017 clinical practice guidelines of the European Society for Vascular Surgery. Eur J Vasc Endovasc Surg. 2018;55:3-81.
14. Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. JACC. 2017;69(11): e71-e126.
15. Bjorck M, Koelemaya M, Acosta S, et al. Management of diseases of mesenteric arteries and veins. Eur J Vasc Endovasc Surg. 2017;53(4):460-510.
16. Gorski L, Hadaway L, Hagle ME, McGoldrick M, Orr M, Doellman D. Infusion therapy standards of practice. J Infusion Nurs. 2016;39:S1-S156.
17. Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm. 2017;14(10):e275-e444.
18. Spyropoulos AC, Al-Badri A, Sherwood MW, Douketis JD. Periprocedural management of patients receiving a vitamin K antagonist or a direct oral anticoagulant requiring an elective procedure or surgery. J Thromb Haemost. 2016;14(5):875-885.
, . Periprocedural bridging management of anticoagulation. Circulation. 2012;126(4):486-490.
20. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e326S-e350S.
21. Sousa-Uva M, Neumann F-J, Ahlsson A, et al; ESC Scientific Document Group. 2018 ESC/EACTS Guidelines on myocardial revascularization. The Task Force on myocardial revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Developed with a special contribution of the European Association for Percutaneous Cardiovascular Interventions (EAPCI). Eur J Cardiothorac Surg. 2019;55(1):4-90.
22. Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes. JACC. 2014;64(24):e139-e228.
23. O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of patients with ST-elevation myocardial infarction. JACC. 2013;61(4):e78-e140.
24. Angiomax [package insert]. Parsippany, NJ: The Medicines Company; March 2016.
25. Sousa-Uva, Head SJ, Milojevic M, et al. 2017 EACTS guidelines on perioperative medication in adult cardiac surgery. Eur J Cardiothorac Surg. 2018;53(1):5-33.
26. Witt DM, Nieuwlaat R, Clark NP, et al. American Society of Hematology 2018 guidelines for the management of venous thromboembolism: optimal management of anticoagulation therapy. Blood Adv. 2018: 2(22):3257-3291
27. Kearon C, Akl EA, Blaivas A, et al. Antithrombotic therapy for VTE disease: Chest guideline and expert panel report. Chest. 2016;149(2):315-352.
28. US Department of Veterans Affairs, Pharmacy Benefits Manager Service. Direct oral anticoagulants criteria for use and algorithm for venous thromboembolism treatment. https://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse.asp. Updated December 2016. [Source not verified]
29. Falck-Ytter Y, Francis CW, Johanson NA, et al. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e278S-e325S.
30. Raja S, Idrees JJ, Blackstone EH, et al. Routine venous thromboembolism screening after pneumonectomy: the more you look, the more you see. J Thorac Cardiovasc Surg. 2016;152(2):524-532.e2.
31. Schünemann HJ, Cushman M, Burnett AE, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: prophylaxis for hospitalized and nonhospitalized patients. Blood Adv. 2018;2(22):3198-3225.
32. Naidu SS, Aronow HD, Box LC, et al. SCAI expert consensus statement: 2016 best practices in the cardiac catheterization laboratory:(endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista; affirmation of value by the Canadian Association of Interventional Cardiology-Association Canadienne de Cardiologie d’intervention). Catheter Cardiovasc Interv. 2016;88(3):407-423.
33. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. JACC. 2011;58(24):e44-e122.
34. Mason PJ, Shah B, Tamis-Holland JE, et al; American Heart Association Interventional Cardiovascular Care Committee of the Council on Clinical Cardiology; Council on Cardiovascular and Stroke Nursing; Council on Peripheral Vascular Disease; and Council on Genomic and Precision Medicine. AHA scientific statement: an update on radial artery access and best practices for transradial coronary angiography and intervention in acute coronary syndrome. Circ Cardiovasc Interv. 2018;11(9):e000035.
35. Rao SV, Tremmel JA, Gilchrist IC, et al; Society for Cardiovascular Angiography and Intervention’s Transradial Working Group. Best practices for transradial angiography and intervention: a consensus statement from the society for cardiovascular angiography and interventions’ transradial working group. Catheter Cardiovasc Interv. 2014;83(2):228-236. 36. Moran JE, Ash SR. Locking solutions for hemodialysis catheters; heparin and citrate: a position paper by ASDIN. Semin Dial. 2008;21(5):490-492.
Heparin is the anticoagulant of choice when a rapid anticoagulant is indicated: Onset of action is immediate when administered IV as a bolus.1 The major anticoagulant effect of heparin is mediated by heparin/antithrombin (AT) interaction. Heparin/AT inactivates factor IIa (thrombin) and factors Xa, IXa, XIa, and XIIa. Heparin is approved for multiple indications, such as venous thromboembolism (VTE) treatment and prophylaxis of medical and surgical patients; stroke prevention in atrial fibrillation (AF); acute coronary syndrome (ACS); vascular and cardiac surgeries; and various interventional procedures (eg, diagnostic angiography and percutaneous coronary intervention [PCI]). It also is used as an anticoagulant in blood transfusions, extracorporeal circulation, and for maintaining patency of central vascular access devices (CVADs).
About 60% of the crude heparin used to manufacture heparin in the US originates in China, derived from porcine mucosa. African swine fever, a contagious virus with no cure, has eliminated about 25% to 35% of China’s pig population, or about 150 million pigs. In July 2019, members of the US House of Representatives Committee on Energy and Commerce sent a letter to the US Food and Drug Administration asking for details on the potential impact of African swine fever on the supply of heparin.2
The US Department of Veterans Affairs (VA) heath care system is currently experiencing a shortage of heparin vials and syringes. It is unclear when resolution of this shortage will occur as it could resolve within several weeks or as late as January 2020.3 Although vials and syringes are the current products that are affected, it is possible the shortage may eventually include IV heparin bags as well.
Since the foremost objective of VA health care providers is to provide timely access to medications for veterans, strategies to conserve unfractionated heparin (UfH) must be used since it is a first-line therapy where few evidence-based alternatives exist. Conservation strategies may include drug rationing, therapeutic substitution, and compounding of needed products using the limited stock available in the pharmacy.4 It is important that all staff are educated on facility strategies in order to be familiar with alternatives and limit the potential for near misses, adverse events, and provider frustration.
In shortage situations, the VA-Pharmacy Benefits Management (PBM) defers decisions regarding drug preservation, processes to shift to viable alternatives, and the best practice for safe transitions to local facilities and their subject matter experts.5 At the VA Tennessee Valley Healthcare System, a 1A, tertiary, dual campus health care system, a pharmacy task force has formed to track drug shortages impacting the facility’s efficiencies and budgets. This group communicates with the Pharmacy and Therapeutics committee about potential risks to patient care and develops shortage briefs (following an SBAR [situation, background, assessment, recommendation] design) generally authored and championed by at least 1 clinical pharmacy specialist and supervising physicians who are field experts. Prior to dissemination, the SBAR undergoes a rapid peer-review process.
To date, VA PBM has not issued specific guidance on how pharmacists should proceed in case of a shortage. However, we recommend strategies that may be considered for implementation during a potential UfH shortage. For example, pharmacists can use therapeutic alternatives for which best available evidence suggests no disadvantage.4 The Table lists alternative agents according to indication and patient-specific considerations that may preclude use. Existing UfH products may also be used for drug compounding (eg, use current stock to provide an indicated aliquot) to meet the need of prioritized patients.4 In addition, we suggest prioritizing current UfH/heparinized saline for use for the following groups of patients4:
- Emergent/urgent cardiac surgery1,6;
- Hemodialysis patients1,7-9 for which the low-molecular-weight heparin (LMWH) dalteparin is deemed inappropriate or the patient is not monitored in the intensive care unit for regional citrate administration;
- VTE prophylaxis for patients with epidurals or chest tubes for which urgent invasive management may occur, recent cardiac or neurosurgery, or for patients with a creatine clearance < 15 mL/min or receiving hemodialysis10-12;
- Vascular surgery (eg, limb ischemia) and interventions (eg, carotid stenting, endarterectomy)13,14;
- Mesenteric ischemia (venous thrombosis) with a potential to proceed to laparotomy15;
- Critically ill patients with arterial lines for which normal saline is deemed inappropriate for line flushing16;
- Electrophysiology procedures (eg, AF ablation)17; and
- Contraindication to use of a long-acting alternative listed in the table or a medical necessity exists for using a rapidly reversible agent. Examples for this category include but are not limited to recent gastrointestinal bleeding, central nervous system lesion, and select neurologic diagnoses (eg, cerebral venous sinus thrombosis with hemorrhage, thrombus in vertebral basilar system or anterior circulation, intraparenchymal hemorrhage plus mechanical valve, medium to large cardioembolic stroke with intracardiac thrombus).
Conclusion
The UfH drug shortage represents a significant threat to public health and is a major challenge for US health care systems, including the Veterans Health Administration. Overreliance on a predominant source of crude heparin has affected multiple UfH manufacturers and products. Current alternatives to UfH include low-molecular-weight heparins, IV direct thrombin inhibitors, and SC fondaparinux, with selection supported by guidelines or evolving literature. However, the shortage has the potential to expand to other injectables, such as dalteparin and enoxaparin, and severely limit care for veterans. It is vital that clinicians rapidly address the current shortage by creating a plan to develop efficient and equitable access to UfH, continue to assess supply and update stakeholders, and select evidence-based alternatives while maintaining focus on efficacy and safety.
Acknowledgments
The authors thank Ashley Yost, PharmD, for her coordination of the multidisciplinary task force assigned to efficiently manage the heparin drug shortage. This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville, Tennessee.
Heparin is the anticoagulant of choice when a rapid anticoagulant is indicated: Onset of action is immediate when administered IV as a bolus.1 The major anticoagulant effect of heparin is mediated by heparin/antithrombin (AT) interaction. Heparin/AT inactivates factor IIa (thrombin) and factors Xa, IXa, XIa, and XIIa. Heparin is approved for multiple indications, such as venous thromboembolism (VTE) treatment and prophylaxis of medical and surgical patients; stroke prevention in atrial fibrillation (AF); acute coronary syndrome (ACS); vascular and cardiac surgeries; and various interventional procedures (eg, diagnostic angiography and percutaneous coronary intervention [PCI]). It also is used as an anticoagulant in blood transfusions, extracorporeal circulation, and for maintaining patency of central vascular access devices (CVADs).
About 60% of the crude heparin used to manufacture heparin in the US originates in China, derived from porcine mucosa. African swine fever, a contagious virus with no cure, has eliminated about 25% to 35% of China’s pig population, or about 150 million pigs. In July 2019, members of the US House of Representatives Committee on Energy and Commerce sent a letter to the US Food and Drug Administration asking for details on the potential impact of African swine fever on the supply of heparin.2
The US Department of Veterans Affairs (VA) heath care system is currently experiencing a shortage of heparin vials and syringes. It is unclear when resolution of this shortage will occur as it could resolve within several weeks or as late as January 2020.3 Although vials and syringes are the current products that are affected, it is possible the shortage may eventually include IV heparin bags as well.
Since the foremost objective of VA health care providers is to provide timely access to medications for veterans, strategies to conserve unfractionated heparin (UfH) must be used since it is a first-line therapy where few evidence-based alternatives exist. Conservation strategies may include drug rationing, therapeutic substitution, and compounding of needed products using the limited stock available in the pharmacy.4 It is important that all staff are educated on facility strategies in order to be familiar with alternatives and limit the potential for near misses, adverse events, and provider frustration.
In shortage situations, the VA-Pharmacy Benefits Management (PBM) defers decisions regarding drug preservation, processes to shift to viable alternatives, and the best practice for safe transitions to local facilities and their subject matter experts.5 At the VA Tennessee Valley Healthcare System, a 1A, tertiary, dual campus health care system, a pharmacy task force has formed to track drug shortages impacting the facility’s efficiencies and budgets. This group communicates with the Pharmacy and Therapeutics committee about potential risks to patient care and develops shortage briefs (following an SBAR [situation, background, assessment, recommendation] design) generally authored and championed by at least 1 clinical pharmacy specialist and supervising physicians who are field experts. Prior to dissemination, the SBAR undergoes a rapid peer-review process.
To date, VA PBM has not issued specific guidance on how pharmacists should proceed in case of a shortage. However, we recommend strategies that may be considered for implementation during a potential UfH shortage. For example, pharmacists can use therapeutic alternatives for which best available evidence suggests no disadvantage.4 The Table lists alternative agents according to indication and patient-specific considerations that may preclude use. Existing UfH products may also be used for drug compounding (eg, use current stock to provide an indicated aliquot) to meet the need of prioritized patients.4 In addition, we suggest prioritizing current UfH/heparinized saline for use for the following groups of patients4:
- Emergent/urgent cardiac surgery1,6;
- Hemodialysis patients1,7-9 for which the low-molecular-weight heparin (LMWH) dalteparin is deemed inappropriate or the patient is not monitored in the intensive care unit for regional citrate administration;
- VTE prophylaxis for patients with epidurals or chest tubes for which urgent invasive management may occur, recent cardiac or neurosurgery, or for patients with a creatine clearance < 15 mL/min or receiving hemodialysis10-12;
- Vascular surgery (eg, limb ischemia) and interventions (eg, carotid stenting, endarterectomy)13,14;
- Mesenteric ischemia (venous thrombosis) with a potential to proceed to laparotomy15;
- Critically ill patients with arterial lines for which normal saline is deemed inappropriate for line flushing16;
- Electrophysiology procedures (eg, AF ablation)17; and
- Contraindication to use of a long-acting alternative listed in the table or a medical necessity exists for using a rapidly reversible agent. Examples for this category include but are not limited to recent gastrointestinal bleeding, central nervous system lesion, and select neurologic diagnoses (eg, cerebral venous sinus thrombosis with hemorrhage, thrombus in vertebral basilar system or anterior circulation, intraparenchymal hemorrhage plus mechanical valve, medium to large cardioembolic stroke with intracardiac thrombus).
Conclusion
The UfH drug shortage represents a significant threat to public health and is a major challenge for US health care systems, including the Veterans Health Administration. Overreliance on a predominant source of crude heparin has affected multiple UfH manufacturers and products. Current alternatives to UfH include low-molecular-weight heparins, IV direct thrombin inhibitors, and SC fondaparinux, with selection supported by guidelines or evolving literature. However, the shortage has the potential to expand to other injectables, such as dalteparin and enoxaparin, and severely limit care for veterans. It is vital that clinicians rapidly address the current shortage by creating a plan to develop efficient and equitable access to UfH, continue to assess supply and update stakeholders, and select evidence-based alternatives while maintaining focus on efficacy and safety.
Acknowledgments
The authors thank Ashley Yost, PharmD, for her coordination of the multidisciplinary task force assigned to efficiently manage the heparin drug shortage. This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System in Nashville, Tennessee.
1. Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119(1):64S-94S.
2. Bipartisan E&C leaders request FDA briefing on threat to U.S. heparin supply [press release]. Washington, DC: House Committee on Energy and Commerce; July 30, 2019. https://energycommerce.house.gov/newsroom/press-releases/bipartisan-ec-leaders-request-fda-briefing-on-threat-to-us-heparin-supply. Accessed September 19, 2019.
3. American Society of Health-System Pharmacists. Drug Shortages. Heparin injection. https://www.ashp.org/Drug-Shortages/Current-Shortages/Drug-Shortages-List?page=CurrentShortages. Accessed September 19, 2019.
4. Reed BN, Fox ER, Konig M, et al. The impact of drug shortages on patients with cardiovascular disease: causes, consequences, and a call to action. Am Heart J. 2016;175:130-141.
5. US Department of Veterans Affairs. Pharmacy Benefits Management Services, Medical Advisory Panel, VISN Pharmacist Executives, The Center For Medication Safety. Heparin supply status: frequently asked questions. PBM-2018-02. https://www.pbm.va.gov/PBM/vacenterformedicationsafety/HeparinandSalineSyringeRecallDuetoContamination_NationalPBMPati.pdf. Published May 3, 2018. Accessed September 11, 2019.
6. Shore-Lesserson I, Baker RA, Ferraris VA, et al. The Society of Thoracic Surgeons, The Society of Cardiovascular Anesthesiologists, and the American Society of ExtraCorporeal Technology: Clinical Practice Guidelines-anticoagulation during cardiopulmonary bypass. Ann Thorac Surg. 2018;105(2):650-662.
7. Soroka S, Agharazii M, Donnelly S, et al. An adjustable dalteparin sodium dose regimen for the prevention of clotting in the extracorporeal circuit in hemodialysis: a clinical trial of safety and efficacy (the PARROT Study). Can J Kidney Health Dis. 2018;5:1-12.
8. Shantha GPS, Kumar AA, Sethi M, Khanna RC, Pancholy SB. Efficacy and safety of low molecular weight heparin compared to unfractionated heparin for chronic outpatient hemodialysis in end stage renal disease: systematic review and meta-analysis. Peer J. 2015;3:e835.
9. Kessler M, Moureau F, and Nguyen P. Anticoagulation in chronic hemodialysis: progress toward an optimal approach. Semin Dial. 2015;28(5):474-489.
10. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e227s-e277S.
11. Kaye AD, Brunk AJ, Kaye AJ, et al. Regional anesthesia in patients on anticoagulation therapies—evidence-based recommendations. Curr Pain Headache Rep. 2019;23(9):67.
12. Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e195S-e226S.
13. Naylor AR, Ricco JB, de Borst GJ, et al. Management of atherosclerotic carotid and vertebral artery disease: 2017 clinical practice guidelines of the European Society for Vascular Surgery. Eur J Vasc Endovasc Surg. 2018;55:3-81.
14. Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. JACC. 2017;69(11): e71-e126.
15. Bjorck M, Koelemaya M, Acosta S, et al. Management of diseases of mesenteric arteries and veins. Eur J Vasc Endovasc Surg. 2017;53(4):460-510.
16. Gorski L, Hadaway L, Hagle ME, McGoldrick M, Orr M, Doellman D. Infusion therapy standards of practice. J Infusion Nurs. 2016;39:S1-S156.
17. Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm. 2017;14(10):e275-e444.
18. Spyropoulos AC, Al-Badri A, Sherwood MW, Douketis JD. Periprocedural management of patients receiving a vitamin K antagonist or a direct oral anticoagulant requiring an elective procedure or surgery. J Thromb Haemost. 2016;14(5):875-885.
, . Periprocedural bridging management of anticoagulation. Circulation. 2012;126(4):486-490.
20. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e326S-e350S.
21. Sousa-Uva M, Neumann F-J, Ahlsson A, et al; ESC Scientific Document Group. 2018 ESC/EACTS Guidelines on myocardial revascularization. The Task Force on myocardial revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Developed with a special contribution of the European Association for Percutaneous Cardiovascular Interventions (EAPCI). Eur J Cardiothorac Surg. 2019;55(1):4-90.
22. Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes. JACC. 2014;64(24):e139-e228.
23. O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of patients with ST-elevation myocardial infarction. JACC. 2013;61(4):e78-e140.
24. Angiomax [package insert]. Parsippany, NJ: The Medicines Company; March 2016.
25. Sousa-Uva, Head SJ, Milojevic M, et al. 2017 EACTS guidelines on perioperative medication in adult cardiac surgery. Eur J Cardiothorac Surg. 2018;53(1):5-33.
26. Witt DM, Nieuwlaat R, Clark NP, et al. American Society of Hematology 2018 guidelines for the management of venous thromboembolism: optimal management of anticoagulation therapy. Blood Adv. 2018: 2(22):3257-3291
27. Kearon C, Akl EA, Blaivas A, et al. Antithrombotic therapy for VTE disease: Chest guideline and expert panel report. Chest. 2016;149(2):315-352.
28. US Department of Veterans Affairs, Pharmacy Benefits Manager Service. Direct oral anticoagulants criteria for use and algorithm for venous thromboembolism treatment. https://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse.asp. Updated December 2016. [Source not verified]
29. Falck-Ytter Y, Francis CW, Johanson NA, et al. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e278S-e325S.
30. Raja S, Idrees JJ, Blackstone EH, et al. Routine venous thromboembolism screening after pneumonectomy: the more you look, the more you see. J Thorac Cardiovasc Surg. 2016;152(2):524-532.e2.
31. Schünemann HJ, Cushman M, Burnett AE, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: prophylaxis for hospitalized and nonhospitalized patients. Blood Adv. 2018;2(22):3198-3225.
32. Naidu SS, Aronow HD, Box LC, et al. SCAI expert consensus statement: 2016 best practices in the cardiac catheterization laboratory:(endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista; affirmation of value by the Canadian Association of Interventional Cardiology-Association Canadienne de Cardiologie d’intervention). Catheter Cardiovasc Interv. 2016;88(3):407-423.
33. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. JACC. 2011;58(24):e44-e122.
34. Mason PJ, Shah B, Tamis-Holland JE, et al; American Heart Association Interventional Cardiovascular Care Committee of the Council on Clinical Cardiology; Council on Cardiovascular and Stroke Nursing; Council on Peripheral Vascular Disease; and Council on Genomic and Precision Medicine. AHA scientific statement: an update on radial artery access and best practices for transradial coronary angiography and intervention in acute coronary syndrome. Circ Cardiovasc Interv. 2018;11(9):e000035.
35. Rao SV, Tremmel JA, Gilchrist IC, et al; Society for Cardiovascular Angiography and Intervention’s Transradial Working Group. Best practices for transradial angiography and intervention: a consensus statement from the society for cardiovascular angiography and interventions’ transradial working group. Catheter Cardiovasc Interv. 2014;83(2):228-236. 36. Moran JE, Ash SR. Locking solutions for hemodialysis catheters; heparin and citrate: a position paper by ASDIN. Semin Dial. 2008;21(5):490-492.
1. Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119(1):64S-94S.
2. Bipartisan E&C leaders request FDA briefing on threat to U.S. heparin supply [press release]. Washington, DC: House Committee on Energy and Commerce; July 30, 2019. https://energycommerce.house.gov/newsroom/press-releases/bipartisan-ec-leaders-request-fda-briefing-on-threat-to-us-heparin-supply. Accessed September 19, 2019.
3. American Society of Health-System Pharmacists. Drug Shortages. Heparin injection. https://www.ashp.org/Drug-Shortages/Current-Shortages/Drug-Shortages-List?page=CurrentShortages. Accessed September 19, 2019.
4. Reed BN, Fox ER, Konig M, et al. The impact of drug shortages on patients with cardiovascular disease: causes, consequences, and a call to action. Am Heart J. 2016;175:130-141.
5. US Department of Veterans Affairs. Pharmacy Benefits Management Services, Medical Advisory Panel, VISN Pharmacist Executives, The Center For Medication Safety. Heparin supply status: frequently asked questions. PBM-2018-02. https://www.pbm.va.gov/PBM/vacenterformedicationsafety/HeparinandSalineSyringeRecallDuetoContamination_NationalPBMPati.pdf. Published May 3, 2018. Accessed September 11, 2019.
6. Shore-Lesserson I, Baker RA, Ferraris VA, et al. The Society of Thoracic Surgeons, The Society of Cardiovascular Anesthesiologists, and the American Society of ExtraCorporeal Technology: Clinical Practice Guidelines-anticoagulation during cardiopulmonary bypass. Ann Thorac Surg. 2018;105(2):650-662.
7. Soroka S, Agharazii M, Donnelly S, et al. An adjustable dalteparin sodium dose regimen for the prevention of clotting in the extracorporeal circuit in hemodialysis: a clinical trial of safety and efficacy (the PARROT Study). Can J Kidney Health Dis. 2018;5:1-12.
8. Shantha GPS, Kumar AA, Sethi M, Khanna RC, Pancholy SB. Efficacy and safety of low molecular weight heparin compared to unfractionated heparin for chronic outpatient hemodialysis in end stage renal disease: systematic review and meta-analysis. Peer J. 2015;3:e835.
9. Kessler M, Moureau F, and Nguyen P. Anticoagulation in chronic hemodialysis: progress toward an optimal approach. Semin Dial. 2015;28(5):474-489.
10. Gould MK, Garcia DA, Wren SM, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e227s-e277S.
11. Kaye AD, Brunk AJ, Kaye AJ, et al. Regional anesthesia in patients on anticoagulation therapies—evidence-based recommendations. Curr Pain Headache Rep. 2019;23(9):67.
12. Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e195S-e226S.
13. Naylor AR, Ricco JB, de Borst GJ, et al. Management of atherosclerotic carotid and vertebral artery disease: 2017 clinical practice guidelines of the European Society for Vascular Surgery. Eur J Vasc Endovasc Surg. 2018;55:3-81.
14. Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. JACC. 2017;69(11): e71-e126.
15. Bjorck M, Koelemaya M, Acosta S, et al. Management of diseases of mesenteric arteries and veins. Eur J Vasc Endovasc Surg. 2017;53(4):460-510.
16. Gorski L, Hadaway L, Hagle ME, McGoldrick M, Orr M, Doellman D. Infusion therapy standards of practice. J Infusion Nurs. 2016;39:S1-S156.
17. Calkins H, Hindricks G, Cappato R, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Heart Rhythm. 2017;14(10):e275-e444.
18. Spyropoulos AC, Al-Badri A, Sherwood MW, Douketis JD. Periprocedural management of patients receiving a vitamin K antagonist or a direct oral anticoagulant requiring an elective procedure or surgery. J Thromb Haemost. 2016;14(5):875-885.
, . Periprocedural bridging management of anticoagulation. Circulation. 2012;126(4):486-490.
20. Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e326S-e350S.
21. Sousa-Uva M, Neumann F-J, Ahlsson A, et al; ESC Scientific Document Group. 2018 ESC/EACTS Guidelines on myocardial revascularization. The Task Force on myocardial revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Developed with a special contribution of the European Association for Percutaneous Cardiovascular Interventions (EAPCI). Eur J Cardiothorac Surg. 2019;55(1):4-90.
22. Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes. JACC. 2014;64(24):e139-e228.
23. O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of patients with ST-elevation myocardial infarction. JACC. 2013;61(4):e78-e140.
24. Angiomax [package insert]. Parsippany, NJ: The Medicines Company; March 2016.
25. Sousa-Uva, Head SJ, Milojevic M, et al. 2017 EACTS guidelines on perioperative medication in adult cardiac surgery. Eur J Cardiothorac Surg. 2018;53(1):5-33.
26. Witt DM, Nieuwlaat R, Clark NP, et al. American Society of Hematology 2018 guidelines for the management of venous thromboembolism: optimal management of anticoagulation therapy. Blood Adv. 2018: 2(22):3257-3291
27. Kearon C, Akl EA, Blaivas A, et al. Antithrombotic therapy for VTE disease: Chest guideline and expert panel report. Chest. 2016;149(2):315-352.
28. US Department of Veterans Affairs, Pharmacy Benefits Manager Service. Direct oral anticoagulants criteria for use and algorithm for venous thromboembolism treatment. https://www.pbm.va.gov/PBM/clinicalguidance/criteriaforuse.asp. Updated December 2016. [Source not verified]
29. Falck-Ytter Y, Francis CW, Johanson NA, et al. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2)(suppl):e278S-e325S.
30. Raja S, Idrees JJ, Blackstone EH, et al. Routine venous thromboembolism screening after pneumonectomy: the more you look, the more you see. J Thorac Cardiovasc Surg. 2016;152(2):524-532.e2.
31. Schünemann HJ, Cushman M, Burnett AE, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: prophylaxis for hospitalized and nonhospitalized patients. Blood Adv. 2018;2(22):3198-3225.
32. Naidu SS, Aronow HD, Box LC, et al. SCAI expert consensus statement: 2016 best practices in the cardiac catheterization laboratory:(endorsed by the Cardiological Society of India, and Sociedad Latino Americana de Cardiologia Intervencionista; affirmation of value by the Canadian Association of Interventional Cardiology-Association Canadienne de Cardiologie d’intervention). Catheter Cardiovasc Interv. 2016;88(3):407-423.
33. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. JACC. 2011;58(24):e44-e122.
34. Mason PJ, Shah B, Tamis-Holland JE, et al; American Heart Association Interventional Cardiovascular Care Committee of the Council on Clinical Cardiology; Council on Cardiovascular and Stroke Nursing; Council on Peripheral Vascular Disease; and Council on Genomic and Precision Medicine. AHA scientific statement: an update on radial artery access and best practices for transradial coronary angiography and intervention in acute coronary syndrome. Circ Cardiovasc Interv. 2018;11(9):e000035.
35. Rao SV, Tremmel JA, Gilchrist IC, et al; Society for Cardiovascular Angiography and Intervention’s Transradial Working Group. Best practices for transradial angiography and intervention: a consensus statement from the society for cardiovascular angiography and interventions’ transradial working group. Catheter Cardiovasc Interv. 2014;83(2):228-236. 36. Moran JE, Ash SR. Locking solutions for hemodialysis catheters; heparin and citrate: a position paper by ASDIN. Semin Dial. 2008;21(5):490-492.
Oral anticoagulant and PPI cotherapy cuts upper GI bleed risk
Background: PPIs reduce gastric acid production, promote ulcer healing, and prevent ulcer recurrence; however, limited evidence is available describing the incidence of anticoagulant-related serious upper GI tract bleeding from the newer non–vitamin K anticoagulants and PPI cotherapy.

Study design: Retrospective cohort.
Setting: Medicare enrollees.
Synopsis: With use of computerized Medicare beneficiaries files, researchers identified 1,643,123 patients with 1,713,183 new episodes of oral anticoagulant treatment between Jan. 1, 2011, and Sept. 30, 2015. This analysis showed that cotherapy with PPIs was associated with a lower incidence of upper GI bleed, with the largest difference associated with dabigatran with an incidence rate ratio of 0.49 (95% CI, 0.52-0.85), followed by warfarin (IRR, 0.65; 95%CI, 0.62-0.69), apixaban (IRR, 0.66; 95% CI, 0.52-0.85), and rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84).
Generalizability was limited by population (Medicare enrollees) and the study excluded prior hospitalizations for GI bleed, as well as switches in anticoagulant therapy during the study period.
Bottom line: PPI cotherapy with oral anticoagulation reduces risk of hospitalization for upper GI bleed.
Citation: Ray WA et al. Association of oral anticoagulants and proton pump inhibitor cotherapy with hospitalization for upper gastrointestinal tract bleeding. JAMA. 2018 Dec 4;320(21):2221-30.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: PPIs reduce gastric acid production, promote ulcer healing, and prevent ulcer recurrence; however, limited evidence is available describing the incidence of anticoagulant-related serious upper GI tract bleeding from the newer non–vitamin K anticoagulants and PPI cotherapy.
Study design: Retrospective cohort.
Setting: Medicare enrollees.
Synopsis: With use of computerized Medicare beneficiaries files, researchers identified 1,643,123 patients with 1,713,183 new episodes of oral anticoagulant treatment between Jan. 1, 2011, and Sept. 30, 2015. This analysis showed that cotherapy with PPIs was associated with a lower incidence of upper GI bleed, with the largest difference associated with dabigatran with an incidence rate ratio of 0.49 (95% CI, 0.52-0.85), followed by warfarin (IRR, 0.65; 95%CI, 0.62-0.69), apixaban (IRR, 0.66; 95% CI, 0.52-0.85), and rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84).
Generalizability was limited by population (Medicare enrollees) and the study excluded prior hospitalizations for GI bleed, as well as switches in anticoagulant therapy during the study period.
Bottom line: PPI cotherapy with oral anticoagulation reduces risk of hospitalization for upper GI bleed.
Citation: Ray WA et al. Association of oral anticoagulants and proton pump inhibitor cotherapy with hospitalization for upper gastrointestinal tract bleeding. JAMA. 2018 Dec 4;320(21):2221-30.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
Background: PPIs reduce gastric acid production, promote ulcer healing, and prevent ulcer recurrence; however, limited evidence is available describing the incidence of anticoagulant-related serious upper GI tract bleeding from the newer non–vitamin K anticoagulants and PPI cotherapy.
Study design: Retrospective cohort.
Setting: Medicare enrollees.
Synopsis: With use of computerized Medicare beneficiaries files, researchers identified 1,643,123 patients with 1,713,183 new episodes of oral anticoagulant treatment between Jan. 1, 2011, and Sept. 30, 2015. This analysis showed that cotherapy with PPIs was associated with a lower incidence of upper GI bleed, with the largest difference associated with dabigatran with an incidence rate ratio of 0.49 (95% CI, 0.52-0.85), followed by warfarin (IRR, 0.65; 95%CI, 0.62-0.69), apixaban (IRR, 0.66; 95% CI, 0.52-0.85), and rivaroxaban (IRR, 0.75; 95% CI, 0.68-0.84).
Generalizability was limited by population (Medicare enrollees) and the study excluded prior hospitalizations for GI bleed, as well as switches in anticoagulant therapy during the study period.
Bottom line: PPI cotherapy with oral anticoagulation reduces risk of hospitalization for upper GI bleed.
Citation: Ray WA et al. Association of oral anticoagulants and proton pump inhibitor cotherapy with hospitalization for upper gastrointestinal tract bleeding. JAMA. 2018 Dec 4;320(21):2221-30.
Dr. Ho is an assistant professor of medicine in the division of general and hospital medicine at UT Health San Antonio and a hospitalist at South Texas Veterans Health Care System.
MONARCH 2: Abemaciclib plus fulvestrant improves overall survival
BARCELONA – Adding the CDK4/6 inhibitor abemaciclib to fulvestrant significantly improves overall survival in hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2–) advanced breast cancer patients who progressed on prior endocrine therapy, according to findings from the phase 3 MONARCH 2 trial.

At a median follow-up of 47.7 months, overall survival – a secondary study endpoint – was 46.7 months in 446 patients randomized to receive abemaciclib and fulvestrant, compared with 37.3 months in 223 patients who received placebo and fulvestrant (hazard ratio, 0.757), George W. Sledge, MD, reported at the European Society for Medical Oncology Congress.
The overall survival benefit was consistent across stratification factors, which included site of metastasis (visceral, bone, or other) and resistance to prior endocrine therapy (primary versus secondary), but it was most pronounced in patients with visceral disease (HR, 0.675) and primary resistance to prior endocrine therapy (HR, 0.686), said Dr. Sledge, a professor of medicine at Stanford (Calif.) Medical Center.
Progression-free survival, the primary study endpoint, was 16.4 and 9.3 months at the previously reported 2-year follow-up in the treatment and placebo groups, respectively, and the current analysis showed progression-free survival to be “highly consistent” with those findings (16.9 vs. 9.3 months; HR, 0.563), he said.
“Of note, and of interest for further follow-up, a landmark analysis at 3 years shows that approximately three times as many patients on the abemaciclib arm remained progression free, compared to the control arm,” he added, also noting that time from randomization to postdiscontinuation chemotherapy was prolonged with abemaciclib, compared with placebo (50.2 vs. 22.1 months; HR, 0.625).
“A highly significant result,” he said.
At the 47.7 month follow-up, 17% and 4% of patients in the treatment and placebo groups, respectively, remained on treatment.
An additional exploratory analysis showed that 5.8% of patients receiving abemaciclib crossed over to another CDK4/6 inhibitor after discontinuation of therapy, compared with 17.0% of those in the placebo group.
“CDK4/6 inhibitors have emerged as standard-of-care treatment for patients with HR+, HER2– breast cancer,” he said, noting that abemaciclib is a selective CDK4/6 inhibitor with continuous, twice-daily oral administration. “It is approved for monotherapy after progression on endocrine therapy and prior chemotherapy in the metastatic setting, and ... in combination with endocrine therapy in the front-line setting and after progression.”
The global, randomized, double-blind MONARCH 2 trial assessed abemaciclib + fulvestrant in women with advanced endocrine therapy–resistant HR+, HER2– advanced breast cancer, including pre- or perimenopausal women with ovarian suppression and postmenopausal women.
Study participants were randomized 2:1 to receive 500 mg of fulvestrant per label instructions plus 150 mg of abemaciclib every 12 hours or placebo.
Treatment-emergent adverse events in MONARCH 2 were consistent with those previously reported in the primary analysis, Dr. Sledge said.
“Continued follow-up of MONARCH 2 is ongoing to further characterize the overall survival benefit and to look at exploratory efficacy and correlative endpoints,” he noted.
Speaking about the findings during a press conference at the meeting, Nadia Harbeck, MD, a professor at Ludwig Maximilians University, Munich, Germany, said they have important practice-changing implications.
“We were always struggling with whether to give these drugs first or second line, and I think now if we consider the first-line survival benefit ... [first-line use] should be standard of care,” she said, referring to CDK4/6 inhibitors and to findings from MONARCH 2 and prior studies of the inhibitors, including the MONALEESA-3 trial presented at the ESMO Congress.
MONALEESA-3 showed similar overall survival results with the CDK4/6 inhibitor ribociclib in postmenopausal women with HR+, HER2– advanced breast cancer.
“The data are highly clinically meaningful; I think they are going to make a huge impact on how we treat breast cancer,” Dr. Harbeck said.
Dr. Sledge has received trial support, research grants, and travel accommodations from Eli Lilly and Company; is a board member for Tessa Therapeutics; and is a consultant for Syndax, Symphogen, and Verseau Therapeutics. Dr. Harbeck disclosed financial relationships with Agendia, Amgen, AstraZeneca, Celgene, Daiichi-Sankyo, Genomic Health, Lilly, MSD, Nanostring, Novartis, Odonate, Pfizer, Roche, Sandoz/Hexal, and Seattle Genetics. She is also director of the West German Study Group and a member of the German AGO Breast Committee.
SOURCE: Sledge G et al., ESMO 2019, Abstract LBA6-PR.
BARCELONA – Adding the CDK4/6 inhibitor abemaciclib to fulvestrant significantly improves overall survival in hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2–) advanced breast cancer patients who progressed on prior endocrine therapy, according to findings from the phase 3 MONARCH 2 trial.
At a median follow-up of 47.7 months, overall survival – a secondary study endpoint – was 46.7 months in 446 patients randomized to receive abemaciclib and fulvestrant, compared with 37.3 months in 223 patients who received placebo and fulvestrant (hazard ratio, 0.757), George W. Sledge, MD, reported at the European Society for Medical Oncology Congress.
The overall survival benefit was consistent across stratification factors, which included site of metastasis (visceral, bone, or other) and resistance to prior endocrine therapy (primary versus secondary), but it was most pronounced in patients with visceral disease (HR, 0.675) and primary resistance to prior endocrine therapy (HR, 0.686), said Dr. Sledge, a professor of medicine at Stanford (Calif.) Medical Center.
Progression-free survival, the primary study endpoint, was 16.4 and 9.3 months at the previously reported 2-year follow-up in the treatment and placebo groups, respectively, and the current analysis showed progression-free survival to be “highly consistent” with those findings (16.9 vs. 9.3 months; HR, 0.563), he said.
“Of note, and of interest for further follow-up, a landmark analysis at 3 years shows that approximately three times as many patients on the abemaciclib arm remained progression free, compared to the control arm,” he added, also noting that time from randomization to postdiscontinuation chemotherapy was prolonged with abemaciclib, compared with placebo (50.2 vs. 22.1 months; HR, 0.625).
“A highly significant result,” he said.
At the 47.7 month follow-up, 17% and 4% of patients in the treatment and placebo groups, respectively, remained on treatment.
An additional exploratory analysis showed that 5.8% of patients receiving abemaciclib crossed over to another CDK4/6 inhibitor after discontinuation of therapy, compared with 17.0% of those in the placebo group.
“CDK4/6 inhibitors have emerged as standard-of-care treatment for patients with HR+, HER2– breast cancer,” he said, noting that abemaciclib is a selective CDK4/6 inhibitor with continuous, twice-daily oral administration. “It is approved for monotherapy after progression on endocrine therapy and prior chemotherapy in the metastatic setting, and ... in combination with endocrine therapy in the front-line setting and after progression.”
The global, randomized, double-blind MONARCH 2 trial assessed abemaciclib + fulvestrant in women with advanced endocrine therapy–resistant HR+, HER2– advanced breast cancer, including pre- or perimenopausal women with ovarian suppression and postmenopausal women.
Study participants were randomized 2:1 to receive 500 mg of fulvestrant per label instructions plus 150 mg of abemaciclib every 12 hours or placebo.
Treatment-emergent adverse events in MONARCH 2 were consistent with those previously reported in the primary analysis, Dr. Sledge said.
“Continued follow-up of MONARCH 2 is ongoing to further characterize the overall survival benefit and to look at exploratory efficacy and correlative endpoints,” he noted.
Speaking about the findings during a press conference at the meeting, Nadia Harbeck, MD, a professor at Ludwig Maximilians University, Munich, Germany, said they have important practice-changing implications.
“We were always struggling with whether to give these drugs first or second line, and I think now if we consider the first-line survival benefit ... [first-line use] should be standard of care,” she said, referring to CDK4/6 inhibitors and to findings from MONARCH 2 and prior studies of the inhibitors, including the MONALEESA-3 trial presented at the ESMO Congress.
MONALEESA-3 showed similar overall survival results with the CDK4/6 inhibitor ribociclib in postmenopausal women with HR+, HER2– advanced breast cancer.
“The data are highly clinically meaningful; I think they are going to make a huge impact on how we treat breast cancer,” Dr. Harbeck said.
Dr. Sledge has received trial support, research grants, and travel accommodations from Eli Lilly and Company; is a board member for Tessa Therapeutics; and is a consultant for Syndax, Symphogen, and Verseau Therapeutics. Dr. Harbeck disclosed financial relationships with Agendia, Amgen, AstraZeneca, Celgene, Daiichi-Sankyo, Genomic Health, Lilly, MSD, Nanostring, Novartis, Odonate, Pfizer, Roche, Sandoz/Hexal, and Seattle Genetics. She is also director of the West German Study Group and a member of the German AGO Breast Committee.
SOURCE: Sledge G et al., ESMO 2019, Abstract LBA6-PR.
BARCELONA – Adding the CDK4/6 inhibitor abemaciclib to fulvestrant significantly improves overall survival in hormone receptor–positive (HR+), human epidermal growth factor receptor 2–negative (HER2–) advanced breast cancer patients who progressed on prior endocrine therapy, according to findings from the phase 3 MONARCH 2 trial.
At a median follow-up of 47.7 months, overall survival – a secondary study endpoint – was 46.7 months in 446 patients randomized to receive abemaciclib and fulvestrant, compared with 37.3 months in 223 patients who received placebo and fulvestrant (hazard ratio, 0.757), George W. Sledge, MD, reported at the European Society for Medical Oncology Congress.
The overall survival benefit was consistent across stratification factors, which included site of metastasis (visceral, bone, or other) and resistance to prior endocrine therapy (primary versus secondary), but it was most pronounced in patients with visceral disease (HR, 0.675) and primary resistance to prior endocrine therapy (HR, 0.686), said Dr. Sledge, a professor of medicine at Stanford (Calif.) Medical Center.
Progression-free survival, the primary study endpoint, was 16.4 and 9.3 months at the previously reported 2-year follow-up in the treatment and placebo groups, respectively, and the current analysis showed progression-free survival to be “highly consistent” with those findings (16.9 vs. 9.3 months; HR, 0.563), he said.
“Of note, and of interest for further follow-up, a landmark analysis at 3 years shows that approximately three times as many patients on the abemaciclib arm remained progression free, compared to the control arm,” he added, also noting that time from randomization to postdiscontinuation chemotherapy was prolonged with abemaciclib, compared with placebo (50.2 vs. 22.1 months; HR, 0.625).
“A highly significant result,” he said.
At the 47.7 month follow-up, 17% and 4% of patients in the treatment and placebo groups, respectively, remained on treatment.
An additional exploratory analysis showed that 5.8% of patients receiving abemaciclib crossed over to another CDK4/6 inhibitor after discontinuation of therapy, compared with 17.0% of those in the placebo group.
“CDK4/6 inhibitors have emerged as standard-of-care treatment for patients with HR+, HER2– breast cancer,” he said, noting that abemaciclib is a selective CDK4/6 inhibitor with continuous, twice-daily oral administration. “It is approved for monotherapy after progression on endocrine therapy and prior chemotherapy in the metastatic setting, and ... in combination with endocrine therapy in the front-line setting and after progression.”
The global, randomized, double-blind MONARCH 2 trial assessed abemaciclib + fulvestrant in women with advanced endocrine therapy–resistant HR+, HER2– advanced breast cancer, including pre- or perimenopausal women with ovarian suppression and postmenopausal women.
Study participants were randomized 2:1 to receive 500 mg of fulvestrant per label instructions plus 150 mg of abemaciclib every 12 hours or placebo.
Treatment-emergent adverse events in MONARCH 2 were consistent with those previously reported in the primary analysis, Dr. Sledge said.
“Continued follow-up of MONARCH 2 is ongoing to further characterize the overall survival benefit and to look at exploratory efficacy and correlative endpoints,” he noted.
Speaking about the findings during a press conference at the meeting, Nadia Harbeck, MD, a professor at Ludwig Maximilians University, Munich, Germany, said they have important practice-changing implications.
“We were always struggling with whether to give these drugs first or second line, and I think now if we consider the first-line survival benefit ... [first-line use] should be standard of care,” she said, referring to CDK4/6 inhibitors and to findings from MONARCH 2 and prior studies of the inhibitors, including the MONALEESA-3 trial presented at the ESMO Congress.
MONALEESA-3 showed similar overall survival results with the CDK4/6 inhibitor ribociclib in postmenopausal women with HR+, HER2– advanced breast cancer.
“The data are highly clinically meaningful; I think they are going to make a huge impact on how we treat breast cancer,” Dr. Harbeck said.
Dr. Sledge has received trial support, research grants, and travel accommodations from Eli Lilly and Company; is a board member for Tessa Therapeutics; and is a consultant for Syndax, Symphogen, and Verseau Therapeutics. Dr. Harbeck disclosed financial relationships with Agendia, Amgen, AstraZeneca, Celgene, Daiichi-Sankyo, Genomic Health, Lilly, MSD, Nanostring, Novartis, Odonate, Pfizer, Roche, Sandoz/Hexal, and Seattle Genetics. She is also director of the West German Study Group and a member of the German AGO Breast Committee.
SOURCE: Sledge G et al., ESMO 2019, Abstract LBA6-PR.
REPORTING FROM ESMO 2019
Inflammatory arthritis induced by ICIs can persist after therapy
according to a new study of long-term outcomes of immune-related adverse events published in Annals of the Rheumatic Diseases.
“This study is one of the largest longitudinal reports to date of patients with ICI-induced IA and the first to evaluate persistence of ICI-induced IA and identify influential factors on outcome,” wrote Tawnie J. Braaten, MD, and coauthors. “Continued clinical and translational investigation on larger longitudinal cohorts will allow for increased understanding of pathophysiology and determination of the best clinical care for patients with ICI-induced IA.”
Dr. Braaten conducted the study at Johns Hopkins University, Baltimore, when she was a postdoctoral fellow there, and she is now in the division of rheumatology at the University of Utah, Salt Lake City.
To determine how long IA can persist after patients cease ICI therapy, along with factors associated with its persistence, the researchers studied 60 patients who were referred to the Johns Hopkins Arthritis Center for IA caused by ICIs. The patients – 32 females and 28 males – had a median follow-up of 9 months after ICI cessation.
Of the 51 patients with 3-month follow-up data, 70.6% had active IA. Of the 41 patients with 6-month follow-up data, 48.8% had active IA. All told, 53.3% of patients had active IA at their last follow-up visit, which occurred anywhere from 1 to 24 months after stopping ICI therapy.
According to univariable analysis, arthritis was less likely to improve in patients with a longer duration of ICI exposure (hazard ratio, 0.93; 95% confidence interval, 0.87-0.99; P = .02), in patients receiving combination ICI therapy (HR, 0.29; 95% CI, 0.12-0.72; P = .008) and in patients with a history of other immune-related adverse events (HR, 0.61; 95% CI, 0.39-0.95; P = .03).
The authors acknowledged their study’s limitations, including a potential selection bias for symptomatic individuals and the possibility that persistent IA sufferers may have pursued follow-up for longer periods of time. In addition, they noted that some patients were omitted from analysis if they were on a blinded clinical trial or had been receiving an investigational immunotherapy agent.
The study was funded via a grant from Bristol-Myers Squibb, an arthritis fellowship award from AbbVie, and additional financial support from the Camille Julia Morgan Arthritis Research and Education Fund, the Jerome L. Greene Foundation, and the National Institutes of Health. The authors reported various conflicts of interest, including receiving honoraria, grants, and research funding from numerous pharmaceutical companies.
SOURCE: Braaten TJ et al. Ann Rheum Dis. 2019 Sep 20. doi: 10.1136/annrheumdis-2019-216109.
according to a new study of long-term outcomes of immune-related adverse events published in Annals of the Rheumatic Diseases.
“This study is one of the largest longitudinal reports to date of patients with ICI-induced IA and the first to evaluate persistence of ICI-induced IA and identify influential factors on outcome,” wrote Tawnie J. Braaten, MD, and coauthors. “Continued clinical and translational investigation on larger longitudinal cohorts will allow for increased understanding of pathophysiology and determination of the best clinical care for patients with ICI-induced IA.”
Dr. Braaten conducted the study at Johns Hopkins University, Baltimore, when she was a postdoctoral fellow there, and she is now in the division of rheumatology at the University of Utah, Salt Lake City.
To determine how long IA can persist after patients cease ICI therapy, along with factors associated with its persistence, the researchers studied 60 patients who were referred to the Johns Hopkins Arthritis Center for IA caused by ICIs. The patients – 32 females and 28 males – had a median follow-up of 9 months after ICI cessation.
Of the 51 patients with 3-month follow-up data, 70.6% had active IA. Of the 41 patients with 6-month follow-up data, 48.8% had active IA. All told, 53.3% of patients had active IA at their last follow-up visit, which occurred anywhere from 1 to 24 months after stopping ICI therapy.
According to univariable analysis, arthritis was less likely to improve in patients with a longer duration of ICI exposure (hazard ratio, 0.93; 95% confidence interval, 0.87-0.99; P = .02), in patients receiving combination ICI therapy (HR, 0.29; 95% CI, 0.12-0.72; P = .008) and in patients with a history of other immune-related adverse events (HR, 0.61; 95% CI, 0.39-0.95; P = .03).
The authors acknowledged their study’s limitations, including a potential selection bias for symptomatic individuals and the possibility that persistent IA sufferers may have pursued follow-up for longer periods of time. In addition, they noted that some patients were omitted from analysis if they were on a blinded clinical trial or had been receiving an investigational immunotherapy agent.
The study was funded via a grant from Bristol-Myers Squibb, an arthritis fellowship award from AbbVie, and additional financial support from the Camille Julia Morgan Arthritis Research and Education Fund, the Jerome L. Greene Foundation, and the National Institutes of Health. The authors reported various conflicts of interest, including receiving honoraria, grants, and research funding from numerous pharmaceutical companies.
SOURCE: Braaten TJ et al. Ann Rheum Dis. 2019 Sep 20. doi: 10.1136/annrheumdis-2019-216109.
according to a new study of long-term outcomes of immune-related adverse events published in Annals of the Rheumatic Diseases.
“This study is one of the largest longitudinal reports to date of patients with ICI-induced IA and the first to evaluate persistence of ICI-induced IA and identify influential factors on outcome,” wrote Tawnie J. Braaten, MD, and coauthors. “Continued clinical and translational investigation on larger longitudinal cohorts will allow for increased understanding of pathophysiology and determination of the best clinical care for patients with ICI-induced IA.”
Dr. Braaten conducted the study at Johns Hopkins University, Baltimore, when she was a postdoctoral fellow there, and she is now in the division of rheumatology at the University of Utah, Salt Lake City.
To determine how long IA can persist after patients cease ICI therapy, along with factors associated with its persistence, the researchers studied 60 patients who were referred to the Johns Hopkins Arthritis Center for IA caused by ICIs. The patients – 32 females and 28 males – had a median follow-up of 9 months after ICI cessation.
Of the 51 patients with 3-month follow-up data, 70.6% had active IA. Of the 41 patients with 6-month follow-up data, 48.8% had active IA. All told, 53.3% of patients had active IA at their last follow-up visit, which occurred anywhere from 1 to 24 months after stopping ICI therapy.
According to univariable analysis, arthritis was less likely to improve in patients with a longer duration of ICI exposure (hazard ratio, 0.93; 95% confidence interval, 0.87-0.99; P = .02), in patients receiving combination ICI therapy (HR, 0.29; 95% CI, 0.12-0.72; P = .008) and in patients with a history of other immune-related adverse events (HR, 0.61; 95% CI, 0.39-0.95; P = .03).
The authors acknowledged their study’s limitations, including a potential selection bias for symptomatic individuals and the possibility that persistent IA sufferers may have pursued follow-up for longer periods of time. In addition, they noted that some patients were omitted from analysis if they were on a blinded clinical trial or had been receiving an investigational immunotherapy agent.
The study was funded via a grant from Bristol-Myers Squibb, an arthritis fellowship award from AbbVie, and additional financial support from the Camille Julia Morgan Arthritis Research and Education Fund, the Jerome L. Greene Foundation, and the National Institutes of Health. The authors reported various conflicts of interest, including receiving honoraria, grants, and research funding from numerous pharmaceutical companies.
SOURCE: Braaten TJ et al. Ann Rheum Dis. 2019 Sep 20. doi: 10.1136/annrheumdis-2019-216109.
FROM ANNALS OF THE RHEUMATIC DISEASES